 Amber G. Bozward
Amber G. Bozward Frazer Warricker
Frazer Warricker Ye H. Oo
Ye H. Oo Salim I. Khakoo
Salim I. Khakoo- 1Centre for Liver and Gastroenterology Research and National Institute for Health Research Biomedical Research Centre (NIHR BRC) Birmingham, Institute of Immunology and Immunotherapy, University of Birmingham, Birmingham, United Kingdom
- 2Centre for Rare Diseases, European Reference Network Centre- Rare Liver, Birmingham, United Kingdom
- 3The School of Clinical and Experimental Sciences, Faculty of Medicine, University of Southampton, Southampton, United Kingdom
- 4NIHR Biomedical Research Centre, The School of Clinical and Experimental Sciences, University of Southampton, Southampton, United Kingdom
- 5Liver Transplant and Hepatobiliary Unit, University Hospital of Birmingham NHS Foundation Trust, Birmingham, United Kingdom
Despite major advances in immunotherapy, hepatocellular carcinoma (HCC) remains a challenging target. Natural Killer (NK) cells are crucial components of the anti-HCC immune response, which can be manipulated for immunotherapeutic benefit as primary targets, modulators of the tumour microenvironment and in synchronising with tumour antigen specific effector CD8 cells for tumour clearance. Regulatory T cells shape the anti-tumour response from effector T cells via multiple suppressive mechanisms. Future research is needed to address the development of novel NK cell-targeted immunotherapy and on restraining Treg frequency and function in HCC. We have now entered a new era of anti-cancer treatment using checkpoint inhibitor (CPI)-based strategies. Combining GMP-NK cell immunotherapy to enhance the frequency of NK cells with CPI targeting both NK and CD8 T cells to release co-inhibitory receptors and enhance the cells anti-tumour immunity of HCC would be an attractive therapeutic option in the treatment of HCC. These therapeutic approaches should now be complemented by the application of genomic, proteomic and metabolomic approaches to understanding the microenvironment of HCC which, together with deep immune profiling of peripheral blood and HCC tissue before and during treatment, will provide the much-needed personalised medicine approach required to improve clinical outcomes for patients with HCC.
The Liver as an Organ of Immunotolerance
The liver is a unique lymphoid organ which plays a key role in the immunological function of the human body. Embryologically, the human liver is derived from the endoderm layer and resides between two venous circulatory systems; the portal vein, receiving venous flow from the gastrointestinal tract and the systemic venous circulation. The liver has a unique immunological environment containing both professional antigen presenting cells, dendritic cells and Kupffer cells (resident hepatic macrophages) as well as non-professional antigen presenting cells (sinusoidal endothelial cells and biliary epithelium) (1). There is also an abundance of natural killer (NK) cells, innate lymphoid cells (ILCs) and innate mucosa associate invariant T (MAIT) cells, all of which play integral roles in the innate immune response of the liver.
The liver is constantly filtering harmful and harmless antigens from the gut acting as an immunological firewall (1). Hepatic tolerance to gut antigens is achieved by a combination of both immune cells and parenchymal cells. The constant exposure to gut-derived bacteria triggers a downregulation of Toll-like receptor 4 (TLR4) on the hepatic sinusoidal endothelial cells (HSEC). Liver-resident dendritic cells (DC) have distinct properties that promote tolerance rather than an immune response. These tolerogenic DCs secrete anti-inflammatory cytokines such as interleukin 10 (IL-10) and transforming growth factor β (TGF-β) to dampen the immune response and can promote T-cell “hyporesponsiveness” (2, 3). Kupffer cells (KCs), known as sinusoidal firewalls, also contribute to hepatic tolerance by continuously phagocytosing the microbial products from the portal vein (4). Hepatic regulatory T cells (Treg) also play a crucial role in maintaining the tolerogenic environment by continuously controlling the cytokine production and proliferation of intrahepatic auto-reactive effector CD4 and CD8 T cells (5).
The liver is known as a “graveyard” of immune cells due to apoptosis of activated lymphocyte populations (6). It can mount an effective immune response to invading pathogens and cancer cells or when there is insult or loss of peripheral self-tolerance in immune-mediated liver injury. The balance between immunity and tolerance is established by competition for primary activation of effector T cells between the liver and its draining lymphoid tissues. For example, naive CD8+ T cells, activated within liver-draining portal lymph nodes are capable of mediating hepatitis, while cells undergoing primary activation within the liver exhibit defective cytotoxic function and do not induce hepatocellular injury (7). The hepatic immune response will depend on the nature of the injury. In acute hepatic injury due to, for example, viral infection or drug-induced liver injury, an innate immune cells infiltrate appears important, with eosinophil or neutrophils and natural killer (NK) cells being the predominant immune cells. Adaptive immune cells are dominant in chronic injury resulting from, for example, chronic hepatitis B and C infection, alcoholic and non-alcoholic steatohepatitis, or autoimmune hepatitis. In the context of HCC, both innate cells such as NK cells and adaptive T cells are involved in auto-tumour immunity. In general, the balance of effector and regulatory T cells determines the outcome of inflammation, either resolution or chronic active hepatitis (8).
Hepatocellular Carcinoma
In 2018, HCC was the sixth most common neoplasm diagnosed globally and was the fourth leading cause of cancer related death. In the vast majority of cases, primary hepatocellular carcinoma (HCC) arises on a background of cirrhosis, driven by chronic inflammation from a number of causes. These include viral (hepatitis B and hepatitis C) and non-viral (non-alcoholic fatty liver disease and alcoholic liver disease). Improved treatments for chronic viral hepatitis, coupled with the global epidemic of obesity imply that in the coming years the global epidemiology of HCC may shift from infectious to non-infectious causes.
The recent observation that hepatocytes within cirrhotic nodules have a higher mutational load than normal liver characterizes cirrhosis as a truly pre-malignant condition (9). Coexistent with this is the immune dysfunction associated with cirrhosis. This is characterized by evidence of peripheral activation of circulating immunocytes exposed to higher than normal levels of bacterial antigens, but a more profound central tolerance. A healthy liver is a tolerogenic organ, however this state is exacerbated by changes in immune sub-populations and their dysfunction in cirrhosis. The combination of an increased hepatic mutational burden together with decreased immune surveillance underpins the development of HCC.
Immune Dysfunction in Cirrhosis
As described above, in a healthy liver, immune system homeostasis is achieved through immunosurveillance of its dual blood supply (portal vein and hepatic artery), protecting the host from microbe-associated molecular patterns and damage-associated molecular patterns (MAMPs and DAMPs respectively) from the gut (10). Concurrently the liver displays features of local immune tolerance to non-pathogenic material and helps mediate the appropriate immune response through the synthesis of pro-inflammatory and anti-inflammatory cytokines (11). The immune system thus plays a decisive role in both the pathogenesis of cirrhosis and subsequent immune dysfunction. Chronic factors including infection, alcohol and obesity, inflict persistent hepatocyte damage leading to fibrosis via hepatic stellate cell (HSC) activation (12). Disease specific alterations may compound these factors and augment the rate of fibrosis progression. Once cirrhosis becomes established, the liver loses its ability to appropriately protect the body from pathogens as a consequence of disordered immune cell activation termed “cirrhosis-associated immune dysfunction”.
In early cirrhosis, changes in the intrahepatic immune compartment are often due to persistent innate immune cell stimulation. As disease progression ensues, ultimately leading to decompensated cirrhosis, immune hyporesponsiveness and increased tolerance develops (13). This is driven by the innate immune system in which long-term exposure of toll-like receptors to bacterial products such as lipopolysaccharide, can lead to a dampened innate immune response (14). Cirrhosis is associated with a multitude of abnormalities in the innate and adaptive arms of the immune system leading to a generalised immune hyporesponsiveness. The reticulo-endothelial system becomes compromised in the context of cirrhosis as a result of fibrotic damage to the sinusoidal vascular space. This leads to capillarisation, porto-systemic shunts and loss of the KC population, which are also dysfunctional, with impaired phagocytosis (10, 15). Consequently, the capacity for clearance of endotoxin and microbes is attenuated, lowering the threshold for bacterial infection. This dysfunction may be exacerbated by a combination of changes in the gut microbiota and increased intestinal permeability (16). Evidence for this persistent immune stimulation is supported by both human disease studies and mouse models (17). Cirrhotic livers possess a reduced ability to synthesise innate immune proteins, such as complement and pattern recognition receptors thus reducing its bactericidal capacity. This is further exacerbated by reduced numbers of MAIT cells which have the capacity to respond directly to bacterial metabolites (18, 19).
Compromised immune function not only occurs at a local level in cirrhosis, but also systemically, and affects many different immune cell sub-populations. Neutrophils are reduced, with impaired chemotaxis and subsequent phagocytosis of bacteria as illustrated by a lower response to peptidoglycan recognition proteins (20). Monocytosis is observed in cirrhotic patients with evidence of cellular dysfunction, such as impaired function of the Fc-γ receptors, which are responsible for the clearance of bacteria (21). In alcohol and hepatitis C virus (HCV) related cirrhosis, a reduced frequency of CD27+ memory B cell function suggests defective antibody function (22). T cell defects have also been noted including T cell lymphopenia affecting both CD4 and CD8 T cells. There is depletion of naïve and memory T cells, although naïve T cells appear to be more profoundly affected putatively related to splenic pooling, and there is also impaired proliferation of peripheral T lymphocytes (23). NK cells are shown to be lower in number in the periphery but also less responsive to cytokine stimuli which hampers their cytotoxic and anti-fibrotic roles (24). Within the intrahepatic compartment, innate lymphoid cells type 2 (ILC-2) cells appear to be the dominant ILC population (25, 26). The observed changes in immune cell function alters the equilibrium of immunosurveillance and immunotolerance within the liver, promoting the latter through relative immune deficiency. Due to the observed microenvironmental shift towards immunotolerance, there is an increase in host vulnerability to tumorigenesis and the occurrence of HCC.
Natural Killer Cells
Natural Killer (NK) cells are a key part of the innate immune response against viruses and tumours, and more recently, have been shown to participate in the adaptive immune response through cross-talk with dendritic cells and T cells. They make up between 5-20% of circulating lymphocytes, but a much larger fraction (~50%) of the intrahepatic lymphocyte compartment. NK cells are usually characterised as CD3-CD56+ lymphocytes and in general do not require priming to initiate anti-viral or anti-tumoral cytotoxic or cytokine secretory effects (27). Recent work has identified that they also have adaptive properties (28), which can be both receptor or cytokine driven. Particular interest has been generated in the utility of cytokine-induced memory NK cells as agents for immunotherapy (29).
Surveillance of diseased cells can be mediated through the ‘missing self’ model (loss of inhibition) or through recognition of stress-induced molecules (gain of activation) (30). The net effects of both of these mechanisms is a change in the balance between activating and inhibitory signals transduced by the NK cell such that activation is favoured. Both these mechanisms may operate in cancer. In the missing-self model, healthy cells which express major histocompatibility complex (MHC) class I are spared from lysis through the engagement of inhibitory receptors on the NK cell surface, such as killer cell immunoglobulin-like receptors (KIR) or CD94:NKG2A. Thus if MHC class I is downregulated, the tonic inhibitory signal to the NK cell is lost and the cell becomes activated (31). Conversely NK cells can be activated by augmenting activating signals. Key activating receptors include the natural cytotoxicity receptors (NKp30, NKp44 and NKp46) and C-type lectin-like receptors, especially NKG2D. In stressed, transformed or infected cells MHC class I is often downregulated and ligands for NKG2D are up-regulated, shifting the NK cell balance towards activation and tumour lysis (31, 32).
The KIR are MHC class I-specific receptors that perform a fundamental role in self-recognition and in functional “licensing” of NK cells. The tuning of the activity of NK cells may be a more dynamic process than previously considered, which is relevant for NK cells within immunosuppressive tumour microenvironments (33, 34), resulting in induced-hyporesponsiveness. The KIR exhibit an extraordinarily high level of diversity at the gene content and allelic levels. In combination with the diversity of MHC class I ligands, the KIR form a complex immunogenetic network, which has been associated with development and outcomes of cancer. The KIR gene family is found on chromosome 19 and comprises 13 functional KIR genes and 2 pseudogenes (35). Two haplotypes KIR-A and KIR-B have been identified with the former having a fixed gene content but substantial allelic diversity, and the latter haplotype displaying variation in gene content and also allelic diversity. There also is substantial diversity in the frequencies of KIR-A and KIR-B haplotypes amongst different human populations (36). This has been proposed to account for some of the diversity noted in anti-cancer responses. For instance, the activating KIR, KIR2DS2, has been associated with protective responses against acute myeloid leukaemia and other solid tumours including HCC (37, 38). Interestingly, KIR2DS2+ NK cells appear to express higher amounts of FcγRIII (CD16), a medium-low affinity IgG receptor essential for antibody-dependent cellular cytotoxicity, providing a potential molecular basis for enhanced protection (39). Improved HCC outcomes have been observed in individuals with different KIR : HLA genotypes including HLA-C group 1, KIR2DS5 and the compound genotypes KIR2DL2: HLA-C group 1, KIR3DS1:HLA-Bw480T and KIR3DS1:HLA-BBw480I (38, 40).
Hepatocellular Carcinoma and Natural Killer Cells
In comparison to other cancers, HCC is relatively cold immunologically with only about 25% having an immune reactive phenotype (41, 42). Individuals with HCC have reduced numbers of NK cells within the periphery and these have lower levels of functionality (43). Within the tumour they are present at low frequencies in contrast to myeloid and other lymphoid cells (44). In HCC accumulation of NK cells in intratumoral tissue, as compared to peritumoural tissue, has increased expression of the activation marker CD49a and an increased CD56bright:CD56dim ratio, demonstrating localised differences in these two subtly distinct microenvironments (45). Individuals with higher frequencies of NK cells and enhanced cytotoxic and cytokine secretory functions have improved overall survival in HCC following liver resection (46–48). Patient survival also positively correlates with the frequencies of both circulating and intratumoral NK cells in HCC (49), and in two separate studies the response to sorafenib was better if a higher frequency of intratumoral NK cells was present (48, 50). Conversely, overall survival is worse in individuals with fewer intratumoral NK cells and a higher proportion of CD56bright to CD56dim NK cells. The CD56bright sub-population are considered less mature and have lower levels of cytotoxicity than the CD56dim subpopulation, indicating that NK cell functionality is important in determining the outcome of HCC.
