 Minah Kim
Minah Kim Petr Tomek*
Petr Tomek*- Auckland Cancer Society Research Centre, Faculty of Medical and Health Sciences, University of Auckland, Auckland, New Zealand
Blockade of the immunosuppressive tryptophan catabolism mediated by indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase (TDO) holds enormous promise for sensitising cancer patients to immune checkpoint blockade. Yet, only IDO1 inhibitors had entered clinical trials so far, and those agents have generated disappointing clinical results. Improved understanding of molecular mechanisms involved in the immune-regulatory function of the tryptophan catabolism is likely to optimise therapeutic strategies to block this pathway. The immunosuppressive role of tryptophan metabolite kynurenine is becoming increasingly clear, but it remains a mystery if tryptophan exerts functions beyond serving as a precursor for kynurenine. Here we hypothesise that tryptophan acts as a rheostat of kynurenine-mediated immunosuppression by competing with kynurenine for entry into immune T-cells through the amino acid transporter called System L. This hypothesis stems from the observations that elevated tryptophan levels in TDO-knockout mice relieve immunosuppression instigated by IDO1, and that the vacancy of System L transporter modulates kynurenine entry into CD4+ T-cells. This hypothesis has two potential therapeutic implications. Firstly, potent TDO inhibitors are expected to indirectly inhibit IDO1 hence development of TDO-selective inhibitors appears advantageous compared to IDO1-selective and dual IDO1/TDO inhibitors. Secondly, oral supplementation with System L substrates such as leucine represents a novel potential therapeutic modality to restrain the immunosuppressive kynurenine and restore anti-tumour immunity.
Introduction
The human immune system can recognise and eradicate tumour cells. Thus, the immunity plays a key role in reducing cancer incidence (1). However, the immunity is a double-edged sword. Elimination of immune-sensitive tumour cells drives evolution of tumours towards the immune-resistant phenotype in a process called cancer immune-editing (2). This, in turn, leads to malignant and clinically apparent cancer. It is of utmost importance to identify and silence tumoural immune escape mechanisms to restore the patient’s anti-tumour immunity. Inhibiting the T-cell regulatory checkpoints, such as the programmed cell death protein 1 (PD-1) axis, by monoclonal antibodies have shown remarkable clinical responses (3). Some patients on the anti-PD-1 inhibitors experience durable tumour regressions but a sizeable fraction of patients do not benefit from these agents (4). This has triggered a search for mechanisms that could be modulated to optimise cancer patients’ responses to immune checkpoint inhibitors (5, 6). One of the key candidates are enzymes IDO1 and TDO that accelerate immunosuppressive tryptophan catabolism along the kynurenine pathway (7).
Initially thought of as a “holy grail” for potentiating cancer immunotherapy, the disappointing outcomes of IDO1 inhibitors in clinical trials (8–10) have generated scepticism (11–13). However, the evidence indicates that the concept of blocking the kynurenine pathway for potentiating immunotherapy is sound and holds true in preclinical models. It is likely that the most optimal therapeutic approaches to silence the kynurenine pathway have not yet been identified. We envision that improved understanding of the immunosuppression induced by the kynurenine pathway will yield insights into optimised therapeutic strategies.
In this perspective, we summarise the current knowledge about the mechanisms mediating immunosuppression by IDO1/TDO, propose a novel immune-regulatory function for the IDO1/TDO’s substrate tryptophan, and discuss the potential impact of this novel function on therapeutic strategies to block the immunosuppressive tryptophan catabolism.
Immunosuppressive Tryptophan Catabolism
Mammals metabolise more than 90% of tryptophan via the kynurenine pathway (KP) (14). The resulting tryptophan metabolites are involved in essential biological processes such as immune regulation, energy metabolism and production of an important enzyme co-factor NAD (15–17). The first and rate limiting step of the KP, tryptophan oxidation, is catalysed by intracellular enzymes IDO1 and TDO (18). There is also a third enzyme called IDO2, but its low catalytic activity suggests that its principal role is unlikely to oxidise tryptophan (19, 20).
Although both IDO1 and TDO catalyse the identical biochemical reaction, their physiological roles differ. IDO1 regulates peripheral immunity (21) and is induced by pro-inflammatory molecules including type I and II interferons, TNF-α, lipopolysaccharide, and prostaglandin E (22–25) in a wide range of cells including myeloid cells, fibroblasts, and cancer cells (26). Due to its inducible nature, IDO1 is absent in most tissues except for the sites where the body is exposed to non-self antigens such as lung, intestine, pregnant placenta, and lymphoid organs (27). In contrast, the evolutionarily older TDO is primarily expressed in the liver where it degrades excess dietary tryptophan (28–30). TDO can be induced by corticosteroids and activated by excess tryptophan (31–33).
The immunosuppressive role of the KP came to the fore in 1998 as a mechanism that confers an allogeneic foetus the ability to evade destruction by the mother’s T-cells (34). Researchers soon realised that the powerful immunosuppressive effect of KP could be co-opted by cancers to escape immune destruction (35, 36). It is now well established that a wide range of different cancer types thrive on accelerated tryptophan catabolism (27, 37–39). However, the mechanisms whereby KP regulates immunity are not completely understood.
Mechanisms Involved in Immunosuppression Mediated by Accelerated Tryptophan Catabolism
As KP is a metabolic pathway, its immune regulatory role has been attributed mainly to tryptophan deprivation and the accumulation of the kynurenine pathway metabolites (Figure 1) (40). However, IDO1 also has a non-enzymatic function in which the enzyme acts as a signalling protein in a non-canonical NF-κB pathway driven by immunosuppressive cytokine TGF-β (41–43). The contribution of each of these mechanisms to the immune regulation is actively discussed in the research community but accumulating evidence suggests that kynurenine is likely the main culprit (44). Increased kynurenine levels have been associated with reduced function of Natural Killer cells (45) and T-cells (46, 47). Mechanistically, kynurenine’s immune regulatory function is primarily linked to a transcription factor called aryl hydrocarbon receptor (AhR). Binding of kynurenine to AhR induces differentiation and activation of immunosuppressive T-regulatory cells (48–53), contributes to the recruitment of tolerogenic myeloid cells such as macrophages (54), and increases expression of the immune checkpoint molecule PD-1 on tumour-specific CD8+ T-cells (Figure 1) (55).
