 Xiaofang Wang
Xiaofang Wang Panpan Yi1*
Panpan Yi1* Yuejin Liang
Yuejin Liang- 1Department of Infectious Diseases, Key Laboratory of Viral Hepatitis of Hunan, Xiangya Hospital, Central South University, Changsha, China
- 2Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX, United States
- 3Institute for Human Infections and Immunity, University of Texas Medical Branch, Galveston, TX, United States
IL-36 is a member of the interleukin 1 cytokine family, which is currently experiencing a renaissance due to the growing understanding of its context-dependent roles and advances in our understanding of the inflammatory response. The immunological role of IL-36 has revealed its profound and indispensable functional roles in psoriasis, as well as in several inflammatory diseases, including inflammatory bowel disease (IBD), systemic lupus erythematosus, rheumatoid arthritis (RA) and cancer. More recently, an increasing body of evidence suggests that IL-36 plays a crucial role in viral, bacterial and fungal infections. There is a growing interest as to whether IL-36 contributes to host protective immune responses against infection as well as the potential implications of IL-36 for the development of new therapeutic strategies. In this review, we summarize the recent progress in understanding cellular expression, regulatory mechanisms and biological roles of IL-36 in infectious diseases, which suggest more specific strategies to maneuver IL-36 as a diagnostic or therapeutic target, especially in COVID-19.
Introduction
Interleukin (IL)-36 is a member of the IL-1 cytokine family. It plays a role in the orchestration of innate and adaptive immunity and appears to have pro-inflammatory activities (1, 2). The IL-36 family includes three agonist ligands (IL-36α, β and γ, previously known as IL-1F6, IL-1F8, IL-1F9), which bind to heterodimeric receptor complexes, the IL-36 receptor (IL-36R, also known as IL-1Rrp2) and co-receptor IL-1 receptor accessory protein (IL-1RAcP) (3). The IL-36 receptor antagonist (IL-36Ra, formerly known as IL-1F5), an antagonist in the IL-36 family, inhibit IL-36-induced inflammation via competing with IL-36 receptor (4). The pro-inflammatory role of IL-36 is well studied in psoriasis (5–9), inflammatory bowel disease (IBD), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and cancer (5, 10–14). IL-36γ is a potential diagnostic marker of psoriatic inflammation (15). The success of treatment using a monoclonal antibody against IL-36 receptor in generalized pustular psoriasis patients highlights the promising potential strategy of blocking the IL-36/IL-36R signaling pathway in clinical therapy (7). IL-38 (previously known as IL-1F10), which shows the highest similarity of percentage amino acid identify with IL-1Ra and IL-36Ra, may act as an IL-36R antagonist (16, 17). Recent study demonstrated the inhibitory function of IL-38 on the phosphorylation of P38 MAPK and the subunit P65 of NF-κB induced by IL-36γ in human keratinocytes and endothelial cells (18). Besides, IL-38 was released from apoptotic cells and restricted human macrophage-dependent induction of IL-17 (19). IL-38 knockout mice had delayed disease resolution with exacerbated IL-17-mediated inflammation, which is reversed by the administration of matured IL-38 in a mouse model of psoriasis (20). Hence, IL-38 is considered an anti-inflammatory factor in the pathologies of autoimmune diseases.
Accumulating evidence suggests that IL-36 is also involved in infectious diseases, especially viral and bacterial infections. Using knockout mice for either IL-36 cytokines or receptor, researchers have revealed that IL-36 plays both protective and pathological roles in distinct animal models of infection (21–23). On one hand, IL-36 is beneficial for pathogen clearance by promoting protective immune responses (21). On the other hand, IL-36 amplifies inflammatory responses, leading to excessive immune infiltration and tissue damage (24). In this review, we first focus on the cellular source and target cells of IL-36, and then highlight the recent advances of the IL-36 research in infectious diseases. At the end, we discuss IL-36 as a potential therapeutic target for COVID-19.
Processing of IL-36 and Downstream Signaling Pathways
Similar to other members of IL-1 family, the inactive precursors of IL-36 require proteolytic and post-translational processing for their maturation and pro-inflammatory activity, respectively (25, 26). Neutrophil granule-derived proteases cathepsin G (Cat G), elastase and proteinase-3 are involved in the processing (25–30). IL-36α is processed and activated by Cat G and elastase respectively via truncating at alanine 4 and lysine 3. IL-36β is selectively stimulated by Cat G through its cleavage at residue arginine 5 (28). IL-36γ can be activated by elastase or proteinase-3 by means of cleavage at the residue valine 15 (28). In addition, IL-36γ also can be cleaved between residues glutamic acid 17 and serine 18 by Cathepsin S (29). Removal of a small number of residues from the N termini of IL-36 increases the biological activity by more than 10,000-fold (26). Similarly, IL-36Ra is cleaved to become mature form by elastase through removal of its N-terminal methionine (30), and the matured IL-36Ra competes with IL-36 cytokines for IL-36 receptor binding to suppress IL-36 activity (26).
Binding of IL-36 agonists (IL-36α, -β and -γ) to IL-36R/IL-1RAcP heterodimer induces inflammatory mediators through MyD88-, MAPK- and NF-κB-dependent signaling pathways (31–33). It is demonstrated that Staphylococcus aureus (S. aureus) exposure drives murine skin inflammation, which is caused by the IL-36R/MyD88-mediated IL-17 (34). IL-36γ stimulation also promoted the expression of NF-κB target genes (TNFAIP3, NFKBIA, NFKB2, CXCL8, and BIRC3) in a MyD88-depenent manner in human epidermal keratinocytes (35). Besides, IL-36 α employed NF-κB and STAT3 for IκBζ induction, and induced several psoriasis-related cytokines and chemokines in psoriatic skin (32). Additionally, activation of IL-36/IL-36R axis enhanced the secretion of IL-6, IL-8, and granulocyte-macrophage colony-stimulating factor by activation of Erk1/2, MAPK and JNK (3), while IL-36Ra suppressed the IL-36 agonist-triggered IL-8 expression (26).
Cellular Source and Function of IL-36
At steady-state, IL-36 is mainly expressed in epithelial cells and fibroblasts (36–41). Several kinds of cells, including epithelial cells, mouse T cells and myeloid cells can respond to IL-36 stimuli (37, 38, 41–43).
Epithelial Cells
IL-36 was predominantly expressed in epithelial cells in experimental colitis, allergic lung inflammation, chronic rhinosinusitis and influenza A virus infection (22, 36, 41, 44, 45) and was upregulated by proinflammatory cytokines, such as IL-17 (41, 46). Reproductive tract epithelial cells also increased IL-36γ and IL-36R expression following treatment with microbial products (47). It is notable that epithelial IL-36 could be expressed as a full-length form and required a cleavage to the biologically active form (41). Epithelial cells can secret inflammatory cytokines in response to IL-36. Subcutaneous injection of IL-36α induced various inflammatory factors including IL-17, IL-20, IL-22, IL-23, interferon (IFN)-γ, TNF-α and KCs (48). IL-17 production by Th17 cells may upregulate all three IL-36 expression from human keratinocytes, creating a feedback loop that drives inflammation and disease development (49). Human keratinocytes were potent sources of chemokines following the exposure of IL-36 cytokines, leading to the recruitment of macrophages, T cells, and neutrophils (43). Notably, human keratinocytes upregulated type I and II IFN-responsive genes in response to IL-36, leading to potent cytokine production (35, 43). These findings indicate that IL-36 is critical for the early regulation of IFN and immune cell recruitment in the skin (50–52). Expression of IL-36R by skin-resident cells (e.g., keratinocytes and fibroblasts), but not the hematopoietic cells (e.g., T cells and DCs) is pivotal for the cutaneous pathology (51). Consistently, using a conditional knockout murine model, Goldstein et al. demonstrated that IL-36R signaling in keratinocytes played a major role in the induction of psoriasis-like dermatitis (52).
Myeloid Cells
Activated neutrophils were considered a source of IL-36 in various diseases such as experimental autoimmune encephalomyelitis (EAE), chronic rhinosinusitis and influenza infection (21, 41, 53), while neutrophils from naïve mice express low levels of IL-36γ (53). Importantly, IL-36R is abundant on murine neutrophils derived from bone marrow, spinal cord and spleen (38, 53). However, IL-36R was not detectable in blood neutrophils in both mice and patients with inflammatory diseases (41, 53). Consistently, healthy human blood neutrophils failed to express IL-36R and did not respond to IL-36 cytokines (43, 50). Interestingly, IL-36R expression on human peripheral neutrophils could be induced by IL-1β, IL-6, and Der p1 (41), suggesting that both IL-36 cytokines and receptor might be inducible on neutrophils by the local inflammatory milieu. However, a study also reported that IL-36-triggered human bronchial epithelial cell can release neutrophil-associated chemokines such as CXCL8, and promote infiltration, activation, and inflammatory activity of neutrophils (54). In addition, IL-36 may active neutrophils and amplify lung inflammation in mice (55)
Dermal macrophages expressed high amounts of IL-36R transcript (37), indicating that the expression of IL-36R might be associated with its anatomical localization and immune microenvironment. IL-36β was as potent as IL-1β in stimulating human M2 macrophages, but not M1 and dermal macrophages (37). In addition, both human M1 macrophages and mouse lung macrophages were reported to produce IL-36 ligands following bacterial infection and LPS exposure (5, 24), indicating that macrophages might be the source of IL-36 similar to IL-1β and IL-33. Bone marrow-derived macrophages have undetectable levels or express much lower IL-36R compared to DCs (38, 41). Both human and mouse DCs were found to express IL-36R and become activated by IL-36 agonists stimulation (41, 43, 50, 56). Human monocyte-derived DCs expressed 6-fold more IL-36R mRNA than their monocyte precursors and accelerated maturation by IL-36α, β and γ (43, 56). IL-36R was also detectable in human Langerhans cells, which responded strongly to IL-36β stimulation (37). Additionally, plasmacytoid DCs (pDCs) can highly express IL-36R (50, 56). These pDCs bound by IL-36 potentiated Toll-like Receptor (TLR)-9 activation and IFN-α production (50).
