 Omar Rafael Alemán
Omar Rafael Alemán Nancy Mora
Nancy Mora Carlos Rosales
Carlos Rosales- Departamento de Inmunología, Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México, Mexico City, Mexico
Human neutrophils express two unique antibody receptors for IgG, the FcγRIIa and the FcγRIIIb. FcγRIIa contains an immunoreceptor tyrosine-based activation motif (ITAM) sequence within its cytoplasmic tail, which is important for initiating signaling. In contrast, FcγRIIIb is a glycosylphosphatidylinositol (GPI)-linked receptor with no cytoplasmic tail. Although, the initial signaling mechanism for FcγRIIIb remains unknown, it is clear that both receptors are capable of initiating distinct neutrophil cellular functions. For example, FcγRIIa is known to induce an increase in L-selectin expression and efficient phagocytosis, while FcγRIIIb does not promote these responses. In contrast, FcγRIIIb has been reported to induce actin polymerization, activation of β1 integrins, and formation of neutrophils extracellular traps (NET) much more efficiently than FcγRIIa. Another function where these receptors seem to act differently is the increase of cytoplasmic calcium concentration. It has been known for a long time that FcγRIIa induces production of inositol triphosphate (IP3) to release calcium from intracellular stores, while FcγRIIIb does not use this phospholipid. Thus, the mechanism for FcγRIIIb-mediated calcium rise remains unknown. Transient Receptor Potential Melastatin 2 (TRPM2) is a calcium permeable channel expressed in many cell types including vascular smooth cells, endothelial cells and leukocytes. TRPM2 can be activated by protein kinase C (PKC) and by oxidative stress. Because we previously found that FcγRIIIb stimulation leading to NET formation involves PKC activation and reactive oxygen species (ROS) production, in this report we explored whether TRPM2 is activated via FcγRIIIb and mediates calcium rise in human neutrophils. Calcium rise was monitored after Fcγ receptors were stimulated by specific monoclonal antibodies in Fura-2-loaded neutrophils. The bacterial peptide fMLF and FcγRIIa induced a calcium rise coming initially from internal pools. In contrast, FcγRIIIb caused a calcium rise by inducing calcium entry from the extracellular medium. In addition, in the presence of 2-aminoethoxydiphenyl borate (2-APB) or of clotrimazole, two inhibitors of TRPM2, FcγRIIIb-induced calcium rise was blocked. fMLF- or FcγRIIa-induced calcium rise was not affected by these inhibitors. These data suggest for the first time that FcγRIIIb aggregation activates TRPM2, to induce an increase in cytoplasmic calcium concentration through calcium internalization in human neutrophils.
introduction
Neutrophils, the most abundant leukocytes in peripheral blood, are considered the first line of defense because these cells arrive first at sites of inflammation or infection (1, 2). Once there, neutrophils display a variety of antimicrobial functions including phagocytosis (3, 4), degranulation, and formation of neutrophil extracellular traps (NET) (5). These functions can be initiated or enhanced by antibodies, in the form of immune complexes, binding to their cognate Fc receptors on the surface of the neutrophil (6). Human neutrophils express constitutively two Fc receptors for IgG, the FcγRIIa (CD32a) and the FcγRIIIb (CD16b). FcγRIIa contains an ITAM (immunoreceptor tyrosine-based activation motif) sequence in its cytoplasmic tail (7), while FcγRIIIb is a glycosylphosphatidylinositol (GPI)-linked receptor, lacking a cytoplasmic tail (8). These two neutrophil antibody receptors are not only structurally different but also have been shown to induce unique cellular responses (9). FcγRIIa is the predominant Fcγ receptor mediating phagocytosis, while the contribution of FcγRIIIb to this response is minimal (10). In contrast, FcγRIIIb signaling to the neutrophil nucleus for nuclear factor activation is more efficient than FcγRIIa signaling (11). In addition, FcγRIIIb is the only Fcγ receptor capable of inducing NET formation (12, 13). Although, these reports indicate that each receptor can activate particular cell responses, the signaling capabilities of each receptor are still incompletely understood.
Early reports clearly showed that both neutrophil Fcγ receptors induce an increase in cytoplasmic Ca2+ concentration ([Ca2+]i) (14, 15). However, the mechanism for this response seems to be different for each receptor (15). FcγRIIa signals via its ITAM sequence to activate Syk (spleen tyrosine kinase). Syk then phosphorylates enzymes such as PI 3-K (phosphatidylinositol 3-kinase) and PLCγ (phospholipase Cγ). PLCγ produces inositoltriphosphate (IP3), and diacylglycerol (DAG). These second messengers cause calcium release from the endoplasmic reticulum (ER), and activation of PKC (protein kinase C), respectively (8, 16, 17). In sharp contrast, FcγRIIIb-mediated increase in [Ca2+]i does not involve IP3 formation (15), and the mechanism used by FcγRIIIb to increase [Ca2+]i is still unknown. Because FcγRIIIb is a GPI-linked receptor lacking a cytoplasmic tail and with no other subunits known to associate with it, its signaling mechanism is only partially described. Recently, it was reported that the signal pathway, activated by FcγRIIIb leading to NET formation, involves Syk, TAK1 (transforming growth factor-β- activated kinase 1), the MEK (ERK kinase)/ERK (extracellular signal-regulated kinase) cascade, activation of PKC, and activation of the nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase complex, which produce reactive oxygen species (ROS) (18). Therefore, it is possible that FcγRIIIb uses some of these signaling molecules to induce an increase in [Ca2+]i.
Transient Receptor Potential Melastatin 2 (TRPM2), a nonselective Ca2+-permeable membrane cation channel, is highly expressed in myeloid cells (19, 20). This receptor is a member of the TRP family of cation-selective channels that are weakly voltage-sensitive and diversely opened by temperature, mechanical force, pH, and oxidative stress (21, 22). TRPM2 can be opened through directly binding with intracellular adenosine diphosphate ribose (ADPR) (23) and can also be indirectly activated under conditions of oxidative stress (24, 25), acidification (26), and elevated intracellular Ca2+ (27). Additionally, in dorsal root ganglion neurons, the activity of TRPM2 is increased by the addition of phorbol 12-myristate 13-acetate (PMA) which leads to activation of PKC (28, 29). Thus, because TRPM2 can be activated by ROS and PKC, two of the second messengers involved in the signaling pathway from FcγRIIIb leading to NET formation (18), it is possible that TRPM2 is used by FcγRIIIb to induce an increase in [Ca2+]i. In order to test this hypothesis, both FcγRIIa and FcγRIIIb were stimulated by specific monoclonal antibodies, and the increase in [Ca2+]i was measured in the presence or absence of pharmacological inhibitors. The neutrophil chemoattractant fMLF (N-formyl-methionil-leucyl-phenylalanine), and also FcγRIIa induced a rapid increase in [Ca2+]i. FcγRIIIb aggregation also induced an increase in [Ca2+]i, but this increase was delayed by several seconds. Despite both, FcγRIIa and FcγRIIIb aggregation-induced ROS production, in the presence of diphenyleneiodonium chloride (DPI), an inhibitor of the NADPH-oxidase complex (30), only the FcγRIIIb-induced increase in [Ca2+]i was reduced. Similarly, in the presence of Gö6976, an inhibitor of PKC (31, 32), only FcγRIIIb-induced increase in [Ca2+]i was reduced. In addition, 2-aminoethoxydiphenyl borate (2-APB) (33–35) and clotrimazole (36–39), inhibitors of TRPM2, reduced FcγRIIIb-induced, but not FcγRIIa-induced increase in [Ca2+]i. These data show for the first time that FcγRIIIb aggregation activates TRPM2 via PKC and ROS for inducing an increase in [Ca2+]i in human neutrophils.
Materials and Methods
Neutrophils
Neutrophils were purified from heparinized peripheral blood collected from adult healthy volunteers following a protocol previously approved by the Bioethics Committee at Instituto de Investigaciones Biomédicas – Universidad Nacional Autónoma de México (UNAM). Neutrophils were purified exactly as previously described (40).
Reagents
Bovine serum albumin (BSA) was from F. Hoffmann-La Roche Ltd. (Mannheim, Germany). Gö6976, a PKC inhibitor (catalog number sc-221684); antibiotic (5Z)-7-Oxozeaenol (LLZ 1640-2), a TAK1 inhibitor (catalog number sc-202055); and 2-aminoethoxydiphenyl borate (2-APB), a TRPM2 inhibitor (catalog number sc-201487) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). 3-(1-methyl-1H-indol-3-yl-methylene)-2-oxo-2,3-dihydro-1H-indole-5-sulfonamide (iSyk), a spleen tyrosine kinase (Syk) inhibitor (catalog number 574711); and fura-2-AM, a calcium indicator (catalog number 344905) were from Calbiochem/EMD Millipore (Billerica, MA, USA). UO126, a MEK inhibitor (catalog number V112A), was from Promega (Madison, WI, USA). Dihydrorhodamine123 (DHR-123) a ROS indicator (catalog number AS-85711), was from Anaspec, Inc (Fremont, CA, USA). The cOmplete™ protease inhibitor cocktail (catalog no. 11697498001) and PhosSTOP™ phosphatase inhibitor cocktail (catalog no. 04906845001) were from Roche Diagnostics (Basel, Switzerland). Diphenyleneiodonium chloride (DPI), an NADPH-oxidase inhibitor (catalog number 300260); phorbol 12-myristate 13-acetate (PMA), a PKC activator (catalog number P8139); N-formyl-Met-Leu-Phe (fMLF), a potent chemotactic peptide (catalog number F6632); pertussis toxin, a G protein-coupled receptor inhibitor (41, 42) (catalog number 516560), clotrimazole, a TRPM2 inhibitor (catalog number C6019), and all other chemicals were from Sigma Aldrich (St. Louis, MO, USA).
The following antibodies were used: anti-human FcγRIIa (CD32a) mAb IV.3 (43) (ATCC® HB-217) was from American Type Culture Collection (Manassas, VA, USA). Anti-human FcγRIIIb (CD16b) mAb 3G8 (44) was donated by Dr. Eric J. Brown (University of California in San Francisco, San Francisco, CA, USA). F(ab’)2 goat anti-mouse IgG (catalog number 115-006-003) was from Jackson Immunoresearch Laboratories, Inc. (West Grove, PA, USA).
Fluorescent Calcium Measurements
Increase of cytosolic calcium concentration was measured by detecting fluorescence changes in neutrophils loaded with Fura 2-AM as previously described (45, 46). Briefly, neutrophils were loaded with 10 μM Fura-2, washed, and resuspended (1.5 x 106 cell/ml) in PBS with 1.5 mM Ca2+and 1.5 mM Mg2+ and kept on ice. Then, fluorescent changes of a 2-ml stirred neutrophil suspension kept at 37°C were monitored with a LS55 spectrofluorimeter (Perking Elmer; Waltham, MA, USA), using 340 and 380 nm excitation wavelengths and 510 nm emission wavelength. Calcium concentrations were calculated as described by Grynkiewicz et al. (47), using the software FL WinLab 4.00.02 (Perking Elmer; Waltham, MA, USA).
For fMLF stimulation, 3 x 106 Fura-2-loaded neutrophils in 2 ml of PBS with Ca2+ and Mg2+ were placed in a spectrofluorimeter cuvette and incubated at 37°C for 2 min. After that, fluorescence changes were recorded for 100 s, then 40 μl of 500 nM fMLF were added (final concentration of 10 nM). For FcγR stimulation, 3 x 106 Fura-2-loaded neutrophils were first resuspended in 100 μl PBS containing 10 μg/ml of the corresponding anti-FcγR antibody, and incubated on ice for 20 min. After one wash in cold PBS, neutrophils were resuspended in 2 ml PBS with Ca2+and Mg2+ and transferred to a spectrofluorimeter cuvette. Fluorescence changes were recorded for 100 s, and then 80 μl of 1.3 mg/ml F(ab’)2 goat anti-mouse IgG (final concentration 52 μg/ml) were added to aggregate the receptors.
In some experiments, Fura-2-loaded neutrophils were resuspended in 2 ml PBS containing 1 mM of EGTA and fluorescence changes detected after various stimuli for 300 s. Then, 40 μl of 100 mM CaCl2 (final concentration 4 mM) were added, and measurements continued until 450 s. Also, in some experiments, neutrophils were incubated for 30 min before stimulation, with the inhibitors LLZ 1640-2 (10 nM), UO126 (50 μM), iSyk (1 μM), DPI (10 μM), Gö6976 (1 μM), 2-APB (5 μM), clotrimazole (10 μM), or the vehicle dimethyl sulfoxide (DMSO) alone. For Pertussis toxin (2 μg/ml), neutrophils were incubated for 75 min before stimulation.
