 Caterina Veroni
Caterina Veroni Francesca Aloisi
Francesca Aloisi- Department of Neuroscience, Istituto Superiore di Sanità, Rome, Italy
The cause and the pathogenic mechanisms leading to multiple sclerosis (MS), a chronic inflammatory disease of the central nervous system (CNS), are still under scrutiny. During the last decade, awareness has increased that multiple genetic and environmental factors act in concert to modulate MS risk. Likewise, the landscape of cells of the adaptive immune system that are believed to play a role in MS immunopathogenesis has expanded by including not only CD4 T helper cells but also cytotoxic CD8 T cells and B cells. Once the key cellular players are identified, the main challenge is to define precisely how they act and interact to induce neuroinflammation and the neurodegenerative cascade in MS. CD8 T cells have been implicated in MS pathogenesis since the 80’s when it was shown that CD8 T cells predominate in MS brain lesions. Interest in the role of CD8 T cells in MS was revived in 2000 and the years thereafter by studies showing that CNS-recruited CD8 T cells are clonally expanded and have a memory effector phenotype indicating in situ antigen-driven reactivation. The association of certain MHC class I alleles with MS genetic risk implicates CD8 T cells in disease pathogenesis. Moreover, experimental studies have highlighted the detrimental effects of CD8 T cell activation on neural cells. While the antigens responsible for T cell recruitment and activation in the CNS remain elusive, the high efficacy of B-cell depleting drugs in MS and a growing number of studies implicate B cells and Epstein-Barr virus (EBV), a B-lymphotropic herpesvirus that is strongly associated with MS, in the activation of pathogenic T cells. This article reviews the results of human studies that have contributed to elucidate the role of CD8 T cells in MS immunopathogenesis, and discusses them in light of current understanding of autoreactivity, B-cell and EBV involvement in MS, and mechanism of action of different MS treatments. Based on the available evidences, an immunopathological model of MS is proposed that entails a persistent EBV infection of CNS-infiltrating B cells as the target of a dysregulated cytotoxic CD8 T cell response causing CNS tissue damage.
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) and one of the world’s most common neurological disorders. The disease is characterized by multifocal inflammatory lesions causing demyelination and neurodegeneration in the brain and spinal cord and leading to the progressive loss of motor, sensory and cognitive functions (1). MS usually has a relapsing remitting course at onset, followed by a secondary progressive phase with gradual worsening of the neurological symptoms; in a minority of patients the disease has a progressive onset. Drugs approved for relapsing remitting MS can reduce clinical relapses and inflammatory disease activity and delay to some extent the progression of neurological deficits (2, 3). However, a definitive cure for MS is still missing.
The etiology of MS involves multiple genetic risk factors acting in concert with environmental exposures (4). Genome-wide association studies (GWAS), over the last decade, have identified more than 230 loci associated with MS susceptibility (5–9). The strongest genetic association for MS maps to the class II region of the human leukocyte antigen (HLA) gene cluster in chromosome 6p21. The ubiquitous herpesvirus Epstein-Barr virus (EBV), cigarette smoking, and vitamin D deficiency are the environmental factors that show the most consistent association with MS (4, 10). Among these, EBV has the strongest impact on the immune system due to its ability to establish a life-long latent infection in B cells, reactivate periodically and induce a very effective immune response that is essential to counteract the virus lymphoproliferative potential (11, 12). The epidemiological data associating EBV infection to MS, the main features of EBV biology and immunology, the compatibility between EBV and MS disease biology, and the potential mechanisms linking EBV to CNS inflammation are discussed in the following sections and are summarized in Table 1.

Table 1 EBV intersection with MS.
At the CNS level, MS is characterized by immune abnormalities that include B-cell activation manifesting as persistent intrathecal immunoglobulin (Ig) synthesis and oligoclonal IgG bands, moderate pleiocytosis and increased levels of pro-inflammatory cytokines and chemokines in the cerebrospinal fluid (CSF) (1). Inflammatory cells (T cells, B cells, plasma cells and myeloid cells) accumulate mainly within the perivascular space of post-capillary venules in the white matter and, to a lesser extent, gray matter, and in the subarachnoid space within the meninges (33). Activation of microglia, the CNS resident innate immune cells, is another prominent feature of CNS inflammation in MS, contributing both to tissue injury and to the healing response (84).
For nearly 40 years, the studies addressing the identity and antigenic specificity of T cells migrating and becoming activated in the CNS have focussed on CD4 T helper (Th) cells and Th subsets (Th1, Th17, Th1/Th17) as executors of putative, still elusive, autoimmune responses towards myelin proteins (85). Quietly, but steadily, evidence has grown for an important role of CD8 T cells and B cells in MS immunopathogenesis. These knowledge advancements have been mostly driven by two sets of data: i) new evidences pointing to EBV as a possible causative agent in MS (16, 17) and a biologically plausible trigger or driver of the recurrent, highly destructive CNS-targeted immune response (49, 66, 86, 87) brought more attention on CD8 T cells as these cells play a key role in antiviral immunity and dominate the CNS immune infiltrate in MS (79); ii) the discovery of B-cell follicle-like structures in the brain meninges and the demonstration of their association with disease severity and cortical damage in patients with progressive MS (36, 37, 88), together with the high efficacy of B-cell depleting monoclonal antibodies in suppressing MS disease activity (89) have fostered investigations on B-lineage cells beyond their role as antibody producing cells (90).
Once the key immune cell players are identified, the main challenge is to define precisely how they act and interact to promote chronic CNS inflammation and the downstream neurodegenerative cascade in MS. This article places a special emphasis on the human genetic, neuropathologic, immunologic and molecular studies that have contributed to elucidate the role of CD8 T cells in MS pathogenesis. The available data are discussed in the context of current understanding of autoimmunity, B-cell and EBV involvement in MS, and of the impact of approved MS treatments on T cells and B cells. By wrapping up this information, a model of MS pathogenesis is proposed that entails defective immune control of EBV in susceptible individuals as an early event leading to establishment of a persistent EBV infection in the CNS and chronic activation of an EBV-specific cytotoxic T cell response as the main determinant of CNS tissue damage. This model challenges the paradigm that MS is an autoimmune disease, paving the way towards new ways to treat and prevent the disease.
MS Risk Genes Linked to CD8 T Cell and B Cell Function
The strongest genetic factor determining MS risk is the HLA-DRB1*15 group of alleles implicating CD4 T helper cells in disease pathogenesis (91). The mechanisms by which the HLA-DRB1*15 haplotype affects MS susceptibility remain to be precisely defined but may include HLA-DRB1*15 hypomethylation causing changes in HLA-DRB1*15 expression on antigen presenting cells (APC) (92) and presentation of self peptides by APC that activate CD4 T cells recognizing antigens expressed in the CNS (72, 93, 94).
MHC class I alleles are also associated with MS suggesting CD8 T cell involvement. HLA-A*0301 increases the risk of MS by about two-fold (95) while HLA-A*0201 confers protection from the disease, halving the risk associated with HLA-DRB1*15 (5, 95). A study in humanized transgenic mice expressing a myelin-specific T cell receptor derived from a CD8 T cell clone from an individual patient along with HLA-A*0301 or HLA-A*0201 showed an opposite effect of these alleles on the development of experimental autoimmune encephalomyelitis, suggesting a potential link between MS genetic risk and CD8 T cell-mediated autoimmune demyelination (96). Of interest, HLA-A*0201 and another MS protective MHC class I allele, HLA-B*44, were recently associated with increased levels of the type 1 interferon (IFN) receptor subunit IFNAR2 in B and T cells and a reduced response to type I IFN stimulation in both cell types (97). These findings suggest that the protective effect of these MHC class I alleles in MS might be mediated by modulation of a key pathway in antiviral immunity.
The association between EBV and MS is the best documented pathogen-chronic disease association and is supported by a large body of seroepidemiological data pointing to an altered virus-host balance before and during the disease (10, 19) (Table 1). There is solid evidence that EBV infection is necessary, though not sufficient, for MS development (14, 98, 99) and that the risk of developing MS is increased in individuals with a history of infectious mononucleosis (15). Higher levels of antibodies to EBV, mainly anti-EBV nuclear antigen-1 (EBNA-1) IgG, but not to other viruses, are present in MS patients compared to healthy subjects and predispose to MS development (10). These evidences have led to investigate whether HLA genes influence MS genetic risk through the control of EBV infection. A suboptimal antibody and cytotoxic response to EBV during primary infection could result in a higher viral load, altering the virus-host immune system balance and increasing the chance for the virus to activate pathologic processes potentially leading to MS, as summarized in Table 1 and discussed below.
Because cytotoxic CD8 T cells are predominantly involved in the killing of virus infected cells and CD4 T cell help is required for CD8 T cell priming, maintenance of CD8 T cell memory and prevention of exhaustion (100, 101), it is likely that both class I- and class II-restricted antigen presentation is involved in EBV control. An interaction between EBV infection and HLA genes associated with MS risk is suggested by the finding that HLA-DRB1*15 carriers with higher anti-EBNA-1 antibody titers have a markedly elevated risk of MS (102–104), which is increased further in the absence of the protective allele HLA-A*0201 (103). MS risk-associated non-MHC genes involved in B and T cell activation, like MYB, CARD11, and CLEC16A were also found associated with higher EBNA-1 IgG levels in MS patients (104).
A recent study in humanized mice (NOD-scid IL2 receptor γ-chain-deficient mice reconstituted with human immune system components) has shown less efficient recognition of EBV-transformed B cell lines by HLA-DR15-restricted CD4 T cell clones, increased CD8 T cell expansion and activation, and higher EBV DNA load in HLA-DR15 donor-reconstituted mice after EBV infection (105). In the same study, HLA-DR15-restricted CD4 T cell clones also recognized myelin basic protein (MBP). These findings suggest that the interaction between EBV and HLA-DR15 can induce a hyperreactive but defective CD8 T-cell response, as well CD4 T cells cross-reactive with self antigens, both events possibly concurring to MS development (105).
Recent analyses of GWAS data implicate innate and adaptive immunity in MS pathogenesis and several of the non-MHC gene susceptibility variants regulate T cell differentiation and function (6–8). These include variants of the genes encoding: the transcription factors eomesodermin (Eomes, also known as T-box brain 2) and T-bet (T-box expressed in T cells, highly homologous to Eomes) which promote the differentiation and activation of type-1 immunity cells (Th1, Tc1, NK cells) implicated in the defense against intracellular pathogens and viruses (106); IFNγ receptor β-chain, involved in the response to IFN-γ, a major product of activated Th1, NK and CD8 T cells; molecules related to the signaling and production of IL12, a cytokine produced by dendritic cells and macrophages with a crucial role in the commitment to Th1 and Tc1 cells; molecules involved in the production and signaling of IL15, a cytokine essential for the development, homeostasis, function and survival of CD8 T cells; the pore-forming protein perforin, a key component of the lytic machinery of cytotoxic lymphocytes. Other MS associated risk variants that are involved in T cell function encode the following molecules: subunits of the IL2 receptor and IL7 receptor that regulate T cell genesis, survival and expansion; CD28 and CTLA4 expressed on T cells and their ligands CD80 and CD86 expressed on antigen presenting cells (APC), which are major regulators of T cell activation and inhibition, respectively; genes involved in CD28 and CTLA4 signaling; CD40 and CD58, both expressed on APC and implicated in APC-T cell interactions; CD69, an early T cell activation marker; CD161, highly expressed in a subset of CD8 T lymphocytes implicated in MS pathogenesis (see below).
GWAS have also identified MS-risk gene variants that encode molecules associated with B cell development and activation (7, 8). These include: CD40, whose signaling is mimicked by the EBV latent protein LMP1 (11); tumor necrosis factor receptor-associated factor adaptor protein 3 (TRAF3) interacting with both CD40 and LMP1 (107); all members of the Fc receptor-like gene family, primarily expressed on B cells; MYB, a transcription factor required for B-cell development; molecules linked to IL6 and IL10 signaling pathways. NFKB1 encoding nuclear factor kappa B (NF-κB), a major regulator of the immune response, also emerged among the genes that increase MS susceptibility. Interestingly, the NF-κB pathway is activated in CSF memory B cells from MS patients (108) and a mutation in the NFKB1 gene has been implicated in a primary immunodeficiency characterized by poor control of EBV and lymphoproliferation (109).
A recent GWAS has identified a variant of the TNFSF13B gene encoding the cytokine B-cell activating factor (BAFF) involved in B-cell activation, differentiation, and survival, that is more common in Sardinia, an Italian region with one of the highest MS prevalence rates worldwide, and is associated with an increased risk of developing MS (110). The TNFSF13B variant (named BAFF-var) leads to increased levels of serum BAFF, immunoglobulins and circulating B cells, particularly memory B cells, the main reservoir for life-long EBV infection, raising the possibility that BAFF-var might favor an altered EBV-immune system balance.
CD8 T Cell Frequency, Phenotype and Expansion in the CNS OF MS Patients
The first hint that CD8 T cells could play a key role in MS came from immunohistochemical studies assessing the frequency and distribution of different T cell subsets in postmortem brain specimens of patients dying during the chronic phase of MS (111–113). These early studies showed that CD8 T cells outnumber CD4 T cells in white matter lesions, perilesional areas and meninges. More recent studies have confirmed predominance of CD8+ cells among T cells in all types of brain areas with inflammatory cell infiltrates (active, chronic active and inactive white lesions and normal appearing white matter), irrespective of disease course (acute, relapsing-remitting or progressive) and extent of immune infiltration (62, 114–116). The numerical data give already valuable information on the T cell population that preferentially migrates and/or reactivates locally in the MS brain. In fact, the CD4:CD8 ratio in the CNS parenchyma of MS patients (mean of 1:3 to 1:5, range from 1:1 to 1:50) is inverse to the CD4:CD8 ratios in the peripheral blood (2:1) and CSF (4-5:1) of MS patients and healthy controls.
CD8 T cells accumulating in brain tissue of MS patients are mainly effector memory T cells (CD45RA-, CD45RO+, CCR7-) that express the activation marker CD69, the costimulatory molecule CD137 and the apoptosis inducing TNF family member CD95L, and produce IFNγ (62, 114, 117). It is less clear whether CD4 and CD8 T cells producing IL17 and GM-CSF are frequent in MS brain lesions, since the available data are contradictory (114, 118–120). A variable but substantial proportion of CNS-infiltrating CD8 T cells express the lytic enzymes granzyme B and perforin (62, 114, 115, 117), their in situ cytotoxic activity being supported by lytic granule polarization, cell surface expression of the degranulation marker CD107a (117, 121) and presence of adjacent cells expressing cleaved caspase 3 (62).
CD8 T cells with a memory phenotype (mainly central memory but also effector memory T cells) are enriched in the CSF of MS patients relatively to the peripheral blood (122–125). CSF effector memory T cells produce predominantly pro-inflammatory cytokines, like IFNγ and TNF, and express granzyme B and perforin (114, 123, 125–127). CSF enrichment of effector memory CD8 T cells is more prominent during early stages of relapse onset MS (125), and increased levels of granzyme B were found in the CSF of MS patients during relapses, suggesting higher CD8 T cell-mediated cytotoxicity (128).
Molecules that are expressed in CSF- and CNS tissue-infiltrating CD8 T cells and have been implicated in memory CD8 T cell migration include: the chemokine receptors CCR5 and CXCR3, that bind CCL5 and CXCL9/10, respectively (123, 129), and are enriched in type-1 immunity cells (106); P-selectin glycoprotein ligand-1 (130); junctional adhesion molecule-like (131); melanoma cell adhesion molecule (132); CD11a, the α chain of the αLβ2 integrin (also known as leukocyte function associated antigen 1) (133); and α4 integrin (114), which is targeted by the MS drug natalizumab preventing lymphocyte entry into the CNS (134).
Preferential activation of CD8 T cells recruited to the CNS is corroborated by several studies showing that CD8 T cells, and to a lesser extent CD4 T cells, proliferate and undergo clonal expansion in the CSF and post-mortem brain tissue of MS patients (80–82, 115, 122, 133, 135). TCR repertoire studies using complementarity-determining region 3 spectratyping have shown that clonally expanded CD8 T cells are present in both MS lesions and normal-appearing white matter (80, 83, 133). The same expanded CD8 T clones were identified in different brain areas, indicating high pervasiveness (83, 133), and in matched biopsy brain, CSF and peripheral blood samples from a few MS patients (81, 133). It was shown that some of the brain-infiltrating CD8 T cell clones persist for several years in the CSF and/or blood of MS patients (81) and that clonal CD8 T cells in the blood show a bias towards a memory phenotype with higher expression of CCR5, CD11a and granzyme B, compared to the non-oligoclonal counterparts (133). Together, these findings suggest a strong, persisting CD8 T cell memory response and ongoing exposure of CD8 T cells to an antigenic stimulus in the periphery and in the CNS. By single-cell RNA sequencing of CSF cells, Beltrán and colleagues (82) recently showed that clonally expanded CD8 T cells can be detected in the CSF of MS discordant monozygotic twins with subclinical neuroinflammation defined by presence of small MRI lesions and CSF alterations, supporting early involvement of activated CD8 T cells in MS immunopathogenesis.
CD8 T Cells in the Peripheral Blood of MS Patients
Changes in the frequency and function of total CD8 T cells or CD8 T cell subsets in the peripheral blood of MS patients have been reported less consistently and appear to be dependent on disease activity, course and duration. Early studies showed reduced CD8 T cell frequencies during relapses and in the progressive phase of MS (136–138). More recently, Pender and colleagues have shown that a deficiency in effector memory and terminally differentiated CD8 T cells is present already in early MS, persists during chronic disease and is associated with a reduced CD8 T cell response to EBV (57, 139). Of interest, studies performed in different control-MS case cohorts have found that expression of the genes encoding Eomes and T-bet, both associated with MS risk, is lower in the peripheral blood of MS patients than in healthy individuals (140, 141). Lower Eomes and T-bet expression was associated with CD56dim NK cells and CD56+ memory CD4 and CD8 T cells (141). Because Eomes and T-bet play a key role in the differentiation and function of type 1 immunity cells (Th1, Tc1 and NK cells), it has been proposed that their reduced expression in MS could be linked to abnormalities in cytotoxic immunity and defective clearance of EBV (141).
An increase in the percentage of CD8 T cells expressing the activation markers CD26 and CD69 was described in patients with clinically isolated syndrome (CIS), a condition predisposing to MS, and MS patients with radiologically active disease compared to patients with inactive disease and healthy individuals (142). In progressive MS, more elevated frequencies of CD8 T cells producing IL4 and IL10 (143), and of both T-bet+ CD4 and CD8 T cells were reported (144).
Antigen Recognition by Pathogenic CD8 T Cells in MS
Self Antigens
Despite decades of intense research to prove the autoimmune etiology of MS, disease-associated autoantigens remain undefined (85, 145). CD4 and CD8 T cells recognizing human myelin protein-derived peptides have been detected in the peripheral blood of both healthy individuals and MS patients (146). However, it is controversial whether T cell reactivity to myelin antigen-derived peptides differs between patients and controls. Some studies have shown that CD8 T cell responses to major myelin proteins [MBP, proteolipid protein (PLP), myelin-associated glycoprotein (MAG), myelin oligodendrocyte glycoprotein (MOG)] (147, 148) or other oligodendrocyte proteins (149) are increased in MS patients compared to healthy subjects whereas other studies report no differences (150–152). However, Sabatino and colleagues (152) found a higher frequency of myelin-specific memory CD8 T cells in the peripheral blood of MS patients compared to controls, suggesting prior exposure to antigen.
Fewer studies have tried to evaluate autoreactivity of intrathecal CD4 and CD8 T cell responses in MS using different experimental approaches. T cells isolated from postmortem brain tissue of a patient with aggressive MS proliferated in response to peptides from different myelin proteins [PLP, MBP, MOG and cyclic nucleotide phosphodiesterase (CNPase)] presented by HLA-DR-matched peripheral blood mononuclear cells (153). Recently, it was reported that short term CD4 and CD8 T cell lines derived from the CSF of patients with CIS and definite MS (154) and from the CSF and brain tissue obtained from chronic MS patients at autopsy (62) showed no reactivity towards several candidate MS associated autoantigens, like myelin (MAG, MBP, MOG, PLP), glial (Kir4.1, S100B) and neuronal (contactin-2, neurofascin) antigens, and alpha B-crystallin presented by autologous EBV-transformed B cell lines or an allogeneic HLA-matched EBV-transformed B cell line stably transduced with these human antigens. Similarly, freshly obtained CSF T cells from MS patients were not activated by autologous dendritic cells pre-loaded with complex candidate autoantigens, like human myelin, brain homogenate, and cell lysates of apoptotically modified oligodendroglial and neuronal cells (50). By in situ pentamer binding, MBP-specific CD8 T cells were not detected among the immune cells accumulating in active white matter lesions and meninges in brain tissue from progressive MS cases (63).
Autoreactive CD8 T cells specific for apoptotic T cell-associated self-epitopes and producing IFNγ or IL17 were found at higher frequency in the peripheral blood of MS patients compared to healthy subjects and in the CSF of MS patients, suggesting a possible mechanism for amplification of the local inflammatory response (151). Recently, Planas and colleagues (155) identified a ubiquitous enzyme, guanosine diphosphate (GDP)-L-fucose as a potential MS autoantigen recognized by a Th2-like CD4 cell clone expanded in MS brain lesions and by CSF-infiltrating CD4 Th1-like cells from HLA-DRB3*-positive patients, and showed crossreactivity with an immunodominant MBP peptide and homologous peptides from gut microbiota. Overall, the paucity of studies and the disparate findings obtained do not allow to draw conclusions on the autoreactivity of CNS-infiltrating CD4 and CD8 T cells in MS.
