 Alexia Carré
Alexia Carré Roberto Mallone
Roberto Mallone- 1Université de Paris, Institut Cochin, CNRS, INSERM, Paris, France
- 2Assistance Publique Hôpitaux de Paris, Service de Diabétologie et Immunologie Clinique, Cochin Hospital, Paris, France
Autoimmune type 1 diabetes (T1D) results from the intricate crosstalk of various immune cell types. CD8+ T cells dominate the pro-inflammatory milieu of islet infiltration (insulitis), and are considered as key effectors of beta-cell destruction, through the recognition of MHC Class I-peptide complexes. The pathways generating MHC Class I-restricted antigens in beta cells are poorly documented. Given their specialized insulin secretory function, the associated granule processing and degradation pathways, basal endoplasmic reticulum stress and susceptibility to additional stressors, alternative antigen processing and presentation (APP) pathways are likely to play a significant role in the generation of the beta-cell immunopeptidome. As direct evidence is missing, we here intersect the specificities of beta-cell function and the literature about APP in other cellular models to generate some hypotheses on APPs relevant to beta cells. We further elaborate on the potential role of these pathways in T1D pathogenesis, based on the current knowledge of antigens presented by beta cells. A better understanding of these pathways may pinpoint novel mechanisms amenable to therapeutic targeting to modulate the immunogenicity of beta cells.
Introduction
Type 1 diabetes (T1D) is an autoimmune disease characterized by the destruction of insulin-producing beta cells. It stems from a complex interplay of innate and adaptive immune cells. CD8+ T cells dominate the immune infiltration of islets and play a prominent role as final effectors of beta-cell loss (1). There is also growing evidence supporting the idea that beta-cell dysfunction is another key driver of T1D pathogenesis (2). The heterogeneity of pancreas histopathology between T1D donors and even across islets from the same pancreas, both in terms of immune infiltrates and residual beta cells, have led to the definition of age-related endotypes (3), in which the component of beta-cell dysfunction may be dominant in adult-onset cases (4, 5). Effector CD8+ T cells recognize MHC Class I (MHC-I)-peptide complexes at the surface of beta cells. The conventional MHC-I antigen processing and presentation (APP) machinery is a multi-step process (Figure 1A) where i) cytosolic proteins of microbial or self-origin are degraded into peptides in a proteasome-mediated manner; ii) the resulting peptides are transported into the endoplasmic reticulum (ER) via the transporter associated with antigen processing (TAP). Here, iii) they are further trimmed by ER aminopeptidase (ERAP)1 prior to iv) loading on nascently-formed MHC-I molecules that are associated to TAP through tapasin; v) the stable peptide-MHC-I complexes are finally translocated to the Golgi complex and to the cell surface (6). This review addresses the specificities of the direct alternative MHC-I APP (aAPP) machinery within beta cells and its potential role in T1D pathogenesis (Figures 1B–D). It therefore focuses on the CD8+ T-cell responses that are triggered by MHC-I APP, notwithstanding the role of other cell types, and particularly of MHC-I and MHC-II APP by professional antigen-presenting cells (APCs) for the priming of naïve T cells. Since the beta-cell APP pathways are poorly if at all documented, we here provide a hypothesis-generating review that bridges the separate bodies of knowledge available for MHC-I APP and beta-cell biology.
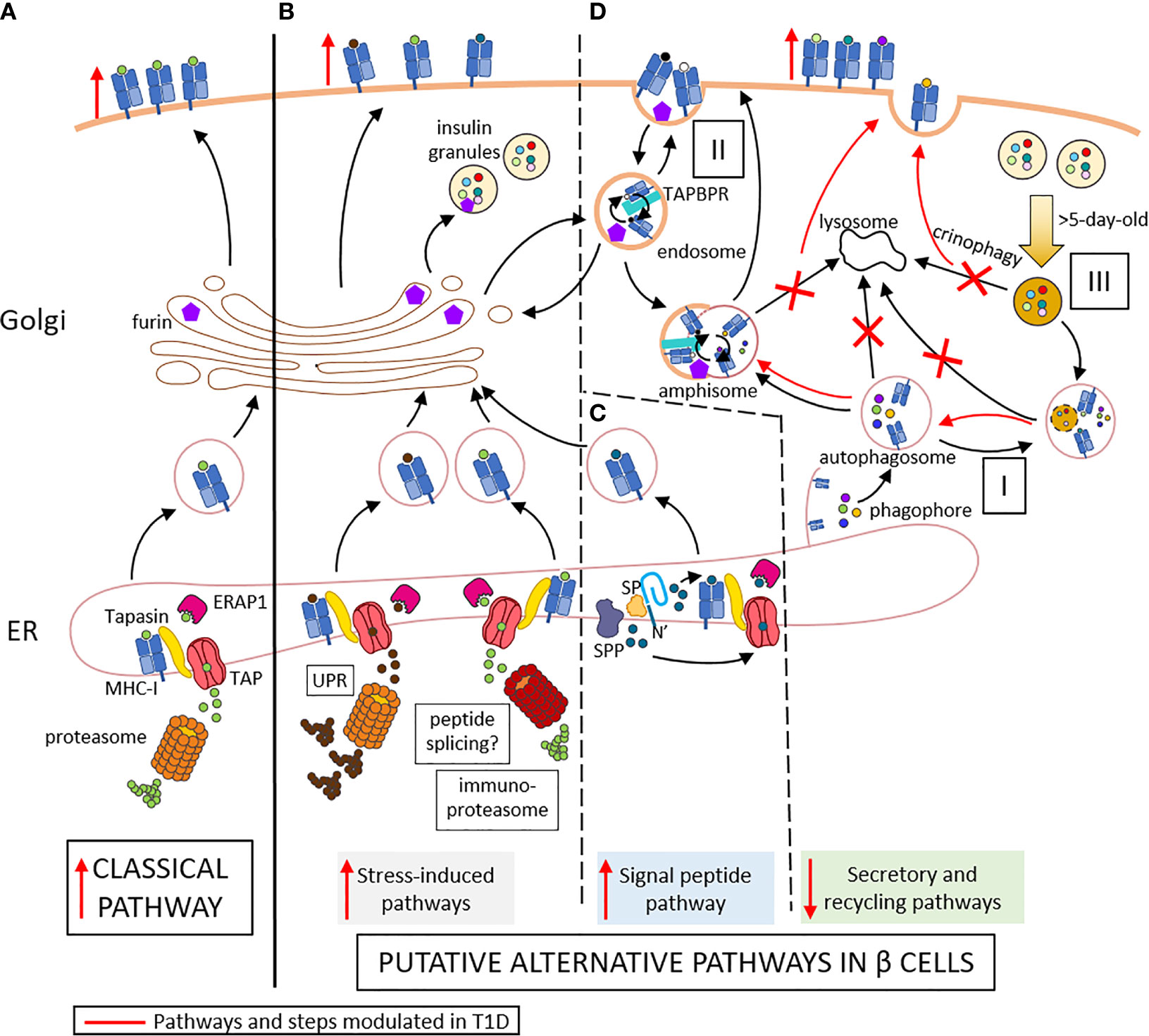
Figure 1 Conventional (A) and putative alternative APP pathways (B–D) in beta cells. (A) In conventional APP pathways, misfolded proteins are degraded by the proteasome into peptides that are transported through TAP into the ER lumen. Here, they are further trimmed by ERAP1 and loaded onto MHC-I molecules that are associated to TAP via tapasin. The peptide-MHC-I complexes are exported to the Golgi and then to the cell surface. (B) Under inflammatory conditions, ER stress induces the UPR, which increases protein degradation and antigen presentation. The immuno-proteasome is also induced, further increasing APP and possibly resulting in alternative peptide splicing events and neo-epitopes generated. (C) Signal peptides are docked within the