Measurement of Reactive Oxygen Species (ROS)
ROS production was assessed by detecting fluoresce changes in neutrophils loaded with dihydrorhodamine 123 (DHR-123). Neutrophils (1 x 106) were resuspended in 100 μl of 15 μM DHR-123 in PBS and incubated for 15 min at 37°C in the dark. Neutrophils were washed with 1 ml PBS, and then resuspended in 100 μl of PBS with the corresponding stimulus as follows. For PMA, neutrophils were resuspended in PBS containing 20 nM of PMA, and then incubated at 37°C in the dark for 45 min. For FcγR stimulation, neutrophils were resuspended in PBS containing 10 μg/ml of the corresponding anti-FcγR antibody, and incubated on ice for 20 min. Next, neutrophils were washed in cold PBS, resuspended in 100 μl of PBS containing 10 μg/ml F(ab’)2 goat anti-mouse IgG, and incubated at 37°C in the dark for 45 min. After incubation, 250 μl cold PBS were added and cells kept on ice for 2 min. Finally, neutrophils were centrifuged and resuspended in 1% paraformaldehyde in PBS at 4°C. Neutrophils were stored cold in the dark until analyzed by flow cytometry using an Attune NxT Flow Cytometer (Thermo Fisher Scientific), with the 485 nm (excitation) and 520 nm (emission) filters. Neutrophils were gated by dot-plot analysis and 10,000 cells were acquired per sample. Data analysis was performed using the Flowjo X software (Tree Star Inc., Ashland, OR, USA).
Statistical Analysis
Quantitative data were expressed as mean ± standard error of mean (SEM). Single variable data were compared by paired-sample Student’s t-tests using the computer program KaleidaGraph® version 4.5.2 for Mac (Synergy Software; Reading, PA, USA). Also, multiple pair-comparisons were performed using Tukey’s test after ordinary one-way analysis of variance (ANOVA). Post hoc differences were considered statistically different at a value p < 0.05.
Results
Fcγ Receptors Induce an Increase in Cytoplasmic Ca2+ Concentration
Neutrophils, the most abundant leukocytes in blood, display important functions for controlling infections, inflammation, and immune responses (2, 48). Several of these functions are initiated by antibodies binding to Fc receptors (6). Although, both Fcγ receptors on human neutrophils (FcγRIIa and FcγRIIIb) are known to activate particular cell responses, the signaling capabilities of each receptor are still incompletely understood (9). About 30 years ago, it was already known that both neutrophil Fcγ receptors induce an increase in cytoplasmic Ca2+ concentration ([Ca2+]i) (15), but important differences were noted. FcγRIIa used the second messenger IP3 to induce release of Ca2+ from an internal pool (15), while the increase in [Ca2+]i induced by FcγRIIIb did not involve IP3 (15). Despite these early studies the mechanism used by FcγRIIIb to mobilize calcium is still unknown. Thus, in order to further explore the mechanisms of [Ca2+]i increase induced by Fcγ receptors, Fura-2-loaded neutrophils were first stimulated with the chemoattractant fMLF. In response to fMLF a rapid almost immediate increase in [Ca2+]i was observed (Figure 1A). After this rise, the [Ca2+]i then returned to basal levels (Figure 1A). Similarly, aggregation of the FcγRIIa with specific monoclonal antibodies also induced a rapid increase in [Ca2+]i (Figure 1B). Also, aggregation of the FcγRIIIb with specific monoclonal antibodies resulted in an increase in [Ca2+]i (Figure 1C). Although, the magnitude of the [Ca2+]i rise was similar with all three stimuli (Figure 1D), the FcγRIIIb-mediated increase in [Ca2+]i was delayed for about 10 to 20 seconds (Figure 1C), suggesting again that the mechanism for calcium mobilization was different between the two Fcγ receptors.
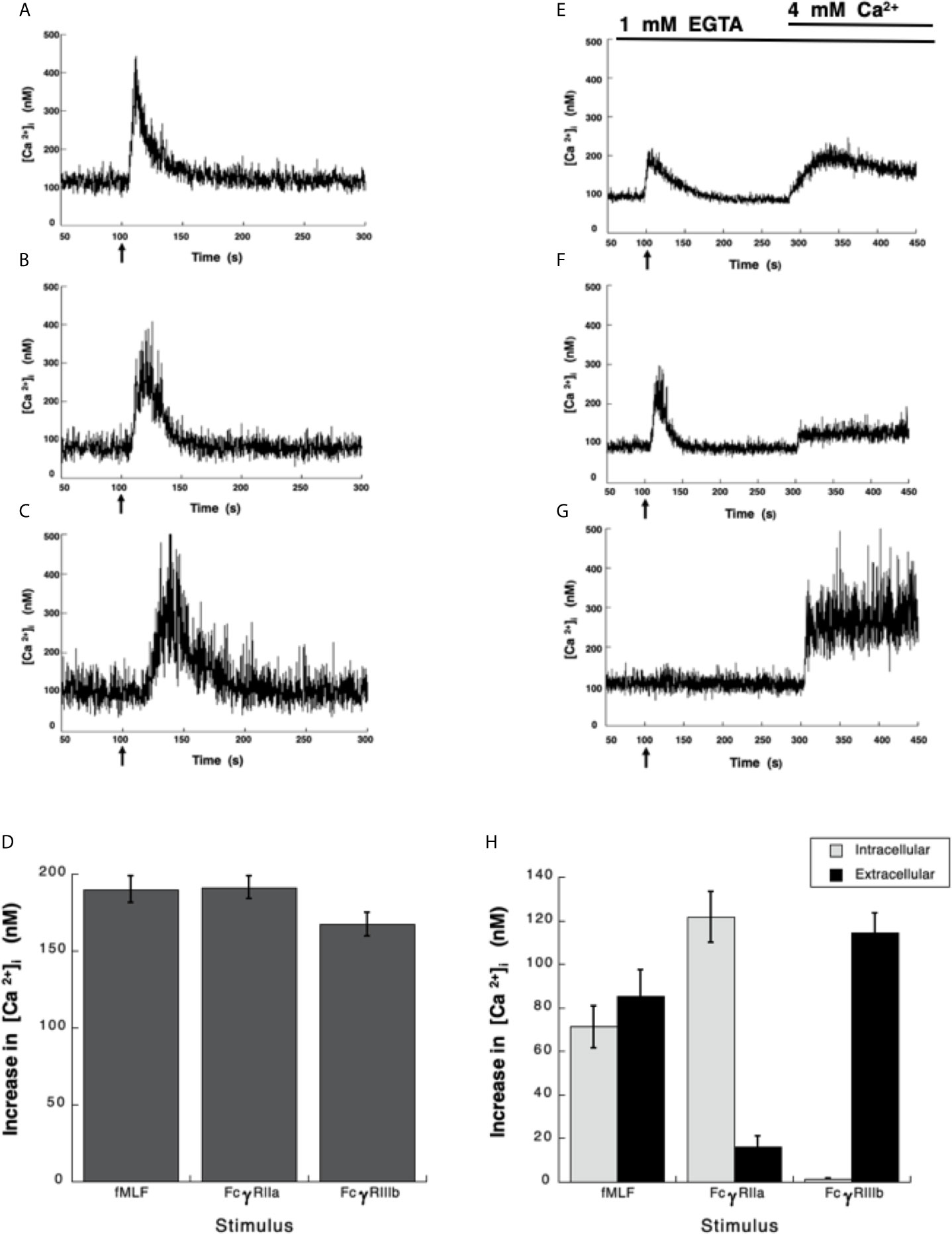
Figure 1 Fcγ receptors induce an increase in [Ca2+]i. (A–D) Fura-2-loaded human neutrophils in PBS with Ca2+ and Mg2+ were stimulated with 10 nM fMLF (A), or were stimulated by aggregating FcγRIIa on the cell membrane with mAb IV.3 (B), or by aggregating FcγRIIIb on the cell membrane with mAb 3G8 (C). (E–H) Fura-2-loaded neutrophils were resuspended in PBS containing 1 mM of EGTA and fluorescence changes detected after stimulating with 10 nM fMLF (E), or by aggregating FcγRIIa (F), or by aggregating FcγRIIIb (G). After 300 seconds, 4 mM Ca2+ was added and measurements continued until 450 seconds. Arrow indicates the moment when the stimulus was added. Changes in cytosolic calcium concentration ([Ca2+]i) were assessed by measuring the variations in fluorescence. Tracings are representative of three experiments with similar results. (D) Increments in [Ca2+]i were calculated by subtracting the baseline value from the maximum value after stimulation. (H) The initial rise in [Ca2+]i represents Ca2+ from intracellular stores, while the rise in [Ca2+]i after addition of 4 mM Ca2+ represents Ca2+ from extracellular medium. Data are mean ± SEM of three independent experiments. Asterisks denote conditions that were statistically different from untreated cells (p < 0.01).
FcγRIIIb Aggregation Induces an Increase in [Ca2+]ivia Ca2+ Entry From Extracellular Medium
It is well established that fMLF-induced increase in [Ca2+]i has two components, one initial release of Ca2+ from internal stores followed by a subsequent influx of this cation from outside the cell by the mechanism known as store-operated calcium entry (SOCE) (49, 50). Similarly, it is believed that Fcγ receptors also display a similar mechanism for an increase in [Ca2+]i (51, 52). Thus, we sought to confirm these ideas by selectively stimulating each Fcγ receptor. Human neutrophils were placed in Ca2+-free medium and then stimulated with fMLF. As expected an initial increase in [Ca2+]i was observed followed by a gradual return to basal levels around 90 seconds later (Figure 1E). At this time and excess of Ca2+ was added to the medium. This led to a second increase in [Ca2+]i, which represents influx of Ca2+ from outside the cell (Figure 1E). Similarly, in Ca2+-free medium aggregation of FcγRIIa induced an increase in [Ca2+]i from internal stores that returned to basal levels after about 90 seconds (Figure 1F). When Ca2+ was restored to the medium a second increase in [Ca2+]i, was also observed (Figure 1F), although this second rise was much smaller. This indicated that similarly to the fMLF, aggregation of FcγRIIa induced a release of Ca2+ from internal stores and then an influx of Ca2+ from outside the cell. In sharp contrast, in Ca2+-free medium aggregation of FcγRIIIb did not induce any increase in [Ca2+]i (Figure 1G), suggesting that no Ca2+ was released from intracellular stores. When Ca2+ was restored to the medium an important increase in [Ca2+]i, was detected (Figure 1G). After fMLF stimulation, the magnitude of the [Ca2+]i rise from internal stores was very similar to the increase in [Ca2+]i from outside the cell (Figure 1H). Each part was about half of the total increase in calcium observed in cells kept in Ca2+-containing medium. For FcγRIIa stimulation, the magnitude of the [Ca2+]i rise from internal stores was similar to the increase in [Ca2+]i induced by fMLF (Figure 1H). The [Ca2+]i rise from extracellular Ca2+ was smaller than the [Ca2+]i rise from internal stores (Figure 1H), suggesting that the major contribution to an increase in [Ca2+]i rise after FcγRIIa engagement was from internal stores. In contrast, the FcγRIIIb-mediated increase in [Ca2+]i was almost exclusively due to influx of extracellular Ca2+ (Figure 1H). These data suggested that contrary to other Fcγ receptors, FcγRIIIb induces only an influx of Ca2+ from outside the cell.
FcγRIIIb-Mediated Increase in [Ca2+]i Is Independent of TAK1 and MEK
After having shown that FcγRIIa and FcγRIIIb induce an increase in [Ca2+]i very differently, we sought to explore the signaling mechanisms that could help explain these differences. Previously, we have reported that FcγRIIIb signaling activates TAK1 and MEK in human neutrophils (18). Thus, we hypothesized that either TAK1 or MEK were required for the increase in [Ca2+]i induced by FcγRIIIb. LLZ 1640-2, a selective TAK1 inhibitor did not affect the increase in [Ca2+]i induced by either FcγRIIa (Figure 2A) nor FcγRIIIb aggregation (Figure 2B). Similarly, UO126, a selective MEK inhibitor did not affect the increase in [Ca2+]i induced by either Fcγ receptor (Figure 2). Additionally, as expected neither LLZ 1640-2 nor UO126 affected the increase in [Ca2+]i induced by fMLF stimulation (Figure 2C). However, since the fMLF receptor is a GPCR (53) treatment with Pertussis toxin completely blocked the increase in [Ca2+]i (Figures 2C, D). In contrast, Pertussis toxin did not affect the increase in [Ca2+]i induced by either Fcγ receptor (Figure 2D). These data strongly suggested, that TAK1 and MEK were not involved in increasing [Ca2+]i after FcγRIIa or FcγRIIIb aggregation in human neutrophils.
