 Anita K. Mehta
Anita K. Mehta Sapana Kadel1
Sapana Kadel1 Madeline G. Townsend
Madeline G. Townsend Madisson Oliwa
Madisson Oliwa Jennifer L. Guerriero
Jennifer L. Guerriero- 1Breast Tumor Immunology Laboratory, Dana-Farber Cancer Institute, Boston, MA, United States
- 2Division of Breast Surgery, Department of Surgery, Brigham and Women's Hospital, Boston, MA, United States
Macrophages are crucial innate immune cells that maintain tissue homeostasis and defend against pathogens; however, their infiltration into tumors has been associated with adverse outcomes. Tumor-associated macrophages (TAMs) represent a significant component of the inflammatory infiltrate in breast tumors, and extensive infiltration of TAMs has been linked to poor prognosis in breast cancer. Here, we detail how TAMs impede a productive tumor immunity cycle by limiting antigen presentation and reducing activation of cytotoxic T lymphocytes (CTLs) while simultaneously supporting tumor cell survival, angiogenesis, and metastasis. There is an urgent need to overcome TAM-mediated immune suppression for durable anti-tumor immunity in breast cancer. To date, failure to fully characterize TAM biology and classify multiple subsets has hindered advancement in therapeutic targeting. In this regard, the complexity of TAMs has recently taken center stage owing to their subset diversity and tightly regulated molecular and metabolic phenotypes. In this review, we reveal major gaps in our knowledge of the functional and phenotypic characterization of TAM subsets associated with breast cancer, before and after treatment. Future work to characterize TAM subsets, location, and crosstalk with neighboring cells will be critical to counteract TAM pro-tumor functions and to identify novel TAM-modulating strategies and combinations that are likely to enhance current therapies and overcome chemo- and immuno-therapy resistance.
Introduction
Macrophages are innate immune cells and play a myriad of important roles such as host defense, tissue homeostasis, and modulating inflammatory responses (1, 2). To perform these functions, immature macrophages with high plasticity respond to microenvironmental cues, causing them to adopt a spectrum of effector function, among which M1-like and M2-like represent extreme polarization states (3–5). Classically activated M1 macrophages exhibit pro-inflammatory behavior by migrating to inflamed tissues, targeting pathogens with the production of reactive oxygen species (ROS), and having high antigen-expressing potential (6–8). Due to their inflammatory behavior, anti-tumor macrophages are commonly called M1 macrophages. These macrophages can be potent effector cells that kill tumor cells and can recruit cytotoxic T lymphocytes (CTLs) to activate adaptive immune responses. On the opposite side of the macrophage polarization spectrum, alternatively activated M2 macrophages secrete anti-inflammatory cytokines to induce immune tolerance and attract T regulatory cells (Tregs) and Th2 T cell subsets capable of protective type 2 responses but devoid of cytotoxic functions. M2 macrophages facilitate canonical tissue repair functions and in cancer are regarded as pro-tumor where they promote tissue remodeling and repair, stimulate angiogenesis with vascular endothelial growth factor (VEGF), and encourage tissue growth with transforming growth factor beta (TGF-β) (9). Therefore, for simplicity, tumor-associated macrophages (TAMs) have been described as either M1-like (anti-tumor) or M2-like (pro-tumor), but it should be recognized that the M1/M2 dichotomy represents idealized polarization states (Figure 1), while in nature, there exists a broad spectrum of macrophage phenotypes, which will be discussed in detail below.
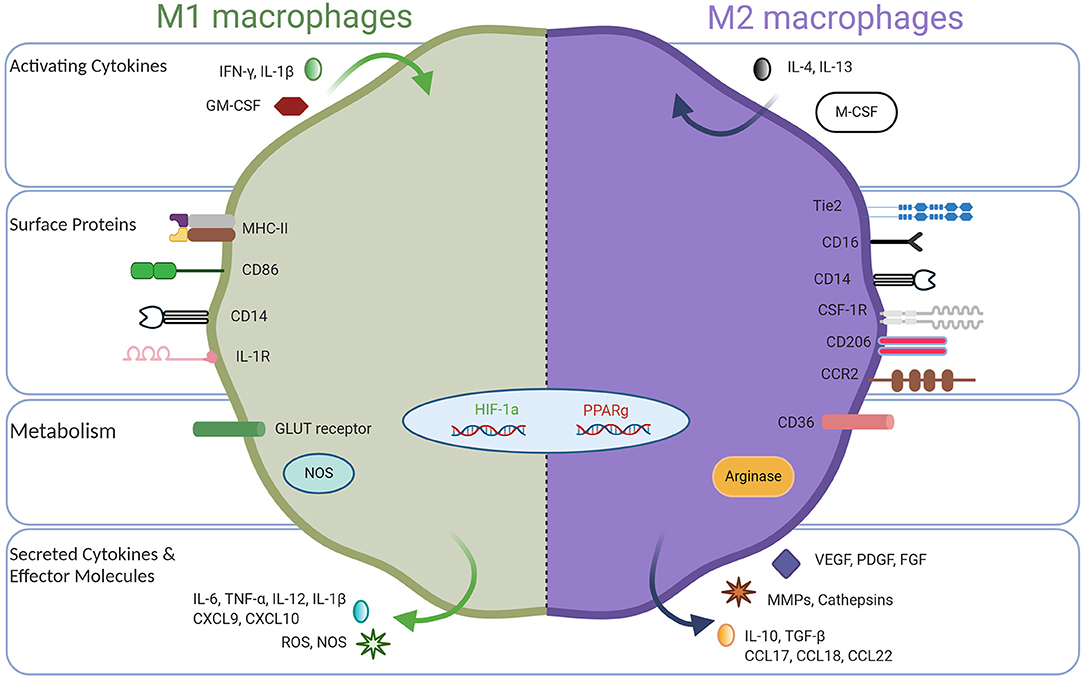
Figure 1. Opposing phenotypes of M1-like and M2-like tumor-associated macrophages.
Tumor Infiltrating Macrophages in Breast Tumors
Recruitment of Monocytes and Macrophages to Breast Tumors
TAMs represent a significant component of the inflammatory infiltrate in breast tumors (10, 11). Tumor-derived growth factors such as chemokines and cytokines facilitate recruitment of monocytes and macrophages into tumors (Figure 2, step 1) (12). One of the best-characterized cytokines responsible for recruiting TAMs into the tumor is chemokine (C-C motif) ligand 2 (CCL2), also known as monocyte chemoattractant protein 1 (MCP-1). CCL2 is expressed by both stromal cells and tumor cells (13) and is associated with poor prognosis in breast cancer (14, 15). Through recruitment of CCR2-expressing monocytes, CCL2 has been shown to promote pulmonary metastasis in mouse models of breast cancer (16). Activation of the CCL2–CCR2 axis promotes CCL3 production from macrophages, enhancing metastatic seeding of breast cancer cells (17). CCL5, also known as Regulated upon Activation, Normal T Cell Expressed and Secreted (RANTES), is another well-known factor that recruits TAMs to the breast tumor. CCL5 is expressed by malignant epithelial cells in breast carcinoma and is associated with advanced disease progression (18–20). Macrophages express high levels of its receptor (CCR5) and respond to CCL5 produced by tumor cells by infiltrating to the TME (21, 22). Importantly, CCL5 has been reported to alter the functionality of TAMs toward a tumor-promoting phenotype in colorectal cancer (23).
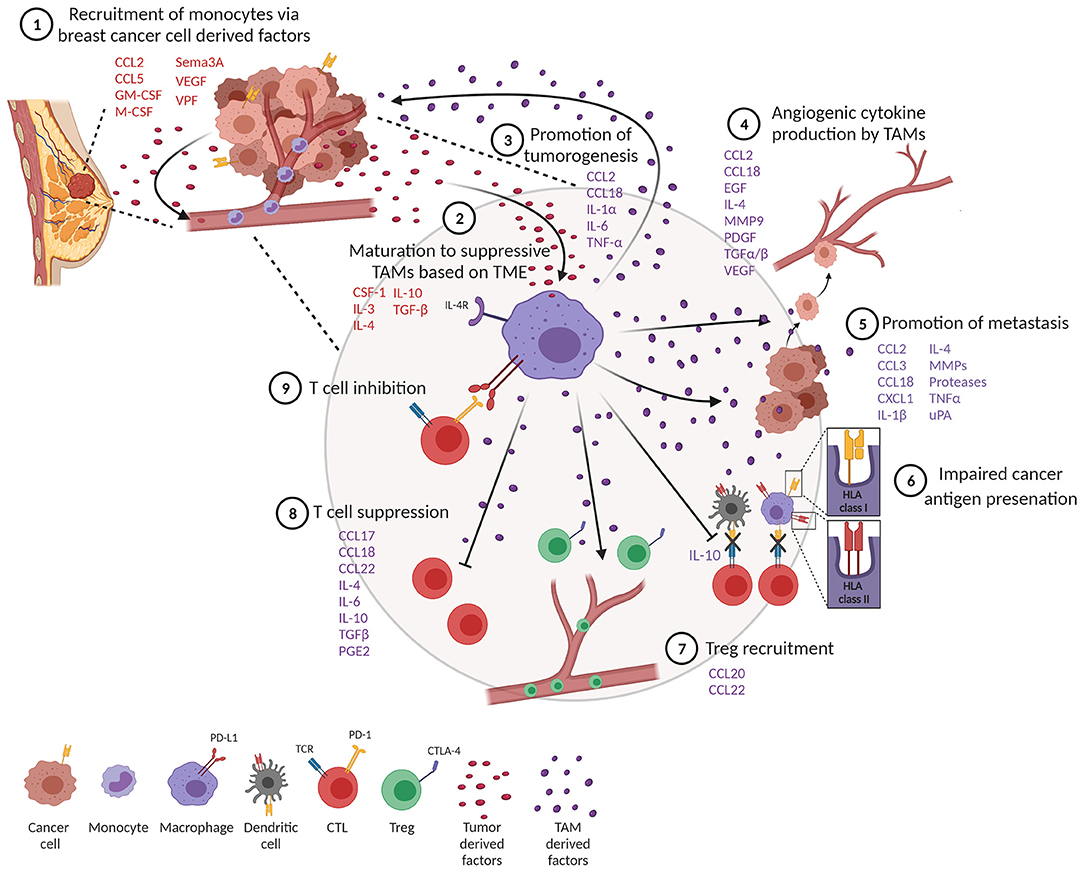
Figure 2. Tumor-associated macrophages (TAMs) impede a productive anti-tumor immune response. (1) Monocytes are recruited to the tumor through tumor-derived factors. (2) One of the tumor monocytes mature to suppressive TAMs. (3) TAMs produce factors that promote tumor cell proliferation and survival. (4) TAMs promote angiogenesis and metastasis (5). (6) TAMs impair productive antigen presentation by dendritic cells and themselves downregulate MHC class I and II molecules. TAMs inhibit T cell function through recruitment of T regulatory cells (Tregs) (7) and suppression (8) and upregulation of co-inhibitory molecules (9).
Several other factors produced by tumor cells help recruit macrophages. Colony-stimulating factor 1 (CSF-1), also known as macrophage colony-stimulating factor (M-CSF), and granulocyte-macrophage colony-stimulating factor (GM-CSF) are other tumor-derived factors produced by breast cancer cells (12, 24, 25). CSF-1 expression is associated with poor prognosis and increased infiltration of CSF-1 receptor (CSF-1R)-expressing macrophages in mouse models of breast cancer (26, 27). Tissue factors important for angiogenesis, such as vascular permeability factor (VPF), are also known to attract monocytes to tumors (28). In line with this, increased VEGF expression in tumor cells correlates with macrophage infiltration in human breast cancer (29). Hypoxia is another well-described factor that alters the activation and accumulation of macrophages in hypoxic regions and prevents migration out of these regions (30–32). Recruitment of TAMs into hypoxic niches occurs in a hypoxia-induced Semaphorin 3A (Sema3A)-dependent manner through VEGF-R1 phosphorylation in a mouse model of breast cancer (33). VEGF signaling and hypoxia also play a crucial role in angiogenesis, an important hallmark of cancer, providing links between angiogenesis and TAMs (34).
