 Yujia Zhai
Yujia Zhai Reza Moosavi
Reza Moosavi Mingnan Chen
Mingnan Chen- Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, UT, United States
Autoimmune diseases, such as multiple sclerosis and type-1 diabetes, are the outcomes of a failure of immune tolerance. Immune tolerance is sustained through interplays between two inter-dependent clusters of immune activities: immune stimulation and immune regulation. The mechanisms of immune regulation are exploited as therapeutic targets for the treatment of autoimmune diseases. One of these mechanisms is immune checkpoints (ICPs). The roles of ICPs in maintaining immune tolerance and hence suppressing autoimmunity were revealed in animal models and validated by the clinical successes of ICP-targeted therapeutics for autoimmune diseases. Recently, these roles were highlighted by the clinical discovery that the blockade of ICPs causes autoimmune disorders. Given the crucial roles of ICPs in immune tolerance, it is plausible to leverage ICPs as a group of therapeutic targets to restore immune tolerance and treat autoimmune diseases. In this review, we first summarize working mechanisms of ICPs, particularly those that have been utilized for therapeutic development. Then, we recount the agents and approaches that were developed to target ICPs and treat autoimmune disorders. These agents take forms of fusion proteins, antibodies, nucleic acids, and cells. We also review and discuss safety information for these therapeutics. We wrap up this review by providing prospects for the development of ICP-targeting therapeutics. In summary, the ever-increasing studies and results of ICP-targeting of therapeutics underscore their tremendous potential to become a powerful class of medicine for autoimmune diseases.
Introduction
The immune system is powerful and versatile in protecting the body: it neutralizes invading pathogens and toxins, detects and destroys infected and malignant cells, and orchestrates the recovery of compromised tissues. As famously said in the movie Spider-Man, “With great power comes great responsibility.” The immune system has the “responsibility” to refrain from excessively attacking and destroying cells and tissues, particularly self and benign cells and tissues. Immunostasis is the immune system’s adaptation to provide restraint. Immunostasis is achieved and maintained through the competition and interplays between two clusters of activities, immune stimulation and immune regulation. The stimulation wing of immunity ensures the immune system is powerful and specific when immunity is needed to decisively eliminate invading pathogens and malignant cells. Stimulation also cultivates immune memory so that the immune system is able to stem the outgrowth of pathogens or malignance faster and more effectively when it encounters pathogens and malignancy again. The regulation wing of immunity, on the other hand, keeps all the traits or strengths of stimulation in check. As put by a Chinese idiom, 过犹不及(Guò-yóu-bù-jí) or in English, “Going beyond the limit is as bad as falling short,” it is extremely important for the immune system and immune stimulation to stay in check because excessive attack, whether in the sense of target ranges, intensity, or duration, will do more harm than benefit to the body. In addition, excessive attack wastes the energy of the immune system, a loss which could prevent the immune system to use its full strength to tackle real threats. Immune regulatory activities may lead to an outcome termed immune tolerance. Indeed, the immune system has diversified and sophisticated mechanisms to achieve and maintain immune tolerance, ranging from central tolerance, peripheral tolerance, to clonal tolerance (1). The failure of immune tolerance, either due to inherited genetic deficiencies or exogeneous stimulants, is the fundamental reason for autoimmune disorders including type 1 diabetes (T1D), multiple sclerosis (MS), rheumatoid arthritis (RA), and so on. Collectively, these diseases affect approximately 12.5% of world population (2).
One relatively newly discovered mechanism to help to maintain immune tolerance is immune checkpoints (ICPs) (3–6). The first ICP, cytotoxic T lymphocyte antigen-4 (CTLA-4), was reported in the early 1990s (7). In the last 20 plus years, ICPs have proven their importance to immune tolerance thanks to extensive research efforts and consequent discoveries around it. ICPs usually required two biomolecular components, a receptor and a ligand. Receptors are mostly expressed on the surface of immune cells (8–13). Ligands, on the other hand, are expressed sometimes by immune cells, and sometimes by non-hemopoietic cells (14–21). When a receptor and a ligand of an ICP engage with each other, that ICP is flipped on and transmits inhibitory signals. Meanwhile, the cells possessing ICP receptors transform into their immune regulative (suppressive) mode. To date, several ICPs have been revealed. Among them, eight ICPs (Figure 1) have been recognized with sufficient knowledge so that experimental therapeutics are developed on the basis of these ICPs. Thus, our review focuses on these eight ICPs: programmed death-1 (PD-1) (22), CTLA-4 (7), B and T cell lymphocyte attenuator (BTLA) (23), T-cell immunoglobulin domain and mucin domain-containing molecule-3 (TIM-3) (24), T cell immunoglobulin and ITIM domain (TIGIT) (25), V-domain Ig suppressor of T cell activation (VISTA) (26), lymphocyte activation gene 3 (LAG-3) (27), and CD200 (28).
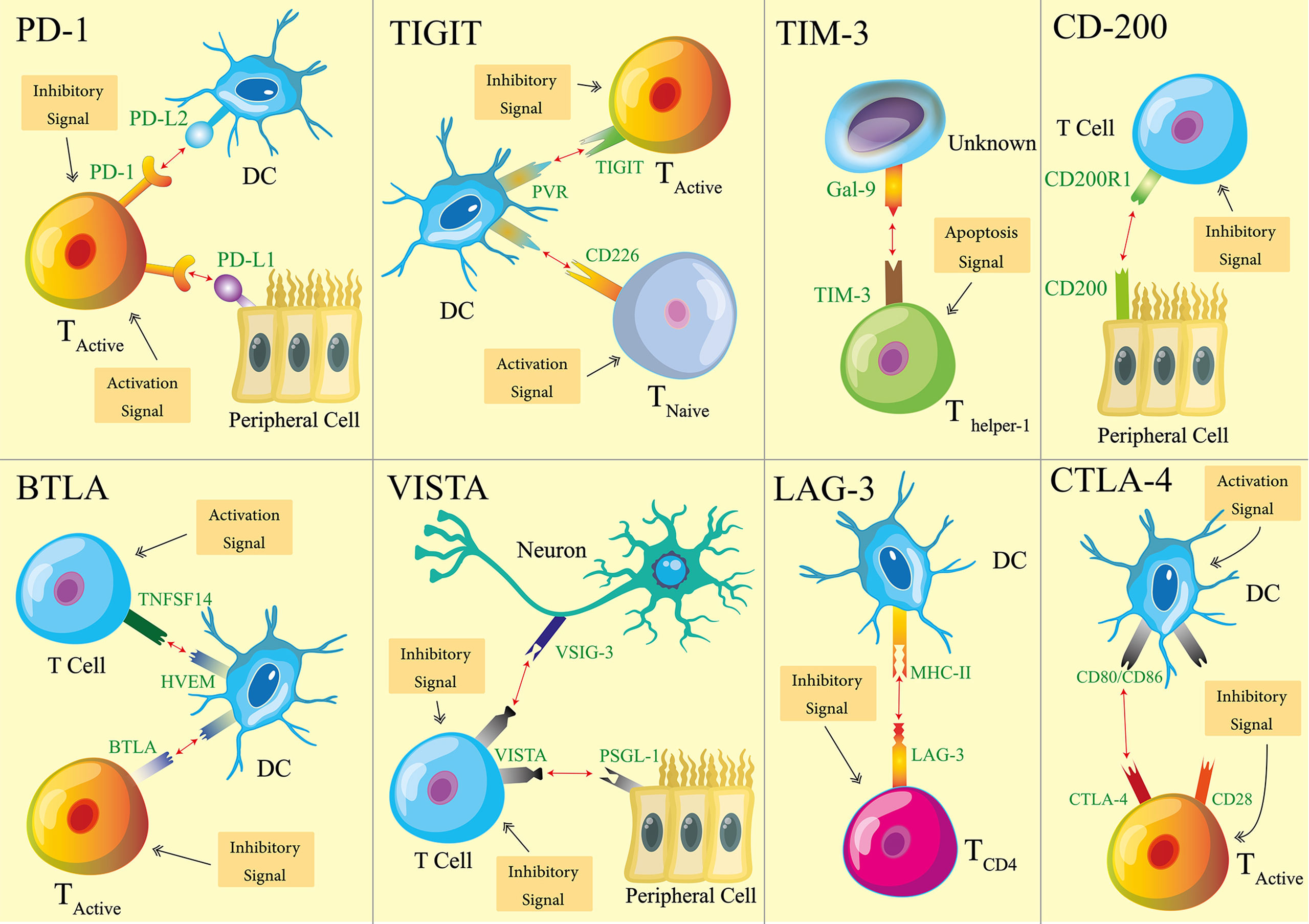
Figure 1 Schematics of eight immune checkpoints (ICPs). For each ICP, one type of cells is used as the representatives that host ICP receptors or ligands. The main immune activation and inhibition implications of ICPs are illustrated with the representative cell types. It is noteworthy that the receptors and ligands may be expressed by additional cell types. The functional implications of ICPs are not limited to what are illustrated here.
The necessity of ICPs in maintaining immune tolerance and preventing autoimmune disorders are underscored by data from animal models. As shown in Table 1, when receptors of ICPs are knocked out in mice, autoimmune disorders emerge and manifest in varied severity and organ-specificity, depending the type of ICPs and the genetic background of the mice. The PD-1 receptor knockout increases the prevalence of T1D among nonobese diabetic (NOD) mice (29), results in the lupus-like phenotype in the C57BL/6 background (30), and causes lethal myocarditis among Murphy Roths Large mice (31). The knockout of the BTLA receptor triggers experimental autoimmune encephalitis (EAE), a murine version of MS (23). The knockout of the TIGIT receptor increase susceptibility to EAE (32). The knockout of the LAG-3 receptor exacerbates T1D among NOD mice (33), and renders B6.SJL mice more susceptible to the hyper production of autoantibodies (34). The most striking and pervasive impact of the ICP knockout comes from the knockout of CTLA-4 ICP. Mice carrying such a knockout experience massive lymphoproliferation and die of multiorgan autoimmune destruction within 4 weeks after their birth (35). Recently, we examined the role of the PD-1 ICP and more specifically, the role of PD-1 positive cells in driving autoimmune disorders including EAE and T1D. The depletion PD-1 positive cells drastically delayed the onset of T1D and promoted the recovery from clinical presentations of EAE (36). These results unambiguously demonstrate the central role of PD-1 positive cells in these autoimmune diseases.
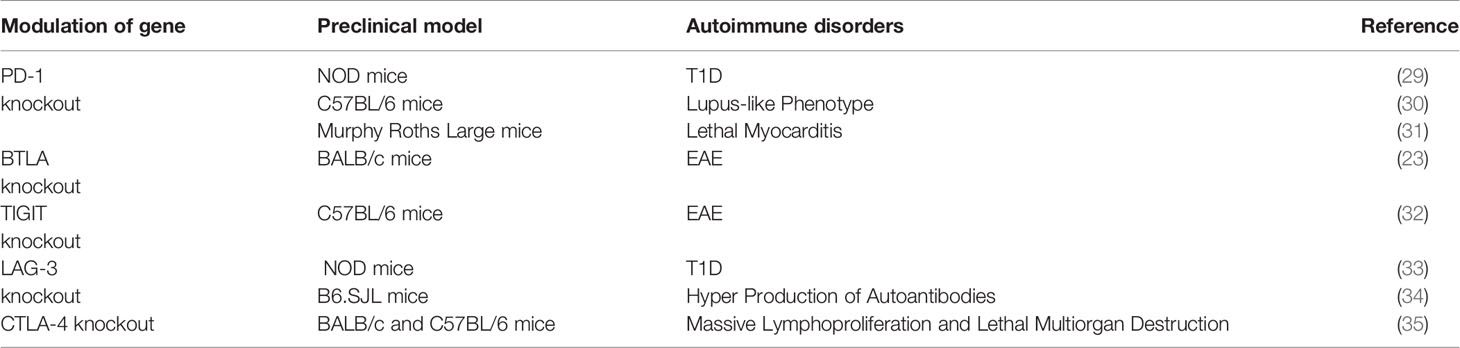
Table 1 Autoimmune disorders caused by the knockout of ICP molecules.