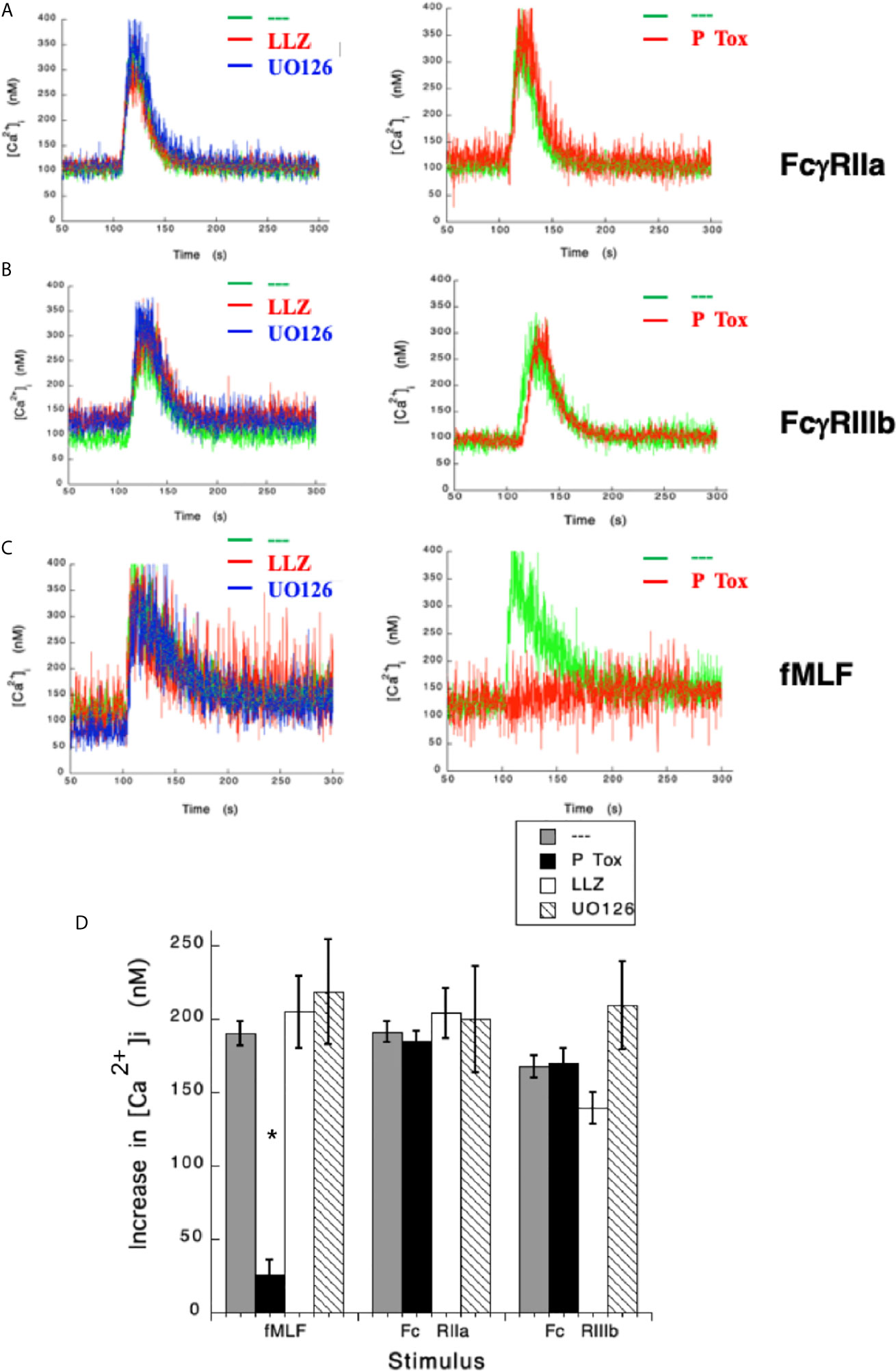
Figure 2 Fcγ receptor-mediated increase in [Ca2+]i is independent of TAK1 and MEK. Fura-2-loaded human neutrophils in PBS with Ca2+ and Mg2+ were left untreated (green line), or treated with 10 nM LLZ 1640-2 (LLZ) a TAK1 inhibitor, or with 50 µM UO126, a MEK inhibitor, or with 2 μg/ml Pertussis toxin (P Tox), before being stimulated by aggregating FcγRIIa (A), or by aggregating FcγRIIIb (B), or with 10 nM fMLF (C). Arrow indicates the moment when the stimulus was added. Changes in cytosolic calcium concentration ([Ca2+]i) were assessed by measuring the variations in fluorescence as described in material and methods. Tracings are representative of three experiments with similar results. (D) Increments in [Ca2+]i were calculated by subtracting the baseline value from the maximum value after stimulation. Data are mean ± SEM of three independent experiments. Asterisks (*) denote conditions that were statistically different from untreated cells (p < 0.01).
Syk, PKC and NADPH-Oxidase Are Involved in FcγRIIIb-Mediated Increase in [Ca2+]i
In the past, we have reported that FcγRIIIb signaling involves Syk, PKC, and NADPH-oxidase (12, 18). Therefore, we explored the possible involvement of these signaling molecules in FcγRIIIb-mediated increase in [Ca2+]i. Treatment with iSyk, a selective Syk inhibitor, reduced the increase in [Ca2+]i induced by either FcγRIIa (Figure 3A) or FcγRIIIb aggregation (Figure 3B). Additionally, Gö6976 a selective PKC inhibitor blocked the increase in [Ca2+]i induced by FcγRIIIb but not by FcγRIIa (Figure 3). Similarly, DPI a selective NADPH-oxidase inhibitor reduced the increase in [Ca2+]i induced by FcγRIIIb but not by FcγRIIa (Figure 3). These data showed that Syk was involved in both FcγRIIa- and FcγRIIIb-mediated increase in [Ca2+]i, and suggested for the first time that FcγRIIIb signaling, but not FcγRIIa signaling to increase [Ca2+]i in human neutrophils involves both PKC and NADPH-oxidase.
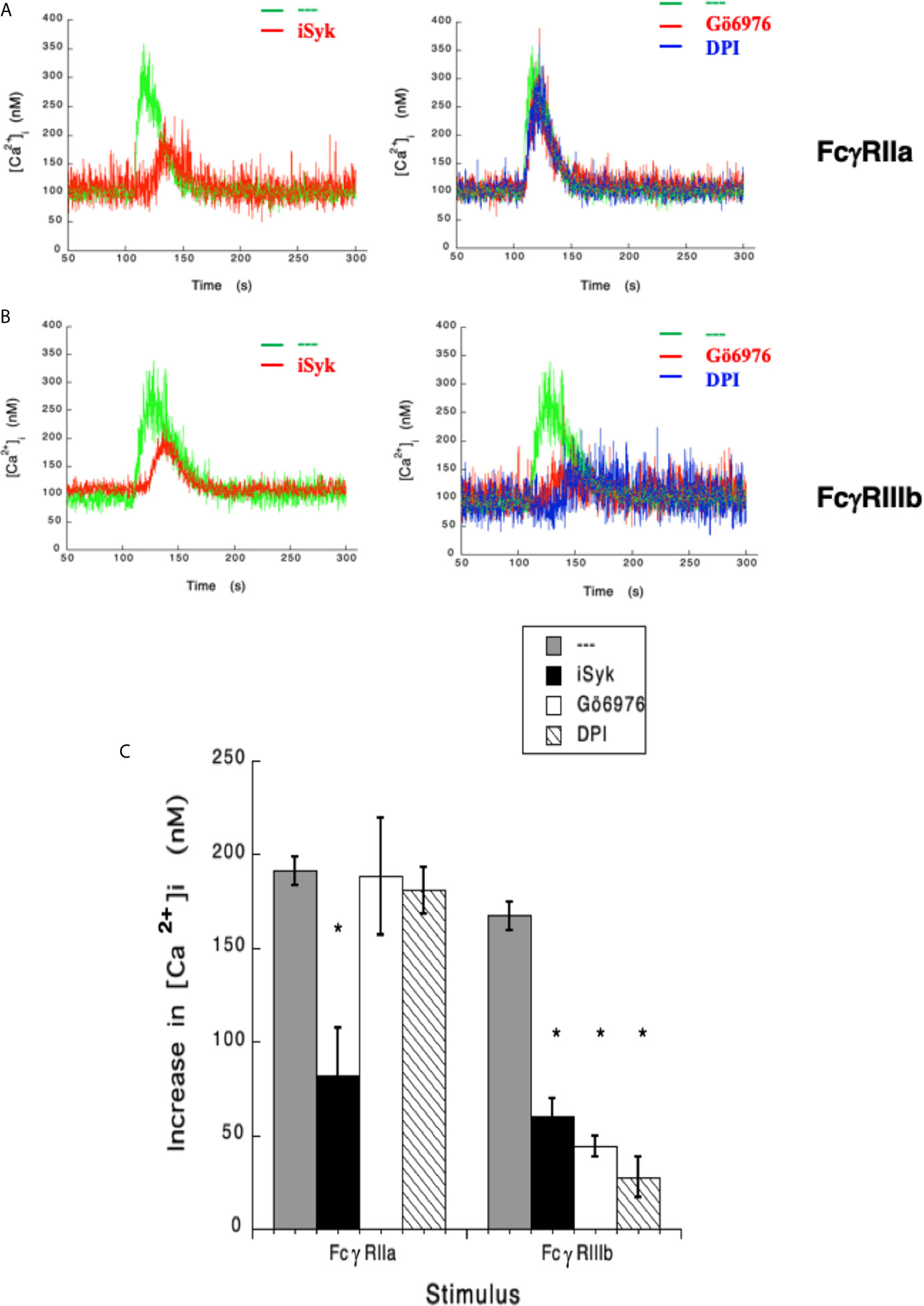
Figure 3 FcγRIIIb-mediated increase in [Ca2+]i. depends on Syk, PKC and NADPH-oxidase. Fura-2-loaded human neutrophils in PBS with Ca2+ and Mg2+ were left untreated (green line), or treated with 1 µM iSyk, a Syk inhibitor; 1 µM Gö6976, a PKC inhibitor; 10 µM DPI, a NADPH-oxidase inhibitor, before being stimulated by aggregating FcγRIIa (A) or FcγRIIIb (B). Arrow indicates the moment when the stimulus was added. Changes in cytosolic calcium concentration ([Ca2+]i) were assessed by measuring the variations in fluorescence as described in material and methods. Tracings are representative of three experiments with similar results. (C) Increments in [Ca2+]i were calculated by subtracting the baseline value from the maximum value after stimulation. Data are mean ± SEM of three independent experiments. Asterisks (*) denote conditions that were statistically different from untreated cells (p < 0.0001).
FcγRIIIb-Induced ROS Production Involves Syk and PKC
It has also been shown that NAPDH-oxidase is an enzymatic complex responsible for ROS production in human neutrophils (54, 55), and PKC is able to induce NAPDH-oxidase activation (56, 57). Because, FcγRIIIb signaling involves both PKC and NADPH-oxidase activation (12, 18), we explored whether FcγRIIIb requires PKC to induce ROS production. Neutrophils stimulated with phorbol 12-myristate 13-acetate (PMA), a strong PKC activator, produced a robust amount of ROS (Figure 4). Similarly, activation of FcγRIIa or FcγRIIIb resulted in significant ROS production, although smaller than the one induced by PMA (Figure 4). When cells were pre-treated with iSyk, a selective Syk inhibitor, PMA-induced ROS production was not affected (Figure 4). In contrast, iSyk reduced ROS production induced by either FcγRIIa or FcγRIIIb (Figure 4). Also, in the presence of Gö6976, a selective PKC inhibitor, PMA-induced ROS production was blocked (Figure 4). Similarly, Gö6976 completely prevented ROS production after aggregation of FcγRIIa or FcγRIIIb (Figure 4). These data suggested that FcγRIIa and FcγRIIIb both activate Syk, leading to PKC activation and ROS production. However, only in FcγRIIIb signaling these molecules are connected to a rise in [Ca2+]i (Figure 3C).
