 Jingtao Qiu
Jingtao Qiu Bowen Wu
Bowen Wu Stuart B. Goodman
Stuart B. Goodman Gerald J. Berry3
Gerald J. Berry3 Jorg J. Goronzy
Jorg J. Goronzy Cornelia M. Weyand
Cornelia M. Weyand- 1Department of Medicine, Stanford University School of Medicine, Stanford, CA, United States
- 2Department of Orthopedic Surgery, Stanford University School of Medicine, Stanford, CA, United States
- 3Department of Pathology, Stanford University School of Medicine, Stanford, CA, United States
Like other autoimmune diseases, rheumatoid arthritis (RA) develops in distinct stages, with each phase of disease linked to immune cell dysfunction. HLA class II genes confer the strongest genetic risk to develop RA. They encode for molecules essential in the activation and differentiation of T cells, placing T cells upstream in the immunopathology. In Phase 1 of the RA disease process, T cells lose a fundamental function, their ability to be self-tolerant, and provide help for autoantibody-producing B cells. Phase 2 begins many years later, when mis-differentiated T cells gain tissue-invasive effector functions, enter the joint, promote non-resolving inflammation, and give rise to clinically relevant arthritis. In Phase 3 of the RA disease process, abnormal innate immune functions are added to adaptive autoimmunity, converting synovial inflammation into a tissue-destructive process that erodes cartilage and bone. Emerging data have implicated metabolic mis-regulation as a fundamental pathogenic pathway in all phases of RA. Early in their life cycle, RA T cells fail to repair mitochondrial DNA, resulting in a malfunctioning metabolic machinery. Mitochondrial insufficiency is aggravated by the mis-trafficking of the energy sensor AMPK away from the lysosomal surface. The metabolic signature of RA T cells is characterized by the shunting of glucose toward the pentose phosphate pathway and toward biosynthetic activity. During the intermediate and terminal phase of RA-imposed tissue inflammation, tissue-residing macrophages, T cells, B cells and stromal cells are chronically activated and under high metabolic stress, creating a microenvironment poor in oxygen and glucose, but rich in metabolic intermediates, such as lactate. By sensing tissue lactate, synovial T cells lose their mobility and are trapped in the tissue niche. The linkage of defective DNA repair, misbalanced metabolic pathways, autoimmunity, and tissue inflammation in RA encourages metabolic interference as a novel treatment strategy during both the early stages of tolerance breakdown and the late stages of tissue inflammation. Defining and targeting metabolic abnormalities provides a new paradigm to treat, or even prevent, the cellular defects underlying autoimmune disease.
Introduction
Rheumatoid arthritis (RA) is a chronic, systemic autoimmune disease characterized by inflammation of the synovial tissue and autoantibody production (1, 2). Typically, the synovial membrane is infiltrated with T cells, B cells and macrophages, eliciting a maladaptive response-to-injury in fibroblast-like synoviocytes (FLS) that hyper-proliferate and destroy cartilage and bone (1, 3). Over the decade-long course of RA, a combination of genetic, epigenetic, and environmental factors contributes to rendering the host susceptible to autoimmunity and the eventual appearance of joint inflammation. An array of immune cells, including macrophages, dendritic cells, mast cells, neutrophils, T cells and B cells, have all been implicated in the disease process and make a pathogenic contribution during early loss of self-tolerance, the subsequent progression to joint inflammation when joint-specific protection factors fail and the final “non-healing wound” when the collective action of immune cells and stromal cells demolishes the joint (1, 4).
Patients diagnosed with RA produce a specific set of autoantibodies, typically reactive against post-translational modifications of proteins (5–7). Such autoantibodies appear decades before clinically apparent disease (8). Reactivity against a broad spectrum of citrullinated proteins, instead of a single autoantigen, has questioned the relevance of classical models of autoimmunity in RA and instead has emphasized the potential role of antigen-nonspecific factors, such as the metabolic control of immune cell function (9, 10). The genetic risk conferred by HLA class II molecules, the accumulation of chronically activated T cells in the diseased joint and the T-cell dependence of autoantibody production all support a critical role of dysfunctional memory T cells as a salient feature of RA. Differentiation and functional commitment of T cells is critically dependent on metabolic adaptations that co-ordinate the biosynthetic and energy demands imposed by chronic activation and the massive generation of cellular offspring (11). It is now clear that T cells not only differentiate into memory T cells, they also can become aberrantly activated, phenotypically unstable, exhausted, or senescent (12, 13). All these different functional states are ultimately interlinked with a specific metabolic program. RA T cells are characterized by a metabolic signature and progress has been made in defining molecular mechanisms underlying the metabolic deviations. Here, we will review how T cell metabolism influences the development of inflammation in the synovial tissue, focusing on three major metabolic pathways: glucose metabolism, glutamine metabolism and lipid metabolism. Ultimately, understanding how energy utilization and metabolite production dictates the cells’ fate outside and inside of the joint will set the stage for the design of novel immunotherapeutic strategies.
Glucose Metabolism as an Arthritogenic Risk Factor
Glucose metabolism, one of the primary metabolic pathways in the body, primarily refers to the process of breaking down glucose into ATP and intermediate metabolites, including glycolysis, aerobic oxidation, and processing in the pentose phosphate pathway (PPP). Glucose metabolism not only provides energy for physical activity but also mediates a variety of physiological processes through the formation of complex signaling networks with metabolic substrates. Glycolysis involves the catalytic conversion of one molecule of glucose into lactic acid, producing two adenosine triphosphate (ATP) molecules, whereas aerobic oxidation catalyzes one molecule of glucose into CO2 and H2O and generates 38 or 36 ATP molecules. Both glycolysis and aerobic oxidation are important ways to generate ATP, but the choice is typically adaptive to the physiological and pathological needs occurring in cells, tissues, and organs. In recent years, data have accumulated identifying glucose metabolism as a key component in the pathogenesis of RA.
Glycolytic Breakdown in Inflammatory Effector Cells
In normal synovial tissues, glycolysis is the primary pathway promoting mitochondrial substrate oxidation of pyruvate. The activity levels of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and lactate dehydrogenase (LDH), the major enzymes of the glycolytic pathway, are increased in RA synovial cells (14). Accordingly, lactate concentrations are high and glucose concentrations are distinctly low in the inflamed joint (15). Hexokinase, which catalyzes the first step in glucose metabolism, enhances the ability of FLS to migrate and invade (16, 17). In RA FLS, the balance between glycolysis and oxidative phosphorylation is shifted toward glycolysis (18). Synovial fibroblasts are key effector cells in the final stages of RA (19), are under considerable metabolic stress and produce competition for energy sources. Synovial tissue with its increased glycolytic activity represents only one “battle ground” of the disease. Disease-relevant cells live and function in other tissue environments, particularly the lymphoid tissues from which they originate (Figure 1).
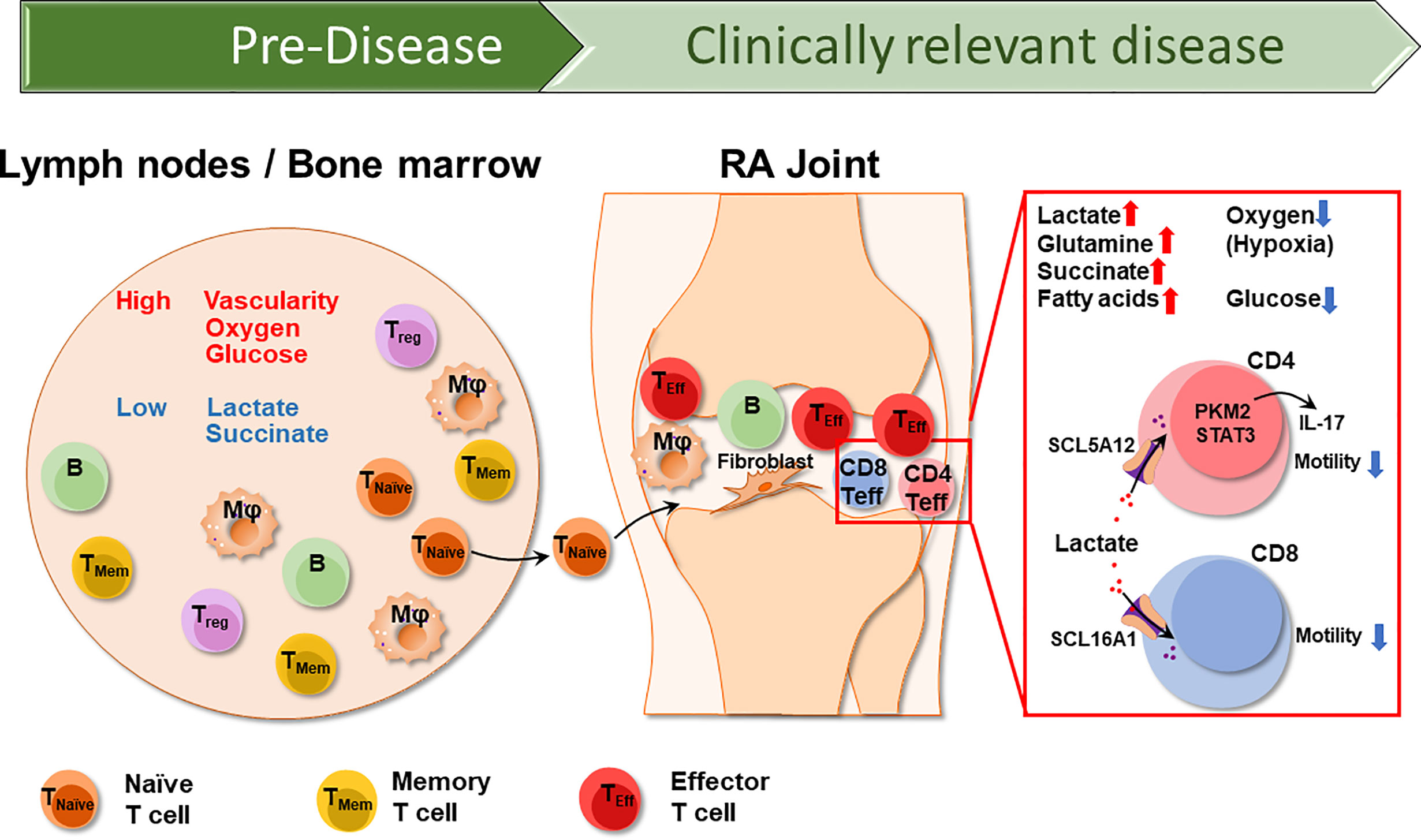
Figure 1 The different Tissue Environments of Rheumatoid Arthritis. Early events in the RA disease process, e.g., the loss of self-tolerance of RA T cells occurs in lymphoid tissues, such as lymph nodes and the bone marrow. Over lifetime, lymphoid tissues remain the home of naïve and memory T cells, which circulate through the peripheral blood where they can be sampled. Lymph nodes are highly vascularized, therefore have access to oxygen and glucose. As RA naïve T cells differentiate into short lived effector T cells (SLEC), they acquire the ability to leave the circulation and invade into the synovial tissue. Synovial effector T cells encounter a different metabolic environment which is hypoxic, low in glucose and high in lactate due to the chronic activation of macrophages and stromal cells. CD4+ effector T cells and CD8+ effector T cells uptake lactate through SCL5A12 and SCL16A1, respectively. Imported lactate promotes IL-17 secretion in CD4+ T cells and inhibits both CD4+ and CD8+ T cell motility, essentially arresting T cells in the lactatehi synovium. T naïve, naïve T cell; TMem, memory T cell; Treg, regulatory T cell; Mφ, macrophage; B, B cell.
Accordingly, naïve CD4+ T cells isolated from RA patient exhibit diminished glycolytic activity (20). Before such naïve CD4+ T cells become pathogenic memory and effector T cells, they utilize glucose in a distinctly different manner than naïve CD4+ T cells from healthy individuals: they avoid glycolytic breakdown into lactate and instead divert glucose into the PPP, driving the accumulation of NADPH and consumption of cellular reactive oxygen species (ROS). With an excess of reducing equivalents, T cells are unable to activate the relevant redox kinases, which allows them to bypass the regulatory checkpoints of the G2/M cell cycle and enter unrestrained proliferation (21). The lack of cellular ROS appears to be a T-cell-specific feature.
Macrophages from RA patients have high mitochondrial activity and can efficiently generate ROS (22–24). In patient-derived macrophages, suppression of GSK-3β fuels mitochondrial activity, enhances ATP synthesis and ROS release. This metabolic constellation causes the cytoplasm-to-nucleus translocation of the glycolytic enzyme pyruvate kinase M2 (PKM2). Functional outcomes include the PKM2-dependent activation of STAT3, boosting the production and secretion of pro-inflammatory cytokines (25), such as IL-6 and IL-1 (Figure 2). Such pro-inflammatory macrophages accompany T cells in the inflamed joint, where both cell types compete for the access to glucose. Naïve CD4+ T cells live in lymphoid organs and it is unlikely that inflammatory macrophages regulate the fate decisions of such T cells prior to their differentiation into memory/effector cells. In the inflamed joint, multiple, functionally diverse macrophage subsets are now recognized (26). In general, inflammatory macrophages are considered to mainly rely on glucose as an energy carrier and resolving/anti-inflammatory macrophages are known to require less glucose and supply their bioenergetic and biosynthetic needs through mitochondrial oxidative phosphorylation (27). Whether synovial macrophage subsets differ in their energy generation and utilization and the metabolic environment they build in their surroundings remains unexplored.
