 Guillermo O. Rangel Rivera1,2,3*
Guillermo O. Rangel Rivera1,2,3* Hannah M. Knochelmann1,2,3
Hannah M. Knochelmann1,2,3 Connor J. Dwyer1
Connor J. Dwyer1 Aubrey S. Smith1,2,3
Aubrey S. Smith1,2,3 Megan M. Wyatt2,3Amalia M. Rivera-Reyes2,3Jessica E. Thaxton1,4Chrystal M. Paulos2,3*
Megan M. Wyatt2,3Amalia M. Rivera-Reyes2,3Jessica E. Thaxton1,4Chrystal M. Paulos2,3*- 1Department of Microbiology and Immunology, Medical University of South Carolina, Charleston, SC, United States
- 2Department of Surgery, Emory University, Atlanta, GA, United States
- 3Department of Microbiology and Immunology, Emory University, Atlanta, GA, United States
- 4Department of Orthopaedics and Physical Medicine, Medical University of South Carolina, Charleston, SC, United States
Emerging reports show that metabolic pathways can be targeted to enhance T cell-mediated immunity to tumors. Yet, tumors consume key metabolites in the host to survive, thus robbing T cells of these nutrients to function and thrive. T cells are often deprived of basic building blocks for energy in the tumor, including glucose and amino acids needed to proliferate or produce cytotoxic molecules against tumors. Immunosuppressive molecules in the host further compromise the lytic capacity of T cells. Moreover, checkpoint receptors inhibit T cell responses by impairing their bioenergetic potential within tumors. In this review, we discuss the fundamental metabolic pathways involved in T cell activation, differentiation and response against tumors. We then address ways to target metabolic pathways to improve the next generation of immunotherapies for cancer patients.
Introduction
It has long been appreciated that glycolysis and mitochondrial respiration work together to satisfy the long-term energetic demands of T cells in the host (1). As T cell survival is often impaired in patients with cancer and chronic infectious disease (1, 2), it is necessary to have an effective metabolic capacity for a productive immune response (1). For example, in patients, one reason T cells do not thrive amidst tumor cells is that they compete for the same energy sources (1). Herein, we review the fundamental metabolic requirements for T cells to survive, proliferate and mount antigen-specific responses in the context of effector and memory responses. We then outline how the harsh tumor microenvironment manipulates T cell metabolism to impair effector functions. Finally, we contemplate emerging data where metabolic manipulations have been performed and have shown promise for augmenting T cell-based immunotherapies for patients with cancer.
A Brief History of T Cell Metabolism
Studies in the mid-20th century first detailed the nutrient requirements for quiescent and activated T cells to survive. It was discovered that energy production and nutrient uptake shifts when a resting T cell is activated via signaling cues (3, 4). In the 1960’s, work by Hedeskov et al. initially described the metabolism of T lymphocytes at the resting state. Surprisingly, resting T cells largely depended on oxidative phosphorylation (OXPHOS) to survive. Additional investigations, published nearly a decade later, uncovered that resting T cells shift from OXPHOS to avid glycolysis and amino acid consumption upon TCR-mediated recognition of antigen (5). While this finding is obvious now, it was unexpected at the time, especially given that exploiting glycolysis for energy was largely thought less efficient than OXPHOS for T cells to generate ATP (3). For many years, these observations remained as descriptive findings of the highly dynamic ways T cells use bioenergetics to thrive. However, from the 1980’s to present day, the significance of bioenergetic requirements for the activation, effector functions and lasting memory of T cell responses against tumors have begun to be elucidated and exploited to improve medicine.
T cells use different metabolic pathways based on their differentiation and memory status (6–8). Figure 1 visually portrays how T cells exploit distinct metabolic pathways throughout their lifetime and during encounters with foreign antigen, such as viruses or transformed cells (9–11). As mentioned, naïve T cells rely on OXPHOS to survive in their resting state (12, 13). However, upon primary exposure to antigen, naïve T cells differentiate into effector cells and use glycolysis to help them effectively secrete cytokines, such as IFN-gamma and TNF-alpha (14–17). Following activation, naïve T cells shift from mostly oxidizing glutamine to lactate (75% of lactate produced from glutamine oxidation) through OXPHOS towards mostly using anaerobic glycolysis and partial glutamine oxidation (67% of all lactate from glucose metabolism, and 33% from glutamine), surprisingly without significantly changing their ATP production (5). After effector T cells encounter an antigen challenge, many of them die (18). However, a few prevail and survive long-term to battle re-infections or tumor relapse (17, 19, 20). These T cells are termed memory T cells. When memory T cells encounter the same antigen, they can more rapidly induce their effector functions to clear the insult (6, 21). These T cells are termed effector memory cells (EM) (22, 23). Effector T cells derived from memory rather than antigen naïve precursors more efficiently produce cytolytic cytokines by improving the coupling of glycolytic enzymes and mitochondrial machinery to rapidly utilize glucose following a secondary encounter with antigen (11, 24). Most effector memory T cells perish, but the few survivors employ OXPHOS to persist (8, 25). Below, we elaborate on the metabolic requirements of T cells at various stages of differentiation.
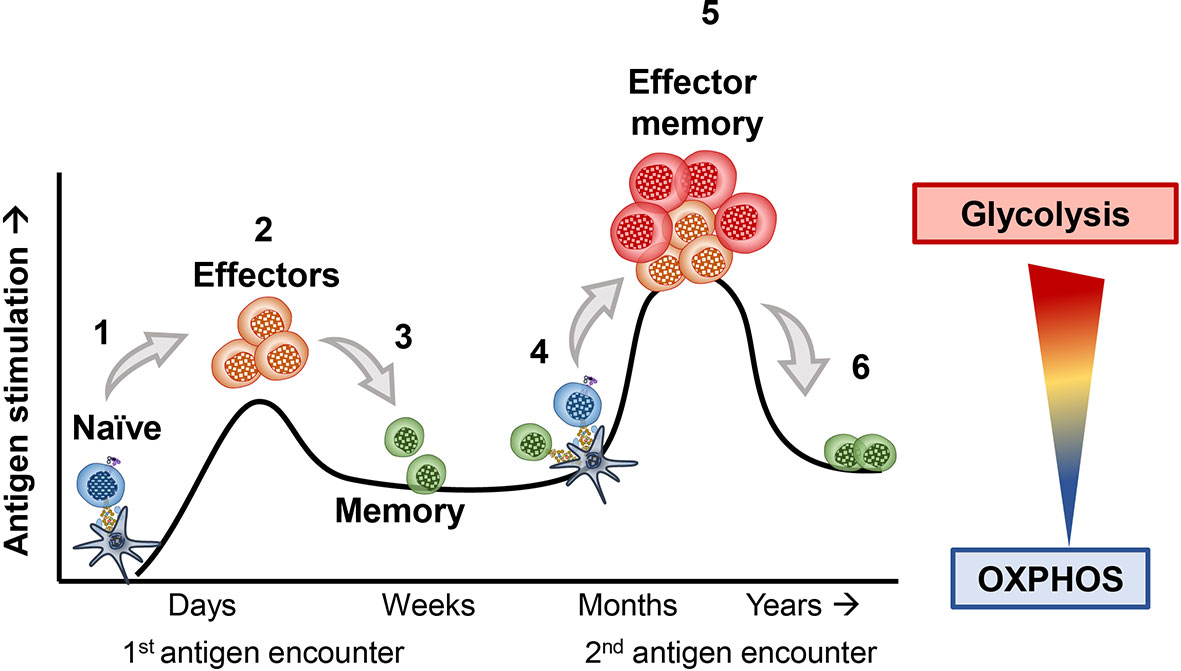
Figure 1 1) Naive T cells breakdown glucose and efficiently break it down through the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) to survive, until they encounter their antigen. 2) Upon a primary exposure to antigen, naïve T cells differentiate into effector T cells. As effectors they shift towards the use of amino acids as well as glucose, both required for their proliferation and cytolytic activity. 3) After clearing their inciting antigen, many effector T cells die. However, a fraction of surviving T cells can form memory T cells, which adapt towards improved mitochondrial biogenesis and OXPHOS. 4) These memory T cells can survive for many months to years until they encounter a similar antigen. 5) If these memory T cells re-encounter the same antigen, they rapidly become effectors and more efficiently engage in glycolysis and amino acid usage to robustly proliferate and secrete cytokines. 6) The T cells that survive maintain their usage of OXPHOS to persist long-term within hosts.
Metabolic Requirements Distinguish Effector and Memory T Cell Subsets
Naïve T Cell Metabolism
Naïve T cells can live for the entire duration of the host’s life. In fact, naïve T cells can be detected in humans as old as 100 years (26). Only after they encounter their respective antigens, do they either become effector T cells that perish or transition into memory T cells that continue to thrive (19). But how do naïve cells remain viable for so long? As in Figure 2A, naïve T cells can only survive when homeostatic cytokines, like interleukin 7 (IL-7) provide signaling cues (27). IL-7 provides the signaling necessary to enable the mechanisms that nurture the survival of naïve T through Akt signaling (28). This pathway, in naïve T cells promotes the translocation of the glucose transporter 1 (Glut1) to take up glucose. Glucose is then broken down into pyruvate, a substrate that enters the mitochondria to activate the synthesis of triacyl glycerol, which serves as a source of lipids that fuels into the fatty acid oxidation (FAO) pathway (28–30). In contrast to this maintenance phase, many nutrients (glucose, glutamine, L-arginine, and other amino acids) are needed to differentiate naïve T cells into the effector phenotype upon antigen encounter (5). We next will detail how activated T cells engage in transcriptional and metabolic changes to license them to proliferate and secrete effector cytokines.