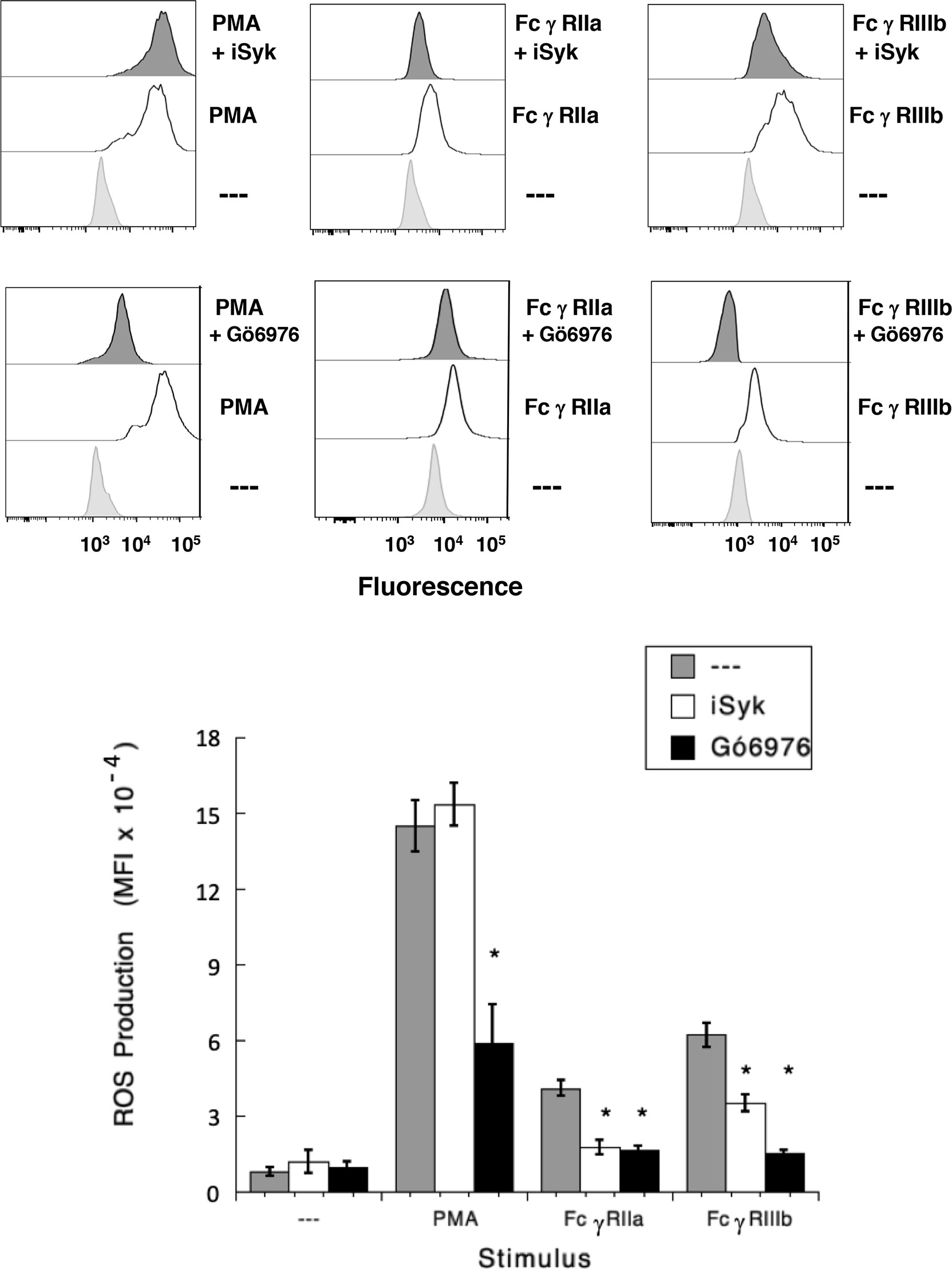
Figure 4 FcγRIIIb-induced ROS production involves Syk and PKC. Reactive Oxygen Species (ROS) production was assessed in dihydrorhodamine 123-loaded neutrophils by detecting fluoresce changes in flow cytometry. (Upper part) Neutrophils were left untreated (light gray), or were stimulated (white) with 20 nM PMA, or by aggregating FcγRIIa, or by aggregating FcγRIIIb. Some neutrophils were previously treated (dark gray) with 1 µM iSyk, a Syk inhibitor; or 1 µM Gö6976, a PKC inhibitor; before being stimulated. (Lower part) Cumulative data (mean ± SEM) of mean fluorescence intensity (MFI) from three independent experiments done in triplicate. Asterisk (*) denote condition that statistically different from untreated cells (p < 0.001).
Transient Potential Receptor Melastatin 2 (TRPM 2) Mediates FcγRIIIb-Induced Increase in [Ca2+]i
Results presented above suggested that FcγRIIIb induces PKC activation and ROS production, conducting to activation of a Ca2+ channel that allows Ca2+ influx into neutrophils. Several ion channels including different TRP family members (19, 21) could be involved in this response (58). In neutrophils several TRP channels were found to be expressed at the mRNA level by RT-PCR, including TRPC6, TRPM2, TRPV1, TRPV2, TRPV5 and TRPV6 (59). Of these channels, TRPM2 was demonstrated to mobilize Ca2+ in granulocytes (60), and to be activated by ROS (24, 27, 61, 62) and by PKC (28, 29). Based on these observations, we then explored whether TRPM2 was involved in FcγRIIIb-mediated increase in [Ca2+]i. Fcγ receptor-mediated increase in [Ca2+]i was evaluated in the presence of 2-APB (35) or clotrimazole (37), two different TRPM2 inhibitors. After FcγRIIa aggregation the increase in [Ca2+]i was not affected by the presence of 2-APB (Figure 5). In contrast, 2-APB efficiently inhibited the FcγRIIIb-mediated increase in [Ca2+]i (Figure 5). Similarly, the FcγRIIa-mediated [Ca2+]i rise was not affected by the presence of clotrimazole (Figure 5). However, clotrimazole also efficiently inhibited the FcγRIIIb-mediated increase in [Ca2+]i (Figure 5). Moreover, 2-APB could not inhibit ROS production induced by PMA (Figure 6) nor by FcγRIIa or FcγRIIIb (Figure 6). These data positioned production of ROS up-stream of TRPM2 activation and suggested that PKC and ROS are required for FcγRIIIb-induced activation of TRPM2 to promote Ca2+ influx in neutrophils (Figure 7).
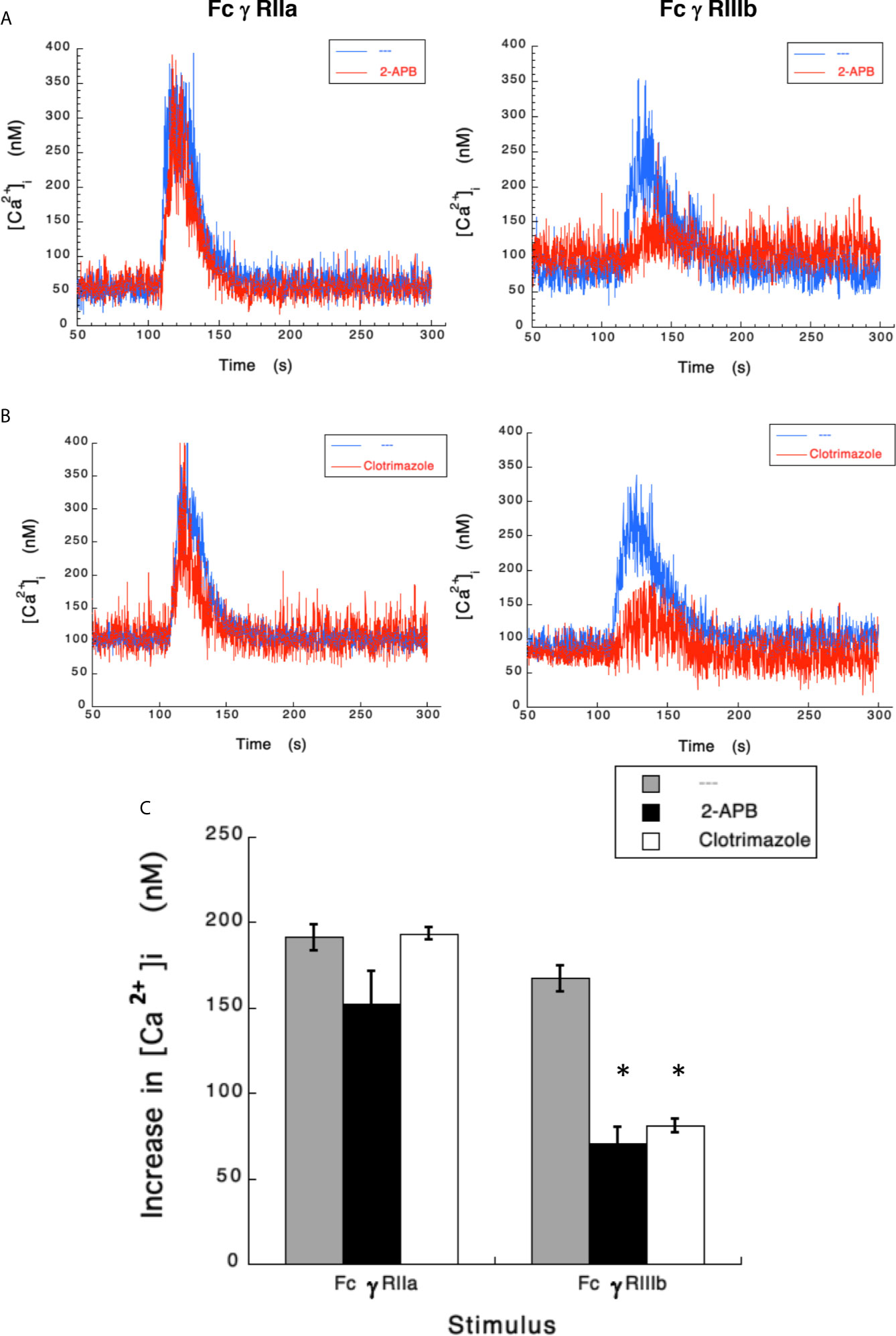
Figure 5 TRPM 2 channel mediates FcγRIIIb-induced increase in [Ca2+]i. Fura-2-loaded human neutrophils in PBS with Ca2+ and Mg2+ were left untreated (blue line), or treated (red line) with 5 µM 2-APB (A) or with 10 µM clotrimazole (B), two TRPM2 inhibitors, before being stimulated by aggregating FcγRIIa or FcγRIIIb. Changes in cytosolic calcium concentration ([Ca2+]i) were assessed by measuring the variations in fluorescence as described in material and methods. Tracings are representative of three experiments with similar results. (C) Increments in [Ca2+]i were calculated by subtracting the baseline value from the maximum value after stimulation. Data are mean ± SEM of three independent experiments. Asterisks (*) denote conditions that were statistically different from untreated cells (p < 0.0008).