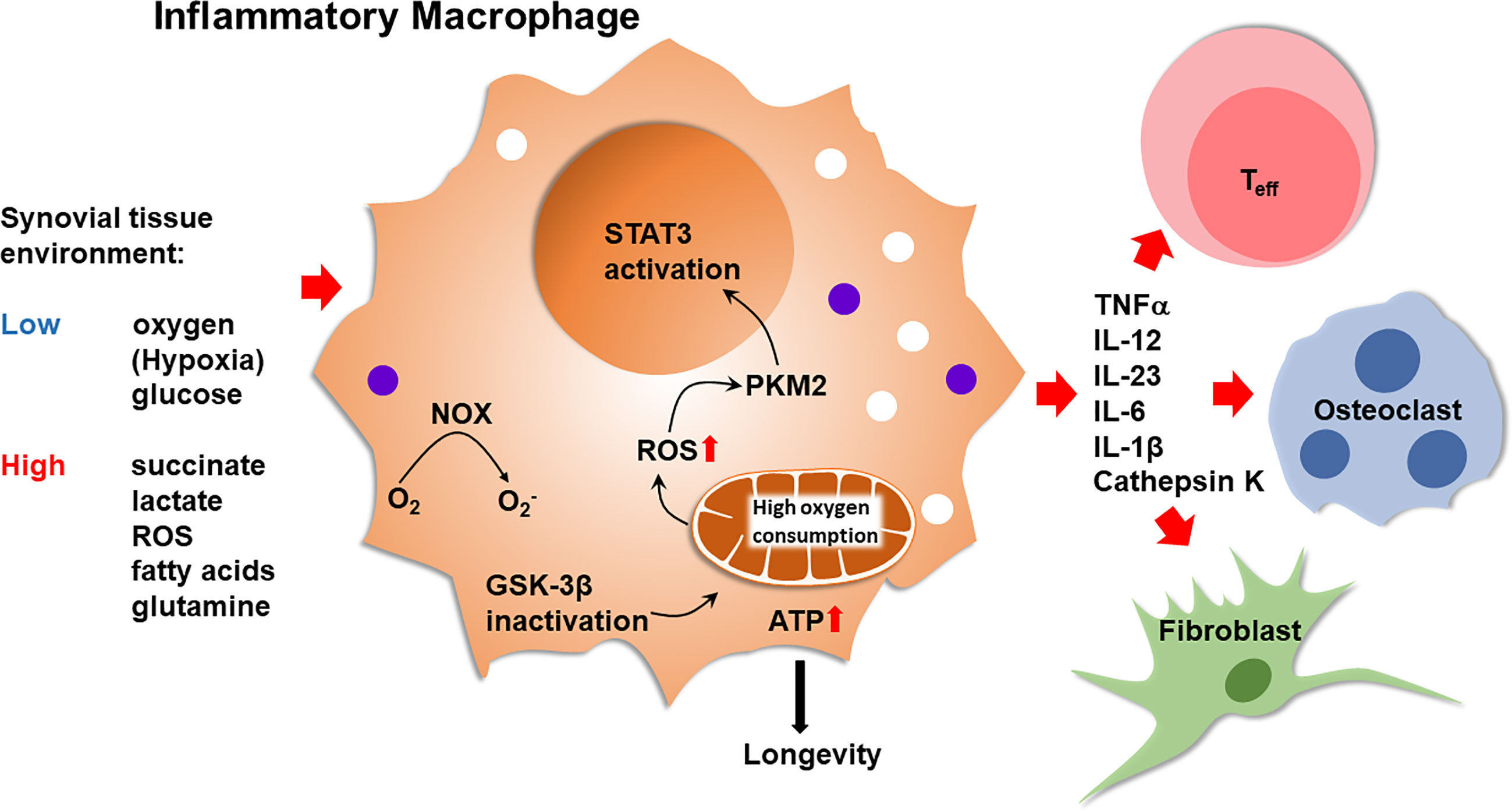
Figure 2 Inflammatory Macrophages in Rheumatoid Synovitis. Bone marrow-derived macrophages that infiltrate into the arthritic joint enter an oxygenlo, glucoselo, ROShi, succinatehi, fatty acidhi, lactatehi tissue environment. Inactivation of glycogen synthase kinase 3β (GSK-3β) in macrophages optimizes pyruvate import, enhancing mitochondrial activity. High oxidative phosphorylation increases ATP production and ROS release. ATP abundance promotes macrophage longevity. ROS facilitate the dimerization of the cytosolic enzyme pyruvate kinase M2 (PKM2), enabling nuclear translocation and STAT3 activation by the kinase. High mitochondrial activity supports the production of TNFα, IL-12, IL-23, IL-6 and IL-1β. Inflammatory macrophages trigger and sustain synovitis by modulating the function of neighboring T effector cells, endothelial cells, osteoclasts and synovial fibroblasts. NOX, NADPH oxidase; Teff, T effector cell.
Oxidative Phosphorylation in Inflammatory Effector Cells
The major metabolic abnormality of RA CD4+ T cells lies in the mitochondria (Figure 3). Mitochondrial failure presents as low ATP generation and reduced release of ROS (28). Production of the mitochondrial intermediate succinate is impaired, and the mitochondrial tricarboxylic acid cycle (TCA) cycle changes direction (29). Unable to convert acetyl-CoA into ATP, RA CD4+ T cells produce excess citrate and transport it out of the mitochondria into the cytosol (29). Here, acetyl-CoA participates in post-translational modification of proteins, shifting the functional commitment of the cells toward mobility and cytokine release (29). Mitochondrial failure in RA T cells results from insufficient repair of mitochondrial DNA, due to loss-of-function of the DNA repair nuclease MRE11A (30). A second dimension in maintaining mitochondrial fitness derives from the misplacement of the energy sensor AMP-activated protein kinase (AMPK) (31). Under physiologic conditions, a decline in ATP would trigger mitochondrial biogenesis via activation of AMPK (32). In RA T cells, this mechanism is paralyzed, forcing the cell to function with damaged mitochondria. The ATPlo status of RA T cells is aggravated by suppressed glycolysis; attributable to the transcriptional repression and functional impairment of the glycolytic enzyme phosphofructokinase (20). Instead, glucose is shunted into the PPP, supporting anabolic metabolism and laying the groundwork for biosynthesis, biomass production and the generation of new cells (21). Together, these data support the concept that irregularities in glucose metabolism are evident during an early stage of RA, when naïve CD4+ T cells deviate from a normal differentiation pattern and make a commitment to become a pro-inflammatory effector cell.
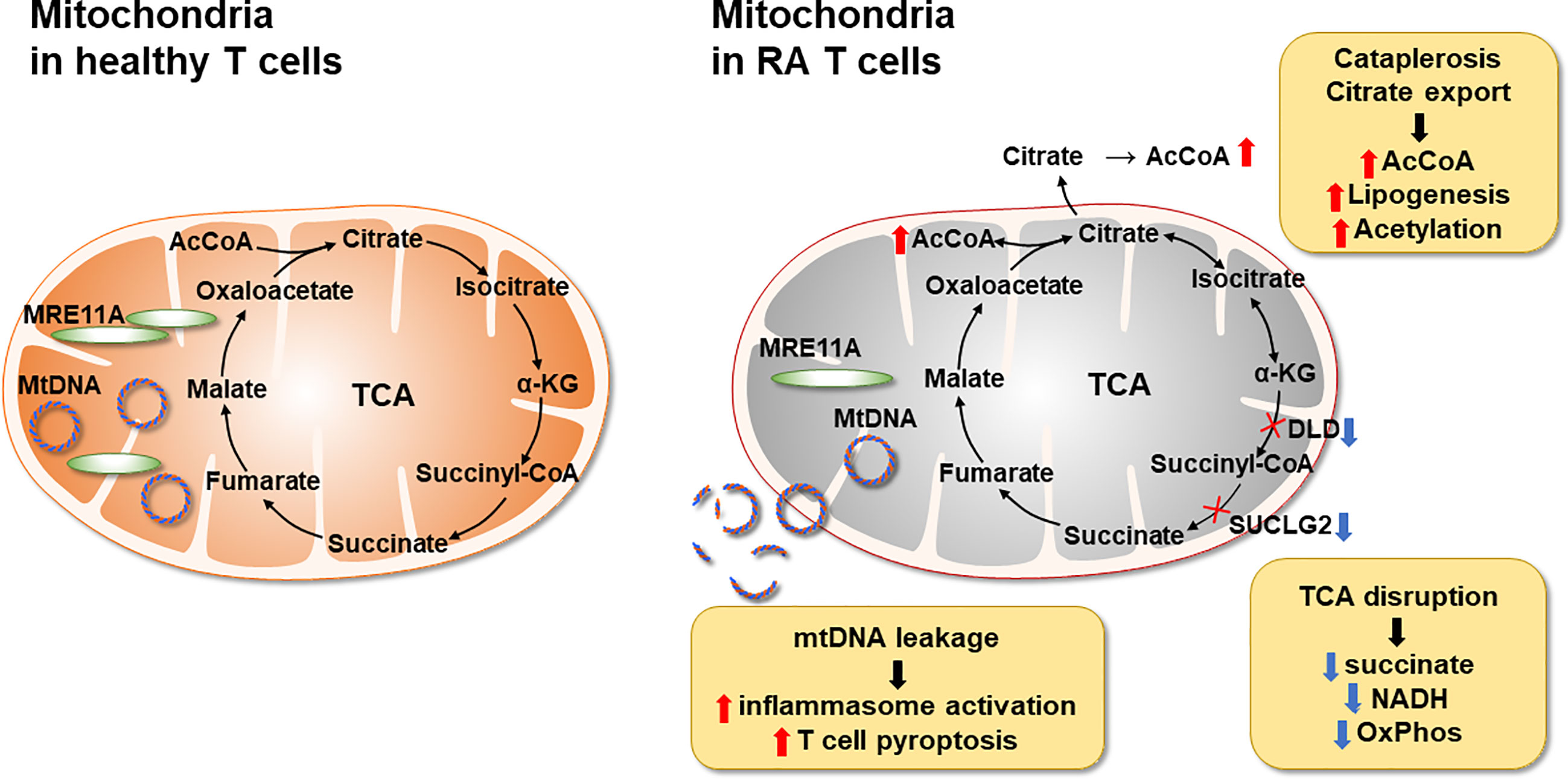
Figure 3 Mitochondrial Defects in T cells in rheumatoid arthritis. RA CD4+ T cells fail to maintain mitochondrial DNA due to the transcriptional repression of the nuclease MRE11A, a limiting factor in DNA double-strand break repair and replication fork protection. Instability of mtDNA causes leakage of mtDNA fragments into the cytosol, triggering activation of the inflammasome and eventually T cell pyroptotic death. Mitochondrial oxidative phosphorylation is further impaired by the disruption of the TCA cycle. Transcriptional repression of succinyl-CoA ligase (SUCLG2) prevents the conversion of a-ketoglutarate into succinate and forces the TCA cycle to change direction. In a cataploretic reaction, mitochondria of RA T cells produce and export excess citrate, creating an acetyl-CoA-rich environment in the cytoplasm. Together with NADPH generated in the hyperactive pentose phosphate pathway surplus acetyl-CoA enters biosynthesis, specifically lipogenesis. Also, excess acetyl-CoA drives protein acetylation, including the acetylation of microtubules. α-KG, α-ketoglutarate; mtDNA, mitochondrial DNA; SUCLG2, Succinate-CoA Ligase GDP-Forming Subunit Beta.
Immunoregulation Through Glucose Uptake
As part of their activation program, T cells switch to high glycolytic flux (33, 34), securing rapid access to ATP, but also to biosynthetic precursor molecules required for cellular expansion. The swift upregulation of glycolysis is linked to the increased expression of the glucose transporter 1 (GLUT1) maximizing glucose uptake by the cell (35). Glucose uptake in T cells undergoing activation is strictly correlated to GLUT1, identifying the transporter as a key determinant in glucose utilization (36, 37). Notably, GLUT1 expression differs among different T cell lineages and is highest in Th17, Th2, and follicular helper T cells (TFH) cells, followed by Th1 cells (38, 39). This difference in expression correlates with differential glycolytic rates in these T cell subsets (38) and identifies GLUT1 as a potential target for immunomodulation. Induction of GLUT1 transcripts is an early event during T cell receptor stimulation and is well known to depend on co-stimulatory signals. Signaling through CD28 and the downstream PI3K/Akt activation determines glucose uptake though GLUT1 upregulation (40). Cross-linking of the T cell receptor alone or in combination with IL-2 appears to have no effect on the level of GLUT1 expression (41, 42). Like T cells, B cells require GLUT1 upregulation for optimal activation responses and similarly, rely on PI3K-dependent signaling (43).
Experimental data suggest that GLUT1-mediated glucose transport may have differential effects in CD4+ and CD8+ T cells. GLUT1 knockout (KO) T cells fail to proliferate in response to immunization with an experimental antigen or in vitro stimulation (44). Similarly, treatment with CG-5, a nonselective small molecule glucose transport inhibitor inhibited T cell proliferation (45). CG-5 blocked Th1 and Th17 polarization and reduced T cell expansion in a mixed lymphocyte reaction. Interestingly, GLUT1neg CD8+ T cells proliferated to a similar extent and expressed equal levels of granzyme B as their wild type counterparts following stimulation. Thus, the cytotoxic potential of CD8+ T cells may not depend on GLUT1. Increased expression of GLUT3 and GLUT6 in GLUT1 KO CD8+ T cells could possibly compensate for the deficiency of glucose uptake by GLUT1 (44). More importantly, immunosuppressive T regulatory cells were not affected by GLUT1 deletion as they rely on fatty acid oxidation rather than glycolysis for their activity (38). GLUT1 expression had major consequences not only for proliferation but also for differentiation of T cells, as exemplified by the deficient induction of IFN-γ and IL-17 in GLUT1lo T cells (45).
The function of GLUT family member in T cells is well studied, but not clear in RA. There are only a few reports on GLUT1, GLUT3 and GLUT4 in RA. Heterozygous deletion of GLUT3 correlates directly with expression levels of GLUT3 and influences glycolysis rates in the human immune system (46), but the frequency of the GLUT3 copy number variant is not different among RA, multiple sclerosis and heathy control, providing no evidence for RA protection in deletion of GLUT3 (46), the study of which demonstrated GLUT3 is not necessary for glucose transfer in patients with RA. However, another study showed the mutual activation between CD4+ T cells and FLS, which resulted in increased proliferation and expression of glucose transporters GLUT1 and GLUT3 in FLS (47). In rat Arthritis model, arthritic rats showed cachexia, reduced adipocyte size, and downregulated GLUT4 in adipocyte membranes (48). Although the function of GLUT4 in pro-inflammatory T cells is unknown, the downregulated membrane GLUT4 confirms its role in autoimmunity.
