 Concetta Quintarelli1,2*
Concetta Quintarelli1,2* Antonio Camera1
Antonio Camera1 Roselia Ciccone1
Roselia Ciccone1 Iside Alessi1
Iside Alessi1 Francesca Del Bufalo1
Francesca Del Bufalo1 Andrea Carai3
Andrea Carai3 Giada Del Baldo1
Giada Del Baldo1 Angela Mastronuzzi1
Angela Mastronuzzi1 Biagio De Angelis1
Biagio De Angelis1- 1Department Onco-Hematology, Cell and Gene Therapy, IRCCS, Bambino Gesù Children’s Hospital, Rome, Italy
- 2Department of Clinical Medicine and Surgery, University of Naples Federico II, Naples, Italy
- 3Neurosurgery Unit, Department of Neurological and Psychiatric Sciences, IRCCS Bambino Gesù Children’s Hospital, Rome, Italy
Although there are several immunotherapy approaches for the treatment of Central Nervous System (CNS) tumors under evaluation, currently none of these approaches have received approval from the regulatory agencies. CNS tumors, especially glioblastomas, are tumors characterized by highly immunosuppressive tumor microenvironment, limiting the possibility of effectively eliciting an immune response. Moreover, the peculiar anatomic location of these tumors poses relevant challenges in terms of safety, since uncontrolled hyper inflammation could lead to cerebral edema and cranial hypertension. The most promising strategies of immunotherapy in neuro-oncology consist of the use of autologous T cells redirected against tumor cells through chimeric antigen receptor (CAR) constructs or genetically modified T-cell receptors. Trials based on native or genetically engineered oncolytic viruses and on vaccination with tumor-associated antigen peptides are also under evaluation. Despite some sporadic complete remissions achieved in clinical trials, the outcome of patients with CNS tumors treated with different immunotherapeutic approaches remains poor. Based on the lessons learned from these unsatisfactory experiences, novel immune-therapy approaches aimed at overcoming the profound immunosuppressive microenvironment of these diseases are bringing new hope to reach the cure for CNS tumors.
Introduction
Brain tumors are the most common solid tumors of children (1, 2). Standard therapy for these diseases includes surgical resection, radiation, and, in selected cases, chemotherapy. Despite aggressive treatment, many patients have a poor long-term outcome or, frequently, develop treatment-related long-term sequelae, including hormone dysfunction, sensory-motor and neurocognitive impairments (3). In the last decades, information on molecular pathways responsible for the development of these tumors has increased as result of extensive knowledge on the genomic, epigenetic, and transcriptomic landscape. This allows for a better tumor classification and provides valuable information for target therapies but does not significantly improve survival for the most aggressive brain tumors. Immunotherapy represents an innovative and attractive approach potentially able to eradicate cancer cells, sparing adjacent normal brain tissue. This peculiarity is relevant for CNS tumors, which cannot be surgically resected due to their location in the brainstem. However, several factors may hinder the efficacy of cell immune therapy in the contest of CNS tumors. Indeed, the brain was historically considered an immune-privileged site due especially to the presence of several physical barriers, including blood–brain barrier (BBB), blood-meningeal barrier, and choroid plexus barrier (also known as blood–cerebrospinal fluid (CSF) barrier) (4, 5). Moreover, the adaptive response to CNS antigens is strongly reduced due to lack of (efficient) lymphatic drainage (6). A recent report describes the presence of lymphatic system in the meninges, allowing T cells to reach the draining cervical lymph nodes for antigen presentation (7). Physiologically, although resting T cells do not cross the BBB, they traffic from meningeal blood vessels into the CSF, where cells can enter the brain parenchyma via the pia mater or choroid plexus. By contrast, activated T cells are able to traverse the BBB trough the capillary tight junctions. For this reason, immunotherapy approaches associated with in vivo or ex vivo activation of T cells may potentially provide a potent tool to facilitate the penetration of the immune system toward the BBB.
Moreover, the development of an effective immunotherapy approach should take into consideration that normal brain parenchyma has evolved to protect itself against an immunologic attack (8), characterized by the paucity of professional antigen-presenting cells (APC) (9) and by the downregulated expression of the major histocompatibility complex (MHC), both features limiting antigen presentation (10).
New evidence shows that the tumor microenvironment (TME) plays a key role in negative modulation of immunotherapeutic approaches.
Glioblastoma (GBM) is one of the best characterized CNS tumors with regard to TME. It is characterised by abundance of cytokines [in particular interleukin-6, interleukin-10, transforming growth factor-beta (TGF-β) and prostaglandin-E] secreted by tumor cells, microglia and/or tumor-associated macrophages (TAMs), exerting strong immunosuppressive activity toward the inhibition of Natural Killer (NK)-cells and T-cells, as well as enhancing T-cell apoptosis, skewing TAM phenotype, and downregulating MHC expression on both tumor cells and APC (11, 12). Moreover, TAM are the most abundant stromal cell type in large number of cancers, including GBM (13) and brain metastases (14), and can represent up to 30–50% of the tumor mass. Indeed, the presence of TAMs, often associated with poor patient prognosis (15), fosters tumor growth, controls metastasis and affects therapeutic response (16, 17).
TME hypoxia is another relevant regulator of immunosuppressive pathways. In particular, TME milieu regulates STAT3 activation associated to hypoxia-inducible factor-1 alpha induction that in turn increase vascular endothelial growth factor (VEGF) expression. This signaling cascade leads to activation of T regulatory cells, and inhibition of APC. For this reason, targeted delivery of cytokines/chemokines has been considered to reduce the negative impact of the TME in the immune-system control of CNS tumors (18–20).
CNS inflammation is another relevant factor to be considered in the context of immunotherapy for CNS tumors. Recent insights into neuro-inflammation indicate that immunocells represent a cell component critical in regulating CNS inflammation. In particular, IL1β, besides its function as key element in inflammation, has a pivotal role in inducing a slow glioma-initiating cell (GIC) transition into a mesenchymal (MES) state (reactive-astrocyte-like cell state), influencing the tumor response to therapy, as well as the development of resistant tumor clones with a peculiar DNA methylation profile (21).
Another relevant factor to be considered in developing an effective immunotherapy for CNS tumor is the strong exhaustion profile induced via continuous TCR/CAR signaling and observed in tumor-infiltrating lymphocytes (TIL) from patients with brain neoplasia, associated to a significant reduction of IL2, IFN-γ and TNFα production (22, 23). In GBM patients, PD-1, LAG-3, TIGIT, and CD39 were found highly expressed on CD8+ TILs (23), supporting the reason to deeply investigate the interplay between PD-1 receptor and its ligand PD-L1 in CNS tumors. In particular, PDL-1 was not only described to be expressed on GBM cells, promoting from one end T cell inhibition and to the other end the invasion of tumor cells in the brain tissue (24, 25) but also on TAMs (26), heightening their immunosuppressive role in the brain TME (27).
These evidences led to the clinical exploration of the use of monoclonal antagonist antibodies directed towards PD1 and PD-L1, with clinical trials enrolling patients with recurrent GBM (24) (Table 1).
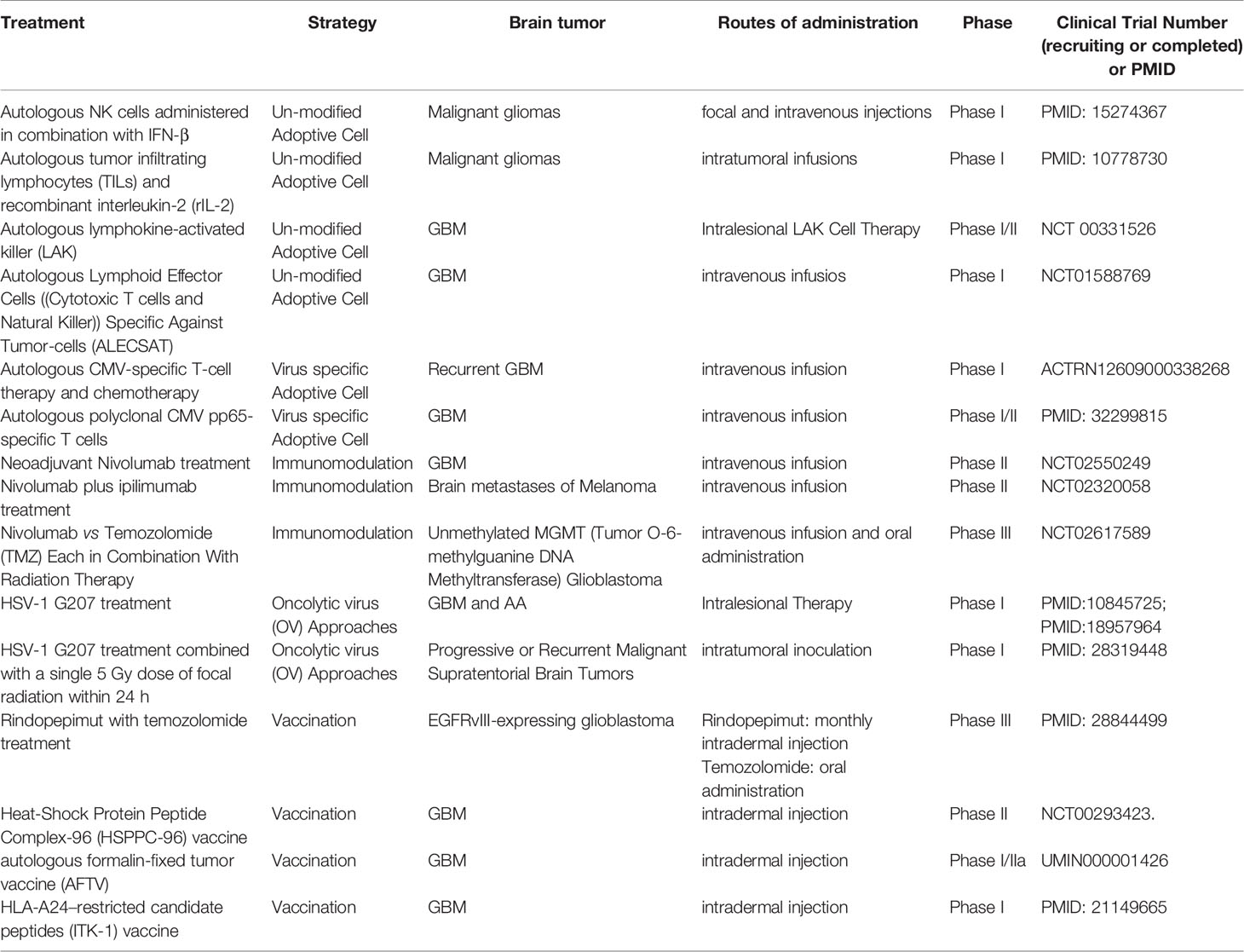
Table 1 Summary of recruiting or completed clinical trials covering brain tumor immuno-therapeutic approaches.
Finally, encouraging results were observed in CNS neoplasia treated with Adoptive Cell Transfer (ACT), of both un-modified or gene-modified immune cells, as well as tumor associated vaccines and oncolytic virus (Figure 1).
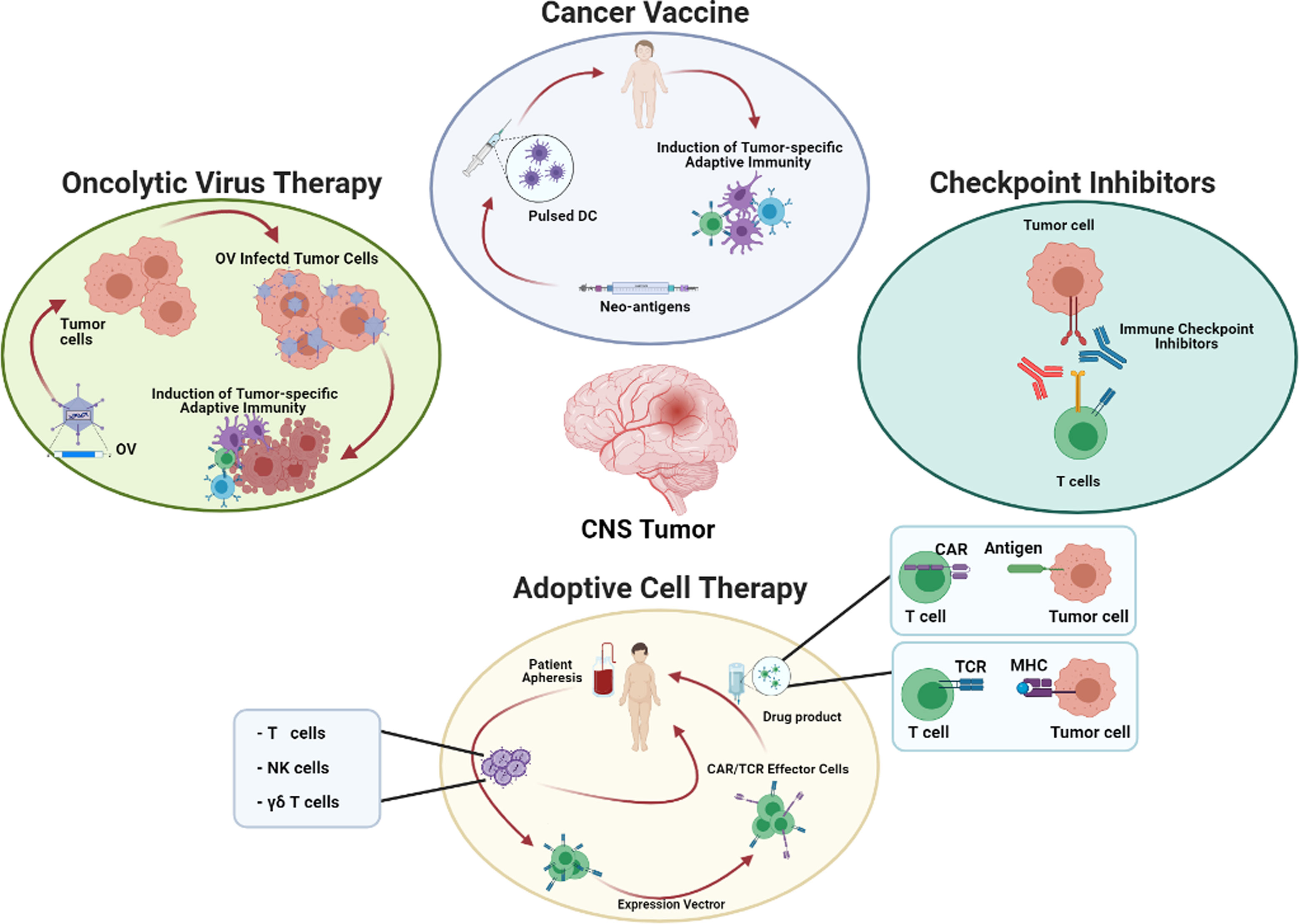
Figure 1 Potential strategies for the treatment of brain tumors. The cartoon summarizes the main immunotherapeutic approaches currently used in clinical trials, subdivided in four major categories: 1) Cancer Vaccine based on Dendritic Cells (DC) pulsed with neoantigens or electropored/infected to transfer neo-antigen genes; 2) Checkpoint Inhibitors based on the use of antibody blocking relevant exhaustion pathway in T cells; 3) Adoptive Cell Therapy based on genetic modification of effector cells with expression vector for TCR or CAR; 4) Oncolytic Virus Therapy based on tumor cell infection, and the consequent activation of adaptive immunity. DC, Dendritic Cells; CAR, Chimeric Antigen Receptor; TCR, T Cell Receptor; MHC, Major histocompatibility complex; NK, Natural Killer cells; OV, Oncolytic virus.