NK cells express multiple activating receptors and therefore can engage many different molecules expressed by tumours. Changes in the balance of expression of activating and inhibitory receptors can determine NK cell function in a “rheostat” model. Thus, in advanced HCC there may be upregulation of the inhibitory receptor NKG2A and this, combined with a reduction in the effector molecules perforin and granzyme B, contributes to a hypofunctionality of NK cells (43, 46). Down-regulation of granzyme B is a feature of intra-tumoral, as opposed to peri-tumoral, NK cells and correlates with expression of IL-10-positive tumour-associated macrophages (51). This immunosuppressive gradient also correlates with expression of exhaustion-associated markers such as PD-1, Tim-3, and Lag-3. In addition to modulating T cell functions these molecules may act as checkpoints for NK cells and are often co-expressed on activated or exhausted NK cells (52). CD96 (TACTILE) is another immunological checkpoint associated with HCC, that in combination with its ligand negatively associates with the outcome of HCC (53). Thus, together this group of checkpoints form potential therapeutic targets for HCC.
A number of activating NK cell receptors have been associated with HCC. Differential splicing of NKp30 leads to preferential generation of an inhibitory isoform that predominates in advanced HCC (54). However, most attention has focussed on NKG2D which engages multiple ligands including MIC-A/B and the ULBP proteins. These stress-induced proteins are express on tumours and may be released into the circulation following proteolytic cleavage, through molecules including ADAM9, which is upregulated in HCC (55). Soluble receptors bind and down-regulate NKG2D on the surface of NK cells thus rendering cells less active. In HCC high levels of soluble ULBP1 is associated with poor survival (56). The observation that in mouse models of HCC, NKG2D can drive tumorigenesis, probably through promotion of the chronic inflammation that leads to mutagenesis, indicates the complexity of the molecular pathology of this disease (57, 58). Nevertheless, in general down-regulation of the NKG2D:NKG2D ligand axis is associated with poorer outcomes (59).
Regulatory T Cells
Sakaguchi and colleagues first described regulatory T cells (Tregs) in the late 1990s (60). Tregs are generated in the thymus. They are a subset of CD4 T cells, expressing high levels of CD25 (the α-chain of the IL-2 receptor) and low levels of IL-7 receptor (CD127). Thus, Tregs are defined by surface receptors as CD4, CD25high, CD127low (61). Tregs are crucial in maintaining peripheral immune tolerance (62). Their phenotype and function are controlled by the transcription factor Foxp3 (63). Tregs represent 2–5% of CD4 T cells in humans and about 10% in rodents.
Tregs are present in the human liver. Our group has previously demonstrated that Tregs reside together with effector CD4 and CD8 T cells, CD11c dendritic cells in both interface and lobular hepatitis areas to control liver inflammation (5). The suppressive function of Treg is reduced in the inflamed microenvironment (64). The main function of Tregs is to control autoreactive effector T cells, thereby maintaining hepatic immune tolerance. We have shown that Treg recruitment via hepatic sinusoids to inflamed human liver is mediated by the chemokine receptor CXCR3 and the integrin VLA-4 on Tregs and the chemokine ligands CXCL9, 10, 11 and VCAM expression on inflamed sinusoids. Following recruitment, post endothelial migration through the fibrous stromal framework occurs via LFA-1 and VLA-4 integrin’s on Treg and cell adhesion molecules ICAM and VCAM on the stroma cells, and subsequently Tregs reside around the area of hepatitis to control liver inflammation (5). Their suppressive function in the human liver is mainly executed via CTLA-4, CD39 or IL-10 dependent mechanisms and low dose IL-2 can upregulate functional CTLA-4 on the surface of Tregs surface via STAT-5 (65, 66).
Tregs have the potential to be plastic towards effector Th1 or Th17 lineages especially in the inflamed human liver and tumour microenvironment (8). The immune response mediated by T lymphocytes plays an important role in anti-tumour immunity. Tregs secrete immunosuppressive cytokines such as IL-10 and IL-35 to suppress the aberrant immune response, while Th1/Treg and Th17/Treg cells can release not only anti-inflammatory cytokines but also proinflammatory Th1 cytokines such as IFNγ, TNFα, and Th17 cytokines IL-17, IL-22 (67, 68). The ratio of Treg to Th17 cells is closely associated with the outcome of many immune mediated diseases and cancers (69).
Hepatocellular Carcinoma (HCC) and Tregs
Multiple studies have reported that the frequency of CD4+CD25++Treg cells in the peripheral blood of HCC patients is significantly higher than in the blood of healthy individuals (70, 71). CD4+CD25++Tregs in advanced (stage III-IV) HCC patients is significantly increased compared to early (stage I-II) HCC patients, implying that the presence of CD4+CD25++Tregs is closely related to tumour progression and enhances the invasiveness and metastasis of HCC (71). In addition, accumulation of Tregs correlates with reduced infiltration of CD8 T cells in HCC tumour regions, and the expression of granzymes and perforin functional molecules is less in tumour-infiltrating CD8 T cells (72). Furthermore, in this study an increased quantity of circulating Treg was associated with a high mortality and reduced survival time of HCC patients.
CD4+CD25++Tregs suppress the anti-tumour immune response either in the draining lymph nodes or in tumour tissue. CD4+CD25++Tregs in the tumour-draining regional lymph node inhibit the proliferation of effector T cells, and Tregs prevent effector T cells from killing tumour cells in the primary tumour tissue (73). It has been shown that the level of Treg cells in cancer tissue was significantly higher than that in adjacent tissues (74). It is thought that Treg cells may decrease the proliferation of effector CD4 and CD8 T lymphocytes in the tumour microenvironment by contact inhibition, subsequently reducing the anti-tumour immune response and resulting in the potential for tumour cells to escape immune surveillance. HCC is one of the most common and aggressive human malignancies and CD4+CD25++Treg promote hepatocellular carcinoma invasion via TGF-β1 dependent mechanisms (75). Furthermore, ICOS+ FOXP3+ Tregs contribute to the immunosuppressive HCC microenvironment and lead to an unfavourable prognosis for HCC patients (76). Thus, removal or reduction of the Treg cell population, or inhibition of CD4+CD25++Treg function in the HCC microenvironment may facilitate the efficacy of tumour immunotherapy (77). In addition, the Th17/Treg ratio is a risk factor for HCC (78). The majority of expanded Tregs do not express CD45RA suggesting that Tregs have a memory phenotype and exponential expansion of these tumour antigen exposed memory Tregs has been a distinct finding in HCC (79).
Treg and NK Cell Interactions in Hepatocellular Carcinoma
The balance of NK cells which provide tumour clearance and Tregs which inhibit tumour immunity may determine the outcome of HCC (Figure 1). A potential role for Tregs in dampening NK cell functions was first suggested in a murine leukaemia model. In this model, the depletion of Tregs by administration of anti‐CD25 mAb before tumour inoculation abolished tumour growth and promoted the generation of cytotoxic cells characterized as NK cells (80). In HCC, the NK cell frequency within the tumour-infiltrating lymphocyte compartment is less than in the non-tumour tissue, and these intratumoral cells demonstrate impaired cytotoxicity and IFN-γ production (43, 81), We have also previously demonstrated that there is a parallel increase in NK cells and Treg in hepatic inflammation (82) suggesting that taming the Treg population will allow NK cells to function more efficiently.
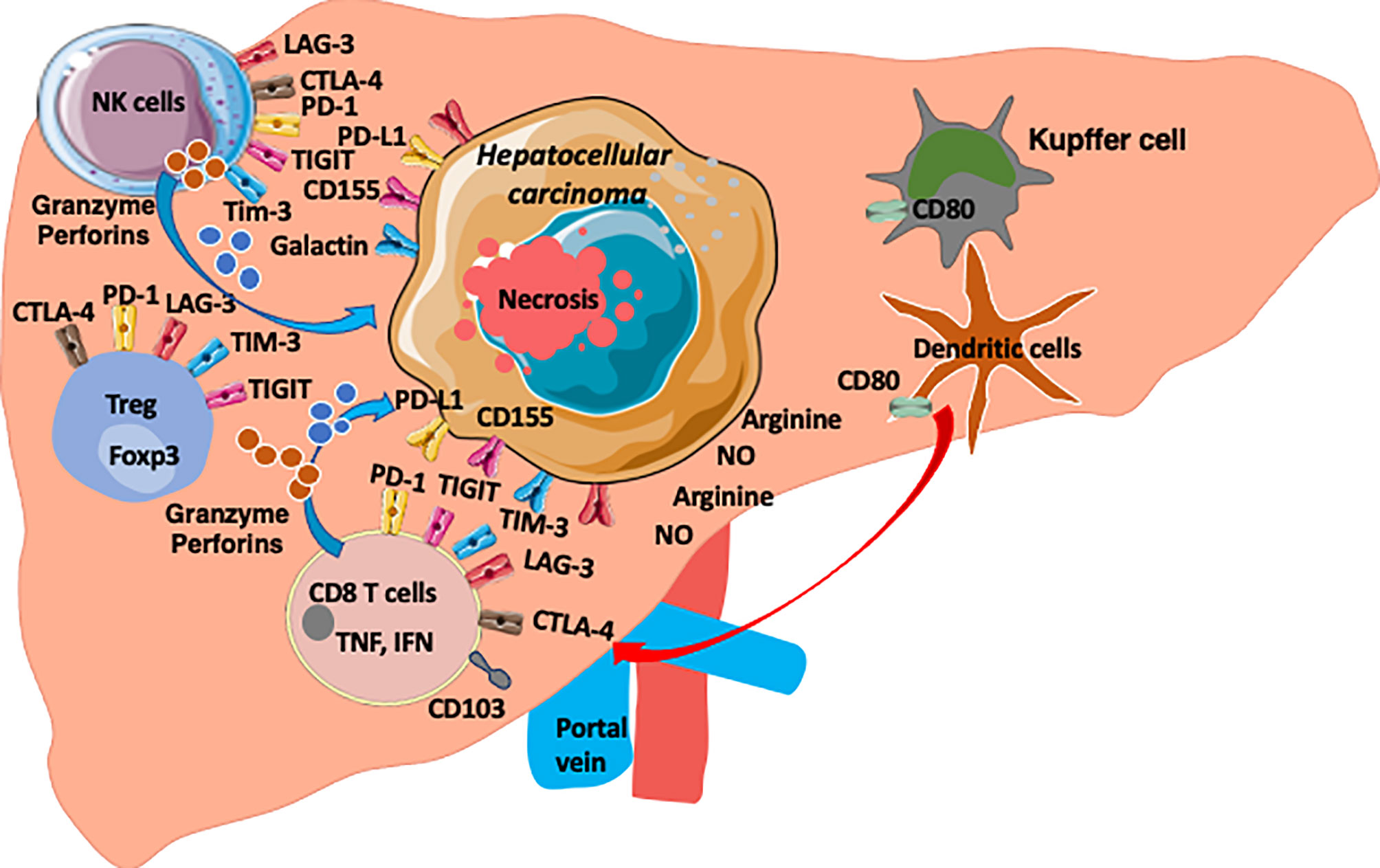
Figure 1 The HCC microenvironment is enriched with anti-tumour CD8 T cells, NK cells and Treg cells. Both T cells and NK cells express checkpoint molecules (PD1, TIGIT, TIM3, LAG3 and CTLA-4) as common targets. HCC express ligands for these checkpoints (PDL1, CD155, Galactin). Arginine and Nitrous oxide are also enriched in HCC environment and the microenvironment is hypoxic environment and enriched with chemokines, cytokines, metabolites and microbial products (8).
Freshly isolated human Tregs can directly inhibit human NK cell cytotoxicity against K562 (83). TGF-β maintains the inhibitory functions of Treg and plays a suppressive role by inhibiting the expansion of NK cells and their cytotoxic functions (84, 85). TGF-β can also have a negative effect by facilitating the onset of tumours due to a reduction of immunosurveillance and anti-cancer responses. Resting NK cells harbour surface expression of TGF‐β receptors, rendering them susceptible to soluble TGF‐β (86). Resting human Tregs express membrane‐bound TGF‐β that is associated with the protein, latency‐associated protein (LAP) (87). When associated with LAP, TGF‐β remains inactive. Membrane‐bound TGF‐β is involved in the inhibitory function of Tregs on NK cells since anti‐TGF‐β blocking antibodies can restore the cytotoxicity IL‐12‐induced IFN‐γ secretion of human NK cells (88). When exposed to TGF‐β or on Treg encounter, Smad signalling in NK cells blunts expression of cytotoxic molecules such as granzyme B and perforin (89). In addition, high levels of TGF-β have been associated with impaired NK cell function and NKG2D expression (90). Therefore, Tregs bearing TGF-β on their membrane can present it directly to NK cells resulting in a reduction in NKG2D expression (88). Activated Tregs can also suppress NK cells responses via an IL-2 mediated mechanism that is crucial for NK cell survival (91). This reduces the capacity of NK cells to secrete IFN‐γ on stimulation with IL‐12 but not on activation by IL‐2 and IL‐15 suggesting the regulation of NK cell control by Tregs is critically dependent on the cytokine milieu in the HCC microenvironment.
The Influence of the Tumour Microenvironment on NK Cell Function
The intrahepatic microenvironment is crucial for both NK and Treg phenotypic stability, functionality, survival and proliferation (8). The HCC microenvironment is an active component of the tumour rather than merely a passive structural support for tumour growth, which changes dynamically and consequently affects HCC behaviour. These immunosuppressive features of HCC are a challenging barrier to clinicians to design effective immunotherapies. The HCC microenvironment is composed of not only growth factors, cytokines, metabolites and chemokines generated by stroma and tumour cells, but also tumour-infiltrating macrophages, myeloid-derived suppressor cells, neutrophils, cancer-associated fibroblasts and regulatory T cells. They all play key roles in the clinical outcome of HCC and success or failure of immunotherapies.
