 Vasilios A. Morikis
Vasilios A. Morikis Alfredo A. Hernandez1
Alfredo A. Hernandez1 Markus Sperandio
Markus Sperandio Scott I. Simon
Scott I. Simon- 1Department of Biomedical Engineering, University of California-Davis, Davis, CA, United States
- 2GlycoMimetics Inc., Rockville, MD, United States
- 3Institute for Cardiovascular Physiology and Pathophysiology, Walter Brendel Center for Experimental Medicine Biomedical Center, Ludwig Maximilians University, Walter Brendel Center, Munich, Germany
Neutrophils are essential to protect the host against invading pathogens but can promote disease progression in sickle cell disease (SCD) by becoming adherent to inflamed microvascular networks in peripheral tissue throughout the body. During the inflammatory response, leukocytes extravasate from the bloodstream using selectin adhesion molecules and migrate to sites of tissue insult through activation of integrins that are essential for combating pathogens. However, during vaso-occlusion associated with SCD, neutrophils are activated during tethering and rolling on selectins upregulated on activated endothelium that line blood vessels. Recently, we reported that recognition of sLex on L-selectin by E-selectin during neutrophil rolling initiates shear force resistant catch-bonds that facilitate tethering to endothelium and activation of integrin bond clusters that anchor cells to the vessel wall. Evidence indicates that blocking this important signaling cascade prevents the congestion and ischemia in microvasculature that occurs from neutrophil capture of sickled red blood cells, which are normally deformable ellipses that flow easily through small blood vessels. Two recently completed clinical trials of therapies targeting selectins and their effect on neutrophil activation in small blood vessels reveal the importance of mechanoregulation that in health is an immune adaption facilitating rapid and proportional leukocyte adhesion, while sustaining tissue perfusion. We provide a timely perspective on the mechanism underlying vaso-occlusive crisis (VOC) with a focus on new drugs that target selectin mediated integrin adhesive bond formation.
Introduction
Sickle cell disease (SCD) is an inherited disease characterized by defects in hemoglobin that distorts red blood cells (RBC) into a “sickle” shape (1–3). Clinical manifestations common to this inherited hemoglobinopathy, chronic hemolysis, and erythrocyte adhesion that can lead to vaso-occlusion (4). Vaso-occlusive crisis (VOC) is defined by clinical episodes of severe ischemic pain due to lack of blood flow resulting in cumulative and irreversible organ damage. As such, the occurrence of VOC is a serious clinical event that is responsible for the increased morbidity and mortality of sickle cell patients. The clinical sequalae associated with VOC often manifests in episodes of pain that vary in intensity and duration as a function of patient comorbidities such as diabetes. Some patients experience only infrequent pain crises each year, while others require acute hospitalization for treatment. Pain management during VOC often includes the prescription of opioids that, while effectively diminishing pain, do not treat the root cause and therefore do not significantly lower the risk of a catastrophic clinical event (5). Thus, treatments to prevent genesis of VOC as opposed to pain management is a primary challenge for scientists aiming to establish effective treatments that target the initiation and block progression (6). In this perspective we review the recent studies highlighting the importance of neutrophil inflammatory recruitment and the role of selectin and integrin adhesive receptors in progression to clinical crisis. We endeavor to illuminate the lessons learned from preclinical studies in mouse models of SCD and how they apply to human disease in order to evaluate the outcome of recent clinical trials that target neutrophil recruitment.
The etiology of VOC involves the innate immune response that enables white blood cells (WBC) predominantly neutrophils to capture on adhesive receptors upregulated on inflamed endothelium. Captured neutrophils become activated during interaction with endothelium at sites of inflamed microvasculature leading to upregulation of additional adhesion molecules on the plasma membrane that serve to recruit additional circulating cells, such as sickle RBC (sRBC) and platelets that aggregate causing rapid occlusion of the microvasculature leading to vessel ischemia (Figure 1) (2, 7, 8). Contributing to microvascular occlusion is a greater capacity for neutrophil extracellular trap (NET) formation characterized by decondensed chromatin enriched in citrullinated histones and bound to granular enzymes (9). The pathophysiology and requirement for pain management directly associated with VOC in patients has been extensively reviewed (2, 4, 6, 10, 11). As such, the focus of this review is to highlight the role of neutrophils in the initiation and propagation of VOC, as well as targeted pharmacological interventions against adhesion molecules critical for neutrophil recruitment and heterotypic adhesion with other blood cells during flow within vessels.
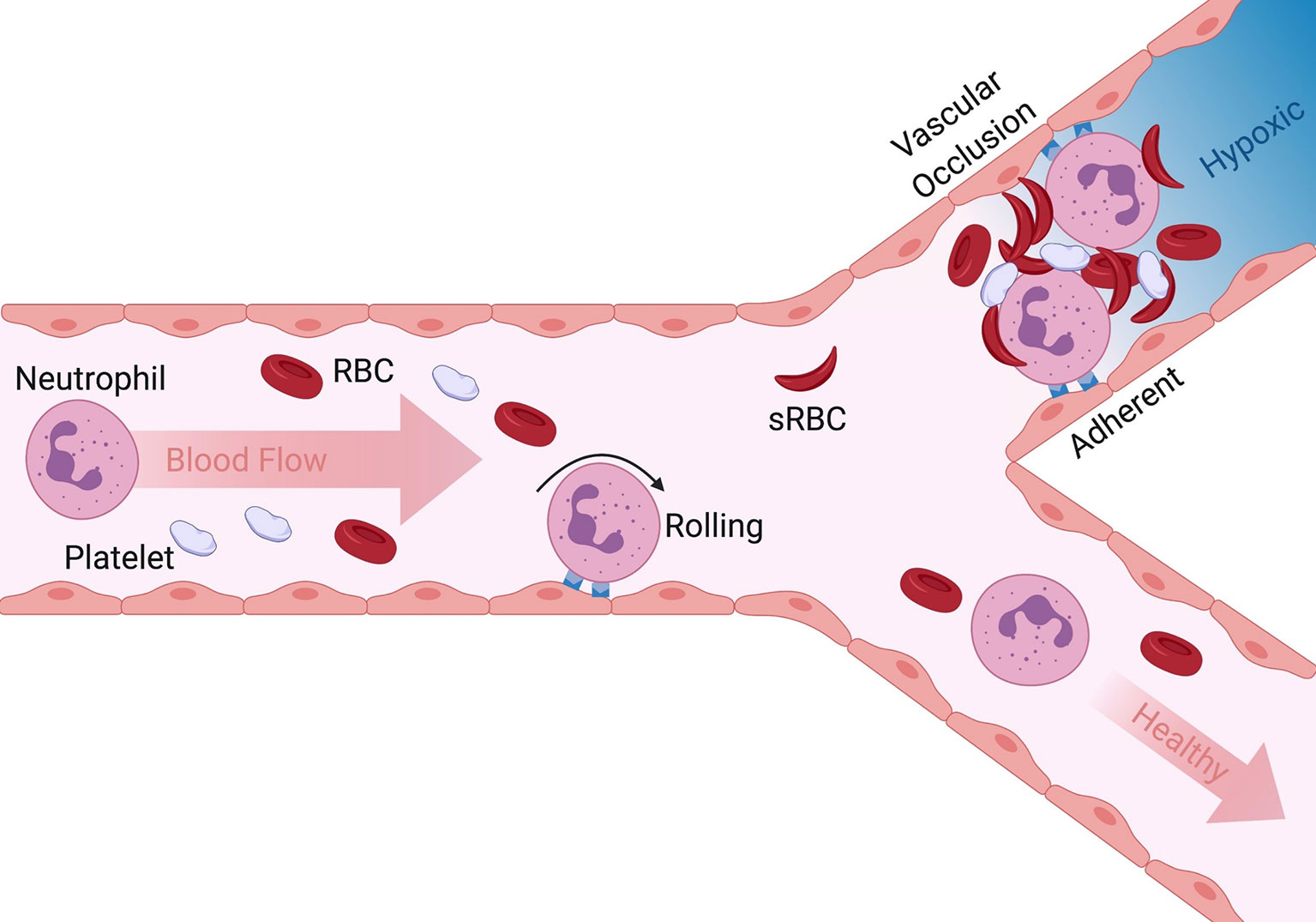
Figure 1 Overview of initiation and progression of VOC in SCD. Neutrophil rolling to arrest on inflamed endothelium is a normal innate immune response to tissue injury. Adherent neutrophils can also capture sickle red blood cells and platelets in post capillary venules leading to vascular occlusion (upper bifurcation). Dramatic reduction in blood flow and lack of oxygen results in hypoxia, episodic painful and often leads to organ failure. When inappropriate neutrophil activation and adhesion is absent and secondary capture of RBC and platelets limited, the vasculature remains perfused and healthy (lower pathway).