ER membrane. While the protein translation continues, the signal peptide is cleaved by the signal peptidase (SP) and further trimmed by the signal peptide peptidase (SPP). The N-terminal cleavage products of the signal peptide are released in the cytosol and can re-enter the ER through TAP for antigen presentation. The C-terminal cleavage products remaining in the ER can be loaded onto MHC-I molecules in a TAP- and proteasome-independent manner. (D) I. Cellular components are engulfed by nascent phagophore at the ER membrane, thus forming autophagosome, likely containing MHC-I molecules. II. Endocytosis of cell surface component forms endosomes, which can contain both MHC-I molecules and TAPBR promoting peptide exchange within endosomes. These vesicles can be directed to the Golgi apparatus, where peptides (free or released from MHC-I) can be trimmed by Golgi enzymes such as furins prior to loading onto MHC-I molecules. Autophagosomes, if not directly fused to lysosomes for degradation of their content, can fuse with endosome to form amphisomes, thus opening another peptide exchange pathway. These vesicles can subsequently be directed to lysosomes for degradation. III. Old insulin granules can be degraded by crinophagy. T1D induces changes (in red) characterized by enhanced ER stress (B), increased insulin production and signal peptide processing (C), and altered vesicle trafficking (D), thus enhancing surface exposure of MHC-I complexes. Red marks indicate changes occuring in T1D: vertical arrows represent the increase or decrease of a step or pathway; bended arrows represent favored events; crosses represent blocked events. Some cellular components were modified from Servier Medical Art (smart.servier.com).
Secreting Insulin: The Achilles’ Heel of Beta Cells?
Like other endocrine cells, beta cells are hormone-secreting cells organized in glands that are highly vascularized, allowing the secretion of their products directly into the bloodstream. These features may influence the APP pathways used by these cells (2). Indeed, beta cells are equipped to sense changes in blood glucose levels and to respond by releasing appropriate amounts of insulin. For this purpose, they need to constantly adapt their secretory response to changes in metabolic and nutritional state. This adaptation is critical compared to that of other tissues because beta cells are long-lived and virtually non-proliferating, hence they can only modulate their function but not their cell numbers. Beta cells accommodate these metabolic variations by intensifying the synthesis of insulin and its precursor proinsulin. Under these intense bio-synthetic rates, misfolded proteins are more likely to accumulate within the ER, activating the unfolded protein response (UPR) (7). The UPR is a natural adaptive response that maintains cellular homeostasis by fulfilling three main tasks: to decrease the protein translation rate, to enhance the synthesis of protein-folding chaperones, and to increase the degradation of misfolded proteins into peptides through the proteasome (7). While part of the free cytosolic peptides generated by this process is further hydrolyzed to single amino acids, another portion is transported to the ER via TAP for the loading onto MHC-I molecules, thus shaping the catalog of MHC-I-bound peptides (immunopeptidome) presented at the beta-cell surface (Figure 1B). Beta cells rely on a well-balanced UPR to ensure their function and survival (8). Thus, beta cells are constantly functioning on the edge between physiological and pathological UPR and are exquisitely sensitive to additional stressors that can flip this delicate balance and lead to a compensatory maladaptive UPR further increasing ER stress prior to apoptosis (9, 10). Metabolic overload can act as an accelerator but is unlikely to drive this UPR transition on its own, as such overload is more common in type 2 diabetes and yet does not usually trigger islet autoimmunity. The environmental factors at play are likely multiple, but their identity and interaction with a susceptible genetic background remain ill-defined, with the possible exception of enteroviral infections (11). Considering the link between the UPR and APP through increased proteasomal degradation of misfolded proteins, the shift from physiological to pathological UPR may modulate the immunopeptidome and the beta-cell visibility to the immune system.