Hepatic stellate cells (HSCs) are the major framework of HCC and can directly promote tumour cell proliferation. Conditioned medium collected from HSCs induce not only proliferation and migration of HCC cells but also promote HCC growth through the activation of NF kappa B and extracellular-regulated kinase (ERK) pathways (92). Cancer-associated fibroblasts are the major cell type within the tumour stroma and play a critical role in tumour-stromal interactions (93). They are activated by TGF-β and are responsible for the synthesis, deposition and remodelling of excessive extracellular matrix thus modulating the biological activities of HCC. HCC cell growth, extravasation and metastatic spread are dependent upon the presence of these fibroblasts. HCC cells can reciprocally stimulate proliferation of tumour associated fibroblasts, suggesting their key role in tumour-stromal interaction (94). Stroma from HCC express several growth factors, including hepatocyte growth factor (HGF), epidermal growth factor (EGF), fibroblast growth factor (FGF) and Wnt family members, stromal-derived factor (SDF)-1α and IL-6 (95).
Additionally, myeloid derived suppressor cells (MDSC) exert multiple mechanisms of immunosuppressive activity in the tumour microenvironment. MDSCs induce differentiation and expansion of Tregs during tumorigenesis; inhibit DCs and NK cells via TGF-β; deprive T cells of essential amino acids such as L-arginine and L-cysteine; and generate the oxidative stress that is associated with HCC progression (96). MDSCs co-cultured with autologous T cells induce an increased number of Tregs, PD-1+-exhausted T cells, and an increase in immunosuppressive cytokine levels in HCC patients (97). MDSCs can also impair NK cell function. In HCC, MDSCs inhibit NK cell cytotoxicity and cytokine release mediated by the NKp30 receptor (98). MDSCs also inhibit TLR-ligand-induced IL-12 production and inhibit the T-cell stimulating activity of DCs in HCC (99). Tumour-associated neutrophils can also recruit macrophages and Treg cells into HCCs to promote their growth, progression, and resistance to sorafenib therapy (100, 101).
Furthermore, immunosuppressive cytokines, extracellular matrix and inflammatory cytokines in the HCC microenvironment can define HCC biology and prognosis. Global gene expression profiling of human HCC indicates that TGF-β gene signatures cluster HCC into two homogeneous groups with early or late TGF-β signatures (102). Importantly the late TGF-β signature is associated with an invasive HCC phenotype and an increased risk of tumour recurrence. MMP1 and TIMP1 were also signature genes in the immature hepatoblast subtypes of HCC that is associated with a poor prognosis (103). Inflammation-associated pathways, gene expression signatures, NF-κB, TNF-α, and IL-6 from the adjacent benign tissue can also predict late recurrence of HCC (104). IL-6, a major pro-inflammatory cytokine, is one of the signature genes in the hepatoblast phenotype signature (103). Osteopontin, secreted from Kupffer or stellate cells in response to inflammatory cytokines, is also associated with metastasis of HCC (105).
The tumour microenvironment also shapes NK cell metabolism and effector functions. Understanding immunometabolic suppression is critical in engineering a new generation of effective natural killer cell-based immunotherapies targeting solid tumours such as HCC. Multiple factors can modulate NK cell metabolism in the tumour microenvironment. HCC exert immunosuppressive effects through a number of mechanisms, a key driver of which is hypoxia. High oxygen consumption by tumour cells can generate hypoxic regions. Hypoxia impairs NK cell effector functions, but also sustains HIF1α, which promotes glycolytic metabolism. Hypoxia fuels the generation of adenosine from the cancer-associated ectoenzymes CD39, expressed on Treg (106), and CD73 on antigen presenting cells. Tumour cells also generate extracellular adenosine through the CD39 and CD73 ectonucleotidases, thus compromising NK cell function through competition for nutrients.
Thus, the interaction between the tumour and stroma interaction generates the microenvironment within HCC tissue and can suppress the effect of surrounding tissues or cell types that stimulate hepatocarcinogenesis, tumour progression, invasion, and metastasis. Sorafenib, an oral multi-kinase inhibitor, which is the most widely used HCC medication, inhibits VEGFR-2/-3 and PDGFR as well as Raf kinase, disrupting tumour-stromal interactions and resulting in decreased cell proliferation and angiogenesis. The efficacy and safety of sorafenib have been demonstrated in Phase III clinical trials, and it is currently the standard of care for patients with advanced stage HCC (104, 107).
Immunotherapeutic Interventions and How they May Enhance NK Function and Tame Treg Suppression
Currently the treatment for HCC is challenging, often due to the stage at which many patients present. Therapeutic options include surgery, transplantation and locoregional therapies can be curative, and systemic therapies with tyrosine kinase inhibitors including sorafenib and lenvantinib result in a modest prolongation of survival. As a result there is much interest in novel therapeutics and therapeutic combinations which have been recently reviewed by Llovet et al. (108).
In terms of immunotherapy, trials of checkpoint inhibitors have been the best studied. The PD-1 checkpoint inhibitors nivolumab and pembrolizumab have been used for the treatment of patients with HCC (109), but only leads to clinical responses of 10-20%. Several factors including the expression of programmed cell death-Ligand 1 (PD-L1), tumour mutational loads, and tumour-infiltration immune cells correlate with patient responses using these medications (110). This relative lack of efficacy implies that combination therapy should be considered and applied to each patient as personalized treatment approaches in HCC. Consistent with this combining a PDL1 inhibitor (Atezolizumab) with an anti-VEGF antibody (Bevacizumab) increased survival for unresectable HCC by approximately six months compared to sorafenib monotherapy (111). As checkpoint inhibitors predominantly target T cells, one attractive approach is to simultaneously target NK cells thus effectively mobilizing two arms of the immune system.
Enhancing NK Cell Frequency and Function
The field of NK cell therapeutics is rapidly growing. The observations of Ruggeri et al. demonstrating that NK cell alloreactivity was beneficial in refractory leukaemia acted as the cornerstone for the development of NK cell-based approaches (112). Initial work focussed on culturing NK cells in vitro and then infusing them, met with success mainly for haematological malignancies rather than solid tumours (113). However, this may be augmented by combining NK cells with monoclonal antibody therapy thus targeting the ADCC function of NK cells, or by genetically modifying NK cells to express chimeric antigen receptors (CAR-NK cells). Both these strategies have reached clinical trials (114, 115). Encouragingly the use of CD19-transduced CAR-NK cells did not result in the increase in cytokine levels associated with the systemic toxicity of CAR-T cell therapies. An alternative strategy is to target inhibitory receptors. As NK cells are held in check by dominant inhibitory signals then they are excellent candidates for having their activity unleashed by blocking these inhibitory receptors using monoclonal antibody therapeutics. These have targeted both the KIR receptors and NKG2A. Importantly, the clinical effects of the anti-NKG2A monoclonal antibody Monalizumab may be related to unleashing both T and NK cells for its anti-tumour effects (116).
In current clinical trials of adoptive tumour immunotherapy, large dosages of NK cells have been used ranging between 5 x 106 to 5 x 107/kg body weight (117). An approach to achieve these large numbers of NK cells is via the enrichment of NK cells from donor-derived leukapheresis products. Multiple protocols have been successfully developed to generate GMP NK cell products through immunomagnetic depletion of T and B cells and positive selection of CD56+ cells (118). The necessity of the NK cell products to be of a high-purity, which requires not only a long manufacturing process and compromises the viability and potency of the NK cell product, combined with the limited availability of autologous leukapheresis products makes the task of obtaining sufficient GMP NK cells from a single leukapheresis challenging. Therefore, an alternative method is to make NK cell products via the expansion of NK cells from PBMCs using feeder cells. An example of this are K562 cells modified with membrane-bound molecules such as IL-15 and 4-1BB ligand which can rapidly expand NK cells from PBMCs by 21.6-fold in 7 days (119). This method also produces NK cell products with purities in the range of 60-70%. To achieve purities needed for allogenic use a further expansion up to 21 days or enrichment of NK cells is still required (117, 119). NK cells can also be derived from induced pluripotent stem cells (IPSCs) and this has the advantage of hugely increasing their availability, as well as the prospect of selecting NK cell populations based on their alloreactivity potential, which may enhance their anti-cancer response (120).
IL-2 can also be used for short term activation of NK cells without feeder cells. IL-2 activation of NK cells can be coupled with CD3+ T-cell and CD19+ B cells depletion to increase the purity of the cell product (121). GMP NK cells have high IFNγ expression upon cultivation with K562 tumour cells and are highly cytotoxicity toward tumour cell lines in vitro (122). These data confirm that NK cells can have high clinical potency and potential for a significant role in tumour immunotherapy, including HCC.
Many companies have developed GMP cell isolation equipment and GMP reagents and have a manufacturing platform which has been utilised for multiple clinical trials utilising NK cell therapy (Figure 2). There are currently only a few MHRA and HTA approved GMP facilities in the UK focusing on GMP T cells and NK cell therapy for patients with both autoimmune diseases and cancer. GMP grade magnetic isolation or GMP cell sorting of clinical grade CD56-positive NK cells and applying these cells as immunotherapy in HCC could be one of the future treatment for these patients.
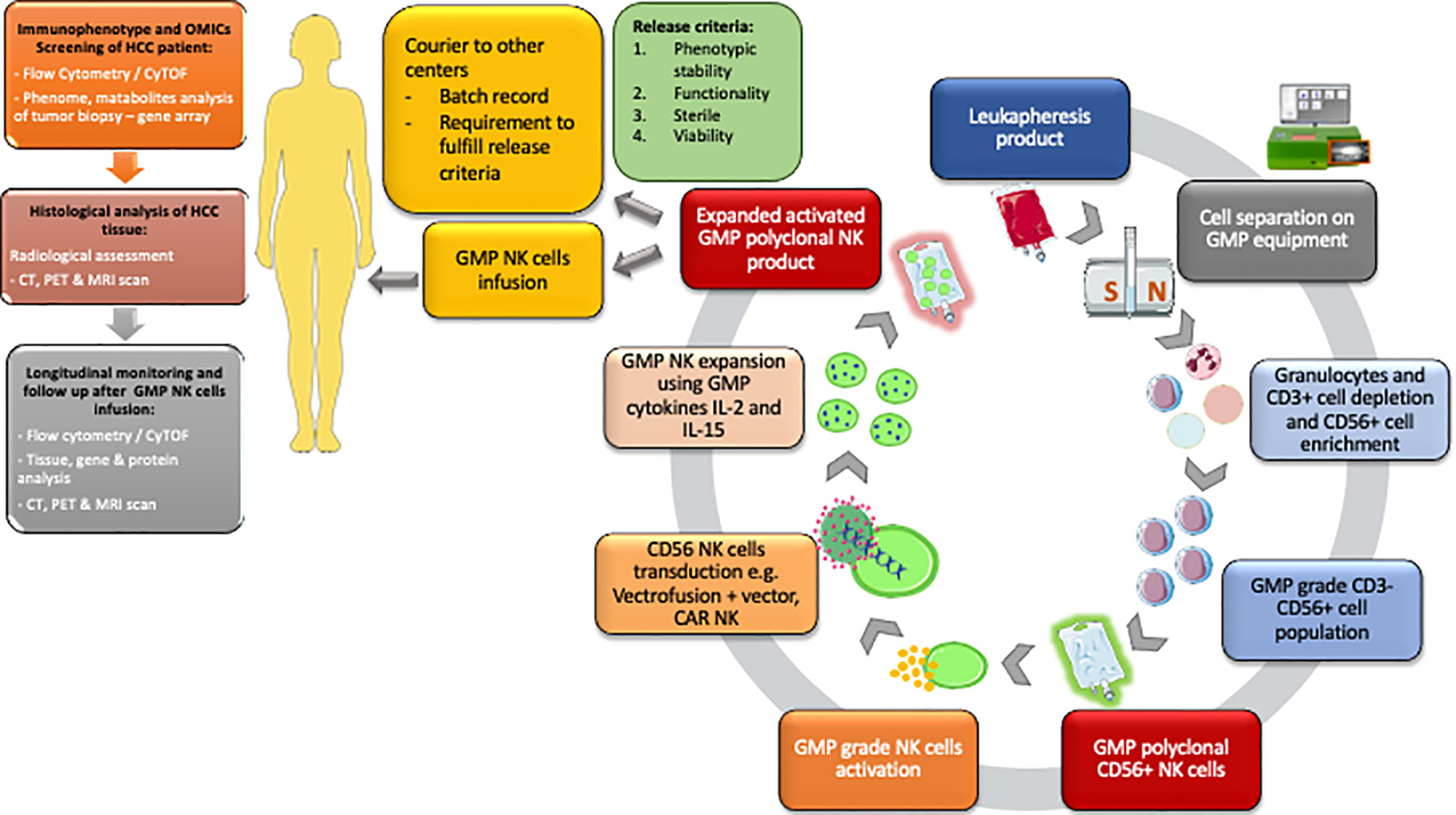
Figure 2 Workflow for the isolation of GMP polyclonal NK product and subsequent pathways from the NK cell isolation to distribution to other centres. GMP NK cell product must fulfil the release criteria according to MHRA criteria. Flow cytometry/CyTOF and histological analysis, CT, PET and MRI scans are performed before cell infusion to help identify individuals for therapy. Following GMP NK cell infusion the patient immune system is monitored using a combination of flow cytometry/CyTOF, tissue analysis and cross-sectional radiological imaging. HCC, Hepatocellular carcinoma; OMICs, genomic proteomic metabolomic; CyTOF, cytometry time of flight.