Taken together, the intensity of glycolysis is highly dependent on active glucose transport, making GLUT1 an excellent marker of glycolytically active T cell populations. Glucose utilization determines T cell proliferation and differentiation, identifying this metabolite as an excellent candidate for metabolic interference. Immunoinhibitory Treg cells are less dependent on glucose uptake and should therefore be less vulnerable to glucose withdrawal. Controlling glucose import may represent an excellent approach to immunomodulation.
Tissue Lactate as an Immunoregulatory Mediator
Glycolysis has a low efficiency in generating energy. The terminal product of the anaerobic oxidation of glucose is lactate. Lactate has long been considered a “waste” by-product of cell metabolism, and it accumulates at sites of high glycolytic activity, such as tumor surroundings or inflamed tissue areas. Clinically, elevated serum lactate levels are a sensitive indicator of stress-induced glycolysis during sepsis (49). Since lactate production acidifies the environment, physiologic lactate concentrations are kept in a narrow range (1.5-3 mM) in blood and tissues of healthy individuals. Lactate can serve as an energy carrier and can be transported across cell membranes by mono-carboxylate transporters and can be reabsorbed by the kidneys to prevent energy loss. However, fueled by extensive glycolysis, lactate can increase up to 10 mM in inflammatory lesions and even higher (30-40 mM) in cancerous tissues (50, 51). Lactate accumulates in the synovial fluid of RA patients (52), reflecting high glucose turnover by cells trapped in the synovial membranes. It has been proposed that synovial lactate measurement may function as a reliable indicator to differentiated subtypes of inflammatory arthritis (53) (Figure 1).
Elegant work has examined the immunomodulatory impact of synovial tissue lactate in RA patients. T cells sense lactate via the expression of specific transporters and RA T cells may even be more reliant on the uptake of energy carriers, given their inherent difficulties of generating ATP in their mitochondria. Lactate uptake by CD4+ T cells has been implicated in inhibiting migratory capability, essentially arresting T cells in the lactatehi microenvironment (Figure 1). Also, lactate availability fostered differentiation of a Th17 subset, further supporting the chronic inflammatory process (49, 54). As reported by Pucino et al., synovial T cells adapt to their milieu by upregulating the lactate transporter SLC5A12. In line with the fundamental difficulties of RA T cells to secure energy production through mitochondrial activity (29, 30), SLC5A12 high-expressing tissue CD4+ T cells appear to be particularly dependent on utilizing alternative energy resources. Nurtured by metabolically highly active macrophages and synovial cells (18, 55) lactate-depending CD4+ T cells will be functionally biased toward IL-17 production, mediated by nuclear PKM2/STAT3 and enhanced fatty acid synthesis (56). Here, tissue-resident T cells display a metabolic signature previously described for pro-inflammatory macrophages that use nuclear PKM2/STAT3 to sustain IL-1 and IL-6 production (57).
Furthermore, tissue lactate not only shapes the functional commitment of CD4+ T cells, but also affects CD8+ effector T cells. Extracellular sodium lactate and lactic acid inhibit the motility of CD4+ and CD8+ T cells, respectively. The selective regulation of T cell mobility is mediated via subtype-specific transporters, SLC5A12 and SLC16A1, selectively expressed on CD4+ and CD8+ T cells, respectively (50). Underlying mechanisms have been defined and show that the lactate-induced inhibition of CD4+ T cell movement results from an interference with glycolysis that is activated when the chemokine CXCL10 engages the chemokine receptor CXCR3. CXCR3 is typically expressed on effector T cells, both CD4+ and CD8+ subtypes (58), emphasizing that the lactate-rich environment modifies the function of differentiated effector T cells as opposed to lymph node-residing naïve T cells that are upstream of synovitis induction (Figure 1). Interestingly, lactate exposure appears to result in a very different outcome for CD4+ and CD8+ effector T cells, stimulating the prior and inhibiting the latter (50). Single cell technology may be helpful in further breaking down the heterogeneous nature of tissue-residing effector T cells (59, 60), enabling the assignment of functional subsets to particularly patterns of nutrient uptake and utilization.
Lactate has drawn broad attention as a metabolite shaping the tissue milieu within and around tumors. A positive correlation between lactate dehydrogenase A (LDH-A), high local lactate levels, and tumor progression has been documented in various tumors (61). Autoimmunity-related tissue inflammation causes less of an acidification, but infiltrating cells may be exposed to similar cues. LDH isoenzymes have been reported to be higher in serum and synovial fluid of RA compared to osteoarthritis patients (62). And, it has long been known that LDH activity is increased in RA synovial tissues compared to healthy controls, emphasizing the dependence of the lesion on glycolytic metabolism (63). In healthy T cells, LDH-A is important in regulating differentiation and lineage assignment. By increasing availability of substrate for acetylation and shifting the T cell epigenome, LDH promotes Th1 commitment and IFN-γ production (64). A recent study has identified LDHA high expression as a feature of all CD8+ T cell subsets in RA patients (65). Inhibition of LDHA with FX11 (LDHA inhibitor) led to reduction in lipogenesis, migration, and proliferation of CD8+ T cells, and lowered CD8+ T cell effector functions (65). LDHA inhibition successfully abrogated the ability of RA CD8+ T cells to sway healthy B cells toward a pro-inflammatory phenotype. The LDHAhi phenotype of peripheral CD8 T cells was maintained in CD8+ T cells from the synovial membrane (65).
In summary, upregulation of glycolysis, and with it the production of lactate, is a marker of cellular activation and growth. The dependence of highly proliferative cells on glucose as an energy carrier leads to localized lactate accumulation and to acidification of the tissue site. During the late stages of the RA disease process, after T cells have arrived in the tissue environment, they are exposed to a lactate-rich milieu. Lactate providing a cellular arrest signal fits well into the concept that T cells build an extra-lymphoid site where they persist, commit to pro-inflammatory effector functions, and promote the building of self-sustained lymphoid architecture (66–68).
Immunoregulation Through the Mitochondrial Intermediate Succinate
By producing NADH, the tricarboxylic acid (TCA) cycle is a central route for oxidative phosphorylation, fueling complex I of the electron transport chain and eventually ATP generation. The TCA cycle fulfills other bio-energetic, biosynthetic, and redox balance requirements and functions as a metabolic hub. One major TCA function is the production of metabolic intermediates, that can be transported out of the mitochondria and participate in cytosol, nuclear and extracellular processes. An abundant TCA metabolite is succinate, which, like lactate, appears in the extracellular milieu, where it can be taken up by surrounding cells. Succinate is a product of myeloid and lymphoid cells, but the succinate receptor GPR91 is selectively expressed on monocytes/macrophages, granulocytes, and dendritic cells (69–71). GPR91 plays a critical role in the development of immune-mediated arthritis, supports the expansion of the Th17 cell population and acts as an overall amplifier of experimental antigen-induced arthritis. GPR91-/- mice show reduced articular hyperalgesia, neutrophil infiltration and inflammatory cytokines in the joint, and reduced frequency of Th17 cells in the draining lymph nodes (71).
Evidence has been provided describing succinate as a signaling molecule in the arthritic joint, with macrophages as the recipients of the stimulatory signals (70) (Figure 2). However, the cellular source of the succinate has not been determined. In general, succinate is recognized as a strong pro-inflammatory stimulator (72). LPS stimulation of macrophages results in abundant succinate production, which stabilizes Hypoxia-inducible factor 1α (HIF-1α) and IL-1β an important downstream target (73). In innate immunity, succinate may be one of the major signaling molecules (72), identifying metabolic activity as a key determinant of regulating the intensity of inflammation. Conversely, in the adaptive immune system, lack of succinate appears to be of particularly relevance in breaking tissue tolerance and causing inflammation (Figure 3). T cells isolated from RA patients are distinctly low in succinate, due to repression of the SUCLG2 gene, which brings the TCA to a halt and shifts from the oxidative to the reductive direction. One outcome is the accumulation of the oncometabolite α-ketoglutarate, feeding the production of citrate and acetyl-CoA. Excess acetyl-CoA imposes a pro-inflammatory effector phenotype through acetylation of the microtubular cytoskeleton (Figure 3). Succinatelow acetylCoAhi T cells rapidly reshape their cellular body, form a rear uropod, become hypermobile and invade into the tissue site (29). In that scenario, succinate supplement could be beneficial, reestablishing directionality of the TCA cycle, supporting the electron transport chain and forcing the T cell into a more tolerant state. Diametrical metabolic states in innate and adaptive immune cells seems to be a consistent finding in RA (55), beyond the role of succinate. Also, ATP and reactive oxygen species are abundant in monocytes/macrophages but scares in T cells. It is currently unknown how hypermetabolic macrophages communicate with hypometabolic T cells and vice versa and what the functional consequences are. But succinate appears to have a context-specific and cell-type specific role extending beyond its function as an energy carrier.
Hypoxia as an Amplifier of Tissue Inflammation
Mitochondrial metabolism ultimately depends on the availability of oxygen, which functions as the final electron acceptor in the electron transport chain. Oxygen pressures are highest in the lung, considerably lower in the blood and reach hypoxic levels in the tissue. Immune cells and tissue stromal cells need to adapt to hypoxic conditions and hypoxia is now recognized as a critical modulator of RA tissue inflammation (74). In line with the concept that reduced mitochondrial fitness is a risk factor for pro-inflammatory behavior, levels of synovial oxygen have been reported to negatively correlate with disease activity. Hypoxia induces a wide spectrum of alterations in mitochondrial structure, dynamics, and mitochondrial DNA (mtDNA) stability, resulting in impaired mitochondrial respiration, excessive production of reactive oxygen species (ROS), loss of ATP, increased oxidative damage and the accumulation of mtDNA mutations (75). One consequence of hypoxia is the activation of the transcription factor HIF-1α, which in turn promotes a gene program designed to enhance the production of glycolytic energy, including glucose transporters and glycolytic enzymes (76, 77). Hypoxic conditions in the rheumatoid joint should favor the survival of T cells that are less dependent on glycolytic breakdown, e.g., T cells with repression of the glycolytic enzyme PFKFB3 (78). PFKFB3lo RA T cells shift glucose toward the PPP (20) and produce biosynthetic precursors even under low oxygen conditions. Cells less dependent on proliferative expansion, such as synovial fibroblasts and macrophages, switch toward glycolysis as an oxygen-independent way of generating ATP.
By sustaining chronic upregulation of HIF-1α, synovial hypoxia provides a feed-forward mechanism amplifying synovitis. HIF-1α increases the activity of lactate dehydrogenase A (LDHA), which converts pyruvate to lactate. The resultant acidic environment promotes fibroblast and immune cell proliferation and persistence. High lactate and low O2 enable the survival of T cells by stabilizing HIF-1α (79, 80). HIF-1α has been reported to be highly expressed in RA synovium (81), identifying this transcription factor as a hallmark of synovial tissue inflammation. Hypoxia further enhances the stabilization of HIF-1α induced by T-cell-receptor-mediated activation of the PI3K-mTOR pathway (82), functioning as a stabilizer of autoimmune tissue inflammation. HIF-1α+ T cells may be particularly adapted to serve as pro-inflammatory effector cells (74). Driven by the hypoxic microenvironment of the joint, FOXP3+ regulatory T cells promptly convert to pathogenic Th17 cells (83), further weakening anti-inflammatory mechanisms. HIF-1α-dependent induction of retinoic acid-receptor-related orphan receptor-γt (RORγt) combined with targeting FOXP3 for degradation is detrimental to Treg cells and fosters Th17 cell generation (84), identifying hypoxia as a potent risk factor for unrelenting inflammation.
Low availability of oxygen equally encourages pathogenic traits of stromal cells. Hypoxia and IL-17 synergize to drive migration and invasion of synovial fibroblasts through MMP2 and MMP9 expression (85). HIF-1α also controls fibroblast IL-33 production, which in turn enhances HIF-1α expression and generates a regulatory cycle that perpetuates RA inflammation (86). Also, cooperation of HIF-1α with intracellular signaling cascades may accelerate pro-inflammatory pathways. Under hypoxic conditions, HIF-1α interacts with Notch-3 and STAT-1 in RA synoviocytes to stabilize and enhance stromal cell inflammation (87).
Glutamine as a Fuel Source for Joint Inflammation
While much of the focus of immunometabolism has been directed to central carbon metabolism, e.g., glycolysis and the TCA cycle, amino acids are now emerging as critical regulators of immunocompetence. Among the 20 amino acids edited by gene codons, glutamine appears particularly important, contributing to bioenergetic as well as biosynthetic processes, while also helping to maintain redox balance. Glutamine is the most abundant and widely used amino acid in the human body. Glutamine is largely anaplerotic and relinquishes both of its amino groups to fuel the TCA cycle. It participates in the inter-organ nitrogen exchange through ammonia (NH3) transport between tissues and is critically involved in maintaining pH stability. Glutamine serves as a carbon and nitrogen donor for nucleotide biosynthesis and is a requirement for nicotinamide adenine dinucleotide phosphate (NADPH) generation. Thus, glutamine is an alternative fuel source, serves as a biosynthetic material, contributes to epigenetic and posttranslational modifications and determines the redox status, identifying this amino acid as a central regulator of immune cell fitness (88, 89).
Glutamine to the Rescue: Maintaining Energy Production in Unfit Mitochondria
Glutamine, and its breakdown product glutamate, have both been identified as critical energy carriers in tumors, and the glutaminolytic pathway is considered a potential therapeutic target to suppress tumor growth (90). Both glucose and glutamine can provide carbons to feed the mitochondrial TCA chain and preference for one over the other may depend on local availability but may also affect the outcome of pathogenic immune responses. Glucose is primarily supplied by the liver, glutamine is synthesized by the muscle (91, 92), implicating these two organ systems in regulating protective and pro-inflammatory immunity. To make glutamine available for ATP generation, T cells utilize the Glutaminase isoenzymes (GLS1 and GLS2). Transcriptomic studies combined with metabolomic studies have defined GLS1 as a marker enzyme of fibroblast-like synoviocytes (FLS) isolated from RA patients (93). Notably, withdrawal of glutamine, but not of glucose, reduced RA-FLS proliferation, suggesting that the amino acid is critical in supporting pannus formation in the arthritic joint. Glucose may be a limiting factor, required to support multiple cell types in the inflamed synovial membrane, while glutamine may be freely accessible, excluding the proliferative FLS from glucose competition. Takahashi et al. have examined which factors can upregulate GLS1 in RA-FLS and found that both, IL-17 and platelet-derived growth factor acted as GLS1 inducers (93). The emerging model suggests that the T cell cytokine IL-17 directs synovial stromal cells to utilize glutamine instead of glucose, implicating metabolic regulation in the coordination of innate and adaptive immune responses in RA.
