 Larissa S. Carnevalli
Larissa S. Carnevalli Hormas Ghadially
Hormas Ghadially Simon T. Barry
Simon T. Barry- Early Oncology, Research and Development, AstraZeneca, Cambridge, United Kingdom
Immunotherapy has transformed cancer treatment by promoting durable clinical responses in a proportion of patients; however, treatment still fails in many patients. Innate immune cells play a key role in the response to immunotherapy. Crosstalk between innate and adaptive immune systems drives T-cell activation but also limits immunotherapy response, as myeloid cells are commonly associated with resistance. Hence, innate cells have both negative and positive effects within the tumor microenvironment (TME), and despite investment in early clinical trials targeting innate cells, they have seen limited success. Suppressive myeloid cells facilitate metastasis and immunotherapy resistance through TME remodeling and inhibition of adaptive immune cells. Natural killer (NK) cells, in contrast, secrete inflammatory cytokines and directly kill transformed cells, playing a key immunosurveillance role in early tumor development. Myeloid and NK cells show reciprocal crosstalk, influencing myeloid cell functional status or antigen presentation and NK effector function, respectively. Crosstalk between myeloid cells and the NK immune network in the TME is especially important in the context of therapeutic intervention. Here we discuss how myeloid and NK cell interactions shape anti-tumor responses by influencing an immunosuppressive TME and how this may influence outcomes of treatment strategies involving drugs that target myeloid and NK cells.
Introduction
Immunotherapy has transformed cancer treatment by harnessing the immune system to target solid and hematological cancers (1), achieving durable responses across multiple tumor types (2). However, only approximately 20% of patients have a durable response, and intrinsic or acquired resistance is often observed in the clinic (3, 4). Therefore, novel combination approaches are needed to expand the therapeutic benefit of these drugs. Currently, several anticancer therapies employing multiple drug modalities and combinatorial approaches are being tested clinically (5), but few have been found to enhance benefit in combination with checkpoint inhibitors.
Because the balance of immunosuppressive versus immunostimulatory cells varies among individual tumors, a major goal of these approaches going forward is to understand immune contexture and segmentation. Most solid tumors include a variety of immunosuppressive cells, such as regulatory T cells, polymorphonuclear (PMN) myeloid-derived suppressor cells (MDSCs), mononuclear MDSCs (M-MDSCs), tumor-associated macrophages (TAMs) [as defined by Bronte et al., (6)] and neutrophils that can suppress effector CD8+ T cells and NK cells (7).
Two cell types of the innate immune system that shape the tumor microenvironment (TME) and can initiate anti-tumor immune responses are natural killer (NK) cells and cells of the myeloid lineage, including immunosuppressive PMN-MDSCs, M-MDSCs, and immune-activating macrophages, dendritic cells, and neutrophils. Although the individual roles of these cell types in the anti-tumor immune response have been extensively studied [for review, see Neophytou et al., (8)] the ways in which interactions between these cell types affect immune responses is only just emerging. Consideration of the interplay between NK cells and suppressive myeloid cells could give new insight into the effects of therapies combining PD-1/PD-L1 and CTLA-4 blockade in the clinic and may also have an impact in early-stage cancers and hematological diseases.
The most advanced therapies are those that modulate myeloid cells, depleting or inhibiting recruitment or promoting reprogramming to activate or de-repress tumoricidal mechanisms (9), but these modalities have been disappointing in the clinic (Table 1). These trials include inhibitors of CSF1R, CCR2, CXCR2, CXCR4, and most recently, PI3Kγ (10, 11). Although many of these modalities have been tested clinically, few have passed beyond phase 2 studies, due to either lack of efficacy or associated adverse effects. It is important to gain insight into the mechanism of action and biomarker changes associated with efficacy in order to refine therapeutic strategies for myeloid-targeting agents and to identify patients who could benefit from these therapies as monotherapies or in combination with immune checkpoint blockade.
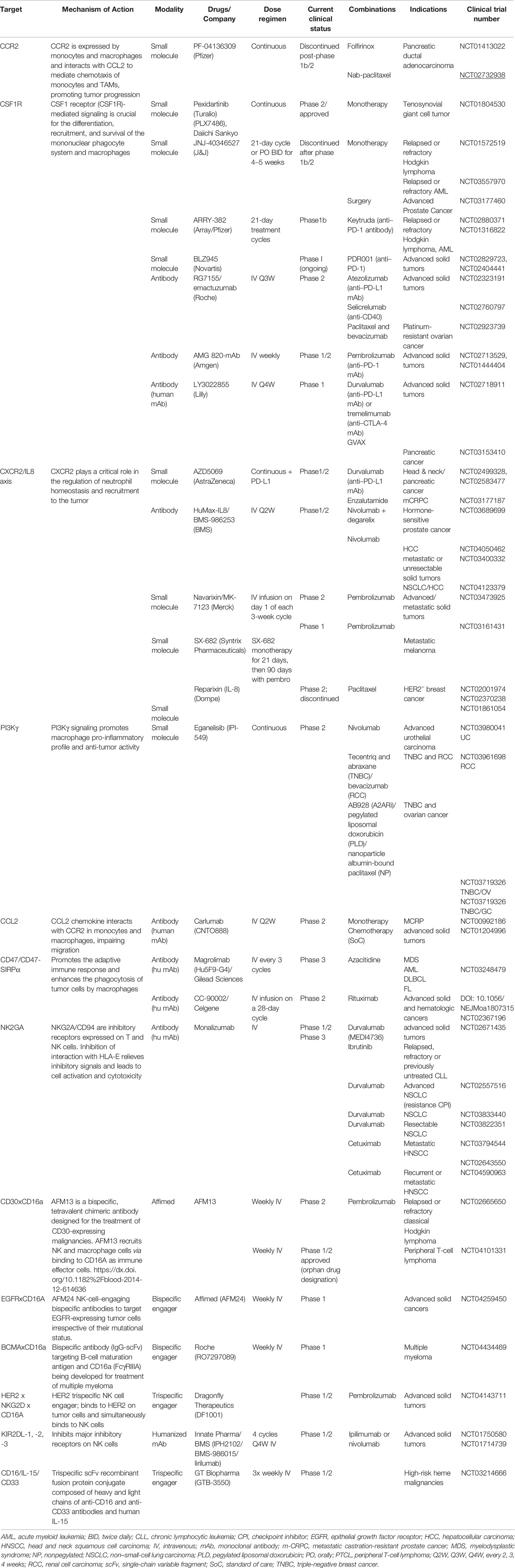
Table 1 Myeloid and NK target therapies tested in clinical studies.
n contrast, only a few drugs targeting NK biology to reverse NK tumor immune tolerance have been progressed to clinical trials (Table 1). These therapies include the anti–KIR2DL-1, -2, and -3 antibody IPH2102/BMS-986015 (lirilumab), the anti-NKG2A antibody IPH2201 (monalizumab), and the anti-CD16 innate cell engager AFM13.