To further add complexity to TAMs, there are at least three lineages of macrophages that arise at different stages of development and persist into adulthood (35, 36). Each tissue in the body is composed of 5–20% tissue-resident macrophages, which are yolk sac-derived and seeded during embryogenesis. During homeostatic adaptations, such as tumorigenesis, macrophages of different phenotypes can be recruited from the monocyte reservoirs of blood, spleen, and bone marrow (37) and from resident progenitors or through local proliferation (38, 39). One report documented the loss of resident macrophages and a concomitant increase in monocyte-derived TAMs in a breast cancer model (40). Tissue macrophages have vastly different transcriptional profiles between tissues, suggesting that the macrophages at metastatic sites may differ from the primary tumor site and may need to be targeted differently (36). It is currently unknown how the ontogeny of TAMs influences primary and metastatic breast cancer and the extent to which TAM origin is important for clinical outcome.
Differentiation and Maturation of TAMs in Breast Tumors
TAM differentiation and polarization is regulated by the tumor microenvironment (TME) and results in a heterogeneous population of cells, but the subset diversity is only just beginning to be understood. Tumors are generally poorly vascularized and lack nutrients, causing recruited monocytes to differentiate into mature wound-repairing macrophages. To an infiltrating monocyte, the tumor is a “wound” that needs repair, and therefore, TAMs polarize to an M2-like phenotype driven by CSF-1, IL-3, IL-4, IL-10, and TGF-β (Figure 2, step 2) (41, 42). TAMs subsequently show pro-tumor functions by promoting tumor cell survival, proliferation, angiogenesis, and dissemination (42–47). Whether or not differentiated macrophages are able to undergo M1/M2 repolarization after adopting a mature phenotype in vivo is still under debate, or if instead new monocytes recruited to the TME acquire their phenotype and remain in that state (41, 48–50). At least ex vivo M1-like human macrophages can be repolarized to M2-like macrophages upon exposure to M2 cytokines (51) and that M2-like macrophages are reprogrammed to express M1-like genes following exposure to TLR ligands or IFN-γ (52, 53).
Pro-Tumor Functions of Macrophages in Breast Tumors
Promotion of Tumorigenesis
The contribution of TAMs to cancer progression and outcomes is multifaceted due to their wide spectrum of phenotypes. In vitro and in vivo studies have shown that macrophages are involved in a feedback loop between tumor cells at invasive fronts of breast tumors, where macrophages provide factors that enhance tumorigenesis. TAMs can directly promote the proliferation of tumor cells by secreting growth factors and inflammatory mediators such as CCL2, IL-1α, IL-6, and TNF-α (Figure 2, step 3) (54). Importantly, TNF-α secreted by TAMs leads to the activation of NF-κB in tumor cells, preventing tumor cell death and enhancing tumor cell invasion (55). In that regard, tumor cells recruit and activate M2-like macrophages, which then produce M2-related cytokines, such as CCL18, which causes breast cancer cells to elongate, lose contact inhibition, and increase vimentin expression. In a humanized murine model, anti-GM-CSF and anti-CCL18 both reversed the epithelial–mesenchymal transition (EMT) state of cancer cells, inhibiting metastasis. This concept was further confirmed with human breast tumors, revealing that high GM-CSF expression significantly corresponds to CCL18-expressing macrophages, cancer cell EMT and metastasis, and poor clinical outcome (56).
Facilitation of Angiogenesis
Angiogenesis is a complex, multistep process of forming new vasculature that facilitates tumor growth and progression (31). Macrophages are a major contributor of angiogenesis through the secretion of angiogenic cytokines (Figure 2, step 4) (57). TAMs secrete a key angiogenic cytokine, VEGF-A, directly, or indirectly by secreting matrix metalloproteinase 9 (MMP9) that activates latent forms of VEGF-A (29, 58, 59). TAMs also produce other pro-angiogenic factors from the fibroblast growth family that includes TGF-α, TGF-β, EGF, and PDGF (12, 60). Further supporting the role of macrophages in tumor angiogenesis, studies have shown that increased infiltration of TAMs promote angiogenesis whereas TAM depletion inhibits the angiogenic switch (61, 62). TAMs also contribute to angiogenesis by producing CCL18, which promotes angiogenesis both in vitro and in vivo (63). In addition to angiogenesis, CCL18 production from TAMs has also been associated with tumor invasiveness and metastasis in breast cancer (64). Furthermore, a subset of angiopoietin receptor Tie2-expressing monocytes, also known as TEMs, promote tumor angiogenesis (65, 66). In murine models, TEMs were initially identified as leukocytes expressing Tie2, CD11b, and CD45 in the peripheral blood and Tie2, CD11b, and Sca-1 in mammary tumors (65, 67). Additionally, TEMs also show a unique surface marker profile consisting of Tlr4, Mrc1, Il4ra, and CD163, which differentiate them from TAMs in murine mammary tumors (68). In humans, TEMs express CD45, CD11b, CD11c, CCR5, CD33, M-CSF-1R, and CD13 but lack expression of CD62L and CCR2 in circulating blood. Interestingly, TEMs from tumors of primary invasive breast carcinoma patients display different markers associated with antigen presentation, such as HLA-DR, CD80, CD86, and CD1a in addition to CD14, Tie-2, and VEGFR-1, which suggests their role in tumor-specific immune responses (69, 70). TEMs are also capable of secreting large amounts of IL-10 and VEGF, promoting a pro-angiogenic and immunosuppressive environment that inhibits tumor-specific T cell responses in breast tumors (70).
Promotion of Metastasis
TAMs play a major role in tumor progression and metastasis by producing various matrix proteolytic enzymes including matrix metalloproteases (MMPs) and urokinase (uPA; Figure 2, step 5) (71, 72). This allows for the local invasion and attachment of cancer cells to other adjacent epithelial cells and the extracellular matrix (ECM) through the formation of adherens junctions. Next, the multistep process of invasion-metastasis occurs with intravasation of cancer cells to blood and lymphatic vessels, escape of cancer cells to distant tissues, and, finally, the growth of small lesions in tumors (34, 73). Direct visualization of macrophages using intravital multiphoton imaging showed that perivascular macrophages are important for the intravasation of cancer cells in the mammary tumor (74). In line with this, the genetic ablation of CSF-1 reduced macrophage density and slowed tumor progression and metastasis in the MMTV-PyMT model of breast cancer (75). CXCL1 secretion from TAMs is known to promote breast cancer migration and epithelial–mesenchymal transition in both mouse and human breast cells (76). Furthermore, a study showed that macrophages were required for breast tumor metastases, and Ets-2 transcription factor in TAMs was important for promoting angiogenesis and growth of both primary tumors and lung metastases (77). In another study, macrophage depletion using clodronate-encapsulated liposomes in an ovarian tumor reduced metastasis in ovarian cancer (78). In the MMTV-PyMT mouse model of breast cancer, TAMs show high levels of cathepsin protease activity, which promotes tumor invasion and angiogenesis (79). It is also reported that IL-4 released from tumor cells and T cells induces cathepsin protease activity in TAMs to promote tumor invasion (79).
Importantly, tissue-resident macrophages at metastatic sites may vary greatly. For example, hypoxic primary breast tumor environments release lysyl oxidase (LOX), triggering NFACTc1-driven osteoclastogenesis, which increases bone resorption and thereby creates a metastatic niche for circulating tumor cells. Bisphosphonate treatment inhibits bone osteoclasts, which could be a potential combination therapeutic for patients with high-LOX primary breast tumors (80). The ability for therapeutics to reach sites of metastasis should also be considered when targeting TAMs. For example, in a murine breast cancer model, local pulmonary administration of CSF-1R inhibition allowed the drug to overcome lung physiological barriers of administration better than oral administration, significantly enhancing the M1/M2 ratio at a much lower dose. Therefore, orally administered medications may not effectively reach the lung-resident macrophages due to mucous layers, enzyme degradation, or mucociliary clearance; therefore, locally administered routes specific to lung metastatic sites should be considered (81).
Non-productive Antigen Presentation
Detrimental to the TME, protumor TAMs can become less efficient antigen-presenting cells, subsequently causing these cells to produce lower levels of pro-inflammatory cytokines and become less tumoricidal or able to activate T cells (82–84). TAMs that express pro-tumor features such as the IL-4R and arginase activity express low levels of MHCII (85). Subsets of TAMs characterized by lower expression of MHCII have been shown to be less effective in antigen presentation (85–87). In general, macrophages are less efficient in processing internalized antigens compared to conventional DCs, and macrophages in tumors appear to be greatly restricted in their antigen-presenting capacity (85, 86). Importantly, IL-10 derived from TAMs can also inhibit DC antigen presentation, further dampening tumor immunity (Figure 2, step 6) (88).
Suppression of T Cell Function
TAMs release anti-inflammatory cytokines that promote an immune-suppressive tumor microenvironment through the recruitment of Tregs, which suppresses effector T cell activation (Figure 2, step 7). For example, CCL22 and CCL20 secreted by TAMs induce an immunosuppressive microenvironment by recruitment of Tregs in ovarian and colorectal cancer (89, 90). In addition, TAM-derived chemokines CCL17, CCL18, CCL22, IL-4, IL-10, TGF-β, and prostaglandin-E2 (PGE2) have all been well-characterized in their ability to suppress T cell functions (Figure 2, step 8) (91–97). As a third way to inhibit T cell function, blocking B7-H4 on macrophages improves macrophage-mediated T cell activation (98), and at the heart of T cell activation as it relates to immunotherapy, TAMs induce immunosuppression through the expression of ligands that bind to PD-1 and CTLA-4 molecules on T cells, which inhibits T cell activation (Figure 2, step 9) (99).
Macrophage Metabolism as a Barrier to Anti-Tumor Immune Responses
Macrophage metabolism is a critical feature of the macrophage phenotype. Previous work has revealed that M2-like macrophages have a distinct metabolic profile compared to M1-like macrophages (100). In vitro, the metabolic profile of macrophages activated with IL-4 (M2-like stimuli) is distinct from those activated with lipopolysaccharide (LPS) + IFN-γ (M1-like stimuli) (100). Studies have revealed that IL-4-stimulated macrophages depend on fatty acid oxidation (FAO) known as β-oxidation (101) and glutamine metabolism for the production of key metabolic intermediates such as α-ketoglutarate and succinate (100), to fuel the oxidative tricarboxylic acid cycle (TCA) cycle. IL-4 activates glutamine catabolism in macrophages, and interestingly, glutamine deprivation or inhibition of N-glycosylation decreased M2 polarization and production of the chemokine CCL22 (100). However, macrophages stimulated with LPS and IFN-γ utilize glycolysis and fatty acid synthesis (FAS) (102). Some early work has revealed that enhanced uptake of glucose through the induction of glucose transporter 1 (GLUT1) is a key feature of LPS-activated macrophages (103). Furthermore, A[1,2-13C2] glucose tracer-based metabolomics approach, coupled with mass isotopomer distribution analysis of the newly formed metabolites, revealed that stimulated macrophages are highly glycolytic cells. The expression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK2), which regulates fructose-2,6-bisphosphate concentration and the glycolytic flux, was found to be the key molecular switch between the two phenotypic states and was independent of hypoxia-inducible factor 1α (HIF-1α) activation (104).
