 Fernanda O. Novais
Fernanda O. Novais Camila Farias Amorim
Camila Farias Amorim Phillip Scott
Phillip Scott- 1Department of Microbial Infection and Immunity, College of Medicine, The Ohio State University, Columbus, OH, United States
- 2Department of Pathobiology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA, United States
Cutaneous leishmaniasis exhibits a wide spectrum of clinical presentations from self-resolving infections to severe chronic disease. Anti-parasitic drugs are often ineffective in the most severe forms of the disease, and in some cases the magnitude of the disease can result from an uncontrolled inflammatory response rather than unrestrained parasite replication. In these patients, host-directed therapies offer a novel approach to improve clinical outcome. Importantly, there are many anti-inflammatory drugs with known safety and efficacy profiles that are currently used for other inflammatory diseases and are readily available to be used for leishmaniasis. However, since leishmaniasis consists of a wide range of clinical entities, mediated by a diverse group of leishmanial species, host-directed therapies will need to be tailored for specific types of leishmaniasis. There is now substantial evidence that host-directed therapies are likely to be beneficial beyond autoimmune diseases and cancer and thus should be an important component in the armamentarium to modulate the severity of cutaneous leishmaniasis.
Introduction
Cutaneous leishmaniasis is caused by several different species of protozoa transmitted by sand flies, and has a variety of clinical forms, ranging from self-healing lesions to chronic disfiguring mucosal disease (1, 2). There is no vaccine for the disease, and drug treatment is not always effective (3, 4). Moreover, in some forms of leishmaniasis the magnitude of the disease appears to be due to the uncontrolled inflammatory response at the cutaneous site of infection. It is clear that new therapeutic approaches are needed, and host-directed therapies to either enhance protective immune responses or to ameliorate excessive cutaneous inflammation represent novel therapeutic strategies worthy of pursuit.
Host-directed therapies for infectious diseases are designed to either amplify protective immune responses, divert non-protective immune responses towards protective responses, or block pathologic immune responses (5). Fortunately, our in-depth understanding of both protective and pathologic immune responses and identification of agents that can be used clinically to influence immune responses has revolutionized treatment of a wide range of diseases. While many of these new treatments are for non-communicable diseases, repurposing such treatments for infectious diseases, such as cutaneous leishmaniasis is advantageous, as their safety and efficacy profiles have often already been established.
In order to be successful, host-directed therapies must not overstimulate the immune response, or block protective immune responses necessary to control the pathogen. These are not theoretical possibilities. For example, checkpoint blockade has revolutionized cancer treatment, but some patients develop adverse events associated with these treatments, including cytokine storms that can be lethal (6, 7). Similarly, anti-inflammatory treatments run the risk of increased susceptibility to infections. Thus, the key to using host-directed therapy with infectious diseases is to lessen the chances of adverse events by defining the mechanisms mediating protection as well as those promoting immunopathologic responses associated with the disease. In cutaneous leishmaniasis there is a good understanding of the protective mechanisms, and thus one strategy is to promote those responses. Here we will review the host-directed therapies that could be used to enhance protection in patients. Many of the studies discussed focus on murine models where potential host-directed therapies can be assessed prior to initiation of clinical trials with patients.
We will also discuss what we know about destructive inflammation seen in patients with chronic cutaneous leishmaniasis and identify potential targets for therapies to promote disease resolution.
Spectrum of Clinical Presentations in Cutaneous Leishmaniasis
A challenging aspect in lessening disease in cutaneous leishmaniasis is the variety of clinical presentations associated with the infection. The type of clinical presentation is driven by the nature of the immune response invoked, which is influenced by both host genetics and the specific species or strain of the parasite causing the infection (1, 2). Following infection by a sand fly, patients develop a small nodule which progresses to an ulcerated lesion that will eventually heal in several months. However, in some cases, the lesions fail to resolve, or the parasites spread to many cutaneous sites without any evidence of control, a form of leishmaniasis known as diffuse cutaneous leishmaniasis (DCL) (8–10). These patients fail to develop a delayed-type hypersensitivity response or a strong IFN-γ response, and thus parasite burdens in the lesions are extremely high (9, 10). Histologically, these lesions appear as masses of macrophages with large numbers of intracellular parasites, and few infiltrating lymphocytes (10). It is clear that enhancing a protective immune response would be important for this disease.
At the other end of the spectrum, parasites can spread to the naso-oropharyngeal mucosa and cause extensive damage mediated by an uncontrolled immune response. This disease, termed mucosal leishmaniasis, is most often caused by L. braziliensis parasites and is refractory to anti-parasitic treatment. While the parasites are largely controlled by the immune response, there is a large infiltration of inflammatory cells into the lesions, suggesting that the damage is due to an overexuberant inflammatory response rather than uncontrolled parasite growth (11). While mucosal leishmaniasis is the most severe form of the disease at the inflammatory end of the spectrum, single lesions in patients infected by L. braziliensis can also be chronic, resistant to drug treatment, and associated with a severe inflammatory response with a low parasite burden in the lesions.
Patients who fail to develop a protective Th1 cell response develop disease, often in spite of a strong antibody response. This is most clearly observed in DCL patients (9). In contrast, patients with a strong Th1 cell response also develop severe disease, but in this case due to inflammation rather than massive parasite numbers (11). This spectrum is not unique to cutaneous leishmaniasis. For example, in another cutaneous disease, leprosy, the disease ranges from lepromatous leprosy in which there is an absence of a strong T cell response and no control of the bacteria to tuberculoid leprosy in which bacteria are scarce, and the immune response causes disease (12, 13). Unfortunately, drug treatment for cutaneous leishmaniasis patients with severe disease at either end of the spectrum can be ineffective, which provides support for considering alternative treatment strategies (8, 10, 14). However, what is clearly evident is that any host-directed therapy will need to take into consideration where a patient is on this immunologic spectrum.