In the present review, we analyze several aspects that limit efficacy of adoptive cell therapy in patients with CNS tumors and present strategies to overcome these hurdles.
Non-Engineered Immune Cell Approaches
Several groups proved that CNS tumors can be targeted by conventional α/β T, NK and γ/δ T cells, although their activity is significantly modulated by the TME.
NK cells play a relevant role in immunity against tumors, without requiring antigen priming or HLA restriction for displaying their activity (28). These advantages are particularly relevant for CNS cancers where the antigenic landscape is extremely heterogeneous, limiting the application of adoptive T-cells. As previously mentioned, CNS tumor cells often downregulate self-markers like HLA class I, allowing them to evade T-cell mediated lysis; however, this renders them potentially more prone to lysis by NK cells. GBM cells do express several activator ligand for NK receptors, as for example MHC class I polypeptide-related sequence A (MICA) (29) ligand for NKG2D+ present on NK cells, as well as CD155 and CD112 recognized by TIGIT and CD96 receptors (30). Indeed, it has been proved that NK cells exert lytic activity toward Medulloblastoma (MB) and GBM cell lines, both in in vitro (31–33) and in vivo models (31, 34, 35). Nevertheless, NK cell activity needs to be optimized in order to overcome inhibition exerted by tumor intrinsic and TME-associated factors. In particular, a pivotal trial (Table 1) has been conducted to test safety and efficacy of autologous NK cells infused in combination with IFN-β to improve NK-cell-mediated cytotoxicity (36). Four weeks after infusion, this led to either stable disease or a measurable response without severe toxicity. These results are encouraging, although no complete response was achieved. For this reason, new strategies were considered, both in the pre-clinical and the clinical setting, to improve NK-cell mediated antitumor activity (31, 37), including their genetic modification by CAR (38, 39). Unlike NK cells, cell therapies with autologous TILs are HLA-restricted, and are based on the tumor recognition toward the T-cell receptor (TCR). TILs specific for somatic mutations have demonstrated significant clinical responses in both primary and metastatic cancers (40–42). Indeed, TILs, locally infused through an Ommaya reservoir and combined with IL2, has been investigated in glioma patients (43), resulting in no significant (Grade 3 or 4) complications, the best response being represented by partial response in three out of six patients.
Although these studies provided evidence that non-engineered immune cells may traffic to CNS and exert measurable antitumor activity, technical issues, mainly associated to difficulties to isolate and expand TILs from tiny fractions of primary brain tumors, have hampered the wide clinical application for this approach in CNS tumors.
Some groups reported that antigens and DNA from human cytomegalovirus (CMV) can be detected in GBM tissues but not in surrounding healthy tissue (44–46). This observation provided the rationale for hypothesizing alternative virus-specific ACT approaches for CNS tumors. However, the clinical relevance of this evidence is still controversial (47, 48). Indeed, it was also proved that the frequency of pp65-specific T cells in CMV-seropositive GBM patients was decreased in comparison to healthy donors (49). Although CMV is not classified as an oncogenic virus, its role in modulation of cell proliferation, angiogenesis, and immune evasion is well known (50). In the context of brain neoplasia, human CMV increases glioblastoma neurosphere proliferation and p-STAT3 levels, these findings suggested the existence of an association between CMV infection and STAT3-dependend modulation in glioma formation/progression, together with tumor suppressor mutations (51). Consistent with these findings, CMV seropositivity correlates with worse prognosis in GBM patients (52). This discovery provides the basis of viral antigen targeting immune therapies in GBM. In this context, besides the incidental observation in one patient of a robust CMV pp65-specific CD8+ cell response after injection of autologous dendritic cells pulsed with an autologous tumor lysate (53), the use of CMV-specific T-cell therapy did not show a significant efficacy in a Phase I pivotal study for GBM patients (54), or in a larger cohort of patients treated at MD Anderson Cancer Center (55).
These preliminary clinical data support the hypothesis that the development of effective immunotherapeutic strategies for CNS tumors requires a robust understanding of factors regulating the activity of the effector cells in the CNS tumor lesions. Several groups proved that un-modified immune cells therapy is significantly potentiated when administered in concomitance to hematopoietic stem and progenitor cells (HPSC). Mitchel’s group (56, 57) elegantly proved, in MB and GBM animal models, that transfer of HSPCs (in particular, CCR2+ HSCs) (57) with concomitant cell therapy approach led to the production of activated Dendric Cells (DCs) (CD86+CD11c+MHC-II+ cells). These intratumoral DCs largely replaced abundant host myeloid-derived suppressor cells. This mechanism was strictly related to the T-cell-released IFNγ that leads to the differentiation of HSPCs into DCs, activation of T cells and rejection of intracranial tumors (56). In line with these findings, it should also be mentioned that macromoleculs such cytokines, can reprogram the TME, but systemically delivered cytokines have limited access to the CNS.
Engineering T Cells To Improve Adoptive Cell Therapy in CNS Neoplasia
RNA-modified T cells have been considered in order to deliver cytokines directly into brain tumors to bypass the hurdles to access the CNS. In a GBM murine model, T cells modified with GM-CSF RNA delivered this cytokine into brain tumors, significantly prolonging overall survival of the animals (18). In particular, GM-CSF recruited DCs in to the tumors, induced their differentiation and maturation, leading to activation of TILs (58).
T Cell Receptor (TCR) Redirected T Cells to Target CNS Tumor Cells
TCR-based adoptive T-cell therapies allow the genetic redirection of T cells on specific tumor targets in a HLA-restricted fashion (59). TCR-based ACT has produced preliminary promising clinical results in extracranial solid tumor (60, 61) and in acute leukemia (62), whereas in brain tumors, the clinical field is still largely unexplored. At the preclinical level, several tumor antigens have been evaluated for TCR approaches in CNS neoplasms, also considering the evidence that tumor-associated antigen (TAA)-reactive T-cells could be detected in patients with glioma (63), suggesting that in this type of cancer the intratumor DCs can process TAA, leading to the emergence of tumor-specific T cells. Between the different categories of TAAs (64), cancer/testis antigens (CTA) are overexpressed in most human cancers due to promoter demethylation in tumor elements (65, 66), whereas their expression in healthy tissues is restricted to germ cells that, lacking the major histocompatibility complex (MHC), are not targeted by T cells.
Among the members of the CTA family, PRAME (antigen preferentially expressed in melanoma) is expressed in leukaemia (67), some solid tumors (68), with a restricted expression in healthy tissues (69). In brain tumors, the CTA PRAME is detectable in about 80% of MB tissues, independently from the molecular and histopathologic subgroups (70–72), with its high expression being correlated with patient dismal overall survival (72). We recently showed that PRAME-TCR T cells are capable of killing MB cells, in vitro as well as in murine xenograft in vivo models (72), representing an innovative, effective strategy for patients with MB, in the absence of significant toxicity.
NY-ESO-1 (New York oesophageal squamous cell carcinoma 1) is a CTA expressed in ovarian cancer (22.6%), oesophageal cancer (25.3%) and lung cancer (28.6%) (73). Discordant data were reported for NY-ESO-1 expression in glioma; most cases, were present at a very low level (63, 74), while the DNA‐demethylating agent, as 5‐aza‐2′‐deoxycytidine, markedly reactivated its expression in GBM but not in normal human cells (74). For this reason, demethylating agents have been administered in combination with ACT approaches targeting NY-ESO-1 (75). These preclinical studies led to a clinical investigation of the administration of T cells expanded ex vivo through the stimulation with APCs (represented by CD4+ T cells exposed to DNA-demethylating agent to induce the expression of high level of CTA). Among the 25 GBM patients enrolled in the Phase I clinical trial (NCT01588769), tumor regression was observed in three patients (76).
Another category of relevant TAA is represented by neoantigens generated by genetic mutation in cancer cells. One example is represented by H3.3 K27M (lysine 27 to methionine substitution in histone H3) mutation, present in 70% of DIPG patients and associated to poor overall survival (77, 78). HLA-A*02-restricted epitope from H3.3 K27M mutated protein was used to generate tumor specific T cell clone with high affinity for the selected peptide. From this T cell clone, TCR α- and β-chains were sequenced to generate H3.3K27M specific TCR, that transferred in polyclonal T cells was able to specifically redirect effector gene modified T cells toward H3.3K27M+ glioma cells in vitro, but also when adoptively transferred into intracranial glioma xenografts mice (79).
Tumor-specific-TCR engineered T cells represent an interesting approach, allowing to target proteins with a wide and specific expression in tumor cells even when their location is subcellular. This feature enables the ability to largely broaden the range of the targetable antigens, in regard to those suitable to be recognized by standard antibody-based immune-strategy. High affinity/avidity TCRs are described to recognize target cells even at low epitope densities (80). Moreover, considering the high cellular and molecular heterogeneity in CNS tumors (81), we could also take into consideration the development of TCRs targeting multiple antigens to avoid the rapid subclone tumor selection due to the targeting of a single peptide. Regardless of these great features and versatility, the efficacy of TCR T-cells is strictly related to the HLA-peptide presentation, which is the major reason for treatment failure in those tumors characterized by HLA down-regulation (82, 83). To overcome this issue, we have recently showed that IFN-γ treatment can re-establish HLA expression on brain tumor cells (72).
CAR T-Cell Approaches in CNS Tumors
T cells genetically modified with CAR emerged in recent years as a promising immunotherapeutic approach, which could mediate a non-MHC-restricted anti-tumor response (84). CARs are artificial receptors composed of a region targeting a specific antigen linked, through an intracytoplasmic domain, to the T-cell activation domain CD3zeta chain (first-generation CAR). The target specific region can comprise a single-chain variable fragment (scFv) obtained from a specific monoclonal antibody, as well as a receptor domain binding a specific ligand expressed on tumor cells.
The intracytoplasmic domain can be enriched by either one or two costimulatory molecules (i.e. CD28, OX40, 4-1BB, or other) to generate second- or third-generation CARs, respectively (85). Clinical trials based on autologous CAR T-cells achieved unprecedented results for the treatment of haematological neoplasia. Two CD19-specific CAR T-cell products, namely Kymriah® (Novartis) and Yescarta® (Kite Pharma), have been recently approved by the US Regulatory Agency Food and Drug Administration and subsequently by the European Medicines Agency for treating B cell ALL and diffuse large B-cell lymphoma (DLBCL), respectively (86). The therapeutic efficacy of CAR T-cells beyond hematopoietic cancers is less clear to date, there is limited documented information on antitumor activity against solid neoplasms. Several reports on CAR.CD19 T cells have proven that the genetically modified cells can cross the BBB (87, 88). A patient with primary refractory DLBCL involving the brain parenchyma who achieved complete remission after CAR.CD19 T-cell infusion, in the absence of cytokine release syndrome or neurotoxic effects, provides strong support that CAR T cells have the capacity to penetrate the CNS (89). Moreover, in the context of CNS, intrathecal and intraventricular administration of CAR T-cells was tested in patients with GBM. The treatment was well tolerated, without cytokine release syndrome or severe neurotoxicity (90–92).
