 John R. Bethea
John R. Bethea Roman Fischer
Roman Fischer- 1Department of Biology, Drexel University, Philadelphia, PA, United States
- 2Institute of Cell Biology and Immunology, University Stuttgart, Stuttgart, Germany
- 3Stuttgart Research Center Systems Biology, University of Stuttgart, Stuttgart, Germany
Chronic neuropathic pain (CNP) is caused by a lesion or disease of the somatosensory nervous system. It affects ~8% of the general population and negatively impacts a person's level of functioning and quality of life. Its resistance to available pain therapies makes CNP a major unmet medical need. Immune cells have been shown to play a role for development, maintenance and recovery of CNP and therefore are attractive targets for novel pain therapies. In particular, in neuropathic mice and humans, microglia are activated in the dorsal horn and peripheral immune cells infiltrate the nervous system to promote chronic neuroinflammation and contribute to the initiation and progression of CNP. Importantly, immunity not only controls pain development and maintenance, but is also essential for pain resolution. In particular, regulatory T cells, a subpopulation of T lymphocytes with immune regulatory function, and macrophages were shown to be important contributors to pain recovery. In this review we summarize the interactions of the peripheral immune system with the nervous system and outline their contribution to the development and recovery of pain.
Introduction
Chronic pain is defined as a more than 12 weeks lasting pain that is characterized by irregular somatosensory processing in the peripheral nervous system (PNS) or the central nervous system (CNS) (1). Chronic neuropathic pain (CNP) is a specific form of chronic pain that is caused by damage to or disease of the somatosensory nervous system (2, 3) and affects up to 8% of the general population (4). CNP can result as a consequence of a large number of medical conditions, such as injuries to the PNS or CNS, metabolic, autoimmune or neurodegenerative diseases as well as cancer and chemotherapy (4, 5). In this review, we will mainly focus on CNP due to peripheral nerve injuries and discuss results from chemotherapy-induced pain. Individuals suffering from CNP exhibit stimulus-independent pain that is often characterized by abnormal sensations or hypersensitivity in the affected area. Patients often describe the pain as a burning and/or stabbing sensation (6). Allodynia, pain elicited by a usually non-painful stimulus, and hyperalgesia, an increased pain response due to painful stimulus, are frequent symptoms described by CNP patients (7). CNP can have a dramatic impact on a person's level of functioning and quality of life and is resistant to conservative pain management (7). Therefore, effective therapies that either prevent or reverse CNP are critical for both public health and clinical practice (5).
Research from the last two decades has demonstrated that a robust neuroimmune response and bidirectional signaling between the sensory and immune system contribute to development and maintenance of CNP. Indeed, increased levels of soluble pro-inflammatory mediators and recruitment of immune cells to the site of nerve injury, the dorsal root ganglion (DRG) and the spinal cord after PNS/CNS injury are well-characterized in rodent models of CNP (8–15). In addition, immune cells also contribute to recovery of CNP (16–18), indicating that modulation of immunity has therapeutic potential to treat CNP. Therefore, a detailed understanding of the contribution of immune responses to the development, maintenance and resolution of CNP may result in novel therapeutic approaches that are superior to current pain-relieving therapies.
Development and Maintenance of CNP is Controlled by CD4+ Effector T Cells
T cells were identified as important contributors to CNP development and maintenance in animal models and human patients. Early on, the general importance of T cells for pain development was demonstrated by several independent studies showing that immunodeficient mice without functional lymphocytes in contrast to control mice do not develop pain hypersensitivity after nerve injury (9, 19, 20). Reconstitution of immunodeficient nude rats (9), and mice (19) with CD4+ T helper cells resulted in reestablishment of the pain phenotype, suggesting that pro-inflammatory T helper cells promote pain responses. T cells were shown to infiltrate into the lumbar spinal cord post-injury (19). Similar, the Th1 cytokine interferon-γ (IFN-γ) is upregulated in the dorsal horn after nerve injury and functional IFN-γ signaling is required for full development of neuropathic hypersensitivity (20), indicating that Th1 responses in the spinal cord contribute to pain hypersensitivity. The important role of T cell infiltration for development of pain was further confirmed by a recent study demonstrating that intrathecal injection of a T cell receptor (TCR) specific antibody that depletes functional CD4+ T cells resulted in alleviation of mechanical allodynia, indicating that CNS infiltrating T cells directly contribute to pain responses. Once treatment was terminated, mechanical allodynia returned to levels comparable to control mice, presumably due to repopulation of functional T cells in the CNS (21).
Next to IFN-γ expressing Th1 cells, IL17-expressing T helper cells were detected in peripheral nerves, indicating that Th17 cells may also contribute to pain development (22). Indeed, intrathecal and intraneural injection of recombinant IL-17A induced pain hypersensitivity (23), indicating a role of IL-17A for pain development. A recent study further confirmed that IL-17A regulates neuron-glial communications, synaptic transmission, and neuropathic pain after chemotherapy (24). Additional mechanistic data showed that DRG-infiltrating T lymphocytes release leukocyte elastase after nerve injury. Confirming that leukocyte elastase promotes T cell-dependent pain responses, adoptive transfer of leukocyte elastase deficient T cells did not restore pain development in immunodeficient Rag2−/− mice (25). This indicated that T cells may also directly contribute to nerve damage.