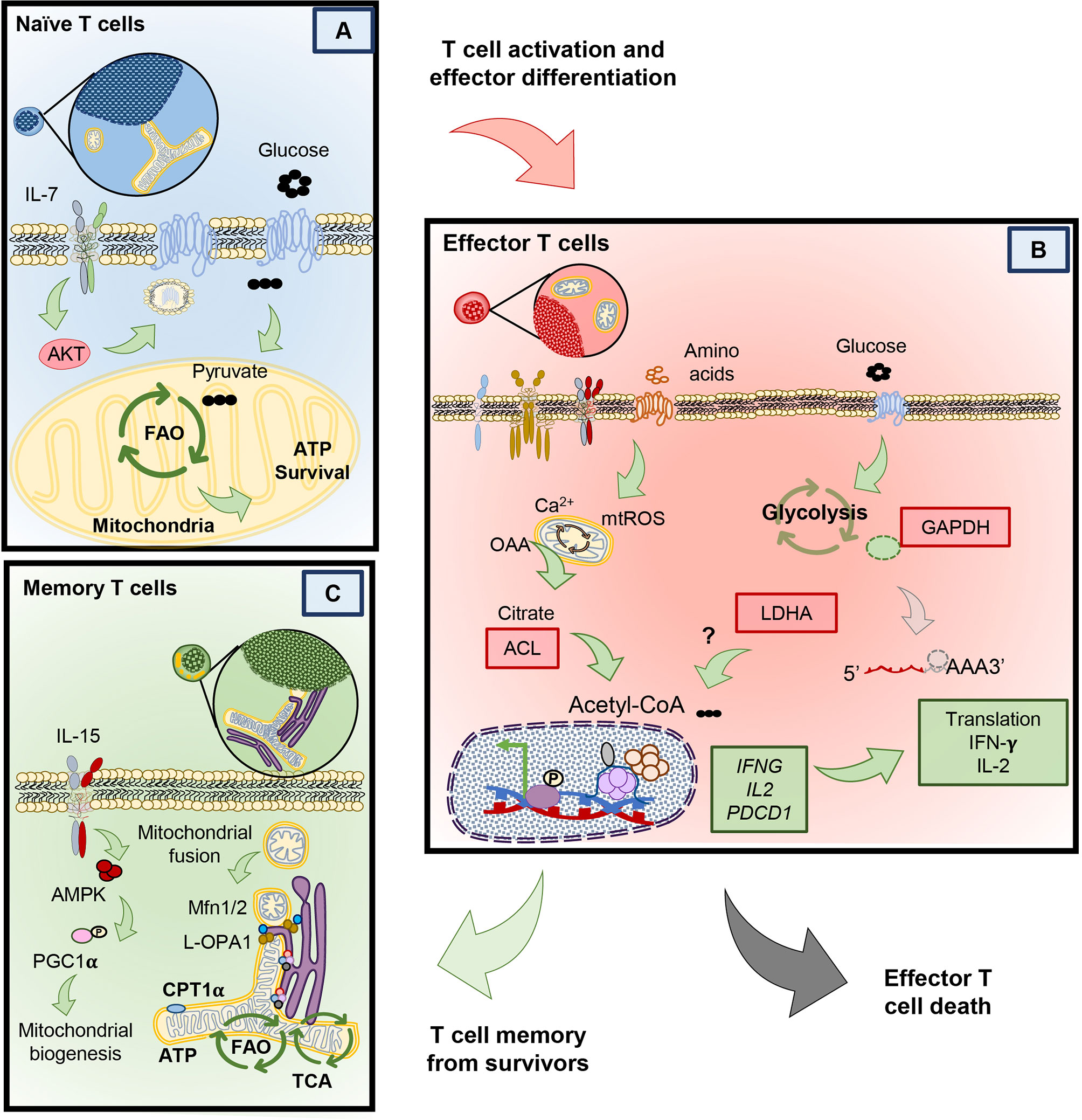
Figure 2 Different metabolic programs between naïve, memory and effector T cells. (A) Naïve T cells rely on the full oxidation of glucose through OXPHOS, in the absence of TCR stimulation. (B) Upon T cell stimulation T cells undergo protein and transcriptional changes in metabolism that allow the sustained activity of glycolysis and other amino acid uptake and usage. Glycolysis by products in effector T cells mediate changes that help sustain effector cytokine release and cytolytic function. Effector T cells that clear antigen either die or contract to form memory T cells. (C) Compared to effectors, memory T cells possess an enhanced metabolic profile dependent on mitochondrial biogenesis, mitochondrial fusion and reliance on fatty acid oxidation.
Glycolysis in Effector T Cell Function
Naïve T cells become activated upon TCR engagement with an antigen presented via the major histocompatibility complex (MHC) on antigen presenting cells (signal 1). However naïve T cells require a second signal via costimulatory molecules (signal 2) to become fully activated and proliferate (31–33). Along with these two signals, cytokines in the host play a key role in fine tuning the fate and metabolic profile of naïve T cells into either an effector or memory phenotype (Figure 2B) (12, 34–36). Effector T cells secrete cytotoxic cytokines, such as interferon-γ, TNF-α and granzyme B. These cytokines and cytotoxic molecules destroy cancer cells or clear viruses (37–39). T cells require many metabolic resources to mediate clearance of these foreign antigens. However, instead of engaging in the highly energetically favorable OXPHOS pathway, effector T cells use Warburg metabolism to proliferate and to produce cytokines (24, 40, 41). Warburg metabolism, initially discovered as an important pathway for the survival of malignant cells, is characterized by an extraordinary ability to breakdown glucose by anaerobic glycolysis and amino acids such as glutamine (as in Figure 2B) (42, 43). In contrast to naïve T cells, effectors break down glucose to pyruvate and lactate with minimal engagement of mitochondrial respiration (44–46).
Although the metabolic adaptations T cells undergo when activated may appear obvious given the increased energetic demand to proliferate and synthesize proteins, recent findings suggest that these changes are tightly coupled to T cell differentiation and acquisition of effector function. Interestingly, two key enzymes in the anaerobic glycolysis pathway— GAPDH and LDHA—are critical in regulating cytokine production in T cells. Glyceraldehyde 3-phosphate Dehydrogenase (GAPDH), aside from its role in metabolizing glucose, can directly bind the mRNA of key cytokines such as IL-2, IFNG and TNFA in CD4+ effector T cells to prevent their protein translation in the absence of glucose (right side of Figure 2B) (41). In contrast to preventing the direct protein translation of cytokines by GAPDH in CD4+ T cells, CD8+ T cells instead utilize lactate dehydrogenase (LDHA), the key enzyme in the conversion of pyruvate into lactate for anaerobic glycolysis, to enforce effector gene expression via histone acetylation (46, 47). Genetic loss of LDHA prevents acetylation at the promoters of effector genes such as IFNG and PDCD1, without compromising proliferation (41, 48). Although glucose is a critical metabolite for T cell function, the enzymes involved in anaerobic glycolysis are also tightly coupled to effector function in both CD4+ and CD8+ T cells (49). A potential mechanism explaining the functional effect of using glycolysis to promote effector functions may lie in production of citrate downstream of glucose breakdown. Citrate is shuttled from the mitochondria where it is converted into acetyl-coA by the action of cytosolic ATP Citrate Lyase (ACL) (50). ACL is an enzyme that is upregulated in both CD4+ and CD8+ effector T cells that can translocate from the cytoplasm to the nucleus and has the ability to directly acetylate histones of effector gene promoters (49). Thus, glycolysis regulates effector T cell functionality; while CD4+ T cells moonlight GAPDH to regulate cytokine translation, both CD4+ and CD8+ T cells use acetyl co-A to regulate histone acetylation of effector genes (left side of Figure 2B).
In addition to glucose, amino acids are critical for T cell proliferation and function. For example, glutamine breakdown in T cells is required for their proliferation but not for their cytokine production (48, 51). In fact, in the absence of L-Glutamine, T cells are unable to proliferate but can still secrete cytokines (48, 52). These findings suggest synergism from the breakdown of glucose and amino acids (such as glutamine) for T cell proliferation and effector functions. How other amino acids regulate T cell function under nutritional stress remains poorly understood, but is likely to be essential for immunity to tumors as we will discuss in later sections.
Because malignant cells use the same nutrients as effector T cells, they compete form them to thrive. Deprivation of glucose or glutamine in the tumor microenvironment vastly impairs T cell proliferation, function and survival (53–57). Although there may be recent findings that suggest inhibiting glutamine metabolism in the tumor may benefit T cells while impairing tumor metabolism (58). Often this tug of war forces effector T cells to use alternative carbon sources to survive (59–61). It is now clear that T cells use glycolysis to sustain their inflammatory potential, not only as a means to an end, but also as a regulatory component in T cell immunity.
Although metabolic changes permit effector T cells to become highly inflammatory, they come at the price of compromising their mitochondrial quality and capacity to self-renew (36, 62). However, the small T cell fraction that survive the initial antigen encounter acquire a different set of metabolic adaptations to prevail longer-term (63). Next, we discuss how changes to mitochondrial metabolism and morphology impact the development of memory T cells and their recall capacity.
Mitochondrial Properties of Effector and Memory T Cells
Memory T cells develop after a primary antigen challenge, persisting from the pool of lymphocytes with specific metabolic adaptations permitting self-renewal and survival long-term (Figure 2C) (64). Given the vast differences in function comparing effector and memory cells, alteration of mitochondrial networking and morphology is critical to fulfill the metabolic needs of these T cells. For example, mitochondria are recruited to the immune synapse after an antigen encounter in effector cells following cleavage from mitochondrial-endoplasmic reticulum (ER) contact sites to enable calcium influx and T cell activation (Figure 2B) (65). In contrast, as memory T cells develop they re-organize their mitochondria to associate tightly with the ER, a feature lacking in terminal effector and naïve T cells (66), which provides a pool of mitochondria primed to sustain aerobic glucose metabolism (67), directly enhancing IFN-gamma production during a secondary response to antigen.
Further, remodeling of mitochondrial morphology is critical for the specialized metabolic needs of effector versus memory T cells. In effector cells, mitochondrial fragmentation, also called fission, produces mitochondria with loose cristae and poorly efficient electron transport but high capacity to buffer calcium (68–71). This morphological and functional change enables the production of reactive oxygen species (ROS) and upregulation of anaerobic glycolysis, needed for the expression of NFAT, a transcription factor required for T cell activation (70, 72–74). In contrast to effectors, memory T cells adapt their mitochondrial morphology for cell-intrinsic usage of lipids and FAO (25). Memory T cells undergo mitochondrial fusion to protect against DNA damage from accumulated ROS to sustain survival under nutritional restriction (71, 75). Cells that acquire the tubular network of fused mitochondria produce less ROS, have tight cristae arrangement and electron transport complexes in close proximity to each other, indicating efficient mitochondrial respiration (70, 76). For example, spare respiratory capacity (SRC) and ATP production is elevated in memory T cells, indicating that they shift towards OXPHOS metabolism with reduced mtROS (36).
Due to these robust differences in metabolic state, manipulation of mitochondrial properties is an active area of research to direct T cells to specific phenotypes. Mitochondrial respiration can be driven by many different types of fuel. For example, IL-7 and IL-15 support the survival of memory T cells, in part, by inducing mitochondrial biogenesis and allowing utilization of alternative substrates to glucose for FAO, such as long chain fatty acids and triacylglycerols (Figure 2C) (11, 35). Seminal work by the Pearce group and others demonstrated that spare respiratory capacity and FAO was key for the development of T cell memory (8, 11, 36). Importantly, memory T cell formation could be induced by AMP-dependent-Protein Kinase (AMPK) activity via metformin, an FDA approved drug for diabetes (8).
AMPK is a serine threonine kinase responsive to AMP production or energy depletion and has critical function in the development of memory T cells without compromising a primary antigen challenge (77, 78). Mitochondrial respiration and memory formation are compromised in T cells deficient in the catalytic subunit of AMPK (63). In fact, AMPK is a critical regulator of the mitochondrial biogenesis transcription factor, peroxisome proliferator-activated coactivator 1α (PGC1α), which bolsters mitochondrial formation (Figure 2C) (79, 80). The importance of mitochondrial biogenesis and function has been recently highlighted by studies showing that either induction of PGC1α through 4-1BB signaling or genetic its overexpression in T cells enhances memory formation against tumors (81, 82). Based on our understanding of how metabolism and mitochondrial homeostasis changes through a T cell’s lifetime, under nutrient competent environments, we next discuss how T cell metabolism is altered in the tumor.
