 Lucas H. Armitage
Lucas H. Armitage Mark A. Wallet
Mark A. Wallet Clayton E. Mathews
Clayton E. Mathews- 1Department of Pathology, Immunology, and Laboratory Medicine, University of Florida, Gainesville, FL, United States
- 2Immuno-Oncology at Century Therapeutics, LLC, Philadelphia, PA, United States
Protein tyrosine phosphatase, non-receptor type 22 (PTPN22) regulates a panoply of leukocyte signaling pathways. A single nucleotide polymorphism (SNP) in PTPN22, rs2476601, is associated with increased risk of Type 1 Diabetes (T1D) and other autoimmune diseases. Over the past decade PTPN22 has been studied intensely in T cell receptor (TCR) and B cell receptor (BCR) signaling. However, the effect of the minor allele on PTPN22 function in TCR signaling is controversial with some reports concluding it has enhanced function and blunts TCR signaling and others reporting it has reduced function and increases TCR signaling. More recently, the core function of PTPN22 as well as functional derangements imparted by the autoimmunity-associated variant allele of PTPN22 have been examined in monocytes, macrophages, dendritic cells, and neutrophils. In this review we will discuss the known functions of PTPN22 in human cells, and we will elaborate on how autoimmunity-associated variants influence these functions across the panoply of immune cells that express PTPN22. Further, we consider currently unresolved questions that require clarification on the role of PTPN22 in immune cell function.
Introduction
Almost 1.6 million Americans have Type 1 Diabetes (T1D), an autoimmune disease that results in destruction of the insulin producing β cells in the pancreas and eventually requires exogenous insulin (1). T1D shows familial clustering and concordance rates between monozygotic twins is over 50% indicating that T1D has a strong genetic component (2, 3). It is estimated that up to 88% of the phenotypic variance is due to genetic factors such as predisposing or protective human leukocyte antigen (HLA) haplotypes and SNP-tagged variants (4–6). Of the genetic component of T1D risk, the HLA region, encoding the major histocompatibility complex (MHC) proteins, accounts for approximately 50% of heritable risk (7). The MHC class I (MHC-I) proteins are expressed on all nucleated cells and present antigenic peptides to CD8+ T cells while the MHC class II (MHC-II) proteins are primarily expressed on APC subsets and present antigen only to CD4+ T cells. The HLA Class II genes, encoding MHC-II, are the major contributing factor of HLA to risk with the DR3 (DRB1*03:01), DR4 (DRB1*04:01/02/04/05/08), DQ8 (DQA1*03:01-DQB1*03:02/04), and DQ2 (DQA1*05:01-DQB1*02:01) haplotypes conferring the greatest risk (7, 8). Indeed, the DR3/4 diplotype confers the greatest risk for T1D development (9, 10). These haplotypes increase risk in a synergistic manner and current research shows they have augmented ability to present T1D autoantigens to T cells, possibly due to alterations in the critical amino acids in the peptide binding pocket involved in which peptides are presented (10–12).
Although the HLA region contributes the bulk of genetic risk for T1D, there have been over 60 non-HLA genetic loci identified that have variants associated with enhanced or reduced risk of T1D (4, 13–22). Of these non-HLA loci, a non-synonymous SNP in PTPN22 has one of the highest reported odds ratios, ~2, and has been repeatedly confirmed across multiple studies and populations (4, 13, 15, 23–25). Protein tyrosine phosphatase, non-receptor type 22 (PTPN22) is a negative regulator of T cell receptor (TCR) and B cell receptor (BCR) signaling (26, 27). The diabetes-associated SNP in PTPN22 (rs2476601) affects TCR and BCR signaling as well as other adaptive and innate immune cell processes (27–39). The following sections will elaborate the known functions of PTPN22 and its autoimmune-linked/diabetogenic, missense SNP in human cells and how this might contribute to the pathogenesis of T1D. While the primary focus of this review is on human biology, we will emphasize specific areas of murine Ptpn22 research, where relevant, to highlight key similarities and differences between species.
Genetic Variation in PTPN22
Protein tyrosine phosphatase, non-receptor type 22 (PTPN22) is expressed in leukocytes and is well-known as a negative regulator of TCR and BCR signaling (26, 27). In non-activated T cells PTPN22 directly complexes with C-src tyrosine kinase (Csk) (32, 40, 41). This interaction is enhanced by phosphorylation of PTPN22 on Ser751 by PKCa. Further, phosphorylation of this residue increases the half-life of PTPN22 by protecting the enzyme from K48-linked ubiquitination and preventing recruitment of PTPN22 to the plasma membrane (42). During leukocyte activation PTPN22 is recruited to the plasma membrane to limit proximal immune cell receptor signaling. Here PTPN22 interacts with and dephosphorylates Grb2 (43), VCP (44), Vav (32, 44), Zap70 (32, 44), Lck (26, 32, 44), TCRζ (44), CD3ϵ (44), c-CBL (45), EB1 (46), and the p85 subunit of PI3K (47) to downregulate NFAT and reduce IL-2 production and secretion. However, PTPN22 also acts a regulator of other signaling networks (i.e., interferon γ receptor signaling, LFA-1 signaling, and TLR4 signaling) in monocytes, macrophages, dendritic cells, and neutrophils (29, 32, 35). There are multiple non-synonymous SNPs in PTPN22 associated with increased risk or decreased risk of autoimmune diseases (Table 1). The minor allele at rs56048322, PTPN22K750N, influences PTPN22 splicing and appears to cause CD4+ T cell hyporesponsiveness that increases risk for T1D (48). The minor allele at rs33996649, PTPN22R263Q, is a loss-of-function variant with diminished phosphatase capacity that reduces the risk of both SLE (49) and RA (50) (Table 1). Here we will examine rs2476601. The minor allele has a thymine substituted for a cytosine at nucleotide 1858, PTPN22C1858T, and encodes a tryptophan instead of an arginine at amino acid 620, PTPN22R620W (Table 1). It was first linked to T1D by Bottini et al. in 2004 (51) and the association between rs2476601 and T1D was quickly replicated (52). This SNP has also been associated with increased risk for multiple autoimmune diseases including rheumatoid arthritis (RA) (28), systemic lupus erythematosus (SLE) (53), Graves’ disease (52, 54), myasthenia gravis (55), primary Sjogren’s syndrome (56), generalized vitiligo (57), Addison’s disease (58), and alopecia areata (59) strongly suggesting PTPN22 regulates immunity.

Table 1 Single nucleotide polymorphisms in human PTPN22, their analogous mutations in mice, and their disease associations.
The SNP, rs2476601, lies in the proline-rich c-terminal domain of PTPN22 and interrupts some protein-protein interactions (e.g., interactions with CSK, TRAF3, and PAD4) (30, 35, 51). This is well illustrated in a recent review article (60). To determine the function of the common or major allotype of PTPN22, namely PTPN22620R, diverse approaches including knock down or overexpression of PTPN22 in primary human cells or human cell lines and knock down/out of Ptpn22, the mouse orthologue of PTPN22, in mice and mouse cell lines, have been used. To study the altered function of the minor allotype of PTPN22, PTPN22620W, researchers have again utilized many techniques including comparative studies in primary cells from human PTPN22620W donors vs. PTPN22620R donors, overexpression of PTPN22620W vs. PTPN22620R in primary human cells and human cell lines, transgenic expression of human PTPN22620W vs. PTPN22620R in Ptpn22−/− mice, and introduction of a mutation that is analogous to PTPN22620W in the mouse orthologue, PEP619W. Notably, this SNP is also associated with protection from Mycobacterium tuberculosis, an infection primarily controlled by T cells and T cell-activated macrophages (61–64). PTPN22 has been described as a negative regulator of multiple stages of danger signal recognition, from the process of T and B cell education, throughout initial detection of microbes, and then T and B cell effector functions. Thus, genetic variation that confers beneficial immunity to a globally-relevant pathogen (M. tuberculosis) might lower the threshold for danger signal responses. In murine models of T1D, lack of key macrophage/CD4+ T cell effector molecules (e.g., CD154 and CD40) but not all (e.g., IFNγ and IFNγR) prevents autoimmunity in T1D-prone NOD mice (65–67). We propose that the T1D-associated risk allotype of PTPN22 permits excessive innate and adaptive immune signaling in response to aseptic and/or septic stress/danger signals, in turn, driving a type IV delayed hypersensitivity response against pancreatic β cell antigens. The end result is insulin deficient diabetes mellitus. Herein we review the findings that support a pan-leukocyte role for PTPN22 in immune regulation. For the purpose of this review, we will examine the known roles for PTPN22 in innate and adaptive leukocyte signaling pathways and functions in humans as well as supporting data from mouse models. Where data is available we will also discuss how the minor allotype of PTPN22, PTPN22620W, influences signaling pathways as well as cellular functions and how these alterations may contribute to the development of T1D.
PTPN22 Expression
PTPN22 is expressed in most types of human leukocytes, including CD4+ T cells, CD8+ T cells, B cells, NK cells, monocytes, macrophages, dendritic cells, and neutrophils. Of these cells, PTPN22 has the highest expression in activated naïve CD8+ and CD4+ T cells, followed by NK cells and B cells, with lower levels in monocytes (28, 68). While the non-synonymous SNP at rs2476601 changes the amino acid sequence, the allelic difference does not modify PTPN22 expression in most lymphocyte subsets. Peripheral blood mononuclear cells (PBMCs) from PTPN22620R/W donors expressed PTPN22 mRNA equally from both alleles and this did not vary with gender (69). Upon anti-CD3/anti-CD28 stimulation of PBMCs (simulated activation of the TCR/CD3/CD28 complex), PTPN22 mRNA expression increased and this rise in expression was equally attributed to both alleles (69). Similarly, PTPN22 expression levels in PMBC-derived DCs and PBMC are the same in PTPN22620R/W and PTPN22620R/R donors (35).
There are, however, exceptions; PTPN22620W/W donors had 9% lower PTPN22 expression in naïve CD4+ T cells compared to PTPN22620R/R donors but there were no additional differences in PTPN22 expression in other T cell subsets (47). There is a report showing that PTPN22620W is more susceptible to calpain-1-mediated degradation and that the PTPN22620W protein is less expressed in naïve and memory T cells compared to PTPN22620R (70); yet, this has been disputed by later studies that observed the antibody used to detect PTPN22 had a higher affinity for PTPN22620R versus PTPN22620W (35, 40, 71). PTPN22 mRNA and protein expression in freshly-differentiated macrophages (so-called M0 or non-polarized macrophages) from PTPN22620R/W and PTPN22620W/W donors was lower than that of PTPN22620R/R donors (38). After M1 polarization of these macrophages (treatment with lipopolysaccharide and IFN-γ to mimic an inflamed septic environment), mRNA and protein expression of PTPN22 was higher in PTPN22620R/W and PTPN22620W/W donors than PTPN22620R/R donors but there was no difference in M2 polarized macrophages (treatment with IL-4 and IL-13 to generate so-called “alternatively activated macrophages”) (38). For macrophages, these findings are suggestive of a relationship between PTPN22 allotype and PTPN22 expression in the context of microbial infections wherein type 1 CD4+ T helper response (TH1) typified by IFN-γ secretion occur – for example, mycobacterial infections. Overall, allelic differences at rs2476601 have modest effect on the expression of PTPN22 in human cells that might be associated with observed immune phenomena (e.g., altered susceptibility to mycobacterial infections), but many questions remain unanswered and causality is merely speculative until more complex studies can be completed. While PTPN22 expression is only modestly influenced by allele, the function of PTPN22 is measurably altered by rs2476601.
Regulation of T Cell Function by PTPN22 Allotypes
The majority of studies focused on PTPN22 have investigated how the PTPN22620 allotypes influence the composition of the T and B cell compartments and intracellular signaling in T cells and B cells. PTPN22 allotypes have minor effects on T cell composition across immune compartments in humans; there are no differences in total T cells, total CD4+ or CD8+ T cells, or CD4+ or CD8+ effector memory T cells when comparing PTPN22620R/W donors to PTPN22620R/R donors (72). Most studies report no differences in most CD4+ T cells subsets (i.e., TH1, TH17, TH1TH17, TFH) (73). However, PTPN22620W/W donors had slightly-increased FOXP3+CD4+ regulatory T cells(Tregs) (7.94% vs. 6.76%) compared to donors with the common PTPN22620R/R allotype (74, 75). It has been reported that PTPN22620R/W donors have increased memory CD4+ T cells when compared to PTPN22620R/R donors (about 50% vs. 41% respectively) with a concomitant decrease in naïve CD4+ T cells (76). EOMES is a T box transcription factor that drives IFNγ secretion by CD4+ T cells (73). PTPN22620W/W donors exhibited increased EOMES+CD4+ T cells compared to PTPN22620R/R donors (~7% vs. ~5%) again with an accompanying decrease in naïve CD4+ T cells (73). It is unclear whether PTPN22 genotype influences naïve CD4+ T cell frequency (72, 73, 76). Two studies have reported a trend toward decreased naïve CD4+ T cells in PTPN22620W/W donors (73, 76) while a third study reported no difference in naïve CD4+ T cells when examining PTPN22 genotype (72). The study that reported no difference had a low number of subjects (3 in each group) and no subjects that were homozygous for the minor allele (72). The two studies that have reported a difference included more participants [13 per group (73) or ≥22 per group (76)] and included a group homozygous for the minor allele. Differences in study populations may explain the inconsistencies. A study with a larger cohort of all three genotypes (i.e., PTPN221858C/C, PTPN221858C/T, and PTPN221858T/T) may be better powered to address whether PTPN22 genotype influences naïve CD4+ T cell frequency.
Impact of PTPN22 Allotypes on TCR Signaling
While PTPN22 allotypes have a minor impact on T cell compartment composition, a significant impact on signal transduction in human T cells has been observed. In primary T cells, PTPN22620R is a negative regulator of TCR (26, 28, 43, 77, 78) (Figure 1A) and lymphocyte function-associated antigen 1 (LFA-1) (32) signaling (Figure 2) while it is a positive regulator of in vitro T regulatory cell (Treg) induction (33). In T cells, PTPN22620R has been shown to directly interact with Grb2 (43), VCP (44), Vav (32, 44), Zap70 (32, 44), Lck (26, 32, 44), TCRζ (44), CD3ϵ (44), c-CBL (45), CSK (32, 40, 41), EB1 (46), and the p85 subunit of PI3K (47). Studies do not agree whether PTPN22620W is a gain-of-function or loss-of-function variant in human TCR signaling but there is compelling evidence for both views (Figures 1B, C) (40, 41, 47, 70, 72, 76, 79–81). PTPN22620W is a loss-of-function variant in LFA-1 signaling (Figure 2) (32). PTPN22620W has not been studied in the context of Treg induction in humans, however activated Tregs (aTregs) from PTPN22620W/W donors have a reduced capacity to inhibit IFNγ secretion from other T cells compared to those from PTPN22620R/R donors (47).
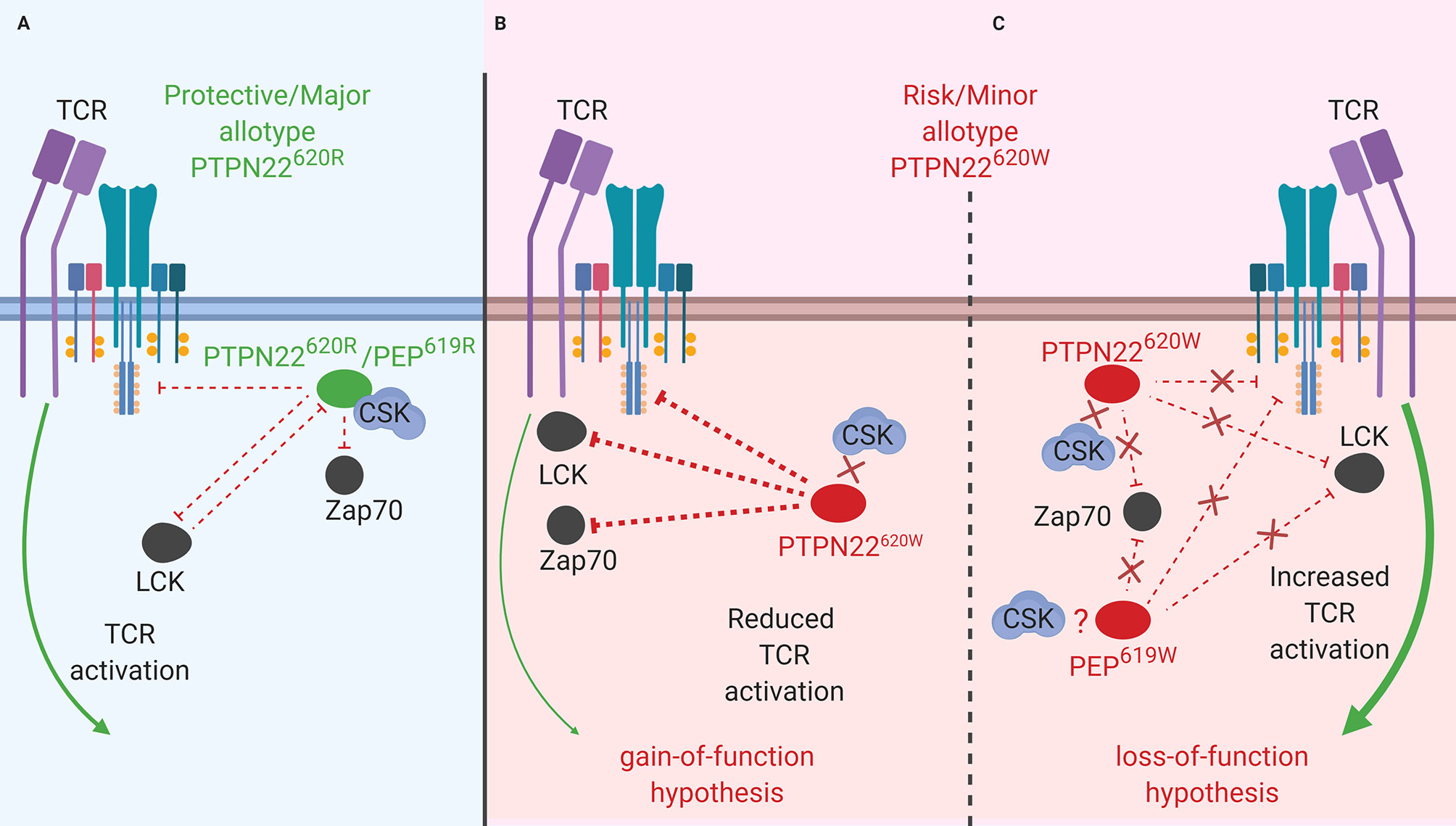
Figure 1 PTPN22 function in T cells. (A) PTPN22620R and PEP619R are negative regulators of TCR signaling in T cells where they dephosphorylate/deactivate signaling intermediates and reduce signaling from the TCR to the nucleus. (B) The PTPN22620W gain-of-function hypothesis. In this scenario, PTPN22620W is more active and dephosphorylates signaling intermediates at an increased rate compared to PTPN22620R. This blunts TCR signaling compared to PTPN22620R and reduces T cell response. (C) The PTPN22620W and PEP619W loss-of-function hypothesis. In this scenario, PTPN22620W/PEP619W are less efficient at dephosphorylating TCR signaling intermediates compared to PTPN22620R/PEP619R. This allows more signal from the TCR to reach the nucleus and increases T cell response to TCR stimulation.