To date, the concept of modulating NK-myeloid cell interactions to relieve tumor immunosuppression is underexplored. However, further consideration of NK-myeloid cell interactions in the TME and periphery may provide insights into both innate and adaptive immune anti-tumor responses. Here we discuss possible mechanisms that can attenuate or enhance a productive immune response through innate cell–mediated responses and the consequence for activation of effector cell types in the TME.
NK Cell Biology and the TME
NK cells are large, granular lymphocytes that can kill target cells without previously encountering an antigen. NK cells also produce proinflammatory cytokines like interferon-alpha (IFN-α), tumor necrosis factor-alpha (TNFα), and granulocyte macrophage–colony-stimulating factor (GM-CSF), as well as chemokines such as CCL1, CCL3, CCL4, CCL5, CCL22, and CXCL8. Their activity is regulated by a balance of signals from activating and inhibitory receptors (12). Most of the inhibitory receptors bind to major histocompatibility complex (MHC) class I–like proteins, which enable NK cells to detect the downregulation of MHC class I molecules on target cells. Activating receptors, on the other hand, bind a variety of molecules, some of which are derived from pathogens such as CMV protein pp65, which is recognized by NKp30 (13), or are induced by cell stress, transformation, or infection (e.g., MICA/B and ULBP1-6, the ligands of NKG2D) (14). NK cells not only play an important role as a first line of defense against viral, bacterial, and fungal infections (15, 16), but are also important in tumor immuno-editing (17), tumor development (18), and control of metastasis (19–21).
Under nonpathological conditions, NK cells and myeloid cell subtypes crosstalk through multiple mechanisms. NK cells interact with macrophages and dendritic cells through both soluble factors, such as IL-12, IL-15, IL-27, and IL-18, and cell-to-cell contact (22–24). These interactions can induce maturation of NK cells, cytotoxicity, and cytokine release. Reciprocally, NK cell–derived cytokines can drive stimulation of macrophages. Pathogen-induced upregulation of ligands for activating NK-cell receptors can result in the elimination of monocytes and macrophages by NK cells (25), as well as the killing of immature but not mature dendritic cells in vitro (26), a process thought to limit the generation of potentially tolerogenic dendritic cells.
Myeloid Cells in the TME Influence NK Function
Tumor-derived myeloid cells are plastic and heterogeneous and have both positive and negative roles in anti-tumor immunity. There are two main subsets of suppressive myeloid cells in tumors, PMN-MDSCs and M-MDSCs (6, 27, 28). Monocytes, M-MDSCs, and TAMs are abundant in solid tumors (29) and are associated with poor prognosis (30, 31). M-MDSCs support tumor progression through both immune-mediated mechanisms and mechanisms not directly associated with immune suppression (32). Macrophages and monocytic MDSCs isolated from mouse murine and human solid tumors can directly suppress T‐cell responses (29, 33) and NK-cell cytotoxicity (34). M-MDSCs are implicated in the recruitment of T regulatory cells and inhibition of T-cell cytotoxicity and have been shown to inhibit NK cell function in vitro and in vivo (35). Normally, neutrophils respond to tissue damage and defend against pathogens (36), but in the TME, tumor-associated neutrophils or PMN-MDSCs express various cytokines, including CCL2 and CCL17, depending on their immunosuppressive or immune-activating state, and can degranulate to release various types of bioactive molecules (37, 38). The formation of neutrophil extracellular traps can convert dormant cancer cells, drive aggressive lung metastases in mice (39), and accelerate hepatocellular cancer (40) in patients and in mouse models (41).
NK Cells in the TME
The TME not only shapes the adaptive immune response but also has profound effects on NK cells, which in many tumors are functionally distinct. Anti-tumor NK effector mechanisms such as cytotoxicity and secretion of pro-inflammatory cytokines are impaired due to low expression of effector molecules perforin and granzyme in patients with lung adenocarcinoma (42), downmodulation of activating receptors NKG2D or NKp30 in gastric cancer (43), and upregulation of inhibitory receptors like NKG2A in cervical cancer (44).
NK cells in tumors also acquire pro-angiogenic and pro-tumor functions, including the secretion of vascular endothelial growth factor (VEGF) (45), angiogenin, and MMP9 (46, 47). Indeed, NK cells play an important role in the menstrual cycle and establishing the placenta (48). The induction of some of these phenotypic features have been attributed to immune-modulatory molecules present in the TME, such as indoleamine-pyrrole 2,3-dioxygenase and tumor growth factor-beta (TGF-β), which can be secreted by MDSCs (49). NK cells in which STAT5 has been silenced express VEGF-A at a level sufficient to promote the growth of murine syngeneic tumors (50). NK cells with a pro-angiogenic phenotype have been identified in non–small-cell lung cancer (47) and colorectal cancer (51). However, it is not clear whether there is a meaningful or broad contribution of these potentially pro-angiogenic NK cells to drive tumor progression or whether they represent the primary angiogenic drive. In one study, genetic inactivation of VEGF in myeloid cells prevented tumor growth and chemotherapy-induced cachexia in B16 and LLC mouse tumor models (52). This study also suggested that increased levels of circulating chemerin by the tumor endothelium improved NK-cell recruitment to the tumor site, suggesting that an indirect mechanism of targeting myeloid cells affects NK recruitment and function. It would be important to understand whether pharmacological interventions would have a similar effect and whether this is a dominant mechanism.
Some of the factors that are known to contribute to functional impairment of NK cells, such as hypoxia, are tumor intrinsic, whereas others are secreted by tumor-associated cells, in particular MDSCs and TAMs. In mouse models, one such mechanism is induction of NK-cell scavenger receptor expression, which is involved in lipid metabolism. Uptake of MDSC-derived factors leads to lipid accumulation and functional impairment (53).
NK cells have also been implicated in anti-tumor immune responses after checkpoint blockade. PD-1 is expressed on about 25% of NK cells in some healthy donors, usually at low levels (54), but has been found to be expressed at substantial levels in patients with ovarian cancer (54); digestive cancers, including esophageal, liver, colorectal, and gastric cancers (55); multiple myeloma (56); Kaposi sarcoma (57); and renal cell carcinoma (58). However, infiltrating NK cells in non–small-cell lung cancer do not express PD-1 (59), although a recent systematic study using multiple methods to detect PD-1 protein and mRNA concluded that NK cells showed only minimal expression of PD-1 in primary human tumor samples of round-cell sarcoma and colorectal cancer, as well as in multiple mouse tumor models (60). Despite these findings, several studies have reported upregulation of PD-1 expression on NK cells in various mouse models (61, 62), and although blockade of PD-1/PD-L1 interaction has been shown to enhance activity of NK cells in vitro and in animal models (63), this is suggested to be mediated mainly through expression of PD-L1 by NK cells (64). Moreover, it is not clear how NK cells contribute to anti-tumor responses in patients. NK cells have also been implicated in playing a role in response to treatment with agonistic anti-CD137/4-1BB antibodies. CD137 is upregulated by Fc receptor cross-linking on NK cells (65) and in patients after treatment with monoclonal antibodies (66). CD137 ligation contributes to activation in vitro (67) and in humanized mice (68) but reports that enhances antibody-dependent cell cytotoxicity have been retracted (69, 70).