The demonstration that B-cell depletion with anti-CD20 monoclonal antibodies is highly effective in reducing CNS inflammation in relapsing remitting MS (89, 156) has fostered investigations on the pathogenic role of B cells in MS, particularly on their ability to promote T cell activation through antigen presentation and cytokine production. In the peripheral blood of MS patients B cells exhibit abnormal pro-inflammatory cytokine production, which can be induced by TLR9 ligand CpG-DNA or IFNγ, and promote T cell and myeloid cell activation (157, 158). A population of activated Tbet+ CXCR3+ memory B cells has been identified that is enriched in CSF (108), meninges and brain tissue of MS patients, is induced by IFNγ and TLR9 signals and has been linked to EBV load in the peripheral blood of MS patients (159, 160).
A role for B cells as APC capable of stimulating autoreactive T cell responses in MS has been proposed in several studies (Figure 1A). Earlier work performed by van Noort and colleagues suggested that EBV-induced expression of the small heat shock protein alpha B-crystallin in B cells and HLA-DR-restricted presentation of this protein may activate autoreactive proinflammatory CD4 T cells that recognize stress-induced alpha B-crystallin in oligodendrocytes and astrocytes in MS lesions (77, 161). A series of studies by Hølmoy and colleagues has shown that B cells from the CSF of MS patients can activate T cells that recognize specific antigenic determinants (idiotopes) from their B cell receptors, suggesting a potential mechanism for intrathecal B cell-T cell interactions promoting CNS inflammation (162). Recently, Jelcic and colleagues (94) have shown that memory B cells from HLADR15+ MS patients activate CD4 T cells in the absence of exogenous antigen and identified a peptide of RAS guanyl-releasing protein 2 (RASGRP2) as a self-peptide responsible for CD4 T cell activation. Because RASGRP2 is also expressed in cortical gray matter neurons (94), it has been proposed that presentation of RASGRP2 by B cells activates CD4 T cells that recognize RASGRP2 in the brain, likely presented by HLA-DR+ CNS APC such as macrophages and microglia. Following this study, Wang and colleagues (72) showed that B cells from HLA-DR15+ MS patients can present self-peptide fragments derived from DR2a and DR2b that activate CD4 T cells and identified memory CD4 T cells in the CSF of MS patients that respond to DR2a or DR2b self-peptides presented by B cells and cross-react with RASGRP2, MBP and peptides from EBV and a commensal gut bacterium. These findings suggest that EBV could trigger potential autoreactive T cells in MS through molecular mimicry between EBV peptides presented by the infected peripheral B cells and self peptides presented by B cells themselves and other APC within the CNS (Figure 1A).
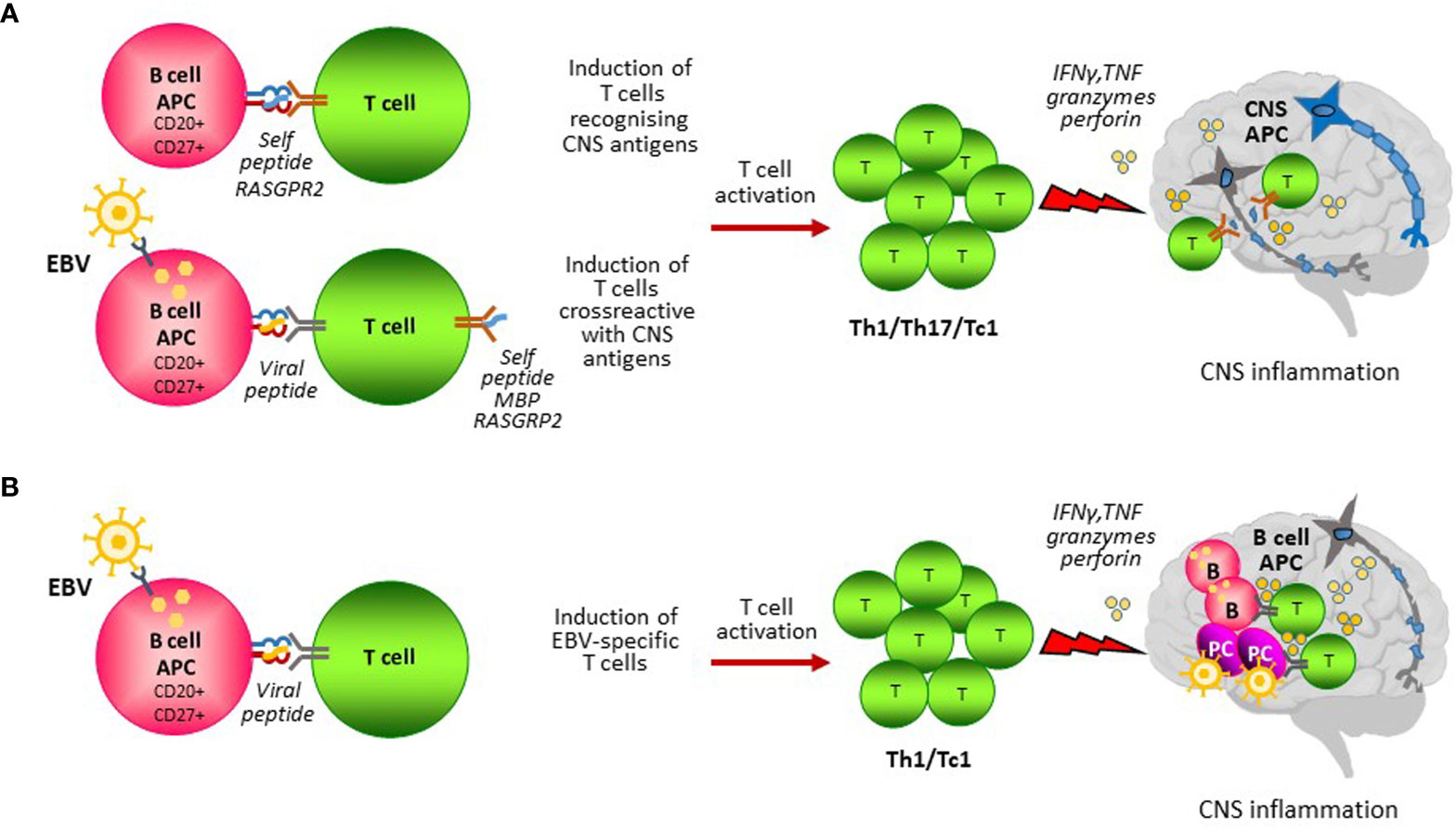
Figure 1 B cell antigen presentation in MS. B cells could contribute to the activation of pathogenic T cells through presentation of self and non-self antigens. (A) CD27+ CD20+ memory B cells could present self-peptides from proteins that are expressed in the CNS leading to the induction of autoreactive T cells. Infection of B cells with EBV induces EBV-specific T cells that exert continuous immune surveillance and are essential for virus-host homeostasis. EBV infected B cells could induce autoreactive T cells by presenting EBV peptides sharing similarities with peptides from CNS self-antigens (i.e. MBP, RASGPR2) (molecular mimicry). Autoreactive T cells homing to the CNS would recognize their target antigen on local antigen presenting cells (APC) and become reactivated causing CNS inflammation and tissue injury. (B) EBV infected CD27+ CD20+ memory B cells induce EBV-specific T cells that migrate in the CNS to counteract an abnormal EBV infection brought inside the CNS by circulating infected B cells. In this model, B cells would act as APC both in the periphery and in the CNS to stimulate a detrimental antiviral immune response causing CNS inflammation and tissue injury.
Over the years, cross-reactive T cells capable of recognizing EBV and myelin antigens have been identified in several studies. Wucherpfennig and Strominger (73) showed that some HLA-DR15-restricted MBP-specific T-cell clones from MS patients also recognize peptides from EBV DNA polymerase and other viral proteins. CD4 T cells cross-reacting with EBV DNA polymerase and MBP peptides were also detected in the CSF of an individual patient (74). Furthermore, Lünemann and colleagues (75) found that a small percentage of IFNγ-producing CD4 T cells specific for the EBV latent protein EBNA-1 cross-recognize MBP in both MS patients and healthy controls, their frequency being higher in MS patients. Despite these intriguing findings, it remains to be demonstrated that CNS autoreactive CD4 T cells contribute to MS pathology. To date, no data are available on CD8 T cells cross-reactive for candidate myelin antigens and MS-associated pathogens.
EBV Antigens
EBV itself has long been suspected to be the antigenic driver of the immunopathological response targeting the brain and spinal cord in MS (163, 164), raising the possibility that B cells participate in MS pathogenesis through presentation of highly immunogenic viral antigens. A chronic latent EBV infection and bursts of viral reactivation within the CNS could explain several MS pathological features, such as persistent intrathecal B-cell activation, lesion reactivation and predominance of activated cytotoxic CD8 T cells in CNS immune infiltrates (87) (Table 1). However, this issue remains highly debated. On the one hand, several studies have reported complete absence or rarity of EBV infected cells in postmortem CNS tissue from MS patients using PCR-based techniques for EBV DNA or RNA or in situ hybridization for the untranslated EBV small RNA (EBER) (62, 165–169). On the other hand, since 2007 our group has repeatedly documented a high frequency of EBV infected B cells and plasma cells in postmortem brain tissue mainly from patients with progressive MS (53, 117, 170–173), but also patients with relapsing-remitting and acute MS (117) and patients with relapsing remitting MS dying because of fatal MS relapses after natalizumab interruption (121, 174). Using EBER in situ hybridization and immunohistochemical techniques to detect a large array of EBV latent (EBNA2, LMP1, LMP2A) and lytic proteins (BZLF1, BMRF1, BFRF1, p160, gp350/220), we have provided evidence for a dysregulated, predominantly latent EBV infection in a large proportion (40 to 80%) of CNS-infiltrating B cells, including those forming ectopic B-follicle-like structures in the meninges, and for viral reactivation in up to 20% of plasmablasts/plasma cells present in active WM lesions and at the periphery of meningeal B cell follicles. EBV latent and, less frequently, lytic transcripts were detected in immune infiltrates microdissected from the MS brain (120). Importantly, we have shown that EBV dysregulation in the CNS is characteristic of MS and is not observed in patients with other infectious and non-infectious inflammatory neurological diseases (117, 170), excluding non-specific seeding of EBV infected B cells into the CNS during MS. Abnormal EBV infection in the MS brain was recently reported in independent studies (175–177). Discrepancies across studies may be explained by differences in sample selection and methods or tools to detect EBV (178, 179).
EBV RNA has not been detected in CSF B cells and plasma cells (108, 168) or has been detected in a minority of CSF cell samples from MS patients (180). Small differences in PBMC-associated EBV DNA or RNA levels between MS patients and healthy controls were also reported (49, 180, 181). Altogether, these findings suggest that EBV dysregulation in MS might be predominantly confined to the CNS tissue and, possibly, cervical lymph nodes (172).
In the last 20 years, several studies have examined and compared the T cell response to EBV in the peripheral blood of MS patients and control subjects. With a few exceptions (182, 183), quantitative and qualitative differences in the EBV-specific CD4 and CD8 T cell response, but not in the T cell response to other herpesviruses (typically human cytomegalovirus), have been described for MS patients. Lünemann and colleagues (49) found an increased frequency and broadened epitope specificity of memory CD4 T cells with a Th1 phenotype recognizing the EBV latent protein EBNA-1 in MS patients compared to healthy controls. CD8 T cells recognizing EBV transformed B-cell lines, EBV latent proteins, like EBNA3A and LMP2, or pooled peptides from EBV latent and lytic proteins were found at higher frequency in the peripheral blood of patients with MS compared to healthy controls (23, 54, 184). Expansions of EBV-specific CD4 and CD8 T cells have been associated with active disease at the radiological and clinical level in some studies (53, 55, 56), suggesting a link between activation of anti-EBV immunity and CNS inflammation. However, it has also been reported that EBV-specific CD8 T cells from MS patients display a dysfunctional phenotype, like reduced IFNγ production and cytotoxic activity (55, 58, 185), and that the frequency and functionality of EBV-specific CD4 and CD8 T cells progressively decreases with increasing disease duration (54, 55, 58). A plausible explanation is that T-cell exhaustion during MS is the result of a persistently active, poorly controlled EBV infection.
Presentation of EBV peptides by HLA-E, a non-classical MHC class I molecule engaging CD94/NKG2A (inhibitory receptor) or CD94/NKG2C (activating receptor) on NK cells and the TCR of cytotoxic CD8 T cells, was also investigated in MS patients (186). This study shows increased HLA-E restricted recognition of an EBV lytic protein (BZLF1)-derived peptide by CD8 T cells in MS patients compared to healthy controls, a finding suggesting altered immune control of EBV.
The T cell response towards EBV has been investigated also in the CSF and brain tissue of MS patients. EBNA-1-specific CD4 T cells (51) and CD4 T cells recognizing EBV-infected B lymphoblastoid cell lines (50, 52) have been detected in the CSF of patients with MS and patients with other inflammatory and non-inflammatory neurologic diseases. Instead, CD8 T cells recognizing peptides from EBV latent and lytic proteins, but not CMV proteins, were found at higher frequency in the CSF of patients with MS compared to patients with other inflammatory and non-inflammatory CNS diseases (59). Using high-throughput sequencing of TCR-β chains in CSF and blood, Lossius and colleagues (60) also found selective enrichment of EBV-reactive CD8 T cells mainly recognizing EBV lytic proteins, but not CD8 T cells recognizing influenza A virus, in the CSF of MS patients compared to patients with other inflammatory neurological diseases. Another study showed enhanced CD4 and CD8 T cell reactivity towards autologous EBV-transformed B cell lines in the CSF of patients with clinically isolated syndrome and MS compared to neurological controls and reported selective recognition of EBV lytic proteins by oligoclonal CSF CD8 T cells (61). The reported differences in the intrathecal EBV-specific CD8 T cell response between MS patients and patients with other inflammatory neurological diseases indicate that accumulation of EBV-specific CD8 T cells in the CSF during MS results from an antigen-driven pathologic process and is not the consequence of non-specific recruitment due to ongoing CNS inflammation.
Using T cells recovered from white matter lesions of brain samples obtained from MS patients at autopsy, strong CD8, not CD4 T cell responses were generated towards autologous EBV-infected B lymphoblastoid cell lines, although the precise target antigen was not identified (62). Stainings performed in our laboratory using the in situ pentamer technique and HLA class I-matched postmortem brain tissue from progressive MS cases have revealed enrichment of CD8 T cells specific for a broad range of EBV latent and lytic protein-derived peptides, but not CD8 T cells recognizing CMV and influenza A virus peptides, in active white matter lesions and in the meninges (63). It was also shown that EBV-specific CD8 T cells accumulating in the MS brain express membrane CD107a, indicative of a cytotoxic phenotype, and adhere to B cells and EBV infected cells, suggesting specific recognition of their target antigen (63). Although these results support a role for EBV-infected B cells as local APC sustaining a detrimental antiviral immune response (Figure 1B), their presence in the MS brain at end-stage disease indicates that the CD8 T cell response fails to get rid of the infection. It is known that EBV adopts a wide range of strategies to compromise both innate and adaptive immunity, including MHC class I and class II downregulation, interference with antigen presentation, and induction of immunosuppressive molecules and immune checkpoint inhibitors (187–189). EBV infected B cells and plasma cells by virtue of abundant expression of latent and/or lytic proteins, viral microRNAs and small noncoding RNAs with immune evasion properties (190) may dispel virus specific T cells which, instead of killing the target cells, would go awry and damage the nearby CNS cells.
Collectively, the few studies exploring anti-EBV immunity in the CNS of MS patients suggest that EBV-specific T cells get access to the cerebral compartment and that skewed intrathecal CD8 T cell responses towards EBV could contribute to CNS inflammation and tissue damage. Future research should aim at better defining the antigenic targets, phenotype, function and evolution of CD4 and CD8 T cell responses to EBV in the blood and in the CNS of MS patients. These studies should help in defining the complex functional versus dysfunctional or exhausted signature of EBV-specific T cells in MS, and verify possible associations with the degree of CNS inflammation which varies by patient, is more prominent at MS onset and decreases as the disease progresses. This information could prove useful for immune monitoring of patients during treatment.
Cytotoxic CD8 T Cell Interactions in the MS Brain
Although most CD4 and CD8 T cells accumulate within the CNS connective tissues (perivascular space in the neural parenchyma and subarachnoid space in the meninges), some T cells, mainly CD8 T cells in actively demyelinating white matter lesions and at the edge of chronic active white matter lesions, cross the outer glia limitans membrane of the perivascular space and enter the neural parenchyma, suggesting increased tissue invasiveness (111, 112, 114–116).
Under inflammatory conditions, all CNS resident cells (neurons, oligodendrocytes, astrocytes, microglia) can be induced to express MHC class I molecules and present peptides derived from endogenous antigens (self or viral antigens), thus becoming potential targets for CD8 T cells (191–193). Microglia, the myeloid immunocompetent cells of the CNS can also take up, process and cross-present exogenous antigen to CD8 T cells (194). If CNS cells, particularly oligodendrocytes, were directly targeted by CD8 T cells, one would expect to observe intraparenchymal CD8 T cells interacting with and displaying immunological synapses and cytotoxic activity towards these cells. To date, there is scant evidence for such an interaction (195) or for preferential interaction of CD8 T cells with any particular neural cell type in MS brain lesions (62). Rather, in the MS brain CD8 T cells with polarized perforin were shown more consistently to interact with CD163+ mononuclear phagocytes in both the perivascular space and the parenchyma in active lesions (116, 196). CNS-infiltrating dendritic cells contacting CD8 T cells and surrounded by proliferating lymphocytes, most likely CD8 T cells, were also detected within the perivascular cuffs in active MS lesions (135). These findings implicate recruited and local myeloid APC in CD8 T cell reactivation within the MS brain, possibly through phagocytosis of cell debris and antigen cross-presentation.
We have repeatedly visualized CD8 T cells that adhere to, form immunological synapses with and polarize perforin or granzyme B towards B cells, plasma cells and cells expressing EBV lytic proteins in white matter and grey matter lesions and in the inflamed meninges in postmortem brain tissue from progressive MS patients (53, 63, 117, 173). CD8 T cell interactions with B cells or EBV infected cells were also observed in multiple actively demyelinating WM lesions in the brain of relapsing remitting patients who died because of fatal relapses after natalizumab interruption (121, 174). As mentioned above, the visualization of EBV-specific CD8 T cells contacting B cells and EBV-infected cells in the MS brain is highly suggestive of local viral antigen presentation (63).
CD8 T Cell Activation and CNS Injury
Neurologic deficits in patients with MS are mainly due to demyelination, axonal damage and neuronal loss. Numerous studies have highlighted the key contribution of CD8 T cells to these pathological features. Earlier histopathological studies of post-mortem brain samples from MS patients found that acutely injured or transected axons are more frequent immediately after disease onset as compared to chronic disease stages, and that the extent of axonal damage positively correlates with the number of CNS-infiltrating CD8 T cell and macrophages/microglia (197, 198).
The generation of transgenic mice with focused expression of neoantigens on neurons, oligodendrocytes and astrocytes has allowed to study the detrimental effects of autoimmune responses mediated by cytotoxic CD8 T cells against specific brain cell components (193). In vitro studies have shown that neurons can be damaged by CD8 T cells both directly, when induced to express MHC class I molecules and present cognate antigen (199–201), and indirectly, when CD8 T cells are activated following recognition of their cognate antigen on other antigen presenting cells (i.e., oligodendrocytes) and cause collateral bystander damage (202). In models of virus-induced demyelination and viral encephalitis, IFNγ (203–206) and perforin (207) released by activated, virus-specific CD8 T cells are critical for inducing neurodegeneration and demyelination. Likewise, blockade of granzyme B production by CD8 T cells in vitro and in the model of experimental autoimmune encephalomyelitis reduces axonal injury and neuronal death (208–210).
IFNγ has a key role in the activation of microglia/macrophages, including activation of macrophage oxidative metabolism and antimicrobial activity (211), and is a potent inducer of NAPDH oxidases (212, 213). Excessive IFNγ production can lead to uncontrolled superoxide generation by microglia/macrophages, which is thought to be the major driving force for demyelination and neurodegeneration in the MS brain (214, 215). Activated CD8 T cells also produce TNF that can cause direct injury to oligodendrocytes and neurons or prevent remyelination (216–218). Collectively, human neuropathological and experimental data support persistent CD8 T cell-mediated cytotoxic activity in combination with a deleterious ‘cytokine storm’ and oxidative stress as major determinants of neural cell damage in MS.
CD8 T Cell Subsets in MS
Different subpopulations of CD8 T cells have been implicated in MS pathogenesis based on studies in brain tissue, CSF and peripheral blood of patients. The most extensively studied CD8 T cell subsets are CD8 mucosal-associated invariant T (MAIT) cells, CD8 tissue-resident memory (Trm) cells and regulatory CD8 T cells.
Several studies have described MS-associated alterations in a subset of CD8 T cells expressing high levels of the natural killer receptor protein 1a/CD161, producing IL-17 and co-expressing for the large part the semi-invariant Vα7.2 TCR identifying MAIT cells. MAIT cells are a unique innate-like T-cell population that is restricted to the MHC-related protein 1 (MR1) and is mainly activated by bacteria but also by proinflammatory cytokines, like IL12 and IL18 (219). A higher frequency of circulating CD8+ CD161high TCR-Va7.2+ MAIT cells was found in relapsing remitting adult MS (220) and pediatric onset MS (221). Other studies reported a decrease in CD8 MAIT cell frequency in relapsing remitting (222, 223) and primary progressive MS (224, 225) or no differences between MS patients and healthy donors in MAIT frequency, phenotype and activation potential (226). CD8 T cells with MAIT cell-related features were also found in CNS lesions at a seemingly low frequency (220, 223, 226, 227). Despite MAIT-like cells could play a role in exacerbating chronic inflammatory processes due to their ability to migrate into inflamed tissues and produce pro-inflammatory cytokines and lytic enzymes, their role in MS pathology remains obscure. A distinct subset of CD8+ T cells expressing intermediate levels of CD161, with characteristics of effector cytotoxic cells and capable of secreting IFNγ, GM-CSF, IL-17 and IL-22, were recently found to accumulate in the CSF and in brain lesions of MS patients (228).