Metabolic regulation is a major difference between the extreme states of macrophage polarization and related to their downstream effector function. M1 macrophages preferentially depend on glycolysis, whereas M2 macrophages facilitates ATP production through the oxidative tricarboxylic acid cycle (TCA) coupled with oxidative phosphorylation (OXPHOS) (105–107). Previous work shows that M1 macrophages heavily depend on glycolysis to fulfill their energy demands; however, glycolysis only generates two ATP molecules per unit of glucose whereas the TCA cycle and OXPHOS generates 36 ATP molecules (108). It is speculated that M1 macrophages produce enough energy and intermediate metabolites for quick execution of effector functions like cytokine production and microbial killing (109, 110). Indeed, the metabolic differences between M1 and M2 macrophages are critical for their effector function. M1 and M2 macrophages display major differences in L-arginine metabolism via nitric oxide synthase (NOS) or arginase, respectively, which ultimately impact their functional outcome. NOS2 is activated through proinflammatory cytokines such as IFNγ, TNFα, IL-1β, and LPS (111) and has been well-characterized to metabolize L-arginine to L-citrulline and cytotoxic nitric oxide (NO; a pro-inflammatory mediator responsible for anti-tumor immune response) (112, 113). The expression of arginase is induced by Th2 cytokines such as IL-4 and IL-13 that activate the signal transducer and activator of transcription 6 (STAT6), which acts with STAT3 and CCAAT/enhancer binding protein β (C/EBPβ) to bind to the arginase 1 enhancer locus. Additionally, GM-CSF, TGFβ, prostaglandin E2 (PGE2), cyclic adenosine monophosphate (cAMP), and TLR agonists can induce the expression of arginase 1 (111). In M2 macrophages, arginase catalyzes L-arginine to urea and L-ornithine (113). In M2 (wound repairing) macrophages, ornithine production promotes cell proliferation and repairs tissue damage through generation of polyamines and collagen, which are important for wound repair, but highly immunosuppressive in the TME (114–117) and involved in mediating TME remodeling (118). Recent studies have challenged this dichotomy of macrophage metabolism. A transcriptomic and metabolomics-based analysis of macrophages stimulated either with LPS or Pam3CysSK4 [P3C; a toll-like receptor (TLR) 2 agonist] revealed divergent metabolic responses such as TCA, OXPHOS, and lipid metabolism pathways between both stimuli, which regulated cytokine production and phagocytosis. Unlike LPS, the TLR2 ligand P3C induced a complex metabolic rewiring by upregulating both glycolysis and OXPHOS (119).
The Role of the TME on Mediating TAM Metabolism
The intricate communication between TAMs and the TME has been well-studied (36, 120–123), and the metabolic interactions between TAMs and cancer cells have been recently reviewed (124). There are growing efforts to understand the crosstalk between metabolic pathways, their metabolites, and the intracellular signaling in macrophages that in turn affects the epigenetic and transcriptional profile, which ultimately controls macrophage plasticity (100, 105, 125–127). One recent study showed that lactic acid production from tumor cells was an essential component in driving the M2-like polarization of TAMs by upregulating ARG1 and vascular endothelial growth factor A (VEGFA), with the effect of lactic acid being dependent on HIF-1α (128). Another study showed that lactate released from cancer cells acts as ligand for G protein-coupled receptor 132 (Gpr132, a pH sensor) that converts macrophages to an immunosuppressive M2-like phenotype. Importantly, genetic deletion of Gpr132 impaired M2-like macrophage phenotype with reduced tumor burden and metastasis in a preclinical model of breast cancer (129). In line with this, in areas of hypoxia, TAMs upregulate HIF-1α, which enables migration and survival (130), suggesting a feedback loop and signaling between tumor cells and TAMs that regulate their metabolic phenotype. Tumor-derived lactic acid has also been shown to induce the activation of the angiopoietin receptors (Tie1 and Tie2) and AXL receptors on TAMs. These receptors have been associated with pro-angiogenic and immunosuppressive phenotypes (131, 132). Similarly, macrophages treated with conditioned media from thyroid cancer cells underwent functional reprogramming. This effect was partly mediated by tumor cell-derived lactate that induced upregulation of cytokine production through an AKT1/mTOR-dependent increase in aerobic glycolysis. Immunohistochemistry (IHC) analysis confirmed the increase in glycolytic enzymes and lactate receptor in TAMs from human tumors (133).
Tumor cell-derived long-chain fatty acids including oleic acid have been shown to reprogram mitochondrial respiration in TAMs and subsequently induce polarization to an immune-suppressive phenotype through activation of the mTORC signaling pathway (134). In another study, tumor cell-derived succinate resulted in TAM polarization toward a pro-tumor phenotype through a succinate receptor (SUCNR1) and was dependent on the PI3K/HIF1α signaling pathway that resulted in enhanced metastasis (135). Similarly, prostaglandin E2 (PGE2), a prostanoid lipid synthesized by tumor cells, has been reported to polarize TAMs toward a pro-tumor phenotype through the cyclic AMP-responsive element binding (CREB) pathway (136, 137). These studies indicate that there is an intricate relationship between the factors secreted by the TME that dictates the outcome of macrophage phenotype and function. Additional studies to evaluate the differences of how the TME at different metastatic sites regulates TAM metabolism are warranted. An in-depth understanding of the crosstalk between TAMs and TME will be critical to design therapeutic strategies to target TAMs at both the primary and metastatic sites.
Clinical Significance of Tams in Breast Tumors
TAM infiltration is associated with poor prognosis, in a variety of cancers, including breast cancer (61, 92, 138, 139). This finding has been corroborated in a meta-analysis where TAM density significantly correlated with poor survival in patients with breast cancer (140). In an analysis of 11,000 breast tumors, the immune cell type that correlated most significantly with poor clinical outcome in estrogen receptor-positive (ER+) breast tumors was the presence of TAMs (141, 142). Clinically, the presence of TAMs is associated with metastasis (44) and poor survival (45–47) and has been shown to induce endocrine therapy resistance in ER+ breast cancer cells in vitro and in vivo through NF-κB- and IL-6-dependent signaling pathways (143). Importantly, a higher fraction of M1-like TAMs in ER+ breast cancer correlated with a higher pathological complete response (pCR) rate as well as prolonged disease-free survival (DFS) and overall survival (OS) (142). In a cohort of 40 HER2+ breast cancer patients who received trastuzumab, an anti-HER2 therapy, a high number of M1-like macrophages (iNOS+) were significantly associated with improved survival whereas high expression of M2-like macrophages (CD163+) were associated with worse prognosis (144). Additionally, high numbers of iNOS+ M1-like macrophages combined with high numbers of CD8+ T cells were significantly associated with improved survival, and this combined marker predicted a patient's ability to remain progression-free without trastuzumab after initially responding to therapy (144). This is in line with other reports demonstrating low TAM and high CD8+ T cell populations are associated with better recurrence-free survival in patients with invasive breast cancer (145).
Relative Numbers of Macrophages Across Breast Tumor Subsets
Evidence for differential TAM regulation between breast tumor subsets comes from ER+ and triple-negative breast cancer (TNBC) cell lines co-cultured with macrophages. Tumor-induced phenotypic drift toward M2-like phenotype has been shown in differentiating bone marrow-derived macrophages (BMDMs) in breast cancer cell-conditioned media (146). Additionally, macrophages exposed to a TNBC cell line upregulated CCL2, reinforcing the concept that macrophage infiltration is a vicious feedback loop. In contrast, ER+ breast cancer cells co-cultured with macrophages were more broadly pro-inflammatory, secreting CXCL10, IL-2RA, and IL-3 (147). Furthermore, macrophage infiltration by breast cancer subtype has been explored using gene expression data with adjacent normal tissue as a baseline, which reported lower macrophage scores in ER+ tumors, especially ER+/HER2– subsets, in comparison to ER– tumors (148). A recent study examined immune-rich ER+ and immune-rich TNBC tumors based on microarray expression scores and compared relative fractions of immune cells. Although TNBC had a higher overall macrophage count, ER+ tumors had a higher relative percentage of M2-like macrophages and upregulation of TGF-β expression, which are both indicators of poor prognosis (149). TGF-β and IL-4 have been shown to upregulate YKL-39, a chitinase-like protein secreted by TAMs, which facilitates monocyte recruitment and angiogenesis. Importantly, YKL-39 expression levels were six times higher in patients with metastases than without metastases after neoadjuvant chemotherapy (NAC), independent of tumor stage or grade (150). YKL-39 has also been shown to be elevated in estrogen receptor 1 (ESR1) mutant metastatic breast cancer (151) and has been shown to be regulated by the androgen receptor in prostate cancer (152). These differences highlight unique targeted therapies, such as targeting the TGF-β pathway or reprogramming M2-like macrophages into M1-like macrophages, as having high potential, especially in ER-positive breast cancer.
Immunohistochemistry (IHC) has revealed that TNBC has significantly more tumor-infiltrating T cells and macrophages compared to non-TNBCs (153). To further understand the extent of macrophage infiltration in TNBC, immune infiltrates in 41 TNBC tumors were characterized using multiplexed ion beam imaging by time of flight (MIBI-TOF) (154). The fraction of infiltrating immune cells ranged widely from 1 to 91% of cells in the analysis fields selected from each tumor. On average, macrophages were among the most abundant immune cell in the tumor region, accounting for 25% of the immune population. Other immune populations also significantly contributed to the immune landscape, including the following: CD8+ (19%), CD4+ (15%), regulatory T cell (1%), B-cells (11%), and NK cells (<1%). However, these numbers varied greatly between patients. In fact, macrophages comprised as high as 100% of the immune population in tumors with low immune infiltration or as low as 5% in more immunologically abundant TNBC tumors. Our group has recently investigated macrophage abundance in TNBC using multiplex cyclic immunofluorescence (CyCIF) (155–158). We also found that TAM infiltration can vary greatly between tumors (Figure 3). In addition, we recently reported that TNBC harboring mutations in the breast cancer gene 1 (BRCA1) has a nearly 10-fold increase of TAMs compared to BRCA1-wildtype TNBC (159). This work highlights that tumor cell intrinsic mutations, such as BRCA mutations, may play a key role in regulating the TME. Indeed, the current and other studies have shown that BRCA-deficient cancer cells have an increase in cytosolic DNA leading to STING pathway activation and secretion of type 1 interferon-related cytokines such as CXCL9 and CXCL10 that can recruit immune cells, including macrophages, to the tumor (160, 161). Another work has confirmed an increase in CD68+ macrophages in TNBC compared to other subsets, and interestingly, breast tumors with high frequency of c-Myb-positive cells, identified through mRNA levels in breast cancer patients from public datasets, were correlated with a lower density of CD68+ macrophages, which was also found within the molecular subtypes (162).
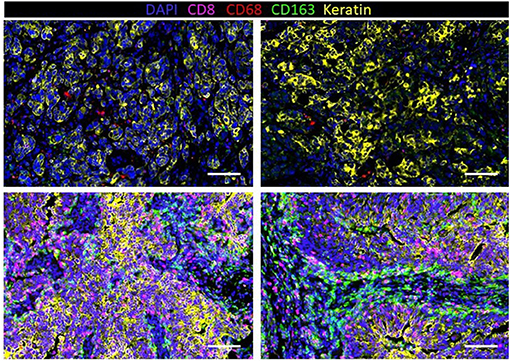
Figure 3. Infiltration of macrophages (CD68 and CD163) and T cells (CD8) in human triple-negative breast cancer (TNBC) with a low density of immune cells (top row) and high density (bottom row).
Relative Numbers of Macrophages in Breast Cancer Progression
Not only does macrophage abundance vary between breast cancer subtypes but also with the stage of cancer progression. In preclinical models, macrophages have been shown to orchestrate early breast cancer dissemination and metastasis in a mouse model of HER2+ breast cancer, where CCL2 produced by both cancer cells and myeloid cells recruited CD206+/Tie2+ macrophages to propagate the disease (163). Similar results were seen in the MMTV-PyMT murine model of luminal B breast cancer, where blocking CCL2 prevented recruitment of TAMs to breast tumors, reduced metastasis, and prolonged the survival of mice (16). Other preclinical studies have shown a significant correlation between CSF-1 and breast cancer metastasis using the MMTV-PyMT model (26).