Lymphocytes
T cells are most likely not the main source of IL-36 cytokines, but can respond to IL-36 due to their expression of IL-36R. Unlike the receptors of other IL-1 family members, such as IL-33R and IL-1R, whose expression are upregulated during T cell activation, IL-36R expression is detectable on naïve T cells, but is negligible in differentiated Th cells (14, 57). It is reported that IL-36γ synergized with IL-12 to facilitate Th1 differentiation, but suppressed Th17 differentiation in vitro in murine experiments (36, 38). Interestingly, IL-36γ inhibited Foxp3-expression in murine regulatory T cell development through the IL-36R/MyD88/NF-κBp50 axis, while concomitantly promoted the differentiation of Th9 and Th22 cells (58, 59). Therefore, IL-36 plays a critical role in mouse T cell differentiation.
Whether IL-36 have effect on human T cells is still unclear. It is reported that IL-36 may induced IFN-γ production in human CD3+ lymphocytes in vitro (14, 56). Penha et al. found the IL-36R expression on CD4+ T cells in the human blood and intestines, and IL-36β stimulation promoted CD4+ T cell proliferation in vitro (42). On the contrary, other researchers reported that IL-36R transcripts were undetectable in blood CD4+ T cells from healthy donors, and IL-36 failed to effect on resting or activated human T cells (43). Similarly, no obvious colocalization of IL-36R with human T cells in nasal polyps (41). Further study is needed to elucidate the regulation of IL-36R as well as the role of IL-36 in human T cell activation and differentiation. Similar to mouse CD4+ T cells, mouse effector CD8+ T cells increased IFN-γ production by IL-36γ stimuli (14, 60), and this process required IL-12 or IL-2 synergy (60). In addition, IL-36γ promoted IFN-γ production in vitro by murine NK cells and γδ T cells, which were able to express IL-36R (14). IL-36R mRNA was undetectable in mouse B cells in a previous study (38); Resident B cells and plasma cells in inflamed human tissues were found to express IL-36α (61). CD138+ and CD79α+ plasma cells were identified as the cellular sources of IL-36α in the synovial tissues and psoriatic skin in patients, respectively (62). How IL-36 regulates B cell functions is still not understood.
Other Cell Types
IL-36R mRNA has been detected in mouse astrocytes and microglia in the brain, but not in primary neurons (39, 40). However, IL-36 cytokines were dispensable for microglia activation and disease development of EAE (39, 40, 53). Increased IL-36α γ expression was also observed in murine hepatocytes following IL-1β/TNF-α/IFN-γ stimulation (12, 63), indicating that IL-36 may play a role in liver diseases. In addition, IL-36β has a pro-inflammatory effect on human synovial fibroblasts and articular chondrocytes in RA, suggesting the potential role of IL-36 in inflammatory responses of autoimmune diseases (64).
IL-36 in Infectious Diseases
There is mounting evidence for the crucial role of IL-36 in infectious diseases via regulation of type I IFN, induction of inflammatory cytokines, recruitment of immune cells, modulation of immune cell activation and differentiation, and maintenance of mucosal integrity and barrier function (Figure 1). In this section, we focus on the functional roles of IL-36 in various infectious diseases (Table 1).
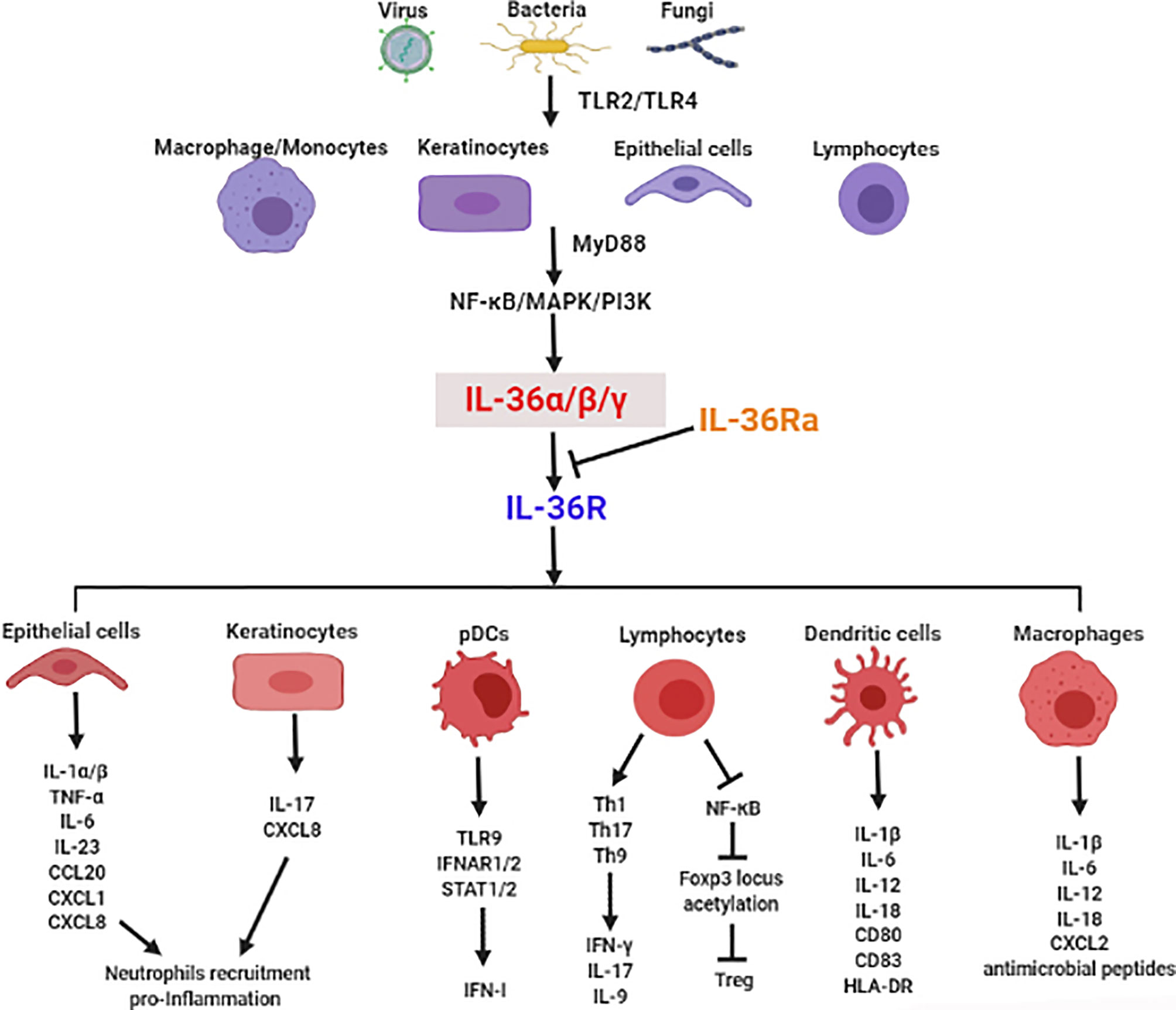
Figure 1 The crucial role of IL-36 in induction of inflammatory cytokines, recruitment of immune cells, modulation of immune cell activation and differentiation, and maintenance of mucosal integrity and barrier function in infectious diseases.
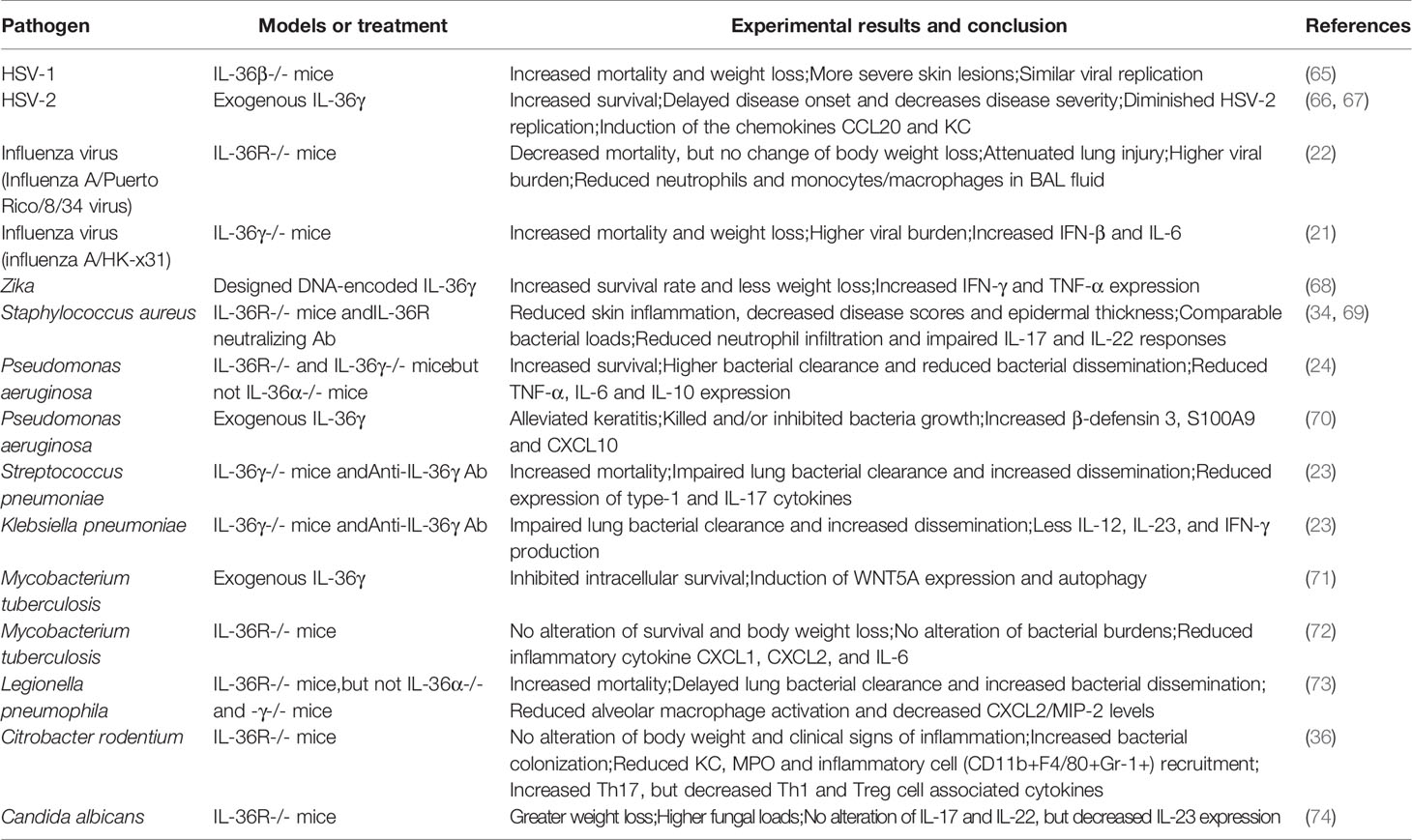
Table 1 The functional role of IL-36 in infectious diseases.
