 Meriem Sekheri
Meriem Sekheri Amira Othman
Amira Othman János G. Filep
János G. Filep- 1Department of Pathology and Cell Biology, University of Montreal, Montreal, QC, Canada
- 2Department of Biomedical Sciences, University of Montreal, Montreal, QC, Canada
- 3Research Center, Maisonneuve-Rosemont Hospital, Montreal, QC, Canada
Neutrophils act as the first line of cellular defense against invading pathogens or tissue injury. Their rapid recruitment into inflamed tissues is critical for the elimination of invading microorganisms and tissue repair, but is also capable of inflicting damage to neighboring tissues. The β2 integrins and Mac-1 (CD11b/CD18, αMβ2 or complement receptor 3) in particular, are best known for mediating neutrophil adhesion and transmigration across the endothelium and phagocytosis of microbes. However, Mac-1 has a broad ligand recognition property that contributes to the functional versatility of the neutrophil population far beyond their antimicrobial function. Accumulating evidence over the past decade has demonstrated roles for Mac-1 ligands in regulating reverse neutrophil transmigration, lifespan, phagocytosis-induced cell death, release of neutrophil extracellular traps and efferocytosis, hence extending the traditional β2 integrin repertoire in shaping innate and adaptive immune responses. Understanding the functions of β2 integrins may partly explain neutrophil heterogeneity and may be instrumental to develop novel therapies specifically targeting Mac-1-mediated pro-resolution actions without compromising immunity. Thus, this review details novel insights on outside-in signaling through β2 integrins and neutrophil functional heterogeneity pertinent to the resolution of inflammation.
Introduction
Neutrophils are the first line of cellular defense against invading pathogens or tissue injury. Rapid recruitment of neutrophils into infected or injured tissues is critical for the elimination of invading microorganisms and tissue repair (1). Ideally, once the pathogens are cleared, cessation of neutrophil recruitment and removal of emigrated neutrophils from the inflamed site will assure timely resolution of inflammation and return to homeostasis (2–4). Aberrant neutrophil accumulation or removal from the inflamed area inflicts damage to the surrounding tissue (2). Indeed, neutrophil-driven tissue injury has been recognized as a common mechanism underlying a wide variety of pathologies, including atherosclerosis, respiratory, autoimmune and neurodegenerative diseases, arthritis, sepsis and cancer (5, 6). Since neutrophils are also involved in the resolution of inflammation (7, 8), the balance between their protective and deleterious actions will likely determine the outcome of the inflammatory response.
The β2 integrins LFA-1 (leukocyte function antigen 1, CD11a/CD18) and Mac-1 (CD11b/CD18, αMβ2 or complement receptor 3) are best known for mediating neutrophil adhesion and transmigration across the activated endothelium and phagocytosis of microbes (9–11). Historically, LFA-1 and Mac-1 have been considered pro-inflammatory for reduced expression or function of β2 integrins causes rare immunodeficiency syndromes, leukocyte adhesion deficiency syndromes (LAD types I-III), characterized by recurrent infections (12, 13). The binding of Mac-1 and LFA-1 to their endothelial counter-ligand ICAM-1 or matrix components generates survival cues for neutrophils (14, 15). Mac-1 may also contribute to sustained inflammation by enhancing the function of heterologous receptors such as Toll-like receptors and Fcγ receptors through modulating intracellular signaling (16, 17). Accumulating data indicates that Mac-1 can bind a variety of ligands (18). This broad ligand recognition property contributes to the functional versatility of the neutrophil population and shapes innate and adaptive immune responses far beyond their antimicrobial functions. In this review, we will focus on recent advances on outside-in signaling through β2 integrins and neutrophil functional heterogeneity during homeostasis and diseases. We also examine how targeting β2 integrin signaling could be exploited for facilitating the resolution of inflammation.
Beta 2 Integrin Activation and Ligand Binding
The β2 integrins, composed of a common β2 (CD18) subunit complexed with unique α subunits (CD11a-d), are a family of myeloid cell-specific adhesion molecules with LFA-1 (leukocyte function antigen 1, CD11a/CD18) and Mac-1 (CD11b/CD18, αMβ2 or complement receptor 3) being the most studied members. β2 integrin ligand binding relies on conformational changes in their ectodomain (19, 20). Ligation of G-protein-coupled receptors or heterologous receptors generates intracellular signals that shift the resting bent/closed β2 integrin conformation (low affinity for ligands) to an extended (E+) and then a high-affinity conformation with an “open” headpiece (H+) (canonical “switchblade” model) (19, 21). Spatiotemporal integrin activation is governed by inside-out (i.e. activation of ligand binding function of integrins) and outside-in signaling cascades (i.e. cellular responses evoked by ligand binding to integrins) and involves inhibitory proteins and activator complexes, such as talin, kindlins, cytohesin-1 and integrin-linked kinase, interacting with the cytoplasmic tail of the β subunit (13, 22, 23). The Src kinase-associated phosphoprotein 2 (Skap2), which regulates actin polymerization and binding of talin-1 and kindlin-3 to the β2 integrin cytoplasmic domain, is indispensable for β2 integrin activation (24). Loss of Skap2 function causes a LAD-like phenotype in mice (24). Mac-1 has two spatially distinct binding sites, the αI-domain and the lectin-like domain (25). The αMI-domain recognizes sequence patterns (consisting of a core of basic residues flanked by hydrophobic residues), rather than specific amino-acid sequence(s) (18) with over 30 structurally unrelated ligands, including ICAM-1, fibrinogen, complement 3b (iC3b), various granule proteins and heparane sulfate (25). The interaction between the αMI-domain and cationic proteins is mediated mostly by hydrophobic contacts independently of divalent cations (26). The lectin-like domain binds β-glucans present in the fungal cell wall (27, 28). Table 1 lists selected Mac-1 ligands and their main biological actions.
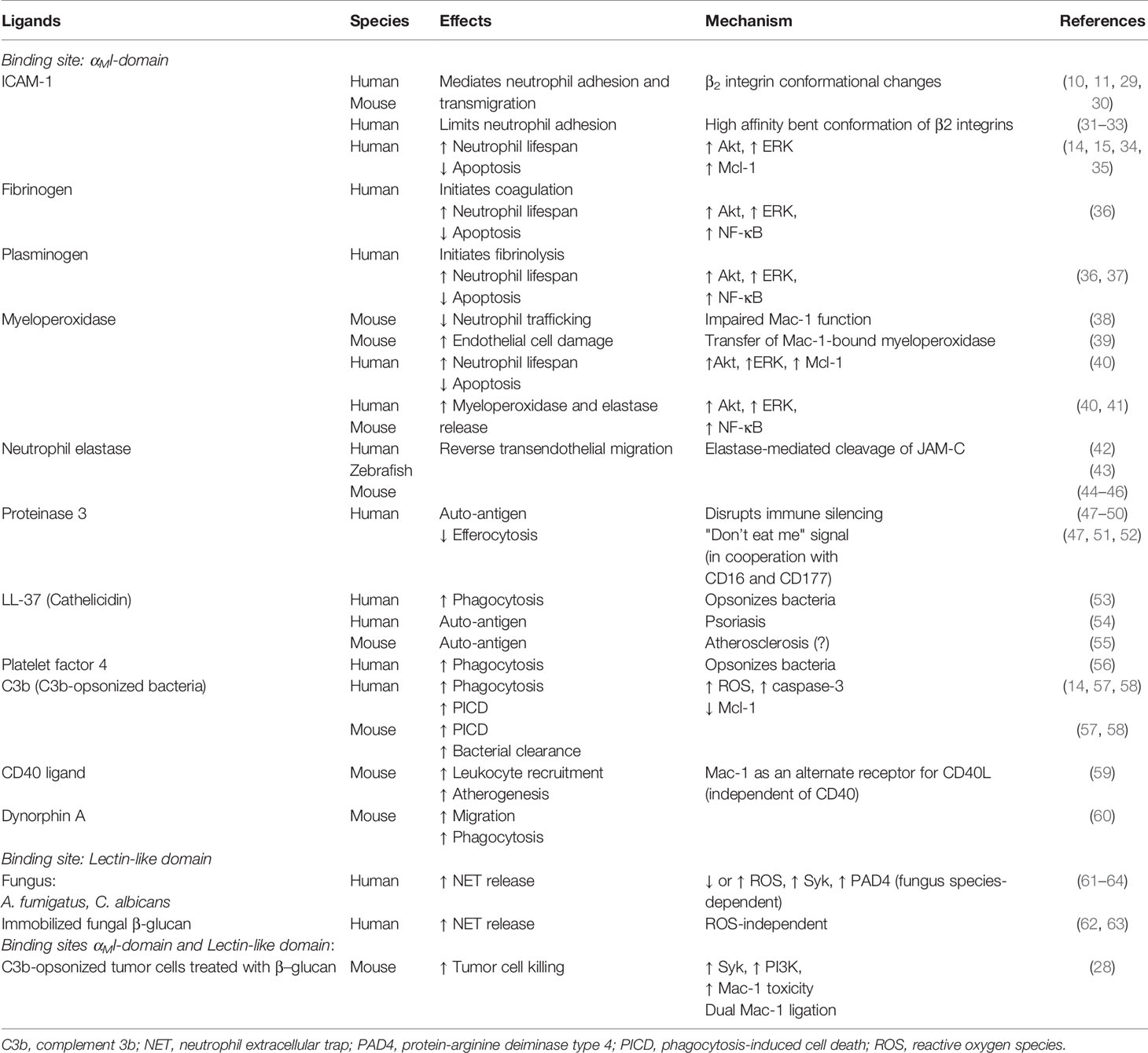
Table 1 Selected Mac-1 (CD11b/CD18) ligands and their actions.
