 Khalil Khalaf1,2*
Khalil Khalaf1,2* Doris Hana1,2
Doris Hana1,2 Jadzia Tin-Tsen Chou1,2Chandpreet Singh1,2Andrzej Mackiewicz1,2
Jadzia Tin-Tsen Chou1,2Chandpreet Singh1,2Andrzej Mackiewicz1,2 Mariusz Kaczmarek1,2
Mariusz Kaczmarek1,2- 1Department of Cancer Diagnostics and Immunology, Greater Poland Cancer Center, Poznań, Poland
- 2Department of Cancer Immunology, Poznan University of Medical Sciences, Poznań, Poland
The tumor microenvironment (TME) is a complex and ever-changing “rogue organ” composed of its own blood supply, lymphatic and nervous systems, stroma, immune cells and extracellular matrix (ECM). These complex components, utilizing both benign and malignant cells, nurture the harsh, immunosuppressive and nutrient-deficient environment necessary for tumor cell growth, proliferation and phenotypic flexibility and variation. An important aspect of the TME is cellular crosstalk and cell-to-ECM communication. This interaction induces the release of soluble factors responsible for immune evasion and ECM remodeling, which further contribute to therapy resistance. Other aspects are the presence of exosomes contributed by both malignant and benign cells, circulating deregulated microRNAs and TME-specific metabolic patterns which further potentiate the progression and/or resistance to therapy. In addition to biochemical signaling, specific TME characteristics such as the hypoxic environment, metabolic derangements, and abnormal mechanical forces have been implicated in the development of treatment resistance. In this review, we will provide an overview of tumor microenvironmental composition, structure, and features that influence immune suppression and contribute to treatment resistance.
Introduction
Experimental observations of tumorigenesis show that tumor cells transition from being transformed and benign to an invasive malignant state. This process is the result of genome instability, in which cells lose their ability to fully differentiate and mature, resulting in the loss of contact inhibition (1).
Various studies have shown that a large majority of cancer-related deaths were attributed to distant metastasis (2, 3). Stephen Paget was the first to hypothesize on what he described as his “seed and soil” theory. In this hypothesis, tumor cells with metastatic potential (i.e. the seed) were inclined to migrate towards specific sites that nurtured and enhanced growth sites (i.e. the soil). This is the earliest publication hypothesizing the importance of the “tumor microenvironment” (TME) in the development of metastases (4). It is known from extensive literature that the metastatic cascade starts with tumor cell dissociation from the cancer niche, followed by extravasation into capillary and lymphatic systems and along nerves, all the while evading immune surveillance. This process culminates in the invasion of distant sites (5). However, metastatic potential develops long before the tumor is ever detected. In the initial stages of primary tumor formation, the accumulation of both genetic and genomic instabilities lead to the development of phenotypic variants with metastatic capacity (6). Furthermore, these variants have the ability to resist apoptosis and circumvent immune defenses by using various soluble factors that are released by malignant and non-malignant tumor-supporting cells (7). These variants in combination with said soluble factors constitute what is now known as the TME.
Additionally, the TME induces chemotherapeutic resistance through acquired or de novo mechanisms. In acquired multi-drug resistance (MDR), the expression of ATP-binding cassettes (ABCs), oncogene activation, and tumor-suppressor gene deregulation are achieved via cellular crosstalk and cell-to-TME-matrix interaction. Previously-exposed cancer cells acquire phenotypic changes that lead to resistance to subsequent therapy (8). On the other hand, in de novo resistance, it has been shown that after exposure to therapy, stromal tissue within the TME provides refuge to a subpopulation of cancer cells and renders them chemo-resistant by inducing stemness (9).
The vast arsenal that is weaponized by the TME in the course of neoplastic disease is currently the topic of great research interest, and the available literature is daunting for researchers and practicing oncologists alike. This review aims to present an overview of the cells and structure of the TME, and its unique characteristics that induce drug resistance and metastasis that remain significant challenges in the treatment of cancer.
The Tumor Microenvironment and Its Role in Immune Surveillance
Stem cells (SCs) are unspecified cells with the ability to differentiate into multiple cell types to maintain tissue homeostasis. They reside in a specific microenvironment called a stem cell niche, which consists of and is sustained by different soluble factors (10). Tissue homeostasis is balanced and maintained in a way that prevents SC depletion and overactive proliferation. This is achieved by choosing alternate fates: the SC is selected for senescence (i.e. death), or self-renewal (i.e. proliferation) through interactions with other cells and molecular signals within the microenvironment (11). Just like the stem cell niche of healthy tissues, the tumor microenvironment (TME) is very heterogeneous and is a complex component of solid tumors. The TME comprises a diverse cellular and acellular milieu in which cancer stem cells (CSCs) develop and thrive, and various stromal and immune cells are recruited to form and maintain this self-sustained environment (12). Stromal and tumor cell crosstalk has been recognized as crucial for the promotion of a well-organized TME, leading to effective immune evasion, ECM remodeling, and angiogenesis (7).
Cells and Components of the TME Involved in Suppression of the Anti-Tumor Response
Stromal Cells
Vascular and Lymphatic Endothelial Cells
Neo-angiogenesis is promoted by both tumor and endothelial cells (ECs). Both vascular and lymphatic systems are implicated in early metastasis, with soluble VEGFA promoting vascular EC proliferation, while VEGFC, VEGFD, and VEGFR-3 promoting lymphatic EC proliferation (13, 14).
Tumor angiogenic vessels are either derived from endothelial progenitor cells or from existing vessels that propagate to feed growing tumors (15). The ECs present within the TME possess abnormal pericytes and pericyte coverage which enables leaks between tight junctions. This directly leads to the systemic circulation of tumor cells, i.e. presence of CTCs, thus increasing the tumor’s metastatic potential (16). Hypoxia triggers stromal release of VEGF. Subsequent activation of VEGF-2 receptors on adjacent ECs promotes their migration to the region of hypoxia and production of hypoxia-inducible factors 1 (HIF-1) and 2 (HIF-2) (17) ultimately leading to EC proliferation, migration and maturation (18, 19). The result is tumor endothelial anergy, the cellular non-response to pro-inflammatory stimulation (i.e. IFN-γ, TNF-α, and IL-1). As vital gatekeepers of the TME, tumor endothelial cells (TECs) are the primary barrier to immune-stimulatory cells which promote the loss of anti-cancer immunity (20–23), TME-derived cytokines such as VEGF, ET1, FGF-2, and EGFL7 function to inhibit tumor endothelial ability to upregulate the expression of chemoattractants (i.e. CXCL7, CXCL10, IL-6, and CCL2) and adhesion molecules (ICAM1 and VCAM1), consequently promoting immunosuppression and tumor progression (20, 23–26). Additionally, TECs were shown to promote regulatory T cell (Treg) accumulation via up-regulation of the lymphatic and vasculature endothelial receptor 1 (CLEVER-1); an abundance of CLEVER-1-positive macrophages support immunosuppression. It has been reported that tumor-induced CLEVER-1 expression in both macrophages and endothelial cell populations was required to support the growth of melanoma, and that the chief driver was the diminished expression of vascular E- and P-selectin, and accumulation of Tregs and M2 macrophages in the tumors induced by CLEVER-1 (27, 28).
ECs can selectively upregulate T cell inhibitory receptors including: IDO1, TIM3, B7-H3, B7-H4, PD-L1 and PD-L2 (29–32) along with other soluble inhibitory molecules such as: TGF-β, IL-6, IL-10, and PGE2 (33–35), thus maintaining a constant inflammatory state within the TME. ECs may also express apoptosis-inducing molecules such as TNF-related apoptosis-inducing ligand (TRAIL) and FasL which were shown to selectively extinguish effector T cells, sparing Tregs (36–40). Thus the tumor vasculature inhibits immune cell extravasation in the tumor bed and promotes the immunosuppressive state, and is one of the main modulators in immune resistance (41–43).
Mesenchymal Stem Cells
As important contributors to the TME, mesenchymal stem cells (MSCs) harvested from different tissues have demonstrated varying expression levels of factors that contribute to embryonic stem cell pluripotency, such as SOX-2, NANOG and OCT-4 (44, 45). MSCs are dispatched by a series of paracrine signaling pathways in response to injury, and either differentiate on-site to replenish damaged tissue with their cell multipotency (10, 46) or activate various trophic factors necessary to activate local SCs specific to the tissue (47), for the purposes of wound healing. The TME continuously recruits MSCs by generating constant inflammation, similar to that seen in wound healing, and is thus about to remodel itself perpetually (48, 49). Thus MSCs are able to populate the TME with other crucial cells such as pericytes and fibroblasts with their multipotency (50). In addition to the aforementioned, MSCs are involved in other cancer-promoting mechanisms. MSCs release specific molecules such as epidermal growth factors (EGFs) (51), IL-8/IL-6 cytokines (52) and CXCL1/2/12 chemokines (53) which directly act on cancer cells in a paracrine fashion and increase cellular proliferation by induction of phenotypic modification. In another immune suppressor mechanism, MSCs were shown to suppress both adaptive and innate immunity by directly inhibiting CD4 and CD8 T cell proliferation (54). A third mechanism includes stimulation of TLRs3/4 present on MSCs, inducing production of CXCL10, IL-8 and IL-6 which are crucial for T cell suppression (55). Furthermore, via adhesion to Th17 via CCL20, MSCs are capable of inducing T cell differentiation to Tregs thus suppressing both innate and adaptive immunity (56). MSCs also promote tumor revascularization by a) secreting various angiogenic factors such as EGF and VEGF, which are responsible for recruitment of ECs for vasculature maturation (57) or b) by converting into endothelial-like cells to modulate neo-angiogenesis (58). MSCs have also been shown to possess the ability to differentiate into tumor stromal progenitor cells, such as cancer-associated fibroblasts (CAFs), which further enhance the development and sustenance of the TME (59). MSCs have been demonstrated to be involved in the production of inflammatory chemokine CCL5, which is responsible for metastatic potential in breast cancer (60). MSCs are capable of impeding all immune responses through interactions with every cell in the immune system, directly or via soluble immune secretomes (40) such as:
prostaglandin E2 (PGE2) - PGE2 suppresses IL-2 formation and T cell function. The literature also suggests that PGE2 regulates the balance between different helper T cell (Th) configurations and responses, solely inhibiting Th1 IFN-γ production (61). PGE2-suppression of Th1 results from its ability to repress IL-12 production in dendritic cells (DCs), and monocytes (62, 63). Additionally, PGE2 is required for the development of myeloid-derived suppressor cells (MDSCs) (64) and tumor-associated macrophages (TAMs) (65). MDSCs express high levels of COX2, a major source of PGE2. The positive feedback between COX2 and PGE2 promotes MDSC stability, and leads to the production of additional MDSC-associated suppressive mediators (64). HIF-1-α also mediates and likely initiates a signaling cascade in PGE2-mediated MDSC development (66).
Indoleamine 2,3-dioxegynase (IDO) - Cells expressing IDO can suppress immunity by catabolizing tryptophan (Trp) and other indole compounds (67). Potent IDO inducers IFN-I and IFN-II are produced at sites of inflammation. IDO is also expressed by DCs, resulting in DC conversion to tolerogenic antigen-presenting cells (APCs) that suppress effector T cells (Teff) whilst promoting Tregs. Non-catalytic signaling induces TGF-β release by a subset of DCs, leading to tolerance. Tolerogenic IDO promotes tumorigenesis by allowing cancer cells to evade immune surveillance. Some cells express IDO1 genes which deplete Trp and generate bioactive catabolites such as kynurenines (Kyn). This is sensed by a population of immune cells, leading to suppression of innate and adaptive immunity (68).
Nitric oxide (NO) - a pleiotropic and short-lived radical which has pathophysiological functions. Produced by MSCs, NO is responsible for mediating T cell-dependent immunosuppression (69). MSCs express compounds such as arginase, β2 integral, Gr-1 granulocyte marker, and inducible nitric oxide synthase (iNOS) which converts L-arginine into urea and L-ornithine (70). This hints at a potential synergy between arginase and iNOS which would result in superoxide (O2-). O2- then may react with NO to produce peroxinitrite (ONOO-) as well as other reactive nitrogen intermediates which induce T cell apoptosis (71). Another immunosuppressive mechanism is the high NO-concentration impairment of IL-2-R-induced signaling. This leads to the activation of Janus kinases 1 (JAK1) and 2 (JAK2), with signal transducer and activator of transcription factor 5 (STAT5) (72).
Cancer-Associated Fibroblasts
Cancer-associated fibroblasts (CAFs) produce key proteins such as periostin and Tenascin-C which are necessary for tumor support and metastasis (73, 74). Their expression in the TME changes the predominant cell type in the stromal tissue as well as the modulator of the ECM. It has been shown that CAFs placed with normal prostate cells in vitro induces rapid cell growth and alters prostate cell histology (75). The histological changes may be the result of CAFs’ ability to induce epithelial-to-mesenchymal transition (EMT) via upregulation of TGF-β, which modifies cellular cytoskeleton architecture, cyclin-dependent kinases, and decreases the potency of immune surveillance (76). This subsequently enables cellular migration and invasion, and induces the development of pluripotent tumor cells (77, 78). This is evident with the demonstration that growth factors such as CCL2 and hepatocyte growth factor (HGF) induced CSC renewal and stemness of cancer cells in both breast (79) and hepatocellular carcinoma (80). Another mechanism of stemness is the upregulation of the NF-κB signaling pathway. This prompts continuous secretion of pro-inflammatory cytokines such as IL-6 and IL-8; this constant inflammatory milieu induces EMT (81). The importance of IL-6 has been previously elucidated. Increased expression of IL-6 in myeloma cells induces activation of the JAK2-STAT3 pathway (82) and increases expression of Bcl-Xc which correlates with resistance to therapy (83). In early TME development, the ECM is reconstructed in a stiffened manner (84); the elastin component of the ECM is cross-linked with collagen in the presence of lysyl oxidase (LOX) and these are both produced by CAFs (85). CAFs are also responsible for the secretion of fibroblast growth factor-2 (FGF-2), an essential signaling molecule responsible for angiogenesis (86), and expresses stromal-derived factor-1 (SDF-1) which induces metastasis in breast cancer by acting as a chemotactic factor for circulating ECs (87). The role of Chi3L1, a non-enzymatic chitinase-3 like-protein 1, has also been studied. Regulated by the ECM, it binds to heparin, hyaluronic acid, and chitin, and is synthesized by a variety of cells including tumor cells, fibroblast-like cells, smooth muscle cells, chondrocytes, macrophages, neutrophils, and synoviocytes (88). Genetic targeting of CAF-derived Chi3L1 in fibroblasts has attenuated recruitment and reprogrammed macrophages to an M2 phenotype, which promotes a Th1 phenotype in the TME (89). Additionally, the TAM polarization to the M2 phenotype was shown to be induced by high expression of TGF-β (90). In an ex vivo model of oral squamous cell carcinoma, CAFs promoted the development of an M2-like phenotype from CD14 myeloid cells after induction by IL-10, TGF-β, and ARG1. This ultimately suppresses T cell proliferation (91). With their reciprocal interactions, TAMs and CAFs are central immunosuppressive players in the TME. Notably, CAFs recruit macrophages through the expression of stromal cell-derived factor 1 (SDF-1/CXCL12). SDF-1 magnifies M2 polarization of macrophages mirroring high production of immunosuppressive cytokine IL-10 (92–94).
Pericytes
Arising from differentiated mesenchymal precursors, pericytes are recruited when cancer cells overexpress platelet-derived growth factor beta (PDGF-β) (95) in both healthy and neoplastic tissues alike. They exhibit many tumor-supporting mechanisms including the release of EC-attracting soluble factors, which rapidly induces revascularization of the TME (96). In addition to their angiogenic properties, pericytes express the cluster of differentiation (CD) markers of MSCs. Their potential for multipotency contributes to metastatic processes by generating other stromal cells for the TME (97). Furthermore, pericytes have been shown to induce immune suppression through secretion of various soluble factors including prostaglandin E2 (PG-E2), TGF-β and nitric oxide (98). Pericytes are capable of regulating T cell trafficking and modulation. Pericytes of the TME were shown to express PD-L1 and PD-L2, responsible for T cell exhaustion (99). Retinal pericytes, too, exhibit immunosuppressive properties. When pericytes were cultured with activated T cells, production of IFN-γ and TNF-α decreased. Pericytes coexpressing CD248, CD90, and PDGFR isolated from human gliomas were able to inhibit cytotoxic T cell (CTL) proliferation, and thus induce immunosuppression within the TME (98, 100). Additionally, pericytes from normal brain tissue and malignant gliomas secrete immunosuppressive factors such as: PGE2, TGF-β, and NO, previously shown to inhibit anti-tumor response and suppressed mitogen-activated T cell activity (98). Pericytes produce growth factors, chemokines, cytokines, and adhesion molecules which regulate the microenvironmental ability to evade immune surveillance.
Cancer Stem Cells
The majority of cancer cells arise from cancer stem cells (CSCs) that express surface markers similar to that of stem cells (SCs), such as CD44, CD90 and CD133. It is uncertain whether CSCs arise from non-SCs or from somatic SCs (101, 102). The tumorigenic potential of CSCs was shown when leukemia-initiating SCs from AML patients were transplanted into severe combined immunodeficiency (SCID) mice, which later developed AML (103). In another study, CSCs and a non-CSC counterpart were injected into immunodeficient mice; only the CSC-injected mice were able to repopulate parental tumor cells (104). The theory of CSC is further supported by their discovery in breast, brain, colon, hematopoietic and lung cancer (101, 105). As the architects of the TME, CSCs are able to self-renew and drive the pathophysiologic mechanisms of carcinogenesis aided by various non-cancerous cells (101). CSCs possess both plasticity and immunomodulatory features capable of evading immune surveillance, thus they are the most distinguished malignant cell unit implicated in primary cancer or in resistance to immunotherapy. Bidirectional release of cytokines, cell-to-cell communication via extracellular vesicles, and fusion of CSCs with fusogenic stromal cells are mechanistic immunomodulatory properties of CSCs. Recent studies suggest that CSCs are pivotal players in immune escape: due to their immunomodulating nature, they are capable of cellular dormancy whilst evading immunosurveillance (106, 107). The tumor niche consists of intratumor immune cell populations which interact with CSCs and affect their functional status (108, 109). Undergoing cell-to-cell fusion (a process which generates tumor cell hybrids under pathological conditions) with various sorts of microenvironmental fusogenic cells such as: fibroblasts, macrophages, MDSCs and MSCs, the tumor niche contributes to the formation of aberrant cells that possess SC-like properties and are correlated with tumor initiation, progression, and metastatic potential (110, 111). CSC-related immune escape mechanisms are further complicated by epigenetic perturbations (112). Epigenetic modifications of differentiated cancer cells and CSCs can lead to expression modifications in immune-related genes. This domino effect impacts antigen presentation, processing, and immune evasion. For example, re-expression may be possible through demyelinating agents, allowing for immunotherapeutic applications (113). CSCs contribute to metastasis and tumor heterogeneity, implying their capacity for resistance to chemo-, radio-, and immunotherapies, and more besides (114). The principal limitation of efficacious anti-CSC treatment is the challenge in recognizing CSC-characteristic biomarkers.
Immune Cells
With regards to carcinogenesis, immune cells possess dual action dependent upon various chemokine expressions within the TME. It has been previously shown that Tregs, M2 macrophages and T-helper 2 cells (Th2) support tumorigenesis while NK cells, antigen-presenting cells (APCs), cytotoxic T cells (Tcs, CTLs) and M1 macrophages are protective against tumor development. High expression of chemokines such as CXC (1-16), with their respective CXC receptors (CXC-R), attracts various cancer-supporting immune cells that have been shown to be responsible for poorer prognosis in colorectal cancer (115).
Macrophages possess critical phagocytic properties in the adaptive and innate systems. Tumor-associated macrophages (TAMs) are derived from CCR2 inflammatory monocytes, and are classified as either pro-inflammatory (M1) anti-cancer cells through the production of IL-1 and tumor-necrosis factor alpha (TNF-α) (116) or anti-inflammatory (M2) cancer-supporting cells through the production of immunosuppressive cytokines such as IL-10 (117). M2 macrophages have been linked to progression in colon, renal cell and breast carcinomas (118–120) via multiple mechanisms. Primarily, anti-inflammatory cytokines and chemokines cause immune suppression by inhibiting T cells and NK cells (121, 122); chemokines CCL5, CCL20 and CCL22 recruit Tregs and activate their inhibitory actions via production of IL-10 and TGF-β1 (123). Secondly, angiogenesis is induced by the release of signaling protein WNT7B, which targets ECs for stimulation of VEGF (124). This produces another major angiogenic factor called pro-matrix metalloproteinase 9 (proMMP9) (125). M2 macrophages also facilitate carcinogenesis and metastasis through the production of CCL18 (126) and the nuclear factor- κB/FAK pathway (127) leading to induction, migration, invasion, and the EMT. Lastly, TNF is a product of both activated macrophages and the cells of the TME; in addition to being an anti-cancer cytokine, it has been implicated in the inflammatory process necessary for tumor growth (128). The role of STAT3 as a mediator between TAMs and tumor cells has been elucidated, showing that STAT3 activation inhibited Th1 subtype differentiation by blocking the expression of immune-stimulatory mediators (129).
Like TAMs, tumor associated neutrophils (TANs) demonstrate two subtypes: the N1 TAN phenotype which possesses anti-tumor action, and the N2 TAN phenotype which has tumor-support activity (130). Sustained inflammation induces an IL-8-dependent neutrophil chemotaxis within the TME (131). As previously described, TGF-β was shown to be highly expressed within the TME, inducing a generalized immunosuppressive state; additionally it was shown to polarize TANs into the N2 phenotype (130). N2 TANs sustain inflammation within the TME by releasing genotoxic elements such as NO and ROS (131). Tumor models have shown that N2-TAN-mediated immune suppression was achieved through various mechanisms: 1) production of TNF-α and NO to induce T cell apoptosis (132); 2) inhibition of T cell proliferation through modulation of PD-1/PD-L1 signaling and release of arginase (133); 3) N2-TAN expression of TGF-β, and 4) production of CCL17, shown to recruit Tregs to further induce an immunosuppressive state (134).
T cells, part of the adaptive immune system, prevent tumor growth through lytic action and the production of IFN-γ-dependent cell-cycle arrest (135). After lysis, the cell component is phagocytosed and expressed on APCs, exposing them to maturing lymphocytes and resulting in tumor suppression (136). Tregs impede the immune response by expressing various cytokines against anti-tumor cells. It has been shown that when the Treg-to-CD8 ratio is high in hepatic (137) and breast carcinoma (138), this results in uncontrolled progression and worse prognosis. Th2 is yet another cell responsible for promoting the necessary inflammatory state within the TME, and it has since been proposed as an agent in tumor progression. Countering the anti-tumor activity of Th1, Th2 has been associated with poorer prognosis when detected (128). Its differentiation is driven by thymic stromal lymphopoietin (TSLP), an IL-17-like cytokine produced in response to TNF-α and IL-1-β from TAMs and TME stromal cells (139).
APCs are innate cells which process and display antigens bound to major histocompatibility complexes (MHCs) to naïve T cells to induce cytotoxicity. They are categorized into professional (dendritic cells; DCs) and non-professional (fibroblast) APCs. It has been previously shown that because of the presence of IL-6 and granulocyte-colony stimulating factors (G-CSF), APCs of the TME lack the co-stimulatory receptor B7 and cannot stimulate T cell cytotoxicity. This alters the differentiation of APC to mature cells (140). Additionally, various signals within the TME induces differentiation of granulocytes to immunosuppressive cells such as TAMs and tumor-associated neutrophils (TANs) (141).
NK cells are an important innate component responsible for destroying tumor cells and preventing the progression of tumorigenesis. In the immunocompetent, NK cells select out the APCs with improper expression of MHC-I and retain a pool of competent APCs (142). However NK activation is greatly inhibited within the TME due to excess production of TGF-β and other anti-inflammatory cytokines and chemokines (143). Microarray analysis of extra-tumoral and intra-tumoral NK cells in the lung tumor microenvironment demonstrated upregulation of cytotoxic gene expression, and intra-tumoral NK cells were associated with better prognosis (144).
B cells are most common in draining lymph nodes. They have been shown to infiltrate tumor margins and have been associated with proper antibody response in ovarian and breast carcinomas (145, 146). On the other hand, B cells have been shown to differentiate into another tumor-associated cell. An IL-10-secreting B cell named Breg (147) promoted metastasis of breast cancer (148) and it has been shown to be implicated in inflammation-induced squamous cell carcinoma through the secretion of TNF-α in animal models (149). It should be noted that this B cell was non-infiltrating – that is, present only in the surrounding tissue – thus further studies are warranted to determine if they behave the same way in human cancers.
Little is known of myeloid-derived suppressor cells (MDSCs). They are identified as immature myeloid cells that are upregulated in cancer and other inflammatory processes (150). Their phenotype is variable and their characterization is difficult. MDSCs can also differentiate into TAMs, as they both possess immunosuppressive markers such as CD115 and F4/80 (151). It has been shown that MDSCs are able to directly suppress CD8 cells by producing nitric oxide synthase-2 (NOS-2) and arginase (ARG-1) (71). Another immunosuppressive mechanism exhibited by MDSCs is their positive effect on T cell differentiation into cancer-supporting Tregs (152).
Dendritic cells (DCs), the so-called professional APCs, are among the first cells to appear during inflammatory states. Varying subsets of DC maturation have been observed in the TME (153); this typically comprises of only a few mature DCs and is associated with better prognosis (154). Generally, the previously described immunosuppressive states impair DC maturation and activation (155). As stated, the DC maturational stage is crucial for normal function. Multiple subsets have been identified including anti-tumor classical DCs with high CD8 and NK cell-activation activity (156); while plasmacytoid DCs (157, 158) and monocyte-differentiating DCs have either immune-supportive or immune-suppressive actions (153, 159). The known immune suppressor PD-L1 is highly expressed within the TME. Tumor derived factors directly increase the expression of PD-L1 in DCs and MDSCs, further inhibiting anti-tumor immunity (160).
Cancer-Associated Adipocytes
Adipocytes are known, key contributors to the TME and are proposed to be involved in the metastatic process, angiogenesis, and resistance to apoptosis (161). Cancer-associated adipocytes (CAAs) are a broad grouping of the following: intratumoral adipocytes, peritumoral adipocytes, recruited adipocytes, and de novo differentiation of MSCs into adipocytes or adipocyte-like cells that store large amounts of energy-rich lipids (162). It has been shown that mature adipocytes incubated with breast cancer cells induced phenotypic change of adipocytes into fibroblast-like cells that contributed to the expansion of CAFs, well-known immune suppressors (163). CAAs can influence the TME through direct contact with adjacent cells or in a paracrine manner through the production of adipokines, hormones and proinflammatory cytokines (i.e. CCL6, CCL2, CCL5, MMP, VEGF, TNF-α, insulin and leptin, to name a few) to facilitate cancer invasion and immune resistance (164). CAAs were shown to possess dysfunctional proinflammatory features that support the TME (165). CCL2 and CCL5 released from CAAs were shown to recruit and promote M2 polarization of macrophages (166). Furthermore, the high concentration of TNF-α and IL-6 mediated JAK2/STAT3 pathway activation to induce phenotypic change into breast cancer cells with SC properties (164). The important adipokine leptin was also shown to make use of the JAK-STAT3 pathway to induce cancer stemness and evade immune surveillance (167). In cachectic mice, phenotypic change in white adipocytes with overexpression of uncoupling protein 1 (UP-1) induced their differentiation into brown adipocytes with fibroblastic characteristics (168). Furthermore, signaling proteins within the TME (i.e. IL-6, exosomal contents, and parathyroid hormone related peptide PTHrP) were shown to promote phenotypic variations into brown adipocytes (169). Because PD-L1 is strongly expressed on brown adipocytes, PTHrP has been linked to tumor invasion and metastasis. Phenotypic variations leading to the differentiation from white to brown adipose tissue appears to be another immunosuppressive mechanism (170).