Skin and Mucosal Barriers
IL-36 was first identified as an inducible inflammatory cytokine in mouse keratinocytes following herpes simplex virus type 1 (HSV-1) infection (75). IL-36β-deficient mice developed more severe secondary zosteriform lesions and succumbed more frequently to HSV-1 infection (65). IL-36γ treatment protected mice from lethal intravaginal challenge, as evidenced by limited vaginal viral replication, delayed disease onset, decreased disease severity, and significantly increased survival (66). Further analysis demonstrated that IL-36β promoted type I IFN production through upregulation of IFN-α receptor expression and activation of the STAT signaling pathway in animal model (76). Indeed, IL-36 also promoted type I IFN in IL-36R+ pDC (50). Therefore, these studies indicate that IL-36 plays a critical role in innate immunity by boosting type I IFN signaling, inducing pro-inflammatory cytokines, and attracting innate immune cells, such as neutrophils.
Using a murine epicutaneous infection model, Nakagawa and Liu et al. found that S. aureus induced IL-1 and IL-36α from keratinocytes via secretion of S. aureus-expressed phenol-soluble modulin α, leading to the induction of IL-17 and recruitment of neutrophils in the skin (34, 69). Interestingly, IL-36α may not only regulate Th17 cell activity, but also modulate IL-17-production by γδ T cells and type 3 innate lymphoid cells (ILC3) (34, 69). Skin inflammation was dependent on IL-1R and IL-36R signals as well as their signaling adaptor MyD88. Satoh et al. also demonstrated that Cutibacterium acnes can induce IL-36γ through NF-κB in keratinocytes and subsequently IL-8, leading to cutaneous neutrophilia (77).
Fungal infection can induce IL-36 expression in epithelial cells and human PBMC (74, 78, 79). In oral candidiasis, IL-36α, β and γ transcript levels were all increased in the tongue of the sublingually challenged mice at 2 days post-injection (74). Candida albicans (C. albicans) infection resulted in increased IL-36 cytokines in human oral epithelial cells via NF-κB, MAPK and PI3K-dependent pathways (74). IL-36R-deficient mice were susceptible to acute oral candidiasis as evidenced by higher fungal loads and greater body weight loss, indicating the protective role of IL-36 in C. albicans infection (74).
Lung
Influenza virus infection can trigger epithelial cell-derived IL-36 cytokines (22, 46, 80), which activated NF-κB signaling and increased inflammatory cytokines (e.g. IL-6 and IL-8) in the lung (46). However, the role of IL-36 in influenza virus infection is incompletely understood. Aoyagi et al. reported that IL-36R-deficient mice were protected from influenza virus-induced lung injury and mortality accompanied by reduced lymphocyte activation, accumulation of myeloid cells, pro-inflammatory cytokine and chemokine production (e.g., IL-6, IL-17, CXCL1, and CXCL10) and permeability of the alveolar-epithelial barrier (22). However, IL-36γ was upregulated in the lungs and played a protective role in severe H1N1 and H3N2 influenza infection via modulating macrophage polarization and activity (21). Lack of IL-36γ resulted in increased viral titers, higher levels of IL-6, and more severe pathology in the lungs (21). Interestingly, macrophages in IL-36γ-deficient mice exhibited an M2-like phenotype and were likely to undergo apoptosis by infection, whereas adoptive transfer of WT alveolar macrophages protected IL-36γ-deficient mice against influenza infection (21). The reason for the discrepancies from the studies using IL-36R- and IL-36γ-deficient mice are not known at present. Different animal models and interfering strategies, such as neutralizing antibodies, should be used to further confirm these results.
The role of IL-36 in Mycobacterium tuberculosis (M. tuberculosis) has been documented in several studies. M. tuberculosis infection induced IL-36γ expression in human macrophages in vitro, and in the lungs of infected mice in vivo (72, 81). Its expression was induced through microbial ligands, which triggered host TLR and MyD88-dependent pathways, and was further amplified by endogenous IL-1β and IL-18 (81). Increased IL-36γ transcriptional expression was also observed in the plasma and bronchoalveolar lavage (BAL) samples of patients with Pseudomonas aeruginosa (P. aeruginosa)- or Streptococcus pneumoniae (S. pneumoniae)-induced acute respiratory distress syndrome (ARDS) (23, 24). Animal studies revealed that IL-36 signaling pathway may play a protective role in the lung with bacterial infection. The induction of IL-36 contributed to antimicrobial peptide production and M. tuberculosis growth restriction through promoting the accumulation of Liver X Receptor and modulating cholesterol biosynthesis and efflux (82). Activation of autophagy in macrophages was considered another hallmark by IL-36γ in restricting M. tuberculosis growth (71). However, IL-36R deficiency showed negligible impact on M. tuberculosis infection in mice, as demonstrated by similar survival rates and bacterial loads (72). Additionally, IL-36γ-deficient mice were more susceptible to S. pneumoniae infection, as evidenced by increased mortality, ameliorated lung bacterial clearance and increased bacterial dissemination, which might be due to the reduced type-1 cytokine expression and impaired lung macrophage M1 polarization (23). Similarly, the protective effect of IL-36γ was also demonstrated in a Klebsiella pneumoniae (K. pneumoniae) mouse model (23). Interestingly, Sequeira et al. revealed that microbiota Bacteroidetes protected against K. pneumoniae colonization (83) via IL-36 signals and macrophages (83). In addition, administration of Legionella pneumophila to IL-36R-deficient mice resulted in more severe disease as evidenced by higher mortality, delayed lung bacterial clearance, increased bacterial dissemination to the spleen, and impaired innate immune responses compared to that in infected wild-type mice (73). In contrast, IL-36R-/- and IL-36γ-/-, but not IL-36α-/-, mice were resistant to during P. aeruginosa infection, as demonstrated by the reduction of bacterial burden, pro-inflammatory cytokine production and lung injury. Further investigation is needed to determine the role of IL-36 in intracellular bacterial infection using various interfering methods, such as IL-36 cytokine knockout mice and neutralizing antibodies.
Gut
Clinical evidence showed that ulcerative colitis patients had higher IL-36α in the colonic mucosa (36). Lack of IL-36R resulted in defective recovery following DSS-induced damage and impaired closure of colonic mucosal biopsy wounds due to the profound reduction of IL-22 (84). Interestingly, IL-36 can also regulate Treg/Th9 balance and the IL-23/IL-22 network in model of colitis induced by oxazolone, indicating that IL-36γ has multiple functions in modulating antigen-presenting cell function and in regulating T cell differentiation in a mouse model (58, 59). Russel et al. reported that infection with Citrobacter rodentium resulted in reduced CD11b+F4/80+Gr-1+ inflammatory cell recruitment, imbalanced Th1/Th17 responses and increased bacterial colonization of the colon in IL-36R-/- mice (36). Accordingly, suppressed Th17, but enhanced Th1 differentiation was observed in vitro by IL-36α supplement (36, 57). However, since IL-36 is necessary for IL-22 production in DSS-induced colitis, it is not clear whether IL-36 differently regulates Th17 and Th22 differentiation in vivo among various animal models of gastrointestinal dysregulation.
Other Organs
Although IL-36 has been detected in hepatocytes (12, 63), the function of IL-36 in the liver remains unclear. Higher levels of IL-36α were observed in chronic hepatitis B virus (HBV) patients compared with that in healthy individuals (85). The positive correlation between IL-36α and HBV-DNA titers may indicate the potential involvement of IL-36 in antiviral immunity during chronic infection (85). Additionally, hepatitis C virus infection significantly increased the production of IL-36Ra but not IL-36 agonist ligands in human monocytes, leading to reduced NK cell activation (86). Further research is needed to dissect the role of IL-36 in liver resident cells (e.g., kupffer cells, hepatic stellate cells and sinusoidal endothelial cells) as well as in different liver disease models.
In addition to lung infections, IL-36α and IL-36γ were also upregulated in the mouse cornea in early responses to P. aeruginosa challenge (70). Exogenous IL-36γ treatment enhanced corneal innate immunity and alleviated P. aeruginosa keratitis. The protective role of IL-36γ required S100A9 and was partially dependent on the CXCL10/CXCR3 axis (70). On the contrary, IL-36Ra treatment exacerbated the outcome of P. aeruginosa keratitis (70).
Louis et al. reported that a truncated IL-36γ-encoded plasmid can act as a potent adjuvant for a DNA-encoded Zika virus (ZIKV) vaccine. Immunization with truncated IL-36γ promoted antiviral T cell responses and protected mice from ZIKV challenge (68). Moreover, co-delivery of truncated IL-36γ can also enhance antiviral immunity against HIV and influenza DNA vaccines (68). Besides, both in vivo and in vitro studies have proved that IL-36 treatment reduced HSV-2 replication in a lethal genital infection model and in human vaginal epithelial cells (66, 67). The absence of IL-36γ led to reduced mature neutrophil recruitment to the vaginal microenvironment at early times in HSV-2 infection (66). These findings set the stage for IL-36 in infectious diseases and shed light on IL-36 in the next generation of vaccines.
IL-36 as a Therapeutic Target of COVID-19
Although several vaccines have been issued for the emergency use authorization for the prevention of coronavirus disease 2019 (COVID-19), intensive efforts are underway to investigate the immunopathology of this infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The majority of patients with COVID-19 are asymptomatic or mild flu symptoms, but in some individuals, who are critically ill with COVID-19, it can develop into severe pneumonia and life-threating ARDS. The members of IL-1 family including IL-1β and IL-33 may contribute to the inflammation and antiviral immune regulation in COVID-19. In severe cases of COVID-19 patients, increased IL-1α and IL-1β have been detected (87, 88). SARS2-CoV-2 may facilitate IL-1β activation and maturation, leading to the cytokine storm together with other pro-inflammatory mediators such as IL-6 and TNF-α (89). Blockage of IL-1 signals using IL-1 receptor antagonist Anakinra might be associated with clinical improvement in patients (87, 88). The alarmin cytokine IL-33 may also play a detrimental role in severe COVID-19 cases through expanding the pathogenic T cells, inducing hyperinflammation, and promoting the pro-fibrotic type 2 innate immune cells (90).