Tissue-resident memory T cells (Trm) are a T cell subset that, unlike central memory and effector memory T cells, do not recirculate but populate permanently various tissues, including the brain, where they provide a first line of defense against spread of viral infections (229, 230). Trm cells are characterized by high levels of CD103 and CD69, low levels of CD62 and CD27, and rapid production of granzyme B, among several cytotoxic effector molecules. Recently, it was shown that a large proportion of CNS-infiltrating CD8 T cells in postmortem brain samples of patients with progressive MS display the phenotype of Trm cells. Combining the results of different studies the CD8 Trm cells characterized in MS brain lesions are CD69+, CD103+ or CD103-, S1PR1-, CCR7- and CXCR6+ (62, 115, 116). In a study analyzing CSF cells from MS-discordant monozygotic MS twin pairs, clonally expanded CD8 T cells showing characteristics of activated Trm cells were found not only in patients with definite MS but also in the cotwins with prodromal (subclinical) neuroinflammation defined by presence of small MRI lesions and CSF alterations (82). Taken together, these studies indicate that CD8 Trm cells are involved in both the early and chronic stages of MS. Because Trm cells persist at sites of prior viral infection representing an autonomous cytotoxic barrier (231), their presence in the MS brain is compatible with a role in contrasting a persistent and dysregulated EBV infection of B cells accumulating in white matter lesions and in the meninges (53, 117, 120, 170–173). Despite being identified for their protective function, Trm cells can turn into drivers of tissue damage in the context of chronic infections (232).
CD8 T cells have also been identified as potential immunoregulatory cells that can act by direct killing of putative pathogenic T cells and/or through production of immunosuppressive molecules (233). Non cytotoxic CD25+ FoxP3+ CD8 T cells producing TGF-β and IL10 and suppressing autoreactive CD4 T cell activation were identified in the peripheral blood of MS patients and healthy subjects (234). A lower percentage of circulating Foxp3+ CD8 T cells was found in relapsing than in remitting patients with MS and in controls (235). An immunoregulatory role for HLA-E restricted CD8 T cell subsets in MS has also been hypothesized (236). Furthermore, CNS antigen-specific CD8 regulatory T cells with cytolytic activity towards autoreactive CD4 T cells were identified in the peripheral blood of MS patients and their activity was deficient during MS relapse (237). Since the pathogenic effector T cell subsets in MS have not yet been identified, the functional relevance of regulatory deficits in the CD8 T cell compartment remains unclear.
CD8 T Cells in Other Neuroinflammatory Diseases and Analogies with MS
In addition to MS, CD8 T cells have been implicated in the pathogenesis of several neurological immune-mediated diseases with defined or unknown etiology. Studying and comparing different neurological diseases characterized by prominent CD8 T cell accumulation and activation in the CNS can provide clues about still elusive pathogenic mechanisms and new therapeutic options, such as drugs limiting access of pathogenic CD8 T cells to the CNS.
Rasmussen encephalitis is a rare brain disorder with unknown etiology mainly affecting children; it is characterized by progressive unihemispheric atrophy and drug-resistant epilepsy. In Rasmussen encephalitis, CD8 T cells dominate the brain immune infiltrates and display cytotoxic activity towards neurons and astrocytes (238, 239). Brain-infiltrating CD8 T cells, and to a lesser extent CD4 T cells, undergo clonal expansion and produce IFNγ, TNF and granzyme B (240), suggesting local reactivation. The target antigens of T cells expanding in the brain of patients with Rasmussen encephalitis have not been identified yet, but viral antigens have long been suspected. A recent study combining single cell RNA-seq with TCR Vβ chain sequencing in resected brain tissue suggests that clonal T cells could recognize CMV epitopes (241).
Susac syndrome is a rare neuroinflammatory disease with CNS endotheliopathy affecting mainly young adults (242). In CNS biopsies of patients with Susac syndrome, CD8 T cells dominate the immune infiltrates, adhere to and show cytotoxic activity towards CNS microvessels (243). It has been shown that oligoclonal expansion of terminally differentiated activated cytotoxic CD8 T cells occurs in the CNS of Susac syndrome patients (243) but the antigens driving the immune attack causing endotheliopathy remain unknown.
Cytotoxic CD8 T cells have also been implicated in neuronal cell death in autoantibody-associated encephalitides, particularly those with antibodies to intracellular antigens, which include non-paraneoplastic (like glutamic acid decarboxylase 65 encephalitis) and paraneoplastic conditions (244). Paraneoplastic encephalitides are rare neurological disorders that develop in cancer patients in whom autoimmunity in the nervous system is triggered by ectopic expression of neuronal proteins (such as Hu, Yo and Ma2) in cancer cells. However, no studies have definitively shown which antigens are recognized by CD8 T cells accumulating in the CNS in antibody-associated encephalitides.
In virus-induced encephalitis, like herpes simplex virus encephalitis and cytomegalovirus encephalitis, CD8 T cells dominate the CNS immune infiltrates and the immune attack is directed towards viral antigens presented by the infected neural cells (245). Progressive multifocal leukoencephalopathy (PML) is a CNS demyelinating disease caused by reactivation of JC virus (JCV), a polyomavirus that usually establishes a persistent, asymptomatic infection. PML can develop in immunocompromised patients and in MS patients treated with natalizumab as a consequence of reduced immune surveillance in the CNS (246). The main mechanism of CNS tissue damage is due to lytic infection of oligodendrocytes by JCV. Prominent infiltration of CD8 T cells and clonal expansion of activated CD8 effector T cells specific for JCV large T antigen have been detected in the CNS of patients with MS who develop natalizumab-associated PML (247).
Human T-lymphotropic virus type 1 (HTLV-1)-associated myelopathy or tropical spastic paraparesis (HAM/TSP) is an infrequent complication of HTLV-1 infection causing progressive neurological disability and chronic pain (248). The clinical presentation and pathophysiology of HAM/TSP is similar to the progressive forms of MS. Histopathologically, HAM/TSP is characterized by perivascular lymphocytic infiltration, loss of myelin and axons, and reactive astrocytosis in the spinal cord. HTLV-1 is a retrovirus that infects T cells, mainly CD4 T cells, providing activating and proliferating signals, and is the etiological agent of adult T cell leukemia. In HAM/TSP, circulating, activated HTLV-1-infected CD4 T cells invade the CNS and trigger a cytotoxic immune response towards HTLV-1 antigens presented by the infected CD4 T cells which promotes detrimental CNS inflammation leading to demyelination and neurodegeneration (249). CD8 T cells specific for the dominant HTLV-1 antigen, Tax protein, accumulate in the CSF (250) and in spinal cord lesions of patients with HAM/TSP (251). Within the CNS tissue, Tax-specific CD8 T cells express granzyme B, perforin and IFNγ and contact Tax-expressing HTLV-1-infected cells (251).
HAM/TSP provides an immunopathological model of virus-driven CNS inflammation and bystander neural cell damage that shows several analogies with the postulated EBV-driven immunopathological model of MS (Figure 2). Of major interest are the tropism of HTLV-1 and EBV for different lymphocyte populations (T cells and B cells, respectively), their ability to induce proliferation and activation of the infected lymphocytes and exploit their mobility to spread within the host, and the induction of strong cytotoxic responses that counteract the oncogenic potential of both viruses (Figure 2). To colonize the host, EBV establishes a growth transforming latent infection of B cells and some of the viral proteins expressed during this phase mimic the B-cell activating signals triggered by BCR stimulation and interaction with T cells (11). Activated EBV infected B cells also upregulate adhesion molecules and chemokine receptors that may favor their migration into tissues (159, 252, 253). In infectious mononucleosis, which increases MS risk by about two fold as compared to asymptomatic primary EBV infection, up to 50% of the circulating memory B cells are infected with EBV (254). It is hypothesized that, in a few susceptible individuals, following symptomatic or subclinical primary infection, activated EBV infected B cells are more likely to cross the brain barriers and spread throughout the CNS, similarly to HTLV-1 infected CD4 T cells in HAM/TSP. Establishment of an intrathecal viral infection could be favored by a predisposing genetic background or any other condition affecting permanently or transiently immune surveillance and leading to defective virus control. Due to its particular anatomy, the CNS would hinder complete eradication of the infectious agent, turning into an ‘extralymphatic viral sanctuary’ (255) (Figure 3). The reduced ability of cytotoxic T cells to clear the infection would enable the preferential expansion of the infected cells in this organ, thereby explaining the CNS localization and persistence of EBV infected B cells in MS and of HTLV-1 infected CD4 T cells in HAM/TSP.
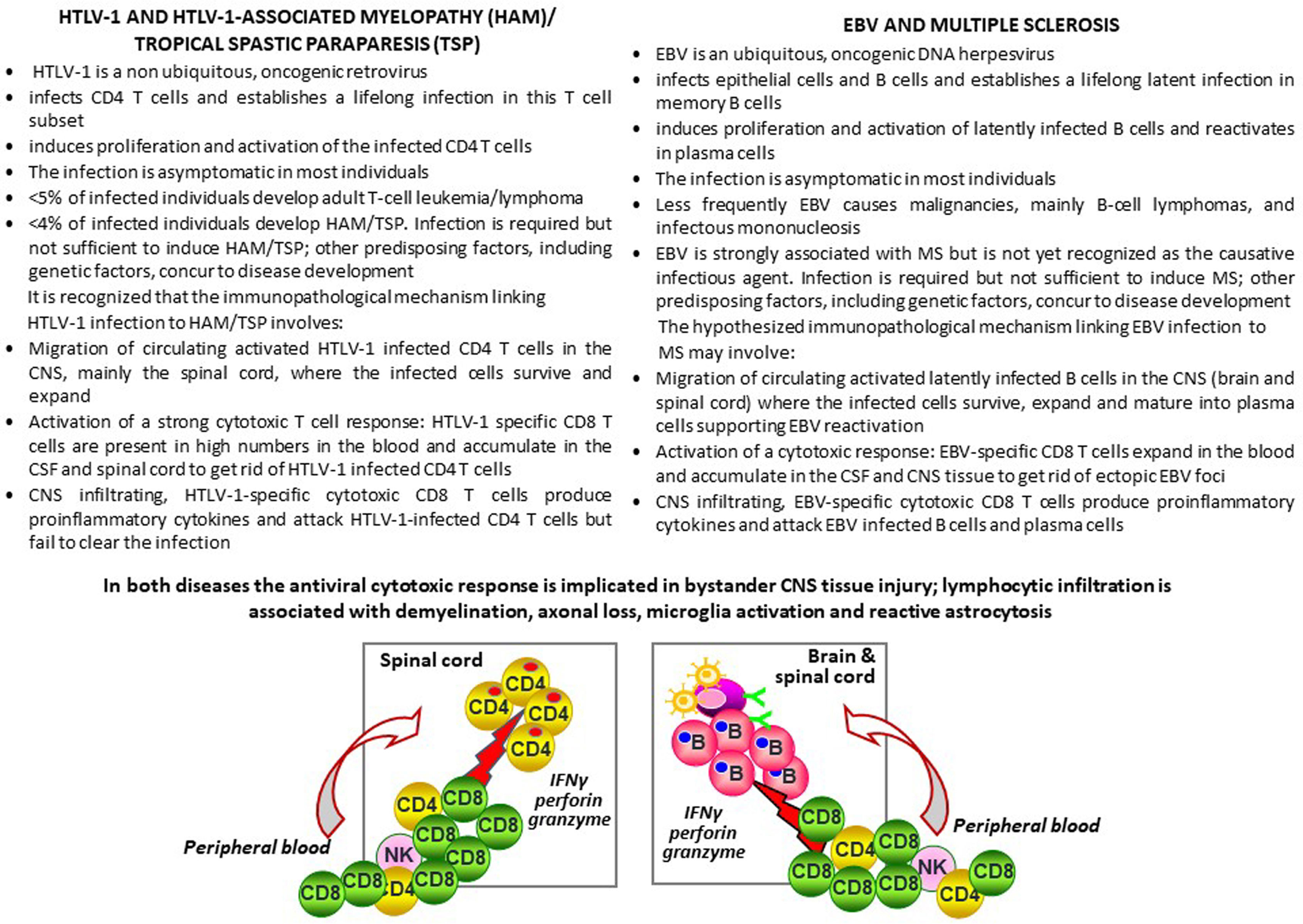
Figure 2 HTLV-1 associated myelopathy/tropical spastic paraparesis and multiple sclerosis: Two chronic CNS inflammatory diseases, two viruses, a common immunopathologic mechanism? The text of this figure summarizes the tropism, biology and pathogenic potential of HTLV-1 and EBV and their association with HAM/TSP and MS, respectively. The HTLV-1-mediated immunopathological model of HAM/TSP is presented vis-a-vis the hypothesized EBV-mediated immunopathological model of MS. The left side of the sketch depicts the migration of HTLV-1-infected CD4 T cells and the activation of a cytotoxic response towards HTLV-1-infected CD4 T cells in the spinal cord in HAM/TSP, leading to production of the pro-inflammatory cytokine IFNγ and the lytic enzymes granzyme B and perforin, which play a key role in bystander tissue injury. On the right side of the sketch, a similar virus-driven immunopathological mechanism involving EBV-infected B cells and EBV-specific CD8 T cells is proposed for MS.
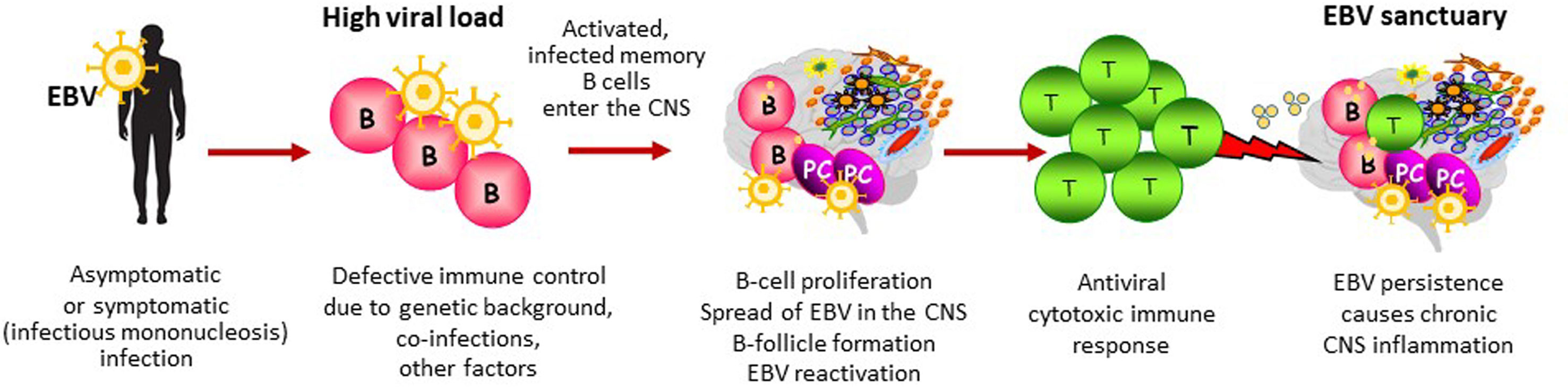
Figure 3 EBV-driven immunopathological model of MS. This figure depicts the main steps potentially leading to establishment of an abnormal EBV infection in the CNS and the ensuing immunopathological response. In individuals at risk of developing MS, defective immune control of the virus could be determined by a high viral load at primary infection (infectious mononucleosis), genetic influences on immune system function, coincident infections and/or any other environmental factor affecting the host’s immune system status. In susceptible individuals, EBV-infected memory B cells could elude immune control and seed into the CNS where they would expand favouring EBV persistence and periodic EBV reactivation (the CNS as an EBV sanctuary). Though activated in the periphery, CNS-homing EBV specific T cells do not clear the virus and become exhausted over time due to persistent, abnormal viral reactivation. The protective antiviral immune response turns into a dysfunctional immune response that promotes CNS inflammation and causes collateral neural cell damage.
CD8 T Cell and B Cell Changes Induced by MS Treatments
Over 15 disease modifying immunotherapies (DMTs) with different efficacy and mechanisms of action are available for the treatment of MS. The newer lymphocyte-targeting drugs are associated with better control of disease activity and long-lasting benefits in MS patients compared to first generation immunomodulatory therapies (interferon beta and glatiramer acetate) (256).
The multitude of treatment options for MS has offered the unique opportunity to get insights into the contribution of different lymphocyte populations to disease pathogenesis. At present, all DMTs approved for MS induce quantitative and/or qualitative changes in T cells but none specifically targets T cells or T cell subsets. Early clinical trials aiming at depleting T cells using anti-CD3 or anti-CD4 monoclonal antibodies did not yield beneficial effects in MS patients. Anti-CD3 monoclonal antibody had major toxic side effects (257). Anti-CD4 monoclonal antibody was less toxic but did not reduce radiological disease activity (258). This result suggests that CD4 T cells are not the best target to reduce CNS inflammation but does not necessarily argue against a role for CD4 T cells in driving CNS inflammation; incomplete CD4 T cell depletion (259) as well as persistence of other pathogenic T cell and APC subsets should be considered as alternative explanations for failure of anti-CD4 therapy in MS. CD8 depleting monoclonal antibodies have been tested in experimental models to block autoimmunity (260), but drugs that target the total CD8 T-cell population are not desirable because lack of specificity would result in enhanced susceptibility to infections. Since the breakthrough finding that the B-cell depleting drug rituximab drastically reduced inflammatory brain lesions and clinical relapses in MS patients (89), B cells began to be considered both as key players in MS pathogenesis and a major target for MS disease control. In the last years, it has become increasingly evident that all DMTs effectively suppressing MS relapses also modulate B cell immunity, leading to focus on B cell-T cell interactions as a potential mechanism driving MS activity (259, 261).
Table 2 summarizes the effects of licensed DMTs on T and B cells in MS patients. Among low to moderately effective drugs, IFN-β and glatiramer acetate are extensively used as first-line therapies for MS and have broad, still poorly understood immunomodulatory effects. These drugs act mainly by shifting CD4 and CD8 T cells from a pro-inflammatory towards an anti-inflammatory phenotype and by potentiating immune regulatory networks, without affecting significantly T cell numbers. Studies on the effects of IFN-β and glatiramer acetate on B cells are scanty (Table 2). Dimethyl fumarate reduces relapses in MS through a still unknown mechanism. This drug causes variable lymphopenia, with partial decrease of circulating memory CD4 T cells, CD8 T cells and B cells, and reduction in CSF leukocyte counts (313). Teriflunomide inhibits pyrimidine synthesis and reduces T cell and B cell proliferation, causing moderate T and B cell depletion in the peripheral blood and CSF (314). Interestingly, a decrease in anti-EBV antibody titers was recently observed in MS patients treated with teriflunomide (315), suggesting potential interference with EBV infection. Consistent with this, it has been shown that teriflunomide inhibits the growth of EBV-transformed B cells and lytic EBV reactivation (316).
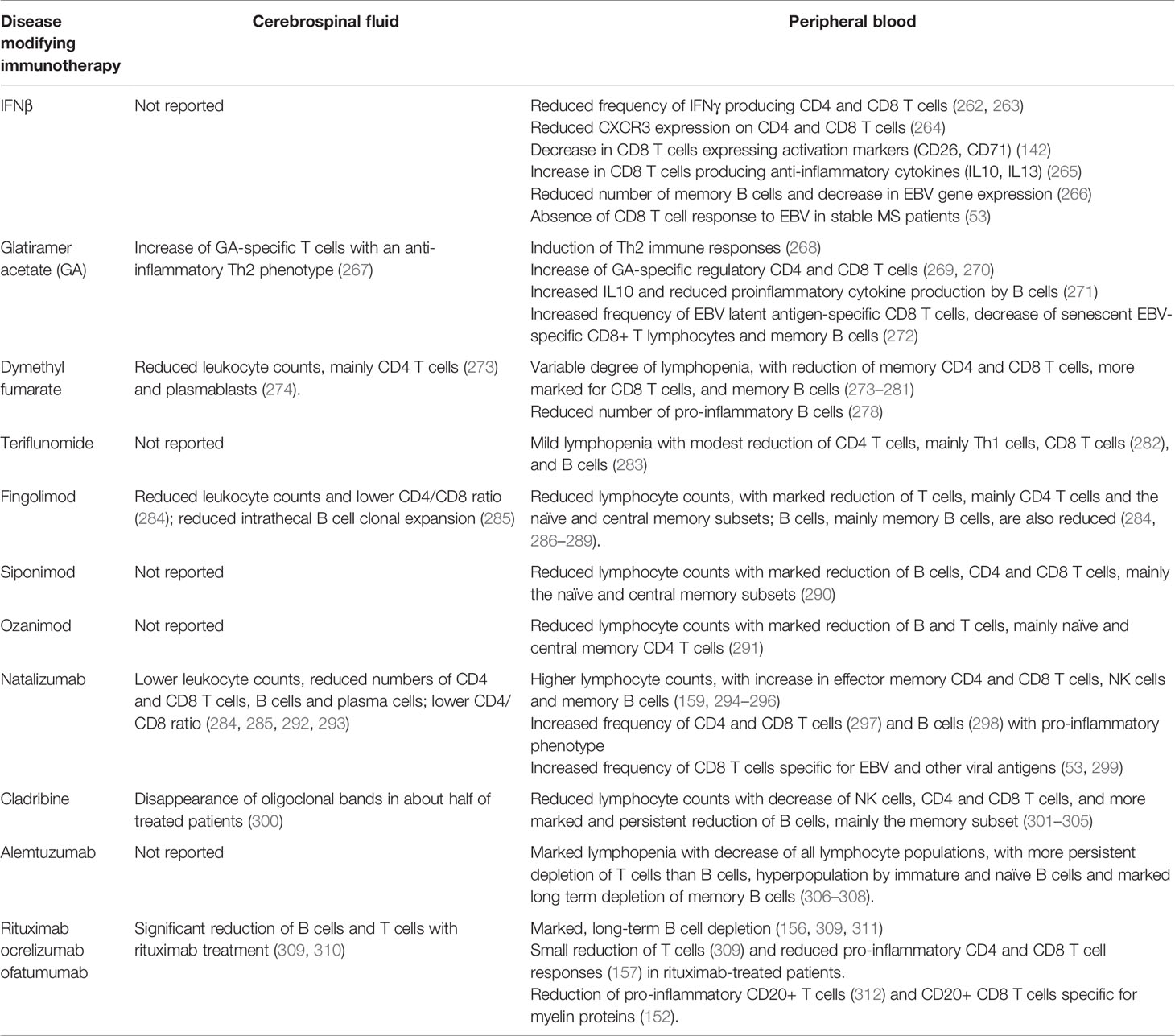
Table 2 Effects of MS therapies on T cells and B cells in the cerebrospinal fluid and peripheral blood of patients with multiple sclerosis.