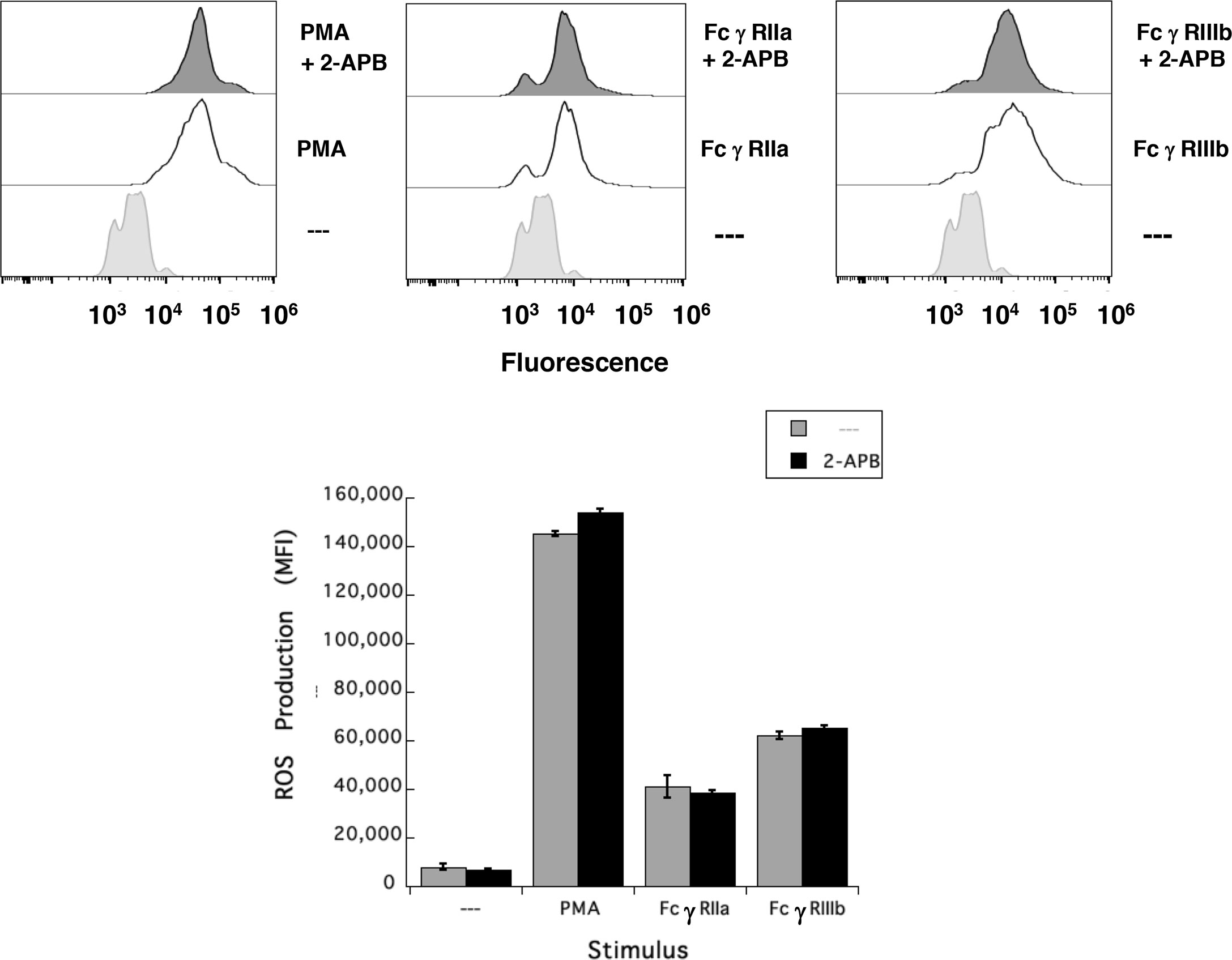
Figure 6 TRPM2 is not required for FcγRIIIb-induced ROS production. Reactive Oxygen Species (ROS) production was assessed in dihydrorhodamine 123-loaded neutrophils by detecting fluoresce changes by flow cytometry. Neutrophils were previously left untreated (—), or were treated with the 5 µM 2-APB, a TRPM2 inhibitor, before being stimulated with 20 nM PMA, or by aggregating FcγRIIa, or by aggregating FcγRIIIb. Data are mean ± SEM of mean fluorescence intensity (MFI) from three independent experiments done in triplicates.
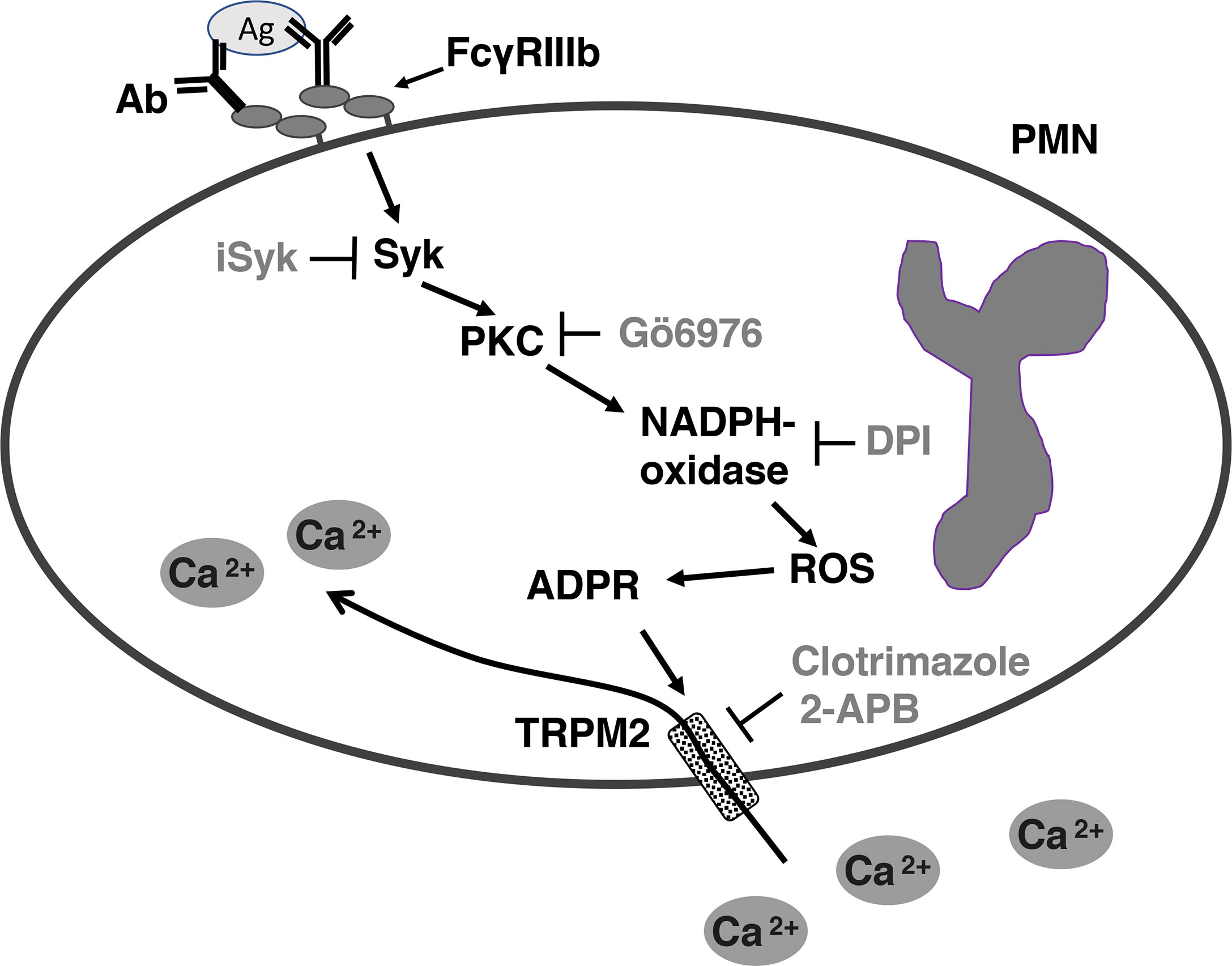
Figure 7 Model for FcγRIIIb-mediated increase in [Ca2+]i in human neutrophils (PMN). After aggregation of FcγRIIIb by antibody (Ab)/antigen (Ag) immune complexes, on the plasma membrane of neutrophils (PMN), spleen tyrosine kinase (Syk) gets activated, leading to protein kinase C (PKC) activation. PKC is then required for nicotinamide adenine dinucleotide phosphate oxidase (NADPH-oxidase) activation. NADPH-oxidase, an enzymatic complex assembled on a membrane (not shown), in turn produces reactive oxygen species (ROS), which induce adenosine diphosphate ribose (ADPR) finally leading to activation of transient receptor potential melastatin 2 (TRPM2) channels on the plasma membrane. Activated TRMP2 allow influx of extracellular Ca2+ into the cell. iSyk, an inhibitor of Syk; Gö6976, an inhibitor of PKC; DPI, an inhibitor of NADPH-oxidase; 2-APB, an inhibitor of TRPM2; Clotrimazole, another inhibitor of TRPM2.
Discussion
Neutrophils represent the most abundant leukocytes in blood and are considered the first line of defense against invading microorganisms because they are the first leukocytes to arrive at sites of inflammation and infection (63–65). At affected sites neutrophils display several antimicrobial functions (66) including degranulation, production of reactive oxygen species (ROS) (67, 68), phagocytosis (4) and the formation of neutrophil extracellular traps (NET) (5). In addition to these innate immune functions, neutrophils also participate in modulating the adaptive immune response (2). Initiation of these multiple cellular functions involves numerous receptors triggering a myriad of intracellular signaling pathways (52). In many of these pathways, changes in intracellular Ca2+ concentration ([Ca2+]i) are a prerequisite for neutrophil activation (52, 69). However, how changes in [Ca2+]i induced by the multitude of receptors expressed on these cells, control neutrophil activation and function remains only partially understood (58, 70, 71). In the case of antibody-mediated neutrophil responses (8, 9), it has been found that both human Fcγ receptors, FcγRIIa and FcγRIIIb, are capable of inducing an increase in [Ca2+]i (45, 72, 73). However, important differences on how each receptor mobilizes Ca2+ were reported since almost 30 years ago. While, FcγRIIa requires 1,4,5-inositol triphosphate (IP3) production for an increase in [Ca2+]i, the FcγRIIIb-mediated increase in [Ca2+]i is independent of IP3 (15). Now in this report, we show for the first time, that in human neutrophils stimulation of FcγRIIIb leads to TRPM2 activation to mediate an increase in [Ca2+]i.
Changes in [Ca2+]i are fundamental for the activation process of neutrophils (52, 69), and consequently Ca2+ fluxes for neutrophil responses are finely regulated in terms of temporal and spatial organization (58). In resting conditions, [Ca2+]i in neutrophils is around 100 nM, a level 10 000-fold lower than the concentration in the extracellular medium (74). Upon stimulation of neutrophils via various receptors such as G-protein coupled receptors (GPCRs) (75), integrins (76), or Fcγ receptors (45, 72) there is a rapid increase in [Ca2+]i caused by the release of Ca2+ from intracellular stores and/or by influx of extracellular Ca2+. Engagement of these receptors leads to activation of phospholipase C (PLC). GPCRs mainly activate the PLCβ2 and PLCβ3 (75), while integrins and Fcγ receptors activate PLCγ1 and PLCγ2 (52, 76). PLC in turn act on phosphatidylinositol 4,5 bisphosphate (PIP2) to generate diacylglycerol (DAG) and IP3. Binding of IP3 to its cognate receptor (IP3R), which also functions as a nonselective Ca2+ channel, localized on the membrane of the endoplasmic reticulum (ER), leads to a release of Ca2+ into the cytoplasm (41, 77). The initial rapid release of Ca2+ from the ER is followed by influx of Ca2+ across the plasma membrane. This influx is induced by the drop in Ca2+ levels inside the ER in a process that is known as store-operated calcium entry (SOCE) (78, 79). The mechanism for SOCE involves [Ca2+]i sensing proteins such as stromal interaction molecule 1 (STIM1), which migrates from the ER to the plasma membrane when intracellular stores are discharged (78). At the plasma membrane STIM1 gets together with the Ca2+ channel protein Orai1 (71, 80), which allows influx of extracellular Ca2+. In addition, it has been observed that after receptor stimulation there is also a Ca2+ influx that is dependent on receptor occupation by agonists and relatively store independent. This other mechanism of Ca2+ entry is known as receptor-operated calcium entry (ROCE) (81, 82). The molecular mechanisms controlling these two components of Ca2+ influx are still not resolved (83, 84).
Although the main mechanism for increasing [Ca2+]i in neutrophils is primarily mediated via IP3-dependent Ca2+ release from intracellular stores followed by SOCE activation of Orai1 channels (16), there is evidence that additional ion channels via ROCE are also involved in calcium influx into these cells (58). This has been clearly demonstrated for fMLF stimulation. After the initial increase in [Ca2+]i a second influx of extracellular Ca2+is observed. This influx is composed predominantly by Orai1 channels which are selective for Ca2+, and also by other non-selective channels that allow entry of both Ca2+ and strontium cations (Sr2+) (50). In phagocytes, the nature of Ca2+ channels mediating ROCE is just beginning to be identified.
An interesting group of nonselective ion channels that may participate in ROCE mechanisms in phagocytes is the superfamily of transient receptor potential (TRP) channels (85). TRP channels are expressed in many cell types and participate in multitude of physiological and pathological processes, such as cell proliferation, differentiation, and death (21). They are particularly important as biosensors of environmental and cellular stimuli including heat, mechanical force (pressure), oxidative (redox) status, and pH (21, 22, 86). The family of TRP channels is divided in six subfamilies: the ankyrin (TRPA), the canonical (TRPC), the melastatin (TRPM), the mucolipin (TRPML), the polycystin (TRPP) and the vanilloid (TRPV) subfamilies. Human and murine neutrophils only express members of the TRPC, TRPM and TRPV subfamilies (58), but their role in ROCE is unclear (81). In mast cells, a ROCE mechanism was described after antigen stimulation. The channel involved allowed influx of both external Ca2+ and Sr2+ to support degranulation, and was identified as TRPC5 (canonical transient receptor potential channel 5) (87). These reports opened the possibility that TRP channels may contribute to changes in [Ca2+]i after Fcγ receptor engagement in human neutrophils.
