 Alexandra Y. Kreins
Alexandra Y. Kreins Paola Bonfanti
Paola Bonfanti E. Graham Davies
E. Graham Davies- 1Great Ormond Street Institute of Child Health, University College London, London, United Kingdom
- 2Department of Immunology, Great Ormond Street Hospital for Children NHS Foundation Trust, London, United Kingdom
- 3Epithelial Stem Cell Biology & Regenerative Medicine Laboratory, The Francis Crick Institute, London, United Kingdom
- 4Institute of Immunity & Transplantation, University College London, London, United Kingdom
Inborn errors of thymic stromal cell development and function lead to impaired T-cell development resulting in a susceptibility to opportunistic infections and autoimmunity. In their most severe form, congenital athymia, these disorders are life-threatening if left untreated. Athymia is rare and is typically associated with complete DiGeorge syndrome, which has multiple genetic and environmental etiologies. It is also found in rare cases of T-cell lymphopenia due to Nude SCID and Otofaciocervical Syndrome type 2, or in the context of genetically undefined defects. This group of disorders cannot be corrected by hematopoietic stem cell transplantation, but upon timely recognition as thymic defects, can successfully be treated by thymus transplantation using cultured postnatal thymic tissue with the generation of naïve T-cells showing a diverse repertoire. Mortality after this treatment usually occurs before immune reconstitution and is mainly associated with infections most often acquired pre-transplantation. In this review, we will discuss the current approaches to the diagnosis and management of thymic stromal cell defects, in particular those resulting in athymia. We will discuss the impact of the expanding implementation of newborn screening for T-cell lymphopenia, in combination with next generation sequencing, as well as the role of novel diagnostic tools distinguishing between hematopoietic and thymic stromal cell defects in facilitating the early consideration for thymus transplantation of an increasing number of patients and disorders. Immune reconstitution after the current treatment is usually incomplete with relatively common inflammatory and autoimmune complications, emphasizing the importance for improving strategies for thymus replacement therapy by optimizing the current use of postnatal thymus tissue and developing new approaches using engineered thymus tissue.
Introduction
Uniquely among hematopoietic stem cell derived lineages, T-cells require a second site for their development, namely the thymus. Thus, primary immunodeficiency disorders (PIDs) leading to T-cell deficiency may be a result of either hematopoietic lineage defects or defects of thymus stromal development or function. The latter includes athymia associated with DiGeorge syndrome (DGS), known as complete DGS (cDGS), and other monogenic defects resulting in failure of thymic stromal development. While hematopoietic stem cell transplantation (HSCT) from a matched family donor has been shown to promote survival in around two-thirds of cases of cDGS, an otherwise lethal condition, the quality of immune reconstitution was poor with low numbers of naïve T-cells and a restricted T-cell repertoire (1, 2). The survival after HSCT using alternative donors was very poor (1). It is logical that corrective therapies for these children should instead involve providing them with thymic tissue to facilitate the development of their normal hematopoietic precursor cells into mature T-cells. HSCT and thymus cell replacement treatments entered the clinic at very different times for both biological and historical reasons. The immunological function of the thymus was only understood four years after the first HSCT had already been reported in patients (3–5). HSCT research started as early as 1949 as a result of the recognition of the effect of radiations on the bone marrow. In addition, bone marrow is a liquid tissue and therefore relatively easy to harvest and to re-infuse with or without ex vivo treatment. Instead, the thymus is a primary lympho-epithelial organ, located in the thorax where access is difficult. The relatively late discovery in 1963 of the thymus as the site of T-cell development (5) resulted in a significant delay in our understanding of the thymic stroma. Furthermore, the complexity of its structure requires an interdisciplinary approach for consideration as to how to reconstitute its function. Transplantation of fetal thymus tissue to correct cDGS was first attempted more than fifty years ago but met with limited success (6–8). More recently the use of postnatal thymus tissue obtained from infant donors undergoing cardiac surgery for congenital heart disease was pioneered by Louise Markert at Duke University (9). Since that time, in more than one hundred cases treated at two centers worldwide, Duke University Hospital in the United States and Great Ormond Street Hospital in the United Kingdom, this has been shown to offer a better rate of survival and quality of immune reconstitution (10–12). This article will review the defects of thymus development focusing on those leading to athymia, the current approach to replacing thymus function through allografting with postnatal thymic tissue, and the future prospects for engineered thymus preparations with the ultimate aim of using a product of autologous origin.
Thymus Development and Function
Thymus organogenesis has extensively been investigated in vertebrates, in particular in the mouse and chick. Early in embryogenesis a series of pharyngeal arches and pouches, known collectively as the pharyngeal apparatus, can be recognized. This includes inner endodermal and outer ectodermal regions associated with mesodermal tissue and neural crest cells (13, 14). From this structure, the thymus and the, developmentally closely allied, parathyroid glands develop, specifically in the region of the ventral part of the 3rd pharyngeal pouch (15–17). This early development is under the control of multiple transcription factors including TBX1, HOXA3, PAX1, PAX9, EYA1, SIX1 and SIX4 (18, 19). Following this, FOXN1 becomes the predominant and enduring regulatory factor promoting thymus development, growth and maintenance of the thymus throughout life (20–22). These mechanisms seem largely conserved between mouse and human (23). From an early embryonic stage (7-8 weeks in humans) hematopoietic cells start to arrive in the thymus (23). The early thymus-seeding precursors originate initially from the aorta-gonad-mesonephros region (24–26) and later from the fetal liver (27) and the bone marrow. These thymus-seeding cells are necessary for the ongoing development of a functional thymus through a process of lymphostromal crosstalk (28, 29). The cellular interactions involved in embryonic thymic development have recently been studied in further detail using single-cell RNA sequencing in both mice (30) and humans (26, 31). The early thymic precursors maintain multipotency before differentiating down a T-cell committed pathway following interaction with NOTCH ligands (32) and other environmental signals.
The cellular composition of the thymus comprises hematopoietic cells and thymus stromal cells (31). The former are predominantly T-cells at various stages of development (thymocytes) but importantly, in addition to B-cells and macrophages, also include different types of dendritic cells (DC), some arising de novo in the thymus and others having migrated in (33). The antigen presenting capability of these DCs is very important in the process of negative selection as part of thymopoiesis (see below). Furthermore self-renewing lymphoid precursor cells have been identified in immunodeficient mice after transplantation of human thymus (34), suggesting the presence of lymphoid progenitors capable of long-term survival and T-cell development in postnatal human thymus. The thymus stroma comprises thymic epithelial cells (TEC), mesenchymal cells, fibroblastic cells and vascular endothelial cells. TECs are the hallmark cells of the thymus being classed broadly into two groups with distinct functions, cortical (cTEC) and medullary (mTEC). The cellular complexity of the thymic stroma with higher TEC heterogeneity is becoming increasingly evident (31, 35, 36). In the mouse, a common bipotent precursor for cTEC and mTEC compartments has long been described (37–39). Recent characterization of expanding human mTECs and cTECs, which lose their compartmental signature, is in keeping with this (35). Finally, the whole structure is supported by a scaffold of extracellular matrix (ECM), comprising, among other things, collagen and fibronectin, which facilitates thymocyte migration and cellular interaction (40–42). Microscopically, the thymus comprises three zones with distinct functions: subcapsular, cortical and medullary.
The process of thymopoiesis, the development of mature T-cells in the thymus, depends on serial steps taking place in distinct thymic microenvironments. This is well understood in the mouse and has been described elsewhere (29). Briefly, hematopoietic precursor cells leave the bone marrow and enter the thymus through the cortico-medullary junction and then pass into the cortex where they become committed to the T-cell lineage. The different stages of their development are characterized by the expression of cell surface molecules, including CD4 and CD8. The first thymic stage is the immature CD4-CD8- double negative (DN) stage. Thymocytes then start to rearrange their T-cell receptors (TCRs), generating double positive (DP) thymocytes, expressing both CD4 and CD8. As they migrate from the cortex to the medulla, these DP thymocytes undergo positive and negative selection, respectively resulting in the elimination of non-reactive and self-reactive cells. Subsequently, the cells will egress from the thymus as functional single positive (SP) CD4+ or CD8+ recent thymic emigrants (RTEs) migrating to the periphery. While there are differences in T-cell development between humans and mice (43–46), the murine model continues to offer fundamental insight into this process and some of these developmental stages have been reproduced during the in vitro differentiation of human hematopoietic stem cells (HSCs) in co-culture systems with Notch ligand-expressing murine stromal cells (47–49). Additionally, novel molecular tools, such as high-throughput RNA sequencing, efficient high-precision genome editing and the possibility to use HSCs and induced pluripotent stem cells (iPSCs) from certain PID patients, are further facilitating the study of human T-cell development in new ex vivo (50–52) and in vivo models (53, 54).
It is lymphostromal crosstalk consisting of MHC-TCR interactions between antigen-presenting cells (APCs) and developing thymocytes (Figure 1A), that supports thymopoiesis with positive and negative selection resulting in the generation of self-tolerant T-cells with a diverse TCR repertoire. During positive selection thymocytes undergo proliferation driven by their interaction with self-peptides expressed in association with major histocompatibility complex (MHC) molecules on cTECs and possibly other cells including thymocytes (55, 56) and fibroblasts (57, 58), though the physiological role of fibroblasts in this process has been disputed by others (59). Evidence suggests that the specificity of the MHC driving this is not critical to this process (60). The positively selected and expanded DP T-cells then undergo negative selection. During a first wave of negative selection, autoreactive DP thymocytes are eliminated in the cortex upon association with MHC-bound self-antigens presented by DCs (61, 62). Remaining thymocytes then mature into SP cells and migrate into the medullary zone where T-cell receptor rearrangement is completed. In the medulla, SP T-cells undergo a second, predominant wave of negative selection. Tissue restricted (self) antigens (TRAs) promiscuously generated in mTECs under the influence of the autoimmune regulator (AIRE) are presented to these T-cells in the context of self MHC expressed on the mTECs and DCs or on other cell types. T-cells with receptors recognizing self TRAs will either be deleted or develop down the pathway of regulatory T-cells (Tregs). Recently, a role for medullary fibroblasts has also been reported in central immune tolerance, specifically through lymphotoxin β-mediated signaling (63). Altogether, these processes lead to T-cell central tolerance to self in order to avoid autoimmunity.