Nutritional Tug of War: T Cells vs the Tumor Microenvironment
Nutrient Competition
It has long been appreciated that the cytotoxic potential of CD8+ (CTL) T cells is impaired in the tumor (83). Emerging reports reveal that tumors and activated T cells share common metabolic programs to survive, thus setting the stage for a continuous battle (or tug of war) for nutrients (40, 42, 84). Several lines of evidence support this notion as tumors with gain-of-function mutations in enzymes involved in glycolysis have increased resistance to T cell mediated immunity. This feature presides independent of checkpoint inhibitory receptor expression (84). For example, in renal cell carcinoma, Glut1 expression in tumors is inversely correlated to CD8+ T cell infiltration and cytolytic capacity (43). Moreover, solid tumors are composed of heterogenous populations with differing metabolic adaptations that outcompete T cells in consuming glutamine, glucose and amino acids (Figure 3A). Within hypoxic regions, tumors use glucose and glutamine via the action of HIF-1α, a hypoxia inducible transcription factor, critical for maintaining glucose and glutamine breakdown under oxygen stress (84, 85). The same mechanism that allows tumors to thrive can further hinder the anti-tumor potential of T cells as hypoxia sensed by prolyl-hydroxylase (PHD) proteins can prevent T cell protection against metastatic lesions in the lungs by downregulating glycolysis genes (86). Because of the heterogenous nature of the tumor mass, areas of hypoxia allow for the development of highly glycolytic tumor regions that contribute to the acidic tumor microenvironment (TME) (87, 88). This contribution can be attributed, in part, to lactate secretion, which relies in proton co-transporters and can be detrimental to T cell activation (89, 90). Lactate must be exported out of the cell along with H+ ions to maintain homeostasis and to sustain glycolysis (91). When exported by tumor cells, lactate hinders T cell activation by altering the gradient across lactate transporters, thereby preventing recycling of glycolytic byproducts and preventing glycolysis in T and NK cells (87–89, 92). Lactate and proton build up leads to acidification (pH <6.4) of the tumor, in turn blunting T cell effector functions (93, 94). Furthermore, recent evidence suggests that lactate can serve as a substrate to promote immunosuppressive populations of regulatory T cells (Tregs) present in the TME (95, 96).
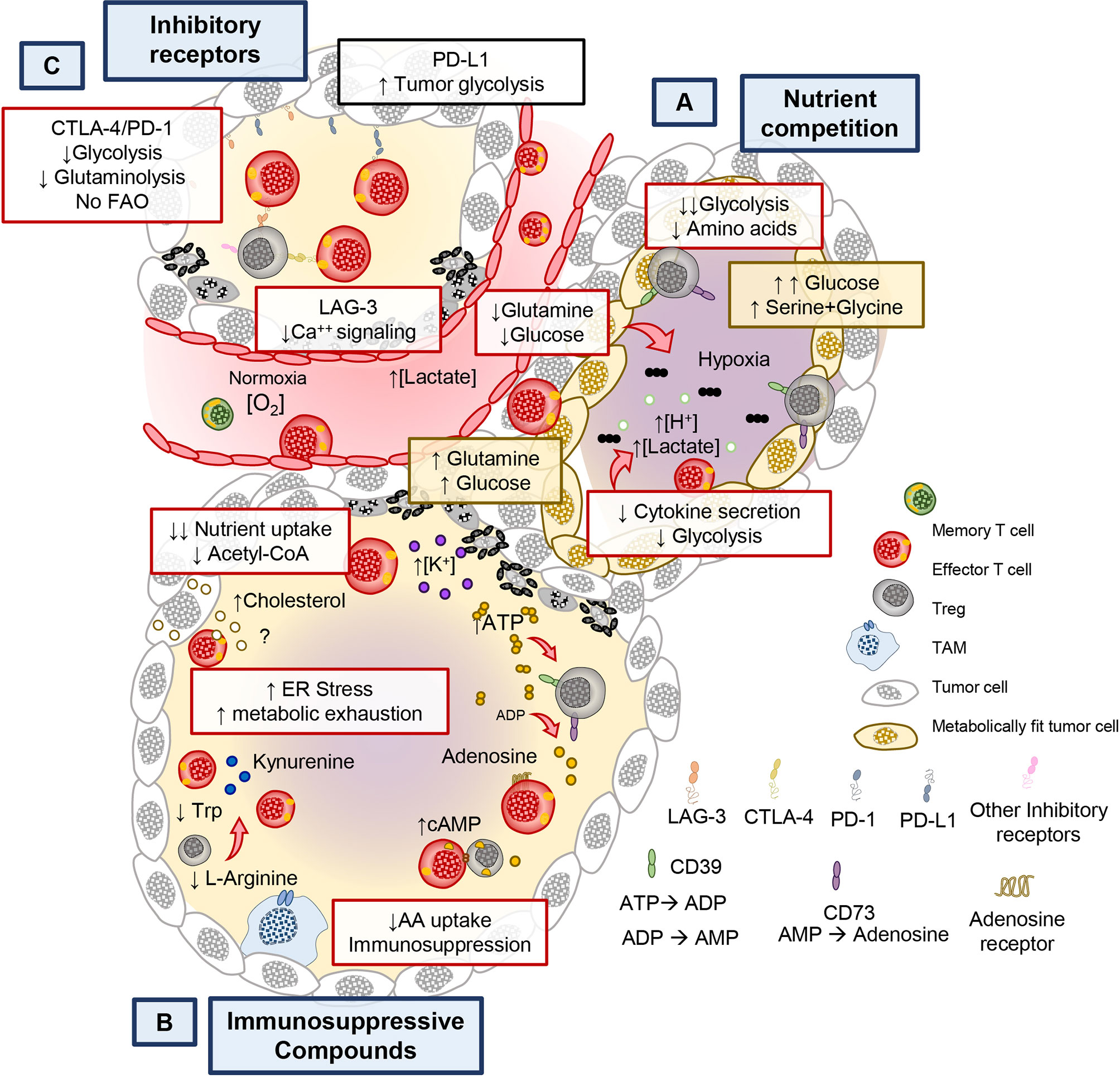
Figure 3 Metabolic and immunological checkpoints that hinder T cell mediated tumor immunity. (A) Tumors can adapt their metabolism in response to nutritional stress to better compete and scavenge for glucose and amino acids to suppress T cell bioenergetics. (B) Chronic stimulation in the tumor bed leads to the expression of immune checkpoint receptors such as PD-1/PD-L1, CTLA-4, LAG-3, and they exert negative metabolic functions in T cells. (C) Furthermore, Ionic imbalances, oxygen availability, and metabolites impact the function of T cells. By products of immunosuppressive immune cells, cell debris and tumor metabolites create the conditions that contribute to the metabolic exhaustion of tumor specific T cells.
Metabolism of Intratumoral Tregs
Regulatory T cells (Tregs) can directly and indirectly blunt cytotoxic CD8+ T cell response against the tumors (97). Similarly to conventional inflammatory CD4 T cells, Tregs can induce the glycolytic machinery upon TCR engagement, however Tregs complement their metabolism by inducing fatty acid biosynthesis and oxidative phosphorylation which allows them to survive longer than their inflammatory counterparts (98). In fact, Tregs rely on the expression and function of the electron transport chain complex III to sustain their suppressive function, as deletion of components of complex III leads to fatal autoimmunity within 25 days in mice and promote tumor immunity in B16 melanoma tumors after inducible deletion (99). The reliance on fatty acid metabolism and the respiratory chain provides Tregs with a metabolic advantage to thrive within tumors as they have scarce levels of glucose available and produce high lactate levels, a metabolic state that not only blunts cytotoxic activity but also provides an alternative fuel source to tumor infiltrating Tregs (100). As noted with deletion of complex III, a targetable vulnerability of tumor infiltrating Tregs exists and can be exploited to destabilize their suppressive function. Indeed, Tregs stability can be perturbed when CTA-4 blockade is used in glycolysis impaired tumors, through metabolic reprograming of Tregs towards glycolysis and a skewing towards an inflammatory phenotype, a process that is inhibited when tumors have high glycolytic capacity (101). This finding has tremendous potential for translation into the clinic, as it can be targeted using pharmaceutical agents.
Immunosuppressive Compounds
Tumors indirectly deprive effector CD8+ and helper CD4+ T cells of the metabolic nutrients required for their function and survival. One example of this scarcity of nutrients for immune cells is driven by the accumulation of potassium [K+] in the interstitial fluid of the tumor, which acts to suppress transporters for amino acid and glucose in T cells (Figure 3B) (102). Nutrient deprivation depletes the nucleocytosolic pools of acetyl CoA in T cells, preventing the acetylation of the IFNG promoter and therefore impairing their production of IFNγ (49, 103). This pathway plays a key role in modulating the epigenetic landscape of effector T cells.
Another mechanism of indirect nutrient deprivation is mediated by the byproducts of suppressive Treg cells, tumor cells, and other suppressive immune cells within the TME. Tregs produce adenosine in tumors by CD39/CD73-mediated catalysis (ATP → ADP → Adenosine, as shown in Figure 3B. Adenosine is a suppressive molecule that binds to adenosine receptors (A2AR) on cytotoxic T cells and suppresses their function via reducing NfkB signaling (104) or by inducing suppressive function on regulatory T cells (105). Furthermore, tumor metabolic byproducts, such as cholesterol, can induce metabolic stress in T cells. Specifically, tumor derived cholesterol induces ER stress which prevents the ability of T cells to secrete cytokines. Furthermore, the ER stress response promotes the factor XBP-1 which can directly increase PD-1, TIM-3, and LAG-3 expression, important immunosuppressive molecules that mediate T cell exhaustion (106). Not only do tumors secrete immunosuppressive molecules, but also other immune cells take up nutrients that are beneficial to T cells and can produce immunosuppressive metabolites. For example, M2 type macrophages in the tumor consume L-arginine in an arginase-1 dependent manner and can deplete tryptophan by breaking it down into immunosuppressive kynurenine derivatives through indoleamine-2,3-oxygenase (IDO) (56). These are just a few of the mechanisms that drive metabolic cross talk between tumors and immune cells. Thus, many byproducts of cellular metabolism synergize in the tumor to suppress T cells from fulfilling their potential to eradicate tumors and are likely to also play an obstacle in the growth of TILs from tumor biopsies (Figure 3B). In addition to this metabolic tug of war, the effector functions of T cells are limited by inhibitory receptor on tumors and immunosuppressive host elements, such as myeloid and Tregs cells.
Immune Checkpoints in T Cell Metabolism
Tumors evade the immune system in order to survive in the host. Tumors do this in many ways, as depicted in Figure 3C. One such mechanism is by promoting T cell exhaustion (107). T cells that become exhausted had a reduced capacity to survive, proliferate and secrete cytokines (108). T cell dysfunction is marked by the progressive acquisition of inhibitory receptors (IRs), including programmed cell death protein 1 (PD-1), lymphocyte activation gene-3 (LAG-3), cytotoxic T-lymphocyte association protein 4 (CTLA4), and T cell immunoglobulin and mucin domain-containing protein 3 (TIM3) (109–111). These IRs alter T cell responses against tumors in part by perturbing their metabolism.
T cell function and proliferation are compromised via immune checkpoint inhibitory pathways in the tumor (15, 112). PD-1 impairs effector function by downregulating glycolysis and increasing the FAO rate limiting enzyme CPT1α, a feature that supports T cell persistence in the tumor but prevents their cytotoxic potential (112). Although mitochondrial FAO supports T cell persistence but not function, PD-1+ T cells exhibit markedly decreased mitochondrial respiration (Figure 3C) (112, 113). Furthermore, Akt signaling is elevated in tumor infiltrating lymphocytes experience, which potent inhibits PGC1α, a key regulator of mitochondrial biogenesis (81, 113). This data suggests that part of the suppressive mechanism of PD-1 and chronic antigen stimulation is attributed to their negative effect on T cell mitochondrial biogenesis, substrate utilization and glycolytic capacity (Figure 3C). Conversely, PD-L1 on tumor cells enhances glucose uptake, further depriving T cells use of this critical energy substrate (84). Collectively this body work suggests that PD-1/PD-L1 blockade can bolster T cell glycolysis to support their antitumor activity (84, 114).