Under physiological conditions, the beta cells maintain consistent intracellular stores of insulin granules to allow their rapid exocytosis when the glycemia rises. While insulin granules are found by thousands in individual beta cells (12), not all are secreted. Indeed, the remarkable functional plasticity of beta cells includes recycling pathways to dispose of old (>5 days) or excess secretory granules. Commonly, insulin granules are degraded through macro-autophagy, by which they are engulfed by autophagosomes prior to degradation in lysosomes. Another endocrine-specific recycling pathway is a specialized macro-autophagy process named crinophagy, by which insulin granules fuse directly with lysosomes, thus generating so-called crinosomes (Figure 1D). While the role of these different types of autophagy has been extensively studied in type 2 diabetes, investigations in T1D are scanty. One recent study (13) reported defective macro-autophagy and diminished crinophagy in the beta cells of non-obese diabetic (NOD) mice compared to non-diabetic NOD littermates and non-obese resistant (NOR) mice. These conclusions were confirmed in T1D pancreas specimens and were based on the lack of co-localization of the lysosomal marker Lamp1 with either the granule marker proinsulin or the autophagosome marker LC3, thus suggesting a defect in the late stage of autophagy, when granules/autophagosomes fuse with lysosomes. An alternative explanation is that this reduced crinophagy may simply reflect a limited accumulation of old granules due to a compensatory higher insulin exocytosis rate in remaining beta cells. Interestingly, this study also reported a parallel increase of autophagosomes in insulin-positive cells of pancreata from autoantibody-positive non-diabetic donors in comparison to both control and T1D donors, whose autophagosome content was instead similar. As autophagy helps to prevent oxidative damage and promote cell survival, we can hypothesize that the beta-cell stress imposed by the inflammatory milieu of insulitis triggers an increase in their autophagy rate (14), prior to, and likely independently of, overt hyperglycemia. Interestingly, lysosomal autophagy can also degrade mitochondria (15). This process, known as mitophagy, is also part of stress responses (16), and is regulated by CLEC16A, whose genetic locus encodes variants associated with T1D risk (17). Pancreatic CLEC16A knockout mice harbor a defect in both mitochondrial and granule turnover (16), pointing to an intersection between the mitophagy and crinophagy pathway. It is thus possible that the altered granule disposal observed in stressed beta cells may also reflect overloading of this pathway by mitochondrial substrates. Its modulation by the CLEC16A genetic background may exacerbate the effect of stress on aAPPs. This feature also emphasizes the current view of lysosomes as dynamic structures that interact with different intracellular organelles and are continuously consumed and re-formed rather than just being end-stage degradation hubs (15).
Given this notion of impaired autophagy in T1D (13), what is the fate of the autophagosomes and insulin granules that are not degraded by lysosomes? In non-professional APCs, surface peptide-loaded MHC-I molecules seem to be recycled by clathrin-independent mechanisms, forming endosomes (18, 19). Whether and how these endosomes are degraded by lysosomes or targeted toward an early or late recycling pathway is unclear. Nonetheless, peptide exchange for MHC-I loading is likely to occur within the endosome. The TAP binding protein related (TAPBPR) chaperone is currently held as the main candidate for this role (20), and its preference for a slightly acidic to neutral pH is in the range of that found in endosomes. Another possibility is the encounter between endosomes and autophagosomes prior to recycling (21), forming so-called amphisomes (Figure 1D). Hence, the autophagosomes that are not degraded by lysosomes may be more likely to fuse with endosomes and gain access to the cell surface. Considering that the autophagosome membrane is thought to derive from the ER and can thus harbor MHC-I molecules amenable to peptide loading (22), this process may provide an aAPP pathway. Similarly, insulin granules that are not degraded by lysosomes could indirectly participate in APP through engulfment by autophagosomes. Collectively, these data suggest that alternative autophagy-derived pathways for MHC-I loading could be operational in beta cells and participate in the recycling processes needed for disposing of insulin granules. While these pathways are active in resting beta cells, they may be further enriched in T1D as autophagy is impaired, possibly providing novel disease-enhanced pathways.
Besides insulin, secretory granules contain other proteins that we reported to dominate the beta-cell-specific immunopeptidome targeted by islet autoimmunity. First, a large fraction of MHC-I-bound peptides recovered from a human beta-cell line and primary islets originate from these granule proteins (23), which is not unexpected given the abundance and fast turnover of these proteins in beta cells (24). Second and most important however, these granule-derived peptides were also prominent targets recognized by circulating CD8+ T cells (23, 25). More recently, we found that H2-Kd-restricted peptides derived from the murine orthologues of some of these proteins, namely secretogranin-5, urocortin-3 and proconvertase-2, are also recognized by islet-infiltrating CD8+ T cells in prediabetic NOD mice (25). Moreover, CD8+ T cells recognizing these peptides were diabetogenic upon in-vivo transfer into NOD/scid recipients (25). Besides their localization in granules, it is noteworthy that these novel antigens (together with chromogranin A and others) (26) share several other features with insulin. First, they are all soluble proteins that are released along with insulin during granule exocytosis, a feature that could endow beta cells with a unique capability of sensitizing T cells at distance following APP of these proteins by extra-pancreatic APCs (2, 27). Second, they are all produced as precursors (pro-proteins) and subsequently undergo intermediate processing, first in the ER to cleave the signal peptide (see below), and then in immature granules through proconvertases, carboxypeptidase E (CPE) and furins that lead to their bioactive products (Figure 2). Intriguingly, reduced levels of proconvertases and CPE along with impaired proconvertase activity and proinsulin processing in beta cells has been repeatedly reported for T1D, even before clinical onset (4, 28–32). These enzymatic defects could lead to increase protein misfolding, which might feed the proteasomal recycling pathway and further fuel the APP machinery. It is also possible that granule protein byproducts may accumulate in the ER, the Golgi or the cytosol and thus become accessible for processing by other enzymes and for loading onto MHC-I molecules, either directly, through retrograde transport to the ER, or indirectly, via TAP. Of further note, proconvertases themselves as well as (pro)cathepsins are also synthesized as inactive precursors and subsequently activated, often through auto-enzymatic reactions. We can thus speculate that ER stress and impaired proconvertase activity may also negatively impact these processes since their earliest steps of ER export and self-activation.