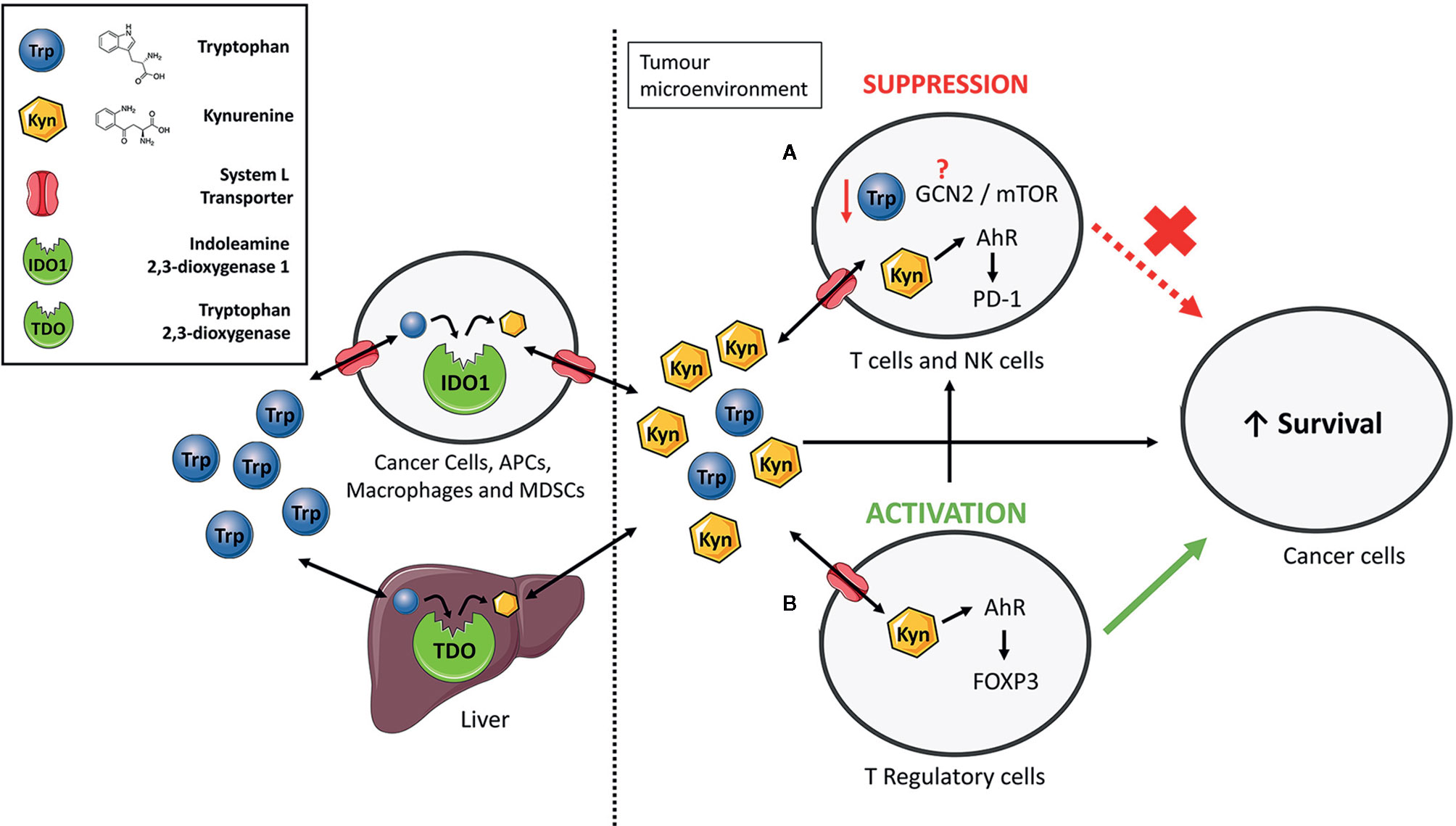
Figure 1 The immunosuppressive functions of IDO1/TDO-mediated tryptophan catabolism. Extrahepatic and hepatic cells express indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase (TDO) to consume tryptophan and give rise to numerous bioactive metabolites such as kynurenine. Elevated expression of IDO1 or TDO, such as in cancer, increases the relative kynurenine levels while reducing tryptophan content. Kynurenine enters cells via System L transporters. (A) Increased kynurenine levels inhibit proliferation of T-cells and natural killer (NK) cells by interacting with aryl hydrocarbon receptor (AhR) to express programmed cell death protein 1 (PD-1). Previous studies have suggested the involvement of the general control non-deprepressible-2 (GCN2) kinase and mammalian target of rapamycin (mTOR) in proliferation inhibition but the exact mechanism through which this occurs still remains unresolved. (B) Kynurenine induces differentiation of naïve CD4+ T-cells to immunosuppressive T-regulatory cells by activation of AhR and induction of the FoxP3 transcription factor. Taken together, an immune suppressed tumour microenvironment is created that promotes survival of cancer cells.
In contrast, the role of tryptophan deprivation in immune regulation is somewhat controversial. An earlier study has demonstrated that tryptophan deprivation inhibits proliferation and induces apoptosis in T-cells (56). But these experiments have been mostly carried out in the dish where tryptophan can get depleted. As tumours accelerating tryptophan catabolism still contain sufficient levels of tryptophan (57–59), it is likely that tumoural tryptophan levels cannot reach levels sufficiently low to activate the stress response pathways to low amino acid levels such as the general control non-derepressible-2 (GCN2) and the mammalian target of rapamycin (mTOR) (Figure 1). The key protagonist involved in regulation of T-cell responses to tryptophan deprivation was initially suspected to be the GCN2 kinase (60). GCN2 mediates conserved stress response pathway to amino acid deprivation in eukaryotes and was shown to be activated in T-cells in response to tryptophan deprivation (60, 61). However, this finding was recently challenged by two studies. Sonner and colleagues demonstrated no difference in the level of immune responses between the GCN2-proficient and GCN2-deficient T-cells against B16 melanomas (59). Similarly, tryptophan deprived T-cells ceased proliferation even in the absence of the GCN2 gene (62). The unlikely role of GCN2 as a low tryptophan sensor in immune cells is further corroborated by studies questioning the canonical role of GCN2 as a sensor of amino acid deficiency in mammals. GCN2 stress response pathway has been widely accepted as a mechanism for maintenance of amino acid homeostasis by controlling the feeding behaviour of omnivores (63). However, more recent studies challenged this paradigm as no significant difference in feeding behaviour was observed between the GCN2-deficient and GCN2-proficient mice that were amino acid-deprived (64). Complementary to GCN2, the mTOR senses amino acid sufficiency (65) and was proposed as a mediator of cellular stress response to low tryptophan levels. Metz and colleagues reported the repression of mTOR kinase activity in tryptophan-deprived HeLa cells, which eventually led to cell cycle arrest and apoptosis (66). It is not yet understood if mTOR could sense low tryptophan levels in immune cells.
The collective evidence accumulated to date tends to favour the conclusion that tryptophan serves primarily as a kynurenine precursor rather than inducing stress by its deprivation in vivo (67). However, a recent study by Schramme and colleagues provides a clue to a new immune regulatory function of tryptophan (57). In this study, elevated (5 to 10-fold) systemic tryptophan levels reaching 500 µM in TDO2- knockout mice overturned tumoural immune suppression induced by IDO1. Consequently, anti-PD1 immune checkpoint therapy alone was sufficient to impede the growth of IDO1-proficient MC38 colon tumours in these TDO2-knockout mice (57). That is a striking observation but how can it be explained mechanistically? How can elevation in circulating tryptophan levels overcome IDO1-mediated immunosuppression in the tumour? We posit that elevated tryptophan levels reverse the immunosuppression by outcompeting kynurenine for entry into T-cells through a shared amino acid transporter.