Interestingly, MHC class II knockout (k/o) mice that lack MHC class II-restricted T helper cells displayed an impaired chronification of mechanical allodynia after peripheral nerve injury (26), indicating the general importance of CD4+ T helper cells for pain chronification in rodents. This was confirmed by a recent study showing that MHC class II-restricted CD4+ T helper cells contribute to the transition from acute to chronic mechanical allodynia in a rat model of peripheral nerve injury (21). Clinical studies have shown that a human major histocompatibility complex (MHC) class II gene polymorphism (DQB1*03:02 HLA haplotype) is associated with an increased risk to develop CNP after inguinal hernia surgery and lumbar disc herniation (27). The importance of MHC class II genes for CNP development indicates that, similar to the animal models, CD4+ Th cells contribute to CNP development in human patients. The relevance of MHCII-restricted T helper cells as an important trigger for chronic hypersensitivity after nerve injuries has been recently discussed by Ding et al. (28), where they indicate that there is a growing body of clinical evidences showing that increased blood Th cell numbers and changes in subset patterns are correlated with neuropathic pain intensities after nerve injuries. Other clinical data indicate that an emergent T-helper 2 profile with high interleukin-6 levels correlates with the appearance of bortezomib-induced neuropathic pain (29). Interestingly, Luchting et al. (30) found a disrupted Th17/Treg balance with significantly increased anti-inflammatory Tregs and decreased pro-inflammatory Th17 cells in patients suffering from chronic unspecific low back pain compared to healthy controls, indicating an anti-inflammatory T cell shift in the patients they analyzed. Altogether, these clinical data indicate that there is a considerable impact of the T cell compartment in neuropathic pain. However, the role of pro- and anti-inflammatory T cell subsets in CNP patients seems to differ depending on the pain condition of the patients.
T Cell Subsets Contribute To Pain Recovery
In 2004 Moalem et al. (9) showed that adoptive transfer of Th2 polarized T helper cells in athymic nude rats further reduced pain hypersensitivity, indicating that anti-inflammatory responses mediated by Th2 cells may counteract inflammatory processes that promote pain development and maintenance. Indeed, higher circulating levels of the anti-inflammatory interleukins IL-10 and IL-4 were detected in patients with painless neuropathy compared to patients with painful neuropathy and controls (31), indicating that anti-inflammatory responses may be necessary to control pain development. Indeed, Leger et al. (32) demonstrated that glatiramer acetate treatment, an approved MS therapy, inhibited microglia activation and increased IL-10 and IL-4 expressing T cells in the dorsal horn after peripheral nerve injury resulting in alleviation of neuropathic allodynia. This indicates that next to their pathologic role for development and maintenance of CNP, T cells may also contribute to the resolution of pain.
CD8+ T Cells Contribute To Pain Recovery
Whereas, CD4+ Th1 cells promote pain development and maintenance, several reports indicate that CD8+ T cells may contribute to pain resolution. Krukowski et al. (33) showed that chemotherapy-induced mechanical hypersensitivity was prolonged in T-cell-deficient Rag1−/− mice compared to wild type mice. Adoptive transfer of CD8+, but not CD4+ T cells to neuropathic Rag1−/− mice restored pain resolution. Mechanistically, this study indicated that CD8+ T Cells and endogenous IL-10 were required for resolution of CNP (33). Other studies have also reported a role of IL-10 for pain resolution and functional recovery after peripheral nerve injury (34), indicating the general importance of anti-inflammatory responses for pain resolution. Laumet et al. (35) recently confirmed that resolution of chemotherapy-induced mechanical allodynia is dependent on presence of CD8+ T cells. They showed that adoptive transfer of CD8+ T cells from naïve wildtype mice to T-cell-deficient neuropathic Rag2−/− mice failed to promote pain resolution. In contrast, adoptive transfer of cisplatin-educated CD8+ T cells prevented the development of chemotherapy-induced CNP (35). Importantly, this T cell education appeared to be independent of antigen recognition by the T cell receptor because cisplatin-educated CD8+ T cells did also promote pain resolution in a model of paclitaxel-induced CNP and reconstitution of T cell deficient mice with ovalbumin-specific CD8+ T cells also restored CNP resolution (35). This study indicates that CD8+ T cells need to be activated to acquire the capacity to promote resolution of CNP, but their therapeutic activity seems to be independent of their antigen-specific education. However, the role of CD8+ T cells for CNP seems to be complex. Using a model of paclitaxel-induced CNP, Liu et al. (36) showed that blocking of functional CD8+ T cells at the level of the spinal cord and the DRG, reversed chemotherapy induced mechanical hypersensitivity. Similar, adoptive transfer of CD8+ T cells exacerbated neuropathic pain in this model (36), suggesting that cytotoxic T cells contribute to pain progression.