If the rheumatoid joint is a glutamine rich environment, then breakdown products derived from glutamine could potentially have pro-inflammatory functions. Through a series of enzymes, T cells convert glutamine into glutamate, which then is transformed into α-Ketoglutarate (α-KG) (94). The oncometabolite α-KG has versatility and has access to multiple cellular compartments, including the mitochondria, the cytosol, and the nucleus (95–97). α-KG can directly enter the TCA cycle, where it can be metabolized through oxidation and reduction. Conversion into succinate is a rate-limiting step in oxidative phosphorylation. Transformation into citrate feeds carbons into lipid metabolism. By functioning as a precursor for glutamine, α-KG also takes a center position in maintaining the redox balance of the cell. Given the enrichment in glutamate, discussions have focused on whether glutamatergic signaling has a role in the pathophysiology of RA (98). Measurements in collagen-induced arthritis (CIA) have shown a marked increase of glutamate in the synovial fluid. Glutamate functioned as an arthritogenic effector molecule by driving proliferation of synovial fibroblasts. Further support for a direct contribution of glutamate in the disease process came from blocking studies, applying Memantine, an N-methyl-D-aspartate (NMDA) ionotropic glutamate receptor antagonist. This receptor antagonist had anti-inflammatory efficacy by upregulating CD4+CD25+ regulatory T cells in the spleen (99). These data suggest that glutamate is not only a stimulatory neurotransmitter, but it also has immunoregulatory functions. To better understand how glutamate promotes disease, it would be important to know the cellular origin and the glutamate receptor expression profile of innate, adaptive, and stromal cells participating in synovitis.
A particularly appealing concept is the idea that the tolerance-breaking effect of glutamate is a consequence of its function as a neurotransmitter. Support for this concept has come from studies applying the non-competitive NMDA ionotropic glutamate receptors antagonist ketamine intrathecally (100). When injected intrathecally into animals with antigen-induced arthritis, arthritis severity is reduced, the density of inflammatory cells in the joints is lowered and joint destruction is halted (100). Therapeutic application would require a more precise understanding of which cells are signal-sending and which cells are signal-receiving.
Pumping in Amino Acids: Glutamine Transporters as Disease-Promoting Molecules
Mechanistic studies have attempted to define upstream mediators that trigger the enhancement of glutaminolysis in T cells. During T cell activation, glutamine transporters and the components of the glutaminolysis machinery are upregulated through a MYC-dependent pathway (101). Compared to the resting state, activated T cells increase their glutamine uptake 5-10-fold. Several amino acid transporters facilitate glutamine uptake in T cells; e.g., the sodium-coupled neutral amino acid transporters of the SLC38 gene family (102). Upon T cell activation, transcription of SLC38A1 and SLC38A2 is activated in a coordinated fashion. Interestingly, SLC38A1 and SLC38A2 protein is stored in intracellular vesicles from where it is rapidly transferred to the cell surface (103). Early events after T cell receptor triggering, such as membrane-proximal signaling and induction of T cell activation markers are unaffected by the withdrawal of glutamine. Conversely, late processes, such as T cell clonal expansion and cytokine release are highly sensitive to the lack of glutamine, predicting that glutamine is partially important for pro-inflammatory effector functions of T cells.
Several transporters ensure sufficient glutamine uptake in T cells undergoing activation, including SLC1A5 (104). Naïve CD4+ T cells rely upon SLC1A5 to fulfill their need for glutamine. In line with the concept that T cell activation and differentiation require metabolic adaptation, SLC1A5 couples TCR and CD28 signals to ultimately activate the mTORC1 pathway and enable the cell to undergo clonal expansion (105). SLC1A5 expression seems to be a prerequisite for the lineage commitment of both Th1 and Th17 cells (105), placing it high on the list of potential upstream regulators of the pathogenic differentiation of short-lived effector T cells encountered in RA patients (31). No information is currently available how a specific tissue microenvironment affects metabolically regulated aspects of T cell differentiation. Considering that T cell pathology in RA occurs both in lymphoid tissues as well as the inflamed joint, it would be important to know whether differences exist for glutamine availability, to which degree glutamine substitutes as an energy carrier under conditions of glucose deprivation and how glutamine interferes with the pathogenic mTORC activation driving disease-relevant effector functions.
Lipids as a Catalyst for Autoimmune Joint Inflammation
Like glucose and amino acids, lipids are an essential nutrient resource for protective and pathogenic immune cells. Lipids are a high-impact energy source, affecting multiple biological processes, including the production and the storage of energy (106), the assembly and functionality of cellular membranes (107), gene regulation of metabolic pathways (108) and post-translational modifications in intracellular signaling pathways (109). Intracellular lipid concentrations are regulated both by nutritional uptake and by ongoing biosynthesis. Fatty acid synthesis involves the creation of fatty acids from acetyl-CoA and NADPH through the action of enzymes called fatty acid synthases (FAS), through which simple lipids are bio-synthesized and sequentially elongated through addition of acetyl-CoA (110).
Disordered Lipid Profiles in Inflammatory Disease
Numerous autoimmune diseases, including RA, psoriasis, and systemic lupus erythematosus, in which autoreactive CD4+ T cells participate in inflammatory tissue lesions share the accumulation of intracellular lipid droplets in such T cells (111–113). These autoimmune diseases are associated with elevation of serum triglycerides and cholesterol (113–115). Whether this dyslipidemia is causative for inflammation or a consequence of persistent inflammatory activity remains unresolved. Evidence for a disease-relevant role of lipids has come from reports that lowering blood lipid levels by diet or drug treatment may improve symptoms, including T cell-dependent autoantibody responses (116–118). Nevertheless, it remains speculative whether modification of lipid profiles could reestablish tissue tolerance and revert pathogenic immunity.
Lipids accumulating in the synovial fluid of RA patients have been analyzed through liquid chromatography mass spectrometry, and more than 70 different components from different lipid classes have been detected (119). Among the broad spectrum of lipids found in synovial fluid, maresin 1, lipoxin A4 and resolvin D5 were associated with RA and 5S,12S-6E,8Z,10E,14Z-dihydroxyeicosatetraenoic acid was identified as a marker of lipoxygenase activity. Nevertheless, lipidomic studies have not yielded any unexpected insights. In early studies more than 50 years ago, increased amounts of phospholipid, cholesterol, and neutral lipids were described, but synovial fluid lipid levels were not predictive of severity of synovitis, questioning a direct involvement of lipids in driving arthritis (120, 121).
The abnormal cholesterol profile in patients with preclinical RA and early RA is typical of metabolic syndrome: normal or mildly elevated total cholesterol, LDL cholesterol and triglycerides, associated with decreased HDL cholesterol levels (122, 123). The cholesterol biosynthesis pathway appears to be a key regulator of controlling CD4+ T cell commitment to inflammatory versus anti-inflammatory effector status. Inhibition of the cholesterol biosynthesis pathway produces a specific block in immune resolution, defined as a significant decrease of T cell IL-10 production (124). Thus, anti-inflammatory response in CD4+ T cells may be particularly dependent on sterol metabolism.
Cytoplasmic Lipid Droplets As Immunoregulatory Organelles
Intracellular lipids, however, play a quintessential role in how immune cells differentiate into effector cells. Indeed, the activation of the T cell receptor is coordinated with up-regulation of genes involved in the biosynthesis of cholesterol and fatty acids (125). This anabolic program is regulated by specialized transcription factors, the sterol regulatory element-binding proteins (SREBP) (125). Without SREBP signaling, T effector cells could not blast and could not undergo clonal expansion (125). T cell activation is associated with massive cellular proliferation, imposing high biosynthetic demands, particularly when it comes to the generation of bio-membranes. Besides their essential role as biosynthetic precursor, lipids also serve a non-replaceable role a bioenergetic materials, specifically in the induction of T memory cells, which are known to dependent on mitochondrial fatty acid β-oxidation (126). Notably, T memory cells, unlike T effector cells, do not uptake extracellular palmitate (126), delineating a stringent association between T cell subtype and preferences for fatty acid substrates. How different T cell subpopulations acquire and mediate their taste for select lipids is not entirely clear. Different fatty acid transporters are expressed on the membrane of distinct T cell subsets. In the case of memory T cells, survival is impaired in the presence of the FAS inhibitor C75 (127), indicating that the fitness of T memory cells depends on de novo fatty acid biosynthesis.
Data that have accumulated over the last 5 years support the concept that lipid metabolism in RA T cells is fundamentally abnormal. Due to the mitochondrial defects (Figure 3), RA T cells lack the flexibility to use lipids as an energy carrier. Instead, RA T cells commit fatty acids to the lipogenesis program to support the biomass building program. Excess lipids that cannot be metabolized are stored as cytoplasmic lipid droplets (29, 128). Fatty acids that are not committed to the building of cellular offspring need to be safely packed and stored. Notably, tissue-residing T cells in rheumatoid synovitis accumulate cytoplasmic lipid droplets (128). Currently, there is no evidence that the lipogenesis>>lipolysis status of RA T cells is a primary defect, but rather appears to be a consequence of abnormal mitochondrial function and rerouting of glucose into the biosynthetic PPP. Reversal of the TCA cycle and the export of citrate out of the mitochondria leads to the accumulation of acetyl-CoA in the cytoplasm (129, 130). Fatty acid synthesis is further facilitated by the availability of NADPH, a cofactor required for lipogenesis and available due to the commitment to the PPP instead of glycolysis (131). Surplus in fatty acids has direct impact on the pathogenic effector functions of RA T cells, as fatty acids can be integrated into phospholipids, creating building blocks for cell membranes. Data on the composition of cellular and subcellular membranes in RA T cells are currently not available, but structural analysis has yielded insights on how these autoimmunity-prone cells adapt to their metabolic programming (132). Colocalization studies of the cytoskeletal marker F-actin and the membrane marker cortactin have demonstrated membrane extrusions indicative of rapid membrane turnover and structural flexibility (133, 134). Membrane ruffling occurs in cells loaded with cytoplasmic lipid droplets and translates into high mobility in 3-dimensional matrix and rapid transition from the blood to the tissue (128). Acetyl-rich RA T cells easily change their cellular shape, building a uropod known to promote cellular motion (29). Studies applying metabolic inhibitors have placed the redirection of glucose and lipids upstream of membrane ruffling and podosome formation (Figure 4) (128). Nevertheless, the question arises whether the loss of a circular cellular shape has metabolic implications by itself. Analysis of the content of the uropod in RA T cells has revealed the displacement of mitochondria into a perinuclear position (29). The mitochondrial rearrangement is rooted in the hyperacetylation of cytoskeletal proteins, specifically tubulin, which propels mitochondria away from the periphery close to the nuclear membrane. The proximity of ROS-producing mitochondria to the nucleus and to chromosomes placed near the nuclear membrane enables mitochondria to impose gene expression changes that by itself can mediate information from the metabolic hub of the cell to the “cockpit”. Essentially, metabolic rewiring may function as a feed forward mechanism, profoundly altering programs that oversee cellular behavior and adaptive immunity.
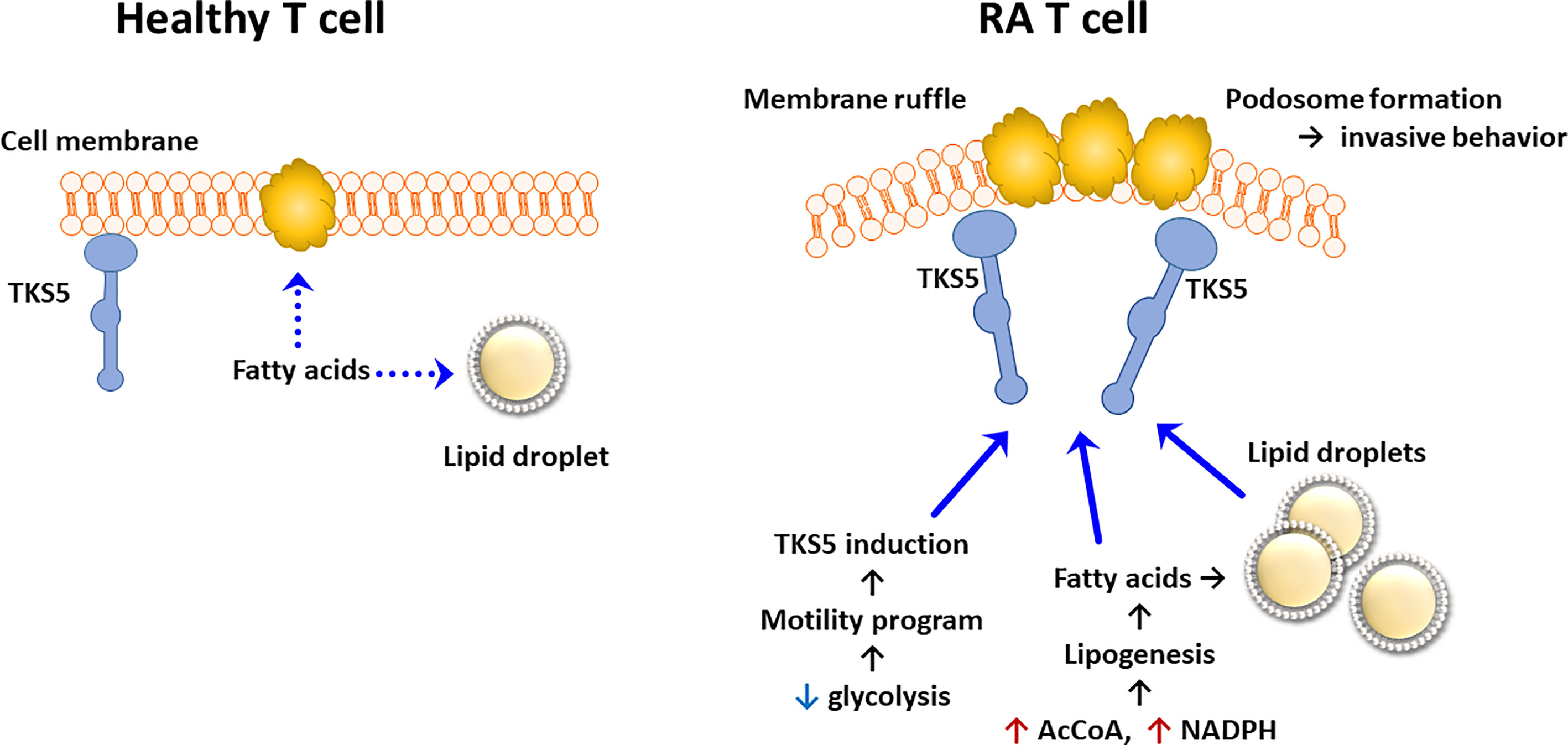
Figure 4 Pathogenic Lipogenesis in RA T cells. Metabolic rewiring in RA T cells, including the shunting of glucose to the pentose phosphate pathway, the export of mitochondrial citrate and the suppression of mitochondrial lipid oxidation, all favor lipogenesis over lipolysis. As a result, RA T cells deposit lipid droplets in the cytoplasm and are rich in biosynthetic precursors for membrane lipids. Lowered glycolytic activity upregulates a module of motility genes, including the scaffolding protein TKS5. As part of their pro-inflammatory phenotype, lipogenesis-biased TKS5hi RA T cells spontaneously form actin- and cortactin-rich membrane ruffles, equipping them tissue invasion and mobility in extracellular matrix. AcCoA, acetyl-CoA.