Limiting Neutrophil Trafficking Into Tissues
Neutrophils exit the circulation at the sites of inflammation through the classical adhesion cascade (10). The molecular mechanisms mediating and governing this multistep process as well as organ-specific differences have been described in detail (10, 29, 30, 65). In general, β2 integrins play vital roles in neutrophil arrest on the activated endothelium under flow (10), transmigration through endothelial cells (66), chemotaxis (67) and neutrophil swarming (68). Counter-ligand-specific binding forces of LFA-1 and Mac-1 imply diverse roles for β2 integrins in neutrophil recruitment (69) and determine the direction of neutrophil migration along the activated endothelium (70). Fully activated E+H+ β2 integrins bind ICAM-1 expressed on the opposing cells in trans and arrest neutrophil rolling (31). Studies with human neutrophils in microfluidic chambers identified high-affinity, bent conformation (E-H+) β2 integrins, which face each other to form oriented nanoclusters (32) and bind ICAM-1 in cis to inhibit neutrophil rolling and consequently neutrophil adhesion to the endothelium (31). Activated β2 integrins may also restrict neutrophil recruitment during acute bacterial infections, for pharmacological inhibition of high-affinity β2 integrins or genetic deletion of talin-1 or kindlin-3 was found to enhance neutrophil trafficking with modest impairment of phagocytosis during Pseudomonas aeruginosa-pneumonia in mice (71). Another potential inhibitory signal is the interaction of the αI-domain of Mac-1 in the bent state with the sialylated ectodomain of the IgG receptor FcγRIIA in cis, leading to reduced FcγRIIA affinity to IgG and subsequently decreased neutrophil recruitment to immune complexes deposited in the vessel wall (33). Disruption of this interaction may increase neutrophil recruitment in autoimmune diseases.
Neutrophils from myeloperoxidase knockout mice display increased surface expression of Mac-1 and a pro-migratory phenotype in a murine model of ischemia-reperfusion-induced liver damage (38). Hence, myeloperoxidase may impair Mac-1 function and subsequently limit neutrophil trafficking into ischemic tissues. Neutrophil-derived myeloperoxidase was reported to protect mice from endotoxin-induced inflammation and mortality (72), though the involvement of β2 integrins in these actions remains to be investigated. On the other side, cell contact-dependent, Mac-1-mediated transfer of myeloperoxidase from neutrophils to endothelial cells can disrupt normal endothelial function (39), leading to endothelial inflammation that underlies atherosclerosis and vasculitis. Following neutrophil adhesion to the endothelium, gelatinase granules translocate to the cell surface and externalize the phospholipid-binding protein annexin A1 (73). Annexin A1 promotes the detachment of adhering leukocytes presumably through inhibiting CCL5-induced switch in β2 integrin conformation, and reducing α4β1 integrin clustering and activation (74, 75). Hence, annexin A1 may function as an endogenous stop signal for neutrophil trafficking (76).
Reverse Transendothelial Migration
In addition to moving from the vascular lumen to the extravascular tissue, neutrophils also exhibit reverse motility through the endothelium, known as reverse transendothelial migration (TEM) both in vitro (42) and in vivo (44, 45). This neutrophil reverse TEM response is relatively prevalent under conditions of ischemia-reperfusion injury, which is associated with reduced expression of junctional adhesion molecule C (JAM-C) at endothelial cell junctions (45, 46). Pharmacological blockade or genetic deletion of JAM-C enhances the frequency of neutrophil reverse TEM in mouse cremaster venules (45). Under ischemia-reperfusion, locally generated LTB4, likely produced by the neutrophils themselves (68), induces elastase release from neutrophils through the LTB4 receptor BLT1 (44). Activated Mac-1 binds neutrophil elastase (77) and JAM-C (78), thereby acting as a molecular “bridge” to facilitate elastase-mediated cleavage of JAM-C and consequently reverse TEM (44) (Figure 1). The importance of Mac-1-bound elastase is further highlighted by the failure of exogenous neutrophil elastase to cleave JAM-C (44). Reversely migrated neutrophils display a phenotype (ICAM-1high, CXCR1low) distinct from tissue-resident or circulating neutrophils and increased capacity to produce superoxide (42, 45). At present, the functional implications of neutrophils undergoing reverse TEM remain unclear. Reverse TEM might facilitate the removal of neutrophils from inflamed tissues, thereby promoting the resolution of inflammation (43, 79). Alternatively, re-entry of a small subset of activated neutrophils into the blood circulation could contribute to spreading a local inflammatory response, ultimately leading to distant organ damage (44, 45). This notion is supported by the association between the percentage of ICAM-1high neutrophils and the severity of lung inflammation in the mouse cremaster ischemia-reperfusion model (45).