Tregs Control Immunity to Promote Pain Resolution
Another important T cell subset that plays a role for pain recovery are regulatory T cells (Tregs), immunomodulatory T lymphocytes that control the activity of innate and adaptive immune cells (37). After peripheral nerve injury, Tregs are recruited to the site of injury, the DRG and the spinal cord (16, 17, 38). Systemic expansion of Tregs was shown to alleviate peripheral CNP following nerve injury and experimental autoimmune neuritis-associated central CNP (16). Similar, anti-CD25 antibody-dependent depletion of CD25+ cells prolonged mechanical hypersensitivity after peripheral nerve injury (16, 18), indicating a role of CD25+ Tregs for pain recovery. In another study DEREG mice, where FoxP3-expressing Tregs can be depleted by injection of diphtheria toxin, were used to specifically assess Treg contribution to pain recovery. Indeed, following toxin application DEREG mice developed increased mechanical pain hypersensitivity after peripheral nerve injury (17), confirming that Tregs are important for pain recovery. The analgesic role of Tregs was further confirmed in a model of chemotherapy-induced CNP, where adoptive transfer of a population of CD4+CD25+ T cells, which largely are composed of Tregs, alleviated CNP (36).
In a recent study it was shown that nerve-infiltrating Tregs suppress the development of neuropathic pain mainly through the inhibition of the Th1 response by CD4+ T helper cells following nerve injury (38). This resulted indirectly in reduced neuronal damage and neuroinflammation at the level of the sensory ganglia. The authors further identified IL-10 signaling as an intrinsic mechanism by which Treg cells counteract neuropathic pain development (38). Indeed, in a previous study the neuroprotective effect of IL-10 secretion by CNS infiltrating Tregs was demonstrated in an ischemia model (39). Another recent study by Duffy et al. (40) showed that adoptive transfer of activated Tregs or intrathecal delivery of the Treg cytokine IL-35 alleviated spontaneous and facial stimulus-evoked pain behaviors in mice with experimental autoimmune encephalomyelitis (EAE). The effects of intrathecal IL-35 therapy were dependent on presence of Tregs and associated with reduced monocyte infiltration in the trigeminal afferent pathway and upregulated IL-10 expression in CNS-infiltrating lymphocytes (40). Interestingly, intrathecal injection of plasmids encoding IL-10 at the onset of clinical EAE suppressed disease development and alleviated pain behaviors (41, 42), indicating that the upregulated IL-10 expression observed in the study of Duffy et al. (40) may be responsible for the observed pain alleviating effect of Tregs.
All these data point toward a critical role of IL-10 for pain resolution. Indeed, additional research showed that central activation of anti-inflammatory cytokines such as IL-10 and TGFβ suppresses allodynia after peripheral nerve injury (43) and in a model of chemotherapy-induced neuropathic pain (44). Mechanistically, in vitro studies showed that in addition to its master anti-inflammatory role, IL-10 reverses voltage-gated sodium currents to reduce neuronal excitability (45), indicating a possible immune cell independent mechanism of IL-10 mediated pain recovery. Therefore, therapies that promote Treg activity and IL-10 signaling in the CNS may prove beneficial for pain therapies.
Over the last decade, we and others showed that tumor necrosis factor receptor 2 (TNFR2) is critical for Treg function and that selective agonism of TNFR2 results in Treg expansion and is therapeutic in inflammatory conditions such as experimental arthritis (46, 47) and graft vs. host disease (48). We recently demonstrated that treatment of neuropathic mice with a TNFR2 agonist promoted long-term pain recovery after peripheral nerve injury (18) and in neuropathic EAE mice (49). Mechanistically, our study revealed that systemic TNFR2 agonist application promoted expansion of Tregs resulting in alleviation of peripheral and central inflammation. We further detected increased Treg and IL-10 levels in the spinal cord after TNFR2 agonist treatment (18), indicating that Treg-mediated IL-10 signaling in the CNS may contribute to the pain alleviating effect of TNFR2 agonists too. Interestingly, next to their anti-inflammatory functions, Tregs were shown to directly promote tissue regeneration in the CNS (50). Confirming, we observed upregulation of various proteins associated with neuroregeneration after TNFR2 agonist treatment in neuropathic mice (18), indicating that TNFR2-dependent expansion of Tregs may promote pain recovery not only by modulation of immunity, but also via enhanced tissue regeneration. Indeed, previous studies showed that TNFR2 directly contributes to neuroprotection (51–53).
Macrophages Contribute to The Development of CNP
Next to T lymphocytes, peripheral monocytes that differentiate into macrophages upon tissue infiltration were shown to play a role for pain development. These cells were shown to infiltrate around injured sensory neurons and in the DRG. Inhibition of monocyte infiltration into the DRG prevented the development of pain hypersensitivity in rodent models of CNP (54, 55), indicating the importance of peripheral macrophages for pain development. In a model of chemotherapy-induced CNP, nerve infiltrating monocytes were activated by the chemokine fractalkine (CX3CL1) resulting in the production of reactive oxygen species that in turn activated the receptor TRPA1 in sensory neurons and evoked the pain response (56). Additional data indicate an interaction between the fractalkine receptor CX3CR1 and the chemokine receptor CCR2 in monocytes that may constitute an underlying mechanism for persistent chemotherapy-induced pain (57).