Tryptophan and Kynurenine: Transporter Competitors
IDO1 and TDO are intracellular enzymes; hence they require the cells to import tryptophan from the extracellular space. Import of large amino acids such as tryptophan typically occurs through the transporter called System L (68, 69). System L transporters are heterodimeric transmembrane proteins comprising a glycoprotein heavy chain (CD98) and a catalytic light chain (LAT1 or LAT2). Kynurenine can also be transported into cells through the System L transporters (70). It has been suggested that the transporters are bidirectional and can exchange tryptophan for kynurenine in cancer cells (71). This would explain the ability of cancer cells to siphon tryptophan from the microenvironment and enrich it with kynurenine to create an immunosuppressive milieu. As System L transports a broad range of amino acids, the transporter substrates compete with each other, and the probability of interacting with the transporter depends on the relative levels of the specific amino acid and their respective affinity for the transporter (72).
This observation indicates that the relative ratio of tryptophan to kynurenine will influence the amount of kynurenine entering T-cells. Therefore, tryptophan concentration can be viewed as a rheostat that modulates kynurenine entry into T-cells and the resulting immunosuppression (Figure 2). This concept is supported by literature evidence. The vacancy of System L transporter influences the ability of kynurenine to enter the CD4+ T-cells and activate AhR (73). Further, both low tryptophan levels and kynurenine accumulation appear to be pivotal for immunosuppression mediated by tryptophan catabolism (47), and tryptophan supplementation reverses the proliferation arrest of IDO1-mediated tryptophan deprivation in T-cells (60, 74). Tryptophan acting as a rheostat of kynurenine-mediated immunosuppression has two important therapeutic implications. Firstly, it can aid to resolve the conundrum whether the IDO1-selective, TDO-selective or dual IDO1/TDO inhibitors would be the most optimal therapeutic agents to block KP. Secondly, it can serve as a basis for a new approach to silence kynurenine-mediated immunosuppression by oral supplementation with System L transporter substrates. We will discuss these two areas in the following section.
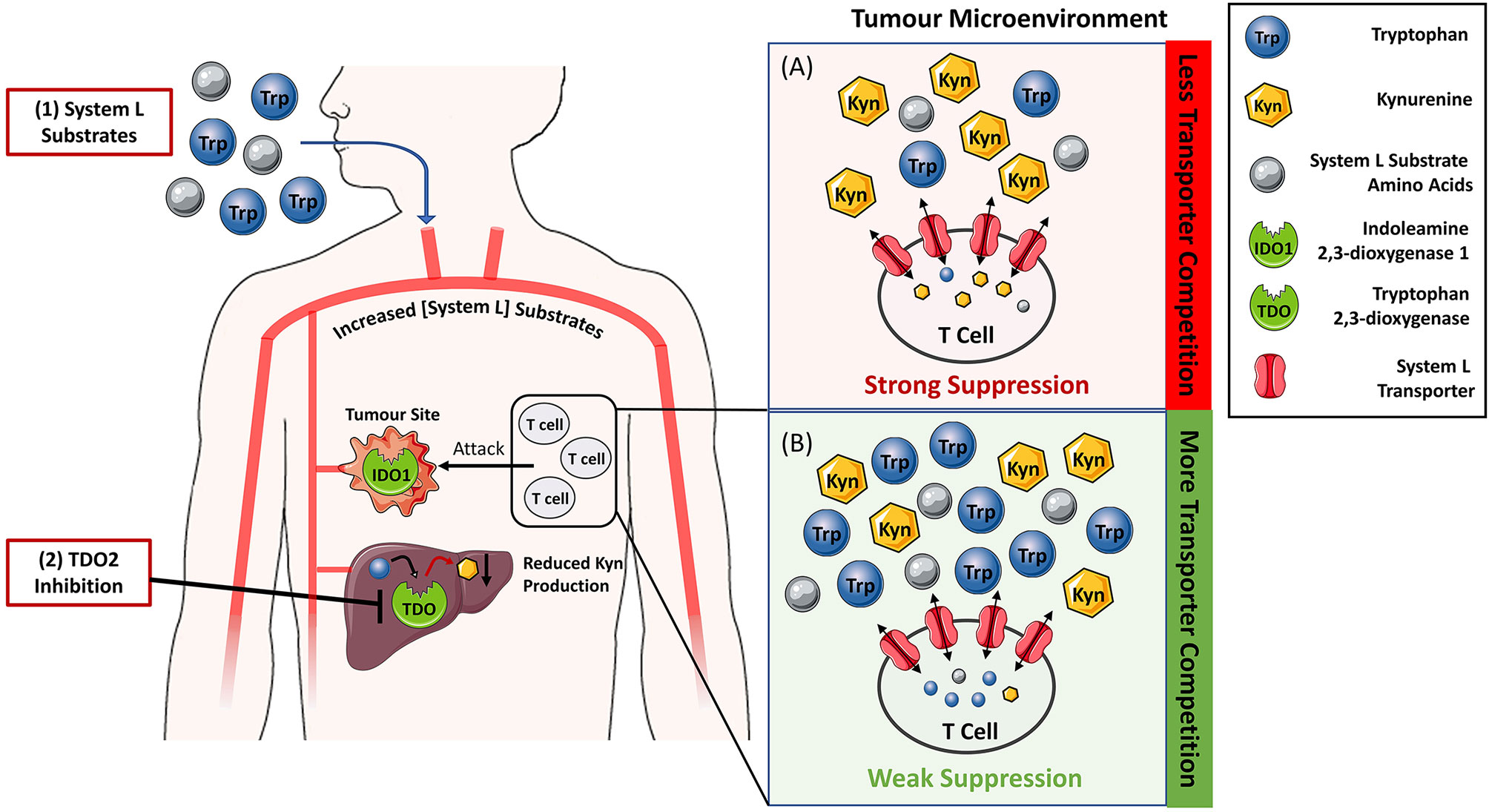
Figure 2 Reversing IDO1/TDO-mediated immunosuppression by increasing the levels of System L transporter substrates to limit kynurenine entry into T-cells. (A) In tumour microenvironment rich in indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase 2 (TDO), the tryptophan to kynurenine ratio is typically low leading to the suppression of T-cell activity and tumour killing. Blockade of TDO enzymatic activity by small molecule inhibitors (2) and/or supplementation with System L substrates such as leucine (1) is expected to increase their blood levels. Hence, the ratio of System L substrates to kynurenine in the tumour microenvironment (B) will also be increased. Elevated System L substrate levels competitively inhibit kynurenine entry into T cells so that T cell suppression is reduced.
Discussion
Efficient Silencing of Kynurenine Pathway May Require Only TDO-Selective Inhibitors
Many cancer types co-opt IDO1, TDO or both enzymes (27, 39, 75) to accelerate tryptophan catabolism and escape immune destruction. Upregulation of IDO1 or elevated kynurenine levels associate with poor patient outcomes (37, 76–79) and resistance to immune checkpoint therapy such as anti-PD-1 inhibitors (80, 81). Moreover, anti-PD-L1 therapy promotes tryptophan catabolism as a consequence of IDO1 upregulation by IFN-γ secreted by re-invigorated tumour-infiltrating lymphocytes (82). These observations provide a strong mechanistic rationale for combining the tryptophan catabolism blockade with immune checkpoint inhibitors and potentially other cancer immunotherapies. Targeting the KP also offers certain advantages over targeting cell surface immune-regulatory molecules such as PD-1. Proteins on the KP including IDO1 and TDO are intracellular enzymes (40, 83) that harbour active sites easily targetable with inexpensive and non-immunogenic small molecules. Such molecules are stable on storage, can be administered orally, and penetrate into the brain. This is in contrast to immune checkpoint molecules such as PD-1 that do not catalyse a biochemical reaction hence lack an active site, and typically require costly antibodies for therapeutic modulation. Due to their size and instability, antibodies need to be administered by injection and cannot pass the blood brain barrier. Of note, small molecules that disrupt PD-1/PD-L1 interaction are currently in development (84, 85).