CTLA-4 is a member of the immunoglobulin family on APCs and tumors that antagonizes CD28 on T cells. CTLA-4 activation on T cells suppresses their function and nutrient acquisition (115–118). Moreover, CTLA-4 downregulates the glutamine transporters (SNAT1, SNAT2) and Glut1, ultimately diminishing the bioenergetic potential of T cells in the tumor (112). LAG-3 also impaired T cell activation and proliferation (111, 119). It has been reported that LAG-3 specifically perturbs calcium influx downstream of CD3/TCR signaling, in turn preventing the differentiation of naïve T cells into effectors (120). As checkpoint blockade mediates remarkable responses in patients with a wide variety of malignancies (121, 122), it is critical to understand how IRs regulate T cell biology. Expanding our knowledge on these mechanisms will inform intelligent design of tumor immunotherapies.
T cells face many challenges to sustain effective immunity to tumors. However, it has become evident that modulating the nutritional demands of the tumor is key for sustaining proper anti-tumor T cell potential. Below we highlight the most exciting findings demonstrating how metabolically manipulating T cell ex vivo for adoptive immunotherapy can enhance and improve future immunotherapies.
Modulating Metabolism to Enhance Adoptive T Cell Therapy
Introduction to Adoptive T Cell Transfer Therapy
Some patients become resistant to checkpoint inhibition therapy. Consequently, many investigators are trying alternative therapeutic approaches that can prevent resistance or relapse, including the transfer of tumor specific T cells. Cellular therapies, such as autologous tumor infiltrating lymphocytes (TIL) or engineered chimeric antigen receptor (CAR) T cell approaches have demonstrated great potential in mediating long-lasting responses against tumors (2). Generally, whether TIL or CAR adoptive T cell therapies rely on three basic principles, a) conditioning of host with nonmyeloablative chemotherapy or total body irradiation, b) growth of T cells to large therapeutic doses and 3) treatment post-transfer with high dose IL-2 (123). This therapeutic approach holds the promise of vastly improving cancer treatment, especially for tumors rich in neoantigens, as reported for epithelial cancers such as ovarian and triple negative breast cancer (124–126). However, two major limitations for this approach are the ability to generate enough tumor specific T cells for infusion into patients and the capacity of the infused T cell products to persist long-term.
In situations where naturally arising TILs cannot be generated from a patient, gene therapy has opened the door for synthesizing T cells by directing them against tumors with chimeric antigen receptors (CAR). CD19-specific CAR T cells, designed to recognize B cell malignancies, have mediated long-lasting responses in some patients that have exhausted all other treatment options (127–129). The efficacy of these CAR T cells resulted in FDA approval of three different CD19-CAR T cell preparations thus far: two with CD28 costimulatory domains (axicabtagene ciloleucel and brexacabtagene autoleucel), and one with 4-1BB as a costimulatory domain (tisagenlecleucel). Although both TIL and CAR therapy have shown promise, sustaining prolonged and durable responses in all patients remains a challenge. Yet, the manipulation of T cells in an ex vivo setting provides a unique opportunity to specifically empower T cells with antitumor properties, including remodeling their metabolism, without indirect effects on the tumor. Herein we describe new advances in how TIL, CAR and TCR-based cellular therapies have been improved by altering both T cell and tumor bioenergetics.
Metabolic Reprogramming in the Design of CAR T Cells
CAR T cell construct design has evolved to include many flavors of signaling domains, kill switches, switch receptors and regulatory functions. These factors in CAR design have been reported to exquisitely control T cell functionality and selectivity against tumor targets, as reviewed previously (130). Results from early trials of CD28ζ and 4-1BBζ CAR T cells made functional differences between these cells apparent; while the CD28ζ CAR had high incidence of cytokine release syndrome and persistence on the order of months (131), the 41BBζ CAR T was able to persist on the order of years after treatment (132) and exhibited lower rates of T cell exhaustion (133). Early on, understanding of these differences was unclear; however, the June lab discovered a mechanism relating these functional differences to effects of the costimulatory domain on mitochondrial function and bioenergetics (82). 4-1BB signaling enhanced T cell bioenergetics by directly upregulating PGC1α, a transcription factor that promotes increased mitochondrial biogenesis and OXPHOS of T cells (82), supporting their long-term persistence (Figure 4A). In contrast, CD28ζ CAR T cells were highly glycolytic and were driven to a terminal effector phenotype (82). Further, reports have also shown that strong and chronic signaling from the CAR domain impaired T cell persistence and function due to impaired mitochondrial metabolism (133). Given these results, it is clear that the costimulatory domains incorporated into CAR T cell designs have functional and metabolic consequences which could be harnessed based on the needs of the patient. 4-1BB is part of the tumor necrosis factor related super family (TNFRSF), which consists of many other members that can be expressed in T cells such as ICOS, OX40, GITR, and CD27. Although their signaling mechanisms are known, whether they affect metabolic fitness or could empower CAR T cell persistence in patients is an active area of study.
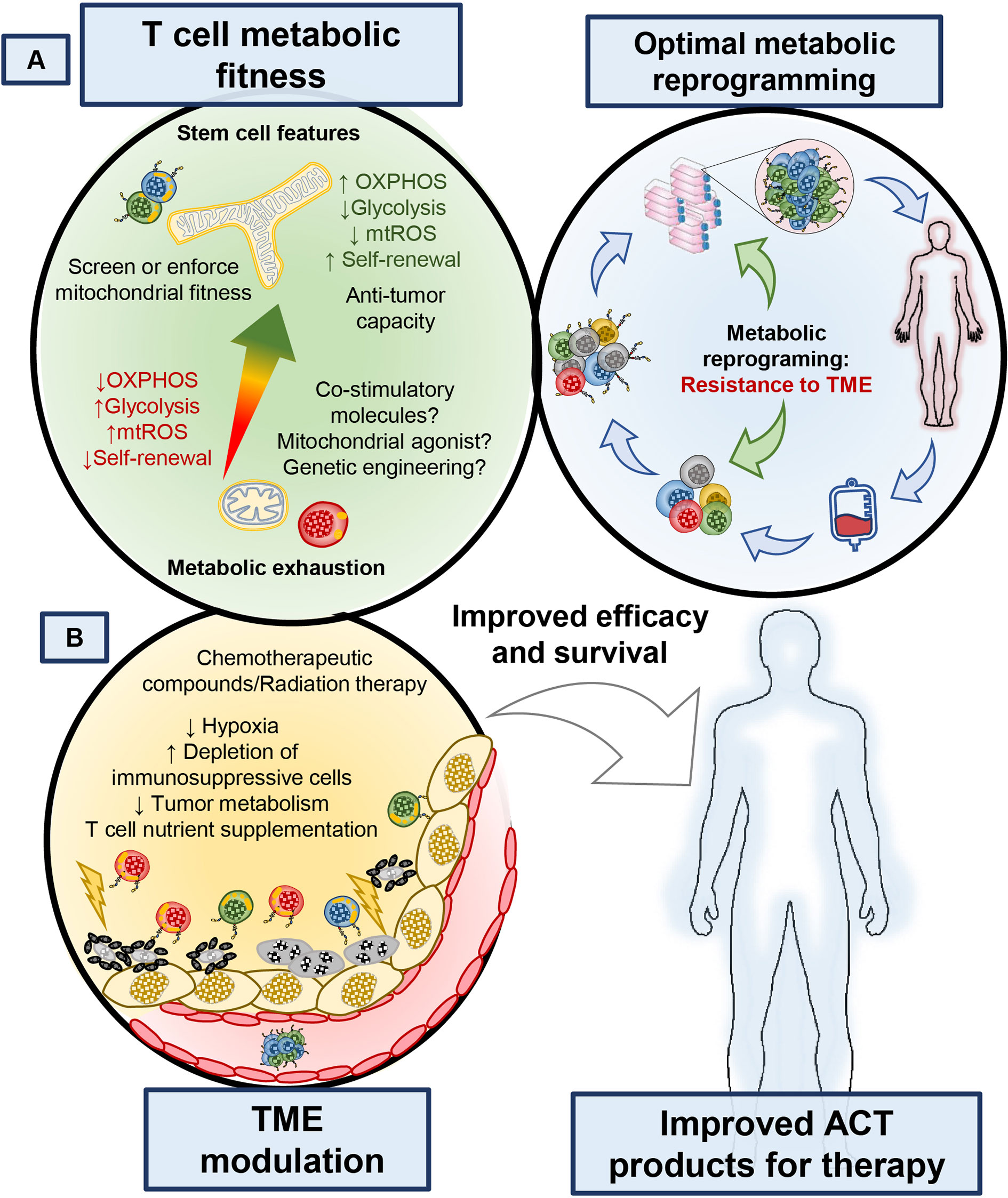
Figure 4 Manipulating the ability of T cells to withstand metabolic stress or altering the metabolism of tumors can enhance the therapeutic potential of T cell-based therapies. (A) Identifying markers that identify metabolically competent T cells, as well as understanding how small molecule compounds, biologics or receptor ligands could improve T cell metabolism will bring new targets to improve the efficacy of T cell products. (B) Better understanding of how the tumor microenvironment is affected by current therapies could provide new avenues to target both T cell and tumor metabolism to bolster immunotherapies. Enriching metabolically fit T cells during T cell isolation from whole blood or including metabolism modulating agents during TIL and CAR T cell expansion or altering CAR design of T cells could improve the survival of patients treated with cellular therapies.
Cytokine Priming and Metabolic Re-Programming
The generation of TIL products is possible through the use of high dose IL-2 in tumor digests. Current protocols promote the proliferation of T cells from tumor biopsies and can yield billions of cells after weeks to months of manufacturing. However, TIL products expanded in vitro are fully differentiated and show features of senescence, which impairs their persistence and antitumor capacity (2, 108). In contrast, T cells generated with central or stem-cell memory properties in vitro have increased potential for antitumor immunity (20, 39, 134). It has long been appreciated that priming T cells with the common γ chain cytokines IL-7, IL-15 or IL-21, can generate and sustain memory T cells and have shown promise in preclinical models of adoptive T cell therapy (135). In fact, expansion of TIL from patient biopsies using a combination of the common γ chain cytokines have yielded less differentiated T cells with improved stemness features, however whether they synergize in combination to improve T cell bioenergetics remains to be fully elucidated (136, 137). Compared to IL-2 conditioning, expanding T cells with IL-15 vastly improves mitochondrial fitness, prevents overt T cell differentiation and improves tumor immunity (12, 36). Furthermore, when compared with IL-15, IL-21 appears to be most effective at preventing T cell differentiation prior to ACT and promotes greater tumor immunity than IL-15 (138); however, whether IL-21 alters T cell metabolism in a similar manner as IL-15 remains to be determined. Recently two independent reports showed that targeting IL-21 directly to T cells rather than systemic delivery in combination with PD-1 therapy improved tumor immunity. They also showed that systemic delivery of soluble IL-21 did not improve the therapeutic efficacy of PD-1 blockade, however when the anti-PD-1 antibody was fused to IL-21 synergetic improvement in tumor immunity was noticed (139, 140). There are currently many efforts to translate the use of single and combinations of these cytokines to expand TIL and CAR products for ACT, as well as novel ways to incorporate cytokine releasing switches in CAR constructs and fusion proteins. However, these studies highlight a need to better understand how and when to use these modulatory cytokines, as they may compromise T cell growth or functionality due to their effect on other immune cells within tumors.