Extracellular Matrix
The extracellular matrix (ECM) contributes the largest component of the TME and is composed of proteins such as collagen, proteoglycans, hyaluronic acid and laminins (171). The ECM is crucial for the maintenance of the TME and the induction of metastasis. Aside from acting as a physical cellular scaffold, it is responsible for cellular adhesion and migration out of the TME. It stores various soluble factors such as angiogenic factors and chemokines that induce a continuous inflammatory state, resulting in expansion of the cellular repertoire (172). The continuous inflammatory state exacerbates the conversion of stromal fibroblasts into myofibroblasts (173) which in turn deposit large amounts of growth factors and ECM proteins, inducing contraction and increasing stiffness (174). Newly-deposited ECM proteins are acted upon by CAF enzymes such as LOX to further stiffen the ECM; stored growth factors are subsequently released to amplify the circuitry between the tumor cells and its ECM. This eventually contributes to metastasis and ensures ECM resistance to treatment (175). The ECM can influence the recruitment of immune cells into the TME. For instance, the ECM can drive PI3K/AKT (pro-survival pathway) activation, which facilitates CSC immune evasion (176). ECM proteins can also recruit immunosuppressive cells such as Tregs and TAMs which were shown to promote CSC survival while blocking anti-tumorigenic immune cell (i.e. CTL) recruitment (177–179). The ECM is capable of impairing the proliferation and activation of T cells, which are responsible for eliminating CSCs (180). The composition of the ECM also plays a crucial role in modulating the state of tumor infiltrating immune cells. For example, M2 polarization of macrophages is achieved in a periostin-rich or stiff collagen-rich ECM (181). After recruitment, CSC survival signaling pathways such as Src, STAT3/SOX2, Hedgehog, and NF-κB are activated by the M2 macrophages, leading to inhibition of T cell proliferation and activation through type I collagen-dependent fusion of LAIR receptors while sequestering T cell proliferation growth factors (177). In addition to the aforementioned, neutrophils and TAMs are capable of selectively recognizing the EMC in order to promote cancer growth as they are recruited to the microenvironment (182, 183). This implies the ability of the ECM to modulate immune surveillance in the CSC microenvironment.
An increase in metabolic stress and hypoxia leads to poor diffusion in ECM-rich tumors, ultimately up-regulating immunosuppressive factors such as: CCL22, CCL18, TGF-β, IL-10, VEGF-B, and PGE2 (184–186). TGF-β in particular acts as a suppressor of CD8 CTLs and NK cells in the TME by attracting Tregs and functioning as an M2-polarizing agent for macrophages (186–188). Both of these phenomena negatively regulate infiltration and activity of CTLs (189). In addition, T cells are suppressed by VEGF-A which recruits Tregs that express NRP1 (a coreceptor of VEGF) (190, 191).
Micro-RNA Deregulation
Micro-RNAs (miRNAs, miRs) are an endogenously-expressed class of non-coding single-stranded RNA fragments that are involved in gene expression modulation. By targeting mRNA at the post-transcriptional level, they may act as tumor suppressors or oncogenes (192). When deregulated they are associated with tumorigenesis and metastatic development (193). Oncogenic miR-21 overexpression in CAFs has been associated with tumor aggressiveness via induction of angiogenesis and treatment resistance (194, 195). MiR-155 and miR-210 over-secretion in cancer cells has been shown to induce the transition of MSCs and normal fibroblasts to CAFs, thus reinforcing the TME (196, 197). Overexpression of miR-17-to-92 may lead to downregulation of tumor suppressor genes, much like with oncogenes, inhibiting apoptosis via various pathways (198). Other types of miRNAs include the tumor suppressors comprising of miR-126, whose main function is to suppress MSC recruitment in the TME by inhibiting SDF-1 and CCL2. When downregulated, miR-126 has been shown to promote breast cancer metastasis by inducing fibroblast recruitment and EMT (199). MiRNAs represents another hurdle to consider when evaluating TME defenses. Cancer cell-derived immune modulatory miRNAs regulate a multitude of immune components such as CTLs, Tregs, NK cells, DCs, and MDSCs via intracellular communication (i.e. micro vesicles and exosomes). Cancer-derived miRNAs have been implicated in various mechanisms to induce immune evasion. This is achieved through the modulation of expression profiles using histone modification and DNA methylation (200). It has been shown that these epigenetic pathways occur simultaneously and act on each other, i.e. DNA methylation- or histone acetylation-induced deregulation of miRNAs, and vice versa (201, 202).
miRNAs: Modulating Antigen Processing and Presentation in Cancer
A number of miRNAs interrupt MHC-I and antigen-processing machinery (APM) components in cancer cells (Table 1). A study of nasopharyngeal cancer cells indicated that miR-9 targeted a multitude of APM constituents such as β2-microglobulin, low molecular weight polypeptide subunits LMP10, LMP9, LMP8, and transporter associated with antigen processing 1 (TAP1). MiR-9 has the potential to downregulate MHC-I molecules (i.e. HLA-H, HLA-B, HLA-C, and HLA-F) and its overexpression in cancer cells enhances immune tolerance in the TME (203). MiR-346 is an endoplasmic reticulum (ER) stress-associated miRNA which regulates the immune response by indirectly suppressing the IFN by targeting adenylate uridylate-rich elements (AREs) on the 3’-UTR region of mRNA transcripts resulting in termination; or by directly phosphorylating TAP1, resulting in interference of MHC-I signaling pathways (206, 207). Likewise, miR-125a-5p in esophageal adenocarcinoma cells bind to the 3’-UTR of TAP2 mRNA resulting in interference with antigen presentation (204). Proteomic screening of miR-27 decreased cell surface expression of MHC-I expression, promoting cancer progression; miR-27-a-induced MHC-I downregulation depended on calreticulin suppression (an essential calcium-binding protein which regulates gene transcription) (208).

Table 1 Cancer antigen processing and presentation, regulated by miRNAs.
miRNAs Targeting HLA-G
This non-classical MHC-I molecule has immune inhibitory function, and it can be hijacked by cancer cells to escape immune attack. When HLA-G binds to NK cells and CTLs, the effector cell cytotoxicity is suppressed (209). HLA-G expression is elevated in cancers such as endometrial, breast, melanoma, gastric, hepatocellular, lung, and colorectal carcinoma (210, 211). The increase in HLA-G expression correlates to the loss of regulatory HLA-G-targeting miRNAs such as miR-152, miR-148a, and miR-148b (212, 213). For instance, the oncogenic estrogenic G-protein-coupled estrogen receptor 1 (GPER) signaling pathway is known to decrease miR-148 levels in breast cancer cells, contributing to cancer immune evasion (214).
miRNAs Associated With Immune Checkpoint Ligands
Immune checkpoint signaling is determined by factors including pre-existing inflammation of the oncogenic signaling pathway. Studies indicate that an increase in PD-L1 expression on numerous cancer cells was achieved by a loss of miR-138, miR-34a, miR-191-5p, miR-148-3p, miR-873, miR-479-5p, miR-195-5p, and miR-3609. A decrease in miR-383 was shown to profoundly elevate PD-L1 expression on cervical cancer cells (215–220). In contrast, PD-L1 expression is promoted by miR-18a via SOX6, WNK2, and PTEN signaling pathways. After induction of PD-L1 expression, various pathways (Wnt/beta-catenin, ERK, and PI3K-AKT) were activated, ultimately leading to PD-L1 transcription (221).
Phenotypic Variations Induced by miRNAs
MHC-I chain-related molecule A and B (MICA, MICB) (222), and UL16-binding protein (ULBP) are stress-induced ligands which are recognized by the presence of NKG2D present on CTLs and NK cells (223). NKG2D is responsible for maintaining cancer immune surveillance, and is downregulated in cancer cells, resulting in cancer cell immune escape at the post-transcriptional level (224). Alternatively, it has been shown that various miRNAs directly target the ULBP2 3’-UTR, and overexpression of these miRNAs lead to downregulation of ULBP expression. Such miRNAs include miR-34a, miR-34c in malignant melanoma and miR-519a-3p in breast cancer (202, 225).
MiRNA mimics can inhibit receptor expression hereby diminishing tumor cell recognition by NK cells. MiRNAs function at the post-transcriptional level of gene expression, and both miRNAs and IFN-γ downregulate expression of the MICA ligand. MiRNA targets MICA/B mRNA by directly binding to the 3’UTR of the target gene, causing mRNA degradation or translation repression. MiRNAs that target MICA include miR-93, miR-106b, miR-106a, miR-373, miR-20a in hepatocellular carcinoma (HCC), miR-519a-3p, miR-20a in breast cancer, and miR-125b in multiple myeloma (202, 226, 227). MiRNAs that target MICB include miR-20a in breast cancer (228), and miR-302c and miR-520c in multiple cancers (228, 229). This leads to immune escape of malignant cells.
miRNAs Relative to Cancer Cell Metabolites
Tryptophan (Trp) is an example of a metabolite responsible for maintaining the function of tumor infiltrating lymphocytes (TILs). The rate limiting enzyme of Trp metabolism, converting Trp to 3-hydroxyanthranilic acid and kynurenine, is IDO1 (230). Increase in IDO1 expression with concurrent decrease in Trp leads to dysfunctional Teffs, permitting cancer immune evasion (231). The downregulation of miR-218 and subsequent elevation of IDO1 has been shown to safeguard cervical cancer cells from immune attack (232).
Cancer Cell-Derived miRNAs Which Regulate Immune Evasion Through Vehicles or Exosomes
Cancer-derived miRNAs are capable of exhibiting extracellular bio-activities through microvesicles or exosomes as well as modulate the expression profile within cancer cells (233). Cancer-derived miRNAs can be transferred via exosome to several TILs in order to mold an immunosuppressive microenvironment. CAFs are immune cells regulated by cancer cell-derived exosomal miRNAs. They can be reprogrammed by various miRNAs to induce tumor progression (234). MiRNAs released into the TME by CAFs function as paracrine stimuli for the activation of adjacent fibroblasts and cancer cells. Exogenous overexpression of miRNAs leads to fibroblast-to-CAF-like cell conversion, resulting in immune suppression (235). Some examples of CAF-derived miRNAs include miR-21 and miR-1247-3p in HCC, and miR-27a in gastric cancer (236–238).
Notably, MDSCs are another class of immune cells which are regulated by cancer cell-derived exosomal miRNAs; miR-17-5p (breast cancer) and miR-20a (in several cancers) (239, 240) promote the STAT3-mediated suppressive function of MDSCs. Additionally, miR-21 and miR-155 show associations with STAT3 activation through the phosphatase and tensin homolog (PTEN) target, along with SHIP1, leading to MDSC expansion in both granulocytic and monocytic subpopulations (241).
TAMs are also immune cells derived from exosomal miRNAs, and can be activated through two pathways: M1 (classical pathway), and M2 (alternative pathway); two perform different regulatory functions in the TME (242). Several miRNAs engage in the polarization into M2 macrophages, which inhibits immune surveillance. For instance, miR-21 regulates TAM through IFN-γ/STAT1 and PTEN to promote M2 polarization, increasing tumor cell migration while decreasing PD-L1 expression and M1 polarization (243, 244). Additionally, miR-324 in colon cancer targets CUEDC2, which regulates TAM to increase pro-inflammatory cytokine production as well as further increase tumorigenesis (245).
Exosomes
These extracellular micro-vesicles contain components of proteins, lipids and genetic materials of the parent cell (246), and are potent signaling molecules within the TME. Exosomes arising from both malignant and non-malignant cells have been shown to be involved in tumorigenesis, therapy resistance, and immune resistance (247).
Homotypic transfer of exosome refers to signal transfer between cancer cells. Glioblastoma cell exosomes were shown to induce change in wild-type cells via transfer of the oncogenic protein epidermal growth factor receptor 3 protein (EGFR-v-III) (248). Similarly, it has been shown that exosomes from breast cancer cell lines and breast cancer patients, which contain miRNA machinery, were able to induce malignant transformation in normal cells (249). Another study showed that exosomes arising from pancreatic adenocarcinoma were able to modify the NOTCH-1 pathway and inhibit cellular death (250). Homotypic exosome transfer promotes cancer progression via the oncogenic pathway.
Heterotypic transfer of exosome, as previously described with regards to tumor growth and dissemination, is widely dependent upon its TME. Cellular crosstalk between the TME and either internal or external components is crucial for TME survival; this is achieved through multiple signaling networks such as paracrine and juxtacrine pathways (251). A study was conducted to spatially separate the TME. The complexity of the system was observed, with the authors demonstrating a vast cellular heterogeneity that consisted of six interacting layers of cells (252). Heterotypic transfer of exosome not only supports tumor growth but also elicits cellular resistance to various therapies as well as the harsh conditions within the TME (247).
Cancer Cells
Aside from the immunosuppressive TME, cancer cells themselves when exposed to CTLs were shown to evade immune surveillance by modifying intrinsic mechanisms. These include expression downregulation of tumor associated antigens (TAAs), expression upregulation of PD-L1/2, and mutation induction within the antigen-binding machinery (β2-macroglobulin and HLA) and extrinsic pro-apoptotic genes such as CASP8 (253, 254). In addition, it was recently shown that clonal expansion of TAAs strongly correlated with the intensity of the immunogenic response (255). In analyzing the tumor genomic landscape, two mechanisms for TAA loss were observed:
1-immune-mediated elimination of TAAs presented by immune cells, followed by outgrowth of the remainder following “Darwinian evolutionary theory”;
2- acquisition of one or more genetic alterations, resulting in TAA loss and subsequent expansion of resistant clones (256). In determining how the EMT may contribute to immune escape, a study demonstrated that after prolonged exposure of breast carcinoma cells to CTLs, expression to TNF-α or via stable expression of SNAIL was increased. Protection from CTL-mediated lysis was linked to the activation of the autophagy pathway, which led to the survival of cells through dormancy (257). Impairment of CTL-mediated lysis was evident in another study in which breast carcinoma cells elicited increased TGF-β expression by silencing the Wnt1-inducible signaling pathway protein 2 (WISP2), which resulted in stemness (258, 259). Autophagy was not evident in the resistant phenotype; however inhibition of TGF-βwas able to induce EMT reversal thus rendering cancer cells more sensitive to CTLs (260). This suggests that chief developmental pathways utilizing TGF-β are fundamental in mediating immune resistance to CTLs. It is evident that along the EMT spectrum, several mesenchymal cancer cell variants have the potential to engage further mechanisms of resistance.
Tumor hypoxia is a significant parameter, as a driver of the EMT, tumor immune escape, and heterogeneity (261). Cells derived from a lung adenocarcinoma model were induced by hypoxia, and demonstrated a shift towards mesenchymal phenotypes. Only some cells underwent the EMT thus promoting cancer cell heterogeneity (262). Hypoxic stress leads to the emergence of cancer subclones, and analysis of these cells showed an increased tendency to resist CTL-mediated lysis. Of note, the resistance mechanism is suspected to be independent of E-cadherin-CD103 interaction. This is because TGF-β inhibition minimized cellular resistance to CTL-mediated killing without causing any changes to the E-cadherin expression in mesenchymal cancer cells (262).
CTLs primarily utilize the perforin/granzyme pathway to demolish target cells. When the perforin pathway is activated, further counter-mechanisms such as Fas or TRAIL are engaged at the cancer cell surface to induce T cell apoptosis (263). The pancreatic carcinoma model was used and given an EMT inducer in the form of the novel tumor antigen Brachyury. The cancer cells showed decreased susceptibility to CTL-mediated killing compared with control. Target cancer cells were co-cultured with CTLs, and poor killing was observed under experimental conditions. This was due to defective caspase-dependent apoptotic cell death despite immune antigenicity (264, 265).
Additionally, defects in the APM – correlated to immune-proteasome deficiency – was found to be common among cancers with a greater mesenchymal profile, and ultimately affected T cell-mediated cytotoxicity (266). Manipulation of cell-to-cell interactions and immunological synapses (IS) has been linked to immune resistance.
The IS involves interactions between immune killer cells (NK cells and CTLs) with their APCs or targeted cancer cells necessary to achieve maturation and production of TNF-α and IFN-γ, and their lytic functions (267–269) IS formation in T cells is regulated by cytoskeletal elements (i.e. actin), interaction of MHC-TCR, and the integration of integrin-based signals, generated when integral molecules (lymphocyte function-associated antigen 1, LFA-1) on the T cell interact with ICAM-1. Integrins undergo conformational changes through phosphorylation cascades (i.e. phosphotyrosine kinase activation linking integrins to the actin cytoskeleton) during peptide-MHC/TCR ligation. The actin cytoskeleton polymerizes at the edges of the active synapses, causing an increase in synaptic diameter size and immune cell flattening (270). This phenomenon leads to the emergence of T cell receptor (TCR) microclusters. These clusters merge at the center of the IS zone and are referred to as the central supramolecular cluster. In contrast, microclusters found at the periphery of the synapse join to form a highly contractile zone called the peripheral supramolecular activation cluster (271).
Mechanical forces brought about at the synapse via intercellular adhesion also play a role in rearranging the actin cytoskeleton and regulating adhesion-based signals. IS and its relationship with NK cells abide by similar rules, differing only in that NK cells express 2B4, DNAM1 and NKG2D receptors, rather than TCR. These receptors also regulate signaling activity and the changes in the integrin-actin network at different points of NK cell cytotoxicity. Numerous genetic aberrations have been shown to alter various stages in CTL and NK cell cytotoxicity, F-actin/microtubule networking, and cellular recognition which ultimately leads to NK or T cell disorders, resulting immunodeficiency (272, 273). These examples highlight the role of the operational IS in an appropriate and effective immune response.
The establishment of the IS and activation cascades relies on heterophilic interactions between ICAM-1 and integrins on target cells; the loss of ICAM-1 can be expected to impede IS formation. Moreover, manipulation of the actin network through changes in mechanical forces renders a significant effect on the IS and the lytic commitment (274, 275). The discomposure of the actin network in certain cells will either render them more resistant or more susceptible to CTL-mediated lysis (276).
See Figure 1 for a diagrammatic summary of the major pathways that promote immune resistance, and immune and treatment resistance.
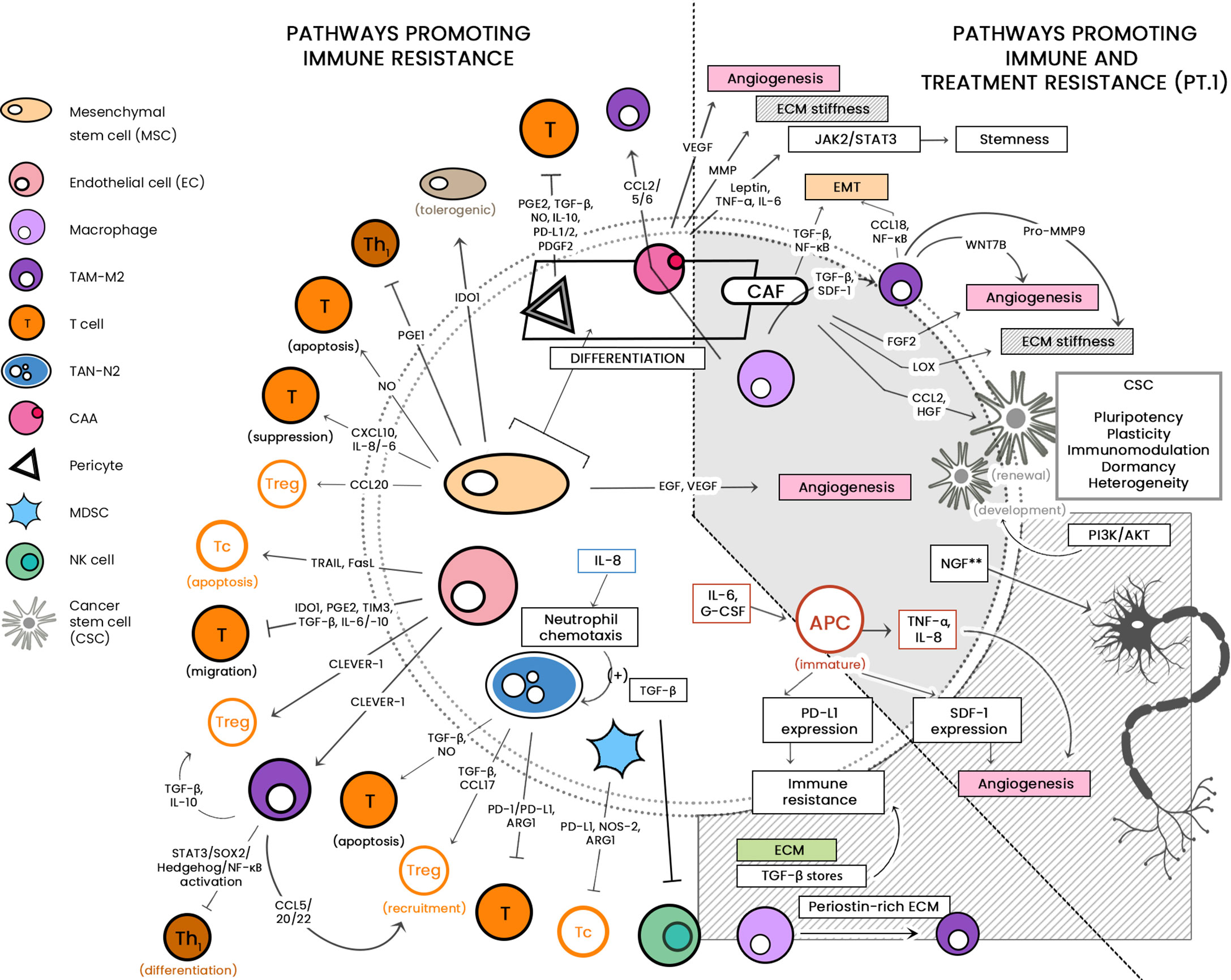
Figure 1 The large cellular repertoire of the tumor microenvironment (TME) is depicted in this diagram. Through the release of soluble factors, the presented cellular entities are seen to be involved in: 1) immune suppression, by either inducing apoptosis or inhibiting anti-tumor activity; and 2) both immune/drug resistance by stiffening the extracellular matrix, inducing epithelial-to-mesenchymal transition (EMT) and induction of stemness. CCL, C-C motif chemokine ligand; PG-E2, Prostaglandin E2; TGF-ß, Transforming growth factor beta; NO, nitric oxide; IL, interleukin; IDO-1, indoleamine 2,3-dioxygenase 1; TRAIL, TNF-related apoptosis-inducing ligand; Fas-L, Fas-ligand; TIM-3, T-cell immunoglobulin and mucin-domain containing-3; CLEVER-1, lymphatic endothelial and vascular endothelial receptor-1; PD-1/-L1, programmed cell death protein 1/ligand 1; Arg-1, arginase; NOS, nitric oxide synthase; JAK/STAT, Janus kinase/signal transducer and activator of transcription; ECM, extracellular matrix; CSC, cancer stem cell; PI3K/AKT, phosphatidylinositol 3 kinase/protein kinase B; NGF, neurotrophic growth factor; TNF-α, tumor necrosis factor alpha; SDF-1, stromal-derived factor-1; EGF, epidermal growth factor; VEGF, vascular endothelial growth factor. **associated with treatment resistance; mechanism as-yet-unknown.
Characteristics of the TME and Its Effects on Treatment Resistance
The Tachyphylactic TME
Epigenetics: The Link to Treatment Resistance
To unleash, hijack, and restrict cellular plasticity, CSCs play a chief and fundamental role in epigenetics. In cancers, one of the most habitually mutated gene classes are epigenetic regulators, resulting in this characteristic uncontrolled cellular self-renewal. Epigenetic regulator mutations lead to oncogenic cellular reprogramming during cancer initiation. CSCs are either promoted or inhibited by the epigenetic mechanisms that integrate the cell-extrinsic (microenvironmental signaling), or cell-intrinsic (subclonal mutations) effects that establish intratumoral heterogeneity. Over time, CSCs generate self-renewing subclones with diverse fitness, whilst environmental changes are able to act on their genetic heterogeneity and modulate their phenotype. Further discussion on the CSC mechanistic roles and implications now focuses on how cellular plasticity can be affected by manipulation of DNA methylation and chromatin. In addition to the previously described role of miRNA, the following sections will shed light on epigenetic DNA methylation and histone modification leading to the development of CSCs, followed by the role of CSCs in drug resistance (277).
Pathways Involved in CSC Development
Wnt/β-Catenin Signaling Pathway
β-catenin is transcription co-activator regulated by the WNT gene family, and is mainly involved in embryonic development, adult homeostasis, and, if highly expressed, various cancers (278, 279). Physiologically, the absence of WNT signaling keeps β-catenin at low levels through the ubiquitin-proteasome system (UPS). β-catenin is recruited into a destruction complex consisting of the adenomatous polyposis coli gene (APC gene) and Axin. This promotes the phosphorylation of β-catenin by glycogen synthase kinase 3β(GSK-3 β) and casein kinase 1 (CK1), which tags β-catenin and subsequently goes through UPS. Stabilization of β-catenin occurs with Wnt ligand binding to Frizzled receptors, allowing the degradation complex to be inactivated via low density lipoprotein receptor-related protein 5/6 (LDR5/6) and Disheveled. β-catenin accumulates and translocates into the nucleus where it couples with T cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors to induce transcription of WNT target genes, Cyclin D-1 (CCND1), c-MYC, and Jun. β-catenin plays a crucial role in the self-renewal and differentiation of CSCs (278, 280, 281). The anomalous activation of Wnt/β-catenin is either through genetic alterations such as mutations in CTNNB1, the APC gene and AXIN genes, or through epigenetic modulation (282–284).
In breast and colorectal cancers, aberrant Wnt/β-catenin pathway activation is carried out by DNA methylation in the promoter region and silencing of multiple Wnt inhibitors such as Wnt inhibitory factor 1 (WIF-1), AXIN2, Secreted frizzled-related protein 1 (SFRP-1), and Dickkopf-related protein 1 (DKK1) (285–287).
Histone modifications are also implemented in the deregulation of the Wnt/β-catenin pathway in cancer. Decreased acetylation of H3K16 and increased H3K27 trimethylation, along with the recruitment of Sirtuin 1 (SirT1), enhancer of zeste homolog 2 (EZH2) and suppressor of zeste 12 protein homolog (Suz12) (components of polycomb repressor complex 2, PCR2) to the DKK1 promoter inhibits the expression of the DKK1 Wnt antagonist (288). Bivalent histone modifications, activating H3K4me3 and repressing H3K27me3 histone marks at its locus, are implemented in colorectal cancer by regulating Disheveled-binding antagonist of β-catenin 3 (DACT3). In turn, DACT3 expression in colorectal cancer lines is decreased, with overexpression of Wnt/β-catenin and CSC induction (289).
Hedgehog-Signaling Pathway
As an important mediator of embryogenesis and tissue homeostasis, Hedgehog (Hh) signaling has been shown to maintain SC and regulate the proliferation of progenitor cells (290). In the absence of the Sonic Hedgehog ligand (SHH), inhibition of Smoothened (Smo) protein by Patched receptor (PTCH-1) activates kinesin family member 7 (Kif7) and suppressor of fused homolog (SUFU), resulting in sequestration of Gli proteins which function as transcription factors. Moreover, upon binding of SHH to PTCH-1, Smo activates Hh signaling by releasing Gli protein back to the nucleus and exerting transcription of target genes (291). The implication of Hh mutation-induced signaling alterations in SCs has been well-documented in medulloblastoma and basal cell carcinoma. The upregulation of SHH within hair follicles or the interfollicular dermis in basal cell carcinoma was shown to contribute to tumorigenesis (292, 293). Moreover, granule neuron progenitors, identified as the medulloblastoma cell of origin, were seen to possess high levels of Hh signaling activity (294). In addition to genetic mutations, epigenetic factors were also seen to impact Hh-signaling. The chromatin remodeling protein SNF5 was seen to directly alter Hh signaling by interacting with Gli, resulting in the downregulation of PTCH-1 and resultant loss of the Hh inactivation feedback loop (295). Furthermore, it has been shown that hypomethylation of the SHH promoter allowed NF-κB to bind to the promoter, resulting in higher expression of SHH in gastric and breast cancer cells (296). Overexpression of SHH has been linked to CSC renewal and cancer aggressiveness (297).