Experimental models of cutaneous leishmaniasis have been critical for understanding the disease, and important in defining the mechanisms associated with T cell subset development. For example, infection of mice with Leishmania major helped define the factors driving CD4 Th1 and CD4 Th2 cell development and maintenance (15–17). These studies established the critical role of IFN-γ produced by CD4 T cells in protection, and the lack of a protective role for antibodies. In contrast, infection of BALB/c mice with L. major results in an uncontrolled infection, which is in part due to the development of a Th2 response. While these uncontrolled infections mimic some aspects of DCL (or visceral leishmaniasis), the role of IL-4 in promoting increased disease in patients is less clear than in murine models (18). Many studies have been done with L. major, but these do not represent the whole breadth of disease patterns that can be seen with other species of Leishmania. For example, while C57BL/6 mice resolve disease following infection with L. major, lesions induced by either L. amazonensis or L. mexicana infections do not resolve (19, 20). In these cases, susceptibility is linked with the failure to develop a strong Th1 response, rather than a Th2 response. Leishmania strain differences can also influence disease outcome. For example, the L. major Seidman strain causes a non-healing infection in C57BL/6 mice in spite of the development of a Th1 response (21, 22). Although all murine models have their limitations, many of these different host-parasite models are useful to assess host-directed therapies that can enhance immune responses. In contrast, fewer models have been available that mimic the excessive inflammatory responses associated with patients infected with L. braziliensis parasites (see below).
Enhancing Protection in Cutaneous Leishmaniasis by Host-Directed Therapies
Leishmania parasites replicate in myeloid cells, including macrophages, monocytes and dendritic cells. Control of the parasites is dependent upon activation of these cells by IFN-γ, leading to increased production of nitric oxide and/or reactive oxygen species, although the role of these molecules may vary depending upon the host and the parasite species (23–26). The primary source of IFN-γ that leads to protection in cutaneous leishmaniasis is the CD4 T cell, although CD8 T cells and NK cells can also contribute to protection (27–29). Once an infection has resolved, resident memory CD4 Th1 cells in the skin, central memory CD4 T cells and circulating effector CD4 Th1 cells maintained by persistent parasites provide protection against a secondary challenge (30, 31). Since resident memory Th1 cells can be maintained in the absence of persistent parasites, they are a good target for vaccine development. While we understand how the immune response can control these parasites, there are multiple mechanisms that can block or lessen the development of protective responses, which is why lesions often take so long to resolve. Defining these barriers to protection can provide targets for host-directed therapies in patients in whom limited Th1 responses develop.
A reasonable first line approach to promote healing is treatment with agents that directly increase protective immunity (Figure 1). One can define protective immunity in both experimental models and humans as the ability to protect against the development of disease, which may not lead to complete elimination of the parasites. While this protection may require IFN-γ, as discussed above it is also clear that IFN-γ by itself does not always lead to lack of disease.
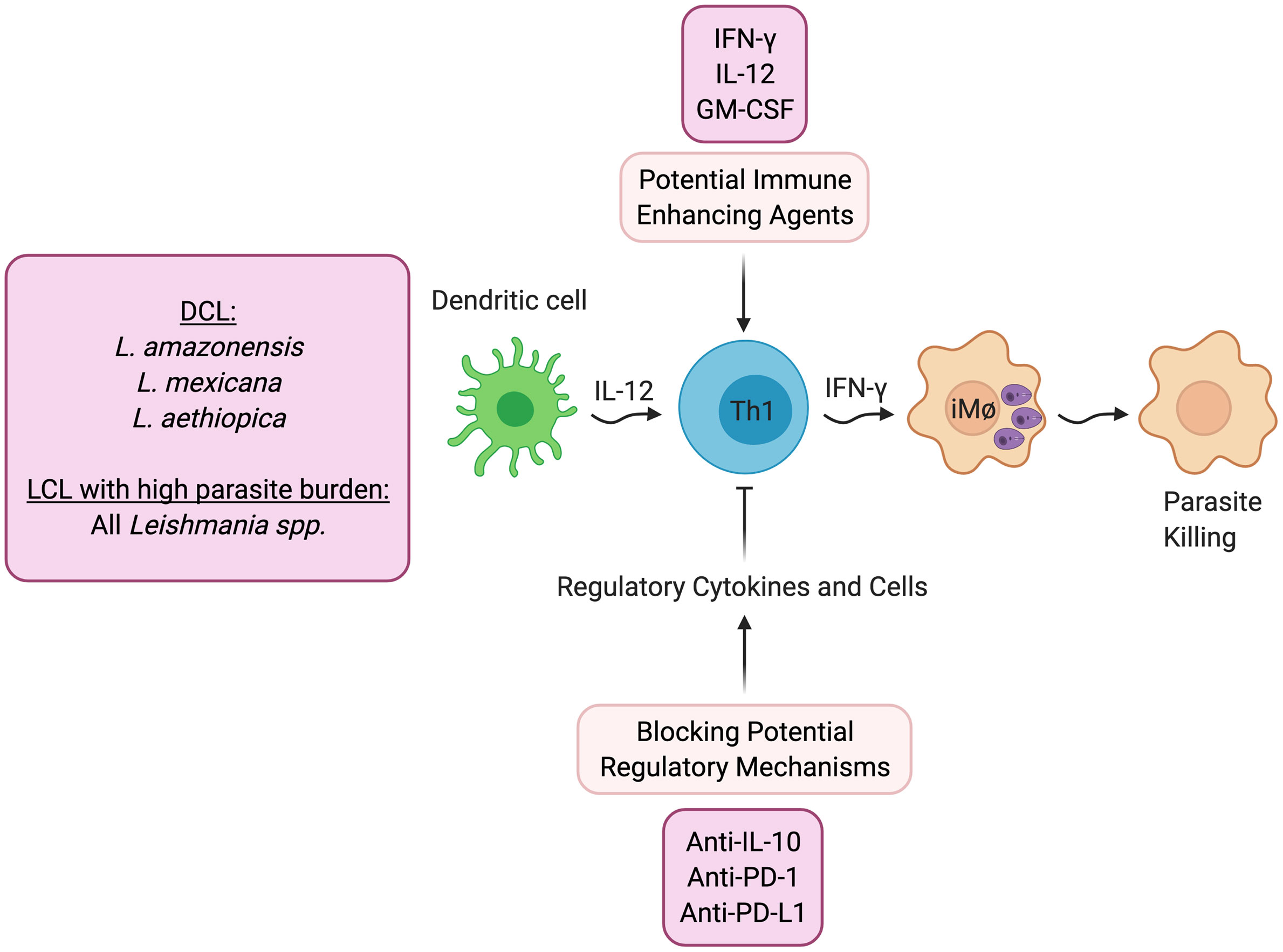
Figure 1 Host directed therapies that promote better parasite control. DCL- Diffuse cutaneous leishmaniasis; LCL- Localized cutaneous leishmaniasis.