In patients with COVID-19, airway epithelial cells showed an average three-fold increase in expression of the SARS-CoV-2 entry receptor angiotensin-converting enzyme-2 ACE2 (91). Notably, bronchial epithelial cell ACE2 expression was correlated with IL-36β in bronchoalveolar lavage in asthma cohorts (92). Moreover, human basal lung epithelial cells exposed to poly(I:C) exhibited significant increase in protein concentrations of IL-36γ (55). SARS-CoV-2 viral RNA and viral nucleocapsid protein can be detected in gastrointestinal tissues from the patients (93, 94). This might be due to the highly expressed ACE2 in human gastrointestinal epithelial cells (95). IL-36γ was predominantly detected in human intestinal epithelium (44), and induced expression of chemokines, GM-CSF and IL-6 (44). Therefore, IL-36 may contribute to the ACE2 regulation and intestinal inflammation in COVID-19 patients. In addition, vasculopathy and lymphoid infiltrate of the superficial and deep dermis is main cutaneous manifestations in COVID-19 patient (96–99). It was reported that ACE2 and SARS-CoV-2 RNA can be detected in the blood vessels (100, 101), whereas IL-36γ and IL-36R also expressed in human dermal microvascular endothelial cells (HDMEC) (18, 102, 103). It is likely that SARS-CoV-2 infection in endothelia cells may induce IL-36 secretion, leading to leukocytes infiltration and skin symptoms in COVID-19 patients. Furthermore, high expression of ACE2 was also found in keratinocytes (104), which can increase IL-36 expression by Poly I:C stimuli (80, 105). Besides, IL-36 upregulated ACE2 expression in human keratinocytes according to publicly available RNAseq data (106). SARS-CoV-2 infection may promote IL-36 production from keratinocytes and exacerbate skin lesion. These findings suggest that IL-36 might be a potential biomarker of disease severity in COVID-19.
Profound pulmonary infiltration of myeloid cells including neutrophils and macrophages/monocytes have been found in COVID-19 patients with severe clinical progression (107–109). Local IL-36 may drive these myeloid cell recruitment and activation, resulting in pulmonary hyper-inflammation (37, 41, 55). Moreover, infiltrated neutrophils may produce high concentrations of neutrophil extracellular traps (NETs) (110–112), and induce lung epithelial cell death in COVID-19 patients (110). In addition, IL-36 can induce IL-6 and IL-8 expression and further increase inflammatory responses (3), while IL-1β and IL-6 are capable of inducing IL-36 expression (81). This proinflammatory positive loop may also contribute to immunopathogenesis of COVID-19. IL-36 was upregulated in the lungs after influenza virus infection (22, 46), and led to inflammatory cytokines production (22). GM-CSF, which is rapidly produced by pathogenic Th1 cells in COVID-19, can act with other inflammatory cytokines to form a cascade signature of inflammatory monocytes with high IL-6 expression (113). Importantly, IL-36 increases the secretion of GM-CSF by activation of Erk1/2, MAPK and JNK (3). IL-36γ cooperated with poly(I:C) in human macrophages also promoted GM-CSF expression (55). These findings indicate that IL-36 may contribute to the induction of IL-6-producing monocytes through GM-CSF. Moreover, IL-36, as a strong inducer of murine Th1 cells (14, 57), may play a role in human Th1 differentiation (56), and exacerbate lung pathogenesis by enhancing pathogenic Th1 responses and the following cytokine storm. In addition to Th1 responses, IL-36 is also a key regulator in IL-17 responses through regulating not only adaptive Th cells, but also γδ T cells and ILC3 (34, 69). Notably, elevated IL-17 levels have been reported in patients infected with coronavirus, including SARS-CoV, MERS and SAR-CoV-2 (89). Blockage of IL-36 signals may lead to proposals for a therapeutic approach to COVID-19 through modulating proinflammatory IL-17 responses.
The application of IL-1 receptor antagonist Anakinra has shown the potential therapeutic effect in COVID-19 patients (87, 88, 114). In addition to the agonistic ligands, IL-36Ra acts as an antagonist for IL-36 signaling pathway and may reduce IL-36-driven inflammation via competing with their receptor IL-36R. Additionally, IL-38, the newest member of IL-36 family, can downregulate poly (I:C)-induced IL-6, CCL5, and IL-1β expressions in bronchial epithelial cells, indicating the anti-inflammatory role of IL-38 in viral infection (115). Notably, it is reported that IL-38 increased significantly in influenza and COVID-19 patients and may function as a suppressor cytokine that inhibits IL-1, IL-6 and TNF-α in COVID patients (116, 117). Significant efforts are undergoing to develop neutralizing antibody targeting the IL-36R signaling axis for the therapy of IL-36-mediated diseases. Antibody-mediated blockade of IL-36R signaling reverses established fibrosis in chronic intestinal inflammation in mice (118) Chimeric antibodies MAB92 and MAB04, binding primarily to domain-2 of the human and mouse IL-36R proteins respectively, have been demonstrated to inhibit skin inflammation (69, 119). Anti-mouse IL-36R mAb M616 specific for murine IL-36R is also under experimental trials (120). Importantly, a single dose of BI 655130, a monoclonal antibody against the IL-36 receptor, reduced the severity of generalized pustular psoriasis in patients (7). Therefore, application of IL-36Ra, IL-38 and IL-36R mAbs might be a promising therapeutic way in COVID-19 patients via inhibiting IL-36-mediated hyperinflammation (Figure 2).
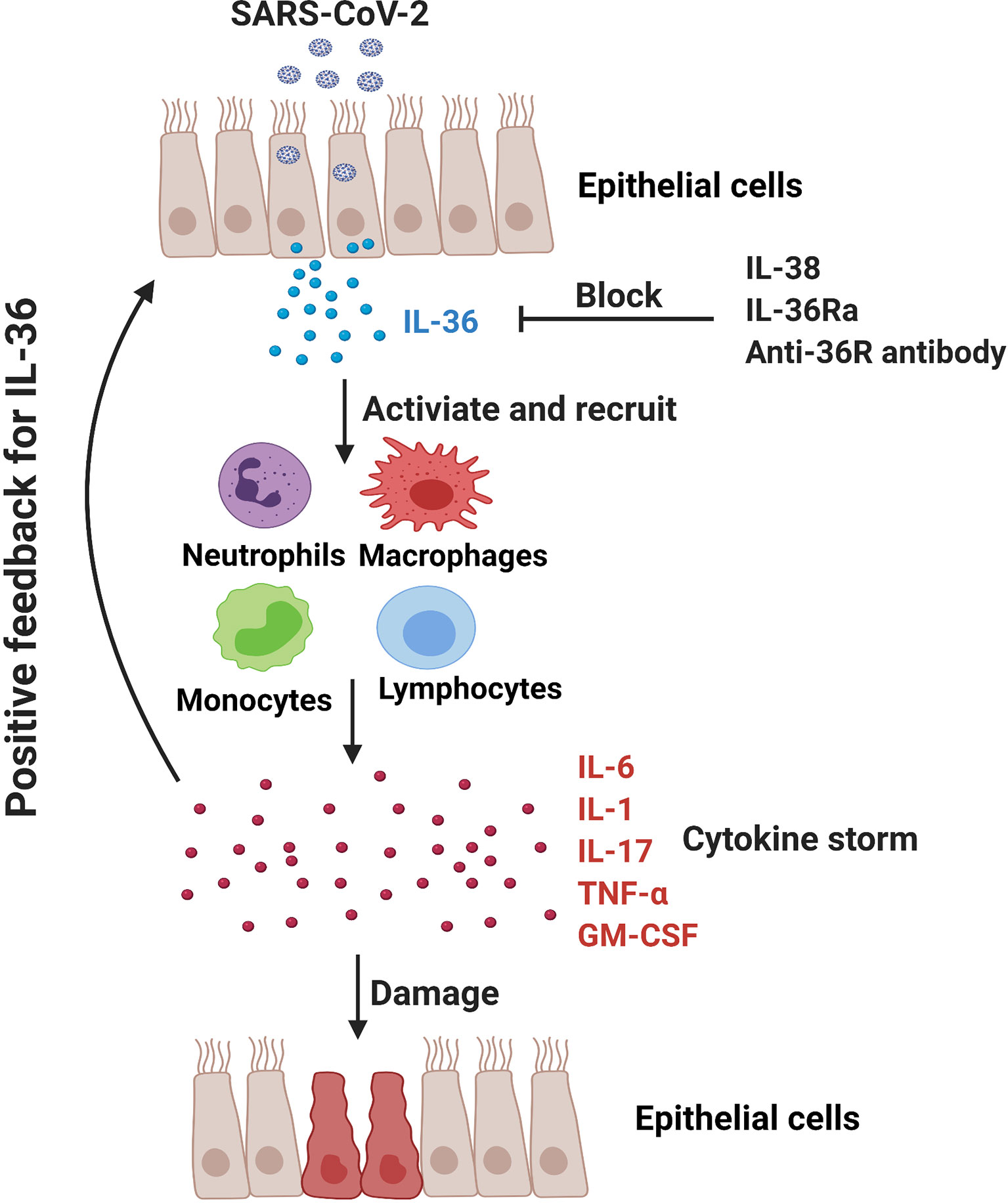
Figure 2 In COVID-19 patients, SARS-CoV-2 may promote hyperinflammation in the lung and exacerbate tissue damage. IL-36-activated inflammatory immune cells (e.g., monocytes, macrophages, neutrophils and pathogenic T cells) produce IL-6, IL-1, IL-17, TNF-α and GM-CSF to further amplify IL-36 responses. IL-36Ra and IL-38, as the natural antagonistic mediators in IL-36 family might be a promising therapeutic target for COVID-19 via inhibiting IL-36 signaling pathway and alleviating pulmonary hyperinflammation.