Suppressing Treg Frequency and Function in HCC
Restraining Tregs in HCC is important for many reasons. Anti-tumour T cell responses are severely compromised in advanced HCC patients through multiple immunosuppressive pathways comprising Tregs, PD-1+ T effector cells, and inhibitory cytokines (72, 97, 123). In HCC, expression of PD-1 is increased on CD8 memory T effector cells and interaction with its ligand PD-L1 on HCC cells blocks signalling, proliferation, and cytokine secretion of anti-tumour CD8 T cells (124, 125). The association between the infiltration of CD8 T cells in HCC and patient survival has been well recognised (126, 127). Granzyme B, perforin and IFNγ secretion by CD8 T cells generates potent anti-tumour activity but high expression of PD-1 on exhausted T cells contributes to ineffective effector T cell function, and selective in vitro depletion of these immunosuppressive cells results in improvement of T effector cell function in HCC patients (128, 129).
Activated Tregs also have the ability to inhibit effector T cells via contact-dependent interactions between checkpoint molecules and their ligands including PD-1 with PD-L1, Tim-3 with galectin-9, CTLA-4 and GITR. Tregs also contribute to the strongly immunosuppressive HCC microenvironment by releasing the inhibitory cytokines TGF-β and IL-10 (130, 131). The mechanism of inhibition of anti-tumour effector T cells by Treg involves several molecular pathways: 1) Tregs may inhibit proliferation and cytokine secretion of T effector cells by IL-10, adenosine production from CD39 on its surface and IL-35, which can be reversed by adding neutralising antibodies (132, 133); 2) via the PD-1/PD-L1 pathway to suppress anti-tumoral immunity in HCC (134); and also via 3) the co-inhibitory molecule CTLA-4 (135).
Suppressing Tregs and Boosting NK Cells in HCC
Combination of these approaches by inhibiting Tregs and enhancing NK cells is an exciting option which has not been tried before (Figure 3). One of the potential approaches would be sequential manipulation by administering Basilizumab (anti-CD25) to deplete CD4pos CD25high Treg cells followed by GMP NK cells either via a peripheral route or direct administration along with transarterial chemoembolization (TACE) as GMP-NK TACE therapy. This would prevent systemic depletion of Treg thus reducing the potential for inducing autoimmunity. With the remarkable success of chimeric antigen receptor (CAR)- engineered technology, developing CAR-engineered NK (CAR-NK) cells for cancer therapy could offer some significant advantages, including better safety by a lack or minimal cytokine release syndrome, multiple different mechanisms for inducing cytotoxic activity including checkpoint inhibition, and off-the-shelf manufacturing. CAR-NK cells could also have better infiltration into solid tumour such as HCC and overcome the resistant tumour microenvironment.
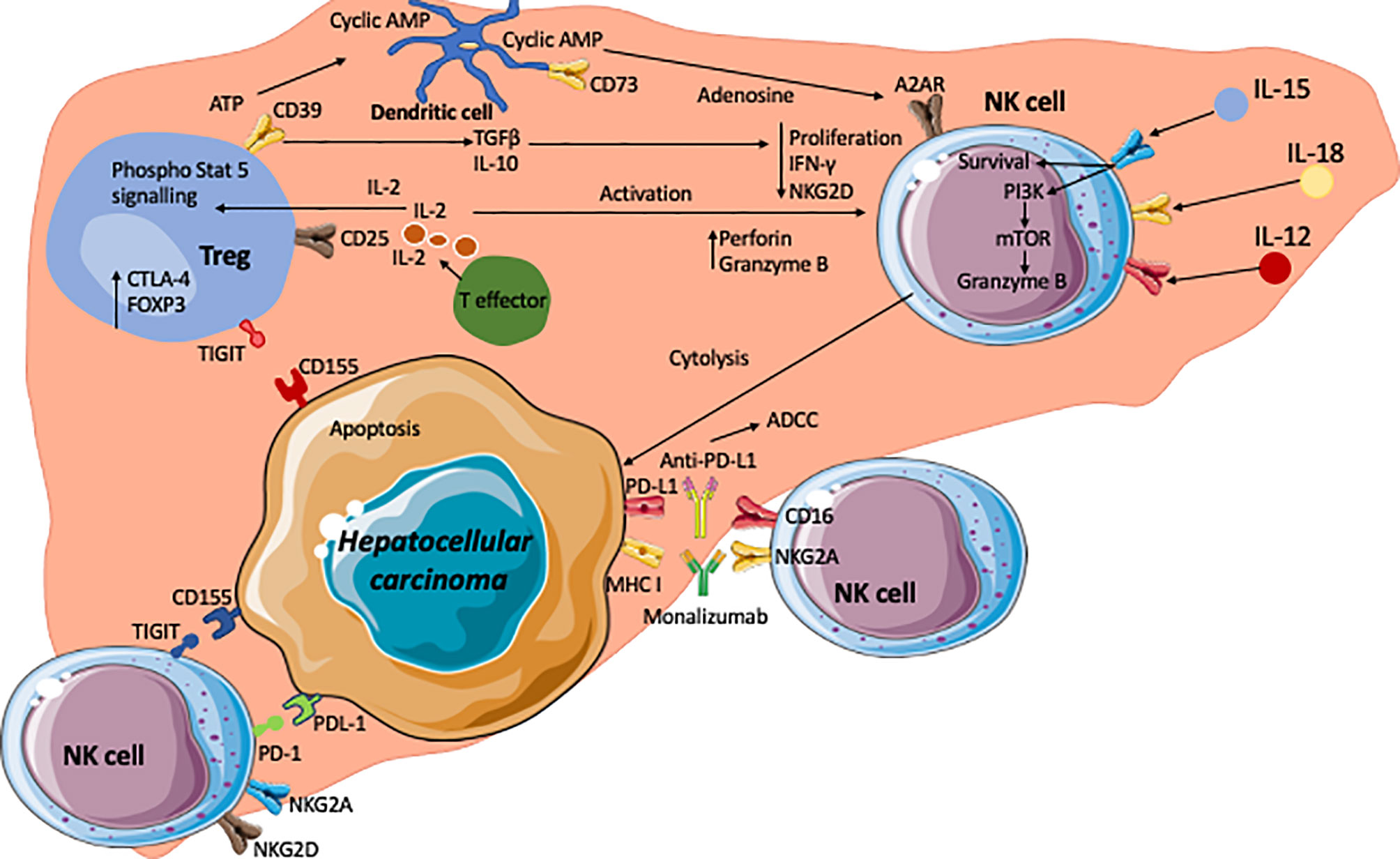
Figure 3 The hepatocellular carcinoma microenvironment and crosstalk of NK, Treg and HCC with the key signalling cascades. Effector T cells are the source of interleukin-2 (IL-2) which acts on the IL-2α receptor in CD25 on regulatory T cells. This leads to phosphorylation of STAT5 which subsequently upregulates Treg functional molecule CTLA-4 and the transcription factor Foxp3. The tumour microenvironment is enriched with adenosine triphosphate (ATP). CD39 on Tregs generates cyclic AMP from ATP. CD73 expressed on intrahepatic Tregs subsequently generate immunosuppressive adenosine from the cyclic AMP. Adenosine act on A2AR on NK cells which leads to the suppression of NK cell function. In addition, immunosuppressive cytokines TGFβ and IL-10 are secreted by Tregs and these cytokines lead to reductions in proliferation, IFNγ production and expression of NKG2D. Conversely, high concentrations of IL-2 lead to an increase in perforin and granzyme B expression on NK cells. IL-12, IL-15 and IL-18 act on their corresponding receptors on NK cells and leading to enhanced cell survival, and granzyme B production via PI3Kinase and mTOR pathways. NK cells express inhibitory receptors such as TIGIT and PD-1, which interact with corresponding ligands CD155 (PVR) and PDL-1 expressed on tumour cells. Inhibition of these receptors with check point inhibitors could lead to unleashing of NK cell cytotoxic activity to tumour cells via secretion of granzymes, perforin, IFNγ and TNFα. Monoclonal antibodies against inhibitory receptors such as immune checkpoint inhibitor monalizumab are developed to block MHC-I ligands and NKG2A interactions and enhance NK cell cytotoxicity towards cancer cells. CD16 receptors on NK cells allows them to carry out ADCC targeting such molecules as PD-L1.
Targeting intratumoral Treg cells may offer a therapeutic direction to modulate the tumour microenvironment. The combination of nivolumab with the Treg-depleting anti-CCR4 antibody, mogamulizumab has been explored by Doi et al. (136). In their proof of concept study this combination provided anti-tumour activity and can thus be a potentially effective option in cancer immunotherapy. A recent study found that CD36 was selectively upregulated in intratumoral Treg cells as a central metabolic modulator. They genetically ablated CD36 in Treg cells resulting in suppressed tumour growth, a decrease in intratumoral Treg cells and enhanced antitumour activity (137). In addition, a recent report identified widespread HLA-E expression in tumour samples, with levels correlating to those of NKG2A. This is of importance as one mechanism of tumour resistance to immune cells is mediated by the expression of peptide-loaded HLA class I molecule (HLA-E) in tumour cells. HLA-E suppresses NK cell activity via ligation of inhibitory receptor, NKG2A (138). Furthermore, blockade of NKG2A results in the enhancement of tumour immunity by promoting both NK and CD8+ T cell effector functions in mice and humans. As described above, monalizumab is a humanised anti-NKG2A antibody which has been shown to enhance the activity of NK cells against various tumour cells and rescue CD8+ T cell function in combination with blockade of the PD-x axis (116).
The Potential for Future Combination Immunotherapy in HCC
Checkpoint Therapy to Enhance T and NK Cell Function
Both T cells and NK cells express co-inhibitory molecules or checkpoint inhibitors, which can be targeted using CPI to unleash potent anti-tumour immunity by recovering both NK and T cell function (116, 139). These include PD-1, CTLA-4, TIGIT and LAG-3. The mechanism of action of PD-1 involves the engagement of its ligands PD-L1 and PD-L2 to deliver inhibitory signals that regulate the balance between T cell exhaustion, tolerance and immunopathology (140). Tumour cells expressing PD-1 ligands on their surface use the PD-1 pathway to attenuate tumour immunity and facilitate tumour progression (141). PD-1/PD-L1 has been recognised to play a role of critical importance in immune escape in HCC after the successful treatment of nivolumab in patients with advanced HCC, achieving an objective response rate of 15-20% (142). Furthermore, elevated PD-L1 expression in HCC significantly correlates to poor survival and tumour aggressiveness (143–146). Blockade of the PD-1 and PD-L1 interaction using monoclonal antibodies produces durable clinical responses in patients with diverse advanced tumour types (147).
CTLA-4 outcompetes the co-stimulatory molecule CD28 for binding to B7-1/CD80 or B7-2/CD86 expressed on the surface of antigen presenting cells, including tumour infiltrating dendritic cells. This is due to its higher affinity for CD80/CD86 as compared to CD28. As a result of this, CTLA-4 negatively regulates T-cell activation, inactivating T lymphocytes in the G1 phase. CD8 T lymphocytes are able to exert their anti-tumour cytotoxic effects via the secretion of TNFα and IFNγ leading to the apoptosis of tumour cells (148), when the T-cell receptor (TCR) binds its cognate peptide:MHC antigen expressed on tumour cells (149–153). CTLA-4 blockade can then provide long-lasting tumour remission due to its impact on memory T cells response to cancer (154). After blocking CTLA-4, an increase in the number and breadth of protective T cells can be seen in the blood as evidenced by TCR V-β analysis (154). However, its role in the anti-tumour NK cell response requires further study.
TIGIT (T cell immunoglobulin and ITM domain) is an inhibitory receptor and is expressed on activated T cells and can also be found on NK cells as well as memory T cells, a subset of Treg cells as well as follicular T helper (Tfh) cells (155–160). TIGIT is recognised for its protective role in autoimmune diseases as well as cancer. To date, tumour associated lymphocytes expressing TIGIT have been shown to exist in acute myeloid leukaemia, non-small cell lung cancer, colo-rectal carcinoma and melanoma (161–163). TIGIT is a key checkpoint inhibitor in anti-tumour responses and thus presents a promising target for future immunotherapies (161). TIGIT binds to its ligand PVR or CD155 on the tumour cells with a much higher affinity than its activating counterpart CD226 (DNAM-1), thereby inhibiting the interaction between CD226 and CD155 which is widely expressed on tumour cells (158–160, 164). CD226 promotes cytotoxicity and enhances anti-tumour responses (165, 166), whereas TIGIT, which outcompetes CD226, negatively regulates anti-tumour responses (167). After TIGIT/CD155 ligation, TIGIT’s immunoglobulin tail tyrosine-like motif becomes phosphorylated at Tyr225 and binds to cytosolic adapter Grb2. This in turn leads to NK cell immunosuppression and dysfunction, including downregulation of IFN-γ production (168). It is now recognised that TIGIT expression on tumour-infiltrating NK cells is associated with tumour progression and was also linked to functional immune exhaustion (139). The role of TIGIT in liver cancer is still under investigation. Recent research has shown that survival and prognosis of HCC patients is positively correlated with NK cell numbers in blood and tumour tissue (169). Tumour progression of these HCC patients was associated with dysfunction of the tumour-infiltrating NK cells (170), and exhausted tumour-infiltrating NK cells correlate with poor clinical outcome for HCC patients. Importantly, this NK cell exhaustion was reversed by manipulating the TIGIT pathway (171). Thus, the clinical application of anti-TIGIT CPI immunotherapy will enhance the NK cell anti-tumour immune response and is a promising new approach towards treating HCC.