Compared to normal breast tissue, macrophage numbers are significantly higher in ductal carcinoma in situ (DCIS) and remain higher through progression to invasive ductal carcinoma (IDC) (164). TAMs from human breast cancer have been shown to have a distinct transcriptomic signature from macrophages in healthy breast tissue, which is enriched in more aggressive forms of breast cancer (165). Additionally, CD68+ macrophages (166, 167), especially those defined by authors as M2 macrophages (CD68+PCNA+), have been shown to be elevated in high-grade compared to low-grade DCIS (167). Recent work by Gil Del Alcazar et al. compared immune cell infiltration in HER2+ and TNBC as a function of disease progression by comparing DCIS to IDCs (168). IDC had higher numbers of macrophages compared to DCIS. Interestingly, in DCIS, higher fractions of CD8+ T cells were associated with a significantly higher frequency of macrophages. Gene expression profiling revealed that Th1 and Th2 cells were enriched in HER2+ IDCs, while TNBC IDCs had enrichment of Th17 cells and T regulatory cells (Tregs). For TNBC tumors, but not HER2+ tumors, the transition from DCIS to IDCs correlated with an increased number of TILs, but fewer were in the activated state, indicative of T cell exhaustion in advanced stages of TNBC. Macrophages were not examined in this context, indicating that further characterization is needed to fully understand how macrophages participate in different stages of cancer progression.
Location of Macrophages in Breast Tumors
In addition to breast cancer subtype and disease state, the location of macrophages within the TME may be a predictor of their function and correlate with clinical outcome. However, the field has not yet come to a consensus on the extent of region-specific TAM behavior as a prognostic factor. Generally, macrophages in the stroma are associated with angiogenesis, immunosuppression, and cancer cell migration. In contrast, macrophages in the cancer nest tend to be more heterogeneous across cancer types and are correlated with reduced overall survival (OS) and recurrence-free survival (RFS) in breast cancer (169). Gwak et al. found stromal, intratumoral, and total TAMs to all have similar prognostic value, while Merdeck et al. reported stromal TAMs, but not tumor nest TAMs, are significantly correlated with tumor size and high tumor grade (45, 170). Recently, CD68 and CD163 were evaluated in both tumor nests and tumor stroma in 60 specimens of invasive breast cancer. High numbers of CD68+ TAMs in the tumor stroma were significantly associated with larger tumor size and positive nodal metastasis, whereas high numbers of CD163+ TAMs in the tumor stroma were significantly associated with positive vascular invasion, nodal metastasis, and molecular subtypes (171). Macrophage behavior may be modulated by breast cancer subtype and TME location, which in turn may regulate tumor behavior with stimulatory and inhibitory signals. For example, in HER2+ and basal-like subtypes, macrophages concentrate in invasive fronts and use TGF-β signaling to thicken the extracellular matrix (ECM), contributing to breast tumor metastasis (172). Therefore, not only are macrophages shaped by their location within the TME but they also reciprocally influence the surrounding TME composition. The significance of histologic localization of TAMs and the degree of TAM infiltration add additional layers of complexity to targeting TAMs in breast tumors for anti-cancer therapy.
Macrophage Characterization After Therapy
The characterization of TAMs before and after therapy may provide insight to how TAMs may change with treatment and play a role in drug resistance and metastasis. Increases in macrophages have been reported after chemotherapy in both preclinical and clinical settings (145). In a primarily ER+ patient cohort, neoadjuvant chemotherapy increased the percentage of CSF-1R+ macrophages (173). In line with these findings, Waks et al. examined changes in TAM populations after neoadjuvant chemotherapy in ER+ breast cancer, reporting an influx of CD68 macrophages with a larger proportion associating with an M2-like phenotype (174). These findings are in line with preclinical studies showing chemotherapy significantly increased F4/80+ macrophages in chemotherapy-sensitive tumors, but not chemotherapy-resistant tumors, indicating that macrophages are recruited to the site of chemotherapy-induced apoptosis and play a role in drug response (175). These changes in the TME could be implicated in mechanisms of chemotherapy resistance.
Macrophages as a Barrier to Chemotherapy and Immunotherapy
While the heterogeneity of TAM populations is still being deconvoluted, clinical evidence suggests that macrophages can contribute to shortcomings of chemo- and hyphenate immuno-therapy. High TAM density has been shown to be an independent poor prognostic marker in breast cancer patients, especially for HR-positive breast cancer (170). Importantly, macrophages have been shown to contribute to reduced efficacy of chemotherapy. Shree et al. discovered that in vitro cathepsin-expressing BMDMs shield mammary cancer cells from paclitaxel-induced cell death. They further demonstrated that tumors from MMTV-PyMT mice treated with paclitaxel had increased TAM infiltration, and cathepsin inhibition in combination with paclitaxel increased long-term survival (176). Similarly, tamoxifen-resistant ER-positive and HER2-negative clinical samples had a higher density of CD163+ macrophage populations and increased expression of EGFR than tamoxifen-sensitive samples, which positively correlated with tumor size and metastasis (177). Furthermore, macrophages can disrupt T cell infiltration, which immunotherapies rely on to mediate efficacy. For example, in lung cancer patients, macrophage exclusion of CD8+ T cells from tumor nests correlated with poor response to anti-PD-1 therapy. When macrophages were depleted with anti-CSF-1R therapy, CD8 T cells successfully infiltrated the tumor to interact with malignant cells and delay tumor progression (178). Also, importantly, in preclinical breast cancer models, when macrophages are depleted using anti-CSF-1R therapy (145, 178) or their phenotype was converted to an anti-tumor phenotype (179), anti-PD-1 therapy induced potent anti-tumor immunity. This highlights the deleterious effects of TAMs in tumors and the importance of targeting both innate and adaptive immune cells to achieve the full potential of immunotherapy and a durable anti-cancer immune response.
Targeting TAMs for Anti-Cancer Therapy
Both preclinical and clinical strategies to target the tumor-promoting functions of TAMs in cancer are being developed. These approaches have been reviewed in great detail and include inhibiting the recruitment of macrophages to tumors by blocking the CCL2–CCR2 or CCR5–CCL5 axes, depleting TAMs by blocking CSF-1 or CSF-1R; blocking macrophage “checkpoint inhibitors” such as CD47/SIRP1α, PD-1/PD-L1, MHCI/LILRB1, and CD24/Siglec-10; and suppressing macrophages' pro-tumor activity (inhibition of TGF-β or VEGF) (36, 180–184). Depletion or inhibition of macrophages using CCL2, CSF-1, and CSF-1R inhibitors has been shown to be effective against both mouse and human tumors (16, 36, 145, 185). Importantly, a recent study showed that CCL2 inhibition as a monotherapy led to more metastasis when the therapy was discontinued, which was driven in an IL-6- and VEGF-dependent manner (186). This study challenges the use of CCL2 as a monotherapy and highlights the need to understand the tumor microenvironment composition for successful anti-metastatic therapy.
CSF-1R is a promising target to address TAMs therapeutically, as high expression of CSF-1 or CSF-1R predicts cancer progression and mortality (187). Blockade of CSF-1R has been shown to decrease TAM infiltration, which subsequently results in the increase in CD8+ T cells and improves response to chemotherapy (95, 145). In a phase Ib study with advanced solid tumors, the combination of pexidartinib, a CSF-1R inhibitor, and paclitaxel was well-tolerated, and the combination showed reduced macrophage infiltration in the tumor microenvironment (188). However, in another phase I a/b study, emactuzumab, a monoclonal antibody against CSF-1R, showed reduction in immunosuppressive TAMs but did not demonstrate clinical benefit alone or in combination with paclitaxel (189). These studies suggest that a careful evaluation of the TME is important before deciding which patients would best benefit from CSF-1R therapy. Other caveats to anti-CSF-1R therapies include reports showing that inhibition of CSF-1R signaling can promote breast cancer metastasis (190).
To enhance anti-CSF-1R therapies, combining anti-CSF-1R with complementary chemotherapy and agents that enhance T cell function may markedly improve outcomes. In that regard, a recent study showed that addition of a CD40 agonist before anti-CSF-1R therapy induced a short-lived hyperactivated macrophage state that was enough to generate an effective T cell response in ICB-resistant tumors (191). Additionally, we have recently shown that CSF-1R inhibition leads to a significant reduction in TAMs and when combined with PARP inhibitor therapy results in an increase in overall survival, with some mice experiencing tumor-free survival for at least 1 year (159). Studies with CSF-1R signaling antagonists, combined with the drug paclitaxel or carboplatin, showed enhanced tumor control and reduced metastasis in preclinical models of breast cancer. Importantly, blockade of CSF-1 signaling also enhanced anti-tumor immunity and cytotoxic T cell infiltration to chemotherapy (145). The blockade of the CCR5–CCL5 axis, which decreased macrophage infiltration in tumors, is another exciting therapeutic target with ongoing clinical trials for breast cancer (192).
An alternative strategy is to convert pro-tumor TAMs to an anti-tumor phenotype. CD40 agonists (193), PI3Kγ inhibitors (194), CD47 inhibitors (195), and a class IIa HDAC inhibitor (179) have all been shown to reduce primary and metastatic murine breast tumors (179) and have emerged as novel modalities to convert TAMs to anti-tumor macrophages. In addition, other strategies have been shown to convert TAMs to an M1 phenotype and include Bruton's tyrosine kinase (BTK) inhibitors (196), TLR agonists (197), STAT3 inhibitors (198), IL-1Ra inhibitors (199), and LILRB2 inhibitors (200) Taken together, strategies to deplete or inhibit suppressive TAM functions or activate anti-tumor TAMs combined with chemotherapy and/or immunotherapy may have a great potential for the treatment of breast cancer patients. However, while many of these compounds in preclinical and clinical development are now filling our toolbox with TAM-targeting strategies, it will likely be necessary to further elucidate the complexity of TAM subsets including their ontogeny and phenotype for successful therapeutic targeting (Figure 4).
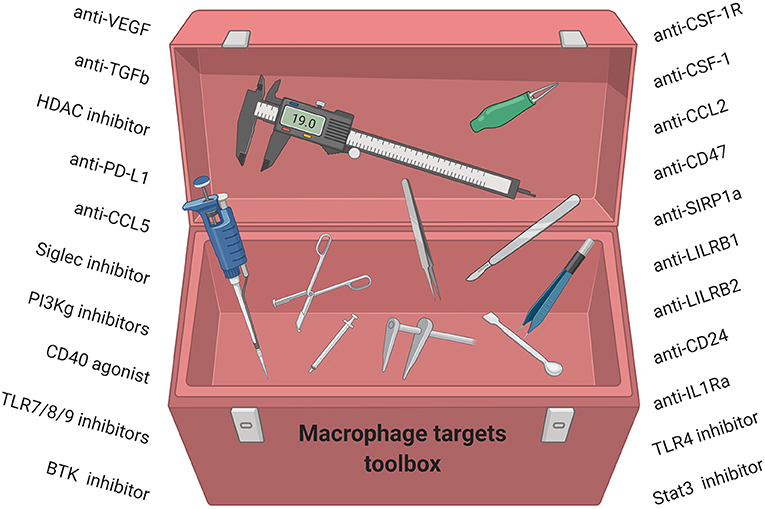
Figure 4. Macrophage-targeting strategies for anti-cancer therapy have started to fill our toolbox. We now need to understand how these compounds work, which subsets of TAMs they modulate, and which breast cancer patients will benefit.