Different approaches with CAR T-cells have been evaluated for the treatment of patients with GBM and/or MB, also in phase I/II clinical trials, by targeting one or more of the following targets (Table 2):
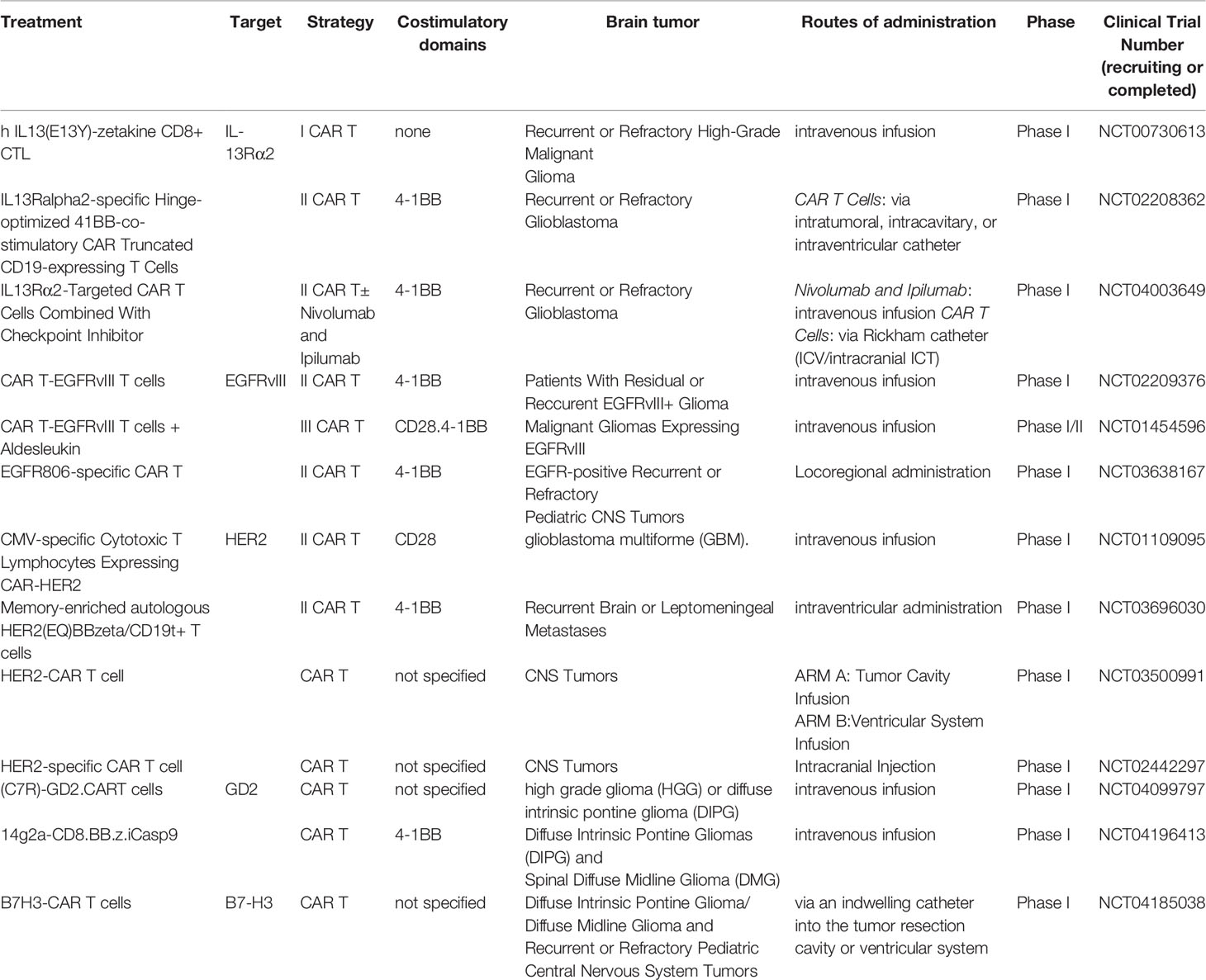
Table 2 Summary of recruiting or completed clinical trials related to the use of CAR-T cells in the treatment of brain tumors.
Interleukin-13 receptor alpha2 (IL-13Rα2) is over-expressed in up to 75% of the patients with GBM but not in normal brain (2, 93, 94). Both first and second-generation CARs, have been successfully used to target IL-13Rα2 (90, 92). This CAR recognizes with high affinity IL13Rα2 via a membrane-tethered IL-13 ligand and starts cytolytic killing (95). Moreover, targeting of IL-13Rα2 positive tumors by CAR T cell approach has been also explored in Phase I clinical trial (90, 92), proving the safety and feasibility of intracranial administration of IL-13Rα2-CAR T cells, administered toward multiple routes of infusion. These trials, pioneers of the field, were extremely relevant since underlying that in the majority of the cases, a patient’s response (whereas partial and transient) was observed in the absence of serious adverse events (90). In the path to optimize the approach and reach more impact on patient outcome, T cell population enriched in central memory cells (91) were considered during the manufacturing process as well as the CAR design was optimized by incorporating 4-1BB (CD137) as a costimulatory domain (96), to achieve a prolonged in vivo survival.
Finally, an innovative and intriguing approach to enhance CAR. IL-13Rα2 T cell antitumor activity toward self-renewing GBM stem cells (GSCs), has been recently discovered by the application of whole-genome clustered randomly interspersed short palindromic repeats (CRISPR)-knockout screen applied to both CAR T cells and GSCs (97). In particular, TLE4 or IKZF2 gene targeting were associated to an increased and long-term efficacy of CAR T cells in this model, with a significant reduction in exhaustion signal of the T cells, whereas knockout of RELA or NPLOC4 GSCs were associated to the increased responsiveness to CAR T cell control (97). Confirmation papers are highly desirable to assess whether the same gene editing could potentially optimize the activity of CAR T lymphocytes characterized by antigenic specificity other than IL-13Rα2, in a CNS tumor context other than GSC.
Epidermal growth factor receptor (EGFR/ErbB1/HER1) belongs to the tyrosine kinase receptor family, also including ErbB2, ErbB3, and ErbB4 (98). Overexpression of EGFR was described in many solid tumor types and associated with cancer cell resistance to chemotherapy (99, 100). In GBM, EGFR amplification is mostly associated with gene rearrangements associated to deletion of particular exons, or portions of exon, and leading to the generation of EGFR variants, that include deletion at N-terminal regional (EGFRvI), deletion at exons 14-15 (EGFRvII), deletion at exons 2–7 (EGFRvIII), deletion at exons 25–27 (EGFRvIV), deletion at exons 25–28 (EGFRvV). The EGFR-encoding gene is overexpressed in almost 50% of GBM patients, 25–64% of which present EGFRvIII (101, 102), making this oncogenic protein an optimal therapeutic target for CAR T cells (103). In a Phase I study (NCT02209376) conducted at the University of Pennsylvania and California (USA), 10 adult patients (45–76 years) affected by EGFRvIII-expressing recurrent GBM were treated with CAR. EGFRvIII T cells, incorporating 4-1BB as costimulatory domain (104). Patients received intravenous infusion of the product, without evidence of EGFR-directed “off-tumor, on-target” toxicity or systemic CRS. However, magnetic resonance imaging (MRI) show the lack of a marked tumor regression in the enrolled patients. Only one patient had residual stable disease for 18 months (104). This study underlined the difficulties of the clinical translation of EGFRvIII targeting, due to the intra-tumoral heterogeneity of the antigen expression, leading to clonal selection of the antigen-negative tumor cells. The antigen-escape phenomenon is mostly observed in highly heterogeneous tumors when a single antigen is targeted. This outcome seems to be related neither to the vector platform nor to the CAR design, since similar results have been observed in a Phase I pilot trial on 18 patients with GBM treated using autologous T cells genetically modified with a lentivirus carrying CAR.EGFRvIII/CD28.4.1bb (105). To optimize the CAR T cell therapy, a combinatory approach has been suggested. In particular, PD-1 checkpoint blockade achieved by either antibody blocking (106) or CRISPR-Cas9 (107–109) gene editing has been used in combination with CAR.EGFRvIII T-cells, both in vitro and in vivo models. To minimize “on-target, off-tumor” toxicity, Ravanpay et al. (110), proposed a second-generation CAR.EGFR designed using the scFv derived from the monoclonal antibody mAb806, specific for EGFR epitope present also in the EGFRvIII. Therefore, the construct binds to EGFR subjected to gene amplification, with no recognition of wild-type EGFR expressed on astrocytes (110). Recently, a Phase I study testing locoregional infusion of CAR.EGFR806-CAR T-cells has been opened to enrol paediatric patients with EGFR-positive recurrent/refractory CNS tumors (NCT03638167).
Choi et al. (111) proposed a CAR.EGFRvIII T-cells secreting EGFR-bispecific T-cell engager (BiTE) to circumvent antigen escape. In mouse models of GBM, they showed that CART.BiTE cells were able to secrete BiTEs locally in the brain tumor, redirecting non-specific bystander T cells against tumors which eliminated efficacy heterogeneous tumors more efficiently, compared to the use of a conventional CAR T cell approach (111). This group elegantly combined two therapeutic tools (CAR and BiTE), which are typically considered competitive technologies, which instead could be applied in a synergic manner.
The human epidermal growth factor receptor 2 (HER2) has been described to be involved in cancer cell growth and metastasis, thus representing an attractive target for immunotherapy (112). The primary mechanism of HER2 activation in these cancers is the gene amplification leading to the overexpression of the HER2 on the cell membrane. The second mechanism recognized to drive the protein activation is related to the occurrence of activating mutations (113). HER2 is overexpressed in approximately 51% Wilms tumor, 44% bladder cancer, 26% pancreatic carcinoma and 25% breast carcinoma (114). ErbB2/HER2 mutation was also detected in 8–41% GBMs (115). Primary GBM stem cells are targeted by CAR.HER2 T, inducing regression of patient derived tumors in the animal model (116). In open-label Phase I dose-escalation trial, Ahmed et al. (117) used virus (CMV, Epstein–Barr Virus, and Adenovirus) specific T cells (VSTs), expressing a CAR.HER2.CD28.ζ construct to treat 16 HER2-positive relapsed/refractory GBMs. CAR-modified VSTs show persistence after the infusion for up to 12 months, using molecular DNA evaluation. Whereas the majority of treated patients relapsed early after the treatment, the overall response rate was of 50%, with the median OS of 11.1 months from the CAR VSTs infusion (117).
Lastly, Zhang et al., in a preclinical model targeting HER2, proposed a different platform to treat GBM patients, based on NK-92 cell line transduced with CAR.HER2.CD28.ζ construct. In the in vitro and in vivo models, they show a potent and specific lytic activity of the CAR NK-92 cell line toward GBM (118).
Disialoganglioside GD2 is a ganglioside (119) of unknown function, with limited expression in normal tissues and high expression in to neuroblastoma (120), melanoma (121), some sarcomas (122) and H3-K27M-mutated diffuse midline gliomas (DMG) (123). Intravenous anti-GD2 antibodies have already become the standard of care for high risk neuroblastoma patients (124, 125). Although it has been reported that a normal brain express low level of GD2 (126), in pre-clinical models (127) and clinical trials exploring the safety and efficacy of GD2-CAR T-cell whose scFv being derived from 14g2a monoclonal antibody (including our academic clinical trial NCT03373097), no cases of significant neurotoxicity were reported (128, 129). Mount et al. showed, that intravenously administered CAR.GD2 T-cells are able to cross the BBB and clear patient-derived H3K27M-mutated DMG tumors (123). Shum et al. showed that CAR.GD2 T cells also engineered to express IL-7 receptor (GD2-CAR.C7R) promotes CAR T cells persistence, proliferation and anti-tumor activity in an orthotopic GBM xenograft model (130). These encouraging studies have fastened the clinical translation of CAR.GD2 T cells in patients with brain tumors (NCT04099797, NCT04196413).
B7-H3/CD276 is a type I transmembrane protein belonging to the B7/CD28 superfamily, found in a wide variety of normal tissues and cells, including T and B cells, endothelial cells, resting fibroblasts, osteoblasts. Recently, several studies have proved the overexpression of CD276 on a variety of TME cells, as well as hematologic and solid tumors, including leukemia (131), prostate cancer (132), melanoma (133), neuroblastoma (134), osteosarcoma (135), and CNS tumors (136).
Many pediatric CNS tumors express B7-H3; in particular, high B7-H3 expression has been found in the following tumors: atypical teratoid/rhabdoid tumors (ATRT), ependymomas (all grades), MB, CNS embryonal tumors, choroid plexus tumors (CPTs), meningioma and craniopharyngiomas (136, 137).
In MB, WNT subtype expresses the highest B7-H3, while SHH subtype show the lowest one (136). However, low-grade gliomas and germ cell tumors show a negligible difference of B7-H3 expression respect to normal brain (136).
Majzner et al., recently reported that CAR.B7-H3 T cells incorporating 4-1BB as costimulatory domain, showed a potent efficacy in in vivo xenograft models, including MB (138). Loco-regional delivery of CAR.B7-H3 T cells also exerted significant activity over the systemic infusion for the treatment of CNS malignancies (139). In this study, CAR.B7-H3 T-cells used for intra-cerebroventricular or intra-tumoral administration, mediated a significant antitumor effects in the brain ATRT xenograft mouse model, with a reduced toxicity relative a significant diminished systemic levels of inflammatory cytokines as opposed to intravenously administered CAR T cells (139). The Seattle Children’s Hospital has started a clinical trial in 2019 based on CAR.B7-H3 T Cell based locoregional infusion in DIPG/DMG patients and recurrent/refractory paediatric CNS tumors (NCT04185038).