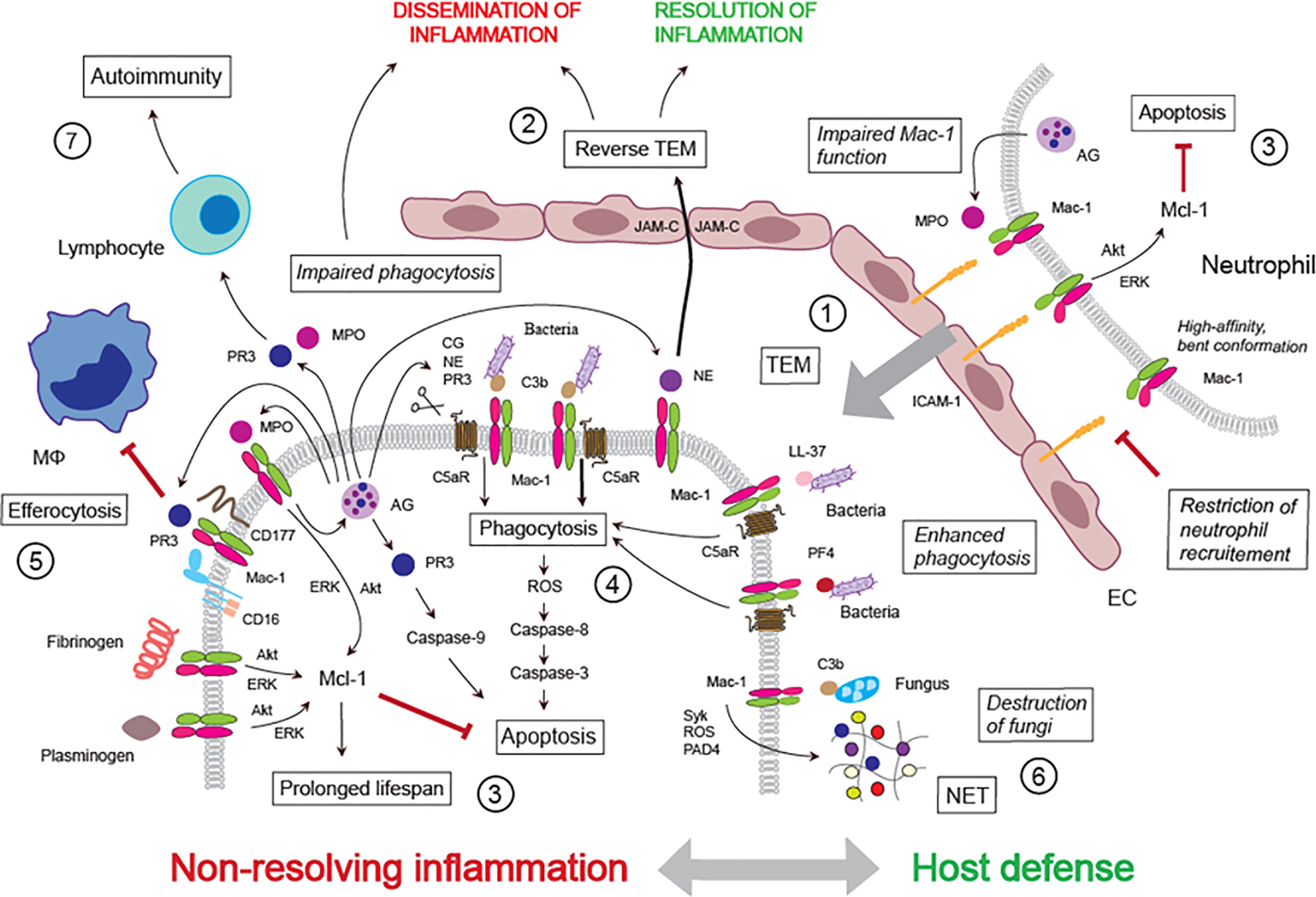
Figure 1 Mac-1 ligand repertoire shapes host defense and non-resolving inflammation. ① Transendothelial migration: Mac-1, together with LFA-1, mediates neutrophil adherence to the activated endothelium and transmigration. Conformational changes in Mac-1 (high affinity, bent conformation) and MPO impairment of Mac-1 function may limit neutrophil trafficking. ② Reverse TEM: Mac-1-bound NE direct neutrophil reverse TEM through binding to and cleaving JAM-C. ③ Neutrophil lifespan: Ligation of Mac-1 with ICAM-1, fibrinogen, plasminogen or MPO generates survival signals for neutrophils through delaying constitutive apoptosis. MPO induces MPO release from the azurophilic granule, thereby forming a feed-forward loop. ④ Phagocytosis: Phagocytosis of complement C3b-opsonized bacteria induces PICD followed by efferocytosis. Cleavage of C5aR (CD88) by NE, PR3 or cathepsin G (released from the azurophilic granule) alters the Mac-1/C5aR ratio, impairs phagocytosis, bacterial clearance and PICD. ⑤ Inhibition of efferocytosis: PR3 bound to Mac-1(in association with CD16 and CD177) inhibits efferocytosis. ⑥ NET release: C3b-opsonized fungus or immobilized fungal β-glucan, which cannot be phagocytosed, evokes release of NET, leading to extracellular killing of the pathogen. ⑦ Autoimmunity: PR3 and MPO (presented by Mac-1 and/or NET) may induce autoimmunity.
Extending Neutrophil Lifespan and Suppression of Apoptosis
Circulating neutrophils have a short lifespan (80, 81), though some reports estimated that their lifespan to be 5.4 days (82). Neutrophils have increased, albeit variable lifetimes upon activation and in healthy and inflamed tissues (4, 6, 83, 84). Blood neutrophils die by constitutive apoptosis. This cell death program renders neutrophils unresponsive to extracellular stimuli and ensures their timely removal from the inflammatory sites by macrophages via efferocytosis, thereby limiting their potentially harmful actions to the host (2, 85, 86). Extended neutrophil lifespan through suppressed apoptosis is observed in patients with chronic inflammation, for example, acute coronary syndrome (87), asthma (88) or sepsis (89), and is associated with increased disease severity. Consistently, studies in experimental models documented that delaying neutrophil apoptosis can adversely affect the outcome of inflammation (40, 90, 91).
During transendothelial migration and at sites of inflammation, neutrophils receive pro-survival cues that extend their lifespan by delaying intrinsic apoptosis (4, 85, 86). Neutrophil adherence to the Mac-1 endothelial counter-ligand ICAM-1induces activation of the PI3k/Akt and MAPK/ERK pathways (34, 35), leading to suppression of caspase-3 activity through preserving the anti-apoptotic protein Mcl-1, a key regulator of neutrophil survival (92). Suppression of apoptosis by the Mac-1 ligands fibrinogen and plasminogen also depends on signaling through Akt and ERK as well as activation of NF-κB (36). Engagement of both Mac-1 subunits with soluble ligands is essential for the generation of pro-survival cues, whereas adhesion per se is not a prerequisite (37). Another ligand for Mac-1 is myeloperoxidase, a granule protein implicated in pathogen killing and inflicting tissue damage (93–96). Myeloperoxidase binding to Mac-1on human neutrophils leads to activation of the PI3K/Akt, p38 MAPK, MAPK/ERK and NF-κB pathways (40, 41) and rescues neutrophil from apoptosis (40). Myeloperoxidase upregulates Mac-1 expression and induces myeloperoxidase release from the primary granules (40, 41), thereby forming an autocrine/paracrine feed-forward loop to amplify the inflammatory response (40) (Figure 1). Increased plasma myeloperoxidase levels were detected in patients with acute coronary syndromes or sepsis and were associated with disease severity (41). Dissociation of myeloperoxidase into monomers with diminished biological activities may represent a mechanism to limit neutrophil responses to this protein (97).
Phagocytosis and Phagocytosis-Induced Cell Death
In contrast to Mac-1 ligation-generated survival signals, outside-in signaling through Mac-1 could also generate pro-apoptosis cues. Thus, phagocytosis of complement C3b-opsonized bacteria or necrotic cells accelerates neutrophil apoptosis, also known as phagocytosis-induced cell death (PICD) (98, 99). The antibiotic peptide LL-37 and platelet factor 4 were also reported to opsonize bacteria and promote Mac-1-mediated phagocytosis (53, 56). Higher levels of Mac-1 expression on neutrophils from female vs. male mice may partly explain an innate sex bias in neutrophil bactericidal killing (100). Phagocytosis is initiated by lateral clustering of Mac-1 (101) and governed by a delicate balance between Mac-1 and the complement C5a receptor (C5aR or CD88) (102, 103). Mac-1-mediated phagocytosis evokes ROS formation through activation of NADPH oxidase, which is thought to mediate bacterial killing in the phagolysosomes (94). ROS, presumably hydroxyl radicals and H2O2, activate caspase-8 and caspase-3, thereby countering survival signals generated by ligation of Mac-1and promoting PICD (4, 14, 98). Release of the granule enzymes, neutrophil elastase, proteinase 3 and cathepsin G can impair phagocytosis by cleaving C5aR, though their involvement appears to be context-dependent (57, 104) (Figure 1). For example, TLR9 activation augments Mac-1 expression and reduces C5aR expression through inducing the release of neutrophil elastase and proteinase 3, resulting in defective phagocytosis in human neutrophils and prolongation of lung injury in mice (57). Reduced neutrophil C5aR expression is a common finding in patients with sepsis (105, 106) and may explain neutrophil unresponsiveness to C5a in sepsis (103).