Therapeutic Targeting of TAM Metabolism for Anti-Cancer Therapy
The metabolic programming of TAMs is complex, and the underlying molecular mechanisms and crosstalk between tumor cells and stroma remain to be characterized. An in-depth analysis of these metabolic circuits may facilitate better appreciation for the functional fates of macrophages, including their pro- vs. anti-tumor phenotype. This important information would further support the clinical application of targeting TAM metabolism for anti-cancer therapy. There is some insight of the potential of this strategy from several recent publications that utilized other immune cell types including Tregs. Recently, Tregs were shown to activate the sterol regulatory element-binding protein 1 (SREBP1)-mediated fatty acid synthesis pathway in TAMs. SREBP1 induced M2-TAM metabolic fitness, mitochondrial integrity, and survival (201). Pharmacological inhibition of de novo fatty acid synthesis using a SREBP1 inhibitor, fatostatin, showed anti-tumor immunity when combined with ICB (anti-PD-1) in a B16 melanoma preclinical tumor model (201). Our group recently reported that PARP inhibition directly modulated macrophage metabolism by shunting glycolysis and inducing a dependence on lipid metabolism, which generated an immunosuppressive TME by inhibiting T cell function and thereby contributed to PARP inhibitor resistance (159). The use of fatostatin in combination with PARP inhibition and macrophage modulation significantly enhanced the overall survival of mice bearing brca1-deficeint TNBC (159). In line with our findings, inhibition of PARP induced upregulation of lipogenic genes by modulating the transcription factor specificity protein 1 (Sp1), which leads to the accumulation of lipid droplets in the liver (202). Similarly, a study suggested that genetic deletion as well as pharmacologic inhibition of PARP induced the expression of ATP-binding cassette transporters (ABCA1) and cholesterol efflux in macrophages (203). These studies highlight the role of both Tregs and PARP inhibitors in regulating macrophage lipid metabolism. Further molecular understanding on the mechanisms of how PARP inhibitors regulate TAM metabolism would provide future opportunity for promising therapeutic strategies.
In a preclinical syngeneic model of pancreatic ductal adenocarcinoma (PDAC), a TLR9 agonist, CpG oligodeoxynucleotide, induced a metabolic state that required fatty acid oxidation and shunting of TCA intermediates for de novo lipid biosynthesis. This shift in central carbon metabolism activated highly phagocytic macrophage that could overcome the CD47 “don't-eat-me” signals on tumor cells to mediate an antitumor response (204). Macrophages cultured with PDAC-conditioned media compared to normal pancreatic cells had higher levels of vascular network formation, enhanced metastatic potential, increased levels of EMT, and a pronounced glycolytic signature. Inhibiting hexokinase II (HK2) with 2-deoxyglucose (2DG) inhibited glycolysis and reversed the pro-tumor TAM phenotype, highlighting the therapeutic potential of modulating TAM metabolism for anti-cancer therapy (205). Molecular metabolic control of TAMs has been demonstrated in vitro by inhibiting glutamine synthetase (GS). In human monocytes, GS expression activates an M2-like phenotype, which is reversed through pharmacological inhibition of GS by methionine sulfoximine (MSO). Inhibition of GS resulted in production of succinate, a critical regulator of the pro-inflammatory response, and enhanced glucose flux through glycolysis. Importantly, in ex vivo studies, GS restored T cell recruitment. In vivo, genetic deletion of macrophage-specific GS reduced metastasis in a preclinical mouse model of lung cancer (206). Taken together, precisely targeting the metabolic rewiring of TAMs may re-educate their phenotype and may overcome TAM-associated immunosuppression.
Beyond M1 And M2 Phenotypes and Next Steps
To date, characterizing the diversity of macrophage subsets has been difficult, as researchers have relied on a limited number of macrophage markers, and gene expression profiling has been done in bulk tissue or total macrophage populations, which preclude detection of unique subsets. Although gene expression data is high throughput, it inherently lacks the ability to determine spatial relationships, precise cellular function, or biochemical analysis at a single-cell level. Another consideration for leukocyte-containing samples is the canonical expression of RNase, which could potentially degrade RNA transcripts and interfere with single-cell analysis sample quality. Likewise, protein analysis should be used to support gene expression data, but is comparably tedious and low yield. The intracellular glycoprotein CD68 is widely used in clinical studies as a TAM marker, but it also detects other cell types such as some lymphoid and non-hematopoietic cells (207, 208) and does not identify TAM phenotype or functional status. The scavenger receptor CD163 has also been used to identify some TAM populations and has been shown to associate with early recurrence and reduced survival in breast cancer patients (209). CD68 and CD163 have been broadly used to show that high TAM density correlates with a worse clinical outcome in breast cancer but do not predict their functional phenotype (122, 140, 210). As high-resolution techniques uncover the heterogeneity and plasticity of macrophage populations, researchers will need to look beyond the binary M1/M2 nomenclature and incorporate an extensive panel of markers in their analysis. Indeed, TAMs have high plasticity and can express both M1-like and M2-like phenotypes simultaneously, creating a need for more nuanced categorizations of macrophages (169). Multiplex IF such as MIBI-TOF and CyCIF have the unique advantage of analyzing a relatively large number of proteins, while maintaining spatial context of the tumor. These technologies will be invaluable in the next era of TAM studies, in which distinct phenotypes will need to be pursued.
Immune-focused mass cytometry has shed light on the diversity of TAMs in breast cancer, where in a study of 144 breast cancer patients, 19 distinct subsets of myeloid cells were identified, which clustered into five broader categories (211). These clusters included CD14-expressing monocytes, early immigrant macrophages, tissue-resident macrophages, TAMs, and myeloid-derived suppressor cells, with each group containing several additional subsets of myeloid cells. The distribution of the 19 clusters was distinct regarding the location within the tumor microenvironment and the subtype of breast cancer. For example, ER– tumors had more PD-L1+ TAMs than ER+ tumors, while luminal B tumors contained a higher proportion of PD-L1+ TAM subsets than luminal A. Although the sample set was predominantly from ER+ tumors, the results expose high-resolution categorizations of macrophages that could shed light on current therapeutic barriers. This work highlights the complexity of macrophages and tumors and is the right step forward; however, it is yet to be understood what the functional significance of these subsets are for clinical outcomes (211). Assays that test the functional ability of macrophages are warranted including high-throughput efferocytosis assays or gene signatures that may predict enhanced efferocytosis. The abovementioned studies have contributed to understanding the immune profile related to macrophages in breast cancer subtypes using bulk and single-cell RNA-sequencing, single- and dual-color IHC, multiplex spatial analysis such as MIBI-TOF and CyCIF, as well as flow cytometry, which compile data with different perspectives and limitations. As researchers continue to assess the breast TME, it is critical to acknowledge the strengths and limitations of different methods regarding their resolution, accuracy, and ability to represent tumor heterogeneity.
TAMs play a major role in tumor progression and metastasis and promote a highly suppressive TME that may limit breast cancer therapy. Therefore, modulating TAMs in combination with chemo- and/or hyphenated immuno-therapy will be critical to achieve maximum tumor reduction and elimination. Thus, it is imperative to characterize TAM subsets and their location. In addition, differences among breast tumor subtypes and how TAMs change after therapy will be important to characterize, along with differences in tissue-resident macrophages at metastatic sites. To develop effective macrophage-targeting therapies for the treatment of breast cancer, it is critical to have a precise understanding of the unique macrophage populations in each subset of breast cancer, as well as their location within different regions of the tumor. In addition, when considering the variety of influences that macrophages have on the TME, it is apparent that the surrounding TME will also need to be considered. Therefore, it is crucial to assess the number of infiltrating macrophages, their phenotype and function, as well as their spatial location to other cells in the TME. Expanding beyond M1 and M2 macrophage nomenclature will enhance the field of cancer immunology by providing better understanding of the tumor environment for rationale design of immunotherapy strategies including future development of macrophage-targeting therapies (Figure 4).
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
JG was supported by the Dana-Farber/Harvard Cancer Center (DF/HCC) Specialized Program of Research Excellence (SPORE) in Breast Cancer P50 CA1685404 Career Enhancement Award, The Susan G. Komen Foundation Career Catalyst Award CCR18547597, The Terri Brodeur Breast Cancer Foundation, and The Ludwig Center at Harvard.
Conflict of Interest
JG is a consultant for Glaxo-Smith Kline (GSK), Codagenix, Verseau, Kymera, and Array BioPharma and receives sponsored research support from GSK, Array BioPharma, and Eli Lilly.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Images were created with BioRender.com.
References
1. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. (2013) 496:445–55. doi: 10.1038/nature12034
2. Okabe Y, Medzhitov R. Tissue biology perspective on macrophages. Nat Immunol. (2016) 17:9–17. doi: 10.1038/ni.3320
3. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. (2005) 5:953–64. doi: 10.1038/nri1733
4. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
5. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. (2014) 40:274–88. doi: 10.1016/j.immuni.2014.01.006
6. Chavez-Galan L, Olleros ML, Vesin D, Garcia I. Much more than M1 and M2 macrophages, there are also CD169(+) and TCR(+) macrophages. Front Immunol. (2015) 6:263. doi: 10.3389/fimmu.2015.00263
7. Kapellos TS, Bonaguro L, Gemünd I, Reusch N, Saglam A, Hinkley ER, et al. Human monocyte subsets and phenotypes in major chronic inflammatory diseases. Front Immunol. (2019) 10:2035. doi: 10.3389/fimmu.2019.02035
8. Haloul M, Oliveira ERA, Kader M, Wells JZ, Tominello TR, El Andaloussi A, et al. mTORC1-mediated polarization of M1 macrophages and their accumulation in the liver correlate with immunopathology in fatal ehrlichiosis. Sci. Rep. (2019) 9:14050. doi: 10.1038/s41598-019-50320-y
9. Jayasingam SD, Citartan M, Thang TH, Mat Zin AA, Ang KC, Ch'ng ES. Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: technicalities and challenges in routine clinical practice. Front Oncol. (2019) 9:1512. doi: 10.3389/fonc.2019.01512
10. O'Sullivan C, Lewis CE. Tumour-associated leucocytes: friends or foes in breast carcinoma. J Pathol. (1994) 172:229–35. doi: 10.1002/path.1711720302
11. De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. (2013) 23:277–86. doi: 10.1016/j.ccr.2013.02.013
12. Leek RD, Harris AL. Tumor-associated macrophages in breast cancer. J Mammary Gland Biol Neoplasia. (2002) 7:177–89. doi: 10.1023/A:1020304003704
13. Ueno T, Toi M, Saji H, Muta M, Bando H, Kuroi K, et al. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin Cancer Res. (2000) 6:3282–9.
14. Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. (2012) 72:2768–79. doi: 10.1158/0008-5472.CAN-11-3567
15. Goede V, Brogelli L, Ziche M, Augustin HG. Induction of inflammatory angiogenesis by monocyte chemoattractant protein-1. Int J Cancer. (1999) 82:765–70. doi: 10.1002/(SICI)1097-0215(19990827)82:5<765::AID-IJC23>3.0.CO;2-F
16. Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. (2011) 475:222–5. doi: 10.1038/nature10138
17. Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. (2015) 212:1043–59. doi: 10.1084/jem.20141836
18. Luboshits G, Shina S, Kaplan O, Engelberg S, Nass D, Lifshitz-Mercer B, et al. Elevated expression of the CC chemokine regulated on activation, normal T cell expressed and secreted (RANTES) in advanced breast carcinoma. Cancer Res. (1999) 59:4681–7.
19. Soria G, Ben-Baruch A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. (2008) 267:271–85. doi: 10.1016/j.canlet.2008.03.018
20. Bieche I, Lerebours F, Tozlu S, Espie M, Marty M, Lidereau R. Molecular profiling of inflammatory breast cancer: identification of a poor-prognosis gene expression signature. Clin Cancer Res. (2004) 10:6789–95. doi: 10.1158/1078-0432.CCR-04-0306
21. An G, Wu F, Huang S, Feng L, Bai J, Gu S, et al. Effects of CCL5 on the biological behavior of breast cancer and the mechanisms of its interaction with tumor-associated macrophages. Oncol Rep. (2019) 42:2499–511. doi: 10.3892/or.2019.7344
22. Walens A, DiMarco AV, Lupo R, Kroger BR, Damrauer JS, Alvarez JV. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. Elife. (2019) 8:e43653. doi: 10.7554/eLife.43653
23. Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, et al. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by Anti-CCR5 therapy in cancer patients. Cancer Cell. (2016) 29:587–601. doi: 10.1016/j.ccell.2016.03.005
24. Fu YX, Cai JP, Chin YH, Watson GA, Lopez DM. Regulation of leukocyte binding to endothelial tissues by tumor-derived GM-CSF. Int J Cancer. (1992) 50:585–8. doi: 10.1002/ijc.2910500416
25. Scholl SM, Pallud C, Beuvon F, Hacene K, Stanley ER, Rohrschneider L, et al. Anti-colony-stimulating factor-1 antibody staining in primary breast adenocarcinomas correlates with marked inflammatory cell infiltrates and prognosis. J Natl Cancer Inst. (1994) 86:120–6. doi: 10.1093/jnci/86.2.120
26. Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. (2001) 193:727–40. doi: 10.1084/jem.193.6.727
27. Aharinejad S, Abraham D, Paulus P, Abri H, Hofmann M, Grossschmidt K, et al. Colony-stimulating factor-1 antisense treatment suppresses growth of human tumor xenografts in mice. Cancer Res. (2002) 62:5317–24.