CD70 is a type II transmembrane protein, ligand for CD27. While activated T/B lymphocytes and a subset of mature dendritic cells express high level of CD70 expression, several hematologic and solid tumors, including GBM, constitutively overexpress CD70 (140–142). Indeed, CD70 expression is an independent predictor of poor OS for patients with low-grade gliomas (LGG) and GBM, correlating with chemokine-mediated immune modulation, tumor aggressiveness and immunosuppression via tumor-associated M2 macrophage recruitment/activation in GBM (142). Linchun et al., reported that CAR.CD70 T cells potently induce lytic activity against CD70+ gliomas, both in vitro and in vivo models (143). In order to increase the intra-tumoral T-cell migration, the same group integrated CXCR1 and CXCR2 chemokine receptors into the CD70-CAR approach (144). Moreover, to overcome the intra-patient variability, for which antigen heterogeneous expression on the tumor cells may result in a non-complete eradication of the tumor, and inter-patient variability, for which tumor cells from different patients may express different antigen patterns (145), bi-specific [CD70 and B7-H3 (146) or HER2 and IL-13Rα2 (147)], or even trivalent (targeting at the same time HER2, IL1-3Rα2 and EphA2) (148) CAR approaches have been developed, also in combination with DNA-demethylating agent azacytidine to diminish the occurrence of relapse after CAR T-cell administration in the animal xenograft models of Group 3 MB and ependymoma, associated to antigenic escape secondary to epigenetic silencing (149).
CD-133 also called Prominin-1, is a pentaspan membrane glycoprotein. Several human malignancies show OS inversely correlating with CD133 expression (150). Indeed, CD133+ cancer stem cells are known markers of chemo- and radio-resistance in GBM (151). Recently, Vora et al. provided evidences that, in a preclinical model, CAR.CD133 T cells, with CD28 costimulation, represent a therapeutic option to target self-renewing, chemo-radio resistant CD133+ brain tumor-initiating cells (151).
Chondroitin sulfate proteoglycan 4 (CSPG4) is a type I transmembrane protein with a central role in tumor progression and metastasis (152). Many types of solid tumors overexpress CSPG4, barely detectable in normal tissues (153–155). In preclinical models, CSPG4-directed CAR T-cell therapy efficiently inhibits GBM-derived neurospheres growth in both in vitro, as well as in vivo xenograft patient-derived GBM orthotopic mouse model (156). Clinical studies need to be conducted to study the safety and real therapeutic activity of the proposed innovative CAR approaches.
Immunomodulation
Approaches to optimize endogenous T-cell immune responses, including Immune Checkpoint Inibitors (ICIs) (157, 158), oncolytic viruses (159), and tumor neoantigen vaccines (160) have shown evidence of bioactivity against brain tumors, also in clinical trials (Figure 1 and Table 1).
Immune Checkpoint Inhibitors (ICIs)
Although immune checkpoint inhibition using antibodies against CTLA-4, PD-1 or PD-L1 has shown considerable tumor regression in several solid tumors, CNS tumors remain refractory to these treatments (161). ICI administration in melanoma patients with CNS metastases has shown a durable anti‐tumor response (162). This positive outcome could be explained considering that melanoma has a high tumor mutational burden (TMB) and that TMB has been clearly correlated to ICI response in treated patients. In line with these observations, also a subset of GBMs characterized by high TMB, show a durable remission after ICIs (157, 158). However, the majority of brain tumors are characterized by low TMB, this last explaining a low overall responsiveness to ICIs (163). It has been suggested that radiation and chemotherapy therapies in newly diagnosed CNS tumors could increase the TMB and therefore, favor the response to ICIs (164), as also proved in an animal model (165). However, recent phase III trials (CheckMate-143 and CheckMate-498) failed to prove the OS improvement in GBM patients receiving PD-1 monotherapy (166). Nevertheless, these studies remain important as they confirm that CNS tumors are accessible to the immune system, and were associated with some clinical benefit, including the induction of the so-called “abscopal effect”. This phenomenon is linked to the recognition and elimination of tumor cells outside the irradiation field that are induced by the immune system when ICI are administered (167). Thus, further clinical studies are required to define the optimal application of ICI in GBM patients, considering the peculiarity of the target tumors. In particular, several reasons could explain this unsatisfactory outcome, including the lack of prominent T cell infiltrates especially observed in to mismatch repair-deficient gliomas (168), the paucity of DC in the brain TME playing a key role in the recruitment of T cells (57, 169–172), and the reduced ability of monoclonal antibodies to cross effectively the BBB (173, 174). Several attempts are under evaluation to circumvent this latter aspect, for example, by the delivery of ICI by nanoparticles (175), to deliver small interfering RNAs (siRNAs) targeting EGFR and PD-L1 (176). In a combinatory point of view, it has been suggested that brain delivery of nanoparticle can be potentiated by low-dose radiation that altering the tumor structure allows for the enhanced nanoparticles delivery in tumor-associated macrophage-dependent fashion (177). In regard to the issue of a reduced contribution of DC in the brain TME, the role of APC in controlling response after ICI is strongly suggested by several evidences. Indeed, as in the model of skin carcinoma in which it was proved that T cell response to ICI is associated to the expansion of T cells that have entered de-novo the tumor instead of the re-activation of pre-existing highly-exhausted TIL (178, 179). It could also be plausible that the clinical response to ICI may be associated to the newly recruitment of T cells to the tumor site that are induced by the signal generated from activated APCs. Further investigations to study the clinical efficacy of combinatory therapy between CARs and ICI are ongoing, including a clinical trial in GBM patients (NCT03726515) that will evaluate the combination of CAR.EGFRvIII T cells in association with pembrolizumab (PD-1 inhibitor).
Oncolytic Virus (OV) Approaches
OV with their ability to selectively replicate in tumor cells, has been described as potent antitumor approach (3). GBM was shown to be responsive to OVs in preclinical models and clinical trial (180–182). To overcome the viral exclusion exerted by the BBB, the OVs can be intratumorally injected, or the approach can be based on parvovirus that is known to have good CNS penetration (183). Pivotal trials have established the safety of OVs in GBM, but their efficacy has been modest to date (184). Innovative approaches are focusing on targeted OVs and combinatory therapy. GBM stem cells utilize autophagy modulation as main resistance mechanism to OVs (185). Indeed, glioma oncolysis is significantly reduced when Beclin-I (autophagy inducer) is downregulated in tumor cells, and tamoxifen drug, upon activation of apoptosis in OV infected cells via BAX/PUMA pathway, is able to re-establish the tumor sensitivity to oncolytic infection (185). Oncolytic HSV-1 (oHSV) G207 has been the most comprehensively investigated virus in human brain tumors (186–190). Phase I studies in adult patients with recurrent GBM provided data of safety for intratumoral injections of single (186) or double doses of G207 dosages (187) also combined with a single 5 Gy dose of focal radiation (188). Based on the safety observed in the trials on adult patients, a paediatric clinical trial (3–18 years old) in recurrent/progressive supratentorial malignant brain tumors has been opened (189).
The oncolytic adenovirus DNX-2401 (Delta-24-RGD) has been infused by single intratumoral injection in patients with GBM. Amongst the 37 treated patients, 20% experienced long term survival (>3 years from treatment), with three patients showing a significant tumor reduction (≥95%). These relevant clinical results were also corroborated by the observation of DNX-2401 replication and spreading within the tumor (191). The same oncolytic adenovirus DNX-2401 has been also used to treat pediatric subjects with DIPG, showing improvement in OS and quality of life (192). Lastly, Rat H-1 parvovirus (H-1PV) in recurrent glioblastoma patients has shown the ability to trigger specific T-cell responses (193).
Vaccine-Based Approaches
The immune system can eradicate malignant cells via recognition of TAAs. Ideal TAAs have an expression restricted to tumor cells, being negligible in surrounding healthy tissue. In CNS tumors, tumor antigens can be categorized into three main classes; (i) antigens with an aberrant expression; (ii) mutated oncogenes and (iii) neo-antigens. One valid example is represented by Rindopepimut, an EGFRvIII-targeted vaccine, which tested in a clinical trial, failed to show survival improvement (194). Several other vaccines are in development with several promising candidates (195).
HSPPC-96, a vaccine based on heat-shock tumor peptides, has been applied to recurrent GBM in a phase II trial, showing 6-month survival rate of 90% (196). A different vaccination approach has been developed for AFTV, which is based on formalin-fixed tumor sample fragments, infused intradermal in GBM patients. This approach allows to reach a 3-year survival rate in 38% of the treated patients (197).
Autologous DCs have been also considered in the vaccination prospective. As previously underlined, CNS TME is characterized by a marked absence of infiltrating APC. For this reason, adoptive transfer of T-cells alone could not be substantially beneficial for patients with CNS tumors. Several approaches aimed at overcoming this CNS tumor limitation have been described. Current evidence deriving from phase I and II trials suggests that DC vaccination may activate an immune response against CNS tumors (198–202). Although more than 500 GBM patients have been treated with DC vaccination and clinical results have been encouraging, there is not robust evidence of clinical efficacy, because of either the non-controlled design of the studies or the low patient numbers. One attempt has been carried out by German clinical centers participating to the randomized controlled phase II trial GlioVax (mature DCs loaded with tumor lysate) (203). This trial has been designed to enroll 136 GBM patients into two cohorts (1): radio/temozolomide “gold standard therapy” and (2) DC vaccination plus standard therapy. Data coming from this trial will clarify the impact of DC vaccination on patient outcome.
Combination Approaches
It is increasingly evident to researchers that a combination of different therapeutic approaches is needed to efficiently treat patients with high immunosuppressive brain tumors. Saha et al., showed that the triple combination of an OV expressing IL-12 with two immune checkpoint inhibitors targeting PD1 and CTLA-4, can eradicate glioma in two mouse models (204), reducing Tregs, increasing CD8+ T cells and inducing M1-like polarization in TAMs. Moreover, in a in vivo model of Glioma, Tang et al., proposed a complex therapy based on a combinatory approach of an adoptive cell transfer of tumor-specific CD8+ cells (to increase adoptive T cell targeting of the tumor), rapamycin (to enhance antigen presentation by DCs), celecoxib (to modulate the TME inflammation and immunosuppression), and intratumoral injection of IL15Rα-IL15-armed oncolytic poxviruses (to boost T cell activation) (205). In tumor-bearing mice, the combinatory approach was safe and able to promote longer survival of tumor-specific cytotoxic T cells, in respect to the administration of only CD8+ T cells (205). Pivotal clinical trials need to be designed and carried out to corroborate the safety and efficacy of the described combinatory approach.
Conclusions
The recent advances in immunotherapies, coupled with the essential insights in the understanding of neuro-immunology, are creating innovative opportunities to treat CNS cancer. To achieve durable antitumor effect, it is likely that combinatorial regimens will be required and based on 1) increase of CNS delivery; 2) “multi-valent” tumor targeting; 3) targeting both tumor cells and the CNS TME. The road to identify an effective cure for brain tumors still appears long and filled with pitfalls, but the deep knowledge of these tumors and their microenvironment will lead in the near future to increasingly personalized treatment, possibly changing the natural history of these diseases.
Author Contributions
CQ rearranged the final version. BDA coordinated the coauthor’s contribution and rearranged the final version. All authors contributed to the article and approved the submitted version.
Funding
We would like to thank all agencies supporting our work on immunotherapy, in particular the Italian Health Ministry [for GR-2013-02359212, (CQ), GR-2016-02364546- (BDA), CAR T RCR-2019-23669115]; AIRC; Ricerca Corrente (CQ, BDA); All the charities that support our research and clinical translation, including “Raffaele Passarelli” Onlus, “Il laboratorio di Chiara,” “Fondazione Heal,” “IRENE” Onlus, “Oscar’s Angels Italia,” and “Martina e la sua luna.”
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
A special mention to Prof. Franco Locatelli, to which we are indebted for the critical revision of the manuscript and his helpful suggestions. We would like to thank Dr. Megan Eckley for the English editing.