As would be expected, treatment with IFN-γ has shown increased control in patients who are refractory to standard treatment (32, 33), and experimentally, IL-12 can promote healing even after lesions have developed when given in conjunction with an anti-parasitic drug (34, 35). In addition, clinical trials have been done with GM-CSF, in which topical treatment was found to promote increased healing (36, 37). Similarly, topical treatment with the TLR7 agonist imiquimod has shown increased healing rates (38), although there have been mixed results in clinical trials (39).
Alternatively, another potential therapeutic approach would be to block pathways that downregulate protective immunity (Figure 1). DCL patients fail to generate a protective IFN-γ response, and the pathology seen in these individuals is due to uncontrolled parasite growth in macrophages in the skin. While IL-4 blockade of protective responses can contribute to the uncontrolled Leishmania replication in experimental models, IL-4 appears to be less important in DCL patients (9) or indeed in any form of human leishmaniasis. Instead, a recent study suggests that DCL patients exhibit an overwhelming B cell response, and little evidence of either a Th1 or Th2 response (9). In contrast, IL-10 plays a critical role in promoting susceptibility to L. major in BALB/c mice, suggesting that blocking IL-10 might increase protective responses. Consistent with this possibility are studies in visceral leishmaniasis patients who can also develop uncontrolled infections. In these patients IL-10, rather than IL-4, has been linked with susceptibility. Importantly, a study with splenic aspirates from visceral leishmaniasis (VL) patients demonstrated that blockade of IL-10 enhanced control of the parasites (40), which provides the experimental foundation for a host-directed therapy where IL-10 would be blocked in VL patients (41). Experimentally, other regulatory cytokines have been shown to block protective Th1 responses in cutaneous leishmaniasis. For example, TGF-β inhibits protection in L. amazonensis infected mice (42), and IL-27 promotes IL-10 responses and increased susceptibility (43). Thus, blocking these regulatory pathways might promote better protective responses.
The role of inhibitory receptors in modulating the outcome of infectious diseases is an area of active investigation, since checkpoint blockade is effective in promoting control of cancer (44). One might predict, therefore, that blocking this regulatory pathway might be protective in cutaneous leishmaniasis as well. However, to date the experimental results in leishmaniasis are unclear. A study with arginase-deficient L. major in mice unable to resolve their infections found that anti-PD-1 monoclonal antibody promoted healing. However, blockade of PD-1 or PD-L1 in L. amazonensis infected mice (45) or infection of PD-L1 knockout mice with L. mexicana (46), had minimal effects on parasite control. A recent study found that T cells with an exhausted phenotype were present in the blood and lesions of L. braziliensis patients, and blocking PD-1 signaling in circulating T cells from patients enhanced their proliferation and production of IFN-γ (47). Clearly, more studies need to be done to understand the role of PD-1/PD-L1, as well as other checkpoint molecules, in cutaneous leishmaniasis.
Controlling Immunopathology in Cutaneous Leishmaniasis by Host-Directed Therapies
Enhancing Th1 responses directly or blocking pathways that lessen Th1 responses will not be effective for every type of cutaneous leishmaniasis. This is particularly true for patients at the immunopathologic end of the spectrum who develop chronic lesions in spite of their ability to generate a strong Th1 response. This clinical presentation is best exemplified by L. braziliensis infections, where chronic lesions are associated with a strong Th1 response and few parasites. While IFN-γ and TNF are important for macrophage activation and parasite control, in excess both cytokines can be associated with pathologic immune responses and it is possible that a poorly regulated Th1 response leading to high levels of IFN-γ and TNF contributes to this chronic inflammation. Moreover, since blocking TNF is a successful host-directed therapy for patients with rheumatoid arthritis, it is reasonable to consider its role in blocking pathology in cutaneous leishmaniasis. In support, a recent study suggests that TNF in L. mexicana infections promotes T cell exhaustion (48). While clinical trials have not yet been done with humanized monoclonal antibodies against TNF, the drug pentoxifylin, which blocks TNF, has been used in L. braziliensis, but with mixed results (49–51).
The optimal pathway to target in patients at the inflammatory end of the spectrum would be one that is not associated with protection. Notably, studies in L. braziliensis patients uncovered a major pathway leading to disease that was independent of protective immune responses. These studies found that cytolysis by CD8 T cells correlated with increased pathology in cutaneous leishmaniasis patients (23, 52–61). Importantly, these studies were followed up with the demonstration that patients who eventually fail drug therapy can be identified prior to treatment based upon expression level of genes associated with cytotoxicity (59).
The identification of CD8 T cells as drivers of disease was initially confusing, since CD8 T cells were protective in models of cutaneous leishmaniasis. For example, infection of CD8 deficient mice with low doses of L. major leads to susceptibility (28). The protective role of CD8 T cells appears to be mediated primarily by promoting Th1 responses in the draining lymph nodes (27, 28). This paradox was resolved by the finding that CD8 T cells in the lesions made little IFN-γ, but were instead cytolytic (53, 54, 56). The mechanisms involved in the differential function of CD8 T cells in the draining lymph nodes and cutaneous lesions has yet to be understood, although one factor may involve the lack of local signals in the lesions that would promote IFN-γ production by CD8 T cells (62). These results raised the question of how cytolytic CD8 T cells promote disease in cutaneous leishmaniasis. Based upon other infections, one might predict that killing of Leishmania-infected cells would lead to better parasite control. However, the evidence suggests that instead of killing the parasites, lysing the infected cell results in parasite dissemination, which then go on to infect other cells (54). Thus, cytolysis may be one mechanism that promotes metastasis in patients.