Modulation of Efferocytosis
Detection and prompt disposal of apoptotic cells, including neutrophils, generally promote an anti-inflammatory pro-resolution response at the tissue level and immunological tolerance. The molecular mechanisms include numerous “find-me” and “eat-me” signals that underpin the recognition and subsequent phagocytosis of apoptotic cells by macrophages and dendritic cells (107). Intriguingly, proteinase 3 is expressed on the plasma membrane in association with its partners Mac-1, FcγRIIIb (CD16) and CD177 at a very early stage of apoptosis (51, 108) and functions as a “don’t eat me” signal that delays uptake of apoptotic neutrophils (52) (Figure 1). Neutrophils lacking CD177, the putative receptor for proteinase 3, express membrane proteinase 3 and respond to proteinase 3-ANCA, suggesting a critical role for Mac-1 and/or FcγRIIIb (47). However, further studies are required to establish how Mac-1 could modulate efferocytosis and contribute to the pathogenesis of ANCA-associated vasculitides. Mac-1 was also reported to support macrophage fusion, leading to the formation of multinucleated giant cells in the inflamed mouse peritoneum (109). The function of these cells remains to be investigated.
Induction of Rapid NET Release
Neutrophils can release extracellular traps (NET) to immobilize and kill harmful bacterial, fungal and viral pathogens in the extracellular space when phagocytosis is not feasible (110–112). The classical pathway of NET extrusion involves activation of NADPH oxidase via the Raf-MEK-ERK and p38 MAPK pathways, myeloperoxidase- and elastase-mediated cleavage of histones and protein-arginine deiminase 4 (PAD4)-mediated chromatin decondensation, eventually leading to extrusion of a DNA scaffold studded with citrullinated histones and cytotoxic granular proteins (113, 114). A more rapid or “vital” NET release occurs in response to Staphylococcus aureus, Candida albicans, Aspergillus fumigatus and Leishmania promastigotes independently of ROS in the presence of matrix and without compromising neutrophil viability (61, 115, 116) or by selective extrusion of mitochondrial DNA (117). Mac-1 recognition of Candida hyphae, the invasive filamentous forms of C. albicans that are too large to be phagocytosed, or immobilized fungal β-glucan triggers rapid NET release and initiates respiratory burst, which is then suppressed by binding of Mac-1 to the extracellular matrix (62, 63) (Figure 1). Mac-1 also triggers NETosis to Aspergillus living conidia through ROS generation downstream to activation of the Src kinase Syk and PI3k-δ, but independently of PAD4 (64). Platelet binding to neutrophils ensuing NET release is mediated either by LFA1 along liver sinusoid in sepsis (118) or Mac-1 along the vascular endothelium during sterile lung injury in mice (119). Hence, it is plausible that the context of NET-inducing stimuli would activate different signaling pathways for NET extrusion. Similar to neutrophil recruitment, a crosstalk between kindlin-3 and β2 integrins is required for NET release in mice (120). Of note, yeast β-glucan was reported to enhance killing of iC3b-opsonized tumor cells through activation of the Syk-PI3K signaling pathway, indicating dual Mac-1 ligation (28) (Table 1). Whether the cytotoxic action involves NET formation remains to be explored.
Autoimmunity
Many neutrophil granule proteins are recognized self-antigens in autoimmunity. Myeloperoxidase and proteinase 3 are target antigens in different forms of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitides (48, 49), whereas the antimicrobial protein LL-37 is an autoantigen in psoriasis (54). Externalization of these molecules, together with other well-known antigens, such as double-stranded DNA and histones, through aberrant NET formation has been implicated in triggering a systemic autoimmune response in susceptible individuals (49). Myeloperoxidase might trigger autoimmunity during uncontrolled inflammation in mice (121), though it is unclear whether this involves β2 integrins and/or NET formation. Proteinase 3 was found to bind directly to Mac-1 (51) or form a complex with the glycosylphosphatidylinositol (GPI)-anchored neutrophil-specific receptor NB1 (CD177) expressed on the surface of a subpopulation of human neutrophils (122) (Figure 1). While surface plasmon resonance analysis indicated direct interaction of NB1 with both LFA1 and Mac-1, only Mac-1 functions as an adaptor for NB1-mediated proteinase 3-ANCA-induced neutrophil activation (123). Proteinase 3 expressed on the surface of apoptotic neutrophils disrupts immune silencing associated with efferocytosis through plasmocytoid dendritic cell-driven generation of Th9/Th2 cells and Th17 response, consistent with promoting systemic necrotizing vasculitis (50). Recent data identified the cathelicidin protein CRAMP (a truncated form of the mouse homolog of hCAP18) as a potential auto-antigen in ApoE-deficient mice (55). Although LL-37 was found to bind to Mac-1 (53), the relevance of this interaction to atherosclerosis remains to be investigated.
Therapeutic Targeting β2 Integrins to Promote Resolution of Inflammation
In the light of their functional significance in shaping neutrophil responses, β2 integrins appear to be attractive therapeutic targets. However, prolonged global blockade of β2 integrins may have limited usefulness because of the potential of development of LAD-like symptoms. Attractive alternative approaches may be targeting β2 integrin conformation or ligand-specific signaling circuits by specialized pro-resolving mediators (SPMs) without compromising the ability of neutrophils to contain the microbial invasion.
The currently available drugs (monoclonal antibodies or small molecules) inhibit the ligand-binding site and block a broad repertoire of β2 integrin functionality (124). Although the beneficial actions of genetic deletion and pharmacological blockade of β2 integrins have been documented in a number of neutrophil-driven inflammatory models (6, 29, 124), conventional anti-β2 integrin blockade lacks specificity and inhibits phagocytosis, promotes apoptosis, and potentiates bacteremia and bacterial sepsis (125). Development of the anti-M7 monoclonal antibody that specifically inhibits Mac-1 interaction with its ligand CD40L without interfering with other β2 integrin ligands (59, 125) opens a new avenue for ligand-targeted anti-Mac-1 therapy. An alternative strategy will be developing allosteric inhibitors that stabilize β2 integrins in the high affinity bent conformation to block neutrophil adherence (31, 71) or to prevent the deleterious effects of immune complex-evoked neutrophil accumulation (33). This might be achieved by selectively targeting discrete glycan motifs present on Mac-1 (126). Thus, plant lectins were shown to reduce Mac-1-mediated adhesion, trans-epithelial migration and ROS production, while enhancing phagocytosis and neutrophil apoptosis (126). Intriguingly, the activation of Mac-1 with the small molecule agonists leukadherins was reported to reduce leukocyte trafficking, arterial narrowing and renal dysfunction, while increasing leukocyte adherence to the endothelium in murine models (127). Leukadherin-1 promotes macrophage polarization toward a pro-inflammatory phenotype through activating microRNA Let7a, thereby driving anti-tumor immunity (128).
SPMs include protein and lipid mediators that are mobilized and/or synthesized during the resolution phase of inflammation. For example, annexin A1 is mobilized from the cytoplasm pool to the cell surface and signals through the lipoxin A4/formyl-peptide receptor 2 (ALX/FPR2) to induce detachment of adhered neutrophils (74, 129). The family of lipid SPMs consists of lipoxins, resolvins, protectins and maresins (3, 130, 131). These lipids act through specific receptors and exhibit cell-specific properties, however, their primary targets are myeloid cells (3, 132). In general, lipid SPMs prevent up-regulation of Mac-1 expression and inhibit β2-integrin-mediated neutrophil adhesion, transendothelial migration and consequently tissue accumulation [signaling pathways and networks are mapped into the searchable Atlas of Inflammation Resolution (133)]. For example, lipoxin A4 mobilizes annexin A1 to form an endogenous anti-inflammation loop to limit neutrophil trafficking into inflammatory loci (134). Aspirin triggered 15-epi-lipoxin A4, acting through ALX/FPR2, disrupts the myeloperoxidase-centered self-amplifying loop and redirects neutrophil to apoptosis (58), and enhances phagocytosis of bacteria by restoring the balance between Mac-1 and C5aR expression in human neutrophils (57). Consistently, 15-lipoxin A4 accelerates the resolution of inflammation in mouse models of myeloperoxidase (58) or E. coli-induced acute lung injury (57).