Neutrophil capture on inflamed endothelium is initiated by Sialyl Lewisx (sLex) bearing selectin ligands on cells in flow (12–15). Ligand recognition by selectins is an early step in the inflammatory process mediating leukocyte rolling and platelet capture, as such selectins are a primary target for therapeutics to prevent propagation of VOC. Early studies on the specific role of cellular adhesion molecules that induce VOC were carried out in mice due to the availability of genetic knockout models of WBC adhesion via selectins and integrins. However, human and mouse genomes have diverged in the past 75 million years and as such selectin function differs between the mouse and human innate immune response (Table 1). While selectin targeting has been a major focus in designing VOC treatments, there is a need to better understand how selectins participate in precipitating neutrophil recruitment leading to VOC in more accurate models of human SCD in order to design informed therapeutic treatments.

Table 1 Differences between mouse and human selectins.
Selectin Function and Bond Mechanics in Vaso-Occlusive Crisis in Humans
To understand the etiology of VOC in terms of the initiation and development of a congested microvascular bed, selectin function during neutrophil recruitment must be examined. In humans three selectins participate in forming stable but transient adhesive bonds with carbohydrate structures between cells in interactions characterized by fast kinetics that allow cell adhesion to proceed under the shear forces of blood flow (21, 22). Expression of endothelial E-selectin requires de novo protein synthesis, and is upregulated within hours of cytokine activation at inflamed sites experiencing disturbed blood flow or focal tissue insult (23, 24). P-selectin is preformed and stored in Weibel Palade Bodies (WPB) of endothelial cells and in α-granules of circulating platelets and is rapidly mobilized from these storage sites by merging with the plasma membrane where it participates in tethering and rolling of leukocytes and platelets on inflamed endothelium (25, 26). Leukocyte expressed L-selectin is a glycoprotein that not only binds sLex as its primary carbohydrate recognition motif expressed by PSGL-1 on neutrophils, but it also presents sLex to facilitate neutrophil homing and subsequent activation within inflamed venules (27, 28). E-selectin binding to sLex supports capture and rolling of human, but not murine neutrophils thereby providing a key event for subsequent mechanosignaling of integrin activation that mediates leukocyte arrest even in absence of chemokine signaling (29). Neutrophil homotypic adhesion is observed as secondary capture of a neutrophil from the blood stream by a rolling or arrested neutrophil via L-selectin binding of PSGL-1 between cells (Figure 2A) (29–31). While E-selectin and P-selectin both function in the early capture and adhesion of leukocytes to the vascular endothelium, there are some distinct differences. All selectins share a similar structure characterized by a lectin binding domain, epidermal growth factor domain, a variable number of short consensus repeats (9 for P-selectin, 6 for E-selectin, and 2 for L-selectin), a transmembrane domain, and a cytosolic tail (13). Despite the similarities in structure, the binding kinetics and capacity to form durable bonds that mechanotransduce activation of integrins are quite different (22, 32). P-selectin projects the furthest above the endothelial surface and is thought to provide the initial interaction between leukocytes in the free stream through recognition of PSGL-1, although it also can bind additional ligands including sulfated polysaccharides (33). However, P-selectin does not mechanotransduce activation of integrin on bound leukocytes in the same manner as E-selectin binding to L-selectin. E-selectin forms longer-lived shear resistant bonds with L-selectin compared with P-selectin, that is independent of recognition of its other cognate ligands (i.e. PSGL-1, CD44). L-selectin appears to be unique on human neutrophils due to its recognition by E-selectin and capacity to actively condense into bond clusters that mechanotransduce signals leading to β2-integrin activation and adhesion (29). This function is attributed to E-selectin dependent formation of a high-affinity complex with sLex under precise hydrodynamic conditions to form a catch-bond with L-selectin (34). A molecular model of E-selectin binding sLex under tension predicts that it adopts an extended conformation, which was verified by small-angle X-ray scattering of it bound to purified ligand (34). Extended E-selectin/sLex complexes form shear resistant bonds characterized by adhesive lifetimes that are long lived (~ 500 msec) at sufficiently high hydrodynamic shear stress. Whereas at low shear rates (i.e. < 1 dyne/cm2) associated with microvascular regions of slow blood flow and ischemia, adhesive bonds are less durable and dissociate more rapidly. This phenomenon is denoted catch-bond behavior (34–38). E-selectin binding to sLex results in co-localization of L-selectin and PSGL-1 on the trailing edge of neutrophils, a process associated with β2-integrin activation and neutrophil arrest (26, 29). A stepwise process depicted in Figure 2 illustrates how neutrophils tethered on endothelial P-selectin, may facilitate E-selectin and L-selectin to engage and mechanically signal cell activation and arrest. Selectins are a prime target for inhibition of the inflammatory response that initiates VOC, with the rationale that blocking the adhesive steps in the vaso-occlusive process will prevent propagation of congestion and decrease the duration and intensity of painful VOC in patients with SCD.2
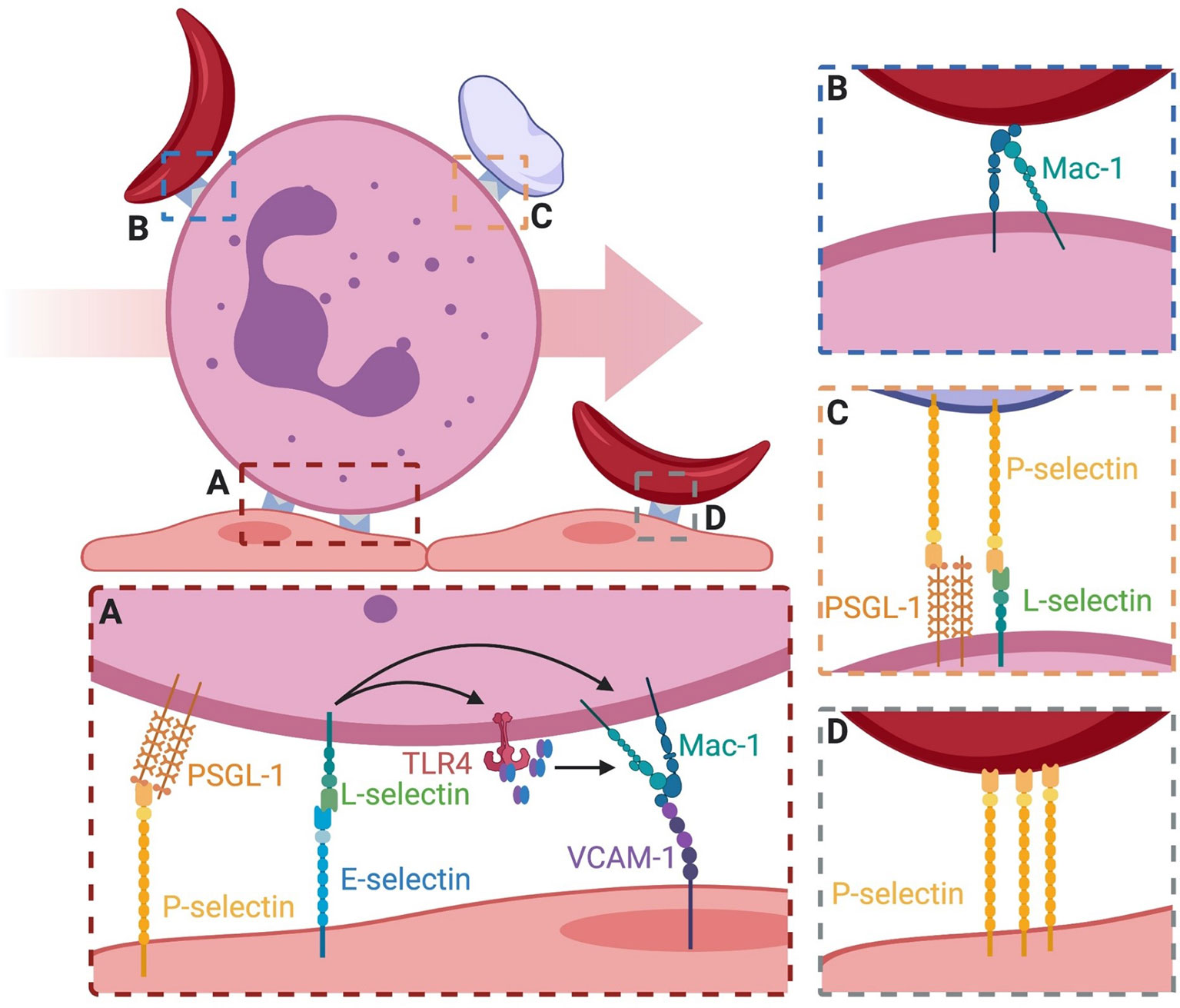
Figure 2 Binding events that lead to vasculature occlusion on inflamed endothelium. (A) Neutrophil rolling is mediated by P-selectin and E-selectin binding PSGL-1 and L-selectin. Signaling through E-selectin/L-selectin bonds results in (1) release of MRP8/14 and endogenous activation of surface TLR4 which primes integrin for activation and (2) direct signaling of transition to a high-affinity integrin state capable of binding ligand. High affinity integrin is capable of stable bond formation to VCAM-1 and ICAM-1 on endothelial to mediate neutrophil arrest. (B) Secondary capture of sRBC to adherent neutrophils is mediated via Mac-1. The mechanism of capture is an active area of research. (C) Secondary capture of platelets to adherent neutrophils is mediated through platelet surface P-selectin and its binding to PSGL-1 and L-selectin. (D) Direct engagement of sRBC to the surface of endothelial cells has been attributed to P-selectin, similar to secondary capture via Mac-1.