As the pathologic role for CD8 T cells is difficult to ascertain in standard experimental models of cutaneous leishmaniasis new models to define the mechanisms leading to CD8 T cell mediated pathology needed to be created. The most straightforward model was the adoptive transfer of CD8 T cells into RAG mice followed by infection with Leishmania (28, 54). Importantly, RAG mice infected with L. braziliensis do not develop substantial lesions over many weeks of infection, in spite of a large number of parasites present at the site of infection (54). These results, and previous studies in RAG mice (63), demonstrate the critical role T cells play in developing ulcerated lesions. RAG mice receiving CD4 and CD8 T cells developed small lesions and controlled parasite replication. In contrast, RAG mice that received CD8 T cells alone and were infected with L. braziliensis developed severe uncontrolled lesions (54). Surprisingly, the number of parasites in infected RAG mice and RAG mice that received CD8 T cells was the same, highlighting the critical role for CD8 T cells in immunopathology. This CD8 T cell dependent pathology required the cytotoxic molecule perforin, but not IFN-γ, since transfer of perforin deficient T cells to RAG mice failed to induce pathology, while IFN-γ -/- CD8 T cells did (54). In a complementary model, bystander cytolytic CD8 T cells were also found to promote increased disease, as mice that had resolved an infection with lymphocytic choriomeningitis virus (LCMV) developed more severe disease than mice that had not previously been infected with LCMV in response to Leishmania challenge weeks after viral clearance (64, 65). In this model, LCMV specific NKG2D expressing CD8 T cells were recruited to the cutaneous lesions non-specifically and mediated killing of targets expressing NKG2D ligands that were upregulated on cells in the lesions due to inflammation. The relevance of bystander CD8 T cells to human leishmaniasis is suggested by the finding that lesions from patients who have been infected with Toxoplasma contained Toxoplasma specific CD8 T cells (66). Thus, studies in both experimental models, as well as gene transcriptional analysis of lesions from patients, identified an immunopathologic pathway dependent upon cytolysis in cutaneous leishmaniasis.
The transcriptional analysis of lesions from patients provided clues as to how cytolysis might promote increased disease (23, 67). Not only were genes associated with cytolysis upregulated in lesions, but those associated with inflammasome activation, including NLRP3, Caspase 1 and IL-1β, were similarly upregulated. The immunopathologic pathway hypothesized from gene transcriptional analysis of lesions was confirmed using the experimental models of CD8 T cell mediated disease described above. Thus, CD8 T cell mediated disease could be blocked by inhibitors of NLRP3, such as MCC950 and glyburide, or blockade of IL-1β with the IL-1 receptor antagonist anakinra or with anti- IL-1β antibody treatment (56) (Figure 2). The pathologic role for IL-1β is not limited to situations where there is uncontrolled CD8 T cell mediated cytolysis. Others have shown that inflammasome-dependent IL-1β mediates the severe disease seen with a virulent L. major strain, and IL-1β administration can exacerbate disease following L. major and L. amazonensis infection (22, 68, 69). In addition, IL-1 serum levels correlate with increased disease severity in L. mexicana patients (70), and more serious disease was reported in mice lacking the natural inhibitor of IL-1β signaling (IL-1RA) (71). In the L. amazonensis model IL-1β was found to promote resistance, although these mice fail to resolve with or without IL-1β (72). IL-1β has many roles in the immune response, but the pathologic role of IL-1β in cutaneous leishmaniasis appears to be when the cytokine is in excess. Notably, because both the inflammasome and IL-1β are associated with many chronic diseases, including autoimmune diseases, cancer and cardiovascular diseases, a number of inhibitors designed to block this pathway are in clinical use or are in clinical trials that can be tested in cutaneous leishmaniasis.
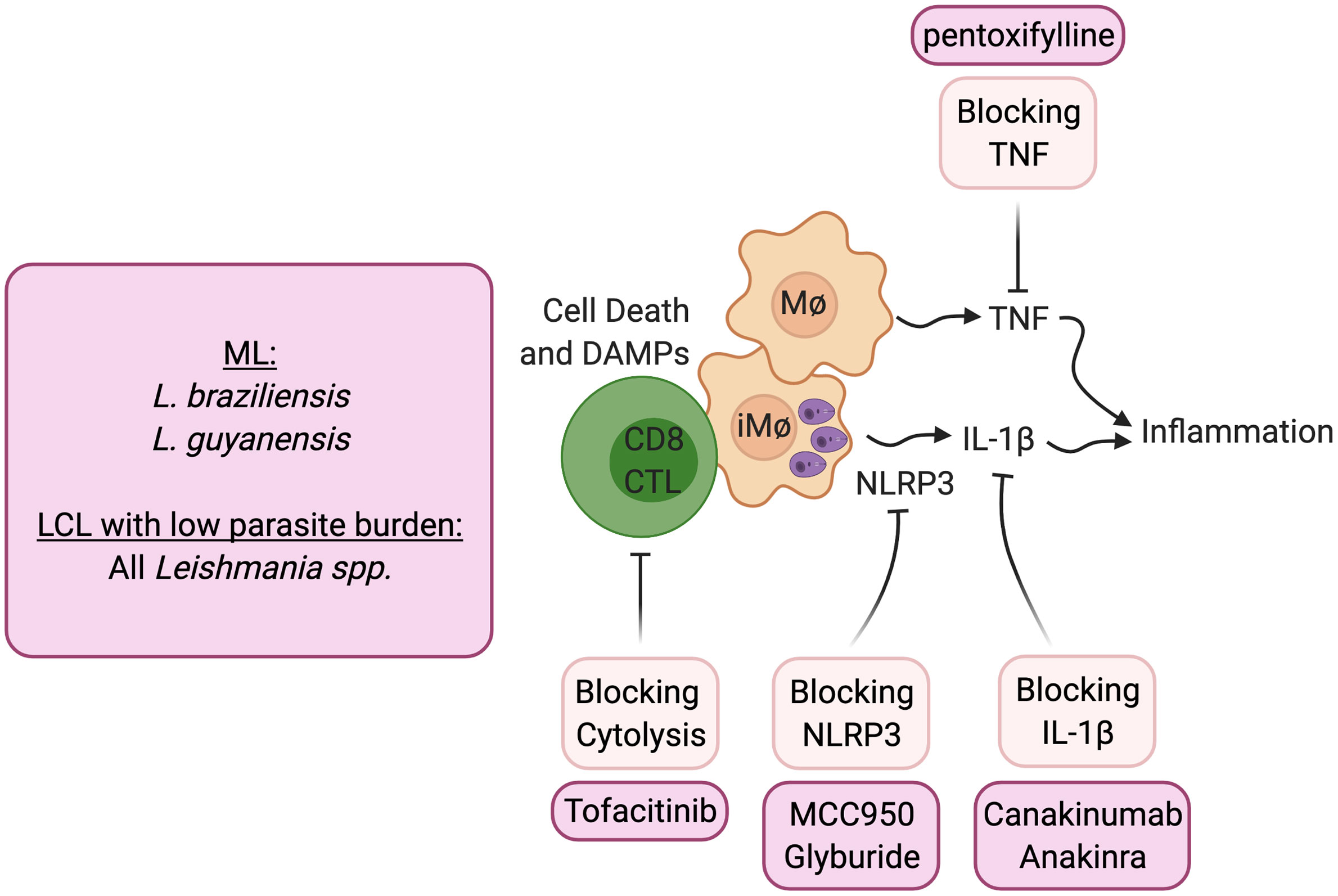
Figure 2 Host directed therapies that block immunopathologic mediated cytolysis. ML, mucosal leishmaniasis; LCL, Localized cutaneous leishmaniasis.