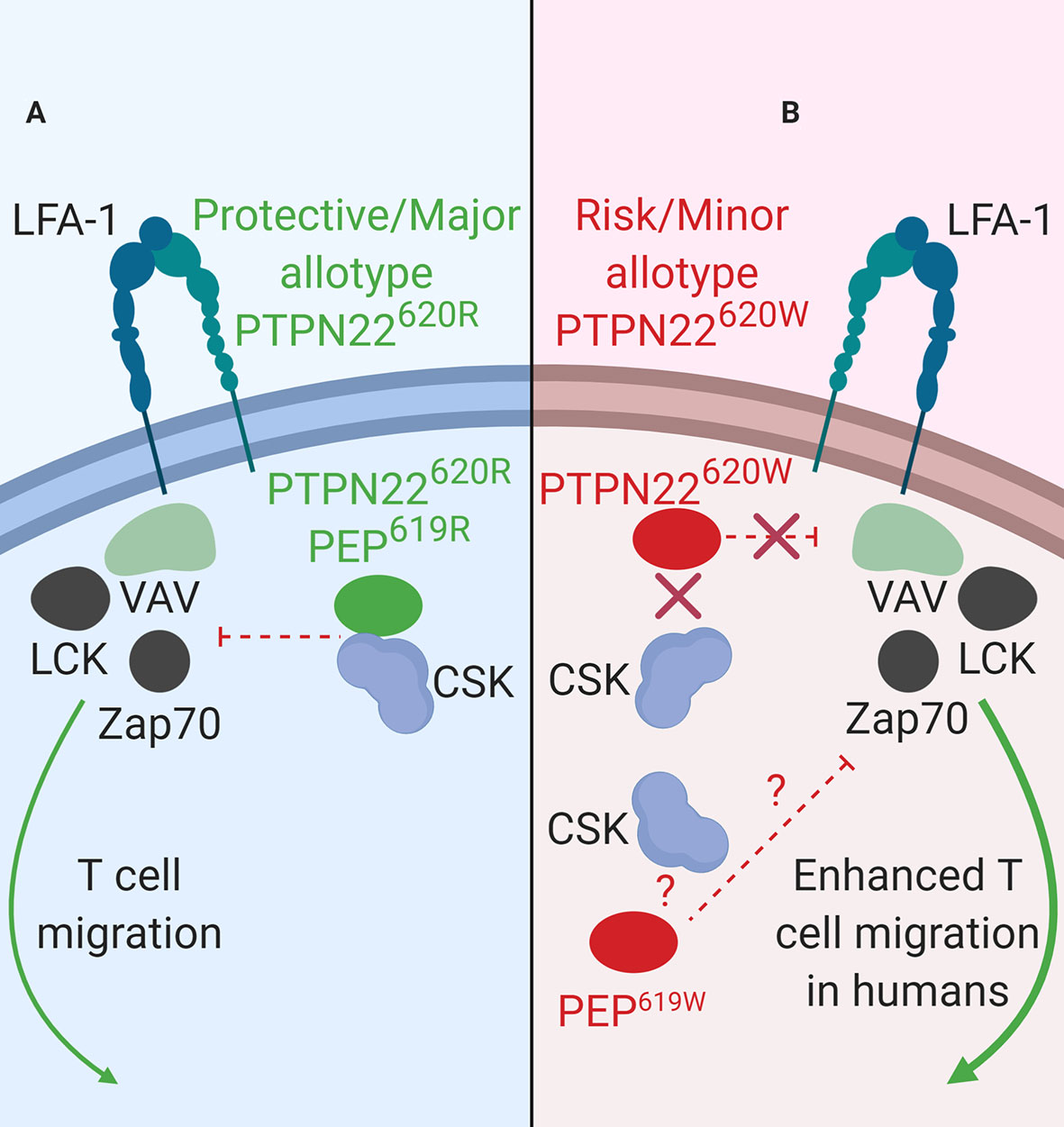
Figure 2 PTPN22 function in LFA-1 signaling. (A) PTPN22620R and PEP619R are negative regulators of LFA-1 signaling in T cells. Upon LFA-1 binding of ICAM-1, PTPN22620R and PEP619R associate with CSK, and are recruited to the leading edge in an LCK-dependent manner where they dephosphorylate PTPN22 substrates and inhibit LFA-1 signaling. (B) PTPN22620W is a loss-of-function variant in LFA-1 signaling. Upon LFA-1 binding of ICAM-1, PTPN22620W does not associate with CSK and is not recruited to the leading edge. This prevents PTPN22620W from interacting with its substrates and inhibiting LFA-1 signaling. PEP619W has not been studied in this context.
PTPN22 is a known negative regulator of TCR signaling (Figure 1A) (82). To investigate the function of PTPN22 in human T cells many studies have utilized the T-cell acute lymphoblastic leukemia cell line, Jurkat (26, 28, 43). This has allowed dissection of the influence of PTPN22 on proximal TCR signaling. In Jurkat T cells, it has been shown that PTPN22 negatively regulates activation of JNK2 (26) and LCK (26), and transcriptional activity driven by NF-κB (28), CD28 response element/NF-IL2B AP-1 (43), NFAT/AP-1 (26), c-fos (26), and c-jun (26) downstream of the TCR. CRISPR/Cas9 mediated knockout of PTPN22 in Jurkat T cells revealed that PTPN22 negatively regulates TCR-driven IL-2 and CD69 expression especially in the context of weak antigen stimulation (83). CRISPR/Cas9 mediated knockout of PTPN22 in primary CD4+ T cells supports that PTPN22 is a negative regulator of TCR signaling (78). These studies also revealed how PTPN22 achieves negative regulation of TCR signaling. PTPN22 cooperates with CSK to inhibit initial TCR signaling (Figure 1A) (26). In resting T cells, PTPN22 is associated with CSK and upon TCR stimulation, this complex dissociates at a rate that parallels dephosphorylation of PTPN22 substrates (40).
While PTPN22620R is a negative regulator of TCR signaling, the effect of the SNP on function of PTPN22620W remains controversial. It is currently debated whether PTPN22620W is a gain-of-function variant that reduces response to TCR stimulation (Figure 1B) or a loss-of-function variant that allows enhanced TCR signaling (Figure 1C). The most studied hypothesis is that PTPN22620W is a gain-of-function variant that suppresses TCR signaling (Figure 1B) (40, 41, 47, 70, 72, 76, 79, 80). These studies have shown that PTPN22620W reduces signaling through the TCR and is associated with significantly reduced IL-2 secretion (72, 79), calcium mobilization (72, 76, 79), and IFNγ production from CD4+ T cells (Table 2) (79). There is also evidence that the PTPN22620W allotype drives enhanced skewing of CD4+ T cells to EOMES+ TH1 cells (47, 73). PTPN22620W is also associated with reduced expression of CD25, lower proliferation, and decreased IL-10 secretion by CD4+ memory T cells (76, 79) (Table 2). In concordance with this, in vivo TCR stimulation in the form of a trivalent influenza vaccine resulted in reduced induction of an influenza virus-specific CD4+ T cell response in PTPN22620R/W subjects compared to PTPN22620R/R subjects (Table 2) (80). Another indication of reduced influenza virus-specific CD4+ T cell induction is the impairment of anti-flu antibody affinity maturation. Antibody affinity maturation relies on activation of CD4+ T follicular helper (TFH) cells; PTPN22620R/W subjects had reduced affinity maturation compared to PTPN22620R/R subjects implying they had reduced activation of TFH cells or reduced activation of anti-flu B cells (80).
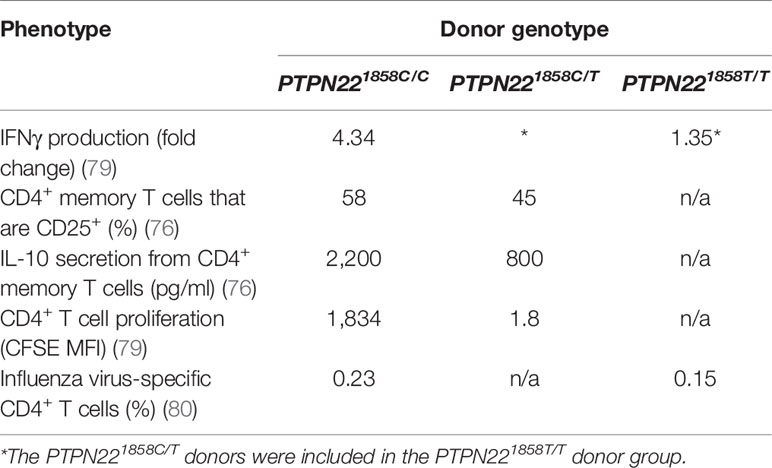
Table 2 PTPN22 genotype influence on TCR-related phenotypes.
Studies tracing the proximal events following TCR stimulation agree that PTPN22620W is a gain-of-function variant in primary T cells (Figure 1B). Overexpression of PTPN22620W decreases NFAT/AP-1-driven luciferase transcription more than overexpression of PTPN22620R (72). In primary T cells from donors with PTPN22620W/W TCR stimulation resulted in lower TCRζ-chain phosphorylation and increased ERK, AKT, and PI3K p85 activation compared to PTPN22620R/R donor T cells (Table 3) (47, 70). These studies offer a molecular mechanism for the difference in function of PTPN22620R and PTPN22620W centered on reduced interactions of CSK and LCK with PTPN22620W.

Table 3 PTPN22 genotype influence on proximal TCR signaling events.
As noted above, in resting T cells PTPN22620R is associated with CSK and upon TCR stimulation this complex dissociates at a rate that parallels dephosphorylation of PTPN22 substrates (Figure 1A) (26, 40). Simultaneously, PTPN22 is phosphorylated at Ser751 by PKCα which enhances the CSK/PTPN22 interaction and restricts PTPN22 activity to allow appropriate TCR signaling (42). PTPN22620W interacts with CSK to a lesser extent than PTPN22620R (immunoprecipitation of PTPN22620R pulls down 2.9 fold more CSK than PTPN22620W), and is more available to dephosphorylate PTPN22 substrates at the initiation of TCR signaling (28, 40, 41, 51). Both PTPN22620R and PTPN22620W are subject to phosphorylation at Ser751 by PKCα, however this only seems to inhibit PTPN22620R activity, by enhancing its association with CSK, while it does not inhibit PTPN22620W or enhance PTPN22620W/CSK interactions (42). Similarly, PTPN22620R is associated with LCK to a greater degree than PTPN22620W and this appears to be CSK-dependent (41). LCK phosphorylates PTPN22 on an inhibitory Y536 residue (41). PTPN22620R has more phosphorylated Y536 residues and is less active than PTPN22620W in Jurkat cells at rest and upon TCR stimulation (41). This may also explain why the in vitro phosphatase activity of PTPN22620W is 50% higher compared to PTPN22620R when the two allotypes of PTPN22 are purified from mammalian cells. When purified from insect cells, where this post-translational modification is absent, the phosphatase activity is equal among the two allotypes (41, 72). In conclusion, PTPN22620W is a more potent inhibitor of TCR signaling than PTPN22620R because PTPN22620W is more available to interact with PTPN22 substrates due to reduced sequestration by CSK. Further, PTPN22620W is more active due to reduced association with its own negative regulator, LCK, and consequent reduced phosphorylation at an inhibitory tyrosine residue (Figure 1B).
While evidence that PTPN22620W is a gain-of-function variant remains compelling, sufficient results exists to argue that PTPN22620W could be a loss-of-function variant (Figure 1C) (70, 81). These studies have observed that T cells from healthy PTPN22620W/W donors expand more upon TCR stimulation than those from healthy PTPN22620R/R donors (70). Further, when CSK is co-expressed with PTPN22620W in Jurkat T Cells, higher calcium fluxes are measured than when CSK is co-expressed with PTPN22620R (81). A study found that the PTPN221858T allele enhances expression of a dominant negative isoform, PTPN22.6, that increases signaling through the TCR (Figure 1C) (84). The authors offered a hypothesis that reconciles human data showing that PTPN22620W is a gain-of-function; PTPN22620W allows chronic signaling through the TCR that drives T cell exhaustion, causing T cells from PTPN22620R/W and PTPN22620W/W donors to be less responsive to stimulation through the TCR—a finding reported by most studies. This is supported by evidence that expression of PD-1, a marker of T cell exhaustion, is enhanced on CD4+ Teff and Tregs in healthy PTPN22620W/W donors compared to healthy PTPN22620R/R donors (74). Furthermore, the reduced calcium flux seen in PTPN22620R/W donors was most notable in memory CD4+ T cells with no difference observed in naïve CD4+ T cells; this could indicate that the experienced population is exhausted (76). While it is not certain whether PTPN22620W is a gain-of-function or loss-of-function variant in human TCR signaling, it is clear that the mouse orthologue of PTPN22620W, PEP619W, is a loss-of-function variant in mouse TCR signaling.
Data from mouse models support the role of PTPN22/PEP as a negative regulator of TCR signaling (Figure 1A). Overexpression of PEP in the mouse antigen specific T cell line, BI-141, reduced TCR-mediated phosphorylation of ZAP70, c-Cbl, and the CD3 ζ-chain (77). Overexpression of PEP also reduced IL-2 secretion from these cells (77). C57BL/6J mice with a genetic ablation of Ptpn22 (B6.Cg-Ptpn22tm2Achn/J commonly referred to as C57BL/6-Ptpn22−/− mice) as well as NOD mice with doxycycline-induced knockdown of PEP [NOD-Tg(tetO-RNAi : Ptpn22,UBC-tetR,-GFP)P2Kslr commonly referred to as NOD-Ptpn22KD] starting at birth have an accumulation of effector/memory CD4+ and CD8+ T cells in secondary lymphoid organs. This phenotype is thought to be a product of increased TCR signaling in the absence of PEP (85–88). Similar to humans harboring PTPN22620W, PEP619W knock-in C57BL/6 mice (C57BL/6-Ptpn22tm1.1Kas commonly referred to as C57BL/6-PEP619W) exhibited an expansion of CD4+ memory T cells compared to unaltered C57BL/6 mice that carry the PEP619R allele (70, 71). In C57BL/6-PEP619W mice there was also a marked expansion of the total effector/memory T cell pool and T cells from these mice exhibited increased IL-2 secretion, increased calcium mobilization, enhanced/prolonged tyrosine-phosphorylation of ZAP-70 and Lck, and increased ex vivo expansion of T cells compared to C57BL/6 mice (70, 71, 86). While the R619W conversion in PEP appears to be a loss-of-function variant with respect to TCR signaling (Figure 1C), controversy exists regarding the human autoimmunity risk allotype, PTPN22620W, with regard to gain-of-function or loss-of-function TCR signaling (Figures 1B, C). Despite this ongoing lack of clarity for PTPN22620W in human TCR signaling, evidence clearly supports that PTPN22620W is a loss-of-function variant in LFA-1 signaling in T cells.
Impact of PTPN22 Allotype on LFA-1 Signaling in T Cells
LFA-1 is fundamentally important to general leukocyte trafficking. Loss of LFA-1 causes the life-threatening disease known as leukocyte adhesion deficiency (LAD) resulting in uncontrolled microbial infections (89). LFA-1 is also critical in T cell activation and migration (90). In human T cells, PTPN22 inhibits LFA-1 signaling (Figure 2) (32). T cells treated with PTPN22 targeting small interfering RNA (siRNA) exhibited increased ICAM-1 (LFA-1 ligand)-induced phosphorylation of LCK, ZAP70, ERK1/2, and Vav compared to cells treated with a non-targeting control (NTC) siRNA. There was also an increase in ICAM-1-induced motility in cells treated with the PTPN22 targeting siRNA (32). The autoimmune associated variant, PTPN22620W, is a loss-of-function variant in LFA-1 signaling (Table 2B). Similar to what was observed with knockdown of PTPN22, human T cells from PTPN22620R/W and PTPN22620W/W donors have enhanced LFA-1 induced signaling (pERK1/2 fold change over unstimulated; PTPN22620W/W ~35 vs. PTPN22620R/W ~25 vs. PTPN22620R/R ~20) and adhesion (mean # of T cells adhered to LFA-1 coated slide at 8 min under shear flow; PTPN22620W/W ~32 vs. PTPN22620R/R~24) compared to T cells from PTPN22620R/R donors. At rest, PTPN22620R and PTPN22620W are aggregated near the plasma membrane of T cells. Upon engagement of ICAM-1 with LFA-1, PTPN22620R leaves these aggregates, associates with CSK, and is recruited to the leading edge of migrating cells in an LCK-dependent manner where it dephosphorylates PTPN22 substrates to inhibit LFA-1 signaling (Figure 2A). In contrast, PTPN22620W stays more clustered and is less recruited to the leading edge resulting in less PTPN22-mediated negative regulation of LFA-1 signaling (Figure 2B) (32).
As observed in human T cells, PEP negatively regulates mouse T cell responses to ICAM-1 stimulation (Figure 2A). T cells from C57BL/6-Ptpn22−/− mice displayed enhanced ERK1/2 phosphorylation (pERK1/2 fold change over-unstimulated; Ptpn22−/− ~12 vs. Ptpn22+/+ ~8) after ICAM-1 stimulation and adhered better to ICAM-1 coated glass slides under shear flow (mean # of T cells adhered at 8 min; Ptpn22−/− ~55 vs. Ptpn22+/+ ~30) compared to Ptpn22-intact mouse T cells (32). C57BL/6-Ptpn22−/− mouse T cells also had increased LFA-1 induced IFNγ secretion and were better at forming T cell-DC conjugates compared to Ptpn22-intact T cells (86). PEP and PTPN22 are both negative regulators of LFA-1 signaling in mice and humans (Figure 2A). PTPN22620W is a loss-of-function variant in humans while it is not known how the PEP 619R to W conversion affects mouse LFA-1 signaling (Figure 2B). While the molecular mechanisms behind PTPN22’s influence on receptor-proximal signaling in T cells (i.e., activation and mobilization) are well studied, PTPN22 has also been shown to influence Treg induction and function however the mechanism is less resolved.