Inhibiting Signaling Pathways to Improve T Cell Therapies
Engagement of TCR, costimulatory molecules and cytokines mediate many internal cascades that contribute to T cell differentiation. These signals promote immunological memory; however, depending on the type and strength, these signals can also drive to T cell differentiation and exhaustion (141). An attractive approach is to use pharmacologic inhibitors against signaling cascades downstream of these signaling cues (Figure 4A) to generate antitumor T cells with durable memory traits. Canonical signaling downstream of T cell activation turns on the PI3K/ATK/mTOR signaling pathway and leads to T cell effector differentiation (142). This signaling axis is critical for rewiring metabolism to enable growth, protein translation and function in all proliferating cells, thus using compounds that target components of this pathway is a sensible approach to modulate T cell biology to improve their anti-tumor potential. Our lab and others have shown that targeting the delta subunit of PI3K, which is expressed specifically in lymphocytes, generates T cells with a less differentiated state (143), including murine and human antitumor CD8+ CTLs or CD4+ Th17 cells (144, 145). In fact, Dwyer et al. reported that blocking the PI3Kδ or PI3Kγ subunits were most advantageous for the production of highly effective anti-tumor T cells compared to those treated with drugs that inhibited both PI3Kδ and PI3Kγ subunits (146). Although is predicted that PI3K inhibition should dampen glucose metabolism and reciprocally improve T cell mitochondrial function it is still to be explored if selectively inhibiting the delta or gamma subunit have lasting effects on T cell metabolism reprograming or mitochondrial fitness (146). Downstream of PI3K, Akt blockade was also found to increase T cell stemness as well as FAO metabolism without perturbation of glycolysis upon restimulation (147, 148). Furthermore, inhibition of mTOR with rapamycin generates T cells that resemble a rare stem-memory like T cell population with enhanced survival capacity, mitochondrial respiration and lasting persistence in hosts (149–151). These findings reveal an interesting approach in modulating T cell differentiation and metabolism, which endow T cells with enhanced tumor-killing capacity. However, the question is raised as to which approach is most effective and what mechanisms govern the efficacy of this therapeutic inhibition during ACT expansion. A potential mechanism is that inhibiting the PI3K/Akt/mTOR pathway leads to enhanced autophagy, which is a well described homeostatic process involved promoting T cell memory and mitochondrial fitness (152, 153). This idea is further strengthened by a new report shown an important role for T cell intrinsic mitochondrial regulation by autophagy as an important part sustained immunity against tumors (154).
Another clue as to how blocking signaling cascades may overlap to improve T cell therapies was recently identified by the Restifo group (155). They used a multi-phenotype CRISPR screen to identify more than 25 targets downstream of T cell activation. They identified the stress response p38 MAP kinase as a key driver involved in preventing T cell mediated tumor immunity (155). This finding reinforces previous studies that elegantly demonstrated that ER stress, a target regulated by p38, impairs intratumoral T cell protein translation of cytotoxic molecules and regulates mitochondrial and T cell exhaustion (156–159). Nonetheless, the current efforts exploring inhibition of these key signaling pathways in vitro may provide TIL and CAR T cell with enhanced bioenergetics, persistence and anti-tumor capacity during their expansion. A potential benefit of using inhibitors of these key proliferation pathways is their effect on tumor and other suppressive immune cells, such as Tregs (160, 161) and myeloid cells (162) as they are sensitive to PI3K inhibition and may enhance the expansion of TIL. Based on current the literature, inhibition of growth and differentiation pathways such as the PI3K/AKT/mTOR signaling axis directly alters the development and metabolic programing of T cells in vitro, which improves their bioenergetics and persistence in vivo (Figure 4A).
Exploiting Nutritional Stress in Vitro for Beneficial Metabolic Adaptations
Recent reports show that T cells expanded in the presence of metabolic stress are surprisingly better at delaying tumor growth (103, 163). Although this finding is counterintuitive, this discovery may be explained by the ability of T cells to adapt to scarce environments by upregulating alternative sources of fuel through metabolic adaptations. For example, Sukumar et al. found that depriving T cells of glucose in vitro increased the number of less differentiated CTLs and supported their stem and central memory profile. These glucose-starved T cells regained potent effector functions in the tumor when infused into mice (163). Moreover, these cells upregulated AMPK activity, known to enhance mitochondrial respiration and fatty acid usage, and mediated robust regression of melanoma compared to conventionally cultured T cells (163). Most recently this finding has been supported by transient glucose restriction which improves T cell immunity against tumors via increased pentose phosphate pathway activity (164). Note that this study is not diminishing the importance of glucose for T cell survival and effector functions. Instead, it highlights the unexpected finding that the biology of T cells can be altered simply by transiently denying them this metabolite in vitro, an adaptation likely to be driven by mitochondrial compensation (62). In fact, T cells engineered to overexpress the gluconeogenesis enzyme phosphoenolpyruvate carboxykinase (PCK) can improve antitumor immunity, putatively by increasing the pool of glucose available to enter glycolysis and other ancillary pathways like the pentose phosphate pathway (165).
Glucose availability is a requirement for T cell mediated immunity in vivo, so how does depriving T cells of glucose in vitro enhance tumor immunity? An explanation could be provided by recent work showing that nutritional deprivation can be a double-edged sword depending on the context. In this work, priming T cells in vitro with high potassium concentrations lead to metabolic reprogramming to increase Acetyl Co-A Synthase (ACCS1) which enhanced mitochondrial respiration, conferred stem memory qualities in T cell and enhanced tumor immunity in vivo (49, 153). This data revealed that priming T cells in vitro with nutritional deprivation can improve antitumor activity, in part due to improved bioenergetic plasticity in a similar fashion as depriving glucose in vitro. Pressuring T cells to undergo metabolic adaptations that allow survival under cellular stress, such as promoting mitochondrial biogenesis, enhancing mitochondrial respiration, or enhancing ancillary pathways such as PPP or gluconeogenesis can benefit tumor control.
Nutritional Support for Anti-tumor T Cells
Collectively, this rich body of work on T cell metabolism highlights the need for T cells to adapt and use alternative fuel sources to thrive in the harsh tumor microenvironment. So, the question is posed, which fuels are most effective at supporting T cell antitumor activity? Recent work suggests that select amino acids and nucleotides may contribute. For example, supplementation of L-arginine in vitro and in vivo improves T cell tumoricidal activity by enhancing their memory formation and mitochondrial respiration (166). Additionally, supplementing inosine (a nucleoside capable of entering the central carbon pathways of glycolysis and the pentose phosphate pathway) under glucose deprivation enhanced the ability of T cells to clear tumors in mice (59). Identifying unique metabolites to augment cancer immunotherapy is attractive, as they can be delivered directly into T cell cultures or in vivo without overt expected side effects. A recent report suggests that highly therapeutic CD26high T cells might have those properties (167), as CD26 docks adenosine deaminase (ADA), which cleaves suppressive adenosine [produced by tumors and Tregs (105)] into inosine an important precursor for nucleotide synthesis and feedback into one carbon metabolism (59). This idea is particularly attractive given how potent CD26high T cells are at ablating large tumors, and suggest that ADA-induced inosine might play a role in their potency. Furthermore, methionine is a metabolite that enters the central carbon cycle and is a required amino acid for supporting T cells with effector properties (60, 61). In vivo, supplementing T cells with surplus L-arginine, inosine or potentially methionine could be an attractive way to enhance tumor immunity by providing alternative fuels for T cells exogenously (Figure 4B).
Direct Mitochondrial Agonists to Enhance Anti-tumor T Cells
Modulating nutrients to directly fuel T cells within tumors may not be the most efficient way to help their support their bioenergetic needs. Instead, directly stimulating the mitochondrial function of T cells using pharmacologic agonists might be more effective (Figure 4B). In fact, several reports have shown that small molecule agonists of AMPK, mTOR and PPARα or γ in combination with immune checkpoint blockade (ICB) therapy can promote mitochondrial function in T cells, leading to a positive immune response against tumors (168). For example, PD-L1 blockade in combination with an agonist of peroxisome proliferator–activated receptor γ and co-activator of PGC1α was remarkably effective at mediating curative responses in mice with melanoma (169). This approach is supported by a recent study revealing that T cell mitochondrial function is a marker for responsiveness in patients treated with ICB (168). Thus, it is likely that mitochondrial T cell health and regulation may play an important role in patient responses to immunotherapy (62, 68). Also, survival cues (such as AMPK, PKA and the Sirtuin family of acetylating enzymes) regulate T cell metabolism under energetic stress. This action improves T cell survival via bolstering mitochondrial biogenesis (PGC1a), remodeling (fused mitochondria) and recycling (i.e. mitochondrial autophagy) (70, 81, 154). However, direct perturbation of the tumor itself is also likely to modulate metabolites that promote T cell immunity (Figure 4B). Many investigators are thus focused on combining radiation or chemotherapy with ICB to help patients, but exactly how these therapies impact the nutrient tug of war between tumor and T cells is still up for debate as well as whether these approaches can be exploited for the expansion of TIL and CAR T cells.
Manipulation of Tumor Cell Metabolism
New insights into the metabolic requirements for tumors has sparked interest in manipulating their metabolism to improve immunity (54, 102). Nonetheless, there is limited but promising data regarding the benefit of combining current chemotherapeutic strategies or inhibition of tumor metabolism. Although glucose availability is a key determinant of T cell response, the heterogenous nature of the tumor warrants exploration of multiple targets (55, 57).
Preventing tumors from using the nutrients they need to survive can provide an advantage for T cell effector functions. For example, targeting the lactate dehydrogenase (LDHA), an enzyme that converts pyruvate to lactate and regeneration of NAD+ in tumors, improves T and NK cell function (47, 170, 171). Additionally, altering the hypoxic tumor environment can improve the therapeutic potential of ICB and adoptive transfer therapies, given the critical role of HIF1-α in altering the metabolic requirements of tumors under oxygen stress (172). For example, metformin plus PD-1 therapy enhanced the antitumor capacity of endogenous T cells in mice, in part by reducing the hypoxic nature of the melanoma (173). Another approach to target the tumor to augment immunity has been the neutralization of the highly acidic TME with sodium bicarbonate or other proton pump inhibitors prior to ICB or ACT (174). Combination of tumor metabolism inhibition and chemotherapeutic regiments may relieve the nutritional tug of war between tumors and T cells (93, 175). One promising strategy is to block glutamine metabolism within tumors, as this can also empower T cell immunity, a remarkable feat for single chemotherapy agents (58). Finally, another example of targeting the tumor to augment outcomes is found in pre-conditioning patients with systemic cisplatin to enhance T cell immunity at a secondary tumor site following radiotherapy, also known as the abscopal effect (176, 177). Thus, identifying FDA approved chemotherapeutics that alter tumor metabolism to augment the therapeutic potential of immunotherapies will be key to improve current therapeutic approaches (Figure 4).