Lymphocyte activation gene-3 (LAG-3) belongs to the immunoglobulin superfamily and is expressed on tumour infiltrating lymphocytes (TILs) (172), NK cells (173), B cells (174) and DCs (175). LAG-3 has a high binding capacity to MHC II (173). Current data suggests that modulating LAG-3 can impact autoimmunity, cancer and chronic viral infection (164). Fibrinogen-like protein 1 (FGL1) is a new major ligand for LAG-3 and it has recently been demonstrated that blocking the FGL1-LAG-3 pathways results in the stimulation of tumour immunity and inhibits tumour growth (176). Furthermore, co-expression of LAG-3 and PD-1 on TILs has been observed in several mouse tumour models. One of the first pre-clinical cancer models using anti-LAG-3 demonstrated enhanced activation of tumour-specific T cells at the tumour site and disruption of tumour growth, especially when used in combination with anti-PD-1 (177). Currently human studies involving LAG-3 as a target are usually performed in combination with PD-x axis blockade, providing a wider checkpoint blockade than monotherapy.
The use of monoclonal antibodies targeting CTLA-4, PD-1 and PD-L1 has seen excellent success, especially in settings such as melanoma and non-small-cell lung cancer. However, most HCC patients have underlying cirrhosis, and there is a concern about the risk of decompensation related to CPI-induced immune mediated hepatitis. This appears relatively rare occurrence during checkpoint inhibitor therapy for HCC with grade ≥3 side effects affected under 5% of patients (142, 178, 179). Nivolumab and pembrolizumab have now received approval from the FDA as second-line treatments for advanced HCC based on both the Checkmate 040 (142) and Keynote 224 (178) clinical trials. Although subsequent phase III trials have failed to show statistically significant data for survival improvement in either first-line (nivolumab vs. sorafenib) or second-line (pembrolizumab vs. placebo) setting (179, 180). These therapeutics now have potential to be combined with anti-VEGF therapies (111), however there remains an unmet clinical need for investigating combinatorial blockade targeting the novel inhibitory receptors, such as TIGIT and LAG-3.
Combination of GMP NK Infusion and Check Point Inhibitors in HCC
NK-cell therapy in cancer has made significant progress in the past decade with many milestones. It has been shown that NK-cell alloreactivity can eliminate the risk of leukaemia relapse and graft rejection as well as protecting against graft-versus-host disease in transplant patients (112). High-risk myelodsysplastic syndrome (MDS) patients have been shown to be responsive to NK-cell therapy due to the infused donor NK cells causing a reduction in high-risk clones and a less pronounced host immune activation (181). The infusion of NK cells has also been successful in patients with refractory acute myeloid leukaemia achieving remission in one third of patients (113). A recent phase I trial investigating the use of NK-cell therapy in combination with trastuzumab in HER2-positive cancer patients demonstrated that the therapy was well tolerated and that target engagement and anti-tumour activity was seen in the patients (115). CAR-NK cell infusion is also an exciting and promising therapy for cancer patients with a recent phase I and II trial showing that the majority (8 out of 11) patients responded to the treatment, of which 7 patients had complete disease remission (114). Recent developments suggested that NK cells derived from induced pluripotent stem cells (iPSCs) produce inflammatory cytokines and exert strong cytotoxicity against a variety of hematologic and solid tumours. iPSC-derived NK cells were also found to recruit T cells and cooperate with T cells and anti-PD-1 antibody, subsequently enhancing inflammatory cytokine production and promoting tumour lysis (120). Additionally, NK-CAR-iPSC-NK cells have been shown to significantly inhibit tumour growth resulting in prolonged survival in an ovarian cancer xenograft model (182).
Anti-PD-1, anti-PD-L1 and anti-CTLA-4 monoclonal antibodies also enhance NK cell tumour trafficking and release cytokines against tumours whilst simultaneously supressing Treg function (183–186). Anti-CTLA-4 monoclonal antibodies have also been shown to induce the release of TNFα against tumour cells via CD16 binding to antibody-bound tumour cells, and simultaneously to induce Treg inactivation (187–189). Blocking TIGIT and its ability to exploit both T cell and NK cell responses is also a strategy that is currently being explored by multiple pharmaceutical companies undergoing phase I/II clinical trials. Experimental drugs such as tiragolumab (anti-TIGIT) is currently undergoing phase I trials in various cancers (https://clinicaltrials.gov/ct2/show/NCT02913313) in combination with atezolizumab and nivolumab. Specific clinical trials looking at patients with HCC include looking at the use of SRF388 which is a fully human IgG1 antibody against IL-27 that decreases the expression of inhibitory immune checkpoint receptors (https://clinicaltrials.gov/ct2/show/NCT04374877?term=TIGIT+HCC&draw=2&rank=).
Thus, combining the massive increase in CPI therapy with the growth in adoptive NK cell therapeutics indicates that there is now a great potential to generate novel therapeutic combinations that target both CD8 T cells, NK cells and Tregs generating a holistic approach to cancer immunotherapy. Such an approach could simultaneously positively modulate the tumour microenvironment and directly cytotoxicity against tumours. In particular, the combination of augmenting both CD8 T cells and NK cells means that tumours that down-regulate MHC class I to avoid CD8 T cell mediated lysis, lose an important inhibitory signal for NK cells, and thus render themselves susceptible to NK cell killing. However, new therapeutic combinations have the potential to exacerbate toxicity, especially those related to autoimmunity and so these approaches need caution. In general, though NK cell therapeutics have proven relatively safe with little systemic toxicity observed, so these combinations may be more advantageous than those that target solely T cells. Nevertheless as with all therapeutic combinations it is important that future studies are carefully monitored for signs of unexpected toxicity.
Conclusion
HCC remains a challenge for clinicians and researchers to approach in partnership. The recent development of multiple new immunotherapies including monoclonal antibodies and cell products present new opportunities for the clinician to treat HCC. However, understanding the immunological microenvironment in which HCC occurs will be key to successfully combining these. Fundamental research is required to unpick the different intrahepatic immunological microenvironments on which HCC occurs. Understanding these on a personalized basis will be the key to selecting the optimal combination of immunotherapies for each patient.
Author Contributions
AB and FW contributed equally to this work. All authors contributed to the article and approved the submitted version.
Funding
We received funding from Sir Jules Thorn Trust Biomedical Research Grant, Transbioline Innovative Medicine Initiative Research Grant, Queen Elizabeth Hospital Birmingham Charity, Medical Research Foundation - G1002552 from the The Medical Research Council (grants M019829 and S009338), CRUK (“HUNTER” accelerator award) the NIHR Birmingham BRC, and the NIHR Southampton BRC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All figures were created using ServierMedical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License, https://smart.servier.com.
References
1. Oo YH, Shetty S, Adams DH. The Role of Chemokines in the Recruitment of Lymphocytes to the Liver. Dig Dis (2010) 28:31–44. doi: 10.1159/000282062
2. McCuskey R, Reilly F. Hepatic Microvasculature: Dynamic Structure and its Regulation. Semin Liver Dis (1993) 13:1–12. doi: 10.1055/s-2007-1007333
3. Arii S, Imamura M. Physiological Role of Sinusoidal Endothelial Cells and Kupffer Cells and Their Implication in the Pathogenesis of Liver Injury. J Hepatobiliary Pancreat Surg (2000) 7:40–8. doi: 10.1007/s005340050152
4. Balmer ML, Slack E, De Gottardi A, Lawson MAE, Hapfelmeier S, Miele L, et al. The Liver may Act as a Firewall Mediating Mutualism Between the Host and its Gut Commensal Microbiota. Sci Transl Med (2014) 6:1–11. doi: 10.1126/scitranslmed.3008618
5. Oo YH, Weston CJ, Lalor PF, Curbishley SM, Withers DR, Reynolds GM, et al. Distinct Roles for CCR4 and CXCR3 in the Recruitment and Positioning of Regulatory T Cells in the Inflamed Human Liver. J Immunol (2010) 184:2886–98. doi: 10.4049/jimmunol.0901216
6. Bertolino P, Bowen DG, Benseler V. T Cells in the Liver: There is Life Beyond the Graveyard. Hepatology (2007) 45:1580–2. doi: 10.1002/hep.21786
7. Bowen DG, Zen M, Holz L, Davis T, McCaughan GW, Bertolino P. The Site of Primary T Cell Activation is a Determinant of the Balance Between Intrahepatic Tolerance and Immunity. J Clin Invest (2004) 114:701–12. doi: 10.1172/JCI200421593
8. Osei-Bordom D, Bozward AG, Oo YH. The Hepatic Microenvironment and Regulatory T Cells. Cell Immunol (2020) 357:104195. doi: 10.1016/j.cellimm.2020.104195
9. Brunner SF, Roberts ND, Wylie LA, Moore L, Aitken SJ, Davies SE, et al. Somatic Mutations and Clonal Dynamics in Healthy and Cirrhotic Human Liver. Nature (2019) 574:538–42. doi: 10.1038/s41586-019-1670-9
10. Jenne CN, Kubes P. Immune Surveillance by the Liver. Nat Immunol (2013) 14:996–1006. doi: 10.1038/ni.2691
11. Racanelli V, Rehermann B. The Liver as an Immunological Organ. Hepatology (2006) 43:54–62. doi: 10.1002/hep.21060
12. Albillos A, Lario M, Álvarez-Mon M. Cirrhosis-Associated Immune Dysfunction: Distinctive Features and Clinical Relevance. J Hepatol (2014) 61:1385–96. doi: 10.1016/j.jhep.2014.08.010
13. Irvine KM, Ratnasekera I, Powell EE, Hume DA. Casuses and Consequences of Innate Immune Dysfucntion in Cirrhosis. Front Immunol (2019) 10:293. doi: 10.3389/fimmu.2019.00293
14. Ifrim D, Quintin J, Joosten L, Jacobs C, Jansen T, Jacobs L. Trained Immunity or Tolerance: Opposing Functional Programs Induced in Human Monocytes After Engagement of Various Pattern Recognition Receptors. Clin Vaccine Immunol (2014) 21:535–45. doi: 10.1128/CVI.00688-13
15. Seki E, Schwabe RF. Hepatic Inflammation and Fibrosis: Functional Links and Key Pathways. Hepatology (2015) 61:1066–79. doi: 10.1002/hep.27332
16. Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, et al. Alterations of the Human Gut Microbiome in Liver Cirrhosis. Nature (2014) 513:59–64. doi: 10.1038/nature13568
17. Tranah TH, Edwards LA, Schnabl B, Shawcross DL. Targeting the Gut-Liver-Immune Axis to Treat Cirrhosis. Gut (2020) 70:1–13. doi: 10.1136/gutjnl-2020-320786
18. Foldi I, Tornai T, Tornai D, Sipeki N, Vitalis Z, Tornai I, et al. Lectin-Complement Pathway Molecules are Decreased in Patients With Cirrhosis and Constitute the Risk of Bacterial Infections. Liver Int (2017) 37:1023–31. doi: 10.1111/liv.13368
19. Niehaus CE, Strunz B, Cornillet M, Falk CS, Schnieders A, Maasoumy B, et al. Mait Cells are Enriched and Highly Functional in Ascites of Patients With Decompensated Liver Cirrhosis. Hepatology (2020) 72:1378–93. doi: 10.1002/hep.31153
20. Fiuza C, Salcedo M, Clemente G, Tellado JM. In Vivo Neutrophil Dysfunction in Cirrhotic Patients With Advanced Liver Disease. J Infect Dis (2000) 182:526–33. doi: 10.1086/315742
21. Gomez F, Ruiz P, Schreiber A. Impaired Function of Macrophage Fcγ Receptors and Bacterial Infection in Alcoholic Cirrhosis. N Engl J Med (1994) 331:1122–28. doi: 10.1056/NEJM199410273311704
22. Doi H, Iyer TK, Carpenter E, Li H, Chang KM, Vonderheide RH, et al. Dysfunctional B-Cell Activation in Cirrhosis Resulting From Hepatitis C Infection Associated With Disappearance of CD27-Positive B-Cell Population. Hepatology (2012) 55:709–19. doi: 10.1002/hep.24689
23. Lario M, Muñoz L, Ubeda M, Borrero MJ, Martínez J, Monserrat J, et al. Defective Thymopoiesis and Poor Peripheral Homeostatic Replenishment of T-Helper Cells Cause T-Cell Lymphopenia in Cirrhosis. J Hepatol (2013) 59:723–30. doi: 10.1016/j.jhep.2013.05.042
24. Tian Z, Chen Y, Gao B. Natural Killer Cells in Liver Disease. Hepatology (2013) 57:1654–62. doi: 10.1002/hep.26115
25. Jeffery HC, McDowell P, Lutz P, Wawman RE, Roberts S, Bagnall C, et al. Human Intrahepatic ILC2 are IL-13positive Amphiregulinpositive and Their Frequency Correlates With Model of End Stage Liver Disease Score. PLoS One (2017) 12:1–16. doi: 10.1371/journal.pone.0188649
26. Forkel M, Berglin L, Kekäläinen E, Carlsson A, Svedin E, Michaëlsson J, et al. Composition and Functionality of the Intrahepatic Innate Lymphoid Cell-Compartment in Human Nonfibro. Eur J Immunol (2017) 47:1280–94. doi: 10.1002/eji.201646890