The importance of selectins in VOC was rigorously studied by Hidalgo et. al., who first reported in a humanized sickle cell mouse model that E-selectin mediated activation of β2-integrins at the leading edge of a neutrophil promotes transition from rolling to arrest in an inflamed microvascular bed (39). In this model of acute lethal VOC, a proinflammatory state was induced in post capillary venules by injection of tumor necrosis factor-α (TNF-α). This resulted in capture of sRBC by activated αMβ2 integrin (macrophage-1 antigen, Mac-1) clustered on the leading edge of arrested neutrophils. Genetic deletion of E-selectin (Sele-/-) or P-selectin (Selp-/-) provided equivalent inhibition of neutrophil capture and reduced RBC/WBC aggregates at early time points within 3 hours of TNF-α stimulation. However, E-selectin knockouts exhibited reduced neutrophil arrest and RBC/WBC aggregates were undetected at later time points of 3-5 hours post stimulation. In contrast, P-selectin knockouts had a spike in the occurrence of RBC/WBC aggregates at 3-5 hours post TNF-α stimulation, resulting in VOC equivalent to control WT mice experiencing a VOC. Significant was the observation that E-selectin knockout mice maintained low levels of secondary capture of RBC that maintained higher rates of blood flow (39). One may conclude that inhibition of P-selectin effectively delayed VOC progression within the initial hours of an inflammatory insult, however, the presence of E-selectin that maintains expression on endothelium elicits sustained neutrophil-RBC recruitment that is responsible for long term VOC. Of note, another important difference between the human and the mouse system concerns the transcriptional regulation of P-selectin. While stimulation of endothelial cells with the proinflammatory cytokine TNF-α leads to the transcriptional upregulation of endothelial P-selectin in the mouse system, endothelial P-selectin expression is downregulated following stimulation with TNF-α in human microvasculature. This has been elegantly demonstrated in the TNF- α stimulated cremaster muscle of mice lacking mouse P-selectin, but expressing human P-selectin supporting the concept that P-selectin in humans might be less important in the pathogenesis of VOC progression than E-selectin (19).
E-selectin mediates recruitment by engaging L-selectin on human neutrophils during rolling which results in arrest via activation of LFA-1 to bind with ICAM-1 expressed on inflamed endothelium (29). Secondary capture of RBCs can also be observed in parallel plate flow channels that mimic the hydrodynamic conditions observed in normal versus inflamed microcirculation. In such a model, it was confirmed that human neutrophil capture and RBC/WBC heterotypic aggregates are enhanced in sickle cell patients (Figure 2B) (40). It is noteworthy that antibodies that block E-selectin mediated activation of Mac-1 on neutrophils, also blocks heterotypic RBC/WBC aggregation. This directly correlates with increased survival compared to isotype nonblocking control antibody (41). Platelet/WBC aggregates are also significantly reduced in Mac-1 knockout mice during an induced VOC event in mice (41). Thus, both E-selectin and Mac-1 are implicated as primary capture molecules initiating and propagating VOC. However, β2-integrins including Mac-1 and LFA-1 are not strategic targets for treatment of VOC in SCD, as leukocytes require these integrins to maintain immune surveillance and to combat infection. Complete blockade of integrin subunit function can be dangerous for high risk immunodeficient patients as realized in clinical trials of the CD11a blocker Efalizumab that was associated with viral meningitis and bacterial sepsis in a subset of patients, which triggered it’s rapid withdrawal from the market in 2009 (42). Another strategy is to target selectins in order to reduce integrin activation and neutrophil capture. In this manner, integrin activation is maintained via alternate pathways such as by chemokine receptor ligation, thereby preserving neutrophil and T cell function necessary to combat infections.
There is evidence that soluble E-selectin levels in serum are higher in SCD patients than in healthy subjects and can increase further during acute VOC in SCD patients (43). Kato et al. measured the level of E-selectin, P-selectin, VCAM-1, and ICAM-1 in the plasma of 160 patients suffering from VOC over a four-year period (44). E-selectin, P-selectin, and the integrin ligand VCAM-1 were all significantly increased in patients suffering from SCD compared to controls. A significant finding was that patients with high levels of soluble E-selectin directly correlated with elevated risk of mortality, while those with high levels of soluble P-selectin did not significantly correlate with increased mortality. This implicates neutrophil binding to E-selectin as a primary means of Mac-1 activation and binding to its ligands as a predominant feature that drives neutrophil adhesion and progression of VOC.
E-selectin and P-selectin recognize a variety of ligands on the neutrophil plasma membrane. L-selectin and PSGL-1 are prominent among these since they can function not only to tether neutrophils to inflamed post capillary venules, but allow function as mechanosignaling receptors that activate rapid cell arrest via high affinity β2-integrin bond formation in shear flow (12). Recruitment of human neutrophils on inflamed endothelium is supported by the preferential recognition of L-selectin by E-selectin and PSGL-1 by P-selectin, respectively (22, 45–47). Studies of neutrophil recruitment using vascular mimetic microfluidic channels under defined hydrodynamic shear on a substrate of recombinant E-selectin revealed that L-selectin and PSGL-1 co-localize into clusters on neutrophil microvilli and spontaneously activate β2-integrin bond formation on ICAM-1 (48). Recently, it was reported that formation of catch-bonds between E-selectin and L-selectin effectively induce the extension of β2-integrin that supports its binding to ICAM-1 during neutrophil deceleration that can prime the transition to a high-affinity state (29). A mechanism has been identified that involves the calcium-modulating proteins myeloid related protein-8 (MRP8) and MRP14 (also known as MRP8/14, S100A8/S100A9, and calprotectin). MRP8/14 can modulate myeloid cell function by binding Toll-like receptor-4 to induce calcium signaling and cytoskeletal reorganization (49–51). A complex of MRP8/14 released from leukocytes in mouse and human models of SCD are known to contribute to vascular inflammation and tissue injury. Supporting such a mechanism is the observation that MRP8/14-/- mice exhibited reduced neutrophil accumulation and lesion severity, implicating it in regulation of neutrophil recruitment (52). This mechanism is associated with acute inflammation, as neutrophils captured on E-selectin secrete MRP8/14 which then binds TLR4 on the same cell to initiate integrin extension (29, 49). Blocking TLR4 binding by treatment with antibody, or inhibiting E-selectin/L-selectin bond formation under shear by addition of the sLex mimetic Rivipansel, blocked the release of MRP8/14 and significantly reduced integrin extension and transient bond formation with ICAM-1 (29). These data reveal a pathway by which E-selectin/L-selectin bonds induce the release of MRP8/14 that bind TLR4 and in an autocrine manner signal conformational extension of LFA-1. This along with signaling via engaged and clustered L-selectin promotes long-lived high-affinity integrin bonds and the efficient arrest of rolling neutrophils in the microcirculation (22, 45, 53). This early step in the multistep process of neutrophil recruitment facilitates deceleration via E-selectin mediated rolling that upregulates high-affinity LFA-1 and subsequently Mac-1. When blocked with small molecule antagonists or anti-L-selectin antibody, LFA-1/Mac-1 mediated neutrophil recruitment and secondary capture observed in VOC is prevented (Figures 2A, B).