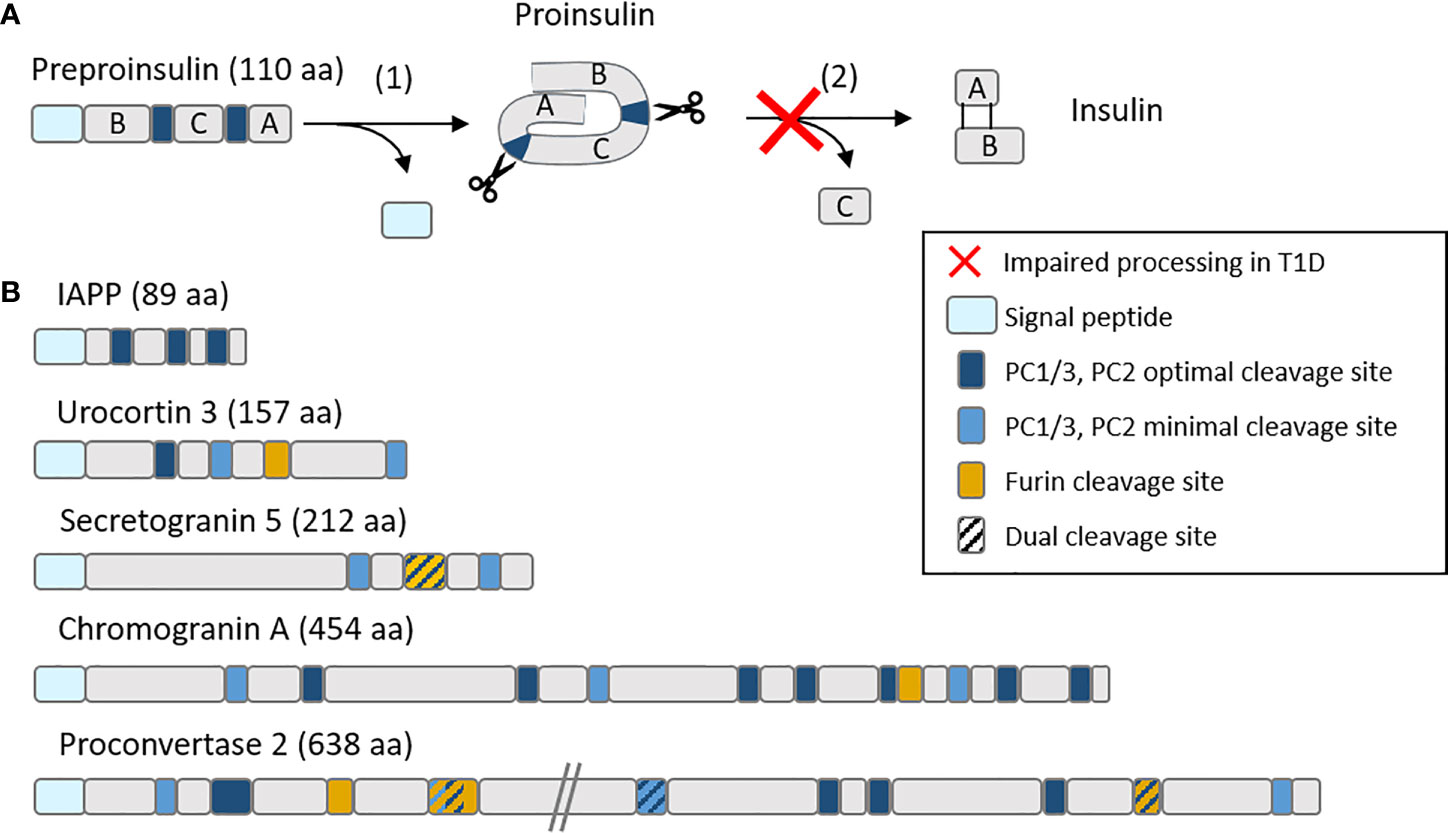
Figure 2 Schematic representation of (A) the insulin maturation process and (B) mapping of proconvertase cleavage sites within insulin granule proteins. (A) Insulin is translated as a preprohormone comprising a signal peptide (light blue), followed by the B chain, C peptide and A chain. The first processing step (1) consists in the cleavage of the signal peptide, followed by (2) the cleavage by proconvertases of the C peptide and formation of disulfide bonds between the A and B chain. In T1D, this second step is often impaired, leading to proinsulin accumulation. (B) PC1/3 and PC2 optimal (RR, KR) and minimal (KK, RK) cleavage sites along with furin cleavage sites (R-X-X-R) (where X is any amino acid) are mapped on major insulin granules proteins. aa, amino acids.
An unsolved conundrum is how the T-cell targeting of granule antigens that are mostly shared with other endocrine cells such as alpha cells leads to an autoimmune response that is exquisitely beta-cell-specific. This question is even more important in light of recent reports suggesting that MHC–I upregulation is more prominent in alpha cells, even before clinical T1D (33). Protective factors at play in alpha cells may include their lower biosynthetic rate, the narrower dynamic range of their secretory response (34), their lower susceptibility to ER stress-induced apoptosis (35), and their higher expression of non-classical, inhibitory MHC-I molecules, i.e. human leukocyte antigen (HLA)-E (36). Moreover, autophagy seems less important for the maintenance of alpha-cell metabolism (37) than it is for beta cells (38), possibly making autophagy-related APP pathways less active.
Neo-Epitope Generation in Beta Cells
The large stores of insulin granules and the functional adaptability to metabolic changes of beta cells point to a remarkably active translation machinery, which enhances the odds of neo-epitope generation. Neo-epitopes are unconventional peptides such as post-translational modified sequences, defective ribosomal products (DRiPs), alternative mRNA splicing products and proteasomal peptide splicing products (i.e. resulting from the fusion of two non-adjacent regions of a protein). We reported that some of these products contribute to the MHC-I peptidome of beta cells (23, 25, 26). Very little is however known about how such peptides are generated and whether the required pathways are active in beta cells and/or professional APCs. In spite of their tissue specificity, the overall quantitative contribution of neo-epitopes to the beta-cell immunopeptidome appears to be minor, as only a limited number of MHC-I-restricted neo-epitopes has been reported to date (26). A possible scenario is that these neo-epitopes might be symptomatic of early beta-cell dysfunction, e.g. altered granule bio-synthesis/disposal, and become visible to T cells via the uptake of granules or of their exocytosed content by APCs (2). The combination of generation mechanisms favored by early beta-cell dysfunction and of recognition mechanisms favored by T cells that have escaped thymic deletion may endow them with a crucial role in the onset of islet autoimmunity.