Highly effective DMTs for MS aim at blunting the CNS inflammatory milieu by preventing the access of pathogenic lymphocytes to the CNS, inhibiting lymphocyte activation, or eliminating the pathogenic immune repertoire to reset the immune system (317). Fingolimod, siponimod and ozanimod are structural analogues of sphingosine that functionally antagonize sphingosine 1-phosphate (S1P) receptor-1 expressed on lymphocytes inhibiting their egress from secondary lymphoid organs. The therapeutic effect of these oral drugs is likely due to sequestration of pathogenic lymphocytes into secondary lymphoid organs (318) and is associated with a marked decrease in circulating naïve and central memory T cells, memory B cells and CSF leukocytes (Table 2). Natalizumab, a humanized monoclonal antibody (mAb) against the α4 subunit of the α4β1 and α4β7 integrins, prevents migration of circulating leukocytes into the CNS thereby excluding entry of pathogenic lymphocytes (134). This is reflected by a significant reduction of both T cells and B cells in the CSF and an increase of all lymphocyte subsets in the peripheral blood, particularly memory effector T cells, NK cells and B cells (Table 2). The reestablishment of lymphocyte migration to the CNS after natalizumab or fingolimod discontinuation may lead to a “rebound effect”, with an increase in the number of relapses and substantial disease reactivation on MRI (319). The rebound effect may suggest that the pathogenic T and B cells have accumulated in the periphery during drug treatment and/or that they receive a stronger antigenic stimulus within the CNS environment after treatment interruption. The rare cases of fatal rebounds after natalizumab withdrawal have multiple active lesions and prominent immune infiltrates populated by B cells, CD4 and CD8 T cells in the CNS (320–322). In two MS cases of fatal post-drug rebound without evidence of JC virus infection in the CNS, massive EBV reactivation and presence of CNS infiltrating EBV-specific CD8 T cells were observed, leading to propose that this condition might represent an EBV-associated immune reconstitution inflammatory syndrome (121, 174).
The lymphocyte depleting anti-CD52 monoclonal antibody alemtuzumab and cladribine are administered as single or short treatment cycles and have long term clinical and immunodepleting effects (259). Alemtuzumab targets the cell-surface glycoprotein CD52 and causes profound depletion of CD4, CD8 T cells and B cells in MS patients (323). After treatment, CD4 and CD8 T cell depletion can persist for one year or longer; B cell recovery is more rapid and the repopulating B-cell pool consists mainly of transitional and naive B cells, with a persistent deficiency of memory B cells (306). Cladribine is a synthetic purine nucleoside analogue that predominantly targets lymphocytes, disrupting DNA synthesis and repair and leading to cell death. In MS patients cladribine induces modest CD4 and CD8 T cell depletion and a more marked and persistent reduction in circulating B cells, mainly memory B cells, which could be the main mechanism underlying its efficacy (301, 302). A preliminary study showing disappearance of CSF oligoclonal Ig bands in about half of MS patients 10 years after cladribine treatment suggests that cladribine also depletes B-cells within the CNS (300).
B-cell depleting anti-CD20 monoclonal antibodies are very effective in treating relapsing MS (89, 156, 311) and exhibit some clinical benefits in active primary progressive MS (324, 325). Rituximab (administered off-label for MS treatment), ocrelizumab and ofatumumab selectively target and cause long-lasting depletion of circulating CD20+ B cells, sparing antibody producing plasma cells and very early B cell precursors (326). As observed with alemtuzumab and cladribine, B cell repopulation by transitional and naïve B cells occurs months later after infusion of rituximab while memory B cell depletion is more persistent (312). Circulating T cells are scarcely affected by anti-CD20 monoclonal antibodies (309). However, it has been shown that rituximab causes a significant reduction of both B cells and T cells in the CSF (309, 310) and of pro-inflammatory T cell responses in the peripheral blood of MS patients (157), suggesting that B cells control T cell activation. Although CD20 is a hallmark cell surface marker for B cells, a small fraction of CD4 and CD8 T cells expresses low levels of CD20. CD20+ T cells with a proinflammatory phenotype are increased in the peripheral blood and CSF of MS patients, can be stimulated by myelin antigens and are reduced by anti-CD20 administration (152, 312, 327).
Because circulating CD4 and CD8 T cells are almost completely depleted by alemtuzumab, partially depleted by cladribine and scarcely affected by anti-CD20 monoclonal antibodies, it could be argued that T cell depletion is not a necessary requirement for effective and durable MS control. Conversely, profound and persistent depletion of circulating memory B cells is a common feature associated with the high therapeutic efficacy of these drugs (259). Because memory B cells are the site of EBV persistence, the long-term memory B cell depletion caused by anti-CD20 monoclonal antibodies, cladribine and alemtuzumab implies that these drugs are actually deleting the main B-cell reservoir of EBV. It is worth noting that rituximab is an effective therapeutic option for EBV+ post-transplant lymphoproliferative diseases and is used in combination with chemotherapy drugs to treat EBV+ B lymphomas (328). In MS, B cell depleting drugs could lower the viral load and hence the burden of an EBV-driven immunopathological response. It is of relevance that widespread, active EBV infection in the brain and spinal cord of a patient with primary progressive MS was accompanied by profound EBV deregulation and marked lymphoproliferation in a deep cervical lymph node, but not in a pulmonary lymph node (172). Although preliminary, this finding points to CNS-draining lymph nodes as the primary site where an immunopathological response targeting intracerebral EBV infection is stimulated. Depletion of EBV infected B cells in CNS-draining lymph nodes could explain the high efficacy of anti-CD20 monoclonal antibodies in reducing MS activity despite their low access to the CNS (329).
Specific interference with B cell activation and maturation by targeting the Bruton´s tyrosine kinase (BTK), an enzyme involved in B cell receptor signaling, represents a promising therapeutic approach for MS that may more selectively remove pathogenic B cells (330). BTK inhibitors are small-molecule drugs that enter the CNS and could target also CNS-infiltrating B cells (331). Interestingly, BCR signaling activates EBV lytic infection and some BTK inhibitors are able to block EBV reactivation in B cells, raising the possibility that these drugs may reduce the pool of EBV infected cells by preventing new infection events (332).
In MS, CD8 T cells have attracted attention not only as key pathogenic effector cells and drug targets, but also for their therapeutic potential. Based on the hypothesis that MS results from a defective CD8 T cell control of EBV infection leading to accumulation of EBV-infected autoreactive B cells in the CNS (66), the feasibility and safety of treating progressive MS patients with autologous EBV-specific T cell therapy was recently assessed in a phase I clinical trial (333). Autologous EBV-specific T cell lines were generated by stimulation of patient peripheral mononuclear blood cells with CD8 T cell epitopes from EBV latent proteins (EBNA1, LMP1, LMP2A) and infused to the patients with the aim to eliminate EBV infected cells and restore the EBV-immune system balance. Autologous EBV-specific T cell therapy was well tolerated without serious adverse events and reduction in disability rate and fatigue was reported in some patients (333). However, larger clinical trials must be performed to establish if adoptive immunotherapy targeting EBV infected B cells can ameliorate MS (334).
Concluding Remarks
A growing body of evidence from epidemiological, genetic, immunological, neuropathological, molecular and clinical studies in MS ties together CD8 T cells, B cells and EBV. The results of the most recent epidemiological studies confirm the link between EBV infection and increased MS risk, reinforcing a causal association (335–339). It is conceivable that the genetic complexity of MS [including 200 autosomal variants outside the MHC locus, one on the X chromosome and 32 MHC alleles affecting MS risk (8)] might reflect the numerous pathways that EBV, a very complex virus with nearly 100 genes encoded by its genome (11), can exploit to alter the virus-host immune system balance and manifest its pathogenicity. Accordingly, MS risk genes are over-represented as target sites for the EBV transcription factor EBNA2, an activator of cellular and viral genes with a key role in the initial phases of EBV infection (340–342). A recent study in humanized animal models (105) stresses the need to investigate in more detail the interactions between specific MS susceptibility genes and immune control of EBV infection that may predispose to disease development (343). Because MHC class II functions as co-receptor for EBV entry into B cells (189), it would be important to assess if MHC class II alleles associated with increased or reduced risk for MS differentially affect the susceptibility of B cells to EBV infection. Along this line, a preliminary study has shown that HLA-DRB1*15:01 acts as a coreceptor for EBV infection of a B cell line (344).
To date, it is still unknown to what extent persistent intrathecal B cell activation in MS is due to B cells that mature and persist in the CNS, migrate from the periphery, or both. Intrathecal B cell activation could be, at least in part, the manifestation of B cell expansion and maturation induced by a persistent EBV infection in the CNS, but this remains to be demonstrated. The presence of proliferating B cells and the high frequency of EBV-infected B cells and plasma cells in the meningeal ectopic B-cell follicles found in chronic MS points in this direction (36, 53, 117, 170, 171). Understanding whether EBV is involved also in abnormal B cell activation outside the CNS, possibly in CNS-draining lymph nodes, and why current MS treatments do not affect intrathecal B cell activation (21), is critical for defining further the mechanism of action of current MS therapies and for developing drugs that more effectively target the ‘hidden’ pathogenic B-cell component.
An important role for CD8 T cells in MS immunopathogenesis is supported by preferential enrichment, expansion and cytotoxic effector phenotype of this T cell population in the CNS. Several studies suggest that EBV could be a major target of the CD8 T cell response in the MS brain (53, 59–63, 117). The identification of CNS-recruited CD8 T cells with a Trm-like phenotype is consistent with local antiviral immune surveillance (62, 82, 115, 116). However, more research is needed to substantiate an EBV-driven immunopathological model of MS. In particular, it is necessary to better characterize both the virological and immunological aspects of EBV infection in MS patients. At the clinical level, cytotoxic immune cell subsets could be valid biomarkers of treatment efficacy, as indicated by a recent study showing that expansion of CD8+ NK cells in the peripheral blood of MS patients is associated with reduced relapse risk (65). It is foreseen that integration of single cell technologies, like single cell transcriptomics and multiparameter spectral flow cytometry, will soon provide a comprehensive and accurate picture of disease relevant immune cell subsets in the CSF and peripheral blood of MS patients, allowing to monitor their frequency and function during treatment.
A model of MS pathogenesis tying together CD8 T cells, B cells and EBV also offers the opportunity of interpreting the broad range of immunological changes underlying the therapeutic effect of the numerous drugs approved for MS. Most MS treatments reducing CNS inflammation and tissue damage have an impact on both T cells and B cells. Although depletion of circulating B cells appears to be a common denominator associated with DMT efficacy in MS (259), it is still impossible to unequivocally link changes in either or both lymphocyte populations to the therapeutic effect. Furthermore, the impact of DMTs on different immune cell subsets in CNS and lymphoid tissues of MS patients remains largely unknown. If presentation of EBV antigens by the infected B cells had a role in MS pathogenesis, it should be assumed that drugs depleting B cells and T cells act by reducing both the antigenic stimulus and the effector arm of the immunopathologic response. Drugs acting selectively on the B cell population would eliminate the primary antigenic drive and consequently prevent the activation of the T cell effector arm. To understand if there is a relationship between EBV status, immune response to EBV and MS disease amelioration after selective B-cell depletion, antiviral cytotoxic immunity should be monitored over time in patients treated with anti-CD20 monoclonal antibodies.
Finally, the concept that MS is a rare neurological complication of a very common infection offers the rationale for clinical trials to test the safety and efficacy of antiviral drugs (345, 346) and EBV-specific adoptive T cell therapy (333, 334) aimed at improving EBV control and restore the EBV-host balance, and for the implementation of a vaccine to reduce the pathogenic potential of EBV (347, 348). It is reasonable to foresee that a better knowledge of CD8 T cell-B cell-EBV interactions in MS patients will translate into a new generation of diagnostic and prognostic biomarkers, antiviral therapeutics and preventive interventions for MS.
Author Contributions
CV wrote some sections of the manuscript, FA designed, wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work has been supported by intramural funding (Ricerca Corrente of Istituto Superiore di Sanità, n. BFC1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Reich DS, Lucchinetti CF, Calabresi PA. Multiple Sclerosis. N Engl J Med (2018) 378:169–80. doi: 10.1056/NEJMra1401483
2. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple Sclerosis. Lancet (2018) 391:1622–36. doi: 10.1016/S0140-6736(18)30481-1
3. Dobson R, Giovannoni G. Multiple Sclerosis - A Review. Eur J Neurol (2019) 26:27–40. doi: 10.1111/ene.13819
4. Olsson T, Barcellos LF, Alfredsson L. Interactions Between Genetic, Lifestyle and Environmental Risk Factors for Multiple Sclerosis. Nat Rev Neurol (2017) 13:25–36. doi: 10.1038/nrneurol.2016.187
5. Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, et al. International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2. Genetic Risk and a Primary Role for Cell-Mediated Immune Mechanisms in Multiple Sclerosis. Nature (2011) 476:214–9. doi: 10.1038/nature10251
6. International Multiple Sclerosis Genetics Consortium. Analysis of Immune-Related Loci Identifies 48 New Susceptibility Variants for Multiple Sclerosis. Nat Genet (2013) 45:1353–60. doi: 10.1038/ng.2770
7. International Multiple Sclerosis Genetics Consortium. Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk. Cell (2018) 175:1679–87.e7. doi: 10.1016/j.cell.2018.09.049
8. International Multiple Sclerosis Genetics Consortium. Multiple Sclerosis Genomic Map Implicates Peripheral Immune Cells and Microglia in Susceptibility. Science (2019) 365(6460):eaav7188. doi: 10.1126/science.aav7188
9. International Multiple Sclerosis Genetics Consortium. A Systems Biology Approach Uncovers Cell-Specific Gene Regulatory Effects of Genetic Associations in Multiple Sclerosis. Nat Commun (2019) 10(1):2236. doi: 10.1038/s41467-019-09773-y
10. Ascherio A, Munger KL, Lünemann JD. The Initiation and Prevention of Multiple Sclerosis. Nat Rev Neurol (2012) 8:602–12. doi: 10.1038/nrneurol.2012.198
11. Thorley-Lawson DA. EBV Persistence–Introducing the Virus. Curr Top Microbiol Immunol (2015) 390:151–209. doi: 10.1007/978-3-319-22822-8_8
12. Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. The Immunology of Epstein-Barr Virus-Induced Disease. Annu Rev Immunol (2015) 33:787–821. doi: 10.1146/annurev-immunol-032414-112326
13. Balfour HH Jr, Dunmire SK, Hogquist KA. Infectious Mononucleosis. Clin Transl Immunol (2015) 4(2):e33. doi: 10.1038/cti.2015.1
14. Levin LI, Munger KL, O’Reilly EJ, Falk KI, Ascherio A. Primary Infection With the Epstein-Barr Virus and Risk of Multiple Sclerosis. Ann Neurol (2010) 67:824–30. doi: 10.1002/ana.21978
15. Jacobs BM, Giovannoni G, Cuzick J, Dobson R. Systematic Review and Meta-Analysis of the Association Between Epstein-Barr Virus, Multiple Sclerosis and Other Risk Factors. Mult Scler (2020) 26:1281–97. doi: 10.1177/1352458520907901
16. Levin LI, Munger KL, Rubertone MV, Peck CA, Lennette ET, Spiegelman D, et al. Temporal Relationship Between Elevation of Epstein-Barr Virus Antibody Titers and Initial Onset of Neurological Symptoms in Multiple Sclerosis. JAMA (2005) 293:2496–500. doi: 10.1001/jama.293.20.2496
17. DeLorenze GN, Munger KL, Lennette ET, Orentreich N, Vogelman JH, Ascherio A. Epstein-Barr Virus and Multiple Sclerosis: Evidence of Association From a Prospective Study With Long-Term Follow-Up. Arch Neurol (2006) 63:839–44. doi: 10.1001/archneur.63.6.noc50328
18. Sundström P, Juto P, Wadell G, Hallmans G, Svenningsson A, Nyström L, et al. An Altered Immune Response to Epstein-Barr Virus in Multiple Sclerosis: A Prospective Study. Neurology (2004) 62:2277–82. doi: 10.1212/01.wnl.0000130496.51156.d7
19. Ascherio A, Munger KL. Environmental Risk Factors for Multiple Sclerosis. Part I: The Role of Infection. Ann Neurol (2007) 61:288–99. doi: 10.1002/ana.21117
20. Haahr S, Höllsberg P. Multiple Sclerosis is Linked to Epstein-Barr Virus Infection. Rev Med Virol (2006) 16:297–310. doi: 10.1002/rmv.503
21. Greenfield AL, Dandekar R, Ramesh A, Eggers EL, Wu H, Laurent S, et al. Longitudinally Persistent Cerebrospinal Fluid B Cells Can Resist Treatment in Multiple Sclerosis. JCI Insight (2019) 4(6):e126599. doi: 10.1172/jci.insight.126599
22. Jarius S, Eichhorn P, Franciotta D, Petereit HF, Akman-Demir G, Wick M, et al. The MRZ Reaction as a Highly Specific Marker of Multiple Sclerosis: Re-Evaluation and Structured Review of the Literature. J Neurol (2017) 264:453–66. doi: 10.1007/s00415-016-8360-4
23. Cepok S, Zhou D, Srivastava R, Nessler S, Stei S, Büssow K, et al. Identification of Epstein-Barr Virus Proteins as Putative Targets of the Immune Response in Multiple Sclerosis. J Clin Invest (2005) 115:1352–60. doi: 10.1172/JCI23661
24. Bray PF, Luka J, Bray PF, Culp KW, Schlight JP. Antibodies Against Epstein-Barr Nuclear Antigen (EBNA) in Multiple Sclerosis CSF, and Two Pentapeptide Sequence Identities Between EBNA and Myelin Basic Protein. Neurology (1992) 42:1798–804. doi: 10.1212/wnl.42.9.1798
25. Rand KH, Houck H, Denslow ND, Heilman KM. Epstein-Barr Virus Nuclear Antigen-1 (EBNA-1) Associated Oligoclonal Bands in Patients With Multiple Sclerosis. J Neurol Sci (2000) 173:32–9. doi: 10.1016/s0022-510x(99)00298-1
26. Wang Z, Kennedy PG, Dupree C, Wang M, Lee C, Pointon T, et al. Antibodies From Multiple Sclerosis Brain Identified Epstein-Barr Virus Nuclear Antigen 1 & 2 Epitopes Which Are Recognized by Oligoclonal Bands. J Neuroimmune Pharmacol (2020) 18:1–14. doi: 10.1007/s11481-020-09948-1
27. Cepok S, Jacobsen M, Schock S, Omer B, Jaekel S, Böddeker I, et al. Patterns of Cerebrospinal Fluid Pathology Correlate With Disease Progression in Multiple Sclerosis. Brain (2001) 124:2169–76. doi: 10.1093/brain/124.11.2169
28. Cepok S, Rosche B, Grummel V, Vogel F, Zhou D, Sayn J, et al. Short-Lived Plasma Blasts are the Main B Cell Effector Subset During the Course of Multiple Sclerosis. Brain (2005) 128:1667–76. doi: 10.1093/brain/awh486
29. Eggers EL, Michel BA, Wu H, Wang SZ, Bevan CJ, Abounasr A, et al. Clonal Relationships of CSF B Cells in Treatment-Naive Multiple Sclerosis Patients. JCI Insight (2017) 2(22):e92724. doi: 10.1172/jci.insight.92724
30. Colombo M, Dono M, Gazzola P, Roncella S, Valetto A, Chiorazzi N, et al. Accumulation of Clonally Related B Lymphocytes in the Cerebrospinal Fluid of Multiple Sclerosis Patients. J Immunol (2000) 164:2782–9. doi: 10.4049/jimmunol.164.5.2782
31. Stern JN, Yaari G, Vander Heiden JA, Church G, Donahue WF, Hintzen RQ, et al. B Cells Populating the Multiple Sclerosis Brain Mature in the Draining Cervical Lymph Nodes. Sci Transl Med (2014) 6(248):248ra107. doi: 10.1126/scitranslmed.3008879
32. Palanichamy A, Apeltsin L, Kuo TC, Sirota M, Wang S, Pitts SJ, et al. Immunoglobulin Class-Switched B Cells Form An Active Immune Axis Between CNS and Periphery in Multiple Sclerosis. Sci Transl Med (2014) 6(248):248ra106. doi: 10.1126/scitranslmed.3008930
33. Lassmann H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front Immunol (2019) 9:3116. doi: 10.3389/fimmu.2018.03116
34. Prineas JW, Wright RG. Macrophages, Lymphocytes, and Plasma Cells in the Perivascular Compartment in Chronic Multiple Sclerosis. Lab Invest (1978) 38:409–21.
35. Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H, et al. Inflammatory Cortical Demyelination in Early Multiple Sclerosis. N Engl J Med (2011) 365:2188–97. doi: 10.1056/NEJMoa1100648
36. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of Ectopic B-Cell Follicles With Germinal Centers in the Meninges of Patients With Secondary Progressive Multiple Sclerosis. Brain Pathol (2004) 14:164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x
37. Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, et al. Meningeal B-Cell Follicles in Secondary Progressive Multiple Sclerosis Associate With Early Onset of Disease and Severe Cortical Pathology. Brain (2007) 130:1089–104. doi: 10.1093/brain/awm038
38. Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, et al. A Gradient of Neuronal Loss and Meningeal Inflammation in Multiple Sclerosis. Ann Neurol (2010) 68:477–93. doi: 10.1002/ana.22230
39. Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, et al. Meningeal Inflammation Is Widespread and Linked to Cortical Pathology in Multiple Sclerosis. Brain (2011) 134:2755–71. doi: 10.1093/brain/awr182
40. Münz C. Cytotoxicity in Epstein Barr Virus Specific Immune Control. Curr Opin Virol (2021) 46:1–8. doi: 10.1016/j.coviro.2020.07.011
41. Long HM, Meckiff BJ, Taylor GS. The T-Cell Response to Epstein-Barr Virus - New Tricks From an Old Dog. Front Immunol (2019) 10:2193. doi: 10.3389/fimmu.2019.02193
42. Larsen PD, Bloomer LC, Bray PF. Epstein-Barr Nuclear Antigen and Viral Capsid Antigen Antibody Titers in Multiple Sclerosis. Neurology (1985) 35:435–8. doi: 10.1212/wnl.35.3.435
43. Lünemann JD, Tintoré M, Messmer B, Strowig T, Rovira A, Perkal H, et al. Elevated Epstein-Barr Virus-Encoded Nuclear Antigen-1 Immune Responses Predict Conversion to Multiple Sclerosis. Ann Neurol (2010) 67:159–69. doi: 10.1002/ana.21886
44. Zivadinov R, Cerza N, Hagemeier J, Carl E, Badgett D, Ramasamy DP, et al. Humoral Response to EBV Is Associated With Cortical Atrophy and Lesion Burden in Patients With MS. Neurol Neuroimmunol Neuroinflamm (2016) 3:e190. doi: 10.1212/NXI.0000000000000190
45. Buljevac D, van Doornum GJ, Flach HZ, Groen J, Osterhaus AD, Hop W, et al. Epstein-Barr Virus and Disease Activity in Multiple Sclerosis. J Neurol Neurosurg Psychiatry (2005) 76:1377–81. doi: 10.1136/jnnp.2004.048504
46. Farrell RA, Antony D, Wall GR, Clark DA, Fisniku L, Swanton J, et al. Humoral Immune Response to EBV in Multiple Sclerosis Is Associated With Disease Activity on MRI. Neurology (2009) 73:32–8. doi: 10.1212/WNL.0b013e3181aa29fe
47. Kvistad S, Myhr KM, Holmøy T, Bakke S, Beiske AG, Bjerve KS, et al. Antibodies to Epstein-Barr Virus and MRI Disease Activity in Multiple Sclerosis. Mult Scler (2014) 20:1833–140. doi: 10.1177/1352458514533843
48. Gieß RM, Pfuhl C, Behrens JR, Rasche L, Freitag E, Khalighy N, et al. Epstein-Barr Virus Antibodies in Serum and DNA Load in Saliva Are Not Associated With Radiological or Clinical Disease Activity in Patients With Early Multiple Sclerosis. PloS One (2017) 12(4):e0175279. doi: 10.1371/journal.pone.0175279
49. Lünemann JD, Edwards N, Muraro PA, Hayashi S, Cohen JI, Münz C, et al. Increased Frequency and Broadened Specificity of Latent EBV Nuclear Antigen-1-Specific T Cells in Multiple Sclerosis. Brain (2006) 129:1493–506. doi: 10.1093/brain/awl067
50. Wuest SC, Mexhitaj I, Chai NR, Romm E, Scheffel J, Xu B, et al. A Complex Role of Herpes Viruses in the Disease Process of Multiple Sclerosis. PloS One (2014) 9(8):e105434. doi: 10.1371/journal.pone.0105434
51. Lossius A, Vartdal F, Holmøy T. Vitamin D Sensitive EBNA-1 Specific T Cells in the Cerebrospinal Fluid of Patients With Multiple Sclerosis. J Neuroimmunol (2011) 240-241:87–96. doi: 10.1016/j.jneuroim.2011.09.011
52. Holmøy T, Vartdal F. Cerebrospinal Fluid T Cells From Multiple Sclerosis Patients Recognize Autologous Epstein-Barr Virus-Transformed B Cells. J Neurovirol (2004) 10:52–6. doi: 10.1080/13550280490261671
53. Angelini DF, Serafini B, Piras E, Severa M, Coccia EM, Rosicarelli B, et al. Increased CD8+ T Cell Response to Epstein-Barr Virus Lytic Antigens in the Active Phase of Multiple Sclerosis. PloS Pathog (2013) 9(4):e1003220. doi: 10.1371/journal.ppat.1003220
54. Jilek S, Schluep M, Meylan P, Vingerhoets F, Guignard L, Monney A, et al. Strong EBV-Specific CD8+ T-Cell Response in Patients With Early Multiple Sclerosis. Brain (2008) 131:1712–21. doi: 10.1093/brain/awn108
55. Pender MP, Csurhes PA, Burrows JM, Burrows SR. Defective T-Cell Control of Epstein-Barr Virus Infection in Multiple Sclerosis. Clin Transl Immunol (2017) 6(1):e126. doi: 10.1038/cti.2016.87
56. Latham LB, Lee MJ, Lincoln JA, Ji N, Forsthuber TG, Lindsey JW. Antivirus Immune Activity in Multiple Sclerosis Correlates With MRI Activity. Acta Neurol Scand (2016) 133:17–24. doi: 10.1111/ane.12417
57. Pender MP, Csurhes PA, Lenarczyk A, Pfluger CM, Burrows SR. Decreased T Cell Reactivity to Epstein-Barr Virus Infected Lymphoblastoid Cell Lines in Multiple Sclerosis. J Neurol Neurosurg Psychiatry (2009) 80:498–505. doi: 10.1136/jnnp.2008.161018
58. Jilek S, Schluep M, Harari A, Canales M, Lysandropoulos A, Zekeridou A, et al. HLA-B7-Restricted EBV-Specific Cd8+ T Cells Are Dysregulated in Multiple Sclerosis. J Immunol (2012) 188:4671–80. doi: 10.4049/jimmunol.1103100
59. Jaquiéry E, Jilek S, Schluep M, Meylan P, Lysandropoulos A, Pantaleo G, et al. Intrathecal Immune Responses to EBV in Early MS. Eur J Immunol (2010) 40:878–87. doi: 10.1002/eji.200939761
60. Lossius A, Johansen JN, Vartdal F, Robins H, Jūratė Šaltytė B, Holmøy T, et al. High-Throughput Sequencing of TCR Repertoires in Multiple Sclerosis Reveals Intrathecal Enrichment of EBV-Reactive CD8+ T Cells. Eur J Immunol (2014) 44:3439–52. doi: 10.1002/eji.201444662
61. van Nierop GP, Mautner J, Mitterreiter JG, Hintzen RQ, Verjans GM. Intrathecal CD8 T-Cells of Multiple Sclerosis Patients Recognize Lytic Epstein-Barr Virus Proteins. Mult Scler (2016) 22:279–91. doi: 10.1177/1352458515588581
62. van Nierop GP, van Luijn MM, Michels SS, Melief MJ, Janssen M, Langerak AW, et al. Phenotypic and Functional Characterization of T Cells in White Matter Lesions of Multiple Sclerosis Patients. Acta Neuropathol (2017) 134:383–401. doi: 10.1007/s00401-017-1744-4
63. Serafini B, Rosicarelli B, Veroni C, Mazzola GA, Aloisi F. Epstein-Barr Virus-Specific CD8 T Cells Selectively Infiltrate the Brain in Multiple Sclerosis and Interact Locally With Virus-Infected Cells: Clue for a Virus-Driven Immunopathological Mechanism. J Virol (2019) 93(24):e00980–19. doi: 10.1128/JVI.00980-19
64. Mimpen M, Smolders J, Hupperts R, Damoiseaux J. Natural Killer Cells in Multiple Sclerosis: A Review. Immunol Lett (2020) 222:1–11. doi: 10.1016/j.imlet.2020.02.012
65. McKinney EF, Cuthbertson I, Harris KM, Smilek DE, Connor C, Manferrari G, et al. A CD8(+) NK Cell Transcriptomic Signature Associated With Clinical Outcome in Relapsing Remitting Multiple Sclerosis. Nat Commun (2021) 12(1):635. doi: 10.1038/s41467-020-20594-2
66. Pender MP. Infection of Autoreactive B Lymphocytes With EBV, Causing Chronic Autoimmune Diseases. Trends Immunol (2003) 24:584–8. doi: 10.1016/j.it.2003.09.005
67. Willis SN, Stathopoulos P, Chastre A, Compton SD, Hafler DA, O’Connor KC. Investigating the Antigen Specificity of Multiple Sclerosis Central Nervous System-Derived Immunoglobulins. Front Immunol (2015) 6:600. doi: 10.3389/fimmu.2015.00600
68. Tracy SI, Kakalacheva K, Lünemann JD, Luzuriaga K, Middeldorp J, Thorley-Lawson DA. Persistence of Epstein-Barr Virus in Self-Reactive Memory B Cells. J Virol (2012) 86:12330–40. doi: 10.1128/JVI.01699-12
69. Mameli G, Cossu D, Cocco E, Masala S, Frau J, Marrosu MG, et al. Epstein-Barr Virus and Mycobacterium Avium Subsp. Paratuberculosis Peptides are Cross Recognized by Anti-Myelin Basic Protein Antibodies in Multiple Sclerosis Patients. J Neuroimmunol (2014) 270:51–5. doi: 10.1016/j.jneuroim.2014.02.013
70. Tengvall K, Huang J, Hellström C, Kammer P, Biström M, Ayoglu B, et al. Molecular Mimicry Between Anoctamin 2 and Epstein-Barr Virus Nuclear Antigen 1 Associates With Multiple Sclerosis Risk. Proc Natl Acad Sci USA (2019) 116:16955–60. doi: 10.1073/pnas.1902623116
71. Jog NR, McClain MT, Heinlen LD, Gross T, Towner R, Guthridge JM, et al. Epstein Barr Virus Nuclear Antigen 1 (EBNA-1) Peptides Recognized by Adult Multiple Sclerosis Patient Sera Induce Neurologic Symptoms in a Murine Model. J Autoimmun (2020) 106:102332. doi: 10.1016/j.jaut.2019.102332
72. Wang J, Jelcic I, Mühlenbruch L, Haunerdinger V, Toussaint NC, Zhao Y, et al. HLA-DR15 Molecules Jointly Shape An Autoreactive T Cell Repertoire in Multiple Sclerosis. Cell (2020) 183(5):1264–81.e20. doi: 10.1016/j.cell.2020.09.054
73. Wucherpfennig KW, Strominger JL. Molecular Mimicry in T Cell-Mediated Autoimmunity: Viral Peptides Activate Human T Cell Clones Specific for Myelin Basic Protein. Cell (1995) 80:695–705. doi: 10.1016/0092-8674(95)90348-8
74. Holmøy T, Kvale EØ, Vartdal F. Cerebrospinal Fluid CD4+ T Cells From a Multiple Sclerosis Patient Cross-Recognize Epstein-Barr Virus and Myelin Basic Protein. J Neurovirol (2004) 10:278–83. doi: 10.1080/13550280490499524
75. Lünemann JD, Jelcić I, Roberts S, Lutterotti A, Tackenberg B, Martin R, et al. EBNA1-Specific T Cells From Patients With Multiple Sclerosis Cross React With Myelin Antigens and Co-Produce IFN-Gamma and IL-2. J Exp Med (2008) 205:1763–73. doi: 10.1084/jem.20072397
76. Tangye SG, Palendira U, Edwards ES. Human Immunity Against EBV-lessons From the Clinic. J Exp Med (2017) 214:269–83. doi: 10.1084/jem.20161846
77. van Noort JM, Bajramovic JJ, Plomp AC, van Stipdonk MJ. Mistaken Self, A Novel Model That Links Microbial Infections With Myelin-Directed Autoimmunity in Multiple Sclerosis. J Neuroimmunol (2000) 105:46–57. doi: 10.1016/s0165-5728(00)00181-8
78. Sotgiu S, Pugliatti M, Contu S, Sanna A, Sgaramella E, VanNoort JM, et al. Alpha B-Crystallin Is Not a Dominant Peripheral T-Cell Autoantigen in Multiple Sclerosis Amongst Sardinians. Eur J Neurol (2003) 10:583–6. doi: 10.1046/j.1468-1331.2003.00652.x
79. Salou M, Nicol B, Garcia A, Laplaud DA. Involvement of CD8 (+) T Cells in Multiple Sclerosis. Front Immunol (2015) 6:604. doi: 10.3389/fimmu.2015.00604
80. Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal Expansions of CD8(+) T Cells Dominate the T Cell Infiltrate in Active Multiple Sclerosis Lesions as Shown by Micromanipulation and Single Cell Polymerase Chain Reaction. J Exp Med (2000) 192:393–404. doi: 10.1084/jem.192.3.393
81. Skulina C, Schmidt S, Dornmair K, Babbe H, Roers A, Rajewsky K, et al. Multiple Sclerosis: Brain-Infiltrating CD8+ T Cells Persist as Clonal Expansions in the Cerebrospinal Fluid and Blood. Proc Natl Acad Sci USA (2004) 101:2428–33. doi: 10.1073/pnas.0308689100
82. Beltrán E, Gerdes LA, Hansen J, Flierl-Hecht A, Krebs S, Blum H, et al. Early Adaptive Immune Activation Detected in Monozygotic Twins With Prodromal Multiple Sclerosis. J Clin Invest (2019) 129:4758–68. doi: 10.1172/JCI128475
83. Junker A, Ivanidze J, Malotka J, Eiglmeier I, Lassmann H, Wekerle H, et al. Multiple Sclerosis: T-Cell Receptor Expression in Distinct Brain Regions. Brain (2007) 130:2789–99. doi: 10.1093/brain/awm214
84. Butovsky O, Weiner HL. Microglial Signatures and Their Role in Health and Disease. Nat Rev Neurosci (2018) 19:622–35. doi: 10.1038/s41583-018-0057-5
85. Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The Search for the Target Antigens of Multiple Sclerosis, Part 1: Autoreactive CD4+ T Lymphocytes as Pathogenic Effectors and Therapeutic Targets. Lancet Neurol (2016) 15:198–209. doi: 10.1016/S1474-4422(15)00334-8
86. Giovannoni G, Cutter GR, Lunemann J, Martin R, Münz C, Sriram S, et al. Infectious Causes of Multiple Sclerosis. Lancet Neurol (2006) 5:887–94. doi: 10.1016/S1474-4422(06)70577-4
87. Salvetti M, Giovannoni G, Aloisi F. Epstein-Barr Virus and Multiple Sclerosis. Curr Opin Neurol (2009) 22:201–6. doi: 10.1097/WCO.0b013e32832b4c8d
88. Franciotta D, Salvetti M, Lolli F, Serafini B, Aloisi F. B Cells and Multiple Sclerosis. Lancet Neurol (2008) 7:852–8. doi: 10.1016/S1474-4422(08)70192-3
89. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-Cell Depletion With Rituximab in Relapsing-Remitting Multiple Sclerosis. N Engl J Med (2008) 358:676–88. doi: 10.1056/NEJMoa0706383
90. Li R, Patterson KR, Bar-Or A. Reassessing B Cell Contributions in Multiple Sclerosis. Nat Immunol (2018) 19:696–707. doi: 10.1038/s41590-018-0135-x
91. Canto E, Oksenberg JR. Multiple Sclerosis Genetics. Mult Scler (2018) 24:75–9. doi: 10.1177/1352458517737371
92. Kular L, Liu Y, Ruhrmann S, Zheleznyakova G, Marabita F, Gomez-Cabrero D, et al. DNA Methylation as a Mediator of HLA-DRB1*15:01 and a Protective Variant in Multiple Sclerosis. Nat Commun (2018) 9(1):2397. doi: 10.1038/s41467-018-04732-5
93. Wucherpfennig KW, Sette A, Southwood S, Oseroff C, Matsui M, Strominger JL, et al. Structural Requirements for Binding of an Immunodominant Myelin Basic Protein Peptide to DR2 Isotypes and for its Recognition by Human T Cell Clones. J Exp Med (1994) 179:279–90. doi: 10.1084/jem.179.1.279
94. Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, et al. Memory B Cells Activate Brain-Homing, Autoreactive CD4(+) T Cells in Multiple Sclerosis. Cell (2018) 175(1):85–100.e23. doi: 10.1016/j.cell.2018.08.011
95. Fogdell-Hahn A, Ligers A, Grønning M, Hillert J, Olerup O. Multiple Sclerosis: A Modifying Influence of HLA Class I Genes in an HLA Class II Associated Autoimmune Disease. Tissue Antigens (2000) 55:140–8. doi: 10.1034/j.1399-0039.2000.550205.x
96. Friese MA, Jakobsen KB, Friis L, Etzensperger R, Craner MJ, McMahon RM, et al. Opposing Effects of HLA Class I Molecules in Tuning Autoreactive CD8+ T Cells in Multiple Sclerosis. Nat Med (2008) 14:1227–35. doi: 10.1038/nm.1881
97. Lundtoft C, Pucholt P, Imgenberg-Kreuz J, Carlsson-Almlöf J, Eloranta ML, Syvänen AC, et al. Function of Multiple Sclerosis-Protective HLA Class I Alleles Revealed by Genome-Wide Protein-Quantitative Trait Loci Mapping of Interferon Signalling. PloS Genet (2020) 16(10):e1009199. doi: 10.1371/journal.pgen.1009199
98. Dobson R, Kuhle J, Middeldorp J, Giovannoni G. Epstein-Barr-Negative MS: A True Phenomenon? Neurol Neuroimmunol Neuroinflamm (2017) 4(2):e318. doi: 10.1212/NXI.0000000000000318
99. Abrahamyan S, Eberspächer B, Hoshi MM, Aly L, Luessi F, Groppa S, et al. Complete Epstein-Barr Virus Seropositivity in a Large Cohort of Patients With Early Multiple Sclerosis. J Neurol Neurosurg Psychiatry (2020) 91:681–6. doi: 10.1136/jnnp-2020-322941
100. Castellino F, Germain RN. Cooperation Between CD4+ and CD8+ T Cells: When, Where, and How. Annu Rev Immunol (2006) 24:519–40. doi: 10.1146/annurev.immunol.23.021704.115825
101. Kanev K, Zehn D. Origin and Fine-Tuning of Effector CD8 T Cell Subpopulations in Chronic Infection. Curr Opin Virol (2020) 46:27–35. doi: 10.1016/j.coviro.2020.10.003
102. De Jager PL, Simon KC, Munger KL, Rioux JD, Hafler DA, Ascherio A. Integrating Risk Factors: HLA-DRB1*1501 and Epstein-Barr Virus in Multiple Sclerosis. Neurology (2008) 70:1113–8. doi: 10.1212/01.wnl.0000294325.63006.f8
103. Sundqvist E, Sundström P, Lindén M, Hedström AK, Aloisi F, Hillert J, et al. Epstein-Barr Virus and Multiple Sclerosis: Interaction With HLA. Genes Immun (2012) 13:14–20. doi: 10.1038/gene.2011.42
104. Kreft KL, Van Nierop GP, Scherbeijn SMJ, Janssen M, Verjans GMGM, Hintzen RQ. Elevated EBNA-1 Igg in MS Is Associated With Genetic MS Risk Variants. Neurol Neuroimmunol Neuroinflamm (2017) 4(6):e406. doi: 10.1212/NXI.0000000000000406
105. Zdimerova H, Murer A, Engelmann C, Raykova A, Deng Y, Gujer C, et al. Attenuated Immune Control of Epstein-Barr Virus in Humanized Mice Is Associated With the Multiple Sclerosis Risk Factor HLA-DR15. Eur J Immunol (2021) 51:64–75. doi: 10.1002/eji.202048655
106. Annunziato F, Romagnani C, Romagnani S. The 3 Major Types of Innate and Adaptive Cell-Mediated Effector Immunity. J Allergy Clin Immunol (2015) 135:626–35. doi: 10.1016/j.jaci.2014.11.001
107. Bishop GA, Stunz LL, Hostager BS. TRAF3 as a Multifaceted Regulator of B Lymphocyte Survival and Activation. Front Immunol (2018) 9:2161. doi: 10.3389/fimmu.2018.02161
108. Ramesh A, Schubert RD, Greenfield AL, Dandekar R, Loudermilk R, Sabatino JJ Jr, et al. A Pathogenic and Clonally Expanded B Cell Transcriptome in Active Multiple Sclerosis. Proc Natl Acad Sci USA (2020) 117:22932–43. doi: 10.1073/pnas.2008523117
109. Boztug H, Hirschmugl T, Holter W, Lakatos K, Kager L, Trapin D, et al. Nf-κb1 Haploinsufficiency Causing Immunodeficiency and EBV-driven Lymphoproliferation. J Clin Immunol (2016) 36:533–40. doi: 10.1007/s10875-016-0306-1
110. Steri M, Orrù V, Idda ML, Pitzalis M, Pala M, Zara I, et al. Overexpression of the Cytokine BAFF and Autoimmunity Risk. N Engl J Med (2017) 376:1615–26. doi: 10.1056/NEJMoa1610528
111. Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological Analysis of T Lymphocyte Subsets in the Central Nervous System in Chronic Progressive Multiple Sclerosis. J Neurol Sci (1983) 62:219–32. doi: 10.1016/0022-510x(83)90201-0
112. Hauser SL, Aubert C, Burks JS, Kerr C, Lyon-Caen O, de The G, et al. Analysis of Human T-Lymphotrophic Virus Sequences in Multiple Sclerosis Tissue. Nature (1986) 322:176–7. doi: 10.1038/322176a0
113. Woodroofe MN, Bellamy AS, Feldmann M, Davison AN, Cuzner ML. Immunocytochemical Characterisation of the Immune Reaction in the Central Nervous System in Multiple Sclerosis. Possible Role for Microglia in Lesion Growth. J Neurol Sci (1986) 74:135–52. doi: 10.1016/0022-510x(86)90100-0
114. Ifergan I, Kebir H, Alvarez JI, Marceau G, Bernard M, Bourbonnière L, et al. Central Nervous System Recruitment of Effector Memory CD8+ T Lymphocytes During Neuroinflammation Is Dependent on α4 Integrin. Brain (2011) 134:3560–77. doi: 10.1093/brain/awr268
115. Machado-Santos J, Saji E, Tröscher AR, Paunovic M, Liblau R, Gabriely G, et al. The Compartmentalized Inflammatory Response in the Multiple Sclerosis Brain Is Composed of Tissue-Resident CD8+ T Lymphocytes and B Cells. Brain (2018) 141:2066–82. doi: 10.1093/brain/awy151
116. Fransen NL, Hsiao CC, van der Poel M, Engelenburg HJ, Verdaasdonk K, Vincenten MCJ, et al. Tissue-Resident Memory T Cells Invade the Brain Parenchyma in Multiple Sclerosis White Matter Lesions. Brain (2020) 143:1714–30. doi: 10.1093/brain/awaa117
117. Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein-Barr Virus Infection in the Multiple Sclerosis Brain. J Exp Med (2007) 204:2899–912. doi: 10.1084/jem.20071030
118. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, et al. Expression of GM-CSF in T Cells is Increased in Multiple Sclerosis and Suppressed by IFN-β Therapy. J Immunol (2015) 194:5085–93. doi: 10.4049/jimmunol.1403243
119. Serafini B, Rosicarelli B, Veroni C, Zhou L, Reali C, Aloisi F. Rorγt Expression and Lymphoid Neogenesis in the Brain of Patients With Secondary Progressive Multiple Sclerosis. J Neuropathol Exp Neurol (2016) 75:877–88. doi: 10.1093/jnen/nlw063
120. Veroni C, Serafini B, Rosicarelli B, Fagnani C, Aloisi F. Transcriptional Profile and Epstein-Barr Virus Infection Status of Laser-Cut Immune Infiltrates From the Brain of Patients With Progressive Multiple Sclerosis. J Neuroinflamm (2018) 15(1):18. doi: 10.1186/s12974-017-1049-5
121. Serafini B, Zandee S, Rosicarelli B, Scorsi E, Veroni C, Larochelle C, et al. Epstein-Barr Virus-Associated Immune Reconstitution Inflammatory Syndrome as Possible Cause of Fulminant Multiple Sclerosis Relapse After Natalizumab Interruption. J Neuroimmunol (2018) 319:9–12. doi: 10.1016/j.jneuroim.2018.03.011
122. Jacobsen M, Cepok S, Quak E, Happel M, Gaber R, Ziegler A, et al. Oligoclonal Expansion of Memory CD8+ T Cells in Cerebrospinal Fluid From Multiple Sclerosis Patients. Brain (2002) 125:538–50. doi: 10.1093/brain/awf059
123. Giunti D, Borsellino G, Benelli R, Marchese M, Capello E, Valle MT, et al. Phenotypic and Functional Analysis of T Cells Homing Into the CSF of Subjects With Inflammatory Diseases of the CNS. J Leukoc Biol (2003) 73:584–90. doi: 10.1189/jlb.1202598
124. Kivisäkk P, Mahad D, Callahan MK, Sikora K, Trebst C, Tucky B, et al. Expression of CCR7 in Multiple Sclerosis: Implications for CNS Immunity. Ann Neurol (2004) 55:627–38. doi: 10.1002/ana.20049
125. Jilek S, Schluep M, Rossetti AO, Guignard L, Le Goff G, Pantaleo G, et al. CSF Enrichment of Highly Differentiated CD8+ T Cells in Early Multiple Sclerosis. Clin Immunol (2007) 123:105–13. doi: 10.1016/j.clim.2006.11.004
126. Wallström E, Khademi M, Andersson M, Olsson T. Increased Numbers of Mononuclear Cells From Blood and CSF Expressing Interferon-Gamma mRNA in Multiple Sclerosis Are From Both the CD4+ and the CD8+ Subsets. Eur J Neurol (2000) 7:71–6. doi: 10.1046/j.1468-1331.2000.00027.x
127. Somma P, Ristori G, Battistini L, Cannoni S, Borsellino G, Diamantini A, et al. Characterization of CD8+ T Cell Repertoire in Identical Twins Discordant and Concordant for Multiple Sclerosis. J Leukoc Biol (2007) 81:696–710. doi: 10.1189/jlb.0906584
128. Malmeström C, Lycke J, Haghighi S, Andersen O, Carlsson L, Wadenvik H, et al. Relapses in Multiple Sclerosis Are Associated With Increased CD8+ T-Cell Mediated Cytotoxicity in CSF. J Neuroimmunol (2008) 196:159–65. doi: 10.1016/j.jneuroim.2008.03.001
129. Kivisäkk P, Trebst C, Liu Z, Tucky BH, Sørensen TL, Rudick RA, et al. T-Cells in the Cerebrospinal Fluid Express a Similar Repertoire of Inflammatory Chemokine Receptors in the Absence or Presence of CNS Inflammation: Implications for CNS Trafficking. Clin Exp Immunol (2002) 129:510–8. doi: 10.1046/j.1365-2249.2002.01947.x
130. Battistini L, Piccio L, Rossi B, Bach S, Galgani S, Gasperini C, et al. CD8+ T Cells From Patients With Acute Multiple Sclerosis Display Selective Increase of Adhesiveness in Brain Venules: A Critical Role for P-Selectin Glycoprotein Ligand-1. Blood (2003) 101:4775–82. doi: 10.1182/blood-2002-10-3309
131. Alvarez JI, Kébir H, Cheslow L, Charabati M, Chabarati M, Larochelle C, et al. JAML Mediates Monocyte and CD8 T Cell Migration Across the Brain Endothelium. Ann Clin Transl Neurol (2015) 2:1032–7. doi: 10.1002/acn3.255
132. Larochelle C, Lécuyer MA, Alvarez JI, Charabati M, Saint-Laurent O, Ghannam S, et al. Melanoma Cell Adhesion Molecule-Positive CD8 T Lymphocytes Mediate Central Nervous System Inflammation. Ann Neurol (2015) 78:39–53. doi: 10.1002/ana.24415
133. Salou M, Garcia A, Michel L, Gainche-Salmon A, Loussouarn D, Nicol B, et al. Expanded CD8 T-Cell Sharing Between Periphery and CNS in Multiple Sclerosis. Ann Clin Transl Neurol (2015) 2:609–22. doi: 10.1002/acn3.199
134. Steinman L. The Discovery of Natalizumab, a Potent Therapeutic for Multiple Sclerosis. J Cell Biol (2012) 199:413–6. doi: 10.1083/jcb.201207175
135. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Capello E, Mancardi GL, et al. Dendritic Cells in Multiple Sclerosis Lesions: Maturation Stage, Myelin Uptake, and Interaction With Proliferating T Cells. J Neuropathol Exp Neurol (2006) 65:124–41. doi: 10.1097/01.jnen.0000199572.96472.1c
136. Bach MA, Phan-Dinh-Tuy F, Tournier E, Chatenoud L, Bach JF, Martin C, et al. Deficit of Suppressor T Cells in Active Multiple Sclerosis. Lancet (1980) 2:1221–3. doi: 10.1016/s0140-6736(80)92480-0
137. Compston A. Lymphocyte Subpopulations in Patients With Multiple Sclerosis. J Neurol Neurosurg Psychiatry (1983) 46:105–14. doi: 10.1136/jnnp.46.2.105
138. Thompson AJ, Brazil J, Whelan CA, Martin EA, Hutchinson M, Feighery C. Peripheral Blood T Lymphocyte Changes in Multiple Sclerosis: A Marker of Disease Progression Rather Than of Relapse? J Neurol Neurosurg Psychiatry (1986) 49:905–12. doi: 10.1136/jnnp.49.8.905
139. Pender MP, Csurhe PA, Pfluger CM, Burrows SR. Deficiency of CD8+ Effector Memory T Cells Is an Early and Persistent Feature of Multiple Sclerosis. Mult Scler (2014) 20:1825–32. doi: 10.1177/1352458514536252
140. Parnell GP, Gatt PN, Krupa M, Nickles D, McKay FC, Schibeci SD, et al. The Autoimmune Disease-Associated Transcription Factors EOMES and TBX21 Are Dysregulated in Multiple Sclerosis and Define a Molecular Subtype of Disease. Clin Immunol (2014) 151:16–24. doi: 10.1016/j.clim.2014.01.003
141. McKay FC, Gatt PN, Fewings N, Parnell GP, Schibeci SD, Basuki MA, et al. The Low EOMES/TBX21 Molecular Phenotype in Multiple Sclerosis Reflects CD56+ Cell Dysregulation and Is Affected by Immunomodulatory Therapies. Clin Immunol (2016) 163:96–107. doi: 10.1016/j.clim.2015.12.015
142. Jensen J, Langkilde AR, Frederiksen JL, Sellebjerg F. Cd8+ T Cell Activation Correlates With Disease Activity in Clinically Isolated Syndromes and is Regulated by Interferon-Beta Treatment. J Neuroimmunol (2006) 179:163–72. doi: 10.1016/j.jneuroim.2006.06.024
143. Killestein J, Den Drijver BF, van der Graaff WL, Uitdehaag BM, Polman CH, Van Lier RA. (Intracellular Cytokine Profile in T-Cell Subsets of Multiple Sclerosis Patients: Different Features in Primary Progressive Disease. Mult Scler (2001) 7:145–50. doi: 10.1177/135245850100700302
144. Frisullo G, Plantone D, Marti A, Iorio R, Damato V, Nociti V, et al. Type 1 Immune Response in Progressive Multiple Sclerosis. J Neuroimmunol (2012) 249:112–6. doi: 10.1016/j.jneuroim.2012.04.019
145. Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The Search for the Target Antigens of Multiple Sclerosis, Part 2: CD8+ T Cells, B Cells, and Antibodies in the Focus of Reverse-Translational Research. Lancet Neurol (2016) 15:317–31. doi: 10.1016/S1474-4422(15)00313-0
146. Nicol B, Salou M, Laplaud DA, Wekerle H. The Autoimmune Concept of Multiple Sclerosis. Presse Med (2015) 44:e103–12. doi: 10.1016/j.lpm.2015.02.009
147. Crawford MP, Yan SX, Ortega SB, Mehta RS, Hewitt R, Price DA, et al. High Prevalence of Autoreactive, Neuroantigen-Specific CD8+ T Cells in Multiple Sclerosis Revealed by Novel Flow Cytometric Assay. Blood (2004) 103:4222–31. doi: 10.1182/blood-2003-11-4025
148. Zang YC, Li S, Rivera VM, Hong J, Robinson RR, Breitbach WT, et al. Increased CD8+ Cytotoxic T Cell Responses to Myelin Basic Protein in Multiple Sclerosis. J Immunol (2004) 172:5120–27. doi: 10.4049/jimmunol.172.8.5120
149. Niland B, Banki K, Biddison WE, Perl A. CD8+ T Cell-Mediated HLA-A*0201-Restricted Cytotoxicity to Transaldolase Peptide 168-176 in Patients With Multiple Sclerosis. J Immunol (2005) 175:8365–78. doi: 10.4049/jimmunol.175.12.8365
150. Berthelot L, Laplaud DA, Pettré S, Ballet C, Michel L, Hillion S, et al. Blood Cd8+ T Cell Responses Against Myelin Determinants in Multiple Sclerosis and Healthy Individuals. Eur J Immunol (2008) 38:1889–99. doi: 10.1002/eji.200838023
151. Lolli F, Martini H, Citro A, Franceschini D, Portaccio E, Amato MP, et al. Increased CD8+ T Cell Responses to Apoptotic T Cell-Associated Antigens in Multiple Sclerosis. J Neuroinflamm (2013) 10:94. doi: 10.1186/1742-2094-10-94
152. Sabatino JJ, Wilson MR, Calabresi PA, Hauser SL, Schneck JP, Zamvil SS. Anti-CD20 Therapy Depletes Activated Myelin-Specific CD8(+) T Cells in Multiple Sclerosis. Proc Natl Acad Sci USA (2019) 116:25800–807. doi: 10.1073/pnas.1915309116
153. Montes M, Zhang X, Berthelot L, Laplaud DA, Brouard S, Jin J, et al. Oligoclonal Myelin-Reactive T-Cell Infiltrates Derived From Multiple Sclerosis Lesions Are Enriched in Th17 Cells. Clin Immunol (2009) 130:133–4. doi: 10.1016/j.clim.2008.08.030
154. van Nierop GP, Janssen M, Mitterreiter JG, van de Vijver DA, de Swart RL, Haagmans BL, et al. Intrathecal CD4(+) and CD8(+) T-Cell Responses to Endogenously Synthesized Candidate Disease-Associated Human Autoantigens in Multiple Sclerosis Patients. Eur J Immunol (2016) 46:347–53. doi: 10.1002/eji.201545921
155. Planas R, Santos R, Tomas-Ojer P, Cruciani C, Lutterotti A, Faigle W, et al. GDP-L-Fucose Synthase Is a CD4+ T Cell-Specific Autoantigen in DRB3*02:02 Patients With Multiple Sclerosis. Sci Transl Med (2018) 10(462):eaat4301. doi: 10.1126/scitranslmed.aat4301
156. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B, et al. Ocrelizumab Versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N Engl J Med (2017) 376:221–34. doi: 10.1056/NEJMoa1601277
157. Bar-Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C, et al. Abnormal B-Cell Cytokine Responses a Trigger of T-Cell-Mediated Disease in MS? Ann Neurol (2010) 67:452–61. doi: 10.1002/ana.21939
158. Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, et al. Proinflammatory GM-CSF-Producing B Cells in Multiple Sclerosis and B Cell Depletion Therapy. Sci Transl Med (2015) 7(310):310ra166. doi: 10.1126/scitranslmed.aab4176
159. van Langelaar J, Rijvers L, Janssen M, Wierenga-Wolf AF, Melief MJ, Siepman TA, et al. Induction of Brain-Infiltrating T-Bet-Expressing B Cells in Multiple Sclerosis. Ann Neurol (2019) 86:264–78. doi: 10.1002/ana.25508
160. van Langelaar J, Wierenga-Wolf AF, Samijn JPA, Luijks CJM, Siepman TA, van Doorn PA, et al. The Association of Epstein-Barr Virus Infection With CXCR3(+) B-Cell Development in Multiple Sclerosis: Impact of Immunotherapies. Eur J Immunol (2021) 51:626–33. doi: 10.1002/eji.202048739
161. van Sechel AC, Bajramovic JJ, van Stipdonk MJ, Persoon-Deen C, Geutskens SB, van Noort JM. EBV-Induced Expression and HLA-DR-Restricted Presentation by Human B Cells of Alpha B-Crystallin, A Candidate Autoantigen in Multiple Sclerosis. J Immunol (1999) 162:129–35.
162. Høglund RA, Bremel RD, Homan EJ, Torsetnes SB, Lossius A, Holmøy T. CD4+ T Cells in the Blood of MS Patients Respond to Predicted Epitopes From B Cell Receptors Found in Spinal Fluid. Front Immunol (2020) 11:598. doi: 10.3389/fimmu.2020.00598
163. Warner HB, Carp RI. Multiple Sclerosis and Epstein-Barr Virus. Lancet (1981) 2(8258):1290. doi: 10.1016/s0140-6736(81)91527-0
164. Warner HB, Carp RI. Multiple Sclerosis Etiology–An Epstein-Barr Virus Hypothesis. Med Hypotheses (1988) 1925:93–7. doi: 10.1016/0306-9877(88)90024-2
165. Opsahl ML, Kennedy PG. An Attempt to Investigate the Presence of Epstein Barr Virus in Multiple Sclerosis and Normal Control Brain Tissue. J Neurol (2007) 254:425–30. doi: 10.1007/s00415-006-0316-7
166. Willis SN, Stadelmann C, Rodig SJ, Caron T, Gattenloehner S, Mallozzi SS, et al. Epstein-Barr Virus Infection is Not a Characteristic Feature of Multiple Sclerosis Brain. Brain (2009) 132:3318–28. doi: 10.1093/brain/awp200
167. Peferoen LA, Lamers F, Lodder LN, Gerritsen WH, Huitinga I, Melief J, et al. Epstein Barr Virus is Not a Characteristic Feature in the Central Nervous System in Established Multiple Sclerosis. Brain (2010) 133(Pt 5):e137. doi: 10.1093/brain/awp296
168. Sargsyan SA, Shearer AJ, Ritchie AM, Burgoon MP, Anderson S, Hemmer B, et al. Absence of Epstein-Barr Virus in the Brain and CSF of Patients With Multiple Sclerosis. Neurology (2010) 74:1127–35. doi: 10.1212/WNL.0b013e3181d865a1
169. Torkildsen Ø, Stansberg C, Angelskår SM, Kooi EJ, Geurts JJ, van der Valk P, et al. Upregulation of Immunoglobulin-Related Genes in Cortical Sections From Multiple Sclerosis Patients. Brain Pathol (2010) 20:720–9. doi: 10.1111/j.1750-3639.2009.00343.x
170. Serafini B, Severa M, Columba-Cabezas S, Rosicarelli B, Veroni C, Chiappetta G, et al. Epstein-Barr Virus Latent Infection and BAFF Expression in B Cells in the Multiple Sclerosis Brain: Implications for Viral Persistence and Intrathecal B-Cell Activation. J Neuropathol Exp Neurol (2010) 69:677–93. doi: 10.1097/NEN.0b013e3181e332ec
171. Serafini B, Muzio L, Rosicarelli B, Aloisi F. Radioactive in Situ Hybridization for Epstein-Barr Virus-Encoded Small RNA Supports Presence of Epstein-Barr Virus in the Multiple Sclerosis Brain. Brain (2013) 136(Pt 7):e233. doi: 10.1093/brain/aws315
172. Serafini B, Rosicarelli B, Aloisi F, Stigliano E. Epstein-Barr Virus in the Central Nervous System and Cervical Lymph Node of a Patient With Primary Progressive Multiple Sclerosis. J Neuropathol Exp Neurol (2014) 73:729–31. doi: 10.1097/NEN.0000000000000082
173. Magliozzi R, Serafini B, Rosicarelli B, Chiappetta G, Veroni C, Reynolds R, et al. B-Cell Enrichment and Epstein-Barr Virus Infection in Inflammatory Cortical Lesions in Secondary Progressive Multiple Sclerosis. J Neuropathol Exp Neurol (2013) 72:29–41. doi: 10.1097/NEN.0b013e31827bfc62
174. Serafini B, Scorsi E, Rosicarelli B, Rigau V, Thouvenot E, Aloisi F. Massive Intracerebral Epstein-Barr Virus Reactivation in Lethal Multiple Sclerosis Relapse After Natalizumab Withdrawal. J Neuroimmunol (2017) 307:14–7. doi: 10.1016/j.jneuroim.2017.03.013
175. Tzartos JS, Khan G, Vossenkamper A, Cruz-Sadaba M, Lonardi S, Sefia E, et al. Association of Innate Immune Activation With Latent Epstein-Barr Virus in Active MS Lesions. Neurology (2012) 78:15–23. doi: 10.1212/WNL.0b013e31823ed057
176. Hassani A, Corboy JR, Al-Salam S, Khan G. Epstein-Barr Virus is Present in the Brain of Most Cases of Multiple Sclerosis and May Engage More Than Just B Cells. PloS One (2018) 13(2):e0192109. doi: 10.1371/journal.pone.0192109
177. Moreno MA, Or-Geva N, Aftab BT, Khanna R, Croze E, Steinman L, et al. Molecular Signature of Epstein-Barr Virus Infection in MS Brain Lesions. Neurol Neuroimmunol Neuroinflamm (2018) 5(4):e466. doi: 10.1212/NXI.0000000000000466
178. Aloisi F, Serafini B, Magliozzi R, Howell OW, Reynolds R. Detection of Epstein-Barr Virus and B-Cell Follicles in the Multiple Sclerosis Brain: What You Find Depends on How and Where You Look. Brain (2010) 133(Pt 12):e157. doi: 10.1093/brain/awq223
179. Lassmann H, Niedobitek G, Aloisi F, Middeldorp JM. Neuropromise EBV Working Group. Epstein-Barr Virus in the Multiple Sclerosis Brain: A Controversial Issue–Report on a Focused Workshop Held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain (2011) 134:2772–86. doi: 10.1093/brain/awr197
180. Veroni C, Marnetto F, Granieri L, Bertolotto A, Ballerini C, Repice AM, et al. Immune and Epstein-Barr Virus Gene Expression in Cerebrospinal Fluid and Peripheral Blood Mononuclear Cells From Patients With Relapsing-Remitting Multiple Sclerosis. J Neuroinflamm (2015) 12:132. doi: 10.1186/s12974-015-0353-1
181. Cocuzza CE, Piazza F, Musumeci R, Oggioni D, Andreoni S, Gardinetti M, et al. Quantitative Detection of Epstein-Barr Virus DNA in Cerebrospinal Fluid and Blood Samples of Patients With Relapsing-Remitting Multiple Sclerosis. PloS One (2014) 9(4):e94497. doi: 10.1371/journal.pone.0094497
182. Gronen F, Ruprecht K, Weissbrich B, Klinker E, Kroner A, Hofstetter HH, et al. Frequency Analysis of HLA-B7-Restricted Epstein-Barr Virus-Specific Cytotoxic T Lymphocytes in Patients With Multiple Sclerosis and Healthy Controls. J Neuroimmunol (2006) 180:185–92. doi: 10.1016/j.jneuroim.2006.08.008
183. Lindsey JW, Hatfield LM. Epstein-Barr Virus and Multiple Sclerosis: Cellular Immune Response and Cross-Reactivity. J Neuroimmunol (2010) 229:238–42. doi: 10.1016/j.jneuroim.2010.08.009
184. Höllsberg P, Hansen HJ, Haahr S. Altered CD8+ T Cell Responses to Selected Epstein-Barr Virus Immunodominant Epitopes in Patients With Multiple Sclerosis. Clin Exp Immunol (2003) 132:137–43. doi: 10.1046/j.1365-2249.2003.02114.x
185. Cencioni MT, Magliozzi R, Nicholas R, Ali R, Malik O, Reynolds R, et al. Programmed Death 1 is Highly Expressed on CD8(+) CD57(+) T Cells in Patients With Stable Multiple Sclerosis and Inhibits Their Cytotoxic Response to Epstein-Barr Virus. Immunology (2017) 152:660–76. doi: 10.1111/imm.12808
186. Jørgensen PB, Livbjerg AH, Hansen HJ, Petersen T, Höllsberg P. Epstein-Barr Virus Peptide Presented by HLA-E Is Predominantly Recognized by CD8(bright) Cells in Multiple Sclerosis Patients. PloS One (2012) 7(9):e46120. doi: 10.1371/journal.pone.0046120
187. Ressing ME, van Leeuwen D, Verreck FA, Gomez R, Heemskerk B, Toebes M, et al. Interference With T Cell Receptor-HLA-DR Interactions by Epstein-Barr Virus gp42 Results in Reduced T Helper Cell Recognition. Proc Natl Acad Sci USA (2003) 100:11583–8. doi: 10.1073/pnas.2034960100
188. Ressing ME, van Gent M, Gram AM, Hooykaas MJ, Piersma SJ, Wiertz EJ. Immune Evasion by Epstein-Barr Virus. Curr Top Microbiol Immunol (2015) 391:355–81. doi: 10.1007/978-3-319-22834-1_12
189. Li Q, Cohen JI. Epstein-Barr Virus and the Human Leukocyte Antigen Complex. Curr Clin Microbiol Rep (2019) 6:175–81. doi: 10.1007/s40588-019-00120-9
190. Albanese M, Tagawa T, Buschle A, Hammerschmidt W. MicroRNAs of Epstein-Barr Virus Control Innate and Adaptive Antiviral Immunity. J Virol (2017) 91(16):e01667–16. doi: 10.1128/JVI.01667-16
191. Jurewicz A, Biddison WE, Antel JP. MHC Class I-Restricted Lysis of Human Oligodendrocytes by Myelin Basic Protein Peptide-Specific CD8 T Lymphocytes. J Immunol (1998) 160:3056–9.