Notch Signaling Pathway
The Notch signaling pathway is a highly-conserved cell signaling system that plays a major role in the regulation of embryonic development. It also regulates cellular proliferation and differentiation amongst a vast range of cell types and stages of cell maturation. Its cell-dependent signaling consists of the binding of ligands Jagged-1/-2 or Delta1-4, which triggers cleavage of the Notch intracellular domain (NICD) by γ-secretase, followed by release into the cytoplasm (298). This allows for modulation of SC differentiation and self-renewal, crucial for the survival and maintenance of neural stem cells (NSCs) (299).
In multiple myeloma, epigenetic histone acetylation causes overexpression of Jagged-2 ligand (300). Histone acetylation is governed by histone deacetylase (HDAC), and the recruitment of HDACs to the promoter regions is usually carried out by nuclear co-repressor silencing mediator of retinoic acid and thyroid hormone receptor (SMRT). In multiple myeloma, decreased levels of SMRT reduces HDAC recruitment to the Jagged-2 promoter, which in turn increases histone acetylation and increases Notch ligand transcription, ultimately resulting in overactivation of the Notch signaling pathway.
Serine-threonine kinase receptor-associated protein (STRAP) promotes tumorigenesis and stemness by stabilizing intracellular fragment of NOTCH3 (ICN3), notable in colorectal cancer. STRAP inhibits histone methylation of H3k27 at the HES5 and HES1 promoters, leading to gene overactivation and inducing treatment resistance (301).
Cancer Stem Cells: Drivers of Therapy Resistance
CSCs and EMT-induced heterogeneity convey resistance to chemotherapeutic agents such as cisplatin, gemcitabine, and 5-fluorouracil (5-FU) (302, 303). Pancreatic cell lines exhibiting resistance to gemcitabine expressed high ZEB1 and low E-cadherin, thus acquiring great migratory ability (304). Tumor cell response to therapy may largely be due to epigenetic modulations.
With the enhanced expression of drug efflux transporters such as multidrug resistance-associated protein 1 (MRP1) and ATP-binding cassette sub-family G member 2 (ABCG2), drug resistance is increasingly observed (305–307). Transporter expression is regulated by various pathways and mechanisms, and deregulation results in protein enrichment and drug efflux. Notch signaling upregulates MRP1 expression and is responsible for CSC drug resistance (308, 309). The modification of histones (decreased HDAC1, and increased H3K4 tri-methylation, H3S10 phosphorylation, and histone H3 acetylation) leads to upregulation of ABCG2 expression. Along with decreased H3K9 tri-methylation, this allows for chromatin remodeling protein Brahma-related gene 1 (Brg1) and RNA polymerase II to gain access to the promotor, ultimately activating ABCG2 transcription (310). As a result of aberrant epigenetic modifications, physiologic SCs are susceptible to deregulation that facilitates tumor progression and invasion. Epigenetic regulation of signaling pathways is thus a potential target for anti-CSC therapy.
Heterogeneity is omnipresent in mammalian cells, and fundamental with regard to CSCs (311). The complicated picture of CSC heterogeneity involves dynamic cell populations capable of undergoing spontaneous state transitions; spontaneous switches from non-SCs to stem-like cells was observed in a study of breast cancer cells where plasticity was regulated by ZEB1 (312, 313). CSC heterogeneity and plasticity in various cancers varies from patient to patient, but phenotypically distinct CSC markers may be identified depending on the tumor genotype (311, 314). Non-CSCs and CSCs in breast cancer exhibited a dynamic equilibrium that was maintained by cytokine-mediated crosstalk among marked populations. This suggests that cancers have reversible phenotypic plasticity and do not solely depend on genetic variation (315, 316). Colorectal cancer studies have provided compelling evidence demonstrating CSC plasticity and tumor progression. The Wnt target gene LGR5 acts as a functional CRC marker. Anti-cancer drug therapy resulted in the conversion of LGR5+ into LGR5- cells, while in the absence of the drug, LGR5- reverted back to LGR5+ (317). CSCs have been shown to overcome DNA damage induced by radio/chemotherapy. Furthermore, they acquire resistance through overactivation of DNA repair mechanisms such as the expression of excision nucleotide repair protein ERCC1 and overexpression of cell cycle checkpoints (318, 319). CSCs have also been shown to inactivate cell cycle gene expression as well as apoptosis-inducing genes such as p53 and c-MYC, creating so-called “undruggable phenotypes” (320). The activation of autophagy pathways after exposure to cytotoxic agents induces apoptosis; unfortunately, this mechanism is a double-edged sword and has been shown to instead enable CSCs and a heterogeneous subpopulation of cancer cells to tolerate the cytotoxic agents and TME-induced stress. These cells enter a state of dormancy and degrade key transcription factors (i.e. p53) to prolong cellular survival until TME conditions become favorable for growth and proliferation (321, 322).
Hypoxia: The Master Regulator of Cellular Heterogeneity
Hypoxia develops as a result of malignant cell overgrowth relative to their angiogenic requirements. To elicit cellular viability and progression, tumor-associated cells increase secretion of hypoxia-inducible factors (HIFs), mainly HIF-1-α and HIF-2-α, which in turn regulates angiogenesis in a chaotic manner (323). Under normal conditions, HIF-1-α is kept in check by hydroxylase enzymes which are dependent upon intracellular oxygen concentration. They are ubiquitinated and degraded after tumor suppressor protein von Hippel-Lindau complex formation. In hypoxic conditions, hydroxylation is diminished resulting in the overexpression of HIFs (324). The resultant chaotic blood vessel formation leads to irregular oxygen delivery and decrease in oxygen perfusion leads to necrosis (325). Drug distribution varies greatly between well-perfused and hypoxic areas, and effective cancer therapy requires efficient tumoral penetration; this patchy blood vessel distribution unfortunately results in tumor cell survival (326). HIF-1-α has been shown to upregulate various transcription factors (i.e. ZEB1/2, TWIST and SNAIL) that reduce E-cadherin expression, which results in EMT (327). Additionally, HIF-1-α activates focal adhesion kinase and steroid receptor coactivator (FAK-Src) which also decreases E-cadherin, promoting the EMT and VEGF-dependent angiogenesis and drug resistance by formation of SC-like phenotypic variants resistant to chemotherapy (328, 329). Intra-tumoral hypoxia induces a harsh environment that is crucial for cellular heterogeneity. The reprogramming of cellular phenotypes and metabolism drives adaptation and enhances signaling pathways leading to treatment resistance. As such, it is associated with poor prognosis (330). In a recent study, different patients with the same cancer type were shown to possess different inter- and intra-tumoral phenotypes. Very low oxygen concentrations correlates with an increase in mutational load in individual cells, and in varying the degree of hypoxia in each patient, alternations in tumor suppressors and oncogenes as such Myc, PTEN, and TP53 was elicited (331). HIF-1-α was shown to be a key player in the regulation of multiple metabolic pathways (i.e. amino acid metabolism, lipid metabolism, glycogenesis, and the TCA cycle) which ensures cancer cell sustenance and resistance to treatment (332, 333). A robust understanding of the HIF-1-α expression pathomechanism is required before we may implement effective therapeutic regimens.
Metabolism of the TME
Lactate Metabolism
Metabolic reprogramming occurs when it is necessary to increase cellular proliferation under hypoxic conditions. It has been shown that cancer cells increase metabolism of reactive oxygen species, lactate, lipids, amino acids, glutamine and glucose (334). Under normoxic conditions, normal cells general energy through oxidative phosphorylation, while cancer cells employ lactate metabolism and glycolysis. It was previously shown that tumor cell production of lactate occurs via: 1) glycolysis using lactate dehydrogenase (LDH), which converts pyruvate into lactate, bypassing the TCA cycle; and 2) glutaminolysis which forms various metabolites, including lactate and pyruvate, allowing the cell to hijack the TCA cycle and utilize glucose-derived metabolism for better efficiency (335, 336). As glucose concentration within the TME is scanty, numerous tumor types (i.e. lung adenocarcinoma, pancreatic adenocarcinoma, and more) have shown very high expression of lactate dehydrogenase which is known to induce the EMT (337). Furthermore, a high-lactate TME has been shown to reprogram TME cells. The high lactate environment prevents the proliferation of cytotoxic and effector T cells while promoting immunosuppressive Tregs (338); it has also been shown to induce M2 polarization of TAMs, subsequently leading to recruitment of other Tregs to enhance the protection of the TME. High lactate content promotes survival of hypoxic cells by inducing angiogenesis (339). Glutaminolysis provides a source of nitrogen, carbon, and energy to fuel the stromal and cancer cells (340). A recent study pointed out the importance of glutamine metabolism, demonstrating that breast cancer cells used the pyruvate metabolite within the TME to effect ECM remodeling, inducing cancer cell stemness and resistance to anti-tumor agents (341, 342). The role of lactate in treatment resistance has been well documented. After irradiation of non-small cell lung cancer (NSCLC), mice xenografts showed resistance within six weeks (343). The importance of lactate as a key molecule in resistance mechanisms has been further elucidated in epidermal growth factor receptor (EGFR) and tyrosine kinase- (TK) targeted therapies. These treatment modalities prompted cancer cell lactate production, which directed TME cells to produce hepatocyte growth factor (HGF), ultimately resulting in EMT and resistance (344). Lactate metabolism was shown to increase DNA repair mechanisms by exploiting DNA-dependent protein kinases (DNA-PK), rendering cells resistant to cisplatin and doxorubicin (345).
Lipid Metabolism
Most neoplasms of organs and tissues are associated with adipocytes. The high rate of cellular proliferation demands abundant fuel via a process called lipid metabolic reprogramming. Lipid metabolism reprogramming has been correlated with resistance to conventional chemotherapeutic agents. Lipid and lipoproteins result from either catabolic processes or de novo synthesis (346). De novo fatty acid synthesis – lipogenesis – is controlled by the upregulation of lipogenic enzymes, and several crucial lipogenic enzymes such as fatty acid synthase (FASN), acetyl co-A carboxylase, stearoyl-CoA-desaturase-1 (SCD-1) and ATP citrate lyase are highly expressed in most neoplastic cells (347). High lipogenic enzyme concentration is correlated to invasion and worse prognosis (348). Upregulation of the prominent enzyme FASN is complex. It may be mediated by various growth factors, such as EGFR, HER2, steroid hormone receptors-androgen receptors, estrogen receptors and progesterone receptors; release is induced by the harsh conditions of the TME, or may result from post-translational miRNA modifications (349). Another key contributor in lipogenesis is SCD1, which is upregulated by growth factors (i.e. EGFR, PDGF, TGF-β) within the TME, and has been associated with treatment resistance and worse prognosis (350, 351). Various studies showed that inhibiting FASN and SCD1 action in lipogenesis led to tumor regression and improved responsiveness to prior therapeutic resistance (352). Another means by which various cancers may derive energy metabolites is via lipolysis. Overexpression of fatty acid-binding protein-4 (FABP-4), which induces lipolysis, has been shown to contribute to rapid tumor growth, metastasis in ovarian cancer and resistance to carboplatin (353). CAAs provide cancer cells with exogenous free fatty acids through cancer cell phenotypic expression of surface fatty acid translocase (CD36) through the fatty acid beta-oxidation (FAO) pathway (162). The CD36+ subpopulation have been shown to be more aggressive and resistant to treatment (354). In another study of radiotherapy-resistant breast cancer cells and breast cancer SCs, carnitine-palmitoyl-transferase-1a-and-2 (CPT1a/2), a known contributor to the FAO pathway was shown to be highly expressed. When CPT was knocked out by genetic editing techniques, this rendered previously-resistant cells sensitive to radiotherapy (355). The TME demonstrates atypical lipid metabolism for cell membrane formation and production of energy (356). Lipid metabolism has been linked to cancer growth, recurrence (357) and CD8 T cell exhaustion via the upregulation of programmed-cell death protein-1 (PD-1) (358) resulting in post-chemotherapy evasion of immune surveillance. The derangements of lipid metabolism are especially crucial for CSCs as the high ectopic metabolism of lipids has been linked to CSC formation, self-renewal and pluripotency (359). In obese breast cancer patients, sustained elevation of IL-6 and FGF-2 was observed. Obese mouse breast cancer xenografts also showed resistance to anti-VEGF therapy; the pathomechanism is hypothesized to be the constant release of proinflammatory cytokines by adipocytes. IL-6 and FGF-2 blockade restored sensitivity of cancer cells to anti-VEGF therapy (360). The association between drug resistance and lipid metabolism reprogramming has been well-documented in the literature (Table 2).
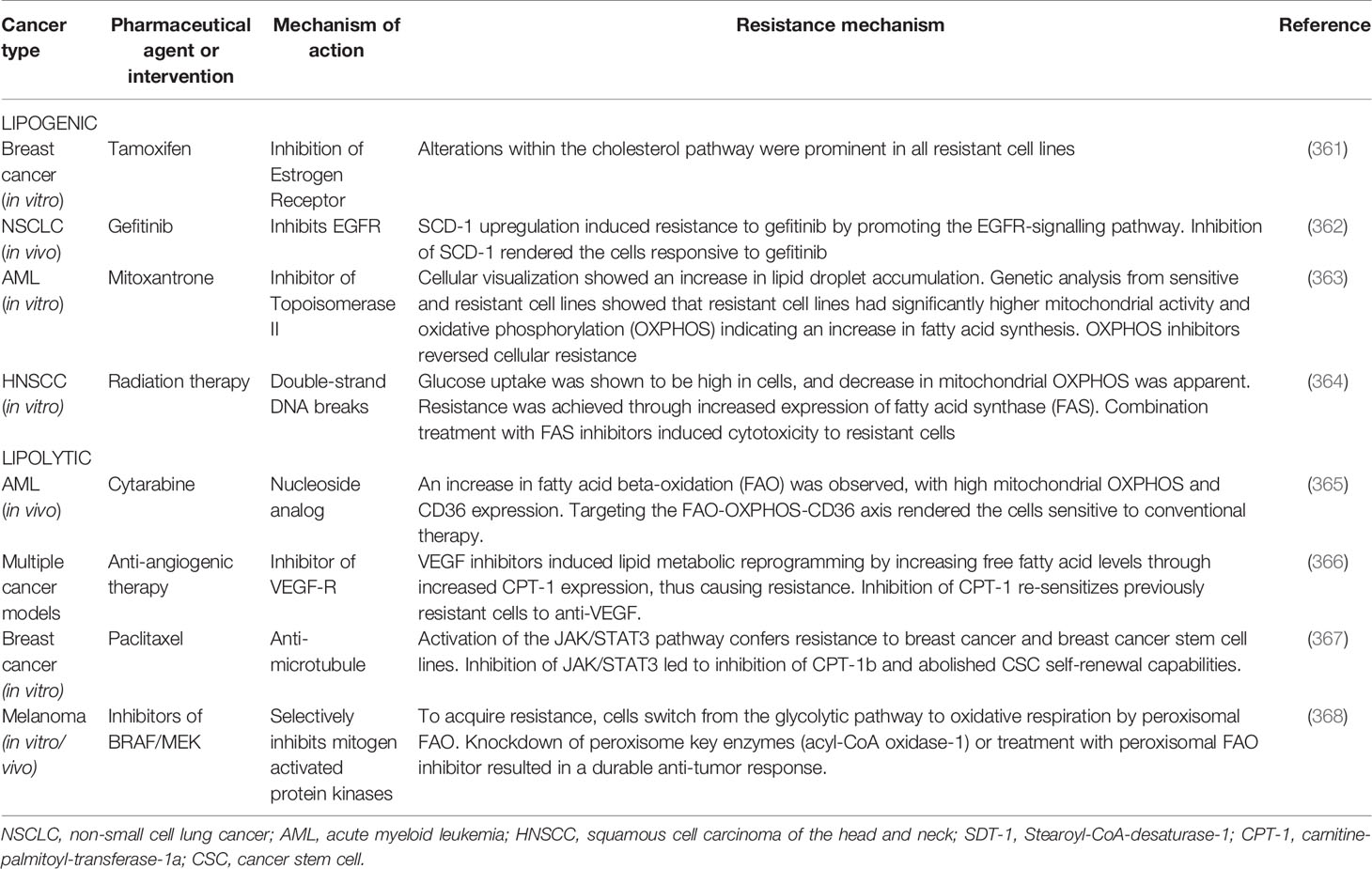
Table 2 Pharmaceutical agents or medical interventions for which a TME-regulated resistance mechanism has been described.
Reactive Oxygen Species Metabolism
Reactive oxygen species (ROS) elevation is closely related to cancer severity due to its influence over tumor immunity, tumorigenesis, and cellular reprogramming (369). Under hypoxic conditions, HIFs are activated by local mitochondrial ROS, and are therefore implicated in angiogenesis (370). ROS are produced by various cells within the TME, inducing activation of the KRAS pathway and promoting tumorigenesis (369). ROS were shown to play a critical role in the activation of TAMs, MDSCs and CAFs, enhancing their immunosuppressive roles (123, 371). Therapy resistance remains the most challenging barrier in cancer treatment. The pioneer in cancer metabolism, Otto Warburg first observed that cancer cells rely on glycolysis rather than oxidative phosphorylation, and this shift from oxidative to reductive metabolism is now termed the “Warburg effect” (336). Although glycolysis is considered an inefficient mode of energy production, ATP can be provided to cells at a faster and safer rate compared to the TCA cycle, which induces more stress via ROS formation (372). Upregulation of the glycolytic pathway aids cellular proliferation by shunting metabolites (glycine, serine, alanine) and nucleotides to the pentose phosphate pathway (PPP) (373). Transketolase, a key enzyme in the PPP was shown to increase pyrimidine synthesis and induce resistance to gemcitabine (374). ROS are the consequence of aerobic metabolism, and the major sources are peroxisomes, mitochondria and NADPH oxidase. Under physiologic conditions, redox homeostasis with low levels of ROS is maintained through fluctuations in generation and elimination processes, as an elevation in ROS is detrimental and leads to cell death. In cancer cells, metabolic derangement and oncogenic signaling induces high production of ROS (375, 376). Mitochondria are susceptible to ROS-induced oxidative damage, which usually results in elevation of NADPH oxidase expression. This in turn favors glycolysis and decreases the intrinsic production of ROS (377, 378).
- ROS-mediated maintenance of glycolysis: Pyruvate kinase (PK) is a rate limiting enzyme of the glycolytic pathway, and appears in two isoforms termed M1 and M2. PKM1 has high kinase activity and is present in physiologic conditions whereas PKM2 exhibits low pyruvate kinase activity which prevents its entrance in the TCA cycle; PKM2 is highly expressed in cancer cells (379). PKM2 was shown to activate HIF-1-α-related genes (i.e. LDHA, SLC2A1) after hypoxia-induced anti-angiogenic therapy (380). Furthermore, the low activity of PKM2 induces glutathione reduction in order to counter the effects of ROS accumulation after ROS-producing therapies (381). Another important glycolytic enzyme termed the “housekeeping gene”, glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) is upregulated in tumors and is associated with cancer aggressiveness (382). GAPDH maintains glycolysis by redirecting metabolites to the PPP in order to induce an increase in NADPH. A study showed that changes in glucose concentration enhanced NADPH oxidase-dependent ROS production, leading to resistance to doxorubicin (383). Upregulation of glycolysis has been shown to enhance DNA repair mechanisms after chemo- or radiation therapy (384). Inhibition of the glycolytic pathway re-sensitized cells to previously resistant drugs (385, 386).
- ROS-mediated activation of oncogenic signals:
Adenosine monophosphate protein kinase (AMPK), a key element of tumor suppression that prevents the Warburg effect was shown to possess tumor-supporting actions, inducing metabolic variations to sustain the ROS-damaged cellular mechanism (387). Apart from its angiogenic functions, HIF-1-α induces the expression of glycolysis-associated genes (i.e. GLUT1/3, hexokinases, and PKM2) to maintain glycolysis and inhibit the TCA cycle (388). The “guardian of the genome”, p53, functions to maintain genome integrity after DNA-induced damage. It has also been shown that p53 acts as a negative regulator of the Warburg effect (389). ROS-induced damage impairs p53 activity and prevents apoptosis (390). Furthermore, ROS metabolism has been linked to treatment-associated metabolic disturbances (391). Chemo- and radiation therapy induce cancer cell death via ROS production, and ROS production has been shown to induce the activation of oncogenic signaling pathways (NF-κB and PI3/Akt) which ensures cell survival against the ROS onslaught (392). Well-documented drug efflux mechanisms induce MDR (ABC transporters, i.e. P-glycoprotein) (393, 394). Eventually, TME cells acclimate to ROS and become resistant to ROS-eliciting drugs by producing antioxidants or increasing efflux of cytotoxic agents (395, 396).
Acidic TME
As a result of hypoxia and high lactate, TME niches are acidic. This harsh environment induces oncogene activation, and cellular metabolism shifts to adapt (397). Compared to normal cells, cancer cells possess a high intracellular pH which promotes proliferation and inhibits apoptosis, and maintains a low extracellular pH in a “reversed pH gradient” (398, 399). The acidic niche acts synergistically with the effects of lactate by inducing TAM M2 polarization, and inhibiting the cytotoxicity of infiltrating T cells (400). These effects support cellular development (401) and regulate immune surveillance. The acidic niche has been shown to induce invasiveness and the EMT in melanoma (402), neuroblastoma (399, 403) and breast carcinoma cells (404). The pH gradient between intra- and extracellular spaces forms a physical barrier to weak-base chemotherapy, preventing proper drug uptake and distribution through physiological resistance or “ion trapping phenomenon”. Ionization of weak-base agents within the acidic extracellular environment prevents them from traversing this barrier (405, 406).
In contrast, weak acids exhibit high intracellular permeability. For example, paclitaxel, a non-ionizable agent, was not impeded by this physiologic barrier, showing how the ion trapping hypothesis may be relevant in future treatment modalities (407). This has prompted researchers to alkalinize appropriate modalities or treatment combinations prior to administration. Low pH brought about by hypoxia and low perfusion was shown to induce epigenetic modifications, mainly in p53, preventing apoptosis and increasing activity of P-glycoprotein in order to induce MDR (408, 409). It has been previously reported that the acidic TME was involved in cellular protection against irradiation (410). In an investigation of radio- and/or chemo-resistance, a study showed that the acidic niche functions to induce cellular dormancy by arresting the cell cycle at G2/M phase (411). Finally, another mechanism of treatment resistance depends on the genomic instability generated by acidic milieu, which induces phenotypic variations that lead to cellular stemness (412).
Immune Micro-Environment Variability Between Primary and Secondary Tumors
As previously described, the vast cellular repertoire within the TME contributes to immune suppression. Secretion of soluble factors within the TME prevents active immune surveillance from entering the tumor; these are known as “cold tumors” – low-immune infiltrates that enhance proliferation, migration and invasion. 90% of cancer-related deaths occur in the metastatic stage because of the inefficient localization of micro-metastatic niches and therapeutic failure (413). A study utilized deep learning was conducted to detect micro-metastatic niches, and this innovative technique enabled metastatic analysis of mice with metastatic lung, pancreatic and breast cancers that may potentially be treatable. Antibody-targeted treatment applied to visible metastatic nodules was also distributed to the micro-niches in close vicinity. This approach provides the means to identify micro-niches distributed throughout the body for the purposes of improving treatment efficacy (414). The TME-induced heterogeneity was more evident in another study that showed discrepancies in the cellular and immune repertoire within primary and metastatic lesions. This suggests yet another therapeutic resistance mechanism (415). In addition to immune suppression, the microenvironmental repertoire of immune cells has been implicated in treatment resistance. As stated earlier, a large portion of the TME consists of bone marrow-derived myeloid cells which are modulated by both physical and biochemical signals that cause them to differentiate. Myeloid cells include TAMs, TANs and MDSCs which were all shown to induce chemo- and radio-therapeutic resistance through a variety of mechanisms.
TAMs are the predominant myeloid cell type within the TME, and their differentiation into the M2 phenotype is an important factor in treatment resistance. An influx of TAMs is observed after the initiation of therapy (416). TAMs were shown to be key players in chemotherapy resistance, producing various inflammatory mediators such as TNF-α, MMP, cathepsin and TGF-β. They are also commonly described as EMT transducers, degrading and synthesizing denser ECM, which ultimately leads to treatment resistance (416, 417). Another resistance mechanism is via TAM production of signaling factors such as FGF-2, IL-8 and VEGF for angiogenesis (418, 419). TAMs were shown to sustain an elevated level STAT3 activation, which has been associated with chemo- and radiotherapy resistance. Elevated STAT3 inhibits apoptosis via upregulation of anti-apoptotic proteins bcl-2 and IAP (420). Similarly, TAM overexpression of EGFs such as milk fat globule EGF-8 (MFG-E8) was shown to induce overactivation of the Sonic Hedgehog and STAT3 pathways in CSCs, resulting in treatment resistance to cisplatin (418, 421).
As previously described, microenvironmental recruitment of TANs results in a high likelihood of N2 polarization. In addition to their immune modulatory effects, they have been implicated in ECM remodeling and the EMT through the secretion of proteins such as HGF, MMP and oncostatin-M (422, 423). Similar to TAMs, TANs also induced angiogenesis, promoting treatment resistance via secretion of Bv8, MMP9 and VEGF (424). Additionally, HCC xenografts showed an increase in TAN activity and an increase in the expression of chemokines such as CCL2 and CCL17, which serve to attract Tregs and macrophages to the TME, thus inducing resistance to sorafenib. Pharmacologic inhibition of the PI3K-AKT pathway was shown to decrease levels of CCL2 and CCL17 chemokines and re-sensitize cells to sorafenib (425).
MDSCs are a major determinant of immunogenicity. Through the production of TGF-β, MDSCs induce polarization of TAMs and TANs into their respective tumor-supporting subtypes (150). IL-10 oversecretion by MDSCs was shown to inhibit anti-tumor activity by preventing macrophage activation and DC maturation (426, 427). The receptor tyrosine kinase inhibitor sunitinib malate was shown to not only reduce the suppressive function of MDSCs, but also decrease the expression of Fox-p3, TGF-β and IL-10, inducing a significant increase in anti-tumor activity (428). An in vivo study of multiple myeloma xenografts showed neutrophil accumulation in the bone marrow in the course of disease. The accumulation of MDSCs and Tregs is thought to be a result of cancer expression of stem-cell factor ckit ligand (429). The resistance of multiple myeloma to melphalan and doxorubicin is due to the immunosuppressive actions of MDSCs, mediated by soluble factors, and it is hypothesized that targeting the MDSCs would enhance chemotherapeutic efficacy in this cancer (430). Furthermore, reprogramming the TME immune repertoire induces a better anti-tumor activity and more robust response to chemotherapy (431).
As described above, CAFs are one of the key mediators of ECM stiffness and myeloid cell differentiation. CAFs differentiate from various stromal cells of the TME. Despite advancements in oncological treatments, the prognosis of solid tumors such as HCC and pancreatic ductal adenocarcinoma remains poor. Firstly, CAF-dependent secretions promote ECM rigidity, which prevents effective drug penetration. Secondly, CAF-derived miRNAs previously shown to induce immune suppression were also shown to induce treatment resistance. Ovarian cancer cells showed downregulation of programmed cell death 4 (PDCD-4) in the presence of CAF-derived overexpression of miR-182. MiRNA-182 alterations of PDCD-4 expression rendered the cancer cells resistant to chemotherapy (432). Cisplatin-based therapy was administered to patients with esophageal cancer; high levels of CAF-derived miR-27a were subsequently observed. MiR-27a-dependent transformation of fibroblasts into CAFs resulted in optimal production of TGF-β, and is thought to be the mechanism of therapy resistance. Inhibition of TGF-β subsequently re-sensitized the cells to cisplatin (433). Thirdly, CAF-derived exosomal release promotes cancer aggressiveness and therapy resistance. This occurs when the EMT is induced by modulating Wnt-PCP autocrine signaling, which is further involved in cellular polarity via JNK and ROCK pathways (434). CAF-derived exosomes were shown to induce therapy resistance in breast cancer cells via juxtacrine and paracrine signaling of STAT-1 and NOTCH-3 pathways (435). Additionally, STAT-1 and NOTCH-3 have been associated with the maintenance of cancer cell stemness, which has been further associated with oxaliplatin and 5-FU resistance in colorectal cancer (436). Within the TME, CAFs were shown to hyperactivate the Wnt/β pathway, which in turn induces the expression of ABC and P-glycoprotein (437, 438). Overactivation of the Wnt pathway not only results in chemo- and radiotherapy resistance, it has also been shown to reduce intracellular ROS through the overexpression of COX-2 and aldehyde dehydrogenase (439, 440).