Humoral vs. Cell-Cell Interaction Crosstalk Between NK and Myeloid Cells
In the TME, cell-cell interactions and humoral responses build an anti-tumor immune response; therefore, it is important to consider how different therapeutic approaches can affect these interactions. The role of myeloid cells in the TME has been extensively studied, and a number of therapeutics have been developed to target these cells. Both neutrophil/PMN-MDSC and macrophage/M-MDSC–like myeloid cells can influence both T-cell and NK-cell activation and play both positive and negative roles in tumor growth and metastatic progression.
During infection, macrophages can modulate NK function either through direct cell-to-cell contact or through secretion of soluble mediators such as IL-18, IL-12, and TGF-β (25) (Figure 1A). CD56bright NK cells accumulate in inflammatory lesions in the presence of IL-12, IL-15, and IL-18 and engage with CD14+ monocytes in a reciprocal activation loop, amplifying the inflammatory response by increasing TNFα production by monocytes and IFNγ by NK cells (71). In vitro, appropriately activated myeloid cells can also facilitate activation of NK cells via cell-cell interactions, enhancing CD69 expression and secretion of IFNγ in co-cultures (71, 72). In contrast, monocytes and macrophages isolated from hepatocellular carcinoma patient samples (34) and gastric cancer (73) tumors can induce NK-cell dysfunction via direct cell-cell interaction and indirectly, e.g., through soluble TGF-β signaling. In other studies, macrophages and monocytes isolated from hepatocellular carcinoma samples expressed high levels of CD48, driving NK-cell dysfunction. This effect was attenuated by blocking the NK-cell CD48 receptor 2B4 (34). Macrophage or M-MDSC secreted factors can have direct and indirect effects on myeloid and NK-cell crosstalk. Soluble TGFβ modulates NK-cell function via activating receptors NKG2D and CD16 antibody-dependent cell-mediated cytotoxicity in tumors by impairing cytotoxicity potential in vivo and in co-culture experiments with acute myeloid leukemia and colon cancer models (74, 75). Conversely, IL-15 plays a role in maintaining NK activation to suppress tumor escape and metastasis (76). Other secreted factors may act indirectly; these include tumor-derived prostaglandin-E2, which induces MDSCs and inhibits NK-cell function in melanoma samples (77). Restoring NK-cell function by co-targeting immunosuppressive myeloid cells may be an important therapeutic strategy to prevent tumor immune escape (Figure 1A).
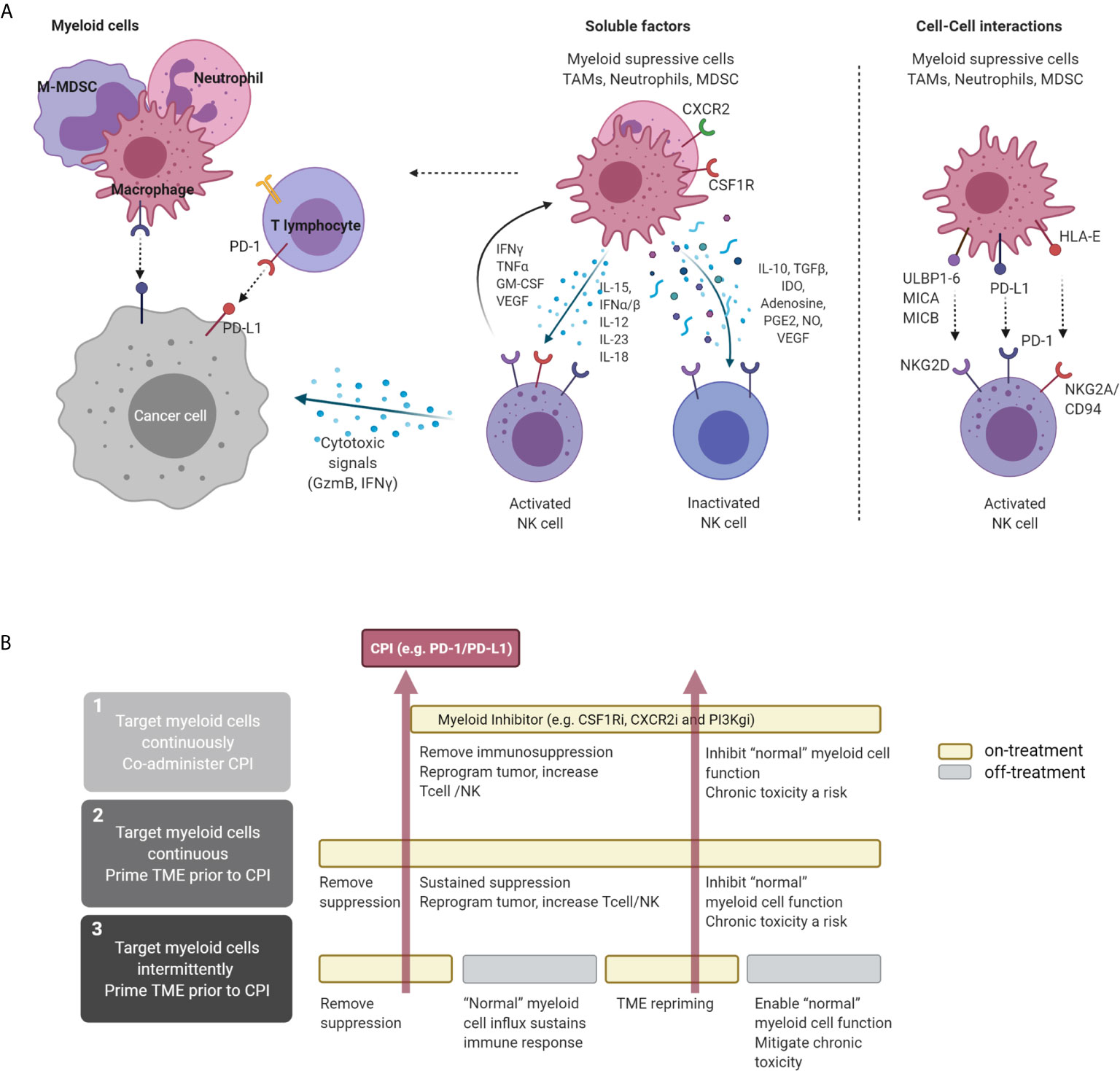
Figure 1 Direct and indirect interaction of NK and myeloid cells in the TME and therapeutic concepts. (A) Interaction of NK and myeloid cells in the TME. NK cells can directly target tumor cells via cytolytic granules, independent of antigen recognition. Macrophages with immunostimulatory properties can independently induce tumor-cell killing through antigen presentation and production of pro-inflammatory cytokines. In the TME, myeloid cells, including TAMs, M-MDSCs, neutrophils, and PMN-MDSCs, can secrete a variety of soluble factors that inhibit NK activation and therefore suppress NK-mediated cytotoxicity. Cytokines secreted by NK cells (e.g., IFNγ, TNFα, and GM-CSF) can stimulate macrophages, driving a pro-inflammatory activated state. These two cell types can also interact at the receptor level, where myeloid cell-surface ligand and NK receptors interact, attenuating downstream signaling, e.g., NKG2D. (B) Proposed therapeutic approaches targeting myeloid-cell subsets in the TME and proposed alternative treatment sequences that can be explored to maximize immune-mediated anti-tumor response. CPI, checkpoint inhibitor. Figure created with BioRender.com.