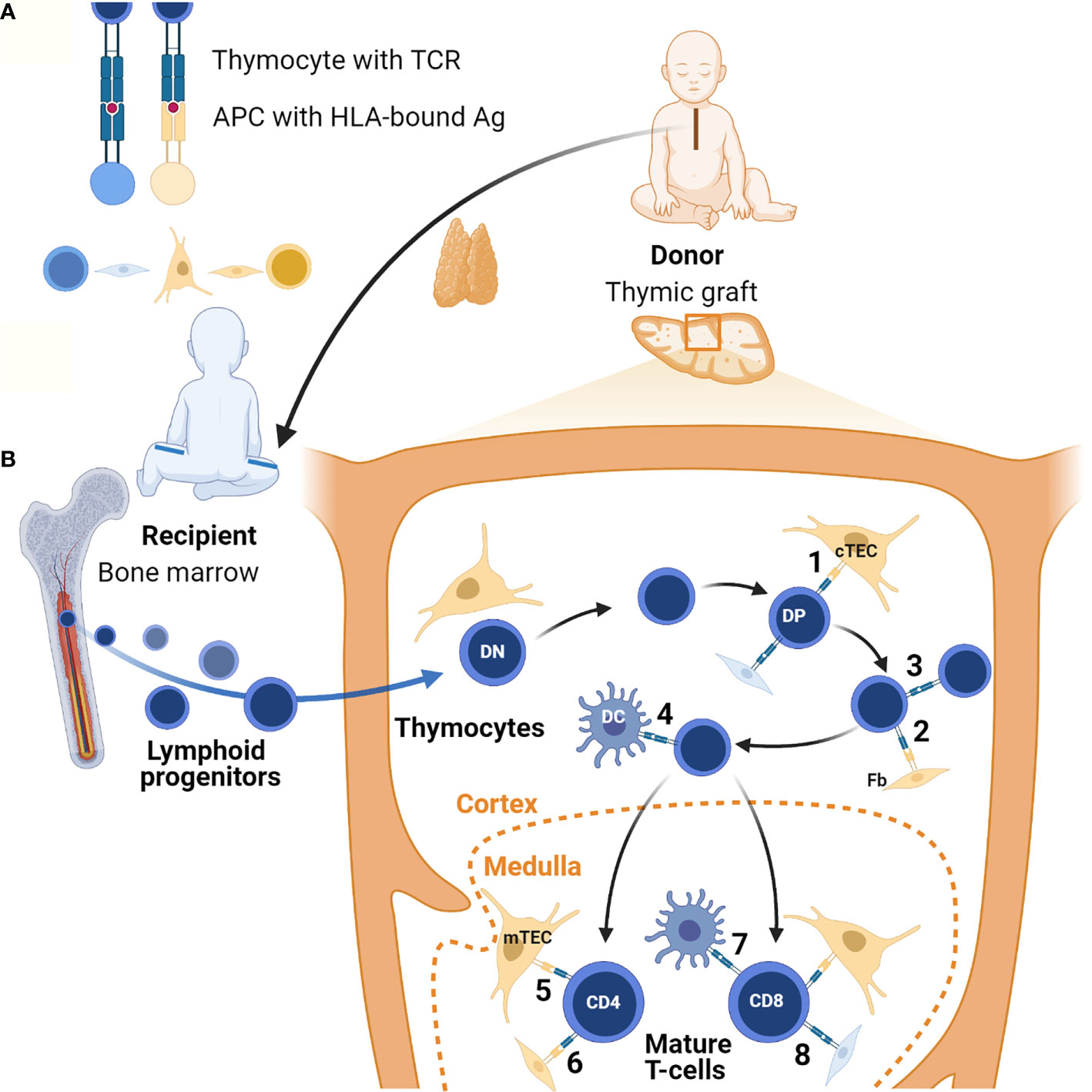
Figure 1 Lymphostromal crosstalk in thymic grafts. (A) Developing thymocytes communicate with antigen (Ag)-presenting cells (APC) through HLA-TCR interactions. APCs include both thymic stromal cells and hematopoietic cells. In thymic grafts these APCs can be host-derived (in blue) or donor-derived (in orange). The thymus tissue donor and the athymic patient are not HLA-matched. (B) After thymus transplantation, lymphoid progenitors migrate from the recipient’s bone marrow into the HLA-mismatched thymic graft. In the thymic cortex developing thymocytes undergo positive selection of competent T-cells through low-affinity engagement of their TCR with HLA-bound antigens. In the case of serendipitous, partial HLA-matching between donor and recipient, traditional antigen-presentation by cTECs may occur (1). Donor-derived fibroblasts (Fb) possibly also contribute to this (2). In theory, host-derived APCs of hematopoietic origin in the graft may also contribute to positive selection, for example through direct thymocyte-thymocyte interactions (3). Positively selected DP thymocytes then undergo negative selection to eliminate autoreactive T-cells. In the thymic graft this may also start in the cortex thanks to direct HLA-TCR mediated interactions with DCs of host origin (4). In normal thymus tissue generation of self-tolerance predominantly takes place in the medulla. If there is partial HLA-matching between the donor and the recipient, donor-derived mTECs and possibly fibroblasts can contribute to this in thymic grafts (5 and 6). Host-derived hematopoietic cells, in particular DCs, may also play a role (7). Additionally, it is possible that upon transplantation chimeric thymic stroma develops with stromal cells of host origin, both in the cortex and the medulla (8). Figure created with BioRender.com.
Abnormalities of Thymic Stromal Development and Function
Failure of development of the thymus can result from defects in the above described processes. The resulting immunodeficiency may vary from none through a mild infection susceptibility and predisposition to autoimmune disease to a severe combined immunodeficiency (SCID) picture with complete athymia and a susceptibility to life threatening opportunistic infections (64). There are a number of different causes of thymic hypoplasia and aplasia, including DGS and other monogenic defects, which are described in the section below and summarized in Table 1. Athymic patients have a profound T-cell lymphopenia and require life-saving corrective therapy.
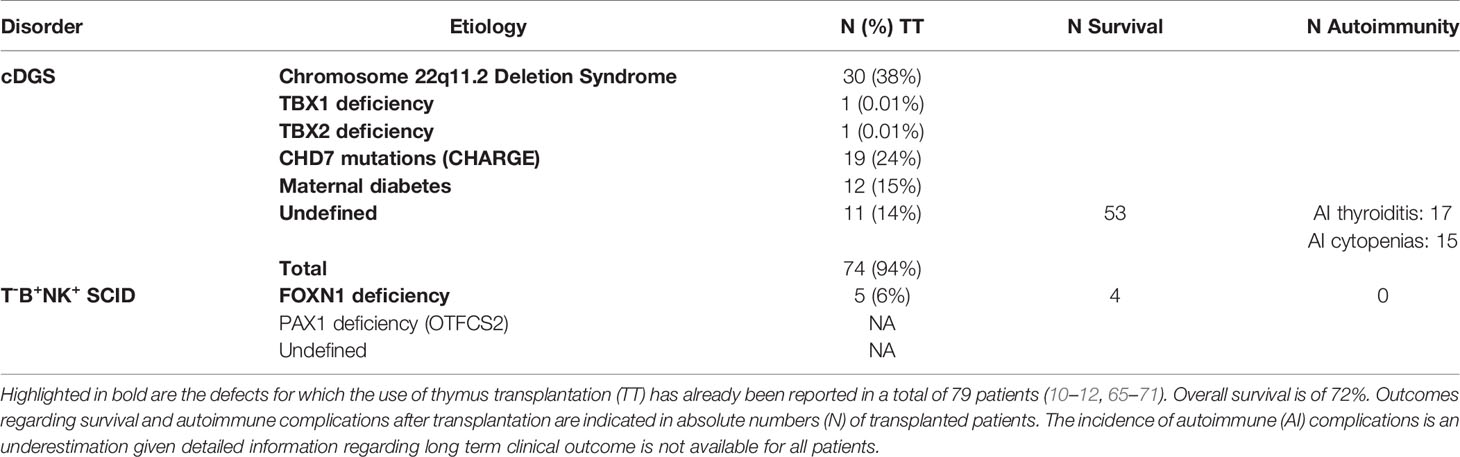
Table 1 Disorders associated with congenital athymia.
To date few genetic defects resulting in malfunction rather than failure of development of the thymus stroma have been identified. Deficiency of AIRE predisposes to multiple manifestations. Biallellic deleterious mutations in AIRE result in autoimmune polyglandular syndrome type 1 (APS1) also known by the acronym APECED (autoimmune polyendocrinopathy, candidiasis, ectodermal dysplasia) (72, 73). Hypoparathyroidism and adrenal insufficiency are the most common endocrinopathies, but APECED’s clinical picture is broader than previously recognized, including significant non-endocrine manifestations (74, 75). It has also been found that single allele AIRE mutations with a dominant-negative effect can lead to a milder predisposition to autoimmune disease (76, 77). Secondary AIRE dysfunction resulting in impaired central tolerance also plays a role in other PIDs, such as in patients with hypomorphic RAG mutations and PRKDC mutations (78–81). Similarly, impaired AIRE signaling has been proposed in early onset common variable immunodeficiency due to heterozygous NFKB2 mutations, for which autoimmune features, including alopecia, hypopituitarism and serum autoantibodies, have been reported alongside a susceptibility to infections and hypogammaglobulinemia (82–85). Most of these patients have mutations in a domain of the full length NFKB2, called p100 degron (84), preventing its phosphorylation and subsequent degradation upon activation of the non-canonical NF-κB pathway (84, 86). A recent murine study showed that mutations in p100 degron underlie autoimmunity through AIRE-independent defects of thymic tolerance, which are largely mediated by changes in the thymic epithelium, including thymic medullary hypoplasia (87).
There may also be thymic stromal malfunction in patients with TTC7A defects with severe enteropathy and immune deficiency. TTC7A regulates the dynamics of the actin cytoskeleton in lymphocytes. It is also abundantly expressed on TECs (88, 89). The impact of TTC7A mutations on TECs has not yet been investigated, but post-mortem analysis of thymus tissue from one patient showed a disrupted thymic architecture with dysplastic changes and blurred cortico-medullary demarcation, yet preserved Hassall’s bodies. Several of these patients underwent HSCT (89, 90). The development of naïve T-cells after HSCT in one of the patients reported suggests that thymic function could be restored upon repopulation of the thymus with HSCT donor-derived thymocytes (89). This would be consistent with the notion that thymic dysfunction in these patients is mainly secondary to lymphodepletion rather than a primary TEC defect.
Congenital Athymia in the Context of cDGS
Most commonly, impaired thymic development is associated with DGS. A triad of congenital heart disease (CHD), hypoparathyroidism and thymic hypoplasia are the hallmark features of the clinical phenotype of DGS patients (91, 92). DGS has been described in a heterogeneous group of disorders, both genetic and environmental, with variable clinical penetrance. A minority of DGS patients are diagnosed with cDGS, which is characterized by thymic aplasia and/or absent thymic function with absolute T-cell counts below 50x106/L and absent mitogen responses (64). B-cell counts are within normal ranges, but T-cell mediated help for antibody production is impaired. cDGS patients thus display a combined immunodeficiency (CID) phenotype (93). Over time, a significant number of cDGS patients, develop atypical features mediated by inflammation causing erythroderma, enteropathy and lymphadenopathy. This Omenn-like picture is due to oligoclonal expansion of dysfunctional T-cells of memory phenotype (94, 95). In atypical cDGS, absolute T-cell counts may be high, but the proportion of naïve T-cells remains negligible (<5%), TCR spectratyping as well as the repertoire of the V beta TCR chains are abnormal, and mitogen responses are impaired. cDGS patients, typical and atypical, usually suffer from severe infections during infancy and, in absence of treatment by allogeneic thymus transplantation or hematopoietic stem cell transplantation (HSCT), cDGS is fatal in the first two years of life. In contrast, patients with partial DGS (pDGS), have higher absolute T-cell counts and display milder degrees of immunodeficiency, including variable susceptibility to infections and autoimmunity. Those most affected require supportive measures such as antibiotic prophylaxis (92).
cDGS is the most frequent etiology of congenital athymia. This may have a number of different causes (Table 1). The most common is chromosome 22q11.2 deletion syndrome (22q11.2DS) (10, 12). 22q11.2DS occurs approximately in 0.25 in 1000 births (96, 97), of which 1.5% display cDGS with athymia (98). Most deletions at chromosome 22q11.2 result in haploinsufficiency of TBX1, a transcription factor regulating key events in early thymus embryogenesis (99, 100). 22q11.2DS patients represent more than a third of the patients who underwent thymus transplantation to date (10, 12). The second most frequent disorder in both cohorts of thymus transplanted cDGS patients is CHARGE syndrome (10, 12). CHARGE syndrome is mainly due to CHD7 variants and is found in about 1 in 8500 to 1 in 10000 births (101–103). The proportion of these infants who have cDGS is unknown, but a reported series of 280 cases did not mention immunodeficiency which is consistent with the proportion being below 1% (104). The third most frequent etiology of athymia, accounting for 10-20% of transplanted cDGS patients, is embryopathy secondary to poorly controlled maternal diabetes (10). The incidence with which this complication occurs again remains unascertained. Less frequent etiologies of DGS include other monogenic disorders and chromosomal microdeletions, as well as exposure to other fetal toxins (105, 106). Pathogenic variants in TBX1 and TBX2 have been reported in few patients, including some suffering from thymic hypoplasia and aplasia (12, 65, 107, 108). It is unclear whether any of the patients with partial chromosome 10p monosomy (109) suffer from complete athymia. In a report of 32 cases, 28% had a partial immunodeficiency phenotype but none had a picture compatible with athymia (110). FOXI3 haploinsufficiency secondary to microdeletions at chromosome 2p11.2 was recently described (111), but whether this may cause complete thymic aplasia is not clear as detailed immunophenotyping of thymic output has not been published. Regardless of the underlying pathogenesis, DGS patients often have associated clinical features, such as CHD, hypoparathyroidism, typical facial dysmorphism, velopharyngeal anomalies and feeding difficulties, which prompt physicians to their diagnosis. As there are no predictive clinical factors to reliably identify the small proportion of DGS patients with athymia, it is important that upon diagnosis of DGS, patients are screened for possible severe immunodeficiency.