Blocking CD8 T cell cytotoxicity, an initiator of this pathway, could be another important target in lessening pathology. IL-15 is a potential target for such treatment, as it is highly expressed in lesions of human cutaneous leishmaniasis patients and promotes the expression of granzyme B dependent CD8 T cell cytotoxicity. Tofacitinib is a small molecule inhibitor of janus kinase (Jak)3 which is required for IL-15 signaling (73). It is currently being used clinically to treat certain types of arthritis under the trade name Xeljanz, and experimentally treats alopecia areata by blocking NKG2D dependent cytolysis (74). In experimental Leishmania models of CD8 T cell mediated pathology, systemic and topical treatment with tofacitinib blocked pathology (75). Notably, tofacitinib did not alter protective Th1 responses or parasite control. Thus, local targeting of CD8 T cell-mediated cytotoxicity can be a safe strategy to block immunopathologic responses locally while sparing protective responses.
Conclusions
Host-directed therapies hold great promise for lessening the more severe forms of cutaneous leishmaniasis. The ease of monitoring the efficacy of host-directed therapies in cutaneous diseases is a significant advantage to such treatments, and particularly important is the potential to develop topical treatments that may reduce untoward systemic responses. While in many diseases host-directed therapies are administered systemically, for those that might be used in cutaneous leishmaniasis it will be important to test whether topical application might be effective. One successful experimental example is the treatment with tofacitinib, which we found was as effective at controlling disease given topically as given systemically (75).
It is evident that care must be taken in the development of such therapies, as there remains the potential for blocking a pathway critical for control of Leishmania. Importantly, all of these therapies should be used in conjunction with standard anti-parasite drug treatment which lessens the risk of unchecked Leishmania multiplication. While increased susceptibility to other pathogens might remain, the short treatment period required would also lessen this risk. Finally, a practical consideration for developing therapies for neglected tropical diseases, such as cutaneous leishmaniasis, is the cost of treatment. Clearly the utility of any new host-directed therapy will depend on cost. However, identification of the targets for a successful host-directed therapy is the first step and can provide the rationale for a search for cheaper alternative treatments targeting the same immunologic pathways.
With the seeming endless development of new approaches to modulate the immune response with cytokines, small molecule inhibitors, humanized monoclonal antibodies, and drugs directed against immune targets, there is a growing interest in applying host-directed therapies to infectious diseases. Cutaneous diseases, such as leishmaniasis, can clearly benefit from such treatments. However, the key to success will be a continued focus on understanding the mechanisms leading to protective and pathologic responses in the skin, where many unanswered questions remain to be addressed. Most studies of cutaneous leishmaniasis have focused on systemic responses, or those occurring in local lymph nodes, and have ignored the unique aspects of the skin. Differences in cell types, metabolism, oxygen levels, and temperature can influence the outcome of cutaneous leishmaniasis, but have been little studied in this disease. Further, the skin directly interacts with the external environment and the skin microbiome can have significant effects on the outcome of infection (76, 77). It is fair to say that the success of host-directed therapies for cutaneous leishmaniasis will depend upon a better understanding of the skin, and for leishmaniasis we have just “scratched the surface” in that arena.
Author Contributions
FN and PS contributed to the conception and design of the review. FN, PS, and CA contributed to aspects of the review and writing. All authors contributed to the article and approved the submitted version.
Funding
The authors were supported funding from NIH grants R01-AI-150606 and R01-AI-149456.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Scott P, Novais FO. Cutaneous leishmaniasis: immune responses in protection and pathogenesis. Nat Rev Immunol (2016) 16:581–92. doi: 10.1038/nri.2016.72
2. Kaye P, Scott P. Leishmaniasis: complexity at the host-pathogen interface. Nat Rev Microbiol (2011) 9:604–15. doi: 10.1038/nrmicro2608
3. Carvalho AM, Guimarães LH, Costa R, Saldanha MG, Prates I, Carvalho LP, et al. Impaired Th1 response is associated therapeutic failure in patients with cutaneous leishmaniasis caused by Leishmania braziliensis. J Infect Dis (2020) 223:527–35. doi: 10.1093/infdis/jiaa374
4. Unger A, O’Neal S, Machado PRL, Guimarães LH, Morgan DJ, Schriefer A, et al. Association of treatment of American cutaneous leishmaniasis prior to ulcer development with high rate of failure in northeastern Brazil. Am J Trop Med Hyg (2009) 80:574–9. doi: 10.4269/ajtmh.2009.80.574
5. Kaufmann SHE, Dorhoi A, Hotchkiss RS, Bartenschlager R. Host-directed therapies for bacterial and viral infections. Nat Rev Drug Discovery (2018) 17:35–56. doi: 10.1038/nrd.2017.162
6. Burke KP, Grebinoski S, Sharpe AH, Vignali DAA. Understanding adverse events of immunotherapy: A mechanistic perspective. J Exp Med (2021) 218:1. doi: 10.1084/jem.20192179
7. Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol (2019) 16:563–80. doi: 10.1038/s41571-019-0218-0
8. Convit J, Kerdel-Vegas F. Disseminated cutaneous leishmaniasis; innoculation to laboratory animals, electron microscopy and fluorescent antibodies studies. Arch Dermatol (1965) 91:439–47. doi: 10.1001/archderm.1965.01600110025007
9. Christensen SM, Belew AT, El-Sayed NM, Tafuri WL, Silveira FT, Mosser DM. Host and parasite responses in human diffuse cutaneous leishmaniasis caused by L. amazonensis. PloS Negl Trop Dis (2019) 13:e0007152. doi: 10.1371/journal.pntd.0007152
10. Barral A, Costa JM, Bittencourt AL, Barral-Netto M, Carvalho EM. Polar and subpolar diffuse cutaneous leishmaniasis in Brazil: clinical and immunopathologic aspects. Int J Dermatol (1995) 34:474–9. doi: 10.1111/j.1365-4362.1995.tb00613.x
11. Saldanha MG, Pagliari C, Queiroz A, Machado PRL, Carvalho L, Scott P, et al. Tissue damage in human cutaneous leishmaniasis: correlations between inflammatory cells and molecule expression. Front Cell Infect Microbiol (2020) 10:355. doi: 10.3389/fcimb.2020.00355
12. Sansonetti P, Lagrange PH. The immunology of leprosy: speculations on the leprosy spectrum. Rev Infect Dis (1981) 3:422–69. doi: 10.1093/clinids/3.3.422
13. Ridley DS, Jopling WH. A classification of leprosy for research purposes. Lepr Rev (1962) 33:119–28. doi: 10.5935/0305-7518.19620014
14. García-Bustos MF, González-Prieto G, Pániz-Mondolfi AE, Parodi C, Beckar J, Monroig S, et al. Risk factors for antimony treatment failure in American Cutaneous Leishmaniasis in Northwestern-Argentina. PloS Negl Trop Dis (2021) 15:e0009003. doi: 10.1371/journal.pntd.0009003
15. Coffman RL, Varkila K, Scott P, Chatelain R. Role of cytokines in the differentiation of CD4+ T-cell subsets in vivo. Immunol Rev (1991) 123:189–207. doi: 10.1111/j.1600-065X.1991.tb00611.x
16. Locksley RM, Scott P. Helper T-cell subsets in mouse leishmaniasis: induction, expansion and effector function. Immunol Today (1991) 12:A58–61. doi: 10.1016/S0167-5699(05)80017-9
17. Sacks D, Noben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol (2002) 2:845–58. doi: 10.1038/nri933
18. Sacks D, Anderson C. Re-examination of the immunosuppressive mechanisms mediating non-cure of Leishmania infection in mice. Immunol Rev (2004) 201:225–38. doi: 10.1111/j.0105-2896.2004.00185.x
19. Alexander J, Kaye PM. Immunoregulatory pathways in murine leishmaniasis: different regulatory control during Leishmania mexicana mexicana and Leishmania major infections. Clin Exp Immunol (1985) 61:674–82.