In phagocytic cells, it is clear that changes in [Ca2+]i are important for antibody-mediated cell responses such as phagocytosis (45, 72) and ROS production (67). It is generally accepted that Fcγ receptors activate a signaling cascade that involves Src family kinase-mediated phosphorylation of an ITAM sequence (7) in the cytoplasmic portion of the receptor (or its associated γ chains) (6, 8). The phosphorylated ITAM becomes a docking site for Syk, which in turn activates PLCγ1 to produce IP3 (17, 52). This pathway is certainly the one used by FcγRIIa to induce Ca2+ release from the ER (15). Then, a SOCE mechanism composed by STIM1 and Orai1 proteins is activated to generate a further influx of Ca2+ from outside the cell (16, 71). This influx of extracellular Ca2+ was found to be important for the intraphagosomal production of ROS during phagocytosis of opsonized yeast particles (51). We indeed found that selective aggregation of FcγRIIa produces an increase in [Ca2+]i resulting from release of Ca2+ from intracellular stores (Figure 1). For FcγRIIa it is clear that increases in [Ca2+]i involve IP3-mediated Ca2+ release from intracellular stores, followed by a SOCE mechanism that allows Ca2+ entry from outside the cell (71, 88).
For FcγRIIIb, elucidating the mechanism for initiating an increase in [Ca2+]i has been more complicated, since this receptor is expressed exclusively on human neutrophils, and it is a glycosylphosphatidylinositol (GPI)-linked receptor, lacking transmembrane and cytoplasmic domains (8, 9). Despite that initial steps in signaling for this receptor remain a mystery, it is clear that it can trigger signaling pathways leading to different neutrophil responses including activation of integrins (89), activation of transcription factors (11), and induction of NET formation (12, 13). In addition, FcγRIIIb induces an increase in [Ca2+]i (14, 45, 90), which is independent of IP3 (15). For a long time, it has been assumed that the initial rise in [Ca2+]i must be due to release of Ca2+ from intracellular stores by another mechanism that is independent of IP3. However, no such mechanism has yet been found. Sphingosine 1 phosphate (S1P), the product of sphingosine kinase (SK) is considered to be a mediator for changes in [Ca2+]i. In neutrophils, S1P formation was dependent on ER store depletion, and inhibition of SK resulted in a reduction of Ca2+ influx (91). Also, in glioblastoma cells it was reported that S1P could activate the TRP channel TRPC1, leading to Ca2+ influx (92). In both cases, S1P was found to mediate Ca2+ entry into the cells and not release of Ca2+ from intracellular stores. Therefore, it is unlikely that S1P mediates release of Ca2+ from intracellular stores in neutrophils.
Now, we report that when neutrophils were in Ca2+-free medium selective aggregation of FcγRIIIb did not cause any increase in [Ca2+]i (Figure 1). This result implied that the increase in [Ca2+]i was due to an influx of Ca2+ from outside the cell. This idea was confirmed when Ca2+ was restored in to the medium (Figure 1). This finding is in complete agreement with the lack of IP3 production when FcγRIIIb is engaged on neutrophils (15). It also pointed to the idea that a membrane ion channel was activated in response to FcγRIIIb aggregation. In previous reports, we described that FcγRIIIb triggers a signaling cascade that involves Syk, TAK1, MEK-ERK for induction of NETosis (18). In addition, we also reported that activation of PKC and production of ROS are important for NET formation (12). Thus, we explored whether these molecules could be involved in FcγRIIIb-mediated increase in [Ca2+]i. Indeed, we found that PKC and ROS are required for the influx of Ca2+ induced by FcγRIIIb (Figure 3).
Based on this, we turned our attention to TRP channels which are non-selective ion channels allowing transport of Ca2+ (85, 93), and capable (some of them) to sense the redox status in the cell (86, 94). Human neutrophils express the TRP channels: TRPC1, TRPC3, TRPC4, TRPC6, TRPM2, TRPV1, TRPV2, TRPV4, TRPV5, and TRPV6 (58, 59). Among these receptors, only TRPM2 (transient receptor potential melastatin type 2 cation channel; previously also named as TRPC7 or LRPC2) (20, 58) is also known to be activated by PKC (28, 29), and ROS (24, 95). In addition, TRPM2 has been found to be involved in several immune functions, including clearance of bacteria (96–98), NET formation by murine neutrophils (99), activation of NLRP3 inflammasome and secretion of interleukin-1β (100, 101), and dendritic cell maturation and chemotaxis (102). We confirmed that TRPM2 is responsible for FcγRIIIb-mediated rise in [Ca2+]i when two inhibitors, 2-APB (33–35, 95) and clotrimazole (36–39) completely blocked FcγRIIIb-mediated rise in [Ca2+]i (Figure 5). Both inhibitors 2-APB and clotrimazole have been used to block TRPM2 in many cell systems, however they are not specific inhibitors of this channel. Therefore, the possibility remains that other TRP channels may be involved in FcγRIIIb-mediated Ca2+ influx. Recently, novel and potentially more specific TRPM2 inhibitors have been reported (103, 104). It would be interesting to use these novel inhibitors to confirm our conclusions. However, we feel confident that TRPM2 is in fact the channel involved because it is the only TRP channel member on human neutrophils that is activated by PKC and ROS. In fact, the mechanism of activation involves adenosine diphosphate ribose (ADPR) binding to the C-terminal domain of the receptor, which presents strong homology to the human nucleotide diphosphate linked moiety X type (Nudix) hydrolase motif 9 (NUDT9) (23, 105). The NADPH-oxidase, like the mitochondrial oxidase, is a molecular complexes vectorially arranged on a membrane such that it accepts an electron from NADPH in the cytosol and transfers it across the membrane reducing oxygen to an oxygen radical (55, 68). The NADPH-oxidase is usually assembled on the phagosomal membrane, to generate ROS into the phagosome, or on the plasma membrane to generate extracellular ROS. Since, oxidative stress induces intracellular accumulation of ADPR it would also be interesting to confirm that aggregation of FcγRIIIb indeed causes an accumulation of ADPR. It is also important to establish whether ADPR is indeed coming from activation of the NADPH-oxidase or from mitochondria in response to oxidative stress. TRPM2 gating requires in addition to ADPR, binding of Ca2+ (61, 106, 107). This requirement for full opening of the channel has been very nicely revealed through analysis of cryo-electron microscopy structures of human and zebrafish TRPM2 [reviewed in (108, 109)]. In the case, of FcγRIIIb-mediated TRPM2 activation, we do not know if the basal [Ca2+]i is sufficient to support the full opening of the channel. In future experiments, we will eliminate intracellular calcium with BAPTA to further explore the mechanism for Ca2+ entry in neutrophils after FcγRIIIb engagement. Also, we will look at the functional consequence of the calcium rise on particular FcRIIIb-mediated neutrophil responses.
The involvement of PKC for inducing activation of TRPM2 comes from indirect studies using PMA (28, 29). In this report, we also demonstrated that indeed PKC is involved in FcγRIIIb-mediated TRPM2 activation using the specific PKC inhibitor Gö6976 (Figure 3). However, the particular isoform of PKC that is required for this function remains undetermined. Human neutrophils expressed PKC isoforms from each group of PKC enzymes (110). The inhibitor Gö6976 has specificity for Ca2+-dependent PKC isozymes alpha and beta (31). The most likely PKC isoform involved in this response may be PKCβ, since this isoform is an upstream mediator of NADPH-oxidase activation and was reported to be involved in NET formation (111); and we have also reported that FcγRIIIb is the main Fc receptor involved in NETosis (12). Future experiments will determine whether PKCβ is actually required for FcγRIIIb-mediated TRPM2 activation. Tremendous advances have taken place in the field of Ca2+ signaling in neutrophils in recent years. However, the literature in this area of research is rather controversial, as human and murine neutrophils and even human HL-60 cells do not always express the same ion channels on their membrane (58, 88). Thus, there is difficulty in integrating the different findings between species, and between cell lines and primary cells. Still, this field has a bright future since there are still many open questions on how calcium signals regulate neutrophil functions (70).
In conclusion, we have found that FcγRIIIb does not induce Ca2+ release from intracellular stores but it does activate, via PKC and ROS, the TRPM2 channel on the plasma membrane for inducing an influx of extracellular Ca2+ into human neutrophils (Figure 7).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Bioethics Committee at Instituto de Investigaciones Biomédicas – Universidad Nacional Autónoma de México (UNAM). The participants provided their written informed consent to participate in this study.
Author Contributions
OA performed most of the experiments and analyzed data. NM performed calcium measurements. CR designed the research, mentored other authors, performed statistical analysis, prepared figures, organized the references, and wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
Research was supported by grant 254434 from Consejo Nacional de Ciencia y Tecnología (CONACyT), Mexico, and by grant PAPIIT IN202520 from Dirección General de Asuntos del Personal Académico, Universidad Nacional Autónoma de México (UNAM), Mexico.
Conflict of Interest
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Liew PX, Kubes P. The Neutrophil’s Role During Health and Disease. Physiol Rev (2019) 99:1223–48. doi: 10.1152/physrev.00012.2018
2. Rosales C. Neutrophils At the Crossroads of Innate and Adaptive Immunity. J Leukoc Biol (2020) 108:377–96. doi: 10.1002/JLB.4MIR0220-574RR
3. Rosales C, Uribe-Querol E. Phagocytosis: A Fundamental Process in Immunity. Biomed Res Int (2017) 2017:9042851. doi: 10.1155/2017/9042851
4. Uribe-Querol E, Rosales C. Phagocytosis: Our Current Understading of a Universal Biological Process. Front Immunol (2020) 11:1066. doi: 10.3389/fimmu.2020.01066
5. Papayannopoulos V. Neutrophil Extracellular Traps in Immunity and Disease. Nat Rev Immunol (2018) 18:134–47. doi: 10.1038/nri.2017.105
6. Rosales C, Uribe-Querol E. Antibody - Fc Receptor Interactions in Antimicrobial Functions. Curr Immunol Rev (2013) 9:44–55. doi: 10.2174/1573395511309010006
7. Underhill DM, Goodridge HS. The Many Faces of ITAMs. Trends Immunol (2007) 28:66–73. doi: 10.1016/j.it.2006.12.004