28. Clauss M, Gerlach M, Gerlach H, Brett J, Wang F, Familletti PC, et al. Vascular permeability factor: a tumor-derived polypeptide that induces endothelial cell and monocyte procoagulant activity, and promotes monocyte migration. J Exp Med. (1990) 172:1535–45. doi: 10.1084/jem.172.6.1535
29. Leek RD, Hunt NC, Landers RJ, Lewis CE, Royds JA, Harris AL. Macrophage infiltration is associated with VEGF and EGFR expression in breast cancer. J Pathol. (2000) 190:430–6. doi: 10.1002/(SICI)1096-9896(200003)190:4<430::AID-PATH538>3.0.CO;2-6
30. Turner L, Scotton C, Negus R, Balkwill F. Hypoxia inhibits macrophage migration. Eur J Immunol. (1999) 29:2280–7. doi: 10.1002/(SICI)1521-4141(199907)29:07<2280::AID-IMMU2280>3.0.CO;2-C
31. Leek RD, Landers RJ, Harris AL, Lewis CE. Necrosis correlates with high vascular density and focal macrophage infiltration in invasive carcinoma of the breast. Br J Cancer. (1999) 79:991–5. doi: 10.1038/sj.bjc.6690158
32. Grimshaw MJ, Balkwill FR. Inhibition of monocyte and macrophage chemotaxis by hypoxia and inflammation–a potential mechanism. Eur J Immunol. (2001) 31:480–9. doi: 10.1002/1521-4141(200102)31:2<480::AID-IMMU480>3.0.CO;2-L
33. Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. (2013) 24:695–709. doi: 10.1016/j.ccr.2013.11.007
34. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
35. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
36. Guerriero JL. Macrophages: the road less traveled, changing anticancer therapy. Trends Mol Med. (2018) 24:472–89. doi: 10.1016/j.molmed.2018.03.006
37. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. (2010) 327:656–61. doi: 10.1126/science.1178331
38. Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. (2011) 332:1284–8. doi: 10.1126/science.1204351
39. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. (2012) 336:86–90. doi: 10.1126/science.1219179
40. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science. (2014) 344:921–5. doi: 10.1126/science.1252510
41. Italiani P, Boraschi D. From monocytes to M1/m2 macrophages: phenotypical vs. functional differentiation. Front Immunol. (2014) 5:514. doi: 10.3389/fimmu.2014.00514
42. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. (2009) 86:1065–73. doi: 10.1189/jlb.0609385
43. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. (2010) 141:39–51. doi: 10.1016/j.cell.2010.03.014
44. Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. (2006) 124:263–6. doi: 10.1016/j.cell.2006.01.007
45. Medrek C, Ponten F, Jirstrom K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer. (2012) 12:306. doi: 10.1186/1471-2407-12-306
46. Tymoszuk P, Charoentong P, Hackl H, Spilka R, Muller-Holzner E, Trajanoski Z, et al. High STAT1 mRNA levels but not its tyrosine phosphorylation are associated with macrophage infiltration and bad prognosis in breast cancer. BMC Cancer. (2014) 14:257. doi: 10.1186/1471-2407-14-257
47. Lan C, Huang X, Lin S, Huang H, Cai Q, Wan T, et al. Expression of M2-polarized macrophages is associated with poor prognosis for advanced epithelial ovarian cancer. Technol Cancer Res Treat. (2013) 12:259–67. doi: 10.7785/tcrt.2012.500312
48. Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS ONE. (2010) 5:e9539. doi: 10.1371/journal.pone.0009539
49. Das A, Sinha M, Datta S, Abas M, Chaffee S, Sen CK, et al. Monocyte and macrophage plasticity in tissue repair and regeneration. Am J Pathol. (2015) 185:2596–606. doi: 10.1016/j.ajpath.2015.06.001
50. Rigamonti E, Zordan P, Sciorati C, Rovere-Querini P, Brunelli S. Macrophage plasticity in skeletal muscle repair. Biomed Res Int. (2014) 2014:560629. doi: 10.1155/2014/560629
51. Italiani P, Mazza EM, Lucchesi D, Cifola I, Gemelli C, Grande A, et al. Transcriptomic profiling of the development of the inflammatory response in human monocytes in vitro. PLoS ONE. (2014) 9:e87680. doi: 10.1371/journal.pone.0087680
52. Mylonas KJ, Nair MG, Prieto-Lafuente L, Paape D, Allen JE. Alternatively activated macrophages elicited by helminth infection can be reprogrammed to enable microbial killing. J Immunol. (2009) 182:3084–94. doi: 10.4049/jimmunol.0803463
53. Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. (2005) 175:342–9. doi: 10.4049/jimmunol.175.1.342
54. Nandi B, Shapiro M, Samur MK, Pai C, Frank NY, Yoon C, et al. Stromal CCR6 drives tumor growth in a murine transplantable colon cancer through recruitment of tumor-promoting macrophages. Oncoimmunology. (2016) 5:e1189052. doi: 10.1080/2162402X.2016.1189052
55. Lee CH, Wu CL, Shiau AL. Toll-like receptor 4 signaling promotes tumor growth. J Immunother. (2010) 33:73–82. doi: 10.1097/CJI.0b013e3181b7a0a4
56. Su S, Liu Q, Chen J, Chen J, Chen F, He C, et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell. (2014) 25:605–20. doi: 10.1016/j.ccr.2014.03.021
57. Zumsteg A, Christofori G. Corrupt policemen: inflammatory cells promote tumor angiogenesis. Curr Opin Oncol. (2009) 21:60–70. doi: 10.1097/CCO.0b013e32831bed7e
58. Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. (2004) 114:623–33. doi: 10.1172/JCI200422087
59. Chun TH, Sabeh F, Ota I, Murphy H, McDonagh KT, Holmbeck K, et al. MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J Cell Biol. (2004) 167:757–67. doi: 10.1083/jcb.200405001
60. O'Sullivan C, Lewis CE, Harris AL, McGee JO. Secretion of epidermal growth factor by macrophages associated with breast carcinoma. Lancet. (1993) 342:148–9. doi: 10.1016/0140-6736(93)91348-P
61. Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. (1996) 56:4625–9.
62. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. (2006) 66:11238–46. doi: 10.1158/0008-5472.CAN-06-1278
63. Lin L, Chen YS, Yao YD, Chen JQ, Chen JN, Huang SY, et al. CCL18 from tumor-associated macrophages promotes angiogenesis in breast cancer. Oncotarget. (2015) 6:34758–73. doi: 10.18632/oncotarget.5325
64. Chen J, Yao Y, Gong C, Yu F, Su S, Chen J, et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell. (2011) 19:541–55. doi: 10.1016/j.ccr.2011.02.006
65. De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. (2005) 8:211–26. doi: 10.1016/j.ccr.2005.08.002
66. Lewis CE, De Palma M, Naldini L. Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res. (2007) 67:8429–32. doi: 10.1158/0008-5472.CAN-07-1684
67. De Palma M, Venneri MA, Roca C, Naldini L. Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nat Med. (2003) 9:789–95. doi: 10.1038/nm871
68. Pucci F, Venneri MA, Biziato D, Nonis A, Moi D, Sica A, et al. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood. (2009) 114:901–14. doi: 10.1182/blood-2009-01-200931
69. Venneri MA, De Palma M, Ponzoni M, Pucci F, Scielzo C, Zonari E, et al. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood. (2007) 109:5276–85. doi: 10.1182/blood-2006-10-053504
70. Ibberson M, Bron S, Guex N, Faes-van't Hull E, Ifticene-Treboux A, Henry L, et al. TIE-2 and VEGFR kinase activities drive immunosuppressive function of TIE-2-expressing monocytes in human breast tumors. Clin Cancer Res. (2013) 19:3439–49. doi: 10.1158/1078-0432.CCR-12-3181
71. Hagemann T, Wilson J, Kulbe H, Li NF, Leinster DA, Charles K, et al. Macrophages induce invasiveness of epithelial cancer cells via NF-kappa B JNK. J Immunol. (2005) 175:1197–205. doi: 10.4049/jimmunol.175.2.1197
72. Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol. (2011) 21:228–37. doi: 10.1016/j.tcb.2010.12.002
73. Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. (2010) 70:5649–69. doi: 10.1158/0008-5472.CAN-10-1040
74. Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. (2007) 67:2649–56. doi: 10.1158/0008-5472.CAN-06-1823
75. Lin EY, Gouon-Evans V, Nguyen AV, Pollard JW. The macrophage growth factor CSF-1 in mammary gland development and tumor progression. J Mammary Gland Biol Neoplasia. (2002) 7:147–62. doi: 10.1023/A:1020399802795
76. Wang N, Liu W, Zheng Y, Wang S, Yang B, Li M, et al. CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-kappaB/SOX4 signaling. Cell Death Dis. (2018) 9:880. doi: 10.1038/s41419-018-0876-3
77. Zabuawala T, Taffany DA, Sharma SM, Merchant A, Adair B, Srinivasan R, et al. An ets2-driven transcriptional program in tumor-associated macrophages promotes tumor metastasis. Cancer Res. (2010) 70:1323–33. doi: 10.1158/0008-5472.CAN-09-1474
78. Robinson-Smith TM, Isaacsohn I, Mercer CA, Zhou M, Van Rooijen N, Husseinzadeh N, et al. Macrophages mediate inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res. (2007) 67:5708–16. doi: 10.1158/0008-5472.CAN-06-4375
79. Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. (2010) 24:241–55. doi: 10.1101/gad.1874010
80. Cox TR, Rumney RMH, Schoof EM, Perryman L, Høye AM, Agrawal A, et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature. (2015) 522:106–10. doi: 10.1038/nature14492
81. Alhudaithi SS, Almuqbil RM, Zhang H, Bielski ER, Du W, Sunbul FS, et al. Local targeting of lung-tumor-associated macrophages with pulmonary delivery of a CSF-1R inhibitor for the treatment of breast cancer lung metastases. Mol Pharm. (2020) 17:4691–703. doi: 10.1021/acs.molpharmaceut.0c00983
82. Watson GA, Lopez DM. Aberrant antigen presentation by macrophages from tumor-bearing mice is involved in the down-regulation of their T cell responses. J Immunol. (1995) 155:3124–34.
83. Handel-Fernandez ME, Cheng X, Herbert LM, Lopez DM. Down-regulation of IL-12, not a shift from a T helper-1 to a T helper-2 phenotype, is responsible for impaired IFN-gamma production in mammary tumor-bearing mice. J Immunol. (1997) 158:280–6.
84. Dinapoli MR, Calderon CL, Lopez DM. The altered tumoricidal capacity of macrophages isolated from tumor-bearing mice is related to reduce expression of the inducible nitric oxide synthase gene. J Exp Med. (1996) 183:1323–9. doi: 10.1084/jem.183.4.1323
85. Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. (2010) 70:5728–39. doi: 10.1158/0008-5472.CAN-09-4672
86. Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res. (2012) 4:376–89.