Monocytes/macrophages were shown to act synergistically with microglia to initiate hypersensitivity and promote the transition from acute to chronic pain after peripheral nerve injury (58). A recent study demonstrated that DRG macrophages, but not macrophages that had infiltrated at the site of injury contribute to initiation and maintenance of mechanical hypersensitivity (59). Indeed, depletion of DRG macrophages, but not at the site of injury, prevented the development of pain and reversed ongoing nerve injury-induced hypersensitivity (59). Macrophages that invade the DRG, release excitatory agents that generate ectopic activity in sensory neurons thereby contributing to neuropathology responsible for pain development (60, 61).
Macrophages Alleviate Pain via The Opioid System and Anti-Inflammatory Responses
Macrophage infiltration into the nerve is an essential step to allow nerve regeneration. In particular, anti-inflammatory/reparative M2 macrophages have been indicated to play a role for repair processes after nerve injury (62). Indeed, perineural transplantation of M2 macrophages resulted in attenuated neuropathy-induced mechanical hypersensitivity (63, 64). Similar, injection of IL-4, a cytokine responsible for M2 macrophage differentiation, at the site of nerve injury promoted repolarization of macrophages into an anti-inflammatory M2 state, and ameliorated mechanic and thermal hypersensitivity (65). Recently, it was shown that local sympathectomy relieves chemotherapy-induced allodynia in mice via anti-inflammatory responses. Depletion of monocytes/macrophages and blockade of transforming growth factor-β (TGF-β) signaling reversed the relief of mechanical allodynia by sympathectomy (66). Importantly, TGF-β induces M2-like macrophage polarization (67), indicating that TGF-β-induced M2 macrophage polarization might be responsible for the therapeutic effect of local sympathectomy in the aforementioned study.
Interestingly, cultured M2 macrophages contained and released higher amounts of opioid peptides (63). Similar, a recent study demonstrated that IL-4 application at injured nerves shifted macrophage polarization from a proinflammatory M1 to an anti-inflammatory M2 phenotype. These M2 macrophages continuously synthesized opioid peptides. IL-4 administration further resulted in a long-lasting attenuation of neuropathy-induced mechanical hypersensitivity after discontinuing treatment. Confirming the importance of M2 macrophage-secreted opioids, IL-4-induced analgesia was decreased after neutralizing opioid peptides or blocking opioid receptors at the injured nerves (68). These studies indicate that M2 polarized macrophages may regulate pain perception by modulation of the opioid system. A key observation of our study showing that TNFR2 agonist treatment promotes long-lasting pain recovery was a repolarization of CNS-infiltrating macrophages into an anti-inflammatory M2-like phenotype (18). However, the contribution of M2 polarized macrophages in the spinal cord to pain alleviation and a potential role of the opioid system is not clear yet.
Immune-Mediated Sex Differences Impact CNP Development
Next to social and psychological factors, functional differences in the immune system contribute to a higher female prevalence for CNP development (69, 70). Using rodent models of injury-induced CNP, it was shown that male and female mice use different immune cells to initiation and maintain CNP. In particular, microglia were shown to be the driver of male neuroinflammation and CNP, whereas T cells primarily drive neuroinflammation and CNP in females (71). These differences seem to be dependent on cell populations, differences in suppression by hormones, and disparate cellular responses in males and females (72). Interestingly, in the absence of adaptive immune cells, e.g., in Rag1−/− mice, female mice use the male, glial-dependent pathway (71). Since sex differences may impact the effectivity of analgesic therapeutics, the different impact of immune cells to pain responses needs to be considered in therapeutics development. Indeed, we observed different responsiveness of male and female mice in a preclinical trial for a novel TNF modulating compound (73). Therefore, it is important to study the therapeutic responses in males and females during preclinical evaluation, in particular if they address T cell or microglial responses. Therapies based on Treg modulation work across sexes (18) indicating that they may interfere with microglial and adaptive immune cell contribution to CNP development and maintenance.
Conclusion and Outlook
The contribution of immunity to development and maintenance of CNP are well-established and a complex interaction of different immune cells contributes to CNP development (Figure 1). Over the last years a growing body of literature on the protective and regenerative role of the immune system for pain has been published, including contributions of CD8+ T cells, Tregs and M2 macrophages (Figure 1). Modulation of immune responses, e.g., by targeting inflammatory or anti-inflammatory mediators of peripheral immune cells, therefore is a promising therapeutic approach to alleviate neuropathic pain (Table 1). Concluding, a detailed understanding of immune-mediated tissue regeneration in pain may promote the development of novel immunotherapies for pain alleviation and ultimately may translate into novel non-opioid therapies.