The necessity of ICPs for immunostasis and immune tolerance is also supported by human data. First and foremost, a therapeutic that reinforces the CTLA-4 ICP, Abatacept (Orencia®), has been approved to treat adult RA. From another perspectives, when the CTLA-4 and PD-1 ICPs are blocked in cancer patients, some patients experienced adverse autoimmune syndromes such as arthralgia, arthritis, Sjögren’s syndrome, and encephalitis (37, 38). The blockade inhibits the function of ICPs, which unleashes the attacking power of immune cells that express the corresponding ICP receptors. Thus, the fact that the ICP blockade induces autoimmune disorders in human proves the suppressive function of ICPs in autoimmunity. Interestingly, the blockade of the CTLA-4 ICP causes significantly more prevalent toxicity than the blockade of PD-1 ICP (39), which echoes the observations of the ICP knockout data from animal models.
With the recognition of the critical role of ICPs in immune tolerance and autoimmune diseases, autoimmune disease therapeutics that target ICPs have already been widely explored. These therapeutics can be generally classified into two categories in terms of strategies: one, enhancement of ICPs; and two, depletion of immune cells that express ICP receptors. We will summarize the therapeutics in these two categories, with an emphasis on their design, mode of action, and efficacy. Before we dive into these therapeutics, we will first provide mechanistic information for eight ICPs that have been exploited as therapeutic targets. Table 2 summarizes the receptors and ligands of these ICPs.
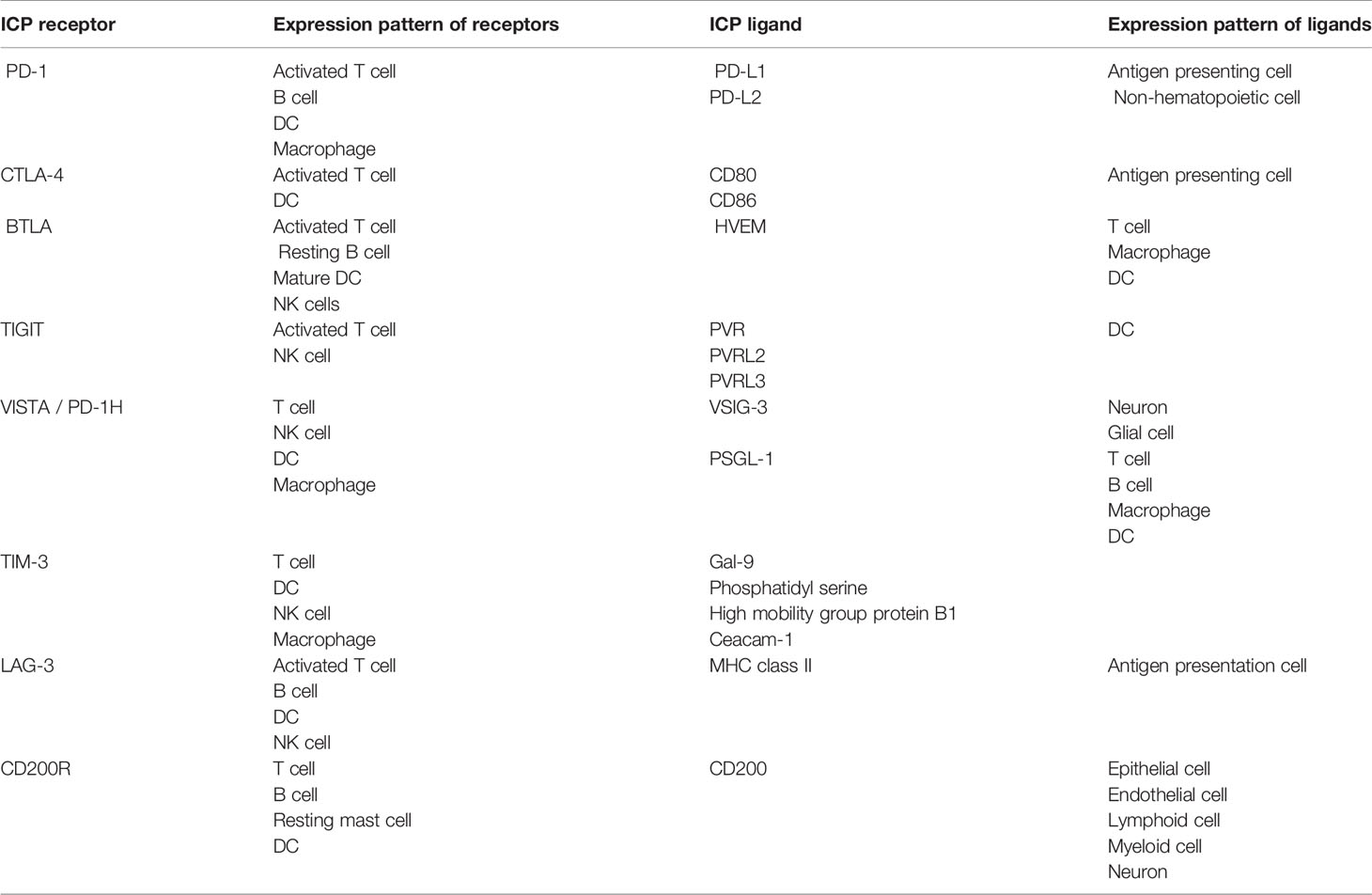
Table 2 Receptors and ligands of immune checkpoints (ICPs).
Mechanisms of Common Immune Checkpoints
For the PD-1 ICP, its receptor, PD-1 is primarily expressed on T cells, B cells, dendritic cells (DCs), and macrophages after their activation. PD-1 has two ligands, PD-L1 and PD-L2. PD-L1 is expressed on antigen presenting cells (APCs) including DCs, macrophages, and B cells (14). PD-L1 is also expressed on non-hematopoietic cells, such as vascular endothelial cells, epithelial cells, fibroblastic reticular cells, and pancreatic islet cells (15). PD-L2, similar to PD-L1, is expressed on both APCs and nonhematopoietic cells such as respiratory tract epithelial cells (16). Regarding PD-1 on T cells, the engagement of PD-1 with its ligands confers inhibitory signals to the T cells that express PD-1. The activation of the PD-1 ICP dampens the proliferation, cytokine secretion, as well as the cytotoxicity of T cells. Regarding PD-1 on human macrophages, an elevated expression of PD-1 by these cells leads to an increase of the IL-10 level and decreases the IL-12 level in blood, a sign of immune self-regulation on macrophages (40, 41). Lastly, PD-1 positive APCs show a weaker ability than PD-1 negative APCs in terms of promoting the differentiation of CD4 T cells into T regulatory cells (Tregs) (42).
CTLA-4 is an ICP receptor primarily expressed on DCs and activated T cells including memory T cells and Tregs (8). It has two ligands, (B7-1) and CD86 (B7-2), mainly expressed on APCs. CD80 and CD86 also bind with a co-stimulation receptor, CD28, on T cells. Although CTLA-4 has been shown to have an intracellular working mechanism where binding with its ligands triggers downstream signaling (43, 44), the literature on CTLA-4 primarily focuses on its extracellular working mechanism. There therapeutic development around the CTLA-4 ICP also primarily leverages the extracellular mechanism. Extracellularly, CTLA-4 executes its immunosuppressive function by competing with CD28 for access to CD80 and CD86. One outcome of this competition is weakened proliferation and cytokine secretion by T cells (45). When CTLA-4 on Tregs binds with CD80 and CD86 on DCs, the DCs show reduced antigen-presenting capacity (46). CTLA-4 expressed on Tregs also augments the suppression effect of Tregs by extending the interaction time between Tregs and effector cells (47). Further, when mature DCs are treated with agonistic anti-CTLA-4 antibodies, the production of IL-8 and IL-12 by the DCs reduces by two thirds, and the antigen presentation ability of DCs decreases to approximately one fourth of the normal level. The treatment of the anti-CTLA-4 antibodies also weakened the capacity of the DCs to stimulate T cell proliferation by 50% (48, 49).
For the BTLA ICP, its receptor, BTLA, belongs to the CD28 family. BTLA is mainly expressed on activated T cells but also on resting B cells, mature DCs, and natural killer (NK) cells (9). The ligand of BTLA is the herpesvirus entry mediator (HVEM) that is expressed on resting T cells, macrophages, and immature DCs (17). In addition to BTLA, HVEM also binds with four other molecules: CD160 (expressed on effector T cells and NK cells), tumor necrosis factor superfamily member 14 (TNFSF14, also termed as LIGHT or CD258, expressed on activated T cells and immature DCs), lymphotoxin-α (expressed on T cells, B cells, and NK cells), and HSV-1 glycoprotein D (expressed on herpes simplex virus infected cells), independently. Indeed, HVEM is a “bidirectional switch” of immune activation (50–53). When HVEM binds with BTLA or CD160, immune inhibitory signals are transmitted (HVEM may also be referred to as a ligand of the CD160 ICP in this sense); when HVEM binds with TNFSF14, lymphotoxin-α, or HSV-1 glycoprotein D, immune stimulatory signals are transmitted. The engagement of HVEM by BTLA delivers a diminishing effect on immune stimulation. The engagement also dampens T cell proliferation and B cell activation, reduces the number of CD8+ DCs, and lessens the secretion of cytokines such as TNF-α, IFN-γ, IL-2, and IL-4 by these cells (52, 54–57).
Regarding the TIGIT ICP, the receptor, TIGIT, is expressed on Tregs, memory T cells, effector T cells, and NK cells after these cells are activated (10, 11). Up to now, TIGIT has been found to interact with three molecules: poliovirus receptor (PVR, also termed as CD155), PVRL2, and PVRL3. The immunological implication of the interactions between TIGIT and PVRL2 or PVRL3 remains elusive. The functional implication of the interaction between TIGIT and PVR is, however, much clearer and intriguing. PVR is found on DCs and B cells and has a high binding affinity to TIGIT. In addition to TIGIT, PVR also interacts with CD226 on naive T cells. When PVR binds with TIGIT on activated T cells, coinhibitory signals are transmitted (58, 59). The binding also dampens the proliferation and activation of T cells and promotes the generation of tolerogenic DCs (10). In contrast, when PVR binds with CD226 on naive T cells, costimulatory signals are delivered (58, 59). In this sense, PVR is a “bidirectional switch” like HVEM in the BTLA ICP. As for TIGIT on NK cells, its ligation with the PVR on B cells leads to a reduction of cytokine secretion by the NK cells. Also, the knockout of TIGIT from NK cells leads to a reduction in the IFN-γ secretion from the cells from 500 to 300 pg/ml in cell culture medium (60).