192. Giuliani F, Goodyer CG, Antel JP, Yong VW. Vulnerability of Human Neurons to T Cell-Mediated Cytotoxicity. J Immunol (2003) 171:368–79. doi: 10.4049/jimmunol.171.1.368
193. Saxena A, Martin-Blondel G, Mars LT, Liblau RS. Role of CD8 T Cell Subsets in the Pathogenesis of Multiple Sclerosis. FEBS Lett (2011) 585:3758–63. doi: 10.1016/j.febslet.2011.08.047
194. Moseman EA, Blanchard AC, Nayak D, McGavern DB. T Cell Engagement of Crosspresenting Microglia Protects the Brain From a Nasal Virus Infection. Sci Immunol (2020) 5(48):eabb1817. doi: 10.1126/sciimmunol.abb1817
195. Saikali P, Antel JP, Newcombe J, Chen Z, Freedman M, Blain M, et al. NKG2D-Mediated Cytotoxicity Toward Oligodendrocytes Suggests a Mechanism for Tissue Injury in Multiple Sclerosis. J Neurosci (2007) 27:1220–8. doi: 10.1523/JNEUROSCI.4402-06.2007
196. Konjevic Sabolek M, Held K, Beltrán E, Niedl AG, Meinl E, Hohlfeld R, et al. Communication of CD8(+) T Cells With Mononuclear Phagocytes in Multiple Sclerosis. Ann Clin Transl Neurol (2019) 6:1151–64. doi: 10.1002/acn3.783
197. Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Brück W. Acute Axonal Injury in Multiple Sclerosis. Correlation With Demyelination and Inflammation. Brain (2000) 123:1174–83. doi: 10.1093/brain/123.6.1174
198. Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Brück W. Acute Axonal Damage in Multiple Sclerosis Is Most Extensive in Early Disease Stages and Decreases Over Time. Brain (2002) 125:2202–12. doi: 10.1093/brain/awf235
199. Medana I, Martinic MA, Wekerle H, Neumann H. Transection of Major Histocompatibility Complex Class I-Induced Neurites by Cytotoxic T Lymphocytes. Am J Pathol (2001) 159:809–15. doi: 10.1016/S0002-9440(10)61755-5
200. Scheikl T, Pignolet B, Dalard C, Desbois S, Raison D, Yamazaki M, et al. Cutting Edge: Neuronal Recognition by CD8 T Cells Elicits Central Diabetes Insipidus. J Immunol (2012) 188:4731–5. doi: 10.4049/jimmunol.1102998
201. Sauer BM, Schmalstieg WF, Howe CL. Axons are Injured by Antigen-Specific CD8(+) T Cells Through a MHC Class I- and Granzyme B-Dependent Mechanism. Neurobiol Dis (2013) 59:194–205. doi: 10.1016/j.nbd.2013.07.010
202. Sobottka B, Harrer MD, Ziegler U, Fischer K, Wiendl H, Hünig T, et al. Collateral Bystander Damage by Myelin-Directed CD8+ T Cells Causes Axonal Loss. Am J Pathol (2009) 175:1160–6. doi: 10.2353/ajpath.2009.090340
203. Murray PD, Pavelko KD, Leibowitz J, Lin X, Rodriguez M. CD4(+) and CD8(+) T Cells Make Discrete Contributions to Demyelination and Neurologic Disease in a Viral Model of Multiple Sclerosis. J Virol (1998) 72:7320–9. doi: 10.1128/JVI.72.9.7320-7329.1998
204. Dandekar AA, Anghelina D, Perlman S. Bystander CD8 T-Cell-Mediated Demyelination Is Interferon-Gamma-Dependent in a Coronavirus Model of Multiple Sclerosis. Am J Pathol (2004) 164:363–9. doi: 10.1016/s0002-9440(10)63126-4
205. Howe CL, Ure D, Adelson JD, LaFrance-Corey R, Johnson A, Rodriguez M. CD8+ T Cells Directed Against a Viral Peptide Contribute to Loss of Motor Function by Disrupting Axonal Transport in a Viral Model of Fulminant Demyelination. J Neuroimmunol (2007) 188:13–21. doi: 10.1016/j.jneuroim.2007.04.005
206. Kreutzfeldt M, Bergthaler A, Fernandez M, Brück W, Steinbach K, Vorm M, et al. Neuroprotective Intervention by Interferon-γ Blockade Prevents CD8+ T Cell-Mediated Dendrite and Synapse Loss. J Exp Med (2013) 210:2087–103. doi: 10.1084/jem.20122143
207. Deb C, Lafrance-Corey RG, Zoecklein L, Papke L, Rodriguez M, Howe CL. Demyelinated Axons and Motor Function Are Protected by Genetic Deletion of Perforin in a Mouse Model of Multiple Sclerosis. J Neuropathol Exp Neurol (2009) 68:1037–48. doi: 10.1097/NEN.0b013e3181b5417e
208. Wang T, Allie R, Conant K, Haughey N, Turchan-Chelowo J, Hahn K, et al. Granzyme B Mediates Neurotoxicity Through a G-Protein-Coupled Receptor. FASEB J (2006) 20:1209–11. doi: 10.1096/fj.05-5022fje
209. Hu L, Wang T, Gocke AR, Nath A, Zhang H, Margolick JB, et al. Blockade of Kv1.3 Potassium Channels Inhibits Differentiation and Granzyme B Secretion of Human CD8+ T Effector Memory Lymphocytes. PloS One (2013) 8(1):e54267. doi: 10.1371/journal.pone.0054267
210. Haile Y, Carmine-Simmen K, Olechowski C, Kerr B, Bleackley RC, Giuliani F. Granzyme B-inhibitor serpina3n Induces Neuroprotection In Vitro and In Vivo. J Neuroinflamm (2015) 12:157. doi: 10.1186/s12974-015-0376-7
211. Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of Interferon-Gamma as the Lymphokine That Activates Human Macrophage Oxidative Metabolism and Antimicrobial Activity. J Exp Med (1983) 158:670–89. doi: 10.1084/jem.158.3.670
212. Casbon AJ, Long ME, Dunn KW, Allen LA, Dinauer MC. Effects of IFN-γ on Intracellular Trafficking and Activity of Macrophage NADPH Oxidase Flavocytochrome B558. J Leukoc Biol (2012) 92:869–82. doi: 10.1189/jlb.0512244
213. Hodny Z, Reinis M, Hubackova S, Vasicova P, Bartek J. Interferon Gamma/NADPH Oxidase Defense System in Immunity and Cancer. Oncoimmunology (2015) 5(2):e1080416. doi: 10.1080/2162402X.2015.1080416
214. Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, et al. NADPH Oxidase Expression in Active Multiple Sclerosis Lesions in Relation to Oxidative Tissue Damage and Mitochondrial Injury. Brain (2012) 135:886–99. doi: 10.1093/brain/aws012
215. Schuh C, Wimmer I, Hametner S, Haider L, Van Dam AM, Liblau RS, et al. Oxidative Tissue Injury in Multiple Sclerosis is Only Partly Reflected in Experimental Disease Models. Acta Neuropathol (2014) 128:247–66. doi: 10.1007/s00401-014-1263-5
216. Agresti C, D’Urso D, Levi G. Reversible Inhibitory Effects of Interferon-Gamma and Tumour Necrosis Factor-Alpha on Oligodendroglial Lineage Cell Proliferation and Differentiation In Vitro. Eur J Neurosci (1996) 8:1106–16. doi: 10.1111/j.1460-9568.1996.tb01278.x
217. Denic A, Wootla B, Rodriguez M. Cd8(+) T Cells in Multiple Sclerosis. Expert Opin Ther Targets (2013) 17:1053–66. doi: 10.1517/14728222.2013.815726
218. Magliozzi R, Howell OW, Durrenberger P, Aricò E, James R, Cruciani C, et al. Meningeal Inflammation Changes the Balance of TNF Signalling in Cortical Grey Matter in Multiple Sclerosis. J Neuroinflamm (2019) 16:259–019-1650-x. doi: 10.1186/s12974-019-1650-x
219. Toubal A, Nel I, Lotersztajn S, Lehuen A. Mucosal-Associated Invariant T Cells and Disease. Nat Rev Immunol (2019) 19:643–57. doi: 10.1038/s41577-019-0191-y
220. Annibali V, Ristori G, Angelini DF, Serafini B, Mechelli R, Cannoni S, et al. CD161(High)CD8+T Cells Bear Pathogenetic Potential in Multiple Sclerosis. Brain (2011) 134:542–54. doi: 10.1093/brain/awq354
221. Mexhitaj I, Nyirenda MH, Li R, O’Mahony J, Rezk A, Rozenberg A, et al. Abnormal Effector and Regulatory T Cell Subsets in Paediatric-Onset Multiple Sclerosis. Brain (2019) 142:617–32. doi: 10.1093/brain/awz017
222. Miyazaki Y, Miyake S, Chiba A, Lantz O, Yamamura T. Mucosal-Associated Invariant T Cells Regulate Th1 Response in Multiple Sclerosis. Int Immunol (2011) 23:529–35. doi: 10.1093/intimm/dxr047
223. Willing A, Leach OA, Ufer F, Attfield KE, Steinbach K, Kursawe N, et al. CD8⁺ MAIT Cells Infiltrate Into the CNS and Alterations in Their Blood Frequencies Correlate With IL-18 Serum Levels in Multiple Sclerosis. Eur J Immunol (2014) 44:3119–28. doi: 10.1002/eji.201344160
224. Acquaviva M, Bassani C, Sarno N, Dalla Costa G, Romeo M, Sangalli F, et al. Loss of Circulating CD8+ CD161(High) T Cells in Primary Progressive Multiple Sclerosis. Front Immunol (2019) 10:1922. doi: 10.3389/fimmu.2019.01922
225. Ammitzbøll C, von Essen MR, Chow HH, McWilliam O, Holm Hansen R, Sellebjerg F. MAIT Cell Subtypes in Multiple Sclerosis. J Neuroimmunol (2020) 339:577117. doi: 10.1016/j.jneuroim.2019.577117
226. Salou M, Nicol B, Garcia A, Baron D, Michel L, Elong-Ngono A, et al. Neuropathologic, Phenotypic and Functional Analyses of Mucosal Associated Invariant T Cells in Multiple Sclerosis. Clin Immunol (2016) 166-167:1–11. doi: 10.1016/j.clim.2016.03.014
227. Held K, Bhonsle-Deeng L, Siewert K, Sato W, Beltrán E, Schmidt S, et al. αβ T-Cell Receptors From Multiple Sclerosis Brain Lesions Show MAIT Cell-Related Features. Neurol Neuroimmunol Neuroinflamm (2015) 2(4):e107. doi: 10.1212/NXI.0000000000000107
228. Nicol B, Salou M, Vogel I, Garcia A, Dugast E, Morille J, et al. An Intermediate Level of CD161 Expression Defines a Novel Activated, Inflammatory, and Pathogenic Subset of CD8(+) T Cells Involved in Multiple Sclerosis. J Autoimmun (2018) 88:61–74. doi: 10.1016/j.jaut.2017.10.005
229. Steinbach K, Vincenti I, Kreutzfeldt M, Page N, Muschaweckh A, Wagner I, et al. Brain-Resident Memory T Cells Represent an Autonomous Cytotoxic Barrier to Viral Infection. J Exp Med (2016) 213:1571–87. doi: 10.1084/jem.20151916
230. Smolders J, Heutinck KM, Fransen NL, Remmerswaal EBM, Hombrink P, Ten Berge IJM, et al. Tissue-Resident Memory T Cells Populate the Human Brain. Nat Commun (2018) 9(1):4593. doi: 10.1038/s41467-018-07053-9
231. Paik DH, Farber DL. Anti-Viral Protective Capacity of Tissue Resident Memory T Cells. Curr Opin Virol (2020) 46:20–6. doi: 10.1016/j.coviro.2020.09.006
232. Steinbach K, Vincenti I, Merkler D. Resident-Memory T Cells in Tissue-Restricted Immune Responses: For Better or Worse? Front Immunol (2018) 9:2827. doi: 10.3389/fimmu.2018.02827
233. Sinha S, Boyden AW, Itani FR, Crawford MP, Karandikar NJ. CD8(+) T-Cells as Immune Regulators of Multiple Sclerosis. Front Immunol (2015) 6:619. doi: 10.3389/fimmu.2015.00619
234. Correale J, Villa A. Role of CD8+ CD25+ Foxp3+ Regulatory T Cells in Multiple Sclerosis. Ann Neurol (2010) 67:625–38. doi: 10.1002/ana.21944
235. Frisullo G, Nociti V, Iorio R, Plantone D, Patanella AK, Tonali PA, et al. Cd8(+)Foxp3(+) T Cells in Peripheral Blood of Relapsing-Remitting Multiple Sclerosis Patients. Hum Immunol (2010) 71:437–41. doi: 10.1016/j.humimm.2010.01.024
236. Pannemans K, Broux B, Goris A, Dubois B, Broekmans T, Van Wijmeersch B, et al. HLA-E Restricted CD8+ T Cell Subsets Are Phenotypically Altered in Multiple Sclerosis Patients. Mult Scler (2014) 20:790–801. doi: 10.1177/1352458513509703
237. Baughman EJ, Mendoza JP, Ortega SB, Ayers CL, Greenberg BM, Frohman EM, et al. Neuroantigen-Specific CD8+ Regulatory T-Cell Function Is Deficient During Acute Exacerbation of Multiple Sclerosis. J Autoimmun (2011) 36:115–24. doi: 10.1016/j.jaut.2010.12.003
238. Bien CG, Bauer J, Deckwerth TL, Wiendl H, Deckert M, Wiestler OD, et al. Destruction of Neurons by Cytotoxic T Cells: A New Pathogenic Mechanism in Rasmussen’s Encephalitis. Ann Neurol (2002) 51:311–18. doi: 10.1002/ana.10100
239. Bauer J, Elger CE, Hans VH, Schramm J, Urbach H, Lassmann H, et al. Astrocytes Are a Specific Immunological Target in Rasmussen’s Encephalitis. Ann Neurol (2007) 62:67–80. doi: 10.1002/ana.21148
240. Al Nimer F, Jelcic I, Kempf C, Pieper T, Budka H, Sospedra M, et al. Phenotypic and Functional Complexity of Brain-Infiltrating T Cells in Rasmussen Encephalitis. Neurol Neuroimmunol Neuroinflamm (2017) 5(1):e419. doi: 10.1212/NXI.0000000000000419
241. Chang JW, Reyes SD, Faure-Kumar E, Lam SK, Lawlo MW, Leventer RJ, et al. Clonally Focused Public and Private T Cells in Resected Brain Tissue From Surgeries to Treat Children With Intractable Seizures. Front Immunol (2021) 12:664344. doi: 10.3389/fimmu.2021.664344
242. Kleffner I, Dörr J, Ringelstein M, Gross CC, Böckenfeld Y, Schwindt W, et al. Diagnostic Criteria for Susac Syndrome. J Neurol Neurosurg Psychiatry (2016) 87:1287–95. doi: 10.1136/jnnp-2016-314295
243. Gross CC, Meyer C, Bhatia U, Yshii L, Kleffner I, Bauer J, et al. Cd8(+) T Cell-Mediated Endotheliopathy is a Targetable Mechanism of Neuro-Inflammation in Susac Syndrome. Nat Commun (2019) 10(1):5779. doi: 10.1038/s41467-019-13593-5
244. Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke I, Graus F, et al. Immunopathology of Autoantibody-Associated Encephalitides: Clues for Pathogenesis. Brain (2012) 135:1622–38. doi: 10.1093/brain/aws082
245. Laukoter S, Rauschka H, Tröscher AR, Köck U, Saji E, Jellinger K, et al. Differences in T Cell Cytotoxicity and Cell Death Mechanisms Between Progressive Multifocal Leukoencephalopathy, Herpes Simplex Virus Encephalitis and Cytomegalovirus Encephalitis. Acta Neuropathol (2017) 133:613–27. doi: 10.1007/s00401-016-1642-1
246. Cortese I, Reich DS, Nath A. Progressive Multifocal Leukoencephalopathy and the Spectrum of JC Virus-Related Disease. Nat Rev Neurol (2021) 17:37–51. doi: 10.1038/s41582-020-00427-y
247. Jelcic I, Jelcic I, Kempf C, Largey F, Planas R, Schippling S, et al. Mechanisms of Immune Escape in Central Nervous System Infection With Neurotropic JC Virus Variant. Ann Neurol (2016) 79:404–18. doi: 10.1002/ana.24574
248. Bangham CR, Araujo A, Yamano Y, Taylor GP. Htlv-1-Associated Myelopathy/Tropical Spastic Paraparesis. Nat Rev Dis Primers (2015) 1:15012. doi: 10.1038/nrdp.2015.12
249. Nozuma S, Jacobson S. Neuroimmunology of Human T-lymphotropic Virus Type 1-Associated Myelopathy/Tropical Spastic Paraparesis. Front Microbiol (2019) 10:885. doi: 10.3389/fmicb.2019.00885
250. Nagai M, Yamano Y, Brennan MB, Mora CA, Jacobson S. Increased HTLV-I Proviral Load and Preferential Expansion of HTLV-I Tax-Specific CD8+ T Cells in Cerebrospinal Fluid From Patients With HAM/TSP. Ann Neurol (2001) 50:807–12. doi: 10.1002/ana.10065
251. Matsuura E, Kubota R, Tanaka Y, Takashima H, Izumo S. Visualization of HTLV-1-Specific Cytotoxic T Lymphocytes in the Spinal Cords of Patients With HTLV-1-Associated Myelopathy/Tropical Spastic Paraparesis. J Neuropathol Exp Neurol (2015) 74:2–14. doi: 10.1097/NEN.0000000000000141
252. Rincon J, Prieto J, Patarroyo M. Expression of Integrins and Other Adhesion Molecules in Epstein-Barr Virus-Transformed B Lymphoblastoid Cells and Burkitt’s Lymphoma Cells. Int J. Cancer (1992) 51:452–8. doi: 10.1002/ijc.2910510319
253. Nakayama T, Fujisawa R, Izawa D, Hieshima K, Takada K, Yoshie O. Human B Cells Immortalized With Epstein-Barr Virus Upregulate CCR6 and CCR10 and Downregulate CXCR4 and CXCR5. J Virol (2002) 76:3072–7. doi: 10.1128/jvi.76.6.3072-3077.2002
254. Hochberg D, Souza T, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. Acute Infection With Epstein-Barr Virus Targets and Overwhelms the Peripheral Memory B-Cell Compartment With Resting, Latently Infected Cells. J Virol (2004) 78:5194–204. doi: 10.1128/jvi.78.10.5194-5204.2004
255. Recher M, Lang KS, Navarini A, Hunziker L, Lang PA, Fink K, et al. Extralymphatic Virus Sanctuaries as a Consequence of Potent T-Cell Activation. Nat Med (2007) 13:1316–23. doi: 10.1038/nm1670
256. McGinley MP, Goldschmidt CH, Rae-Grant AD. Diagnosis and Treatment of Multiple Sclerosis: A Review. JAMA (2021) 325:765–79. doi: 10.1001/jama.2020.26858
257. Weinshenker BG, Bass B, Karlik S, Ebers GC, Rice GP. An Open Trial of OKT3 in Patients With Multiple Sclerosis. Neurology (1991) 41:1047–52. doi: 10.1212/wnl.41.7.1047
258. van Oosten BW, Lai M, Hodgkinson S, Barkhof F, Miller DH, Moseley IF, et al. Treatment of Multiple Sclerosis With the Monoclonal Anti-CD4 Antibody Cm-T412: Results of a Randomized, Double-Blind, Placebo-Controlled, MR-Monitored Phase II Trial. Neurology (1997) 49:351–7. doi: 10.1212/wnl.49.2.351