Treg Induction and T Cell Suppression by aTreg
PTPN22 positively regulates in vitro induced Treg (iTreg) differentiation in human T cells. Primary naive T cells (CD4+CD127+CD25−) from PTPN22620R/R healthy donors and PTPN22620R/R donors with T1D were subjected to PTPN22 knockdown with antisense oligonucleotides. Differentiation of iTregs via treatment with IL-2/TGF-β1/αCD3/αCD28 was reduced with PTPN22 knockdown compared to control oligonucleotide transfected cells (% of CD4 T cells that are CD25+FoxP3+; PTPN22 knockdown resulted in ~20% iTreg vs. control ~40%) (33). No direct clinical studies have shown how PTPN22620 allotypes influence iTreg differentiation; however, healthy PTPN22620W/W donors have increased CD4+ Tregs compared to healthy PTPN22620R/R donors (7.94% vs. 6.76%) implying that PTPN22620W might potentiate iTreg development (74, 75). Although PTPN22620W/W donors have slightly more CD4+ Tregs, these Tregs exhibit a reduced capacity to inhibit IFNγ secretion from conventional T cells compared to those from PTPN22620R/R donors (47, 76).
As observed in humans, PEP also influences Treg development in mice; C57BL/6-Ptpn22−/− mice and NOD-Ptpn22KD mice had increased numbers of Tregs (87, 88). Data from C57BL/6-Ptpn22−/− mice and NOD-Ptpn22KD mice provided evidence that deficiency of PEP reduces the TCR signal strength required for in vitro induction of iTregs (91, 92). The iTreg induction can be accomplished by stimulating naïve FoxP3-CD4+ T cells with a combination of agonistic anti-CD3 and anti-CD28 targeting antibodies in the presence of TGF-β (87). Lower levels of stimulation with reduced concentrations of anti-CD3 antibodies increased in vitro iTreg induction in Ptpn22−/− cells compared to Ptpn22-intact cells. Increased concentrations of anti-CD3 resulted in elevated stimulation and decreased iTreg induction in Ptpn22−/− cells compared to Ptpn22-intact cells. At levels of TCR-stimulation that drive optimal in vitro iTreg induction in parental C57BL/6 mice, C57BL/6-Ptpn22−/− had reduced iTreg induction (87). Much like PTPN22620W humans, aged C57BL/6-PEP619W mice had increased Tregs compared to C57BL/6 mice. However, young C57BL/6-PEP619W mice exhibited no increase in Tregs. Tregs from young C57BL/6-PEP619W mice exhibited no differences in suppressive activity when compared to C57BL/6 mice, however Tregs from aged mice were not assessed. This difference may be due to the age of the mice, however, it remains to be seen if Tregs from older C57BL/6-PEP619W mice exhibit the same defect in suppression as human Tregs (71). It is clear that PTPN22 plays multiple roles in human T cells and that the diabetogenic allotype of PTPN22, PTPN22620W, alters these roles; how might the altered function of PTPN22620W in T cells impact T1D development?
PTPN22 in T Cells and Impact on T1D
T1D is generally considered a T cell mediated disease where CD8+ T cells are the major islet infiltrating immune cells (93, 94). The SNP in PTPN22, rs2476601, is associated with increased risk for T1D, reduced age at onset (95), and reduced residual β cell function at diagnosis (96). This SNP affects T cell function. PTPN22 is a negative regulator of TCR (26, 28, 43, 77) and LFA-1 (32) signaling and influences aTreg suppressive capacity (Figures 1 and 2) (47). In T cells, the effect of the T1D-risk variant PTPN22620W on TCR-induced signaling is currently unresolved with data supporting both gain-of-function and loss-of-function hypotheses (40, 41, 47, 70, 72, 76, 79, 80). In contrast, PTPN22620W has been characterized as a loss-of-function variant in LFA-1-induced signaling because it is not available to interact with its substrates (32). Adaptive Tregs (aTregs) from PTPN22620W/W donors have reduced capacity to suppress IFNγ secretion from conventional T cells (47). The enhanced LFA-1-induced signaling and motility, and the reduced capacity of aTregs to suppress IFNγ secretion from conventional T cells seen in PTPN22620R/W and PTPN22620W/W humans could help explain why rs2476601 is associated with increased overall risk of T1D development. The seemingly small magnitudes of reported biochemical, phenotypic, and functional effects of PTPN22620W in human T cells are surprising for a genetic variation that ranks near the top of the list for T1D genetic risk. We ask ourselves, “How could such minor fluctuations contribute to a life-threatening pathology?” The answer might lie in the thymus - the immune tissue where developing thymocytes (soon to be T cells) are exquisitely sensitive to the strength and duration of nascent TCR signaling. If PTPN22620W is a gain-of-function variant in TCR signaling, the PTPN22620W variant might impair the process of negative selection whereby autoreactive thymocytes are normally eliminated upon strong TCR signaling. Thus, effectively blinded to the fact that a given TCR is recognizing a self-antigen (e.g., insulin), autoreactive T cells might survive and escape into the periphery (72). More autoreactive T cells in the periphery would lead to increased autoreactive T cells surveying tissues, including the pancreas, and more opportunities for an autoimmune reaction to occur.
The alternate scenario postulates that thymic selection is more or less unaffected, and that the biologic effects of PTPN22620W manifest in the periphery. If PTPN22620W is a loss-of-function variant in TCR signaling, circulating T cells would be more sensitive to TCR ligation and this could explain the genesis of autoreactive T cell activation and thus autoimmunity. Both intrathymic and peripheral scenarios would be complicated by enhanced LFA-1-induced signaling (enhancing T cell migration) and reduced capacity of aTregs to suppress IFNγ secretion from activated T cells that could result in enhanced T cell infiltration into tissues (i.e., islets of Langerhans) as well as secretion of more IFNγ, thus creating a more inflammatory local environment. For T cells, additional new work will be needed to understand how thymic development and intra-islet T cell function is modulated by PTPN22 variants. Is there a single dominant mechanism at fault for autoimmune risk, or is this a case of death by a thousand cuts—multiple subtle effects which alone appear innocuous but together add up to complete destruction of a vital tissue? If the story weren’t complicated enough, T cells alone might not be the culprit of T1D. Autoantibodies produced by B cells are a prevalent feature and remain the gold standard biomarker of T1D progression. While it is hypothesized that autoantibodies are not pathogenic in human T1D, B cells are thought to play an important role as antigen specific APCs. It is known that depletion of B cells with Rituximab can delay disease progression (97). Additionally, many of the other rs2476601-asocciated autoimmune diseases are characterized by production of autoantibodies (e.g., RA, SLE, etc.). As such, many studies have focused on the effect of the PTPN22620R versus PTPN22620W in human B cells.
Regulation of B Cell Function by PTPN22 Allotypes
PTPN22 has been studied extensively in human B cells. Unlike the minor difference observed in the T cell compartment, PTPN22620W has a profound impact on B cell composition (described in detail below) (76). PTPN22 also impacts signal transduction in human B cells where it functions as a negative regulator of BCR signaling and BCR-induced apoptosis (34). Because PTPN22 influences BCR signaling and BCR-induced apoptosis, it also influences the central and peripheral B cell tolerance checkpoints (27, 76, 98–100).
Impact of PTPN22 Allotype on BCR Signaling
PTPN22 functions to dampen BCR signaling as well as BCR-induced apoptosis (Figure 3B). PTPN22 is overexpressed in primary chronic B lymphocytic leukemia (CLL) cells (34). CLL cells express functional BCRs and have been characterized for ligand-dependent signaling. PTPN22 depletion in CLL cells increased soluble-αIgM (simulated strong BCR signaling) induced apoptosis (34). Knockdown of PTPN22 also resulted in increased soluble αIgM-induced phosphorylation of LYN, SYK, BLNK, PKCδ, ERK, JNK, and p38 MAPK and reduced soluble-αIgM-induced phosphorylation of AKT, GSK3, and FOXO (34). PTPN22620W is a gain-of-function variant that acts to further blunt BCR signaling (Figure 3B). In heterozygous PTPN22620R/W donors there is reduced BCR-induced calcium flux compared to PTPN22620R/R donors (27, 76, 98). Heterozygous donors also had reduced phosphorylation of the BCR-proximal signaling components, SYK, PLCγ2 (MFI phospho-PLCγ2-Y759; PTPN22620R/W ~700 vs. PTPN22620R/R ~950), and AKT compared to PTPN22620R/R donors (27, 76, 98). In PTPN22620R/W donors there is also reduced total phosphorylated tyrosine in resting (% of CD27+ B cells that are phospho-tyrosine+; PTPN22620R/W ~4% vs. PTPN22620R/R ~8%) and BCR-activated memory B cells compared to PTPN22620R/R donors (27). Inhibition of PTPN22 in B cells of PTPN22620R/W donors increased SYK, PLCγ2, and AKT phosphorylation to levels equivalent to those of B cells from PTPN22620R/R donors (27, 98). Signaling through the BCR can also induce B cell expansion. However, it is not clear how PTPN22620W affects BCR-induced expansion of B cells; different studies have shown conflicting results (27, 70). Overall, PTPN22620W is more effective at regulating BCR signaling (Figure 3B).
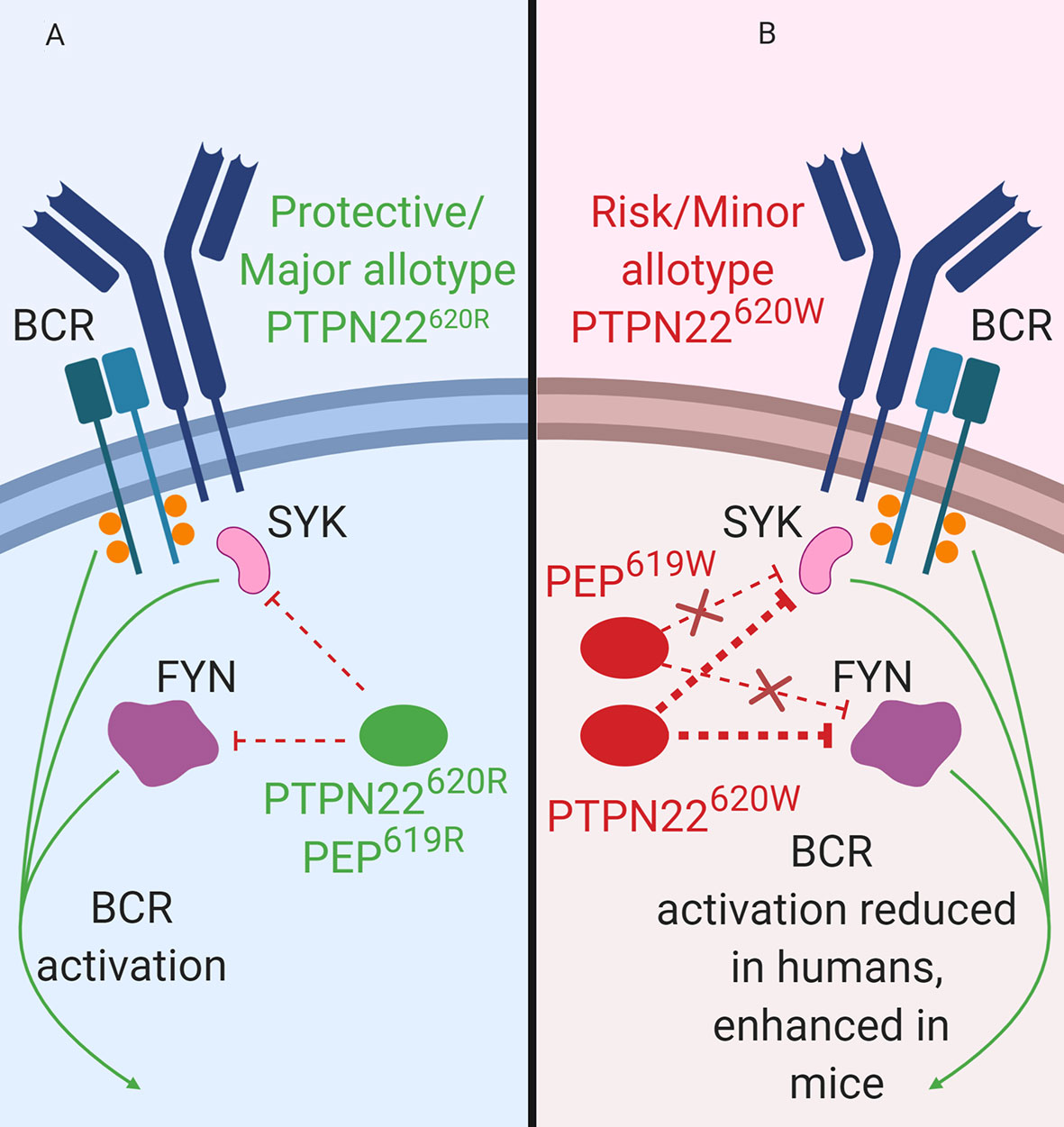
Figure 3 PTPN22 function in B cells. (A) PTPN22620R and PEP619R are negative regulators of BCR signaling in B cells. (B) PTPN22620W is a gain-of-function variant with respect to BCR signaling and leads to blunted BCR signaling. PEP619W is a loss-of-function variant with respect to BCR signaling and leads to enhanced BCR signaling.
Consistent with data from human B cells, PEP is also a negative regulator of BCR signaling in mice (Figure 3). Silencing of PEP via doxycycline-induced expression of a Ptpn22-targeting siRNA in NOD mice (the NOD-Ptpn22KD mice) increased B cell response to anti-IgM/anti-CD40 stimulation (88). Additionally, silencing of PEP resulted in the increased proliferation, robust expression of CD25 and CD69, and elevated phosphorylation of PLCγ2 (88). These results have been replicated via Ptpn22 knockout in other mouse strains (81, 101, 102). Unlike PTPN22620W humans, C57BL/6-PEP619W mice had a phenotype similar to C57BL/6-Ptpn22−/− mice, with increased anti-IgM induced B cell activation, increased anti-IgM induced proliferation, and increased phosphorylation of PLCγ2 compared to C57BL/6 mice (70, 71, 103). While PTPN22620W is a gain-of-function variant in human BCR signaling, PEP619W is a loss-of-function variant with respect to BCR signaling in mice (Figure 3B).
Regulation of B Cell Gene Expression and B Cell Expression of Surface Receptors by PTPN22
PTPN22620W alters gene expression and immune receptor levels in B cells. Naïve B cells from both PTPN22620R/W and PTPN22620W/W donors had significantly upregulated IL4R, IL13R, IL17R, and IL21R mRNA expression (genes involved in B cell proliferation/differentiation) and significantly upregulated genes in the BCR, CD40, and TLR activating pathways compared to those from PTPN22620R/R donors (100). PTPN22620W differentially affects expression of other genes with SNPs associated with T1D and other autoimmune diseases (BLK, PTPN2, CD40, TRAF1, CD19, SLAM, IRF5) (100). The surface expression of BAFFR, CD40, and SLAMF6 was enhanced in PTPN22620R/W and PTPN22620W/W donors compared to PTPN22620R/R donors (Table 4) (100, 103). Naïve B cells from PTPN22620R/W and PTPN22620W/W donors were more responsive to CD40L stimulation with an increased percent of B cells expressing CD69 and CD25 than those from PTPN22620R/R donors (100). CpG stimulation of PBMC for 4 days resulted in greater expansion of IgM+ memory B cells (CD19+CD27+IgM+) and IgM- Plasma cells (CD19+CD27hiIgM-) in PTPN22620R/W patients with T1D compared to PTPN22620R/R patients with T1D and in healthy control PTPN22620R/W donors compared to healthy control PTPN22620R/R donors (99). The combination of increased BAFFR, CD40, and SLAMF6 surface levels and the increased expression of IL4R, IL13R, IL17R, IL21R, as well as genes belonging to the CD40, TLR, and BCR activation pathways may explain the enhanced CpG-induced expansion of IgM+ memory B cells and IgM- Plasma cells in PBMCs seen in PTPN22620R/W and PTPN22620W/W donors compared to PTPN22620R/R donors. Importantly, this phenomenon is present in both PTPN22620R/W and PTPN22620W/W donors implying that the effects of PTPN22620W are either dominant or co-dominant.
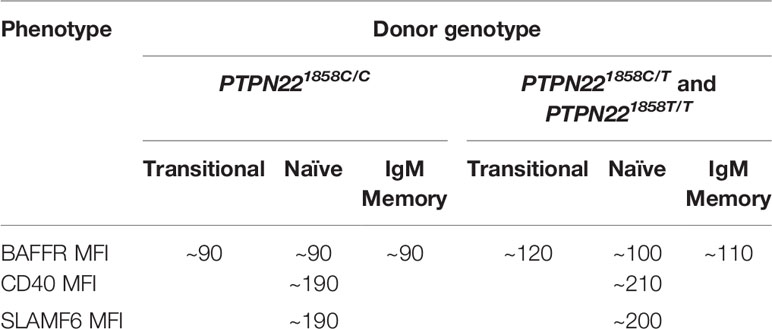
Table 4 PTPN22 genotype influence on B cell surface receptor expression.
Unlike humans, there was decreased CD40 and BAFFR surface expression on total splenocytes with decreased CD40 and BAFFR on immature B cells and increased CD40 on T2 B cells of C57BL/6-PEP619W mice compared to C57BL/6 mice (103). Tnfrsf13c (BAFFR) mRNA levels were enhanced in immature B cells and Cd40 mRNA levels were enhanced in T2 B cells of C57BL/6-PEP619W mice compared to C57BL/6 mice (103). Taken together we see that PTPN22 and PEP affect expression of costimulatory molecules in B cells of both humans and mice however the effects of the R to W conversion are not consistent when comparing humans to mice.
B Cell Tolerance Checkpoints and Composition
PTPN22620W alters the central and peripheral B cell tolerance checkpoints as well as the composition of the B cell compartment in humans (76, 98–100). Central B cell tolerance is mediated via clonal deletion or receptor editing to remove autoreactive or polyreactive B cells from the bone marrow before they enter the periphery (e.g., spleen, blood, lymph nodes, tissues) (104). Central tolerance results in a large reduction of polyreactive and autoreactive B cells and is readily apparent when comparing the bone marrow to the spleen and blood; 40%–70% of early immature B cells are polyreactive and 50%–75% are autoreactive in the bone marrow while 5%–10% of transitional B cells are polyreactive and 30%–50% are autoreactive in the periphery (104, 105). A common method for determining if B cells are autoreactive is to assess their response to human epithelial type 2 (HEp-2) cells. HEp-2 cells express a large array of self-antigens and HEp-2 reactive B cells are considered autoreactive (106). Healthy PTPN22620R/W and PTPN22620W/W donors had an increased proportion of polyreactive and HEp-2-reactive new emigrant/transitional B cells (CD20+CD10+CD21loIgMhiCD27−: 25%–30% of new emigrant/transitional B cells were polyreactive and ~50% were HEp-2-reactive) compared to healthy PTPN22620R/R donors (8%–10% of new emigrant/transitional B cells were polyreactive and ~30% were HEp-2 reactive) (100, 107). Most studies agreed that transitional B cells (CD19+CD27−CD24hiCD38hi) were increased in healthy PTPN22620R/W and PTPN22620W/W donors compared to healthy PTPN22620R/R donors (percentage of total B cells that are transitional; PTPN22620W/W and PTPN22620R/W ~5% vs. PTPN22620R/R ~2.5%), although not all studies observe this effect (98, 99, 108). The increased numbers of transitional B cells and polyreactive/HEp-2-reactive new emigrant/transitional B cells in healthy PTPN22620R/W and PTPN22620W/W donors indicates that the central B cell tolerance checkpoint is altered by PTPN22620W. Ergo, the autoimmune-linked allotype allows more polyreactive and autoreactive B cells to escape central tolerance and proceed into the periphery. B cells that enter the periphery will go through another round of selection to remove or inactivate autoreactive cells.