For the TIM-3 ICP, its receptor, TIM-3, is expressed on T cells, DCs, NK cells, and macrophages. There are at least four ligands identified for TIM-3: Galectin-9 (Gal-9), Phosphatidyl serine (61) high mobility group protein B1 (62) and Ceacam-1 (63). Among these ligands, Gal-9 is the best known and is ubiquitously expressed in the lymph nodes, spleen (18), and liver. Functional implications of the TIM-3 activation on Th1 cell, CD8 T cells, NK cells, macrophages, and DCs have been reported. The ligation of TIM-3 on Th1 cells by Gal-9 on hepatocytes decreases the secretion of cytokines such as IFN-γ and TNF-α by Th1 cells. The ligation also promotes apoptosis of Th1 cells (64), which is accompanied by an influx of calcium (65). The ligation of TIM-3 on CD8 T cells by Gal-9 leads to an inhibition of TCR signaling through the co-localization of TIM-3 and receptor phosphatases (66). For NK cells, it was proven that the blockade of TIM-3 on these cells boost the IFN-γ production by the cells (67). As for macrophages, the ligation of TIM-3 on these cells by Gal-9 on hepatocytes inhibits the production of IL-2 and IFN-γ by the macrophages (68). Last, the enhancement of the TIM-3 ICP on DCs by agonist antibody inhibits the activation and maturation of the DCs (12).
As for the LAG-3 ICP, its receptor, LAG-3, is expressed on activated T cells. It is also expressed on B cells, DCs, and NK cells. The ligand of LAG-3 is MHC class II on APCs. It was found that the ligation between LAG-3 and MHC II suppressed the proliferation of Th cells (12). In addition, Tregs are able to inhibit the activation and maturation of DCs through the LAG-3 ICP (69). And, the knockout of LAG-3 reduces the likelihood for CD4 T cells to differentiate into Tregs (70). As for NK cells, the LAG-3 knockout compromises natural killer activity in a mouse model (71). Lastly and interestingly, the PD-1 and LAG-3 ICPs show synergy with each other. Mice with the single knockout of PD-1 only show minimal autoimmune sequelae, such as a lupus-like condition. Mice with the single knockout of LAG-3 do not develop any autoimmune disorders within the first year after birth (72). In contrast, mice with the double knockouts of LAG-3 and PD-1 have lethal autoimmune disorders, including myocarditis and pancreatitis (73). In addition, two studies showed that cancer patients who were resistant to a single PD-1 ICP blockade therapy responded to the dual blockade therapy involving LAG-3 and PD-1 ICPs (74, 75).
CD200R1 is an ICP receptor that is expressed on resting macrophages, DCs, plasma cells, memory B cells, and T cells. T helper cells express a greater level of CD200R1 than cytotoxic T lymphocytes and naive T cells (19). The ligand of the ICP, CD200, is expressed on epithelial cells, endothelial cells, lymphoid cells, myeloid cells, neurons, and mesenchymal stem cells (19, 20). For macrophages and lymphocytes, the activation of the CD200 ICP boosts their production of TGF-β and IL-10, while decreasing their production of TNF-α, IL-1, IL-6, and INF-γ (20, 76–78). In addition, the activation of this ICP inhibits the activation of lymphocytes and macrophages, while promoting CD4 T cells to differentiate into Tregs (76–78).
VISTA is the newest ICP among the ones we discuss and was identified in 2011 (26). Its receptor, VISTA, is constitutively expressed on most immune cells except for B cells (13). VISTA is also known as a PD-1 homolog. The knockout of VISTA lowers the fraction of Tregs in the lung and spleen from 20% to approximately 10% (79). Since Tregs are critical to immune tolerance (80, 81), the reduction of Tregs implicates the importance of the VISTA ICP in maintaining immune tolerance. The VISTA ICP suppresses the proliferation and differentiation of T cells, as well as their production of cytokines such as IL-17 (82). The activation of the VISTA ICP results in the inhibition of the production of inflammatory cytokines and chemokines by DCs and macrophages, such as IP-10 and MCP-1 (83). To date, two ligands were reported for VISTA, V-set and immunoglobulin domain containing 3 (VSIG-3) and P-selectin glycoprotein ligand-1 (PSGL-1). VSIG-3 is expressed restrictively on neurons and glial cells in the brain and on Sertoli cells in the testis (21). Activation of the VISTA ICP by VSIG-3 retards the proliferation and cytokine secretion of T cells, such as the secretion of IFN-γ, IL-2, and IL-17 (84). PSGL-1 is expressed on many types of immune cells including T cells, B cells, DCs, and macrophages. It is also expressed on platelets and endothelial cells. The biological implications of the ligation PSGL-1 to VISTA remain elusive to date.
ICP-Targeted Experimental Therapeutics for Autoimmune Diseases
The reported, ICP-targeted therapeutics may be divided into two classes based on their intended working mechanisms. The first class includes most of the reported therapeutics and shares a working mechanism—enhancing the ICP. This class takes the form of recombinant proteins, monoclonal antibodies, nucleic acids, and engineered cells (Figure 2). The second class embraces a more straightforward idea that ICP receptors or at least some ICP receptors on immune cells may be employed as biomarkers for pathogenic immune cells in autoimmune diseases. A corresponding working mechanism is the depletion of those ICP receptor positive cells. Currently, there is only one reported therapeutic in the second group. Below, we will recount these ICP-targeted therapeutics in the order of working mechanisms and forms. Table 3 summarized these therapeutics and the disease models in which they are tested. We emphasize “experimental” in our section title because there is only one ICP-targeted drug, Abatacept, approved for use in clinics, which indicates both the challenges and opportunities in the development these therapeutics.
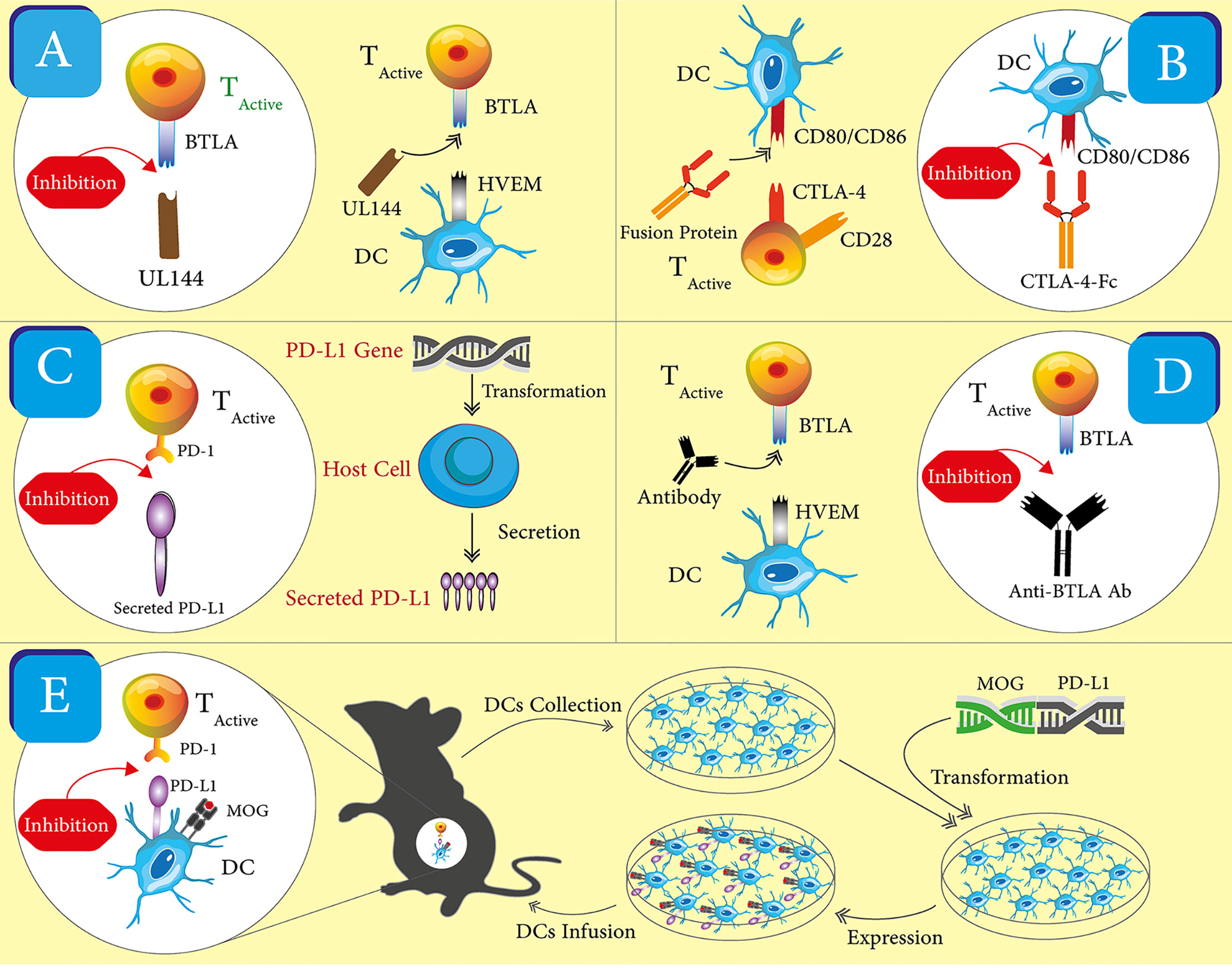
Figure 2 Schematics for the different forms of ICP-targeting therapeutics. One representative for each form of therapeutics was shown. (A) Viral proteins. UL144 is used to engage with BTLA and activate the corresponding ICP, which enhances immune inhibitory signals. (B) Soluble ligand and receptors. A fusion of CTLA-4 and Fc is used to engage with CD86/CD86 and activate the CTLA-4 ICP, which enhance immune inhibitory signals. (C) Nucleic acids. A coding gene of PD-L1 is used to increase the expression of PD-L1 in host cells. The increased expression strengthens the PD-1 ICP and immune inhibitory signals. (D) Antibodies. An anti-BTLA antibody is used as an agonist to enhance the BTLA ICP, which amplifies immune inhibitory signals. (E) Cells. DCs are collected and transfected with the coding genes of PD-L1 and MOG peptide. These engineered DCs have the enhanced expression of PD-L1 and MOG peptides. After these DCs are transferred back into mice, they promote immune inhibitory signals in vivo through the PD-1 ICP and the presentation of the MOG peptide to T cells.