27. Kiessling R. Natural Killer Cells in the Mouse. Role Prod Histocompat Gene Complex Immune Responses (1976) 77–85. doi: 10.1016/b978-0-12-401660-6.50012-6
28. Adams NM, Grassmann S, Sun JC. Clonal Expansion of Innate and Adaptive Lymphocytes. Nat Rev Immunol (2020) 20:694–707. doi: 10.1038/s41577-020-0307-4
29. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-Induced Memory-Like Natural Killer Cells Exhibit Enhanced Responses Against Myeloid Leukemia. Sci Transl Med (2016) 8:357ra123. doi: 10.1126/scitranslmed.aaf2341
30. Ljunggren H-G, Kärre K. In Search of the ‘Missing Self’: MHC Molecules and NK Cell Recognition. Immunol Today (1990) 11:237–44. doi: 10.1016/0167-5699(90)90097-S
31. Peng H, Wisse E, Tian Z. Liver Natural Killer Cells: Subsets and Roles in Liver Immunity. Cell Mol Immunol (2016) 13:328–36. doi: 10.1038/cmi.2015.96
32. Pende D, Falco M, Vitale M, Cantoni C, Vitale C, Munari E, et al. Killer Ig-like Receptors (KIRs): Their Role in NK Cell Modulation and Developments Leading to Their Clinical Exploitation. Front Immunol (2019) 10:1179. doi: 10.3389/fimmu.2019.01179
33. Goodridge JP, Jacobs B, Saetersmoen ML, Clement D, Hammer Q, Clancy T, et al. Remodeling of Secretory Lysosomes During Education Tunes Functional Potential in NK Cells. Nat Commun (2019) 10:1–15. doi: 10.1038/s41467-019-08384-x
34. Goodson-Gregg FJ, Krepel SA, Anderson SK. Tuning of Human NK Cells by Endogenous HLA-C Expression. Immunogenetics (2020) 72:205–15. doi: 10.1007/s00251-020-01161-x
35. Hsu KC, Liu X-R, Selvakumar A, Mickelson E, O’Reilly RJ, Dupont B. Killer Ig-Like Receptor Haplotype Analysis by Gene Content: Evidence for Genomic Diversity With a Minimum of Six Basic Framework Haplotypes, Each With Multiple Subsets. J Immunol (2002) 169:5118–29. doi: 10.4049/jimmunol.169.9.5118
36. Blunt MD, Khakoo SI. Activating Killer Cell Immunoglobulin-Like Receptors: Detection, Function and Therapeutic Use. Int J Immunogenet (2020) 47:1–12. doi: 10.1111/iji.12461
37. Cooley S, Trachtenberg E, Bergemann TL, Saeteurn K, Klein J, Chap TL, et al. Donors With Group B KIR Haplotypes Improve Relapse-Free Survival After Unrelated Hematopoietic Cell Transplantation for Acute Myelogenous Leukemia. Blood (2009) 113:726–32. doi: 10.1182/blood-2008-07-171926
38. Cariani E, Pilli M, Zerbini A, Rota C, Olivani A, Zanelli P, et al. HLA and Killer Immunoglobulin-Like Receptor Genes as Outcome Predictors of Hepatitis C Virus-Related Hepatocellular Carcinoma. Clin Cancer Res (2013) 19:5465–73. doi: 10.1158/1078-0432.CCR-13-0986
39. Gras Navarro A, Kmiecik J, Leiss L, Zelkowski M, Engelsen A, Bruserud Ø, et al. NK Cells With KIR2DS2 Immunogenotype Have a Functional Activation Advantage to Efficiently Kill Glioblastoma and Prolong Animal Survival. J Immunol (2014) 193:6192–206. doi: 10.4049/jimmunol.1400859
40. López-Vázquez A, Rodrigo L, Martínez-Borra J, Pérez R, Rodríguez M, Fdez-Morera JL, et al. Protective Effect of the HLA-Bw4I80 Epitope and the Killer Cell Immunoglobulin-Like Receptor 3DS1 Gene Against the Development of Hepatocellular Carcinoma in Patients With Hepatitis C Virus Infection. J Infect Dis (2005) 192:162–5. doi: 10.1086/430351
41. Llovet J, Montal R, Sia D, Finn RS. Molecular Therapies and Precision Medicine for Hepatocellular Carcinoma. Nat Rev Clin Oncol (2018) 15:599–616. doi: 10.1038/s41571-018-0073-4
42. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity (2018) 48:812–30.e14. doi: 10.1016/j.immuni.2018.03.023
43. Cai L, Zhang Z, Zhou L, Wang H, Fu J, Zhang S, et al. Functional Impairment in Circulating and Intrahepatic NK Cells and Relative Mechanism in Hepatocellular Carcinoma Patients. Clin Immunol (2008) 129:428–37. doi: 10.1016/j.clim.2008.08.012
44. Carrega P, Bonaccorsi I, Di Carlo E, Morandi B, Paul P, Rizzello V, et al. CD56 Bright Perforin Low Noncytotoxic Human NK Cells Are Abundant in Both Healthy and Neoplastic Solid Tissues and Recirculate to Secondary Lymphoid Organs Via Afferent Lymph. J Immunol (2014) 192:3805–15. doi: 10.4049/jimmunol.1301889
45. Sun H, Liu L, Huang Q, Liu H, Huang M, Wang J, et al. Accumulation of Tumor-Infiltrating cd49a Nk Cells Correlates With Poor Prognosis for Human Hepatocellular Carcinoma. Cancer Immunol Res (2019) 7:1535–46. doi: 10.1158/2326-6066.CIR-18-0757
46. Cariani E, Pilli M, Barili V, Porro E, Biasini E, Olivani A, et al. Natural Killer Cells Phenotypic Characterization as an Outcome Predictor of HCV-linked HCC After Curative Treatments. Oncoimmunology (2016) 5:1–9. doi: 10.1080/2162402X.2016.1154249
47. Larsen S, Gao Y, Basse PH. NK Cells in the Tumor Microenvironment. Crit Rev (2014) 19:91–105. doi: 10.1615/CritRevOncog.2014011142
48. Wu M, Mei F, Liu W, Jiang J. Comprehensive Characterization of Tumor Infiltrating Natural Killer Cells and Clinical Significance in Hepatocellular Carcinoma Based on Gene Expression Profiles. BioMed Pharmacother (2020) 121:109637. doi: 10.1016/j.biopha.2019.109637
49. Chew V, Chen J, Lee D, Loh E, Lee J, Lim KH, et al. Chemokine-Driven Lymphocyte Infiltration: An Early Intratumoural Event Determining Long-Term Survival in Resectable Hepatocellular Carcinoma. Gut (2012) 61:427–38. doi: 10.1136/gutjnl-2011-300509
50. Pinyol R, Montal R, Bassaganyas L, Sia D, Takayama T, Chau GY, et al. Molecular Predictors of Prevention of Recurrence in HCC With Sorafenib as Adjuvant Treatment and Prognostic Factors in the Phase 3 STORM Trial. Gut (2019) 68:1065–75. doi: 10.1136/gutjnl-2018-316408
51. Chew V, Lai L, Pan L, Lim CJ, Li J, Ong R, et al. Delineation of an Immunosuppressive Gradient in Hepatocellular Carcinoma Using High-Dimensional Proteomic and Transcriptomic Analyses. Proc Natl Acad Sci U S A (2017) 114:E5900–9. doi: 10.1073/pnas.1706559114
52. Vacca P, Munari E, Tumino N, Moretta F, Pietra G, Vitale M, et al. Human Natural Killer Cells and Other Innate Lymphoid Cells in Cancer: Friends or Foes? Immunol Lett (2018) 201:14–9. doi: 10.1016/j.imlet.2018.11.004
53. Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology (2019) 70:168–83. doi: 10.1002/hep.30347
54. Mantovani S, Oliviero B, Lombardi A, Varchetta S, Mele D, Sangiovanni A, et al. Deficient Natural Killer Cell NKp30-Mediated Function and Altered NCR3 Splice Variants in Hepatocellular Carcinoma. Hepatology (2019) 69:1165–79. doi: 10.1002/hep.30235
55. Kohga K, Takehara T, Tatsumi T, Ishida H, Miyagi T, Hosui A, et al. Sorafenib Inhibits the Shedding of Major Histocompatibility Complex Class I–related Chain A on Hepat. Hepatology (2010) 51:1264–73. doi: 10.1002/hep.23456
56. Easom NJW, Marks M, Jobe D, Gillmore R, Meyer T, Maini MK, et al. ULBP1 Is Elevated in Human Hepatocellular Carcinoma and Predicts Outcome. Front Oncol (2020) 10:971. doi: 10.3389/fonc.2020.00971
57. Sheppard S, Guedes J, Mroz A, Zavitsanou AM, Kudo H, Rothery SM, et al. The Immunoreceptor NKG2D Promotes Tumour Growth in a Model of Hepatocellular Carcinoma. Nat Commun (2017) 8:1–13. doi: 10.1038/ncomms13930
58. Sheppard S, Ferry A, Guedes J, Guerra N. The Paradoxical Role of NKG2D in Cancer Immunity. Front Immunol (2018) 9:1808. doi: 10.3389/fimmu.2018.01808
59. Zhang J, Xu Z, Zhou X, Zhang H, Yang N, Wu Y, et al. Loss of Expression of MHC Class I-related Chain A (MICA) is a Frequent Event and Predicts Poor Survival in Patients With Hepatocellular Carcinoma. Int J Clin Exp Pathol (2014) 7:3123–31.
60. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (1995) 1:1151–64.
61. Liu W, Putnam AL, Xu-yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 Expression Inversely Correlates With FoxP3 and Suppressive Function of Human CD4+ T Reg Cells. J Exp Med (2006) 203:1701–11. doi: 10.1084/jem.20060772
62. Walker LSK, Abbas AK. The Enemy Within: Keeping Self-Reactive T Cells At Bay in the Periphery. Nat Rev Immunol (2002) 2:11–9. doi: 10.1038/nri701
63. Hori S, Nomura T, Sakaguchi S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. J Immunol (2003) 299:1057–61. doi: 10.1126/science.1079490
64. Chen YY, Jeffery HC, Hunter S, Bhogal R, Birtwistle J, Braitch MK, et al. Human Intrahepatic Regulatory T Cells are Functional, Require IL-2 From Effector Cells for Survival, and are Susceptible to Fas Ligand-Mediated Apoptosis. Hepatology (2016) 64:138–50. doi: 10.1002/hep.28517
65. Jeffery HC, Jeffery LE, Lutz P, Corrigan M, Webb GJ, Hirschfield GM, et al. Low-Dose Interleukin-2 Promotes STAT-5 Phosphorylation, Treg Survival and CTLA-4-Dependent Function in Autoimmune Liver Diseases. Clin Exp Immunol (2017) 188:394–411. doi: 10.1111/cei.12940
66. Oo YH, Adams DH. Regulatory T Cells and Autoimmune Hepatitis: What Happens in the Liver Stays in the Liver. J Hepatol (2014) 61:973–5. doi: 10.1016/j.jhep.2014.08.005
67. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A Distinct Lineage of CD4 T Cells Regulates Tissue Inflammation by Producing Interleukin 17. Nat Immunol (2005) 6:1133–41. doi: 10.1038/ni1261
68. Littman DR, Rudensky AY. Th17 and Regulatory T Cells in Mediating and Restraining Inflammation. Cell (2010) 140:845–58. doi: 10.1016/j.cell.2010.02.021
69. Kleinewietfeld M, Hafler DA. The Plasticity of Human Treg and Th17 Cells and its Role in Autoimmunity. Semin Immunol (2013) 25:305–12. doi: 10.1016/j.smim.2013.10.009
70. Ormandy L, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased Populations of Regulatory T Cells in Peripheral Blood of Patients With Hepatocellular Carcinoma. Cancer Res (2005) 65:2457–64. doi: 10.1158/0008-5472.CAN-04-3232
71. Lan YT, Fan XP, Fan YC, Zhao J, Wang K. Change in the Treg/Th17 Cell Imbalance in Hepatocellular Carcinoma Patients and its Clinical Value. Medicine (United States) (2017) 96:0–5. doi: 10.1097/MD.0000000000007704
72. Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, et al. Increased Regulatory T Cells Correlate With CD8 T-Cell Impairment and Poor Survival in Hepatocellular Carcinoma Patients. Gastroenterology (2007) 132:2328–39. doi: 10.1053/j.gastro.2007.03.102
73. Zou W. Immunosuppressive Networks in the Tumour Environment and Their Therapeutic Relevance. Nat Rev Cancer (2005) 5:263–74. doi: 10.1038/nrc1586
74. Zhou J, Ding T, Pan W, Zhu LY, Li A, Zheng L. Increased Intratumoral Regulatory T Cells are Related to Intratumoral Macrophages and Poor Prognosis in Hepatocellular Carcinoma Patients. Int J Cancer (2009) 125:1640–8. doi: 10.1002/ijc.24556
75. Shi C, Chen Y, Chen Y, Yang Y, Bing W, Qi J. CD4+ CD25+ Regulatory T Cells Promote Hepatocellular Carcinoma Invasion Via TGF-β1-Induced Epithelial–Mesenchymal Transition. Onco Targets Ther (2018) Volume 12:279–89. doi: 10.2147/OTT.S172417
76. Tu JF, Ding YH, Ying XH, Wu FZ, Zhou XM, Zhang DK, et al. Regulatory T Cells, Especially ICOS+ FOXP3+ Regulatory T Cells, are Increased in the Hepatocellular Carcinoma Microenvironment and Predict Reduced Survival. Sci Rep (2016) 6:2–9. doi: 10.1038/srep35056
77. Yu SZ, Wang Y, Hou J, Li WY, Wang X, Xiang LCL, et al. Tumor-Infiltrating Immune Cells in Hepatocellular Carcinoma: Tregs is Correlated With Poor Overall Survival. PloS One (2020) 15:1–13. doi: 10.1371/journal.pone.0231003
78. Li K, Liu H, Guo T. Th17/Treg Imbalance is an Indicator of Liver Cirrhosis Process and a Risk Factor for HCC Occurrence in HBV Patients. Clin Res Hepatol Gastroenterol (2017) 41:399–407. doi: 10.1016/j.clinre.2016.12.004
79. Yoshizawa K, Abe H, Kubo Y, Kitahara T, Aizawa R, Matsuoka M, et al. Expansion of CD4+CD25+FoxP3+ Regulatory T Cells in Hepatitis C Virus-Related Chronic Hepatitis, Cirr.Pdf. Hepatol Res (2010) 40:179–87. doi: 10.1111/j.1872-034X.2009.00587.x
80. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor Cells Convert Immature Myeloid Dendritic Cells Into TGF-β-Secreting Cells Inducing CD4 +CD25 + Regulatory T Cell Proliferation. J Exp Med (2005) 202:919–29. doi: 10.1084/jem.20050463
81. Wu Y, Kuang DM, Pan WD, Le WY, XM L, Wang D, et al. Monocyte/Macrophage-Elicited Natural Killer Cell Dysfunction in Hepatocellular Carcinoma is Mediated by CD48/2B4 Interactions. Hepatology (2013) 57:1107–16. doi: 10.1002/hep.26192
82. Jeffery HC, Braitch MK, Bagnall C, Hodson J, Jeffery LE, Wawman RE, et al. Changes in Natural Killer Cells and Exhausted Memory Regulatory T Cells With Corticosteroid Therapy in Acute Autoimmune Hepatitis. Hepatol Commun (2018) 2:421–36. doi: 10.1002/hep4.1163
83. Ghiringhelli F, Ménard C, Martin F, Zitvogel L. The Role of Regulatory T Cells in the Control of Natural Killer Cells: Relevance During Tumor Progression. Immunol Rev (2006) 214:229–38. doi: 10.1111/j.1600-065X.2006.00445.x
84. Rook AH, Kehrl JH, Wakefield LM, Roberts AB, Sporn MB, Burlington DB, et al. Effects of Transforming Growth Factor Beta on the Functions of Natural Killer Cells: Depressed Cytolytic Activity and Blunting of Interferon Responsiveness. J Immunol (1986) 136:3916–20.