Peripheral B cell tolerance results in anergy or clonal deletion via apoptosis that is dependent on caspase-3 activation and is triggered by strong signaling though the BCR (98). This results in the reduction of autoreactive peripheral B cells. There are more autoreactive transitional B cells than autoreactive naïve mature B cells due to the peripheral B cell tolerance checkpoint; 30%–50% of transitional B cells are autoreactive while 10%–30% of naïve mature B cells are autoreactive (105). To simulate strong BCR signaling in naïve B cells, anti-IgM is used to crosslink the BCRs; this is similar to encountering a multivalent self-antigen during peripheral B cell tolerance and will cause some naïve B cells to undergo apoptosis. After 12 h of anti-IgM treatment, significantly fewer naïve B cells from PTPN22620R/W donors had begun the process of apoptosis by cleaving/activating caspase-3 when compared to PTPN22620R/R donors (% of naïve B cells with cleaved/active caspase-3; PTPN22620R/W ~10% vs. PTPN22620R/R ~18%) (98). Basal levels of the anti-apoptotic protein, Bcl-2, were higher in transitional B cells from PTPN22620R/W donors compared to PTPN22620R/R donors (Normalized BCL-2 MFI; PTPN22620R/W ~20 vs. PTPN22620R/R ~12) with no alteration in the pro-apoptotic protein, Bim (98). Healthy PTPN22620W/W and PTPN22620R/W donors had increased frequencies of polyreactive and HEp-2-reactive mature naïve B cells (CD20+CD10−CD21+IgM+CD27−). In these donors ~30% of mature naïve B cells were polyreactive and ~45% were HEp-2-reactive. In contrast, healthy PTPN22620R/R donors had ~10% polyreactive mature naïve B cells were and ~20% HEp-2-reactive (100). A unique subset of autoreactive anergic B cells (naïve IgD+ B cells [BND]: CD19+CD27−IgD+IgM−) are cells in the periphery thought to be anergic due to low chronic antigen stimulation through the BCR (109). BND cells were increased in healthy PTPN22620R/W donors compared to healthy PTPN22620R/R donors (% of CD19+ B cells that are BND cells; PTPN22620R/W ~3% vs. PTPN22620R/R ~2%) (98). PTPN22620R/W donors had a lower percentage of memory B cells compared to PTPN22620R/R donors (% of CD19+ B cells that are CD27+; PTPN22620R/W ~35% vs. PTPN22620R/R ~45%) (76). The reduced caspase-3 activation, increased levels of Bcl-2, increased frequencies of BND cells, HEp-2-reactive mature naïve B cells, and polyreactive mature naïve B cells, and decreased frequency of mature B cells found in PTPN22620R/W and PTPN22620W/W donors indicates that PTPN22620W alters the peripheral B cell tolerance checkpoint (98, 100, 107). The increase in autoreactive/polyreactive new emigrant/transition B cells, all transitional B cells, BND cells, and decrease in memory B cells was also seen when comparing T1D donors regardless of genotype to healthy PTPN22620R/R donors and this may represent a common B cell phenotype present in T1D patients (98, 100). Currently, it is thought that the blunting of BCR signaling by the gain-of-function PTPN22620W allotype leads to reduced negative selection and is responsible for the alterations seen in central and peripheral B cell tolerance mechanisms (76, 98, 100, 107). These B cell phenotypes are observed in both patients with autoimmunity and healthy controls that encode PTPN22620W.
C57BL/6-Ptpn22−/− as well as other strains of Ptpn22 knockout mice exhibit an altered B cell compartment. Deletion of Ptpn22 increased age-associated B cells (ABCs), plasma cells, autoantibodies, as well as germinal center activity and size when compared to Ptpn22-intact mice. However, germinal center size and activity appears to be partially dependent on an alteration in T follicular helper cells (81, 101, 102). Unlike humans harboring PTPN22620W, alterations in the B cell compartment of the loss-of-function PEP619W variant in mice is attributed to altered positive B cell selection due to enhanced BCR signaling (103). C57BL/6-PEP619W mice have increased splenic transitional 1 B cells, increased age-dependent B cells (ABCs), increased class-switched B cells, increased germinal center B cells, and less mature recirculating B cells when compared to C57BL/6 mice (103). Like humans however, the enhanced positive selection leads to increased self-reactive B cells, increased autoantibody titers, and reduced apoptosis of T1 B cells in C57BL/6-PEP619W when compared to C57BL/6 mice (70, 71, 103). The similarities between the B cell compartments of Ptpn22−/− mouse strains and C57BL/6-PEP619W mice implies that PEP619W is a loss-of-function variant in mice with respect to its effects on B cell positive selection while PTPN22620W decreases human B cell negative selection.
While PEP619W mice do not display the same central B cell tolerance phenotype as humans heterozygous or homozygous for PTPN22620W, immunodeficient NOD.Cg-Prkdcscid.Il2rgtm1Wjl (NSG) mice engrafted with human CD34+ hematopoietic stem cells (HSCs) from either PTPN22620R/W, PTPN22620W/W donors, or with HSCs overexpressing PTPN22620W phenocopy humans that are heterozygous or homozygous for PTPN22620W. These PTPN22620W HSC engrafted NSG mice display an increased proportion of polyreactive and HEp-2-reactive new emigrant/transitional B cells when compared to NSG mice engrafted with HSCs from PTPN22620R/R donors or HSCs overexpressing PTPN22620R (100, 107). Importantly, inhibition of PTPN22 in NSG mice engrafted with PTPN22620W HSCs reduced polyreactive and HEp2-reactive new emigrant B cells to the same levels as NSG mice engrafted with PTPN22620R HSCs indicating that PTPN22 is the main driver of this difference (107). The increased numbers of transitional B cells and polyreactive/HEp-2-reactive new emigrant/transitional B cells in healthy PTPN22620R/W and PTPN22620W/W donors and in PTPN22620W HSC engrafted NSG mice indicates that the central B cell tolerance checkpoint is altered by PTPN22620W. This alteration allows more polyreactive and autoreactive B cells to escape central tolerance and proceed into the periphery. Overall, PEP619W is a loss-of-function variant in mice with respect to its effects on B cell positive selection while PTPN22620W decreases human B cell negative selection; both of these alterations result in more autoreactive B cells with increased autoantibody titers.
PTPN22 in B Cells and Impact on T1D
Autoantibodies produced by B cells are a prevalent feature of T1D and remain the gold standard biomarker of islet autoimmunity and T1D progression (110). The SNP in PTPN22, rs2476601, is associated with increased risk of persistent islet autoimmunity (i.e., autoantibodies directed against insulin, GAD65, or IA-2) (111). While the role of pathogenesis of human T1D remains controversial, the importance of B cells has been demonstrated in preclinical models and clinical trials. Depletion of B cells pauses the loss of β cell function in some patients with recent onset T1D and can prevent or reverse disease in NOD mice (97, 112). B cells are not only capable of producing antibodies, they also act as APCs to present antigen to T cells in a process called linked recognition (113). In linked recognition, B cells uptake antigen recognized by the BCR, process it, and load peptides derived from the antigen on MHC-II to present to CD4+ T cells (113). These responding CD4+ T cells must have already encountered antigen and been activated by other APCs in the periphery before they can provide T cell help to the B cells. The T cell help initiates class-switching in germinal centers, while the B cells provide co-stimulatory signals to the T cells capable of enhancing in-progress T cell responses (114). In NOD mice, it is thought that B cells primarily enhance autoreactive T cell function as APCs and through the production of pro-inflammatory cytokines (115). PTPN22620R/W and PTPN22620W/W donors have increased B cell surface expression of CD40, SLAMF6, and BAFFR (Table 4), as well as B cell mRNA expression of IL4R, IL13R, IL17R, IL21R compared to PTPN22620R/R donors. PTPN22620R is a negative regulator of BCR signaling and PTPN22620W is a gain-of-function variant that reduces signaling through the BCR. This reduction in BCR signaling alters central and peripheral B cell tolerance allowing more autoreactive and polyreactive B cells into the periphery. The increased surface expression of CD40, SLAMF6, and BAFFR (Table 4), as well as B cell mRNA expression of IL4R, IL13R, IL17R, IL21R could enhance clonal expansion of B cells, differentiation into plasma cells, class switching, and cell survival in PTPN22620R/W and PTPN22620W/W humans (116–121). Increased SLAMF6 and CD40 expression on B cells could also enhance/prolong B cell-T cell interactions leading to more T cell and B cell activation in PTPN22620R/W and PTPN22620W/W humans. The combination of these phenotypes could lead to increased class switching of autoreactive B cells and increased survival of autoreactive and polyreactive B cells. These autoreactive/polyreactive B cells could go on to increase or simply sustain activation of autoreactive T cells. The increased/sustained activation of autoreactive T cells by autoreactive/polyreactive B cells could explain why rs2476601 is associated with increased risk of persistent islet autoimmunity (111) and why treatment with a B cell depleting therapy (rituximab) can delay loss of, but not restore, the c-peptide response in patients with recent onset T1D (97).
While adaptive immune cells are integral for targeting and destroying β cells, they are not the only cells implicated in development of T1D. The innate arm of the immune system is generally required to initiate antigen-specific responses by T and B cells. Monocytes, macrophages, and dendritic cells (DCs) are all APCs capable of initiating these potent immune responses in inflammatory contexts.
PTPN22 Allotypes in Monocytes, Macrophages, and Dendritic Cells
Monocytes, macrophages, and DCs are innate immune cells that are a part of the front-line sentinels that sense (via conserved PRRs such as TLRs and nucleic acid sensors) and eliminate invading microbes. While the function of PTPN22620R and altered function of PTPN22620W have been extensively examined in T and B cells, the roles of these allotypes in monocytes, macrophages, and DCs have been less studied. In human DCs and macrophages, PTPN22620R is a positive regulator of TLR4-induced Type 1 interferon (T1-IFN) production while PTPN22620W is less effective at driving TLR4- and TLR7/8-induced T1-IFN production (35, 122). In macrophages, PTPN22620R is a positive regulator of NLRP3 inflammasome activation and PTPN22620W is a gain-of-function variant leading to more NLRP3 activation and subsequent IL-1β release (36, 37). In monocytes, PTPN22620R negatively regulates NOD2-induced autophagy (39) and regulates IFNγ-induced signaling (29) while PTPN22620W has not been studied in the regulation of NOD2-induced autophagy or IFNγ-induced signaling. When examining the polarization of macrophages, PTPN22620R is a negative regulator of IL-23/IL-12 production following M1 induction (IFNγ/LPS treatment) while PTPN22620W is a gain-of-function variant that reduces IL-21/IL-12 production following M1 polarization. PTPN22620R is a positive regulator of IL-10 expression following M2 induction (IL-4/IL-13 treatment) and PTPN22620W does not alter this (38). As these previous studies illustrate, PTPN22 plays diverse roles in monocytes, macrophages, and DCs and the 620R to W conversion alters function in many aspects.
TLR-Induced Type 1 Interferons
PTPN22620R associates with TRAF3 following LPS stimulation and promotes T1-IFN production while PTPN22620W does not (Figure 4A) (35). This effect is not limited to TLR4 stimulation, plasmacytoid dendritic cells (pDCs) from PTPN22620W/W and PTPN22620R/W patients with SLE have reduced IFNα production following R848 (TLR7/8 agonist) stimulation compared to PTPN22620R/R patients (PTPN22620R/W+PTPN22620W/W; ~35% pDCs IFNα2+ with gMFI of ~250 vs. PTPN22620R/R; 45% pDCs IFNα2+ with gMFI of ~500) (122). STAT1 phosphorylation, a marker of interferon receptor signaling, is significantly reduced by about 50% in PBMCs from PTPN22620R/W donors after LPS treatment when compared to PTPN22620R/R donors. T1-IFN-inducible genes (IRF7, MX1, and ISG15) were also significantly reduced by about 50% in PBMC-derived DCs from PTPN22620R/W donors compared to PTPN22620R/R donors, probably due to reduced production of T1-IFNs. TRAF3 is an adaptor protein that links TLR4 and TLR7/8 signaling to induction of T1-IFNs. PTPN22 co-immunoprecipitated TRAF3 from human monocyte derived DCs (moDCs). In transgenic C57BL/6-Ptpn22−/− mice expressing either human PTPN22620R or PTPN22620W, PTPN22620R associated with TRAF3 and promoted its poly-ubiquitination and subsequent induction of Ifnb1 while PTPN22620W did not. C57BL/6-Ptpn22−/− mice expressing human PTPN22620W had reduced LPS-induced T1-IFN production [~50% of Ifnb1 from bone marrow-derived dendritic cells (BMDCs), and ~50% of Ifnb1/Ifna4 from bone marrow-derived macrophages (BMMΦ)] compared to those expressing human PTPN22620R (35).
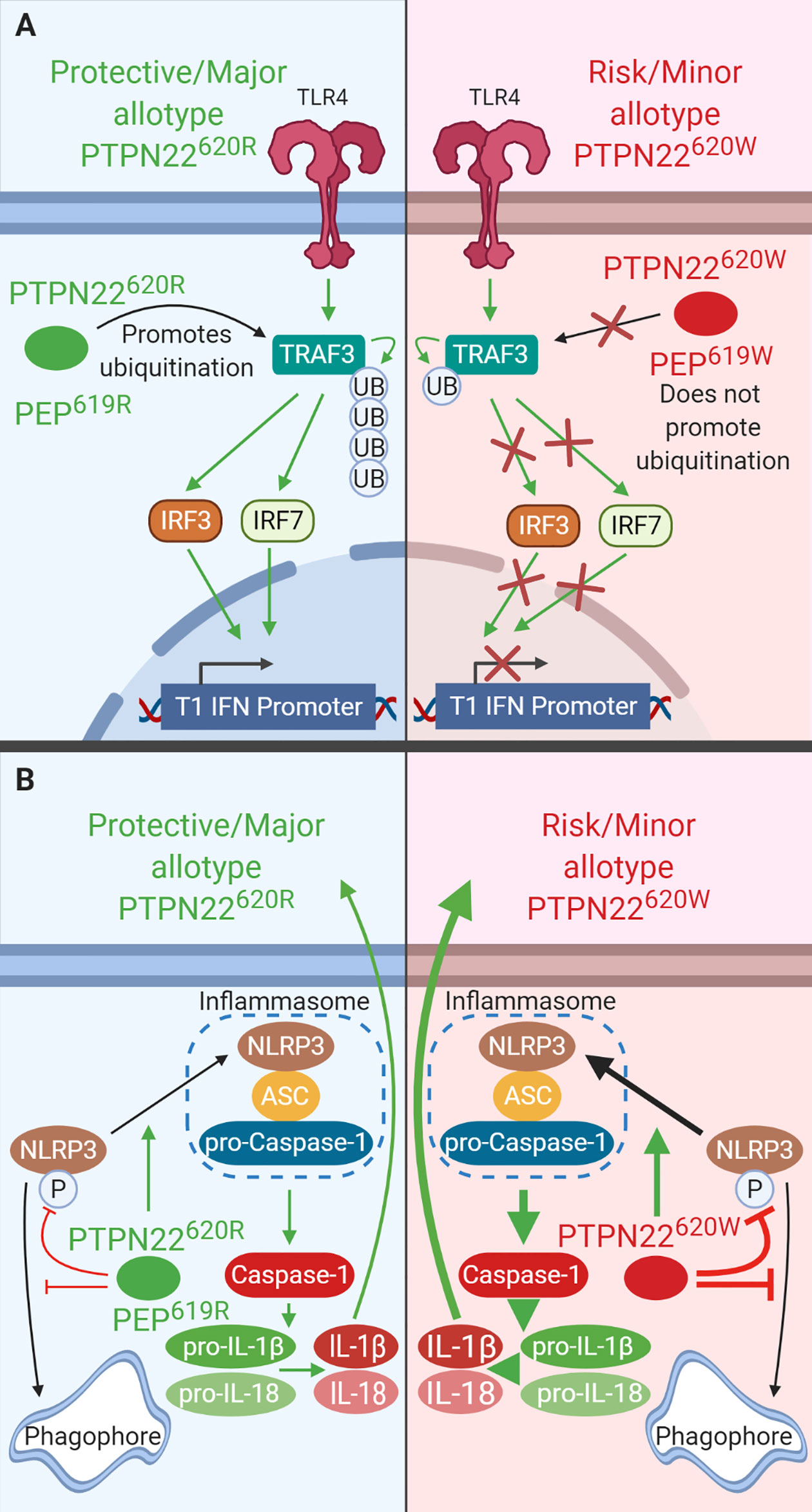
Figure 4 PTPN22 regulates TLR-induced T1-IFN secretion and NLRP3 inflammasome activation in macrophages and DCs. (A) PTPN22620R and PEP619R promote T1-IFN secretion in response to TLR-agonists by interacting with TRAF3 and promoting its autoubiquitination and subsequent induction of T1-IFN. PTPN22620W and PEP620W do not interact with TRAF3 and fail to support TLR-induced T1-IFN production. (B) PTPN22620R and PEP619R promote NLRP3 inflammasome activation by dephosphorylating NLRP3 and preventing its sequestration into the autophagosome. PTPN22620W is a gain-of-function variant that has enhanced capacity to dephosphorylate NLRP3. This leads to increased NLRP3 inflammasome activation.
Like PTPN22620W in humans and transgenic mice, BMMΦ from C57BL/6-Ptpn22−/− mice had impaired TLR4-induced T1-IFN (Ifnb1 and Ifna4 mRNA production were ~50% less) and decreased TLR4- and TLR3-induced IFN-β production (~60% less) compared to WT BMMΦ (Figure 4A). BMMΦ from C57BL/6 mice reconstituted with PEP227S, a phosphatase-inactive mutant, restored TLR-induced Ifnb1 expression indicating that the phosphatase activity of PEP is not required in this process. C57BL/6-Ptpn22−/− BMMΦ have reduced K63-linked polyubiquitination of TRAF3 following LPS stimulation compared to WT BMMΦ. These data are not confined to mouse BMMΦ, pDCs from C57BL/6-Ptpn22−/− mice and BXSB/MpJ-Ptpn22−/− mice had fewer pDCs making IFNα (~50% reduction) and the pDCs that were making IFNα made less than pDCs from WT mice (again ~50% reduction) (102). Also like PTPN22620W humans, C57BL/6-PEP619W mice had significantly reduced TLR-7-driven T1-IFN serum levels following injection of R848 compared to C57BL/6 mice (~3 ng/ml in C57BL/6-PEP619W mice vs. 5 ng/ml in C57BL/6 mice) (122). The combined data from mice and humans shows that both PTPN22620W and PEP619W are loss-of-function variants with respect to TLR-induced T1-IFN resulting in reduced T1-IFN following TLR stimulation (Figure 4A) (35). TLR stimulation does not only induce T1-IFN, it is also capable of priming the NLRP3 inflammasome for subsequent activation following an inflammatory stimulus such as murmamyldipeptide (MDP), an aganoist of nucleotide-binding oligomerization domain-containing protein (NOD2) that is a component of bacterial cell walls. The role of PEP/PTPN22 allotypes in NLRP3 inflammasome may also impact autoimmunity.