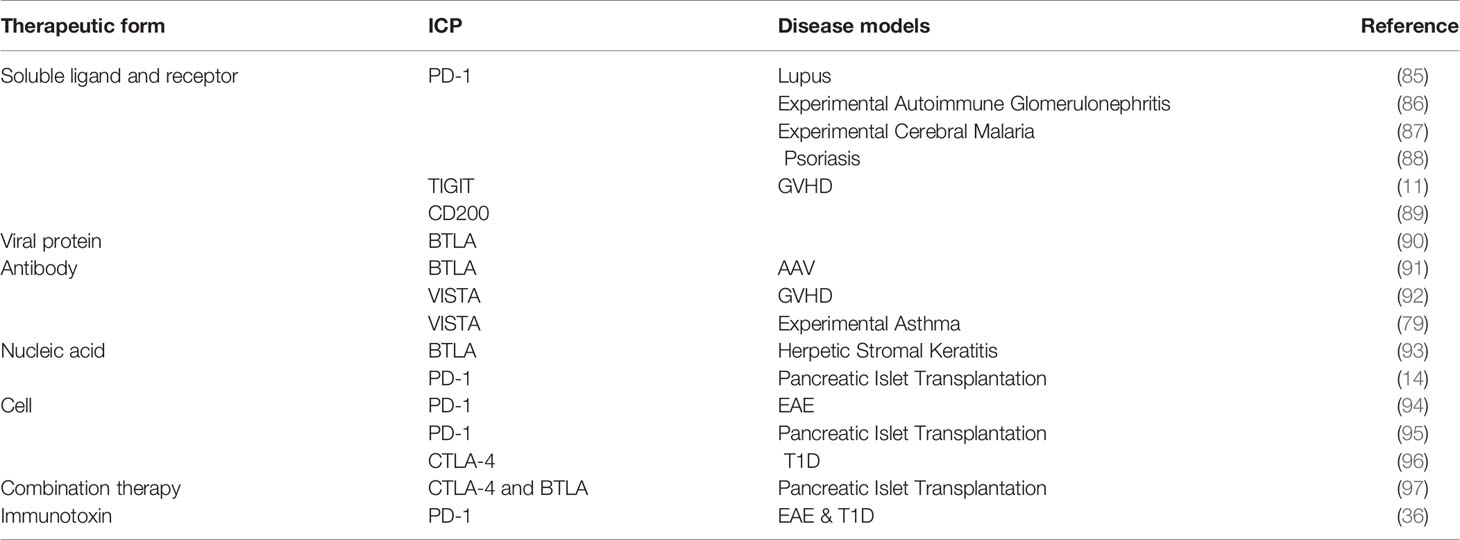
Table 3 Experimental therapeutics to enhance ICPs.
Enhancement of ICPs
Soluble ICP Ligands and Receptors
Soluble ICP ligands are used to enhance the activity of an ICP. For example, soluble PD-L1 was used to engage with PD-1 and flick on the PD-1 ICP. On the other hand, soluble ICP receptors were used in a more fascinating manner. Taking the TIGIT ICP as an example, TIGIT, as a receptor, engages with its cognate receptor, CD155, on DCs (10). The engagement dampens the activation signaling mediated by CD226 because the activation signaling depends on the interaction between CD155 and CD266 while TIGIT’s “occupation” of CD155 prevents the interaction (58, 59). Soluble TIGIT also has the ability to prevent the interaction between CD155 and CD266.
The developers of these soluble ICP ligands and receptors have put them on a proven protein “Noah’s Ark”, the fragment crystallizable region of an antibody (Fc). When fused with Fc, proteins usually assume longer in vivo half-lives. Fc also helps to immobilize Fc fusion proteins on the cell surface of APCs through binding with Fc receptors on these cells (98). These fusion proteins are often produced as secreted proteins using a mammalian cell expression system. One example of such production procedure is that Wang and coworkers generated an expression plasmid carrying the coding genes for the mouse PD-L1 extracellular domain and the mouse lgG1 Fc (87). Then, they transfected FreeStyle 293 cells with the plasmid. Lastly, they purified fusion proteins from the supernatant of the cell culture.
The most successful soluble ICP receptor-based therapeutic is Abatacept. Abatacept is a fusion protein that consists of the extracellular domain of human CTLA-4 and the Fc of human IgG1 (99). Compared to CD28, Abatacept has a greater affinity to CD80 and CD86. Abatacept is able to inhibit the CD28-mediated stimulation signaling through competition for CD80 and CD86. The therapeutic has a plasma half-life of 14.7 days in patients in the dosing range of 0.5 to 50 mg/kg (99). Abatacept was approved to treat RA. Arthritis patients were evaluated based on a 50% improvement in the number of tender and swollen joints (American College of Rheumatology, ACR50) after treatments. Among Abatacept-treated patients, 21.8% of reached ACR50 after 6 months; in contrast, only 3.8% of placebo-treated patients reached ACR50 (100). The same study also proved that this medicine is safe: adverse events happened to 79.5% of the Abatacept-treated patients and 71.4% of the placebo-treated patients; but there is no increase in serious infections among Abatacept-treated patients when the infections of Abatacept-treated and placebo-treated patients were compared (100). In 2011, a derivative of Abatacept, Belatacept, was also approved to be used as an immunosuppressive agent for kidney transplantation.
Besides Abatacept, other PD-L1-Fc fusions have also been reported (86–88, 101). The efficacy of these fusions was demonstrated in various autoimmune disease models, including experimental autoimmune glomerulonephritis and lupus. Zhou et al. found that PD-L1-Fc significantly reduced the auto-antibody production and tubular proteinosis in murine lupus models according to histological assessments. The treatment of PD-L1-Fc extended the survival of treated mice from an average of 45 to 60 weeks. When PD-L1-Fc was used in a prophylactical manner, it completely prevented proteinuria (101). Reynolds and coworkers showed the treatment of PD-L1-Fc reduced the glomerular infiltration of T cells by half in a mouse experimental autoimmune glomerulonephritis model (86). It is noteworthy that PD-L1-Fc fusions have also demonstrated their ability to weaken immune responses in inflammation and infection models. A study reported by Wang et al. showed that PD-L1-Fc reduced the number of CTLs by 75% and alleviated the disruption of the blood–brain barrier that was caused by over-reactive CD8 T cells in the mouse experimental cerebral malaria model (87). The treatment also increased the survival rate of the mice from 10 to 60% by 10 days after infection. Kim and coworkers demonstrated that the treatment of PD-L1-Fc reduced the production of IL-17A by T cells by 75% and alleviated psoriasis inflammation in imiquimod-treated mice (88). Together, these data suggest that PD-L1-Fc is able to suppress immune responses in both autoimmune disease and inflammation models.
The TIGIT-Fc fusion is an example of soluble receptors that are used to compete for its cognate receptors. The fusion consists of the extracellular domain of mouse TIGIT and human lgG3 Fc domain. The TIGIT-Fc was found to inhibit the activation of macrophages and increase their secretion of IL-10, an anti-inflammatory cytokine, by three times (102). Further, in the mouse acute GVHD model, the treatment of TIGIT-Fc increases the survival rate from ~5 to ~30%. Among the mice that have already shown the GVHD symptoms, TIGIT-Fc prolongs their survival time from shorter than 30 days to longer than 40 days (11). Together, these data validate the idea that TIGIT-Fc can be used to suppress immune responses.
There was also an exploration of adding a cytokine into the fusion proteins that consist of ICP ligands and the Fc (89). Gorczynski and coworkers generated an immunosuppressive protein that includes CD200, Fc, and TGF-β. There is a Glycine (n = 6) linker between the Fc and TGF-β (93). The fusion protein, CD200FcGly6TGF-β; was able to bind with TGF-β receptors on T cells and CD200R1 on APCs. The immunosuppression effect of the protein was observed both in vitro and in vivo. Specifically, the treatment of CD200FcGly6TGF-β boosted the percentage of Tregs in the spleen from ~3% to more than 10%.
Viral Proteins
Some viral proteins mimic the biological functions of ICP receptors or ligands and are hence explored as an “Off” switch for undesirable immune responses (90, 103). UL144, a cytomegalovirus protein, is an ortholog of HVEM, a ligand of the BTLA ICP (104). However, in contrast to HVEM that interacts with not only BTLA but also CD160, TNFSF14, lymphotoxin-α, and HSV-1 glycoprotein D (50), UL144 only binds with BTLA. Thus, while HVEM is a bidirectional switch depending on its binding partners, UL144 is dedicated to activate the BTLA ICP (105). In addition, UL144 is approximately three times more potent than HVEM in suppressing the proliferation and activation of T cells, despite the fact that UL144 binds five times weaker to BTLA than HVEM (90). Further, the binding selectivity of UL144 was attributed to its N-terminal cysteine-rich domain 1 (CRD1) and the CRD2 loop (106). Sedy and coworkers leveraged the binding selectivity insight of UL144 and engineered a tetra-mutant of HVEM that carried four site mutations, S58R, G68T, L70W, and L90A. The tetra-mutant of HVEM not only assumed the binding selectivity just like UL144 but also acquired a 10-fold stronger affinity to BTLA compared with the wild type (90). The tetra-mutant exhibited a significant inhibitory effect to the SHP-1-sensitive, type I interferon signaling pathway of B cells and NK cells. It also effectively inhibited TCR-mediated antigen signaling.
Monoclonal Antibodies
Monoclonal antibodies are one of the most common forms of therapeutics in general. They have specific binding target and activity, and possess desired safety and pharmacokinetics properties. Their popularity is also backed by robust antibody engineering and production technology, which ensure reproducible and economically sensible products. Thanks to these advantages, monoclonal antibodies are also a popular form of therapeutics that enhance ICPs even though they were not examined exclusively in autoimmune disease models. Monoclonal antibodies are developed as agonists of ICP receptors to enhance immune regulation. It is noteworthy that along with the development of ICP-enhancing antibodies, there are successful developments of monoclonal antibodies as ICP blockers. These blockers are developed to facilitate immune stimulation and are now exclusively used in boosting anti-cancer immune responses. Seven of such ICP blockers have been approved by the FDA for cancer therapy. This development outcome, on the one hand, highlights the feasibility to develop antibody-based therapeutics to target ICPs, but on the other hand, alerts us to distinguish ICP-blockade and -enhancement antibodies.
An anti-BTLA antibody was found to have the ICP agonistic effect in the antineutrophil cytoplasmic antibody-associated vasculitis (AAV) model (91). This antibody suppressed T-cell proliferation by 32% and IL-17A secretion by 30% in the AAV model. It is worth mentioning that IL-17A is a key driver in pathogenesis of AAV and that the knockout of IL-17A protects mice from developing AAV (107, 108). IMP761, a humanized anti-LAG-3 antibody, showed immunosuppressive effect in a delayed-type hypersensitivity model of cynomolgus macaque that was induced by tuberculin. IMP761 decreased the infiltration of inflammatory T cells into the DTH site to one third of the control level. In vitro, IMP761 reduced the proliferation and activation of self-antigen-induced T cells with a mean inhibition rate of 50% (109).
The immunosuppressive effect of ICP agonist antibody was also demonstrated in the graft-versus-host disease (GVHD) and asthma models. Dallas and coworkers showed the treatment of an anti-VISTA antibody extended the survival of mice of the GVHD model. Treated mice were able to live up to 18 months with no sign of GVHD, infection, or cancer (92). Meanwhile, the treatment reduced the accumulation of T cells in the spleen and liver by eight and five times, respectively. When an anti-VISTA antibody was used to treat OVA-induced asthma in mice, the accumulation of eosinophils in bronchoalveolar lavage fluid of these mice was reduced to one third of the untreated, control level (79). According to the histological analysis, the mucus production also decreased significantly in the antibody-treated mice. Data from these aforementioned animal models reinforces the point that agonistic antibodies of ICPs could be a powerful tool to enhance ICPs and suppress autoimmunity and other undesired immune responses.
It is noteworthy that the sub-classification of agonist antibodies influences their efficacy (110). lgG is the most popular class of therapeutic antibodies due to its stability and long half-life. Among the subclasses of IgG, IgG2 appears to be the most effective subclass of agonists (95, 111–113). When three subclasses anti-CD200R antibodies, lgG1, lgG2, and lgG4, were compared, lgG2 showed 2-fold greater efficacy than both lgG1 and lgG4 (110). This result suggests the importance of considering the subclasses and the Fc structure of antibodies when designing antibody therapeutics targeting ICPs.