Conclusions and Perspectives
The accumulated evidence during the past decade indicates that IL-36 plays a fascinating role in systemic inflammatory diseases and cancer (36, 51, 121). The genetic deficiency of IL-36Ra leads to generalized pustular psoriasis (GPP), while IL-36Ra was considered an effective treatment of psoriasis diseases (106). Inhibition of IL-36R with a single dose of BI 655130 monoclonal antibody reduced the severity of GPP in patients (7). Interestingly, direct intra-tumoral delivery of IL-36 mRNA led to robust anticancer responses in a broad range of tumor microenvironments (122). These studies highlight the clinical therapeutic potential of IL-36 in inflammatory diseases and cancer.
Several aspects of IL-36 are less understood and remain somewhat controversial. It is still not clear what the distribution of IL-36R is in immune cells, especially in humans (41, 42). Whether the receptor is inducible by other host factors or pathogens is still not well understood.
IL-36 may be more than just a general inflammatory marker but a pathogenic sensor due to its location at epithelial/environmental interface and its release and activation by pathogenic damage (123). It is less well elucidated what the crucial bioactive forms of IL-36 in vivo are or how they are generated in each infectious disease condition. Moreover, it is striking that the different isoforms of IL-36 are expressed differently under physiological as well as pathological conditions, and have different functions in the development of infection. Further investigations are needed to elucidate the molecular mechanisms underlying their biological functions, especially in COVID-19. In terms of clinical implications, future study of the functions of the IL-36/IL-36R pathway in disease pathogenesis may facilitate the development of therapeutics targeting these cytokines for the treatment of infectious diseases.
Author Contributions
XW wrote the manuscript. PY and YL wrote and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China 81800506 and Natural Science Foundation of Hunan Province of China 2019JJ40494 to PY. The NIH AI153586 and the UTMB IHII Data Acquisition Grant to YL.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Jiaren Sun and Dr. Sherry Haller for manuscript revision. All figures are created with BioRender.com.
References
1. Garlanda C, Dinarello CA, Mantovani A. The Interleukin-1 Family: Back to the Future. Immunity (2013) 39(6):1003–18. doi: 10.1016/j.immuni.2013.11.010
2. Sims JE, Smith DE. The IL-1 Family: Regulators of Immunity. Nat Rev Immunol (2010) 10(2):89–102. doi: 10.1038/nri2691
3. Towne JE, Garka KE, Renshaw BR, Virca GD, Sims JE. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 Signal Through IL-1Rrp2 and IL-1racp to Activate the Pathway Leading to NF-Kappab and Mapks. J Biol Chem (2004) 279(14):13677–88. doi: 10.1074/jbc.M400117200
4. Mulero JJ, Pace AM, Nelken ST, Loeb DB, Correa TR, Drmanac R, et al. IL1HY1: A Novel Interleukin-1 Receptor Antagonist Gene. Biochem Biophys Res Commun (1999) 263(3):702–6. doi: 10.1006/bbrc.1999.1440
5. Boutet MA, Bart G, Penhoat M, Amiaud J, Brulin B, Charrier C, et al. Distinct Expression of Interleukin (IL)-36alpha, Beta and Gamma, Their Antagonist IL-36Ra and IL-38 in Psoriasis, Rheumatoid Arthritis and Crohn’s Disease. Clin Exp Immunol (2016) 184(2):159–73. doi: 10.1111/cei.12761
6. Todorovic V, Su Z, Putman CB, Kakavas SJ, Salte KM, McDonald HA, et al. Small Molecule IL-36gamma Antagonist as a Novel Therapeutic Approach for Plaque Psoriasis. Sci Rep (2019) 9(1):9089. doi: 10.1038/s41598-019-45626-w
7. Bachelez H, Choon SE, Marrakchi S, Burden AD, Tsai TF, Morita A, et al. Inhibition of the Interleukin-36 Pathway for the Treatment of Generalized Pustular Psoriasis. N Engl J Med (2019) 380(10):981–3. doi: 10.1056/NEJMc1811317
8. Li M, Han J, Lu Z, Li H, Zhu K, Cheng R, et al. Prevalent and Rare Mutations in IL-36RN Gene in Chinese Patients With Generalized Pustular Psoriasis and Psoriasis Vulgaris. J Invest Dermatol (2013) 133(11):2637–9. doi: 10.1038/jid.2013.267
9. Johnston A, Xing X, Guzman AM, Riblett M, Loyd CM, Ward NL, et al. IL-1F5, -F6, -F8, and -F9: A Novel IL-1 Family Signaling System That is Active in Psoriasis and Promotes Keratinocyte Antimicrobial Peptide Expression. J Immunol (2011) 186(4):2613–22. doi: 10.4049/jimmunol.1003162
10. Nishida A, Hidaka K, Kanda T, Imaeda H, Shioya M, Inatomi O, et al. Increased Expression of Interleukin-36, a Member of the Interleukin-1 Cytokine Family, in Inflammatory Bowel Disease. Inflammation Bowel Dis (2016) 22(2):303–14. doi: 10.1097/MIB.0000000000000654
11. Gresnigt MS, van de Veerdonk FL. Biology of IL-36 Cytokines and Their Role in Disease. Semin Immunol (2013) 25(6):458–65. doi: 10.1016/j.smim.2013.11.003
12. Pan QZ, Pan K, Zhao JJ, Chen JG, Li JJ, Lv L, et al. Decreased Expression of Interleukin-36alpha Correlates With Poor Prognosis in Hepatocellular Carcinoma. Cancer Immunol Immunother (2013) 62(11):1675–85. doi: 10.1007/s00262-013-1471-1
13. Chen F, Qu M, Zhang F, Tan Z, Xia Q, Hambly BD, et al. IL-36 s in the Colorectal Cancer: Is Interleukin 36 Good or Bad for the Development of Colorectal Cancer? BMC Cancer (2020) 20(1):92. doi: 10.1186/s12885-020-6587-z
14. Wang X, Zhao X, Feng C, Weinstein A, Xia R, Wen W, et al. IL-36gamma Transforms the Tumor Microenvironment and Promotes Type 1 Lymphocyte-Mediated Antitumor Immune Responses. Cancer Cell (2015) 28(3):296–306. doi: 10.1016/j.ccell.2015.07.014
15. Berekmeri A, Latzko A, Alase A, Macleod T, Ainscough JS, Laws P, et al. Detection of IL-36gamma Through Noninvasive Tape Stripping Reliably Discriminates Psoriasis From Atopic Eczema. J Allergy Clin Immunol (2018) 142(3):988–991 e4. doi: 10.1016/j.jaci.2018.04.031
16. Bensen JT, Dawson PA, Mychaleckyj JC, Bowden DW. Identification of a Novel Human Cytokine Gene in the Interleukin Gene Cluster on Chromosome 2q12-14. J Interferon Cytokine Res (2001) 21(11):899–904. doi: 10.1089/107999001753289505
17. van de Veerdonk FL, Stoeckman AK, Wu G, Boeckermann AN, Azam T, Netea MG, et al. IL-38 Binds to the IL-36 Receptor and has Biological Effects on Immune Cells Similar to IL-36 Receptor Antagonist. Proc Natl Acad Sci USA (2012) 109(8):3001–5. doi: 10.1073/pnas.1121534109
18. Mercurio L, Morelli M, Scarponi C, Eisenmesser EZ, Doti N, Pagnanelli G, et al. IL-38 has an Anti-Inflammatory Action in Psoriasis and its Expression Correlates With Disease Severity and Therapeutic Response to Anti-IL-17A Treatment. Cell Death Dis (2018) 9(11):1104. doi: 10.1038/s41419-018-1143-3
19. Mora J, Schlemmer A, Wittig I, Richter F, Putyrski M, Frank AC, et al. Interleukin-38 is Released From Apoptotic Cells to Limit Inflammatory Macrophage Responses. J Mol Cell Biol (2016) 8(5):426–38. doi: 10.1093/jmcb/mjw006
20. Han Y, Mora J, Huard A, da Silva P, Wiechmann S, Putyrski M, et al. IL-38 Ameliorates Skin Inflammation and Limits IL-17 Production From Gammadelta T Cells. Cell Rep (2019) 27(3):835–846 e5. doi: 10.1016/j.celrep.2019.03.082
21. Wein AN, Dunbar PR, McMaster SR, Li ZT, Denning TL, Kohlmeier JE. IL-36gamma Protects Against Severe Influenza Infection by Promoting Lung Alveolar Macrophage Survival and Limiting Viral Replication. J Immunol (2018) 201(2):573–82. doi: 10.4049/jimmunol.1701796
22. Aoyagi T, Newstead MW, Zeng X, Kunkel SL, Kaku M, Standiford TJ. IL-36 Receptor Deletion Attenuates Lung Injury and Decreases Mortality in Murine Influenza Pneumonia. Mucosal Immunol (2017) 10(4):1043–55. doi: 10.1038/mi.2016.107
23. Kovach MA, Singer B, Martinez-Colon G, Newstead MW, Zeng X, Mancuso P, et al. IL-36gamma is a Crucial Proximal Component of Protective Type-1-Mediated Lung Mucosal Immunity in Gram-Positive and -Negative Bacterial Pneumonia. Mucosal Immunol (2017) 10(5):1320–34. doi: 10.1038/mi.2016.130
24. Aoyagi T, Newstead MW, Zeng X, Nanjo Y, Peters-Golden M, Kaku M, et al. Interleukin-36gamma and IL-36 Receptor Signaling Mediate Impaired Host Immunity and Lung Injury in Cytotoxic Pseudomonas Aeruginosa Pulmonary Infection: Role of Prostaglandin E2. PloS Pathog (2017) 13(11):e1006737. doi: 10.1371/journal.ppat.1006737
25. Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, et al. IL-33 is Processed Into Mature Bioactive Forms by Neutrophil Elastase and Cathepsin G. Proc Natl Acad Sci USA (2012) 109(5):1673–8. doi: 10.1073/pnas.1115884109
26. Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, et al. Interleukin-36 (IL-36) Ligands Require Processing for Full Agonist (IL-36alpha, IL-36beta, and IL-36gamma) or Antagonist (IL-36Ra) Activity. J Biol Chem (2011) 286(49):42594–602. doi: 10.1074/jbc.M111.267922
27. Clancy DM, Sullivan GP, Moran HBT, Henry CM, Reeves EP, McElvaney NG, et al. Extracellular Neutrophil Proteases are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity But Poor Effectors of Microbial Killing. Cell Rep (2018) 22(11):2937–50. doi: 10.1016/j.celrep.2018.02.062
28. Henry CM, Sullivan GP, Clancy DM, Afonina IS, Kulms D, Martin SJ. Neutrophil-Derived Proteases Escalate Inflammation Through Activation of IL-36 Family Cytokines. Cell Rep (2016) 14(4):708–22. doi: 10.1016/j.celrep.2015.12.072
29. Ainscough JS, Macleod T, McGonagle D, Brakefield R, Baron JM, Alase A, et al. Cathepsin s is the Major Activator of the Psoriasis-Associated Proinflammatory Cytokine IL-36gamma. Proc Natl Acad Sci USA (2017) 114(13):E2748–57. doi: 10.1073/pnas.1620954114
30. Macleod T, Doble R, McGonagle D, Wasson CW, Alase A, Stacey M, et al. Neutrophil Elastase-Mediated Proteolysis Activates the Anti-Inflammatory Cytokine IL-36 Receptor Antagonist. Sci Rep (2016) 6:24880. doi: 10.1038/srep24880
31. Nguyen TT, Niyonsaba F, Ushio H, Akiyama T, Kiatsurayanon C, Smithrithee R, et al. Interleukin-36 Cytokines Enhance the Production of Host Defense Peptides Psoriasin and LL-37 by Human Keratinocytes Through Activation of Mapks and NF-Kappab. J Dermatol Sci (2012) 68(1):63–6. doi: 10.1016/j.jdermsci.2012.07.010
32. Muller A, Hennig A, Lorscheid S, Grondona P, Schulze-Osthoff K, Hailfinger S, et al. Ikappabzeta is a Key Transcriptional Regulator of IL-36-Driven Psoriasis-Related Gene Expression in Keratinocytes. Proc Natl Acad Sci USA (2018) 115(40):10088–93. doi: 10.1073/pnas.1801377115
33. Debets R, Timans JC, Homey B, Zurawski S, Sana TR, Lo S, et al. Two Novel IL-1 Family Members, IL-1 Delta and IL-1 Epsilon, Function as an Antagonist and Agonist of NF-Kappa B Activation Through the Orphan IL-1 Receptor-Related Protein 2. J Immunol (2001) 167(3):1440–6. doi: 10.4049/jimmunol.167.3.1440
34. Liu H, Archer NK, Dillen CA, Wang Y, Ashbaugh AG, Ortines RV, et al. Staphylococcus Aureus Epicutaneous Exposure Drives Skin Inflammation Via IL-36-Mediated T Cell Responses. Cell Host Microbe (2017) 22(5):653–666 e5. doi: 10.1016/j.chom.2017.10.006
35. Swindell WR, Beamer MA, Sarkar MK, Loftus S, Fullmer J, Xing X, et al. RNA-Seq Analysis of IL-1B and IL-36 Responses in Epidermal Keratinocytes Identifies a Shared Myd88-Dependent Gene Signature. Front Immunol (2018) 9:80. doi: 10.3389/fimmu.2018.00080
36. Russell SE, Horan RM, Stefanska AM, Carey A, Leon G, Aguilera M, et al. IL-36alpha Expression is Elevated in Ulcerative Colitis and Promotes Colonic Inflammation. Mucosal Immunol (2016) 9(5):1193–204. doi: 10.1038/mi.2015.134
37. Dietrich D, Martin P, Flacher V, Sun Y, Jarrossay D, Brembilla N, et al. Interleukin-36 Potently Stimulates Human M2 Macrophages, Langerhans Cells and Keratinocytes to Produce Pro-Inflammatory Cytokines. Cytokine (2016) 84:88–98. doi: 10.1016/j.cyto.2016.05.012
38. Vigne S, Palmer G, Lamacchia C, Martin P, Talabot-Ayer D, Rodriguez E, et al. IL-36R Ligands are Potent Regulators of Dendritic and T Cells. Blood (2011) 118(22):5813–23. doi: 10.1182/blood-2011-05-356873
39. Wang P, Meinhardt B, Andre R, Renshaw BR, Kimber I, Rothwell NJ, et al. The Interleukin-1-Related Cytokine IL-1F8 is Expressed in Glial Cells, But Fails to Induce IL-1beta Signalling Responses. Cytokine (2005) 29(6):245–50. doi: 10.1016/j.cyto.2004.12.002
40. Berglof E, Andre R, Renshaw BR, Allan SM, Lawrence CB, Rothwell NJ, et al. IL-1Rrp2 Expression and IL-1F9 (IL-1H1) Actions in Brain Cells. J Neuroimmunol (2003) 139(1-2):36–43. doi: 10.1016/s0165-5728(03)00130-9
41. Wang H, Li ZY, Jiang WX, Liao B, Zhai GT, Wang N, et al. The Activation and Function of IL-36gamma in Neutrophilic Inflammation in Chronic Rhinosinusitis. J Allergy Clin Immunol (2018) 141(5):1646–58. doi: 10.1016/j.jaci.2017.12.972
42. Penha R, Higgins J, Mutamba S, Barrow P, Mahida Y, Foster N. IL-36 Receptor is Expressed by Human Blood and Intestinal T Lymphocytes and is Dose-Dependently Activated Via IL-36beta and Induces CD4+ Lymphocyte Proliferation. Cytokine (2016) 85:18–25. doi: 10.1016/j.cyto.2016.05.023
43. Foster AM, Baliwag J, Chen CS, Guzman AM, Stoll SW, Gudjonsson JE, et al. IL-36 Promotes Myeloid Cell Infiltration, Activation, and Inflammatory Activity in Skin. J Immunol (2014) 192(12):6053–61. doi: 10.4049/jimmunol.1301481
44. Scheibe K, Backert I, Wirtz S, Hueber A, Schett G, Vieth M, et al. IL-36R Signalling Activates Intestinal Epithelial Cells and Fibroblasts and Promotes Mucosal Healing In Vivo. Gut (2017) 66(5):823–38. doi: 10.1136/gutjnl-2015-310374
45. Ramadas RA, Ewart SL, Medoff BD, LeVine AM. Interleukin-1 Family Member 9 Stimulates Chemokine Production and Neutrophil Influx in Mouse Lungs. Am J Respir Cell Mol Biol (2011) 44(2):134–45. doi: 10.1165/rcmb.2009-0315OC
46. Chustz RT, Nagarkar DR, Poposki JA, Favoreto S Jr., Avila PC, Schleimer RP, et al. Regulation and Function of the IL-1 Family Cytokine IL-1F9 in Human Bronchial Epithelial Cells. Am J Respir Cell Mol Biol (2011) 45(1):145–53. doi: 10.1165/rcmb.2010-0075OC
47. Winkle SM, Throop AL, Herbst-Kralovetz MM. IL-36gamma Augments Host Defense and Immune Responses in Human Female Reproductive Tract Epithelial Cells. Front Microbiol (2016) 7:955. doi: 10.3389/fmicb.2016.00955
48. Blumberg H, Dinh H, Dean C Jr., Trueblood ES, Bailey K, Shows D, et al. IL-1RL2 and its Ligands Contribute to the Cytokine Network in Psoriasis. J Immunol (2010) 185(7):4354–62. doi: 10.4049/jimmunol.1000313
49. Carrier Y, Ma HL, Ramon HE, Napierata L, Small C, O’Toole M, et al. Inter-Regulation of Th17 Cytokines and the IL-36 Cytokines In Vitro and In Vivo: Implications in Psoriasis Pathogenesis. J Invest Dermatol (2011) 131(12):2428–37. doi: 10.1038/jid.2011.234
50. Catapano M, Vergnano M, Romano M, Mahil SK, Choon SE, Burden AD, et al. IL-36 Promotes Systemic IFN-I Responses in Severe Forms of Psoriasis. J Invest Dermatol (2020) 140(4):816–826 e3. doi: 10.1016/j.jid.2019.08.444
51. Tortola L, Rosenwald E, Abel B, Blumberg H, Schafer M, Coyle AJ, et al. Psoriasiform Dermatitis is Driven by IL-36-Mediated DC-Keratinocyte Crosstalk. J Clin Invest (2012) 122(11):3965–76. doi: 10.1172/JCI63451
52. Goldstein JD, Bassoy EY, Caruso A, Palomo J, Rodriguez E, Lemeille S, et al. IL-36 Signaling in Keratinocytes Controls Early IL-23 Production in Psoriasis-Like Dermatitis. Life Sci Alliance (2020) 3(6):e202000688. doi: 10.26508/lsa.202000688
53. Bozoyan L, Dumas A, Patenaude A, Vallieres L. Interleukin-36gamma is Expressed by Neutrophils and can Activate Microglia, But has No Role in Experimental Autoimmune Encephalomyelitis. J Neuroinflamm (2015) 12:173. doi: 10.1186/s12974-015-0392-7
54. Zhang J, Yin Y, Lin X, Yan X, Xia Y, Zhang L, et al. IL-36 Induces Cytokine IL-6 and Chemokine CXCL8 Expression in Human Lung Tissue Cells: Implications for Pulmonary Inflammatory Responses. Cytokine (2017) 99:114–23. doi: 10.1016/j.cyto.2017.08.022
55. Koss CK, Wohnhaas CT, Baker JR, Tilp C, Przibilla M, Lerner C, et al. IL36 is a Critical Upstream Amplifier of Neutrophilic Lung Inflammation in Mice. Commun Biol (2021) 4(1):172. doi: 10.1038/s42003-021-01703-3
56. Mutamba S, Allison A, Mahida Y, Barrow P, Foster N. Expression of IL-1Rrp2 by Human Myelomonocytic Cells is Unique to Dcs and Facilitates DC Maturation by IL-1F8 and IL-1F9. Eur J Immunol (2012) 42(3):607–17. doi: 10.1002/eji.201142035