Metabolic Networks as Therapeutic Targets
Immunomodulatory therapies currently applied in RA have metabolic “side effects” (Table 1). It is difficult to say whether the metabolic consequences induced by these therapeutics are primary or secondary. Obviously, effective control of systemic inflammation may leave a metabolic footprint, even if the pathways of intracellular metabolic control are ignored by the therapeutic. Some of these immunosuppressive medications may directly interfere with glycolysis, mitochondrial function, lipogenesis, and amino acid metabolism, but mechanistic understanding of such effects remains scarce. The gold standard of RA management, Methotrexate (MTX), is a potent antimetabolite drug that targets the processing of purine (155) and folic acid (156) by inhibiting dihydrofolate reductase. MTX, corticosteroids, sulfasalazine, leflunomide and cyclosporine A function as immunomodulators by inhibiting pro-inflammatory Th1 and Th2-dependet immunity (157). However, their metabolic impact is not always protective. Corticosteroids as well as nonsteroidal anti-inflammatory drugs have metabolic consequences that enhance cardiovascular risk (158).

Table 1 Metabolic effects of currently used therapies in rheumatoid arthritis.
Anti-TNF-α drugs have become a cornerstone in managing RA, particularly in patients resistant to traditional therapeutic approaches (159, 160). Anti-TNF-α treatment decreases glycolytic activity in RA synovium, likely through its potent anti-inflammatory effect (161). Blockade of IL-6 appears to have primarily a systemic are metabolic impact. Anti-IL-6 receptor treatment leads to weight gain and modified fat distribution and gain in muscle mass suggests that blocking IL-6 might be efficient in treating sarcopenia associated with RA (151). Anti-IL-6 receptor treatment has been reported to decreases oxidative stress in RA leucocytes (152). Inhibitors of the JAK-STAT pathway (e.g. Tofacitinib) display powerful anti-inflammatory effects and significantly decrease mitochondrial membrane potential, mitochondrial mass and ROS production in RA synovial fibroblasts (162). In an elegant study, McGarry et al. have provided evidence that tofacitinib directly modulates mitochondrial function, such as regulating key mitochondrial genes, increasing oxidative phosphorylation, enhancing ATP production while diminishing glycolytic flow and key glycolytic genes (162).
Capitalizing on the understanding of how fuel selection guides cellular behavior and on the concept that fuel determines function, multiple novel therapeutic targets emerge. Besides the central role of glucose itself (163), metabolic intermediates such as lactate (53) and succinate (70) appear to be critically important in regulating tissue inflammation. Much will be learned from therapeutic approaches in cancer therapy that rely on disrupting bioenergetic needs to the tumor and metabolic vulnerabilities in anti-tumor immune responses (164). Unselected approaches, such as shutting off glucose consumption, amino acid utilization or lipid import are likely going to fail, as all organ systems are dependent on energy supply from these energy carriers (165) and more sophisticated strategies are needed to manipulate cellular metabolism.
Conclusion
Autoimmune diseases, such as RA, begin with T cells losing self-tolerance and providing help to auto-antibody producing B cells. The root cause is believed to be the recognition of autoantigen and the failure of mechanisms that sort out autoreactive T cells. Recent data have questioned this simplified model and have added metabolic signals as decisive determinants in T cell fate and behavior. Based on the concept that “fuel feeds function”, the metabolic networks of disease-inducing T cells have been defined and are being explored as liabilities that may be targeted to re-engineer “bad” T cells into “good” T cells. An additional layer of metabolic control is introduced by the metabolic cues provided by the tissue environment in which cells live, die, proliferate, and communicate. RA CD4+ T cells lose tolerance decades before joint inflammation begins, while living in lymphoid organs, such as lymph nodes and the bone marrow. Once such T cells differentiate into short-lived effector cells (SLEC) instead of long-lived memory cells, they function as tissue-infiltrating and pro-inflammatory effector cells.
Here are the hallmarks of altered immunometabolic regulation in RA:
● RA T cells are characterized by abnormal glycolysis and inappropriate lipogenesis (Figure 5).
● The underlying defect is a malfunctioning mitochondrion. Lacking DNA repair of the mitochondrial genome leads to DNA instability and cytosolic leakage. Triggering of the inflammasome gives rise to T cell pyroptosis, a process that by itself functions as a strong instigator of tissue inflammation.
● Insufficient mtDNA damage repair undermines OxPhos and ATP generation. The mitochondrial TCA cycle is disrupted due to transcriptional repression of SUCLG2, impairing the conversion of α-KG into succinate. Unprocessed α-KG leads to reversal of the TCA cycle and results in the accumulation and the export of citrate.
● This cataploretic step promotes acetyl-CoA accumulation in the cytoplasm and favors acetylation of cytoplasmic proteins. Hyperacetylation of microtubules stiffens the cytoskeleton, fundamentally changing the cellular shape and the distribution of subcellular organelles. The most significant consequence of the hyperacetylate state is transfiguration of the cell away from a circular shape toward rear-front polarization and uropod formation. The resulting cell is hypermobile, swiftly transmigrating from the blood to the tissue.
● Besides the change in migratory behavior and invasiveness, RA T cells differentiate into pro-inflammatory cytokine producers, favoring IFN-γ and IL-17 as effector cytokines. The commitment to Th1 and Th17 differentiation results from the persistent activation of mTORC1, a consequence of the misplacement of the energy sensor AMPK. Lacking a myristoylation tail, AMPK in RA T cells fails to anchor on the lysosomal surface and thus forgoes the suppression of mTORC1 (Figure 6). Unopposed mTORC1 activation encourages RA T cells to proliferate and build daughter cells despite the low ATP state. The proliferation program of SLECs, however, is supported by the shunting of glucose from bioenergetic to biosynthetic utilization. Instead of breaking glucose down into ATP and pyruvate, RA T cells shift glucose into the PPP and generate NADPH plus biosynthetic precursors. Ongoing lipogenesis provides building blocks for membranes and organelles and the cells’ reductive state promotes fatty acid synthesis.
● Once transitioned into the inflamed joint, RA T cells encounter a hypoxic, lactate-rich, glucose- low environment. The chronic activation of stromal cells and invading immune cells depletes the tissue of glucose and acidifies the environment with lactate. Synovial T cells have been shown to uptake lactate, supplementing their energy supply but also signaling the cells to undergo arrest and persist in the tissue niche.
● Most metabolic abnormalities in RA T cells are present in the naïve population and sustained in tissue-residing memory T cells, placing metabolic dysregulation upstream of the joint. These data classify the metabolic malfunction, at least in part, as an “original sin”, leading to autoimmunity.
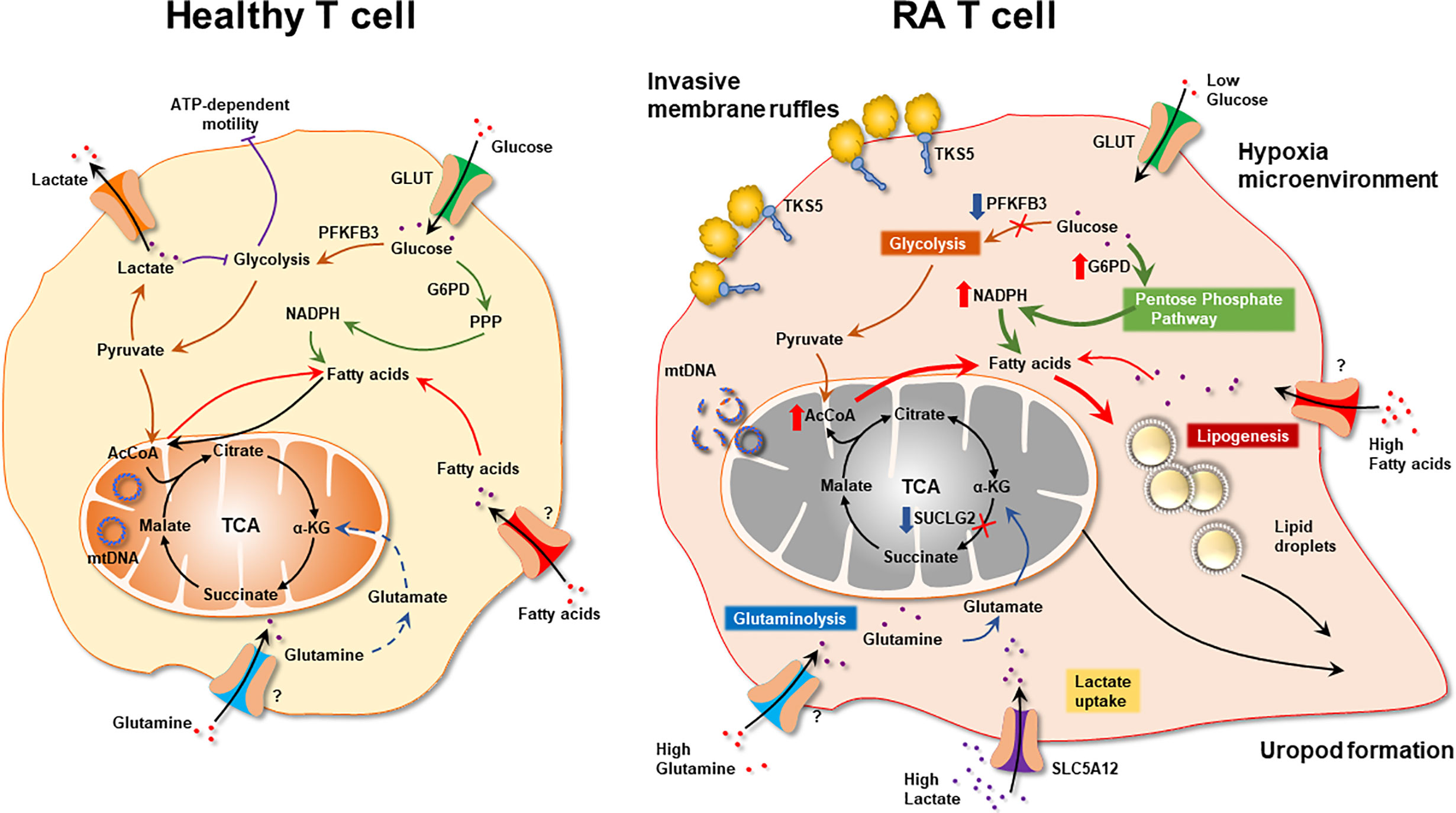
Figure 5 The Metabolic Signature of Autoreactive T cells in Rheumatoid Arthritis. Schematic diagram of metabolic pathways in health and RA T cells. Healthy T cell: Low-proliferating healthy T cells (naïve, memory) rely on mitochondrial energy production. When differentiating into highly replicating effector T cells, bioenergetic and biosynthetic needs are fulfilled by engaging glycolysis. RA T cells: Biased toward biomass generation, RA T cells shunt glucose into the pentose phosphate pathway and produce NADPH. Mitochondria with unrepaired mtDNA utilize relatively low amounts of oxygen and produce low concentrations of ROS. Mitochondrial succinate production is suppressed due to low activity of Succinate-CoA ligase and a-ketoglutarate is converted into citrate. Exported citrate supplies cytoplasmic acetyl-CoA pools and promotes lipogenesis. Surplus lipids sustain the generation of invasive membrane ruffles and of cellular uropods. With low ATP production, RA T cells require external energy sources, such as glutamine and lactate to meet basic bioenergetic needs. By diverting energy away from ATP production toward assembly of biosynthetic precursor molecules, RA T nourish the emergence and the survival of replicating effector T cells that are tissue-invasive and pro-inflammatory. α-KG, α-ketoglutarate; mtDNA, mitochondrial DNA.