References
1. Pollack IF, Agnihotri S, Broniscer A. Childhood Brain Tumors: Current Management, Biological Insights, and Future Directions. J Neurosurg Pediatr (2019) 23:261–73. doi: 10.3171/2018.10.PEDS18377
2. Sharma P, Debinski W. Receptor-Targeted Glial Brain Tumor Therapies. Int J Mol Sci (2018) 19(11):3326. doi: 10.3390/ijms19113326
3. Cripe TP, Chen CY, Denton NL, Haworth KB, Hutzen B, Leddon JL, et al. Pediatric Cancer Gone Viral. Part I: Strategies for Utilizing Oncolytic Herpes Simplex Virus-1 in Children. Mol Ther Oncolytics (2015) 21–9. doi: 10.1038/mto.2015.15
4. Lyon JG, Mokarram N, Saxena T, Bellamkonda RV. Engineering Challenges for Brain Tumor Immunotherapy. Adv Drug Delivery Rev (2017) 114:19–32. doi: 10.1016/j.addr.2017.06.006
5. Liddelow SA. Development of the Choroid Plexus and Blood-CSF Barrier. Front Neurosci (2015) 9:1–32. doi: 10.3389/fnins.2015.00032
6. Engelhardt B, Carare RO, Bechmann I, Flugel A, Laman JD, Weller RO. Vascular, Glial, and Lymphatic Immune Gateways of the Central Nervous System. Acta Neuropathol (2016) 132:317–38. doi: 10.1007/s00401-016-1606-5
7. Schlager C, Korner H, Krueger M, Vidoli S, Haberl M, Mielke D, et al. Effector T-Cell Trafficking Between the Leptomeninges and the Cerebrospinal Fluid. Nature (2016) 530:349–53. doi: 10.1038/nature16939
8. Kipnis J. Multifaceted Interactions Between Adaptive Immunity and the Central Nervous System. Science (2016) 353:766–71. doi: 10.1126/science.aag2638
9. Heimberger AB, Sampson JH. Immunotherapy Coming of Age: What Will It Take to Make It Standard of Care for Glioblastoma? Neuro Oncol (2011) 13:3–13. doi: 10.1093/neuonc/noq169
10. Chandramohan V, Mitchell DA, Johnson LA, Sampson JH, Bigner DD. Antibody, T-Cell and Dendritic Cell Immunotherapy for Malignant Brain Tumors. Future Oncol (2013) 9:977–90. doi: 10.2217/fon.13.47
11. Brown NF, Carter TJ, Ottaviani D, Mulholland P. Harnessing the Immune System in Glioblastoma. Br J Cancer (2018) 119:1171–81. doi: 10.1038/s41416-018-0258-8
12. Razavi SM, Lee KE, Jin BE, Aujla PS, Gholamin S, Li G. Immune Evasion Strategies of Glioblastoma. Front Surg (2016) 3:11. doi: 10.3389/fsurg.2016.00011
13. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. Csf-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat Med (2013) 19:1264–72. doi: 10.1038/nm.3337
14. Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, et al. Analysis of Tumour- and Stroma-Supplied Proteolytic Networks Reveals a Brain-Metastasis-Promoting Role for Cathepsin S. Nat Cell Biol (2014) 16:876–88. doi: 10.1038/ncb3011
15. Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The Role of Human Glioma-Infiltrating Microglia/Macrophages in Mediating Antitumor Immune Responses. Neuro Oncol (2006) 8:261–79. doi: 10.1215/15228517-2006-008
16. Joyce JA, Pollard JW. Microenvironmental Regulation of Metastasis. Nat Rev Cancer (2009) 9:239–52. doi: 10.1038/nrc2618
17. Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer Cell (2017) 31:326–41. doi: 10.1016/j.ccell.2017.02.009
18. Pohl-Guimaraes F, Yang C, Dyson KA, Wildes TJ, Drake J, Huang J, et al. Rna-Modified T Cells Mediate Effective Delivery of Immunomodulatory Cytokines to Brain Tumors. Mol Ther (2019) 27:837–49. doi: 10.1016/j.ymthe.2018.10.007
19. Beug ST, Beauregard CE, Healy C, Sanda T, St-Jean M, Chabot J, et al. Publisher Correction: Smac Mimetics Synergize With Immune Checkpoint Inhibitors to Promote Tumour Immunity Against Glioblastoma. Nat Commun (2018) 9:16231. doi: 10.1038/ncomms16231
20. Tahtinen S, Kaikkonen S, Merisalo-Soikkeli M, Gronberg-Vaha-Koskela S, Kanerva A, Parviainen S, et al. Favorable Alteration of Tumor Microenvironment by Immunomodulatory Cytokines for Efficient T-Cell Therapy in Solid Tumors. PloS One (2015) 10:e0131242. doi: 10.1371/journal.pone.0131242
21. Niklasson M, Bergstrom T, Jarvius M, Sundstrom A, Nyberg F, Haglund C, et al. Mesenchymal Transition and Increased Therapy Resistance of Glioblastoma Cells is Related to Astrocyte Reactivity. J Pathol (2019) 249:295–307. doi: 10.1002/path.5317
22. Mohme M, Schliffke S, Maire CL, Runger A, Glau L, Mende KC, et al. Immunophenotyping of Newly Diagnosed and Recurrent Glioblastoma Defines Distinct Immune Exhaustion Profiles in Peripheral and Tumor-infiltrating Lymphocytes. Clin Cancer Res (2018) 24:4187–200. doi: 10.1158/1078-0432.CCR-17-2617
23. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-Cell Exhaustion Signatures Vary With Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res (2018) 24:4175–86. doi: 10.1158/1078-0432.CCR-17-1846
24. Litak J, Mazurek M, Grochowski C, Kamieniak P, Rolinski J. Pd-L1/Pd-1 Axis in Glioblastoma Multiforme. Int J Mol Sci (2019) 20(21):1–16. doi: 10.3390/ijms20215347
25. Hennequin C, Bossard N, Servagi-Vernat S, Maingon P, Dubois J-B, Datchary J, et al. Ten-Year Survival Results of a Randomized Trial on Irradiation of Internal Mammary Nodes After Mastectomy. Int J Radiat Oncol Biol Phys (2013) 86:860–6. doi: 10.1016/j.ijrobp.2013.03.021
26. Antonios JP, Soto H, Everson RG, Moughon D, Orpilla JR, Shin NP, et al. Immunosuppressive Tumor-Infiltrating Myeloid Cells Mediate Adaptive Immune Resistance Via a PD-1/PD-L1 Mechanism in Glioblastoma. Neuro Oncol (2017) 19:796–807. doi: 10.1093/neuonc/now287
27. Zhu Z, Zhang H, Chen B, Liu X, Zhang S, Gao M. Pd-L1-Mediated Immunosuppression in Glioblastoma Is Associated With the Infiltration and M2-Polarization of Tumor-Associated Macrophages. Front Immunol (2020) 11:588552. doi: 10.3389/fimmu.2020.588552
28. Sivori S, Meazza R, Quintarelli C, Carlomagno S, Della Chiesa M, Falco M, et al. Nk Cell-Based Immunotherapy for Hematological Malignancies. J Clin Med (2019) 8:1–35. doi: 10.3390/jcm8101702
29. Friese MA, Platten M, Lutz SZ, Naumann U, Aulwurm S, Bischof F, et al. MICA/NKG2D-Mediated Immunogene Therapy of Experimental Gliomas. Cancer Res (2003) 63:8996–9006.
30. Gao J, Zheng Q, Xin N, Wang W, Zhao C. CD155, An Onco-Immunologic Molecule in Human Tumors. Cancer Sci (2017) 108:1934–8. doi: 10.1111/cas.13324
31. Powell AB, Yadavilli S, Saunders D, Van Pelt S, Chorvinsky E, Burga RA, et al. Medulloblastoma Rendered Susceptible to NK-Cell Attack by TGFbeta Neutralization. J Transl Med (2019) 17:321. doi: 10.1186/s12967-019-2055-4
32. Castriconi R, Dondero A, Negri F, Bellora F, Nozza P, Carnemolla B, et al. Both CD133+ and CD133- Medulloblastoma Cell Lines Express Ligands for Triggering NK Receptors and Are Susceptible to NK-Mediated Cytotoxicity. Eur J Immunol (2007) 37:3190–6. doi: 10.1002/eji.200737546
33. Fernandez L, Portugal R, Valentin J, Martin R, Maxwell H, Gonzalez-Vicent M, et al. In Vitro Natural Killer Cell Immunotherapy for Medulloblastoma. Front Oncol (2013) 3:94. doi: 10.3389/fonc.2013.00094
34. Friese MA, Wischhusen J, Wick W, Weiler M, Eisele G, Steinle A, et al. RNA Interference Targeting Transforming Growth Factor-Beta Enhances NKG2D-Mediated Antiglioma Immune Response, Inhibits Glioma Cell Migration and Invasiveness, and Abrogates Tumorigenicity In Vivo. Cancer Res (2004) 64:7596–603. doi: 10.1158/0008-5472.CAN-04-1627
35. Alizadeh D, Zhang L, Brown CE, Farrukh O, Jensen MC, Badie B. Induction of Anti-Glioma Natural Killer Cell Response Following Multiple Low-Dose Intracerebral CpG Therapy. Clin Cancer Res (2010) 16:3399–408. doi: 10.1158/1078-0432.CCR-09-3087
36. Ishikawa E, Tsuboi K, Saijo K, Harada H, Takano S, Ohno T. Autologous Natural Killer Cell Therapy for Human Recurrent Malignant Glioma. Anticancer Res (2004) 24:1861–71.
37. Kmiecik J, Zimmer J, Chekenya M. Natural Killer Cells in Intracranial Neoplasms: Presence and Therapeutic Efficacy Against Brain Tumours. J Neurooncol (2014) 116:1–9. doi: 10.1007/s11060-013-1265-5
38. Nakazawa T, Murakami T, Natsume A, Nishimura F, Morimoto T, Matsuda R, et al. Khyg-1 Cells With EGFRvIII-Specific Car Induced a Pseudoprogression-Like Feature in Subcutaneous Tumours Derived From Glioblastoma-Like Cells. Anticancer Res (2020) 40:3231–7. doi: 10.21873/anticanres.14304
39. Murakami T, Nakazawa T, Natsume A, Nishimura F, Nakamura M, Matsuda R, et al. Novel Human Nk Cell Line Carrying Car Targeting Egfrviii Induces Antitumor Effects in Glioblastoma Cells. Anticancer Res (2018) 38:5049–56. doi: 10.21873/anticanres.12824
40. Choi BD, Maus MV, June CH, Sampson JH. Immunotherapy for Glioblastoma: Adoptive T-Cell Strategies. Clin Cancer Res (2019) 25:2042–8. doi: 10.1158/1078-0432.CCR-18-1625
41. Hinrichs CS, Rosenberg SA. Exploiting the Curative Potential of Adoptive T-Cell Therapy for Cancer. Immunol Rev (2014) 257:56–71. doi: 10.1111/imr.12132
42. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
43. Quattrocchi KB, Miller CH, Cush S, Bernard SA, Dull ST, Smith M, et al. Pilot Study of Local Autologous Tumor Infiltrating Lymphocytes for the Treatment of Recurrent Malignant Gliomas. J Neurooncol (1999) 45:141–57. doi: 10.1023/A:1006293606710
44. Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, et al. Human Cytomegalovirus Infection and Expression in Human Malignant Glioma. Cancer Res (2002) 62:3347–50.
45. Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE, et al. Sensitive Detection of Human Cytomegalovirus in Tumors and Peripheral Blood of Patients Diagnosed With Glioblastoma. Neuro Oncol (2008) 10:10–8. doi: 10.1215/15228517-2007-035
46. Lucas KG, Bao L, Bruggeman R, Dunham K, Specht C. The Detection of CMV pp65 and IE1 in Glioblastoma Multiforme. J Neurooncol (2011) 103:231–8. doi: 10.1007/s11060-010-0383-6
47. Lau SK, Chen YY, Chen WG, Diamond DJ, Mamelak AN, Zaia JA, et al. Lack of Association of Cytomegalovirus With Human Brain Tumors. Mod Pathol (2005) 18:838–43. doi: 10.1038/modpathol.3800352
48. Sabatier J, Uro-Coste E, Pommepuy I, Labrousse F, Allart S, Tremoulet M, et al. Detection of Human Cytomegalovirus Genome and Gene Products in Central Nervous System Tumours. Br J Cancer (2005) 92:747–50. doi: 10.1038/sj.bjc.6602339
49. Ghazi A, Ashoori A, Hanley PJ, Brawley VS, Shaffer DR, Kew Y, et al. Generation of Polyclonal CMV-Specific T Cells for the Adoptive Immunotherapy of Glioblastoma. J Immunother (2012) 35:159–68. doi: 10.1097/CJI.0b013e318247642f
50. Naucler CS, Geisler J, Vetvik K. The Emerging Role of Human Cytomegalovirus Infection in Human Carcinogenesis: A Review of Current Evidence and Potential Therapeutic Implications. Oncotarget (2019) 10:4333–47. doi: 10.18632/oncotarget.27016
51. Price RL, Song J, Bingmer K, Kim TH, Yi JY, Nowicki MO, et al. Cytomegalovirus Contributes to Glioblastoma in the Context of Tumor Suppressor Mutations. Cancer Res (2013) 73:3441–50. doi: 10.1158/0008-5472.CAN-12-3846
52. Foster H, Piper K, DePledge L, Li HF, Scanlan J, Jae-Guen Y, et al. Human Cytomegalovirus Seropositivity Is Associated With Decreased Survival in Glioblastoma Patients. Neurooncol Adv (2019) 1:vdz020. doi: 10.1093/noajnl/vdz020
53. Prins RM, Cloughesy TF, Liau LM. Cytomegalovirus Immunity After Vaccination With Autologous Glioblastoma Lysate. N Engl J Med (2008) 359:539–41. doi: 10.1056/NEJMc0804818
54. Schuessler A, Smith C, Beagley L, Boyle GM, Rehan S, Matthews K, et al. Autologous T-cell Therapy for Cytomegalovirus as a Consolidative Treatment for Recurrent Glioblastoma. Cancer Res (2014) 74:3466–76. doi: 10.1158/0008-5472.CAN-14-0296
55. Weathers SP, Penas-Prado M, Pei BL, Ling X, Kassab C, Banerjee P, et al. Glioblastoma-Mediated Immune Dysfunction Limits CMV-Specific T Cells and Therapeutic Responses: Results From a Phase I/II Trial. Clin Cancer Res (2020) 26:3565–77. doi: 10.1158/1078-0432.CCR-20-0176
56. Wildes TJ, Grippin A, Dyson KA, Wummer BM, Damiani DJ, Abraham RS, et al. Cross-Talk Between T Cells and Hematopoietic Stem Cells During Adoptive Cellular Therapy for Malignant Glioma. Clin Cancer Res (2018) 24:3955–66. doi: 10.1158/1078-0432.CCR-17-3061
57. Flores CT, Wildes TJ, Drake JA, Moore GL, Dean BD, Abraham RS, et al. Lin(-)CCR2(+) Hematopoietic Stem and Progenitor Cells Overcome Resistance to PD-1 Blockade. Nat Commun (2018) 9:4313. doi: 10.1038/s41467-018-06182-5
58. Dranoff G. Gm-CSF-Based Cancer Vaccines. Immunol Rev (2002) 188:147–54. doi: 10.1034/j.1600-065X.2002.18813.x
59. Rath JA, Arber C. Engineering Strategies to Enhance TCR-Based Adoptive T Cell Therapy. Cells (2020) 9:1–34. doi: 10.3390/cells9061485
60. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor Regression in Patients With Metastatic Synovial Cell Sarcoma and Melanoma Using Genetically Engineered Lymphocytes Reactive With NY-ESO-1. J Clin Oncol (2011) 29:917–24. doi: 10.1200/JCO.2010.32.2537
61. Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. Ny-ESO-1-Specific TCR-Engineered T Cells Mediate Sustained Antigen-Specific Antitumor Effects in Myeloma. Nat Med (2015) 21:914–21. doi: 10.1038/nm.3910
62. Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, et al. T Cell Receptor Gene Therapy Targeting WT1 Prevents Acute Myeloid Leukemia Relapse Post-Transplant. Nat Med (2019) 25:1064–72. doi: 10.1038/s41591-019-0472-9
63. Liu Z, Poiret T, Persson O, Meng Q, Rane L, Bartek J Jr., et al. Ny-ESO-1- and Survivin-Specific T-Cell Responses in the Peripheral Blood From Patients With Glioma. Cancer Immunol Immunother (2018) 67:237–46. doi: 10.1007/s00262-017-2066-z
64. Zhang JG, Eguchi J, Kruse CA, Gomez GG, Fakhrai H, Schroter S, et al. Antigenic Profiling of Glioma Cells to Generate Allogeneic Vaccines or Dendritic Cell-Based Therapeutics. Clin Cancer Res (2007) 13:566–75. doi: 10.1158/1078-0432.CCR-06-1576
65. Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/Testis Antigens, Gametogenesis and Cancer. Nat Rev Cancer (2005) 5:615–25. doi: 10.1038/nrc1669
66. De Smet C, Loriot A. DNA Hypomethylation in Cancer: Epigenetic Scars of a Neoplastic Journey. Epigenetics (2010) 5:206–13. doi: 10.4161/epi.5.3.11447
67. Quintarelli C, Dotti G, De Angelis B, Hoyos V, Mims M, Luciano L, et al. Cytotoxic T Lymphocytes Directed to the Preferentially Expressed Antigen of Melanoma (PRAME) Target Chronic Myeloid Leukemia. Blood (2008) 112:1876–85. doi: 10.1182/blood-2008-04-150045
68. Xu Y, Zou R, Wang J, Wang ZW, Zhu X. The Role of the Cancer Testis Antigen PRAME in Tumorigenesis and Immunotherapy in Human Cancer. Cell Prolif (2020) 53:e12770. doi: 10.1111/cpr.12770
69. Wadelin F, Fulton J, McEwan PA, Spriggs KA, Emsley J, Heery DM. Leucine-Rich Repeat Protein PRAME: Expression, Potential Functions and Clinical Implications for Leukaemia. Mol Cancer (2010) 9:226. doi: 10.1186/1476-4598-9-226
70. Boon K, Edwards JB, Siu IM, Olschner D, Eberhart CG, Marra MA, et al. Comparison of Medulloblastoma and Normal Neural Transcriptomes Identifies a Restricted Set of Activated Genes. Oncogene (2003) 22:7687–94. doi: 10.1038/sj.onc.1207043
71. Vulcani-Freitas TM, Saba-Silva N, Cappellano A, Cavalheiro S, Toledo SR. PRAME Gene Expression Profile in Medulloblastoma. Arq Neuropsiquiatr (2011) 69:9–12. doi: 10.1590/S0004-282X2011000100003
72. Orlando D, Miele E, De Angelis B, Guercio M, Boffa I, Sinibaldi M, et al. Adoptive Immunotherapy Using Prame-Specific T Cells in Medulloblastoma. Cancer Res (2018) 78:3337–49. doi: 10.1158/0008-5472.CAN-17-3140
73. Ishihara M, Kageyama S, Miyahara Y, Ishikawa T, Ueda S, Soga N, et al. Mage-A4, NY-ESO-1 and SAGE mRNA Expression Rates and Co-Expression Relationships in Solid Tumours. BMC Cancer (2020) 20:606. doi: 10.1186/s12885-020-07098-4
74. Natsume A, Wakabayashi T, Tsujimura K, Shimato S, Ito M, Kuzushima K, et al. The DNA Demethylating Agent 5-Aza-2’-Deoxycytidine Activates NY-ESO-1 Antigenicity in Orthotopic Human Glioma. Int J Cancer (2008) 122:2542–53. doi: 10.1002/ijc.23407
75. Everson RG, Antonios JP, Lisiero DN, Soto H, Scharnweber R, Garrett MC, et al. Efficacy of Systemic Adoptive Transfer Immunotherapy Targeting NY-ESO-1 for Glioblastoma. Neuro Oncol (2016) 18:368–78. doi: 10.1093/neuonc/nov153
76. Kirkin AF, Dzhandzhugazyan KN, Guldberg P, Fang JJ, Andersen RS, Dahl C, et al. Adoptive Cancer Immunotherapy Using DNA-Demethylated T Helper Cells as Antigen-Presenting Cells. Nat Commun (2018) 9:785. doi: 10.1038/s41467-018-03217-9
77. Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, et al. K27M Mutation in Histone H3.3 Defines Clinically and Biologically Distinct Subgroups of Pediatric Diffuse Intrinsic Pontine Gliomas. Acta Neuropathol (2012) 124:439–47. doi: 10.1007/s00401-012-0998-0
78. Jones C, Baker SJ. Unique Genetic and Epigenetic Mechanisms Driving Paediatric Diffuse High-Grade Glioma. Nat Rev Cancer (2014) 14:1–23. doi: 10.1038/nrc3811
79. Chheda ZS, Kohanbash G, Okada K, Jahan N, Sidney J, Pecoraro M, et al. Novel and Shared Neoantigen Derived From Histone 3 Variant H3.3K27M Mutation for Glioma T Cell Therapy. J Exp Med (2018) 215:141–57. doi: 10.1084/jem.20171046
80. Campillo-Davo D, Flumens D, Lion E. The Quest for the Best: How TCR Affinity, Avidity, and Functional Avidity Affect TCR-Engineered T-Cell Antitumor Responses. Cells (2020) 9:1–8. doi: 10.3390/cells9071720
81. Gularyan SK, Gulin AA, Anufrieva KS, Shender VO, Shakhparonov MI, Bastola S, et al. Investigation of Inter- and Intratumoral Heterogeneity of Glioblastoma Using Tof-Sims. Mol Cell Proteomics (2020) 19:960–70. doi: 10.1074/mcp.RA120.001986
82. Dersh D, Holly J, Yewdell JW. Author Correction: A Few Good Peptides: MHC Class I-Based Cancer Immunosurveillance and Immunoevasion. Nat Rev Immunol (2020) 20:644. doi: 10.1038/s41577-020-00445-3
83. Dersh D, Phelan JD, Gumina ME, Wang B, Arbuckle JH, Holly J, et al. Genome-Wide Screens Identify Lineage- and Tumor-Specific Genes Modulating Mhc-I- and MHC-II-Restricted Immunosurveillance of Human Lymphomas. Immunity (2021) 54:116–131 e10. doi: 10.1016/j.immuni.2020.11.002
84. Wu L, Wei Q, Brzostek J, Gascoigne NRJ. Signaling From T Cell Receptors (TCRs) and Chimeric Antigen Receptors (Cars) on T Cells. Cell Mol Immunol (2020) 17:600–12. doi: 10.1038/s41423-020-0470-3
85. Golubovskaya V, Wu L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers (Basel) (2016) 8:1–12. doi: 10.3390/cancers8030036
86. Fournier C, Martin F, Zitvogel L, Kroemer G, Galluzzi L, Apetoh L. Trial Watch: Adoptively Transferred Cells for Anticancer Immunotherapy. Oncoimmunology (2017) 6:e1363139. doi: 10.1080/2162402X.2017.1363139
87. Rubinstein JD, Krupski C, Nelson AS, O’Brien MM, Davies SM, Phillips CL. Chimeric Antigen Receptor T Cell Therapy in Patients With Multiply Relapsed or Refractory Extramedullary Leukemia. Biol Blood Marrow Transplant (2020) 26:e280–5. doi: 10.1016/j.bbmt.2020.07.036
88. Abbasi A, Peeke S, Shah N, Mustafa J, Khatun F, Lombardo A, et al. Axicabtagene Ciloleucel CD19 CAR-T Cell Therapy Results in High Rates of Systemic and Neurologic Remissions in Ten Patients With Refractory Large B Cell Lymphoma Including Two With HIV and Viral Hepatitis. J Hematol Oncol (2020) 13:1. doi: 10.1186/s13045-019-0838-y
89. Abramson JS, McGree B, Noyes S, Plummer S, Wong C, Chen YB, et al. Anti-CD19 Car T Cells in CNS Diffuse Large-B-Cell Lymphoma. N Engl J Med (2017) 377:783–4. doi: 10.1056/NEJMc1704610
90. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor Cd8+ T Cells in Patients With Recurrent Glioblastoma. Clin Cancer Res (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
91. Wang X, Berger C, Wong CW, Forman SJ, Riddell SR, Jensen MC. Engraftment of Human Central Memory-Derived Effector CD8+ T Cells in Immunodeficient Mice. Blood (2011) 117:1888–98. doi: 10.1182/blood-2010-10-310599
92. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma After Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
93. Debinski W, Obiri NI, Powers SK, Pastan I, Puri RK. Human Glioma Cells Overexpress Receptors for Interleukin 13 and Are Extremely Sensitive to a Novel Chimeric Protein Composed of Interleukin 13 and Pseudomonas Exotoxin. Clin Cancer Res (1995) 1:1253–8. doi: 10.1038/nm.3910
94. Sattiraju A, Solingapuram Sai KK, Xuan A, Pandya DN, Almaguel FG, Wadas TJ, et al. IL13RA2 Targeted Alpha Particle Therapy Against Glioblastomas. Oncotarget (2017) 8:42997–3007. doi: 10.18632/oncotarget.17792
95. Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D’Apuzzo M, et al. Stem-Like Tumor-Initiating Cells Isolated From IL13Ralpha2 Expressing Gliomas are Targeted and Killed by IL13-Zetakine-Redirected T Cells. Clin Cancer Res (2012) 18:2199–209. doi: 10.1158/1078-0432.CCR-11-1669
96. Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, et al. Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-Tumor Efficacy Against Glioblastoma. Mol Ther (2018) 26:31–44. doi: 10.1016/j.ymthe.2017.10.002
97. Wang D, Prager BC, Gimple RC, Aguilar B, Alizadeh D, Tang H, et al. Crispr Screening of CAR T Cells and Cancer Stem Cells Reveals Critical Dependencies for Cell-Based Therapies. Cancer Discovery (2020) 11(5):1192–211. doi: 10.1158/2159-8290.CD-20-1243
98. Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D, Lavi S, et al. A Hierarchical Network of Interreceptor Interactions Determines Signal Transduction by Neu Differentiation Factor/Neuregulin and Epidermal Growth Factor. Mol Cell Biol (1996) 16:5276–87. doi: 10.1128/MCB.16.10.5276
99. Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M. The Epidermal Growth Factor Receptor: From Development to Tumorigenesis. Differentiation (2007) 75:770–87. doi: 10.1111/j.1432-0436.2007.00238.x
100. Mansour AM, Abdelrahim M, Laymon M, Elsherbeeny M, Sultan M, Shokeir A, et al. Epidermal Growth Factor Expression as a Predictor of Chemotherapeutic Resistance in Muscle-Invasive Bladder Cancer. BMC Urol (2018) 18:100. doi: 10.1186/s12894-018-0413-9
101. Saikali S, Avril T, Collet B, Hamlat A, Bansard JY, Drenou B, et al. Expression of Nine Tumour Antigens in a Series of Human Glioblastoma Multiforme: Interest of EGFRvIII, Il-13Ralpha2, gp100 and TRP-2 for Immunotherapy. J Neurooncol (2007) 81:139–48. doi: 10.1007/s11060-006-9220-3
102. Feldkamp MM, Lala P, Lau N, Roncari L, Guha A. Expression of Activated Epidermal Growth Factor Receptors, Ras-Guanosine Triphosphate, and Mitogen-Activated Protein Kinase in Human Glioblastoma Multiforme Specimens. Neurosurgery (1999) 45:1442–53. doi: 10.1097/00006123-199912000-00034
103. Rutkowska A, Stoczynska-Fidelus E, Janik K, Wlodarczyk A, Rieske P. EGFR(VIII): An Oncogene With Ambiguous Role. J Oncol (2019) 2019:1092587. doi: 10.1155/2019/1092587
104. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A Single Dose of Peripherally Infused EGFRvIII-Directed Car T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients With Recurrent Glioblastoma. Sci Transl Med (2017) 9. doi: 10.1126/scitranslmed.aaa0984
105. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-Transduced T Cells Targeting EgfrvIII in Patients With Glioblastoma. J Immunother (2019) 42:126–35. doi: 10.1097/CJI.0000000000000260
106. Song Y, Liu Q, Zuo T, Wei G, Jiao S. Combined Antitumor Effects of anti-EGFR Variant III CAR-T Cell Therapy and PD-1 Checkpoint Blockade on Glioblastoma in Mouse Model. Cell Immunol (2020) 352:104112. doi: 10.1016/j.cellimm.2020.104112
107. Zhu H, You Y, Shen Z, Shi L. Egfrviii-CAR-T Cells With PD-1 Knockout Have Improved Anti-Glioma Activity. Pathol Oncol Res (2020) 26:2135–41. doi: 10.1007/s12253-019-00759-1
108. Choi BD, Yu X, Castano AP, Darr H, Henderson DB, Bouffard AA, et al. Crispr-Cas9 Disruption of PD-1 Enhances Activity of Universal EGFRvIII Car T Cells in a Preclinical Model of Human Glioblastoma. J Immunother Cancer (2019) 7:304. doi: 10.1186/s40425-019-0806-7
109. Nakazawa T, Natsume A, Nishimura F, Morimoto T, Matsuda R, Nakamura M, et al. Effect of CRISPR/Cas9-Mediated Pd-1-Disrupted Primary Human Third-Generation Car-T Cells Targeting EGFRvIII on In Vitro Human Glioblastoma Cell Growth. Cells (2020) 9. doi: 10.3390/cells9040998
110. Ravanpay AC, Gust J, Johnson AJ, Rolczynski LS, Cecchini M, Chang CA, et al. Egfr806-Car T Cells Selectively Target a Tumor-Restricted EGFR Epitope in Glioblastoma. Oncotarget (2019) 10:7080–95. doi: 10.18632/oncotarget.27389
111. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T Cells Secreting BiTEs Circumvent Antigen Escape Without Detectable Toxicity. Nat Biotechnol (2019) 37:1049–58. doi: 10.1038/s41587-019-0192-1
112. Meric-Bernstam F, Hung MC. Advances in Targeting Human Epidermal Growth Factor Receptor-2 Signaling for Cancer Therapy. Clin Cancer Res (2006) 12:6326–30. doi: 10.1158/1078-0432.CCR-06-1732
113. Vranic S, Beslija S, Gatalica Z. Targeting HER2 Expression in Cancer: New Drugs and New Indications. Bosn J Basic Med Sci (2020) 21(1):1–4. doi: 10.17305/bjbms.2020.4908
114. Menard S, Casalini P, Campiglio M, Pupa S, Agresti R, Tagliabue E. HER2 Overexpression in Various Tumor Types, Focussing on its Relationship to the Development of Invasive Breast Cancer. Ann Oncol (2001) 12 Suppl 1:S15–9. doi: 10.1093/annonc/12.suppl_1.S15
115. Pearson JRD, Regad T. Targeting Cellular Pathways in Glioblastoma Multiforme. Signal Transduct Target Ther (2017) 2:17040. doi: 10.1038/sigtrans.2017.40
116. Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-Specific T Cells Target Primary Glioblastoma Stem Cells and Induce Regression of Autologous Experimental Tumors. Clin Cancer Res (2010) 16:474–85. doi: 10.1158/1078-0432.CCR-09-1322
117. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. Her2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
118. Zhang C, Burger MC, Jennewein L, Genssler S, Schonfeld K, Zeiner P, et al. Erbb2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J Natl Cancer Inst (2016) 108. doi: 10.1093/jnci/djv375
119. Nazha B, Inal C, Owonikoko TK. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front Oncol (2020) 10:1000. doi: 10.3389/fonc.2020.01000
120. Schulz G, Cheresh DA, Varki NM, Yu A, Staffileno LK, Reisfeld RA. Detection of Ganglioside GD2 in Tumor Tissues and Sera of Neuroblastoma Patients. Cancer Res (1984) 44:5914–20.