Another aspect is the central role that the constitutive proteasome, and its inflammation-induced counterpart, the immunoproteasome, play in the generation of some of these neo-epitopes (39). It is noteworthy that in-vitro exposure of beta cells to interferon (IFN)-γ induces the expression of the immunoproteasome (40) which is known to enhance proteolysis, whereas immunoproteasome-deficient cells accumulate reactive oxygen species (ROS) (41). The immunoproteasome deficiency has also been investigated by in-vivo knockout in mice (42), which led to a CD8+ T-cell-mediated multi-organ autoimmunity comprising insulitis and CD8+ T cells reactive to an IGRP beta-cell peptide with low affinity H-2Kb binding. The authors hypothesized that the constitutive proteasome alone could fail at generating enough high-affinity MHC-I binders. This possibility is supported by other studies highlighting that the immunoproteasome uses the same cleavage sites as the constitutive proteasome, hence impacting the relative quantity rather than the quality of the peptides produced (39, 43, 44). Defective immunoproteasome activity and the resulting decrease in peptide output could thus allow weak-binding immunogenic peptides usually outcompeted by stronger binders to be processed and presented. Conversely, the inflammatory insulitis microenvironment of T1D may induce the well-known MHC-I upregulation (45), but also immunoproteasome expression, thus further enriching the immunopeptidome and favoring strong MHC binders. The higher binding affinity of these peptides may synergize with MHC-I upregulation to increase their availability for T-cell recognition. This increase in beta-cell visibility to T cells may provide one explanation to our recent observation that beta-cell-reactive CD8+ T cells circulate at similar frequencies in T1D and healthy donors (23, 46). This observation suggests that the difference between the ‘benign’ autoimmunity of healthy donors and the progressive autoimmunity of T1D patients may lie not only in their T-cell repertoire, but also in their beta-cell vulnerability (2).
Pathways for Signal Peptide Epitopes
Insulin is a protein hormone that is initially synthesized as a preprohormone. Preproinsulin is linearly composed of a signal peptide, as most secretory and membrane proteins, a B chain, a C peptide and an A chain (Figure 2A). Once the signal peptide is translated, it binds the signal recognition particle that docks to its receptor within the ER membrane and thus allows the direct translocation of newly synthesized insulin into the ER (Figure 1C). As the translation continues, the signal peptide is cleaved by signal peptidase (SP) and undergoes intramembrane proteolysis by the signal peptide peptidase (SPP) (47) while the nascent proinsulin is released in the ER. Proinsulin is further cleaved by endopeptidases that remove the C peptide, and folded by chaperones that favor the formation of two disulfide bonds between the B and the A chain, leading to mature insulin stored into granules until its exocytosis. The biosynthesis and secretion of insulin are two distinct and thoroughly regulated processes. Secretion is triggered by a higher glucose concentration (48, 49), meaning that insulin biosynthesis is continually active even at normal glucose concentrations and that most regulation occurs at the secretion stage. This feature has two consequences for APP. First, high numbers of free signal peptides are continually cleaved and released in the cytosol and ER. Second, this high bio-synthetic rate leads to a high number of misfolded insulin molecules, estimated to represent up to 20% of insulin production in normal, unstressed, beta cells (7).
The fate of post-cleavage signal peptides is poorly described. The most studied instance is that of non-classical HLA-E molecules, which present signal-peptide sequences derived from classical HLA-A, -B and -C molecules (50). Surface peptide-loaded HLA-E complexes serve as a marker of proper MHC-I expression and prevent NK-cell activation and killing (51). Even though the presentation of HLA signal sequences by HLA-E is altered when TAP or tapasin are inhibited or knocked-out (52, 53), most reports suggest that the MHC-I presentation of signal peptides is a TAP- and proteasome-independent process (47, 54, 55). The presentation of signal-peptide-derived sequences is also increasingly recognized for classical MHC-I molecules, as described for tumor epitopes (56) and beta-cell peptides (23). We identified several HLA-A2-restricted sequences presented by beta cells and originating from the signal peptide of insulin granule proteins such as insulin, urocortin-3 (UCN3), islet amyloid polypeptide (IAPP) and proconvertase 2 (PCKS2), although the latter was not recognized by CD8+ T cells (23). Interestingly, preproinsulin epitopes restricted for HLA-A2 (23) and other MHC-I variants (57) map to the short (24 aa) signal peptide region. Focused investigations have therefore been conducted to characterize the generation of preproinsulin signal peptides for MHC-I presentation (57, 58). Using K562 cells transfected with a single MHC-I allele and insulin gene, different signal peptide products were found bound to each variant tested, and the fate of insulin signal peptide sequences was dependent on their ER intramembrane location. Indeed, after SPP cleavage within the ER membrane, N-terminal peptides are more likely to be released in the cytosol (Figure 1C). These cytosolic free peptides can follow the conventional APP route and be translocated to the ER through TAP transport and subsequently loaded on nascent MHC-I molecules after ERAP1 trimming. On the opposite, upon SPP cleavage, C-terminal peptides are directly released in the ER lumen and loaded on MHC-I molecules, in a TAP- and proteasome-independent manner (57, 58). It is unknown how a peptide can be loaded into MHC-I molecules without TAP and whether ERAP1 trimming is always required, as is the case for a PPI15-24 peptide (59). TAPBPR is the chaperone that likely replaces TAP, as tapasin and TAPBPR are mutually exclusive in their binding to MHC-I (55). One of the enzymes suspected to assist peptide trimming in the trans-Golgi network (TGN) is furin. Furin is a proconvertase that is known to traffic from the TGN to the cell surface via the vesicular pathway, where it can cleave peptides and proteins (60). Besides being involved in the maturation of secreted proteins such as those of the granule, furin has been shown to process antigens and direct them to the secretory route for MHC-I presentation in a TAP-independent manner (61). Similarly to another proconvertase from the same protein family (PC7) that has been shown to rescue unstable HLA-B51 complexes (62), furin could be crucial in post-ER stabilization of peptide-MHC-I binding. Indeed, resident TGN enzymes like furin could ensure an allele-dependent stability of these complexes against the destabilizing effect of the slightly acidic pH of the TGN. The mono-allelic HLA-I-expressing cells used in these studies may however introduce some bias and might not be representative of (multi-allelic) human beta cells. Consistent with their functional pH range, furin could also be involved in peptide exchanges occurring in the endosomes.