Nucleic Acids
Genes that encode ICP receptors and ligands are used as therapeutics to suppress immunity. These genes are able to increase the production of receptors and ligands inside the body. Plasmids and virus are used as vectors to deliver the genes and transfect cells in murine models. To date, this gene therapy approach has achieved an increased expression of receptors and ligands as well as immunosuppression in the animal models of inflammation and organ transplantation. It could be effective in autoimmune disease models.
A plasmid that carries a coding gene of BTLA was used to treat herpetic stromal keratitis (93). According to immunohistological analysis, this treatment decreased the infiltration of CD4 T cells into infected corneas by half. The treatment also reduced IFN-γ positive cells in murine corneas and draining lymph nodes by seven and two times, respectively. Further, splenocytes collected from the treated mice produced less IFN-γ (0.4 ng/ml versus 1.4 ng/ml of the control group). The result of IFN-γ is a sign of weakened Th1 responses. Ultimately, the treatment with the BTLA plasmid lowered the incidence rate and the severity of corneal lesions (the clinical score decreased from ~4 to less than 1, even 0).
An adenovirus was constructed to deliver the coding gene of PD-L1 by Li and coworkers (14). The adenovirus was preferred for gene delivery due to their high transfection efficiency and low toxicity. This PD-L1 adenovirus was tested in a pancreatic islet transplantation model using mice with diabetes. The treatment with the virus resulted in a high-level presence of soluble PD-L1 in mice for at least 28 days (21 mg/ml in caudal vein blood). The treatment also prolonged the survival time of islet grafts from an average of 8 to 28 days. In addition, the treatment lowered blood glucose levels (from >20.0 mmol/L to <11.1 mmol/L), another indicator that the treatment helped to protect islet grafts.
Cells
Since ligands of ICPs are intrinsically expressed in hematopoietic and non-hematopoietic cells to ignite ICPs, it is a reasonable strategy to supplement cells that express the ligands to the body in order to strengthen ICPs. DCs express multiple types of ICP ligands (12, 14, 16, 46, 84). and hence were exploited to carry out the task. Hirata and coworkers used this approach to prevent EAE (94). They engineered DCs that co-expressed both PD-L1 and a myelin oligodendrocyte glycoprotein-derived antigenic peptide (MOG, p35-55). The mice that received a transfer of these dual expression DCs experienced the significantly reduced severity of EAE as compared to untreated mice or mice that were treated with DCs expressing the MOG peptide only. The mean clinical score reduced from 3.0 to less than 1.0. Treatment with the PD-L1/MOG peptide dual expression DCs also abolished T cell infiltration into spinal cords. As a prophylactic measure, the dual expression DCs prevented the development of EAE and maintained a mean clinical score under 1.0. It is worth mentioning that PD-L1 and the MOG peptide are preferred to be expressed by the same DCs according to data. When PD-L1 and the MOG peptide are expressed in two separate DC populations, and when the combination of the two populations was used to treat mice with EAE, the combination therapy did not deliver the same level of efficacy as the dual expression DCs. The result underscores the necessity of co-expression of PD-L1 and the MOG peptide in the same DCs for this therapeutic strategy.
In addition to DCs, other types of cells were also engineered and utilized to ameliorate autoimmune diseases. Pancreatic β cells were engineered to express the scFv of anti-CTLA-4 antibody (96). In a coculture experiment containing T cells and the engineered β-cells, the β-cells inhibited the level of T cell proliferation by half. The same research group also generated transgenic NOD mice that specifically express the scFv of anti-CTLA-4 antibody in β cells. The expression was induced by insulin. It was found that, at 21-weeks old, the diabetes incidence rate for the wile type NOD mice was 59%, while the rate for the CTLA-4 expression NOD mice was only 7.4%.
The cell engineering strategy was also employed to protect transplanted organ allografts. Wen et al. generated pancreatic β-cells that overexpress PD-L1 (95). They treated streptozotocin-induced diabetic mice by intraperitoneal injection of allogenic PD-L1-overexpressing β-cells. The engineered β-cell allograft had much longer survival than the wildtype β-cell allograft, more than 60 days versus fewer than 20 days. Their analysis further showed that the engineered β-cells lowered lymphocyte proliferation by half, increased the apoptosis rate of lymphocytes from ~40 to ~60%, and reduced the secretion of IFN-γ by lymphocytes from ~25 to ~10 pg/ml.
Combination of Therapeutics
Since some of the aforementioned therapeutic strategies have complementary working mechanisms, it is reasonable to explore combination therapies that consist of multiple of strategies. One reported effort is the combination of soluble ICP molecules and antibodies (97). Truong et al. designed a combination that included an anti-BTLA antibody and a CTLA-4-Fc fusion and used the combination to foster immune tolerance toward islet allografts in mice (97). This combinational treatment led to indefinite allograft survival (>100 days) through attenuating CD4 and CD8 T-cell mediated immune rejections. In contrast, the monotherapy of the anti-BTLA antibody or the CTLA-4-Fc fusion only delivered moderate survival benefits; the islet allografts survived for 20–30 days.
Depletion of ICP Receptor Positive Immune Cells
Existing data on the PD-1 ICP point to the importance of PD-1 positive cells in the pathogenesis of multiple autoimmune diseases including T1D, MS, and SLE (29, 114–122). Further, a correlation was reported between the pathogenic potential and the PD-1 expression of autoreactive lymphocytes (123). Thus, it is plausible to use PD-1 as a biomarker of autoreactive immune cells in autoimmune diseases. We recently created an immunotoxin that is able to specifically ablate PD-1 positive cells (36). The depletion of PD-1 positive cells effectively ameliorated autoimmune attacks. Specifically, the depletion treatment delayed the onset age of the spontaneous T1D in NOD mice from 19 to 29 weeks. The depletion also enabled mice to recover from the advanced stage of EAE. What makes this depletion treatment even more desirable is that the treated mice maintained healthy adaptive immunity, evidenced by their full-strength humoral and cellular immune responses to vaccinations. The healthy adaptive immunity was protected after the depletion of PD-1 positive cell because naive lymphocytes, which are PD-1-negative (PD-1−), are spared by the depletion (15, 124–126). These naive lymphocytes can be activated, mount normal immune responses, and maintain healthy adaptive immunity after the depletion.
Safety of ICP Related Autoimmune Therapies
Therapeutics for autoimmune diseases, in principle, should exert an immunosuppressive effect. Often, such suppression happens to not only autoimmunity but also healthy immunity, leading to immune deficiency and undesirable side effects. Patients who take these therapeutics long term are prone to opportunistic infections and endure a greater risk of malignancy. Thus, it is critical to evaluate whether autoimmune disease therapeutics cause unacceptable immune deficiency during the therapeutic development. Such a requirement should apply to ICP-based therapeutics as well.
According to reported data of ICP-based therapeutics, these therapeutics appear devoid of serious side effects. None of the cited studies in this review revealed serious side effects. Given that the efficacy evaluation of these therapeutics normally requires long-term studies, it is reasonable to assume these therapeutics do not cause severe side effects within a reasonably long period of time. In our recent study, we continuously applied the treatment of PD-1 positive cell depletion to NOD mice and observed these mice for 17 weeks. We did not observe any serious side effects on the mice during the entire study except for hyperglycemia at the end phase of the study (36). In Zhou’s study of PD-L1 fusion proteins, treated mice were followed for more than 60 weeks after treatment. No severe side effect was detected (101). In addition, Abatacept was proven safe for clinical use through clinical trials (127). These results may serve as an indication that these ICP therapeutics are devoid of severe side effects when treating autoimmune diseases.
In addition to the aforementioned passive observations of side effects, some studies of ICP-based therapeutics included an active assessment of protective immunity after treatments. In the above-mentioned PD-1 positive cell depletion study, we evaluated blood cell counts and humoral and cellular immunity two days after the depletion. Our data showed that treated mice have a normal level of B cell, CD4 T cell, and CD8 T cell counts in blood and spleens (36). These treated mice developed normal antibody and cytotoxic T lymphocyte (CTL) responses toward vaccinations. In a study of PD-L1-Fc proteins, the administration of PD-L1-Fc did not alter the percentages of peripheral CD4 and CD8 T cells, nor the population of FoxP3 positive cells (101). However, the treatment of PD-L1-Fc reduced the fractions of antibody-secreting cells among peripheral mononuclear cells, splenic cells, and bone marrow cells to one third of the normal levels. Together, these results of active assessments suggest that the impact of ICP-based therapeutics on protective immunity may be dependent on the mode of action of these therapeutics. Meanwhile, while the side effects of ICP therapeutics are generally mild, it is difficult to draw general conclusions about their safety because of their distinct working mechanisms. Meanwhile, these results are promising because they suggest the possibility to mitigate side effects of ICP therapeutics by employing therapeutics with different mode of actions.
The Prospect of ICPs as a Class of Therapeutic Target
ICPs have promising prospects as targets for autoimmune disease therapeutics. Such a point of view is backed by various facts and reasonings that we will state below. These facts and reasonings may be categorized into four aspects, clinical successes, mechanistical support, preclinical successes, and safety assurance of ICP-based therapeutics.
First and foremost, there is already an FDA-approved drug targeting an ICP. Abatacept targets the CTLA-4 ICP and was approved as a treatment of a type of RA that does not respond to disease-modifying anti-rheumatic drugs and anti-TNF-α. Besides Abatacept, an anti-PD-1 agonist antibody (Celgene: CC-90006) has been tested clinically in psoriasis patients since 2016 (NCT03337022). In all, although there is only one FDA-approved ICP drug thus far, the clinical progress still underscores the feasibility of developing autoimmune disease therapeutics targeting ICPs.
Second, there exists a wide range of data supporting ICPs as the targets for autoimmune disease therapeutics from a mechanistic aspect. Data from the murine models demonstrated the critical relationship between the dysfunction of ICPs and the pathogenesis of autoimmune diseases (23, 29–36, 128). Further, clinical data revealed the correlation between autoimmune diseases and the deficiency of ICP signaling in humans (129–133). These data, together, show the important role of ICPs in the maintenance of immune tolerance and support the notion that molecules that restore or enhance ICP signaling could be effective therapeutics to prevent, alleviate, or cure autoimmune diseases.
Third, the experimental therapeutics we recounted in Section 3 showed solid efficacy in various animal models of autoimmune disease and inflammation (Table 3). These therapeutics are able to suppress autoimmunity and inflammation. These preclinical results validate ICPs as an appealing therapeutic target for the treatment of autoimmune diseases. In addition, these results confirmed that fairly diversified methods and agents can be used to leverage on ICPs to dampen autoimmunity.
Last, experimental therapeutics that target ICPs showed relatively mild and potentially acceptable side effects. As summarized in Section 4, no severe side effect was reported for the wide range of therapeutics we reviewed. Although one therapeutic of one ICP was shown to impair humoral responses, another therapeutic targeting the same ICP but having a different mode of action did not compromise humoral responses. While more systemic studies are required to draw a general conclusion on the safety of the therapeutics that target ICPs, the safety data to date point to a positive prospective for these therapeutics.
Overall, the development of ICP-targeted, autoimmune disease therapeutics is still in its infancy and trails behind the development of ICP-targeted cancer therapeutics: fewer drugs were approved for clinical use; fewer publications are present. The underdevelopment of autoimmune disease therapeutics could be attributed to multifaceted factors: greater research efforts and investment in cancer therapy development, the urgency to develop cancer therapeutics, the higher safety requirements for autoimmune disease therapeutics, and so on. The underdevelopment reality, however, also provides a broader horizon to explore ICPs as therapeutic targets and utilize the targets. There is a great development space to explore. Given the aforementioned prospects of this class of therapeutics, it is reasonable to believe that ICPs as therapeutic targets will yield more successes in the future.