Although the molecular interplay between treatment resistance and immune suppression is not yet fully elucidated, these novel resistance mechanisms induced by TAMs, TANs, MDSCs and CAFs may present the future for targeted therapy.
Mechanical TME
The importance of ECM remodeling as a result of mechanical changes has been well established. Multiple studies demonstrate that tension accumulated in the TME induces an increase in metabolism for: 1) rapid proliferation (441); 2) mobility and structural changes that regulate invasion (442), and 3) immune evasion, acquired epigenetic modification by miRNA, and stress-induced signaling that induces resistance to therapy, which collectively constitute the most threatening aspect of cancer cell dormancy (443, 444). It has been shown that physical signals can alter cellular behavior beyond the traits of CSCs (445). The TME – with dense interstitial matrix, abnormal blood and lymphatic vessels, and increased stromal pressure – is physically distinct from normal tissue (446). Physical signals of the TME include increased matrix stiffness, solid stress and interstitial fluid pressure. Operating in tandem, these physical factors contribute to treatment resistance (445, 447).
Increased ECM Stiffness
ECM composition determines its rigidity. As described previously, the ECM provides crucial biochemical and structural support for the TME and is comprises of two components: 1) polysaccharides, which assemble into proteoglycans; this forms a gel-like structure in which fibrillar proteins embed; and 2) fibrillar proteins (fibronectins, laminins, collagen and elastin) which function as ligands for cell adhesion molecules (448). ECM proteins are produced by mesenchymal cells and the constant restructuring of the ECM is modulated by hormones, growth factors, cytokines and extracorporeal factors which influence homeostasis, repair mechanisms and morphology (449). A key aspect of ECM remodeling focuses on mesenchymal cell (i.e. fibroblast)-mediated proteolysis and re-synthesis, which is dependent on the activity of MMPs and LOX, respectively. During re-synthesis, CAFs express high levels of LOX which cross-links collagen and elastin thus increasing the rigidity of the ECM. Another stiffening mechanism, previously described, is the constant inflammatory state of the TME that induces fibroblastic transformation into myelofibroblasts. The level of desmoplastic reconstruction is positively associated with treatment resistance and worse prognosis (450). A meta-analysis showed that the level of ECM stiffness was positively correlated with cancer cell genomic instability. Three hypotheses were proposed by the authors: 1) stiffness induces DNA damage during cellular migration; 2) tumor invasion of a densely-packed environment results in the selection of more aggressive phenotypes; and 3) stiffness enhances proliferation (451). It was hypothesized that a shift from the physiologic basement membrane to a collagen-rich, dense and rigid ECM is a key factor in therapy resistance (452).
Cellular stiffness within the TME is exerted via transmembrane proteins, mainly integrins. Integrins exhibit dual function when exposed to stress:
1. as messengers that interact with intracellular-signaling pathways (kinases such as FAK/Src, MAPK, ROCK, JNK) and anti-apoptotic oncogenes (e.g. the YAP/TAZ/HIPPO pathway) delivering mechanical signals from surrounding cells to the transcription apparatus of the nucleus (453, 454). This activates integrin and kinase overexpression, inducing phenotypic variation and EMT (455);
2. physically connects to actin components of the cytoskeleton via linker proteins (e.g. vinculin, α-actinin and talin), signaling molecules (FAK, Src), and adapter proteins (Paxillin, senescent cell-antigen-like containing domain 1, PINCH-1) to modify cytoskeletal contractile forces and thereby inducing the EMT (456, 457).
The modification of the nuclear envelope, with regard to cancer cell progression has been described in a number of studies (458). The nuclear envelope mainly consists of the nuclear pore complex and lamins, which link the nuclear and cellular cytoskeletons, and both were shown to be greatly modulated by cancer cells (458). A mechano-sensor, the nuclear envelope converts and transmits signals to the nucleus, thus dictating nuclear deformability (459). This parameter in turn regulates cellular plasticity and invasion of dense tissue (460). Nucleus-cytoskeleton interactions influence nuclear stiffness, which impacts chromatin rearrangement, transcription of previously repressed genes, and change in cellular polarity. These interactions are shown to support resistance to therapy and facilitate the metastatic process (461).
Multiple cancer models have established how ECM rigidity influences chemotherapeutic resistance and cancer proliferation. Breast cancer is resistant to sorafenib, a result that is positively correlated with collagen concentration and degree of stiffness. Furthermore, triple negative breast cancer (TNBC) cells exhibit resistance as a result of overexpression of β1-integrin dependent activation of the JNK pathway (462). Moreover, another study cultured TNBC cells in varying degrees of ECM stiffness before exposing them to doxorubicin, and doxorubicin efficacy is seen to be negatively correlated with ECM stiffness. Nuclear translocation of YAP in those cells appears to be the primary driver of the EMT (463). Another well-studied entity is HCC which presents with extensive fibrosis. HCC resistance to paclitaxel, cisplatin, 5-FU and sorafenib is shown to be positively correlated with ECM stiffness (462, 464). High stiffness-ECM was seen to induce HCC dormancy, with expression of SC markers such as CD133, CXCR4 and NANOG (465). Furthermore, ECM stiffness has been shown to mediate HCC stemness and resistance to oxaliplatin. The resistance mechanism appears to depend on integrin expression in response to ECM-mediated stiffness, which in turn upregulates phosphorylation of the Akt/mTOR pathway that is crucial for self-renewal (466). These investigations show how ECM stiffness mediates treatment resistance, utilizing a cascade of signals that originate from cell-cell and cell-ECM connections, and are a potential target to mitigate treatment resistance.
Growth-Induced Solid Stress
Solid stress arises from mechanical (shear, compressive, and tensile) forces exerted by the elastic and solid components of the TME. Rapid cellular proliferation, infiltration, and ECM deposition leads to the ready accumulation of solid stress in the TME, and becomes a significant barrier to effective drug delivery. Furthermore, solid stress collapses vessels and initiates cellular dormancy; after conventional treatments deplete sensitive cancer cells, these dormant cells with stemness are reawakened and nourished by the blood vessels (467–469). Solid stress induces hypoxic failure of chemo- and radiotherapy delivery, while hypoxia-mediated HIF-1-α has the capacity to induce EMT and encourage the development of cells with SC phenotypes (470). Moreover, a study showed that solid stress induced the upregulation of ECM adhesion molecules and the formation of “leader cells”. These are capable of coordinating cellular migration, resulting in cellular invasion and metastasis (471). A boost in leader cell phenotypes has been observed following exposure to conventional treatments (472). Demonstrating high transcriptional plasticity, leader cells have been shown to possess CSC-like properties with resistance to chemo- and radiotherapy (473).
Interstitial Fluid Pressure
High interstitial fluid pressure (IFP) is also dependent on ECM stiffness. This is caused by hypoxia-induced angiogenesis and impaired vessel function, which has been associated with resistance to targeted therapy, chemo-, and radiation therapy (474, 475). IFP increase in tumors has not been fully explained but it is thought to occur after leakage in defective vessels, followed by high protein deposition, contributing to ECM rigidity (476). This was particularly apparent in pancreatic ductal adenocarcinoma, where it was observed that high hyaluronan content collapsed vessels and decreased cytotoxic drug distribution. This mechanical resistance was reversed after enzymatic breakdown of the stroma (477).
The mechanical changes within tumors render otherwise effective chemo- and radiotherapeutic approaches ineffective. In addition to the physical barrier to therapy, the stiffened ECM was shown to induce the EMT and the development of cellular dormancy. As was described in these three mechanisms of resistance, early combination therapies targeting the cancer type and aspects of the physical blockade could increase efficacy and prevent the development of therapy-resistant variants.
TME Innervation
It has been well established that cancer invasion can occur into or around nerve routes via perineural invasion (PNI) (478); this has been associated with pain and poor prognosis (479). It has been shown that PNI induces the release of factors necessary for tumor growth (480), and consequently, the cells of the TME were seen to induce an adrenergic neuronal cell phenotype which supports metastasis in pulmonary (481), ovarian (482) and pancreatic cancers (483, 484). Prostate cancer studies have shown that cancer cells express neurotrophic growth factor (NGF), which attracts nerve fibers toward the TME to promote tumor invasion and metastasis (485). Additionally, denervation has been shown to suppress tumorigenesis, further denoting the importance of innervation (486). Although apparently significant, neurotransmitter concentration in serum was not sufficiently elevated, and it is now thought that perhaps this increased concentration is diverted towards the TME. Additionally, it has been shown that astrocytomas were able to resist treatment modalities by forming a tight microenvironment covered by a microtubular network resistant to radiotherapy (487), and expressing phenotypic changes within the TME that induced stemness, which resulted in chemotherapeutic resistance (488).
See Figure 2 for a diagrammatic summary of the major pathways that promote treatment resistance, and immune and treatment resistance.
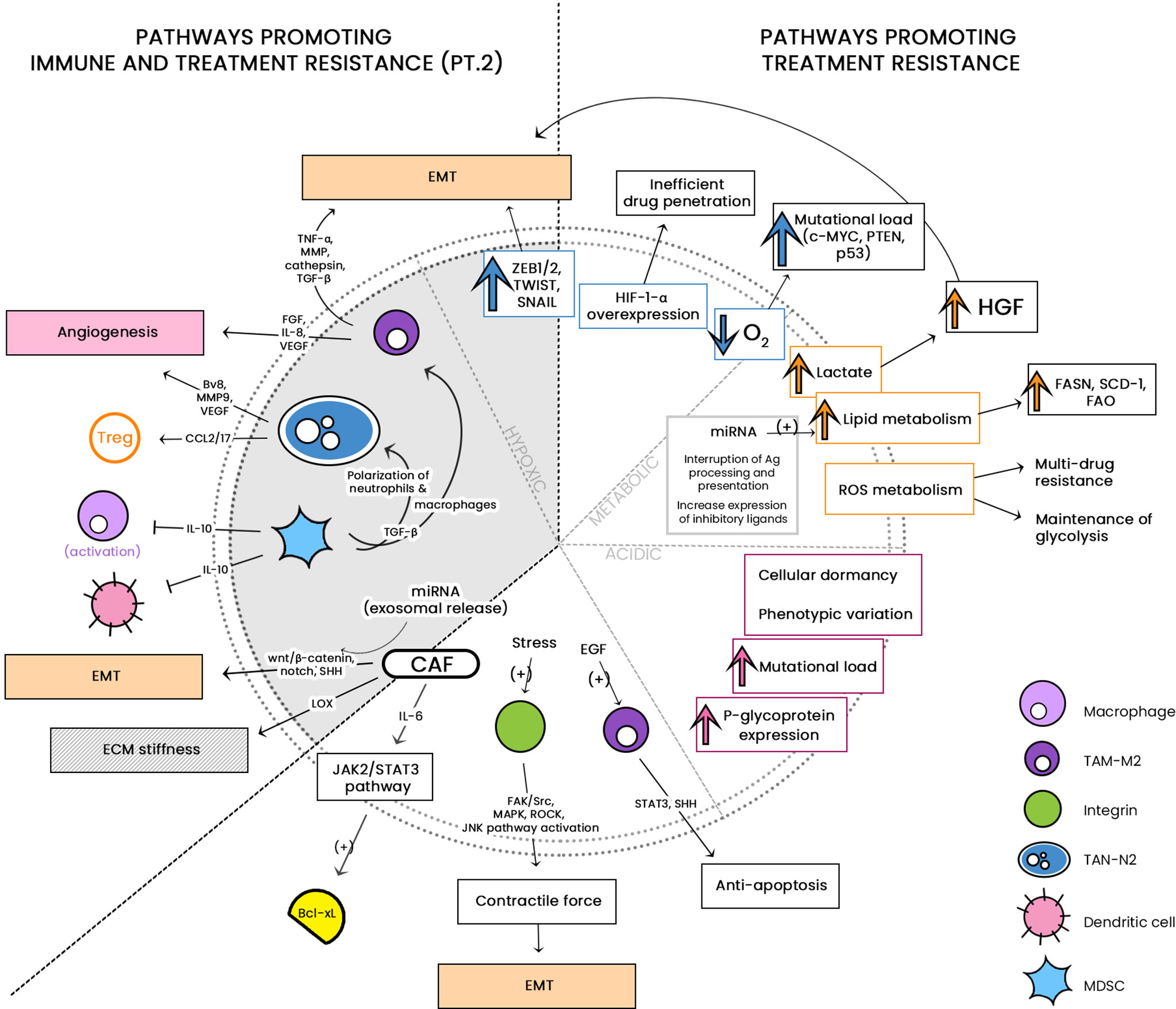
Figure 2 Alterations in the tumor microenvironment (TME) induce modifications in metabolic pathways and mechanical stress. These alterations have been shown to induce drug resistance by amplifying cell-to-cell and cell-to-ECM crosstalk, activating protective pathways and inducing phenotypic variations, in addition to biochemical signaling and resistance to apoptosis. Moreover, soluble factors released by tumor-supporting immune cells have been shown to induce both immune and drug resistance via induction of epithelial-to-mesenchymal transition (EMT), extracellular matrix (ECM) stiffness and angiogenesis. TGF-ß, Transforming growth factor- ß; LOX, lysyl oxidase; TNF- α, tumor necrosis factor alpha; ROS, reactive oxygen species; ECM, extracellular matrix; HGF, hepatocyte growth factor; HIF-1-α, hypoxia inducible factor-1-alpha; VEGF, vascular endothelial growth factor; CCL, C-C motif chemokine ligand; MAPK, mitogen activated protein kinase; EMT, epithelial-to-mesenchymal transition; IL, interleukin; JNK, c-Jun-N-terminal kinase; JAK/STAT, Janus kinase/Signal transducer and activator of transcription; ROCK, Rho-associated protein kinases; FAK/Src, Focal adhesion kinase/src family kinase; FASN, fatty acid synthase; Bv8, prokineticin 1; FGF, fibroblast growth factor; FAO, fatty acid oxidation; SCD-1, stearoyl-CoA-desaturase-1; MMP, matrix metalloproteinase.
Conclusion
Within the complex microenvironment of the TME, immune progenitors are encouraged to differentiate into regulatory T cells, M2 macrophages and MDSCs, amongst others, rather than fulfilling their tumor-suppressive roles as fully mature immune cells. The interaction between cellular components and soluble factors of the TME efficiently nurtures immune evasion and suppression, drug resistance, and promotes malignancy. Cellular crosstalk via both paracrine and juxtracrine signaling coordinates key elements that define cancer stemness, extracellular matrix remodeling, and the recruitment of non-malignant tumor-supporting cells. In addition to immune resistance, therapy resistance within the TME is achieved through various physical and biochemical factors that induce the EMT and modulate epigenetic changes that result in the formation of CSCs. In this review, it is evident that landmark research has elucidated these dysfunctional immune components with increasing clarity. Many of these components are now targets of promising drug therapies currently undergoing investigation, and these ground-breaking new discoveries continue to pave the way for new treatment modalities in the fight against cancer.
Author Contributions
Conceptualisation, KK; Investigation and Resources, KK, DH, JC, CS; Writing - Original Draft Preparation, KK, DH, JC, CS; Writing - Review and Editing, KK, JC, MK; Visualisation, KK, JC, MK; Graphics, JC, KK; Supervision, AM, MK; and Project Administration, AM, MK. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic Alterations During Colorectal-Tumor Development. N Engl J Med (1988) 319:525–32. doi: 10.1056/NEJM198809013190901
2. Chaffer CL, Weinberg RA. A Perspective on Cancer Cell Metastasis. Science (2011) 331:1559–64. doi: 10.1126/science.1203543
3. Dillekås H, Rogers MS, Straume O. Are 90% of Deaths From Cancer Caused by Metastases? Cancer Med (2019) 8:5574–6. doi: 10.1002/cam4.2474
4. Paget S. The Distribution of Secondary Growths In Cancer Of the Breast. Lancet (1889) 133:571–3. doi: 10.1016/S0140-6736(00)49915-0
5. Seyfried TN, Huysentruyt LC. On the Origin of Cancer Metastasis. Crit Rev Oncog (2013) 18:43–73. doi: 10.1615/CritRevOncog.v18.i1-2.40
6. Fidler IJ, Talmadge JE. Evidence That Intravenously Derived Murine Pulmonary Melanoma Metastases can Originate From the Expansion of a Single Tumor Cell. Cancer Res (1986) 46:5167–71.
7. Hanahan D, Coussens LM. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
8. Castells M, Thibault B, Delord J-P, Couderc B. Implication of Tumor Microenvironment in Chemoresistance: Tumor-Associated Stromal Cells Protect Tumor Cells From Cell Death. Int J Mol Sci (2012) 13:9545–71. doi: 10.3390/ijms13089545
9. Dauer P, Nomura A, Saluja A, Banerjee S. Microenvironment in Determining Chemo-Resistance in Pancreatic Cancer: Neighborhood Matters. Pancreatology (2017) 17:7–12. doi: 10.1016/j.pan.2016.12.010
10. Hernández R, Jiménez-Luna C, Perales-Adán J, Perazzoli G, Melguizo C, Prados J. Differentiation of Human Mesenchymal Stem Cells Towards Neuronal Lineage: Clinical Trials in Nervous System Disorders. Biomol Ther (2020) 28:34–44. doi: 10.4062/biomolther.2019.065
11. Martinez-Agosto JA, Mikkola HKA, Hartenstein V, Banerjee U. The Hematopoietic Stem Cell and its Niche: A Comparative View. Genes Dev (2007) 21:3044–60. doi: 10.1101/gad.1602607
12. Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, et al. Origins of the Tumor Microenvironment: Quantitative Assessment of Adipose-Derived and Bone Marrow-Derived Stroma. PLoS One (2012) 7:e30563. doi: 10.1371/journal.pone.0030563
13. Achen MG, McColl BK, Stacker SA. Focus on Lymphangiogenesis in Tumor Metastasis. Cancer Cell (2005) 7:121–7. doi: 10.1016/j.ccr.2005.01.017
14. Chen J-C, Chang Y-W, Hong C-C, Yu Y-H, Su J-L. The Role of the VEGF-C/VEGFRs Axis in Tumor Progression and Therapy. Int J Mol Sci (2012) 14:88–107. doi: 10.3390/ijms14010088
15. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
16. Carmeliet P, Jain RK. Principles and Mechanisms of Vessel Normalization for Cancer and Other Angiogenic Diseases. Nat Rev Drug Discov (2011) 10:417–27. doi: 10.1038/nrd3455
17. Wong BW, Kuchnio A, Bruning U, Carmeliet P. Emerging Novel Functions of the Oxygen-Sensing Prolyl Hydroxylase Domain Enzymes. Trends Biochem Sci (2013) 38:3–11. doi: 10.1016/j.tibs.2012.10.004
18. Skuli N, Liu L, Runge A, Wang T, Yuan L, Patel S, et al. Endothelial Deletion of Hypoxia-Inducible factor-2alpha (Hif-2alpha) Alters Vascular Function and Tumor Angiogenesis. Blood (2009) 114:469–77. doi: 10.1182/blood-2008-12-193581
19. Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber H-P, et al. Loss of HIF-1Alpha in Endothelial Cells Disrupts a Hypoxia-Driven VEGF Autocrine Loop Necessary for Tumorigenesis. Cancer Cell (2004) 6:485–95. doi: 10.1016/j.ccr.2004.09.026
20. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B Receptor Mediates the Endothelial Barrier to T Cell Homing to Tumors and Disables Immune Therapy. Nat Med (2008) 14:28–36. doi: 10.1038/nm1699
21. Kambayashi T, Laufer TM. Atypical MHC Class II-Expressing Antigen-Presenting Cells: Can Anything Replace a Dendritic Cell? Nat Rev Immunol (2014) 14:719–30. doi: 10.1038/nri3754
22. Goveia J, Rohlenova K, Taverna F, Treps L, Conradi L-C, Pircher A, et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell (2020) 37::21–36.e13. doi: 10.1016/j.ccell.2019.12.001
23. De Sanctis F, Ugel S, Facciponte J, Facciabene A. The Dark Side of Tumor-Associated Endothelial Cells. Semin Immunol (2018) 35:35–47. doi: 10.1016/j.smim.2018.02.002
24. Flati V, Pastore LI, Griffioen AW, Satijn S, Tomato E, D’Alimonte I, et al. Endothelial Cell Anergy Is Mediated by bFGF Through the Sustained Activation of P38-MAPK and NF-κb Inhibition. Int J Immunopathol Pharmacol (2006) 19:761–73. doi: 10.1177/039463200601900406
25. Delfortrie S, Pinte S, Mattot V, Samson C, Villain G, Caetano B, et al. Egfl7 Promotes Tumor Escape From Immunity by Repressing Endothelial Cell Activation. Cancer Res (2011) 71:7176–86. doi: 10.1158/0008-5472.CAN-11-1301
26. Nagl L, Horvath L, Pircher A, Wolf D. Tumor Endothelial Cells (Tecs) as Potential Immune Directors of the Tumor Microenvironment – New Findings and Future Perspectives. Front Cell Dev Biol (2020) 8:766. doi: 10.3389/fcell.2020.00766
27. Shetty S, Weston CJ, Oo YH, Westerlund N, Stamataki Z, Youster J, et al. Common Lymphatic Endothelial and Vascular Endothelial Receptor-1 Mediates the Transmigration of Regulatory T Cells Across Human Hepatic Sinusoidal Endothelium. J Immunol (2011) 186:4147–55. doi: 10.4049/jimmunol.1002961
28. Hollmén M, Figueiredo CR, Jalkanen S. New Tools to Prevent Cancer Growth and Spread: A ‘Clever’ Approach. Br J Cancer (2020) 123:501–9. doi: 10.1038/s41416-020-0953-0
29. Krambeck AE, Thompson RH, Dong H, Lohse CM, Park ES, Kuntz SM, et al. B7-H4 Expression in Renal Cell Carcinoma and Tumor Vasculature: Associations With Cancer Progression and Survival. Proc Natl Acad Sci (2006) 103:10391–6. doi: 10.1073/pnas.0600937103
30. Riesenberg R, Weiler C, Spring O, Eder M, Buchner A, Popp T, et al. Expression of Indoleamine 2,3-Dioxygenase in Tumor Endothelial Cells Correlates With Long-Term Survival of Patients With Renal Cell Carcinoma. Clin Cancer Res (2007) 13:6993–7002. doi: 10.1158/1078-0432.CCR-07-0942
31. Huang D, Ding Y, Zhou M, Rini BI, Petillo D, Qian C-N, et al. Interleukin-8 Mediates Resistance to Antiangiogenic Agent Sunitinib in Renal Cell Carcinoma. Cancer Res (2010) 70:1063–71. doi: 10.1158/0008-5472.CAN-09-3965
32. Mazanet MM, Hughes CCW. B7-H1 Is Expressed by Human Endothelial Cells and Suppresses T Cell Cytokine Synthesis. J Immunol (2002) 169:3581–8. doi: 10.4049/jimmunol.169.7.3581
33. Mulligan JK, Young MRI. Tumors Induce the Formation of Suppressor Endothelial Cells In Vivo. Cancer Immunol Immunother (2009) 59:267. doi: 10.1007/s00262-009-0747-y
34. Casós K, Siguero L, Fernández-Figueras M-T, León X, Sardá M-P, Vila L, et al. Tumor Cells Induce COX-2 and mPGES-1 Expression in Microvascular Endothelial Cells Mainly by Means of IL-1 Receptor Activation. Microvasc Res (2011) 81:261–8. doi: 10.1016/j.mvr.2011.01.006
35. Taflin C, Favier B, Baudhuin J, Savenay A, Hemon P, Bensussan A, et al. Human Endothelial Cells Generate Th17 and Regulatory T Cells Under Inflammatory Conditions. Proc Natl Acad Sci (2011) 108:2891–6. doi: 10.1073/pnas.1011811108
36. Sata M, Walsh K. Tnfα Regulation of Fas Ligand Expression on the Vascular Endothelium Modulates Leukocyte Extravasation. Nat Med (1998) 4:415–20. doi: 10.1038/nm0498-415
37. Yu JS, Lee PK, Ehtesham M, Samoto K, Black KL, Wheeler CJ. Intratumoral T Cell Subset Ratios and Fas Ligand Expression on Brain Tumor Endothelium. J Neurooncol (2003) 64:55–61. doi: 10.1007/BF02700020
38. Motz GT, Santoro SP, Wang L-P, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor Endothelium FasL Establishes a Selective Immune Barrier Promoting Tolerance in Tumors. Nat Med (2014) 20:607–15. doi: 10.1038/nm.3541
39. Lanitis E, Irving M, Coukos G. Targeting the Tumor Vasculature to Enhance T Cell Activity. Curr Opin Immunol (2015) 33:55–63. doi: 10.1016/j.coi.2015.01.011
40. Wang M, Yuan Q, Xie L. Mesenchymal Stem Cell-Based Immunomodulation: Properties and Clinical Application. Stem Cells Int (2018) 2018:e3057624. doi: 10.1155/2018/3057624
41. Ley K, Kansas GS. Selectins in T-cell Recruitment to Non-Lymphoid Tissues and Sites of Inflammation. Nat Rev Immunol (2004) 4:325–36. doi: 10.1038/nri1351
42. Weber C, Fraemohs L, Dejana E. The Role of Junctional Adhesion Molecules in Vascular Inflammation. Nat Rev Immunol (2007) 7:467–77. doi: 10.1038/nri2096
43. Ala A, Dhillon AP, Hodgson HJ. Role of Cell Adhesion Molecules in Leukocyte Recruitment in the Liver and Gut. Int J Exp Pathol (2003) 84:1–16. doi: 10.1046/j.1365-2613.2003.00235.x
44. Greco SJ, Liu K, Rameshwar P. Functional Similarities Among Genes Regulated by OCT4 in Human Mesenchymal and Embryonic Stem Cells. Stem Cells Dayt Ohio (2007) 25:3143–54. doi: 10.1634/stemcells.2007-0351
45. Riekstina U, Cakstina I, Parfejevs V, Hoogduijn M, Jankovskis G, Muiznieks I, et al. Embryonic Stem Cell Marker Expression Pattern in Human Mesenchymal Stem Cells Derived From Bone Marrow, Adipose Tissue, Heart and Dermis. Stem Cell Rev Rep (2009) 5:378–86. doi: 10.1007/s12015-009-9094-9
46. Hocking AM, Gibran NS. Mesenchymal Stem Cells: Paracrine Signaling and Differentiation During Cutaneous Wound Repair. Exp Cell Res (2010) 316:2213–9. doi: 10.1016/j.yexcr.2010.05.009
47. Saud B, Malla R, Shrestha K. A Review on the Effect of Plant Extract on Mesenchymal Stem Cell Proliferation and Differentiation. Stem Cells Int (2019) 2019:7513404. doi: 10.1155/2019/7513404
48. Komarova S, Kawakami Y, Stoff-Khalili MA, Curiel DT, Pereboeva L. Mesenchymal Progenitor Cells as Cellular Vehicles for Delivery of Oncolytic Adenoviruses. Mol Cancer Ther (2006) 5:755–66. doi: 10.1158/1535-7163.MCT-05-0334
49. Ren C, Kumar S, Chanda D, Chen J, Mountz JD, Ponnazhagan S. Therapeutic Potential of Mesenchymal Stem Cells Producing Interferon-Alpha in a Mouse Melanoma Lung Metastasis Model. Stem Cells Dayt Ohio (2008) 26:2332–8. doi: 10.1634/stemcells.2008-0084
50. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan Impairs Vascular Function and Drug Delivery in a Mouse Model of Pancreatic Cancer. Gut (2013) 62:112–20. doi: 10.1136/gutjnl-2012-302529
51. Yan X, Fu C, Chen L, Qin J, Zeng Q, Yuan H, et al. Mesenchymal Stem Cells From Primary Breast Cancer Tissue Promote Cancer Proliferation and Enhance Mammosphere Formation Partially Via EGF/EGFR/Akt Pathway. Breast Cancer Res Treat (2012) 132:153–64. doi: 10.1007/s10549-011-1577-0
52. Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, et al. Breast Cancer Stem Cells Are Regulated by Mesenchymal Stem Cells Through Cytokine Networks. Cancer Res (2011) 71:614–24. doi: 10.1158/0008-5472.CAN-10-0538
53. Rhodes LV, Antoon JW, Muir SE, Elliott S, Beckman BS, Burow ME. Effects of Human Mesenchymal Stem Cells on ER-Positive Human Breast Carcinoma Cells Mediated Through ER-SDF-1/CXCR4 Crosstalk. Mol Cancer (2010) 9:295. doi: 10.1186/1476-4598-9-295
54. Xu G, Zhang L, Ren G, Yuan Z, Zhang Y, Zhao RC, et al. Immunosuppressive Properties of Cloned Bone Marrow Mesenchymal Stem Cells. Cell Res (2007) 17:240–8. doi: 10.1038/cr.2007.4
55. Liotta F, Angeli R, Cosmi L, Filì L, Manuelli C, Frosali F, et al. Toll-Like Receptors 3 and 4 are Expressed by Human Bone Marrow-Derived Mesenchymal Stem Cells and Can Inhibit Their T-Cell Modulatory Activity by Impairing Notch Signaling. Stem Cells Dayt Ohio (2008) 26:279–89. doi: 10.1634/stemcells.2007-0454
56. Ghannam S, Pène J, Moquet-Torcy G, Torcy-Moquet G, Jorgensen C, Yssel H. Mesenchymal Stem Cells Inhibit Human Th17 Cell Differentiation and Function and Induce a T Regulatory Cell Phenotype. J Immunol Baltim Md 1950 (2010) 185:302–12. doi: 10.4049/jimmunol.0902007
57. Chen L, Tredget EE, Wu PYG, Wu Y. Paracrine Factors of Mesenchymal Stem Cells Recruit Macrophages and Endothelial Lineage Cells and Enhance Wound Healing. PLoS One (2008) 3:e1886. doi: 10.1371/journal.pone.0001886
58. Oswald J, Boxberger S, Jørgensen B, Feldmann S, Ehninger G, Bornhäuser M, et al. Mesenchymal Stem Cells Ban be Differentiated Into Endothelial Cells In Vitro. Stem Cells Dayt Ohio (2004) 22:377–84. doi: 10.1634/stemcells.22-3-377
59. Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, et al. Mesenchymal Stem Cell Transition to Tumor-Associated Fibroblasts Contributes to Fibrovascular Network Expansion and Tumor Progression. PLoS One (2009) 4:e4992. doi: 10.1371/journal.pone.0004992
60. Aldinucci D, Colombatti A. The Inflammatory Chemokine CCL5 and Cancer Progression. Mediators Inflammation (2014) 2014:292376. doi: 10.1155/2014/292376
61. Betz M, Fox BS. Prostaglandin E2 Inhibits Production of Th1 Lymphokines But Not of Th2 Lymphokines. J Immunol Baltim Md 1950 (1991) 146:108–13.