Collectively, the considerable translation rate of insulin and other granule proteins may provide a major source of aAPP pathways already within resting beta cells, through the release of high amounts of signal peptides and further protein processing.
Concluding Remarks and Future Directions
Beta cells are very active cells with a specialized secretory activity. On one hand, this activity requires unique recycling pathways to maintain homeostasis. On the other, it is a burden that makes beta cells more vulnerable to additional ER stress. These properties may enhance the contribution of MHC-I aAPP pathways in beta cells. Although direct evidence is missing, data from other cellular models make this possibility plausible and invite further investigations. Gaps in knowledge that need to be filled include: 1) the relative contribution of each APP pathway to the beta-cell immunopeptidome under basal and stressed conditions; 2) how these pathways are modulated in the pro-inflammatory environment of insulitis; and 3) whether the same antigenic peptides are generated by professional APCs, a required step to prime naïve T cells in draining lymph nodes before their homing to the pancreas.
Author Contributions
AC and RM wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Work in our Laboratory is supported by The Leona M. and Harry B. Helmsley Charitable Trust (1901–03689), JDRF (2-SRA-2016-164-Q-R), the Agence Nationale de la Recherche (ANR-19-CE15-0014-01), the Fondation pour la Recherche Medicale (EQU20193007831), and the Innovative Medicines Initiative 2 Joint Undertaking under grant agreements 115797 and 945268 (INNODIA and INNODIA HARVEST), which receive support from the EU Horizon 2020 program, the European Federation of Pharmaceutical Industries and Associations, JDRF, and The Leona M. & Harry B. Helmsley Charitable Trust.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr Loredana Saveanu (Université de Paris, Centre de Recherche sur l’Inflammation, INSERM U1149, CNRS ERL8252) for her critical reviewing of the manuscript and expert advice.
References
1. Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest (2017) 127:2881–91. doi: 10.1172/JCI94549
2. Mallone R, Eizirik DL. Presumption of innocence for beta cells: why are they vulnerable autoimmune targets in type 1 diabetes? Diabetologia (2020) 63:1999–2006. doi: 10.1007/s00125-020-05176-7
3. Battaglia M, Ahmed S, Anderson MS, Atkinson MA, Becker D, Bingley PJ, et al. Introducing the Endotype Concept to Address the Challenge of Disease Heterogeneity in Type 1 Diabetes. Diabetes Care (2020) 43:5–12. doi: 10.2337/dc19-0880
4. Leete P, Oram RA, McDonald TJ, Shields BM, Ziller C, TIGI study team, et al. Studies of insulin and proinsulin in pancreas and serum support the existence of aetiopathological endotypes of type 1 diabetes associated with age at diagnosis. Diabetologia (2020) 63:1258–67. doi: 10.1007/s00125-020-05115-6
5. Carré A, Richardson SJ, Larger E, Mallone R. Presumption of guilt for T cells in type 1 diabetes: lead culprits or partners in crime depending on age of onset? Diabetologia (2021) 64:15–25. doi: 10.1007/s00125-020-05298-y
6. Blum JS, Wearsch PA, Cresswell P. Pathways of Antigen Processing. Annu Rev Immunol (2013) 31:443–73. doi: 10.1146/annurev-immunol-032712-095910
7. Sun J, Cui J, He Q, Chen Z, Arvan P, Liu M. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol Aspects Med (2015) 42:105–18. doi: 10.1016/j.mam.2015.01.001
8. Engin F, Yermalovich A, Nguyen T, Ngyuen T, Hummasti S, Fu W, et al. Restoration of the unfolded protein response in pancreatic β cells protects mice against type 1 diabetes. Sci Transl Med (2013) 5:211ra156. doi: 10.1126/scitranslmed.3006534
9. Eizirik DL, Pasquali L, Cnop M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat Rev Endocrinol (2020) 16:349–62. doi: 10.1038/s41574-020-0355-7
10. Eizirik DL, Miani M, Cardozo AK. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia (2013) 56:234–41. doi: 10.1007/s00125-012-2762-3
11. Craig ME, Kim KW, Isaacs SR, Penno MA, Hamilton-Williams EE, Couper JJ, et al. Early-life factors contributing to type 1 diabetes. Diabetologia (2019) 62:1823–34. doi: 10.1007/s00125-019-4942-x
12. Dean PM. Ultrastructural morphometry of the pancreatic beta-cell. Diabetologia (1973) 9:115–9. doi: 10.1007/BF01230690
13. Muralidharan C, Conteh AM, Marasco MR, Crowder JJ, Kuipers J, de Boer P, et al. Pancreatic beta cell autophagy is impaired in type 1 diabetes. Diabetologia (2021) 64:865–77. doi: 10.1007/s00125-021-05387-6
14. Marasco MR, Conteh AM, Reissaus CA, Cupit JE, Appleman EM, Mirmira RG, et al. Interleukin-6 Reduces β-Cell Oxidative Stress by Linking Autophagy With the Antioxidant Response. Diabetes (2018) 67:1576–88. doi: 10.2337/db17-1280
15. Trivedi PC, Bartlett JJ, Pulinilkunnil T. Lysosomal Biology and Function: Modern View of Cellular Debris Bin. Cells (2020) 9:1131. doi: 10.3390/cells9051131
16. Soleimanpour SA, Gupta A, Bakay M, Ferrari AM, Groff DN, Fadista J, et al. The Diabetes Susceptibility Gene Clec16a Regulates Mitophagy. Cell (2014) 157:1577–90. doi: 10.1016/j.cell.2014.05.016