20. Afonso LC, Scott P. Immune responses associated with susceptibility of C57BL/10 mice to Leishmania amazonensis. Infect Immun (1993) 61:2952–9. doi: 10.1128/IAI.61.7.2952-2959.1993
21. Anderson CF, Mendez S, Sacks DL. Nonhealing infection despite Th1 polarization produced by a strain of Leishmania major in C57BL/6 mice. J Immunol (2005) 174:2934–41. doi: 10.4049/jimmunol.174.5.2934
22. Charmoy M, Hurrell BP, Romano A, Lee SH, Ribeiro-Gomes F, Riteau N, et al. The Nlrp3 inflammasome, IL-1β, and neutrophil recruitment are required for susceptibility to a nonhealing strain of Leishmania major in C57BL/6 mice. Eur J Immunol (2016) 46:897–911. doi: 10.1002/eji.201546015
23. Novais FO, Carvalho LP, Passos S, Roos DS, Carvalho EM, Scott P, et al. Genomic profiling of human Leishmania braziliensis lesions identifies transcriptional modules associated with cutaneous immunopathology. J Invest Dermatol (2015) 135:94–101. doi: 10.1038/jid.2014.305
24. Glennie ND, Volk SW, Scott P. Skin-resident CD4+ T cells protect against Leishmania major by recruiting and activating inflammatory monocytes. PloS Pathog (2017) 13:e1006349. doi: 10.1371/journal.ppat.1006349
25. Roma EH, Macedo JP, Goes GR, Gonçalves JL, de CW, Cisalpino D, et al. Impact of reactive oxygen species (ROS) on the control of parasite loads and inflammation in Leishmania amazonensis infection. Parasit Vectors (2016) 9:193. doi: 10.1186/s13071-016-1472-y
26. Carneiro MBH, Roma EH, Ranson AJ, Doria NA, Debrabant A, Sacks DL, et al. NOX2-Derived Reactive Oxygen Species Control Inflammation during Leishmania amazonensis Infection by Mediating Infection-Induced Neutrophil Apoptosis. J Immunol (2018) 200:196–208. doi: 10.4049/jimmunol.1700899
27. Uzonna JE, Joyce KL, Scott P. Low dose Leishmania major promotes a transient T helper cell type 2 response that is down-regulated by interferon gamma-producing CD8+ T cells. J Exp Med (2004) 199:1559–66. doi: 10.1084/jem.20040172
28. Belkaid Y, Von Stebut E, Mendez S, Lira R, Caler E, Bertholet S, et al. CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J Immunol (2002) 168:3992–4000. doi: 10.4049/jimmunol.168.8.3992
29. Scharton-Kersten T, Afonso LC, Wysocka M, Trinchieri G, Scott P. IL-12 is required for natural killer cell activation and subsequent T helper 1 cell development in experimental leishmaniasis. J Immunol (1995) 154:5320–30.
30. Glennie ND, Yeramilli VA, Beiting DP, Volk SW, Weaver CT, Scott P. Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J Exp Med (2015) 212:1405–14. doi: 10.1084/jem.20142101
31. Peters NC, Pagán AJ, Lawyer PG, Hand TW, Henrique Roma E, Stamper LW, et al. Chronic parasitic infection maintains high frequencies of short-lived Ly6C+CD4+ effector T cells that are required for protection against re-infection. PloS Pathog (2014) 10:e1004538. doi: 10.1371/journal.ppat.1004538
32. Kolde G, Luger T, Sorg C, Sunderkotter C. Successful treatment of cutaneous leishmaniasis using systemic interferon-gamma. Dermatol (Basel) (1996) 192:56–60. doi: 10.1159/000246316
33. Falcoff E, Taranto NJ, Remondegui CE, Dedet JP, Canini LM, Ripoll CM, et al. Clinical healing of antimony-resistant cutaneous or mucocutaneous leishmaniasis following the combined administration of interferon-gamma and pentavalent antimonial compounds. Trans R Soc Trop Med Hyg (1994) 88:95–7. doi: 10.1016/0035-9203(94)90518-5
34. Heinzel FP, Schoenhaut DS, Rerko RM, Rosser LE, Gately MK. Recombinant interleukin 12 cures mice infected with Leishmania major. J Exp Med (1993) 177:1505–9. doi: 10.1084/jem.177.5.1505
35. Nabors GS, Afonso LC, Farrell JP, Scott P. Switch from a type 2 to a type 1 T helper cell response and cure of established Leishmania major infection in mice is induced by combined therapy with interleukin 12 and Pentostam. Proc Natl Acad Sci USA (1995) 92:3142–6. doi: 10.1073/pnas.92.8.3142
36. Santos JB, de Jesus AR, Machado PR, Magalhães A, Salgado K, Carvalho EM, et al. Antimony plus recombinant human granulocyte-macrophage colony-stimulating factor applied topically in low doses enhances healing of cutaneous Leishmaniasis ulcers: a randomized, double-blind, placebo-controlled study. J Infect Dis (2004) 190:1793–6. doi: 10.1086/424848
37. Almeida RP, Brito J, Machado PL, DE Jesus AR, Schriefer A, Guimarães LH, et al. Successful treatment of refractory cutaneous leishmaniasis with GM-CSF and antimonials. Am J Trop Med Hyg (2005) 73:79–81.