87. Reeves E, James E. Antigen processing and immune regulation in the response to tumours. Immunology. (2017) 150:16–24. doi: 10.1111/imm.12675
88. Mittal SK, Roche PA. Suppression of antigen presentation by IL-10. Curr Opin Immunol. (2015) 34:22–7. doi: 10.1016/j.coi.2014.12.009
89. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. (2004) 10:942–9. doi: 10.1038/nm1093
90. Liu J, Zhang N, Li Q, Zhang W, Ke F, Leng Q, et al. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS ONE. (2011) 6:e19495. doi: 10.1371/journal.pone.0019495
91. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J Biomed Sci. (2019) 26:78. doi: 10.1186/s12929-019-0568-z
92. Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. (2002) 196:254–65. doi: 10.1002/path.1027
93. Annacker O, Asseman C, Read S, Powrie F. Interleukin-10 in the regulation of T cell-induced colitis. J Autoimmun. (2003) 20:277–9. doi: 10.1016/S0896-8411(03)00045-3
94. Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. (2005) 8:369–80. doi: 10.1016/j.ccr.2005.10.012
95. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. (2014) 26:623–37. doi: 10.1016/j.ccell.2014.09.006
96. Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. (2012) 188:21–8. doi: 10.4049/jimmunol.1101029
97. Kim OH, Kang GH, Noh H, Cha JY, Lee HJ, Yoon JH, et al. Proangiogenic TIE2(+)/CD31 (+) macrophages are the predominant population of tumor-associated macrophages infiltrating metastatic lymph nodes. Mol Cells. (2013) 36:432–8. doi: 10.1007/s10059-013-0194-7
98. Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. (2006) 203:871–81. doi: 10.1084/jem.20050930
99. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
100. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
101. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. (2006) 4:13–24. doi: 10.1016/j.cmet.2006.05.011
102. Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS, et al. Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol. (2012) 92:829–39. doi: 10.1189/jlb.1111537
103. Fukuzumi M, Shinomiya H, Shimizu Y, Ohishi K, Utsumi S. Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1. Infect Immun. (1996) 64:108–12. doi: 10.1128/IAI.64.1.108-112.1996
104. Rodríguez-Prados JC, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. (2010) 185:605–14. doi: 10.4049/jimmunol.0901698
105. Galván-Peña S, O'Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. (2014) 5:420. doi: 10.3389/fimmu.2014.00420
106. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
107. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. (2013) 38:633–43. doi: 10.1016/j.immuni.2013.04.005
108. Saha S, Shalova IN, Biswas SK. Metabolic regulation of macrophage phenotype and function. Immunol Rev. (2017) 280:102–11. doi: 10.1111/imr.12603
109. Nagy C, Haschemi A. Time and demand are two critical dimensions of immunometabolism: the process of macrophage activation and the pentose phosphate pathway. Front Immunol. (2015) 6:164. doi: 10.3389/fimmu.2015.00164
110. O'Neill LA. A metabolic roadblock in inflammatory macrophages. Cell Rep. (2016) 17:625–6. doi: 10.1016/j.celrep.2016.09.085
111. Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front Immunol. (2017) 8:93. doi: 10.3389/fimmu.2017.00093
112. Yang Z, Ming X-F. Functions of arginase isoforms in macrophage inflammatory responses: impact on cardiovascular diseases and metabolic disorders. Front Immunol. (2014) 5:533. doi: 10.3389/fimmu.2014.00533
113. Geeraerts X, Bolli E, Fendt S-M, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol. (2017) 8:289. doi: 10.3389/fimmu.2017.00289
114. Modolell M, Corraliza IM, Link F, Soler G, Eichmann K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur J Immunol. (1995) 25:1101–4. doi: 10.1002/eji.1830250436
115. Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. Enhancer-mediated control of macrophage-specific arginase I expression. J Immunol. (2004) 172:7565–73. doi: 10.4049/jimmunol.172.12.7565
116. Hayes CS, Shicora AC, Keough MP, Snook AE, Burns MR, Gilmour SK. Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer Immunol Res. (2014) 2:274–85. doi: 10.1158/2326-6066.CIR-13-0120-T
117. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. (2016) 17:684–96. doi: 10.1016/j.celrep.2016.09.008
118. Squadrito ML, Pucci F, Magri L, Moi D, Gilfillan GD, Ranghetti A, et al. miR-511-3p modulates genetic programs of tumor-associated macrophages. Cell Rep. (2012) 1:141–54. doi: 10.1016/j.celrep.2011.12.005
119. Lachmandas E, Boutens L, Ratter JM, Hijmans A, Hooiveld GJ, Joosten LA, et al. Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat Microbiol. (2016) 2:16246. doi: 10.1038/nmicrobiol.2016.246
120. Guerriero JL. Macrophages: Their Untold Story in T Cell Activation and Function. Int Rev Cell Mol Biol. (2019) 342:73–93. doi: 10.1016/bs.ircmb.2018.07.001
121. Franklin RA, Li MO. Ontogeny of tumor-associated macrophages and its implication in cancer regulation. Trends Cancer. (2016) 2:20–34. doi: 10.1016/j.trecan.2015.11.004
122. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. (2015) 27:462–72. doi: 10.1016/j.ccell.2015.02.015
123. DeNardo DG, Johansson M, Coussens LM. Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev. (2008) 27:11–8. doi: 10.1007/s10555-007-9100-0
124. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. (2019) 30:36–50. doi: 10.1016/j.cmet.2019.06.001
125. O'Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. (2016) 213:15–23. doi: 10.1084/jem.20151570
126. Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB, et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest. (2015) 125:4334–48. doi: 10.1172/JCI81676
127. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
128. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
129. Chen P, Zuo H, Xiong H, Kolar MJ, Chu Q, Saghatelian A, et al. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci USA. (2017) 114:580–5. doi: 10.1073/pnas.1614035114
130. Burke B, Giannoudis A, Corke KP, Gill D, Wells M, Ziegler-Heitbrock L, et al. Hypoxia-induced gene expression in human macrophages: implications for ischemic tissues and hypoxia-regulated gene therapy. Am J Pathol. (2003) 163:1233–43. doi: 10.1016/S0002-9440(10)63483-9
131. Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. (2010) 70:5270–80. doi: 10.1158/0008-5472.CAN-10-0012
132. Ruan GX, Kazlauskas A. Lactate engages receptor tyrosine kinases Axl, Tie2, and vascular endothelial growth factor receptor 2 to activate phosphoinositide 3-kinase/Akt and promote angiogenesis. J Biol Chem. (2013) 288:21161–72. doi: 10.1074/jbc.M113.474619
133. Arts RJ, Plantinga TS, Tuit S, Ulas T, Heinhuis B, Tesselaar M, et al. Transcriptional and metabolic reprogramming induce an inflammatory phenotype in non-medullary thyroid carcinoma-induced macrophages. Oncoimmunology. (2016) 5:e1229725. doi: 10.1080/2162402X.2016.1229725
134. Wu H, Han Y, Rodriguez Sillke Y, Deng H, Siddiqui S, Treese C, et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol Med. (2019) 11:e10698. doi: 10.15252/emmm.201910698
135. Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol Cell. (2020) 77:213–27.e215. doi: 10.1016/j.molcel.2019.10.023
136. Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M. CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci USA. (2015) 112:15642–47. doi: 10.1073/pnas.1519644112
137. Na YR, Jung D, Yoon BR, Lee WW, Seok SH. Endogenous prostaglandin E2 potentiates anti-inflammatory phenotype of macrophage through the CREB-C/EBP-β cascade. Eur J Immunol. (2015) 45:2661–71. doi: 10.1002/eji.201545471
138. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. (2004) 4:71–8. doi: 10.1038/nrc1256
139. Mahmoud SM, Lee AH, Paish EC, Macmillan RD, Ellis IO, Green AR. Tumour-infiltrating macrophages and clinical outcome in breast cancer. J Clin Pathol. (2012) 65:159–63. doi: 10.1136/jclinpath-2011-200355
140. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE. (2012) 7:e50946. doi: 10.1371/journal.pone.0050946
141. Ali HR, Chlon L, Pharoah PD, Markowetz F, Caldas C. Patterns of immune infiltration in breast cancer and their clinical implications: a gene-expression-based retrospective study. PLoS Med. (2016) 13:e1002194. doi: 10.1371/journal.pmed.1002194
142. Bense RD, Sotiriou C, Piccart-Gebhart MJ, Haanen J, van Vugt M, de Vries EGE, et al. Relevance of tumor-infiltrating immune cell composition and functionality for disease outcome in breast cancer. J Natl Cancer Inst. (2017) 109:djw192. doi: 10.1093/jnci/djw192
143. Castellaro AM, Rodriguez-Baili MC, Di Tada CE, Gil GA. Tumor-associated macrophages induce endocrine therapy resistance in ER+ breast cancer cells. Cancers. (2019) 11:189. doi: 10.3390/cancers11020189
144. Honkanen TJ, Tikkanen A, Karihtala P, Mäkinen M, Väyrynen JP, Koivunen JP. Prognostic and predictive role of tumour-associated macrophages in HER2 positive breast cancer. Sci Rep. (2019) 9:10961. doi: 10.1038/s41598-019-47375-2
145. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. (2011) 1:54–67. doi: 10.1158/2159-8274.CD-10-0028
146. Sousa S, Brion R, Lintunen M, Kronqvist P, Sandholm J, Mönkkönen J, et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. (2015) 17:101. doi: 10.1186/s13058-015-0621-0
147. Hollmén M, Roudnicky F, Karaman S, Detmar M. Characterization of macrophage–cancer cell crosstalk in estrogen receptor positive and triple-negative breast cancer. Sci Rep. (2015) 5:9188. doi: 10.1038/srep09188
148. Quigley DA, Tahiri A, Luders T, Riis MH, Balmain A, Borresen-Dale AL, et al. Age, estrogen, and immune response in breast adenocarcinoma and adjacent normal tissue. Oncoimmunology. (2017) 6:e1356142. doi: 10.1080/2162402X.2017.1356142
149. O'Meara T, Marczyk M, Qing T, Yaghoobi V, Blenman K, Cole K, et al. Immunological differences between immune-rich estrogen receptor–positive and immune-rich triple-negative breast cancers. JCO Precision Oncol. (2020) 4:767–79. doi: 10.1200/PO.19.00350
150. Liu T, Larionova I, Litviakov N, Riabov V, Zavyalova M, Tsyganov M, et al. Tumor-associated macrophages in human breast cancer produce new monocyte attracting and pro-angiogenic factor YKL-39 indicative for increased metastasis after neoadjuvant chemotherapy. Oncoimmunology. (2018) 7:e1436922. doi: 10.1080/2162402X.2018.1436922
151. Williams MM, Spoelstra NS, Arnesen S, O'Neill KI, Christenson JL, Reese J, et al. Steroid hormone receptor and infiltrating immune cell status reveals therapeutic vulnerabilities of ESR1 mutant breast cancer. Cancer Res. (2021) 81:732–46. doi: 10.1158/0008-5472.CAN-20-1200
152. Jeet V, Tevz G, Lehman M, Hollier B, Nelson C. Elevated YKL40 is associated with advanced prostate cancer (PCa) and positively regulates invasion and migration of PCa cells. Endocr Relat Cancer. (2014) 21:723–37. doi: 10.1530/ERC-14-0267
153. Lu X, Yang R, Zhang L, Xi Y, Zhao J, Wang F, et al. Macrophage colony-stimulating factor mediates the recruitment of macrophages in triple negative breast cancer. Int J Biol Sci. (2019) 15:2859–71. doi: 10.7150/ijbs.39063
154. Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell. (2018) 174:1373–87.e1319. doi: 10.1016/j.cell.2018.08.039
155. Lin JR, Fallahi-Sichani M, Chen JY, Sorger PK. Cyclic Immunofluorescence (CycIF), a highly multiplexed method for single-cell imaging. Curr Protoc Chem Biol. (2016) 8:251–64. doi: 10.1002/cpch.14