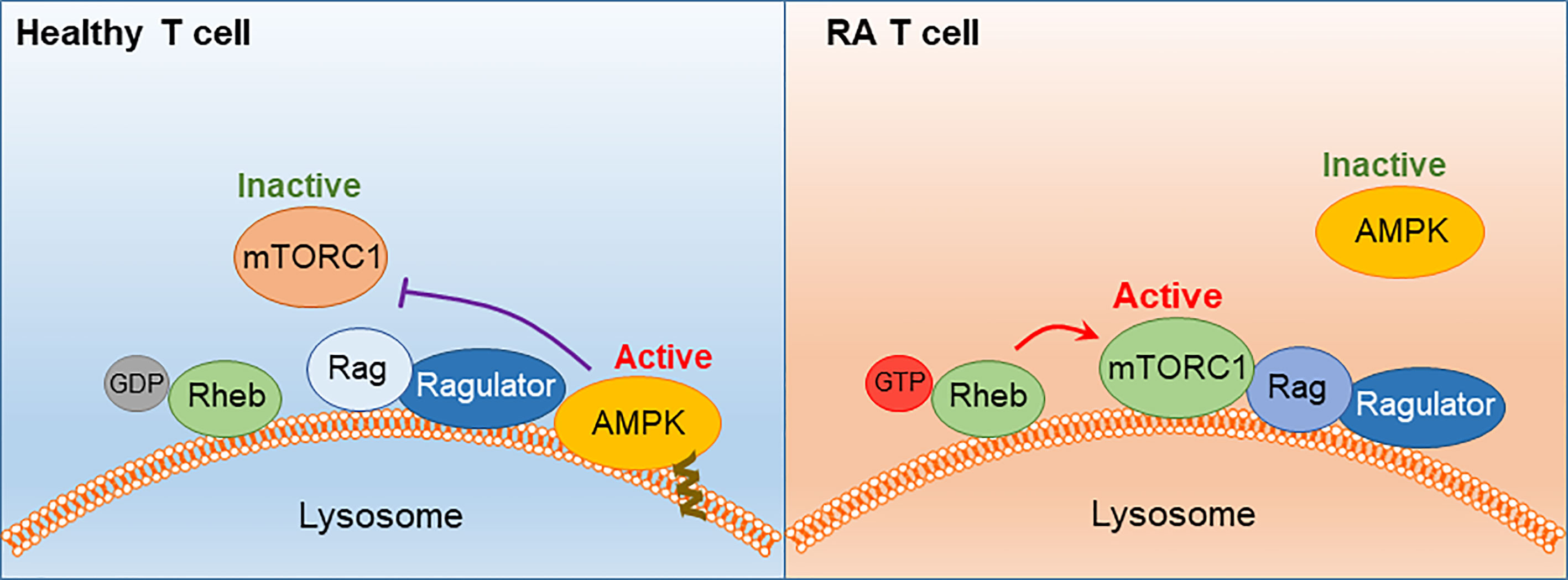
Figure 6 AMPK signaling failure and unopposed mTORC1 activation in RA T cells. In healthy T cells, AMPK senses the cell’s energy state by trafficking to the lysosomal surface, where it is anchored in the lysosomal membrane through a myristoylated tail. AMPK interacts with the Ragulator-Rag complex, resulting in dissociation and inactivation of mTORC1. In RA T cells, a defect in protein myristoylation leads to mistrafficking and cytosolic retention of AMPK. mTORC1 is retained at the lysosomal surface, where Rheb-GDP converts into Rheb-GTP and switches on the kinase activity of mTORC1. mTORC1 activation persists in RA T cells despite low ATP availability.
Based on the novel paradigm that metabolic programming determines the risk for inappropriate immune function, new therapeutic interventions can be developed. Ideally, a dietary approach can be designed to direct the flow of energy carriers and that of intracellular and extracellular metabolites. Alternatives include small molecules designed to finetune glycolysis, glutaminolysis and lipogenesis. Metabolic intervention may eventually be the most elegant way to manipulate the energy sensor mTORC1, which is critically involved in misleading T cells and turning them into multiplying and self-aggressive effector cells.
Author Contributions
All authors contributed to concept development. JQ, BW, GB, SG, JG and CW wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institutes of Health (R01 AR042527, R01 HL117913, R01 AI108906, R01 HL142068, and P01 HL129941 to CW and R01 AI108891, R01 AG045779, U19 AI057266, and R01 AI129191 to JG. Merit Review Award I01 BX001669 from the United States (U.S.) Department of Veterans Affairs to JG. JQ and BW were supported by the Encrantz Family Discovery Fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Weyand CM, Goronzy JJ. The immunology of rheumatoid arthritis. Nat Immunol (2020) 22:10–18. doi: 10.1038/s41590-020-00816-x
2. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 365(23):2205–19. doi: 10.1056/NEJMra1004965
3. Bustamante MF, Garcia-Carbonell R, Whisenant KD, Guma M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res Ther (2017) 19(1):110. doi: 10.1186/s13075-017-1303-3
4. McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet (2017) 389(10086):2328–37. doi: 10.1016/S0140-6736(17)31472-1
5. Carubbi F, Alunno A, Gerli R, Giacomelli R. Post-Translational Modifications of Proteins: Novel Insights in the Autoimmune Response in Rheumatoid Arthritis. Cells (2019) 8(7):657. doi: 10.3390/cells8070657
6. Conigliaro P, Chimenti MS, Triggianese P, Sunzini F, Novelli L, Perricone C, et al. Autoantibodies in inflammatory arthritis. Autoimmun Rev (2016) 15(7):673–83. doi: 10.1016/j.autrev.2016.03.003
7. van Herwijnen MJ, Wieten L, van der Zee R, van Kooten PJ, Wagenaar-Hilbers JP, Hoek A, et al. Regulatory T cells that recognize a ubiquitous stress-inducible self-antigen are long-lived suppressors of autoimmune arthritis. Proc Natl Acad Sci U S A (2012) 109(35):14134–9. doi: 10.1073/pnas.1206803109
8. Steiner G, Smolen J. Autoantibodies in rheumatoid arthritis and their clinical significance. Arthritis Res (2002) 4 Suppl 2:S1–5. doi: 10.1186/ar551
9. Perl A. Review: Metabolic Control of Immune System Activation in Rheumatic Diseases. Arthritis Rheumatol (2017) 69(12):2259–70. doi: 10.1002/art.40223
10. Weyand CM, Goronzy JJ. Immunometabolism in early and late stages of rheumatoid arthritis. Nat Rev Rheumatol (2017) 13(5):291–301. doi: 10.1038/nrrheum.2017.49
11. Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol (2014) 14(7):435–46. doi: 10.1038/nri3701
12. Weyand CM, Wu B, Goronzy JJ. The metabolic signature of T cells in rheumatoid arthritis. Curr Opin Rheumatol (2020) 32(2):159–67. doi: 10.1097/BOR.0000000000000683
13. Weyand CM, Yang Z, Goronzy JJ. T-cell aging in rheumatoid arthritis. Curr Opin Rheumatol (2014) 26(1):93–100. doi: 10.1097/BOR.0000000000000011
14. Henderson B, Bitensky L, Chayen J. Glycolytic activity in human synovial lining cells in rheumatoid arthritis. Ann Rheum Dis (1979) 38(1):63–7. doi: 10.1136/ard.38.1.63
15. Ciurtin C, Cojocaru VM, Miron IM, Preda F, Milicescu M, Bojinca M, et al. Correlation between different components of synovial fluid and pathogenesis of rheumatic diseases. Rom J Intern Med (2006) 44(2):171–81.
16. Bustamante MF, Oliveira PG, Garcia-Carbonell R, Croft AP, Smith JM, Serrano RL, et al. Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann Rheum Dis (2018) 77(11):1636–43. doi: 10.1136/annrheumdis-2018-213103
17. de Oliveira PG, Farinon M, Sanchez-Lopez E, Miyamoto S, Guma M. Fibroblast-Like Synoviocytes Glucose Metabolism as a Therapeutic Target in Rheumatoid Arthritis. Front Immunol (2019) 10:1743. doi: 10.3389/fimmu.2019.01743
18. Garcia-Carbonell R, Divakaruni AS, Lodi A, Vicente-Suarez I, Saha A, Cheroutre H, et al. Critical Role of Glucose Metabolism in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Arthritis Rheumatol (2016) 68(7):1614–26. doi: 10.1002/art.39608
19. Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature (2019) 570(7760):246–51. doi: 10.1038/s41586-019-1263-7
20. Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med (2013) 210(10):2119–34. doi: 10.1084/jem.20130252
21. Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med (2016) 8(331):331ra38. doi: 10.1126/scitranslmed.aad7151
22. Gelderman KA, Hultqvist M, Pizzolla A, Zhao M, Nandakumar KS, Mattsson R, et al. Macrophages suppress T cell responses and arthritis development in mice by producing reactive oxygen species. J Clin Invest (2007) 117(10):3020–8. doi: 10.1172/JCI31935
23. King MR, Ismail AS, Davis LS, Karp DR. Oxidative stress promotes polarization of human T cell differentiation toward a T helper 2 phenotype. J Immunol (2006) 176(5):2765–72. doi: 10.4049/jimmunol.176.5.2765
24. Zeisbrich M, Yanes RE, Zhang H, Watanabe R, Li Y, Brosig L, et al. Hypermetabolic macrophages in rheumatoid arthritis and coronary artery disease due to glycogen synthase kinase 3b inactivation. Ann Rheum Dis (2018) 77(7):1053–62. doi: 10.1136/annrheumdis-2017-212647
25. Veale DJ, Orr C, Fearon U. Cellular and molecular perspectives in rheumatoid arthritis. Semin Immunopathol (2017) 39(4):343–54. doi: 10.1007/s00281-017-0633-1
26. Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat Med (2020) 26(8):1295–306. doi: 10.1038/s41591-020-0939-8
27. Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The Metabolic Signature of Macrophage Responses. Front Immunol (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
28. Weyand CM, Goronzy JJ. Immunometabolism in the development of rheumatoid arthritis. Immunol Rev (2020) 294(1):177–87. doi: 10.1111/imr.12838
29. Wu B, Qiu J, Zhao TV, Wang Y, Maeda T, Goronzy IN, et al. Succinyl-CoA Ligase Deficiency in Pro-inflammatory and Tissue-Invasive T Cells. Cell Metab (2020) 32(6):967–980 e5. doi: 10.1016/j.cmet.2020.10.025
30. Li Y, Shen Y, Jin K, Wen Z, Cao W, Wu B, et al. The DNA Repair Nuclease MRE11A Functions as a Mitochondrial Protector and Prevents T Cell Pyroptosis and Tissue Inflammation. Cell Metab (2019) 30(3):477–492 e6. doi: 10.1016/j.cmet.2019.06.016
31. Wen Z, Jin K, Shen Y, Yang Z, Li Y, Wu B, et al. N-myristoyltransferase deficiency impairs activation of kinase AMPK and promotes synovial tissue inflammation. Nat Immunol (2019) 20(3):313–25. doi: 10.1038/s41590-018-0296-7
32. Ke R, Xu Q, Li C, Luo L, Huang D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol Int (2018) 42(4):384–92. doi: 10.1002/cbin.10915
33. Almeida L, Lochner M, Berod L, Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol (2016) 28(5):514–24. doi: 10.1016/j.smim.2016.10.009
34. O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol (2016) 16(9):553–65. doi: 10.1038/nri.2016.70
35. Cretenet G, Clerc I, Matias M, Loisel S, Craveiro M, Oburoglu L, et al. Cell surface Glut1 levels distinguish human CD4 and CD8 T lymphocyte subsets with distinct effector functions. Sci Rep (2016) 6:24129. doi: 10.1038/srep24129
36. Kavanagh Williamson M, Coombes N, Juszczak F, Athanasopoulos M, Khan MB, Eykyn TR, et al. Upregulation of Glucose Uptake and Hexokinase Activity of Primary Human CD4+ T Cells in Response to Infection with HIV-1. Viruses (2018) 10(3):114. doi: 10.3390/v10030114
37. Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol (2015) 6:1. doi: 10.3389/fimmu.2015.00001
38. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
39. Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, et al. mTORC1 and mTORC2 Kinase Signaling and Glucose Metabolism Drive Follicular Helper T Cell Differentiation. Immunity (2016) 45(3):540–54. doi: 10.1016/j.immuni.2016.08.017
40. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood (2010) 115(23):4742–9. doi: 10.1182/blood-2009-10-249540
41. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16(6):769–77. doi: 10.1016/S1074-7613(02)00323-0
42. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol (2008) 180(7):4476–86. doi: 10.4049/jimmunol.180.7.4476
43. Doughty CA, Bleiman BF, Wagner DJ, Dufort FJ, Mataraza JM, Roberts MF, et al. Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood (2006) 107(11):4458–65. doi: 10.1182/blood-2005-12-4788
44. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi: 10.1016/j.cmet.2014.05.004
45. Li W, Qu G, Choi SC, Cornaby C, Titov A, Kanda N, et al. Targeting T Cell Activation and Lupus Autoimmune Phenotypes by Inhibiting Glucose Transporters. Front Immunol (2019) 10:833. doi: 10.3389/fimmu.2019.00833
46. Simpfendorfer KR, Li W, Shih A, Wen H, Kothari HP, Einsidler EA, et al. Influence of genetic copy number variants of the human GLUT3 glucose transporter gene SLC2A3 on protein expression, glycolysis and rheumatoid arthritis risk: A genetic replication study. Mol Genet Metab Rep (2019) 19:100470. doi: 10.1016/j.ymgmr.2019.100470
47. Petrasca A, Phelan JJ, Ansboro S, Veale DJ, Fearon U, Fletcher JM. Targeting bioenergetics prevents CD4 T cell-mediated activation of synovial fibroblasts in rheumatoid arthritis. Rheumatology (Oxford) (2020) 59(10):2816–28. doi: 10.1093/rheumatology/kez682
48. Jurcovicova J, Stofkova A, Skurlova M, Baculikova M, Zorad S, Stancikova M. Alterations in adipocyte glucose transporter GLUT4 and circulating adiponectin and visfatin in rat adjuvant induced arthritis. Gen Physiol Biophys (2010) 29(1):79–84. doi: 10.4149/gpb_2010_01_79
49. Pucino V, Bombardieri M, Pitzalis C, Mauro C. Lactate at the crossroads of metabolism, inflammation, and autoimmunity. Eur J Immunol (2017) 47(1):14–21. doi: 10.1002/eji.201646477
50. Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D’Acquisto F, et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PloS Biol (2015) 13(7):e1002202. doi: 10.1371/journal.pbio.1002202
51. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
52. Gobelet C, Gerster JC. Synovial fluid lactate levels in septic and non-septic arthritides. Ann Rheum Dis (1984) 43(5):742–5. doi: 10.1136/ard.43.5.742
53. Pucino V, Cucchi D, Mauro C. Lactate transporters as therapeutic targets in cancer and inflammatory diseases. Expert Opin Ther Targets (2018) 22(9):735–43. doi: 10.1080/14728222.2018.1511706
54. Certo M, Marone G, de Paulis A, Mauro C, Pucino V. Lactate: Fueling the fire starter. Wiley Interdiscip Rev Syst Biol Med (2020) 12(3):e1474. doi: 10.1002/wsbm.1474
55. Weyand CM, Zeisbrich M, Goronzy JJ. Metabolic signatures of T-cells and macrophages in rheumatoid arthritis. Curr Opin Immunol (2017) 46:112–20. doi: 10.1016/j.coi.2017.04.010
56. Pucino V, Certo M, Bulusu V, Cucchi D, Goldmann K, Pontarini E, et al. Lactate Buildup at the Site of Chronic Inflammation Promotes Disease by Inducing CD4(+) T Cell Metabolic Rewiring. Cell Metab (2019) 30(6):1055–1074 e8. doi: 10.1016/j.cmet.2019.10.004
57. Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med (2016) 213(3):337–54. doi: 10.1084/jem.20150900
58. Ruth JH, Rottman JB, Katschke KJ Jr, Qin S, Wu L, LaRosa G, et al. Selective lymphocyte chemokine receptor expression in the rheumatoid joint. Arthritis Rheum (2001) 44(12):2750–60. doi: 10.1002/1529-0131(200112)44:12<2750::AID-ART462>3.0.CO;2-C
59. Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol (2019) 20(7):928–42. doi: 10.1038/s41590-019-0378-1
60. Stephenson W, Donlin LT, Butler A, Rozo C, Bracken B, Rashidfarrokhi A, et al. Single-cell RNA-seq of rheumatoid arthritis synovial tissue using low-cost microfluidic instrumentation. Nat Commun (2018) 9(1):791. doi: 10.1038/s41467-017-02659-x
61. Liu N, Luo J, Kuang D, Xu S, Duan Y, Xia Y, et al. Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2alpha-mediated tumor progression. J Clin Invest (2019) 129(2):631–46. doi: 10.1172/JCI123027
62. Pejovic M, Stankovic A, Mitrovic DR. Lactate dehydrogenase activity and its isoenzymes in serum and synovial fluid of patients with rheumatoid arthritis and osteoarthritis. J Rheumatol (1992) 19(4):529–33.