38. Miranda-Verástegui C, Llanos-Cuentas A, Arévalo I, Ward BJ, Matlashewski G. Randomized, double-blind clinical trial of topical imiquimod 5% with parenteral meglumine antimoniate in the treatment of cutaneous leishmaniasis in Peru. Clin Infect Dis (2005) 40:1395–403. doi: 10.1086/429238
39. Berbert TRN, de Mello TFP, Wolf Nassif P, Mota CA, Silveira AV, Duarte GC, et al. Pentavalent Antimonials Combined with Other Therapeutic Alternatives for the Treatment of Cutaneous and Mucocutaneous Leishmaniasis: A Systematic Review. Dermatol Res Pract (2018) 2018:9014726. doi: 10.1155/2018/9014726
40. Gautam S, Kumar R, Maurya R, Nylén S, Ansari N, Rai M, et al. IL-10 neutralization promotes parasite clearance in splenic aspirate cells from patients with visceral leishmaniasis. J Infect Dis (2011) 204:1134–7. doi: 10.1093/infdis/jir461
41. Faleiro RJ, Kumar R, Bunn PT, Singh N, Chauhan SB, Sheel M, et al. Combined immune therapy for the treatment of visceral leishmaniasis. PloS Negl Trop Dis (2016) 10:e0004415. doi: 10.1371/journal.pntd.0004415
42. Barral-Netto M, Barral A, Brownell CE, Skeiky YA, Ellingsworth LR, Twardzik DR, et al. Transforming growth factor-beta in leishmanial infection: a parasite escape mechanism. Science (1992) 257:545–8. doi: 10.1126/science.1636092
43. Anderson CF, Stumhofer JS, Hunter CA, Sacks D. IL-27 regulates IL-10 and IL-17 from CD4+ cells in nonhealing Leishmania major infection. J Immunol (2009) 183:4619–27. doi: 10.4049/jimmunol.0804024
44. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
45. da Fonseca-Martins AM, Ramos TD, Pratti JES, Firmino-Cruz L, Gomes DCO, Soong L, et al. Immunotherapy using anti-PD-1 and anti-PD-L1 in Leishmania amazonensis-infected BALB/c mice reduce parasite load. Sci Rep (2019) 9:20275. doi: 10.1038/s41598-019-56336-8
46. Liang SC, Greenwald RJ, Latchman YE, Rosas L, Satoskar A, Freeman GJ, et al. PD-L1 and PD-L2 have distinct roles in regulating host immunity to cutaneous leishmaniasis. Eur J Immunol (2006) 36:58–64. doi: 10.1002/eji.200535458
47. Garcia De Moura R, Covre LP, Fantecelle CH, Gajardo VAT, Cunha CB, Stringari LL, et al. PD-1 blockade modulates functional activities of exhausted-like T cell in patients with cutaneous leishmaniasis. Front Immunol (2021), 1–12. doi: 10.3389/fimmu.2021.632667
48. González-Tafoya E, Diupotex M, Zamora-Chimal J, Salaiza-Suazo N, Ruiz-Remigio A, Becker I. TNF contributes to T-cell exhaustion in chronic L. mexicana infections of mice through PD-L1 up-regulation. Cell Immunol (2020) 358:104196. doi: 10.1016/j.cellimm.2020.104196
49. Machado PRL, Lessa H, Lessa M, Guimarães LH, Bang H, Ho JL, et al. Oral pentoxifylline combined with pentavalent antimony: a randomized trial for mucosal leishmaniasis. Clin Infect Dis (2007) 44:788–93. doi: 10.1086/511643
50. Brito G, Dourado M, Guimarães LH, Meireles E, Schriefer A, de Carvalho EM, et al. Oral Pentoxifylline Associated with Pentavalent Antimony: A Randomized Trial for Cutaneous Leishmaniasis. Am J Trop Med Hyg (2017) 96:1155–9. doi: 10.4269/ajtmh.16-0435
51. Lessa HA, Machado P, Lima F, Cruz AA, Bacellar O, Guerreiro J, et al. Successful treatment of refractory mucosal leishmaniasis with pentoxifylline plus antimony. Am J Trop Med Hyg (2001) 65:87–9. doi: 10.4269/ajtmh.2001.65.87
52. Brodskyn CI, Barral A, Boaventura V, Carvalho E, Barral-Netto M. Parasite-driven in vitro human lymphocyte cytotoxicity against autologous infected macrophages from mucosal leishmaniasis. J Immunol (1997) 159:4467–73.