While E-selectin and P-selectin mediate the capture of individual neutrophils, it is important to highlight heterotypic adhesive interactions in the genesis of VOC. Secondary capture of free-flowing neutrophils, platelets and erythrocytes with adherent WBC are implicated in various disease states such as atherosclerotic plaque formation and VOC (54, 55). Homotypic neutrophil-neutrophil adhesion is mediated by L-selectin on one cell and PSGL-1 on the adjacent cell (Figures 2B, C). This results in strings of rolling neutrophils aligned along the direction of shear flow, which is thought to be relevant in the pathogenesis of VOC. Cell aggregation leading to microvascular congestion is also mediated by neutrophil-platelet interactions. Activated platelets express P-selectin and can bind free flowing neutrophils, thereby amplifying the surface area and the likelihood of capture on inflamed endothelium or on adherent neutrophils (Figure 2C) (56, 57). Adherent platelets help orchestrate diapedesis of neutrophils by Mac-1 binding to platelet GPIIb/IIIa in a fibrinogen dependent manner (58). Cellular crosstalk between platelet P-selectin binding to neutrophil PSGL-1 and Mac-1/fibrinogen/GPIIb/IIIa supports the efficient trafficking of neutrophils to sites of inflammation. Taken together, this implicates Mac-1 and P-selectin as adhesion receptors that may play a central role in the heterotypic platelet adhesive events that occur downstream of E-selectin in the progression of VOC. While the importance of selectins and their ligands cannot be overstated, there are key differences in function between human and mouse models that must be appreciated in order to efficiently develop effective therapeutics to prevent VOC.
Of Mice and Men, What Are the Lessons for Treatment of VOC
Much of the knowledge base surrounding E-selectin function stems from transgenic mouse models, which indicate that rolling via PSGL-1 elicits a distinct mechanosignal that leads to integrin mediated neutrophil arrest. A key difference in mouse E-selectin ligands versus humans stems from fucosyltransferase-9/7 function, which is critical for assembly of the ligands that decorate glycoproteins supporting neutrophil rolling on E-selectin in human microvasculature, but not mice (Table 1) (18–20). These fucosyltransferases function to modify O- and N-linked carbohydrates on L-selectin to express the E-selectin ligand sLex (18, 20, 59). This adaption in humans enables the sLex on L-selectin to be recognized by E-selectin directly, which does not occur for mouse L-selectin. The observation that mouse E-selectin plays a critical role in mechanosignaling during rolling in inflamed venules is accounted for by the finding that recognition of sLex on PSGL-1 results in its co-clustering with L-selectin, which in turn elicits outside-in signaling of integrin activation (27). Knockout mice lacking L-selectin do not possess the capacity for direct outside-in signaling of integrin activation, yet they can still roll via endothelial selectins. This difference between human and mouse E-selectin ligands may account for the spontaneous vaso-occlusion observed in humans and not mouse microvasculature (60). It is clear that in humans the E-selectin/L-selectin signaling complex represents a primary step in neutrophil adhesion even in the absence of cytokine or chemokine. Targeting this mechanosignaling pathway by antagonizing selectin catch-bond formation could provide a strategic tipping point for therapeutic targets to prevent development of VOC in SCD patients.
Another difference in the regulation of selectin expression between mouse and humans consists in downregulation of human and upregulation of mouse P-selectin expression during TNF-α stimulated inflammation (19). Evidence supporting a primary role for P-selectin in SCD was demonstrated in a mouse model lacking P-selectin that showed a protective effect in pulmonary VOC (61). To further illuminate the contribution of P-selectin in human VOC, the next logical step would be to generate sickle cell mice lacking mouse P-selectin but edited to express human P-selectin. Such a hybrid mouse line could be used to reveal a species difference in P-selectin function.
Targeted Therapeutics Against Leukocyte and Platelet Activation in Vaso-Occlusive Crisis
Mouse models of SCD have been indispensable for dissecting the molecular determinants of cytokine driven and selectin mediated vascular congestion, including to test selectin targeted drug treatments (16, 62). In the case of the small molecule sLex mimetic Rivipansel® (GMI-1070), we tested its capacity to antagonize the rolling and activation of neutrophils in blood over time of treatment in a Phase-3 clinical trial for treatment of VOC (29). Employing real-time immunofluorescence imaging of neutrophils in microfluidic channels, we examined the molecular dynamics of selectin-ligand receptor engagement during the transition from cell tethering and rolling to arrest on recombinant E-selectin and ICAM-1 ex-vivo. Rivipansel was found to block E-selectin recognition of sLex on L-selectin and antagonize the formation of catch-bonds and outside-in signaling of integrin mediated cell arrest in human SCD patient blood. GSnP-6, a novel PSGL-1 mimetic that blocks its recognition by E-selectin and P-selectin, effectively doubled the rolling velocity of neutrophils, but had no effect on L-selectin outside-in signaling of β2-integrin or the frequency of neutrophil rolling to arrest (29, 63). This differential glycomimetic inhibition of neutrophil rolling versus outside-in activation of integrins via selectins reveals a novel mechanosignaling circuit. E-selectin transmitted tension on L-selectin induces clustering and signaling within sites of focal adhesion, which is markedly different than that observed in mouse neutrophils. PSGL-1 is the primary ligand for E-selectin mediated rolling in mouse as compared with L-selectin on human neutrophils. To determine the therapeutic efficacy of antagonists directed at sLex recognition by selectins in treatment of VOC, humanized P-selectin mouse models and/or ex-vivo assays could be used to quantify MRP8/14 release and TLR4 signaling of integrin activation.
Improving patient quality of life through preventative treatments of VOC occurrence or decreasing the time to crisis resolution are critical goals for long-term care of patients with SCD. Current standards for clinical treatment serve to modify the pathobiology of the disease through blood transfusions and dosing with hydroxyurea which serves to increase circulating Fetal Hemoglobin (HbF) and diminish the polymerization of intracellular sickle cell hemoglobin (HbS) in adults and pediatric populations (59, 60, 63–66). Studies by Frenette et al. demonstrated the importance of elevated plasma heme in a humanized SCD mouse model. In fact, unstimulated neutrophils isolated from SCD patients rapidly begin forming NETs compared to almost no NET formation in age-matched healthy controls, even when under treatment with hydroxyurea (67). This elevated level of heme induces arrested neutrophils to release NETs that have been associated with hypothermia and rapid death. Modulation of plasma heme does alter the NET release and subsequent survival in this mouse model, leaving the underlying issue of the role of elevated neutrophil arrest efficiency and the onset of vaso-occlusion (8, 9). While allogenic hematopoietic stem cell transplants (HSCT) and gene therapies are currently under investigation for their potential long-term curative effects by reversing the defect in hemoglobin function, these have proven to be costly and currently have limited availability for wide spread treatment of SCD.
Activated platelets contribute to vaso-occlusion by participating in intercellular adhesion leading to formation of cell aggregates (6, 10). Pharmacological inhibition of platelet activation has been achieved using receptor antagonists of P2Y12R, the chemoreceptor involved in ADP-stimulated activation of platelets including platelet degranulation (41, 68). Clinical assessment of P2Y12R antagonists including Prasugrel and Ticagrelor have shown limited efficacy in adolescents and young adults with modest reduction of platelet activity and frequency of VOC (69, 70). These interventions have been ineffective in decreasing the frequency of pediatric VOC, pointing to a more significant role for platelet function on the occurrence of VOC in young adults. Directly targeting platelet adhesion through blocking GPIIbIIIa via administration of eptifbatide has been evaluated in a small cohort of subjects undergoing VOC (71). Treatment with eptifbatide did not diminish time to VOC resolution, but did successfully inhibit platelet and leukocyte aggregation, release into circulation, as well as diminished surface expression of P-selectin and serum measures of inflammation such as CD40L, sVCAM-1, sICAM-1, sICAM-3, MIP1α, and TNFα (71, 72).