Conclusion
ICPs are an important mechanism to maintain immunotolerance and suppress autoimmunity. Evidences from preclinical studies and clinical trials have shown that an abolishment or a blockade of ICPs leads to the loss of immune tolerance and autoimmune disorders. To date, there are eight ICPs that have been mostly studied and reported. Distinct to other reviews of ICPs that focus on their utilizations in cancer immunotherapy (85, 134, 135), this review focuses on autoimmune disease therapeutics targeting ICPs. These therapeutics may be categorized into two groups according to their working mechanisms: the first group shared a working mechanism of enhancing ICPs; the second group utilized ICP receptors as biomarkers for pathogenic immune cells of autoimmune diseases and treated the diseases by depleting ICP-receptor positive immune cells. The ICP-based autoimmune disease therapeutics, in general, have acceptable safety profiles although more clinical and long-term evaluations are warranted. Given the impact of the ICPs on immune tolerance and the current development status of ICP-based therapeutics, it is deemed that there is a great space to develop this line of therapeutics, and this type of therapeutics holds promise to transform autoimmune disease therapy.
Author Contributions
YZ and MC contributed the idea of this manuscript. All authors contributed to the article and approved the submitted version.
Funding
MC is supported by the funding from the NIAID and NMSS. This work was supported by the NIH, 1R01AI139535 and National Multiple Sclerosis Society, GR-1807-31630.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Murphy K. Autoimmunity and transplantation. In: Janeway’s Immunobiology, 8th Edition. New York City, USA: Garland Science (2012). p. 201.
2. Lerner A, Jeremias P, Matthias T. The World Incidence and Prevalence of Autoimmune Diseases is Increasing. Int J Celiac Dis (2015) 3(4):151–5. doi: 10.12691/ijcd-3-4-8
3. Bour-Jordan H, Esensten JH, Martinez-Llordella M, Penaranda C, Stumpf M, Bluestone JA. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/ B7 family. Immunol Rev (2011) 241(1):180–205. doi: 10.1111/j.1600-065X.2011.01011.x
4. De Sousa Linhares A, Leitner J, Grabmeier-Pfistershammer K, Steinberger P. Not All Immune Checkpoints Are Created Equal. Front Immunol (2018) 9:1909. doi: 10.3389/fimmu.2018.01909
5. Wilde B, Hua F, Dolff S, Jun C, Cai X, Specker C, et al. Aberrant expression of the negative costimulator PD-1 on T cells in granulomatosis with polyangiitis. Rheumatol (Oxford England) (2012) 51(7):1188–97. doi: 10.1093/rheumatology/kes034
6. Steiner K, Moosig F, Csernok E, Selleng K, Gross WL, Fleischer B, et al. Increased expression of CTLA-4 (CD152) by T and B lymphocytes in Wegener’s granulomatosis. Clin Exp Immunol (2001) 126(1):143–50. doi: 10.1046/j.1365-2249.2001.01575.x
7. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity (1994) 1(9):793–801. doi: 10.1016/S1074-7613(94)80021-9
8. Chambers CA, Kuhns MS, Allison JP. Cytotoxic T lymphocyte antigen-4 (CTLA-4) regulates primary and secondary peptide-specific CD4(+) T cell responses. Proc Natl Acad Sci USA (1999) 96(15):8603–8. doi: 10.1073/pnas.96.15.8603
9. Zeng J, Bao J, Wang H, Zhang H, Zhang J, Wang W, et al. [Construction and identification of mouse BTLA lentiviral expression vector]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi Chin J Cell Mol Immunol (2013) 29(3):261–4.
10. Gao J, Zheng Q, Xin N, Wang W, Zhao C. CD155, an onco-immunologic molecule in human tumors. Cancer Sci (2017) 108(10):1934–8. doi: 10.1111/cas.13324
11. Zhang D, Hu W, Xie J, Zhang Y, Zhou B, Liu X, et al. TIGIT-Fc alleviates acute graft-versus-host disease by suppressing CTL activation via promoting the generation of immunoregulatory dendritic cells. Biochim Biophys Acta Mol Basis Dis (2018) 1864(9 Pt B):3085–98. doi: 10.1016/j.bbadis.2018.06.022
12. Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol (Baltimore Md 1950) (2013) 190(9):4899–909. doi: 10.4049/jimmunol.1300271
13. Ohno T, Zhang C, Kondo Y, Kang S, Furusawa E, Tsuchiya K, et al. The immune checkpoint molecule VISTA regulates allergen-specific Th2-mediated immune responses. Int Immunol (2018) 30(1):3–11. doi: 10.1093/intimm/dxx070
14. Li T, Ma R, Zhu JY, Wang FS, Huang L, Leng XS. PD-1/PD-L1 costimulatory pathway-induced mouse islet transplantation immune tolerance. Transplant Proc (2015) 47(1):165–70. doi: 10.1016/j.transproceed.2014.10.043
15. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
16. Momtaz P, Postow MA. Immunologic checkpoints in cancer therapy: focus on the programmed death-1 (PD-1) receptor pathway. Pharmacogenom Personalized Med (2014) 7:357–65. doi: 10.2147/PGPM.S53163
17. Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell (1996) 87(3):427–36. doi: 10.1016/S0092-8674(00)81363-X
18. Wada J, Kanwar YS. Identification and characterization of galectin-9, a novel beta-galactoside-binding mammalian lectin. J Biol Chem (1997) 272(9):6078–86. doi: 10.1074/jbc.272.9.6078
19. Caserta S, Nausch N, Sawtell A, Drummond R, Barr T, Macdonald AS, et al. Chronic infection drives expression of the inhibitory receptor CD200R, and its ligand CD200, by mouse and human CD4 T cells. PloS One (2012) 7(4):e35466. doi: 10.1371/journal.pone.0035466
20. Li Y, Zhang D, Xu L, Dong L, Zheng J, Lin Y, et al. Cell-cell contact with proinflammatory macrophages enhances the immunotherapeutic effect of mesenchymal stem cells in two abortion models. Cell Mol Immunol (2019) 16(12):908–20. doi: 10.1038/s41423-019-0204-6
21. Harada H, Suzu S, Hayashi Y, Okada S. BT-IgSF, a novel immunoglobulin superfamily protein, functions as a cell adhesion molecule. J Cell Physiol (2005) 204(3):919–26. doi: 10.1002/jcp.20361
22. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11(11):3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
23. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol (2003) 4(7):670–9. doi: 10.1038/ni944
24. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415(6871):536–41. doi: 10.1038/415536a
25. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
26. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med (2011) 208(3):577–92. doi: 10.1084/jem.20100619
27. Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med (1992) 176(2):327–37. doi: 10.1084/jem.176.2.327
28. Wright GJ, Puklavec MJ, Willis AC, Hoek RM, Sedgwick JD, Brown MH, et al. Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity (2000) 13(2):233–42. doi: 10.1016/S1074-7613(00)00023-6
29. Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci USA (2005) 102(33):11823–8. doi: 10.1073/pnas.0505497102
30. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11(2):141–51. doi: 10.1016/S1074-7613(00)80089-8
31. Wang J, Okazaki IM, Yoshida T, Chikuma S, Kato Y, Nakaki F, et al. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int Immunol (2010) 22(6):443–52. doi: 10.1093/intimm/dxq026
32. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol (Baltimore Md 1950) (2011) 186(3):1338–42. doi: 10.4049/jimmunol.1003081
33. Bettini M, Szymczak-Workman AL, Forbes K, Castellaw AH, Selby M, et al. Cutting edge: accelerated autoimmune diabetes in the absence of LAG-3. J Immunol (Baltimore Md 1950) (2011) 187(7):3493–8. doi: 10.4049/jimmunol.1100714
34. Jha V, Workman CJ, McGaha TL, Li L, Vas J, Vignali DA, et al. Lymphocyte Activation Gene-3 (LAG-3) negatively regulates environmentally-induced autoimmunity. PloS One (2014) 9(8):e104484. doi: 10.1371/journal.pone.0104484
35. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3(5):541–7. doi: 10.1016/1074-7613(95)90125-6
36. Zhao P, Wang P, Dong S, Zhou Z, Cao Y, Yagita H, et al. Depletion of PD-1-positive cells ameliorates autoimmune disease. Nat Biomed Engineer (2019) 3(4):292–305. doi: 10.1038/s41551-019-0360-0
37. Tocut M, Brenner R, Zandman-Goddard G. Autoimmune phenomena and disease in cancer patients treated with immune checkpoint inhibitors. Autoimmun Rev (2018) 17(6):610–6. doi: 10.1016/j.autrev.2018.01.010
38. Williams TJ, Benavides DR, Patrice K-A, Dalmau JO, de Ávila AL, Le DT, et al. Association of Autoimmune Encephalitis With Combined Immune Checkpoint Inhibitor Treatment for Metastatic Cancer. JAMA Neurol (2016) 73(8):928–33. doi: 10.1001/jamaneurol.2016.1399
39. De Velasco G, Je Y, Bossé D, Awad MM, Ott PA, Moreira RB, et al. Comprehensive Meta-analysis of Key Immune-Related Adverse Events from CTLA-4 and PD-1/PD-L1 Inhibitors in Cancer Patients. Cancer Immunol Res (2017) 5(4):312–8. doi: 10.1158/2326-6066.CIR-16-0237
40. Cho HY, Choi EK, Lee SW, Jung KO, Seo SK, Choi IW, et al. Programmed death-1 receptor negatively regulates LPS-mediated IL-12 production and differentiation of murine macrophage RAW264.7 cells. Immunol Lett (2009) 127(1):39–47. doi: 10.1016/j.imlet.2009.08.011
41. Said EA, Dupuy FP, Trautmann L, Zhang Y, Shi Y, El-Far M, et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat Med (2010) 16(4):452–9. doi: 10.1038/nm.2106
42. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206(13):3015–29. doi: 10.1084/jem.20090847
43. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, et al. Reversal of the TCR stop signal by CTLA-4. Sci (N Y NY) (2006) 313(5795):1972–5. doi: 10.1126/science.1131078
44. Lingel H, Wissing J, Arra A, Schanze D, Lienenklaus S, Klawonn F, et al. CTLA-4-mediated posttranslational modifications direct cytotoxic T-lymphocyte differentiation. Cell Death Differ (2017) 24(10):1739–49. doi: 10.1038/cdd.2017.102
45. Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell (1992) 71(7):1065–8. doi: 10.1016/S0092-8674(05)80055-8
46. Alissafi T, Banos A, Boon L, Sparwasser T, Ghigo A, Wing K, et al. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J Clin Invest (2017) 127(7):2789–804. doi: 10.1172/JCI92079