NLRP3 and IL-1β
PTPN22620R positively regulates activation of NLRP3 and subsequent release of IL-1β (Figure 4B). PTPN22620W is a gain-of-function variant that potentiates NLRP3 activity (Figure 4B) (36, 37). PTPN22 dephosphorylates NLPR3 at Y861 which prevents it from being sequestered into phagophores and degraded via autophagy (36, 37). PTPN22 knockdown in THP-1 macrophages primed with ultrapure LPS (upLPS) led to increased NLRP3 phosphorylation and increased NLRP3 sequestration in autophagosomes, with a concomitant reduction in IL-1β secretion ranging from about 50% with MDP treatment and up to 80% with monosodium urate (MSU) treatment (36, 37). In support of this, inhibiting autophagy restored IL-1β secretion from PTPN22 knockdown THP-1 cells (37). PTPN22620W is a gain-of-function variant and is better able to dephosphorylate NLRP3 and prevents its sequestration into phagophores and subsequent degradation (Figure 4B). PTPN22620W has an enhanced capacity to dephosphorylate NLRP3 in a cell free system compared to PTPN22620R (36). When moDCs from PTPN22620R/W donors were primed with ultrapure LPS and treated with monosodium urate (MSU) cleaved caspase-1 was increase by 500% and produced 300% more mature IL-1β compared to PTPN22620R/R donors (36).
Much like THP-1 cells with PTPN22 knockdown, C57BL/6-Ptpn22−/− mice exhibited a 50% reduction in MDP-, MSU-, and ATP-induced IL-1β secretion from BMDCs compared to those of Ptpn22-competent mice and this effect was abrogated by inhibition of autophagy (Figure 4B) (37). This is due to the catalytic activity of PTPN22. In C57BL/6-Ptpn22−/− BMDCs expressing the catalytically dead human PTPN22 (PTPN22263Q) the same effect was observed. Similar to moDCs from PTPN22620R/W donors, BMDCs from C57BL/6-PEP619W mice have less NLRP3 in autophagosomes upon upLPS/MSU treatment and over 50% increased IL-1β secretion compared to C57BL/6 mice (36, 37). The same was seen when comparing BMMΦ from C57BL/6-PEP619W mice with C57BL/6 mice (36). Taken together, these data demonstrate that PTPN22620W and PEP619W are gain-of-function variants with respect to NLRP3 dephosphorylation and enhance NLRP3-inflammasome activation and mature IL-1β release. While signaling via NOD2 is capable of activating the NLRP3 inflammasome following priming with LPS, it also induces autophagy and cytokine secretion.
NOD2-Induced Autophagy and Cytokine Secretion
PTPN22 is a negative regulator of NOD2-induced autophagy (Figure 5A). Knockdown of PTPN22 via shRNA in THP-1 monocytes enhanced NOD2-induced LC3B-II, a cleaved and activated form of LC3B indicative of autophagosome formation. There was also a decrease in p62 protein levels consistent with enhanced autolysosome activity. Knockdown of PTPN22 via shRNA in THP-1 monocytes also led to enhanced JNK, p38, NF-κB-p65, and NF-κB-p50, activation downstream of NOD2 while reducing ERK activation. Enhanced NOD2-induced IL-6 and TNF mRNA expression and IL-6, IL-8, and TNF secretion were also seen with PTPN22 knockdown (36, 39). In addition, the reduction in PTPN22 resulted in decreased NOD2-induced ICAM1, NOD2, T-bet, and IFN-γ mRNA expression as well as reduced IFN-γ secretion (39). Interestingly, the variant in PTPN22 is associated with reduced risk of Crohn’s disease while loss-of-function mutations in NOD2 are associated with increased risk of Crohn’s disease (123, 124). This could indicate that the T1D-risk allotype (PTPN22620W) enhances NOD2 activity to suppress gastrointestinal pathology, however, more studies are necessary to clarify how PTPN22620W alters NOD2 response compared to PTPN22620R. These PTPN22 knockdown studies indicate that PTPN22 negatively regulates NOD2-induced autophagy, IL-6, IL-8, and TNF production while positively regulating NOD2-induced ICAM1, NOD2, and IFN-γ production.
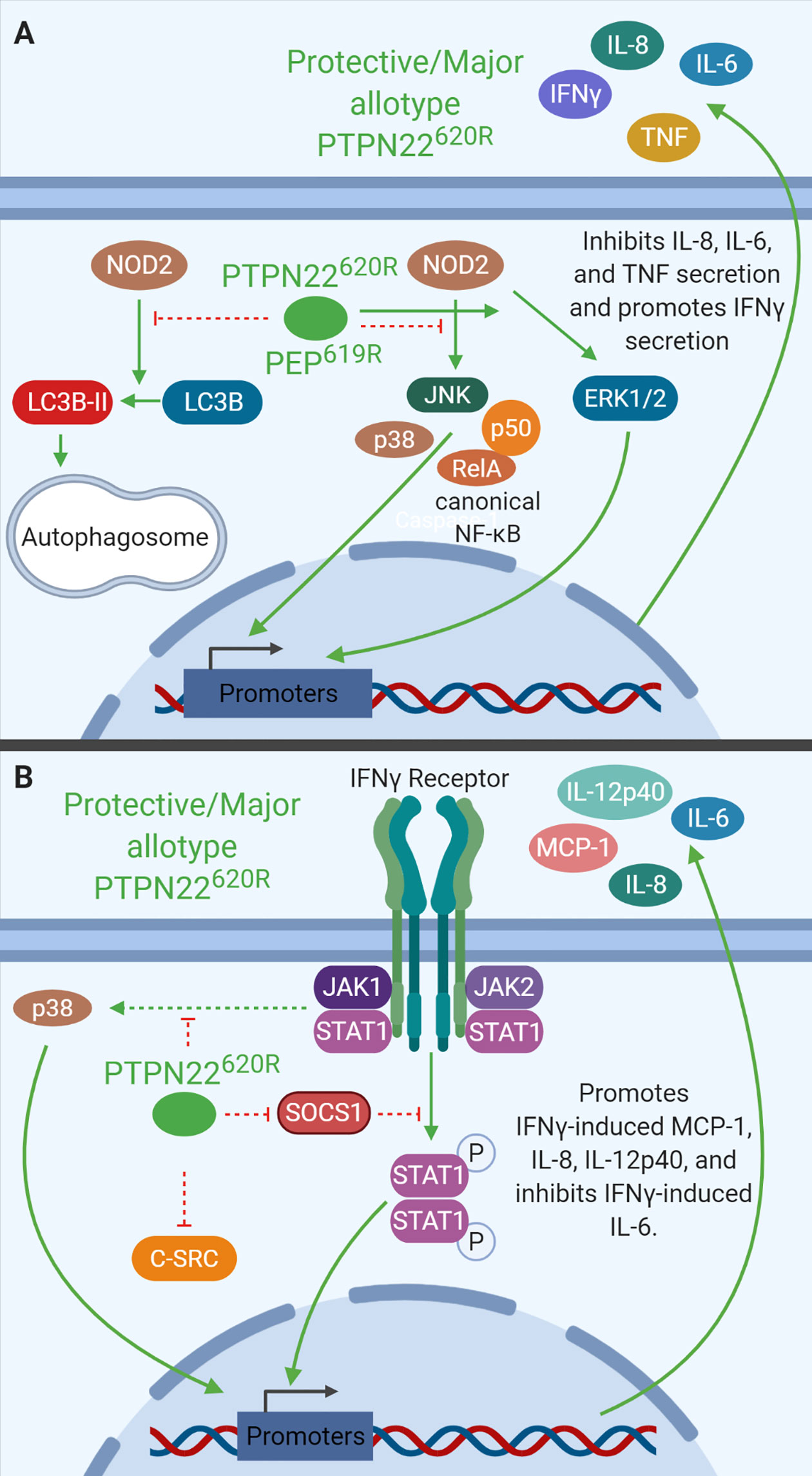
Figure 5 PTPN22 regulates NOD2 and IFNγ signaling in monocytes. (A) PTPN22620R and PEP619R negatively regulate NOD2-induced autophagy and NOD2-induced IL-8, IL-6, and TNF secretion while promoting NOD2-induced IFNγ secretion. (B) PTPN22620R negatively regulates p38, SOCS1, and C-SRC activation downstream of the IFNγR. PTPN22620R promotes IFNγ-induced MCP-1, IL-8, and IL-12p40, while it inhibits IFNγ-induced IL-6.
Like PTPN22 knockdown in THP-1 cells, C57BL/6-Ptpn22−/− mice demonstrate that PEP is a negative regulator of NOD2-induced cytokine secretion in BMDCs of mice (Figure 5A). BMDCs from Ptpn22−/− mice treated with MDP had increased p38, NF-κB p65, and NF-κB p50 phosphorylation, and decreased ERK phosphorylation compared to Ptpn22-competent BMDCs. MDP-treated BMDCs from Ptpn22−/− mice had increased levels of IL6 and TNF but decreased levels of NOD2, ICAM-1, and IFNγ mRNA compared to Ptpn22 competent BMDCs (39). MDP-treated Ptpn22−/− BMDCs had enhanced IL-6, IL-8, and TNF secretion compared to Ptpn22-intact BMDCs (39). These data closely mirror data from PTPN22 knockdown THP-1 cells and demonstrate that PTPN22620R and PEP619R are negative regulators of NOD2-induced autophagy and cytokine secretion (Figure 5A). PTPN22 does not only influence signaling downstream of TLRs and other pattern recognition receptors in monocytes, macrophages, and DCs, it also influences cytokine secretion and signaling in response to IFNγ.
IFNγ Receptor Signaling
PTPN22 regulates IFN-γ receptor (IFNγR) signaling in human monocytes (Figure 5B). PTPN22 knockdown in THP-1 monocytes followed by treatment with IFNγ induced increased SOCS1 phosphorylation and activity and reduced protein levels of SOCS3 compared to control siRNA transfected cells. PTPN22 pulls down with SOCS1, suggesting that PTPN22 may be responsible for dephosphorylating and inactivating SOCS1 when it is present. In agreement with this, PTPN22 knockdown reduced activation (phosphorylation) of known SOCS1 targets, Jak1, STAT1, and STAT3 in response to IFNγ. It also reduced subsequent production of ICAM1 (~70% reduced), NOD2 (~15% reduced), and T-bet mRNA (~40% reduced) when compared to control siRNA transfected cells. Knockdown of PTPN22 also decreased IFNγ-induced MCP-1 (~70% reduced), IL-8 (~50% less), and IL12p40 (~75% reduced) secretion (29). These data indicate that PTPN22 is a positive regulator of STAT1 and STAT3 activation following IFNγ treatment. Activation of STAT1 and subsequent gene induction is the most well characterized portion of IFNγR signaling, however, the signaling cascade activated by the IFNγR includes many other signaling molecules. Treatment with IFNγ also induces signaling via p38 MAPK and Src. Upon knockdown of PTPN22 in THP-1 monocytes, IFNγ-induced p38 MAPK activation and subsequent IL-6 mRNA expression and protein production were enhanced compared to control siRNA transfected cells. This suggests that PTPN22 is negatively regulating p38 MAPK activation downstream of the IFNγR. It is unknown how PTPN22 regulates p38 MAPK activation downstream of the IFNγR, however, there are several plausible targets. Current literature indicates that p38 MAPK is activated by the IFNγR via a signaling cascade involving JAK2, Pyk2, MEKK4, MEK6, and finally p38 MAPK (125, 126). Pyk2, MEKK4, and p38 MAPK are attractive potential targets of PTPN22 because they are all activated by phosphorylation on a tyrosine residue. At this time, more targeted research is necessary to define the PTPN22 target(s) in this pathway. Similarly, PTPN22 knockdown induced basal Src phosphorylation that increased after IFNγ treatment; however, in control siRNA transfected cells there was no basal Src phosphorylation nor was there IFNγ-induced Src phosphorylation. This indicates that PTPN22 negatively regulates basal Src activation and IFNγR-induced Src activation (29). While PTPN22 influences response to IFNγ treatment alone it also influences macrophage cytokine secretion following polarization in response to IFNγ/LPS or IL-4/IL-13 treatment.
Macrophage Polarization
In primary MDMs, PTPN22 is a negative regulator of IL-12 and IL-23 production following M1 polarization (Figure 6A) and a positive regulator of IL-10 production following M2 polarization (Figure 6B). PTPN22 knockdown in MDMs led to increased IL-23 (~60% more) and IL-12 (~30% more) secretion upon IFNγ/LPS treatment (M1 polarization) and decreased IL-10 expression (~50% less) following IL-4/IL-13 treatment (M2 polarization). PTPN22620W appears to be a gain-of-function negative regulator of IL-12 and IL-23 production following M1 polarization (Figure 6A). M1 polarized macrophages from PTPN22620W/W donors expressed significantly less IL-12, IL-1β, and IL-6 than those from PTPN22620R/R donors. It is thought that this gain-of-function phenotype is due to enhanced expression of PTPN22620W upon M1 polarization. M1 polarized macrophages from PTPN22620W/W donors expressed significantly more PTPN22 than those from PTPN22620R/R donors. PTPN22620W and PTPN22620R are comparable positive regulators of M2 polarization with no differences in IL-10 expression following IL-4/IL-13 treatment (Figure 6B) (38).
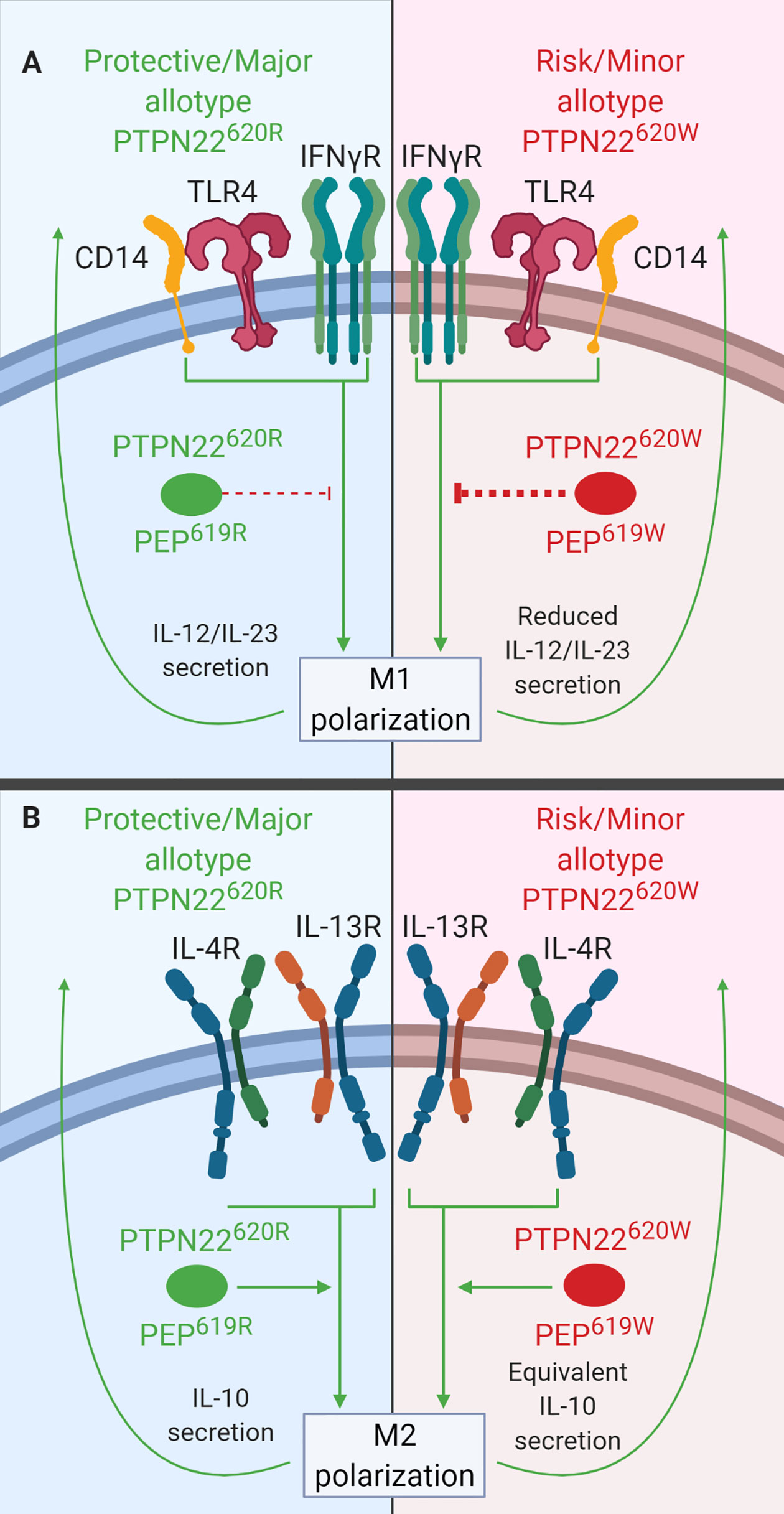
Figure 6 PTPN22 regulates macrophage polarization. (A) PTPN22620R and PEP619R inhibit cytokine secretion from M1 macrophages. Upon M1 polarization of PTPN22620W or PEP619W macrophages, there is more PTPN22620W and PEP619W present and the enhanced expression leads to an increased capacity to inhibit cytokine secretion from M1 macrophages. (B) PTPN22620R and PTPN22620W promote cytokine secretion from M2 macrophages equivalently.