Conclusions
Unlike other integrins, the β2 integrin Mac-1 has two spatially distinct binding sites and exhibits broad ligand recognition specificity and numerous neutrophil responses. A novel aspect of Mac-1 bioactivity is that its conformations and ligands contribute to neutrophil functional plasticity and heterogeneity. The classical view of β2 integrins does not accommodate all aspects of their role in neutrophil biology. Their role in regulating neutrophil reverse transmigration, lifespan, phagocytosis-induced cell death, NET formation and efferocytosis extend the β2 integrin repertoire in shaping innate and adaptive immunity and may partly explain neutrophil heterogeneity. Understanding the functions of β2 integrins may be instrumental to develop novel therapies specifically targeting pro-resolution actions without compromising immunity.
Author Contributions
MS, AO, and JF conceived, designed, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Canadian Institutes of Health Research (MOP-97742 and MOP-102619) (to JF).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AG, azurophilic granule; C5aR, complement 5a receptor; CG, cathepsin G; EC, endothelial cell; JAM-C, junctional adhesion molecule-C; MΦ, macrophage; MPO, myeloperoxidase; NE, neutrophil elastase; NET, neutrophil extracellular traps; PICD, phagocytosis-induced cell death; PR3, proteinase 3; TEM, transendothelial migration.
References
1. Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol (2014) 15:602–11. doi: 10.1038/ni.2921
2. Nathan C, Ding A. Nonresolving inflammation. Cell (2010) 140:871–82. doi: 10.1016/j.cell.2010.02.029
3. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest (2018) 128:2657–69. doi: 10.1172/JCI97943
4. Filep JG, Ariel A. Neutrophil heterogeneity and fate in inflamed tissues: implications for the resolution of inflammation. Am J Physiol Cell Physiol (2020) 319:C510–32. doi: 10.1152/ajpcell.00181.2020
5. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11:519–31. doi: 10.1038/nri3024
6. Liew PX, Kubes P. The neutrophil’s role during health and disease. Physiol Rev (2019) 99:1223–48. doi: 10.1152/physrev.00012.2018
7. Jones HR, Robb CT, Perretti M, Rossi AG. The role of neutrophils in inflammation resolution. Semin Immunol (2016) 28:137–45. doi: 10.1016/j.smim.2016.03.007
8. Potey PMD, Rossi AG, Lucas CD, Dorward DA. Neutrophils in the initiation and resolution of acute pulmonary inflammation: understanding biological function and therapeutic potential. J Pathol (2019) 247:672–85. doi: 10.1002/path.5221
9. Ross GD. Regulation of the adhesion versus cytotoxic functions of Mac-1/CR3/alphaMbeta2- integrin glycoprotein. Crit Rev Immunol (2000) 20:197–222. doi: 10.1615/CritRevImmunol.v20.i3.20
10. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7:678–89. doi: 10.1038/nri2156
11. Filippi MD. Neutrophil transendothelial migration: updates and new perspectives. Blood (2019) 133:2149–56. doi: 10.1182/blood-2018-12-844605
12. Harris ES, Weyrich AS, Zimmerman GA. Lessons from rare maladies: leukocyte adhesion deficiency syndromes. Curr Opin Hematol (2013) 20:16–25. doi: 10.1097/MOH.0b013e32835a0091
13. Fagerholm SC, Gunther C, Llort Asens M, Savinko T, Uotila LM. Beta2-integrins and interacting proteins in leukocyte trafficking, immune suppression, and immunodeficiency. Front Immunol (2019) 10:254. doi: 10.3389/fimmu.2019.00254
14. Mayadas TN, Cullere X. Neutrophil β2 integrins: moderators of life and death decisions. Trends Immunol (2005) 26:388–95. doi: 10.1016/j.it.2005.05.002
15. El Kebir D, Filep JG. Modulation of neutrophil apoptosis and the resolution of inflammation through β2 integrins. Front Immunol (2013) 4:1–5. doi: 10.3389/fimmu.2013.00060
16. Han C, Jin Y, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol (2010) 11:734–42. doi: 10.1038/ni.1908
17. Rosetti F, Mayadas TN. The many faces of Mac-1 in autoimmune disease. Immunol Rev (2016) 269:175–93. doi: 10.1111/imr.12373
18. Podolnikova NP, Podolnikov AV, Haas TA, Lishko VK, Ugarova TP. Ligand recognition specificity of leukocyte integrin αMβ2 (Mac-1, CD11b/CD18) and its functional consequences. Biochemistry (2015) 54:1408–20. doi: 10.1021/bi5013782
19. Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol (2007) 25:619–47. doi: 10.1146/annurev.immunol.25.022106.141618
20. Fan Z, Ley K. Leukocyte arrest: biomechanics and molecular mechanisms of β2 integrin activation. Biorheology (2015) 52:353–77. doi: 10.3233/BIR-15085
21. Alon R, Feigelson SW. Chemokine-triggered leukocyte arrest: force-regulated bi-directional integrin activation in quantal adhesive contacts. Curr Opin Cell Biol (2012) 24:670–6. doi: 10.1016/j.ceb.2012.06.001
22. Kim C, Ye F, Grinsberg MH. Regulation of integrin activation. Annu Rev Cell Dev Biol (2011) 27:321–45. doi: 10.1146/annurev-cellbio-100109-104104
23. El Azreq MA, Garceau V, Bourgoin SG. Cytohesin-1 regulates fMLF-mediated activation and functions of the β2 integrin Mac-1 in human neutrophils. J Leukoc Biol (2011) 89:823–36. doi: 10.1189/jl.0410222
24. Boras M, Volmering S, Bokemeyer A, Rossaint J, Block H, Bardel B, et al. Skap2 is required for β2 integrin-mediated neutrophil recruitment and functions. J Exp Med (2017) 214:851–74. doi: 10.1084/jem.20160647
25. Yakubenko VP, Lishko VK, Lam SC-T, Ugarova TP. A molecular basis for integrin alphaMbeta2 ligand binding promiscuity. J Biol Chem (2002) 277:48635–42. doi: 10.1074/jbc.M208877200
26. Feng W, Nguyen H, Shen D, Deng H, Jiang Z, Podolnikova N, et al. Structural characterization of the interaction between the αMI-domain of the integrin Mac-1 (αMβ2) and the cytokine pleiotrophin. Biochemistry (2021) 60:182–93. doi: 10.1021/acs.biochem.0c00700
27. O`Brian XM, Reichner JS. Neutrophil integrins and matrix ligands and NET release. Front Immunol (2016) 7:363:363. doi: 10.3389/fimmu.2016.00363
28. Li B, Allendorf DJ, Hansen R, Marroquin J, Ding C, Cramer DE, et al. Yeast beta-glucan amplifies phagocyte killing of iC3b-opsonized tumor cells via complement receptor 3-Syk-phosphatidylinositol 3-kinase pathway. J Immunol (2006) 177:1661–9. doi: 10.4049/jimmunol.177.3.1661
29. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13:159–75. doi: 10.1038/nri3399
30. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity (2014) 41:694–707. doi: 10.1016/j.immuni.2014.10.008 2014.