Summary and Perspectives
It has 70 years since the first studies on T cell metabolism (178). The importance of T cell bioenergetics and its effect on immunity are gaining a new level of appreciation today and are being explored by multiple investigators. Yet, many key questions remain unanswered about how T cell metabolism impacts immunotherapy. For example, how do expression of inhibitory receptors and co-stimulatory molecules [such as LAG-3, TIM3, ICOS and other TNFRSF receptors (i.e. OX40, 4-1BB] impact T cell and tumor metabolism? Moreover, how do “suffering” T cells preconditioned under nutritional deficits gain antitumor activity in vivo? Insights into these mechanisms will be critical to design optimal therapies as mono- or combination approaches.
Herein, we have highlighted the myriad of ways metabolism is emerging as a major target for next generation immunotherapies. While the optimal therapeutic approach is unclear, promising strategies include targeting the tumor/immune axis either altogether or as individual branches. Chemotherapy and irradiation as preconditioning agents hinder the tumor directly, permit release of antigens and host immune activation. After effective tumor priming, administration of potent immune activating agents can help overcome immune evasion by the tumor. These immune therapies include checkpoint inhibitors, costimulatory agonists, and adoptively transferred T cells, each have the potential to harness a metabolic advantage for antitumor immune cells. Additionally, direct administration of agents which alter nutrient plasticity or promote metabolic adaptation of T cells over tumors could also synergize. However, as highlighted by the effect of tumor glycolysis and CTLA-4 blockade and the ability of other immunosuppressive cells to benefit from metabolism modulators there is always a possibility that fostering a metabolic advantage for the T cells in the tumor could also benefit the immunosuppressive microenvironment of the tumor, thus defining the timing and sequence of intervention is a challenge that needs to be addressed. One advantage of adoptive T cell therapy as an alternative, is the flexibility to manipulate the T cell directly obviating the challenge of competing immunosuppressive cells and the tumor. However, there are challenges that remain in designing optimal methods to reliably and potently alter the metabolism and function of ex vivo expanded T cells. This body of work suggest that by using inhibitors of key differentiation/stress pathways or conditioning with cytokines or co-receptors that improve metabolic function, we could provide the competitive advantage needed to ablate tumors long-term in patients. Regardless of the specific method, metabolic rewiring is likely to play a significant role in eliciting durable and long-lasting immunity in tumors resistant to conventional therapies.
Author Contributions
GR and CP designed, wrote and edited the manuscript. HK, CD, AS, MW, AR and JT provided feedback and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the NIH Training grant T32 5T32GM008716-19 and MUSC CGS Provost Scholarship to GR. NIH Training grant T32 GM08716 and NCI F30 243307 to HK. NIH training grant T32 AI132164-01 to CD. Hollings Cancer Center Graduate Fellowship to AS. NIH R50 CA233186 to MW. NIH R01 CA175061 and R01 CA208514 grants, KL2 South Carolina Clinical and Translational Research grant UL1 TR000062, ACS-IRG grant 016623-004 and MUSC Start-up funds to CP.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the researchers and clinicians for their dedication and passion to improve cancer immunotherapy, and the patients whose sacrifice and courage inspire us to move research forward. We also greatly appreciate the researchers that have contributed to this body of knowledge.
References
1. Allison KE, Coomber BL, Bridle BW. Metabolic reprogramming in the tumour microenvironment: a hallmark shared by cancer cells and T lymphocytes. Immunology (2017) 152:175–84. doi: 10.1111/imm.12777
2. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
3. Roos D, Loos JA. Changes in the carbohydrate metabolism of mitogenically stimulated human peripheral lymphocytes. II. Relative importance of glycolysis and oxidative phosphorylation on phytohaemagglutinin stimulation. Exp Cell Res (1973) 77:127–35. doi: 10.1016/0014-4827(73)90561-2
4. Ardawi MS, Newsholme EA. Glutamine metabolism in lymphocytes of the rat. Biochem J (1983) 212:835–42. doi: 10.1042/Bj2120835
5. Bental M, Deutsch C. Metabolic changes in activated T cells: an NMR study of human peripheral blood lymphocytes. Magn Reson Med (1993) 29:317–26. doi: 10.1002/mrm.1910290307
6. Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell (2002) 111:837–51. doi: 10.1016/S0092-8674(02)01139-X
7. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity (2007) 27:670–84. doi: 10.1016/j.immuni.2007.09.006
8. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7. doi: 10.1038/nature08097
9. Gett AV, Hodgkin PD. Cell division regulates the T cell cytokine repertoire, revealing a mechanism underlying immune class regulation. Proc Natl Acad Sci U S A (1998) 95:9488–93. doi: 10.1073/pnas.95.16.9488
10. Youngblood B, Hale JS, Ahmed R. T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology (2013) 139:277–84. doi: 10.1111/imm.12074
11. van der Windt GJ, O’Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci U S A (2013) 110:14336–41. doi: 10.1073/pnas.1221740110
12. Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood (2013) 121:573–84. doi: 10.1182/blood-2012-05-431718
13. Cho BK, Wang C, Sugawa S, Eisen HN, Chen J. Functional differences between memory and naive CD8 T cells. Proc Natl Acad Sci U S A (1999) 96:2976–81. doi: 10.1073/pnas.96.6.2976
14. Gudmundsdottir H, Wells AD, Turka LA. Dynamics and requirements of T cell clonal expansion in vivo at the single-cell level: effector function is linked to proliferative capacity. J Immunol (1999) 162:5212–23.
15. Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, et al. Role of PD-1 during effector CD8 T cell differentiation. Proc Natl Acad Sci U S A (2018) 115:4749–54. doi: 10.1073/pnas.1718217115
16. Wherry EJ, Blattman JN, Ahmed R. Low CD8 T-cell proliferative potential and high viral load limit the effectiveness of therapeutic vaccination. J Virol (2005) 79:8960–8. doi: 10.1128/JVI.79.14.8960-8968.2005
17. Badovinac VP, Porter BB, Harty JT. Programmed contraction of CD8(+) T cells after infection. Nat Immunol (2002) 3:619–26. doi: 10.1038/ni804
18. Smith CA, Williams GT, Kingston R, Jenkinson EJ, Owen JJ. Antibodies to CD3/T-cell receptor complex induce death by apoptosis in immature T cells in thymic cultures. Nature (1989) 337:181–4. doi: 10.1038/337181a0
19. Veiga-Fernandes H, Walter U, Bourgeois C, McLean A, Rocha B. Response of naive and memory CD8+ T cells to antigen stimulation in vivo. Nat Immunol (2000) 1:47–53. doi: 10.1038/76907
20. Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci U S A (2009) 106:17469–74. doi: 10.1073/pnas.0907448106
21. Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naïve cells. Nat Immunol (2001) 2:415–22. doi: 10.1038/87720
22. Lanzavecchia A, Sallusto F. Progressive differentiation and selection of the fittest in the immune response. Nat Rev Immunol (2002) 2:982–7. doi: 10.1038/nri959
23. Smith NL, Patel RK, Reynaldi A, Grenier JK, Wang J, Watson NB, et al. Developmental Origin Governs CD8(+) T Cell Fate Decisions during Infection. Cell (2018) 174:117–30.e114. doi: 10.1016/j.cell.2018.05.029
24. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol (2013) 14:1064–72. doi: 10.1038/ni.2687
25. O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity (2014) 41:75–88. doi: 10.1016/j.immuni.2014.06.005
26. Nasi M, Troiano L, Lugli E, Pinti M, Ferraresi R, Monterastelli E, et al. Thymic output and functionality of the IL-7/IL-7 receptor system in centenarians: implications for the neolymphogenesis at the limit of human life. Aging Cell (2006) 5:167–75. doi: 10.1111/j.1474-9726.2006.00204.x
27. Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell (2000) 6:683–92. doi: 10.1016/S1097-2765(00)00066-6
28. Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood (2008) 111:2101–11. doi: 10.1182/blood-2007-06-096297
29. Edinger AL, Thompson CB. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell (2002) 13:2276–88. doi: 10.1091/mbc.01-12-0584
30. Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem (2006) 281:37372–80. doi: 10.1074/jbc.M608372200
31. Borowski AB, Boesteanu AC, Mueller YM, Carafides C, Topham DJ, Altman JD, et al. Memory CD8+ T cells require CD28 costimulation. J Immunol (2007) 179:6494–503. doi: 10.4049/jimmunol.179.10.6494
32. Vezys V, Penaloza-MacMaster P, Barber DL, Ha SJ, Konieczny B, Freeman GJ, et al. 4-1BB signaling synergizes with programmed death ligand 1 blockade to augment CD8 T cell responses during chronic viral infection. J Immunol (2011) 187:1634–42. doi: 10.4049/jimmunol.1100077
33. Kundig TM, Shahinian A, Kawai K, Mittrucker HW, Sebzda E, Bachmann MF, et al. Duration of TCR stimulation determines costimulatory requirement of T cells. Immunity (1996) 5:41–52. doi: 10.1016/S1074-7613(00)80308-8
34. Richer MJ, Pewe LL, Hancox LS, Hartwig SM, Varga SM, Harty JT. Inflammatory IL-15 is required for optimal memory T cell responses. J Clin Invest (2015) 125:3477–90. doi: 10.1172/JCI81261
35. Cui G, Staron MM, Gray SM, Ho PC, Amezquita RA, Wu J, et al. IL-7-Induced Glycerol Transport and TAG Synthesis Promotes Memory CD8+ T Cell Longevity. Cell (2015) 161:750–61. doi: 10.1016/j.cell.2015.03.021
36. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
37. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2006) 439:682–7. doi: 10.1038/nature04444
38. Nowacki TM, Kuerten S, Zhang W, Shive CL, Kreher CR, Boehm BO, et al. Granzyme B production distinguishes recently activated CD8(+) memory cells from resting memory cells. Cell Immunol (2007) 247:36–48. doi: 10.1016/j.cellimm.2007.07.004
39. Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS, et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res (2011) 17:5343–52. doi: 10.1158/1078-0432.CCR-11-0503
40. Cham CM, Gajewski TF. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol (2005) 174:4670–7. doi: 10.4049/jimmunol.174.8.4670
41. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
42. Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
43. Singer K, Kastenberger M, Gottfried E, Hammerschmied CG, Buttner M, Aigner M, et al. Warburg phenotype in renal cell carcinoma: high expression of glucose-transporter 1 (GLUT-1) correlates with low CD8(+) T-cell infiltration in the tumor. Int J Cancer (2011) 128:2085–95. doi: 10.1002/ijc.25543
44. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol (2008) 180:4476–86. doi: 10.4049/jimmunol.180.7.4476
45. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
46. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science (2016) 354:481–4. doi: 10.1126/science.aaf6284
47. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
48. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol (2010) 185:1037–44. doi: 10.4049/jimmunol.0903586
49. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science (2009) 324:1076–80. doi: 10.1126/science.1164097
50. Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha NH, et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science (2019) 363. doi: 10.1126/science.aau0135
51. Yaqoob P, Calder PC. Glutamine requirement of proliferating T lymphocytes. Nutrition (1997) 13:646–51. doi: 10.1016/S0899-9007(97)83008-0
52. Horig H, Spagnoli GC, Filgueira L, Babst R, Gallati H, Harder F, et al. Exogenous glutamine requirement is confined to late events of T cell activation. J Cell Biochem (1993) 53:343–51. doi: 10.1002/jcb.240530412
53. Sukumar M, Kishton RJ, Restifo NP. Metabolic reprograming of anti-tumor immunity. Curr Opin Immunol (2017) 46:14–22. doi: 10.1016/j.coi.2017.03.011
54. Luengo A, Gui DY, Vander Heiden MG. Targeting Metabolism for Cancer Therapy. Cell Chem Biol (2017) 24:1161–80. doi: 10.1016/j.chembiol.2017.08.028
55. Reina-Campos M, Moscat J, Diaz-Meco M. Metabolism shapes the tumor microenvironment. Curr Opin Cell Biol (2017) 48:47–53. doi: 10.1016/j.ceb.2017.05.006
56. Mazzone M, Menga A, Castegna A. Metabolism and TAM functions-it takes two to tango. FEBS J (2018) 285:700–16. doi: 10.1111/febs.14295
57. Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab (2018) 27:977–87.e974. doi: 10.1016/j.cmet.2018.02.024
58. Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science (2019) 366:1013–21. doi: 10.1126/science.aav2588
59. Wang T, Gnanaprakasam JNR, Chen X, Kang S, Xu X, Sun H, et al. Inosine is an alternative carbon source for CD8(+)-T-cell function under glucose restriction. Nat Metab (2020) 2:635–47. doi: 10.1038/s42255-020-0219-4
60. Chisolm DA, Weinmann AS. Connections Between Metabolism and Epigenetics in Programming Cellular Differentiation. Annu Rev Immunol (2018) 36:221–46. doi: 10.1146/annurev-immunol-042617-053127
61. Sinclair LV, Howden AJ, Brenes A, Spinelli L, Hukelmann JL, Macintyre AN, et al. Antigen receptor control of methionine metabolism in T cells. Elife (2019) 8. doi: 10.7554/eLife.44210
62. Sedlackova L, Korolchuk VI. Mitochondrial quality control as a key determinant of cell survival. Biochim Biophys Acta Mol Cell Res (2019) 1866:575–87. doi: 10.1016/j.bbamcr.2018.12.012
63. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vazquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity (2015) 42:41–54. doi: 10.1016/j.immuni.2014.12.030
64. Youngblood B, Davis CW, Ahmed R. Making memories that last a lifetime: heritable functions of self-renewing memory CD8 T cells. Int Immunol (2010) 22:797–803. doi: 10.1093/intimm/dxq437
65. Quintana A, Schwindling C, Wenning AS, Becherer U, Rettig J, Schwarz EC, et al. T cell activation requires mitochondrial translocation to the immunological synapse. Proc Natl Acad Sci U S A (2007) 104:14418–23. doi: 10.1073/pnas.0703126104
66. Bantug GR, Fischer M, Grahlert J, Balmer ML, Unterstab G, Develioglu L, et al. Mitochondria-Endoplasmic Reticulum Contact Sites Function as Immunometabolic Hubs that Orchestrate the Rapid Recall Response of Memory CD8(+) T Cells. Immunity (2018) 48:542–55.e546. doi: 10.1016/j.immuni.2018.02.012
67. Rieusset J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: an update. Cell Death Dis (2018) 9:388. doi: 10.1038/s41419-018-0416-1
68. Wu S, Zhou F, Zhang Z, Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J (2011) 278:941–54. doi: 10.1111/j.1742-4658.2011.08010.x
69. Quintana A, Hoth M. Mitochondrial dynamics and their impact on T cell function. Cell Calcium (2012) 52:57–63. doi: 10.1016/j.ceca.2012.02.005
70. Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell (2016) 166:63–76. doi: 10.1016/j.cell.2016.05.035
71. Rambold AS, Pearce EL. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol (2018) 39:6–18. doi: 10.1016/j.it.2017.08.006
72. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science (2011) 334:358–62. doi: 10.1126/science.1207385
73. Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep (2007) 8:939–44. doi: 10.1038/sj.embor.7401062
74. Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A (2008) 105:15803–8. doi: 10.1073/pnas.0808249105
75. Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A (2011) 108:10190–5. doi: 10.1073/pnas.1107402108
76. Wang D, Wang J, Bonamy GM, Meeusen S, Brusch RG, Turk C, et al. A small molecule promotes mitochondrial fusion in mammalian cells. Angew Chem Int Ed Engl (2012) 51:9302–5. doi: 10.1002/anie.201204589
77. Garcia D, Shaw RJ. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol Cell (2017) 66:789–800. doi: 10.1016/j.molcel.2017.05.032
78. Mayer A, Denanglaire S, Viollet B, Leo O, Andris F. AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. Eur J Immunol (2008) 38:948–56. doi: 10.1002/eji.200738045
79. Ito K, Ito K. Metabolism and the Control of Cell Fate Decisions and Stem Cell Renewal. Annu Rev Cell Dev Biol (2016) 32:399–409. doi: 10.1146/annurev-cellbio-111315-125134
80. Jeninga EH, Schoonjans K, Auwerx J. Reversible acetylation of PGC-1: connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene (2010) 29:4617–24. doi: 10.1038/onc.2010.206
81. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity (2016) 45:374–88. doi: 10.1016/j.immuni.2016.07.009
82. Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
83. Almeida L, Lochner M, Berod L, Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol (2016) 28:514–24. doi: 10.1016/j.smim.2016.10.009
84. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
85. Guillaumond F, Leca J, Olivares O, Lavaut MN, Vidal N, Berthezene P, et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A (2013) 110:3919–24. doi: 10.1073/pnas.1219555110
86. Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA, et al. Oxygen Sensing by T Cells Establishes an Immunologically Tolerant Metastatic Niche. Cell (2016) 166:1117–31.e1114. doi: 10.1016/j.cell.2016.07.032
87. Broer S. Lactate transportation is required for lymphocyte activation. Nat Chem Biol (2005) 1:356–7. doi: 10.1038/nchembio1205-356
88. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
89. Murray CM, Hutchinson R, Bantick JR, Belfield GP, Benjamin AD, Brazma D, et al. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat Chem Biol (2005) 1:371–6 doi: 10.1038/nchembio744.
90. Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest (2013) 123:3685–92. doi: 10.1172/JCI69741
91. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer (2004) 4:891–9. doi: 10.1038/nrc1478
92. Sommer F, Bischof S, Rollinghoff M, Lohoff M. Demonstration of organic anion transport in T lymphocytes. L-lactate and fluo-3 are target molecules. J Immunol (1994) 153:3523–32.
93. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res (2012) 72:2746–56. doi: 10.1158/0008-5472.CAN-11-1272
94. van Stipdonk MJ, Hardenberg G, Bijker MS, Lemmens EE, Droin NM, Green DR, et al. Dynamic programming of CD8+ T lymphocyte responses. Nat Immunol (2003) 4:361–5. doi: 10.1038/ni912
95. Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab (2017) 25:1282–93.e1287. doi: 10.1016/j.cmet.2016.12.018
96. Comito G, Iscaro A, Bacci M, Morandi A, Ippolito L, Parri M, et al. Lactate modulates CD4(+) T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene (2019) 38:3681–95. doi: 10.1038/s41388-019-0688-7
97. Szymczak-Workman AL, Delgoffe GM, Green DR, Vignali DA. Cutting edge: regulatory T cells do not mediate suppression via programmed cell death pathways. J Immunol (2011) 187:4416–20. doi: 10.4049/jimmunol.1100548
98. Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci U S A (2018) 115:E6546–55. doi: 10.1073/pnas.1720113115
99. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I, et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature (2019) 565:495–9. doi: 10.1038/s41586-018-0846-z
100. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature (2021). doi: 10.1038/s41586-020-03045-2
101. Zappasodi R, Serganova I, Cohen IJ, Maeda M, Shindo M, Senbabaoglu Y, et al. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature (2021). doi: 10.1038/s41586-021-03326-4
102. DeVita VT Jr, Chu E. A history of cancer chemotherapy. Cancer Res (2008) 68:8643–53. doi: 10.1158/0008-5472.CAN-07-6611
103. Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature (2016) 537:539–43. doi: 10.1038/nature19364
104. Ohta A, Madasu M, Subramanian M, Kini R, Jones G, Chouker A, et al. Hypoxia-induced and A2A adenosine receptor-independent T-cell suppression is short lived and easily reversible. Int Immunol (2014) 26:83–91. doi: 10.1093/intimm/dxt045
105. Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol (2014) 5:304. doi: 10.3389/fimmu.2014.00304
106. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab (2019) 30:143–56.e145. doi: 10.1016/j.cmet.2019.04.002
107. Ando M, Ito M, Srirat T, Kondo T, Yoshimura A. Memory T cell, exhaustion, and tumor immunity. Immunol Med (2020) 43:1–9. doi: 10.1080/25785826.2019.1698261
108. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114:1537–44. doi: 10.1182/blood-2008-12-195792
109. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207:2187–94. doi: 10.1084/jem.20100643
110. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
111. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2012) 72:917–27. doi: 10.1158/0008-5472.CAN-11-1620
112. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
113. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity (2016) 45:358–73. doi: 10.1016/j.immuni.2016.07.008
114. Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol (2008) 38:2438–50. doi: 10.1002/eji.200838289
115. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1:405–13 doi: 10.1084/jem.183.6.2541.
116. Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med (1996) 183:2541–50. doi: 10.1084/jem.183.6.2541
117. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al. A new member of the immunoglobulin superfamily–CTLA-4. Nature (1987) 328:267–70. doi: 10.1038/328267a0
118. Gribben JG, Freeman GJ, Boussiotis VA, Rennert P, Jellis CL, Greenfield E, et al. CTLA4 mediates antigen-specific apoptosis of human T cells. Proc Natl Acad Sci U S A (1995) 92:811–5. doi: 10.1073/pnas.92.3.811
119. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med (1990) 171:1393–405. doi: 10.1084/jem.171.5.1393
120. Hannier S, Tournier M, Bismuth G, Triebel F. CD3/TCR complex-associated lymphocyte activation gene-3 molecules inhibit CD3/TCR signaling. J Immunol (1998) 161:4058–65.