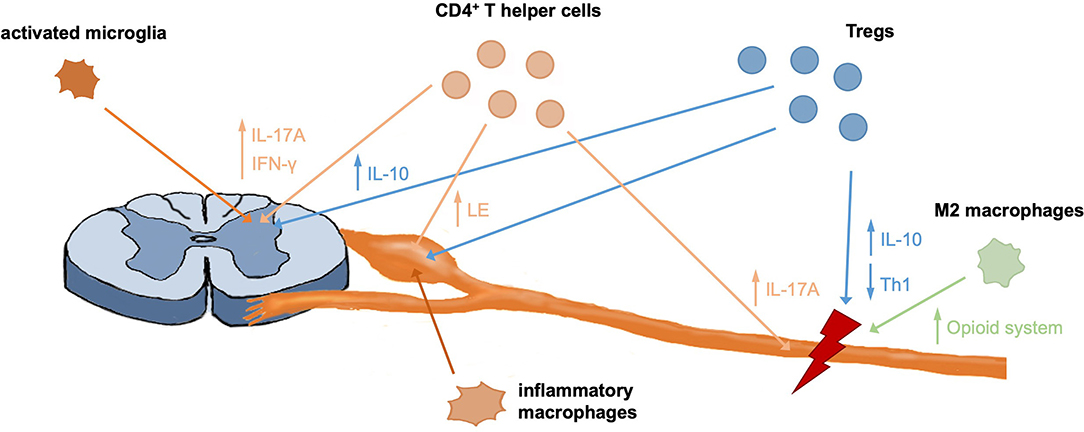
Figure 1. Contribution of different immune cells to pain development and recovery. CD4+ T helper cells were shown to infiltrate the spinal cord, DRG and injured nerve, where they contribute to pain responses through different mechanisms. Further, DRG invading macrophages were shown to be important mediators of pain. Like T effector cells, Tregs infiltrate the nerve, DRG, and spinal cord in neuropathic mice and contribute to immunomodulation and tissue regeneration through different mechanisms, including secretion of anti-inflammatory IL-10. M2 macrophages were shown to initiate analgesic responses in the nerve through upregulation of the endogenous opioid system and anti-inflammatory responses.

Table 1. Overview of peripheral immune cell contribution to pain development and recovery.
Author Contributions
RF wrote the review and generated the figure. JB reviewed and revised the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
RF was supported by a grant from the Michael J. Fox Foundation (MJFF18334) and JB was supported by grants from the NIH (R01NS051709, R01NS111761, R01NS106908, R01NS096971).
Conflict of Interest
JB and RF are named inventors on patent applications covering the use of TNFR2 agonists. RF is a named inventor on patent applications covering the TNFR2 agonist technology.
References
1. Fasick V, Spengler RN, Samankan S, Nader ND, Ignatowski TA. The hippocampus and TNF: Common links between chronic pain and depression. Neurosci Biobehav Rev. (2015) 53:139–59. doi: 10.1016/j.neubiorev.2015.03.014
2. Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. (2009) 32:1–32. doi: 10.1146/annurev.neuro.051508.135531
3. Jensen TS, Baron R, Haanpää M, Kalso E, Loeser JD, Rice ASC, et al. A new definition of neuropathic pain. Pain. (2011) 152:2204–5. doi: 10.1016/j.pain.2011.06.017
4. Murphy KL, Bethea JR, Fischer R. Multiple Sclerosis: Perspectives in Treatment and Pathogenesis: Neuropathic Pain in Multiple Sclerosis—Current Therapeutic Intervention and Future Treatment Perspectives. Brisbane, QLD: Codon Publications. (2017). doi: 10.15586/codon.multiplesclerosis.2017.ch4
5. Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, et al. Neuropathic pain. Nat Rev Dis Primers. (2017) 3:17002. doi: 10.1038/nrdp.2017.2
6. Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. (1999) 353:1959–64. doi: 10.1016/S0140-6736(99)01307-0
7. Jensen TS, Finnerup NB. A brief history of pain. Lancet Neurol. (2014) 13:872. doi: 10.1016/S1474-4422(14)70187-5
8. Sweitzer SM, Hickey WF, Rutkowski MD, Pahl JL, DeLeo JA. Focal peripheral nerve injury induces leukocyte trafficking into the central nervous system: potential relationship to neuropathic pain. Pain. (2002) 100:163–70. doi: 10.1016/S0304-3959(02)00257-9
9. Moalem G, Xu K, Yu L. T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience. (2004) 129:767–77. doi: 10.1016/j.neuroscience.2004.08.035
10. Moalem G, Grafe P, Tracey DJ. Chemical mediators enhance the excitability of unmyelinated sensory axons in normal and injured peripheral nerve of the rat. Neuroscience. (2005) 134:1399–411. doi: 10.1016/j.neuroscience.2005.05.046
11. Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev. (2006) 51:240–64. doi: 10.1016/j.brainresrev.2005.11.004
12. Austin PJ, Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol. (2010) 229:26–50. doi: 10.1016/j.jneuroim.2010.08.013
13. Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol. (2014) 14:217–31. doi: 10.1038/nri3621
14. Chavan SS, Ma P, Chiu IM. Neuro-immune interactions in inflammation and host defense: Implications for transplantation. Am J Transplant. (2018) 18:556–63. doi: 10.1111/ajt.14515
15. Laumet G, Ma J, Robison AJ, Kumari S, Heijnen CJ, Kavelaars A. T cells as an emerging target for chronic pain therapy. Front Mol Neurosci. (2019) 12:216. doi: 10.3389/fnmol.2019.00216
16. Austin PJ, Kim CF, Perera CJ, Moalem-Taylor G. Regulatory T cells attenuate neuropathic pain following peripheral nerve injury and experimental autoimmune neuritis. Pain. (2012) 153:1916–31. doi: 10.1016/j.pain.2012.06.005
17. Lees JG, Duffy SS, Perera CJ, Moalem-Taylor G. Depletion of Foxp3+ regulatory T cells increases severity of mechanical allodynia and significantly alters systemic cytokine levels following peripheral nerve injury. Cytokine. (2015) 71:207–14. doi: 10.1016/j.cyto.2014.10.028