Pro-inflammatory macrophages, such as IL-12–secreting macrophages (9, 78), that promote NK function in infection and mouse tumor models highlight the importance of understanding the difference between specific myeloid phenotypes and their influence on NK activation and function (79). Some myeloid-targeting therapies rely on cell depletion mechanisms, whereas others attempt to block recruitment or to reprogram these cells into a pro-inflammatory anti-tumor state (9). This is an important consideration when developing therapies, given the high plasticity of myeloid cell types and multiple cell interactions, including activating and suppressive impacts on T- and NK-cell effector functions.
Most studies have focused on the effects of myeloid-cell inhibitors (e.g., CSF1R inhibitors) on primary tumors, but not much information is available in the context of metastasis. CSF1R inhibition reprograms the TME to increase responses to chemotherapy and checkpoint inhibitors and to decrease metastatic spread (80). In some studies, inhibition of CSF1R depleted tumor-associated macrophages but unexpectedly promoted metastasis in 4T1 orthotopic syngeneic models. In one report, CSF1R inhibition reduced the number of NK cells due to a decrease in IL-15, a T-cell and NK-cell survival factor secreted by myeloid cells (22). Moreover, dosing exogenous IL-15 during CSF1Ri treatment restored NK-cell numbers and metastasis control. Genetic ablation of IL-15 in mice and in Th2-polarized CD4 T cells has been found to promote the formation of M2 macrophages that are thought to contribute to metastasis formation (81). NK cells may control the seeding of circulating tumor cells due to crosstalk with myeloid cells, a process that is affected when tumors are treated with depleting CSF1R antibodies (82). It is interesting to contrast this finding with the observation that neutrophils or PMN-MDSCs promoted metastasis in this model (83). Other therapeutic approaches can influence this axis. Blocking CD39 activity in myeloid cells has been shown to improve control of metastases via NK-cell effector function (84). CD39 expression by myeloid cells, but not NK cells, was required for efficacy, suggesting that blockade of CD39 on myeloid cells limits the impact of eATP in driving intratumor myeloid pyroptosis or the release of IL-18, both of which have been shown to stimulate NK-cell effector function (84).
CCL2 (MCP1), which interacts with CCR2, is an alternative mechanism that influences macrophage-related myeloid recruitment to tumors and subsequent tumor progression (85). Inhibition of CCL2 has been shown to limit early metastatic processes in breast cancer; however, after cessation of therapy, increased metastatic spread is observed due to enhanced recruitment of monocytes to micrometastatic lesions in breast (86, 87) and lung (88, 89) metastasis mouse tumor models. Importantly, it has been suggested that combined inhibition of CCL2 and IL-6, a cytokine expressed by myeloid cells, reduced metastasis and improved survival in prostate cancer (90). Targeting CCR2 also reduces tumor progression associated with an influx of T cells in preclinical glioma (91) and pancreatic models (92).
Although macrophages can contribute to anti-tumor immunity, both monocyte-derived MDSCs and TAMs can also promote cancer initiation, stimulate angiogenesis, and suppress anti-tumor immunity during malignant progression. Pro-inflammatory, or “anti-tumor”, macrophages contribute to an anti-tumor response by producing pro-inflammatory cues such as IFNγ and IL-12 secretion or by acquiring an antimicrobial and tumoricidal phenotype (93, 94). Therefore, “reprogramming” macrophages into an anti-tumor and proinflammatory state is an attractive strategy to tip the balance on tumor immunity. Targeting STAT3 (95) or PI3Kγ signaling has been shown preclinically to change the TME in tumors by remodeling suppressive macrophages into proinflammatory macrophages. Selective targeting of PI3Kγ signaling in combination with checkpoint blockade is thought to promote reprogramming of macrophages into a pro-inflammatory state, leading to cytotoxic T-cell–mediated anti-tumor response in preclinical mouse models (96, 97). The combination of PI3Kγ with PD-1 blockade is currently under investigation in clinical trials and recently received FDA Fast Track designation in urothelial cancers (ClinicalTrials.gov NCT03980041).
In normal physiological processes, neutrophil depletion impairs NK-cell maturation, function, and homeostasis (98). The role of neutrophils and PMN-MDSCs in cancer has been extensively studied, and these cells play an important role in facilitating tumor progression. In various tumor models, targeting or depleting neutrophils or PMN-MDSCs reduces metastasis in both autochthonous models of pancreas (99), colon cancer (100), breast (101), and metastatic syngeneic models (83, 102). This metastatic process may be through γδT-cell–orchestrated suppression of CD8 T cells by modified neutrophils (101). However, there is evidence that immunosurveillance of metastatic 4T1 cells by NK cells is inhibited by interaction with CD11B+/Ly6G+ neutrophils (most likely PMN-MDSCs), increasing residence time for metastatic tumor cells arriving at the lung and enabling extravasation and establishment of the metastatic niche (83). Soluble factors such as IL-17, granulocyte-CSF (G-CSF) (101), and TGFβ signaling (100, 103) play pivotal roles in establishing this suppressive network. The crosstalk between neutrophils and PMN-MDSCs is not a one-way process. In MCA205-Luc2 tumors, depletion of NK cells with antibodies or CXCR3 blockade has been shown to promote tumor growth due to reduced IFNγ and upregulation of IL-17A and VEGF-A, modifying the TME and recruitment of suppressive neutrophils of PMN-MDSCs (104).
Therapeutic targeting of CXCR2 (or IL-8) inhibits neutrophil-granulocytic myeloid cells or PMN-MDSCs, leading to suppression of metastasis in mouse models of pancreatic cancer (99) and colorectal cancer (100), as well as in metastatic syngeneic models 4T1 and B16F10 (105). In preclinical efficacy studies, CXCR2 inhibition resulted in an influx of T cells (99, 100, 105, 106); however, the impact on the broader immune environment, including NK biology, has not been explored. Although CXCR2 blockade inhibits recruitment of granulocytic myeloid cells to the tumor, it may also inhibit NK recruitment. CXCR1 and CXCR2 are highly expressed by cytotoxic CD56dim NK cells (107), and increasing CXCR2 expression on NK cells promotes recruitment to tumors that overexpress CXCR2 ligands (108). Importantly, the CXCR2 ligand CXCL8 is secreted within the TME of melanoma-infiltrated lymph nodes and may play a role in the efficient recruitment of highly cytotoxic NK cells (109). Because it has been suggested that chronic combined inhibition of both CXCR2 and CSF1R can increase the efficacy of checkpoint inhibition in syngeneic models (110), understanding the potential impact of comprehensive myeloid suppressor cell inhibition on NK-cell activity should be considered.