Congenital Athymia in the Context of Nude SCID and OTFCS2
More infrequently than in DGS, athymia has been described in T-B+NK+ SCID. In classical Nude SCID, athymia is associated with alopecia totalis and nail dystrophy (112). This is an autosomal recessive (AR) disorder due to bi-allelic loss-of-function (LOF) mutations in FOXN1, a critically important transcription factor expressed in epithelial cells of the thymus, skin and nail matrix (113). Nude SCID has been reported in a small number of patients in whom FOXN1 deficiency underlies an impaired development of the thymic epithelial stroma with subsequent impaired T-cell differentiation (66, 67, 114–116). Their profound T-cell lymphopenia is marked by very low counts of naïve T-cells and recent thymic emigrants, as well as impaired T-cell proliferation responses. A handful of Nude SCID patients have been treated by HSCT, of which three, receiving transplantation from a matched sibling donor, have been reported (67, 116–118). Only one patient survived, but without regeneration of CD4+ naïve T-cells (117).
T-B+NK+ SCID has recently been described in another rare AR disorder, otofaciocervical syndrome type 2 (OTFCS2), which is caused by PAX1 deficiency and is characterized by syndromic features, including facial dysmorphism, ear and vertebral anomalies (119, 120). PAX1 is a transcription factor with a key role during thymus embryogenesis. In OTFCS2 patients from four unrelated families, bi-allelic LOF mutations in PAX1 resulted in severe T-cell lymphopenia due to altered thymus development (120). Four of these patients underwent HSCT before identification and characterization of their genetic defect, but despite full donor engraftment, T-cell immunity remained impaired. Naïve T-cells did not develop, consistent with absent thymopoiesis. Rather than treatment by HSCT, we would thus recommend referral of FOXN1-/- and PAX1-/- SCID patients for thymus transplantation.
Congenital Athymia in the Context of Undefined, Novel Defects
The incidence of congenital athymia among profoundly T-cell lymphopenic patients is not known. Even though it is rare, it is becoming increasingly apparent that a non-negligible proportion of SCID patients are affected by genetically defined and undefined thymic stromal cell defects. In the Italian SCID cohort, for example, genetically defined thymic disorders were found in 6% of the patients (121). This is almost certainly an underestimation of athymic cases, as a significant number of patients with inborn errors of immunity, including SCID, do not have a genetic diagnosis despite the expanding access to next-generation sequencing (NGS) (122, 123). In a recent cohort of SCID patients, 7% of the patients did not have a genetic variant in any of the known SCID genes (124). Additionally, timely diagnosis of thymic defects is complicated by the clinical variability among affected patients and in practice, not infrequently, inborn errors of thymic development and function are missed or are only recognized after a failed attempt at T-cell reconstitution using HSCT. In such cases, thymus transplantation can be successful as a second procedure particularly if it is possible to use a thymus matching for any MHC alleles (also called human leukocyte antigen (HLA) alleles in humans) in the recipient which were mismatched with the original HSCT donor to reduce the risk that the procedure might facilitate graft-versus-host disease (GVHD) mediated by the HSCT donor cells (125). However, such “rescue” second treatments will not always be feasible and depend on the absence of severe post-HSCT morbidities. It is important therefore to discriminate between patients with hematopoietic defects, who require a HSCT, and patients with thymic defects so that, where feasible, thymus transplantation should be considered as the preferred option. The progressive introduction of newborn screening (NBS) programs for SCID in a growing number of countries should facilitate this. NBS is based on the measure of T-cell receptor excision circles (TRECs) from dried blood spots by quantitative PCR and identifies infants with low T-cell numbers, including those with non-SCID T-cell lymphopenia (126, 127). NBS is essential to improving the outcome for patients by starting prophylactic measures early on, while completing immunological and genetic investigations with the aim of proceeding with the appropriate curative treatment.
Genetically undefined T-B+NK+ SCID and non-SCID T-lymphopenia pose a serious therapeutic dilemma, as the underlying defects are either intrinsic to HSC differentiation, or HSC-extrinsic when secondary to impaired T-cell differentiation in the thymus. Existing in vitro T-cell differentiation tools that assess the intrinsic potential of HSC to differentiate into mature T-cells have been useful to address this challenge. T-cell differentiation has been pursued in ex vivo culture models since 2005 and makes use of co-culture systems or functionalized microbeads able to provide Notch Delta-like ligand (DLL)-1 or DLL-4 to HSC differentiating toward T-cell fate (128–130). We have taken advantage of a monolayer co-culture system of patients’ bone marrow-derived CD34+ cells together with OP9/DL1 stromal cells and growth factors (49). Recently, two groups reported the use of artificial thymic organoids aggregating DLL4-expressing stromal cell lines (OP9/DLL4 and MS5-hDLL4) to facilitate the differentiation of CD34+ cells, isolated either from the patients’ bone marrow aspirate (52) or from their peripheral blood (131). Using these three-dimensional tools, they have shown that CD34+ cells of two cDGS patients, one with a TBX1 mutation and the other with a deletion at chromosome 22q11.2, had the potential to differentiate into CD3+ mature T-cells, in line with their disorder not affecting the HSC. Whereas, in contrast, they observed early developmental blocks when attempting the differentiation of CD34+ cells from SCID patients with known HSC-intrinsic defects. With the propagation of NBS programs and NGS, these tools are destined to play an essential role in assisting physicians making therapeutic decisions for undefined T-cell lymphopenia. Importantly, CD34+ cells from patients carrying RAG mutations and from one patient with a IL2RG mutation could also be differentiated in vitro into T-cell stages beyond the expected developmental block, emphasizing that further work is required to optimize the use of these assays in therapeutic management.
NBS for SCID was commenced in parts of the United States in 2008. The North American experience so far highlights that even when a thymic stromal defect is genetically and functionally confirmed early, evaluating the best therapeutic approach remains difficult in the case of rare, novel defects. This is the case, for example, in regards to the management of infants with selective T-cell lymphopenia and low TREC levels at birth who have been diagnosed with hypomorphic heterozygous FOXN1 mutations since the introduction of NBS (132, 133). None displayed alopecia totalis and nail dystrophy was seen in approximately half the reported cases only. Immunological follow up showed that in most individuals, absolute CD4+ T-cell counts normalized over time, while CD8+ T-cell counts typically remained below the normal range. Most patients either displayed mild immune deficiency, mainly with recurrent respiratory infections, or remained largely asymptomatic. Yet 1 in 8 of the reported cases with FOXN1 heterozygosity suffered a more severe degree of immune deficiency. An additional recent report highlights that a spectrum of clinical manifestations may be associated with different mutations (67). Disease severity likely depends on the residual FOXN1 protein activity. In three patients treatment was attempted with HSCT before the genetic diagnosis was made but their T-cell immunity remained impaired (132), suggesting that thymus transplantation, and not HSCT, should be considered in carefully selected FOXN1+/- patients with more severe clinical phenotypes. In the mouse model, a gene dosage effect was documented for Foxn1 expression in TECs for all of the heterozygote variants. Three patients with compound heterozygosity for LOF mutations also displayed more severe clinical phenotype (67, 133). While most FOXN1+/- patients can be successfully managed conservatively, a better understanding of the mechanisms of action of these mutations and of their impact on FOXN1 function is necessary to be able to predict which cases should at least be considered for potentially corrective treatment.
Thymus Transplantation for cDGS
The process of thymus transplantation using cultured, postnatal thymus tissue has not changed much since it was first developed (9).
Preparation of Thymus and Transplantation
Donor thymus tissue is obtained with parental informed consent from otherwise healthy infants undergoing cardiac surgery via median sternotomy for congenital heart disease. Removal of thymus tissue is necessary to facilitate surgery in these patients. There are increasing concerns over the potential long term consequences of very early total thymectomy (134–136) and surgeons should where possible conserve some of the thymus tissue. The preparatory process has been previously described (9).The removed thymus requires culturing for 13-20 days before transplantation in order to deplete thymocytes while preserving the thymic stroma. This process has been shown to be consistent between slices of the same thymus and between different thymus cultures (137). The lymphodepletion is incomplete with some lymphoid cell preservation showing a relative enrichment of viable CD4+ SP cells (12, 137). The stromal cells in contrast retain over 90% estimated viability (12, 137). Dependent on satisfactory histological evaluation and negative screening for infectious agents the cultured thymus slices are then implanted into the quadriceps muscles as previously described (138). It is important to note that the thymus donor is not tissue type matched with the recipient.
Patients
Eligibility for thymus transplantation requires a diagnosis of cDGS based on clinical features of DGS, with or without a genetic diagnosis, associated with either extreme T-cell lymphopenia (in typical cDGS) or T-cells showing a restricted repertoire with very low naïve T-cells (< 50x10^6/L) (in atypical cDGS, often with Omenn–like features). Among transplanted cDGS patients reported in both series the proportion showing atypical features varied between one third and two thirds (10, 12). Patients with other causes of athymia can also, appropriately, be treated with thymus transplantation (discussed later in this review). They may also have an atypical (Omenn-like) phenotype (66, 114, 116).
Prior to transplantation it is important to treat or correct other co-morbidities such as congenital heart defects which might affect the clinical stability of the patient after thymus transplantation. Those with the atypical phenotype require immunosuppression while the remainder are transplanted without conditioning. This immunosuppressive treatment involves Cyclosporin A (CSA) and antithymocyte globulin (ATG) with methylprednisolone given prior to the transplantation of the allograft. The CSA is then continued post-transplantation until evidence of thymic output is achieved.
Outcome
Overall survival has been reported as around 75% in transplanted cDGS cases (Table 1). Infection, often acquired prior to transplantation, was the most frequent cause of death (10–12). Following transplantation, the recipient’s bone marrow-derived hematopoietic precursors repopulate the thymic graft. It takes a minimum of four months and often longer before T-cell numbers increase with evidence of thymic output. Evidence of developing thymopoeisis can however be obtained from biopsies of transplanted tissue from as early as two months (139). Typically, the absolute numbers of T-cells achieved do not reach the normal age-related range and neither do the absolute numbers of naïve T-cells and TRECs, however there is a diverse T-cell receptor repertoire and normal T-cell proliferation in response to mitogens and normal HLA-restricted specific antigen responses (10–12). An analysis of variables associated with the procedure and the outcome in terms of T-cell and naïve T-cell numbers showed no effect of thymus dose, use of immunosuppression or chance partial HLA-matching between donor and recipient (140). ABO blood group incompatibility was associated with lower T-cell counts at 1-2 years post-transplantation but not at 3 years (11). In our unit, selected donors are blood group compatible with the athymic recipients. Even though there is no matching for HLA tissue type, the recipients’ T-cells have been shown to become tolerized to donor specific HLA antigens (141, 142). The mechanisms by which a HLA-mismatched thymic graft achieves self-tolerance remain to be determined. It is likely that host-derived hematopoietic cells, in particular DCs, can migrate into the graft and therefore contribute to HLA-TCR mediated interactions with developing thymocytes undergoing negative and positive selection. It is also possible that, akin to what has been observed after kidney and lung transplantation (143, 144), host stromal cells develop in the graft, resulting in a chimeric thymus including host fibroblasts which may contribute to direct lymphostromal interactions. Additionally if there is some serendipitous partial HLA-matching, direct TEC-thymocytes interactions may occur, regardless of host or donor origin of the cells. These different hypotheses are summarized in Figure 1B.