18. Fischer R, Sendetski M, del Rivero T, Martinez GF, Bracchi-Ricard V, Swanson KA, et al. TNFR2 promotes Treg-mediated recovery from neuropathic pain across sexes. Proc Natl Acad Sci USA. (2019) 116:17045–50. doi: 10.1073/pnas.1902091116
19. Cao L, DeLeo JA. CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection-induced neuropathic pain. Eur J Immunol. (2008) 38:448–58. doi: 10.1002/eji.200737485
20. Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci. (2009) 29:14415–22. doi: 10.1523/JNEUROSCI.4569-09.2009
21. Du B, Ding YQ, Xiao X, Ren HY, Su BY, Qi JG. CD4+ αβ T cell infiltration into the leptomeninges of lumbar dorsal roots contributes to the transition from acute to chronic mechanical allodynia after adult rat tibial nerve injuries. J Neuroinflammation. (2018) 15:81. doi: 10.1186/s12974-018-1115-7
22. Kleinschnitz C, Hofstetter HH, Meuth SG, Braeuninger S, Sommer C, Stoll G. T cell infiltration after chronic constriction injury of mouse sciatic nerve is associated with interleukin-17 expression. Exp Neurol. (2006) 200:480–5. doi: 10.1016/j.expneurol.2006.03.014
23. Kim CF, Moalem-Taylor G. Interleukin-17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury in mice. J Pain. (2011) 12:370–83. doi: 10.1016/j.jpain.2010.08.003
24. Luo H, Liu HZ, Zhang WW, Matsuda M, Lv N, Chen G, et al. Interleukin-17 regulates neuron-glial communications, synaptic transmission, and neuropathic pain after chemotherapy. Cell Rep. (2019) 29:2384–97.e5. doi: 10.1016/j.celrep.2019.10.085
25. Vicuña L, Strochlic DE, Latremoliere A, Bali KK, Simonetti M, Husainie D, et al. The serine protease inhibitor SerpinA3N attenuates neuropathic pain by inhibiting T cell-derived leukocyte elastase. Nat Med. (2015) 21:518–23. doi: 10.1038/nm.3852
26. Sweitzer S. The differential role of spinal MHC class II and cellular adhesion molecules in peripheral inflammatory versus neuropathic pain in rodents. J Neuroimmunol. (2002) 125:82–93. doi: 10.1016/S0165-5728(02)00036-X
27. Dominguez CA, Kalliomäki M, Gunnarsson U, Moen A, Sandblom G, Kockum I, et al. The DQB1 *03:02 HLA haplotype is associated with increased risk of chronic pain after inguinal hernia surgery and lumbar disc herniation. Pain. (2013) 154:427–33. doi: 10.1016/j.pain.2012.12.003
28. Ding YQ, Luo H, Qi JG. MHCII-restricted T helper cells: an emerging trigger for chronic tactile allodynia after nerve injuries. J Neuroinflammation. (2020) 17:3. doi: 10.1186/s12974-019-1684-0
29. Mangiacavalli S, Corso A, Amici M, de Varettoni M, Alfonsi E, Lozza A, et al. Emergent T-helper 2 profile with high interleukin-6 levels correlates with the appearance of bortezomib-induced neuropathic pain. Br J Haematol. (2010) 149:916–8. doi: 10.1111/j.1365-2141.2010.08138.x
30. Luchting B, Rachinger-Adam B, Heyn J, Hinske LC, Kreth S, Azad SC. Anti-inflammatory T-cell shift in neuropathic pain. J Neuroinflammation. (2015) 12:12. doi: 10.1186/s12974-014-0225-0
31. Uçeyler N, Rogausch JP, Toyka KV, Sommer C. Differential expression of cytokines in painful and painless neuropathies. Neurology. (2007) 69:42–9. doi: 10.1212/01.wnl.0000265062.92340.a5
32. Leger T, Grist J, D'Acquisto F, Clark AK, Malcangio M. Glatiramer acetate attenuates neuropathic allodynia through modulation of adaptive immune cells. J Neuroimmunol. (2011) 234:19–26. doi: 10.1016/j.jneuroim.2011.01.005
33. Krukowski K, Eijkelkamp N, Laumet G, Hack CE, Li Y, Dougherty PM, et al. CD8+ T cells and endogenous IL-10 are required for resolution of chemotherapy-induced neuropathic pain. J Neurosci. (2016) 36:11074–83. doi: 10.1523/JNEUROSCI.3708-15.2016
34. Siqueira Mietto B, Kroner A, Girolami EI, Santos-Nogueira E, Zhang J, David S. Role of IL-10 in resolution of inflammation and functional recovery after peripheral nerve injury. J Neurosci. (2015) 35:16431–42. doi: 10.1523/JNEUROSCI.2119-15.2015
35. Laumet G, Edralin JD, Dantzer R, Heijnen CJ, Kavelaars A. Cisplatin educates CD8+ T cells to prevent and resolve chemotherapy-induced peripheral neuropathy in mice. Pain. (2019) 160:1459–68. doi: 10.1097/j.pain.0000000000001512