62. van der Pouw Kraan TC, Boeije LC, Smeenk RJ, Wijdenes J, Aarden LA. Prostaglandin-E2 Is a Potent Inhibitor of Human Interleukin 12 Production. J Exp Med (1995) 181:775–9. doi: 10.1084/jem.181.2.775
63. Kaliński P, Hilkens CM, Snijders A, Snijdewint FG, Kapsenberg ML. IL-12-Deficient Dendritic Cells, Generated in the Presence of Prostaglandin E2, Promote Type 2 Cytokine Production in Maturing Human Naive T Helper Cells. J Immunol Baltim Md 1950 (1997) 159:28–35.
64. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive Feedback Between PGE2 and COX2 Redirects the Differentiation of Human Dendritic Cells Toward Stable Myeloid-Derived Suppressor Cells. Blood (2011) 118:5498–505. doi: 10.1182/blood-2011-07-365825
65. Heusinkveld M, de Vos van Steenwijk PJ, Goedemans R, Ramwadhdoebe TH, Gorter A, Welters MJP, et al. M2 Macrophages Induced by Prostaglandin E2 and IL-6 From Cervical Carcinoma Are Switched to Activated M1 Macrophages by CD4+ Th1 Cells. J Immunol Baltim Md 1950 (2011) 187:1157–65. doi: 10.4049/jimmunol.1100889
66. Jung Y-J, Isaacs JS, Lee S, Trepel J, Neckers L. Il-1beta-mediated Up-Regulation of HIF-1alpha Via an NFkappaB/COX-2 Pathway Identifies HIF-1 as a Critical Link Between Inflammation and Oncogenesis. FASEB J (2003) 17:2115–7. doi: 10.1096/fj.03-0329fje
67. Munn DH, Mellor AL. Indoleamine 2,3 Dioxygenase and Metabolic Control of Immune Responses. Trends Immunol (2013) 34:137–43. doi: 10.1016/j.it.2012.10.001
68. Mellor AL, Munn DH. Creating Immune Privilege: Active Local Suppression That Benefits Friends, But Protects Foes. Nat Rev Immunol (2008) 8:74–80. doi: 10.1038/nri2233
69. Hobbs AJ, Higgs A, Moncada S. Inhibition of Nitric Oxide Synthase as a Potential Therapeutic Target. Annu Rev Pharmacol Toxicol (1999) 39:191–220. doi: 10.1146/annurev.pharmtox.39.1.191
70. Bronte V, Zanovello P. Regulation of Immune Responses by L-Arginine Metabolism. Nat Rev Immunol (2005) 5:641–54. doi: 10.1038/nri1668
71. Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L-Arginine Metabolism in Myeloid Cells Controls T-Lymphocyte Functions. Trends Immunol (2003) 24:302–6. doi: 10.1016/s1471-4906(03)00132-7
72. Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-Derived Nitric Oxide Regulates T Cell Activation Via Reversible Disruption of the Jak3/STAT5 Signaling Pathway. J Immunol Baltim Md 1950 (1998) 160:5729–34.
73. Kikuchi Y, Kashima TG, Nishiyama T, Shimazu K, Morishita Y, Shimazaki M, et al. Periostin Is Expressed in Pericryptal Fibroblasts and Cancer-Associated Fibroblasts in the Colon. J Histochem Cytochem (2008) 56:753–64. doi: 10.1369/jhc.2008.951061
74. De Wever O, Nguyen Q-D, Van Hoorde L, Bracke M, Bruyneel E, Gespach C, et al. Tenascin-C and SF/HGF Produced by Myofibroblasts In Vitro Provide Convergent Pro-Invasive Signals to Human Colon Cancer Cells Through RhoA and Rac. FASEB J (2004) 18:1016–8. doi: 10.1096/fj.03-1110fje
75. Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-Associated Fibroblasts Direct Tumor Progression of Initiated Human Prostatic Epithelium. Cancer Res (1999) 59:5002–11. doi: 10.1186/bcr138
76. Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, et al. Reconstruction of Functionally Normal and Malignant Human Breast Tissues in Mice. Proc Natl Acad Sci (2004) 101:4966–71. doi: 10.1073/pnas.0401064101
77. Ribatti D, Tamma R, Annese T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl Oncol (2020) 13:100773. doi: 10.1016/j.tranon.2020.100773
78. Zhou B, Chen W-L, Wang Y-Y, Lin Z-Y, Zhang D-M, Fan S, et al. A Role for Cancer-Associated Fibroblasts in Inducing the Epithelial-to-Mesenchymal Transition in Human Tongue Squamous Cell Carcinoma. J Oral Pathol Med (2014) 43:585–92. doi: 10.1111/jop.12172
79. Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, et al. Ccl2 Mediates Cross-Talk Between Cancer Cells and Stromal Fibroblasts That Regulates Breast Cancer Stem Cells. Cancer Res (2012) 72:2768–79. doi: 10.1158/0008-5472.CAN-11-3567
80. Lau EYT, Lo J, Cheng BYL, Ma MKF, Lee JMF, Ng JKY, et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma Through c-Met/FRA1/HEY1 Signaling. Cell Rep (2016) 15:1175–89. doi: 10.1016/j.celrep.2016.04.019
81. Gascard P, Tlsty TD. Carcinoma-Associated Fibroblasts: Orchestrating the Composition of Malignancy. Genes Dev (2016) 30:1002–19. doi: 10.1101/gad.279737.116
82. Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, et al. Constitutive Activation of Stat3 Signaling Confers Resistance to Apoptosis in Human U266 Myeloma Cells. Immunity (1999) 10:105–15. doi: 10.1016/s1074-7613(00)80011-4
83. Inohara N, Ding L, Chen S, Núñez G. Harakiri, a Novel Regulator of Cell Death, Encodes a Protein That Activates Apoptosis and Interacts Selectively With Survival-Promoting Proteins Bcl-2 and Bcl-X(L). EMBO J (1997) 16:1686–94. doi: 10.1093/emboj/16.7.1686
84. Bonnans C, Chou J, Werb Z. Remodelling the Extracellular Matrix in Development and Disease. Nat Rev Mol Cell Biol (2014) 15:786–801. doi: 10.1038/nrm3904
85. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell (2009) 139:891–906. doi: 10.1016/j.cell.2009.10.027
86. Pietras K, Pahler J, Bergers G, Hanahan D. Functions of Paracrine Pdgf Signaling in the Proangiogenic Tumor Stroma Revealed by Pharmacological Targeting. PLoS Med (2008) 5:e19. doi: 10.1371/journal.pmed.0050019
87. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis Through Elevated SDF-1/CXCL12 Secretion. Cell (2005) 121:335–48. doi: 10.1016/j.cell.2005.02.034
88. Zhao T, Su Z, Li Y, Zhang X, You Q. Chitinase-3 Like-Protein-1 Function and its Role in Diseases. Signal Transduct Target Ther (2020) 5:1–20. doi: 10.1038/s41392-020-00303-7
89. Monteran L, Erez N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front Immunol (2019) 10:1835. doi: 10.3389/fimmu.2019.01835
90. Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of Tumor-Associated Macrophages in Neoplastic Progression and Immune Surveillance. Immunol Rev (2008) 222:155–61. doi: 10.1111/j.1600-065X.2008.00607.x
91. Takahashi H, Sakakura K, Kudo T, Toyoda M, Kaira K, Oyama T, et al. Cancer-Associated Fibroblasts Promote an Immunosuppressive Microenvironment Through the Induction and Accumulation of Protumoral Macrophages. Oncotarget (2017) 8:8633–47. doi: 10.18632/oncotarget.14374
92. Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-Associated Fibroblasts and M2-Polarized Macrophages Synergize During Prostate Carcinoma Progression. Oncogene (2014) 33:2423–31. doi: 10.1038/onc.2013.191
93. Scherz-Shouval R, Santagata S, Mendillo ML, Sholl LM, Ben-Aharon I, Beck AH, et al. The Reprogramming of Tumor Stroma by HSF1 Is a Potent Enabler of Malignancy. Cell (2014) 158:564–78. doi: 10.1016/j.cell.2014.05.045
94. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 From FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes With Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc Natl Acad Sci USA (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
95. Yang W, Wetterskog D, Matsumoto Y, Funa K. Kinetics of Repression by Modified p53 on the PDGF Beta-Receptor Promoter. Int J Cancer (2008) 123:2020–30. doi: 10.1002/ijc.23735
96. Agrawal V, Maharjan S, Kim K, Kim N-J, Son J, Lee K, et al. Direct Endothelial Junction Restoration Results in Significant Tumor Vascular Normalization and Metastasis Inhibition in Mice. Oncotarget (2014) 5:2761–77. doi: 10.18632/oncotarget.1942
97. Courtney J-M, Sutherland BA. Harnessing the Stem Cell Properties of Pericytes to Repair the Brain. Neural Regener Res (2019) 15:1021–2. doi: 10.4103/1673-5374.270301
98. Ochs K, Sahm F, Opitz CA, Lanz TV, Oezen I, Couraud P-O, et al. Immature Mesenchymal Stem Cell-Like Pericytes as Mediators of Immunosuppression in Human Malignant Glioma. J Neuroimmunol (2013) 265:106–16. doi: 10.1016/j.jneuroim.2013.09.011
99. Pober JS. Tellides G. Participation of Blood Vessel Cells in Human Adaptive Immune Responses. Trends Immunol (2012) 33:49–57. doi: 10.1016/j.it.2011.09.006
100. Tu Z, Li Y, Smith DS, Sheibani N, Huang S, Kern T, et al. Retinal Pericytes Inhibit Activated T Cell Proliferation. Invest Ophthalmol Vis Sci (2011) 52:9005–10. doi: 10.1167/iovs.11-8008
101. Beck B, Blanpain C. Unravelling Cancer Stem Cell Potential. Nat Rev Cancer (2013) 13:727–38. doi: 10.1038/nrc3597
102. Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, Terzis AJA. Opinion: The Origin of the Cancer Stem Cell: Current Controversies and New Insights. Nat Rev Cancer (2005) 5:899–904. doi: 10.1038/nrc1740
103. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A Cell Initiating Human Acute Myeloid Leukaemia After Transplantation Into SCID Mice. Nature (1994) 367:645–8. doi: 10.1038/367645a0
104. Zhou B-BS, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-Initiating Cells: Challenges and Opportunities for Anticancer Drug Discovery. Nat Rev Drug Discovery (2009) 8:806–23. doi: 10.1038/nrd2137
105. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc Natl Acad Sci (2003) 100:3983–8. doi: 10.1073/pnas.0530291100
106. Maccalli C, Rasul KI, Elawad M, Ferrone S. The Role of Cancer Stem Cells in the Modulation of Anti-Tumor Immune Responses. Semin Cancer Biol (2018) 53:189–200. doi: 10.1016/j.semcancer.2018.09.006
107. Boyd AS, Rodrigues NP. Stem Cells Cycle Toward Immune Surveillance. Immunity (2018) 48:187–90. doi: 10.1016/j.immuni.2018.02.006
108. Korkaya H, Liu S, Wicha MS. Regulation of Cancer Stem Cells by Cytokine Networks: Attacking Cancer’s Inflammatory Roots. Clin Cancer Res (2011) 17:6125–9. doi: 10.1158/1078-0432.CCR-10-2743
109. Sultan M, Coyle KM, Vidovic D, Thomas ML, Gujar S, Marcato P. Hide-and-Seek: The Interplay Between Cancer Stem Cells and the Immune System. Carcinogenesis (2017) 38:107–18. doi: 10.1093/carcin/bgw115
110. Nagler C, Zänker KS, Dittmar T. Cell Fusion, Drug Resistance and Recurrence Cscs. Adv Exp Med Biol (2011) 714:173–82. doi: 10.1007/978-94-007-0782-5_9
111. Jiang E, Yan T, Xu Z, Shang Z. Tumor Microenvironment and Cell Fusion. BioMed Res Int (2019) 2019:5013592. doi: 10.1155/2019/5013592
112. Pires BRB, Amorim ÍSS DE, Souza LDE, Rodrigues JA, Mencalha AL. Targeting Cellular Signaling Pathways in Breast Cancer Stem Cells and Its Implication for Cancer Treatment. Anticancer Res (2016) 36:5681–91. doi: 10.21873/anticanres.11151
113. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
114. Shibue T, Weinberg RA. Emt, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat Rev Clin Oncol (2017) 14:611–29. doi: 10.1038/nrclinonc.2017.44
115. Verbeke H, Struyf S, Laureys G, Van Damme J. The Expression and Role of CXC Chemokines in Colorectal Cancer. Cytokine Growth Factor Rev (2011) 22:345–58. doi: 10.1016/j.cytogfr.2011.09.002
116. Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
117. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-Associated Macrophages (TAM) as Major Players of the Cancer-Related Inflammation. J Leukoc Biol (2009) 86:1065–73. doi: 10.1189/jlb.0609385
118. Daurkin I, Eruslanov E, Stoffs T, Perrin GQ, Algood C, Gilbert SM, et al. Tumor-Associated Macrophages Mediate Immunosuppression in the Renal Cancer Microenvironment by Activating the 15-Lipoxygenase-2 Pathway. Cancer Res (2011) 71:6400–9. doi: 10.1158/0008-5472.CAN-11-1261
119. Liu J, Zhang N, Li Q, Zhang W, Ke F, Leng Q, et al. Tumor-Associated Macrophages Recruit CCR6+ Regulatory T Cells and Promote the Development of Colorectal Cancer Via Enhancing CCL20 Production in Mice. PLoS One (2011) 6:e19495. doi: 10.1371/journal.pone.0019495
120. Yang M, Chen J, Su F, Yu B, Su F, Lin L, et al. Microvesicles Secreted by Macrophages Shuttle Invasion-Potentiating microRNAs Into Breast Cancer Cells. Mol Cancer (2011) 10:117. doi: 10.1186/1476-4598-10-117
121. Hao N-B, Lü M-H, Fan Y-H, Cao Y-L, Zhang Z-R, Yang S-M. Macrophages in Tumor Microenvironments and the Progression of Tumors. Clin Dev Immunol (2012) 2012:948098. doi: 10.1155/2012/948098
122. Ruffell B, Affara NI, Coussens LM. Differential Macrophage Programming in the Tumor Microenvironment. Trends Immunol (2012) 33:119–26. doi: 10.1016/j.it.2011.12.001
123. Costa NL, Valadares MC, Souza PPC, Mendonça EF, Oliveira JC, Silva TA, et al. Tumor-Associated Macrophages and the Profile of Inflammatory Cytokines in Oral Squamous Cell Carcinoma. Oral Oncol (2013) 49:216–23. doi: 10.1016/j.oraloncology.2012.09.012
124. Yeo E-J, Cassetta L, Qian B-Z, Lewkowich I, Li J, Stefater JA, et al. Myeloid WNT7b Mediates the Angiogenic Switch and Metastasis in Breast Cancer. Cancer Res (2014) 74:2962–73. doi: 10.1158/0008-5472.CAN-13-2421
125. Deryugina EI, Zajac E, Juncker-Jensen A, Kupriyanova TA, Welter L, Quigley JP. Tissue-Infiltrating Neutrophils Constitute the Major In Vivo Source of Angiogenesis-Inducing MMP-9 in the Tumor Microenvironment. Neoplasia N Y N (2014) 16:771–88. doi: 10.1016/j.neo.2014.08.013
126. Korbecki J, Olbromski M, Dzięgiel P. CCL18 in the Progression of Cancer. Int J Mol Sci (2020) 21:7955. doi: 10.3390/ijms21217955
127. Wang H, Wang X, Li X, Fan Y, Li G, Guo C, et al. Cd68(+)Hla-Dr(+) M1-like Macrophages Promote Motility of HCC Cells Via NF-κb/FAK Pathway. Cancer Lett (2014) 345:91–9. doi: 10.1016/j.canlet.2013.11.013
128. Candido J, Hagemann T. Cancer-Related Inflammation. J Clin Immunol (2013) 33(Suppl 1):S79–84. doi: 10.1007/s10875-012-9847-0
129. Yu H, Kortylewski M, Pardoll D. Crosstalk Between Cancer and Immune Cells: Role of STAT3 in the Tumour Microenvironment. Nat Rev Immunol (2007) 7:41–51. doi: 10.1038/nri1995
130. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-beta: “N1” Versus “N2” Tan. Cancer Cell (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
131. Haqqani AS, Sandhu JK, Birnboim HC. Expression of Interleukin-8 Promotes Neutrophil Infiltration and Genetic Instability in Mutatect Tumors. Neoplasia N Y N (2000) 2:561–8. doi: 10.1038/sj.neo.7900110
132. Michaeli J, Shaul ME, Mishalian I, Hovav A-H, Levy L, Zolotriov L, et al. Tumor-Associated Neutrophils Induce Apoptosis of Non-Activated CD8 T-Cells in a Tnfα and NO-dependent Mechanism, Promoting a Tumor-Supportive Environment. Oncoimmunology (2017) 6:e1356965. doi: 10.1080/2162402X.2017.1356965
133. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing Myeloid-Derived Suppressor Cells in Renal Cell Carcinoma Are a Subpopulation of Activated Granulocytes. Cancer Res (2009) 69:1553–60. doi: 10.1158/0008-5472.CAN-08-1921
134. Shaul ME, Fridlender ZG. Neutrophils as Active Regulators of the Immune System in the Tumor Microenvironment. J Leukoc Biol (2017) 102:343–9. doi: 10.1189/jlb.5MR1216-508R
135. Matsushita H, Hosoi A, Ueha S, Abe J, Fujieda N, Tomura M, et al. Cytotoxic T Lymphocytes Block Tumor Growth Both by Lytic Activity and Ifnγ-Dependent Cell-Cycle Arrest. Cancer Immunol Res (2015) 3:26–36. doi: 10.1158/2326-6066.CIR-14-0098
136. Chiang CL-L, Balint K, Coukos G, Kandalaft LE. Potential Approaches for More Successful Dendritic Cell-Based Immunotherapy. Expert Opin Biol Ther (2015) 15:569–82. doi: 10.1517/14712598.2015.1000298
137. Gao Q, Qiu S-J, Fan J, Zhou J, Wang X-Y, Xiao Y-S, et al. Intratumoral Balance of Regulatory and Cytotoxic T Cells Is Associated With Prognosis of Hepatocellular Carcinoma After Resection. J Clin Oncol (2007) 25:2586–93. doi: 10.1200/JCO.2006.09.4565
138. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, et al. Quantification of Regulatory T Cells Enables the Identification of High-Risk Breast Cancer Patients and Those At Risk of Late Relapse. J Clin Oncol (2006) 24:5373–80. doi: 10.1200/JCO.2006.05.9584
139. Protti MP, De Monte L. Cross-Talk Within the Tumor Microenvironment Mediates Th2-Type Inflammation in Pancreatic Cancer. Oncoimmunology (2012) 1:89–91. doi: 10.4161/onci.1.1.17939
140. Bharadwaj U, Li M, Zhang R, Chen C, Yao Q. Elevated Interleukin-6 and G-CSF in Human Pancreatic Cancer Cell Conditioned Medium Suppress Dendritic Cell Differentiation and Activation. Cancer Res (2007) 67:5479–88. doi: 10.1158/0008-5472.CAN-06-3963
141. Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of Tumor-Associated Macrophages and Neutrophils. Proc Natl Acad Sci USA (2012) 109:2491–6. doi: 10.1073/pnas.1113744109
142. Moretta A. Natural Killer Cells and Dendritic Cells: Rendezvous in Abused Tissues. Nat Rev Immunol (2002) 2:957–64. doi: 10.1038/nri956
143. Wilson EB, El-Jawhari JJ, Neilson AL, Hall GD, Melcher AA, Meade JL, et al. Human Tumour Immune Evasion Via TGF-β Blocks Nk Cell Activation But Not Survival Allowing Therapeutic Restoration of Anti-Tumour Activity. PLoS One (2011) 6:e22842. doi: 10.1371/journal.pone.0022842
144. Gillard-Bocquet M, Caer C, Cagnard N, Crozet L, Perez M, Fridman WH, et al. Lung Tumor Microenvironment Induces Specific Gene Expression Signature in Intratumoral NK Cells. Front Immunol (2013) 4:19. doi: 10.3389/fimmu.2013.00019
145. Coronella JA, Telleman P, Kingsbury GA, Truong TD, Hays S, Junghans RP. Evidence for an Antigen-Driven Humoral Immune Response in Medullary Ductal Breast Cancer. Cancer Res (2001) 61:7889–99.