8. Rosales C, Uribe-Querol E. Fc Receptors: Cell Activators of Antibody Functions. Adv Biosci Biotechnol (2013) 4:21–33. doi: 10.4236/abb.2013.44A004
9. Rosales C. Fcγ Receptor Heterogeneity in Leukocyte Functional Responses. Front Immunol (2017) 8:280. doi: 10.3389/fimmu.2017.00280
10. Rivas-Fuentes S, García-García E, Nieto-Castañeda G, Rosales C. Fcγ Receptors Exhibit Different Phagocytosis Potential in Human Neutrophils. Cell Immunol (2010) 263:114–21. doi: 10.1016/j.cellimm.2010.03.006
11. García-García E, Nieto-Castañeda G, Ruiz-Saldaña M, Mora N, Rosales C. FcγRIIA and FcγRIIIB Mediate Nuclear Factor Activation Through Separate Signaling Pathways in Human Neuthophils. J Immunol (2009) 182:4547–56. doi: 10.4049/jimmunol.0801468
12. Alemán OR, Mora N, Cortes-Vieyra R, Uribe-Querol E, Rosales C. Differential Use of Human Neutrophil Fcγ Receptors for Inducing Neutrophil Extracellular Trap Formation. J Immunol Res (2016) 2016:142643. doi: 10.1155/2016/2908034
13. Behnen M, Leschczyk C, Möller S, Batel T, Klinger M, Solbach W, et al. Immobilized Immune Complexes Induce Neutrophil Extracellular Trap Release by Human Neutrophil Granulocytes Via FcγRIIIB and Mac-1. J Immunol (2014) 193:1954–65. doi: 10.4049/jimmunol.1400478
14. Edberg JC, Moon JJ, Chang DJ, Kimberly RP. Differential Regulation of Human Neutrophil FcγRIIa (CD32) and FcγRIIIb (CD16)-Induced Ca2+ Transients. J Biol Chem (1998) 273:8071–9. doi: 10.1074/jbc.273.14.8071
15. Rosales C, Brown EJ. Signal Transduction by Neutrophil IgG Fc Receptors: Dissociation of [Ca+2] Rise From IP3. J Biol Chem (1992) 267:5265–71. doi: 10.1016/S0021-9258(18)42761-5
16. Clemens RA, Lowell CA. Store-Operated Calcium Signaling in Neutrophils. J Leukoc Biol (2015) 98:497–502. doi: 10.1189/jlb.2MR1114-573R
17. Rosales C. Fc Receptor and Integrin Signaling in Phagocytes. Signal Transduct (2007) 7:386–401. doi: 10.1002/sita.200700141
18. Alemán OR, Mora N, Cortes-Vieyra R, Uribe-Querol E, Rosales C. Transforming Growth Factor-β-Activated Kinase 1 is Required for Human FcγRIIIb-Induced Neutrophil Extracellular Trap Formation. Front Immunol (2016) 7:277. doi: 10.3389/fimmu.2016.00277
19. Najder K, Musset B, Lindemann O, Bulk E, Schwab A, Fels B. The Function of TRP Channels in Neutrophil Granulocytes. Pflugers Arch (2018) 470:1017–33. doi: 10.1007/s00424-018-2146-8
20. Sumoza-Toledo A, Penner R. TRPM2: A Multifunctional Ion Channel for Calcium Signaling. J Physiol (2011) 589:1515–25. doi: 10.1113/jphysiol.2010.201855
21. Flockerzi V, Nilius B. TRPs: Truly Remarkable Proteins. Handb Exp Pharmacol (2014) 222:1–12. doi: 10.1007/978-3-642-54215-2_1
22. Jimenez I, Prado Y, Marchant F, Otero C, Eltit F, Cabello-Verrugio C, et al. TRPM Channels in Human Diseases. Cells (2020) 9:2604. doi: 10.3390/cells9122604
23. Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, et al. ADP-Ribose Gating of the Calcium-Permeable LTRPC2 Channel Revealed by Nudix Motif Homology. Nature (2001) 411:595–9. doi: 10.1038/35079100
24. Syed Mortadza SA, Wang L, Li D, Jiang LH. TRPM2 Channel–Mediated ROS-sensitive Ca2+ Signaling Mechanisms in Immune Cells. Front Immunol (2015) 6:407. doi: 10.3389/fimmu.2015.00407
25. Wang G, Cao L, Liu X, Sieracki NA, Di A, Wen X, et al. Oxidant Sensing by TRPM2 Inhibits Neutrophil Migration and Mitigates Inflammation. Dev Cell (2016) 38:453–62. doi: 10.1016/j.devcel.2016.07.014
26. Du J, Xie J, Yue L. Modulation of TRPM2 by Acidic pH and the Underlying Mechanisms for pH Sensitivity. J Gen Physiol (2009) 134:471–88. doi: 10.1085/jgp.200910254
27. Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, et al. LTRPC2 Ca2+-permeable Channel Activated by Changes in Redox Status Confers Susceptibility to Cell Death. Mol Cell (2002) 9:163–73. doi: 10.1016/S1097-2765(01)00438-5
28. Nazıroğlu M. Activation of TRPM2 and TRPV1 Channels in Dorsal Root Ganglion by NADPH Oxidase and Protein Kinase C Molecular Pathways: A Patch Clamp Study. J Mol Neurosci (2017) 61:425–35. doi: 10.1007/s12031-017-0882-4
29. Nazıroğlu M, Özgül C. Vitamin E Modulates Oxidative Stress and Protein Kinase C Activator (PMA)-Induced TRPM2 Channel Gate in Dorsal Root Ganglion of Rats. J Bioenerg Biomembr (2013) 45:541–9. doi: 10.1007/s10863-013-9524-x
30. Keshari RS, Verma A, Barthwal MK, Dikshit M. Reactive Oxygen Species-Induced Activation of ERK and P38 MAPK Mediates PMA-induced Nets Release From Human Neutrophils. J Cell Biochem (2013) 114:532–40. doi: 10.1002/jcb.24391
31. Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, et al. Selective Inhibition of Protein Kinase C Isozymes by the Indolocarbazole Gö 6976. J Biol Chem (1993) 268:9194–7. doi: 10.1016/S0021-9258(18)98335-3
32. Asehnoune K, Strassheim D, Mitra S, Yeol Kim J, Abraham E. Involvement of PKCalpha/beta in TLR4 and TLR2 Dependent Activation of NF-KappaB. Cell Signal (2005) 17:385–94. doi: 10.1016/j.cellsig.2004.08.005
33. Chung MK, Asgar J, Lee J, Shim MS, Dumler C, Ro JY. The Role of TRPM2 in Hydrogen Peroxide-Induced Expression of Inflammatory Cytokine and Chemokine in Rat Trigeminal Ganglia. Neuroscience (2015) 297:160–9. doi: 10.1016/j.neuroscience.2015.03.067
34. Övey IS, Naziroğlu M. Homocysteine and Cytosolic GSH Depletion Induce Apoptosis and Oxidative Toxicity Through Cytosolic Calcium Overload in the Hippocampus of Aged Mice: Involvement of TRPM2 and TRPV1 Channels. Neuroscience (2015) 284:225–33. doi: 10.1016/j.neuroscience.2014.09.078
35. Togashi K, Inada H, Tominaga M. Inhibition of the Transient Receptor Potential Cation Channel TRPM2 by 2-Aminoethoxydiphenyl Borate (2-APB). Br J Pharmacol (2008) 153:1324–30. doi: 10.1038/sj.bjp.0707675
36. Eisfeld J, Lückhoff A. Trpm2. Handb Exp Pharmacol (2007) 179:237–52. doi: 10.1007/978-3-540-34891-7_14
37. Hill K, McNulty S, Randall AD. Inhibition of TRPM2 Channels by the Antifungal Agents Clotrimazole and Econazole. Naunyn Schmiedebergs Arch Pharmacol (2004) 370:227–37. doi: 10.1007/s00210-004-0981-y
38. Roberge S, Roussel J, Andersson DC, Meli AC, Vidal B, Blandel F, et al. TNF-α-Mediated Caspase-8 Activation Induces ROS Production and TRPM2 Activation in Adult Ventricular Myocytes. Cardiovasc Res (2014) 103:90–9. doi: 10.1093/cvr/cvu112
39. Ham HY, Hong CW, Lee SN, Kwon MS, Kim YJ, Song DK. Sulfur Mustard Primes Human Neutrophils for Increased Degranulation and Stimulates Cytokine Release Via TRPM2/p38 MAPK Signaling. Toxicol Appl Pharmacol (2012) 258:82–8. doi: 10.1016/j.taap.2011.10.010
40. García-García E, Uribe-Querol E, Rosales C. A Simple and Efficient Method to Detect Nuclear Factor Activation in Human Neutrophils by Flow Cytometry. J Vis Exp (2013) 74:e50410. doi: 10.3791/50410
41. Fruman DA, Gamache DA, Ernest MJ. Changes in Inositol 1,4,5-Trisphosphate Mass in Agonist-Stimulated Human Neutrophils. Agents Actions (1991) 34:16–9. doi: 10.1007/BF01993225
42. Krause KH, Schlegel W, Wollheim CB, Andersson T, Waldvogel FA, Lew PD. Chemotactic Peptide Activation of Human Neutrophils and HL-60 Cells. Pertussis Toxin Reveals Correlation Between Inositol Trisphosphate Generation, Calcium Ion Transients, and Cellular Activation. J Clin Invest (1985) 76:1348–54. doi: 10.1172/JCI112109
43. Looney RJ, Abraham GN, Anderson CL. Human Monocytes and U937 Cells Bear Two Distinct Fc Receptors for IgG. J Immunol (1986) 136:1641–7.
44. Fleit HB, Wright SD, Unkeless JC. Human Neutrophil Fc Gamma Receptor Distribution and Structure. Proc Natl Acad Sci USA (1982) 79:3275–9. doi: 10.1073/pnas.79.10.3275
45. Rosales C, Brown EJ. Two Mechanisms for IgG Fc Receptor-Mediated Phagocytosis by Human Neutrophils. J Immunol (1991) 146:3937–44.
46. Rosales C, Brown EJ. Calcium Channel Blockers Nifedipine and Diltiazem Inhibit Ca2+ Release From Intracellular Stores in Neutrophils. J Biol Chem (1992) 267:1443–8. doi: 10.1016/S0021-9258(18)45965-0
47. Grynkiewicz G, Poenie M, Tsien RY. A New Generation of Ca2+ Indicators With Greatly Improved Fluorescence Properties. J Biol Chem (1985) 260:3440–50. doi: 10.1016/S0021-9258(19)83641-4
48. Rosales C. Neutrophil: A Cell With Many Roles in Inflammation or Several Cell Types? Front Physiol (2018) 9:113. doi: 10.3389/fphys.2018.00113
49. Anderson R, Goolam Mahomed A. Calcium Efflux and Influx in F-Met-Leu-Phe (fMLP)-activated Human Neutrophils are Chronologically Distinct Events. Clin Exp Immunol (1997) 110:132–8. doi: 10.1046/j.1365-2249.1997.5051403.x
50. Cai C, Tang S, Wang X, Cai S, Meng X, Zou W, et al. Requirement for Both Receptor-Operated and Store-Operated Calcium Entry in N-formyl-methionine-leucine-phenylalanine-induced Neutrophil Polarization. Biochem Biophys Res Commun (2013) 430:816–21. doi: 10.1016/j.bbrc.2012.11.063
51. Steinckwich N, Schenten V, Melchior C, Bréchard S, Tschirhart EJ. An Essential Role of STIM1, Orai1, and S100A8-A9 Proteins for Ca2+ Signaling and FcγR-Mediated Phagosomal Oxidative Activity. J Immunol (2011) 186:2182–91. doi: 10.4049/jimmunol.1001338
52. Futosi K, Fodor S, Mócsai A. Neutrophil Cell Surface Receptors and Their Intracellular Signal Transduction Pathways. Int Immunopharmacol (2013) 17:638–50. doi: 10.1016/j.intimp.2013.06.034
53. Schepetkin IA, Khlebnikov AI, Kirpotina LN, Quinn MT. Antagonism of Human Formyl Peptide Receptor 1 With Natural Compounds and Their Synthetic Derivatives. Int Immunopharmacol (2016) 37:43–58. doi: 10.1016/j.intimp.2015.08.036
54. Nauseef WM. Identification and Quantitation of Superoxide Anion: Essential Steps in Elucidation of the Phagocyte “Respiratory Burst”. J Immunol (2014) 193:5357–8. doi: 10.4049/jimmunol.1402580
55. Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front Cell Infect Microbiol (2017) 7:373. doi: 10.3389/fcimb.2017.00373
56. Nauseef WM, Volpp BD, McCormick S, Leidal KG, Clark RA. Assembly of the Neutrophil Respiratory Burst Oxidase. Protein Kinase C Promotes Cytoskeletal and Membrane Association of Cytosolic Oxidase Components. J Biol Chem (1991) 266:5911–7. doi: 10.1016/S0021-9258(19)67684-2
57. Verhoeven AJ, Leusen JH, Kessels GC, Hilarius PM, de Bont DB, Liskamp RM. Inhibition of Neutrophil NADPH Oxidase Assembly by a Myristoylated Pseudosubstrate of Protein Kinase C. J Biol Chem (1993) 268:18593–8. doi: 10.1016/S0021-9258(17)46669-5
58. Immler R, Simon SI, Sperandio M. Calcium Signalling and Related Ion Channels in Neutrophil Recruitment and Function. Eur J Clin Invest (2018) 48:e12964. doi: 10.1111/eci.12964
59. Heiner I, Eisfeld J, Lückhoff A. Role and Regulation of TRP Channels in Neutrophil Granulocytes. Cell Calcium (2003) 33:533–40. doi: 10.1016/s0143-4160(03)00058-7
60. Heiner I, Eisfeld J, Halaszovich CR, Wehage E, Jüngling E, Zitt C, et al. Expression Profile of the Transient Receptor Potential (TRP) Family in Neutrophil Granulocytes: Evidence for Currents Through Long TRP Channel 2 Induced by ADP-ribose and NAD. Biochem J (2003) 371:1045–53. doi: 10.1042/BJ20021975