Major effort has been devoted to disable tryptophan catabolism using IDO1 inhibitors (7, 86, 87). Whilst IDO1 inhibitors can boost immunotherapy in mouse cancer models (88–91), the most advanced IDO1 inhibitor Epacadostat (INCB024360) could not potentiate anti-PD1 inhibitor pembrolizumab in a recent Phase III trial involving about 700 advanced melanoma patients (9, 92). Reasons for this negative outcome are unclear and discussed extensively elsewhere (11–13, 93, 94) but poor target engagement, compensatory expression of TDO or IDO2, dose-limiting toxicities or the lack of selection for IDO1-positive patients are likely one of the contributing factors.
Unlike IDO1 inhibitors, no TDO-specific inhibitor has yet reached clinical trials. This is not surprising given the paucity of potent TDO-selective inhibitors. High affinity TDO inhibitors seems to be much more difficult to develop than IDO1 inhibitors. That is likely because the TDO’s active site is less flexible than that of IDO1 and thus cannot accommodate bulky ligands (7, 95). As a consequence, the majority of TDO-specific inhibitors [reviewed in (7, 96)] mimic tryptophan (28, 36, 97–100). Currently, the most promising TDO inhibitors appear to be the derivatives of IDO1-specific clinical candidate Navoximod (100) reported by Genentech, and PF06845102 (57) developed by iTeos Therapeutics. These inhibitors display submicromolar potencies, up to 100-fold TDO selectivity over IDO1, good metabolic stability and ability to raise systemic tryptophan levels in mice (57, 100). PF06845102 has also been shown to potentiate anti-tumour activity of the anti-CTLA4 immune checkpoint inhibitor in a mouse model of colorectal cancer (57). These promising data support further development of next-generation TDO inhibitors.
The hypothesis presented in this perspective suggests that TDO inhibition can be advantageous to IDO1 inhibition. Firstly, TDO inhibition is expected to indirectly inhibit the immunosuppressive action of IDO1 by raising the systemic levels of tryptophan and limiting entry of IDO1-generated kynurenine into T-cells (Figure 2). This concept assumes relatively low contribution of non-enzymatic signalling function of IDO1 to the overall immunosuppression. Secondly, unlike IDO1 inhibitors, TDO inhibitors do not require tumoural TDO expression because TDO is constitutively expressed in the liver. This is clearly demonstrated in studies where TDO2-knockout mice but not IDO1-knockout mice have markedly increased plasma tryptophan levels compared to their respective wild-type counterparts (29, 57). Therefore, TDO inhibition is anticipated to silence kynurenine-mediated immunosuppression in a greater subset of patients. The potential toxicity of hepatic TDO blockade in humans still remains unresolved. However, the absence of serious clinical pathologies of a woman diagnosed with hypertryptophanaemia due to TDO deficiency (101) suggests that TDO inhibition will be well tolerated in humans.
As cancers can express both IDO1 and TDO, the industry has been pursuing the development of dual IDO1/TDO inhibitors. This concept has not yet been supported by strong evidence but some dual inhibitors are in preclinical development or Phase I trials (40, 102, 103) including Navoximod (89) which was originally thought to be an IDO1-selective inhibitor. Similarly to IDO1 inhibitors, we contend that development of dual inhibitors may be unnecessary. However, it cannot be excluded that IDO1 inhibition will be needed to complement TDO blockade. Inhibition of hepatic TDO may not increase tryptophan levels significantly in humans. Further, it is possible that kynurenine enters cells through a transporter other than System L or triggers the immunosuppressive effect in the absence of secretion from the IDO1/TDO-expressing cells.
Can High-Dose Amino Acid Supplementation Reverse Kynurenine Mediated Immunosuppression and Potentiate Immunotherapy?
We propose that any strategy that safely increases the levels of circulating System L substrates to out-compete kynurenine has potential to reverse the IDO1/TDO mediated immunosuppression. One additional possibility to TDO inhibition is oral supplementation with amino acids that are System L substrates (Figure 2) such as leucine, isoleucine, valine, phenylalanine, tyrosine, tryptophan, methionine, or histidine. Whilst tryptophan supplementation emerges as a possibility, it is unlikely to increase systemic tryptophan levels because hepatic TDO efficiently breaks down excess tryptophan. This is consistent with the study of Schramme et al. showing that three-fold increase of tryptophan in the diet from 0.06 to 0.18% did not increase circulating tryptophan levels of the mice (57). Further, tryptophan supplementation (30 mg per mouse) did not significantly impede growth of mouse CT26 colon tumours (104). On the other hand, there are preclinical data showing that leucine, a high affinity substrate of System L, limits System L-mediated entry of kynurenine into brain (105). There are no data available to show if high-dose dietary supplementation with leucine or any other amino acid would translate into improved tumour control or blockade of kynurenine-mediated immunosuppression. However, the above-mentioned study strongly supports the feasibility of limiting kynurenine transport in vivo at leucine doses that are well tolerated by an organism.
The safety of high dose amino acid supplementation raises a potential concern. It is generally assumed that amino acids do not pose serious health hazards as they are natural substances produced endogenously and part of human diet and supplements (106). Perhaps not surprisingly, toxicities associated with high dose amino acid supplementation to mammals differ significantly (107) but leucine appears to be the least toxic amino acid. Oral or intravenous supplementation of leucine (5 g–6 g) increased systemic leucine levels in humans in the absence of overt toxicities (108, 109). Similarly, as stated in the preceding paragraph, elevated leucine levels sufficient to prevent kynurenine transport are well tolerated by mice. This is in contrast to methionine and histidine which, at high doses, appear to be one of the most toxic amino acids to humans (110–112). However, the toxicity of a substance depends on its dose. It is therefore likely that even the seemingly most toxic amino acids may prevent kynurenine entry into T-cells at levels which are well tolerated by an organism. Experimental studies will be necessary to rigorously investigate this concept and determine which amino acids and at what doses will provide therapeutic benefit, if any.
Overall, the therapeutic supplementation with a high dose of amino acids with the intent to inhibit kynurenine-mediated immunosuppression appears like a highly feasible and exciting research prospect. It offers a simple and economical alternative to synthetic drugs inhibiting tryptophan catabolising enzymes or downstream kynurenine targets such as AhR.
Summary
The disappointing outcome of the Phase III trial of Epacadostat (9) has stimulated search for alternative approaches to silence the KP. Such approaches include kynurenine depletion by kynureninase (44), small molecule inhibitors to the kynurenine’s downstream target AhR (81, 94), and perhaps we will see small molecule inhibitors to a recently discovered tryptophan metabolising enzyme IL4I1 that produces AhR agonists (113). This perspective proposes a novel function for the IDO1’s substrate tryptophan that could lead to an additional therapeutic strategy to block KP. We posit that tryptophan acts as a rheostat of kynurenine-mediated immunosuppression, i.e., high tryptophan to kynurenine ratio limits kynurenine’s entry into immune T-cells through the shared System L amino acid transporter.