63. Lindy S, Uitto J, Turto H, Rokkanen P, Vainio K. Lactate dehydrogenase in the synovial tissue in rheumatoid arthritis: total activity and isoenzyme composition. Clin Chim Acta (1971) 31(1):19–23. doi: 10.1016/0009-8981(71)90357-3
64. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science (2016) 354(6311):481–4. doi: 10.1126/science.aaf6284
65. Souto-Carneiro MM, Klika KD, Abreu MT, Meyer AP, Saffrich R, Sandhoff R, et al. Effect of Increased Lactate Dehydrogenase A Activity and Aerobic Glycolysis on the Proinflammatory Profile of Autoimmune CD8+ T Cells in Rheumatoid Arthritis. Arthritis Rheumatol (2020) 72:2050–64. doi: 10.1002/art.41420
66. Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O’Fallon WM, et al. Lymphoid neogenesis in rheumatoid synovitis. J Immunol (2001) 167(2):1072–80. doi: 10.4049/jimmunol.167.2.1072
67. Thurlings RM, Wijbrandts CA, Mebius RE, Cantaert T, Dinant HJ, van der Pouw-Kraan TC, et al. Synovial lymphoid neogenesis does not define a specific clinical rheumatoid arthritis phenotype. Arthritis Rheum (2008) 58(6):1582–9. doi: 10.1002/art.23505
68. Bombardieri M, Lewis M, Pitzalis C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat Rev Rheumatol (2017) 13(3):141–54. doi: 10.1038/nrrheum.2016.217
69. Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol (2008) 9(11):1261–9. doi: 10.1038/ni.1657
70. Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med (2016) 213(9):1655–62. doi: 10.1084/jem.20160061
71. Saraiva AL, Veras FP, Peres RS, Talbot J, de Lima KA, Luiz JP, et al. Succinate receptor deficiency attenuates arthritis by reducing dendritic cell traffic and expansion of Th17 cells in the lymph nodes. FASEB J (2018) 32:fj201800285. doi: 10.1096/fj.201800285
72. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496(7444):238–42. doi: 10.1038/nature11986
73. McGarry T, Biniecka M, Veale DJ, Fearon U. Hypoxia, oxidative stress and inflammation. Free Radic Biol Med (2018) 125:15–24. doi: 10.1016/j.freeradbiomed.2018.03.042
74. Fearon U, Canavan M, Biniecka M, Veale DJ. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat Rev Rheumatol (2016) 12(7):385–97. doi: 10.1038/nrrheum.2016.69
75. Vega RB, Horton JL, Kelly DP. Maintaining ancient organelles: mitochondrial biogenesis and maturation. Circ Res (2015) 116(11):1820–34. doi: 10.1161/CIRCRESAHA.116.305420
76. Peansukmanee S, Vaughan-Thomas A, Carter SD, Clegg PD, Taylor S, Redmond C, et al. Effects of hypoxia on glucose transport in primary equine chondrocytes in vitro and evidence of reduced GLUT1 gene expression in pathologic cartilage in vivo. J Orthop Res (2009) 27(4):529–35. doi: 10.1002/jor.20772
77. Lee YZ, Guo HC, Zhao GH, Yang CW, Chang HY, Yang RB, et al. Tylophorine-based compounds are therapeutic in rheumatoid arthritis by targeting the caprin-1 ribonucleoprotein complex and inhibiting expression of associated c-Myc and HIF-1alpha. Pharmacol Res (2020) 152:104581. doi: 10.1016/j.phrs.2019.104581
78. Yang Z, Goronzy JJ, Weyand CM. The glycolytic enzyme PFKFB3/phosphofructokinase regulates autophagy. Autophagy (2014) 10(2):382–3. doi: 10.4161/auto.27345
79. Lee DC, Sohn HA, Park ZY, Oh S, Kang YK, Lee KM, et al. A lactate-induced response to hypoxia. Cell (2015) 161(3):595–609. doi: 10.1016/j.cell.2015.03.011
80. Makino Y, Nakamura H, Ikeda E, Ohnuma K, Yamauchi K, Yabe Y, et al. Hypoxia-inducible factor regulates survival of antigen receptor-driven T cells. J Immunol (2003) 171(12):6534–40. doi: 10.4049/jimmunol.171.12.6534
81. Gaber T, Haupl T, Sandig G, Tykwinska K, Fangradt M, Tschirschmann M, et al. Adaptation of human CD4+ T cells to pathophysiological hypoxia: a transcriptome analysis. J Rheumatol (2009) 36(12):2655–69. doi: 10.3899/jrheum.090255
82. Nakamura H, Makino Y, Okamoto K, Poellinger L, Ohnuma K, Morimoto C, et al. TCR engagement increases hypoxia-inducible factor-1 alpha protein synthesis via rapamycin-sensitive pathway under hypoxic conditions in human peripheral T cells. J Immunol (2005) 174(12):7592–9. doi: 10.4049/jimmunol.174.12.7592
83. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20(1):62–8. doi: 10.1038/nm.3432
84. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
85. Li G, Zhang Y, Qian Y, Zhang H, Guo S, Sunagawa M, et al. Interleukin-17A promotes rheumatoid arthritis synoviocytes migration and invasion under hypoxia by increasing MMP2 and MMP9 expression through NF-kappaB/HIF-1alpha pathway. Mol Immunol (2013) 53(3):227–36. doi: 10.1016/j.molimm.2012.08.018
86. Hu F, Shi L, Mu R, Zhu J, Li Y, Ma X, et al. Hypoxia-inducible factor-1alpha and interleukin 33 form a regulatory circuit to perpetuate the inflammation in rheumatoid arthritis. PloS One (2013) 8(8):e72650. doi: 10.1371/journal.pone.0072650
87. Gao W, McCormick J, Connolly M, Balogh E, Veale DJ, Fearon U. Hypoxia and STAT3 signalling interactions regulate pro-inflammatory pathways in rheumatoid arthritis. Ann Rheum Dis (2015) 74(6):1275–83. doi: 10.1136/annrheumdis-2013-204105
88. Curi R, Newsholme P, Marzuca-Nassr GN, Takahashi HK, Hirabara SM, Cruzat V, et al. Regulatory principles in metabolism-then and now. Biochem J (2016) 473(13):1845–57. doi: 10.1042/BCJ20160103
89. Cruzat VF, Pantaleao LC, Donato J Jr, de Bittencourt PI Jr, Tirapegui J. Oral supplementations with free and dipeptide forms of L-glutamine in endotoxemic mice: effects on muscle glutamine-glutathione axis and heat shock proteins. J Nutr Biochem (2014) 25(3):345–52. doi: 10.1016/j.jnutbio.2013.11.009
90. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16(10):619–34. doi: 10.1038/nrc.2016.71
91. Neermann J, Wagner R. Comparative analysis of glucose and glutamine metabolism in transformed mammalian cell lines, insect and primary liver cells. J Cell Physiol (1996) 166(1):152–69. doi: 10.1002/(SICI)1097-4652(199601)166:1<152::AID-JCP18>3.0.CO;2-H
92. Varnier M, Leese GP, Thompson J, Rennie MJ. Stimulatory effect of glutamine on glycogen accumulation in human skeletal muscle. Am J Physiol (1995) 269(2 Pt 1):E309–15. doi: 10.1152/ajpendo.1995.269.2.E309
93. Takahashi S, Saegusa J, Sendo S, Okano T, Akashi K, Irino Y, et al. Glutaminase 1 plays a key role in the cell growth of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther (2017) 19(1):76. doi: 10.1186/s13075-017-1283-3
94. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal (2015) 8(396):ra97. doi: 10.1126/scisignal.aab2610
95. Smith CM, Bryla J, Williamson JR. Regulation of mitochondrial alpha-ketoglutarate metabolism by product inhibition at alpha-ketoglutarate dehydrogenase. J Biol Chem (1974) 249(5):1497–505. doi: 10.1016/S0021-9258(19)42910-4
96. Safer B, Williamson JR. Mitochondrial-cytosolic interactions in perfused rat heart. Role of coupled transamination in repletion of citric acid cycle intermediates. J Biol Chem (1973) 248(7):2570–9. doi: 10.1016/S0021-9258(19)44146-X
97. Litwack G, Sears ML, Diamondstone TI. Intracellular distribution of tyrosine-alpha-ketoglutarate transaminase and 4-C-14-hydrocortisone activities during induction. J Biol Chem (1963) 238:302–5. doi: 10.1016/S0021-9258(19)83995-9
98. Hinoi E, Yoneda Y. Possible involvement of glutamatergic signaling machineries in pathophysiology of rheumatoid arthritis. J Pharmacol Sci (2011) 116(3):248–56. doi: 10.1254/jphs.11R03CR
99. Lindblad SS, Mydel P, Hellvard A, Jonsson IM, Bokarewa MI. The N-methyl-d-aspartic acid receptor antagonist memantine ameliorates and delays the development of arthritis by enhancing regulatory T cells. Neurosignals (2012) 20(2):61–71. doi: 10.1159/000329551
100. Boettger MK, Weber K, Gajda M, Brauer R, Schaible HG. Spinally applied ketamine or morphine attenuate peripheral inflammation and hyperalgesia in acute and chronic phases of experimental arthritis. Brain Behav Immun (2010) 24(3):474–85. doi: 10.1016/j.bbi.2009.12.002
101. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35(6):871–82. doi: 10.1016/j.immuni.2011.09.021
102. Mackenzie B, Erickson JD. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch (2004) 447(5):784–95. doi: 10.1007/s00424-003-1117-9
103. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol (2010) 185(2):1037–44. doi: 10.4049/jimmunol.0903586
104. Levring TB, Hansen AK, Nielsen BL, Kongsbak M, von Essen MR, Woetmann A, et al. Activated human CD4+ T cells express transporters for both cysteine and cystine. Sci Rep (2012) 2:266. doi: 10.1038/srep00266
105. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity (2014) 40(5):692–705. doi: 10.1016/j.immuni.2014.04.007
106. Fielding BA, Frayn KN. Lipid metabolism. Curr Opin Lipidol (2003) 14(4):389–91. doi: 10.1097/00041433-200308000-00009
107. van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol (2008) 9(2):112–24. doi: 10.1038/nrm2330
108. Calder PC. Long chain fatty acids and gene expression in inflammation and immunity. Curr Opin Clin Nutr Metab Care (2013) 16(4):425–33. doi: 10.1097/MCO.0b013e3283620616
109. Kendall AC, Pilkington SM, Massey KA, Sassano G, Rhodes LE, Nicolaou A. Distribution of bioactive lipid mediators in human skin. J Invest Dermatol (2015) 135(6):1510–20. doi: 10.1038/jid.2015.41
110. Lodhi IJ, Wei X, Semenkovich CF. Lipoexpediency: de novo lipogenesis as a metabolic signal transmitter. Trends Endocrinol Metab (2011) 22(1):1–8. doi: 10.1016/j.tem.2010.09.002
111. Diani M, Altomare G, Reali E. T cell responses in psoriasis and psoriatic arthritis. Autoimmun Rev (2015) 14(4):286–92. doi: 10.1016/j.autrev.2014.11.012
112. Goodson N, Marks J, Lunt M, Symmons D. Cardiovascular admissions and mortality in an inception cohort of patients with rheumatoid arthritis with onset in the 1980s and 1990s. Ann Rheum Dis (2005) 64(11):1595–601. doi: 10.1136/ard.2004.034777
113. Pietrzak A, Michalak-Stoma A, Chodorowska G, Szepietowski JC. Lipid disturbances in psoriasis: an update. Mediators Inflamm (2010) 2010:13. doi: 10.1155/2010/535612
114. Yuan J, Li LI, Wang Z, Song W, Zhang Z. Dyslipidemia in patients with systemic lupus erythematosus: Association with disease activity and B-type natriuretic peptide levels. BioMed Rep (2016) 4(1):68–72. doi: 10.3892/br.2015.544
115. Steiner G, Urowitz MB. Lipid profiles in patients with rheumatoid arthritis: mechanisms and the impact of treatment. Semin Arthritis Rheum (2009) 38(5):372–81. doi: 10.1016/j.semarthrit.2008.01.015
116. Ghazizadeh R, Tosa M, Ghazizadeh M. Clinical improvement in psoriasis with treatment of associated hyperlipidemia. Am J Med Sci (2011) 341(5):394–8. doi: 10.1097/MAJ.0b013e3181ff8eeb
117. Mercurio V, Lobasso A, Barbieri L, Parrella P, Ciervo D, Liccardo B, et al. Inflammatory, Serological and Vascular Determinants of Cardiovascular Disease in Systemic Lupus Erythematosus Patients. Int J Mol Sci (2019) 20(9):2154. doi: 10.3390/ijms20092154
118. Yu HH, Chen PC, Yang YH, Wang LC, Lee JH, Lin YT, et al. Statin reduces mortality and morbidity in systemic lupus erythematosus patients with hyperlipidemia: A nationwide population-based cohort study. Atherosclerosis (2015) 243(1):11–8. doi: 10.1016/j.atherosclerosis.2015.08.030