Current reports link activation of the anaphylatoxin complement- C5a that activates neutrophils, platelets and endothelial cells with induction of vaso-occlusion in SCD mice (73). Blocking P-selectin inhibits C5a-induced vaso-occlusion in liver and skin venules of SCD mice. Moreover, blocking receptor binding with antibody targeting the C5a receptor antagonizes upregulation of endothelial adhesion molecules including VCAM-1, ICAM-1, and E-selectin in liver and lungs. It is noteworthy that a P-selectin deficient SCD mouse model revealed that chronic P-selectin deficiency attenuated liver ischemia but did not resolve cellular senescence and reduced epithelial cell proliferation necessary to maintain hepatic homeostasis (74). Complement from human sera has also been shown to stimulate expression of E-selectin in porcine endothelial cells (75). Likewise, the most abundant enzyme of the complement pathway, MASP-1 induced expression of TNFα, and upregulation of E-selectin on microvascular endothelium (76). Complement activation leads to venous thrombosis, an alternate mechanism driving vaso- occlusion. Interestingly, E-selectin plays a dominant role in venous thrombosis and a specific and potent antagonist of E-selectin adhesive function (GMI-1271; Uproleselan) blocks venous thrombosis, without the need to infuse anti-coagulants such as heparin (77, 78). In addition, blocking E-selectin with Uproleselan also inhibited deep venous thrombosis in a non-human primate model and showed efficacy in treating 2 patients in a case study of calf vein thrombosis (77, 79). Taken together, it appears that complement activation is a viable target to alleviate acute P-selectin and E-selectin dependent mechanisms of microvasculature inflammation that include thrombosis, but may not provide a long-term solution in context of preserving vital organ function.
Numerous anti-adhesive therapies that target RBC-endothelial and leukocyte-endothelial interactions have been completed or are undergoing clinical trials (Table 2). Of these, selectin mediating leukocyte and erythrocyte adhesion to the endothelium are prominent targets to limit vaso-occlusion in SCD patients (80–82). Improvement to time of VOC resolution or delay in first incidence of VOC appears to be a common efficacious event in pharmacological inhibitors of selectin binding, such as Rivipansel (5, 83), unfractionated heparin (84, 85), intravenous immunoglobulin (86, 87), and Crizanlizumab (88). Of these, Crizanlizumab® which inhibits P-selectin binding to PSGL-1 currently has completed clinical trials in adults and has the most ongoing trials to assess efficacy in pediatric populations as well as subjects with non-HbSS genotypes (Table 2). High dose (5 mg/mL) Crizanlizumab resulted in significant diminishment of the rate of crisis per year and delayed mean time to first or secondary crisis in adults (88). There was, however, no detectable difference in cell hemolysis suggesting that treatment with Crizanlizumab only serves to modify cell adhesion, but not to completely diminish patient systemic inflammation. Antagonism of E-selectin for treatment of VOC using the small molecule Rivipansel did not initially meet the Phase III clinical endpoint of time to readiness for patient discharge. However, subsequent analysis revealed that Rivipansel was efficacious in a subset of patients who were treated earlier from the time of onset of their VOC, with statistical significance demonstrated in both adults and adolescents who received treatment within 26 hours of the initial painful episode of VOC (median difference of 58 hours between Rivipansel and placebo; HR = 0.58; p = 0.03). Importantly, rapid and sustained statistically significant reductions in soluble E-selectin in circulation in this acute setting was observed in the Phase III trial supporting the biological effect of Rivipansel. This raises an important point on the beneficial effects of administering treatment to VOC early in progression and following systemic markers of the capacity of neutrophils to participate in adhesion. Clinical trials on the effects of anti-inflammatory agents such as statins and leukotriene and adenosine receptor inhibitors on resolution of VOC have also been performed (82, 89). Clinical trials assessing the efficacy of simvastatin in adolescents and young adults appear to have a significant effect on occurrence of VOC, as well as a reduction in patient reported pain, and decreased nitric oxide metabolites, human serum C-reactive protein, and soluble cell adhesion molecules ICAM1, VCAM-1, and E-Selectin (89). The data collected over the course of these clinical trials indicates that use of monotherapies targeting selectin expression, activity, and shedding on activated platelet and leukocytes are likely to improve patient quality of life.
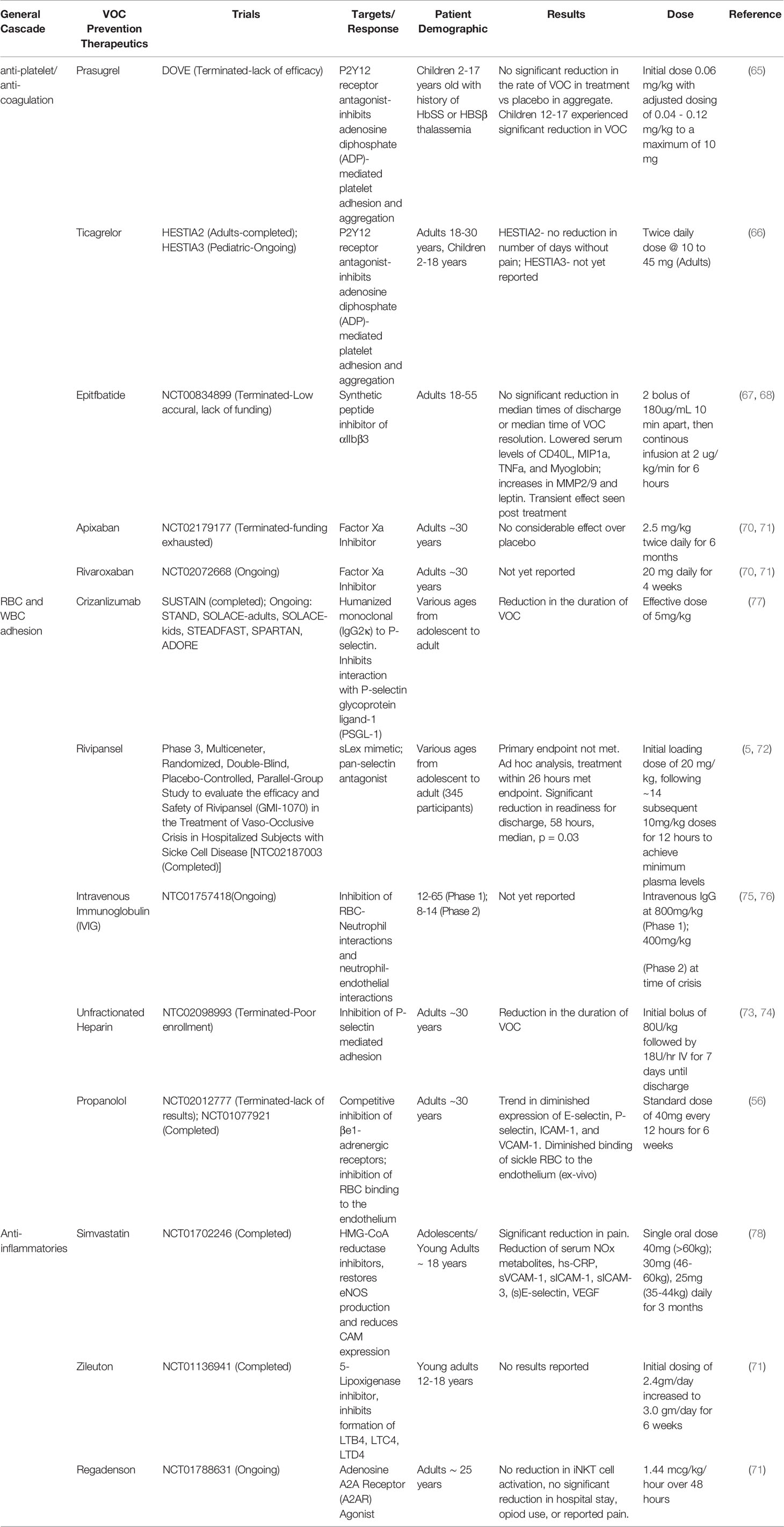
Table 2 Current agents being investigated to treat VOC.