121. Tsuchida T, Saxton RE, Morton DL, Irie RF. Gangliosides of Human Melanoma. J Natl Cancer Inst (1987) 78:45–54. doi: 10.1093/jnci/78.1.45
122. Dobrenkov K, Ostrovnaya I, Gu J, Cheung IY, Cheung NK. Oncotargets GD2 and GD3 are Highly Expressed in Sarcomas of Children, Adolescents, and Young Adults. Pediatr Blood Cancer (2016) 63:1780–5. doi: 10.1002/pbc.26097
123. Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, et al. Potent Antitumor Efficacy of anti-GD2 Car T Cells in H3-K27M(+) Diffuse Midline Gliomas. Nat Med (2018) 24:572–9. doi: 10.1038/s41591-018-0006-x
124. Ladenstein R, Potschger U, Valteau-Couanet D, Luksch R, Castel V, Yaniv I, et al. Interleukin 2 With Anti-GD2 Antibody ch14.18/CHO (Dinutuximab Beta) in Patients With High-Risk Neuroblastoma (HR-NBL1/SIOPEN): A Multicentre, Randomised, Phase 3 Trial. Lancet Oncol (2018) 19:1617–29. doi: 10.1016/S1470-2045(18)30578-3
125. Kushner BH, Cheung IY, Modak S, Basu EM, Roberts SS, Cheung NK. Humanized 3f8 Anti-GD2 Monoclonal Antibody Dosing With Granulocyte-Macrophage Colony-Stimulating Factor in Patients With Resistant Neuroblastoma: A Phase 1 Clinical Trial. JAMA Oncol (2018) 4:1729–35. doi: 10.1001/jamaoncol.2018.4005
127. Quintarelli C, Orlando D, Boffa I, Guercio M, Polito VA, Petretto A, et al. Choice of Costimulatory Domains and of Cytokines Determines CAR T-Cell Activity in Neuroblastoma. Oncoimmunology (2018) 7:e1433518. doi: 10.1080/2162402X.2018.1433518
128. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-Specific T Cells Engineered to Coexpress Tumor-Specific Receptors: Persistence and Antitumor Activity in Individuals With Neuroblastoma. Nat Med (2008) 14:1264–70. doi: 10.1038/nm.1882
129. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor Activity and Long-Term Fate of Chimeric Antigen Receptor-Positive T Cells in Patients With Neuroblastoma. Blood (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
130. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive Signaling From an Engineered Il7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discovery (2017) 7:1238–47. doi: 10.1158/2159-8290.CD-17-0538
131. Guery T, Roumier C, Berthon C, Renneville A, Preudhomme C, Quesnel B. B7-H3 Protein Expression in Acute Myeloid Leukemia. Cancer Med (2015) 4:1879–83. doi: 10.1002/cam4.522
132. Roth TJ, Sheinin Y, Lohse CM, Kuntz SM, Frigola X, Inman BA, et al. B7-H3 Ligand Expression by Prostate Cancer: A Novel Marker of Prognosis and Potential Target for Therapy. Cancer Res (2007) 67:7893–900. doi: 10.1158/0008-5472.CAN-07-1068
133. Tekle C, Nygren MK, Chen YW, Dybsjord I, Nesland JM, Maelandsmo GM, et al. B7-H3 Contributes to the Metastatic Capacity of Melanoma Cells by Modulation of Known Metastasis-Associated Genes. Int J Cancer (2012) 130:2282–90. doi: 10.1002/ijc.26238
134. Castriconi R, Dondero A, Augugliaro R, Cantoni C, Carnemolla B, Sementa AR, et al. Identification of 4Ig-B7-H3 as a Neuroblastoma-Associated Molecule That Exerts a Protective Role From an NK Cell-Mediated Lysis. Proc Natl Acad Sci USA (2004) 101:12640–5. doi: 10.1073/pnas.0405025101
135. Wang L, Zhang Q, Chen W, Shan B, Ding Y, Zhang G, et al. B7-H3 is Overexpressed in Patients Suffering Osteosarcoma and Associated With Tumor Aggressiveness and Metastasis. PloS One (2013) 8:e70689. doi: 10.1371/journal.pone.0070689
136. Maachani UB, Tosi U, Pisapia DJ, Mukherjee S, Marnell CS, Voronina J, et al. B7-H3 as a Prognostic Biomarker and Therapeutic Target in Pediatric Central Nervous System Tumors. Transl Oncol (2020) 13:365–71. doi: 10.1016/j.tranon.2019.11.006
137. Wang Z, Wang Z, Zhang C, Liu X, Li G, Liu S, et al. Genetic and Clinical Characterization of B7-H3 (CD276) Expression and Epigenetic Regulation in Diffuse Brain Glioma. Cancer Sci (2018) 109:2697–705. doi: 10.1111/cas.13744
138. Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, et al. Car T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin Cancer Res (2019) 25:2560–74. doi: 10.1158/1078-0432.CCR-18-0432
139. Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, et al. Locoregionally Administered B7-H3-Targeted Car T Cells for Treatment of Atypical Teratoid/Rhabdoid Tumors. Nat Med (2020) 26:712–9. doi: 10.1038/s41591-020-0821-8
140. Jacobs J, Deschoolmeester V, Zwaenepoel K, Rolfo C, Silence K, Rottey S, et al. Cd70: An Emerging Target in Cancer Immunotherapy. Pharmacol Ther (2015) 155:1–10. doi: 10.1016/j.pharmthera.2015.07.007
141. Held-Feindt J, Mentlein R. CD70/CD27 Ligand, A Member of the TNF Family, is Expressed in Human Brain Tumors. Int J Cancer (2002) 98:352–6. doi: 10.1002/ijc.10207
142. Ge H, Mu L, Jin L, Yang C, Chang YE, Long Y, et al. Tumor Associated CD70 Expression is Involved in Promoting Tumor Migration and Macrophage Infiltration in GBM. Int J Cancer (2017) 141:1434–44. doi: 10.1002/ijc.30830
143. Jin L, Ge H, Long Y, Yang C, Chang YE, Mu L, et al. CD70, a Novel Target of CAR T-Cell Therapy for Gliomas. Neuro Oncol (2018) 20:55–65. doi: 10.1093/neuonc/nox116
144. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-Modified Car T Cells Co-Opt IL-8 for Maximal Antitumor Efficacy in Solid Tumors. Nat Commun (2019) 10:4016. doi: 10.1038/s41467-019-11869-4
145. Caruso H, Heimberger AB. Comment on “Trivalent Car T Cells Overcome Interpatient Antigenic Variability in Glioblastoma”. Neuro Oncol (2018) 20:1003–4. doi: 10.1093/neuonc/noy045
146. Yang M, Tang X, Zhang Z, Gu L, Wei H, Zhao S, et al. Tandem CAR-T Cells Targeting CD70 and B7-H3 Exhibit Potent Preclinical Activity Against Multiple Solid Tumors. Theranostics (2020) 10:7622–34. doi: 10.7150/thno.43991
147. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem Car T Cells Targeting HER2 and IL13Ralpha2 Mitigate Tumor Antigen Escape. J Clin Invest (2016) 126:3036–52. doi: 10.1172/JCI83416
148. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent Car T Cells Overcome Interpatient Antigenic Variability in Glioblastoma. Neuro Oncol (2018) 20:506–18. doi: 10.1093/neuonc/nox182
149. Donovan LK, Delaidelli A, Joseph SK, Bielamowicz K, Fousek K, Holgado BL, et al. Locoregional Delivery of CAR T Cells to the Cerebrospinal Fluid for Treatment of Metastatic Medulloblastoma and Ependymoma. Nat Med (2020) 26:720–31. doi: 10.1038/s41591-020-0827-2
150. Li Z. CD133: A Stem Cell Biomarker and Beyond. Exp Hematol Oncol (2013) 2:17. doi: 10.1186/2162-3619-2-17
151. Vora P, Venugopal C, Salim SK, Tatari N, Bakhshinyan D, Singh M, et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell (2020) 26:32–844.e6. doi: 10.1016/j.stem.2020.04.008
152. Wang X, Wang Y, Yu L, Sakakura K, Visus C, Schwab JH, et al. CSPG4 in Cancer: Multiple Roles. Curr Mol Med (2010) 10:419–29. doi: 10.2174/156652410791316977
153. Wang X, Osada T, Wang Y, Yu L, Sakakura K, Katayama A, et al. CSPG4 Protein as a New Target for the Antibody-Based Immunotherapy of Triple-Negative Breast Cancer. J Natl Cancer Inst (2010) 102:1496–512. doi: 10.1093/jnci/djq343
154. Rivera Z, Ferrone S, Wang X, Jube S, Yang H, Pass HI, et al. CSPG4 as a Target of Antibody-Based Immunotherapy for Malignant Mesothelioma. Clin Cancer Res (2012) 18:5352–63. doi: 10.1158/1078-0432.CCR-12-0628
155. Tsidulko AY, Kazanskaya GM, Kostromskaya DV, Aidagulova SV, Kiselev RS, Volkov AM, et al. Prognostic Relevance of NG2/CSPG4, CD44 and Ki-67 in Patients With Glioblastoma. Tumour Biol (2017) 39:1010428317724282. doi: 10.1177/1010428317724282
156. Pellegatta S, Savoldo B, Di Ianni N, Corbetta C, Chen Y, Patane M, et al. Constitutive and TNFalpha-inducible Expression of Chondroitin Sulfate Proteoglycan 4 in Glioblastoma and Neurospheres: Implications for CAR-T Cell Therapy. Sci Transl Med (2018) 10(435):1–16. doi: 10.1126/scitranslmed.aao2731
157. AlHarbi M, Ali Mobark N, AlMubarak L, Aljelaify R, AlSaeed M, Almutairi A, et al. Durable Response to Nivolumab in a Pediatric Patient With Refractory Glioblastoma and Constitutional Biallelic Mismatch Repair Deficiency. Oncologist (2018) 23:1401–6. doi: 10.1634/theoncologist.2018-0163
158. Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol (2016) 34:2206–11. doi: 10.1200/JCO.2016.66.6552
159. Desjardins A, Gromeier M, Herndon JE, Beaubier 2N, Bolognesi DP, Friedman AH, et al. Recurrent Glioblastoma Treated With Recombinant Poliovirus. N Engl J Med (2018) 379:150–61. doi: 10.1056/NEJMoa1716435
160. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature (2019) 565:234–9.
161. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable Therapeutic Efficacy Utilizing Combinatorial Blockade Against IDO, Ctla-4, and PD-L1 in Mice With Brain Tumors. Clin Cancer Res (2014) 20:5290–301. doi: 10.1158/1078-0432.CCR-14-0514
162. Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. N Engl J Med (2018) 379:722–30. doi: 10.1056/NEJMoa1805453
163. Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, et al. Nivolumab With or Without Ipilimumab in Patients With Recurrent Glioblastoma: Results From Exploratory Phase I Cohorts of CheckMate 143. Neuro Oncol (2018) 20:674–86. doi: 10.1093/neuonc/nox208
164. McGranahan T, Therkelsen KE, Ahmad S, Nagpal S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr Treat Options Oncol (2019) 20:24. doi: 10.1007/s11864-019-0619-4
165. Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 Blockade and Stereotactic Radiation Produce Long-Term Survival in Mice With Intracranial Gliomas. Int J Radiat Oncol Biol Phys (2013) 86:343–9. doi: 10.1016/j.ijrobp.2012.12.025
166. Filley AC, Henriquez M, Dey M. Recurrent Glioma Clinical Trial, CheckMate-143: The Game Is Not Over Yet. Oncotarget (2017) 8:91779–94. doi: 10.18632/oncotarget.21586
167. Morganti S, Curigliano G. Combinations Using Checkpoint Blockade to Overcome Resistance. Ecancermedicalscience (2020) 14:1148. doi: 10.3332/ecancer.2020.1148
168. Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, et al. Mechanisms and Therapeutic Implications of Hypermutation in Gliomas. Nature (2020) 580:517–23. doi: 10.1038/s41586-020-2209-9
169. Pfirschke C, Siwicki M, Liao HW, Pittet MJ. Tumor Microenvironment: No Effector T Cells Without Dendritic Cells. Cancer Cell (2017) 31:614–5. doi: 10.1016/j.ccell.2017.04.007
170. Oba T, Long MD, Keler T, Marsh HC, Minderman H, Abrams SI, et al. Overcoming Primary and Acquired Resistance to Anti-PD-L1 Therapy by Induction and Activation of Tumor-Residing Cdc1s. Nat Commun (2020) 11:5415. doi: 10.1038/s41467-020-19192-z
171. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell (2017) 31:711–723 e4. doi: 10.1016/j.ccell.2017.04.003
172. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of Tumor Rejection With Doublets of CTLA-4, Pd-1/Pd-L1, or IDO Blockade Involves Restored IL-2 Production and Proliferation of CD8(+) T Cells Directly Within the Tumor Microenvironment. J Immunother Cancer (2014) 2:3. doi: 10.1186/2051-1426-2-3
173. Pardridge WM. Drug Transport Across the Blood-Brain Barrier. J Cereb Blood Flow Metab (2012) 32:1959–72. doi: 10.1038/jcbfm.2012.126
174. Stanimirovic DB, Sandhu JK, Costain WJ. Emerging Technologies for Delivery of Biotherapeutics and Gene Therapy Across the Blood-Brain Barrier. BioDrugs (2018) 32:547–59. doi: 10.1007/s40259-018-0309-y
175. Galstyan A, Markman JL, Shatalova ES, Chiechi A, Korman AJ, Patil R, et al. Blood-Brain Barrier Permeable Nano Immunoconjugates Induce Local Immune Responses for Glioma Therapy. Nat Commun (2019) 10:3850. doi: 10.1038/s41467-019-11719-3
176. Erel-Akbaba G, Carvalho LA, Tian T, Zinter M, Akbaba H, Obeid PJ, et al. Radiation-Induced Targeted Nanoparticle-Based Gene Delivery for Brain Tumor Therapy. ACS Nano (2019) 13:4028–40. doi: 10.1021/acsnano.8b08177
177. Miller MA, Chandra R, Cuccarese MF, Pfirschke C, Engblom C, Stapleton S, et al. Radiation Therapy Primes Tumors for Nanotherapeutic Delivery Via Macrophage-Mediated Vascular Bursts. Sci Transl Med (2017) 9:1–24. doi: 10.1126/scitranslmed.aal0225
178. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal Replacement of Tumor-Specific T Cells Following PD-1 Blockade. Nat Med (2019) 25:1251–9. doi: 10.1038/s41591-019-0522-3
179. Callahan MK, Wolchok JD. Recruit or Reboot? How Does Anti-Pd-1 Therapy Change Tumor-Infiltrating Lymphocytes? Cancer Cell (2019) 36:215–7. doi: 10.1016/j.ccell.2019.08.009
180. Rius-Rocabert S, Garcia-Romero N, Garcia A, Ayuso-Sacido A, Nistal-Villan E. Oncolytic Virotherapy in Glioma Tumors. Int J Mol Sci (2020) 21:1–30. doi: 10.3390/ijms21207604
181. Post DE, Sandberg EM, Kyle MM, Devi NS, Brat DJ, Xu Z, et al. Targeted Cancer Gene Therapy Using a Hypoxia Inducible Factor Dependent Oncolytic Adenovirus Armed With Interleukin-4. Cancer Res (2007) 67:6872–81. doi: 10.1158/0008-5472.CAN-06-3244
182. Leber MF, Bossow S, Leonard VH, Zaoui K, Grossardt C, Frenzke M, et al. MicroRNA-sensitive Oncolytic Measles Viruses for Cancer-Specific Vector Tropism. Mol Ther (2011) 19:1097–106. doi: 10.1038/mt.2011.55
183. Geletneky K, Huesing J, Rommelaere J, Schlehofer JR, Leuchs B, Dahm M, et al. Phase I/IIa Study of Intratumoral/Intracerebral or Intravenous/Intracerebral Administration of Parvovirus H-1 (ParvOryx) in Patients With Progressive Primary or Recurrent Glioblastoma Multiforme: ParvOryx01 Protocol. BMC Cancer (2012) 12:99. doi: 10.1186/1471-2407-12-99
184. Wollmann G, Ozduman K, van den Pol AN. Oncolytic Virus Therapy for Glioblastoma Multiforme: Concepts and Candidates. Cancer J (2012) 18:69–81. doi: 10.1097/PPO.0b013e31824671c9
185. Kaverina NV, Kadagidze ZG, Borovjagin AV, Karseladze AI, Kim CK, Lesniak MS, et al. Tamoxifen Overrides Autophagy Inhibition in Beclin-1-deficient Glioma Cells and Their Resistance to Adenovirus-Mediated Oncolysis Via Upregulation of PUMA and BAX. Oncogene (2018) 37:6069–82. doi: 10.1038/s41388-018-0395-9
186. Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, et al. Conditionally Replicating Herpes Simplex Virus Mutant, G207 for the Treatment of Malignant Glioma: Results of a Phase I Trial. Gene Ther (2000) 7:867–74. doi: 10.1038/sj.gt.3301205
187. Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, et al. Phase Ib Trial of Mutant Herpes Simplex Virus G207 Inoculated Pre- and Post-Tumor Resection for Recurrent GBM. Mol Ther (2009) 17:199–207. doi: 10.1038/mt.2008.228
188. Markert JM, Razdan SN, Kuo HC, Cantor A, Knoll A, Karrasch M, et al. A Phase 1 Trial of Oncolytic HSV-1, G207, Given in Combination With Radiation for Recurrent GBM Demonstrates Safety and Radiographic Responses. Mol Ther (2014) 22:1048–55. doi: 10.1038/mt.2014.22
189. Waters AM, Johnston JM, Reddy AT, Fiveash J, Madan-Swain A, Kachurak K, et al. Rationale and Design of a Phase 1 Clinical Trial to Evaluate Hsv G207 Alone or With a Single Radiation Dose in Children With Progressive or Recurrent Malignant Supratentorial Brain Tumors. Hum Gene Ther Clin Dev (2017) 28:7–16. doi: 10.1089/humc.2017.002
190. Bernstock JD, Bag AK, Fiveash J, Kachurak K, Elsayed G, Chagoya G, et al. Design and Rationale for First-in-Human Phase 1 Immunovirotherapy Clinical Trial of Oncolytic Hsv G207 to Treat Malignant Pediatric Cerebellar Brain Tumors. Hum Gene Ther (2020) 31:1132–9. doi: 10.1089/hum.2020.101
191. Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, et al. Phase I Study of DNX-2401 (Delta-24-Rgd) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J Clin Oncol (2018) 36:1419–27. doi: 10.1200/JCO.2017.75.8219
192. Tejada S, Alonso M, Patino A, Fueyo J, Gomez-Manzano C, Diez-Valle R. Phase I Trial of DNX-2401 for Diffuse Intrinsic Pontine Glioma Newly Diagnosed in Pediatric Patients. Neurosurgery (2018) 83:1050–6. doi: 10.1093/neuros/nyx507
193. Geletneky K, Hajda J, Angelova AL, Leuchs B, Capper D, Bartsch AJ, et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol Ther (2017) 25:2620–34. doi: 10.1016/j.ymthe.2017.08.016
194. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut With Temozolomide for Patients With Newly Diagnosed, EGFRvIII-Expressing Glioblastoma (ACT IV): A Randomised, Double-Blind, International Phase 3 Trial. Lancet Oncol (2017) 18:1373–85. doi: 10.1016/S1470-2045(17)30517-X
195. Winograd EK, Ciesielski MJ, Fenstermaker RA. Novel Vaccines for Glioblastoma: Clinical Update and Perspective. Immunotherapy (2016) 8:1293–308. doi: 10.2217/imt-2016-0059
196. Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, Berger MS, et al. Heat-Shock Protein Peptide Complex-96 Vaccination for Recurrent Glioblastoma: A Phase II, Single-Arm Trial. Neuro Oncol (2014) 16:274–9. doi: 10.1093/neuonc/not203
197. Ishikawa E, Muragaki Y, Yamamoto T, Maruyama T, Tsuboi K, Ikuta S, et al. Phase I/IIa Trial of Fractionated Radiotherapy, Temozolomide, and Autologous Formalin-Fixed Tumor Vaccine for Newly Diagnosed Glioblastoma. J Neurosurg (2014) 121:543–53. doi: 10.3171/2014.5.JNS132392
198. Mitsuya K, Akiyama Y, Iizuka A, Miyata H, Deguchi S, Hayashi N, et al. Alpha-Type-1 Polarized Dendritic Cell-Based Vaccination in Newly Diagnosed High-Grade Glioma: A Phase II Clinical Trial. Anticancer Res (2020) 40:6473–84. doi: 10.21873/anticanres.14669
199. Wen PY, Reardon DA, Armstrong TS, Phuphanich S, Aiken RD, Landolfi JC, et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine Ict-107 in Newly Diagnosed Patients With Glioblastoma. Clin Cancer Res (2019) 25:5799–807. doi: 10.1158/1078-0432.CCR-19-0261
200. Eoli M, Corbetta C, Anghileri E, Di Ianni N, Milani M, Cuccarini V, et al. Expansion of Effector and Memory T Cells Is Associated With Increased Survival in Recurrent Glioblastomas Treated With Dendritic Cell Immunotherapy. Neurooncol Adv (2019) 1:vdz022. doi: 10.1093/noajnl/vdz022
201. Cuccarini V, Aquino D, Gioppo A, Anghileri E, Pellegatta S, Schettino C, et al. Advanced MRI Assessment During Dendritic Cell Immunotherapy Added to Standard Treatment Against Glioblastoma. J Clin Med (2019) 8:1–16. doi: 10.3390/jcm8112007
202. Pellegatta S, Eoli M, Cuccarini V, Anghileri E, Pollo B, Pessina S, et al. Survival Gain in Glioblastoma Patients Treated With Dendritic Cell Immunotherapy is Associated With Increased NK But Not CD8(+) T Cell Activation in the Presence of Adjuvant Temozolomide. Oncoimmunology (2018) 7:1–10. doi: 10.1080/2162402X.2017.1412901
203. Rapp M, Grauer OM, Kamp M, Sevens N, Zotz N, Sabel M, et al. A Randomized Controlled Phase II Trial of Vaccination With Lysate-Loaded, Mature Dendritic Cells Integrated Into Standard Radiochemotherapy of Newly Diagnosed Glioblastoma (GlioVax): Study Protocol for a Randomized Controlled Trial. Trials (2018) 19:293. doi: 10.1186/s13063-018-2659-7
204. Saha D, Martuza RL, Rabkin SD. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell (2017) 32:253–67.e5. doi: 10.1016/j.ccell.2017.07.006
Keywords: CNS, brain tumor, immunotherapy, chimeric antigen receptor, T cell receptor, oncolytic virotherapy, vaccination, immunocheck point inhibitors
Citation: Quintarelli C, Camera A, Ciccone R, Alessi I, Del Bufalo F, Carai A, Del Baldo G, Mastronuzzi A and De Angelis B (2021) Innovative and Promising Strategies to Enhance Effectiveness of Immunotherapy for CNS Tumors: Where Are We? Front. Immunol. 12:634031. doi: 10.3389/fimmu.2021.634031
Received: 26 November 2020; Accepted: 30 April 2021;
Published: 07 June 2021.
Edited by:
Sujith K. Joseph, Baylor College of Medicine, United StatesReviewed by:
Evan Weber, Stanford University, United StatesAmer Najjar, University of Texas MD Anderson Cancer Center, United States
Copyright © 2021 Quintarelli, Camera, Ciccone, Alessi, Del Bufalo, Carai, Del Baldo, Mastronuzzi and De Angelis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Concetta Quintarelli, Y29uY2V0dGEucXVpbnRhcmVsbGlAb3BiZy5uZXQ=