17. Hakonarson H, Grant SFA, Bradfield JP, Marchand L, Kim CE, Glessner JT, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature (2007) 448:591–4. doi: 10.1038/nature06010
18. Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol (2009) 10:597–608. doi: 10.1038/nrm2755
19. Montealegre S, van Endert PM. Endocytic Recycling of MHC Class I Molecules in Non-professional Antigen Presenting and Dendritic Cells. Front Immunol (2019) 9:3098:3098. doi: 10.3389/fimmu.2018.03098
20. Ilca FT, Neerincx A, Hermann C, Marcu A, Stevanović S, Deane JE, et al. TAPBPR mediates peptide dissociation from MHC class I using a leucine lever. eLife (2018) 7:e40126. doi: 10.7554/eLife.40126
21. Jahreiss L, Menzies FM, Rubinsztein DC. The Itinerary of Autophagosomes: From Peripheral Formation to Kiss-and-Run Fusion with Lysosomes. Traffic (2008) 9:574–87. doi: 10.1111/j.1600-0854.2008.00701.x
22. Yorimitsu T, Klionsky DJ. Eating the endoplasmic reticulum: quality control by autophagy. Trends Cell Biol (2007) 17:279–85. doi: 10.1016/j.tcb.2007.04.005
23. Gonzalez-Duque S, Azoury ME, Colli ML, Afonso G, Turatsinze J-V, Nigi L, et al. Conventional and Neo-Antigenic Peptides Presented by β Cells Are Targeted by Circulating Naïve CD8+ T Cells in Type 1 Diabetic and Healthy Donors. Cell Metab (2018) 28:946–60. doi: 10.1016/j.cmet.2018.07.007
24. Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics (2015) 14:658–73. doi: 10.1074/mcp.M114.042812
25. Azoury ME, Tarayrah M, Afonso G, Pais A, Colli ML, Maillard C, et al. Peptides Derived From Insulin Granule Proteins are Targeted by CD8+ T Cells Across MHC Class I Restrictions in Humans and NOD Mice. Diabetes (2020) 69:2678–90. doi: 10.2337/db20-0013
26. James EA, Mallone R, Kent SC, DiLorenzo TP. T-Cell Epitopes and Neo-epitopes in Type 1 Diabetes: A Comprehensive Update and Reappraisal. Diabetes (2020) 69:1311–35. doi: 10.2337/dbi19-0022
27. Wan X, Zinselmeyer BH, Zakharov PN, Vomund AN, Taniguchi R, Santambrogio L, et al. Pancreatic islets communicate with lymphoid tissues via exocytosis of insulin peptides. Nature (2018) 560:107–11. doi: 10.1038/s41586-018-0341-6
28. Sims EK, Chaudhry Z, Watkins R, Syed F, Blum J, Ouyang F, et al. Elevations in the Fasting Serum Proinsulin–to–C-Peptide Ratio Precede the Onset of Type 1 Diabetes. Diabetes Care (2016) 39:1519–26. doi: 10.2337/dc15-2849
29. Rodriguez-Calvo T, Zapardiel-Gonzalo J, Amirian N, Castillo E, Lajevardi Y, Krogvold L, et al. Increase in Pancreatic Proinsulin and Preservation of β-Cell Mass in Autoantibody-Positive Donors Prior to Type 1 Diabetes Onset. Diabetes (2017) 66:1334–45. doi: 10.2337/db16-1343
30. Wasserfall C, Nick HS, Campbell-Thompson M, Beachy D, Haataja L, Kusmartseva I, et al. Persistence of Pancreatic Insulin mRNA Expression and Proinsulin Protein in Type 1 Diabetes Pancreata. Cell Metab (2017) 26:568–75. doi: 10.1016/j.cmet.2017.08.013
31. Sims EK, Bahnson HT, Nyalwidhe J, Haataja L, Davis AK, Speake C, et al. Proinsulin Secretion Is a Persistent Feature of Type 1 Diabetes. Diabetes Care (2019) 42:258–64. doi: 10.2337/dc17-2625
32. Sims EK, Syed F, Nyalwidhe J, Bahnson HT, Haataja L, Speake C, et al. Abnormalities in proinsulin processing in islets from individuals with longstanding T1D. Transl Res (2019) 213:90–9. doi: 10.1016/j.trsl.2019.08.001
33. Benkahla MA, Sabouri S, Kiosses WB, Rajendran S, Quesada-Masachs E, von Herrath M. HLA class I hyper-expression unmasks beta cells but not alpha cells to the immune system in pre-diabetes. J Autoimmun (2020) 119:102628. doi: 10.1016/j.jaut.2021.102628
34. Vieira E, Salehi A, Gylfe E. Glucose inhibits glucagon secretion by a direct effect on mouse pancreatic alpha cells. Diabetologia (2007) 50:370–9. doi: 10.1007/s00125-006-0511-1
35. Marroqui L, Masini M, Merino B, Grieco FA, Millard I, Dubois C, et al. Pancreatic α Cells are Resistant to Metabolic Stress-induced Apoptosis in Type 2 Diabetes. EBioMedicine (2015) 2:378–85. doi: 10.1016/j.ebiom.2015.03.012
36. Wyatt RC, Lanzoni G, Russell MA, Gerling I, Richardson SJ. What the HLA-I!—Classical and Non-classical HLA Class I and Their Potential Roles in Type 1 Diabetes. Curr Diabetes Rep (2019) 19:159. doi: 10.1007/s11892-019-1245-z
37. Himuro M, Miyatsuka T, Suzuki L, Miura M, Katahira T, Goto H, et al. Cellular Autophagy in α Cells Plays a Role in the Maintenance of Islet Architecture. J Endocr Soc (2019) 3:1979–92. doi: 10.1210/js.2019-00075
38. Sheng Q, Xiao X, Prasadan K, Chen C, Ming Y, Fusco J, et al. Autophagy protects pancreatic beta cell mass and function in the setting of a high-fat and high-glucose diet. Sci Rep (2017) 7:16348. doi: 10.1038/s41598-017-16485-0
39. Dalet A, Stroobant V, Vigneron N, Van den Eynde BJ. Differences in the production of spliced antigenic peptides by the standard proteasome and the immunoproteasome. Eur J Immunol (2011) 41:39–46. doi: 10.1002/eji.201040750
40. Lundh M, Bugliani M, Dahlby T, Chou DH-C, Wagner B, Ghiasi SM, et al. The immunoproteasome is induced by cytokines and regulates apoptosis in human islets. J Endocrinol (2017) 233:369–79. doi: 10.1530/JOE-17-0110
41. Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schröter F, et al. Immunoproteasomes Preserve Protein Homeostasis upon Interferon-Induced Oxidative Stress. Cell (2010) 142:613–24. doi: 10.1016/j.cell.2010.07.036