Conclusions
Recurrent painful episodes of VOC severely diminish patient’s quality of life and frequently increase the risk of infections and complication of comorbidities that require emergency intervention. There is a need for therapeutics that target the earliest events leading to VOC progression before development of painful crises and end organ damage that typically call for immediate treatment of pain perception rather than the cause of its onset. Neutrophil rolling on E-selectin expressed in the inflamed microvasculature of patients with SCD can initiate integrin activation and firm arrest. Secondary capture of sRBCs and platelet aggregation leads to vascular congestion and tissue ischemia. Targeting these earliest selectin-dependent events in neutrophil recruitment with antagonists that block both initial tethering and signaling of adhesion appear to be clinically effective at mitigating VOC and painful episodes. First clinical trials are promising and should be accompanied by further experimental studies including novel appropriate murine in vivo models which address the various differences in the regulation of selectin function between mouse and human and ex-vivo analysis of treatment efficacy in inflammatory recruitment especially of neutrophils and sRBC over the course of the disease. Combined, this will shed new light on the role of selectins in the pathogenesis of VOC and hopefully contribute to establishing and refining a potentially beneficial selectin blocking strategy for the treatment of patients suffering from sickle cell disease.
Author Contributions
VM contributed his knowledge on selectin function in humans, helped outline and wrote the first draft of the manuscript. AH contributed his knowledge on drug efficacy in treatment of VOC in SCD and wrote corresponding sections. MS and JM contributed their vast knowledge of selectin function and clinical mechanisms of VOC and wrote the corresponding sections. SS contributed his vast knowledge on selectin function and neutrophil recruitment in human and mouse inflammation, outlined the manuscript, and wrote corresponding sections. All authors contributed to the article and approved the submitted version.
Funding
The work was supported by National Institute of Health grants AI047294 (SIS), AI103687 (SIS), and by the German research foundation collaborative research center (SFB), project B01 (MS).
Conflict of Interest
JM is Senior Vice-President, for Research and Chief Scientific Officer of GlycoMimetics, Inc. He has a financial interest in clinical development of selectin antagonist currently in clinical trials.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Stuart MJ, Nagel RL. Sickle-cell disease. Lancet (2004) 364:1343–60. doi: 10.1016/S0140-6736(04)17192-4
2. Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest (2007) 117:850–8. doi: 10.1172/JCI30920
3. Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, et al. Sickle cell disease. Nat Rev Dis Primers (2018) 4:18010. doi: 10.1038/nrdp.2018.10
4. Uwaezuoke SN, Ayuk AC, Ndu IK, Eneh CI, Mbanefo NR, Ezenwosu OU. Vaso-occlusive crisis in sickle cell disease: current paradigm on pain management. J Pain Res (2018) 11:3141–50. doi: 10.2147/JPR.S185582
5. Telen MJ, Wun T, McCavit TL, De Castro LM, Krishnamurti L, Lanzkron S, et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood (2015) 125:2656–64. doi: 10.1182/blood-2014-06-583351
6. Darbari DS, Sheehan VA, Ballas SK. The Vaso-Occlusive Pain Crisis in Sickle Cell Disease: Definition, Pathophysiology, and Management. Eur J Haematol (2020). doi: 10.1111/ejh.13430
7. Okpala I. Investigational selectin-targeted therapy of sickle cell disease. Expert Opin Investig Drugs (2015) 24:229–38. doi: 10.1517/13543784.2015.963552
8. Nader E, Romana M, Connes P. The Red Blood Cell-Inflammation Vicious Circle in Sickle Cell Disease. Front Immunol (2020) 11:454. doi: 10.3389/fimmu.2020.00454
9. Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, Frenette PS. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood (2014) 123:3818–27. doi: 10.1182/blood-2013-10-529982
10. Ballas SK, Darbari DS. Review/overview of pain in sickle cell disease. Complement Ther Med (2020) 49:102327. doi: 10.1016/j.ctim.2020.102327
11. Veluswamy S, Shah P, Denton CC, Chalacheva P, Khoo MCK, Coates TD. Vaso-Occlusion in Sickle Cell Disease: Is Autonomic Dysregulation of the Microvasculature the Trigger? J Clin Med (2019) 8(10). doi: 10.3390/jcm8101690
12. Simon SI, Hu Y, Vestweber D, Smith CW. Neutrophil tethering on E-selectin activates beta 2 integrin binding to ICAM-1 through a mitogen-activated protein kinase signal transduction pathway. J Immunol (2000) 164:4348–58. doi: 10.4049/jimmunol.164.8.4348
13. Ley K. The role of selectins in inflammation and disease. Trends Mol Med (2003) 9:263–8. doi: 10.1016/S1471-4914(03)00071-6
14. Hidalgo A, Peired AJ, Wild M, Vestweber D, Frenette PS. Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity (2007) 26:477–89. doi: 10.1016/j.immuni.2007.03.011
15. Sperandio M, Gleissner CA, Ley K. Glycosylation in immune cell trafficking. Immunol Rev (2009) 230:97–113. doi: 10.1111/j.1600-065X.2009.00795.x
16. Rocheleau AD, Cao TM, Takitani T, King MR. Comparison of human and mouse E-selectin binding to Sialyl-Lewis(x). BMC Struct Biol (2016) 16:10. doi: 10.1186/s12900-016-0060-x
17. Rocheleau AD, Sumagin R, Sarelius IH, King MR. Simulation and Analysis of Tethering Behavior of Neutrophils with Pseudopods. PloS One (2015) 10:e0128378. doi: 10.1371/journal.pone.0128378
18. Mondal N, Buffone A Jr, Neelamegham S. Distinct glycosyltransferases synthesize E-selectin ligands in human vs. mouse leukocytes. Cell Adh Migr (2013) 7:288–92. doi: 10.4161/cam.24714
19. Liu Z, Miner JJ, Yago T, Yao L, Lupu F, Xia L, et al. Differential regulation of human and murine P-selectin expression and function in vivo. J Exp Med (2010) 207:2975–87. doi: 10.1084/jem.20101545
20. Buffone A Jr, Mondal N, Gupta R, McHugh KP, Lau JT, Neelamegham S. Silencing alpha1,3-fucosyltransferases in human leukocytes reveals a role for FUT9 enzyme during E-selectin-mediated cell adhesion. J Biol Chem (2013) 288:1620–33. doi: 10.1074/jbc.M112.400929
21. Chen S, Springer TA. Selectin receptor-ligand bonds: Formation limited by shear rate and dissociation governed by the Bell model. Proc Natl Acad Sci U S A (2001) 98:950–5. doi: 10.1073/pnas.98.3.950
22. Morikis VA, Simon SI. Neutrophil Mechanosignaling Promotes Integrin Engagement With Endothelial Cells and Motility Within Inflamed Vessels. Front Immunol (2018) 9:2774. doi: 10.3389/fimmu.2018.02774
23. Leeuwenberg JF, Smeets EF, Neefjes JJ, Shaffer MA, Cinek T, Jeunhomme TM, et al. E-selectin and intercellular adhesion molecule-1 are released by activated human endothelial cells in vitro. Immunology (1992) 77:543–9.