For the majority of transplanted patients in either cohort the long term outcomes of the immune recovery have not been described yet. The observations in the small numbers of reported patients with follow up of more than 3 years (up to 8 years) after thymus transplantation show sustained, even if suboptimal, T-cell counts (10, 12, 138, 145), whereas TREC levels, which correlate directly with thymic output, typically decrease and plateau over time (12). In spite of the lower than normal T-cell counts, patients develop the ability to clear both pre-existing and acquired infections. Immunoglobulin replacement therapy and antibiotic prophylaxis can be stopped. Patients can respond to vaccination with antigen-specific cell responses. Regulatory T-cells have been shown to be present in normal proportions of circulating T-cells though in subnormal absolute numbers (because of the relative T-cell lymphopenia) and these cells functioned normally in a CTLA-4 mediated endocytosis assay (12).
Complications
Particularly in children with DGS, there may be multiple system co-morbidities which can complicate the post-transplantation course by jeopardizing the general stability required for successful engraftment of the transplant with repopulation of the new thymus by recipient-derived progenitor T-cells. These complications need to be differentiated from those potentially arising as a direct result of thymus transplantation. Furthermore, the prolonged wait before immune reconstitution is seen, means that transplanted children remain susceptible to infective and possibly to autoimmune complications as a result of their underlying condition rather than the procedure itself. Inflammatory complications may be seen after transplantation and fall into three categories. First, particularly in those not treated with ATG prior to transplantation, oligoclonal T-cell expansions and Omenn-like features may still develop. This requires the institution of CSA treatment. Secondly, with persistent infections, at the time of immune reconstitution, there may be an immune reconstitution inflammatory response (IRIS). This has been seen in those infected with BCG, rotavirus (including vaccine strain), norovirus, Clostridium difficile, and HHV6 ( (12) and unpublished observations). Finally, a small number of patients have been seen in whom a severe systemic IRIS occurred without an underlying identifiable driving pathogen (unpublished). These complications can be life-threatening and typically require the use of high doses of cortico-steroids, often resulting in a delay in the development of thymopoiesis.
Autoimmune complications are relatively common after thymus transplantation (Table 1). Early autoimmunity, prior to immune reconstitution, included hemolytic anemia (12) and renal disease has also been seen at this stage (142). Mostly these patients did well with no recurrence. Later autoimmunity predominantly involves thyroid disease but cytopenias are also common. Out of sixty transplanted patients in one series, thirteen developed thyroid disease and nine had cytopenias (11). In our series the incidence of autoimmunity may be higher than this. In most cases the autoimmune disease is either relatively easily treated, as in the case of thyroid disease, or a lasting remission can be achieved after initial therapy in the case of cytopenias. However in one small series one patient’s hemolysis required ongoing immunosuppressive treatment while one patient died of a cerebral hemorrhage complicating immune thrombocytopenia (12). The frequency of these complications suggests they occur at least in part not because of the background susceptibility as part of DGS, but because of a failure of generation of central tolerance to these particular organ systems after transplantation. The lack of HLA-matching between donor and host may be responsible due to likely impaired lymphostromal crosstalk in the thymic grafts. Coincidental partial HLA-matching within donor-recipient pairs at one or more loci was seen in all patients without autoimmune complications described in the initial report on the UK cohort (12), whereas all patients without any HLA-matching developed autoimmune complications. A trend toward less autoimmunity in a partially HLA-matched setting was not statistically significant, but the number of patients was too small (n=10) to properly address this question. Additionally achieving matching at specific loci or of specific alleles may be of importance.
Thymus Transplantation for Non–DGS Thymic Stromal Defects
Thymus transplantation has also been shown to be the most appropriate treatment for athymia due to FOXN1 deficiency. A successful outcome after thymus transplantation in nude SCID has been reported in 2 patients. Interestingly in one of those patients reconstitution with naïve circulating T-cells was very slow, taking over twelve months (66). The long term follow up of the other patient showed sustained immune recovery, despite decreased thymic output over time (68), similar to what has been observed in transplanted cDGS patients. More recently, we have transplanted three further cases with homozygous or compound heterozygous FOXN1 mutations (67). Two had allograft biopsies confirming the establishment of thymopoeisis of whom, one has done well and one died as a consequence of severe inflammatory complications. The third patient has not developed thymopoeisis after 17 months.
We have also very recently transplanted two athymic OTFCS2 patients, but the outcome for these cases is not yet known.
Thymus replacement therapy has not at the present time been attempted in patients with APS1. The attraction of attempting this would be to halt the inexorable progression to multisystem autoimmune complications seen in these patients. However, such an approach would have additional challenges over and above those normally encountered, including the question of competition for bone marrow precursors between the recipient’s own thymus and the transplanted tissue, the possibility of rejection and the likely presence of pre-established autoimmune problems. Furthermore, recent work has highlighted the potential benefit of early, pre-symptomatic immunomodulatory therapies that may improve the clinical outcome for these patients (73). This makes the case for a radical intervention with thymus transplantation less attractive.
Possible Strategies for Improving the Use of Cultured Postnatal Thymic Tissue
Improving the Pre-Transplantation Thymic Tissue Culture Process
As mentioned above, the lymphodepletion during pre-transplantation tissue culture is incomplete and viable donor T-cells are transplanted with the graft (12, 137). While it is possible that such HLA-mismatched donor T-cells could mediate GVHD, this has not been reported. On the other hand, these cells may be helpful in providing necessary lymphostromal interaction to support initial TEC homeostasis post-thymus transplantation. Whether the self-renewing lymphoid progenitors as well as other donor-derived hematopoietic cells, including dendritic cells and Tregs, survive the culture period is not known. Repercussions on TEC function have been reported with downregulation of AIRE expression in culture (146), though normal AIRE expression has been shown in biopsies taken from 2-3 months after transplantation (12). It is therefore possible that after thymus transplantation early thymopoiesis occurs in the absence of adequate AIRE-mediated negative selection, possibly contributing to failure of early deletion of autoreactive T-cells and post-transplantation autoimmunity. These observations support the need for studies addressing whether modulation of the preparatory culture protocol might improve AIRE expression and potentially reduce autoimmune complications.
Using Cryopreserved Thymic Tissue
Thymus transplantation is a complex procedure which is only offered at 2 centers worldwide. Inevitably, access to the treatment is restricted to patients who have access to the resources to support traveling significant distances, sometimes across continents. In the future, use of pre-prepared, cryopreserved thymus tissue may make it possible for patients to be treated at institutions within their own country. We have shown that the transplantation of cryopreserved and subsequently thawed human thymic tissue into athymic mice was able to support thymopoiesis, achieving reconstitution of T-cells (147). If previously cryopreserved slices can be shown to support thymopoeisis when transplanted into athymic patients, then thymus tissue banks could be used to simplify the logistics of this treatment. It could also facilitate the use of at least partially HLA-matched tissue. In theory at least, matching at >4 loci might allow more efficient thymopoiesis in the graft, resulting in a reduced incidence of autoimmune complications. In line with this, SCID patients undergoing haploidentical HSCT procedures generally develop normal thymopoiesis in the presence of good myeloid engraftment (148).
Future Directions for Thymus Replacement Therapy
Despite the life-saving potential of transplantation of postnatal thymus tissue described above, there are limitations to this approach. These include global accessibility, incomplete immune reconstitution and a relatively high rate of complications. New therapeutic strategies for replacing thymus function might have the potential to overcome these limitations for patients with primary athymia (Table 2), but possibly might also allow such treatments to be used in other clinical scenarios, such as secondary loss of thymus function through intensive chemotherapy or neonatal thymectomy, or to help tolerization after solid organ transplantation. These scenarios have been reviewed elsewhere (150).

Table 2 Future treatment approaches for congenital athymia (in comparison with standard treatment).
In recent years, there has been a constant increase in research output aimed at producing engineered thymic stroma. The importance of the epithelium in the thymus makes it possible to apply expertise and knowledge from the epithelial stem cell field which have been used to develop successful tissue replacement strategies using cultivated stem cells. These regenerative medicine studies have already resulted in translation to the clinic for replacement of non-lymphoid organs such as the skin and the cornea (151–154). However, in contrast to the multi-layered structure of these tissues, TECs form a complex three-dimensional (3D) arrangement, which is essential for their functioning. 3D co-culture systems, referred to as re-aggregate thymus organ cultures (RTOCs), have successfully been developed using thymic stromal cells from mouse embryos (155–157). These RTOCs are able to support initial T-cell differentiation of hematopoietic progenitors in vitro and also in vivo after transplantation into athymic mice. Nevertheless, production of T-cells is still not very efficient; moreover these systems are either mouse or mouse-human hybrids and therefore the clinical applications in the context of thymic regeneration are limited due to the lack of human thymic stroma and absent instructive MHC-II expression. Attempts at culturing TECs have intensified in the last fifteen years (38, 158–160). Two groups reported significant TEC expansion in rodents and demonstrated “stemness” properties of thymic epithelial cells (158, 159).
For clinical translation, it is essential to develop new tools that can deliver TECs within an appropriate 3D structure, capable of supporting thymopoiesis. One group used artificial collagen scaffolds seeded with postnatal, gene-modified murine TECs, able to transiently express Oct4 to promote TEC expansion in vitro and in vivo (161). However, these artificial collagen scaffolds did not show efficacy in supporting thymopoiesis when transplanted into mice. Early attempts using natural ECM from decellularized mouse thymi following repopulation with freshly isolated stromal cells, lacked TEC organization and did not support ex vivo thymopoiesis either (162, 163). Thymus decellularization was achieved by freeze/thaw cycles and detergent-induced cell lysis (164). This method cannot be used to obtain human natural scaffolds, as it cannot be applied to the much larger human thymi. The very recent development of a novel whole-organ perfusion system has made it possible to obtain a natural decellularized ECM from human thymi, despite the lack of major vascular access (35). This has made it possible to obtain an engineered thymus after reconstitution with in vitro expanded human thymic stromal progenitor cells. Of note, clinically relevant numbers of expanded human TECs and other stromal progenitors were achieved within a few weeks. When transplanted into a humanized small animal model, the engineered thymi demonstrated the capacity to re-organize into a recognizable thymus with cortex and medulla, including Hassall’s bodies. This reconstituted human thymus had the ability to support thymopoiesis, both ex vivo and in vivo (35). Having overcome important obstacles, given the possibility of expanding large numbers of thymic stromal cells and of perfusing a whole thymus organ, this approach can be scaled up, thus facilitating translation into the clinic. In the future, this approach may lead to production of a large amount of human thymus stroma for transplantation. This transplanted thymus would be free of donor thymocytes, which may be beneficial, at least for any possible complications mediated by these cells in the currently used model of allogeneic thymus transplantation. The option to bank these stromal cells will also offer the possibility to achieve partial tissue type matching.