36. Liu XJ, Zhang Y, Liu T, Xu ZZ, Park CK, Berta T, et al. Nociceptive neurons regulate innate and adaptive immunity and neuropathic pain through MyD88 adapter. Cell Res. (2014) 24:1374–7. doi: 10.1038/cr.2014.106
37. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
38. Davoli-Ferreira M, Lima KA, de Fonseca MM, Guimarães RM, Gomes FI, Cavallini MC, et al. Regulatory T cells counteract neuropathic pain through inhibition of the Th1 response at the site of peripheral nerve injury. Pain. (2020) 161:1730–43. doi: 10.1097/j.pain.0000000000001879
39. Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. (2009) 15:192–9. doi: 10.1038/nm.1927
40. Duffy SS, Keating BA, Perera CJ, Lees JG, Tonkin RS, Makker PGS, et al. Regulatory T cells and their derived cytokine, interleukin-35, reduce pain in experimental autoimmune encephalomyelitis. J Neurosci. (2019) 39:2326–46. doi: 10.1523/JNEUROSCI.1815-18.2019
41. Sloane E, Ledeboer A, Seibert W, Coats B, van Strien M, Maier SF, et al. Anti-inflammatory cytokine gene therapy decreases sensory and motor dysfunction in experimental multiple sclerosis: MOG-EAE behavioral and anatomical symptom treatment with cytokine gene therapy. Brain Behav Immun. (2009) 23:92–100. doi: 10.1016/j.bbi.2008.09.004
42. Grace PM, Loram LC, Christianson JP, Strand KA, Flyer-Adams JG, Penzkover KR, et al. Behavioral assessment of neuropathic pain, fatigue, and anxiety in experimental autoimmune encephalomyelitis (EAE) and attenuation by interleukin-10 gene therapy. Brain Behav Immun. (2017) 59:49–54. doi: 10.1016/j.bbi.2016.05.012
43. Milligan ED, Langer SJ, Sloane EM, He L, Wieseler-Frank J, O'Connor K, et al. Controlling pathological pain by adenovirally driven spinal production of the anti-inflammatory cytokine, interleukin-10. Eur J Neurosci. (2005) 21:2136–48. doi: 10.1111/j.1460-9568.2005.04057.x
44. Ledeboer A, Jekich BM, Sloane EM, Mahoney JH, Langer SJ, Milligan ED, et al. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain Behav Immun. (2007) 21:686–98. doi: 10.1016/j.bbi.2006.10.012
45. Shen KF, Zhu HQ, Wei XH, Wang J, Li YY, Pang RP, et al. Interleukin-10 down-regulates voltage gated sodium channels in rat dorsal root ganglion neurons. Exp Neurol. (2013) 247:466–75. doi: 10.1016/j.expneurol.2013.01.018
46. Fischer R, Proske M, Duffey M, Stangl H, Martinez GF, Peters N, et al. Selective activation of tumor necrosis factor receptor II induces antiinflammatory responses and alleviates experimental arthritis. Arthritis Rheumatol. (2018) 70:722–35. doi: 10.1002/art.40413
47. Lamontain V, Schmid T, Weber-Steffens D, Zeller D, Jenei-Lanzl Z, Wajant H, et al. Stimulation of TNF receptor type 2 expands regulatory T cells and ameliorates established collagen-induced arthritis in mice. Cell Mol Immunol. (2019) 16:65–74. doi: 10.1038/cmi.2017.138
48. Chopra M, Biehl M, Steinfatt T, Brandl A, Kums J, Amich J, et al. Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J Exp Med. (2016) 213:1881–900. doi: 10.1084/jem.20151563
49. Fischer R, Padutsch T, Bracchi-Ricard V, Murphy KL, Martinez GF, Delguercio N, et al. Exogenous activation of tumor necrosis factor receptor 2 promotes recovery from sensory and motor disease in a model of multiple sclerosis. Brain Behav Immun. (2019) 81:247–59. doi: 10.1016/j.bbi.2019.06.021
50. Dombrowski Y, O'Hagan T, Dittmer M, Penalva R, Mayoral SR, Bankhead P, et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat Neurosci. (2017) 20:674–80. doi: 10.1038/nn.4528
51. Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel ULM. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J Biol Chem. (2004) 279:32869–81. doi: 10.1074/jbc.M311766200
52. Fischer R, Maier O, Siegemund M, Wajant H, Scheurich P, Pfizenmaier K. A TNF receptor 2 selective agonist rescues human neurons from oxidative stress-induced cell death. PLoS ONE. (2011) 6:e27621. doi: 10.1371/journal.pone.0027621
53. Dong Y, Fischer R, Naudé PJW, Maier O, Nyakas C, Duffey M, et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc Natl Acad Sci USA. (2016) 113:12304–9. doi: 10.1073/pnas.1605195113
54. Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, et al. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci USA. (2003) 100:7947–52. doi: 10.1073/pnas.1331358100