Therefore, increasing circulating levels of System L substrates can relieve kynurenine-induced immunosuppression. One way to achieve this is via inhibition of hepatic kynurenine pathway by TDO inhibitors. This supports the development of TDO-selective inhibitors that, unlike IDO1 inhibitors, are not contingent on tumoural TDO expression. Alternatively, kynurenine can be out-competed by therapeutic supplementation of amino acids such as leucine which is a high-affinity System L substrate. Leucine supplementation appears highly feasible. Leucine has low toxicity to mammals and was shown to block kynurenine entry into the mouse brain. If confirmed, we envision the amino acid supplementation strategy will enrich the armamentarium of therapeutic approaches modulating KP, and increase the likelihood of realising the prospect of silencing the KP for cancer immunotherapy.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author Contributions
PT conceived, designed and supervised the study. MK drafted the manuscript and figures. All authors contributed to the article and approved the submitted version.
Funding
PT and MK acknowledge support from Health Research Council New Zealand through the Emerging Researcher First Grants 17/586 awarded to PT. PT acknowledges additional support from Auckland Medical Research Foundation Project grant 1120009 awarded to PT, School of Medical Sciences at the University of Auckland in New Zealand and Auckland Cancer Society Research Centre. The funding bodies had no role in design of this study, decision to publish and preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AhR, Aryl hydrocarbon Receptor; GCN2, General control non-derepressible 2; IDO1, Indoleamine 2,3-dioxygenase 1; KP, Kynurenine Pathway; mTOR, Mammalian target of rapamycin; PD-1, Programmed cell death protein 1; TDO, Tryptophan 2,3-dioxygenase.
References
1. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFN[gamma] and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature (2001) 410:1107–11. doi: 10.1038/35074122
2. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol (2002) 3:991–8. doi: 10.1038/ni1102-991
3. Hoos A. Development of immuno-oncology drugs [mdash] from CTLA4 to PD1 to the next generations, Nature reviews. Drug Discov (2016) 15:235–47. doi: 10.1038/nrd.2015.35
4. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer (2019) 19:133–50. doi: 10.1038/s41568-019-0116-x
5. Bader JE, Voss K, Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol Cell (2020) 78:1019–33. doi: 10.1016/j.molcel.2020.05.034
6. Egen JG, Ouyang W, Wu LC. Human anti-tumor immunity: insights from immunotherapy clinical trials. Immunity (2020) 52:36–54. doi: 10.1016/j.immuni.2019.12.010
7. Dolšak A, Gobec S, Sova M. Indoleamine and tryptophan 2,3-dioxygenases as important future therapeutic targets. Pharmacol Ther (2020), 107746. doi: 10.1016/j.pharmthera.2020.107746
8. Jung KH, LoRusso P, Burris H, Gordon M, Bang Y-J, Hellmann MD, et al. Phase I study of the indoleamine 2,3-Dioxygenase 1 (IDO1) inhibitor Navoximod (GDC-0919) administered with PD-L1 inhibitor (Atezolizumab) in advanced solid tumors. Clin Cancer Res (2019) 25:3220–8. doi: 10.1158/1078-0432.CCR-18-2740
9. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol (2019) 20:1083–97. doi: 10.1016/S1470-2045(19)30274-8
10. Reardon DA, Desjardins A, Rixe O, Cloughesy T, Alekar S, Williams JH, et al. A phase 1 study of PF-06840003, an oral indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor in patients with recurrent malignant glioma. Invest New Drugs (2020) 10:1784–95. doi: 10.1007/s10637-020-00950-1
11. Günther J, Däbritz J, Wirthgen E. Limitations and off-target effects of tryptophan-related ido inhibitors in cancer treatment. Front Immunol (2019) 10:1–8. doi: 10.3389/fimmu.2019.01801
12. Eynde BJV, Baren N, Baurain J-F. Is There a clinical future for ido1 inhibitors after the failure of epacadostat in melanoma? Annu Rev Cancer Biol (2020) 4:241–56. doi: 10.1146/annurev-cancerbio-030419-033635
13. Muller AJ, Manfredi MG, Zakharia Y, Prendergast GC. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin Immunopathol (2018) 41:41–8. doi: 10.1007/s00281-018-0702-0
14. Leklem JE. Quantitative aspects of tryptophan metabolism in humans and other species: a review. Am J Clin Nutr (1971) 24:659–72. doi: 10.1093/ajcn/24.6.659
15. Hopper AT, Campbell BM, Kao H, Pintchovski SA, Staal RGW. Recent developments in targeting neuroinflammation in disease. In: . Annual Reports in Medicinal Chemistry Volume, vol. 47. Elsevier (2012). p. 37–53.
16. Badawy AA. Hypothesis kynurenic and quinolinic acids: The main players of the kynurenine pathway and opponents in inflammatory disease. Med Hypotheses (2018) 118:129–38. doi: 10.1016/j.mehy.2018.06.021
17. Kolodziej LR, Paleolog EM, Williams RO. Kynurenine metabolism in health and disease. Amino Acids (2011) 41:1173–83. doi: 10.1007/s00726-010-0787-9
18. Ball HJ, Jusof FF, Bakmiwewa SM, Hunt NH, Yuasa H. Tryptophan catabolizing enzymes – party of three. Front Immunol (2014) 5:1–10. doi: 10.3389/fimmu.2014.00485
19. Ball HJ, Sanchez-Perez A, Weiser S, Austin CJD, Astelbauer F, Miu J, et al. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene (2007) 396:203–13. doi: 10.1016/j.gene.2007.04.010
20. Metz R, DuHadaway JB, Kamasani U, Laury-Kleintop L, Muller AJ, Prendergast GC. Novel Tryptophan Catabolic Enzyme IDO2 Is the Preferred Biochemical Target of the Antitumor Indoleamine 2,3-Dioxygenase Inhibitory Compound d-1-Methyl-Tryptophan. Cancer Res (2007) 67:7082–7. doi: 10.1158/0008-5472.CAN-07-1872
21. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol (2004) 4:762–74. doi: 10.1038/nri1457
22. Fujigaki S, Saito K, Sekikawa K, Tone S, Takikawa O, Fujii H, et al. Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN-γ-independent mechanism. Eur J Immunol (2001) 31:2313–8. doi: 10.1002/1521-4141(200108)31:8<2313::AID-IMMU2313>3.0.CO;2-S
23. Grant RS. Indoleamine 2,3-Dioxygenase Activity Increases NAD+ Production in IFN-gamma-Stimulated human primary mononuclear cells. Int J Tryptophan Res (2018) 11:1–8. doi: 10.1177/1178646917751636
24. Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J (1991) 5:2516–22. doi: 10.1096/fasebj.5.11.1907934
25. Trabanelli S, Očadlíková D, Ciciarello M, Salvestrini V, Lecciso M, Jandus C, et al. The SOCS3-independent expression of ido2 supports the homeostatic generation of t regulatory cells by human dendritic cells. J Immunol (2014) 192:1231–40. doi: 10.4049/jimmunol.1300720
26. Meireson A, Devos M, Brochez L. IDO expression in cancer: different compartment, different functionality? Front Immunol (2020) 11:1–17. doi: 10.3389/fimmu.2020.531491
27. Théate I, van Baren N, Pilotte L, Moulin P, Larrieu P, Renauld J-C, et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol Res (2015) 3:161–72. doi: 10.1158/2326-6066.CIR-14-0137
28. Salter M, Hazelwood R, Pogson CI, Iyer R, Madge DJ. The effects of a novel and selective inhibitor of tryptophan 2,3-dioxygenase on tryptophan and serotonin metabolism in the rat. Biochem Pharmacol (1995) 49:1435–42. doi: 10.1016/0006-2952(95)00006-L
29. Kanai M, Funakoshi H, Takahashi H, Hayakawa T, Mizuno S, Matsumoto K, et al. Tryptophan 2,3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in mice. Mol Brain (2009) 2:1–16. doi: 10.1186/1756-6606-2-8
30. Mehler AH, Knox WE. The conversion of tryptophan to kynurenine in liver: II. The enzymatic hydrolysis of formylkynurenine. J Biol Chem (1950) 187:431–8. doi: 10.1016/S0021-9258(19)50968-1
31. Li JS, Han Q, Fang J, Rizzi M, James AA, Li J. Biochemical mechanisms leading to tryptophan 2,3-dioxygenase activation. Arch Insect Biochem Physiol (2007) 64:74–87. doi: 10.1002/arch.20159
32. Green AR, Joseph MH, Woods HF. Proceedings: tryptophan metabolism by the isolated perfused rat liver. Br J Pharmacol (1975) 55:253–4. doi: 10.1111/j.1476-5381.1976.tb07660.x
33. Danesch U, Gloss B, Schmid W, Schütz G, Schüle R, Renkawitz R. Glucocorticoid induction of the rat tryptophan oxygenase gene is mediated by two widely separated glucocorticoid-responsive elements. EMBO J (1987) 6:625–30. doi: 10.1002/j.1460-2075.1987.tb04800.x
34. Munn DH, Zhou M, Attwood JT, Bondarev I. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science (1998) 281:1191–3. doi: 10.1126/science.281.5380.1191
35. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9:1269–74. doi: 10.1038/nm934
36. Pilotte L, Larrieu P, Stroobant V, Colau D, Dolušić E, Frédérick R, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci (2012) 109:2497–502. doi: 10.1073/pnas.1113873109
37. Sucher R, Kurz K, Weiss G, Margreiter R, Fuchs D, Brandacher G. IDO-mediated tryptophan degradation in the pathogenesis of malignant tumor disease. Int J Tryptophan Res (2010) 3:113–20. doi: 10.4137/IJTR.S4157
38. van Baren N, Van den Eynde BJ. Tryptophan-degrading enzymes in tumoral immune resistance. Front Immunol (2015) 6:1–9. doi: 10.3389/fimmu.2015.00034
39. Hoffmann D, Dvorakova T, Stroobant V, Bouzin C, Daumerie A, Solvay M, et al. Tryptophan 2,3-dioxygenase expression identified in human hepatocellular carcinoma cells and in intratumoral pericytes of most cancers. Cancer Immunol Res (2019) 8:19–31. doi: 10.1158/2326-6066.CIR-19-0040
40. Platten M, Nollen EAA, Röhrig UF, Fallarino F, Opitz CA. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov (2019) 18:379–401. doi: 10.1038/s41573-019-0016-5
41. Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol (2011) 12:870–8. doi: 10.1038/ni.2077
42. Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature (2014) 511:184–90. doi: 10.1038/nature13323
43. Pallotta MT, Fallarino F, Matino D, Macchiarulo A, Orabona C. AhR-mediated, non-genomic modulation of IDO1 function. Front Immunol (2014) 5:1–6. doi: 10.3389/fimmu.2014.00497
44. Triplett TA, Garrison KC, Marshall N, Donkor M, Blazeck J, Lamb C, et al. Reversal of indoleamine 2,3-dioxygenase–mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat Biotechnol (2018) 36:758–64. doi: 10.1038/nbt.4180
45. Chiesa MD, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood (2006) 108:4118–25. doi: 10.1182/blood-2006-03-006700
46. Rad Pour S, Morikawa H, Kiani NA, Yang M, Azimi A, Shafi G, et al. Exhaustion of CD4+ T-cells mediated by the Kynurenine Pathway in Melanoma. Sci Rep (2019) 9:12150. doi: 10.1038/s41598-019-48635-x
47. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells. J Immunol (2006) 176:6752–61. doi: 10.4049/jimmunol.176.11.6752
48. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature (2011) 478:197–203. doi: 10.1038/nature10491
49. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol (2010) 185:3190–8. doi: 10.4049/jimmunol.0903670
50. Gutierrez-Vazquez C, Quintana FJ. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity (2018) 48:19–33. doi: 10.1016/j.immuni.2017.12.012
51. Brenk M, Scheler M, Koch S, Neumann J, Takikawa O, Hacker G, et al. Tryptophan deprivation induces inhibitory receptors ILT3 and ILT4 on dendritic cells favoring the induction of human CD4+CD25+ Foxp3+ T regulatory cells. J Immunol (2009) 183:145–54. doi: 10.4049/jimmunol.0803277
52. Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8:523–32. doi: 10.1038/nri2343
53. Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci (2010) 107:19961–6. doi: 10.1073/pnas.1014465107
54. Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao C-C, et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci (2019) 22:729–40. doi: 10.1038/s41593-019-0370-y
55. Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell (2018) 33:480–94.e487. doi: 10.1016/j.ccell.2018.02.005
56. Lee GK, Park HJ, Macleod M, Chandler P, Munn DH, Mellor AL. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology (2002) 107:452–60. doi: 10.1046/j.1365-2567.2002.01526.x
57. Schramme F, Crosignani S, Frederix K, Hoffmann D, Pilotte L, Stroobant V, et al. Inhibition of tryptophan-dioxygenase activity increases the antitumor efficacy of immune checkpoint inhibitors. Cancer Immunol Res (2019) 8:32–45. doi: 10.1158/2326-6066.CIR-19-0041
58. Koblish HK, Hansbury MJ, Bowman KJ, Yang G, Neilan CL, Haley PJ, et al. Hydroxyamidine Inhibitors of Indoleamine-2,3-dioxygenase Potently Suppress Systemic Tryptophan Catabolism and the Growth of IDO-Expressing Tumors. Mol Cancer Ther (2010) 9:489–98. doi: 10.1158/1535-7163.MCT-09-0628