Finally, another approach for future thymus replacement therapy is to reprogram the patients’ own cells to generate TECs. Direct reprogramming of mouse fibroblasts by enforced FOXN1 expression produced TECs capable of supporting thymocyte development in the mouse (165). In another approach, by adapting protocols which successfully differentiated human embryonic stem cells into thymic epithelium (166, 167), patient-derived fibroblasts, induced to revert to a pluripotent stage of induced pluripotent stem cells (iPSCs), could be differentiated into thymic stromal progenitors (149). They could mature only upon in vivo transplantation into athymic nude mice and their capacity to fully instruct human HSC has not yet been demonstrated. Once optimized, this approach in combination with gene correction might make the use of autologous engineered thymus tissue possible for athymic patients, in particular in the case of monogenic defects. Potentially, thymic stromal cells induced in this way could be used to populate the decellularized human scaffolds described above. In theory, this approach could reduce the risk of autoimmune complications compared to conventional thymus transplantation.
Concluding Remarks
Inborn errors of thymic stromal cell development and function result in immunodeficiency and autoimmunity, with the most severe thymic defects causing thymic aplasia. For athymic patients with profound T-cell lymphopenia, transplantation of cultured, post-natal thymus tissue is a life-saving treatment, achieving sufficient immune reconstitution to clear and prevent infectious complications. Failure to survive after thymus transplantation is most commonly associated with infectious complications, often in the context of pre-transplantation infections. This is expected to reduce as the use of NBS for T-cell deficiencies becomes more prevalent, facilitating earlier diagnosis and introduction of prophylaxis. Increasing access to NGS, along with the development of new tools to characterize novel thymic defects and distinguish them from primary hematopoietic defects, will also contribute to a growing number of patients and disorders to be considered for thymus transplantation. Given the suboptimal immune reconstitution and the relatively common inflammatory and autoimmune complications after thymus transplantation, there is a need to optimize treatment by developing new approaches to replacing defective thymus function. Use of cryopreserved thymus slices for transplantation may in the future make the treatment more readily available and potentially reduce the rate of complications if partial HLA-matching is confirmed as having an influence on this. In the longer term, the use of engineered thymus tissue is likely to be translated into the clinic. The ultimate aim will be to be able to use autologous cells to generate (gene-corrected) thymus stromal cells for producing engineered tissue for transplantation.
Author Contributions
AYK, PB, and ED all conceived and co-wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
AK and ED are supported by LetterOne & Mikhail Fridman in conjunction with Great Ormond Street Hospital Children’s Charity, The Jeffrey Modell Foundation and the UK National Institute of Health Research and the Great Ormond Street Hospital Biomedical Research Centre (NIHR GOSH BRC). PB is supported by the European Research Council (ERC-Stg No. 639429), the Rosetrees Trust (M362-F1; M553), the London Advanced Therapies (C2N-AT.006), the UCL Therapeutic Acceleration Support Fund, and by the NIHR GOSH BRC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Janda A, Sedlacek P, Honig M, Friedrich W, Champagne M, Matsumoto T, et al. Multicenter survey on the outcome of transplantation of hematopoietic cells in patients with the complete form of DiGeorge anomaly. Blood (2010) 116(13):2229–36. doi: 10.1182/blood-2010-03-275966
2. Land MH, Garcia-Lloret MI, Borzy MS, Rao PN, Aziz N, McGhee SA, et al. Long-term results of bone marrow transplantation in complete DiGeorge syndrome. J Allergy Clin Immunol (2007) 120(4):908–15. doi: 10.1016/j.jaci.2007.08.048
3. Thomas ED, Lochte HL Jr., Lu WC, Ferrebee JW. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Engl J Med (1957) 257(11):491–6. doi: 10.1056/NEJM195709122571102
4. Thomas ED, Lochte HL Jr., Cannon JH, Sahler OD, Ferrebee JW. Supralethal whole body irradiation and isologous marrow transplantation in man. J Clin Invest (1959) 38:1709–16. doi: 10.1172/JCI103949
5. Miller JF. Role of the Thymus in Immunity. Br Med J (1963) 2(5355):459–64. doi: 10.1136/bmj.2.5355.459
6. August CS, Berkel AI, Levey RH, Rosen FS, Kay HE. Establishment of immunological competence in a child with congenital thymic aplasia by a graft of fetal thymus. Lancet (1970) 1(7656):1080–3. doi: 10.1016/S0140-6736(70)92755-8
7. Cleveland WW, Fogel BJ, Brown WT, Kay HE. Foetal thymic transplant in a case of Digeorge’s syndrome. Lancet (1968) 2(7580):1211–4. doi: 10.1016/S0140-6736(68)91694-2
8. Pahwa S, Pahwa R, Incefy G, Reece E, Smithwick E, O’Reilly R, et al. Failure of immunologic reconstitution in a patient with the DiGeorge syndrome after fetal thymus transplantation. Clin Immunol Immunopathol (1979) 14(1):96–106. doi: 10.1016/0090-1229(79)90130-2
9. Markert ML, Kostyu DD, Ward FE, McLaughlin TM, Watson TJ, Buckley RH, et al. Successful formation of a chimeric human thymus allograft following transplantation of cultured postnatal human thymus. J Immunol (1997) 158(2):998–1005.
10. Markert ML, Devlin BH, Alexieff MJ, Li J, McCarthy EA, Gupton SE, et al. Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants. Blood (2007) 109(10):4539–47. doi: 10.1182/blood-2006-10-048652
11. Markert ML, Devlin BH, McCarthy EA. Thymus transplantation. Clin Immunol (2010) 135(2):236–46. doi: 10.1016/j.clim.2010.02.007
12. Davies EG, Cheung M, Gilmour K, Maimaris J, Curry J, Furmanski A, et al. Thymus transplantation for complete DiGeorge syndrome: European experience. J Allergy Clin Immunol (2017) 140(6):1660–70. doi: 10.1016/j.jaci.2017.03.020
13. Graham A. The development and evolution of the pharyngeal arches. J Anat (2001) 199(Pt 1-2):133–41. doi: 10.1046/j.1469-7580.2001.19910133.x
14. Graham A, Richardson J. Developmental and evolutionary origins of the pharyngeal apparatus. Evodevo (2012) 3(1):24. doi: 10.1186/2041-9139-3-24
15. Le Lievre CS, Le Douarin NM. Mesenchymal derivatives of the neural crest: analysis of chimaeric quail and chick embryos. J Embryol Exp Morphol (1975) 34(1):125–54.
16. Gordon J, Bennett AR, Blackburn CC, Manley NR. Gcm2 and Foxn1 mark early parathyroid- and thymus-specific domains in the developing third pharyngeal pouch. Mech Dev (2001) 103(1-2):141–3. doi: 10.1016/S0925-4773(01)00333-1
17. Veitch E, Begbie J, Schilling TF, Smith MM, Graham A. Pharyngeal arch patterning in the absence of neural crest. Curr Biol (1999) 9(24):1481–4. doi: 10.1016/S0960-9822(00)80118-9
18. Gordon J, Manley NR. Mechanisms of thymus organogenesis and morphogenesis. Development (2011) 138(18):3865–78. doi: 10.1242/dev.059998
19. Manley NR, Condie BG. Transcriptional regulation of thymus organogenesis and thymic epithelial cell differentiation. Prog Mol Biol Transl Sci (2010) 92:103–20. doi: 10.1016/S1877-1173(10)92005-X
20. Vaidya HJ, Briones Leon A, Blackburn CC. FOXN1 in thymus organogenesis and development. Eur J Immunol (2016) 46(8):1826–37. doi: 10.1002/eji.201545814
21. Bredenkamp N, Nowell CS, Blackburn CC. Regeneration of the aged thymus by a single transcription factor. Development (2014) 141(8):1627–37. doi: 10.1242/dev.103614
22. Chen L, Xiao S, Manley NR. Foxn1 is required to maintain the postnatal thymic microenvironment in a dosage-sensitive manner. Blood (2009) 113(3):567–74. doi: 10.1182/blood-2008-05-156265
23. Farley AM, Morris LX, Vroegindeweij E, Depreter ML, Vaidya H, Stenhouse FH, et al. Dynamics of thymus organogenesis and colonization in early human development. Development (2013) 140(9):2015–26. doi: 10.1242/dev.087320
24. Ivanovs A, Rybtsov S, Anderson RA, Turner ML, Medvinsky A. Identification of the niche and phenotype of the first human hematopoietic stem cells. Stem Cell Rep (2014) 2(4):449–56. doi: 10.1016/j.stemcr.2014.02.004
25. Zeng Y, He J, Bai Z, Li Z, Gong Y, Liu C, et al. Tracing the first hematopoietic stem cell generation in human embryo by single-cell RNA sequencing. Cell Res (2019) 29(11):881–94. doi: 10.1038/s41422-019-0228-6
26. Zeng Y, Liu C, Gong Y, Bai Z, Hou S, He J, et al. Single-Cell RNA Sequencing Resolves Spatiotemporal Development of Pre-thymic Lymphoid Progenitors and Thymus Organogenesis in Human Embryos. Immunity (2019) 51(5):930–48.e6. doi: 10.1016/j.immuni.2019.09.008
27. Oberlin E, Fleury M, Clay D, Petit-Cocault L, Candelier JJ, Mennesson B, et al. VE-cadherin expression allows identification of a new class of hematopoietic stem cells within human embryonic liver. Blood (2010) 116(22):4444–55. doi: 10.1182/blood-2010-03-272625
28. Takahama Y. Journey through the thymus: stromal guides for T-cell development and selection. Nat Rev Immunol (2006) 6(2):127–35. doi: 10.1038/nri1781
29. Anderson G, Takahama Y. Thymic epithelial cells: working class heroes for T cell development and repertoire selection. Trends Immunol (2012) 33(6):256–63. doi: 10.1016/j.it.2012.03.005
30. Kernfeld EM, Genga RMJ, Neherin K, Magaletta ME, Xu P, Maehr R. A Single-Cell Transcriptomic Atlas of Thymus Organogenesis Resolves Cell Types and Developmental Maturation. Immunity (2018) 48(6):1258–70.e6. doi: 10.1016/j.immuni.2018.04.015
31. Park JE, Botting RA, Dominguez Conde C, Popescu DM, Lavaert M, Kunz DJ, et al. A cell atlas of human thymic development defines T cell repertoire formation. Science (2020) 367(6480). doi: 10.1101/2020.01.28.911115
32. Van de Walle I, Waegemans E, De Medts J, De Smet G, De Smedt M, Snauwaert S, et al. Specific Notch receptor-ligand interactions control human TCR-alphabeta/gammadelta development by inducing differential Notch signal strength. J Exp Med (2013) 210(4):683–97. doi: 10.1084/jem.20121798