31. Fan Z, McArdle S, Marki A, Mikulski Z, Gutierrez E, Engelhardt B, et al. Neutrophil recruitment limited by high-affinity bent β2 integrin binding ligand in cis. Nat Commun (2016) 7:12658. doi: 10.1038/ncomms12658
32. Fan Z, Kiosses WB, Sun H, Oreicchioni M, Ghosheh Y, Zajonc DM, et al. High-affinity bent β2-integrin molecules in arresting neutrophils face each other through binding to ICAMs in cis. Cell Rep (2019) 26:119–30. doi: 10.1016/j.cellrep.2018.12.038
33. Saggu G, Okubo K, Chen Y, Vattepu R, Tsuboi N, Rosetti F, et al. Cis interaction between sialylated FcγRIIA and the αI-domain of Mac-1 limits antibody-mediated neutrophil recruitment. Nat Commun (2018) 9:5058. doi: 10.1038/s41467-018-07506-1
34. Whitlock BB, Gardai S, Fadok V, Bratton D, Henson PM. Differential roles for a(M)b(2) integrin clustering or activation in the control of apoptosis via regulation of Akt and ERK survival mechanisms. J Cell Biol (2000) 151:1305–20. doi: 10.1083/jcb.151.6.1305
35. Yan SR, Sapru K, Issekutz AC. The CD11/CD18 (beta2) integrins modulate neutrophil caspase activation and survival following TNF-alpha or endotoxin-induced transendothelial migration. Immunol Cell Biol (2004) 82:435–46. doi: 10.1111/j.0818-9641.2004.01268.x
36. Rubel C, Gomez S, Fernandez GC, Isturiz MA, Caamaño J, Palermo MS. Fibrinogen-CD11b/CD18 interaction activates the NF-kB pathway and delays apoptosis in human neutrophils. Eur J Immunol (2003) 33:1429–38. doi: 10.1002/eji.200323512
37. Pluskota E, Soloviev DA, Szpak D, Weber C, Plow EF. Neutrophil apoptosis: selective regulation by different ligands of integrin αMβ2. J Immunol (2008) 181:3609–19. doi: 10.4049/jimmunol.181.5.3609
38. Tseng A, Kim K, Li J, Cho J. Myeloperoxidase negatively regulates neutrophil-endothelial cell interactions by impairing αMβ2 integrin function in sterile inflammation. Front Med (Lausanne) (2018) 5:134:134. doi: 10.3389/fmed.2018.00134
39. Jerke U, Rolle S, Purfürst B, Luft FC, Nauseef WM, Kettritz R. β2 integrin-mediated cell-cell contact transfers active myeloperoxidase from neutrophils to endothelial cells. J Biol Chem (2013) 288:12910–9. doi: 10.1074/jbc.M112.434613
40. El Kebir D, József L, Pan W, Filep JG. Myeloperoxidase delays neutrophil apoptosis through CD11b/CD18 integrins and prolongs inflammation. Circ Res (2008) 103:352–9. doi: 10.1161/01.RES.0000326772.76822.7a
41. Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM, et al. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci USA (2005) 102:431–6. doi: 10.1073/pnas.0405193102
42. Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J Leukoc Biol (2006) 79:303–11. doi: 10.1189/jlb.0905496
43. Mathias JR, Perrin BJ, Liu TX, Kanki J, Look AT, Huttenlocher A. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J Leukoc Biol (2006) 80:1281–8. doi: 10.1189/jlb.0506346
44. Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, et al. Leukotriene B4-neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity (2015) 42:1075–86. doi: 10.1016/j.immuni.2015.05.010
45. Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol (2011) 12:761–9. doi: 10.1038/ni.2062
46. Scheiermann C, Colom B, Meda P, Patel NS, Voisin MB, Marrelli A, et al. Junctional adhesion molecule-C mediates leukocyte infiltration in response to ischemia reperfusion injury. Arterioscler Thromb Vasc Biol (2009) 29:1509–15. doi: 10.1161/ATVBAHA.109.187559
47. Witko-Sarsat V, Reuter N, Mouthon L. Interaction of proteinase 3 with its associated partners: implications in the pathogenesis of Wegener`s granulomatosis. Curr Opin Rheumatol (2010) 22:1–7. doi: 10.1097/BOR.0b013e3283331594
48. Jennette JC, Xiao H, Falk RJ. Pathogenesis of vascular inflammation by antineutrophil cytoplasmic antibodies. J Am Soc Nephrol (2006) 17:1235–42. doi: 10.1681/ASN.2005101048
49. Gupta S, Kaplan MJ. The role of neutrophil NETosis in autoimmune and renal diseases. Nat Rev Nephrol (2016) 12:402–13. doi: 10.1038/nrneph.2016.71
50. Millet A, Martin KR, Bonnefoy F, Saas P, Mocek J, Alkan M, et al. Proteinase 3 on apoptotic cells disrupts immune silencing in autoimmune vasculitis. J Clin Invest (2015) 125:4107–21. doi: 10.1172/JCI78182
51. David A, Kacher Y, Specks U, Aviram I. Interaction of proteinase 3 with CD11b/CD18 (beta2 integrin) on the cell membrane of human neutrophils. J Leukoc Biol (2003) 74:551–7:doi: 10.1189/jlb.1202624. doi: 10.1189/jlb.1202624
52. Kantari C, Pederzoli-Ribeil M, Amir-Moazami O, Gausson-Dorey V, Moura IC, Lecomte MC, et al. Proteinase 3, the Wegener autoantigen, is externalized during neutrophil apoptosis: evidence for a functional association with phospholipid scramblase 1 and interference with macrophage phagocytosis. Blood (2007) 110:4086–95. doi: 10.1182/blood-2007-03-080457
53. Lishko VK, Moreno B, Podolnikova NP, Ugarova TP. Identification of human cathelicidin peptide LL-37 as a ligand for macrophage integrin αMβ2 (Mac-1, CD11b/CD18) that promotes phagocytosis by opsonizing bacteria. Res Rep Biochem (2016) 2016:39–55. doi: 10.2147/rrbc.s107070
54. Morizane S, Gallo RL. Antimicrobial peptides in the pathogenesis of psoriasis. J Dermatol (2012) 39:225–30. doi: 10.1111/j.1346-8138.2011.01483.x
55. Mihailovic PM, Lio WM, Yano J, Zhao X, Zhou J, Chyu KY, et al. The cathelicidin protein CRAMP is a potential atherosclerosis self-antigen in ApoE(-/-) mice. PloS One (2017) 12:e0187432. doi: 10.371/journal.pone.0187432
56. Lishko VK, Yakubenko VP, Ugarova TP, Podolnikova NP. Leukocyte integrin Mac-1 (CD11b/CD18, αMβ2, CR3) acts as a functional receptor for platelet factor 4. J Biol Chem (2018) 293:6869–82. doi: 10.1074/jbc.RA117.000515
57. Sekheri M, El Kebir D, Edner N, Filep JG. 15-Epi-LXA4 and 17-epi-RvD1 restore TLR9-mediated impaired neutrophil phagocytosis and accelerate resolution of lung inflammation. Proc Natl Acad Sci USA (2020) 117:7971–80. doi: 10.1073/pnas.1920193117
58. El Kebir D, József L, Pan W, Wang L, Petasis NA, Serhan CN, et al. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med (2009) 180:311–9. doi: 10.1164/rccm.200810-1601OC
59. Wolf D, Hohmann J-D, Wiedermann A, Bledzka K, Blankenbach H, Marchini T, et al. Binding of CD40L to Mac-1’s I-domain involves the EQLKKSKTL motif and mediates leukocyte recruitment and atherosclerosis–but does not affect immunity and thrombosis in mice. Circ Res (2011) 109:1269–79. doi: 10.1161/CIRCRESAHA.111.247684
60. Podolnikova NP, Brothwell JA, Ugarova TP. The opioid peptide dynorphin A induces leukocyte responses via integrin Mac-1 (αMβ2, CD11b/CD18). Mol Pain (2015) 11:33. doi: 10.1186/s12990-015-0027-0
61. Byrd AS, O’Brien XM, Johnson CM, Lavigne LM, Reichner JS. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J Immunol (2013) 190:4136–48. doi: 10.4049/jimmunol.1202671
62. Lavigne LM, O’Brien XM, Kim M, Janowski JW, Albina JE, Reichner JS. Integrin engagement mediates the human polymorphonuclear leukocyte response to a fungal pathogen-associated molecular pattern. J Immunol (2007) 178:7276–82. doi: 10.4049/jimmunol.178.11.7276
63. O’Brien XM, Reichner JS. Neutrophil integrins and matrix ligands and NET release. Front Immunol (2016) 7:363:363. doi: 10.3389/fimmu.2016.00363
64. Silva JC, Rodrigues NC, Thompson-Souza GA, de S Muniz V, Neves JS, Figueiredo RT. Mac-1 triggers neutrophil DNA extracellular trap formation to Aspergillus fumigatus independently of PAD4 histone citrullination. J Leukoc Biol (2020) 107:69–83. doi: 10.1002/JLB.4A0119-009RR
65. Maas SL, Soehnlein O, Viola JR. Organ-specific mechanisms of transendothelial neutrophil migration in the lung, liver, kidney, and aorta. Front Immunol (2018) 9:2739:2739. doi: 10.3389/fimmu.2018.02739
66. Ostermann G, Weber KS, Zernecke A, Schroder A, Weber C. JAM-1 is a ligand of the β2 integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol (2002) 3:151–8. doi: 10.1038/ni755
67. Filippi MD, Harris CE, Meller J, Gu Y, Zheng Y, Williams DA. Localization of Rac2 via the C terminus and aspartic acid 150 specifies superoxide generation, actin polarity and chemotaxis in neutrophils. Nat Immunol (2004) 5:744–51. doi: 10.1038/ni1081
68. Lämmermann T, Afonso PV, Angermann BR, Wang JM, Kastenmüller W, Parent CA, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature (2013) 498:371–5. doi: 10.1038/nature12175 2013.