61. Naziroğlu M. New Molecular Mechanisms on the Activation of TRPM2 Channels by Oxidative Stress and ADP-Ribose. Neurochem Res (2007) 32:1990–2001. doi: 10.1007/s11064-007-9386-x
62. Li J, Gao Y, Bao X, Li F, Yao W, Feng Z, et al. TRPM2: A Potential Drug Target to Retard Oxidative Stress. Front Biosci (Landmark Ed) (2017) 22:1427–38. doi: 10.2741/4551
63. Borregaard N. Neutrophils, From Marrow to Microbes. Immunity (2010) 33:657–70. doi: 10.1016/j.immuni.2010.11.011
64. Mayadas TN, Cullere X, Lowell CA. The Multifaceted Functions of Neutrophils. Annu Rev Pathol (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
65. Fine N, Tasevski N, McCulloch CA, Tenenbaum HC, Glogauer M. The Neutrophil: Constant Defender and First Responder. Front Immunol (2020) 11:571085. doi: 10.3389/fimmu.2020.571085
66. Nauseef WM, Borregaard N. Neutrophils At Work. Nat Immunol (2014) 15:602–11. doi: 10.1038/ni.2921
67. Dupré-Crochet S, Erard M, Nüβe O. ROS Production in Phagocytes: Why, When, and Where? J Leukoc Biol (2013) 94:657–70. doi: 10.1189/jlb.1012544
68. Zeng MY, Miralda I, Armstrong CL, Uriarte SM, Bagaitkar J. The Roles of NADPH Oxidase in Modulating Neutrophil Effector Responses. Mol Oral Microbiol (2019) 34:27–38. doi: 10.1111/omi.12252
69. Nunes P, Demaurex N. The Role of Calcium Signaling in Phagocytosis. J Leukoc Biol (2010) 88:57–68. doi: 10.1189/jlb.0110028
70. Hann J, Bueb JL, Tolle F, Bréchard S. Calcium Signaling and Regulation of Neutrophil Functions: Still a Long Way to Go. J Leukoc Biol (2020) 107:285–97. doi: 10.1002/JLB.3RU0719-241R
71. Demaurex N, Nunes P. The Role of STIM and ORAI Proteins in Phagocytic Immune Cells. Am J Physiol Cell Physiol (2016) 310:C496–508. doi: 10.1152/ajpcell.00360.2015
72. Kimberly RP, Ahlstrom JW, Click ME, Edberg JC. The Glycosyl Phosphatidylinositol-Linked Fc Gamma RIII PMN Mediates Transmembrane Signaling Events Distinct From Fc Gamma RII. J Exp Med (1990) 171:1239–55. doi: 10.1084/jem.171.4.1239
73. Hundt M, Schmidt RE. The Glycosylphosphatidylinositol-Linked Fc Gamma Receptor III Represents the Dominant Receptor Structure for Immune Complex Activation of Neutrophils. Eur J Immunol (1992) 22:811–6. doi: 10.1002/eji.1830220327
74. Bagur R, Hajnóczky G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol Cell (2017) 66:780–8. doi: 10.1016/j.molcel.2017.05.028
75. Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-beta2 and -Beta3 and PI3Kgamma in Chemoattractant-Mediated Signal Transduction. Science (2000) 287:1046–9. doi: 10.1126/science.287.5455.1046
76. Jakus Z, Simon E, Frommhold D, Sperandio M, Mócsai A. Critical Role of Phospholipase Cgamma2 in Integrin and Fc Receptor-Mediated Neutrophil Functions and the Effector Phase of Autoimmune Arthritis. J Exp Med (2009) 206:577–93. doi: 10.1084/jem.20081859
77. Streb H, Irvine RF, Berridge MJ, Schulz I. Release of Ca2+ From a Nonmitochondrial Intracellular Store in Pancreatic Acinar Cells by Inositol-1,4,5-Trisphosphate. Nature (1983) 306:67–9. doi: 10.1038/306067a0
78. Várnai P, Hunyady L, Balla T. STIM and Orai: The Long-Awaited Constituents of Store-Operated Calcium Entry. Trends Pharmacol Sci (2009) 30:118–28. doi: 10.1016/j.tips.2008.11.005
79. Scrimgeour N, Litjens T, Ma L, Barritt GJ, Rychkov GY. Properties of Orai1 Mediated Store-Operated Current Depend on the Expression Levels of STIM1 and Orai1 Proteins. J Physiol (2009) 587:2903–18. doi: 10.1113/jphysiol.2009.170662
80. Lunz V, Romanin C, Frischauf I. STIM1 Activation of Orai1. Cell Calcium (2019) 77:29–38. doi: 10.1016/j.ceca.2018.11.009
81. DeHaven WI, Jones BF, Petranka JG, Smyth JT, Tomita T, Bird GS, et al. TRPC Channels Function Independently of STIM1 and Orai1. J Physiol (2009) 587:2275–98. doi: 10.1113/jphysiol.2009.170431
82. Salmon MD, Ahluwalia J. Pharmacology of Receptor Operated Calcium Entry in Human Neutrophils. Int Immunopharmacol (2011) 11:145–8. doi: 10.1016/j.intimp.2010.11.006
83. Putney JW, Steinckwich-Besançon N, Numaga-Tomita T, Davis FM, Desai PN, D’Agostin DM, et al. The Functions of Store-Operated Calcium Channels. Biochim Biophys Acta Mol Cell Res (2017) 1864:900–6. doi: 10.1016/j.bbamcr.2016.11.028
84. Lopez JJ, Jardin I, Albarrán L, Sanchez-Collado J, Cantonero C, Salido GM, et al. Molecular Basis and Regulation of Store-Operated Calcium Entry. Adv Exp Med Biol (2020) 1131:445–69. doi: 10.1007/978-3-030-12457-1_17
85. Nishida M, Hara Y, Yoshida T, Inoue R, Mori Y. TRP Channels: Molecular Diversity and Physiological Function. Microcirculation (2006) 13:535–50. doi: 10.1080/10739680600885111
86. Sakaguchi R, Mori Y. Transient Receptor Potential (TRP) Channels: Biosensors for Redox Environmental Stimuli and Cellular Status. Free Radic Biol Med (2020) 146:36–44. doi: 10.1016/j.freeradbiomed.2019.10.415
87. Ma HT, Peng Z, Hiragun T, Iwaki S, Gilfillan AM, Beaven MA. Canonical Transient Receptor Potential 5 Channel in Conjunction With Orai1 and STIM1 Allows Sr2+ Entry, Optimal Influx of Ca2+, and Degranulation in a Rat Mast Cell Line. J Immunol (2008) 180:2233–9. doi: 10.4049/jimmunol.180.4.2233
88. Demaurex N, Saul S. The Role of STIM Proteins in Neutrophil Functions. J Physiol (2018) 596:2699–708. doi: 10.1113/JP275639
89. Ortiz-Stern A, Rosales C. FcγRIIIB Stimulation Promotes β1 Integrin Activation in Human Neutrophils. J Leukoc Biol (2005) 77:787–99. doi: 10.1189/jlb.0504310
90. Marois L, Paré G, Vaillancourt M, Rollet-Labelle E, Naccache PH. Fcγ RIIIb Triggers Raft-Dependent Calcium Influx in IgG-mediated Responses in Human Neutrophils. J Biol Chem (2011) 286:3509–19. doi: 10.1074/jbc.M110.169516
91. Itagaki K, Hauser CJ. Sphingosine 1-Phosphate, a Diffusible Calcium Influx Factor Mediating Store-Operated Calcium Entry. J Biol Chem (2003) 278:27540–7. doi: 10.1074/jbc.M301763200
92. Lepannetier S, Zanou N, Yerna X, Emeriau N, Dufour I, Masquelier J, et al. Sphingosine-1-Phosphate-Activated TRPC1 Channel Controls Chemotaxis of Glioblastoma Cells. Cell Calcium (2016) 60:373–83. doi: 10.1016/j.ceca.2016.09.002
93. Gamberucci A, Giurisato E, Pizzo P, Tassi M, Giunti R, McIntosh DP, et al. Diacylglycerol Activates the Influx of Extracellular Cations in T-lymphocytes Independently of Intracellular Calcium-Store Depletion and Possibly Involving Endogenous TRP6 Gene Products. Biochem J (2002) 364:245–54. doi: 10.1042/bj3640245
94. Ogawa N, Kurokawa T, Mori Y. Sensing of Redox Status by TRP Channels. Cell Calcium (2016) 60:115–22. doi: 10.1016/j.ceca.2016.02.009
95. Nazıroğlu M. TRPM2 Cation Channels, Oxidative Stress and Neurological Diseases: Where are We Now? Neurochem Res (2011) 36:355–66. doi: 10.1007/s11064-010-0347-4
96. Knowles H, Heizer JW, Li Y, Chapman K, Ogden CA, Andreasen K, et al. Transient Receptor Potential Melastatin 2 (TRPM2) Ion Channel is Required for Innate Immunity Against Listeria Monocytogenes. Proc Natl Acad Sci USA (2011) 108:11578–83. doi: 10.1073/pnas.1010678108
97. Qian X, Zhao H, Chen X, Li J. Disruption of Transient Receptor Potential Melastatin 2 Decreases Elastase Release and Bacterial Clearance in Neutrophils. Innate Immun (2018) 24:122–30. doi: 10.1177/1753425918759181
98. Zhang Z, Cui P, Zhang K, Chen Q, Fang X. Transient Receptor Potential Melastatin 2 Regulates Phagosome Maturation and is Required for Bacterial Clearance in Escherichia coli sepsis. Anesthesiology (2017) 126:128–39. doi: 10.1097/ALN.0000000000001430
99. Tripathi JK, Sharma A, Sukumaran P, Sun Y, Mishra BB, Singh BB, et al. Oxidant Sensor Cation Channel TRPM2 Regulates Neutrophil Extracellular Trap Formation and Protects Against Pneumoseptic Bacterial Infection. FASEB J (2018) 32:6848–59. doi: 10.1096/fj.201800605
100. Wang L, Negro R, Wu H. TRPM2, Linking Oxidative Stress and Ca2+ Permeation to NLRP3 Inflammasome Activation. Curr Opin Immunol (2020) 62:131–5. doi: 10.1016/j.coi.2020.01.005
101. Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, et al. TRPM2 Links Oxidative Stress to NLRP3 Inflammasome Activation. Nat Commun (2013) 4:1611. doi: 10.1038/ncomms2608
102. Sumoza-Toledo A, Lange I, Cortado H, Bhagat H, Mori Y, Fleig A, et al. Dendritic Cell Maturation and Chemotaxis is Regulated by TRPM2-mediated Lysosomal Ca2+ Release. FASEB J (2011) 25:3529–42. doi: 10.1096/fj.10-178483
103. Cruz-Torres I, Backos DS, Herson PS. Characterization and Optimization of the Novel Transient Receptor Potential Melastatin 2 Antagonist TatM2NX. Mol Pharmacol (2020) 97:102–11. doi: 10.1124/mol.119.117549
104. Luo X, Li M, Zhan K, Yang W, Zhang L, Wang K, et al. Selective Inhibition of TRPM2 Channel by Two Novel Synthesized ADPR Analogues. Chem Biol Drug Des (2018) 91:552–66. doi: 10.1111/cbdd.13119
105. Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, et al. Accumulation of Free ADP-ribose From Mitochondria Mediates Oxidative Stress-Induced Gating of TRPM2 Cation Channels. J Biol Chem (2005) 280:6138–48. doi: 10.1074/jbc.M411446200
106. Fliegert R, Bauche A, Wolf Pérez AM, Watt JM, Rozewitz MD, Winzer R, et al. 2’-Deoxyadenosine 5’-Diphosphoribose is an Endogenous TRPM2 Superagonist. Nat Chem Biol (2017) 13:1036–44. doi: 10.1038/nchembio.2415
107. Yu P, Xue X, Zhang J, Hu X, Wu Y, Jiang LH, et al. Identification of the ADPR Binding Pocket in the NUDT9 Homology Domain of TRPM2. J Gen Physiol (2017) 149:219–35. doi: 10.1085/jgp.201611675
108. Kühn FJP. Structure-Function Relationship of TRPM2: Recent Advances, Contradictions, and Open Questions. Int J Mol Sci (2020) 21:6481. doi: 10.3390/ijms21186481
109. Xia S, Wang L, Fu TM, Wu H. Mechanism of TRPM2 Channel Gating Revealed by Cryo-EM. FEBS J (2019) 286:3333–9. doi: 10.1111/febs.14939
110. Balasubramanian N, Advani SH, Zingde SM. Protein Kinase C Isoforms in Normal and Leukemic Neutrophils: Altered Levels in Leukemic Neutrophils and Changes During Myeloid Maturation in Chronic Myeloid Leukemia. Leuk Res (2002) 26:67–81. doi: 10.1016/s0145-2126(01)00098-4
Keywords: neutrophil, Fc gamma receptor, calcium, reactive oxygen species, PKC (protein kinase C), TRPM2 cation channel
Citation: Alemán OR, Mora N and Rosales C (2021) The Antibody Receptor Fc Gamma Receptor IIIb Induces Calcium Entry via Transient Receptor Potential Melastatin 2 in Human Neutrophils. Front. Immunol. 12:657393. doi: 10.3389/fimmu.2021.657393
Received: 22 January 2021; Accepted: 23 April 2021;
Published: 13 May 2021.
Edited by:
Sonja Vermeren, University of Edinburgh, United KingdomReviewed by:
Lin-Hua Jiang, University of Leeds, United KingdomLlewelyn Roderick, KU Leuven, Belgium
Maurice Bartlett Hallett, Cardiff University, United Kingdom
Copyright © 2021 Alemán, Mora and Rosales. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos Rosales, Y2Fyb3NhbEB1bmFtLm14