33. Lavaert M, Liang KL, Vandamme N, Park JE, Roels J, Kowalczyk MS, et al. Integrated scRNA-Seq Identifies Human Postnatal Thymus Seeding Progenitors and Regulatory Dynamics of Differentiating Immature Thymocytes. Immunity (2020) 52(6):1088–104.e6. doi: 10.1016/j.immuni.2020.03.019
34. Tang Y, Yang YG, Bai O, Xia J, Hu Z. Long-term survival and differentiation of human thymocytes in human thymus-grafted immunodeficient mice. Immunotherapy (2019) 11(10):881–8. doi: 10.2217/imt-2019-0030
35. Campinoti S, Gjinovci A, Ragazzini R, Zanieri L, Ariza-McNaughton L, Catucci M, et al. Reconstitution of a functional human thymus by postnatal stromal progenitor cells and natural whole-organ scaffolds. Nat Commun (2020) 11(1):6372. doi: 10.1038/s41467-020-20082-7
36. Bautista JL, Cramer NT, Miller CN, Chavez J, Berrios DI, Byrnes LE, et al. Single-cell transcriptional profiling of human thymic stroma uncovers novel cellular heterogeneity in the thymic medulla. Nat Commun (2021) 12(1):1096. doi: 10.1038/s41467-021-21346-6
37. Rossi SW, Jenkinson WE, Anderson G, Jenkinson EJ. Clonal analysis reveals a common progenitor for thymic cortical and medullary epithelium. Nature (2006) 441(7096):988–91. doi: 10.1038/nature04813
38. Ucar A, Ucar O, Klug P, Matt S, Brunk F, Hofmann TG, et al. Adult thymus contains FoxN1(-) epithelial stem cells that are bipotent for medullary and cortical thymic epithelial lineages. Immunity (2014) 41(2):257–69. doi: 10.1016/j.immuni.2014.07.005
39. Abramson J, Anderson G. Thymic Epithelial Cells. Annu Rev Immunol (2017) 35:85–118. doi: 10.1146/annurev-immunol-051116-052320
40. Gameiro J, Nagib P, Verinaud L. The thymus microenvironment in regulating thymocyte differentiation. Cell Adh Migr (2010) 4(3):382–90. doi: 10.4161/cam.4.3.11789
41. Halkias J, Melichar HJ, Taylor KT, Robey EA. Tracking migration during human T cell development. Cell Mol Life Sci (2014) 71(16):3101–17. doi: 10.1007/s00018-014-1607-2
42. Savino W, Mendes-Da-Cruz DA, Smaniotto S, Silva-Monteiro E, Villa-Verde DM. Molecular mechanisms governing thymocyte migration: combined role of chemokines and extracellular matrix. J Leukoc Biol (2004) 75(6):951–61. doi: 10.1189/jlb.1003455
43. Spits H. Development of alphabeta T cells in the human thymus. Nat Rev Immunol (2002) 2(10):760–72. doi: 10.1038/nri913
44. Joachims ML, Chain JL, Hooker SW, Knott-Craig CJ, Thompson LF. Human alpha beta and gamma delta thymocyte development: TCR gene rearrangements, intracellular TCR beta expression, and gamma delta developmental potential–differences between men and mice. J Immunol (2006) 176(3):1543–52. doi: 10.4049/jimmunol.176.3.1543
45. Taghon T, Waegemans E, Van de Walle I. Notch signaling during human T cell development. Curr Top Microbiol Immunol (2012) 360:75–97. doi: 10.1007/82_2012_230
46. Famili F, Wiekmeijer AS, Staal FJ. The development of T cells from stem cells in mice and humans. Future Sci OA (2017) 3(3):FSO186. doi: 10.4155/fsoa-2016-0095
47. Schmitt TM, Zuniga-Pflucker JC. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity (2002) 17(6):749–56. doi: 10.1016/S1074-7613(02)00474-0
48. De Smedt M, Hoebeke I, Plum J. Human bone marrow CD34+ progenitor cells mature to T cells on OP9-DL1 stromal cell line without thymus microenvironment. Blood Cells Mol Dis (2004) 33(3):227–32. doi: 10.1016/j.bcmd.2004.08.007
49. Six EM, Benjelloun F, Garrigue A, Bonhomme D, Morillon E, Rouiller J, et al. Cytokines and culture medium have a major impact on human in vitro T-cell differentiation. Blood Cells Mol Dis (2011) 47(1):72–8. doi: 10.1016/j.bcmd.2011.04.001
50. Brauer PM, Pessach IM, Clarke E, Rowe JH, Ott de Bruin L, Lee YN, et al. Modeling altered T-cell development with induced pluripotent stem cells from patients with RAG1-dependent immune deficiencies. Blood (2016) 128(6):783–93. doi: 10.1182/blood-2015-10-676304
51. Themeli M, Chhatta A, Boersma H, Prins HJ, Cordes M, de Wilt E, et al. iPSC-Based Modeling of RAG2 Severe Combined Immunodeficiency Reveals Multiple T Cell Developmental Arrests. Stem Cell Rep (2020) 14(2):300–11. doi: 10.1016/j.stemcr.2019.12.010
52. Bosticardo M, Pala F, Calzoni E, Delmonte OM, Dobbs K, Gardner CL, et al. Artificial thymic organoids represent a reliable tool to study T-cell differentiation in patients with severe T-cell lymphopenia. Blood Adv (2020) 4(12):2611–6. doi: 10.1182/bloodadvances.2020002957
53. Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol (2005) 174(10):6477–89. doi: 10.4049/jimmunol.174.10.6477
54. Wiekmeijer AS, Pike-Overzet K, IJ H, Brugman MH, Wolvers-Tettero IL, Lankester AC, et al. Identification of checkpoints in human T-cell development using severe combined immunodeficiency stem cells. J Allergy Clin Immunol (2016) 137(2):517–26.e3. doi: 10.1016/j.jaci.2015.08.022
55. Choi EY, Jung KC, Park HJ, Chung DH, Song JS, Yang SD, et al. Thymocyte-thymocyte interaction for efficient positive selection and maturation of CD4 T cells. Immunity (2005) 23(4):387–96. doi: 10.1016/j.immuni.2005.09.005
56. Li W, Kim MG, Gourley TS, McCarthy BP, Sant’Angelo DB, Chang CH. An alternate pathway for CD4 T cell development: thymocyte-expressed MHC class II selects a distinct T cell population. Immunity (2005) 23(4):375–86. doi: 10.1016/j.immuni.2005.09.002
57. Pawlowski T, Elliott JD, Loh DY, Staerz UD. Positive selection of T lymphocytes on fibroblasts. Nature (1993) 364(6438):642–5. doi: 10.1038/364642a0
58. Hugo P, Kappler JW, McCormack JE, Marrack P. Fibroblasts can induce thymocyte positive selection in vivo. Proc Natl Acad Sci U S A (1993) 90(21):10335–9. doi: 10.1073/pnas.90.21.10335
59. Lilic M, Santori FR, Neilson EG, Frey AB, Vukmanovic S. The role of fibroblasts in thymocyte-positive selection. J Immunol (2002) 169(9):4945–50. doi: 10.4049/jimmunol.169.9.4945
60. Martinic MM, van den Broek MF, Rulicke T, Huber C, Odermatt B, Reith W, et al. Functional CD8+ but not CD4+ T cell responses develop independent of thymic epithelial MHC. Proc Natl Acad Sci U S A (2006) 103(39):14435–40. doi: 10.1073/pnas.0606707103
61. McCaughtry TM, Baldwin TA, Wilken MS, Hogquist KA. Clonal deletion of thymocytes can occur in the cortex with no involvement of the medulla. J Exp Med (2008) 205(11):2575–84. doi: 10.1084/jem.20080866
62. Daley SR, Hu DY, Goodnow CC. Helios marks strongly autoreactive CD4+ T cells in two major waves of thymic deletion distinguished by induction of PD-1 or NF-kappaB. J Exp Med (2013) 210(2):269–85. doi: 10.1084/jem.20121458
63. Nitta T, Tsutsumi M, Nitta S, Muro R, Suzuki EC, Nakano K, et al. Fibroblasts as a source of self-antigens for central immune tolerance. Nat Immunol (2020) 21(10):1172–80. doi: 10.1038/s41590-020-0756-8
64. Davies EG. Immunodeficiency in DiGeorge Syndrome and Options for Treating Cases with Complete Athymia. Front Immunol (2013) 4:322. doi: 10.3389/fimmu.2013.00322
65. Liu N, Schoch K, Luo X, Pena LDM, Bhavana VH, Kukolich MK, et al. Functional variants in TBX2 are associated with a syndromic cardiovascular and skeletal developmental disorder. Hum Mol Genet (2018) 27(14):2454–65. doi: 10.1093/hmg/ddy146
66. Markert ML, Marques JG, Neven B, Devlin BH, McCarthy EA, Chinn IK, et al. First use of thymus transplantation therapy for FOXN1 deficiency (nude/SCID): a report of 2 cases. Blood (2011) 117(2):688–96. doi: 10.1182/blood-2010-06-292490
67. Giardino G, Sharapova SO, Ciznar P, Dhalla F, Radha Rama Devi A, Islamoglu C, Ikinciogullari A, et al. Expanding the Nude SCID/CID phenotype associated to FOXN1 homozygous, compound heterozygous or heterozygous mutations. J Clin Immunol (2021). doi: 10.1007/s10875-021-00967-y
68. Albuquerque AS, Marques JG, Silva SL, Ligeiro D, Devlin BH, Dutrieux J, et al. Human FOXN1-deficiency is associated with alphabeta double-negative and FoxP3+ T-cell expansions that are distinctly modulated upon thymic transplantation. PLoS One (2012) 7(5):e37042. doi: 10.1371/journal.pone.0037042
69. Stone CA, Jr., Markert ML, Abraham RS, Norton A. A case of atypical, complete DiGeorge syndrome without 22q11 mutation. Ann Allergy Asthma Immunol (2017) 118(5):640–2.e2. doi: 10.1016/j.anai.2017.02.025
70. Yin SM, Ferdman RM, Wang L, Markert ML, Tam JS. Disseminated Mycobacterium kansasii disease in complete DiGeorge syndrome. J Clin Immunol (2015) 35(5):435–8. doi: 10.1007/s10875-015-0171-3
71. Lee JH, Markert ML, Hornik CP, McCarthy EA, Gupton SE, Cheifetz IM, et al. Clinical course and outcome predictors of critically ill infants with complete DiGeorge anomaly following thymus transplantation. Pediatr Crit Care Med (2014) 15(7):e321–6. doi: 10.1097/PCC.0000000000000219
72. Husebye ES, Perheentupa J, Rautemaa R, Kampe O. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J Intern Med (2009) 265(5):514–29. doi: 10.1111/j.1365-2796.2009.02090.x
73. Constantine GM, Lionakis MS. Lessons from primary immunodeficiencies: Autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol Rev (2019) 287(1):103–20. doi: 10.1111/imr.12714
74. Ferre EM, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight (2016) 1(13). doi: 10.1172/jci.insight.88782
75. Ferre EMN, Break TJ, Burbelo PD, Allgauer M, Kleiner DE, Jin D, et al. Lymphocyte-driven regional immunopathology in pneumonitis caused by impaired central immune tolerance. Sci Transl Med (2019) 11(495). doi: 10.1126/scitranslmed.aav5597
76. Bruserud O, Oftedal BE, Wolff AB, Husebye ES. AIRE-mutations and autoimmune disease. Curr Opin Immunol (2016) 43:8–15. doi: 10.1016/j.coi.2016.07.003
77. Colobran R, Gimenez-Barcons M, Marin-Sanchez A, Porta-Pardo E, Pujol-Borrell R. AIRE genetic variants and predisposition to polygenic autoimmune disease: The case of Graves’ disease and a systematic literature review. Hum Immunol (2016) 77(8):643–51. doi: 10.1016/j.humimm.2016.06.002
78. Poliani PL, Facchetti F, Ravanini M, Gennery AR, Villa A, Roifman CM, et al. Early defects in human T-cell development severely affect distribution and maturation of thymic stromal cells: possible implications for the pathophysiology of Omenn syndrome. Blood (2009) 114(1):105–8. doi: 10.1182/blood-2009-03-211029