57. Vigne S, Palmer G, Martin P, Lamacchia C, Strebel D, Rodriguez E, et al. IL-36 Signaling Amplifies Th1 Responses by Enhancing Proliferation and Th1 Polarization of Naive CD4+ T Cells. Blood (2012) 120(17):3478–87. doi: 10.1182/blood-2012-06-439026
58. Harusato A, Abo H, Ngo VL, Yi SW, Mitsutake K, Osuka S, et al. IL-36gamma Signaling Controls the Induced Regulatory T Cell-Th9 Cell Balance Via Nfkappab Activation and STAT Transcription Factors. Mucosal Immunol (2017) 10(6):1455–67. doi: 10.1038/mi.2017.21
59. Ngo VL, Abo H, Maxim E, Harusato A, Geem D, Medina-Contreras O, et al. A Cytokine Network Involving IL-36gamma, IL-23, and IL-22 Promotes Antimicrobial Defense and Recovery From Intestinal Barrier Damage. Proc Natl Acad Sci USA (2018) 115(22):E5076–85. doi: 10.1073/pnas.1718902115
60. Tsurutani N, Mittal P, St Rose MC, Ngoi SM, Svedova J, Menoret A, et al. Costimulation Endows Immunotherapeutic CD8 T Cells With IL-36 Responsiveness During Aerobic Glycolysis. J Immunol (2016) 196(1):124–34. doi: 10.4049/jimmunol.1501217
61. Schmitt V, Hahn M, Kastele V, Wagner O, Wiendl M, Derer A, et al. Interleukin-36 Receptor Mediates the Crosstalk Between Plasma Cells and Synovial Fibroblasts. Eur J Immunol (2017) 47(12):2101–12. doi: 10.1002/eji.201646788
62. Frey S, Derer A, Messbacher ME, Baeten DL, Bugatti S, Montecucco C, et al. The Novel Cytokine Interleukin-36alpha is Expressed in Psoriatic and Rheumatoid Arthritis Synovium. Ann Rheum Dis (2013) 72(9):1569–74. doi: 10.1136/annrheumdis-2012-202264
63. Scheiermann P, Bachmann M, Hardle L, Pleli T, Piiper A, Zwissler B, et al. Application of IL-36 Receptor Antagonist Weakens CCL20 Expression and Impairs Recovery in the Late Phase of Murine Acetaminophen-Induced Liver Injury. Sci Rep (2015) 5:8521. doi: 10.1038/srep08521
64. Magne D, Palmer G, Barton JL, Mezin F, Talabot-Ayer D, Bas S, et al. The New IL-1 Family Member IL-1F8 Stimulates Production of Inflammatory Mediators by Synovial Fibroblasts and Articular Chondrocytes. Arthritis Res Ther (2006) 8(3):R80. doi: 10.1186/ar1946
65. Milora KA, Uppalapati SR, Sanmiguel JC, Zou W, Jensen LE. Interleukin-36beta Provides Protection Against HSV-1 Infection, But Does Not Modulate Initiation of Adaptive Immune Responses. Sci Rep (2017) 7(1):5799. doi: 10.1038/s41598-017-05363-4
66. Gardner JK, Herbst-Kralovetz MM. IL-36gamma Induces a Transient HSV-2 Resistant Environment That Protects Against Genital Disease and Pathogenesis. Cytokine (2018) 111:63–71. doi: 10.1016/j.cyto.2018.07.034
67. Stanfield BA, Rider PJF, Caskey J, Del Piero F, Kousoulas KG. Intramuscular Vaccination of Guinea Pigs With the Live-Attenuated Human Herpes Simplex Vaccine VC2 Stimulates a Transcriptional Profile of Vaginal Th17 and Regulatory Tr1 Responses. Vaccine (2018) 36(20):2842–9. doi: 10.1016/j.vaccine.2018.03.075
68. Louis L, Wise MC, Choi H, Villarreal DO, Muthumani K, Weiner DB. Designed DNA-Encoded IL-36 Gamma Acts as a Potent Molecular Adjuvant Enhancing Zika Synthetic DNA Vaccine-Induced Immunity and Protection in a Lethal Challenge Model. Vaccines (Basel) (2019) 7(2):42. doi: 10.3390/vaccines7020042
69. Nakagawa S, Matsumoto M, Katayama Y, Oguma R, Wakabayashi S, Nygaard T, et al. Staphylococcus Aureus Virulent Psmalpha Peptides Induce Keratinocyte Alarmin Release to Orchestrate IL-17-Dependent Skin Inflammation. Cell Host Microbe (2017) 22(5):667–77.e5. doi: 10.1016/j.chom.2017.10.008
70. Gao N, Me R, Dai C, Seyoum B, Yu FX. Opposing Effects of IL-1Ra and IL-36Ra on Innate Immune Response to Pseudomonas Aeruginosa Infection in C57BL/6 Mouse Corneas. J Immunol (2018) 201(2):688–99. doi: 10.4049/jimmunol.1800046
71. Gao Y, Wen Q, Hu S, Zhou X, Xiong W, Du X, et al. IL-36gamma Promotes Killing of Mycobacterium Tuberculosis by Macrophages Via WNT5A-Induced Noncanonical WNT Signaling. J Immunol (2019) 203(4):922–35. doi: 10.4049/jimmunol.1900169
72. Segueni N, Vigne S, Palmer G, Bourigault ML, Olleros ML, Vesin D, et al. Limited Contribution of IL-36 Versus IL-1 and TNF Pathways in Host Response to Mycobacterial Infection. PloS One (2015) 10(5):e0126058. doi: 10.1371/journal.pone.0126058
73. Nanjo Y, Newstead MW, Aoyagi T, Zeng X, Takahashi K, Yu FS, et al. Overlapping Roles for Interleukin-36 Cytokines in Protective Host Defense Against Murine Legionella Pneumophila Pneumonia. Infect Immun (2019) 87(1):e00583–18. doi: 10.1128/IAI.00583-18
74. Verma AH, Zafar H, Ponde NO, Hepworth OW, Sihra D, Aggor FEY, et al. IL-36 and IL-1/IL-17 Drive Immunity to Oral Candidiasis Via Parallel Mechanisms. J Immunol (2018) 201(2):627–34. doi: 10.4049/jimmunol.1800515
75. Kumar S, McDonnell PC, Lehr R, Tierney L, Tzimas MN, Griswold DE, et al. Identification and Initial Characterization of Four Novel Members of the Interleukin-1 Family. J Biol Chem (2000) 275(14):10308–14. doi: 10.1074/jbc.275.14.10308
76. Wang P, Gamero AM, Jensen LE. IL-36 Promotes Anti-Viral Immunity by Boosting Sensitivity to IFN-Alpha/Beta in IRF1 Dependent and Independent Manners. Nat Commun (2019) 10(1):4700. doi: 10.1038/s41467-019-12318-y
77. Satoh TK, Mellett M, Meier-Schiesser B, Fenini G, Otsuka A, Beer HD, et al. IL-36gamma Drives Skin Toxicity Induced by EGFR/MEK Inhibition and Commensal Cutibacterium Acnes. J Clin Invest (2020) 130(3):1417–30. doi: 10.1172/JCI128678
78. Braegelmann J, Braegelmann C, Bieber T, Wenzel J. Candida Induces the Expression of IL-36gamma in Human Keratinocytes: Implications for a Pathogen-Driven Exacerbation of Psoriasis? J Eur Acad Dermatol Venereol (2018) 32(11):e403–6. doi: 10.1111/jdv.14994
79. Gresnigt MS, Rosler B, Jacobs CW, Becker KL, Joosten LA, van der Meer JW, et al. The IL-36 Receptor Pathway Regulates Aspergillus Fumigatus-Induced Th1 and Th17 Responses. Eur J Immunol (2013) 43(2):416–26. doi: 10.1002/eji.201242711
80. Lian LH, Milora KA, Manupipatpong KK, Jensen LE. The Double-Stranded RNA Analogue Polyinosinic-Polycytidylic Acid Induces Keratinocyte Pyroptosis and Release of IL-36gamma. J Invest Dermatol (2012) 132(5):1346–53. doi: 10.1038/jid.2011.482
81. Ahsan F, Moura-Alves P, Guhlich-Bornhof U, Klemm M, Kaufmann SH, Maertzdorf J. Role of Interleukin 36gamma in Host Defense Against Tuberculosis. J Infect Dis (2016) 214(3):464–74. doi: 10.1093/infdis/jiw152
82. Ahsan F, Maertzdorf J, Guhlich-Bornhof U, Kaufmann SHE, Moura-Alves P. IL-36/LXR Axis Modulates Cholesterol Metabolism and Immune Defense to Mycobacterium Tuberculosis. Sci Rep (2018) 8(1):1520. doi: 10.1038/s41598-018-19476-x
83. Sequeira RP, McDonald JAK, Marchesi JR, Clarke TB. Commensal Bacteroidetes Protect Against Klebsiella Pneumoniae Colonization and Transmission Through IL-36 Signalling. Nat Microbiol (2020) 5(2):304–13. doi: 10.1038/s41564-019-0640-1
84. Medina-Contreras O, Harusato A, Nishio H, Flannigan KL, Ngo V, Leoni G, et al. Cutting Edge: IL-36 Receptor Promotes Resolution of Intestinal Damage. J Immunol (2016) 196(1):34–8. doi: 10.4049/jimmunol.1501312
85. Gong Y, Tingxi Z, Qing L, Guozhen Z, Bing T, Xiaoliang Y, et al. Elevated Production of IL-36alpha in Chronic Hepatitis B Virus-Infected Patients Correlates With Viral Load. Microb Pathog (2017) 113:412–5. doi: 10.1016/j.micpath.2017.11.023
86. Mele D, Mantovani S, Oliviero B, Grossi G, Lombardi A, Mondelli MU, et al. Monocytes Inhibit Hepatitis C Virus-Induced TRAIL Expression on CD56(Bright) NK Cells. J Hepatol (2017) 67(6):1148–56. doi: 10.1016/j.jhep.2017.07.028
87. Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, et al. Interleukin-1 Blockade With High-Dose Anakinra in Patients With COVID-19, Acute Respiratory Distress Syndrome, and Hyperinflammation: A Retrospective Cohort Study. Lancet Rheumatol (2020) 2(6):e325–31. doi: 10.1016/S2665-9913(20)30127-2
88. van de Veerdonk FL, Netea MG. Blocking IL-1 to Prevent Respiratory Failure in COVID-19. Crit Care (2020) 24(1):445. doi: 10.1186/s13054-020-03166-0
89. Costela-Ruiz VJ, Illescas-Montes R, Puerta-Puerta JM, Ruiz C, Melguizo-Rodriguez L. SARS-Cov-2 Infection: The Role of Cytokines in COVID-19 Disease. Cytokine Growth Factor Rev (2020) 54:62–75. doi: 10.1016/j.cytogfr.2020.06.001