59. Sonner JK, Deumelandt K, Ott M, Thomé CM, Rauschenbach KJ, Schulz S, et al. The stress kinase GCN2 does not mediate suppression of antitumor T cell responses by tryptophan catabolism in experimental melanomas. Oncoimmunology (2016) 5:e1240858. doi: 10.1080/2162402X.2016.1240858
60. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity (2005) 22:633–42. doi: 10.1016/j.immuni.2005.03.013
61. Castilho BA, Shanmugam R, Silva RC, Ramesh R, Himme BM, Sattlegger E. Keeping the eIF2 alpha kinase Gcn2 in check. Biochim Biophys Acta (BBA) - Mol Cell Res (2014) 1843:1948–68. doi: 10.1016/j.bbamcr.2014.04.006
62. Van de Velde LA, Guo XJ, Barbaric L, Smith AM, Oguin TH3, Thomas PG, et al. Stress Kinase GCN2 Controls the Proliferative Fitness and Trafficking of Cytotoxic T Cells Independent of Environmental Amino Acid Sensing. Cell Rep (2016) 17:2247–58. doi: 10.1016/j.celrep.2016.10.079
63. Maurin A-C, Jousse C, Averous J, Parry L, Bruhat A, Cherasse Y, et al. The GCN2 kinase biases feeding behavior to maintain amino acid homeostasis in omnivores. Cell Metab (2005) 1:273–7. doi: 10.1016/j.cmet.2005.03.004
64. Leib DE, Knight ZA. Re-examination of Dietary Amino Acid Sensing Reveals a GCN2-Independent Mechanism. Cell Rep (2015) 13:1081–9. doi: 10.1016/j.celrep.2015.09.055
65. Guertin DA, Sabatini DM. Defining the Role of mTOR in Cancer. Cancer Cell (2007) 12:9–22. doi: 10.1016/j.ccr.2007.05.008
66. Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology (2012) 1:1460–8. doi: 10.4161/onci.21716
67. Badawy AAB, Namboodiri AMA, Moffett JR. The end of the road for the tryptophan depletion concept in pregnancy and infection. Clin Sci (2016) 130:1327–33. doi: 10.1042/CS20160153
68. Segawa H, Fukasawa Y, Miyamoto K, Takeda E, Endou H, Kanai Y. Identification and Functional Characterization of a Na+ independent Neutral Amino Acid Transporter with Broad Substrate Selectivity. J Biol Chem (1999) 274:19745–51. doi: 10.1074/jbc.274.28.19745
69. Kudo Y, Boyd CAR. The role of L-tryptophan transport in L-tryptophan degradation by indoleamine 2,3-dioxygenase in human placental explants. J Physiol (2001) 531:417–23. doi: 10.1111/j.1469-7793.2001.0417i.x
70. Sekine A, Okamoto M, Kanatani Y, Sano M, Shibata K, Fukuwatari T. Amino acids inhibit kynurenic acid formation via suppression of kynurenine uptake or kynurenic acid synthesis in rat brain in vitro. Springerplus (2015) 4:48. doi: 10.1186/s40064-015-0826-9
71. Verrey F. System L: heteromeric exchangers of large, neutral amino acids involved in directional transport. Pflugers Arch (2003) 445:529–33. doi: 10.1007/s00424-002-0973-z
72. Hoglund E, Overli O, Winberg S. Tryptophan Metabolic Pathways and Brain Serotonergic Activity: A Comparative Review. Front Endocrinol (Lausanne) (2019) 10:158. doi: 10.3389/fendo.2019.00158
73. Sinclair LV, Neyens D, Ramsay G, Taylor PM, Cantrell DA. Single cell analysis of kynurenine and System L amino acid transport in T cells. Nat Commun (2018) 9:1981. doi: 10.1038/s41467-018-04366-7
74. Silk JD, Lakhal S, Laynes R, Vallius L, Karydis I, Marcea C, et al. IDO induces expression of a novel tryptophan transporter in mouse and human tumor cells. J Immunol (2011) 187:1617–25. doi: 10.4049/jimmunol.1000815
75. Li H, Ning S, Ghandi M, Kryukov GV, Gopal S, Deik A, et al. The landscape of cancer cell line metabolism. Nat Med (2019) 25:850–60. doi: 10.1038/s41591-019-0404-8
76. Ferns DM, Kema IP, Buist MR, Nijman HW, Kenter GG, Jordanova ES. Indoleamine-2,3-dioxygenase (IDO) metabolic activity is detrimental for cervical cancer patient survival. OncoImmunology (2015) 4:e981457. doi: 10.4161/2162402X.2014.981457
77. Weinlich G, Murr C, Richardsen L, Winkler C, Fuchs D. Decreased serum tryptophan concentration predicts poor prognosis in malignant melanoma patients. Dermatology (2007) 214:8–14. doi: 10.1159/000096906
78. Tanizaki Y, Kobayashi A, Toujima S, Shiro M, Mizoguchi M, Mabuchi Y, et al. Indoleamine 2,3-dioxygenase promotes peritoneal metastasis of ovarian cancer by inducing an immunosuppressive environment. Cancer Sci (2014) 105:966–73. doi: 10.1111/cas.12445
79. Masaki A, Ishida T, Maeda Y, Suzuki S, Ito A, Takino H, et al. Prognostic significance of tryptophan catabolism in adult T-cell leukemia/lymphoma. Clin Cancer Res (2015) 21:2830–9. doi: 10.1158/1078-0432.CCR-14-2275
80. Li H, Bullock K, Gurjao C, Braun D, Shukla SA, Bosse D, et al. Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat Commun (2019) 10:4346. doi: 10.1038/s41467-019-12361-9
81. Campesato LF, Budhu S, Tchaicha J, Weng C-H, Gigoux M, Cohen IJ, et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat Commun (2020) 11:4011. doi: 10.1038/s41467-020-17750-z
82. Gomes B, Driessens G, Bartlett D, Cai D, Cauwenberghs S, Crosignani S, et al. Characterization of the Selective Indoleamine 2,3-Dioxygenase-1 (IDO1) Catalytic Inhibitor EOS200271/PF-06840003 Supports IDO1 as a Critical Resistance Mechanism to PD-(L)1 Blockade Therapy. Mol Cancer Ther (2018) 17:2530–42. doi: 10.1158/1535-7163.MCT-17-1104
83. Dounay AB, Tuttle JB, Verhoest PR. Challenges and Opportunities in the Discovery of New Therapeutics Targeting the Kynurenine Pathway. J Med Chem (2015) 58:8762–82. doi: 10.1021/acs.jmedchem.5b00461
84. Guzik K, Tomala M, Muszak D, Konieczny M, Hec A, Błaszkiewicz U, et al. Development of the Inhibitors That Target the PD-1/PD-L1 Interaction—A Brief Look at Progress on Small Molecules, Peptides and Macrocycles. Molecules (2019) 24:2071. doi: 10.3390/molecules24112071
85. Konieczny M, Musielak B, Kocik J, Skalniak L, Sala D, Czub M, et al. Di-bromo-Based Small-Molecule Inhibitors of the PD-1/PD-L1 Immune Checkpoint. J Med Chem (2020) 63:11271–85. doi: 10.1021/acs.jmedchem.0c01260
86. Cheong JE, Ekkati A, Sun L. A patent review of IDO1 inhibitors for cancer. Expert Opin Ther Pat (2018) 28:317–30. doi: 10.1080/13543776.2018.1441290
87. Sun L. Advances in the discovery and development of selective heme-displacing IDO1 inhibitors. Expert Opin Drug Discov (2020) 15(10): 1–10. doi: 10.1080/17460441.2020.1781811