121. Daskivich TJ, Belldegrun A. Words of wisdom. Re: Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. Eur Urol (2015) 67:816–7. doi: 10.1016/j.eururo.2014.12.052
122. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
123. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348:62–8. doi: 10.1126/science.aaa4967
124. Tran E, Robbins PF, Rosenberg SA. ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol (2017) 18:255–62. doi: 10.1038/ni.3682
125. Lee H, Lee M, Seo JH, Gong G, Lee HJ. Changes in Tumor-infiltrating Lymphocytes After Neoadjuvant Chemotherapy and Clinical Significance in Triple Negative Breast Cancer. Anticancer Res (2020) 40:1883–90. doi: 10.21873/anticanres.14142
126. Yossef R, Tran E, Deniger DC, Gros A, Pasetto A, Parkhurst MR, et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight (2018) 3. doi: 10.1172/jci.insight.122467
127. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol (2015) 33:540–9. doi: 10.1200/JCO.2014.56.2025
128. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
129. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5:177ra138. doi: 10.1126/scitranslmed.3005930
130. Knochelmann HM, Smith AS, Dwyer CJ, Wyatt MM, Mehrotra S, Paulos CM. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front Immunol (2018) 9:1740. doi: 10.3389/fimmu.2018.01740
131. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci Trans Med (2014) 6:224ra225. doi: 10.1126/scitranslmed.3008226
132. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
133. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med (2015) 21:581–90. doi: 10.1038/nm.3838
134. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17:1290–7. doi: 10.1038/nm.2446
135. Dwyer CJ, Knochelmann HM, Smith AS, Wyatt MM, Rangel Rivera GO, Arhontoulis DC, et al. Fueling Cancer Immunotherapy With Common Gamma Chain Cytokines. Front Immunol (2019) 10:263. doi: 10.3389/fimmu.2019.00263
136. Liu S, Riley J, Rosenberg S, Parkhurst M. Comparison of common gamma-chain cytokines, interleukin-2, interleukin-7, and interleukin-15 for the in vitro generation of human tumor-reactive T lymphocytes for adoptive cell transfer therapy. J Immunother (2006) 29:284–93. doi: 10.1097/01.cji.0000190168.53793.6b
137. Yang S, Ji Y, Gattinoni L, Zhang L, Yu Z, Restifo NP, et al. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol Immunother (2013) 62:727–36. doi: 10.1007/s00262-012-1378-2
138. Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood (2008) 111:5326–33. doi: 10.1182/blood-2007-09-113050
139. Shen S, Sckisel G, Sahoo A, Lalani A, Otter DD, Pearson J, et al. Engineered IL-21 Cytokine Muteins Fused to Anti-PD-1 Antibodies Can Improve CD8+ T Cell Function and Anti-tumor Immunity. Front Immunol (2020) 11:832. doi: 10.3389/fimmu.2020.00832
140. Li Y, Cong Y, Jia M, He Q, Zhong H, Zhao Y, et al. Targeting IL-21 to tumor-reactive T cells enhances memory T cell responses and anti-PD-1 antibody therapy. Nat Commun (2021) 12:951. doi: 10.1038/s41467-021-21241-0
141. Catakovic K, Klieser E, Neureiter D, Geisberger R. T cell exhaustion: from pathophysiological basics to tumor immunotherapy. Cell Commun Signal (2017) 15:1. doi: 10.1186/s12964-016-0160-z
142. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16:769–77. doi: 10.1016/s1074-7613(02)00323-0
143. Petersen CT, Hassan M, Morris AB, Jeffery J, Lee K, Jagirdar N, et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kdelta inhibitors and VIP antagonists. Blood Adv (2018) 2:210–23. doi: 10.1182/bloodadvances.2017011254
144. Bowers JS, Majchrzak K, Nelson MH, Aksoy BA, Wyatt MM, Smith AS, et al. PI3Kdelta Inhibition Enhances the Antitumor Fitness of Adoptively Transferred CD8(+) T Cells. Front Immunol (2017) 8:1221. doi: 10.3389/fimmu.2017.01221
145. Majchrzak K, Nelson MH, Bowers JS, Bailey SR, Wyatt MM, Wrangle JM, et al. beta-catenin and PI3Kdelta inhibition expands precursor Th17 cells with heightened stemness and antitumor activity. JCI Insight (2017) 2. doi: 10.1172/jci.insight.90547
146. Dwyer CJ, Arhontoulis DC, Rangel Rivera GO, Knochelmann HM, Smith AS, Wyatt MM, et al. Ex vivo blockade of PI3K gamma or delta signaling enhances the antitumor potency of adoptively transferred CD8(+) T cells. Eur J Immunol (2020) 50:1386–9. doi: 10.1002/eji.201948455
147. Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res (2015) 75:296–305. doi: 10.1158/0008-5472.CAN-14-2277
148. Mousset CM, Hobo W, Ji Y, Fredrix H, De Giorgi V, Allison RD, et al. Ex vivo AKT-inhibition facilitates generation of polyfunctional stem cell memory-like CD8(+) T cells for adoptive immunotherapy. Oncoimmunology (2018) 7:e1488565. doi: 10.1080/2162402X.2018.1488565
149. He S, Kato K, Jiang J, Wahl DR, Mineishi S, Fisher EM, et al. Characterization of the metabolic phenotype of rapamycin-treated CD8+ T cells with augmented ability to generate long-lasting memory cells. PLoS One (2011) 6:e20107. doi: 10.1371/journal.pone.0020107
150. Scholz G, Jandus C, Zhang L, Grandclement C, Lopez-Mejia IC, Soneson C, et al. Modulation of mTOR Signalling Triggers the Formation of Stem Cell-like Memory T Cells. EBioMedicine (2016) 4:50–61. doi: 10.1016/j.ebiom.2016.01.019
151. Klebanoff CA, Crompton JG, Leonardi AJ, Yamamoto TN, Chandran SS, Eil RL, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight (2017) 2. doi: 10.1172/jci.insight.95103
152. Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol (2014) 15:1152–61. doi: 10.1038/ni.3025
153. Semenza GL. Mitochondrial autophagy: life and breath of the cell. Autophagy (2008) 4:534–6. doi: 10.4161/auto.5956
154. Yu YR, Imrichova H, Wang H, Chao T, Xiao Z, Gao M, et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat Immunol (2020) 21:1540–51. doi: 10.1038/s41590-020-0793-3
155. Gurusamy D, Henning AN, Yamamoto TN, Yu Z, Zacharakis N, Krishna S, et al. Multi-phenotype CRISPR-Cas9 Screen Identifies p38 Kinase as a Target for Adoptive Immunotherapies. Cancer Cell (2020) 37:818–33. doi: 10.1016/j.ccell.2020.05.004
156. Thaxton JE, Wallace C, Riesenberg B, Zhang Y, Paulos CM, Beeson CC, et al. Modulation of Endoplasmic Reticulum Stress Controls CD4(+) T-cell Activation and Antitumor Function. Cancer Immunol Res (2017) 5:666–75. doi: 10.1158/2326-6066.CIR-17-0081
157. Hurst KE, Lawrence KA, Reyes Angeles L, Ye Z, Zhang J, Townsend DM, et al. Endoplasmic Reticulum Protein Disulfide Isomerase Shapes T Cell Efficacy for Adoptive Cellular Therapy of Tumors. Cells (2019) 8. doi: 10.3390/cells8121514
158. Hurst KE, Lawrence KA, Essman MT, Walton ZJ, Leddy LR, Thaxton JE. Endoplasmic Reticulum Stress Contributes to Mitochondrial Exhaustion of CD8(+) T Cells. Cancer Immunol Res (2019) 7:476–86. doi: 10.1158/2326-6066.CIR-18-0182
159. Hurst KE, Lawrence KA, Robino RA, Ball LE, Chung D, Thaxton JE. Remodeling Translation Primes CD8(+) T-cell Antitumor Immunity. Cancer Immunol Res (2020) 8:587–95. doi: 10.1158/2326-6066.CIR-19-0516
160. Chellappa S, Kushekhar K, Munthe LA, Tjonnfjord GE, Aandahl EM, Okkenhaug K, et al. The PI3K p110delta Isoform Inhibitor Idelalisib Preferentially Inhibits Human Regulatory T Cell Function. J Immunol (2019) 202:1397–405. doi: 10.4049/jimmunol.1701703
161. Ahmad S, Abu-Eid R, Shrimali R, Webb M, Verma V, Doroodchi A, et al. Differential PI3Kdelta Signaling in CD4(+) T-cell Subsets Enables Selective Targeting of T Regulatory Cells to Enhance Cancer Immunotherapy. Cancer Res (2017) 77:1892–904. doi: 10.1158/0008-5472.CAN-16-1839
162. Akhiani AA, Hallner A, Kiffin R, Aydin E, Werlenius O, Aurelius J, et al. Idelalisib Rescues Natural Killer Cells from Monocyte-Induced Immunosuppression by Inhibiting NOX2-Derived Reactive Oxygen Species. Cancer Immunol Res (2020) 8:1532–41. doi: 10.1158/2326-6066.CIR-20-0055
163. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123:4479–88. doi: 10.1172/JCI69589
164. Klein Geltink RI, Edwards-Hicks J, Apostolova P, O’Sullivan D, Sanin DE, Patterson AE, et al. Metabolic conditioning of CD8(+) effector T cells for adoptive cell therapy. Nat Metab (2020) 2:703–16. doi: 10.1038/s42255-020-0256-z
165. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
166. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell (2016) 167:829–42.e813. doi: 10.1016/j.cell.2016.09.031
167. Bailey SR, Nelson MH, Majchrzak K, Bowers JS, Wyatt MM, Smith AS, et al. Human CD26(high) T cells elicit tumor immunity against multiple malignancies via enhanced migration and persistence. Nat Commun (2017) 8:1961. doi: 10.1038/s41467-017-01867-9
168. Chamoto K, Chowdhury PS, Kumar A, Sonomura K, Matsuda F, Fagarasan S, et al. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci U S A (2017) 114:E761–70. doi: 10.1073/pnas.1620433114
169. Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8(+) T Cells and Facilitates Anti-PD-1 Therapy. Cancer Immunol Res (2018) 6:1375–87. doi: 10.1158/2326-6066.CIR-18-0095
170. Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res (2014) 74:908–20. doi: 10.1158/0008-5472.CAN-13-2034
171. Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, et al. Targeting lactate dehydrogenase–a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab (2014) 19:795–809. doi: 10.1016/j.cmet.2014.03.003
172. Hatfield SM, Kjaergaard J, Lukashev D, Belikoff B, Schreiber TH, Sethumadhavan S, et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1alpha-dependent and extracellular adenosine-mediated tumor protection. J Mol Med (Berl) (2014) 92:1283–92. doi: 10.1007/s00109-014-1189-3
173. Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol Res (2017) 5:9–16. doi: 10.1158/2326-6066.CIR-16-0103
174. Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, et al. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res (2016) 76:1381–90. doi: 10.1158/0008-5472.CAN-15-1743
175. Sborov DW, Haverkos BM, Harris PJ. Investigational cancer drugs targeting cell metabolism in clinical development. Expert Opin Investig Drugs (2015) 24:79–94. doi: 10.1517/13543784.2015.960077
176. Kroon P, Frijlink E, Iglesias-Guimarais V, Volkov A, van Buuren MM, Schumacher TN, et al. Radiotherapy and Cisplatin Increase Immunotherapy Efficacy by Enabling Local and Systemic Intratumoral T-cell Activity. Cancer Immunol Res (2019) 7:670–82. doi: 10.1158/2326-6066.CIR-18-0654
177. Hong M, Puaux AL, Huang C, Loumagne L, Tow C, Mackay C, et al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res (2011) 71:6997–7009. doi: 10.1158/0008-5472.CAN-11-1466
Keywords: T cell metabolism, tumor microenvironment, adoptive T cell transfer, immune checkpoint therapy, tumor metabolism, tumor infiltrating lymphocytes
Citation: Rangel Rivera GO, Knochelmann HM, Dwyer CJ, Smith AS, Wyatt MM, Rivera-Reyes AM, Thaxton JE and Paulos CM (2021) Fundamentals of T Cell Metabolism and Strategies to Enhance Cancer Immunotherapy. Front. Immunol. 12:645242. doi: 10.3389/fimmu.2021.645242
Received: 22 December 2020; Accepted: 01 March 2021;
Published: 18 March 2021.
Edited by:
Peng Qu, National Institutes of Health (NIH), United StatesReviewed by:
Yiming Yin, University of Florida, United StatesXiaobin Huang, University of Pennsylvania, United States
Copyright © 2021 Rangel Rivera, Knochelmann, Dwyer, Smith, Wyatt, Rivera-Reyes, Thaxton and Paulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guillermo O. Rangel Rivera, cmFuZ2VsYXJAbXVzYy5lZHU=; Chrystal M. Paulos, Y2hyeXN0YWwubWFyeS5wYXVsb3NAZW1vcnkuZWR1