55. Huang ZZ, Li D, Liu CC, Cui Y, Zhu HQ, Zhang WW, et al. CX3CL1-mediated macrophage activation contributed to paclitaxel-induced DRG neuronal apoptosis and painful peripheral neuropathy. Brain Behav Immun. (2014) 40:155–65. doi: 10.1016/j.bbi.2014.03.014
56. Old EA, Nadkarni S, Grist J, Gentry C, Bevan S, Kim K-W, et al. Monocytes expressing CX3CR1 orchestrate the development of vincristine-induced pain. J Clin Invest. (2014) 124:2023–36. doi: 10.1172/JCI71389
57. Montague K, Simeoli R, Valente J, Malcangio M. A novel interaction between CX3CR1 and CCR2 signalling in monocytes constitutes an underlying mechanism for persistent vincristine-induced pain. J Neuroinflammation. (2018) 15:101. doi: 10.1186/s12974-018-1116-6
58. Peng J, Gu N, Zhou LB, Eyo U, Murugan M, Gan WB, et al. Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat Commun. (2016) 7:12029. doi: 10.1038/ncomms12029
59. Yu X, Liu H, Hamel KA, Morvan MG, Yu S, Leff J, et al. Dorsal root ganglion macrophages contribute to both the initiation and persistence of neuropathic pain. Nat Commun. (2020) 11:264. doi: 10.1038/s41467-019-13839-2
60. Hu P, McLachlan EM. Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience. (2002) 112:23–38. doi: 10.1016/S0306-4522(02)00065-9
61. Hu P, Bembrick AL, Keay KA, McLachlan EM. Immune cell involvement in dorsal root ganglia and spinal cord after chronic constriction or transection of the rat sciatic nerve. Brain Behav Immun. (2007) 21:599–616. doi: 10.1016/j.bbi.2006.10.013
62. Liu P, Peng J, Han GH, Ding X, Wei S, Gao G, et al. Role of macrophages in peripheral nerve injury and repair. Neural Regen Res. (2019) 14:1335–42. doi: 10.4103/1673-5374.253510
63. Pannell M, Labuz D, Celik MÖ, Keye J, Batra A, Siegmund B, et al. Adoptive transfer of M2 macrophages reduces neuropathic pain via opioid peptides. J Neuroinflammation. (2016) 13:262. doi: 10.1186/s12974-016-0735-z
64. Takahashi Y, Hasegawa-Moriyama M, Sakurai T, Inada E. The macrophage-mediated effects of the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone attenuate tactile allodynia in the early phase of neuropathic pain development. Anesth Analg. (2011) 113:398–404. doi: 10.1213/ANE.0b013e31821b220c
65. Kiguchi N, Kobayashi Y, Saika F, Sakaguchi H, Maeda T, Kishioka S. Peripheral interleukin-4 ameliorates inflammatory macrophage-dependent neuropathic pain. Pain. (2015) 156:684–93. doi: 10.1097/j.pain.0000000000000097
66. Tonello R, Xie W, Lee SH, Wang M, Liu X, Strong JA, et al. Local sympathectomy promotes anti-inflammatory responses and relief of paclitaxel-induced mechanical and cold allodynia in mice. Anesthesiology. (2020) 132:1540–53. doi: 10.1097/ALN.0000000000003241
67. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget. (2016) 7:52294–306. doi: 10.18632/oncotarget.10561
68. Celik MÖ, Labuz D, Keye J, Glauben R, Machelska H. IL-4 induces M2 macrophages to produce sustained analgesia via opioids. JCI Insight. (2020) 5:e133093. doi: 10.1172/jci.insight.133093
69. Berkley KJ. Sex differences in pain. Behav Brain Sci. (1997) 20:371–80; discussion 435–513. doi: 10.1017/S0140525X97221485
70. Fillingim RB, King CD, Ribeiro-Dasilva MC, Rahim-Williams B, Riley JL. Sex, gender, and pain: a review of recent clinical and experimental findings. J Pain. (2009) 10:447–85. doi: 10.1016/j.jpain.2008.12.001
71. Sorge RE, Mapplebeck JCS, Rosen S, Beggs S, Taves S, Alexander JK, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. (2015) 18:1081–3. doi: 10.1038/nn.4053
72. Sorge RE, Totsch SK. Sex differences in pain. J Neurosci Res. (2017) 95:1271–81. doi: 10.1002/jnr.23841
Keywords: chronic neuropathic pain, immune cells, T cells, Tregs, recovery, macrophages
Citation: Bethea JR and Fischer R (2021) Role of Peripheral Immune Cells for Development and Recovery of Chronic Pain. Front. Immunol. 12:641588. doi: 10.3389/fimmu.2021.641588
Received: 14 December 2020; Accepted: 03 February 2021;
Published: 22 February 2021.
Edited by:
Michael D. Burton, The University of Texas at Dallas, United StatesReviewed by:
Temugin Berta, University of Cincinnati, United StatesPrapti Mody, The University of Texas at Dallas, United States
Copyright © 2021 Bethea and Fischer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roman Fischer, cm9tYW4uZmlzY2hlckBpemkudW5pLXN0dXR0Z2FydC5kZQ==