79. De Ravin SS, Cowen EW, Zarember KA, Whiting-Theobald NL, Kuhns DB, Sandler NG, et al. Hypomorphic Rag mutations can cause destructive midline granulomatous disease. Blood (2010) 116(8):1263–71. doi: 10.1182/blood-2010-02-267583
80. Cavadini P, Vermi W, Facchetti F, Fontana S, Nagafuchi S, Mazzolari E, et al. AIRE deficiency in thymus of 2 patients with Omenn syndrome. J Clin Invest (2005) 115(3):728–32. doi: 10.1172/JCI200523087
81. Mathieu AL, Verronese E, Rice GI, Fouyssac F, Bertrand Y, Picard C, et al. PRKDC mutations associated with immunodeficiency, granuloma, and autoimmune regulator-dependent autoimmunity. J Allergy Clin Immunol (2015) 135(6):1578–88.e5. doi: 10.1016/j.jaci.2015.01.040
82. Oh H, Ghosh S. NF-kappaB: roles and regulation in different CD4(+) T-cell subsets. Immunol Rev (2013) 252(1):41–51. doi: 10.1111/imr.12033
83. Chen K, Coonrod EM, Kumanovics A, Franks ZF, Durtschi JD, Margraf RL, et al. Germline mutations in NFKB2 implicate the noncanonical NF-kappaB pathway in the pathogenesis of common variable immunodeficiency. Am J Hum Genet (2013) 93(5):812–24. doi: 10.1016/j.ajhg.2013.09.009
84. Klemann C, Camacho-Ordonez N, Yang L, Eskandarian Z, Rojas-Restrepo JL, Frede N, et al. Clinical and Immunological Phenotype of Patients With Primary Immunodeficiency Due to Damaging Mutations in NFKB2. Front Immunol (2019) 10:297. doi: 10.3389/fimmu.2019.00297
85. Lee CE, Fulcher DA, Whittle B, Chand R, Fewings N, Field M, et al. Autosomal-dominant B-cell deficiency with alopecia due to a mutation in NFKB2 that results in nonprocessable p100. Blood (2014) 124(19):2964–72. doi: 10.1182/blood-2014-06-578542
86. Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol (2017) 17(9):545–58. doi: 10.1038/nri.2017.52
87. Wirasinha RC, Davies AR, Srivastava M, Sheridan JM, Sng XYX, Delmonte OM, et al. Nfkb2 variants reveal a p100-degradation threshold that defines autoimmune susceptibility. J Exp Med (2021) 218(2). doi: 10.1084/jem.20200476
88. Lemoine R, Pachlopnik-Schmid J, Farin HF, Bigorgne A, Debre M, Sepulveda F, et al. Immune deficiency-related enteropathy-lymphocytopenia-alopecia syndrome results from tetratricopeptide repeat domain 7A deficiency. J Allergy Clin Immunol (2014) 134(6):1354–64.e6. doi: 10.1016/j.jaci.2014.07.019
89. Chen R, Giliani S, Lanzi G, Mias GI, Lonardi S, Dobbs K, et al. Whole-exome sequencing identifies tetratricopeptide repeat domain 7A (TTC7A) mutations for combined immunodeficiency with intestinal atresias. J Allergy Clin Immunol (2013) 132(3):656–64.e17. doi: 10.1016/j.jaci.2013.06.013
90. Kammermeier J, Lucchini G, Pai SY, Worth A, Rampling D, Amrolia P, et al. Stem cell transplantation for tetratricopeptide repeat domain 7A deficiency: long-term follow-up. Blood (2016) 128(9):1306–8. doi: 10.1182/blood-2016-01-696385
91. McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers (2015) 1:15071. doi: 10.1038/nrdp.2015.71
92. Giardino G, Radwan N, Koletsi P, Morrogh DM, Adams S, Ip W, et al. Clinical and immunological features in a cohort of patients with partial DiGeorge syndrome followed at a single center. Blood (2019) 133(24):2586–96. doi: 10.1182/blood.2018885244
93. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
94. Gennery AR, Slatter MA, Rice J, Hoefsloot LH, Barge D, McLean-Tooke A, et al. Mutations in CHD7 in patients with CHARGE syndrome cause T-B + natural killer cell + severe combined immune deficiency and may cause Omenn-like syndrome. Clin Exp Immunol (2008) 153(1):75–80. doi: 10.1111/j.1365-2249.2008.03681.x
95. Markert ML, Alexieff MJ, Li J, Sarzotti M, Ozaki DA, Devlin BH, et al. Complete DiGeorge syndrome: development of rash, lymphadenopathy, and oligoclonal T cells in 5 cases. J Allergy Clin Immunol (2004) 113(4):734–41. doi: 10.1016/j.jaci.2004.01.766
96. Tezenas Du Montcel S, Mendizabai H, Ayme S, Levy A, Philip N. Prevalence of 22q11 microdeletion. J Med Genet (1996) 33(8):719. doi: 10.1136/jmg.33.8.719
97. Devriendt K, Fryns JP, Mortier G, van Thienen MN, Keymolen K. The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet (1998) 35(9):789–90. doi: 10.1136/jmg.35.9.789-a
98. Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet (1997) 34(10):798–804. doi: 10.1136/jmg.34.10.798
99. Fulcoli FG, Franzese M, Liu X, Zhang Z, Angelini C, Baldini A. Rebalancing gene haploinsufficiency in vivo by targeting chromatin. Nat Commun (2016) 7:11688. doi: 10.1038/ncomms11688
100. Du Q, de la Morena MT, van Oers NSC. The Genetics and Epigenetics of 22q11.2 Deletion Syndrome. Front Genet (2019) 10:1365. doi: 10.3389/fgene.2019.01365
101. Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet (2004) 36(9):955–7. doi: 10.1038/ng1407
102. Janssen N, Bergman JE, Swertz MA, Tranebjaerg L, Lodahl M, Schoots J, et al. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat (2012) 33(8):1149–60. doi: 10.1002/humu.22086
103. Issekutz KA, Graham JM, Jr., Prasad C, Smith IM, Blake KD. An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am J Med Genet A (2005) 133A(3):309–17. doi: 10.1002/ajmg.a.30560
104. Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet (2011) 48(5):334–42. doi: 10.1136/jmg.2010.087106
105. Yutzey KE. DiGeorge syndrome, Tbx1, and retinoic acid signaling come full circle. Circ Res (2010) 106(4):630–2. doi: 10.1161/CIRCRESAHA.109.215319
106. Sulik KK, Johnston MC, Daft PA, Russell WE, Dehart DB. Fetal alcohol syndrome and DiGeorge anomaly: critical ethanol exposure periods for craniofacial malformations as illustrated in an animal model. Am J Med Genet Suppl (1986) 2:97–112. doi: 10.1002/ajmg.1320250614
107. Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet (2003) 362(9393):1366–73. doi: 10.1016/S0140-6736(03)14632-6
108. Hasegawa K, Tanaka H, Higuchi Y, Hayashi Y, Kobayashi K, Tsukahara H. Novel heterozygous mutation in TBX1 in an infant with hypocalcemic seizures. Clin Pediatr Endocrinol (2018) 27(3):159–64. doi: 10.1297/cpe.27.159
109. Daw SC, Taylor C, Kraman M, Call K, Mao J, Schuffenhauer S, et al. A common region of 10p deleted in DiGeorge and velocardiofacial syndromes. Nat Genet (1996) 13(4):458–60. doi: 10.1038/ng0896-458
110. Van Esch H, Groenen P, Fryns JP, Van de Ven W, Devriendt K. The phenotypic spectrum of the 10p deletion syndrome versus the classical DiGeorge syndrome. Genet Couns (1999) 10(1):59–65.
111. Bernstock JD, Totten AH, Elkahloun AG, Johnson KR, Hurst AC, Goldman F, et al. Recurrent microdeletions at chromosome 2p11.2 are associated with thymic hypoplasia and features resembling DiGeorge syndrome. J Allergy Clin Immunol (2020) 145(1):358–67.e2. doi: 10.1016/j.jaci.2019.09.020
112. Frank J, Pignata C, Panteleyev AA, Prowse DM, Baden H, Weiner L, et al. Exposing the human nude phenotype. Nature (1999) 398(6727):473–4. doi: 10.1038/18997
113. Romano R, Palamaro L, Fusco A, Giardino G, Gallo V, Del Vecchio L, et al. FOXN1: A Master Regulator Gene of Thymic Epithelial Development Program. Front Immunol (2013) 4:187. doi: 10.3389/fimmu.2013.00187
114. Pignata C, Fiore M, Guzzetta V, Castaldo A, Sebastio G, Porta F, et al. Congenital Alopecia and nail dystrophy associated with severe functional T-cell immunodeficiency in two sibs. Am J Med Genet (1996) 65(2):167–70. doi: 10.1002/(SICI)1096-8628(19961016)65:2<167::AID-AJMG17>3.0.CO;2-O
115. Adriani M, Martinez-Mir A, Fusco F, Busiello R, Frank J, Telese S, et al. Ancestral founder mutation of the nude (FOXN1) gene in congenital severe combined immunodeficiency associated with alopecia in southern Italy population. Ann Hum Genet (2004) 68(Pt 3):265–8. doi: 10.1046/j.1529-8817.2004.00091.x
116. Chou J, Massaad MJ, Wakim RH, Bainter W, Dbaibo G, Geha RS. A novel mutation in FOXN1 resulting in SCID: a case report and literature review. Clin Immunol (2014) 155(1):30–2. doi: 10.1016/j.clim.2014.08.005
117. Pignata C, Gaetaniello L, Masci AM, Frank J, Christiano A, Matrecano E, et al. Human equivalent of the mouse Nude/SCID phenotype: long-term evaluation of immunologic reconstitution after bone marrow transplantation. Blood (2001) 97(4):880–5. doi: 10.1182/blood.V97.4.880
118. Radha Rama Devi A, Panday NN, Naushad SM. FOXN1 Italian founder mutation in Indian family: Implications in prenatal diagnosis. Gene (2017) 627:222–5. doi: 10.1016/j.gene.2017.06.033
119. Paganini I, Sestini R, Capone GL, Putignano AL, Contini E, Giotti I, et al. A novel PAX1 null homozygous mutation in autosomal recessive otofaciocervical syndrome associated with severe combined immunodeficiency. Clin Genet (2017) 92(6):664–8. doi: 10.1111/cge.13085
120. Yamazaki Y, Urrutia R, Franco LM, Giliani S, Zhang K, Alazami AM, et al. PAX1 is essential for development and function of the human thymus. Sci Immunol (2020) 5(44). doi: 10.1126/sciimmunol.aax1036
121. Cirillo E, Cancrini C, Azzari C, Martino S, Martire B, Pession A, et al. Clinical, Immunological, and Molecular Features of Typical and Atypical Severe Combined Immunodeficiency: Report of the Italian Primary Immunodeficiency Network. Front Immunol (2019) 10:1908. doi: 10.3389/fimmu.2019.01908
122. Thaventhiran JED, Lango Allen H, Burren OS, Rae W, Greene D, Staples E, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature (2020) 583(7814):90–5. doi: 10.1038/s41586-020-2265-1
123. Fusaro M, Rosain J, Grandin V, Lambert N, Hanein S, Fourrage C, et al. Improving the diagnostic efficiency of primary immunodeficiencies with targeted next-generation sequencing. J Allergy Clin Immunol (2020) 147(2):734–7. doi: 10.1016/j.jaci.2020.05.046
124. Dvorak CC, Haddad E, Buckley RH, Cowan MJ, Logan B, Griffith LM, et al. The genetic landscape of severe combined immunodeficiency in the United States and Canada in the current era (2010-2018). J Allergy Clin Immunol (2019) 143(1):405–7. doi: 10.1016/j.jaci.2018.08.027
125. Tyler R, Yates JP, Morna J, Ziad Khatib D, Chen K, Hernandez-Trujillo V, et al. Investigational cultured thymus tissue transplantation (RVT-802) following failed stem cell transplantationin athymic patients. CIS Abstract (2019).
126. Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA (2014) 312(7):729–38. doi: 10.1001/jama.2014.9133
127. Argudo-Ramirez A, Martin-Nalda A, Marin-Soria JL, Lopez-Galera RM, Pajares-Garcia S, Gonzalez de Aledo-Castillo JM, et al. First Universal Newborn Screening Program for Severe Combined Immunodeficiency in Europe. Two-Years’ Experience in Catalonia (Spain). Front Immunol (2019) 10:2406. doi: 10.3389/fimmu.2019.02406
128. La Motte-Mohs RN, Herer E, Zuniga-Pflucker JC. Induction of T-cell development from human cord blood hematopoietic stem cells by Delta-like 1 in vitro. Blood (2005) 105(4):1431–9. doi: 10.1182/blood-2004-04-1293
129. Taqvi S, Dixit L, Roy K. Biomaterial-based notch signaling for the differentiation of hematopoietic stem cells into T cells. J BioMed Mater Res A (2006) 79(3):689–97. doi: 10.1002/jbm.a.30916
130. Shukla S, Langley MA, Singh J, Edgar JM, Mohtashami M, Zuniga-Pflucker JC, et al. Progenitor T-cell differentiation from hematopoietic stem cells using Delta-like-4 and VCAM-1. Nat Methods (2017) 14(5):531–8. doi: 10.1038/nmeth.4258
131. Bifsha P, Leiding JW, Pai SY, Colamartino ABL, Hartog N, Church JA, et al. Diagnostic assay to assist clinical decisions for unclassified severe combined immune deficiency. Blood Adv (2020) 4(12):2606–10. doi: 10.1182/bloodadvances.2020001736
132. Bosticardo M, Yamazaki Y, Cowan J, Giardino G, Corsino C, Scalia G, et al. Heterozygous FOXN1 Variants Cause Low TRECs and Severe T Cell Lymphopenia, Revealing a Crucial Role of FOXN1 in Supporting Early Thymopoiesis. Am J Hum Genet (2019) 105(3):549–61. doi: 10.1016/j.ajhg.2019.07.014
133. Du Q, Huynh LK, Coskun F, Molina E, King MA, Raj P, et al. FOXN1 compound heterozygous mutations cause selective thymic hypoplasia in humans. J Clin Invest (2019) 129(11):4724–38. doi: 10.1172/JCI127565
134. Deya-Martinez A, Flinn AM, Gennery AR. Neonatal thymectomy in children-accelerating the immunologic clock? J Allergy Clin Immunol (2020) 146(2):236–43. doi: 10.1016/j.jaci.2020.02.028
135. Gudmundsdottir J, Soderling J, Berggren H, Oskarsdottir S, Neovius M, Stephansson O, et al. Long-term clinical effects of early thymectomy: Associations with autoimmune diseases, cancer, infections, and atopic diseases. J Allergy Clin Immunol (2018) 141(6):2294–7.e8. doi: 10.1016/j.jaci.2018.01.037
136. Mengrelis K, Kucera F, Shahid N, Watt E, Ross S, Lau CI, et al. T cell phenotype in paediatric heart transplant recipients. Pediatr Transplant (2020), e13930. doi: 10.1111/petr.13930
137. Hale LP, Neff J, Cheatham L, Cardona D, Markert ML, Kurtzberg J. Histopathologic assessment of cultured human thymus. PLoS One (2020) 15(3):e0230668. doi: 10.1371/journal.pone.0230668
138. Markert ML, Sarzotti M, Ozaki DA, Sempowski GD, Rhein ME, Hale LP, et al. Thymus transplantation in complete DiGeorge syndrome: immunologic and safety evaluations in 12 patients. Blood (2003) 102(3):1121–30. doi: 10.1182/blood-2002-08-2545
139. Markert ML, Li J, Devlin BH, Hoehner JC, Rice HE, Skinner MA, et al. Use of allograft biopsies to assess thymopoiesis after thymus transplantation. J Immunol (2008) 180(9):6354–64. doi: 10.4049/jimmunol.180.9.6354
140. Markert ML, Devlin BH, Chinn IK, McCarthy EA, Li YJ. Factors affecting success of thymus transplantation for complete DiGeorge anomaly. Am J Transplant (2008) 8(8):1729–36. doi: 10.1111/j.1600-6143.2008.02301.x
141. Chinn IK, Devlin BH, Li YJ, Markert ML. Long-term tolerance to allogeneic thymus transplants in complete DiGeorge anomaly. Clin Immunol (2008) 126(3):277–81. doi: 10.1016/j.clim.2007.11.009
142. Kreins AY, Junghanns F, Mifsud W, Somana K, Sebire N, Rampling D, et al. Correction of both immunodeficiency and hypoparathyroidism by thymus transplantation in complete DiGeorge syndrome. Am J Transplant (2020) 20(5):1447–50. doi: 10.1111/ajt.15668
143. Mengel M, Jonigk D, Marwedel M, Kleeberger W, Bredt M, Bock O, et al. Tubular chimerism occurs regularly in renal allografts and is not correlated to outcome. J Am Soc Nephrol (2004) 15(4):978–86. doi: 10.1097/01.ASN.0000120369.92378.54
144. Spencer H, Rampling D, Aurora P, Bonnet D, Hart SL, Jaffe A. Transbronchial biopsies provide longitudinal evidence for epithelial chimerism in children following sex mismatched lung transplantation. Thorax (2005) 60(1):60–2. doi: 10.1136/thx.2004.029678
145. Li B, Li J, Devlin BH, Markert ML. Thymic microenvironment reconstitution after postnatal human thymus transplantation. Clin Immunol (2011) 140(3):244–59. doi: 10.1016/j.clim.2011.04.004
146. Li B, Li J, Hsieh CS, Hale LP, Li YJ, Devlin BH, et al. Characterization of cultured thymus tissue used for transplantation with emphasis on promiscuous expression of thyroid tissue-specific genes. Immunol Res (2009) 44(1-3):71–83. doi: 10.1007/s12026-008-8083-4
147. Ross S, Cheung M, Lau CI, Sebire N, Burch M, Kilbride P, et al. Transplanted human thymus slices induce and support T-cell development in mice after cryopreservation. Eur J Immunol (2018) 48(4):716–9. doi: 10.1002/eji.201747193
148. Haddad E, Hoenig M. Hematopoietic Stem Cell Transplantation for Severe Combined Immunodeficiency (SCID). Front Pediatr (2019) 7:481. doi: 10.3389/fped.2019.00481
149. Chhatta AR, Cordes M, Hanegraaf MAJ, Vloemans S, Cupedo T, Cornelissen JJ, et al. De novo generation of a functional human thymus from induced pluripotent stem cells. J Allergy Clin Immunol (2019) 144(5):1416–9.e7. doi: 10.1016/j.jaci.2019.05.042
150. Kreins AY, Graham Davies E. Replacing defective thymus function. Curr Opin Allergy Clin Immunol (2020) 20(6):541–8. doi: 10.1097/ACI.0000000000000695
151. Ronfard V, Vertes AA, May MH, Dupraz A, van Dyke ME, Bayon Y. Evaluating the Past, Present, and Future of Regenerative Medicine: A Global View. Tissue Eng Part B Rev (2017) 23(2):199–210. doi: 10.1089/ten.teb.2016.0291
152. Rama P, Matuska S, Paganoni G, Spinelli A, De Luca M, Pellegrini G. Limbal stem-cell therapy and long-term corneal regeneration. N Engl J Med (2010) 363(2):147–55. doi: 10.1056/NEJMoa0905955
153. Hirsch T, Rothoeft T, Teig N, Bauer JW, Pellegrini G, De Rosa L, et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature (2017) 551(7680):327–32. doi: 10.1038/nature24487
154. Ronfard V, Rives JM, Neveux Y, Carsin H, Barrandon Y. Long-term regeneration of human epidermis on third degree burns transplanted with autologous cultured epithelium grown on a fibrin matrix. Transplantation (2000) 70(11):1588–98. doi: 10.1097/00007890-200012150-00009
155. Anderson G, Jenkinson EJ. Fetal thymus organ culture. CSH Protoc (2007) 2007:pdb prot4808. doi: 10.1101/pdb.prot4808
156. Marshall D, Bagley J, Le P, Hogquist K, Cyr S, Von Schild E, et al. T cell generation including positive and negative selection ex vivo in a three-dimensional matrix. J Hematother Stem Cell Res (2003) 12(5):565–74. doi: 10.1089/152581603322448277
157. Poznansky MC, Evans RH, Foxall RB, Olszak IT, Piascik AH, Hartman KE, et al. Efficient generation of human T cells from a tissue-engineered thymic organoid. Nat Biotechnol (2000) 18(7):729–34. doi: 10.1038/77288
158. Senoo M, Pinto F, Crum CP, McKeon F. p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell (2007) 129(3):523–36. doi: 10.1016/j.cell.2007.02.045
159. Bonfanti P, Claudinot S, Amici AW, Farley A, Blackburn CC, Barrandon Y. Microenvironmental reprogramming of thymic epithelial cells to skin multipotent stem cells. Nature (2010) 466(7309):978–82. doi: 10.1038/nature09269
160. Wong K, Lister NL, Barsanti M, Lim JM, Hammett MV, Khong DM, et al. Multilineage potential and self-renewal define an epithelial progenitor cell population in the adult thymus. Cell Rep (2014) 8(4):1198–209. doi: 10.1016/j.celrep.2014.07.029
161. Bortolomai I, Sandri M, Draghici E, Fontana E, Campodoni E, Marcovecchio GE, et al. Gene Modification and Three-Dimensional Scaffolds as Novel Tools to Allow the Use of Postnatal Thymic Epithelial Cells for Thymus Regeneration Approaches. Stem Cells Transl Med (2019) 8(10):1107–22. doi: 10.1002/sctm.18-0218
162. Fan Y, Tajima A, Goh SK, Geng X, Gualtierotti G, Grupillo M, et al. Bioengineering Thymus Organoids to Restore Thymic Function and Induce Donor-Specific Immune Tolerance to Allografts. Mol Ther (2015) 23(7):1262–77. doi: 10.1038/mt.2015.77
163. Hun M, Barsanti M, Wong K, Ramshaw J, Werkmeister J, Chidgey AP. Native thymic extracellular matrix improves in vivo thymic organoid T cell output, and drives in vitro thymic epithelial cell differentiation. Biomaterials (2017) 118:1–15. doi: 10.1016/j.biomaterials.2016.11.054
164. Tajima A, Pradhan I, Geng X, Trucco M, Fan Y. Construction of Thymus Organoids from Decellularized Thymus Scaffolds. Methods Mol Biol (2019) 1576:33–42. doi: 10.1007/7651_2016_9
165. Bredenkamp N, Ulyanchenko S, O’Neill KE, Manley NR, Vaidya HJ, Blackburn CC. An organized and functional thymus generated from FOXN1-reprogrammed fibroblasts. Nat Cell Biol (2014) 16(9):902–8. doi: 10.1038/ncb3023
166. Parent AV, Russ HA, Khan IS, LaFlam TN, Metzger TC, Anderson MS, et al. Generation of functional thymic epithelium from human embryonic stem cells that supports host T cell development. Cell Stem Cell (2013) 13(2):219–29. doi: 10.1016/j.stem.2013.04.004
Keywords: thymus transplantation, primary immunodeficiency, DiGeorge syndrome, severe combined immunodeficiency (SCID), regenerative medicine, PAX1, FOXN1
Citation: Kreins AY, Bonfanti P and Davies EG (2021) Current and Future Therapeutic Approaches for Thymic Stromal Cell Defects. Front. Immunol. 12:655354. doi: 10.3389/fimmu.2021.655354
Received: 18 January 2021; Accepted: 03 March 2021;
Published: 18 March 2021.
Edited by:
Claudio Pignata, University of Naples Federico II, ItalyReviewed by:
Ana E. Sousa, University of Lisbon, PortugalFrank Staal, Leiden University Medical Center, Netherlands
Anna Villa, National Research Council (CNR), Italy
Copyright © 2021 Kreins, Bonfanti and Davies. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandra Y. Kreins, YS5rcmVpbnNAdWNsLmFjLnVr