24. Keelan ET, Licence ST, Peters AM, Binns RM, Haskard DO. Characterization of E-selectin expression in vivo with use of a radiolabeled monoclonal antibody. Am J Physiol (1994) 266:H278–290. doi: 10.1152/ajpheart.1994.266.1.H279
25. Kiskin NI, Hellen N, Babich V, Hewlett L, Knipe L, Hannah MJ, et al. Protein mobilities and P-selectin storage in Weibel-Palade bodies. J Cell Sci (2010) 123:2964–75. doi: 10.1242/jcs.073593
26. Chase SD, Magnani JL, Simon SI. E-selectin ligands as mechanosensitive receptors on neutrophils in health and disease. Ann BioMed Eng (2012) 40:849–59. doi: 10.1007/s10439-011-0507-y
27. Stadtmann A, Germena G, Block H, Boras M, Rossaint J, Sundd P, et al. The PSGL-1-L-selectin signaling complex regulates neutrophil adhesion under flow. J Exp Med (2013) 210:2171–80. doi: 10.1084/jem.20130664
28. McEver RP. Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res (2015) 107:331–9. doi: 10.1093/cvr/cvv154
29. Morikis VA, Chase S, Wun T, Chaikof EL, Magnani JL, Simon SI. Selectin catch-bonds mechanotransduce integrin activation and neutrophil arrest on inflamed endothelium under shear flow. Blood (2017) 130:2101–10. doi: 10.1182/blood-2017-05-783027
30. Shimaoka M, Xiao T, Liu JH, Yang Y, Dong Y, Jun CD, et al. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell (2003) 112:99–111. doi: 10.1016/S0092-8674(02)01257-6
31. Riese SB, Kuehne C, Tedder TF, Hallmann R, Hohenester E, Buscher K. Heterotropic modulation of selectin affinity by allosteric antibodies affects leukocyte rolling. J Immunol (2014) 192:1862–9. doi: 10.4049/jimmunol.1302147
32. Beste MT, Hammer DA. Selectin catch-slip kinetics encode shear threshold adhesive behavior of rolling leukocytes. Proc Natl Acad Sci USA (2008) 105:20716–21. doi: 10.1073/pnas.0808213105
33. Guyer DA, Moore KL, Lynam EB, Schammel CM, Rogelj S, McEver RP, et al. P-selectin glycoprotein ligand-1 (PSGL-1) is a ligand for L-selectin in neutrophil aggregation. Blood (1996) 88:2415–21. doi: 10.1182/blood.V88.7.2415.bloodjournal8872415
34. Preston RC, Jakob RP, Binder FP, Sager CP, Ernst B, Maier T. E-selectin ligand complexes adopt an extended high-affinity conformation. J Mol Cell Biol (2016) 8:62–72. doi: 10.1093/jmcb/mjv046
35. Sokurenko EV, Vogel V, Thomas WE. Catch-bond mechanism of force-enhanced adhesion: counterintuitive, elusive, but ... widespread? Cell Host Microbe (2008) 4:314–23. doi: 10.1016/j.chom.2008.09.005
36. Snook JH, Guilford WH. The Effects of Load on E-Selectin Bond Rupture and Bond Formation. Cell Mol Bioeng (2010) 3:128–38. doi: 10.1007/s12195-010-0110-6
37. Pereverzev YV, Prezhdo E, Sokurenko EV. The two-pathway model of the biological catch-bond as a limit of the allosteric model. Biophys J (2011) 101:2026–36. doi: 10.1016/j.bpj.2011.09.005
38. Wayman AM, Chen W, McEver RP, Zhu C. Triphasic force dependence of E-selectin/ligand dissociation governs cell rolling under flow. Biophys J (2010) 99:1166–74. doi: 10.1016/j.bpj.2010.05.040
39. Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med (2009) 15:384–91. doi: 10.1038/nm.1939
40. Finnegan EM, Turhan A, Golan DE, Barabino GA. Adherent leukocytes capture sickle erythrocytes in an in vitro flow model of vaso-occlusion. Am J Hematol (2007) 82:266–75. doi: 10.1002/ajh.20819
41. Wun T, Cordoba M, Rangaswami A, Cheung AW, Paglieroni T. Activated monocytes and platelet-monocyte aggregates in patients with sickle cell disease. Clin Lab Haematol (2002) 24:81–8. doi: 10.1046/j.1365-2257.2002.t01-1-00433.x
42. Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu Rev Med (2010) 61:35–47. doi: 10.1146/annurev.med.080708.082655
43. Blum A, Yeganeh S, Peleg A, Vigder F, Kryuger K, Khatib A, et al. Endothelial function in patients with sickle cell anemia during and after sickle cell crises. J Thromb Thrombolysis (2005) 19:83–6. doi: 10.1007/s11239-005-1377-7
44. Kato GJ, Martyr S, Blackwelder WC, Nichols JS, Coles WA, Hunter LA, et al. Levels of soluble endothelium-derived adhesion molecules in patients with sickle cell disease are associated with pulmonary hypertension, organ dysfunction, and mortality. Br J Haematol (2005) 130:943–53. doi: 10.1111/j.1365-2141.2005.05701.x
45. Kuwano Y, Spelten O, Zhang H, Ley K, Zarbock A. Rolling on E- or P-selectin induces the extended but not high-affinity conformation of LFA-1 in neutrophils. Blood (2010) 116:617–24. doi: 10.1182/blood-2010-01-266122
46. Sundd P, Pospieszalska MK, Cheung LS, Konstantopoulos K, Ley K. Biomechanics of leukocyte rolling. Biorheology (2011) 48:1–35. doi: 10.3233/BIR-2011-0579
47. Shao B, Yago T, Setiadi H, Wang Y, Mehta-D’souza P, Fu J, et al. O-glycans direct selectin ligands to lipid rafts on leukocytes. Proc Natl Acad Sci U.S.A. (2015) 112:8661–6. doi: 10.1073/pnas.1507712112
48. Green CE, Pearson DN, Camphausen RT, Staunton DE, Simon SI. Shear-dependent capping of L-selectin and P-selectin glycoprotein ligand 1 by E-selectin signals activation of high-avidity beta2-integrin on neutrophils. J Immunol (Baltimore Md 1950) (2004) 172:7780–90. doi: 10.4049/jimmunol.172.12.7780
49. Pruenster M, Kurz AR, Chung KJ, Cao-Ehlker X, Bieber S, Nussbaum CF, et al. Extracellular MRP8/14 is a regulator of beta2 integrin-dependent neutrophil slow rolling and adhesion. Nat Commun (2015) 6:6915. doi: 10.1038/ncomms7915
50. Hobbs JA, May R, Tanousis K, McNeill E, Mathies M, Gebhardt C, et al. Myeloid cell function in MRP-14 (S100A9) null mice. Mol Cell Biol (2003) 23:2564–76. doi: 10.1128/MCB.23.7.2564-2576.2003
51. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med (2007) 13:1042–9. doi: 10.1038/nm1638
52. Croce K, Gao H, Wang Y, Mooroka T, Sakuma M, Shi C, et al. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation (2009) 120:427–36. doi: 10.1161/CIRCULATIONAHA.108.814582
53. Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ, et al. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood (2012) 119:4275–82. doi: 10.1182/blood-2011-08-373118
54. Eriksson EE, Xie X, Werr J, Thoren P, Lindbom L. Importance of primary capture and L-selectin-dependent secondary capture in leukocyte accumulation in inflammation and atherosclerosis in vivo. J Exp Med (2001) 194:205–18. doi: 10.1084/jem.194.2.205
55. Begandt D, Thome S, Sperandio M, Walzog B. How neutrophils resist shear stress at blood vessel walls: molecular mechanisms, subcellular structures, and cell-cell interactions. J Leukoc Biol (2017) 102:699–709. doi: 10.1189/jlb.3MR0117-026RR
56. Rossaint J, Zarbock A. Platelets in leucocyte recruitment and function. Cardiovasc Res (2015) 107:386–95. doi: 10.1093/cvr/cvv048
57. Rossaint J, Margraf A, Zarbock A. Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Front Immunol (2018) 9:2712. doi: 10.3389/fimmu.2018.02712
58. Weber C, Springer TA. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J Clin Invest (1997) 100:2085–93. doi: 10.1172/JCI119742
59. Weninger W, Ulfman LH, Cheng G, Souchkova N, Quackenbush EJ, Lowe JB, et al. Specialized contributions by alpha(1,3)-fucosyltransferase-IV and FucT-VII during leukocyte rolling in dermal microvessels. Immunity (2000) 12:665–76. doi: 10.1016/S1074-7613(00)80217-4