47. Matheu MP, Othy S, Greenberg ML, Dong TX, Schuijs M, Deswarte K, et al. Imaging regulatory T cell dynamics and CTLA4-mediated suppression of T cell priming. Nat Commun (2015) 6:6219. doi: 10.1038/ncomms7219
48. Wang XB, Fan ZZ, Anton D, Vollenhoven AV, Ni ZH, Chen XF, et al. CTLA4 is expressed on mature dendritic cells derived from human monocytes and influences their maturation and antigen presentation. BMC Immunol (2011) 12:21. doi: 10.1186/1471-2172-12-21
49. Laurent S, Carrega P, Saverino D, Piccioli P, Camoriano M, Morabito A, et al. CTLA-4 is expressed by human monocyte-derived dendritic cells and regulates their functions. Hum Immunol (2010) 71(10):934–41. doi: 10.1016/j.humimm.2010.07.007
50. Ware CF, Sedý JR. TNF Superfamily Networks: bidirectional and interference pathways of the herpesvirus entry mediator (TNFSF14). Curr Opin Immunol (2011) 23(5):627–31. doi: 10.1016/j.coi.2011.08.008
51. Cai G, Freeman GJ. The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol Rev (2009) 229(1):244–58. doi: 10.1111/j.1600-065X.2009.00783.x
52. De Trez C, Schneider K, Potter K, Droin N, Fulton J, Norris PS, et al. The inhibitory HVEM-BTLA pathway counter regulates lymphotoxin receptor signaling to achieve homeostasis of dendritic cells. J Immunol (Baltimore Md 1950) (2008) 180(1):238–48. doi: 10.4049/jimmunol.180.1.238
53. Steinberg MW, Cheung TC, Ware CF. The signaling networks of the herpesvirus entry mediator (TNFRSF14) in immune regulation. Immunol Rev (2011) 244(1):169–87. doi: 10.1111/j.1600-065X.2011.01064.x
54. Oya Y, Watanabe N, Kobayashi Y, Owada T, Oki M, Ikeda K, et al. Lack of B and T lymphocyte attenuator exacerbates autoimmune disorders and induces Fas-independent liver injury in MRL-lpr/lpr mice. Int Immunol (2011) 23(5):335–44. doi: 10.1093/intimm/dxr017
55. Krieg C, Han P, Stone R, Goularte OD, Kaye J. Functional analysis of B and T lymphocyte attenuator engagement on CD4+ and CD8+ T cells. J Immunol (Baltimore Md 1950) (2005) 175(10):6420–7. doi: 10.4049/jimmunol.175.10.6420
56. Liu X, Alexiou M, Martin-Orozco N, Chung Y, Nurieva RI, Ma L, et al. Cutting edge: A critical role of B and T lymphocyte attenuator in peripheral T cell tolerance induction. J Immunol (Baltimore Md 1950) (2009) 182(8):4516–20. doi: 10.4049/jimmunol.0803161
57. Murphy TL, Murphy KM. Slow down and survive: Enigmatic immunoregulation by BTLA and HVEM. Annu Rev Immunol (2010) 28:389–411. doi: 10.1146/annurev-immunol-030409-101202
58. Xu Z, Jin B. A novel interface consisting of homologous immunoglobulin superfamily members with multiple functions. Cell Mol Immunol (2010) 7(1):11–9. doi: 10.1038/cmi.2009.108
59. Gao Y, Cui J, He W, Yue J, Yu D, Cai L, et al. Generation and characterization of polyclonal antibodies against mouse T-cell immunoglobulin and immunoreceptor tyrosine-based inhibitory domain by DNA-based immunization. Transplant Proc (2014) 46(1):260–5. doi: 10.1016/j.transproceed.2013.10.039
60. Li M, Xia P, Du Y, Liu S, Huang G, Chen J, et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-γ production of natural killer cells via β-arrestin 2-mediated negative signaling. J Biol Chem (2014) 289(25):17647–57. doi: 10.1074/jbc.M114.572420
61. Santiago C, Ballesteros A, Martínez-Muñoz L, Mellado M, Kaplan GG, Freeman GJ, et al. Structures of T cell immunoglobulin mucin protein 4 show a metal-Ion-dependent ligand binding site where phosphatidylserine binds. Immunity (2007) 27(6):941–51. doi: 10.1016/j.immuni.2007.11.008
62. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol (2012) 13(9):832–42. doi: 10.1038/ni.2376
63. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2015) 517(7534):386–90. doi: 10.1038/nature13848
64. Liu Y, Ji H, Zhang Y, Shen X, Gao F, He X, et al. Recipient T cell TIM-3 and hepatocyte galectin-9 signalling protects mouse liver transplants against ischemia-reperfusion injury. J Hepatol (2015) 62(3):563–72. doi: 10.1016/j.jhep.2014.10.034
65. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6(12):1245–52. doi: 10.1038/ni1271
66. Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat Med (2012) 18(9):1394–400. doi: 10.1038/nm.2871
67. Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, et al. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol (2015) 29(2):635–41. doi: 10.1016/j.intimp.2015.09.017
68. Ma CJ, Li GY, Cheng YQ, Wang JM, Ying RS, Shi L, et al. Cis association of galectin-9 with Tim-3 differentially regulates IL-12/IL-23 expressions in monocytes via TLR signaling. PloS One (2013) 8(8):e72488. doi: 10.1371/journal.pone.0072488
69. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol (Baltimore Md 1950) (2008) 180(9):5916–26. doi: 10.4049/jimmunol.180.9.5916
70. Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol (Baltimore Md 1950) (2010) 184(11):6545–51. doi: 10.4049/jimmunol.0903879
71. Durham NM, Nirschl CJ, Jackson CM, Elias J, Kochel CM, Anders RA, et al. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PloS One (2014) 9(11):e109080. doi: 10.1371/journal.pone.0109080
72. Miyazaki T, Dierich A, Benoist C, Mathis D. Independent modes of natural killing distinguished in mice lacking Lag3. Sci (N Y NY) (1996) 272(5260):405–8. doi: 10.1126/science.272.5260.405
73. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2012) 72(4):917–27. doi: 10.1158/0008-5472.CAN-11-1620
74. Ascierto PA, Melero I, Bhatia S, Bono P, Sanborn RE, Lipson EJ, et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/ PD-L1 therapy. ASCO Annual Meeting . J Clin Onco (2017) 35. doi: 10.1200/JCO.2017.35.15_suppl.9520
75. Ascierto PA, Bono P, Bhatia S, Melero I, Nyakas MS, Svane IM, et al. Efficacy of BMS-986016, a monoclonal antibody that targets lymphocyte activation gene-3 (LAG-3), in combination with nivolumab in pts with melanoma who progressed during prior anti–PD-1/PD-L1 therapy (mel prior IO) in all-comer and biomarker-enriched populations. Ann Oncol (2017) 28:v611–2. doi: 10.1093/annonc/mdx440.011
76. Fan H, Wang J, Zhou X, Liu Z, Zheng SG. Induction of antigen-specific immune tolerance by TGF-beta-induced CD4+Foxp3+ regulatory T cells. Int J Clin Exp Med (2009) 2(3):212–20.
77. Clark DA, Dmetrichuk JM, McCready E, Dhesy-Thind S, Arredondo JL. Changes in expression of the CD200 tolerance-signaling molecule and its receptor (CD200R) by villus trophoblasts during first trimester missed abortion and in chronic histiocytic intervillositis. Am J Reprod Immunol (N Y NY 1989) (2017) 78(1):e12665. doi: 10.1111/aji.12665
78. Jenmalm MC, Cherwinski H, Bowman EP, Phillips JH, Sedgwick JD. Regulation of myeloid cell function through the CD200 receptor. J Immunol (Baltimore Md 1950) (2006) 176(1):191–9. doi: 10.4049/jimmunol.176.1.191
79. Liu H, Li X, Hu L, Zhu M, He B, Luo L, et al. A crucial role of the PD-1H coinhibitory receptor in suppressing experimental asthma. Cell Mol Immunol (2018) 15(9):838–45. doi: 10.1038/cmi.2017.16
80. Duvernelle C, Freund V, Frossard N. Transforming growth factor-beta and its role in asthma. Pulmonary Pharmacol Ther (2003) 16(4):181–96. doi: 10.1016/S1094-5539(03)00051-8
81. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity (2008) 28(4):546–58. doi: 10.1016/j.immuni.2008.02.017
82. Oliveira P, Carvalho J, Rocha S, Azevedo M, Reis I, Camilo V, et al. Dies1/VISTA expression loss is a recurrent event in gastric cancer due to epigenetic regulation. Sci Rep (2016) 6:34860. doi: 10.1038/srep34860
83. Wang L, Le Mercier I, Putra J, et al. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc Natl Acad Sci USA (2014) 111(41):14846–51. doi: 10.1073/pnas.1407447111
84. Wang J, Wu G, Manick B, Hernandez V, Renelt M, Erickson C, et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology (2019) 156(1):74–85. doi: 10.1111/imm.13001
85. Zhang Q, Vignali DA. Co-stimulatory and Co-inhibitory Pathways in Autoimmunity. Immunity (2016) 44(5):1034–51. doi: 10.1016/j.immuni.2016.04.017
86. Reynolds J, Sando GS, Marsh OB, Salama AD, Evans DJ, Cook HT, et al. Stimulation of the PD-1/PDL-1 T-cell co-inhibitory pathway is effective in treatment of experimental autoimmune glomerulonephritis. Nephrol Dialysis Transplant Off Publ Eur Dialysis Transplant Assoc Eur Renal Assoc (2012) 27(4):1343–50. doi: 10.1093/ndt/gfr529
87. Wang J, Li Y, Shen Y, Liang J, Li Y, Huang Y, et al. PDL1 Fusion Protein Protects Against Experimental Cerebral Malaria via Repressing Over-Reactive CD8(+) T Cell Responses. Front Immunol (2018) 9:3157. doi: 10.3389/fimmu.2018.03157
88. Kim JH, Choi YJ, Lee BH, Song MY, Ban CY, Kim J, et al. Programmed cell death ligand 1 alleviates psoriatic inflammation by suppressing IL-17A production from programmed cell death 1-high T cells. J Allergy Clin Immunol (2016) 137(5):1466–1476.e1463. doi: 10.1016/j.jaci.2015.11.021
89. Gorczynski RM, Chen Z, Shivagnahnam S, Taseva A, Wong K, Yu K, et al. Potent immunosuppression by a bivalent molecule binding to CD200R and TGF-betaR. Transplantation (2010) 90(2):150–9. doi: 10.1097/TP.0b013e3181e2d6a1
90. Ware CF. Targeting lymphocyte activation through the lymphotoxin and LIGHT pathways. Immunol Rev (2008) 223:186–201. doi: 10.1111/j.1600-065X.2008.00629.x
91. Werner K, Dolff S, Dai Y, Ma X, Brinkhoff A, Korth J, et al. The Co-inhibitor BTLA Is Functional in ANCA-Associated Vasculitis and Suppresses Th17 Cells. Front Immunol (2019) 10:2843. doi: 10.3389/fimmu.2019.02843
92. Flies DB, Wang S, Xu H, Chen L. Cutting edge: A monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J Immunol (Baltimore Md 1950) (2011) 187(4):1537–41. doi: 10.4049/jimmunol.1100660
93. Xia L, Zhang S, Zhou J, Li Y. A crucial role for B and T lymphocyte attenuator in preventing the development of CD4+ T cell-mediated herpetic stromal keratitis. Mol Vision (2010) 16:2071–83.