53. Faria DR, Souza PEA, Durães FV, Carvalho EM, Gollob KJ, Machado PR, et al. Recruitment of CD8(+) T cells expressing granzyme A is associated with lesion progression in human cutaneous leishmaniasis. Parasite Immunol (2009) 31:432–9. doi: 10.1111/j.1365-3024.2009.01125.x
54. Novais FO, Carvalho LP, Graff JW, Beiting DP, Ruthel G, Roos DS, et al. Cytotoxic T cells mediate pathology and metastasis in cutaneous leishmaniasis. PloS Pathog (2013) 9:e1003504. doi: 10.1371/journal.ppat.1003504
55. Novais FO, Scott P. CD8+ T cells in cutaneous leishmaniasis: the good, the bad, and the ugly. Semin Immunopathol (2015) 37:251–9. doi: 10.1007/s00281-015-0475-7
56. Novais FO, Carvalho AM, Clark ML, Carvalho LP, Beiting DP, Brodsky IE, et al. CD8+ T cell cytotoxicity mediates pathology in the skin by inflammasome activation and IL-1β production. PloS Pathog (2017) 13:e1006196. doi: 10.1371/journal.ppat.1006196
57. Santos C da S, Boaventura V, Ribeiro Cardoso C, Tavares N, Lordelo MJ, Noronha A, et al. CD8(+) granzyme B(+)-mediated tissue injury vs. CD4(+)IFNγ(+)-mediated parasite killing in human cutaneous leishmaniasis. J Invest Dermatol (2013) 133:1533–40. doi: 10.1038/jid.2013.4
58. Cardoso TM, Machado Á, Costa DL, Carvalho LP, Queiroz A, Machado P, et al. Protective and pathological functions of CD8+ T cells in Leishmania braziliensis infection. Infect Immun (2015) 83:898–906. doi: 10.1128/IAI.02404-14
59. Amorim CF, Novais FO, Nguyen BT, Misic AM, Carvalho LP, Carvalho EM, et al. Variable gene expression and parasite load predict treatment outcome in cutaneous leishmaniasis. Sci Transl Med (2019) 11:eaax4204. doi: 10.1126/scitranslmed.aax4204
60. Campos TM, Novais FO, Saldanha M, Costa R, Lordelo M, Celestino D, et al. Granzyme B produced by natural killer cells enhances inflammatory response and contributes to the immunopathology of cutaneous leishmaniasis. J Infect Dis (2020) 221:973–82. doi: 10.1093/infdis/jiz538
61. Covre LP, Devine O, Garcia de Moura R, Vukmanovic-Stejic M, Dietze R, Rodrigues RR, et al. Compartmentalized cytotoxic immune response leads to distinct pathogenic roles of natural killer and senescent CD8+ T cells in human cutaneous leishmaniasis. Immunology (2020) 159:429–40. doi: 10.1111/imm.13173
62. Novais FO, Wong AC, Villareal DO, Beiting DP, Scott P. CD8+ T Cells Lack Local Signals To Produce IFN-γ in the Skin during Leishmania Infection. J Immunol (2018) 200:1737–45. doi: 10.4049/jimmunol.1701597
63. Soong L, Chang CH, Sun J, Longley BJ, Ruddle NH, Flavell RA, et al. Role of CD4+ T cells in pathogenesis associated with Leishmania amazonensis infection. J Immunol (1997) 158:5374–83.
64. Crosby EJ, Goldschmidt MH, Wherry EJ, Scott P. Engagement of NKG2D on bystander memory CD8 T cells promotes increased immunopathology following Leishmania major infection. PloS Pathog (2014) 10:e1003970. doi: 10.1371/journal.ppat.1003970
65. Crosby EJ, Clark M, Novais FO, Wherry EJ, Scott P. Lymphocytic Choriomeningitis Virus Expands a Population of NKG2D+CD8+ T Cells That Exacerbates Disease in Mice Coinfected with Leishmania major. J Immunol (2015) 195:3301–10. doi: 10.4049/jimmunol.1500855
66. Da-Cruz AM, Oliveira-Neto MP, Bertho AL, Mendes-Aguiar CO, Coutinho SG. T cells specific to leishmania and other nonrelated microbial antigens can migrate to human leishmaniasis skin lesions. J Invest Dermatol (2010) 130:1329–36. doi: 10.1038/jid.2009.428
67. Christensen SM, Dillon LAL, Carvalho LP, Passos S, Novais FO, Hughitt VK, et al. Meta-transcriptome Profiling of the Human-Leishmania braziliensis Cutaneous Lesion. PloS Negl Trop Dis (2016) 10:e0004992. doi: 10.1371/journal.pntd.0004992
68. Xin L, Li Y, Soong L. Role of interleukin-1beta in activating the CD11c(high) CD45RB- dendritic cell subset and priming Leishmania amazonensis-specific CD4+ T cells in vitro and in vivo. Infect Immun (2007) 75:5018–26. doi: 10.1128/IAI.00499-07
69. Voronov E, Dotan S, Gayvoronsky L, White RM, Cohen I, Krelin Y, et al. IL-1-induced inflammation promotes development of leishmaniasis in susceptible BALB/c mice. Int Immunol (2010) 22:245–57. doi: 10.1093/intimm/dxq006
70. Fernández-Figueroa EA, Rangel-Escareño C, Espinosa-Mateos V, Carrillo-Sánchez K, Salaiza-Suazo N, Carrada-Figueroa G, et al. Disease severity in patients infected with Leishmania mexicana relates to IL-1β. PloS Negl Trop Dis (2012) 6:e1533. doi: 10.1371/journal.pntd.0001533
71. Kautz-Neu K, Kostka SL, Dinges S, Iwakura Y, Udey MC, von Stebut E. A role for leukocyte-derived IL-1RA in DC homeostasis revealed by increased susceptibility of IL-1RA-deficient mice to cutaneous leishmaniasis. J Invest Dermatol (2011) 131:1650–9. doi: 10.1038/jid.2011.99
72. Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva ALN, Mineo TWP, et al. Inflammasome-derived IL-1β production induces nitric oxide-mediated resistance to Leishmania. Nat Med (2013) 19:909–15. doi: 10.1038/nm.3221
73. Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discovery (2017) 16:843–62. doi: 10.1038/nrd.2017.201
74. Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med (2014) 20:1043–9. doi: 10.1038/nm.3645
75. Novais FO, Nguyen BT, Scott P. Granzyme B inhibition by tofacitinib blocks the pathology induced by CD8 T cells in cutaneous leishmaniasis. J Invest Dermatol (2020) 141:575–85. doi: 10.1016/j.jid.2020.07.011
76. Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, et al. Compartmentalized control of skin immunity by resident commensals. Science (2012) 337:1115–9. doi: 10.1126/science.1225152
Keywords: cutaneous leishmaniasis, host-directed therapies, skin immunity, immunopathology, cytokines
Citation: Novais FO, Amorim CF and Scott P (2021) Host-Directed Therapies for Cutaneous Leishmaniasis. Front. Immunol. 12:660183. doi: 10.3389/fimmu.2021.660183
Received: 28 January 2021; Accepted: 11 March 2021;
Published: 26 March 2021.
Edited by:
Fabienne Tacchini-Cottier, University of Lausanne, SwitzerlandReviewed by:
Christian Engwerda, Queensland Children’s Medical Research Institute, AustraliaMaria Adelaida Gomez, Centro Internacional de Entrenamiento e Investigaciones Medicas, Colombia
Copyright © 2021 Novais, Amorim and Scott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fernanda O. Novais, RmVybmFuZGEuTm92YWlzQG9zdW1jLmVkdQ==; Phillip Scott, UHNjb3R0QHVwZW5uLmVkdQ==