Like PTPN22 knockdown in human MDMs, splenic macrophages from C57BL/6-Ptpn22−/− mice had increased expression of IL-23 (~200%) and IL-12 (~250%) following M1 polarization (Figure 6A) and decreased expression of IL-10 (~50%) following M2 polarization (Figure 6B) compared to those from Ptpn22-intact mice (38). These Ptpn22−/− splenic macrophages had increased NF-κB activity (~200%) compared to Ptpn22-intact macrophages and this could explain the increase in LPS/IFNγ-induced IL-12 and IL-23. Splenic macrophages from C57BL/6-Ptpn22−/− mice reconstituted in vitro with PEP619R or PEP619W and then polarized to M1 or M2 macrophages had no difference in gene expression. If the level of PEP expression is important in mouse macrophages like the level of PTPN22 expression is in human MDMs, then reconstituting macrophages with the same amount or PEP619R and PEP619W would not capture the effects seen in human MDMs where PTPN22620W and PTPN22620R expression levels are different (38). Like human PTPN22620W M1 macrophages, M1 peritoneal macrophages from C57BL/6-PEP619W mice had lower mRNA levels for the M1 genes, iNOS (~50 fold less) and TNF (~2 fold less), than those from WT mice (127). Overall, these data indicate that PTPN22620W and PEP619W are gain-of-function negative regulators of macrophage cytokine secretion following M1 polarization due to increased PTPN22 expression (Figure 6A). PTPN22 has multiple roles in macrophage polarization and in fact influences diverse functions in monocytes, macrophages, and DCs. The T1D-associated variant of PTPN22, PTPN22620W, influences a large number of these functions and these cellular phenotypes could contribute to the pathogenesis of T1D.
PTPN22 in Monocytes, Macrophages, and DCs and Impact on T1D
Monocytes, macrophages, and DCs are APCs that are all capable of initiating and enhancing adaptive immune responses. The precipitating events that lead to loss of tolerance and the development of T1D are unknown; be it physiological β cell death, viral infection, bacterial infection, or some other initiating event, monocytes, macrophages, and DCs are the cells most likely to sense β cell death/inflammation and initiate the adaptive immune response. After APCs trigger the adaptive immune response, these cells enhance and support the ongoing immune response against β cells. In APCs, PTPN22620R plays a role in signaling downstream of many PRRs [i.e., TLR4 (35), TLR7/8 (122), NOD2 (36, 37, 39)], cytokine receptors [i.e., IL-4R/IL-13R (38), and IFNγR (29, 128)]. PTPN22620W enhances NLRP3 activation and subsequent IL-1β release following priming via TLR4 (LPS) and treatment with a NOD2 agonists (MDP) while dampening the T1-IFN response following TLR4/7/8 stimulation. The combination of these phenotypes renders APCs from PTPN22620R/W and PTPN22620W/W humans more sensitive to NLRP3 activation while dampening their ability to produce T1-IFNs in response to PRR signaling. IL-1β enhances naïve and memory CD4 T cell expansion and this could in turn exacerbate activation of autoreactive CD4 T cells during the initiation of T1D (129). T1-IFNs enhance CD8 T cell activation and support activated T cell survival and are considered a major feature of the diabetic islet microenvironment where they enhance expression of MHC-I on β cells and expression of T cell chemoattractants (e.g., CXCL10) (130–132). Importantly, the T1-IFN phenotype results in a reduction of T1-IFN and not a complete loss. This might reduce the induction of MHC-I and T cell chemoattractants, however, it would not ablate them and in a genetically predisposed individual this may still be more than sufficient to help initiate and sustain T1D especially in combination with enhanced IL-1β production.
Neutrophils
While neutrophils are not essential for T1D pathology (133, 134), they do play a role in other rs2476601-associated autoimmune diseases (e.g., RA, SLE). Thus, it is paramount to consider how PTPN22 influences neutrophil function (135). Importantly, PTPN22 is expressed in neutrophils and PTPN22620 allotype influences neutrophil function. This section will review what is known about the function of PTPN22620R and PTPN22620W in human neutrophils. PTPN22 protein level does not vary when comparing neutrophils from PTPN22620R/R and PTPN22620R/W donors; however, at time of writing, PTPN22620W/W donors have not been assessed for neutrophil PTPN22 content (30). In human neutrophils, PTPN22 plays a role in protein citrullination (30), neutrophil extracellular trap formation (NETosis) (Figure 7A) (30), transmigration across inflamed endothelium (31), and response to N-formyl- Methionine-Leucine-Phenylalanine (fMLP) (Figure 7B) (31). PTPN22620R has been shown to interact with PAD4 in human neutrophils and is a negative regulator of PAD4 activity and NETosis while PTPN22620W is a loss-of-function variant in this process (Figure 7A) (30). PTPN22620W potentiates neutrophil calcium flux and ROS production in response to fMLP stimulation (Figure 7B) as well as transmigration across inflamed epithelium when compared to PTPN22620R (31).
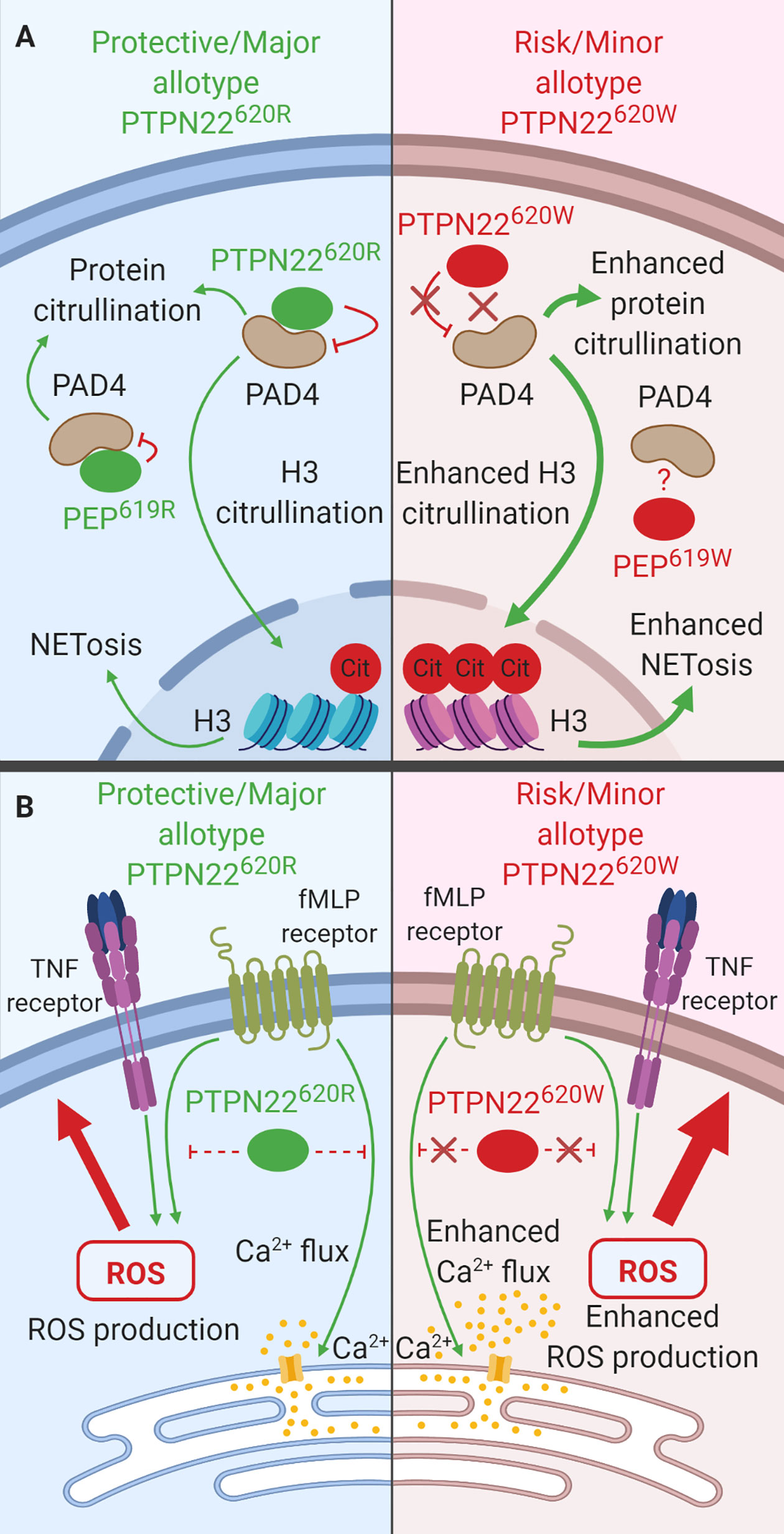
Figure 7 The function of PTPN22 in Neutrophils. (A) PTPN22620R and PEP619R are negative regulators of PAD4 activation and subsequent citrullination of target proteins in neutrophils. PTPN22620W is a loss-of-function variant that potentiates PAD4 activation and citrullination of PAD4 targets. (B) PTPN22620R is a negative regulator of fMLP induced calcium flux and ROS production while PTPN22620W a loss-of-function variant that results in enhanced fMLP-induced calcium flux and ROS.
Protein Citrullination and NETosis
PTPN22 is a negative regulator of protein citrullination and NETosis and PTPN22620W is a loss-of-function variant (30) (Figure 7A). Neutrophils from heterozygous PTPN22620R/W donors displayed a hypercitrullinated protein profile (~4 fold more in PTPN22620R/W neutrophils), they had enhanced citrullination of histone H3, a marker of NETosis (~5 fold more in PTPN22620R/W neutrophils), and they were more prone to NETosis (3%–15% of PTPN22620R/W neutrophils vs. ~2% of PTPN22620R/R neutrophils) compared to those from PTPN22620R/R donors (30, 136). PAD4 co-immunoprecipitated PTPN22 in human neutrophils and PTPN22 allotype influences this interaction; there is a significantly decreased amount of PTPN22 co-immunoprecipitated with PAD4 in heterozygous PTPN22620R/W donors when compared to PTPN22620R/R donors (~66% decreased). The total PTPN22 protein level was the same between donors implying that PTPN22620R interacts with PAD4 more than PTPN22620W. In C57BL/6-Ptpn22−/− mouse macrophages transfected with human PTPN22620R or PTPN22620W expressing constructs, PTPN22620R but not PTPN22620W reduced protein citrullination and co-immunoprecipitated with PAD4 further supporting the lack of association of PTPN22620W with PAD4 (30).
Much like in human neutrophils, PEP in C57BL/6 mouse neutrophils interacts with PAD-4. PEP co-immunoprecipitated with PAD-4. The absence of PEP in C57BL/6 mice enhanced protein citrullination by approximately 100%; however, the enhanced protein citrullination was abrogated in the presence of a catalytically dead PEP indicating that the catalytic activity of PEP is not involved in this process. Unlike in humans, PEP does not specifically impact histone H3 citrullination or NETosis in mouse neutrophils (30). Taken together, these data indicate that PTPN22620R is a negative regulator of protein citrullination and NETosis in human neutrophils and PTPN22620W is a loss-of-function variant (Figure 7A).
Transmigration, ROS Production, and Calcium Flux
PTPN22 plays a role in transmigration across inflamed endothelium, as well as the response to fMLP, a highly chemotactic n-formylated oligopeptide actively released by invading bacteria or passively released by mitochondria of dying host cells (31, 137, 138). Significantly more neutrophils from PTPN22620R/W donors transmigrate across inflamed (TNF treated) endothelium over 2 min than those from PTPN22620R/R donors (PTPN22620R/W = 43 ± 9% vs. PTPN22620R/R = 24 ± 4%). Stimulation of neutrophils from healthy PTPN22620R/W donors with fMLP resulted in increased calcium flux compared to neutrophils from healthy PTPN22620R/R donors (PTPN22620R/W = 0.28 ± 0.02 vs. PTPN22620R/R = 0.24 ± 0.02 Indo-1 ratio). Priming of neutrophils from healthy PTPN22620R/W donors with TNF followed by stimulation with fMLP resulted in significantly increased ROS production (4-fold increase) compared to PTPN22620R/R donors (Figure 7B) (31).
Unlike in humans, PEP does not appear to play a role in transmigration across inflamed endothelium or the response to fMLP in C57BL/6 mice. Ptpn22−/− and Ptpn22-intact mouse neutrophils migrated across TNF-treated endothelium at the same rate (139). Ptpn22−/− and PTPN22-intact neutrophils produce similar amounts of ROS in response to fMLF (also called fMLP) and PMA stimulation, however, they were not primed with TNF like the human neutrophils which may explain why there was no difference in ROS production. Neutrophils from C57BL/6-Ptpn22−/− mice did however exhibit decreased ROS production (~50% reduced) and degranulation (~25% reduced) in response to FcγR and integrin stimulation compared to neutrophils from C57BL/6 mice. These pathways have not been investigated in the context of PTPN22 in humans (139). In human neutrophils, PTPN22620W enhances transmigration across inflamed endothelium, calcium flux in response to fMLP stimulation, and ROS production in response to TNF priming followed by fMLP stimulation.
PTPN22 in Neutrophils and Impact on T1D
Current data indicates that neutrophils most likely do not play a direct role in the pathogenesis of T1D in humans (133, 134) and it is apparent that they do not influence pathogenesis in NOD mice; depletion of neutrophils starting at 4 weeks of age does not impact development of T1D in NOD mice (134). While data indicate that neutrophils do not play a role in human T1D pathogenesis, many neutrophil products (e.g., ROS, NETs, cytokines) are capable of damaging tissues, including pancreatic β cells (140). Neutrophils from PTPN22620R/W donors had enhanced calcium flux and ROS production in response TNF priming followed by treatment with fMLP compared to those from PTPN22620R/R donors. These PTPN22620R/W neutrophils also transmigrated across TNF-inflamed epithelium faster than their PTPN22620R/R counterparts, displayed enhanced protein citrullination, and were more prone to NETosis (30, 31, 136). The combined effects of these phenotypes mean that PTPN22620R/W and PTPN22620W/W patients with T1D could display enhanced neutrophil accumulation in the exocrine pancreas due to enhanced transmigration across inflamed epithelium and increased frequency of these infiltrating neutrophils releasing NETs and producing high amount of ROS. More studies need to be undertaken to understand if neutrophils participate in the pathogenesis of human T1D and if the influence of PTPN22620 allotype effects their participation. Overall, it is clear that PTPN22 plays diverse roles in many cell types that have the potential to influence the pathogenesis of T1D.
Conclusions
PTPN22 acts as a negative regulator of TCR and BCR signaling by preventing weak TCR/BCR ligation from activating T cells or B cells. In addition, PTPN22 functions in diverse signaling pathways in leukocytes. This phosphatase downregulates signaling in the NOD2, IFNγ/LPS, IFNγR, and fMLP receptor signaling pathways. Conversely, PTPN22 positively regulates NLRP3 inflammasome activation, TLR4/7/8 induction of T1-IFN secretion, PAD4 activation, and IL-4/IL-13 signaling. There are several rare genetic variants of PTPN22 in humans that are associated with increased or decreased risk of autoimmune diseases. Also, rs2476601 marks the PTPN22R620W variant that is associated with increased risk for T1D and many other autoimmune diseases (28, 51–59). The 620R->W conversion creates a gain-of-function variant that suppresses TCR/BCR signaling and impacts autoimmunity by increasing the number of autoreactive T cells and B cells that escape central tolerance. Similarly, rs56048322, marks the variant, PTPN22K750N, and is associated with increased risk of T1D. The 750K->N conversion induces alternative splicing of PTPN22 that results in a novel isoform that competes with other PTPN22 isoforms for CSK binding causing T cell hyporesponsiveness and, like rs2476601, could allow more autoreactive T cells to escape central tolerance (48). In contrast, rs33996649, encodes the variant, PTPN22R263Q, which has diminished phosphatase activity and reduces risk for SLE and RA possibly by enhancing T cell central tolerance (49, 50).
The mouse orthologue of PTPN22, Ptpn22 encoding PEP, plays similar roles to human PTPN22 and is even included in one of the insulin-dependent diabetes (Idd) intervals, Idd18.2 (141). While rodent models, especially the NOD mouse, have been integral to furthering our understanding of T1D, the analogous mutation to PTPN22R620W, PEPR619W, is not naturally present in NOD and does not induce the same phenotype as observed in humans. This is not entirely surprising, PTPN22 and PEP are two of the most divergent phosphatase orthologues between humans and mice (38, 142). PTPN22 and PEP share 70% amino acid identity overall and only 61% amino acid identity in the c-terminal domain, where rs2476601 lies (38, 45, 51, 142).
PTPN22 is also expressed in NK cells, monocytes, macrophages, DCs, and neutrophils where it influences diverse signaling pathways (28, 68). The expression of PTPN22 in APCs adds another layer of possible confounding factors when interpretting data in TCR and BCR signaling due to the fact that APCs directly influence T cell and B cell activation. Data describing the influence of PTPN22 on interactions of APCs with T cells in humans is lacking, but there are hints in both human and mouse data that can inform future studies. T cell/macrophage interactions are largely mediated by IFNγ/IFNγR and CD40L/CD40 in an antigen-dependent context (143, 144). PTPN22 knockdown in THP-1 monocytes had diverse effects on the IFNγR signaling pathways. PTPN22 knockdown increased activation of SOCS1, and predictably led to lower activation of JAK1, STAT1, and STAT3, known SOCS1 targets, as well as lower mRNA expression of ICAM-1, NOD2, and T-bet. PTPN22 knockdown also enhanced IFNγR-induced p38 activation and subsequent IL-6 mRNA and protein expression (29). There have not been any studies of the impact of PTPN22 on human CD40 signaling however heterozygous and homozygous PTPN22620W donors have increased CD40 expression on their immature B cells compared to PTPN22620R donors promoting speculation that CD40 signaling would be enhanced in these cells (100, 103). All of these data emphasize the need to elucidate how PTPN22620W influences human macrophage and DC expression of CD40, MHC-II, CD80 and CD86.
At this time, there are more questions than answers pertaining to the influence of rs2476601 on TCR signaling and the interface of the innate and adaptive arms of the immune system. More studies aimed at illucidating the impact PTPN22620W has on TCR signaling and innate immune cell/adaptive immune cell interactions and crosstalk in humans need to be undertaken, especially in light of the conflicting data between mouse PEP619W and human PTPN22620W studies, to answer these questions.
Author Contributions
LA, MW, and CM wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by research grants from the National Institutes of Health UC4 DK104194 (CM), UG3 DK122638 (CM), P01 AI042288 (CM, MW), T32 DK108736 (LA), and the Sebastian Family Endowment for Diabetes Research.
Conflict of Interest
MW is currently employed by Century Therapeutics.
The remaining authors declare that this work was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All figures were created with BioRender.com.
Abbreviations
ABCs, Age-associated B cells; APCs, Antigen presenting cells; aTreg, Activated Treg; BCR, B cell Receptor; BMDC, Bone marrow-derived dendritic cell; BMMΦ, Bone marrow-derived macrophage; BND cells, Naïve IgD+ B cells; CLL, Chronic B lymphocytic leukemia; CSK, C-src tyrosine kinase; DCs, Dendritic cells; fMLF, N-formyl-Methionine-Leucine-Phenylalanine; fMLP, N-formyl-Methionine-Leucine-Phenylalanine; Hep-2 cells, Human epithelial type 2 cells; HLA, Human Leukocyte Antigen; HSCs, CD34+ hematopoietic stem cells; Idd intervals, Insulin-depending diabetes intervals; IFNγR, Interferon gamma receptor; iTreg, Induced Treg; LAD, Leukocyte adhesion deficiency; MDM, Monocyte-derived macrophage; MDP, Muramyldipeptide; MHC, Major Histocompatibility Complex; moDC, Monocyte-derived dendritic cell; MSU, Monosodium urate; NETosis, Neutrophil extracellular trap formation; NOD2, Nucleotide-binding oligomerization domain-containing protein; NTC siRNA, Non-targeting control siRNA; PBMC, Peripheral blood mononuclear cells; pDC, Plasmacytoid dendritic cell; PRR, Pattern recognition receptor; PTPN22, Protein tyrosine phosphatase non-receptor type 22; RA, Rheumatoid arthritis; ROS, Reactive oxygen species; siRNA, Small interfering RNA; SLE, Systemic lupus erythematosus; SNP, Single nucleotide polymorphism; T1D, Type 1 Diabetes; T1-IFN, Type 1 interferon; TCR, T cell receptor; TLR, Toll-like receptor; Treg, T regulatory cell; upLPS, Ultrapure LPS.