85. Bellone G, Aste-Amezaga M, Trinchieri G, Rodeck U. Regulation of NK Cell Functions by TGF-beta 1. J Immunol (1995) 155:1066–73.
86. Dasgupta S, Bhattacharya-Chatterjee M, O’Malley BW, Chatterjee SK. Inhibition of NK Cell Activity Through TGF-β1 by Down-Regulation of NKG2D in a Murine Model of Head and Neck Cancer. J Immunol (2005) 175:5541–50. doi: 10.4049/jimmunol.175.8.5541
87. Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H, et al. TGF-β1 Plays an Important Role in the Mechanism of CD4 + CD25 + Regulatory T Cell Activity in Both Humans and Mice. J Immunol (2004) 172:834–42. doi: 10.4049/jimmunol.172.2.834
88. Ghiringhelli F, Ménard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ Regulatory T Cells Inhibit Natural Killer Cell Functions in a Transforming Growth Factor-β-Dependent Manner. J Exp Med (2005) 202:1075–85. doi: 10.1084/jem.20051511
89. Uhl M, Aulwurm S, Wischhusen J, Weiler M, Ma JY, Almirez R, et al. SD-208, a Novel Transforming Growth Factor β Receptor I Kinase Inhibitor, Inhibits Growth and Invasiveness and Enhances Immunogenicity of Murine and Human Glioma Cells In Vitro and In Vivo. Cancer Res (2004) 64:7954–61. doi: 10.1158/0008-5472.CAN-04-1013
90. Lazarova M, Steinle A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front Immunol (2019) 10:2689. doi: 10.3389/fimmu.2019.02689
91. Littwitz-Salomon E, Akhmetzyanova I, Vallet C, Francois S, Dittmer U, Gibbert K. Activated Regulatory T Cells Suppress Effector NK Cell Responses by an IL-2-mediated Mechanism During an Acute Retroviral Infection. Retrovirology (2015) 12:1–13. doi: 10.1186/s12977-015-0191-3
92. Amann T, Bataille F, Spruss T, Mühlbauer M, Gäbele E, Schölmerich J, et al. Activated Hepatic Stellate Cells Promote Tumorigenicity of Hepatocellular Carcinoma. Cancer Sci (2009) 100:646–53. doi: 10.1111/j.1349-7006.2009.01087.x
93. Kalluri R, Zeisberg M. Fibroblasts in Cancer. Nat Rev Cancer (2006) 6:392–401. doi: 10.1038/nrc1877
94. Fransvea E, Mazzocca A, Antonaci S, Giannelli G. Targeting Transforming Growth Factor (TGF)-βRI Inhibits Activation of β1 Integrin and Blocks Vascular Invasion in Hepatocellular Carcinoma. Hepatology (2009) 49:839–50. doi: 10.1002/hep.22731
95. Bhowmick NA, Neilson EG, Moses HL. Stromal Fibroblasts in Cancer Initiation and Progression. Nature (2004) 432:332–7. doi: 10.1038/nature03096
96. Consonni FM, Porta C, Marino A, Pandolfo C, Mola S, Bleve A, et al. Myeloid-Derived Suppressor Cells: Ductile Targets in Disease. Front Immunol (2019) 10:949. doi: 10.3389/fimmu.2019.00949
97. Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y. Higher Frequencies of GARP+CTLA-4+Foxp3+ T Regulatory Cells and Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma Patients are Associated With Impaired T-Cell Functionality. Cancer Res (2013) 73:2435–44. doi: 10.1158/0008-5472.CAN-12-3381
98. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid Derived Suppressor Cells Inhibit Natural Killer Cells in Patients With Hepatocellular Carcin. Hepatology (2009) 50:799–807. doi: 10.1002/hep.23054
99. Hu CE, Gan J, Zhang RD, Cheng YR, Huang GJ. Up-Regulated Myeloid-Derived Suppressor Cell Contributes to Hepatocellular Carcinoma Development by Impairing Dendritic Cell Function. Scand J Gastroenterol (2011) 46:156–64. doi: 10.3109/00365521.2010.516450
100. Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology (2016) 150:1646–58.e17. doi: 10.1053/j.gastro.2016.02.040
101. Zhou SL, Yin D, Hu ZQ, Bin LC, ZJ Z, HY X, et al. A Positive Feedback Loop Between Cancer Stem-Like Cells and Tumor-Associated Neutrophils Controls Hepatocellular Carcinoma Progression. Hepatology (2019) 70:1214–30. doi: 10.1002/hep.30630
102. Coulouarn C, Factor VM, Thorgeirsson SS. Transforming Growth Factor-β Gene Expression Signature in Mouse Hepatocytes Predicts Clinical Outcome in Human Cancer. Hepatology (2008) 47:2059–67. doi: 10.1002/hep.22283
103. Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, et al. A Novel Prognostic Subtype of Human Hepatocellular Carcinoma Derived From Hepatic Progenitor Cells. Nat Med (2006) 12:410–6. doi: 10.1038/nm1377
104. Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, et al. Gene Expression in Fixed Tissues and Outcome in Hepatocellular Carcinoma. N Engl J Med (2008) 359:1995–2004. doi: 10.1056/NEJMoa0804525
105. Ye QH, Qin LX, Forgues M, He P, Kim JW, Peng AC, et al. Predicting Hepatitis B Virus-Positive Metastatic Hepatocellular Carcinomas Using Gene Expression Profiling and Supervised Machine Learning. Nat Med (2003) 9:416–23. doi: 10.1038/nm843
106. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine Generation Catalyzed by CD39 and CD73 Expressed on Regulatory T Cells Mediates Immune Suppression. J Exp Med (2007) 204:1257–65. doi: 10.1084/jem.20062512
107. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and Safety of Sorafenib in Patients in the Asia-Pacific Region With Advanced Hepatocellular Carcinoma: A Phase III Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Oncol (2009) 10:25–34. doi: 10.1016/S1470-2045(08)70285-7
108. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular Carcinoma. Nat Rev Dis Primers (2021) 7. doi: 10.1038/s41572-020-00240-3
109. Prieto J, Melero I, Sangro B. Immunological Landscape and Immunotherapy of Hepatocellular Carcinoma. Nat Rev Gastroenterol Hepatol (2015) 12:681–700. doi: 10.1038/nrgastro.2015.173
110. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-Driven Biomarkers to Guide Immune Checkpoint Blockade in Cancer Therapy. Physiol Behav (2017) 176:139–48. doi: 10.1016/j.physbeh.2017.03.040
111. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, et al. Atezolizumab Plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med (2020) 382:1894–905. doi: 10.1056/nejmoa1915745
112. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of Donor Natural Killer Cell Aloreactivity in Mismatched Hematopoietic Transplants. Science (80) (2002) 295:2097–100. doi: 10.1126/science.1068440
113. Cooley S, He F, Bachanova V, Vercellotti GM, DeFor TE, Curtsinger JM, et al. First-in-Human Trial of rhIL-15 and Haploidentical Natural Killer Cell Therapy for Advanced Acute Myeloid Leukemia. Blood Adv (2019) 3:1970–80. doi: 10.1182/bloodadvances.2018028332
114. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382:545–53. doi: 10.1056/nejmoa1910607
115. Lee S-C, Shimasaki N, Lim JSJ, Wong A, Yadav K, Yong WP, et al. Phase I Trial of Expanded, Activated Autologous NK-Cell Infusions With Trastuzumab in Patients With HER2-positive Cancers. Clin Cancer Res (2020) 26:4494–502. doi: 10.1158/1078-0432.ccr-20-0768
116. André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor That Promotes Anti-Tumor Immunity by Unleashing Both T and NK Cells. Cell (2018) 175:1731–43.e13. doi: 10.1016/j.cell.2018.10.014
117. Lapteva N, Szmania SM, van Rhee F, Rooney CM. Clinical Grade Purification and Expansion of Natural Killer Cells. Crit Rev Oncog (2014) 1-2:121–32. doi: 10.1615/CritRevOncog.2014010931
118. Koehl U, Brehm C, Huenecke S, Zimmermann SY, Kloess S, Bremm M, et al. Clinical Grade Purification and Expansion of NK Cell Products for an Optimized Manufacturing Protocol. Front Oncol (2013) 3:118. doi: 10.3389/fonc.2013.00118
119. Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of Highly Cytotoxic Human Natural Killer Cells for Cancer Cell Therapy. Cancer Res (2009) 69:4010–7. doi: 10.1158/0008-5472.CAN-08-3712
120. Cichocki F, Bjordahl R, Gaidarova S, Mahmood S, Abujarour R, Wang H, et al. PSC-Derived NK Cells Maintain High Cytotoxicity and Enhance In Vivo Tumor Control in Concert With T Cells and Anti–PD-1 Therapy. Sci Transl Med (2020) 12:568, eaaz5618. doi: 10.1126/scitranslmedaaz5618
121. Skeate R, Singh C, Cooley S, Geller M, Northouse J, Welbig J, et al. Hemolytic Anemia Due to Passenger Lymphocyte Syndrome in Solid Malignancy Patients Treated With Allogeneic Natural Killer Cell Products. Transfusion (2013) 53:419–23. doi: 10.1111/j.1537-2995.2012.03942.x
122. Bröker K, Sinelnikov E, Gustavus D, Schumacher U, Pörtner R, Hoffmeister H, et al. Mass Production of Highly Active Nk Cells for Cancer Immunotherapy in a Gmp Conform Perfusion Bioreactor. Front Bioeng Biotechnol (2019) 7:194. doi: 10.3389/fbioe.2019.00194
123. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Krüger C, Manns MP, et al. A New Population of Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma Patients Induces CD4+CD25+Foxp3+ T Cells. Gastroenterology (2008) 135:234–43. doi: 10.1053/j.gastro.2008.03.020
124. Shi F, Shi M, Zeng Z, Qi RZ, Liu ZW, Zhang JY, et al. PD-1 and PD-L1 Upregulation Promotes CD8+ T-Cell Apoptosis and Postoperative Recurrence in Hepatocellular Carcinoma Patients. Int J Cancer (2011) 128:887–96. doi: 10.1002/ijc.25397
125. Kim HD, Song GW, Park S, Jung MK, Kim MH, Kang HJ, et al. Association Between Expression Level of PD1 by Tumor-Infiltrating CD8+ T Cells and Features of Hepatocellular Carcinoma. Gastroenterology (2018) 155:1936–50.e17. doi: 10.1053/j.gastro.2018.08.030
126. Unitt E, Marshall A, Gelson W, Rushbrook SM, Davies S, Vowler SL, et al. Tumour Lymphocytic Infiltrate and Recurrence of Hepatocellular Carcinoma Following Liver Transplantation. J Hepatol (2006) 45:246–53. doi: 10.1016/j.jhep.2005.12.027
127. Budhu A, Forgues M, Ye QH, Jia HL, He P, Zanetti KA, et al. Prediction of Venous Metastases, Recurrence, and Prognosis in Hepatocellular Carcinoma Based on a Unique Immune Response Signature of the Liver Microenvironment. Cancer Cell (2006) 10:99–111. doi: 10.1016/j.ccr.2006.06.016
128. Brandacher G, Winkler C, Schroecksnadel K, Margreiter R, Fuchs D. Antitumoral Activity of Interferon-gamma Involved in Impaired Immune Function in Cancer Patients. Curr Drug Metab (2006) 7:599–612. doi: 10.2174/138920006778017768
129. Lugade AA, Kalathil S, Miller A, Iyer R, Thanavala Y. High Immunosuppressive Burden in Advanced Hepatocellular Carcinoma Patients: Can Effector Functions be Restored? Oncoimmunology (2013) 2:e24679. doi: 10.4161/onci.24679
130. Wang F, Wan L, Zhang C, Zheng X, Li J, Chen ZK. Tim-3-Galectin-9 Pathway Involves the Suppression Induced by CD4+CD25+ Regulatory T Cells. Immunobiology (2009) 214:342–9. doi: 10.1016/j.imbio.2008.10.007
131. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3 + Regulatory T Cells in the Human Immune System. Nat Rev Immunol (2010) 10:490–500. doi: 10.1038/nri2785
132. Langhans B, Braunschweiger I, Arndt S, Schulte W, Satoguina J, Layland LE, et al. Core-Specific Adaptive Regulatory T-cells in Different Outcomes of Hepatitis C. Clin Sci (2010) 119:97–109. doi: 10.1042/CS20090661
133. Langhans B, Krämer B, Louis M, Nischalke HD, Hüneburg R, Staratschek-Jox A, et al. Intrahepatic IL-8 Producing Foxp3+CD4+ Regulatory T Cells and Fibrogenesis in Chronic Hepatitis C. J Hepatol (2013) 59:229–35. doi: 10.1016/j.jhep.2013.04.011
134. Langhans B, Nischalke HD, Krämer B, Dold L, Lutz P, Mohr R, et al. Role of Regulatory T Cells and Checkpoint Inhibition in Hepatocellular Carcinoma. Cancer Immunol Immunother (2019) 68:2055–66. doi: 10.1007/s00262-019-02427-4
135. Ohue Y, Nishikawa H. Regulatory T (Treg) Cells in Cancer: Can Treg Cells be a New Therapeutic Target? Cancer Sci (2019) 110:2080–9. doi: 10.1111/cas.14069
136. Doi T, Muro K, Ishii H, Kato T, Tsushima T, Takenoyama M, et al. A Phase I Study of the anti-CC Chemokine Receptor 4 Antibody, Mogamulizumab, in Combination With Nivolumab in Patients With Advanced or Metastatic Solid Tumors. Clin Cancer Res (2019) 25:6614–22. doi: 10.1158/1078-0432.CCR-19-1090
137. Wang H, Franco F, Tsui Y, Xie X, Trefny MP, Zappasodi R, et al. CD36-Mediated Metabolic Adaptation Supports Regulatory T Cell Survival and Function in Tumors. Nat Immunol (2020) 21:298–308. doi: 10.1038/s41590-019-0589-5
138. Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking Expression of Inhibitory Receptor NKG2A Overcomes Tumor Resistance to NK Cells. J Clin Invest (2019) 129:2094–106. doi: 10.1172/JCI123955
139. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the Checkpoint Receptor TIGIT Prevents NK Cell Exhaustion and Elicits Potent Anti-Tumor Immunity. Nat Immunol (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
140. Errico A. PD-1-PD-L1 Axis: Efficient Checkpoint Blockade Against Cancer. Nat Rev Clin Oncol (2015) 12:63. doi: 10.1038/nrclinonc.2014.221
141. Topalian SL, Drake CG, Pardoll DM. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
142. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in Patients With Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet (2017) 389:2492–502. doi: 10.1016/S0140-6736(17)31046-2
143. Gao Q, Wang XY, Qiu SJ, Yamato I, Sho M, Nakajima Y, et al. Overexpression of PD-L1 Significantly Associates With Tumor Aggressiveness and Postoperative Recurrence in Human Hepatocellular Carcinoma. Clin Cancer Res (2009) 15:971–9. doi: 10.1158/1078-0432.CCR-08-1608
144. Calderaro J, Rousseau B, Amaddeo G, Mercey M, Charpy C, Costentin C, et al. Programmed Death Ligand 1 Expression in Hepatocellular Carcinoma: Relationship With Clinical and Pathological Features. Hepatology (2016) 64:2038–46. doi: 10.1002/hep.28710
145. Sideras K, Biermann K, Verheij J, Takkenberg BR, Mancham S, Hansen BE, et al. PD-L1, Galectin-9 and CD8+ Tumor-Infiltrating Lymphocytes are Associated With Survival in Hepatocellular Carcinoma. Oncoimmunology (2017) 6:1–11. doi: 10.1080/2162402X.2016.1273309
146. Chen J, Li G, Meng H, Fan Y, Song Y, Wang S, et al. Upregulation of B7-H1 Expression is Associated With Macrophage Infiltration in Hepatocellular Carcinomas. Cancer Immunol Immunother (2012) 61:101–8. doi: 10.1007/s00262-011-1094-3
147. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N Engl J Med (2012) 26:2443–54. doi: 10.1056/nejmoa1200690
148. Zen Y, Yeh MM. Hepatotoxicity of Immune Checkpoint Inhibitors: A Histology Study of Seven Cases in Comparison With Autoimmune Hepatitis and Idiosyncratic Drug-Induced Liver Injury. Mod Pathol (2018) 31:965–73. doi: 10.1038/s41379-018-0013-y
149. Walunas BTL, Christina YB, Bluestone JA. CTLA-4 Ligation Blocks CD28-Dependent T Cell Activation. J Exp Med (1996) 183:2541–50. doi: 10.1084/jem.183.6.2541
150. Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol Cancer Clin Trials (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
151. Grosso JF, Jure-Kunkel MN. CTLA-4 Blockade in Tumor Models: An Overview of Preclinical and Translational Research. Cancer Immun (2013) 13:1–14.