146. Milne K, Köbel M, Kalloger SE, Barnes RO, Gao D, Gilks CB, et al. Systematic Analysis of Immune Infiltrates in High-Grade Serous Ovarian Cancer Reveals CD20, FoxP3 and TIA-1 as Positive Prognostic Factors. PLoS One (2009) 4:e6412. doi: 10.1371/journal.pone.0006412
147. Ran Z, Yue-Bei L, Qiu-Ming Z, Huan Y. Regulatory B Cells and Its Role in Central Nervous System Inflammatory Demyelinating Diseases. Front Immunol (2020) 11:1884. doi: 10.3389/fimmu.2020.01884
148. Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, et al. Tumor-Evoked Regulatory B Cells Promote Breast Cancer Metastasis by Converting Resting CD4+ T Cells to T Regulatory Cells. Cancer Res (2011) 71:3505–15. doi: 10.1158/0008-5472.CAN-10-4316
149. Schioppa T, Moore R, Thompson RG, Rosser EC, Kulbe H, Nedospasov S, et al. B Regulatory Cells and the Tumor-Promoting Actions of TNF-α During Squamous Carcinogenesis. Proc Natl Acad Sci USA (2011) 108:10662–7. doi: 10.1073/pnas.1100994108
150. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated Regulation of Myeloid Cells by Tumours. Nat Rev Immunol (2012) 12:253–68. doi: 10.1038/nri3175
151. Kusmartsev S, Nagaraj S, Gabrilovich DI. Tumor Associated CD8+ T-Cell Tolerance Induced by Bone Marrow Derived Immature Myeloid Cells. J Immunol Baltim Md 1950 (2005) 175:4583–92. doi: 10.4049/jimmunol.175.7.4583
152. Huang B, Pan P-Y, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res (2006) 66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299
153. Laoui D, Keirsse J, Morias Y, Van Overmeire E, Geeraerts X, Elkrim Y, et al. The Tumour Microenvironment Harbours Ontogenically Distinct Dendritic Cell Populations With Opposing Effects on Tumour Immunity. Nat Commun (2016) 7:13720. doi: 10.1038/ncomms13720
154. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell (2014) 26:638–52. doi: 10.1016/j.ccell.2014.09.007
155. Michea P, Noël F, Zakine E, Czerwinska U, Sirven P, Abouzid O, et al. Adjustment of Dendritic Cells to the Breast-Cancer Microenvironment Is Subset Specific. Nat Immunol (2018) 19:885–97. doi: 10.1038/s41590-018-0145-8
156. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A Natural Killer–Dendritic Cell Axis Defines Checkpoint Therapy–Responsive Tumor Microenvironments. Nat Med (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
157. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I Interferon Is Selectively Required by Dendritic Cells for Immune Rejection of Tumors. J Exp Med (2011) 208:1989–2003. doi: 10.1084/jem.20101158
158. Mercier IL, Poujol D, Sanlaville A, Sisirak V, Gobert M, Durand I, et al. Tumor Promotion by Intratumoral Plasmacytoid Dendritic Cells Is Reversed by TLR7 Ligand Treatment. Cancer Res (2013) 73:4629–40. doi: 10.1158/0008-5472.CAN-12-3058
159. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/Inos-Producing Dendritic Cells Mediate Innate Immune Defense Against Bacterial Infection. Immunity (2003) 19:59–70. doi: 10.1016/S1074-7613(03)00171-7
160. Fujimura T, Kambayashi Y, Aiba S. Crosstalk Between Regulatory T Cells (Tregs) and Myeloid Derived Suppressor Cells (MDSCS) During Melanoma Growth. Oncoimmunology (2012) 1:1433–4. doi: 10.4161/onci.21176
161. Wu Q, Li B, Sun S, Sun S. Unraveling Adipocytes and Cancer Links: Is There a Role for Senescence? Front Cell Dev Biol (2020) 8:282. doi: 10.3389/fcell.2020.00282
162. Cao Y. Adipocyte and Lipid Metabolism in Cancer Drug Resistance. J Clin Invest (2019) 129:3006–17. doi: 10.1172/JCI127201
163. Bochet L, Lehuédé C, Dauvillier S, Wang YY, Dirat B, Laurent V, et al. Adipocyte-Derived Fibroblasts Promote Tumor Progression and Contribute to the Desmoplastic Reaction in Breast Cancer. Cancer Res (2013) 73:5657–68. doi: 10.1158/0008-5472.CAN-13-0530
164. Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA. Obesity, Inflammation, and Cancer. Annu Rev Pathol (2016) 11:421–49. doi: 10.1146/annurev-pathol-012615-044359
165. Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, et al. Fat Tissue, Aging, and Cellular Senescence. Aging Cell (2010) 9:667–84. doi: 10.1111/j.1474-9726.2010.00608.x
166. Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
167. Sánchez-Jiménez F, Pérez-Pérez A, de la Cruz-Merino L, Sánchez-Margalet V. Obesity and Breast Cancer: Role of Leptin. Front Oncol (2019) 9:596. doi: 10.3389/fonc.2019.00596
168. Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J, et al. A Switch From White to Brown Fat Increases Energy Expenditure in Cancer-Associated Cachexia. Cell Metab (2014) 20:433–47. doi: 10.1016/j.cmet.2014.06.011
169. Wu Q, Li J, Li Z, Sun S, Zhu S, Wang L, et al. Exosomes From the Tumour-Adipocyte Interplay Stimulate Beige/Brown Differentiation and Reprogram Metabolism in Stromal Adipocytes to Promote Tumour Progression. J Exp Clin Cancer Res CR (2019) 38:223. doi: 10.1186/s13046-019-1210-3
170. Ingram JR, Dougan M, Rashidian M, Knoll M, Keliher EJ, Garrett S, et al. Pd-L1 is an Activation-Independent Marker of Brown Adipocytes. Nat Commun (2017) 8:647. doi: 10.1038/s41467-017-00799-8
171. Henke E, Nandigama R, Ergün S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front Mol Biosci (2020) 6:160. doi: 10.3389/fmolb.2019.00160
172. Tan T-T, Coussens LM. Humoral Immunity, Inflammation and Cancer. Curr Opin Immunol (2007) 19:209–16. doi: 10.1016/j.coi.2007.01.001
173. Wever OD, Demetter P, Mareel M, Bracke M. Stromal Myofibroblasts are Drivers of Invasive Cancer Growth. Int J Cancer (2008) 123:2229–38. doi: 10.1002/ijc.23925
174. Desmoulière A, Guyot C, Gabbiani G. The Stroma Reaction Myofibroblast: A Key Player in the Control of Tumor Cell Behavior. Int J Dev Biol (2004) 48:509–17. doi: 10.1387/ijdb.041802ad
175. Paszek MJ, Weaver VM. The Tension Mounts: Mechanics Meets Morphogenesis and Malignancy. J Mammary Gland Biol Neoplasia (2004) 9:325–42. doi: 10.1007/s10911-004-1404-x
176. Dituri F, Mazzocca A, Giannelli G, Antonaci S. Pi3k Functions in Cancer Progression, Anticancer Immunity and Immune Evasion by Tumors. Clin Dev Immunol (2011) 2011:e947858. doi: 10.1155/2011/947858
177. Lu H, Clauser KR, Tam WL, Fröse J, Ye X, Eaton EN, et al. A Breast Cancer Stem Cell Niche Supported by Juxtacrine Signalling From Monocytes and Macrophages. Nat Cell Biol (2014) 16:1105–17. doi: 10.1038/ncb3041
178. Stahl M, Schupp J, Jäger B, Schmid M, Zissel G, Müller-Quernheim J, et al. Lung Collagens Perpetuate Pulmonary Fibrosis Via CD204 and M2 Macrophage Activation. PLoS One (2013) 8:e81382. doi: 10.1371/journal.pone.0081382
179. Bollyky PL, Wu RP, Falk BA, Lord JD, Long SA, Preisinger A, et al. ECM Components Guide IL-10 Producing Regulatory T-Cell (TR1) Induction From Effector Memory T-Cell Precursors. Proc Natl Acad Sci (2011) 108:7938–43. doi: 10.1073/pnas.1017360108
180. Tomaso TD, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, et al. Immunobiological Characterization of Cancer Stem Cells Isolated From Glioblastoma Patients. Clin Cancer Res (2010) 16:800–13. doi: 10.1158/1078-0432.CCR-09-2730
181. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin Secreted by Glioblastoma Stem Cells Recruits M2 Tumour-Associated Macrophages and Promotes Malignant Growth. Nat Cell Biol (2015) 17:170–82. doi: 10.1038/ncb3090
182. Yakubenko VP, Cui K, Ardell CL, Brown KE, West XZ, Gao D, et al. Oxidative Modifications of Extracellular Matrix Promote the Second Wave of Inflammation Via β2 Integrins. Blood (2018) 132:78–88. doi: 10.1182/blood-2017-10-810176
183. Martinez FO, Gordon S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/P6-13
184. Wei J, Wu A, Kong L-Y, Wang Y, Fuller G, Fokt I, et al. Hypoxia Potentiates Glioma-Mediated Immunosuppression. PLoS One (2011) 6:e16195. doi: 10.1371/journal.pone.0016195
185. Xue X, Shah YM. Hypoxia-Inducible Factor-2α Is Essential in Activating the COX2/mPGES-1/PGE 2 Signaling Axis in Colon Cancer. Carcinogenesis (2013) 34:163–9. doi: 10.1093/carcin/bgs313
186. Schaaf MB, Garg AD, Agostinis P. Defining the Role of the Tumor Vasculature in Antitumor Immunity and Immunotherapy. Cell Death Dis (2018) 9:1–14. doi: 10.1038/s41419-017-0061-0
187. Ostroukhova M, Qi Z, Oriss TB, Dixon-McCarthy B, Rabir R, Ray A. Treg-Mediated Immunosuppression Involves Activation of the Notch-HES1 Axis by Membrane-Bound TGF-β. J Clin Invest (2006) 116:996–1004. doi: 10.1172/JCI26490
188. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. Tgf-β Induces M2-like Macrophage Polarization Via SNAIL-Mediated Suppression of a Pro-Inflammatory Phenotype. Oncotarget (2016) 7:52294–306. doi: 10.18632/oncotarget.10561
189. Ruella M, Klichinsky M, Kenderian SS, Shestova O, Ziober A, Kraft DO, et al. Overcoming the Immunosuppressive Tumor Microenvironment of Hodgkin Lymphoma Using Chimeric Antigen Receptor T Cells. Cancer Discov (2017) 7:1154–67. doi: 10.1158/2159-8290.CD-16-0850
190. Powell J, Mota F, Steadman D, Soudy C, Miyauchi JT, Crosby S, et al. Small Molecule Neuropilin-1 Antagonists Combine Antiangiogenic and Antitumor Activity With Immune Modulation Through Reduction of Transforming Growth Factor Beta (Tgfβ) Production in Regulatory T-Cells. J Med Chem (2018) 61:4135–54. doi: 10.1021/acs.jmedchem.8b00210
191. Gavalas NG, Tsiatas M, Tsitsilonis O, Politi E, Ioannou K, Ziogas AC, et al. VEGF Directly Suppresses Activation of T Cells From Ascites Secondary to Ovarian Cancer Via VEGF Receptor Type 2. Br J Cancer (2012) 107:1869–75. doi: 10.1038/bjc.2012.468
192. Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell (2009) 136:642–55. doi: 10.1016/j.cell.2009.01.035
193. Gangaraju VK, Lin H. MicroRNAs: Key Regulators of Stem Cells. Nat Rev Mol Cell Biol (2009) 10:116–25. doi: 10.1038/nrm2621
194. Kadera BE, Li L, Toste PA, Wu N, Adams C, Dawson DW, et al. MicroRNA-21 in Pancreatic Ductal Adenocarcinoma Tumor-Associated Fibroblasts Promotes Metastasis. PLoS One (2013) 8:e71978. doi: 10.1371/journal.pone.0071978
195. Shah SH, Miller P, Garcia-Contreras M, Ao Z, Machlin L, Issa E, et al. Hierarchical Paracrine Interaction of Breast Cancer Associated Fibroblasts With Cancer Cells Via hMAPK-microRNAs to Drive ER-negative Breast Cancer Phenotype. Cancer Biol Ther (2015) 16:1671–81. doi: 10.1080/15384047.2015.1071742
196. Pang W, Su J, Wang Y, Feng H, Dai X, Yuan Y, et al. Pancreatic Cancer-Secreted miR-155 Implicates in the Conversion From Normal Fibroblasts to Cancer-Associated Fibroblasts. Cancer Sci (2015) 106:1362–9. doi: 10.1111/cas.12747
197. Taddei ML, Cavallini L, Comito G, Giannoni E, Folini M, Marini A, et al. Senescent Stroma Promotes Prostate Cancer Progression: The Role of Mir-210. Mol Oncol (2014) 8:1729–46. doi: 10.1016/j.molonc.2014.07.009
198. Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as Oncogenes and Tumor Suppressors. Dev Biol (2007) 302:1–12. doi: 10.1016/j.ydbio.2006.08.028
199. Zhang Y, Yang P, Sun T, Li D, Xu X, Rui Y, et al. miR-126 and miR-126* Repress Recruitment of Mesenchymal Stem Cells and Inflammatory Monocytes to Inhibit Breast Cancer Metastasis. Nat Cell Biol (2013) 15:284–94. doi: 10.1038/ncb2690
200. Toh TB, Lim JJ, Chow EK-H. Epigenetics in Cancer Stem Cells. Mol Cancer (2017) 16:29. doi: 10.1186/s12943-017-0596-9
201. Suzuki H, Maruyama R, Yamamoto E, Kai M. Epigenetic Alteration and microRNA Dysregulation in Cancer. Front Genet (2013) 4:258. doi: 10.3389/fgene.2013.00258
202. Yi M, Xu L, Jiao Y, Luo S, Li A, Wu K. The Role of Cancer-Derived microRNAs in Cancer Immune Escape. J Hematol OncolJ Hematol Oncol (2020) 13:25. doi: 10.1186/s13045-020-00848-8
203. Gao F, Zhao Z-L, Zhao W-T, Fan Q-R, Wang S-C, Li J, et al. miR-9 Modulates the Expression of Interferon-Regulated Genes and MHC Class I Molecules in Human Nasopharyngeal Carcinoma Cells. Biochem Biophys Res Commun (2013) 431:610–6. doi: 10.1016/j.bbrc.2012.12.097
204. Mari L, Hoefnagel SJM, Zito D, van de Meent M, van Endert P, Calpe S, et al. microRNA 125a Regulates MHC-I Expression on Esophageal Adenocarcinoma Cells, Associated With Suppression of Antitumor Immune Response and Poor Outcomes of Patients. Gastroenterology (2018) 155:784–98. doi: 10.1053/j.gastro.2018.06.030
205. Colangelo T, Polcaro G, Ziccardi P, Pucci B, Muccillo L, Galgani M, et al. Proteomic Screening Identifies Calreticulin as a Mir-27a Direct Target Repressing MHC Class I Cell Surface Exposure In Colorectal Cancer. Cell Death Dis (2016) 7:e2120. doi: 10.1038/cddis.2016.28
206. Chen HL, Gabrilovich D, Tampé R, Girgis KR, Nadaf S, Carbone DP. A Functionally Defective Allele of TAP1 Results in Loss of MHC Class I Antigen Presentation in a Human Lung Cancer. Nat Genet (1996) 13:210–3. doi: 10.1038/ng0696-210
207. Ysla RM, Wilson GM, Brewer G. Assays of Adenylate Uridylate-Rich Element-Mediated Mrna Decay in Cells. Methods Enzymol (2008) 449:47–71. doi: 10.1016/S0076-6879(08)02403-8
208. Raghavan M, Wijeyesakere SJ, Peters LR, Del Cid N. Calreticulin in the Immune System: Ins and Outs. Trends Immunol (2013) 34:13–21. doi: 10.1016/j.it.2012.08.002
209. Carosella ED, Rouas-Freiss N, Tronik-Le Roux D, Moreau P, LeMaoult J. Hla-G: An Immune Checkpoint Molecule. Adv Immunol (2015) 127:33–144. doi: 10.1016/bs.ai.2015.04.001
210. Lin A, Yan W-H. Heterogeneity of HLA-G Expression in Cancers: Facing the Challenges. Front Immunol (2018) 9:2164. doi: 10.3389/fimmu.2018.02164
211. Yan W-H. Human Leukocyte Antigen-G in Cancer: Are They Clinically Relevant? Cancer Lett (2011) 311:123–30. doi: 10.1016/j.canlet.2011.07.019
212. Yang J, Li B, Lu H, Chen Y, Lu C, Zhu R, et al. Serum miR-152, miR-148a, miR-148b, and miR-21 as Novel Biomarkers in Non-Small Cell Lung Cancer Screening. Tumour Biol J Int Soc Oncodev Biol Med (2015) 36:3035–42. doi: 10.1007/s13277-014-2938-1
213. Zhu X, Han T, Wang X, Li Y, Yang H, Luo Y, et al. Overexpression of miR-152 Leads to Reduced Expression of Human Leukocyte Antigen-G and Increased Natural Killer Cell Mediated Cytolysis in JEG-3 Cells. Am J Obstet Gynecol (2010) 202:592.e1–7. doi: 10.1016/j.ajog.2010.03.002
214. Tao S, He H, Chen Q, Yue W. GPER Mediated Estradiol Reduces miR-148a to Promote HLA-G Expression in Breast Cancer. Biochem Biophys Res Commun (2014) 451:74–8. doi: 10.1016/j.bbrc.2014.07.073
215. Ashizawa M, Okayama H, Ishigame T, Thar Min AK, Saito K, Ujiie D, et al. Mirna-148a-3p Regulates Immunosuppression in DNA Mismatch Repair-Deficient Colorectal Cancer by Targeting Pd-L1. Mol Cancer Res MCR (2019) 17:1403–13. doi: 10.1158/1541-7786.MCR-18-0831
216. Gao L, Guo Q, Li X, Yang X, Ni H, Wang T, et al. MiR-873/PD-L1 Axis Regulates the Stemness of Breast Cancer Cells. EBioMedicine (2019) 41:395–407. doi: 10.1016/j.ebiom.2019.02.034
217. Qu F, Ye J, Pan X, Wang J, Gan S, Chu C, et al. MicroRNA-497-5p Down-Regulation Increases PD-L1 Expression in Clear Cell Renal Cell Carcinoma. J Drug Target (2019) 27:67–74. doi: 10.1080/1061186X.2018.1479755
218. Anastasiadou E, Stroopinsky D, Alimperti S, Jiao AL, Pyzer AR, Cippitelli C, et al. Epstein–Barr Virus-Encoded EBNA2 Alters Immune Checkpoint PD-L1 Expression by Downregulating miR-34a in B-Cell Lymphomas. Leukemia (2019) 33:132–47. doi: 10.1038/s41375-018-0178-x
219. Wang Y, Wang L. miR-34a Attenuates Glioma Cells Progression and Chemoresistance Via Targeting PD-L1. Biotechnol Lett (2017) 39:1485–92. doi: 10.1007/s10529-017-2397-z
220. Zhang X, Xu L, Wang F. Hsa_Circ_0020397 Regulates Colorectal Cancer Cell Viability, Apoptosis and Invasion by Promoting the Expression of the miR-138 Targets TERT and PD-L1. Cell Biol Int (2017) 41:1056–64. doi: 10.1002/cbin.10826
221. Zhang T, Chen H, Zhou Y, Dong W, Cai H, Tan W-S. Cooperation of FGF/MEK/ERK and Wnt/β-Catenin Pathway Regulators to Promote the Proliferation and Pluripotency of Mouse Embryonic Stem Cells in Serum- and Feeder-Free Conditions. Bioresour Bioprocess (2019) 6:12. doi: 10.1186/s40643-019-0249-5
222. Baranwal AK, Mehra NK. Major Histocompatibility Complex Class I Chain-Related A (Mica) Molecules: Relevance in Solid Organ Transplantation. Front Immunol (2017) 8:182. doi: 10.3389/fimmu.2017.00182
223. López-Cobo S, Romera-Cárdenas G, García-Cuesta EM, Reyburn HT, Valés-Gómez M. Transfer of the Human NKG2D Ligands UL16 Binding Proteins (ULBP) 1–3 Is Related to Lytic Granule Release and Leads to Ligand Retransfer and Killing of ULBP-recipient Natural Killer Cells. Immunology (2015) 146:70–80. doi: 10.1111/imm.12482
224. Hayakawa Y. Targeting NKG2D in Tumor Surveillance. Expert Opin Ther Targets (2012) 16:587–99. doi: 10.1517/14728222.2012.681378
225. Heinemann A, Zhao F, Pechlivanis S, Eberle J, Steinle A, Diederichs S, et al. Tumor Suppressive MicroRNAs Mir-34a/C Control Cancer Cell Expression of ULBP2, A Stress-Induced Ligand of the Natural Killer Cell Receptor Nkg2d. Cancer Res (2012) 72:460–71. doi: 10.1158/0008-5472.CAN-11-1977
226. Nguyen VHL, Yue C, Du KY, Salem M, O’Brien J, Peng C. The Role of microRNAs in Epithelial Ovarian Cancer Metastasis. Int J Mol Sci (2020) 21:7093. doi: 10.3390/ijms21197093
227. Jiang Y, Luan Y, He D, Chen G. miR-125b Expression Affects Tumor Growth of Multiple Myeloma Via Targeting MKK7. Int J Clin Exp Pathol (2017) 10:8487–94.
228. Shen J, Pan J, Du C, Si W, Yao M, Xu L, et al. Silencing NKG2D Ligand-Targeting miRNAs Enhances Natural Killer Cell-Mediated Cytotoxicity in Breast Cancer. Cell Death Dis (2017) 8:e2740–0. doi: 10.1038/cddis.2017.158
229. Min D, Lv X-b, Wang X, Zhang B, Meng W, Yu F, et al. Downregulation of miR-302c and miR-520c by 1,25(OH)2D3 Treatment Enhances the Susceptibility of Tumour Cells to Natural Killer Cell-Mediated Cytotoxicity. Br J Cancer (2013) 109:723–30. doi: 10.1038/bjc.2013.337
230. Cheong JE, Sun L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy - Challenges and Opportunities. Trends Pharmacol Sci (2018) 39:307–25. doi: 10.1016/j.tips.2017.11.007
231. Mbongue JC, Nicholas DA, Torrez TW, Kim N-S, Firek AF, Langridge WHR. The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity. Vaccines (2015) 3:703–29. doi: 10.3390/vaccines3030703
232. Mellor AL, Lemos H, Huang L. Indoleamine 2,3-Dioxygenase and Tolerance: Where Are We Now? Front Immunol (2017) 8:1360. doi: 10.3389/fimmu.2017.01360
233. Eichmüller SB, Osen W, Mandelboim O, Seliger B. Immune Modulatory Micrornas Involved in Tumor Attack and Tumor Immune Escape. JNCI J Natl Cancer Inst (2017) 109. doi: 10.1093/jnci/djx034
234. Wang Z, Tan Y, Yu W, Zheng S, Zhang S, Sun L, et al. Small Role With Big Impact: miRNAs as Communicators in the Cross-Talk Between Cancer-Associated Fibroblasts and Cancer Cells. Int J Biol Sci (2017) 13:339–48. doi: 10.7150/ijbs.17680
235. Yang F, Ning Z, Ma L, Liu W, Shao C, Shu Y, et al. Exosomal miRNAs and miRNA Dysregulation in Cancer-Associated Fibroblasts. Mol Cancer (2017) 16:148. doi: 10.1186/s12943-017-0718-4
236. Wang J, Guan X, Zhang Y, Ge S, Zhang L, Li H, et al. Exosomal Mir-27a Derived From Gastric Cancer Cells Regulates the Transformation of Fibroblasts Into Cancer-Associated Fibroblasts. Cell Physiol Biochem (2018) 49:869–83. doi: 10.1159/000493218
237. Fang T, Lv H, Lv G, Li T, Wang C, Han Q, et al. Tumor-Derived Exosomal miR-1247-3p Induces Cancer-Associated Fibroblast Activation to Foster Lung Metastasis of Liver Cancer. Nat Commun (2018) 9:191. doi: 10.1038/s41467-017-02583-0
238. Zhou Y, Ren H, Dai B, Li J, Shang L, Huang J, et al. Hepatocellular Carcinoma-Derived Exosomal miRNA-21 Contributes to Tumor Progression by Converting Hepatocyte Stellate Cells to Cancer-Associated Fibroblasts. J Exp Clin Cancer Res (2018) 37:324. doi: 10.1186/s13046-018-0965-2
239. Hossain A, Kuo MT, Saunders GF. Mir-17-5p Regulates Breast Cancer Cell Proliferation by Inhibiting Translation of AIB1 Mrna. Mol Cell Biol (2006) 26:8191–201. doi: 10.1128/MCB.00242-06
240. Wang T, Tao W, Zhang L, Li S. Oncogenic Role of microRNA-20a in Human Multiple Myeloma. OncoTargets Ther (2017) 10:4465–74. doi: 10.2147/OTT.S143612
241. Chen S, Zhang Y, Kuzel TM, Zhang B. Regulating Tumor Myeloid-Derived Suppressor Cells by Micrornas. Cancer Cell Microenviron (2015) 2:e637. doi: 10.1093/jnci/djx034
242. Chen C, Liu J, Luo Y. MicroRNAs in Tumor Immunity: Functional Regulation in Tumor-Associated Macrophages. J Zhejiang Univ Sci B (2020) 21:12–28. doi: 10.1631/jzus.B1900452
243. Mathsyaraja H, Thies K, Taffany DA, Deighan C, Liu T, Yu L, et al. Csf1-ETS2-induced microRNA in Myeloid Cells Promote Metastatic Tumor Growth. Oncogene (2015) 34:3651–61. doi: 10.1038/onc.2014.294
244. Xi J, Huang Q, Wang L, Ma X, Deng Q, Kumar M, et al. miR-21 Depletion in Macrophages Promotes Tumoricidal Polarization and Enhances PD-1 Immunotherapy. Oncogene (2018) 37:3151–65. doi: 10.1038/s41388-018-0178-3
245. Chen Y, Wang S-X, Mu R, Luo X, Liu Z-S, Liang B, et al. Dysregulation of the miR-324-5p-CUEDC2 Axis Leads to Macrophage Dysfunction and is Associated With Colon Cancer. Cell Rep (2014) 7:1982–93. doi: 10.1016/j.celrep.2014.05.007
246. Llorente A, Skotland T, Sylvänne T, Kauhanen D, Róg T, Orłowski A, et al. Molecular Lipidomics of Exosomes Released by PC-3 Prostate Cancer Cells. Biochim Biophys Acta (2013) 1831:1302–9. doi: 10.1016/j.bbalip.2013.04.011
247. Li I, Nabet BY. Exosomes in the Tumor Microenvironment as Mediators of Cancer Therapy Resistance. Mol Cancer (2019) 18:32. doi: 10.1186/s12943-019-0975-5
248. Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, et al. Intercellular Transfer of the Oncogenic Receptor EGFRvIII by Microvesicles Derived From Tumour Cells. Nat Cell Biol (2008) 10:619–24. doi: 10.1038/ncb1725
249. Melo SA, Sugimoto H, O’Connell JT, Kato N, Villanueva A, Vidal A, et al. Cancer Exosomes Perform Cell-Independent microRNA Biogenesis and Promote Tumorigenesis. Cancer Cell (2014) 26:707–21. doi: 10.1016/j.ccell.2014.09.005
250. Ristorcelli E, Beraud E, Mathieu S, Lombardo D, Verine A. Essential Role of Notch Signaling in Apoptosis of Human Pancreatic Tumoral Cells Mediated by Exosomal Nanoparticles. Int J Cancer (2009) 125:1016–26. doi: 10.1002/ijc.24375
251. Quail DF, Joyce JA. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat Med (2013) 19:1423–37. doi: 10.1038/nm.3394
252. Laplane L, Duluc D, Larmonier N, Pradeu T, Bikfalvi A. The Multiple Layers of the Tumor Environment. Trends Cancer (2018) 4:802–9. doi: 10.1016/j.trecan.2018.10.002
253. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and Genetic Properties of Tumors Associated With Local Immune Cytolytic Activity. Cell (2015) 160:48–61. doi: 10.1016/j.cell.2014.12.033
254. O’Donnell JS, Long GV, Scolyer RA, Teng MWL, Smyth MJ. Resistance to PD1/PDL1 Checkpoint Inhibition. Cancer Treat Rev (2017) 52:71–81. doi: 10.1016/j.ctrv.2016.11.007
255. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal Neoantigens Elicit T Cell Immunoreactivity and Sensitivity to Immune Checkpoint Blockade. Science (2016) 351:1463–9. doi: 10.1126/science.aaf1490
256. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of Neoantigen Landscape During Immune Checkpoint Blockade in Non–Small Cell Lung Cancer. Cancer Discov (2017) 7:264–76. doi: 10.1158/2159-8290.CD-16-0828
257. Akalay I, Janji B, Hasmim M, Noman MZ, André F, De Cremoux P, et al. Epithelial-to-Mesenchymal Transition and Autophagy Induction in Breast Carcinoma Promote Escape From T-Cell-Mediated Lysis. Cancer Res (2013) 73:2418–27. doi: 10.1158/0008-5472.CAN-12-2432
258. Ferrand N, Gnanapragasam A, Dorothee G, Redeuilh G, Larsen AK, Sabbah M. Loss of WISP2/CCN5 in Estrogen-Dependent MCF7 Human Breast Cancer Cells Promotes a Stem-Like Cell Phenotype. PLoS One (2014) 9:e87878. doi: 10.1371/journal.pone.0087878
259. Fritah A, Saucier C, De Wever O, Bracke M, Bièche I, Lidereau R, et al. Role of WISP-2/CCN5 in the Maintenance of a Differentiated and Noninvasive Phenotype in Human Breast Cancer Cells. Mol Cell Biol (2008) 28:1114–23. doi: 10.1128/MCB.01335-07
260. Tojo M, Hamashima Y, Hanyu A, Kajimoto T, Saitoh M, Miyazono K, et al. The ALK-5 Inhibitor A-83-01 Inhibits Smad Signaling and Epithelial-to-Mesenchymal Transition by Transforming Growth Factor-Beta. Cancer Sci (2005) 96:791–800. doi: 10.1111/j.1349-7006.2005.00103.x
261. Noman MZ, Janji B, Abdou A, Hasmim M, Terry S, Tan TZ, et al. The Immune Checkpoint Ligand PD-L1 Is Upregulated in EMT-Activated Human Breast Cancer Cells by a Mechanism Involving ZEB-1 and Mir-200. Oncoimmunology (2017) 6:e1263412. doi: 10.1080/2162402X.2016.1263412
262. Terry S, Buart S, Tan TZ, Gros G, Noman MZ, Lorens JB, et al. Acquisition of Tumor Cell Phenotypic Diversity Along the EMT Spectrum Under Hypoxic Pressure: Consequences on Susceptibility to Cell-Mediated Cytotoxicity. Oncoimmunology (2017) 6:e1271858. doi: 10.1080/2162402X.2016.1271858
263. Barry M, Bleackley RC. Cytotoxic T Lymphocytes: All Roads Lead to Death. Nat Rev Immunol (2002) 2:401–9. doi: 10.1038/nri819
264. Palena C, Polev DE, Tsang KY, Fernando RI, Litzinger M, Krukovskaya LL, et al. The Human T-Box Mesodermal Transcription Factor Brachyury Is a Candidate Target for T-Cell-Mediated Cancer Immunotherapy. Clin Cancer Res (2007) 13:2471–8. doi: 10.1158/1078-0432.CCR-06-2353
265. Hamilton DH, Huang B, Fernando RI, Tsang K-Y, Palena C. WEE1 Inhibition Alleviates Resistance to Immune Attack of Tumor Cells Undergoing Epithelial-Mesenchymal Transition. Cancer Res (2014) 74:2510–9. doi: 10.1158/0008-5472.CAN-13-1894
266. Tripathi SC, Peters HL, Taguchi A, Katayama H, Wang H, Momin A, et al. Immunoproteasome Deficiency is a Feature of Non-Small Cell Lung Cancer With a Mesenchymal Phenotype and Is Associated With a Poor Outcome. Proc Natl Acad Sci USA (2016) 113:E1555–64. doi: 10.1073/pnas.1521812113
267. Dustin ML, Long EO. Cytotoxic Immunological Synapses. Immunol Rev (2010) 235:24–34. doi: 10.1111/j.0105-2896.2010.00904.x
268. Dustin ML. What Counts in the Immunological Synapse? Mol Cell (2014) 54:255–62. doi: 10.1016/j.molcel.2014.04.001
269. Dustin ML, Chakraborty AK, Shaw AS. Understanding the Structure and Function of the Immunological Synapse. Cold Spring Harb Perspect Biol (2010) 2:a002311. doi: 10.1101/cshperspect.a002311
270. Comrie WA, Burkhardt JK. Action and Traction: Cytoskeletal Control of Receptor Triggering At the Immunological Synapse. Front Immunol (2016) 7:68. doi: 10.3389/fimmu.2016.00068
271. Yu Y, Smoligovets AA, Groves JT. Modulation of T Cell Signaling by the Actin Cytoskeleton. J Cell Sci (2013) 126:1049–58. doi: 10.1242/jcs.098210
272. Milner JD, Holland SM. The Cup Runneth Over: Lessons From the Ever-Expanding Pool of Primary Immunodeficiency Diseases. Nat Rev Immunol (2013) 13:635–48. doi: 10.1038/nri3493
273. Orange JS. Formation and Function of the Lytic NK-cell Immunological Synapse. Nat Rev Immunol (2008) 8:713–25. doi: 10.1038/nri2381
274. Hamaï A, Meslin F, Benlalam H, Jalil A, Mehrpour M, Faure F, et al. ICAM-1 has a Critical Role in the Regulation of Metastatic Melanoma Tumor Susceptibility to CTL Lysis by Interfering With PI3K/AKT Pathway. Cancer Res (2008) 68:9854–64. doi: 10.1158/0008-5472.CAN-08-0719
275. Anikeeva N, Somersalo K, Sims TN, Thomas VK, Dustin ML, Sykulev Y. Distinct Role of Lymphocyte Function-Associated Antigen-1 in Mediating Effective Cytolytic Activity by Cytotoxic T Lymphocytes. Proc Natl Acad Sci USA (2005) 102:6437. doi: 10.1073/pnas.0502467102
276. Abouzahr S, Bismuth G, Gaudin C, Caroll O, Van Endert P, Jalil A, et al. Identification of Target Actin Content and Polymerization Status as a Mechanism of Tumor Resistance After Cytolytic T Lymphocyte Pressure. Proc Natl Acad Sci USA (2006) 103:1428–33. doi: 10.1073/pnas.0510454103
277. Wainwright EN, Scaffidi P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer (2017) 3:372–86. doi: 10.1016/j.trecan.2017.04.004
278. Shang S, Hua F, Hu Z-W. The Regulation of β-Catenin Activity and Function in Cancer: Therapeutic Opportunities. Oncotarget (2017) 8:33972. doi: 10.18632/oncotarget.15687
279. MacDonald BT, Tamai K, He X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev Cell (2009) 17:9–26. doi: 10.1016/j.devcel.2009.06.016
280. Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, et al. Wnt/β-Catenin Signaling Regulates Telomerase in Stem Cells and Cancer Cells. Science (2012) 336:1549–54. doi: 10.1126/science.1218370
281. Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ, Cordero JB, et al. ROS Production and NF-κb Activation Triggered by RAC1 Facilitate WNT-Driven Intestinal Stem Cell Proliferation and Colorectal Cancer Initiation. Cell Stem Cell (2013) 12:761–73. doi: 10.1016/j.stem.2013.04.006
282. Barker N, Clevers H. Mining the Wnt Pathway for Cancer Therapeutics. Nat Rev Drug Discovery (2006) 5:997–1014. doi: 10.1038/nrd2154
284. Lindvall C, Bu W, Williams BO, Li Y. Wnt Signaling, Stem Cells, and the Cellular Origin of Breast Cancer. Stem Cell Rev (2007) 3:157–68. doi: 10.1007/s12015-007-0025-3
285. Klarmann GJ, Decker A, Farrar WL. Epigenetic Gene Silencing in the Wnt Pathway in Breast Cancer. Epigenetics (2008) 3:59–63. doi: 10.4161/epi.3.2.5899
286. Suzuki H, Watkins DN, Jair K-W, Schuebel KE, Markowitz SD, Chen WD, et al. Epigenetic Inactivation of SFRP Genes Allows Constitutive WNT Signaling in Colorectal Cancer. Nat Genet (2004) 36:417–22. doi: 10.1038/ng1330
287. Koinuma K, Yamashita Y, Liu W, Hatanaka H, Kurashina K, Wada T, et al. Epigenetic Silencing of AXIN2 in Colorectal Carcinoma With Microsatellite Instability. Oncogene (2006) 25:139–46. doi: 10.1038/sj.onc.1209009
288. Hussain M, Rao M, Humphries AE, Hong JA, Liu F, Yang M, et al. Tobacco Smoke Induces Polycomb-Mediated Repression of Dickkopf-1 in Lung Cancer Cells. Cancer Res (2009) 69:3570–8. doi: 10.1158/0008-5472.CAN-08-2807
289. Jiang X, Tan J, Li J, Kivimäe S, Yang X, Zhuang L, et al. DACT3 is an Epigenetic Regulator of Wnt/beta-Catenin Signaling in Colorectal Cancer and Is a Therapeutic Target of Histone Modifications. Cancer Cell (2008) 13:529–41. doi: 10.1016/j.ccr.2008.04.019
290. Beachy PA, Karhadkar SS, Berman DM. Tissue Repair and Stem Cell Renewal in Carcinogenesis. Nature (2004) 432:324–31. doi: 10.1038/nature03100
291. Ingham PW, McMahon AP. Hedgehog Signaling in Animal Development: Paradigms and Principles. Genes Dev (2001) 15:3059–87. doi: 10.1101/gad.938601
292. Sellheyer K. Basal Cell Carcinoma: Cell of Origin, Cancer Stem Cell Hypothesis and Stem Cell Markers. Br J Dermatol (2011) 164:696–711. doi: 10.1111/j.1365-2133.2010.10158.x
293. Youssef KK, Van Keymeulen A, Lapouge G, Beck B, Michaux C, Achouri Y, et al. Identification of the Cell Lineage At the Origin of Basal Cell Carcinoma. Nat Cell Biol (2010) 12:299–305. doi: 10.1038/ncb2031
294. Teglund S, Toftgård R. Hedgehog Beyond Medulloblastoma and Basal Cell Carcinoma. Biochim Biophys Acta (2010) 1805:181–208. doi: 10.1016/j.bbcan.2010.01.003
295. Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B. Germ-Line and Acquired Mutations of INI1 in Atypical Teratoid and Rhabdoid Tumors. Cancer Res (1999) 59:74–9.