References
1. Centers for Disease Control and Prevention. National Diabetes Statistics Report 2020. Atlanta, GA: Centers for Disease Control and Prevention, U.S. Dept of Health and Human Services (2020).
2. Barnett AH, Eff C, Leslie RDG, Pyke DA. Diabetes in identical twins. A study of 200 pairs. Diabetologia (1981) 20:87–93. doi: 10.1007/bf00262007
3. Redondo MJ, Jeffrey J, Fain PR, Eisenbarth GS, Orban T. Concordance for islet autoimmunity among monozygotic twins. N Engl J Med (2008) 359:2849–50. doi: 10.1056/NEJMc0805398
4. Onengut-Gumuscu S, Chen W-M, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet (2015) 47:381–6. doi: 10.1038/ng.3245
5. Cooper JD, Walker NM, Smyth DJ, Downes K, Healy BC, Todd JA. Type I Diabetes Genetics Consortium. Follow-up of 1715 SNPs from the Wellcome Trust Case Control Consortium genome-wide association study in type I diabetes families. Genes Immun (2009) 10(Suppl 1):S85–94. doi: 10.1038/gene.2009.97
6. Hyttinen V, Kaprio J, Kinnunen L, Koskenvuo M, Tuomilehto J. Genetic liability of type 1 diabetes and the onset age among 22,650 young Finnish twin pairs: a nationwide follow-up study. Diabetes (2003) 52:1052–5. doi: 10.2337/diabetes.52.4.1052
7. Noble JA, Valdes AM. Genetics of the HLA region in the prediction of type 1 diabetes. Curr Diabetes Rep (2011) 11:533–42. doi: 10.1007/s11892-011-0223-x
8. Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes (2008) 57:1084–92. doi: 10.2337/db07-1331
9. Inshaw JRJ, Cutler AJ, Crouch DJM, Wicker LS, Todd JA. Genetic Variants Predisposing Most Strongly to Type 1 Diabetes Diagnosed Under Age 7 Years Lie Near Candidate Genes That Function in the Immune System and in Pancreatic β-Cells. Diabetes Care (2020) 43:169–77. doi: 10.2337/dc19-0803
10. Nepom BS, Schwarz D, Palmer JP, Nepom GT. Transcomplementation of HLA genes in IDDM. HLA-DQ alpha- and beta-chains produce hybrid molecules in DR3/4 heterozygotes. Diabetes (1987) 36:114–7. doi: 10.2337/diab.36.1.114
11. van Lummel M, van Veelen PA, Zaldumbide A, de Ru A, Janssen GMC, Moustakas AK, et al. Type 1 diabetes-associated HLA-DQ8 transdimer accommodates a unique peptide repertoire. J Biol Chem (2012) 287:9514–24. doi: 10.1074/jbc.M111.313940
12. Reichstetter S, Kwok WW, Nepom GT. Impaired binding of a DQ2 and DQ8-binding HSV VP16 peptide to a DQA1*0501/DQB1*0302 trans class II heterodimer. Tissue Antigens (1999) 53:101–5. doi: 10.1034/j.1399-0039.1999.530111.x
13. Plagnol V, Howson JMM, Smyth DJ, Walker N, Hafler JP, Wallace C, et al. Genome-wide association analysis of autoantibody positivity in type 1 diabetes cases. PloS Genet (2011) 7:e1002216. doi: 10.1371/journal.pgen.1002216
14. Cooper JD, Smyth DJ, Smiles AM, Plagnol V, Walker NM, Allen JE, et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat Genet (2008) 40:1399–401. doi: 10.1038/ng.249
15. Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet (2007) 39:857–64. doi: 10.1038/ng2068
16. Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature (2007) 447:661–78. doi: 10.1038/nature05911
17. Hakonarson H, Grant SFA, Bradfield JP, Marchand L, Kim CE, Glessner JT, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature (2007) 448:591–4. doi: 10.1038/nature06010
18. Huang J, Ellinghaus D, Franke A, Howie B, Li Y. 1000 Genomes-based imputation identifies novel and refined associations for the Wellcome Trust Case Control Consortium phase 1 Data. Eur J Hum Genet (2012) 20:801–5. doi: 10.1038/ejhg.2012.3
19. Bradfield JP, Qu H-Q, Wang K, Zhang H, Sleiman PM, Kim CE, et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PloS Genet (2011) 7:e1002293. doi: 10.1371/journal.pgen.1002293
20. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet (2009) 41:703–7. doi: 10.1038/ng.381
21. Grant SFA, Qu H-Q, Bradfield JP, Marchand L, Kim CE, Glessner JT, et al. Follow-up analysis of genome-wide association data identifies novel loci for type 1 diabetes. Diabetes (2009) 58:290–5. doi: 10.2337/db08-1022
22. Hakonarson H, Qu H-Q, Bradfield JP, Marchand L, Kim CE, Glessner JT, et al. A novel susceptibility locus for type 1 diabetes on Chr12q13 identified by a genome-wide association study. Diabetes (2008) 57:1143–6. doi: 10.2337/db07-1305
23. Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med (2009) 360:1646–54. doi: 10.1056/NEJMra0808284
24. Tang W, Cui D, Jiang L, Zhao L, Qian W, Long SA, et al. Association of common polymorphisms in the IL2RA gene with type 1 diabetes: evidence of 32,646 individuals from 10 independent studies. J Cell Mol Med (2015) 19:2481–8. doi: 10.1111/jcmm.12642
25. Howson JMM, Walker NM, Smyth DJ, Todd JA. Type I Diabetes Genetics Consortium. Analysis of 19 genes for association with type I diabetes in the Type I Diabetes Genetics Consortium families. Genes Immun (2009) 10(Suppl 1):S74–84. doi: 10.1038/gene.2009.96
26. Gjörloff-Wingren A, Saxena M, Williams S, Hammi D, Mustelin T. Characterization of TCR-induced receptor-proximal signaling events negatively regulated by the protein tyrosine phosphatase PEP. Eur J Immunol (1999) 29:3845–54. doi: 10.1002/(SICI)1521-4141(199912)29:12<3845::AID-IMMU3845>3.0.CO;2-U
27. Arechiga AF, Habib T, He Y, Zhang X, Zhang Z-YZ-Y, Funk A, et al. Cutting edge: the PTPN22 allelic variant associated with autoimmunity impairs B cell signaling. J Immunol (2009) 182:3343–7. doi: 10.4049/jimmunol.0713370
28. Begovich AB, Carlton VEH, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet (2004) 75:330–7. doi: 10.1086/422827
29. Spalinger MR, Lang S, Weber A, Frie P, Fried M, Rogler G, et al. Loss of protein tyrosine phosphatase non-receptor type 22 regulates interferon-γ-induced signaling in human monocytes. Gastroenterology (2013) 144:978–88. doi: 10.1053/j.gastro.2013.01.048
30. Chang H-H, Dwivedi N, Nicholas AP, Ho I-C. The W620 Polymorphism in PTPN22 Disrupts Its Interaction With Peptidylarginine Deiminase Type 4 and Enhances Citrullination and NETosis. Arthritis Rheumatol (Hoboken NJ) (2015) 67:2323–34. doi: 10.1002/art.39215
31. Bayley R, Kite KA, McGettrick HM, Smith JP, Kitas GD, Buckley CD, et al. The autoimmune-associated genetic variant PTPN22 R620W enhances neutrophil activation and function in patients with rheumatoid arthritis and healthy individuals. Ann Rheum Dis (2015) 74:1588–95. doi: 10.1136/annrheumdis-2013-204796
32. Burn GL, Cornish GH, Potrzebowska K, Samuelsson M, Griffié J, Minoughan S, et al. Superresolution imaging of the cytoplasmic phosphatase PTPN22 links integrin-mediated T cell adhesion with autoimmunity. Sci Signal (2016) 9:ra99. doi: 10.1126/scisignal.aaf2195
33. Fousteri G, Jofra T, Debernardis I, Stanford SM, Laurenzi A, Bottini N, et al. The protein tyrosine phosphatase PTPN22 controls forkhead box protein 3 T regulatory cell induction but is dispensable for T helper type 1 cell polarization. Clin Exp Immunol (2014) 178:178–89. doi: 10.1111/cei.12393
34. Negro R, Gobessi S, Longo PG, He Y, Zhang Z-Y, Laurenti L, et al. Overexpression of the autoimmunity-associated phosphatase PTPN22 promotes survival of antigen-stimulated CLL cells by selectively activating AKT. Blood J (2012) 119:6278–87. doi: 10.1182/blood-2012-01-403162
35. Wang Y, Shaked I, Stanford S, Zhou W, Curtsinger J, Mikulski Z, et al. The Autoimmunity-Associated Gene PTPN22 Potentiates Toll-like Receptor-Driven, Type 1 Interferon-Dependent Immunity. Immunity (2013) 39:111–22. doi: 10.1016/j.immuni.2013.06.013
36. Spalinger MR, Kasper S, Gottier C, Lang S, Atrott K, Vavricka SR, et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest (2016) 126:1783–800. doi: 10.1172/JCI83669
37. Spalinger MR, Lang S, Gottier C, Dai X, Rawlings DJ, Chan AC, et al. PTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy-dependent manner. Autophagy (2017) 13:1590–601. doi: 10.1080/15548627.2017.1341453
38. Chang H-H, Miaw S-C, Tseng W, Sun Y-W, Liu C-C, Tsao H-W, et al. PTPN22 modulates macrophage polarization and susceptibility to dextran sulfate sodium-induced colitis. J Immunol (2013) 191:2134–43. doi: 10.4049/jimmunol.1203363
39. Spalinger MR, Lang S, Vavricka SR, Fried M, Rogler G, Scharl M. Protein Tyrosine Phosphatase Non-Receptor Type 22 Modulates NOD2-Induced Cytokine Release and Autophagy. PloS One (2013) 8:e72384. doi: 10.1371/journal.pone.0072384
40. Vang T, Liu WH, Delacroix L, Wu S, Vasile S, Dahl R, et al. LYP inhibits T-cell activation when dissociated from CSK. Nat Chem Biol (2012) 8:437–46. doi: 10.1038/nchembio.916
41. Fiorillo E, Orrú V, Stanford SM, Liu Y, Salek M, Rapini N, et al. Autoimmune-associated PTPN22 R620W variation reduces phosphorylation of lymphoid phosphatase on an inhibitory tyrosine residue. J Biol Chem (2010) 285:26506–18. doi: 10.1074/jbc.M110.111104
42. Yang S, Svensson MND, Harder NHO, Hsieh W-C, Santelli E, Kiosses WB, et al. PTPN22 phosphorylation acts as a molecular rheostat for the inhibition of TCR signaling. Sci Signal (2020) 13:8130. doi: 10.1126/scisignal.aaw8130
43. Hill RJ, Zozulya S, Lu Y-LL, Ward K, Gishizky M, Jallal B. The lymphoid protein tyrosine phosphatase Lyp interacts with the adaptor molecule Grb2 and functions as a negative regulator of T-cell activation. Exp Hematol (2002) 30:237–44. doi: 10.1016/S0301-472X(01)00794-9
44. Wu J, Katrekar A, Honigberg LA, Smith AM, Conn MT, Tang J, et al. Identification of substrates of human protein-tyrosine phosphatase PTPN22. J Biol Chem (2006) 281:11002–10. doi: 10.1074/jbc.M600498200
45. Cohen S, Dadi H, Shaoul E, Sharfe N, Roifman CM. Cloning and characterization of a lymphoid-specific, inducible human protein tyrosine phosphatase, Lyp. Blood (1999) 93:2013–24. doi: 10.1016/S0092-8674(00)81356-2
46. Zhang X, Yu Y, Bai B, Wang T, Zhao J, Zhang N, et al. PTPN22 interacts with EB1 to regulate T-cell receptor signaling. FASEB J (2020) 34:8959–74. doi: 10.1096/fj.201902811RR
47. Vang T, Landskron J, Viken MK, Oberprieler N, Torgersen KM, Mustelin T, et al. The autoimmune-predisposing variant of lymphoid tyrosine phosphatase favors T helper 1 responses. Hum Immunol (2013) 74:574–85. doi: 10.1016/j.humimm.2012.12.017
48. Ge Y, Onengut-Gumuscu S, Quinlan AR, Mackey AJ, Wright JA, Buckner JH, et al. Targeted Deep Sequencing in Multiple-Affected Sibships of European Ancestry Identifies Rare Deleterious Variants in PTPN22 That Confer Risk for Type 1 Diabetes. Diabetes (2016) 65:794–802. doi: 10.2337/db15-0322
49. Orrú V, Tsai SJ, Rueda B, Fiorillo E, Stanford SM, Dasgupta J, et al. A loss-of-function variant of PTPN22 is associated with reduced risk of systemic lupus erythematosus. Hum Mol Genet (2009) 18:569–79. doi: 10.1093/hmg/ddn363
50. Rodríguez-Rodríguez L, Taib WRW, Topless R, Steer S, González-Escribano MF, Balsa A, et al. The PTPN22 R263Q polymorphism is a risk factor for rheumatoid arthritis in Caucasian case-control samples. Arthritis Rheum (2011) 63:365–72. doi: 10.1002/art.30145
51. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet (2004) 36:337–8. doi: 10.1038/ng1323
52. Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JMM, et al. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes (2004) 53:3020–3. doi: 10.2337/diabetes.53.11.3020
53. Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VEH, et al. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet (2004) 75:504–7. doi: 10.1086/423790
54. Skórka A, Bednarczuk T, Bar-Andziak E, Nauman J, Ploski R. Lymphoid tyrosine phosphatase (PTPN22/LYP) variant and Graves’ disease in a Polish population: association and gene dose-dependent correlation with age of onset. Clin Endocrinol (Oxf) (2005) 62:679–82. doi: 10.1111/j.1365-2265.2005.02279.x
55. Vandiedonck C, Capdevielle C, Giraud M, Krumeich S, Jais J-P, Eymard B, et al. Association of the PTPN22*R620W polymorphism with autoimmune myasthenia gravis. Ann Neurol (2006) 59:404–7. doi: 10.1002/ana.20751
56. Vlachogiannis NI, Nezos A, Tzioufas AG, Koutsilieris M, Moutsopoulos HM, Mavragani CP. Increased frequency of the PTPN22W* variant in primary Sjogren’s Syndrome: Association with low type I IFN scores. Clin Immunol (2016) 173:157–60. doi: 10.1016/j.clim.2016.10.015
57. LaBerge GS, Bennett DC, Fain PR, Spritz RA. PTPN22 is genetically associated with risk of generalized vitiligo, but CTLA4 is not. J Invest Dermatol (2008) 128:1757–62. doi: 10.1038/sj.jid.5701233
58. Skinningsrud B, Husebye ES, Gervin K, Løvås K, Blomhoff A, Wolff AB, et al. Mutation screening of PTPN22: association of the 1858T-allele with Addison’s disease. Eur J Hum Genet (2008) 16:977–82. doi: 10.1038/ejhg.2008.33
59. Betz RC, König K, Flaquer A, Redler S, Eigelshoven S, Kortüm A-K, et al. The R620W polymorphism in PTPN22 confers general susceptibility for the development of alopecia areata. Br J Dermatol (2008) 158:389–91. doi: 10.1111/j.1365-2133.2007.08312.x
60. Mustelin T, Bottini N, Stanford SM. The Contribution of PTPN22 to Rheumatic Disease. Arthritis Rheumatol (2019) 71:486–95. doi: 10.1002/art.40790
61. Gomez LM, Anaya J-M, Martin J. Genetic influence of PTPN22 R620W polymorphism in tuberculosis. Hum Immunol (2005) 66:1242–7. doi: 10.1016/j.humimm.2005.11.008
62. Kouhpayeh H, Hashemi M, Hashemi S, Moazeni-Roodi A, Naderi M, Sharifi-Mood B, et al. R620W functional polymorphism of protein tyrosine phosphatase non-receptor type 22 is not associated with pulmonary tuberculosis in Zahedan, southeast Iran. Genet Mol Res (2012) 11:1075–81. doi: 10.4238/2012.April.27.6
63. Lamsyah H, Rueda B, Baassi L, Elaouad R, Bottini N, Sadki K, et al. Association of PTPN22 gene functional variants with development of pulmonary tuberculosis in Moroccan population. Tissue Antigens (2009) 74:228–32. doi: 10.1111/j.1399-0039.2009.01304.x
64. Boechat AL, Ogusku MM, Sadahiro A, dos Santos MC. Association between the PTPN22 1858C/T gene polymorphism and tuberculosis resistance. Infect Genet Evol (2013) 16:310–3. doi: 10.1016/j.meegid.2013.02.019
65. Vaitaitis GM, Waid DM, Yussman MG, Wagner DH. CD40-mediated signalling influences trafficking, T-cell receptor expression, and T-cell pathogenesis, in the NOD model of type 1 diabetes. Immunology (2017) 152:243–54. doi: 10.1111/imm.12761
66. Serreze DV, Post CM, Chapman HD, Johnson EA, Lu B, Rothman PB. Interferon-gamma receptor signaling is dispensable in the development of autoimmune type 1 diabetes in NOD mice. Diabetes (2000) 49:2007–11. doi: 10.2337/diabetes.49.12.2007
67. Serreze DV, Chapman HD, Post CM, Johnson EA, Suarez-Pinzon WL, Rabinovitch A. Th1 to Th2 cytokine shifts in nonobese diabetic mice: sometimes an outcome, rather than the cause, of diabetes resistance elicited by immunostimulation. J Immunol (2001) 166:1352–9. doi: 10.4049/jimmunol.166.2.1352
68. Schmiedel BJ, Singh D, Madrigal A, Valdovino-Gonzalez AG, White BM, Zapardiel-Gonzalo J, et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell (2018) 175:1701–1715.e16. doi: 10.1016/j.cell.2018.10.022
69. Nielsen C, Barington T, Husby S, Lillevang ST. Expression of human PTPN22 alleles. Genes Immun (2007) 8:131–7. doi: 10.1038/sj.gene.6364369
70. Zhang J, Zahir N, Jiang Q, Miliotis H, Heyraud S, Meng X, et al. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet (2011) 43:902–7. doi: 10.1038/ng.904
71. Dai X, James RG, Habib T, Singh S, Jackson S, Khim S, et al. A disease-associated PTPN22 variant promotes systemic autoimmunity in murine models. J Clin Invest (2013) 123:2024–36. doi: 10.1172/JCI66963