88. Spranger S, Koblish H, Horton B, Scherle P, Newton R, Gajewski T. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J Immunother Cancer (2014) 2:3. doi: 10.1186/2051-1426-2-3
89. Tu W, Yang F, Xu G, Chi J, Liu Z, Peng W, et al. Discovery of Imidazoisoindole Derivatives as Highly Potent and Orally Active Indoleamine-2,3-dioxygenase Inhibitors. ACS Med Chem Lett (2019) 10:949–53. doi: 10.1021/acsmedchemlett.9b00114
90. Zhang H, Liu K, Pu Q, Achab A, Ardolino MJ, Cheng M, et al. Discovery of Amino-cyclobutarene-derived Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitors for Cancer Immunotherapy. ACS Med Chem Lett (2019) 10:1530–6. doi: 10.1021/acsmedchemlett.9b00344
91. Watanabe T, Gaedicke S, Guffart E, Firat E, Niedermann G. Adding Indoximod to Hypofractionated Radiotherapy with Anti-PD-1 Checkpoint Blockade Enhances Early NK and CD8+ T-Cell–Dependent Tumor Activity. Clin Cancer Res (2020) 26:945–56. doi: 10.1158/1078-0432.CCR-19-0476
92. Jochems C, Fantini M, Fernando RI, Kwilas AR, Donahue RN, Lepone LM. The IDO1 selective inhibitor epacadostat enhances dendritic cell immunogenicity and lytic ability of tumor antigen-specific T cells. Oncotarget (2016) 7:37762–72. doi: 10.18632/oncotarget.9326
93. Sondak VK, Khushalani NI. Echoes of a failure: what lessons can we learn? Lancet Oncol (2019) 20(8):1037–9. doi: 10.1016/S1470-2045(19)30312-2
94. Opitz CA, Somarribas Patterson LF, Mohapatra SR, Dewi DL, Sadik A, Platten M, et al. The therapeutic potential of targeting tryptophan catabolism in cancer. Br J Cancer (2019) 122:30–44. doi: 10.1038/s41416-019-0664-6
95. Pham KN, Lewis-Ballester A, Yeh S-R. Structural Basis of Inhibitor Selectivity in Human Indoleamine 2,3-Dioxygenase 1 and Tryptophan Dioxygenase. J Am Chem Soc (2019) 141(47):18771–9. doi: 10.1021/jacs.9b08871
96. Kozlova A, Frédérick R. Current state on tryptophan 2,3-dioxygenase inhibitors: a patent review. Expert Opin Ther Pat (2019) 29:11–23. doi: 10.1080/13543776.2019.1556638
97. Dolušié E, Larrieu P, Moineaux L, Stroobant V, Pilotte L, Colau D, et al. Tryptophan 2,3-Dioxygenase (TDO) Inhibitors. 3-(2-(Pyridyl)ethenyl)indoles as Potential Anticancer Immunomodulators. J Med Chem (2011) 54:5320–34. doi: 10.1021/jm2006782
98. Pantouris G, Loudon-Griffiths J, Mowat CG. Insights into the mechanism of inhibition of tryptophan 2,3-dioxygenase by isatin derivatives. J Enzyme Inhib Med Chem (2016) 1–9:70–8. doi: 10.3109/14756366.2016.1170013
99. Pei Z, Mendonca R, Gazzard L, Pastor R, Goon L, Gustafson A, et al. Aminoisoxazoles as Potent Inhibitors of Tryptophan 2,3-Dioxygenase 2 (TDO2). ACS Med Chem Lett (2018) 9:417–21. doi: 10.1021/acsmedchemlett.7b00427
100. Parr BT, Pastor R, Sellers BD, Pei Z, Jaipuri FA, Castanedo GM, et al. Implementation of the CYP Index for the Design of Selective Tryptophan-2,3-dioxygenase Inhibitors. ACS Med Chem Lett (2020) 11:541–9. doi: 10.1021/acsmedchemlett.0c00004
101. Ferreira P, Shin I, Sosova I, Dornevil K, Jain S, Dewey D, et al. Hypertryptophanemia due to tryptophan 2,3-dioxygenase deficiency. Mol Genet Metab (2017) 120:317–24. doi: 10.1016/j.ymgme.2017.02.009
102. Gyulveszi G, Fischer C, Mirolo M, Stern M, Green L, Ceppi M, et al. Abstract LB-085: RG70099: A novel, highly potent dual IDO1/TDO inhibitor to reverse metabolic suppression of immune cells in the tumor micro-environment. AACR (2016) 76(14 Supplement). doi: 10.1158/1538-7445.AM2016-LB-085
103. Gullapalli S, Roychowdhury A, Khaladkar T, Sawargave S, Janrao R, Kalhapure V, et al. EPL-1410, a novel fused heterocycle based orally active dual inhibitor of IDO1/TDO2, as a potential immune-oncology therapeutic. Cancer Res (2018) 78:1701. doi: 10.1158/1538-7445.AM2018-1701
104. Satoh Y, Kotani H, Iida Y, Taniura T, Notsu Y, Harada M. Supplementation of l-arginine boosts the therapeutic efficacy of anticancer chemoimmunotherapy. Cancer Sci (2020) 111:2248–58. doi: 10.1111/cas.14490
105. Walker AK, Wing EE, Banks WA, Dantzer R. Leucine competes with kynurenine for blood-to-brain transport and prevents lipopolysaccharide-induced depression-like behavior in mice. Mol Psychiatry (2019) 24:1523–32. doi: 10.1038/s41380-018-0076-7
106. Roberts A. The Safety and Regulatory Process for Amino Acids in Europe and the United States. J Nutr (2016) 146:2635S–42S. doi: 10.3945/jn.116.234591
107. Garlick PJ. The Nature of Human Hazards Associated with Excessive Intake of Amino Acids. J Nutr (2004) 134:1633S–9S. doi: 10.1093/jn/134.6.1633S
108. Anderson SA, Raiten DJ. Safety of amino acids used as dietary supplements. Bethesda, Md. (9650 Rockville Pike, Bethesda 20814-3998): Life Sciences Research Office, Federation of American Societies for Experimental Biology (1992).
109. Lupton JR, Brooks J, Butte N, Caballero B, Flatt J, Fried S. Dietary reference intakes for energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein, and amino acids. vol. 5. Washington, DC, USA: National Academy Press (2002). p. 589–768.
110. Benevenga N, Steele R. Adverse effects of excessive consumption of amino acids. Annu Rev Nutr (1984) 4:157–81. doi: 10.1146/annurev.nu.04.070184.001105
111. Matsueda S, Niiyama Y. The effects of excess amino acids on maintenance of pregnancy and fetal growth in rats. J Nutr Sci Vitaminol (1982) 28:557–73. doi: 10.3177/jnsv.28.557
112. Benevenga N, Yeh M-H, Lalich J. Growth depression and tissue reaction to the consumption of excess dietary methionine and S-methyl-L-cysteine. J Nutr (1976) 106:1715–20. doi: 10.1093/jn/106.12.1715
Keywords: IDO1, TDO, inhibitors, immunotherapy, tryptophan, kynurenine, AhR, System L
Citation: Kim M and Tomek P (2021) Tryptophan: A Rheostat of Cancer Immune Escape Mediated by Immunosuppressive Enzymes IDO1 and TDO. Front. Immunol. 12:636081. doi: 10.3389/fimmu.2021.636081
Received: 30 November 2020; Accepted: 04 January 2021;
Published: 23 February 2021.
Edited by:
Keqiang Chen, National Cancer Institute at Frederick, United StatesReviewed by:
Haiwei Mou, Cold Spring Harbor Laboratory, United StatesShuo Geng, Virginia Tech, United States
Copyright © 2021 Kim and Tomek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Petr Tomek, cC50b21la0BhdWNrbGFuZC5hYy5ueg==