69. Li N, Yang H, Wang M, Lü S, Zhang Y, Long M. Ligand-specific forces of LFA-1 and Mac-1 in neutrophil adhesion and crawling. Mol Biol Cell (2018) 29:408–18. doi: 10.1091/mbc.E16-12-0827
70. Buffone A, Anderson NR, Hammer DA. Human neutrophils will crawl upstream on ICAM-1 if Mac-1 is blocked. Biophys J (2019) 117:1393–404. doi: 10.1016/j.bpj.2019.08.044
71. Wilson ZS, Ahn LB, Serratelli WS, Belley MD, Lomas-Nera J, Sen M, et al. Activated β2 integrins restrict neutrophil recruitment during murine acute Pseudomonal pneumonia. Am J Respir Cell Mol Biol (2017) 56:620–7. doi: 10.1165/rcmb.2016-0215OC
72. Reber LL, Gillis CM, Starkl P, Jönsson F, Sibilano R, Marichal T, et al. Neutrophil myeloperoxidase diminishes the toxic effects and mortality induced by lipopolysaccharide. J Exp Med (2017) 214:1249–58. doi: 10.1084/jem.20161238
73. Perretti M, Christian H, Wheller SK, Aiello I, Mugridge KG, Morris JF, et al. Annexin 1 is stored within gelatinase granules of human neutrophil and mobilized on the cell surface upon adhesion but not phagocytosis. Cell Biol Int (2000) 24:163–74. doi: 10.1006/cbir.1999.0468
74. Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol (2009) 9:62–70. doi: 10.1038/nri2470
75. Drechsler M, de Jong R, Roissant J, Viola JR, Leoni G, Wang JM, et al. Annexin A1 counteracts chemokine-induced arterial myeloid cell recruitment. Circ Res (2015) 116:827–35. doi: 10.1161/CIRCRESAHA.116.305825
76. Chatterjee BE, Yona S, Rosignoli G, Young RE, Nourshargh S, Flower RJ, et al. Annexin 1-deficient neutrophils exhibit enhanced transmigration in vivo and increased responsiveness in vitro. J Leukoc Biol (2005) 78:639–46. doi: 10.1189/jlb.0405206
77. Cai TQ, Wright SD. Human leukocyte elastase is an endogenous ligand for the integrin CR3 (CD11b/CD18, Mac-1, alpha M beta 2) and modulates polymorphonuclear leukocyte adhesion. J Exp Med (1996) 184:1213–23. doi: 10.1084/jem.184.4.1213
78. Santoso S, Sachs UJ, Kroll H, Linder M, Ruf A, Preissner KT, et al. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J Exp Med (2002) 196:679–91. doi: 10.1084/jem.20020267
79. Nourshargh S, Renshaw SA, Imhof BA. Reverse migration of neutrophils: Where, when, how and why? Trends Immunol (2016) 37:273–86. doi: 10.1016/j.it.2016.03.006
80. Lahoz-Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M, et al. Human neutrophil kinetics: modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood (2016) 127:3431–8. doi: 10.1182/blood-2016-03-700336
81. Tak T, Tesselaar K, Pillay J, Borghans JA, Koenderman L. What’s your age again? Determination Hum neutrophil half-lives revisited J Leukoc Biol (2013) 94:595–601. doi: 10.1189/jlb.1112571
82. Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JA, et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood (2010) 116:625–7. doi: 10.1182/blood-2010-01-259028
83. Ballesteros I, Rubio-Ponce A, Genua M, Lusito E, Kwok I, Fernández-Calvo G, et al. Co-option of neutrophil fates by tissue environments. Cell (2020) 183:1282–97.e18. doi: 10.1016/j.cell.2020.10.003
84. Pfirschke C, Engblom C, Gungabeesoon J, Lin Y, Rickelt S, Zilionis R, et al. Tumor-promoting Ly-6G+ SiglecFhigh cells are mature and long-lived neutrophils. Cell Rep (2020) 32:108164. doi: 10.1016/j.celrep.2020.108164
85. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol (2002) 2:965–75. doi: 10.1038/nri957 2002.
86. Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discovery (2004) 3:401–16. doi: 10.1038/nrd1383
87. Garlichs CD, Eskafi S, Cicha I, Schmeisser A, Walzog B, Raaz D, et al. Delay of neutrophil apoptosis in acute coronary syndromes. J Leukoc Biol (2004) 75:828–35. doi: 10.1189/jlb.0703358
88. Uddin M, Nong G, Ward J, Seumois G, Prince LR, Wilson SJ, et al. Prosurvival activity for airway neutrophils in severe asthma. Thorax (2010) 65:684–9. doi: 10.1136/thx.2009.120741
89. Keel M, Ungethum U, Steckholzer U, Niederer E, Hartung T, Trentz O, et al. Interleukin-10 counterregulates proinflammatory cytokine-induced inhibition of neutrophil apoptosis during severe sepsis. Blood (1997) 90:3356–63. doi: 10.1182/blood.V90.9.3356
90. Jonsson H, Allen P, Peng SL. Inflammatory arthritis requires Foxo3a to prevent Fas ligand-induced neutrophil apoptosis. Nat Med (2005) 11:666–71. doi: 10.1038/nm1248
91. Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med (2006) 12:1056–64. doi: 10.1038/nm1468
92. Dzhagalov I, St. John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood (2007) 109:1620–6. doi: 10.1182/blood-2006-03-013771
93. Klebanoff SJ. Myeloperoxidase: friend or foe. J Leukoc Biol (2005) 77:598–625. doi: 10.1189/jlb.1204697
94. Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev (2007) 219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x
95. Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol (2008) 4:278–86. doi: 10.1038/nchembio.85
96. Arnhold J, Flemmig J. Human myeloperoxidase in innate and acquired immunity. Arch Biochem Biophys (2010) 500:92–106. doi: 10.1016/j.abb.2010.04.008
97. Gorudko IV, Grigorieva DV, Sokolov AV, Shamova EV, Kostevich VA, Kudryavtsev IV, et al. Neutrophil activation in response to monomeric myeloperoxidase. Biochem Cell Biol (2018) 96:592–601. doi: 10.1139/bcb-2017-0290
98. Perskvist N, Long M, Stendahl O, Zheng L. Mycobacterium tuberculosis promotes apoptosis in human neutrophils by activating caspase-3 and altering expression of Bax/Bcl-xL via an oxygen-dependent pathway. J Immunol (2002) 168:6358–65. doi: 10.4049/jimmunol.168.12.6358
99. De Leo FR. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis (2004) 9:399–413. doi: 10.1023/B:APPT.0000031448.64969.fa
100. Pokhrel S, Triplett KD, Daly SM, Joyner JA, Sharma G, Hathaway HJ, et al. Complement receptor 3 contributes to the sexual dimorphism in neutrophil killing of Staphylococcus aureus. J Immunol (2020) 205:1593–600. doi: 10.4049/jimmunol.2000545
101. Freeman SA, Goyette J, Furuya W, Woods EC, Bertozzi CR, Bergmeier W, et al. Integrins form an expanding diffusion barrier that coordinates phagocytosis. Cell (2016) 164:128–40. doi: 10.1016/j.cell.2015.11.048
102. Mollnes TE, Brekke OL, Fung M, Fure H, Christiansen D, Bergseth G, et al. Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood (2002) 100:1869–77.
103. Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol (2005) 23:821–52. doi: 10.1146/annurev.immunol.23.021704.115835
104. Van den Berg CW, Tambourgi DV, Clark HW, Hoong SJ, Spiller OB, McGreal EP. Mechanism of neutrophil dysfunction: Neutrophil serine proteases cleave and inactivate the C5a receptor. J Immunol (2014) 192:1787–95. doi: 10.4049/jimmunol.1301920
105. Conway Morris A, Kefala K, Wilkinson TS, Dhaliwal K, Farrell L, Walsh T, et al. C5a mediates peripheral blood neutrophil dysfunction in critically ill patients. Am J Respir Crit Care Med (2009) 180:19–28. doi: 10.1164/rccm.200812-1928OC
106. Unnewehr H, Rittirsch D, Sarma JV, Zetoune F, Flierl MA, Perl M, et al. Changes and regulation of the C5a receptor on neutrophils during septic shock in humans. J Immunol (2013) 190:4215–25. doi: 10.4049/jimmunol.1200534
107. Poon IKH, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol (2014) 14:166–80. doi: 10.1038/nri3607
108. Durant S, Pederzoli M, Lepelletier Y, Canteloup S, Nusbaum P, Lesavre P, et al. Apoptosis-induced proteinase 3 membrane expression is independent from degranulation. J Leukoc Biol (2004) 75:87–98. doi: 10.1189/jlb.0203079
109. Podolnikova NP, Kushchayeva YS, Wu Y, Faust J, Ugarova TP. The Role of integrins αMβ2 (Mac-1, CD11b/CD18) and αDβ2 (CD11d/CD18) in macrophage fusion. Am J Pathol (2016) 186:2105–16. doi: 10.1016/j.ajpath.2016.04.001
110. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
111. Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A. The balancing act of neutrophils. Cell Host Microbe (2014) 15:526–36. doi: 10.1016/j.chom.2014.04.011
112. Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol (2006) 8:668–76. doi: 10.1111/j.1462-5822.2005.00659
113. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176:231–41. doi: 10.1083/jcb.200606027
114. Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol (2014) 15:703–8. doi: 10.1038/nrm3890
115. Pilsczek FH, Salina D, Poon KKH, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
116. Rochael NC, Guimarães-Costa AB, Nascimento MTC, De Souza-Vieira TS, Oliveira MP, Garcia Souza LF, et al. Classical ROS-dependent and early/rapid ROS-independent release of neutrophil extracellular traps triggered by Leishmania parasites. Sci Rep (2015) 5:18302 doi: 10.1038/srep18302. doi: 10.1038/srep18302
117. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
118. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe (2012) 12:324–33. doi: 10.1016/j.chom.2012.06.011
119. Rossaint J, Herter JM, Van Aken H, Napirei M, Döring Y, Weber C, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood (2014) 123:2573–84. doi: 10.1182/blood-2013-07-516484
120. Xu Z, Cai J, Gao J, White GC2, Chen F, Ma YQ. Interaction of kindlin-3 and β2-integrins differentially regulates neutrophil recruitment and NET release in mice. Blood (2015) 126:373–7. doi: 10.1182/blood-2015-03-636720
121. Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest (2002) 110:955–63. doi: 10.1172/JCI15918
122. Jerke U, Mariono SF, Daumke O, Kettritz R. Characterization of the CD177 interaction with the ANCA antigen proteinase 3. Sci Rep (2017) 7:43328. doi: 10.1038/srep43328
123. Jerke U, Rolle S, Dittmar G, Bayat B, Santoso S, Sporbert A, et al. Complement receptor Mac-1 is an adaptor for NB1 (CD177)-mediated PR3-ANCA neutrophil activation. J Biol Chem (2011) 286:7070–81. doi: 10.1074/jbc.M110.171256
124. Ley K, Rivera-Nieves J, Sandborn WJ, Shattil S. Integrin-based therapeutics: biological basis, clinical use and new drugs. Nat Rev Drug Discovery (2016) 15:173–83. doi: 10.1038/nrd.2015.10
125. Wolf D, Anto-Michel N, Blankenbach H, Wiedermann A, Buscher K, Hohmann JD, et al. A ligand-specific blockade of the integrin Mac-1 selectively targets pathologic inflammation while maintaining protective host-defense. Nat Commun (2018) 9:525. doi: 10.1038/s41467-018-02896-8
126. Kelm M, Lehoux S, Azcutia V, Cummings RD, Nusrat A, Parkos CA, et al. Regulationof neutrophil function by selective targeting of glycan epitopes expressed on the integrin CD11b/CD18. FASEB J (2020) 34:2326–43. doi: 10.1096/fj.201902542R
127. Maiguel D, Faridi MH, Wei C, Kuwano Y, Balla KM, Hernandez D, et al. Small molecule–mediated activation of the integrin CD11b/CD18 reduces inflammatory disease. Sci Signal (2011) 4:ra57. doi:10.1126/scisignal.2001811(2001)(
128. Schmid MC, Khan SQ, Kaneda MM, Pathria P, Shephard R, Louis TL, et al. Integrin CD11b activation drives anti-tumor innate immunity. Nat Commun (2018) 9:5379. doi: 10.1038/s41467-018-07387-4
129. Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, et al. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med (2002) 8:1296–302. doi: 10.1038/nm786
130. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature (2014) 510:92–101. doi: 10.1038/nature13479
131. Serhan CN. Treating inflammation and infection in the 21st century: new hints for decoding resolution mediators and mechanisms. FASEB J (2017) 31:1273–88. doi: 10.1096/fj.201601222R
132. Chiang N, Serhan CN. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol Aspects Med (2017) 58:114–29. doi: 10.1016/j.mam.2017.03.005
133. Serhan CN, Gupta SK, Perretti M, Godson C, Brennan E, Li Y, et al. The Atlas of Inflammation Resolution (AIR). Mol Aspects Med (2020) 74:100894. doi: 10.1016/j.mam.2020.100894
134. Brancaleone V, Dalli J, Bena S, Flower RJ, Cirino G, Perretti M. Evidence for an anti-inflammatory loop centered on polymorphonuclear leukocyte formyl peptide receptor 2/lipoxin A4 receptor and operative in the inflamed microvasculature. J Immunol (2011) 186:4905–14. doi: 10.4049/jimmunol.1003145
Keywords: neutrophils, neutrophil trafficking, phagocytosis-induced cell death, apoptosis, NET formation, immunity, resolution of inflammation, Mac-1 (αMβ2)
Citation: Sekheri M, Othman A and Filep JG (2021) β2 Integrin Regulation of Neutrophil Functional Plasticity and Fate in the Resolution of Inflammation. Front. Immunol. 12:660760. doi: 10.3389/fimmu.2021.660760
Received: 29 January 2021; Accepted: 08 March 2021;
Published: 30 March 2021.
Edited by:
Sonja Vermeren, University of Edinburgh, United KingdomReviewed by:
Jonathan S. Reichner, Rhode Island Hospital, United StatesIoannis Kourtzelis, University of York, United Kingdom
Scott I. Simon, University of California, Davis, United States
Copyright © 2021 Sekheri, Othman and Filep. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: János G. Filep, amFub3MuZy5maWxlcEB1bW9udHJlYWwuY2E=