Taken together, these studies of different myeloid lineages exemplify how depletion of specific subsets of myeloid cells can affect different features of the TME, modulating innate effector-cell activity and promoting tumor progression and metastasis. However, because myeloid cells, and particularly macrophages, play an important role in assisting NK- and T-cell activation, it is important to target the right population of cells. Moreover, given that these are essential cell types, translation to a clinical setting may be limited by tolerability, as observed in studies targeting the CSFR1 axis with antibodies or small molecules (80, 111, 112), which resulted in increased liver enzymes and induction of periorbital edema. Less toxicity was observed when the alternate macrophage regulating receptor CCR2 was targeted (113–115).
Nontargeted therapies, such as chemotherapy, can also deplete myeloid cells from tumor. Paclitaxel-carboplatin treatment was shown to alter circulating and intratumoral myeloid cell populations and to promote anti-tumor responses when combined with vaccination in HPV-16–positive tumors in mice (98). In a phase 2 trial in patients with extensive small-cell lung cancer, it was reported that ipilimumab treatment beginning with the third cycle of paclitaxel-carboplatin treatment produced better clinical outcomes than giving the drugs during cycles 1 to 4 (116). An understanding of the pivotal points in these complex signaling and transcriptional networks that program the myeloid cell phenotypes is essential to guide more effective therapeutic approaches.
Perspective: Impact of Dose and Schedule in Myeloid Target Therapies and Checkpoint Inhibitors
Translation to the clinic of preclinical concepts, which were largely developed using fast-growing subcutaneous in vivo models, presents a challenge. Subcutaneous models are limited because they do not reflect the variations observed in the tissue of residence, and the speed of cell growth in these models does not enable elucidation of the longer-term consequences of the treatment strategy. As shown in Table 1, most clinical studies have taken a standard approach in which the myeloid therapy is co-administered with the checkpoint inhibitor or chemotherapy and then dosing is maintained chronically (80, 111–113, 115). This approach has a number of drawbacks. Myeloid cells exhibit both positive and negative effects on the TME, as described above. Accumulation of myeloid cells in the tumor (macrophage-like and neutrophil-like cells) clearly defines a resistance phenotype, and depletion of macrophages and neutrophils can remodel the TME. In addition to preventing the suppressive crosstalk to immune cell types, including T cells and NK cells, removal of these cells results in remodeling of the stroma and, in some cases, reprogramming of the tumor cell compartment. These changes make the tumor more susceptible to appropriate recruitment of activated immune cells. Hence, it is likely that pretreatment with a myeloid modulation agent prior to treatment with immunotherapy or even chemotherapy would “prime” the TME by reversing the resistant features in the tumor, facilitating more effective stimulation of the immune system. However, once the immune response is progressing, then more “normal” myeloid cells could be required to sustain that response, especially in situations where there is less effective immune recognition of the tumor. Paradoxically, chronic suppression of the myeloid cells may result in attenuation of the immune response in certain situations, mitigating the advantages gained from targeting the suppressive cells. Therefore, therapies that deplete myeloid cells or prevent recruitment to the TME may be more effective with intermittent or sequenced dosing, using the myeloid therapy for a short time prior to treatment to “prime” the TME, but then stopping dosing after introduction of the checkpoint inhibitor to allow the more normal immune response to progress (Figure 1B). These types of intermittent approaches could also mitigate clinical toxicity.
Discussion
The development of cancer immunotherapies, specifically immune checkpoint blockade, has shifted the treatment of cancer by promoting complete and durable responses (117, 118). Immunotherapies focus on enhancing the activities of T cells; however, the complexity of the TME limits the response. The pivotal role of tumor myeloid cells, particularly macrophages, in conditioning the TME and regulating the broader response to host immune response and therapy is broadly appreciated. Unfortunately, the development of targeted therapeutics has only just started to teach us about the complexity of this cross-regulation, particularly in the context of different tumor mutational backgrounds and TMEs, as well as the broader systemic immune response. To enhance success, it is worth considering the positive influence of myeloid cells on the other components of the immune system, such as NK cells, and their role in sustaining persistent T-cell responses. Although myeloid therapies have largely been combined with checkpoint inhibitors and, to a lesser extent, chemotherapeutics, little consideration has been given to combinations with therapies targeting other functional nodes, such as NK cells or stimulators of innate immunity. As we seek to improve responses in patients earlier in disease progression, at the point of metastatic spread, such alternative strategies could become important.
Author Contributions
All authors contributed to the article and approved the submitted version.
Conflict of Interest
Authors are AstraZeneca Plc employees and shareholders.
Acknowledgments
Thanks to Deborah Shuman for comments and revision of the manuscript.
References
1. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
2. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell (2015) 161(2):205–14. doi: 10.1016/j.cell.2015.03.030
3. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
4. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med (2015) 372(21):2018–28. doi: 10.1056/NEJMoa1501824
5. Hoos A. Development of immuno-oncology drugs: from CTLA4 to PD1 to the next generations. Nat Rev Drug Discovery (2016) 15(4):235–47. doi: 10.1038/nrd.2015.35
6. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
7. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer (2020) 20(9):485–503. doi: 10.1038/s41568-020-0281-y
8. Neophytou CM, Pierides C, Christodoulou MI, Costeas P, Kyriakou TC, Papageorgis P. The role of tumor-associated myeloid cells in modulating cancer therapy. Front Oncol (2020) 10:899. doi: 10.3389/fonc.2020.00899
9. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discovery (2018) 17(12):887–904. doi: 10.1038/nrd.2018.169