90. Zizzo G, Cohen PL. Imperfect Storm: Is Interleukin-33 the Achilles Heel of COVID-19? Lancet Rheumatol (2020) 2(12):e779–90. doi: 10.1016/S2665-9913(20)30340-4
91. Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, et al. COVID-19 Severity Correlates With Airway Epithelium-Immune Cell Interactions Identified by Single-Cell Analysis. Nat Biotechnol (2020) 38(8):970–9. doi: 10.1038/s41587-020-0602-4
92. Camiolo M, Gauthier M, Kaminski N, Ray A, Wenzel SE. Expression of SARS-Cov-2 Receptor ACE2 and Coincident Host Response Signature Varies by Asthma Inflammatory Phenotype. J Allergy Clin Immunol (2020) 146(2):315–324 e7. doi: 10.1016/j.jaci.2020.05.051
93. Xiao F, Tang M, Zheng X, Liu Y, Li X, Shan H. Evidence for Gastrointestinal Infection of SARS-Cov-2. Gastroenterology (2020) 158(6):1831–1833 e3. doi: 10.1053/j.gastro.2020.02.055
94. Livanos AE, Jha D, Cossarini F, Gonzalez-Reiche AS, Tokuyama M, Aydillo T, et al. Intestinal Host Response to SARS-Cov-2 Infection and COVID-19 Outcomes in Patients With Gastrointestinal Symptoms. Gastroenterology (2021) S0016-5085(21):00461–3. doi: 10.1053/j.gastro.2021.02.056
95. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J Pathol (2004) 203(2):631–7. doi: 10.1002/path.1570
96. Carrascosa JM, Morillas V, Bielsa I, Munera-Campos M. Cutaneous Manifestations in the Context of SARS-Cov-2 Infection (COVID-19). Actas Dermosifiliogr (2020) 111(9):734–42. doi: 10.1016/j.ad.2020.08.002
97. Colmenero I, Santonja C, Alonso-Riano M, Noguera-Morel L, Hernandez-Martin A, Andina D, et al. SARS-Cov-2 Endothelial Infection Causes COVID-19 Chilblains: Histopathological, Immunohistochemical and Ultrastructural Study of Seven Paediatric Cases. Br J Dermatol (2020) 183(4):729–37. doi: 10.1111/bjd.19327
98. Freeman EE, McMahon DE, Lipoff JB, Rosenbach M, Kovarik C, Desai SR, et al. The Spectrum of COVID-19-Associated Dermatologic Manifestations: An International Registry of 716 Patients From 31 Countries. J Am Acad Dermatol (2020) 83(4):1118–29. doi: 10.1016/j.jaad.2020.06.1016
99. Recalcati S, Barbagallo T, Frasin LA, Prestinari F, Cogliardi A, Provero MC, et al. Acral Cutaneous Lesions in the Time of COVID-19. J Eur Acad Dermatol Venereol (2020) 34(8):e346–7. doi: 10.1111/jdv.16533
100. Monteil V, Kwon H, Prado P, Hagelkruys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-Cov-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell (2020) 181(4):905–913 e7. doi: 10.1016/j.cell.2020.04.004
101. Zulli A, Burrell LM, Buxton BF, Hare DL. ACE2 and AT4R are Present in Diseased Human Blood Vessels. Eur J Histochem (2008) 52(1):39–44. doi: 10.4081/1184
102. Mercurio L, Failla CM, Capriotti L, Scarponi C, Facchiano F, Morelli M, et al. Interleukin (IL)-17/IL-36 Axis Participates to the Crosstalk Between Endothelial Cells and Keratinocytes During Inflammatory Skin Responses. PloS One (2020) 15(4):e0222969. doi: 10.1371/journal.pone.0222969
103. Joo YH, Kim HK, Hak Choi I, Han HM, Lee KJ, Kim TH, et al. Increased Expression of Interleukin 36 in Chronic Rhinosinusitis and its Contribution to Chemokine Secretion and Increased Epithelial Permeability. Cytokine (2020) 125:154798. doi: 10.1016/j.cyto.2019.154798
104. Xue X, Mi Z, Wang Z, Pang Z, Liu H, Zhang F. High Expression of ACE2 on Keratinocytes Reveals Skin as a Potential Target for SARS-Cov-2. J Invest Dermatol (2021) 141(1):206–209 e1. doi: 10.1016/j.jid.2020.05.087
105. Rana AA, Lucs AV, DeVoti J, Blanc L, Papoin J, Wu R, et al. Poly(I:C) Induces Controlled Release of IL-36gamma From Keratinocytes in the Absence of Cell Death. Immunol Res (2015) 63(1-3):228–35. doi: 10.1007/s12026-015-8692-7
106. Mahil SK, Catapano M, Di Meglio P, Dand N, Ahlfors H, Carr IM, et al. An Analysis of IL-36 Signature Genes and Individuals With IL1RL2 Knockout Mutations Validates IL-36 as a Psoriasis Therapeutic Target. Sci Transl Med (2017) 9(411):eaan2514. doi: 10.1126/scitranslmed.aan2514
107. Sanchez-Cerrillo I, Landete P, Aldave B, Sanchez-Alonso S, Sanchez-Azofra A, Marcos-Jimenez A, et al. COVID-19 Severity Associates With Pulmonary Redistribution of CD1c+ Dcs and Inflammatory Transitional and Nonclassical Monocytes. J Clin Invest (2020) 130(12):6290–300. doi: 10.1172/JCI140335
108. Laforge M, Elbim C, Frere C, Hemadi M, Massaad C, Nuss P, et al. Tissue Damage From Neutrophil-Induced Oxidative Stress in COVID-19. Nat Rev Immunol (2020) 20(9):515–6. doi: 10.1038/s41577-020-0407-1
109. Merad M, Martin JC. Pathological Inflammation in Patients With COVID-19: A Key Role for Monocytes and Macrophages. Nat Rev Immunol (2020) 20(6):355–62. doi: 10.1038/s41577-020-0331-4
110. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-Cov-2-Triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J Exp Med (2020) 217(12):e20201129. doi: 10.1084/jem.20201129
111. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
112. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight (2020) 5(11):e138999. doi: 10.1172/jci.insight.138999
113. Yonggang Zhou BF, Zheng X, Wang D, Zhao C, Qi Y, Sun R, et al. Author Notes.: Pathogenic T-Cells and Inflammatory Monocytes Incite Inflammatory Storms in Severe COVID-19 Patients. Natl Sci Rev (2020) 7(6):998–1002. doi: 10.1093/nsr/nwaa041
114. Cauchois R, Koubi M, Delarbre D, Manet C, Carvelli J, Blasco VB, et al. Early IL-1 Receptor Blockade in Severe Inflammatory Respiratory Failure Complicating COVID-19. Proc Natl Acad Sci USA (2020) 117(32):18951–3. doi: 10.1073/pnas.2009017117
115. Sun X, Hou T, Cheung E, Iu TN, Tam VW, Chu IM, et al. Anti-Inflammatory Mechanisms of the Novel Cytokine Interleukin-38 in Allergic Asthma. Cell Mol Immunol (2020) 17(6):631–46. doi: 10.1038/s41423-019-0300-7
116. Conti P, Ronconi G, Caraffa A, Gallenga CE, Ross R, Frydas I, et al. Induction of Pro-Inflammatory Cytokines (IL-1 and IL-6) and Lung Inflammation by Coronavirus-19 (COVI-19 or SARS-Cov-2): Anti-Inflammatory Strategies. J Biol Regul Homeost Agents (2020) 34(2):327–31. doi: 10.23812/CONTI-E
117. Gao X, Chan PKS, Lui GCY, Hui DSC, Chu IM, Sun X, et al. Interleukin-38 Ameliorates Poly(I:C) Induced Lung Inflammation: Therapeutic Implications in Respiratory Viral Infections. Cell Death Dis (2021) 12(1):53. doi: 10.1038/s41419-020-03283-2
118. Scheibe K, Kersten C, Schmied A, Vieth M, Primbs T, Carle B, et al. Inhibiting Interleukin 36 Receptor Signaling Reduces Fibrosis in Mice With Chronic Intestinal Inflammation. Gastroenterology (2019) 156(4):1082–1097 e11. doi: 10.1053/j.gastro.2018.11.029
119. Ganesan R, Raymond EL, Mennerich D, Woska JR Jr., Caviness G, Grimaldi C, et al. Generation and Functional Characterization of Anti-Human and Anti-Mouse IL-36R Antagonist Monoclonal Antibodies. MAbs (2017) 9(7):1143–54. doi: 10.1080/19420862.2017.1353853
120. Conner KP, Pastuskovas CV, Soto M, Thomas VA, Wagner M, Rock DA. Preclinical Characterization of the ADME Properties of a Surrogate Anti-IL-36R Monoclonal Antibody Antagonist in Mouse Serum and Tissues. MAbs (2020) 12(1):1746520. doi: 10.1080/19420862.2020.1746520
121. Yuan ZC, Xu WD, Liu XY, Liu XY, Huang AF, Su LC. Biology of IL-36 Signaling and its Role in Systemic Inflammatory Diseases. Front Immunol (2019) 10:2532. doi: 10.3389/fimmu.2019.02532
122. Hewitt SL, Bai A, Bailey D, Ichikawa K, Zielinski J, Karp R, et al. Durable Anticancer Immunity From Intratumoral Administration of IL-23, IL-36gamma, and OX40L Mrnas. Sci Transl Med (2019) 11(477):eaat9143. doi: 10.1126/scitranslmed.aat9143
Keywords: IL-36, IL-1 family, cytokine, infection, COVID-19
Citation: Wang X, Yi P and Liang Y (2021) The Role of IL-36 in Infectious Diseases: Potential Target for COVID-19? Front. Immunol. 12:662266. doi: 10.3389/fimmu.2021.662266
Received: 05 February 2021; Accepted: 28 April 2021;
Published: 13 May 2021.
Edited by:
Remi Cheynier, U1016 Institut Cochin (INSERM), FranceReviewed by:
Patrick Walsh, Trinity College Dublin, IrelandAndrew Johnston, University of Michigan, United States
Martin Stacey, University of Leeds, United Kingdom
Copyright © 2021 Wang, Yi and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Panpan Yi, c3VuYW5kbGVhZkAxMjYuY29t; Yuejin Liang, eXUybGlhbmdAdXRtYi5lZHU=