296. Cui W, Wang L-H, Wen Y-Y, Song M, Li B-L, Chen X-L, et al. Expression and Regulation Mechanisms of Sonic Hedgehog in Breast Cancer. Cancer Sci (2010) 101:927–33. doi: 10.1111/j.1349-7006.2010.01495.x
297. Duan Z-H, Wang H-C, Zhao D-M, Ji X-X, Song M, Yang X-J, et al. Cooperatively Transcriptional and Epigenetic Regulation of Sonic Hedgehog Overexpression Drives Malignant Potential of Breast Cancer. Cancer Sci (2015) 106:1084–91. doi: 10.1111/cas.12697
298. Andersson ER, Lendahl U. Therapeutic Modulation of Notch Signalling–Are We There Yet? Nat Rev Drug Discovery (2014) 13:357–78. doi: 10.1038/nrd4252
299. Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, et al. Notch Signalling Regulates Stem Cell Numbers In Vitro and In Vivo. Nature (2006) 442:823–6. doi: 10.1038/nature04940
300. Ghoshal P, Nganga AJ, Moran-Giuati J, Szafranek A, Johnson TR, Bigelow AJ, et al. Loss of the SMRT/NCoR2 Corepressor Correlates With JAG2 Overexpression in Multiple Myeloma. Cancer Res (2009) 69:4380–7. doi: 10.1158/0008-5472.CAN-08-3467
301. Jin L, Vu TT, Datta PK. Abstract 1709: STRAP Mediates the Stemness of Human Colorectal Cancer Cells by Epigenetic Regulation of Notch Pathway. Cancer Res (2016) 76:1709–9. doi: 10.1158/1538-7445.AM2016-1709
302. Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE. Development and Characterization of Gemcitabine-Resistant Pancreatic Tumor Cells. Ann Surg Oncol (2007) 14:3629–37. doi: 10.1245/s10434-007-9583-5
303. Shukla S, Meeran SM. Epigenetics of Cancer Stem Cells: Pathways and Therapeutics. Biochim Biophys Acta BBA - Gen Subj (2014) 1840:3494–502. doi: 10.1016/j.bbagen.2014.09.017
304. Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, et al. Epithelial to Mesenchymal Transition Contributes to Drug Resistance in Pancreatic Cancer. Cancer Res (2009) 69:5820–8. doi: 10.1158/0008-5472.CAN-08-2819
305. Chow EK-H, Fan L, Chen X, Bishop JM. Oncogene-Specific Formation of Chemoresistant Murine Hepatic Cancer Stem Cells. Hepatology (2012) 56:1331–41. doi: 10.1002/hep.25776
306. Chung-Pu W, Anna MC, Suresh VA. Reversal of ABC Drug Transporter-Mediated Multidrug Resistance in Cancer Cells: Evaluation of Current Strategies. Curr Mol Pharmacol (2008) 1:93–105.
307. Wang X, Low XC, Hou W, Abdullah LN, Toh TB, Mohd Abdul Rashid M, et al. Epirubicin-Adsorbed Nanodiamonds Kill Chemoresistant Hepatic Cancer Stem Cells. ACS Nano (2014) 8:12151–66. doi: 10.1021/nn503491e
308. Cho S, Lu M, He X, Ee P-LR, Bhat U, Schneider E, et al. Notch1 Regulates the Expression of the Multidrug Resistance Gene ABCC1/MRP1 in Cultured Cancer Cells. Proc Natl Acad Sci (2011) 108:20778–83. doi: 10.1073/pnas.1019452108
309. Kim B, Stephen SL, Hanby AM, Horgan K, Perry SL, Richardson J, et al. Chemotherapy Induces Notch1-Dependent MRP1 Up-Regulation, Inhibition of Which Sensitizes Breast Cancer Cells to Chemotherapy. BMC Cancer (2015) 15:634. doi: 10.1186/s12885-015-1625-y
310. To KKW, Polgar O, Huff LM, Morisaki K, Bates SE. Histone Modifications at the ABCG2 Promoter Following Treatment With Histone Deacetylase Inhibitor Mirror Those in Multidrug-Resistant Cells. Mol Cancer Res (2008) 6:151–64. doi: 10.1158/1541-7786.MCR-07-0175
311. Tang DG. Understanding Cancer Stem Cell Heterogeneity and Plasticity. Cell Res (2012) 22:457–72. doi: 10.1038/cr.2012.13
312. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and Neoplastic Nonstem Cells can Spontaneously Convert to a Stem-Like State. Proc Natl Acad Sci (2011) 108:7950–5. doi: 10.1073/pnas.1102454108
313. Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, et al. Poised Chromatin At the ZEB1 Promoter Enables Breast Cancer Cell Plasticity and Enhances Tumorigenicity. Cell (2013) 154:61–74. doi: 10.1016/j.cell.2013.06.005
314. Chen W, Dong J, Haiech J, Kilhoffer M-C, Zeniou M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int (2016) 2016:e1740936. doi: 10.1155/2016/1740936
315. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible Formation of Breast Cancer Stem Cells and Their Dynamic Equilibrium With Non-Stem Cancer Cells Via IL6 Secretion. Proc Natl Acad Sci (2011) 108:1397–402. doi: 10.1073/pnas.1018898108
316. Jolly MK, Kulkarni P, Weninger K, Orban J, Levine H. Phenotypic Plasticity, Bet-Hedging, and Androgen Independence in Prostate Cancer: Role of Non-Genetic Heterogeneity. Front Oncol (2018) 8:50. doi: 10.3389/fonc.2018.00050
317. Morgan RG, Mortensson E, Williams AC. Targeting LGR5 in Colorectal Cancer: Therapeutic Gold or Too Plastic? Br J Cancer (2018) 118:1410–8. doi: 10.1038/s41416-018-0118-6
318. Tsai L-L, Yu C-C, Lo J-F, Sung W-W, Lee H, Chen S-L, et al. Enhanced Cisplatin Resistance in Oral-Cancer Stem-Like Cells Is Correlated With Upregulation of Excision-Repair Cross-Complementation Group 1. J Dent Sci (2012) 7:111–7. doi: 10.1016/j.jds.2012.03.006
319. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature (2006) 444:756–60. doi: 10.1038/nature05236
320. Kim NH, Kim HS, Li X-Y, Lee I, Choi H-S, Kang SE, et al. A P53/miRNA-34 Axis Regulates Snail1-dependent Cancer Cell Epithelial-Mesenchymal Transition. J Cell Biol (2011) 195:417–33. doi: 10.1083/jcb.201103097
321. Apel A, Zentgraf H, Büchler MW, Herr I. Autophagy-a Double-Edged Sword in Oncology. Int J Cancer (2009) 125:991–5. doi: 10.1002/ijc.24500
322. Smith AG, Macleod KF. Autophagy, Cancer Stem Cells and Drug Resistance. J Pathol (2019) 247:708–18. doi: 10.1002/path.5222
323. Schito L, Semenza GL. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer (2016) 2:758–70. doi: 10.1016/j.trecan.2016.10.016
324. Semenza GL. Defining the Role of Hypoxia-Inducible Factor 1 in Cancer Biology and Therapeutics. Oncogene (2010) 29:625–34. doi: 10.1038/onc.2009.441
325. Wilson WR, Hay MP. Targeting Hypoxia in Cancer Therapy. Nat Rev Cancer (2011) 11:393–410. doi: 10.1038/nrc3064
326. Minchinton AI, Tannock IF. Drug Penetration in Solid Tumours. Nat Rev Cancer (2006) 6:583–92. doi: 10.1038/nrc1893
327. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of Hypoxia in Cancer Therapy by Regulating the Tumor Microenvironment. Mol Cancer (2019) 18:157. doi: 10.1186/s12943-019-1089-9
328. Mitra SK, Schlaepfer DD. Integrin-Regulated FAK–Src Signaling in Normal and Cancer Cells. Curr Opin Cell Biol (2006) 18:516–23. doi: 10.1016/j.ceb.2006.08.011
329. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, et al. Epithelial-to-Mesenchymal Transition Is Not Required for Lung Metastasis But Contributes to Chemoresistance. Nature (2015) 527:472–6. doi: 10.1038/nature15748
330. Qiu G-Z, Jin M-Z, Dai J-X, Sun W, Feng J-H, Jin W-L. Reprogramming of the Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies. Trends Pharmacol Sci (2017) 38:669–86. doi: 10.1016/j.tips.2017.05.002
331. Bhandari V, Li CH, Bristow RG, Boutros PC. Divergent Mutational Processes Distinguish Hypoxic and Normoxic Tumours. Nat Commun (2020) 11:737. doi: 10.1038/s41467-019-14052-x
332. Denko NC. Hypoxia, HIF1 and Glucose Metabolism in the Solid Tumour. Nat Rev Cancer (2008) 8:705–13. doi: 10.1038/nrc2468
333. Semenza GL. Hif-1: Upstream and Downstream of Cancer Metabolism. Curr Opin Genet Dev (2010) 20:51. doi: 10.1016/j.gde.2009.10.009
334. Reina-Campos M, Moscat J, Diaz-Meco M. Metabolism Shapes the Tumor Microenvironment. Curr Opin Cell Biol (2017) 48:47–53. doi: 10.1016/j.ceb.2017.05.006
335. Goto M, Miwa H, Shikami M, Tsunekawa-Imai N, Suganuma K, Mizuno S, et al. Importance of Glutamine Metabolism in Leukemia Cells by Energy Production Through Tca Cycle and by Redox Homeostasis. Cancer Invest (2014) 32:241–7. doi: 10.3109/07357907.2014.907419
336. Warburg O, Wind F, Negelein E. The Metabolism of Tumors in The Body. J Gen Physiol (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
337. Hou X-M, Yuan S-Q, Zhao D, Liu X-J, Wu X-A. LDH-a Promotes Malignant Behavior Via Activation of Epithelial-to-Mesenchymal Transition in Lung Adenocarcinoma. Biosci Rep (2019) 39:BSR20181476. doi: 10.1042/BSR20181476
338. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab (2017) 25:1282–93.e7. doi: 10.1016/j.cmet.2016.12.018
339. Hunt TK, Aslam RS, Beckert S, Wagner S, Ghani QP, Hussain MZ, et al. Aerobically-Derived Lactate Stimulates Revascularization and Tissue Repair Via Redox Mechanisms. Antioxid Redox Signal (2007) 9:1115–24. doi: 10.1089/ars.2007.1674
340. Altman BJ, Stine ZE, Dang CV. From Krebs to Clinic: Glutamine Metabolism to Cancer Therapy. Nat Rev Cancer (2016) 16:619–34. doi: 10.1038/nrc.2016.71
341. Elia I, Rossi M, Stegen S, Broekaert D, Doglioni G, van Gorsel M, et al. Breast Cancer Cells Rely on Environmental Pyruvate to Shape the Metastatic Niche. Nature (2019) 568:117–21. doi: 10.1038/s41586-019-0977-x
342. Nallanthighal S, Heiserman JP, Cheon D-J. The Role of the Extracellular Matrix in Cancer Stemness. Front Cell Dev Biol (2019) 7:86. doi: 10.3389/fcell.2019.00086
343. Quennet V, Yaromina A, Zips D, Rosner A, Walenta S, Baumann M, et al. Tumor Lactate Content Predicts for Response to Fractionated Irradiation of Human Squamous Cell Carcinomas in Nude Mice. Radiother Oncol J Eur Soc Ther Radiol Oncol (2006) 81:130–5. doi: 10.1016/j.radonc.2006.08.012
344. Apicella M, Giannoni E, Fiore S, Ferrari KJ, Fernández-Pérez D, Isella C, et al. Increased Lactate Secretion by Cancer Cells Sustains non-Cell-Autonomous Adaptive Resistance to MET and EGFR Targeted Therapies. Cell Metab (2018) 28:848–65. doi: 10.1016/j.cmet.2018.08.006
345. Wagner W, Ciszewski WM, Kania KD. L- and D-lactate Enhance DNA Repair and Modulate the Resistance of Cervical Carcinoma Cells to Anticancer Drugs Via Histone Deacetylase Inhibition and Hydroxycarboxylic Acid Receptor 1 Activation. Cell Commun Signal CCS (2015) 13:36. doi: 10.1186/s12964-015-0114-x
346. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid Metabolic Reprogramming in Cancer Cells. Oncogenesis (2016) 5:e189–9. doi: 10.1038/oncsis.2015.49
347. Menendez JA, Lupu R. Fatty Acid Synthase and the Lipogenic Phenotype in Cancer Pathogenesis. Nat Rev Cancer (2007) 7:763–77. doi: 10.1038/nrc2222
348. Kuhajda FP. Fatty Acid Synthase and Cancer: New Application of an Old Pathway. Cancer Res (2006) 66:5977–80. doi: 10.1158/0008-5472.CAN-05-4673
349. Mashima T, Seimiya H, Tsuruo T. De Novo Fatty-Acid Synthesis and Related Pathways as Molecular Targets for Cancer Therapy. Br J Cancer (2009) 100:1369–72. doi: 10.1038/sj.bjc.6605007
350. Qin X-Y, Kojima S. Inhibition of Stearoyl-CoA Desaturase-1 Activity Suppressed SREBP Signaling in Colon Cancer Cells and Their Spheroid Growth. Gastrointest Disord (2019) 1:191–200. doi: 10.3390/gidisord1010014
351. Tracz-Gaszewska Z, Dobrzyn P. Stearoyl-Coa Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers (2019) 11:948. doi: 10.3390/cancers11070948
352. Tadros S, Shukla SK, King RJ, Gunda V, Vernucci E, Abrego J, et al. De Novo Lipid Synthesis Facilitates Gemcitabine Resistance Through Endoplasmic Reticulum Stress in Pancreatic Cancer. Cancer Res (2017) 77:5503–17. doi: 10.1158/0008-5472.CAN-16-3062
353. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes Promote Ovarian Cancer Metastasis and Provide Energy for Rapid Tumor Growth. Nat Med (2011) 17:1498–503. doi: 10.1038/nm.2492
354. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS-O, et al. Targeting Metastasis-Initiating Cells Through the Fatty Acid Receptor CD36. Nature (2017) 541:41–5. doi: 10.1038/nature20791
355. Han S, Wei R, Zhang X, Jiang N, Fan M, Huang JH, et al. Cpt1a/2-Mediated FAO Enhancement-a Metabolic Target in Radioresistant Breast Cancer. Front Oncol (2019) 9:1201. doi: 10.3389/fonc.2019.01201
356. Peck B, Schulze A. Lipid Metabolism At the Nexus of Diet and Tumor Microenvironment. Trends Cancer (2019) 5:693–703. doi: 10.1016/j.trecan.2019.09.007
357. Long J, Zhang C-J, Zhu N, Du K, Yin Y-F, Tan X, et al. Lipid Metabolism and Carcinogenesis, Cancer Development. Am J Cancer Res (2018) 8:778–91.
358. Ma L, Hernandez MO, Zhao Y, Mehta M, Tran B, Kelly M, et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell (2019) 36:418–30. doi: 10.1016/j.ccell.2019.08.007
359. Li H, Feng Z, He M-L. Lipid Metabolism Alteration Contributes to and Maintains the Properties of Cancer Stem Cells. Theranostics (2020) 10:7053–69. doi: 10.7150/thno.41388
360. Incio J, Ligibel JA, McManus DT, Suboj P, Jung K, Kawaguchi K, et al. Obesity Promotes Resistance to Anti-VEGF Therapy in Breast Cancer by Up-Regulating IL-6 and Potentially FGF-2. Sci Transl Med (2018) 10:eaag0945. doi: 10.1126/scitranslmed.aag0945
361. Hultsch S, Kankainen M, Paavolainen L, Kovanen R-M, Ikonen E, Kangaspeska S, et al. Association of Tamoxifen Resistance and Lipid Reprogramming in Breast Cancer. BMC Cancer (2018) 18:850. doi: 10.1186/s12885-018-4757-z
362. She K, Fang S, Du W, Fan X, He J, Pan H, et al. SCD1 Is Required for EGFR-Targeting Cancer Therapy Of Lung Cancer Via Re-Activation of EGFR/PI3K/AKT Signals. Cancer Cell Int (2019) 19:103. doi: 10.1186/s12935-019-0809-y
363. Salunkhe S, Mishra SV, Ghorai A, Hole A, Chandrani P, Dutt A, et al. Metabolic Rewiring In Drug Resistant Cells Exhibit Higher OXPHOS and Fatty Acids as Preferred Major Source to Cellular Energetics. Biochim Biophys Acta Bioenerg (2020) 1861:148300. doi: 10.1016/j.bbabio.2020.148300
364. Mims J, Bansal N, Bharadwaj MS, Chen X, Molina AJ, Tsang AW, et al. Energy Metabolism in a Matched Model of Radiation Resistance for Head and Neck Squamous Cell Cancer. Radiat Res (2015) 183:291–304. doi: 10.1667/RR13828.1
365. Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov (2017) 7:716–35. doi: 10.1158/2159-8290.CD-16-0441
366. Iwamoto H, Abe M, Yang Y, Cui D, Seki T, Nakamura M, et al. Cancer Lipid Metabolism Confers Antiangiogenic Drug Resistance. Cell Metab (2018) 28:104–17.e5. doi: 10.1016/j.cmet.2018.05.005
367. Wang T, Fahrmann JF, Lee H, Li Y-J, Tripathi SC, Yue C, et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab (2018) 27:136–50.e5. doi: 10.1016/j.cmet.2017.11.001
368. Shen S, Faouzi S, Souquere S, Roy S, Routier E, Libenciuc C, André F, Pierron G, Scoazec J-Y, Robert C. Melanoma Persister Cells Are Tolerant to BRAF/MEK Inhibitors Via ACOX1-Mediated Fatty Acid Oxidation. Cell Rep (2020) 33:108421. doi: 10.1016/j.celrep.2020.108421
369. Weinberg F, Ramnath N, Nagrath D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers (2019) 11:1191. doi: 10.3390/cancers11081191
370. Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial Reactive Oxygen Species Trigger Hypoxia-Induced Transcription. Proc Natl Acad Sci (1998) 95:11715–20. doi: 10.1073/pnas.95.20.11715
371. Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, Liu Z-G. ROS Play a Critical Role in the Differentiation of Alternatively Activated Macrophages and the Occurrence of Tumor-Associated Macrophages. Cell Res (2013) 23:898–914. doi: 10.1038/cr.2013.75
372. Nakashima RA, Paggi MG, Pedersen PL. Contributions of Glycolysis and Oxidative Phosphorylation to Adenosine 5’-Triphosphate Production in AS-30D Hepatoma Cells. Cancer Res (1984) 44:5702–6.
373. Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth Factor Regulation of Autophagy and Cell Survival in the Absence of Apoptosis. Cell (2005) 120:237–48. doi: 10.1016/j.cell.2004.11.046
374. Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell (2017) 32:71–87.e7. doi: 10.1016/j.ccell.2017.06.004
375. D’Autréaux B, Toledano MB. ROS as Signalling Molecules: Mechanisms That Generate Specificity in ROS Homeostasis. Nat Rev Mol Cell Biol (2007) 8:813–24. doi: 10.1038/nrm2256
376. Panieri E, Santoro MM. ROS Homeostasis and Metabolism: A Dangerous Liason in Cancer Cells. Cell Death Dis (2016) 7:e2253–3. doi: 10.1038/cddis.2016.105
377. Lu W, Hu Y, Chen G, Chen Z, Zhang H, Wang F, et al. Correction: Novel Role of NOX in Supporting Aerobic Glycolysis in Cancer Cells With Mitochondrial Dysfunction and as a Potential Target for Cancer Therapy. PLoS Biol (2017) 15:e1002616. doi: 10.1371/journal.pbio.1002616
378. Bedard K, Krause K-H. The NOX Family of ROS-generating NADPH Oxidases: Physiology and Pathophysiology. Physiol Rev (2007) 87:245–313. doi: 10.1152/physrev.00044.2005
379. Anastasiou D, Yu Y, Israelsen WJ, Jiang J-K, Boxer MB, Hong BS, et al. Pyruvate Kinase M2 Activators Promote Tetramer Formation and Suppress Tumorigenesis. Nat Chem Biol (2012) 8:839–47. doi: 10.1038/nchembio.1060
380. Bluemlein K, Grüning N-M, Feichtinger RG, Lehrach H, Kofler B, Ralser M. No Evidence for a Shift in Pyruvate Kinase PKM1 to PKM2 Expression During Tumorigenesis. Oncotarget (2011) 2:393–400. doi: 10.18632/oncotarget.278
381. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J, Shen M, et al. Inhibition of Pyruvate Kinase M2 by Reactive Oxygen Species Contributes to Cellular Antioxidant Responses. Science (2011) 334:1278–83. doi: 10.1126/science.1211485
382. Rubie C, Kempf K, Hans J, Su T, Tilton B, Georg T, et al. Housekeeping Gene Variability in Normal and Cancerous Colorectal, Pancreatic, Esophageal, Gastric and Hepatic Tissues. Mol Cell Probes (2005) 19:101–9. doi: 10.1016/j.mcp.2004.10.001
383. Seebacher NA, Richardson DR, Jansson PJ. Glucose Modulation Induces Reactive Oxygen Species and Increases P-Glycoprotein-Mediated Multidrug Resistance to Chemotherapeutics. Br J Pharmacol (2015) 172:2557–72. doi: 10.1111/bph.13079
384. Bhatt AN, Chauhan A, Khanna S, Rai Y, Singh S, Soni R, et al. Transient Elevation of Glycolysis Confers Radio-Resistance by Facilitating DNA Repair in Cells. BMC Cancer (2015) 15:335. doi: 10.1186/s12885-015-1368-9
385. Zhao J-G, Ren K-M, Tang J. Overcoming 5-Fu Resistance in Human non-Small Cell Lung Cancer Cells by the Combination of 5-Fu and Cisplatin Through the Inhibition of Glucose Metabolism. Tumour Biol J Int Soc Oncodev Biol Med (2014) 35:12305–15. doi: 10.1007/s13277-014-2543-3
386. Ma S, Jia R, Li D, Shen B. Targeting Cellular Metabolism Chemosensitizes the Doxorubicin-Resistant Human Breast Adenocarcinoma Cells. BioMed Res Int (2015) 2015:453986. doi: 10.1155/2015/453986
387. Vara-Ciruelos D, Russell FM, Hardie DG. The Strange Case of AMPK and Cancer: Dr Jekyll or Mr Hyde?†. Open Biol (2019) 9:190099. doi: 10.1098/rsob.190099
388. Ghanbari Movahed Z, Rastegari-Pouyani M, Mohammadi MH, Mansouri K. Cancer Cells Change Their Glucose Metabolism to Overcome Increased ROS: One Step From Cancer Cell to Cancer Stem Cell? BioMed Pharmacother (2019) 112:108690. doi: 10.1016/j.biopha.2019.108690
389. Maddocks ODK, Vousden KH. Metabolic Regulation by P53. J Mol Med Berl Ger (2011) 89:237–45. doi: 10.1007/s00109-011-0735-5
390. Wu GS. The Functional Interactions Between the MAPK and P53 Signaling Pathways. Cancer Biol Ther (2004) 3:156–61. doi: 10.4161/cbt.3.2.614
391. Bhardwaj V, He J. Reactive Oxygen Species, Metabolic Plasticity, and Drug Resistance in Cancer. Int J Mol Sci (2020) 21:3412. doi: 10.3390/ijms21103412
392. suruo T, Naito M, Tomida A, Fujita N, Mashima T, Sakamoto H, et al. Molecular Targeting Therapy of Cancer: Drug Resistance, Apoptosis and Survival Signal. Cancer Sci (2003) 94:15–21. doi: 10.1111/j.1349-7006.2003.tb01345.x
393. Kuo MT. Redox Regulation of Multidrug Resistance in Cancer Chemotherapy: Molecular Mechanisms and Therapeutic Opportunities. Antioxid Redox Signal (2009) 11:99–133. doi: 10.1089/ars.2008.2095
394. Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the Role of ABC Transporters in Multidrug-Resistant Cancer. Nat Rev Cancer (2018) 18:452–64. doi: 10.1038/s41568-018-0005-8
395. Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, et al. ROS in Cancer Therapy: The Bright Side of the Moon. Exp Mol Med (2020) 52:192–203. doi: 10.1038/s12276-020-0384-2
396. Cort A, Ozben T, Saso L, De Luca C, Korkina L. Redox Control of Multidrug Resistance and Its Possible Modulation by Antioxidants. Oxid Med Cell Longev (2016) 2016:e4251912. doi: 10.1155/2016/4251912
397. Chiche J, Brahimi-Horn MC, Pouysségur J. Tumour Hypoxia Induces a Metabolic Shift Causing Acidosis: A Common Feature in Cancer. J Cell Mol Med (2010) 14:771–94. doi: 10.1111/j.1582-4934.2009.00994.x
398. Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: A Perfect Storm for Cancer Progression. Nat Rev Cancer (2011) 11:671–7. doi: 10.1038/nrc3110
399. Hashim AI, Zhang X, Wojtkowiak JW, Martinez GV, Gillies RJ. Imaging Ph and Metastasis. NMR BioMed (2011) 24:582–91. doi: 10.1002/nbm.1644
400. Gillies RJ, Pilot C, Marunaka Y, Fais S. Targeting Acidity in Cancer and Diabetes. Biochim Biophys Acta Rev Cancer (2019) 1871:273–80. doi: 10.1016/j.bbcan.2019.01.003
401. Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting Lactate-Fueled Respiration Selectively Kills Hypoxic Tumor Cells in Mice. J Clin Invest (2008) 118:3930–42. doi: 10.1172/JCI36843
402. Peppicelli S, Toti A, Giannoni E, Bianchini F, Margheri F, Del Rosso M, et al. Metformin is Also Effective on Lactic Acidosis-Exposed Melanoma Cells Switched to Oxidative Phosphorylation. Cell Cycle Georget Tex (2016) 15:1908–18. doi: 10.1080/15384101.2016.1191706
403. Mazzio EA, Boukli N, Rivera N, Soliman KFA. Pericellular Ph Homeostasis is a Primary Function of the Warburg Effect: Inversion of Metabolic Systems to Control Lactate Steady State in Tumor Cells. Cancer Sci (2012) 103:422–32. doi: 10.1111/j.1349-7006.2012.02206.x
404. Lamonte G, Tang X, Chen JL-Y, Wu J, Ding C-KC, Keenan MM, et al. Acidosis Induces Reprogramming of Cellular Metabolism to Mitigate Oxidative Stress. Cancer Metab (2013) 1:23. doi: 10.1186/2049-3002-1-23
405. Vukovic V, Tannock IF. Influence of Low Ph on Cytotoxicity of Paclitaxel, Mitoxantrone and Topotecan. Br J Cancer (1997) 75:1167–72. doi: 10.1038/bjc.1997.201
406. Gerweck LE, Vijayappa S, Kozin S. Tumor Ph Controls the In Vivo Efficacy of Weak Acid and Base Chemotherapeutics. Mol Cancer Ther (2006) 5:1275–9. doi: 10.1158/1535-7163.MCT-06-0024
407. Saunders DE, Lawrence WD, Christensen C, Wappler NL, Ruan H, Deppe G. Paclitaxel-Induced Apoptosis in MCF-7 Breast-Cancer Cells. Int J Cancer (1997) 70:214–20. doi: 10.1002/(sici)1097-0215(19970117)70:2<214::aid-ijc13>3.0.co;2-i
408. Williams AC, Collard TJ, Paraskeva C. An Acidic Environment Leads to p53 Dependent Induction of Apoptosis in Human Adenoma and Carcinoma Cell Lines: Implications for Clonal Selection During Colorectal Carcinogenesis. Oncogene (1999) 18:3199–204. doi: 10.1038/sj.onc.1202660
409. Lotz C, Kelleher DK, Gassner B, Gekle M, Vaupel P, Thews O. Role of the Tumor Microenvironment in the Activity and Expression of the P-Glycoprotein in Human Colon Carcinoma Cells. Oncol Rep (2007) 17:239–44. doi: 10.3892/or.17.1.239
410. Ohtsubo T, Igawa H, Saito T, Matsumoto H, Park HJ, Song CW, et al. Acidic Environment Modifies Heat- or Radiation-Induced Apoptosis in Human Maxillary Cancer Cells. Int J Radiat Oncol Biol Phys (2001) 49:1391–8. doi: 10.1016/s0360-3016(00)01590-x
411. Ingangi V, Minopoli M, Ragone C, Motti ML, Carriero MV. Role of Microenvironment on the Fate of Disseminating Cancer Stem Cells. Front Oncol (2019) 9:82. doi: 10.3389/fonc.2019.00082
412. Vander Linden C, Corbet C. Therapeutic Targeting of Cancer Stem Cells: Integrating and Exploiting the Acidic Niche. Front Oncol (2019) 9:159. doi: 10.3389/fonc.2019.00159
413. Gupta GP, Massagué J. Cancer Metastasis: Building a Framework. Cell (2006) 127:679–95. doi: 10.1016/j.cell.2006.11.001
414. Pan C, Schoppe O, Parra-Damas A, Cai R, Todorov MI, Gondi G, et al. Deep Learning Reveals Cancer Metastasis and Therapeutic Antibody Targeting in the Entire Body. Cell (2019) 179:1661–76.e19. doi: 10.1016/j.cell.2019.11.013
415. Friebel E, Kapolou K, Unger S, Núñez NG, Utz S, Rushing EJ, et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell (2020) 181:1626–42.e20. doi: 10.1016/j.cell.2020.04.055
416. Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, et al. Macrophages and Cathepsin Proteases Blunt Chemotherapeutic Response in Breast Cancer. Genes Dev (2011) 25:2465–79. doi: 10.1101/gad.180331.111
417. Santoni M, Massari F, Amantini C, Nabissi M, Maines F, Burattini L, et al. Emerging Role of Tumor-Associated Macrophages as Therapeutic Targets in Patients With Metastatic Renal Cell Carcinoma. Cancer Immunol Immunother CII (2013) 62:1757–68. doi: 10.1007/s00262-013-1487-6
418. Jinushi M, Komohara Y. Tumor-Associated Macrophages as an Emerging Target Against Tumors: Creating a New Path From Bench to Bedside. Biochim Biophys Acta (2015) 1855:123–30. doi: 10.1016/j.bbcan.2015.01.002
419. Pepper MS. Role of the Matrix Metalloproteinase and Plasminogen Activator-Plasmin Systems in Angiogenesis. Arterioscler Thromb Vasc Biol (2001) 21:1104–17. doi: 10.1161/hq0701.093685
420. Masuda M, Wakasaki T, Suzui M, Toh S, Joe AK, Weinstein IB. Stat3 Orchestrates Tumor Development and Progression: The Achilles’ Heel of Head and Neck Cancers? Curr Cancer Drug Targets (2010) 10:117–26. doi: 10.2174/156800910790980197
421. Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, et al. Tumor-Associated Macrophages Regulate Tumorigenicity and Anticancer Drug Responses of Cancer Stem/Initiating Cells. Proc Natl Acad Sci USA (2011) 108:12425–30. doi: 10.1073/pnas.1106645108
422. Shamamian P, Schwartz JD, Pocock BJ, Monea S, Whiting D, Marcus SG, et al. Activation of Progelatinase A (Mmp-2) by Neutrophil Elastase, Cathepsin G, and proteinase-3: A Role for Inflammatory Cells in Tumor Invasion and Angiogenesis. J Cell Physiol (2001) 189:197–206. doi: 10.1002/jcp.10014
423. Mm Q, Re R, Rg H, Cr K-P, Cl J. Breast Cancer Cells Stimulate Neutrophils to Produce Oncostatin M: Potential Implications for Tumor Progression. Cancer Res (2005) 65:8896–904. doi: 10.1158/0008-5472.can-05-1734
424. Shojaei F, Singh M, Thompson JD, Ferrara N. Role of Bv8 in Neutrophil-Dependent Angiogenesis in a Transgenic Model of Cancer Progression. Proc Natl Acad Sci USA (2008) 105:2640–5. doi: 10.1073/pnas.0712185105
425. Zhou S-L, Zhou Z-J, Hu Z-Q, Huang X-W, Wang Z, Chen E-B, et al. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology (2016) 150:1646–58.e17. doi: 10.1053/j.gastro.2016.02.040
426. Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-Talk Between Myeloid-Derived Suppressor Cells (MDSC), Macrophages, and Dendritic Cells Enhances Tumor-Induced Immune Suppression. Semin Cancer Biol (2012) 22:275–81. doi: 10.1016/j.semcancer.2012.01.011
427. Beury DW, Parker KH, Nyandjo M, Sinha P, Carter KA, Ostrand-Rosenberg S. Cross-Talk Among Myeloid-Derived Suppressor Cells, Macrophages, and Tumor Cells Impacts the Inflammatory Milieu of Solid Tumors. J Leukoc Biol (2014) 96:1109–18. doi: 10.1189/jlb.3A0414-210R
428. Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, et al. The Novel Role of Tyrosine Kinase Inhibitor in the Reversal of Immune Suppression and Modulation of Tumor Microenvironment for Immune-Based Cancer Therapies. Cancer Res (2009) 69:2514–22. doi: 10.1158/0008-5472.CAN-08-4709
429. Kao J, Ko EC, Eisenstein S, Sikora AG, Fu S, Chen S-H. Targeting Immune Suppressing Myeloid-Derived Suppressor Cells in Oncology. Crit Rev Oncol Hematol (2011) 77:12–9. doi: 10.1016/j.critrevonc.2010.02.004
430. Ramachandran IR, Condamine T, Lin C, Herlihy SE, Garfall A, Vogl DT, et al. Bone Marrow PMN-MDSCs and Neutrophils are Functionally Similar in Protection of Multiple Myeloma From Chemotherapy. Cancer Lett (2016) 371:117–24. doi: 10.1016/j.canlet.2015.10.040
431. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discovery (2011) 1:54–67. doi: 10.1158/2159-8274.CD-10-0028
432. Wang Y-Q, Guo R-D, Guo R-M, Sheng W, Yin L-R. MicroRNA-182 Promotes Cell Growth, Invasion, and Chemoresistance by Targeting Programmed Cell Death 4 (PDCD4) in Human Ovarian Carcinomas. J Cell Biochem (2013) 114:1464–73. doi: 10.1002/jcb.24488
433. Tanaka K, Miyata H, Sugimura K, Fukuda S, Kanemura T, Yamashita K, et al. miR-27 Is Associated With Chemoresistance in Esophageal Cancer Through Transformation of Normal Fibroblasts to Cancer-Associated Fibroblasts. Carcinogenesis (2015) 36:894–903. doi: 10.1093/carcin/bgv067
434. Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E, et al. Exosomes Mediate Stromal Mobilization of Autocrine Wnt-PCP Signaling in Breast Cancer Cell Migration. Cell (2012) 151:1542–56. doi: 10.1016/j.cell.2012.11.024
435. Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, et al. Exosome Transfer From Stromal to Breast Cancer Cells Regulates Therapy Resistance Pathways. Cell (2014) 159:499–513. doi: 10.1016/j.cell.2014.09.051
436. Hu Y, Yan C, Mu L, Huang K, Li X, Tao D, et al. Fibroblast-Derived Exosomes Contribute to Chemoresistance Through Priming Cancer Stem Cells in Colorectal Cancer. PLoS One (2015) 10:e0125625. doi: 10.1371/journal.pone.0125625
437. Bielecka ZF, Czarnecka AM, Solarek W, Kornakiewicz A, Szczylik C. Mechanisms of Acquired Resistance to Tyrosine Kinase Inhibitors in Clear - Cell Renal Cell Carcinoma (Ccrcc). Curr Signal Transduct Ther (2014) 8:218–28. doi: 10.2174/1574362409666140206223014
438. Zhang H, Zhang X, Wu X, Li W, Su P, Cheng H, et al. Interference of Frizzled 1 (FZD1) Reverses Multidrug Resistance in Breast Cancer Cells Through the Wnt/β-Catenin Pathway. Cancer Lett (2012) 323:106–13. doi: 10.1016/j.canlet.2012.03.039
439. Chang HW, Roh J-L, Jeong E-J, Lee S, Kim S-W, Choi S-H, et al. Wnt Signaling Controls Radiosensitivity Via Cyclooxygenase-2-Mediated Ku Expression in Head and Neck Cancer. Int J Cancer (2008) 122:100–7. doi: 10.1002/ijc.23069
440. Cojoc M, Peitzsch C, Kurth I, Trautmann F, Kunz-Schughart LA, Telegeev GD, et al. Aldehyde Dehydrogenase is Regulated by β-Catenin/TCF and Promotes Radioresistance in Prostate Cancer Progenitor Cells. Cancer Res (2015) 75:1482–94. doi: 10.1158/0008-5472.CAN-14-1924
441. Park JS, Burckhardt CJ, Lazcano R, Solis LM, Isogai T, Li L, et al. Mechanical Regulation of Glycolysis Via Cytoskeleton Architecture. Nature (2020) 578:621–6. doi: 10.1038/s41586-020-1998-1
442. Ulrich TA, Pardo EM de J, Kumar S. The Mechanical Rigidity of the Extracellular Matrix Regulates the Structure, Motility, and Proliferation of Glioma Cells. Cancer Res (2009) 69:4167–74. doi: 10.1158/0008-5472.CAN-08-4859
443. Jahanban-Esfahlan R, Seidi K, Manjili MH, Jahanban-Esfahlan A, Javaheri T, Zare P. Tumor Cell Dormancy: Threat or Opportunity in the Fight Against Cancer. Cancers (2019) 11:1207. doi: 10.3390/cancers11081207
444. Liu Y, Lv J, Liang X, Yin X, Zhang L, Chen D, et al. Fibrin Stiffness Mediates Dormancy of Tumor-Repopulating Cells Via a Cdc42-Driven Tet2 Epigenetic Program. Cancer Res (2018) 78:3926–37. doi: 10.1158/0008-5472.CAN-17-3719
445. Yu H, Mouw JK, Weaver VM. Forcing Form and Function: Biomechanical Regulation of Tumor Evolution. Trends Cell Biol (2011) 21:47–56. doi: 10.1016/j.tcb.2010.08.015
446. Pratt SJP, Lee RM, Martin SS. The Mechanical Microenvironment in Breast Cancer. Cancers (2020) 12:1452. doi: 10.3390/cancers12061452
447. Jain RK, Martin JD, Stylianopoulos T. The Role of Mechanical Forces in Tumor Growth and Therapy. Annu Rev BioMed Eng (2014) 16:321–46. doi: 10.1146/annurev-bioeng-071813-105259
448. Kular JK, Basu S, Sharma RI. The Extracellular Matrix: Structure, Composition, Age-Related Differences, Tools for Analysis and Applications for Tissue Engineering. J Tissue Eng (2014) 5:2041731414557112. doi: 10.1177/2041731414557112
449. McLeod CM, Mauck RL. High Fidelity Visualization of Cell-to-Cell Variation and Temporal Dynamics in Nascent Extracellular Matrix Formation. Sci Rep (2016) 6:38852. doi: 10.1038/srep38852
450. Cox TR, Erler JT. Remodeling and Homeostasis of the Extracellular Matrix: Implications for Fibrotic Diseases and Cancer. Dis Model Mech (2011) 4:165–78. doi: 10.1242/dmm.004077
451. Pfeifer CR, Alvey CM, Irianto J, Discher DE. Genome Variation Across Cancers Scales With Tissue Stiffness - an Invasion-Mutation Mechanism and Implications for Immune Cell Infiltration. Curr Opin Syst Biol (2017) 2:103–14. doi: 10.1016/j.coisb.2017.04.005
452. Deville SS, Cordes N. The Extracellular, Cellular, and Nuclear Stiffness, a Trinity in the Cancer Resistome—a Review. Front Oncol (2019) 9:1376. doi: 10.3389/fonc.2019.01376
453. Dhillon AS, Hagan S, Rath O, Kolch W. MAP Kinase Signalling Pathways in Cancer. Oncogene (2007) 26:3279–90. doi: 10.1038/sj.onc.1210421
454. Cobbaut M, Karagil S, Bruno L, Diaz de la Loza MDC, Mackenzie FE, Stolinski M, et al. Dysfunctional Mechanotransduction Through the YAP/TAZ/Hippo Pathway as a Feature of Chronic Disease. Cells (2020) 9:151. doi: 10.3390/cells9010151
455. Wei L, Surma M, Shi S, Lambert-Cheatham N, Shi J. Novel Insights Into the Roles of Rho Kinase in Cancer. Arch Immunol Ther Exp (Warsz) (2016) 64:259–78. doi: 10.1007/s00005-015-0382-6
456. Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and Extracellular Matrix Homeostasis. Nat Rev Mol Cell Biol (2014) 15:802–12. doi: 10.1038/nrm3896
457. Northcott JM, Dean IS, Mouw JK, Weaver VM. Feeling Stress: The Mechanics of Cancer Progression and Aggression. Front Cell Dev Biol (2018) 6:17. doi: 10.3389/fcell.2018.00017
458. Chow K-H, Factor RE, Ullman KS. The Nuclear Envelope Environment and its Cancer Connections. Nat Rev Cancer (2012) 12:196–209. doi: 10.1038/nrc3219
459. Dahl KN, Kahn SM, Wilson KL, Discher DE. The Nuclear Envelope Lamina Network has Elasticity and a Compressibility Limit Suggestive of a Molecular Shock Absorber. J Cell Sci (2004) 117:4779–86. doi: 10.1242/jcs.01357
460. Denais C, Lammerding J. Nuclear Mechanics in Cancer. Adv Exp Med Biol (2014) 773:435–70. doi: 10.1007/978-1-4899-8032-8_20
461. Isermann P, Lammerding J. Nuclear Mechanics and Mechanotransduction in Health and Disease. Curr Biol (2013) 23:R1113–21. doi: 10.1016/j.cub.2013.11.009
462. Nguyen TV, Sleiman M, Moriarty T, Herrick WG, Peyton SR. Sorafenib Resistance and JNK Signaling in Carcinoma During Extracellular Matrix Stiffening. Biomaterials (2014) 35:5749–59. doi: 10.1016/j.biomaterials.2014.03.058
463. Joyce MH, Lu C, James ER, Hegab R, Allen SC, Suggs LJ, et al. Phenotypic Basis for Matrix Stiffness-Dependent Chemoresistance of Breast Cancer Cells to Doxorubicin. Front Oncol (2018) 8:337. doi: 10.3389/fonc.2018.00337
464. Liu C, Liu Y, Xie H-G, Zhao S, Xu X-X, Fan L-X, et al. Role of Three-Dimensional Matrix Stiffness in Regulating the Chemoresistance of Hepatocellular Carcinoma Cells. Biotechnol Appl Biochem (2015) 62:556–62. doi: 10.1002/bab.1302
465. Schrader J, Gordon-Walker TT, Aucott RL, van Deemter M, Quaas A, Walsh S, et al. Matrix Stiffness Modulates Proliferation, Chemotherapeutic Response, and Dormancy in Hepatocellular Carcinoma Cells. Hepatol Baltim Md (2011) 53:1192–205. doi: 10.1002/hep.24108
466. You Y, Zheng Q, Dong Y, Xie X, Wang Y, Wu S, et al. Matrix Stiffness-Mediated Effects on Stemness Characteristics Occurring in HCC Cells. Oncotarget (2016) 7:32221–31. doi: 10.18632/oncotarget.8515
467. Helmlinger G, Netti PA, Lichtenbeld HC, Melder RJ, Jain RK. Solid Stress Inhibits the Growth of Multicellular Tumor Spheroids. Nat Biotechnol (1997) 15:778–83. doi: 10.1038/nbt0897-778
468. Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Cancer Cells Compress Intratumour Vessels. Nature (2004) 427:695–5. doi: 10.1038/427695a
469. Talukdar S, Bhoopathi P, Emdad L, Das S, Sarkar D, Fisher PB. Dormancy and Cancer Stem Cells: An Enigma for Cancer Therapeutic Targeting. Adv Cancer Res (2019) 141:43–84. doi: 10.1016/bs.acr.2018.12.002
470. Jain RK. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell (2014) 26:605–22. doi: 10.1016/j.ccell.2014.10.006
471. Tse JM, Cheng G, Tyrrell JA, Wilcox-Adelman SA, Boucher Y, Jain RK, et al. Mechanical Compression Drives Cancer Cells Toward Invasive Phenotype. Proc Natl Acad Sci USA (2012) 109:911–6. doi: 10.1073/pnas.1118910109
472. Moffitt L, Karimnia N, Stephens A, Bilandzic M. Therapeutic Targeting of Collective Invasion in Ovarian Cancer. Int J Mol Sci (2019) 20:1466. doi: 10.3390/ijms20061466
473. Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective Invasion in Breast Cancer Requires a Conserved Basal Epithelial Program. Cell (2013) 155:1639–51. doi: 10.1016/j.cell.2013.11.029
474. Karakashev SV, Reginato MJ. Progress Toward Overcoming Hypoxia-Induced Resistance to Solid Tumor Therapy. Cancer Manag Res (2015) 7:253–64. doi: 10.2147/CMAR.S58285
475. Broders-Bondon F, Nguyen Ho-Bouldoires TH, Fernandez-Sanchez M-E, Farge E. Mechanotransduction in Tumor Progression: The Dark Side of the Force. J Cell Biol (2018) 217:1571–87. doi: 10.1083/jcb.201701039
476. Heldin C-H, Rubin K, Pietras K, Östman A. High Interstitial Fluid Pressure — An Obstacle in Cancer Therapy. Nat Rev Cancer (2004) 4:806–13. doi: 10.1038/nrc1456
477. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell (2012) 21:418–29. doi: 10.1016/j.ccr.2012.01.007
478. Marchesi F, Piemonti L, Mantovani A, Allavena P. Molecular Mechanisms of Perineural Invasion, a Forgotten Pathway of Dissemination and Metastasis. Cytokine Growth Factor Rev (2010) 21:77–82. doi: 10.1016/j.cytogfr.2009.11.001
479. Coarfa C, Florentin D, Putluri N, Ding Y, Au J, He D, et al. Influence of the Neural Microenvironment on Prostate Cancer. Prostate (2018) 78:128–39. doi: 10.1002/pros.23454
480. Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic Nerve Development Contributes to Prostate Cancer Progression. Science (2013) 341:1236361. doi: 10.1126/science.1236361
481. Melamed R, Rosenne E, Shakhar K, Schwartz Y, Abudarham N, Ben-Eliyahu S. Marginating Pulmonary-NK Activity and Resistance to Experimental Tumor Metastasis: Suppression by Surgery and the Prophylactic Use of a Beta-Adrenergic Antagonist and a Prostaglandin Synthesis Inhibitor. Brain Behav Immun (2005) 19:114–26. doi: 10.1016/j.bbi.2004.07.004
482. Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, et al. Chronic Stress Promotes Tumor Growth and Angiogenesis in a Mouse Model of Ovarian Carcinoma. Nat Med (2006) 12:939–44. doi: 10.1038/nm1447
483. Amit M, Takahashi H, Dragomir MP, Lindemann A, Gleber-Netto FO, Pickering CR, et al. Loss of p53 Drives Neuron Reprogramming in Head and Neck Cancer. Nature (2020) 578:449–54. doi: 10.1038/s41586-020-1996-3
484. Kim-Fuchs C, Le CP, Pimentel MA, Shackleford D, Ferrari D, Angst E, et al. Chronic Stress Accelerates Pancreatic Cancer Growth and Invasion: A Critical Role for Beta-Adrenergic Signaling in the Pancreatic Microenvironment. Brain Behav Immun (2014) 40:40–7. doi: 10.1016/j.bbi.2014.02.019
485. Pundavela J, Demont Y, Jobling P, Lincz LF, Roselli S, Thorne RF, et al. ProNGF Correlates With Gleason Score and Is a Potential Driver of Nerve Infiltration in Prostate Cancer. Am J Pathol (2014) 184:3156–62. doi: 10.1016/j.ajpath.2014.08.009
486. Zhao C-M, Hayakawa Y, Kodama Y, Muthupalani S, Westphalen CB, Andersen GT, et al. Denervation Suppresses Gastric Tumorigenesis. Sci Transl Med (2014) 6:250ra115. doi: 10.1126/scitranslmed.3009569
487. Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J, et al. Brain Tumour Cells Interconnect to a Functional and Resistant Network. Nature (2015) 528:93–8. doi: 10.1038/nature16071
Keywords: TME (tumor microenvironment), HIF - hypoxia inducible factor, CAF, microRNA (miR), MDSC (myeloid-derived suppressor cells), tumor associated macrophage (TAM), Treg - regulatory T cell, TGF - β1
Citation: Khalaf K, Hana D, Chou JT-T, Singh C, Mackiewicz A and Kaczmarek M (2021) Aspects of the Tumor Microenvironment Involved in Immune Resistance and Drug Resistance. Front. Immunol. 12:656364. doi: 10.3389/fimmu.2021.656364
Received: 20 January 2021; Accepted: 27 April 2021;
Published: 27 May 2021.
Edited by:
Sergio Rutella, Nottingham Trent University, United KingdomReviewed by:
Lisa Sevenich, Georg Speyer Haus, GermanyZong Sheng Guo, University of Pittsburgh, United States
Copyright © 2021 Khalaf, Hana, Chou, Singh, Mackiewicz and Kaczmarek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Khalil Khalaf, a2hhbGlsa2hhbGFmMDIxNkBnbWFpbC5jb20=