42. Zaiss DMW, Bekker CPJ, Gröne A, Lie BA, Sijts AJAM. Proteasome immunosubunits protect against the development of CD8 T-cell-mediated autoimmune diseases. J Immunol (2011) 187:2302–9. doi: 10.4049/jimmunol.1101003
43. Mishto M, Liepe J, Textoris-Taube K, Keller C, Henklein P, Weberruß M, et al. Proteasome isoforms exhibit only quantitative differences in cleavage and epitope generation. Eur J Immunol (2014) 44:3508–21. doi: 10.1002/eji.201444902
44. Winter MB, La Greca F, Arastu-Kapur S, Caiazza F, Cimermancic P, Buchholz TJ, et al. Immunoproteasome functions explained by divergence in cleavage specificity and regulation. eLife (2017) 6:e27364. doi: 10.7554/eLife.27364
45. Marroqui L, Dos Santos RS, Op de Beeck A, Coomans de Brachène A, Marselli L, Marchetti P, et al. Interferon-α mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia (2017) 60:656–67. doi: 10.1007/s00125-016-4201-3
46. Culina S, Lalanne AI, Afonso G, Cerosaletti K, Pinto S, Sebastiani G, et al. Islet-reactive CD8+ T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci Immunol (2018) 3:eaao4013. doi: 10.1126/sciimmunol.aao4013
47. Martoglio B, Dobberstein B. Signal sequences: more than just greasy peptides. Trends Cell Biol (1998) 8:410–5. doi: 10.1016/S0962-8924(98)01360-9
48. Alarcón C, Lincoln B, Rhodes CJ. The biosynthesis of the subtilisin-related proprotein convertase PC3, but no that of the PC2 convertase, is regulated by glucose in parallel to proinsulin biosynthesis in rat pancreatic islets. J Biol Chem (1993) 268:4276–80. doi: 10.1016/S0021-9258(18)53606-1
49. Schuit FC, In’t Veld PA, Pipeleers DG. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc Natl Acad Sci USA (1988) 85:3865–9. doi: 10.1073/pnas.85.11.3865
50. Braud V, Jones EY, McMichael A. The human major histocompatibility complex class Ib molecule HLA-E binds signal sequence-derived peptides with primary anchor residues at positions 2 and 9. Eur J Immunol (1997) 27:1164–9. doi: 10.1002/eji.1830270517
51. Braud VM, Allan DSJ, O’Callaghan CA, Söderström K, D’Andrea A, Ogg GS, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature (1998) 391:795–9. doi: 10.1038/35869
52. Lampen MH, Hassan C, Sluijter M, Geluk A, Dijkman K, Tjon JM, et al. Alternative peptide repertoire of HLA-E reveals a binding motif that is strikingly similar to HLA-A2. Mol Immunol (2013) 53:126–31. doi: 10.1016/j.molimm.2012.07.009
53. Celik AA, Kraemer T, Huyton T, Blasczyk R, Bade-Döding C. The diversity of the HLA-E-restricted peptide repertoire explains the immunological impact of the Arg107Gly mismatch. Immunogenetics (2016) 68:29–41. doi: 10.1007/s00251-015-0880-z
54. Wei ML, Cresswell P. HLA-A2 molecules in an antigen-processing mutant cell contain signal sequence-derived peptides. Nature (1992) 356:443–6. doi: 10.1038/356443a0
55. Oliveira CC, van Hall T. Alternative Antigen Processing for MHC Class I: Multiple Roads Lead to Rome. Front Immunol (2015) 6:298:298. doi: 10.3389/fimmu.2015.00298
56. Hage FE, Stroobant V, Vergnon I, Baurain J-F, Echchakir H, Lazar V, et al. Preprocalcitonin signal peptide generates a cytotoxic T lymphocyte-defined tumor epitope processed by a proteasome-independent pathway. Proc Natl Acad Sci USA (2008) 105:10119–24. doi: 10.1073/pnas.0802753105
57. Kronenberg-Versteeg D, Eichmann M, Russell MA, de Ru A, Hehn B, Yusuf N, et al. Molecular Pathways for Immune Recognition of Preproinsulin Signal Peptide in Type 1 Diabetes. Diabetes (2018) 67:687–96. doi: 10.2337/db17-0021
58. Skowera A, Ellis RJ, Varela-Calviño R, Arif S, Huang GC, Van-Krinks C, et al. CTLs are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J Clin Invest (2008) 118:3390–402. doi: 10.1172/JCI35449
59. Thomaidou S, Kracht MJL, van der Slik A, Laban S, de Koning EJ, Carlotti F, et al. β-Cell Stress Shapes CTL Immune Recognition of Preproinsulin Signal Peptide by Posttranscriptional Regulation of Endoplasmic Reticulum Aminopeptidase 1. Diabetes (2020) 69:670–80. doi: 10.2337/db19-0984
60. Thomas G. Furin At The Cutting Edge: From Protein Traffic To Embryogenesis And Disease. Nat Rev Mol Cell Biol (2002) 3:753–66. doi: 10.1038/nrm934
61. Medina F, Ramos M, Iborra S, de León P, Rodríguez-Castro M, Del Val M. Furin-processed antigens targeted to the secretory route elicit functional TAP1-/-CD8+ T lymphocytes in vivo. J Immunol (2009) 183:4639–47. doi: 10.4049/jimmunol.0901356
Keywords: antigen presentation, antigen processing, autophagy, crinophagy, insulin granule, MHC class I, neo-epitopes, signal peptide
Citation: Carré A and Mallone R (2021) Making Insulin and Staying Out of Autoimmune Trouble: The Beta-Cell Conundrum. Front. Immunol. 12:639682. doi: 10.3389/fimmu.2021.639682
Received: 09 December 2020; Accepted: 12 March 2021;
Published: 29 March 2021.
Edited by:
Iñaki Alvarez, Universitat Autònoma de Barcelona, SpainReviewed by:
Howard Davidson, University of Colorado Anschutz Medical Campus, United StatesBruce Verchere, University of British Columbia, Canada
Copyright © 2021 Carré and Mallone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexia Carré, YWxleGlhLmNhcnJlQGluc2VybS5mcg==; Roberto Mallone, cm9iZXJ0by5tYWxsb25lQGluc2VybS5mcg==