72. Vang T, Congia M, Macis MD, Musumeci L, Orrú V, Zavattari P, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet (2005) 37:1317–9. doi: 10.1038/ng1673
73. Chemin K, Ramsköld D, Diaz-Gallo L-M, Herrath J, Houtman M, Tandre K, et al. EOMES-positive CD4+ T cells are increased in PTPN22 (1858T) risk allele carriers. Eur J Immunol (2018) 48:655–69. doi: 10.1002/eji.201747296
74. Ferreira RC, Castro Dopico X, Oliveira JJ, Rainbow DB, Yang JH, Trzupek D, et al. Chronic Immune Activation in Systemic Lupus Erythematosus and the Autoimmune PTPN22 Trp620 Risk Allele Drive the Expansion of FOXP3+ Regulatory T Cells and PD-1 Expression. Front Immunol (2019) 10:2606:2606. doi: 10.3389/fimmu.2019.02606
75. Valta M, Gazali AM, Viisanen T, Ihantola E, Ekman I, Toppari J, et al. Type 1 diabetes linked PTPN22 gene polymorphism is associated with the frequency of circulating regulatory T cells. Eur J Immunol (2019) 50:581–8. doi: 10.1002/eji.201948378
76. Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic Variation in PTPN22 Corresponds to Altered Function of T and B Lymphocytes. J Immunol (2007) 179:4704–10. doi: 10.4049/jimmunol.179.7.4704
77. Cloutier JF, Veillette A. Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med (1999) 189:111–21. doi: 10.1084/jem.189.1.111
78. Anderson W, Thorpe J, Long SA, Rawlings DJ. Efficient CRISPR/Cas9 Disruption of Autoimmune-Associated Genes Reveals Key Signaling Programs in Primary Human T Cells. J Immunol (2019) 203:3166–78. doi: 10.4049/jimmunol.1900848
79. Aarnisalo J, Treszl A, Svec P, Marttila J, Öling V, Simell O, et al. Reduced CD4+T cell activation in children with type 1 diabetes carrying the PTPN22/Lyp 620Trp variant. J Autoimmun (2008) 31:13–21. doi: 10.1016/j.jaut.2008.01.001
80. Crabtree JN, He W, Guan W, Flage M, Miller MS, Peterson EJ. Autoimmune Variant PTPN22 C1858T Is Associated with Impaired Responses to Influenza Vaccination. J Infect Dis (2016) 214:248–57. doi: 10.1093/infdis/jiw126
81. Zikherman J, Hermiston M, Steiner D, Hasegawa K, Chan A, Weiss A. PTPN22 deficiency cooperates with the CD45 E613R allele to break tolerance on a non-autoimmune background. J Immunol (2009) 182:4093–106. doi: 10.4049/jimmunol.0803317
82. Rhee I, Veillette A. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat Immunol (2012) 13:439–47. doi: 10.1038/ni.2246
83. Bray C, Wright D, Haupt S, Thomas S, Stauss H, Zamoyska R. Crispr/Cas Mediated Deletion of PTPN22 in Jurkat T Cells Enhances TCR Signaling and Production of IL-2. Front Immunol (2018) 9:2595:2595. doi: 10.3389/fimmu.2018.02595
84. Chang H-H, Tai T-S, Lu B, Iannaccone C, Cernadas M, Weinblatt M, et al. PTPN22.6, a dominant negative isoform of PTPN22 and potential biomarker of rheumatoid arthritis. PloS One (2012) 7:e33067. doi: 10.1371/journal.pone.0033067
85. Hasegawa K, Martin F, Huang G, Tumas D, Diehl L, Chan AC. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science (2004) 303:685–9. doi: 10.1126/science.1092138
86. Sanchez-Blanco C, Clarke F, Cornish GH, Depoil D, Thompson SJ, Dai X, et al. Protein tyrosine phosphatase PTPN22 regulates LFA-1 dependent Th1 responses. J Autoimmun (2018) 94:45–55. doi: 10.1016/j.jaut.2018.07.008
87. Brownlie RJ, Miosge LA, Vassilakos D, Svensson LM, Cope A, Zamoyska R. Lack of the phosphatase PTPN22 increases adhesion of murine regulatory T cells to improve their immunosuppressive function. Sci Signal (2012) 5:ra87. doi: 10.1126/scisignal.2003365
88. Zheng P, Kissler S. PTPN22 silencing in the NOD model indicates the type 1 diabetes-associated allele is not a loss-of-function variant. Diabetes (2013) 62:896–904. doi: 10.2337/db12-0929
89. Kishimoto TK, Hollander N, Roberts TM, Anderson DC, Springer TA. Heterogeneous mutations in the beta subunit common to the LFA-1, Mac-1, and p150,95 glycoproteins cause leukocyte adhesion deficiency. Cell (1987) 50:193–202. doi: 10.1016/0092-8674(87)90215-7
90. Walling BL, Kim M. LFA-1 in T Cell Migration and Differentiation. Front Immunol (2018) 9:952:952. doi: 10.3389/fimmu.2018.00952
91. Maine CJ, Hamilton-Williams EE, Cheung J, Stanford SM, Bottini N, Wicker LS, et al. PTPN22 alters the development of T regulatory cells in the thymus. J Immunol (2012) 188:5267–75. doi: 10.4049/jimmunol.1200150
92. Nowakowska DJ, Kissler S. Ptpn22 Modifies Regulatory T Cell Homeostasis via GITR Upregulation. J Immunol (2016) 196:2145–52. doi: 10.4049/jimmunol.1501877
93. Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest (2017) 127:2881–91. doi: 10.1172/JCI94549
94. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol (2009) 155:173–81. doi: 10.1111/j.1365-2249.2008.03860.x
95. Kordonouri O, Hartmann R, Badenhoop K, Kahles H, Ilonen J. PTPN22 1858T allele is associated with younger age at onset of type 1 diabetes and is not related to subsequent thyroid autoimmunity. Hum Immunol (2010) 71:731–2. doi: 10.1016/j.humimm.2010.04.002
96. Petrone A, Spoletini M, Zampetti S, Capizzi M, Zavarella S, Osborn J, et al. Immunotherapy Diabetes (IMDIAB) Group. The PTPN22 1858T gene variant in type 1 diabetes is associated with reduced residual beta-cell function and worse metabolic control. Diabetes Care (2008) 31:1214–8. doi: 10.2337/dc07-1158
97. Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med (2009) 361:2143–52. doi: 10.1056/NEJMoa0904452
98. Habib T, Funk A, Rieck M, Brahmandam A, Dai X, Panigrahi AK, et al. Altered B Cell Homeostasis Is Associated with Type I Diabetes and Carriers of the PTPN22 Allelic Variant. J Immunol (2012) 188:487–96. doi: 10.4049/jimmunol.1102176
99. Gianchecchi E, Crinò A, Giorda E, Luciano R, Perri V, Lo Russo A, et al. Altered B Cell Homeostasis and Toll-Like Receptor 9-Driven Response in Type 1 Diabetes Carriers of the C1858T PTPN22 Allelic Variant: Implications in the Disease Pathogenesis. PloS One (2014) 9:e110755. doi: 10.1371/journal.pone.0110755
100. Menard L, Saadoun D, Isnardi I, Ng Y-SS, Meyers G, Massad C, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest (2011) 121:3635–44. doi: 10.1172/JCI45790
101. Maine CJ, Marquardt K, Cheung J, Sherman LA. PTPN22 controls the germinal center by influencing the numbers and activity of T follicular helper cells. J Immunol (2014) 192:1415–24. doi: 10.4049/jimmunol.1302418
102. Maine CJ, Marquardt K, Scatizzi JC, Pollard KM, Kono DH, Sherman LA. The effect of the autoimmunity-associated gene, PTPN22, on a BXSB-derived model of lupus. Clin Immunol (2015) 156:65–73. doi: 10.1016/j.clim.2014.11.003
103. Metzler G, Dai X, Thouvenel CD, Khim S, Habib T, Buckner JH, et al. The Autoimmune Risk Variant PTPN22 C1858T Alters B Cell Tolerance at Discrete Checkpoints and Differentially Shapes the Naive Repertoire. J Immunol (2017) 199:2249–60. doi: 10.4049/jimmunol.1700601
104. Pelanda R, Torres RM. Central B-cell tolerance: where selection begins. Cold Spring Harb Perspect Biol (2012) 4:a007146. doi: 10.1101/cshperspect.a007146
105. Meffre E. The establishment of early B cell tolerance in humans: lessons from primary immunodeficiency diseases. Ann N Y Acad Sci (2011) 1246:1–10. doi: 10.1111/j.1749-6632.2011.06347.x
106. Buchner C, Bryant C, Eslami A, Lakos G. Anti-nuclear antibody screening using HEp-2 cells. J Vis Exp (2014) 88:e51211. doi: 10.3791/51211
107. Schickel J-N, Kuhny M, Baldo A, Bannock JM, Massad C, Wang H, et al. PTPN22 inhibition resets defective human central B cell tolerance. Sci Immunol (2016) 1:aaf7153–aaf7153. doi: 10.1126/sciimmunol.aaf7153
108. Thompson WS, Pekalski ML, Simons HZ, Smyth DJ, Castro-Dopico X, Guo H, et al. Multi-parametric flow cytometric and genetic investigation of the peripheral B cell compartment in human type 1 diabetes. Clin Exp Immunol (2014) 177:571–85. doi: 10.1111/cei.12362
109. Duty JA, Szodoray P, Zheng N-Y, Koelsch KA, Zhang Q, Swiatkowski M, et al. Functional anergy in a subpopulation of naive B cells from healthy humans that express autoreactive immunoglobulin receptors. J Exp Med (2009) 206:139–51. doi: 10.1084/jem.20080611
110. Winter WE, Schatz DA. Autoimmune Markers in Diabetes. Clin Chem (2011) 57:168–75. doi: 10.1373/clinchem.2010.148205
111. Steck AK, Zhang W, Bugawan TL, Barriga KJ, Blair A, Erlich HA, et al. Do non-HLA genes influence development of persistent islet autoimmunity and type 1 diabetes in children with high-risk HLA-DR,DQ genotypes? Diabetes (2009) 58:1028–33. doi: 10.2337/db08-1179
112. Hu CY, Rodriguez-Pinto D, Du W, Ahuja A, Henegariu O, Wong FS, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest (2007) 117:3857–67. doi: 10.1172/JCI32405
113. Ploegh HL. Bridging B cell and T cell recognition of antigen. J Immunol (2007) 179:7193. doi: 10.4049/jimmunol.179.11.7193
114. Chen X, Jensen PE. The role of B lymphocytes as antigen-presenting cells. Arch Immunol Ther Exp (Warsz) (2008) 56:77–83. doi: 10.1007/s00005-008-0014-5
115. Hinman RM, Smith MJ, Cambier JC. B cells and type 1 diabetes …in mice and men. Immunol Lett (2014) 160:128–32. doi: 10.1016/j.imlet.2014.01.010
116. Radomir L, Cohen S, Kramer MP, Bakos E, Lewinsky H, Barak A, et al. T Cells Regulate Peripheral Naive Mature B Cell Survival by Cell-Cell Contact Mediated through SLAMF6 and SAP. J Immunol (2017) 199:2745–57. doi: 10.4049/jimmunol.1700557
117. Rush JS, Hodgkin PD. B cells activated via CD40 and IL-4 undergo a division burst but require continued stimulation to maintain division, survival and differentiation. Eur J Immunol (2001) 31:1150–9. doi: 10.1002/1521-4141(200104)31:4<1150::AID-IMMU1150gt;3.0.CO;2-V
118. Ding BB, Bi E, Chen H, Yu JJ, Ye BH. IL-21 and CD40L synergistically promote plasma cell differentiation through upregulation of Blimp-1 in human B cells. J Immunol (2013) 190:1827–36. doi: 10.4049/jimmunol.1201678
119. Tangye SG, Ferguson A, Avery DT, Ma CS, Hodgkin PD. Isotype switching by human B cells is division-associated and regulated by cytokines. J Immunol (2002) 169:4298–306. doi: 10.4049/jimmunol.169.8.4298
120. Mitsdoerffer M, Lee Y, Jäger A, Kim H-J, Korn T, Kolls JK, et al. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci U S A (2010) 107:14292–7. doi: 10.1073/pnas.1009234107
121. Smulski CR, Eibel H. BAFF and BAFF-Receptor in B Cell Selection and Survival. Front Immunol (2018) 9:2285:2285. doi: 10.3389/fimmu.2018.02285
122. Wang Y, Ewart D, Crabtree JN, Yamamoto A, Baechler EC, Fazeli P, et al. PTPN22 Variant R620W Is Associated With Reduced Toll-like Receptor 7-Induced Type I Interferon in Systemic Lupus Erythematosus. Arthritis Rheumatol (2015) 67:2403–14. doi: 10.1002/art.39211
123. Sharp RC, Abdulrahim M, Naser ES, Naser SA. Genetic variations of PTPN2 and PTPN22: Role in the pathogenesis of Type 1 diabetes and Crohn’s disease. Front Cell Infect Microbiol (2015) 5:95:95. doi: 10.3389/fcimb.2015.00095
124. Comalada M, Peppelenbosch MP. Impaired innate immunity in Crohn’s disease. Trends Mol Med (2006) 12:397–9. doi: 10.1016/j.molmed.2006.07.005
125. Takaoka A, Tanaka N, Mitani Y, Miyazaki T, Fujii H, Sato M, et al. Protein tyrosine kinase Pyk2 mediates the Jak-dependent activation of MAPK and Stat1 in IFN-gamma, but not IFN-alpha, signaling. EMBO J (1999) 18:2480–8. doi: 10.1093/emboj/18.9.2480
126. Halfter UM, Derbyshire ZE, Vaillancourt RR. Interferon-gamma-dependent tyrosine phosphorylation of MEKK4 via Pyk2 is regulated by annexin II and SHP2 in keratinocytes. Biochem J (2005) 388:17–28. doi: 10.1042/BJ20041236
127. Li M, Beauchemin H, Popovic N, Peterson A, D’Hennezel E, Piccirillo CA, et al. The common, autoimmunity-predisposing 620Arg > Trp variant of PTPN22 modulates macrophage function and morphology. J Autoimmun (2017) 79:74–83. doi: 10.1016/j.jaut.2017.01.009
128. Ghazizadeh S, Bolen JB, Fleit HB. Physical and functional association of Src-related protein tyrosine kinases with Fc gamma RII in monocytic THP-1 cells. J Biol Chem (1994) 269:8878–84. doi: 10.1016/S0021-9258(17)37050-3
129. Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, et al. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A (2009) 106:7119–24. doi: 10.1073/pnas.0902745106
130. Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med (1999) 189:521–30. doi: 10.1084/jem.189.3.521
131. Jennings RN, Grayson JM, Barton ES. Type I interferon signaling enhances CD8+ T cell effector function and differentiation during murine gammaherpesvirus 68 infection. J Virol (2014) 88:14040–9. doi: 10.1128/JVI.02360-14
132. Newby BN, Mathews CE. Type I Interferon Is a Catastrophic Feature of the Diabetic Islet Microenvironment. Front Endocrinol (Lausanne) (2017) 8:232:232. doi: 10.3389/fendo.2017.00232
133. Roep BO. β-cells, autoimmunity, and the innate immune system: “Un ménage á trois”? Diabetes (2013) 62:1821–2. doi: 10.2337/db13-0276
134. Thayer TC, Delano M, Liu C, Chen J, Padgett LE, Tse HM, et al. Superoxide production by macrophages and T cells is critical for the induction of autoreactivity and type 1 diabetes. Diabetes (2011) 60:2144–51. doi: 10.2337/db10-1222
135. Wang X, Qiu L, Li Z, Wang X-Y, Yi H. Understanding the Multifaceted Role of Neutrophils in Cancer and Autoimmune Diseases. Front Immunol (2018) 9:2456:2456. doi: 10.3389/fimmu.2018.02456
136. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol (2009) 184:205–13. doi: 10.1083/jcb.200806072
137. Carp H. Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J Exp Med (1982) 155:264–75. doi: 10.1084/jem.155.1.264
138. Marasco WA, Phan SH, Krutzsch H, Showell HJ, Feltner DE, Nairn R, et al. Purification and identification of formyl-methionyl-leucyl-phenylalanine as the major peptide neutrophil chemotactic factor produced by Escherichia coli. J Biol Chem (1984) 259:5430–9. doi: 10.1016/S0021-9258(18)91029-X
139. Vermeren S, Miles K, Chu JY, Salter D, Zamoyska R, Gray M. PTPN22 Is a Critical Regulator of Fcγ Receptor-Mediated Neutrophil Activation. J Immunol (2016) 197:4771–9. doi: 10.4049/jimmunol.1600604
140. Huang J, Xiao Y, Xu A, Zhou Z. Neutrophils in type 1 diabetes. J Diabetes Investig (2016) 7:652–63. doi: 10.1111/jdi.12469
141. Fraser HI, Howlett S, Clark J, Rainbow DB, Stanford SM, Wu DJ, et al. Ptpn22 and Cd2 Variations Are Associated with Altered Protein Expression and Susceptibility to Type 1 Diabetes in Nonobese Diabetic Mice. J Immunol (2015) 195:4841–52. doi: 10.4049/jimmunol.1402654
142. Liu Y, Stanford SM, Jog SP, Fiorillo E, Orrú V, Comai L, et al. Regulation of lymphoid tyrosine phosphatase activity: inhibition of the catalytic domain by the proximal interdomain. Biochemistry (2009) 48:7525–32. doi: 10.1021/bi900332f
143. Stout RD, Bottomly K. Antigen-specific activation of effector macrophages by IFN-gamma producing (TH1) T cell clones. Failure of IL-4-producing (TH2) T cell clones to activate effector function in macrophages. J Immunol (1989) 142:760–5.
Keywords: PTPN22, PTPN22 620Arg > Trp, type 1 diabetes, cell signaling, rs2476601, autoimmunity, leukocytes
Citation: Armitage LH, Wallet MA and Mathews CE (2021) Influence of PTPN22 Allotypes on Innate and Adaptive Immune Function in Health and Disease. Front. Immunol. 12:636618. doi: 10.3389/fimmu.2021.636618
Received: 01 December 2020; Accepted: 18 January 2021;
Published: 25 February 2021.
Edited by:
F. Susan Wong, Cardiff University, United KingdomReviewed by:
Bergithe Eikeland Oftedal, University of Bergen, NorwayMaikel Peppelenbosch, Erasmus Medical Center, Netherlands
Copyright © 2021 Armitage, Wallet and Mathews. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clayton E. Mathews, Y2xheXRvbi5tYXRoZXdzQHBhdGhvbG9neS51ZmwuZWR1