119. Giera M, Ioan-Facsinay A, Toes R, Gao F, Dalli J, Deelder AM, et al. Lipid and lipid mediator profiling of human synovial fluid in rheumatoid arthritis patients by means of LC-MS/MS. Biochim Biophys Acta (2012) 1821(11):1415–24. doi: 10.1016/j.bbalip.2012.07.011
120. Bole GG. Synovial fluid lipids in normal individuals and patients with rheumatoid arthritis. Arthritis Rheum (1962) 5:589–601. doi: 10.1002/art.1780050606
121. Viikari J, Jalava S, Terho T. Synovial fluid lipids in rheumatoid arthritis. Scand J Rheumatol (1980) 9(3):164–6. doi: 10.3109/03009748009098150
122. Liao KP, Playford MP, Frits M, Coblyn JS, Iannaccone C, Weinblatt ME, et al. The association between reduction in inflammation and changes in lipoprotein levels and HDL cholesterol efflux capacity in rheumatoid arthritis. J Am Heart Assoc (2015) 4(2):e001588. doi: 10.1161/JAHA.114.001588
123. Myasoedova E, Crowson CS, Kremers HM, Fitz-Gibbon PD, Therneau TM, Gabriel SE. Total cholesterol and LDL levels decrease before rheumatoid arthritis. Ann Rheum Dis (2010) 69(7):1310–4. doi: 10.1136/ard.2009.122374
124. Perucha E, Melchiotti R, Bibby JA, Wu W, Frederiksen KS, Roberts CA, et al. The cholesterol biosynthesis pathway regulates IL-10 expression in human Th1 cells. Nat Commun (2019) 10(1):498. doi: 10.1038/s41467-019-08332-9
125. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol (2013) 14(5):489–99. doi: 10.1038/ni.2570
126. O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity (2014) 41(1):75–88. doi: 10.1016/j.immuni.2014.06.005
127. Ibitokou SA, Dillon BE, Sinha M, Szczesny B, Delgadillo A, Reda Abdelrahman D, et al. Early Inhibition of Fatty Acid Synthesis Reduces Generation of Memory Precursor Effector T Cells in Chronic Infection. J Immunol (2018) 200(2):643–56. doi: 10.4049/jimmunol.1602110
128. Shen Y, Wen Z, Li Y, Matteson EL, Hong J, Goronzy JJ, et al. Metabolic control of the scaffold protein TKS5 in tissue-invasive, proinflammatory T cells. Nat Immunol (2017) 18(9):1025–34. doi: 10.1038/ni.3808
129. Shi L, Tu BP. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol (2015) 33:125–31. doi: 10.1016/j.ceb.2015.02.003
130. Lv X, Wang F, Zhou P, Ye L, Xie W, Xu H, et al. Dual regulation of cytoplasmic and mitochondrial acetyl-CoA utilization for improved isoprene production in Saccharomyces cerevisiae. Nat Commun (2016) 7:12851. doi: 10.1038/ncomms12851
131. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38(4):633–43. doi: 10.1016/j.immuni.2013.04.005
132. Li Y, Goronzy JJ, Weyand CM. DNA damage, metabolism and aging in pro-inflammatory T cells: Rheumatoid arthritis as a model system. Exp Gerontol (2018) 105:118–27. doi: 10.1016/j.exger.2017.10.027
133. Schnoor M, Stradal TE, Rottner K. Cortactin: Cell Functions of A Multifaceted Actin-Binding Protein. Trends Cell Biol (2018) 28(2):79–98. doi: 10.1016/j.tcb.2017.10.009
134. Beaty BT, Condeelis J. Digging a little deeper: the stages of invadopodium formation and maturation. Eur J Cell Biol (2014) 93(10-12):438–44. doi: 10.1016/j.ejcb.2014.07.003
135. Montesinos MC, Desai A, Delano D, Chen JF, Fink JS, Jacobson MA, et al. Adenosine A2A or A3 receptors are required for inhibition of inflammation by methotrexate and its analog MX-68. Arthritis Rheum (2003) 48(1):240–7. doi: 10.1002/art.10712
136. Zhou J, Chen J, Hu C, Xie Z, Li H, Wei S, et al. Exploration of the serum metabolite signature in patients with rheumatoid arthritis using gas chromatography-mass spectrometry. J Pharm BioMed Anal (2016) 127:60–7. doi: 10.1016/j.jpba.2016.02.004
137. Fairbanks LD, Ruckemann K, Qiu Y, Hawrylowicz CM, Richards DF, Swaminathan R, et al. Methotrexate inhibits the first committed step of purine biosynthesis in mitogen-stimulated human T-lymphocytes: a metabolic basis for efficacy in rheumatoid arthritis? Biochem J (1999) 342( Pt 1):143–52. doi: 10.1042/bj3420143
138. Cronstein BN, Naime D, Ostad E. The antiinflammatory effects of methotrexate are mediated by adenosine. Adv Exp Med Biol (1994) 370:411–6. doi: 10.1007/978-1-4615-2584-4_89
139. Gonzalez-Gay MA, De Matias JM, Gonzalez-Juanatey C, Garcia-Porrua C, Sanchez-Andrade A, Martin J, et al. Anti-tumor necrosis factor-alpha blockade improves insulin resistance in patients with rheumatoid arthritis. Clin Exp Rheumatol (2006) 24(1):83–6.
140. Gonzalez-Gay MA, Gonzalez-Juanatey C, Vazquez-Rodriguez TR, Miranda-Filloy JA, Llorca J. Insulin resistance in rheumatoid arthritis: the impact of the anti-TNF-alpha therapy. Ann N Y Acad Sci (2010) 1193:153–9. doi: 10.1111/j.1749-6632.2009.05287.x
141. Seriolo B, Paolino S, Ferrone C, Cutolo M. Impact of long-term anti-TNF-alpha treatment on insulin resistance in patients with rheumatoid arthritis. Clin Exp Rheumatol (2008) 26(1):159–60; author reply 160.
142. Tam LS, Tomlinson B, Chu TT, Li TK, Li EK. Impact of TNF inhibition on insulin resistance and lipids levels in patients with rheumatoid arthritis. Clin Rheumatol (2007) 26(9):1495–8. doi: 10.1007/s10067-007-0539-8
143. Spanakis E, Sidiropoulos P, Papadakis J, Ganotakis E, Katsikas G, Karvounaris S, et al. Modest but sustained increase of serum high density lipoprotein cholesterol levels in patients with inflammatory arthritides treated with infliximab. J Rheumatol (2006) 33(12):2440–6.
144. Cauza E, Cauza K, Hanusch-Enserer U, Etemad M, Dunky A, Kostner K. Intravenous anti TNF-alpha antibody therapy leads to elevated triglyceride and reduced HDL-cholesterol levels in patients with rheumatoid and psoriatic arthritis. Wien Klin Wochenschr (2002) 114(23-24):1004–7.
145. Stinco G, Piccirillo F, Patrone P. Hypertriglyceridaemia during treatment with adalimumab in psoriatic arthritis. Br J Dermatol (2007) 157(6):1273–4. doi: 10.1111/j.1365-2133.2007.08188.x
146. Sweeney SR, Kavanaugh A, Lodi A, Wang B, Boyle D, Tiziani S, et al. Metabolomic profiling predicts outcome of rituximab therapy in rheumatoid arthritis. RMD Open (2016) 2(2):e000289. doi: 10.1136/rmdopen-2016-000289
147. Kerr G, Aujero M, Richards J, Sayles H, Davis L, Cannon G, et al. Associations of hydroxychloroquine use with lipid profiles in rheumatoid arthritis: pharmacologic implications. Arthritis Care Res (Hoboken) (2014) 66(11):1619–26. doi: 10.1002/acr.22341
148. Penn SK, Kao AH, Schott LL, Elliott JR, Toledo FG, Kuller L, et al. Hydroxychloroquine and glycemia in women with rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol (2010) 37(6):1136–42. doi: 10.3899/jrheum.090994
149. Restrepo JF, Del Rincon I, Molina E, Battafarano DF, Escalante A. Use of Hydroxychloroquine Is Associated With Improved Lipid Profile in Rheumatoid Arthritis Patients. J Clin Rheumatol (2017) 23(3):144–8. doi: 10.1097/RHU.0000000000000502
150. Solomon DH, Garg R, Lu B, Todd DJ, Mercer E, Norton T, et al. Effect of hydroxychloroquine on insulin sensitivity and lipid parameters in rheumatoid arthritis patients without diabetes mellitus: a randomized, blinded crossover trial. Arthritis Care Res (Hoboken) (2014) 66(8):1246–51. doi: 10.1002/acr.22285
151. Tournadre A, Pereira B, Dutheil F, Giraud C, Courteix D, Sapin V, et al. Changes in body composition and metabolic profile during interleukin 6 inhibition in rheumatoid arthritis. J Cachexia Sarcopenia Muscle (2017) 8(4):639–46. doi: 10.1002/jcsm.12189
152. Ruiz-Limon P, Ortega R, Arias de la Rosa I, Abalos-Aguilera MDC, Perez-Sanchez C, Jimenez-Gomez Y, et al. Tocilizumab improves the proatherothrombotic profile of rheumatoid arthritis patients modulating endothelial dysfunction, NETosis, and inflammation. Transl Res (2017) 183:87–103. doi: 10.1016/j.trsl.2016.12.003
153. Fragoso YD, Brooks JB. Leflunomide and teriflunomide: altering the metabolism of pyrimidines for the treatment of autoimmune diseases. Expert Rev Clin Pharmacol (2015) 8(3):315–20. doi: 10.1586/17512433.2015.1019343
154. Costa NT, Scavuzzi BM, Iriyoda TMV, Lozovoy MAB, Alfieri DF, de Medeiros FA, et al. Metabolic syndrome and the decreased levels of uric acid by leflunomide favor redox imbalance in patients with rheumatoid arthritis. Clin Exp Med (2018) 18(3):363–72. doi: 10.1007/s10238-018-0500-y
155. Cronstein BN, Aune TM. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat Rev Rheumatol (2020) 16(3):145–54. doi: 10.1038/s41584-020-0373-9
156. Shea B, Swinden MV, Tanjong Ghogomu E, Ortiz Z, Katchamart W, Rader T, et al. Folic acid and folinic acid for reducing side effects in patients receiving methotrexate for rheumatoid arthritis. Cochrane Database Syst Rev (2013) 5):CD000951. doi: 10.1002/14651858.CD000951.pub2
157. Lina C, Conghua W, Nan L, Ping Z. Combined treatment of etanercept and MTX reverses Th1/Th2, Th17/Treg imbalance in patients with rheumatoid arthritis. J Clin Immunol (2011) 31(4):596–605. doi: 10.1007/s10875-011-9542-6
158. Carrillo-de Sauvage MA, Maatouk L, Arnoux I, Pasco M, Sanz Diez A, Delahaye M, et al. Potent and multiple regulatory actions of microglial glucocorticoid receptors during CNS inflammation. Cell Death Differ (2013) 20(11):1546–57. doi: 10.1038/cdd.2013.108
159. Takata M, Nakagomi T, Kashiwamura S, Nakano-Doi A, Saino O, Nakagomi N, et al. Glucocorticoid-induced TNF receptor-triggered T cells are key modulators for survival/death of neural stem/progenitor cells induced by ischemic stroke. Cell Death Differ (2012) 19(5):756–67. doi: 10.1038/cdd.2011.145
160. Weinblatt ME, van Riel PL. Targeted therapies: summary clinical trials working group. Ann Rheum Dis (2006) 65 Suppl 3:iii89. doi: 10.1136/ard.2006.060939
161. Biniecka M, Canavan M, McGarry T, Gao W, McCormick J, Cregan S, et al. Dysregulated bioenergetics: a key regulator of joint inflammation. Ann Rheum Dis (2016) 75(12):2192–200. doi: 10.1136/annrheumdis-2015-208476
162. McGarry T, Orr C, Wade S, Biniecka M, Wade S, Gallagher L, et al. JAK/STAT Blockade Alters Synovial Bioenergetics, Mitochondrial Function, and Proinflammatory Mediators in Rheumatoid Arthritis. Arthritis Rheumatol (2018) 70(12):1959–70. doi: 10.1002/art.40569
163. Kornberg MD. The immunologic Warburg effect: Evidence and therapeutic opportunities in autoimmunity. Wiley Interdiscip Rev Syst Biol Med (2020) 12(5):e1486. doi: 10.1002/wsbm.1486
164. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
Keywords: T cell, metabolism, autoimmunity, rheumatoid arthritis, mitochondria, glycolysis, glutaminolysis, fatty acid
Citation: Qiu J, Wu B, Goodman SB, Berry GJ, Goronzy JJ and Weyand CM (2021) Metabolic Control of Autoimmunity and Tissue Inflammation in Rheumatoid Arthritis. Front. Immunol. 12:652771. doi: 10.3389/fimmu.2021.652771
Received: 13 January 2021; Accepted: 15 March 2021;
Published: 02 April 2021.
Edited by:
Monica Guma, University of California, San Diego, United StatesReviewed by:
Ursula Fearon, Trinity College Dublin, IrelandGuenter Steiner, Medical University of Vienna, Austria
Copyright © 2021 Qiu, Wu, Goodman, Berry, Goronzy and Weyand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cornelia M. Weyand, Y3dleWFuZEBzdGFuZm9yZC5lZHU=