10. Cassetta L, Kitamura T. Macrophage targeting: opening new possibilities for cancer immunotherapy. Immunology (2018) 155(3):285–93. doi: 10.1111/imm.12976
11. Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C, Breckpot K. Turn Back the TIMe: Targeting Tumor Infiltrating Myeloid Cells to Revert Cancer Progression. Front Immunol (2018) 9:1977. doi: 10.3389/fimmu.2018.01977
12. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol (2019) 16(5):430–41. doi: 10.1038/s41423-019-0206-4
13. Arnon TI, Achdout H, Levi O, Markel G, Saleh N, Katz G, et al. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat Immunol (2005) 6(5):515–23. doi: 10.1038/ni1190
14. Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, et al. NKG2D and its ligands: “one for all, all for one”. Front Immunol (2018) 9:476. doi: 10.3389/fimmu.2018.00476
15. Schmidt S, Tramsen L, Lehrnbecher T. Natural killer cells in antifungal immunity. Front Immunol (2017) 8:1623. doi: 10.3389/fimmu.2017.01623
16. Schmidt S, Ullrich E, Bochennek K, Zimmermann SY, Lehrnbecher T. Role of natural killer cells in antibacterial immunity. Exp Rev Hematol (2016) 9(12):1119–27. doi: 10.1080/17474086.2016.1254546
17. Elboim M, Gazit R, Gur C, Ghadially H, Betser-Cohen G, Mandelboim O. Tumor immunoediting by NKp46. J Immunol (2010) 184(10):5637–44. doi: 10.4049/jimmunol.0901644
18. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet (2000) 356(9244):1795–9. doi: 10.1016/S0140-6736(00)03231-1
19. Kim S, Iizuka K, Aguila HL, Weissman IL, Yokoyama WM. In vivo natural killer cell activities revealed by natural killer cell-deficient mice. Proc Natl Acad Sci USA (2000) 97(6):2731–6. doi: 10.1073/pnas.050588297
20. Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell (2017) 32(2):135–54. doi: 10.1016/j.ccell.2017.06.009
21. Sathe P, Delconte RB, Souza-Fonseca-Guimaraes F, Seillet C, Chopin M, Vandenberg CJ, et al. Innate immunodeficiency following genetic ablation of Mcl1 in natural killer cells. Nat Commun (2014) 5:4539. doi: 10.1038/ncomms5539
22. Santana Carrero RM, Beceren-Braun F, Rivas SC, Hegde SM, Gangadharan A, Plote D, et al. IL-15 is a component of the inflammatory milieu in the tumor microenvironment promoting antitumor responses. Proc Natl Acad Sci USA (2019) 116(2):599–608. doi: 10.1073/pnas.1814642116
23. Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol (2012) 13(8):722–8. doi: 10.1038/ni.2366
24. Ziblat A, Domaica CI, Spallanzani RG, Iraolagoitia XL, Rossi LE, Avila DE, et al. IL-27 stimulates human NK-cell effector functions and primes NK cells for IL-18 responsiveness. Eur J Immunol (2015) 45(1):192–202. doi: 10.1002/eji.201444699
25. Michel T, Hentges F, Zimmer J. Consequences of the crosstalk between monocytes/macrophages and natural killer cells. Front Immunol (2012) 3:403. doi: 10.3389/fimmu.2012.00403
26. Morandi B, Mortara L, Chiossone L, Accolla RS, Mingari MC, Moretta L, et al. Dendritic cell editing by activated natural killer cells results in a more protective cancer-specific immune response. PloS One (2012) 7(6):e39170. doi: 10.1371/journal.pone.0039170
27. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111(8):4233–44. doi: 10.1182/blood-2007-07-099226
28. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol (2008) 181(8):5791–802. doi: 10.4049/jimmunol.181.8.5791
29. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell (2015) 27(4):462–72. doi: 10.1016/j.ccell.2015.02.015
30. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med (2015) 21(8):938–45. doi: 10.1038/nm.3909
31. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PloS One (2012) 7(12):e50946. doi: 10.1371/journal.pone.0050946
32. Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann N Y Acad Sci (2014) 1319:47–65. doi: 10.1111/nyas.12469
33. Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol (2015) 15(2):73–86. doi: 10.1038/nri3789
34. Wu Y, Kuang DM, Pan WD, Wan YL, Lao XM, Wang D, et al. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology (2013) 57(3):1107–16. doi: 10.1002/hep.26192
35. Elkabets M, Ribeiro VS, Dinarello CA, Ostrand-Rosenberg S, Di Santo JP, Apte RN, et al. IL-1beta regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol (2010) 40(12):3347–57. doi: 10.1002/eji.201041037
36. Mollinedo F. Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol (2019) 40(3):228–42. doi: 10.1016/j.it.2019.01.006
37. Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol (2019) 16(10):601–20. doi: 10.1038/s41571-019-0222-4
38. Shaul ME, Levy L, Sun J, Mishalian I, Singhal S, Kapoor V, et al. Tumor-associated neutrophils display a distinct N1 profile following TGFbeta modulation: A transcriptomics analysis of pro- vs. antitumor TANs. Oncoimmunology (2016) 5(11):e1232221. doi: 10.1080/2162402X.2016.1232221
39. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science (2018) 361(6409);eaao4227. doi: 10.1126/science.aao4227
40. Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Res (2016) 76(6):1367–80. doi: 10.1158/0008-5472.CAN-15-1591
41. Masucci MT, Minopoli M, Del Vecchio S, Carriero MV. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front Immunol (2020) 11:1749. doi: 10.3389/fimmu.2020.01749
42. Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell (2017) 169(4):750–65 e17. doi: 10.1016/j.cell.2017.04.014
43. Han B, Mao FY, Zhao YL, Lv YP, Teng YS, Duan M, et al. Altered NKp30, NKp46, NKG2D, and DNAM-1 expression on circulating NK cells is associated with tumor progression in human gastric cancer. J Immunol Res (2018) 2018:6248590. doi: 10.1155/2018/6248590
44. Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell (2018) 175(7):1731–43.e13. doi: 10.1016/j.cell.2018.10.014
45. Levi I, Amsalem H, Nissan A, Darash-Yahana M, Peretz T, Mandelboim O, et al. Characterization of tumor infiltrating natural killer cell subset. Oncotarget (2015) 6(15):13835–43. doi: 10.18632/oncotarget.3453
46. Bruno A, Bassani B, D’Urso DG, Pitaku I, Cassinotti E, Pelosi G, et al. Angiogenin and the MMP9-TIMP2 axis are up-regulated in proangiogenic, decidual NK-like cells from patients with colorectal cancer. FASEB J (2018) 32(10):5365–77. doi: 10.1096/fj.201701103R
47. Bruno A, Focaccetti C, Pagani A, Imperatori AS, Spagnoletti M, Rotolo N, et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia (2013) 15(2):133–42. doi: 10.1593/neo.121758
48. Ribatti D, Tamma R, Crivellato E. Cross talk between natural killer cells and mast cells in tumor angiogenesis. Inflammation Res (2019) 68(1):19–23. doi: 10.1007/s00011-018-1181-4
49. Bruno A, Mortara L, Baci D, Noonan DM, Albini A. Myeloid-derived suppressor cells’ interactions with natural killer cells and pro-angiogenic activities: roles in tumor progression. Front Immunol (2019) 10:771. doi: 10.3389/fimmu.2019.00771