60. Manwani D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood (2013) 122:3892–8. doi: 10.1182/blood-2013-05-498311
61. Bennewitz MF, Tutuncuoglu E, Gudapati S, Brzoska T, Watkins SC, Monga SP, et al. P-selectin-deficient mice to study pathophysiology of sickle cell disease. Blood Adv (2020) 4:266–73. doi: 10.1182/bloodadvances.2019000603
62. Wun T, Styles L, DeCastro L, Telen MJ, Kuypers F, Cheung A, et al. Phase 1 study of the E-selectin inhibitor GMI 1070 in patients with sickle cell anemia. PloS One (2014) 9:e101301. doi: 10.1371/journal.pone.0101301
63. Krishnamurthy VR, Sardar MY, Ying Y, Song X, Haller C, Dai E, et al. Glycopeptide analogues of PSGL-1 inhibit P-selectin in vitro and in vivo. Nat Commun (2015) 6:6387. doi: 10.1038/ncomms7387
64. Charache S, Dover GJ, Moore RD, Eckert S, Ballas SK, Koshy M, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood (1992) 79:2555–65. doi: 10.1182/blood.V79.10.2555.bloodjournal79102555
65. Steinberg MH, Lu ZH, Barton FB, Terrin ML, Charache S, Dover GJ. Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Multicenter Study of Hydroxyurea. Blood (1997) 89:1078–88. doi: 10.1182/blood.V89.3.1078
66. Zimmerman SA, Schultz WH, Davis JS, Pickens CV, Mortier NA, Howard TA, et al. Sustained long-term hematologic efficacy of hydroxyurea at maximum tolerated dose in children with sickle cell disease. Blood (2004) 103:2039–45. doi: 10.1182/blood-2003-07-2475
67. Barbu EA, Dominical VM, Mendelsohn L, Thein SL. Neutrophils remain detrimentally active in hydroxyurea-treated patients with sickle cell disease. PloS One (2019) 14:e0226583. doi: 10.1371/journal.pone.0226583
68. Wun T, Paglieroni T, Rangaswami A, Franklin PH, Welborn J, Cheung A, et al. Platelet activation in patients with sickle cell disease. Br J Haematol (1998) 100:741–9. doi: 10.1046/j.1365-2141.1998.00627.x
69. Heeney MM, Hoppe CC, Abboud MR, Inusa B, Kanter J, Ogutu B, et al. A Multinational Trial of Prasugrel for Sickle Cell Vaso-Occlusive Events. N Engl J Med (2016) 374:625–35. doi: 10.1056/NEJMoa1512021
70. Kanter J, Abboud MR, Kaya B, Nduba V, Amilon C, Gottfridsson C, et al. Ticagrelor does not impact patient-reported pain in young adults with sickle cell disease: a multicentre, randomised phase IIb study. Br J Haematol (2019) 184:269–78. doi: 10.1111/bjh.15646
71. Lee SP, Ataga KI, Zayed M, Manganello JM, Orringer EP, Phillips DR, et al. Phase I study of eptifibatide in patients with sickle cell anaemia. Br J Haematol (2007) 139:612–20. doi: 10.1111/j.1365-2141.2007.06787.x
72. Desai PC, Brittain JE, Jones SK, McDonald A, Wilson DR, Dominik R, et al. A pilot study of eptifibatide for treatment of acute pain episodes in sickle cell disease. Thromb Res (2013) 132:341–5. doi: 10.1016/j.thromres.2013.08.002
73. Vercellotti GM, Dalmasso AP, Schaid TR, Nguyen J, Chen C, Ericson ME, et al. Critical role of C5a in sickle cell disease. Am J Hematol (2019) 94:327–37. doi: 10.1002/ajh.25384
74. Vats R, Kaminski TW, Ju EM, Brzoska T, Tutuncuoglu E, Tejero J, et al. P-selectin deficiency promotes liver senescence in sickle cell disease mice. Blood (2021). doi: 10.1182/blood.2020009779
75. Sølvik U, Haraldsen G, Fiane AE, Boretti E, Lambris JD, Fung M, et al. Human serum-induced expression of E-selectin on porcine aortic endothelial cells in vitro is totally complement mediated. Transplantation (001) 72:1967–73. doi: 10.1097/00007890-200112270-00017
76. Jani PK, Kajdácsi E, Megyeri M, Dobó J, Doleschall Z, Futosi K, et al. MASP-1 induces a unique cytokine pattern in endothelial cells: a novel link between complement system and neutrophil granulocytes. PloS One (2014) 9(1):e87104. doi: 10.1371/journal.pone.0087104
77. Devata S, Angelini DE, Blackburn S, Hawley A, Myers DD, Schaefer JK, et al. Use of GMI-1271, an E-selectin antagonist, in healthy subjects and in 2 patients with calf vein thrombosis. Res Pract Thromb Haemost (2020) 4(2):193–204. doi: 10.1002/rth2.12279
78. Culmer DL, Dunbar ML, Hawley AE, Sood S, Sigler RE, Henke PK, et al. E-selectin inhibition with GMI-1271 decreases venous thrombosis without profoundly affecting tail vein bleeding in a mouse model. Thromb Haemost (2017) 117:1171–81. doi: 10.1160/TH16-04-0323
79. Myers D, Lester P, Adili R, Hawley A, Durham L, Dunivant V, et al. A new way to treat proximal deep venous thrombosis using E-selectin inhibition. J Vasc Surg Venous Lymphat Disord (2020) 8:268–78. doi: 10.1016/j.jvsv.2019.08.016
80. De Castro L, Zennadi R, Jonassaint J, Batchvarova M, Telen M. Effect of Propranolol as Anti-Adhesive Therapy in Sickle Cell Disease. Clin Trans Sci (2012) 5:437–44. doi: 10.1111/cts.12005
81. Christen JR, Bertolino J, Jean E, Camoin L, Ebbo M, Harlé JR, et al. Use of Direct Oral Anticoagulants in Patients with Sickle Cell Disease and Venous Thromboembolism: A Prospective Cohort Study of 12 Patients. Hemoglobin (2019) 43:296–9. doi: 10.1080/03630269.2019.1689997
82. Telen MJ. Beyond hydroxyurea: new and old drugs in the pipeline for sickle cell disease. Blood (2016) 127:810–9. doi: 10.1182/blood-2015-09-618553
83. Chang J, Patton JT, Sarkar A, Ernst B, Magnani JL, Frenette PS. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood (2010) 116:1779–86. doi: 10.1182/blood-2009-12-260513
84. Nelson RM, Cecconi O, Roberts WG, Aruffo A, Linhardt RJ, Bevilacqua MP. Heparin oligosaccharides bind L- and P-selectin and inhibit acute inflammation. Blood (1993) 82:3253–8. doi: 10.1182/blood.V82.11.3253.bloodjournal82113253
85. Qari MH, Aljaouni SK, Alardawi MS, Fatani H, Alsayes FM, Zografos P, et al. Reduction of painful vaso-occlusive crisis of sickle cell anaemia by tinzaparin in a double-blind randomized trial. Thromb Haemost (2007) 98:392–6. doi: 10.1160/Th06-12-0718
86. Chang J, Shi PA, Chiang EY, Frenette PS. Intravenous immunoglobulins reverse acute vaso-occlusive crises in sickle cell mice through rapid inhibition of neutrophil adhesion. Blood (2008) 111:915–23. doi: 10.1182/blood-2007-04-084061
87. Turhan A, Jenab P, Bruhns P, Ravetch JV, Coller BS, Frenette PS. Intravenous immune globulin prevents venular vaso-occlusion in sickle cell mice by inhibiting leukocyte adhesion and the interactions between sickle erythrocytes and adherent leukocytes. Blood (2004) 103:2397–400. doi: 10.1182/blood-2003-07-2209
88. Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med (2017) 376:429–39. doi: 10.1056/NEJMoa1611770
Keywords: vaso-occlusion crises, neutrophil, selectin, integrins, sickle cell disease
Citation: Morikis VA, Hernandez AA, Magnani JL, Sperandio M and Simon SI (2021) Targeting Neutrophil Adhesive Events to Address Vaso-Occlusive Crisis in Sickle Cell Patients. Front. Immunol. 12:663886. doi: 10.3389/fimmu.2021.663886
Received: 03 February 2021; Accepted: 29 March 2021;
Published: 28 April 2021.
Edited by:
Emmanuel Donnadieu, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Lubka T. Roumenina, INSERM U1138 Centre de Recherche des Cordeliers (CRC), FranceRostyslav Bilyy, Danylo Halytsky Lviv National Medical University, Ukraine
Copyright © 2021 Morikis, Hernandez, Magnani, Sperandio and Simon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Scott I. Simon, c2lzaW1vbkB1Y2RhdmlzLmVkdQ==