94. Hirata S, Senju S, Matsuyoshi H, Fukuma D, Uemura Y, Nishimura Y. Prevention of experimental autoimmune encephalomyelitis by transfer of embryonic stem cell-derived dendritic cells expressing myelin oligodendrocyte glycoprotein peptide along with TRAIL or programmed death-1 ligand. J Immunol (Baltimore Md 1950) (2005) 174(4):1888–97. doi: 10.4049/jimmunol.174.4.1888
95. Wen X, Zhu H, Li L, Li Y, Wang M, Liu J, et al. Transplantation of NIT-1 cells expressing pD-L1 for treatment of streptozotocin-induced diabetes. Transplantation (2008) 86(11):1596–602. doi: 10.1097/TP.0b013e31818c6e64
96. Shieh SJ, Chou FC, Yu PN, Lin WC, Chang DM, Roffler SR, et al. Transgenic expression of single-chain anti-CTLA-4 Fv on beta cells protects nonobese diabetic mice from autoimmune diabetes. J Immunol (Baltimore Md 1950) (2009) 183(4):2277–85. doi: 10.4049/jimmunol.0900679
97. Truong W, Plester JC, Hancock WW, Merani S, Murphy TL, Murphy KM, et al. Combined coinhibitory and costimulatory modulation with anti-BTLA and CTLA4Ig facilitates tolerance in murine islet allografts. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surgeons (2007) 7(12):2663–74. doi: 10.1111/j.1600-6143.2007.01996.x
98. Paluch C, Santos AM, Anzilotti C, Cornall RJ, Davis SJ. Immune Checkpoints as Therapeutic Targets in Autoimmunity. Front Immunol (2018) 9:2306. doi: 10.3389/fimmu.2018.02306
99. Vital EM, Emery P. Abatacept. Drugs Today (Barcelona Spain 1998) (2006) 42(2):87–93. doi: 10.1358/dot.2006.42.2.957359
100. Genovese MC, Becker JC, Schiff M, Luggen M, Sherrer Y, Kremer J, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. New Engl J Med (2005) 353(11):1114–23. doi: 10.1056/NEJMoa050524
101. Zhou H, Xiong L, Wang Y, et al. Treatment of murine lupus with PD-LIg. Clin Immunol (Orlando Fla) (2016) 162:1–8. doi: 10.1016/j.clim.2015.10.006
102. Chen X, Lu PH, Liu L, Fang ZM, Duan W, Liu ZL, et al. TIGIT negatively regulates inflammation by altering macrophage phenotype. Immunobiology (2016) 221(1):48–55. doi: 10.1016/j.imbio.2015.08.003
103. Šedý JR, Balmert MO, Ware BC, Smith W, Nemčovičova I, Norris PS, et al. A herpesvirus entry mediator mutein with selective agonist action for the inhibitory receptor B and T lymphocyte attenuator. J Biol Chem (2017) 292(51):21060–70. doi: 10.1074/jbc.M117.813295
104. Cheung TC, Humphreys IR, Potter KG, Norris PS, Shumway HM, Tran BR, et al. Evolutionarily divergent herpesviruses modulate T cell activation by targeting the herpesvirus entry mediator cosignaling pathway. Proc Natl Acad Sci USA (2005) 102(37):13218–23. doi: 10.1073/pnas.0506172102
105. Šedý JR, Bjordahl RL, Bekiaris V, Macauley MG, Ware BC, Norris PS, et al. CD160 activation by herpesvirus entry mediator augments inflammatory cytokine production and cytolytic function by NK cells. J Immunol (Baltimore Md 1950) (2013) 191(2):828–36. doi: 10.4049/jimmunol.1300894
106. Bitra A, Nemčovičová I, Picarda G, Doukov T, Wang J, Benedict CA, et al. Structure of human cytomegalovirus UL144, an HVEM orthologue, bound to the B and T cell lymphocyte attenuator. J Biol Chem (2019) 294(27):10519–29. doi: 10.1074/jbc.RA119.009199
107. Dolff S, Witzke O, Wilde B. Th17 cells in renal inflammation and autoimmunity. Autoimmun Rev (2019) 18(2):129–36. doi: 10.1016/j.autrev.2018.08.006
108. Gan PY, Steinmetz OM, Tan DS, O’Sullivan KM, Ooi JD, Iwakura Y, et al. Th17 cells promote autoimmune anti-myeloperoxidase glomerulonephritis. J Am Soc Nephrol JASN (2010) 21(6):925–31. doi: 10.1681/ASN.2009070763
109. Angin M, Brignone C, Triebel F. A LAG-3–Specific Agonist Antibody for the Treatment of T Cell–Induced Autoimmune Diseases. J Immunol (2020) 204(4):810–8. doi: 10.4049/jimmunol.1900823
110. Grujic O, Stevens J, Chou RY, Weiszmann JV, Sekirov L, Thomson C, et al. Impact of antibody subclass and disulfide isoform differences on the biological activity of CD200R and βklotho agonist antibodies. Biochem Biophys Res Commun (2017) 486(4):985–91. doi: 10.1016/j.bbrc.2017.03.145
111. Irani V, Guy AJ, Andrew D, Beeson JG, Ramsland PA, Richards JS. Molecular properties of human IgG subclasses and their implications for designing therapeutic monoclonal antibodies against infectious diseases. Mol Immunol (2015) 67(2 Pt A):171–82. doi: 10.1016/j.molimm.2015.03.255
112. Schuurman J, Perdok GJ, Gorter AD, Aalberse RC. The inter-heavy chain disulfide bonds of IgG4 are in equilibrium with intra-chain disulfide bonds. Mol Immunol (2001) 38(1):1–8. doi: 10.1016/S0161-5890(01)00050-5
113. Wypych J, Li M, Guo A, Zhang Z, Martinez T, Allen MJ, et al. Human IgG2 antibodies display disulfide-mediated structural isoforms. J Biol Chem (2008) 283(23):16194–205. doi: 10.1074/jbc.M709987200
114. Joller N, Peters A, Anderson AC, Kuchroo VK. Immune checkpoints in central nervous system autoimmunity. Immunol Rev (2012) 248(1):122–39. doi: 10.1111/j.1600-065X.2012.01136.x
115. Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol (2003) 33(10):2706–16. doi: 10.1002/eji.200324228
116. Zhu B, Guleria I, Khosroshahi A, Chitnis T, Imitola J, Azuma M, et al. Differential role of programmed death-ligand 1 [corrected] and programmed death-ligand 2 [corrected] in regulating the susceptibility and chronic progression of experimental autoimmune encephalomyelitis. J Immunol (Baltimore Md 1950) (2006) 176(6):3480–9. doi: 10.4049/jimmunol.176.6.3480
117. Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, Tushima F, et al. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med (2003) 198(1):71–8. doi: 10.1084/jem.20022119
118. Carter LL, Leach MW, Azoitei ML, Cui J, Pelker JW, Jussif J, et al. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol (2007) 182(1-2):124–34. doi: 10.1016/j.jneuroim.2006.10.006
119. Schreiner B, Bailey SL, Shin T, Chen L, Miller SD. PD-1 ligands expressed on myeloid-derived APC in the CNS regulate T-cell responses in EAE. Eur J Immunol (2008) 38(10):2706–17. doi: 10.1002/eji.200838137
120. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol (2013) 14(12):1212–8. doi: 10.1038/ni.2762
121. Hughes J, Vudattu N, Sznol M, Gettinger S, Kluger H, Lupsa B, et al. Precipitation of autoimmune diabetes with anti-PD-1 immunotherapy. Diabetes Care (2015) 38(4):e55–57. doi: 10.2337/dc14-2349
122. Kasagi S, Kawano S, Okazaki T, Honjo T, Morinobu A, Hatachi S, et al. Anti-programmed cell death 1 antibody reduces CD4+PD-1+ T cells and relieves the lupus-like nephritis of NZB/W F1 mice. J Immunol (Baltimore Md 1950) (2010) 184(5):2337–47. doi: 10.4049/jimmunol.0901652
123. Jiang TT, Martinov T, Xin L, Kinder JM, Spanier JA, Fife BT, et al. Programmed Death-1 Culls Peripheral Accumulation of High-Affinity Autoreactive CD4 T Cells to Protect against Autoimmunity. Cell Rep (2016) 17(7):1783–94. doi: 10.1016/j.celrep.2016.10.042
124. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol (1996) 8(5):765–72. doi: 10.1093/intimm/8.5.765
125. Nishimura H, Agata Y, Kawasaki A, Sato M, Imamura S, Minato N, et al. Developmentally regulated expression of the PD-1 protein on the surface of double-negative (CD4-CD8-) thymocytes. Int Immunol (1996) 8(5):773–80. doi: 10.1093/intimm/8.5.773
126. Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol (Baltimore Md 1950) (2002) 169(10):5538–45. doi: 10.4049/jimmunol.169.10.5538
127. Hervey PS, Keam SJ. Abatacept. BioDrugs Clin Immunother Biopharmaceut Gene Ther (2006) 20(1):53–61. discussion 62. doi: 10.2165/00063030-200620010-00004
128. Oki M, Watanabe N, Owada T, Oya Y, Ikeda K, Saito Y, et al. A functional polymorphism in B and T lymphocyte attenuator is associated with susceptibility to rheumatoid arthritis. Clin Dev Immunol (2011) 2011:305656. doi: 10.1155/2011/305656
129. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Sci (N Y NY) (2014) 345(6204):1623–7. doi: 10.1126/science.1255904
130. Greisen SR, Rasmussen TK, Stengaard-Pedersen K, Hetland ML, Hørslev-Petersen K, Hvid M, et al. Increased soluble programmed death-1 (sPD-1) is associated with disease activity and radiographic progression in early rheumatoid arthritis. Scandinavian J Rheumatol (2014) 43(2):101–8. doi: 10.3109/03009742.2013.823517
131. Szmyrka-Kaczmarek M, Kosmaczewska A, Ciszak L, Szteblich A, Wiland P. Peripheral blood Th17/Treg imbalance in patients with low-active systemic lupus erythematosus. Postepy Higieny I Medycyny Doswiadczalnej (Online) (2014) 68:893–8. doi: 10.5604/17322693.1111127
132. Arruda LCM, Lima-Júnior JR, Clave E, Moraes DA, Douay C, Fournier I, et al. Homeostatic proliferation leads to telomere attrition and increased PD-1 expression after autologous hematopoietic SCT for systemic sclerosis. Bone Marrow Transpl (2018) 53(10):1319–27. doi: 10.1038/s41409-018-0162-0
133. Zhou L, Zhang Y, Xu H, Hu L, Zhang C, Sun L, et al. Decreased programmed death-1 expression on the T cells of patients with ankylosing spondylitis. Am J Med Sci (2015) 349(6):488–92. doi: 10.1097/MAJ.0000000000000468
134. Park YJ, Kuen DS, Chung Y. Future prospects of immune checkpoint blockade in cancer: from response prediction to overcoming resistance. Exp Mol Med (2018) 50(8):109. doi: 10.1038/s12276-018-0130-1
Keywords: immune checkpoints, autoimmune diseases, therapeutics, fusion protein, viral protein, nucleic acid, cell
Citation: Zhai Y, Moosavi R and Chen M (2021) Immune Checkpoints, a Novel Class of Therapeutic Targets for Autoimmune Diseases. Front. Immunol. 12:645699. doi: 10.3389/fimmu.2021.645699
Received: 23 December 2020; Accepted: 02 March 2021;
Published: 21 April 2021.
Edited by:
Hyewon Phee, Amgen, United StatesReviewed by:
Bergithe Eikeland, University of Bergen, NorwayLeonard Calabrese, Cleveland Clinic, United States
Copyright © 2021 Zhai, Moosavi and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingnan Chen, TWluZ25hbi5DaGVuQHV0YWguZWR1