50. Gotthardt D, Putz EM, Grundschober E, Prchal-Murphy M, Straka E, Kudweis P, et al. STAT5 Is a Key Regulator in NK Cells and Acts as a Molecular Switch from Tumor Surveillance to Tumor Promotion. Cancer Discovery (2016) 6(4):414–29. doi: 10.1158/2159-8290.CD-15-0732
51. Rocca YS, Roberti MP, Arriaga JM, Amat M, Bruno L, Pampena MB, et al. Altered phenotype in peripheral blood and tumor-associated NK cells from colorectal cancer patients. Innate Immun (2013) 19(1):76–85. doi: 10.1177/1753425912453187
52. Klose R, Krzywinska E, Castells M, Gotthardt D, Putz EM, Kantari-Mimoun C, et al. Targeting VEGF-A in myeloid cells enhances natural killer cell responses to chemotherapy and ameliorates cachexia. Nat Commun (2016) 7:12528. doi: 10.1038/ncomms12528
53. Niavarani SR, Lawson C, Bakos O, Boudaud M, Batenchuk C, Rouleau S, et al. Lipid accumulation impairs natural killer cell cytotoxicity and tumor control in the postoperative period. BMC Cancer (2019) 19(1):823. doi: 10.1186/s12885-019-6045-y
54. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J Allergy Clin Immunol (2017) 139(1):335–46 e3. doi: 10.1016/j.jaci.2016.04.025
55. Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene (2017) 36(44):6143–53. doi: 10.1038/onc.2017.209
56. Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood (2010) 116(13):2286–94. doi: 10.1182/blood-2010-02-271874
57. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vely F, Vivier E, Olive D, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget (2016) 7(45):72961–77. doi: 10.18632/oncotarget.12150
58. MacFarlane AW, Jillab M, Plimack ER, Hudes GR, Uzzo RG, Litwin S, et al. PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection. Cancer Immunol Res (2014) 2(4):320–31. doi: 10.1158/2326-6066.CIR-13-0133
59. Datar I, Sanmamed MF, Wang J, Henick BS, Choi J, Badri T, et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin Cancer Res (2019) 25(15):4663–73. doi: 10.1158/1078-0432.CCR-18-4142
60. Judge SJ, Dunai C, Aguilar EG, Vick SC, Sturgill IR, Khuat LT, et al. Minimal PD-1 expression in mouse and human NK cells under diverse conditions. J Clin Invest (2020) 130(6):3051–68. doi: 10.1172/JCI133353
61. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest (2018) 128(10):4654–68. doi: 10.1172/JCI99317
62. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol (2018) 19(7):723–32. doi: 10.1038/s41590-018-0132-0
63. Concha-Benavente F, Kansy B, Moskovitz J, Moy J, Chandran U, Ferris RL. PD-L1 Mediates Dysfunction in Activated PD-1(+) NK Cells in Head and Neck Cancer Patients. Cancer Immunol Res (2018) 6(12):1548–60. doi: 10.1158/2326-6066.CIR-18-0062
64. Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, et al. The mechanism of anti-PD-L1 antibody efficacy against PD-L1-negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discovery (2019) 9(10):1422–37. doi: 10.1158/2159-8290.CD-18-1259
65. Srivastava RM, Trivedi S, Concha-Benavente F, Gibson SP, Reeder C, Ferrone S, et al. CD137 Stimulation Enhances Cetuximab-Induced Natural Killer: Dendritic Cell Priming of Antitumor T-Cell Immunity in Patients with Head and Neck Cancer. Clin Cancer Res (2017) 23(3):707–16. doi: 10.1158/1078-0432.CCR-16-0879
66. Makkouk A, Sundaram V, Chester C, Chang S, Colevas AD, Sunwoo JB, et al. Characterizing CD137 upregulation on NK cells in patients receiving monoclonal antibody therapy. Ann Oncol Off J Eur Soc Med Oncol (2017) 28(2):415–20. doi: 10.1093/annonc/mdw570
67. Vidard L, Dureuil C, Baudhuin J, Vescovi L, Durand L, Sierra V, et al. CD137 (4-1BB) Engagement Fine-Tunes Synergistic IL-15- and IL-21-Driven NK Cell Proliferation. J Immunol (2019) 203(3):676–85. doi: 10.4049/jimmunol.1801137
68. Ochoa MC, Perez-Ruiz E, Minute L, Onate C, Perez G, Rodriguez I, et al. Daratumumab in combination with urelumab to potentiate anti-myeloma activity in lymphocyte-deficient mice reconstituted with human NK cells. Oncoimmunology (2019) 8(7):1599636. doi: 10.1080/2162402X.2019.1599636
69. Kohrt HE, Houot R, Weiskopf K, Goldstein MJ, Scheeren F, Czerwinski D, et al. Stimulation of natural killer cells with a CD137-specific antibody enhances trastuzumab efficacy in xenotransplant models of breast cancer. J Clin Invest (2012) 122(3):1066–75. doi: 10.1172/JCI61226
70. Kohrt HE, Colevas AD, Houot R, Weiskopf K, Goldstein MJ, Lund P, et al. Targeting CD137 enhances the efficacy of cetuximab. J Clin Invest (2019) 129(6):2595. doi: 10.1172/JCI129689
71. Dalbeth N, Gundle R, Davies RJ, Lee YC, McMichael AJ, Callan MF. CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation. J Immunol (2004) 173(10):6418–26. doi: 10.4049/jimmunol.173.10.6418
72. Haller D, Serrant P, Granato D, Schiffrin EJ, Blum S. Activation of human NK cells by staphylococci and lactobacilli requires cell contact-dependent costimulation by autologous monocytes. Clin Diagn Lab Immunol (2002) 9(3):649–57. doi: 10.1128/CDLI.9.3.649-657.2002
73. Peng LS, Zhang JY, Teng YS, Zhao YL, Wang TT, Mao FY, et al. Tumor-Associated Monocytes/Macrophages Impair NK-Cell Function via TGFbeta1 in Human Gastric Cancer. Cancer Immunol Res (2017) 5(3):248–56. doi: 10.1158/2326-6066.CIR-16-0152
74. Liu C, Yu S, Kappes J, Wang J, Grizzle WE, Zinn KR, et al. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood (2007) 109(10):4336–42. doi: 10.1182/blood-2006-09-046201
75. Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, et al. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PloS One (2018) 13(1):e0191358. doi: 10.1371/journal.pone.0191358
76. Kubo H, Mensurado S, Goncalves-Sousa N, Serre K, Silva-Santos B. Primary Tumors Limit Metastasis Formation through Induction of IL15-Mediated Cross-Talk between Patrolling Monocytes and NK Cells. Cancer Immunol Res (2017) 5(9):812–20. doi: 10.1158/2326-6066.CIR-17-0082
77. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res (2014) 20(15):4096–106. doi: 10.1158/1078-0432.CCR-14-0635
78. Watkins SK, Egilmez NK, Suttles J, Stout RD. IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages in vitro and in vivo. J Immunol (2007) 178(3):1357–62. doi: 10.4049/jimmunol.178.3.1357
79. Narasimhan PB, Eggert T, Zhu YP, Marcovecchio P, Meyer MA, Wu R, et al. Patrolling Monocytes Control NK Cell Expression of Activating and Stimulatory Receptors to Curtail Lung Metastases. J Immunol (2020) 204(1):192–8. doi: 10.4049/jimmunol.1900998
80. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell (2014) 25(6):846–59. doi: 10.1016/j.ccr.2014.05.016
81. Gillgrass A, Gill N, Babian A, Ashkar AA. The absence or overexpression of IL-15 drastically alters breast cancer metastasis via effects on NK cells, CD4 T cells, and macrophages. J Immunol (2014) 193(12):6184–91. doi: 10.4049/jimmunol.1303175
82. Beffinger M, Tallon de Lara P, Tugues S, Vermeer M, Montagnolo Y, Ohs I, et al. CSF1R-dependent myeloid cells are required for NK-mediated control of metastasis. JCI Insight (2018) 3(10):e97792. doi: 10.1172/jci.insight.97792
83. Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, et al. Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discovery (2016) 6(6):630–49. doi: 10.1158/2159-8290.CD-15-1157
84. Yan J, Li XY, Roman Aguilera A, Xiao C, Jacoberger-Foissac C, Nowlan B, et al. Control of Metastases via Myeloid CD39 and NK Cell Effector Function. Cancer Immunol Res (2020) 8(3):356–67. doi: 10.1158/2326-6066.CIR-19-0749
85. Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, et al. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep (2015) 12(2):244–57. doi: 10.1016/j.celrep.2015.06.024
86. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature (2014) 515(7525):130–3. doi: 10.1038/nature13862
87. Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med (2015) 212(7):1043–59. doi: 10.1084/jem.20141836
88. Roblek M, Protsyuk D, Becker PF, Stefanescu C, Gorzelanny C, Glaus Garzon JF, et al. CCL2 Is a Vascular Permeability Factor Inducing CCR2-Dependent Endothelial Retraction during Lung Metastasis. Mol Cancer Res (2019) 17(3):783–93. doi: 10.1158/1541-7786.MCR-18-0530
89. Tu MM, Abdel-Hafiz HA, Jones RT, Jean A, Hoff KJ, Duex JE, et al. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun Biol (2020) 3(1):720. doi: 10.1038/s42003-020-01441-y
90. Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem (2009) 284(49):34342–54. doi: 10.1074/jbc.M109.042671
91. Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc Natl Acad Sci U.S.A. (2020) 117(2):1129–38. doi: 10.1073/pnas.1910856117
92. Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res (2013) 19(13):3404–15. doi: 10.1158/1078-0432.CCR-13-0525
93. Bogdan C, Schleicher U. Production of interferon-gamma by myeloid cells—fact or fancy? Trends Immunol (2006) 27(6):282–90. doi: 10.1016/j.it.2006.04.004
94. Frucht DM, Fukao T, Bogdan C, Schindler H, O’Shea JJ, Koyasu S. IFN-gamma production by antigen-presenting cells: mechanisms emerge. Trends Immunol (2001) 22(10):556–60. doi: 10.1016/S1471-4906(01)02005-1
95. Proia TA, Singh M, Woessner R, Carnevalli L, Bommakanti G, Magiera L, et al. STAT3 Antisense Oligonucleotide Remodels the Suppressive Tumor Microenvironment to Enhance Immune Activation in Combination with Anti-PD-L1. Clin Cancer Res (2020) 26(23):6335–49. doi: 10.1158/1078-0432.CCR-20-1066
96. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature (2016) 539(7629):443–7. doi: 10.1038/nature20554
97. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature (2016) 539(7629):437–42. doi: 10.1038/nature19834
98. Welters MJ, van der Sluis TC, van Meir H, Loof NM, van Ham VJ, van Duikeren S, et al. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci Transl Med (2016) 8(334):334ra52. doi: 10.1126/scitranslmed.aad8307
99. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell (2016) 29(6):832–45. doi: 10.1016/j.ccell.2016.04.014
100. Jackstadt R, van Hooff SR, Leach JD, Cortes-Lavaud X, Lohuis JO, Ridgway RA, et al. Epithelial NOTCH Signaling Rewires the Tumor Microenvironment of Colorectal Cancer to Drive Poor-Prognosis Subtypes and Metastasis. Cancer Cell (2019) 36(3):319–36 e7. doi: 10.1016/j.ccell.2019.08.003
101. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature (2015) 522(7556):345–8. doi: 10.1038/nature14282
102. Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell (2012) 150(1):165–78. doi: 10.1016/j.cell.2012.04.042
103. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell (2009) 16(3):183–94. doi: 10.1016/j.ccr.2009.06.017
104. Ogura K, Sato-Matsushita M, Yamamoto S, Hori T, Sasahara M, Iwakura Y, et al. NK Cells Control Tumor-Promoting Function of Neutrophils in Mice. Cancer Immunol Res (2018) 6(3):348–57. doi: 10.1158/2326-6066.CIR-17-0204
105. Richmond A, Yang J, Yan C, Vilgelm AE, Chen SC, Ayers GD, et al. Targeted Deletion of CXCR2 in Myeloid Cells Alters the Tumor Immune Environment to Improve Antitumor Immunity. Cancer Immunol Res (2020) 9(2):200–13. doi: 10.1158/2326-6066.cir-20-0312
106. Chao T, Furth EE, Vonderheide RH. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol Res (2016) 4(11):968–82. doi: 10.1158/2326-6066.CIR-16-0188
107. Lima M, Leander M, Santos M, Santos AH, Lau C, Queiros ML, et al. Chemokine receptor expression on normal blood CD56(+) NK cells elucidates cell partners that comigrate during the innate and adaptive immune responses and identifies a transitional NK cell population. J Immunol Res (2015) 2015:839684. doi: 10.1155/2015/839684
108. Kremer V, Ligtenberg MA, Zendehdel R, Seitz C, Duivenvoorden A, Wennerberg E, et al. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunother Cancer (2017) 5(1):73. doi: 10.1186/s40425-017-0292-8
109. Ali TH, Pisanti S, Ciaglia E, Mortarini R, Anichini A, Garofalo C, et al. Enrichment of CD56(dim)KIR + CD57 + highly cytotoxic NK cells in tumour-infiltrated lymph nodes of melanoma patients. Nat Commun (2014) 5:5639. doi: 10.1038/ncomms6639
110. Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell (2017) 32(5):654–68 e5. doi: 10.1016/j.ccell.2017.10.005
111. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer (2017) 5(1):53. doi: 10.1186/s40425-017-0257-y
112. Razak AR, Cleary JM, Moreno V, Boyer M, Calvo Aller E, Edenfield W, et al. Safety and efficacy of AMG 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J Immunother Cancer (2020) 8(2):e001006. doi: 10.1136/jitc-2020-001006
113. Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget (2016) 7(19):28697–710. doi: 10.18632/oncotarget.7376
114. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol (2016) 17(5):651–62. doi: 10.1016/S1470-2045(16)00078-4
115. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011) 475(7355):222–5. doi: 10.1038/nature10138
116. Reck M, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol (2013) 24(1):75–83. doi: 10.1093/annonc/mds213
117. Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am Soc Clin Oncol Educ Book (2019) 39:147–64. doi: 10.1200/EDBK_240837
Keywords: immunotherapy, cancer immunotherapy, myeloid cell, NK cell, tumor microenvironment
Citation: Carnevalli LS, Ghadially H and Barry ST (2021) Therapeutic Approaches Targeting the Natural Killer-Myeloid Cell Axis in the Tumor Microenvironment. Front. Immunol. 12:633685. doi: 10.3389/fimmu.2021.633685
Received: 25 November 2020; Accepted: 29 March 2021;
Published: 19 April 2021.
Edited by:
Erik Wennerberg, Institute of Cancer Research (ICR), United KingdomReviewed by:
Hun Sik Kim, University of Ulsan, South KoreaDagmar Gotthardt, University of Veterinary Medicine Vienna, Austria
Copyright © 2021 Carnevalli, Ghadially and Barry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Larissa S. Carnevalli, bGFyaXNzYS5jYXJuZXZhbGxpQGFzdHJhemVuZWNhLmNvbQ==; Simon T. Barry, c2ltb24udC5iYXJyeUBhc3RyYXplbmVjYS5jb20=