152. Linsley PS, Clark EA, Ledbetter JA. T-Cell Antigen CD28 Mediates Adhesion With B Cells by Interacting With Activation Antigen B7/BB-1. Proc Natl Acad Sci U S A (1990) 87:5031–5. doi: 10.1073/pnas.87.13.5031
153. Engelhardt JJ, Sullivan TJ, Allison JP. CTLA-4 Overexpression Inhibits T Cell Responses Through a CD28-B7-Dependent Mechanism. J Immunol (2006) 177:1052–61. doi: 10.4049/jimmunol.177.2.1052
154. Robert L, Tsoi J, Wang X, Emerson R, Homet B, Chodon T, et al. CTLA4 Blockade Broadens the Peripheral T-cell Receptor Repertoire. Clin Cancer Res (2014) 20:2424–32. doi: 10.1158/1078-0432.CCR-13-2648
155. Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG, et al. A Novel Molecular Interaction for the Adhesion of Follicular CD4 T Cells to Follicular DC. Eur J Immunol (2009) 39:695–703. doi: 10.1002/eji.200839116
156. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg Cells Expressing the Coinhibitory Molecule TIGIT Selectively Inhibit Proinflammatory Th1 and Th17 Cell Responses. Immunity (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
157. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting Edge: TIGIT has T Cell-Intrinsic Inhibitory Functions. J Immunol (2011) 186:1338–42. doi: 10.4049/jimmunol.1003081
158. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a Member of the CD28 Family and an Important Modulator of T-cell Function. Eur J Immunol (2011) 41:902–15. doi: 10.1002/eji.201041136
159. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The Interaction of TIGIT With PVR and PVRL2 Inhibits Human NK Cell Cytotoxicity. Proc Natl Acad Sci U S A (2009) 106:17858–63. doi: 10.1073/pnas.0903474106
160. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat Immunol (2009) 10:48–57. doi: 10.1038/ni.1674
161. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8+ T Cell Effector Function. Cancer Cell (2014) 26:923–37. doi: 10.1016/j.ccell.2014.10.018
162. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 Impair Tumor Antigen-Specific CD8+ T Cells in Melanoma Patients. J Clin Invest (2015) 125:2046–58. doi: 10.1172/JCI80445
163. Kong Y, Zhu L, Schell TD, Zhang J, Claxton DF, Ehmann WC, et al. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates With CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin Cancer Res (2016) 22:3057–66. doi: 10.1158/1078-0432.CCR-15-2626
164. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-Inhibitory Receptors With Specialized Functions in Immune Regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
165. Gilfillan S, Chan CJ, Cella M, Haynes NM, Rapaport AS, Boles KS, et al. DNAM-1 Promotes Activation of Cytotoxic Lymphocytes by Nonprofessional Antigen-Presenting Cells and Tumors. J Exp Med (2008) 205:2965–73. doi: 10.1084/jem.20081752
166. Iguchi-Manaka A, Kai H, Yamashita Y, Shibata K, Tahara-Hanaoka S, Honda SI, et al. Accelerated Tumor Growth in Mice Deficient in DNAM-1 Receptor. J Exp Med (2008) 205:2959–64. doi: 10.1084/jem.20081611
167. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MWL, et al. TIGIT Predominantly Regulates the Immune Response Via Regulatory T Cells. J Clin Invest (2015) 125:4053–62. doi: 10.1172/JCI81187
168. Liu S, Zhang H, Li M, Hu D, Li C, Ge B, et al. Recruitment of Grb2 and SHIP1 by the ITT-like Motif of TIGIT Suppresses Granule Polarization and Cytotoxicity of NK Cells. Cell Death Differ (2013) 20:456–64. doi: 10.1038/cdd.2012.141
169. Sun C, Sun HY, Xiao WH, Zhang C, Tian ZG. Natural Killer Cell Dysfunction in Hepatocellular Carcinoma and NK Cell-Based Immunotherapy. Acta Pharmacol Sin (2015) 36:1191–9. doi: 10.1038/aps.2015.41
170. Choi JW, Lee ES, Kim SY, Park S, Oh S, Kang JH, et al. Cytotoxic Effects of Ex Vivo-Expanded Natural Killer Cell-Enriched Lymphocytes (MYJ1633) Against Liver Cancer. BMC Cancer (2019) 19:1–11. doi: 10.1186/s12885-019-6034-1
171. Zheng Q, Xu J, Gu X, Wu F, Deng J, Cai X, et al. Immune Checkpoint Targeting TIGIT in Hepatocellular Carcinoma. Am J Transl Res (2020) 12:3212–24.
172. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG-3 Regulates CD8+ T Cell Accumulation and Effector Function in Murine Self- and Tumor-Tolerance Systems. J Clin Invest (2007) 117:3383–92. doi: 10.1172/JCI31184
173. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a Novel Lymphocyte Activation Gene Closely Related to CD4. J Exp Med (1990) 171:1393–405. doi: 10.1084/jem.171.5.1393
174. Kisielow M, Kisielow J, Capoferri-Sollami G, Karjalainen K. Expression of Lymphocyte Activation Gene 3 (LAG-3) on B Cells is Induced by T Cells. Eur J Immunol (2005) 35:2081–8. doi: 10.1002/eji.200526090
175. Andreae S, Buisson S, Triebel F. MHC Class II Signal Transduction in Human Dendritic Cells Induced by a Natural Ligand, the LAG-3 Protein (CD223). Blood (2003) 102:2130–7. doi: 10.1182/blood-2003-01-0273
176. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-Like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell (2019) 176:334–47.e12. doi: 10.1016/j.cell.2018.11.010
177. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res (2012) 72:917–27. doi: 10.1158/0008-5472.CAN-11-1620
178. Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib (KEYNOTE-224): A non-Randomised, Open-Label Phase 2 Trial. Lancet Oncol (2018) 19:940–52. doi: 10.1016/S1470-2045(18)30351-6
179. Finn RS, Ryoo B-Y, Merle P, Kudo M, Bouattour M, Lim H-Y, et al. Results of KEYNOTE-240: Phase 3 Study of Pembrolizumab (Pembro) vs Best Supportive Care (BSC) for Second Line Therapy in Advanced Hepatocellular Carcinoma (HCC). J Clin Oncol (2019) 37:4004–4004. doi: 10.1200/JCO.2019.37.15_suppl.4004
180. Yau T, Park JW, Finn RS, Cheng A-L, Mathurin P, Edeline J, et al. CheckMate 459: A Randomized, Multi-Center Phase III Study of Nivolumab (NIVO) vs Sorafenib (SOR) as First-Line (1L) Treatment in Patients (Pts) With Advanced Hepatocellular Carcinoma (aHCC). Ann Oncol (2019) 30:v874–5. doi: 10.1093/annonc/mdz394.029
181. Bjorklund AT, Carlsten M, Sohlberg E, Liu LL, Clancy T, Karimi M, et al. Complete Remission With Reduction of High-Risk Clones Following Haploidentical NK-cell Therapy Against MDS and AML. Clin Cancer Res (2018) 24:1834–44. doi: 10.1158/1078-0432.CCR-17-3196
182. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered With Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell (2018) 23:181–92.e5. doi: 10.1016/j.stem.2018.06.002
183. Zhu D, Corral LG, Fleming YW, Stein B. Immunomodulatory Drugs Revlimid® (Lenalidomide) and CC-4047 Induce Apoptosis of Both Hematological and Solid Tumor Cells Through NK Cell Activation. Cancer Immunol Immunother (2008) 57:1849–59. doi: 10.1007/s00262-008-0512-7
184. Hayashi T, Hideshima T, Akiyama M, Podar K, Yasui H, Raje N, et al. Molecular Mechanisms Whereby Immunomodulatory Drugs Activate Natural Killer Cells: Clinical Application. Br J Haematol (2005) 128:192–203. doi: 10.1111/j.1365-2141.2004.05286.x
185. Benson DM, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 Axis Modulates the Natural Killer Cell Versus Multiple Myeloma Effect: A Therapeutic Target for CT-011, a Novel Monoclonal Anti-PD-1 Antibody. Blood (2010) 116:2286–94. doi: 10.1182/blood-2010-02-271874
186. Davies FE, Raje N, Hideshima T, Lentzsch S, Young G, Tai YT, et al. Thalidomide and Immunomodulatory Derivatives Augment Natural Killer Cell Cytotoxicity in Multiple Myeloma. Blood (2001) 98:210–6. doi: 10.1182/blood.V98.1.210
187. Callahan MK, Postow MA, Wolchok JD. Immunomodulatory Therapy for Melanoma: Ipilimumab and Beyond. Clin Dermatol (2013) 31:191–9. doi: 10.1016/j.clindermatol.2012.08.006
188. Laurent S, Queirolo P, Boero S, Salvi S, Piccioli P, Boccardo S, et al. The Engagement of CTLA-4 on Primary Melanoma Cell Lines Induces Antibody-Dependent Cellular Cytotoxicity and TNF-α Production. J Transl Med (2013) 11:1–13. doi: 10.1186/1479-5876-11-108
Keywords: liver, NK cells, regulatory T cells, hepatocellular carcinoma, tumour microenvironment, GMP cell therapy
Citation: Bozward AG, Warricker F, Oo YH and Khakoo SI (2021) Natural Killer Cells and Regulatory T Cells Cross Talk in Hepatocellular Carcinoma: Exploring Therapeutic Options for the Next Decade. Front. Immunol. 12:643310. doi: 10.3389/fimmu.2021.643310
Received: 18 December 2020; Accepted: 12 April 2021;
Published: 30 April 2021.
Edited by:
Ignacio Melero, University of Navarra, SpainReviewed by:
Han Chong Toh, National Cancer Centre Singapore, SingaporeAnil Shanker, Meharry Medical College, United States
Copyright © 2021 Bozward, Warricker, Oo and Khakoo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Salim I. Khakoo, Uy5JLktoYWtvb0Bzb3Rvbi5hYy51aw==; Amber G. Bozward, QS5HLkJvendhcmRAYmhhbS5hYy51aw==
†These authors have contributed equally to this work