156. Lin JR, Fallahi-Sichani M, Sorger PK. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat Commun. (2015) 6:8390. doi: 10.1038/ncomms9390
157. Lin JR, Izar B, Wang S, Yapp C, Mei S, Shah PM, et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. Elife. (2018) 7:e31657. doi: 10.7554/eLife.31657
158. Keenan TE, Li T, Vallius T, Guerriero JL, Tayob N, Kochupurakkal BS, et al. Clinical efficacy and molecular response correlates of the WEE1 inhibitor adavosertib combined with cisplatin in patients with metastatic triple-negative breast cancer (mTNBC). Clin Cancer Res. (2021) 27:983–91. doi: 10.1158/1078-0432.CCR-20-3089
159. Mehta A, Cheney EM, Hartl C, Pantelidou C, Oliwa M, Castrillon JA, et al. Differentiating macrophages are regulated by PARP inhibitors and can be harnessed to overcome PARP-inhibitor resistance in BRCA-associated triple-negative breast cancer. Nat Cancer. (2021). 2:66–82. doi: 10.1038/s43018-020-00148-7
160. Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-dependent innate immune signaling by s-phase-specific DNA damage in breast cancer. J Natl Cancer Inst. (2017) 109:djw199. doi: 10.1093/jnci/djw199
161. Pantelidou C, Sonzogni O, de Oliveira Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP inhibitor efficacy depends on CD8+ T cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. (2019) 9:722–37. doi: 10.1158/2159-8290.Cd-18-1218
162. Volodko N, Gutor T, Petronchak O, Huley R, Dúcka M, Šmarda J, et al. Low infiltration of tumor-associated macrophages in high c-Myb-expressing breast tumors. Sci Rep. (2019) 9:11634. doi: 10.1038/s41598-019-48051-1
163. Linde N, Casanova-Acebes M, Sosa MS, Mortha A, Rahman A, Farias E, et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat Commun. (2018) 9:21. doi: 10.1038/s41467-017-02481-5
164. Hussein MR, Hassan HI. Analysis of the mononuclear inflammatory cell infiltrate in the normal breast, benign proliferative breast disease, in situ and infiltrating ductal breast carcinomas: preliminary observations. J Clin Pathol. (2006) 59:972–7. doi: 10.1136/jcp.2005.031252
165. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. (2019) 35:588–602.e510. doi: 10.1016/j.ccell.2019.02.009
166. Esserman LJ, Kumar AS, Herrera AF, Leung J, Au A, Chen YY, et al. Magnetic resonance imaging captures the biology of ductal carcinoma in situ. J Clin Oncol. (2006) 24:4603–10. doi: 10.1200/JCO.2005.04.5518
167. Campbell MJ, Baehner F, O'Meara T, Ojukwu E, Han B, Mukhtar R, et al. Characterizing the immune microenvironment in high-risk ductal carcinoma in situ of the breast. Breast Cancer Res Treatment. (2017) 161:17–28. doi: 10.1007/s10549-016-4036-0
168. Gil Del Alcazar CR, Huh SJ, Ekram MB, Trinh A, Liu LL, Beca F, et al. Immune escape in breast cancer during in situ to invasive carcinoma transition. Cancer Discov. (2017) 7:1098–115. doi: 10.1158/2159-8290.CD-17-0222
169. Yang M, McKay D, Pollard JW, Lewis CE. Diverse functions of macrophages in different tumor microenvironments. Cancer Res. (2018) 78:5492–503. doi: 10.1158/0008-5472.CAN-18-1367
170. Gwak JM, Jang MH, Kim DI, Seo AN, Park SY. Prognostic value of tumor-associated macrophages according to histologic locations and hormone receptor status in breast cancer. PLoS ONE. (2015) 10:e0125728. doi: 10.1371/journal.pone.0125728
171. Mwafy SE, El-Guindy DM. Pathologic assessment of tumor-associated macrophages and their histologic localization in invasive breast carcinoma. J Egypt Natl Canc Inst. (2020) 32:6. doi: 10.1186/s43046-020-0018-8
172. Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol. (2015) 7:1120–34. doi: 10.1039/c5ib00040h
173. Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM. Leukocyte composition of human breast cancer. Proc Natl Acad Sci USA. (2012) 109:2796–801. doi: 10.1073/pnas.1104303108
174. Waks AG, Stover DG, Guerriero JL, Dillon DA, Barry WT, Gjini E, et al. The immune microenvironment in hormone receptor-positive breast cancer before and after preoperative chemotherapy. Clin Cancer Res. (2019) 25:4644–55. doi: 10.1158/1538-7445.AM2018-4564
175. Cao Q, Yan X, Chen K, Huang Q, Melancon MP, Lopez G, et al. Macrophages as a potential tumor-microenvironment target for noninvasive imaging of early response to anticancer therapy. Biomaterials. (2018) 152:63–76. doi: 10.1016/j.biomaterials.2017.10.036
176. Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. (2011) 25:2465–79. doi: 10.1101/gad.180331.111
177. Xuan QJ, Wang JX, Nanding A, Wang ZP, Liu H, Lian X, et al. Tumor-associated macrophages are correlated with tamoxifen resistance in the postmenopausal breast cancer patients. Pathol Oncol Res. (2014) 20:619–24. doi: 10.1007/s12253-013-9740-z
178. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci USA. (2018) 115:E4041–e50. doi: 10.1073/pnas.1720948115
179. Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. (2017) 543:428–32. doi: 10.1038/nature21409
180. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. (2018) 17:887–904. doi: 10.1038/nrd.2018.169
181. Brown JM, Recht L, Strober S. The promise of targeting macrophages in cancer therapy. Clin Cancer Res. (2017) 23:3241–50. doi: 10.1158/1078-0432.CCR-16-3122
182. Poh AR, Ernst M. Targeting macrophages in cancer: from bench to bedside. Front Oncol. (2018) 8:49. doi: 10.3389/fonc.2018.00049
183. Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. (2019) 40:310–327. doi: 10.1016/j.it.2019.02.003
184. Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X. Tumor-associated macrophages: recent insights and therapies. Front Oncol. (2020) 10:188. doi: 10.3389/fonc.2020.00188
185. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. (2013) 19:1264–72. doi: 10.1038/nm.3337
186. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. (2014) 515:130–3. doi: 10.1038/nature13862
187. Richardsen E, Uglehus RD, Johnsen SH, Busund LT. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. Anticancer Res. (2015) 35:865–74.
188. Wesolowski R, Sharma N, Reebel L, Rodal MB, Peck A, West BL, et al. Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Therap Adv Med Oncol. (2019) 11:1758835919854238. doi: 10.1177/1758835919854238
189. Gomez-Roca CA, Italiano A, Le Tourneau C, Cassier PA, Toulmonde M, D'Angelo SP, et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann Oncol. (2019) 30:1381–92. doi: 10.1093/annonc/mdz163
190. Swierczak A, Cook AD, Lenzo JC, Restall CM, Doherty JP, Anderson RL, et al. The promotion of breast cancer metastasis caused by inhibition of CSF-1R/CSF-1 signaling is blocked by targeting the G-CSF receptor. Cancer Immunol Res. (2014) 2:765–76. doi: 10.1158/2326-6066.CIR-13-0190
191. Hoves S, Ooi CH, Wolter C, Sade H, Bissinger S, Schmittnaegel M, et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J Exp Med. (2018) 215:859–76. doi: 10.1084/jem.20171440
192. Jiao X, Nawab O, Patel T, Kossenkov AV, Halama N, Jaeger D, et al. Recent advances targeting CCR5 for Cancer and its role in immuno-oncology. Cancer Res. (2019) 79:4801–7. doi: 10.1158/0008-5472.CAN-19-1167
193. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. (2011) 331:1612–6. doi: 10.1126/science.1198443
194. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature. (2016) 539:437–42. doi: 10.1038/nature19834
195. Zhang W, Huang Q, Xiao W, Zhao Y, Pi J, Xu H, et al. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPα Axis. Front Immunol. (2020) 11:18. doi: 10.3389/fimmu.2020.00018
196. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov. (2016) 6:270–85. doi: 10.1158/2159-8290.CD-15-0827
197. Feng Y, Mu R, Wang Z, Xing P, Zhang J, Dong L, et al. A toll-like receptor agonist mimicking microbial signal to generate tumor-suppressive macrophages. Nat Commun. (2019) 10:2272. doi: 10.1038/s41467-019-10354-2
198. Yin Z, Ma T, Lin Y, Lu X, Zhang C, Chen S, et al. IL-6/STAT3 pathway intermediates M1/M2 macrophage polarization during the development of hepatocellular carcinoma. J Cell Biochem. (2018) 119:9419–32. doi: 10.1002/jcb.27259
199. Luz-Crawford P, Djouad F, Toupet K, Bony C, Franquesa M, Hoogduijn MJ, et al. Mesenchymal stem cell-derived interleukin 1 receptor antagonist promotes macrophage polarization and inhibits B cell differentiation. Stem Cells. (2016) 34:483–92. doi: 10.1002/stem.2254
200. Chen H-M, van der Touw W, Wang YS, Kang K, Mai S, Zhang J, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest. (2018) 128:5647–62. doi: 10.1172/JCI97570
201. Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T, et al. Treg cells promote the SREBP1-dependent metabolic fitness of tumor-promoting macrophages via repression of CD8(+) T cell-derived Interferon-γ. Immunity. (2019) 51:381–97.e386. doi: 10.1016/j.immuni.2019.06.017
202. Pang J, Cui J, Xi C, Shen T, Gong H, Dou L, et al. Inhibition of Poly(ADP-Ribose) polymerase increased lipid accumulation through SREBP1 modulation. Cell Physiol Biochem. (2018) 49:645–52. doi: 10.1159/000493028
203. Shrestha E, Hussein MA, Savas JN, Ouimet M, Barrett TJ, Leone S, et al. Poly(ADP-ribose) polymerase 1 represses liver X receptor-mediated ABCA1 expression and cholesterol efflux in macrophages. J Biol Chem. (2016) 291:11172–84. doi: 10.1074/jbc.M116.726729
204. Liu M, O'Connor RS, Trefely S, Graham K, Snyder NW, Beatty GL. Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47-mediated 'don't-eat-me' signal. Nat Immunol. (2019) 20:265–75. doi: 10.1038/s41590-018-0292-y
205. Penny HL, Sieow JL, Adriani G, Yeap WH, See Chi Ee P, San Luis B, et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology. (2016) 5:e1191731. doi: 10.1080/2162402X.2016.1191731
206. Palmieri EM, Menga A, Martín-Pérez R, Quinto A, Riera-Domingo C, De Tullio G, et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like Phenotype and inhibits tumor metastasis. Cell Rep. (2017) 20:1654–66. doi: 10.1016/j.celrep.2017.07.054
207. Pulford KA, Sipos A, Cordell JL, Stross WP, Mason DY. Distribution of the CD68 macrophage/myeloid associated antigen. Int Immunol. (1990) 2:973–80. doi: 10.1093/intimm/2.10.973
208. Kaiserling E, Xiao JC, Ruck P, Horny HP. Aberrant expression of macrophage-associated antigens (CD68 and Ki-M1P) by Schwann cells in reactive and neoplastic neural tissue. light- and electron-microscopic findings. Mod Pathol. (1993) 6:463–8.
209. Shabo I, Stål O, Olsson H, Doré S, Svanvik J. Breast cancer expression of CD163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int J Cancer. (2008) 123:780–6. doi: 10.1002/ijc.23527
210. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin's lymphoma. N Engl J Med. (2010) 362:875–85. doi: 10.1056/NEJMoa0905680
Keywords: breast cancer, tumor associated macrophages, immune suppression, T cell inhibition, cancer immunity
Citation: Mehta AK, Kadel S, Townsend MG, Oliwa M and Guerriero JL (2021) Macrophage Biology and Mechanisms of Immune Suppression in Breast Cancer. Front. Immunol. 12:643771. doi: 10.3389/fimmu.2021.643771
Received: 18 December 2020; Accepted: 17 February 2021;
Published: 23 April 2021.
Edited by:
Carlo Bifulco, Providence Portland Medical Center, United StatesReviewed by:
Mario Leonardo Squadrito, Vita-Salute San Raffaele University, ItalyJennifer Richer, University of Colorado, United States
Copyright © 2021 Mehta, Kadel, Townsend, Oliwa and Guerriero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer L. Guerriero, amd1ZXJyaWVyb0Bid2guaGFydmFyZC5lZHU=