259. Baker D, Marta M, Pryce G, Giovannoni G, Schmierer K. Memory B Cells Are Major Targets for Effective Immunotherapy in Relapsing Multiple Sclerosis. EBioMedicine (2017) 16:41–50. doi: 10.1016/j.ebiom.2017.01.042
260. Clement M, Pearson JA, Gras S, van den Berg HA, Lissina A, Llewellyn-Lacey S, et al. Targeted Suppression of Autoreactive CD8(+) T-Cell Activation Using Blocking Anti-CD8 Antibodies. Sci Rep (2016) 6:35332. doi: 10.1038/srep35332
261. van Langelaar J, Rijvers L, Smolders J, van Luijn MM. B and T Cells Driving Multiple Sclerosis: Identity, Mechanisms and Potential Triggers. Front Immunol (2020) 11:760. doi: 10.3389/fimmu.2020.00760
262. Becher B, Giacomini PS, Pelletier D, McCrea E, Prat A, Antel JP. Interferon-Gamma Secretion by Peripheral Blood T-Cell Subsets in Multiple Sclerosis: Correlation With Disease Phase and Interferon-Beta Therapy. Ann Neurol (1999) 45:247–50. doi: 10.1002/1531-8249(199902)45:2<247::AID-ANA16>3.0.CO;2-U
263. Furlan R, Bergami A, Lang R, Brambilla E, Franciotta D, Martinelli V, et al. Interferon-Beta Treatment in Multiple Sclerosis Patients Decreases the Number of Circulating T Cells Producing Interferon-Gamma and Interleukin-4. J Neuroimmunol (2000) 111:86–92. doi: 10.1016/s0165-5728(00)00377-5
264. Sørensen TL, Sellebjerg F. Selective Suppression of Chemokine Receptor CXCR3 Expression by interferon-beta1a in Multiple Sclerosis. Mult Scler (2002) 8:104–7. doi: 10.1191/1352458502ms781oa
265. Dhib-Jalbut S, Sumandeep S, Valenzuela R, Ito K, Patel P, Rametta M. Immune Response During Interferon Beta-1b Treatment in Patients With Multiple Sclerosis Who Experienced Relapses and Those Who Were Relapse-Free in the START Study. J Neuroimmunol (2013) 254:131–40. doi: 10.1016/j.jneuroim.2012.08.012
266. Rizzo F, Giacomini E, Mechelli R, Buscarinu MC, Salvetti M, Severa M, et al. Interferon-β Therapy Specifically Reduces Pathogenic Memory B Cells in Multiple Sclerosis Patients by Inducing a FAS-Mediated Apoptosis. Immunol Cell Biol (2016) 94:886–94. doi: 10.1038/icb.2016.55
267. Hestvik AL, Skorstad G, Price DA, Vartdal F, Holmøy T. Multiple Sclerosis: Glatiramer Acetate Induces Anti-Inflammatory T Cells in the Cerebrospinal Fluid. Mult Scler (2008) 14:749–58. doi: 10.1177/1352458508089411
268. Duda PW, Schmied MC, Cook SL, Krieger JI, Hafler DA. Glatiramer Acetate (Copaxone) Induces Degenerate, Th2-Polarized Immune Responses in Patients With Multiple Sclerosis. J Clin Invest (2000) 105:967–76. doi: 10.1172/JCI8970
269. Karandikar NJ, Crawford MP, Yan X, Ratts RB, Brenchley JM, Ambrozak DR, et al. Glatiramer Acetate (Copaxone) Therapy Induces CD8(+) T Cell Responses in Patients With Multiple Sclerosis. J Clin Invest (2002) 109:641–9. doi: 10.1172/JCI14380
270. Tennakoon DK, Mehta RS, Ortega SB, Bhoj V, Racke MK, Karandikar NJ. Therapeutic Induction of Regulatory, Cytotoxic CD8+ T Cells in Multiple Sclerosis. J Immunol (2006) 176:7119–29. doi: 10.4049/jimmunol.176.11.7119
271. Ireland SJ, Guzman AA, O’Brien DE, Hughes S, Greenberg B, Flores A, et al. The Effect of Glatiramer Acetate Therapy on Functional Properties of B Cells From Patients With Relapsing-Remitting Multiple Sclerosis. JAMA Neurol (2014) 71:1421–8. doi: 10.1001/jamaneurol.2014.1472
272. Guerrera G, Ruggieri S, Picozza M, Piras E, Gargano F, Placido R, et al. EBV-Specific CD8 T Lymphocytes and B Cells During Glatiramer Acetate Therapy in Patients With MS. Neurol Neuroimmunol Neuroinflamm (2020) 7:e876. doi: 10.1212/NXI.0000000000000876
273. Holm Hansen R, Højsgaard Chow H, Christensen JR, Sellebjerg F, von Essen MR. Dimethyl Fumarate Therapy Reduces Memory T Cells and the CNS Migration Potential in Patients With Multiple Sclerosis. Mult Scler Relat Disord (2020) 37:101451. doi: 10.1016/j.msard.2019.101451
274. Høglund RA, Polak J, Vartdal F, Holmøy T, Lossius A. B-Cell Composition in the Blood and Cerebrospinal Fluid of Multiple Sclerosis Patients Treated With Dimethyl Fumarate. Mult Scler Relat Disord (2018) 26:90–5. doi: 10.1016/j.msard.2018.08.032
275. Spencer CM, Crabtree-Hartman EC, Lehmann-Horn K, Cree BA, Zamvil SS. Reduction of CD8(+) T Lymphocytes in Multiple Sclerosis Patients Treated With Dimethyl Fumarate. Neurol Neuroimmunol Neuroinflamm (2015) 2(3):e76. doi: 10.1212/NXI.0000000000000076
276. Longbrake EE, Ramsbottom MJ, Cantoni C, Ghezzi L, Cross AH, Piccio L. Dimethyl Fumarate Selectively Reduces Memory T Cells in Multiple Sclerosis Patients. Mult Scler (2016) 22:1061–70. doi: 10.1177/1352458515608961
277. Lundy SK, Wu Q, Wang Q, Dowling CA, Taitano SH, Mao G, et al. Dimethyl Fumarate Treatment of Relapsing-Remitting Multiple Sclerosis Influences B-Cell Subsets. Neurol Neuroimmunol Neuroinflamm (2016) 3:e211. doi: 10.1212/NXI.0000000000000211
278. Li R, Rezk A, Ghadiri M, Luessi F, Zipp F, Li H, et al. Dimethyl Fumarate Treatment Mediates an Anti-Inflammatory Shift in B Cell Subsets of Patients With Multiple Sclerosis. J Immunol (2017) 198:691–8. doi: 10.4049/jimmunol.1601649
279. Fleischer V, Friedrich M, Rezk A, Bühler U, Witsch E, Uphaus T, et al. Treatment Response to Dimethyl Fumarate Is Characterized by Disproportionate CD8+ T Cell Reduction in MS. Mult Scler (2018) 24:632–41. doi: 10.1177/1352458517703799
280. Mehta D, Miller C, Arnold DL, Bame E, Bar-Or A, Gold R, et al. Effect of Dimethyl Fumarate on Lymphocytes in RRMS: Implications for Clinical Practice. Neurology (2019) 92:e1724–38. doi: 10.1212/WNL.0000000000007262
281. Lückel C, Picard F, Raifer H, Campos Carrascosa L, Guralnik A, Zhang Y, et al. IL-17(+) CD8(+) T Cell Suppression by Dimethyl Fumarate Associates With Clinical Response in Multiple Sclerosis. Nat Commun (2019) 10(1):5722. doi: 10.1038/s41467-019-13731-z
282. Klotz L, Eschborn M, Lindner M, Liebmann M, Herold M, Janoschka C, et al. Teriflunomide Treatment for Multiple Sclerosis Modulates T Cell Mitochondrial Respiration With Affinity-Dependent Effects. Sci Trans Med (2019) 11(490):eaao5563. doi: 10.1126/scitranslmed.aao5563
283. Gandoglia I, Ivaldi F, Laroni A, Benvenuto F, Solaro C, Mancardi G, et al. Teriflunomide Treatment Reduces B Cells in Patients With MS. Neurol Neuroimmunol Neuroinflamm (2017) 4(6):e403. doi: 10.1212/NXI.0000000000000403
284. Kowarik MC, Pellkofer HL, Cepok S, Korn T, Kümpfel T, Buck D, et al. Differential Effects of Fingolimod (FTY720) on Immune Cells in the CSF and Blood of Patients With MS. Neurology (2011) 76:1214–21. doi: 10.1212/WNL.0b013e3182143564
285. Kowarik MC, Astling D, Lepennetier G, Ritchie A, Hemmer B, Owens GP, et al. Differential Effects of Fingolimod and Natalizumab on B Cell Repertoires in Multiple Sclerosis Patients. Neurotherapeutics (2021) 18:364–77. doi: 10.1007/s13311-020-00975-7
286. Mehling M, Brinkmann V, Antel J, Bar-Or A, Goebels N, Vedrine C, et al. FTY720 Therapy Exerts Differential Effects on T Cell Subsets in Multiple Sclerosis. Neurology (2008) 71:1261–7. doi: 10.1212/01.wnl.0000327609.57688.ea
287. Nakamura M, Matsuoka T, Chihara N, Miyake S, Sato W, Araki M, et al. Differential Effects of Fingolimod on B-Cell Populations in Multiple Sclerosis. Mult Scler (2014) 20:1371–80. doi: 10.1177/1352458514523496
288. Blumenfeld S, Staun-Ram E, Miller A. Fingolimod Therapy Modulates Circulating B Cell Composition, Increases B Regulatory Subsets and Production of IL-10 and TGFbeta in Patients With Multiple Sclerosis. J Autoimmun (2016) 70:40–51. doi: 10.1016/j.jaut.2016.03.012
289. Ghadiri M, Rezk A, Li R, Evans A, Giacomini PS, Barnett MH, et al. Pre-Treatment T-Cell Subsets Associate With Fingolimod Treatment Responsiveness in Multiple Sclerosis. Sci Rep (2020) 10(1):356. doi: 10.1038/s41598-019-57114-2
290. Wu Q, Mills EA, Wang Q, Dowling CA, Fisher C, Kirch B, et al. Siponimod Enriches Regulatory T and B Lymphocytes in Secondary Progressive Multiple Sclerosis. JCI Insight (2020) 5(3):e134251. doi: 10.1172/jci.insight.134251
291. Harris S, Tran JQ, Southworth H, Spencer CM, Cree BAC, Zamvil SS. Effect of the Sphingosine-1-Phosphate Receptor Modulator Ozanimod on Leukocyte Subtypes in Relapsing MS. Neurol Neuroimmunol Neuroinflamm (2020) 7(5):e839. doi: 10.1212/NXI.0000000000000839
292. Stüve O, Marra CM, Jerome KR, Cook L, Cravens PD, Cepok S, et al. Immune Surveillance in Multiple Sclerosis Patients Treated With Natalizumab. Ann Neurol (2006) 59:743–7. doi: 10.1002/ana.20858
293. Warnke C, Stettner M, Lehmensiek V, Dehmel T, Mausberg AK, von Geldern G, et al. Natalizumab Exerts a Suppressive Effect on Surrogates of B Cell Function in Blood and CSF. Mult Scler (2015) 21:1036–44. doi: 10.1177/1352458514556296
294. Skarica M, Eckstein C, Whartenby KA, Calabresi PA. Novel Mechanisms of Immune Modulation of Natalizumab in Multiple Sclerosis Patients. J Neuroimmunol (2011) 235:70–6. doi: 10.1016/j.jneuroim.2011.02.010
295. Mellergård J, Edström M, Jenmalm MC, Dahle C, Vrethem M, Ernerudh J. Increased B Cell and Cytotoxic NK Cell Proportions and Increased T Cell Responsiveness in Blood of Natalizumab-Treated Multiple Sclerosis Patients. PloS One (2013) 8(12):e81685. doi: 10.1371/journal.pone.0081685
296. Kaufmann M, Haase R, Proschmann U, Ziemssen T, Akgün K. Real-World Lab Data in Natalizumab Treated Multiple Sclerosis Patients Up to 6 Years Long-Term Follow Up. Front Neurol (2018) 9:1071. doi: 10.3389/fneur.2018.01071
297. Kivisäkk P, Healy BC, Viglietta V, Quintana FJ, Hootstein MA, Weiner HL, et al. Natalizumab Treatment Is Associated With Peripheral Sequestration of Proinflammatory T Cells. Neurology (2009) 72:1922–30. doi: 10.1212/WNL.0b013e3181a8266f
298. Traub JW, Pellkofer HL, Grondey K, Seeger I, Rowold C, Brück W, et al. Natalizumab Promotes Activation and Pro-Inflammatory Differentiation of Peripheral B Cells in Multiple Sclerosis Patients. J Neuroinflamm (2019) 16(1):228. doi: 10.1186/s12974-019-1593-2
299. Jilek S, Jaquiéry E, Hirsch HH, Lysandropoulos A, Canales M, Guignard L, et al. Immune Responses to JC Virus in Patients With Multiple Sclerosis Treated With Natalizumab: A Cross-Sectional and Longitudinal Study. Lancet Neurol (2010) 9:264–72. doi: 10.1016/S1474-4422(10)70006-5
300. Rejdak K, Stelmasiak Z, Grieb P. Cladribine Induces Long Lasting Oligoclonal Bands Disappearance in Relapsing Multiple Sclerosis Patients: 10-Year Observational Study. Mult Scler Relat Disord (2019) 27:117–20. doi: 10.1016/j.msard.2018.10.006
301. Giovannoni G. Cladribine to Treat Relapsing Forms of Multiple Sclerosis. Neurotherapeutics (2017) 14:874–87. doi: 10.1007/s13311-017-0573-4
302. Moser T, Schwenker K, Seiberl M, Feige J, Akgün K, Haschke-Becher E, et al. Long-Term Peripheral Immune Cell Profiling Reveals Further Targets of Oral Cladribine in MS. Ann Clin Transl Neurol (2020) 7:2199–212. doi: 10.1002/acn3.51206
303. Ceronie B, Jacobs BM, Baker D, Dubuisson N, Mao Z, Ammoscato F, et al. Cladribine Treatment of Multiple Sclerosis Is Associated With Depletion of Memory B Cells. J Neurol (2018) 265:1199–209. doi: 10.1007/s00415-018-8830-y
304. Stüve O, Soelberg Soerensen P, Leist T, Giovannoni G, Hyvert Y, Damian D, et al. Effects of Cladribine Tablets on Lymphocyte Subsets in Patients With Multiple Sclerosis: An Extended Analysis of Surface Markers. Ther Adv Neurol Disord (2019) 12:1756286419854986. doi: 10.1177/1756286419854986
305. Comi G, Cook S, Giovannoni G, Rieckmann P, Sørensen PS, Vermersch P, et al. Effect of Cladribine Tablets on Lymphocyte Reduction and Repopulation Dynamics in Patients With Relapsing Multiple Sclerosis. Mult Scler Relat Disord (2019) 29:168–74. doi: 10.1016/j.msard.2019.01.038
306. Baker D, Herrod SS, Alvarez-Gonzalez C, Giovannoni G, Schmierer K. Interpreting Lymphocyte Reconstitution Data From the Pivotal Phase 3 Trials of Alemtuzumab. JAMA Neurol (2017) 74:961–9. doi: 10.1001/jamaneurol.2017.0676
307. Hill-Cawthorne GA, Button T, Tuohy O, Jones JL, May K, Somerfield J, et al. Long Term Lymphocyte Reconstitution After Alemtuzumab Treatment of Multiple Sclerosis. J Neurol Neurosurg Psychiatry (2012) 83:298–304. doi: 10.1136/jnnp-2011-300826
308. Zhang X, Tao Y, Chopra M, Ahn M, Marcus KL, Choudhary N, et al. Differential Reconstitution of T Cell Subsets Following Immunodepleting Treatment With Alemtuzumab (Anti-CD52 Monoclonal Antibody) in Patients With Relapsing-Remitting Multiple Sclerosis. J Immunol (2013) 191:5867–74. doi: 10.4049/jimmunol.1301926
309. Piccio L, Naismith RT, Trinkaus K, Klein RS, Parks BJ, Lyons JA, et al. Changes in B- and T-Lymphocyte and Chemokine Levels With Rituximab Treatment in Multiple Sclerosis. Arch Neurol (2010) 67:707–14. doi: 10.1001/archneurol.2010.99
310. Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab Reduces B Cells and T Cells in Cerebrospinal Fluid of Multiple Sclerosis Patients. J Neuroimmunol (2006) 180:63–70. doi: 10.1016/j.jneuroim.2006.06.029
311. Bar-Or A, Grove RA, Austin DJ, Tolson JM, VanMeter SA, Lewis EW, et al. Subcutaneous Ofatumumab in Patients With Relapsing-Remitting Multiple Sclerosis: The MIRROR Study. Neurology (2018) 90(20):e1805–14. doi: 10.1212/WNL.0000000000005516
312. Palanichamy A, Jahn S, Nickles D, Derstine M, Abounasr A, Hauser SL, et al. Rituximab Efficiently Depletes Increased CD20-Expressing T Cells in Multiple Sclerosis Patients. J Immunol (2014) 193:580–6. doi: 10.4049/jimmunol.1400118
313. Mills EA, Ogrodnik MA, Plave A, Mao-Draayer Y. Emerging Understanding of the Mechanism of Action for Dimethyl Fumarate in the Treatment of Multiple Sclerosis. Front Neurol (2018) 9:5. doi: 10.3389/fneur.2018.00005
314. Bar-Or A, Pachner A, Menguy-Vacheron F, Kaplan J, Wiendl H. Teriflunomide and its Mechanism of Action in Multiple Sclerosis. Drugs (2014) 74:659–74. doi: 10.1007/s40265-014-0212-x
315. Zivadinov R, Ramanathan M, Hagemeier J, Bergsland N, Ramasamy DP, Durfee J, et al. Teriflunomide’s Effect on Humoral Response to Epstein-Barr Virus and Development of Cortical Gray Matter Pathology in Multiple Sclerosis. Mult Scler Relat Disord (2019) 36:101388. doi: 10.1016/j.msard.2019.101388
316. Bilger A, Plowshay J, Ma S, Nawandar D, Barlow EA, Romero-Masters JC, et al. Leflunomide/Teriflunomide Inhibit Epstein-Barr Virus (EBV)-Induced Lymphoproliferative Disease and Lytic Viral Replication. Oncotarget (2017) 8:44266–80. doi: 10.18632/oncotarget.17863
317. Schmierer K, Sørensen PS, Baker D. Highly Effective Disease-Modifying Treatment as Initial MS Therapy. Curr Opin Neurol (2021) 34:286–94. doi: 10.1097/WCO.0000000000000937
318. Roy R, Alotaibi AA, Freedman MS. Sphingosine 1-Phosphate Receptor Modulators for Multiple Sclerosis. CNS Drugs (2021) 35:385–402. doi: 10.1007/s40263-021-00798-w
319. Giovannoni G, Hawkes C, Waubant E, Lublin F. The ‘Field Hypothesis’: Rebound Activity After Stopping Disease-Modifying Therapies. Mult Scler Relat Disord (2017) 15:A1–2. doi: 10.1016/j.msard.2017.06.005
320. Lenhard T, Biller A, Mueller W, Metz I, Schönberger J, Wildemann B. Immune Reconstitution Inflammatory Syndrome After Withdrawal of Natalizumab? Neurology (2010) 75:831–3. doi: 10.1212/WNL.0b013e3181f07362
321. Rigau V, Mania A, Béfort P, Carlander B, Jonquet O, Lassmann H, et al. Lethal Multiple Sclerosis Relapse After Natalizumab Withdrawal. Neurology (2012) 79:2214–6. doi: 10.1212/WNL.0b013e318275979d
322. Larochelle C, Metz I, Lécuyer MA, Terouz S, Roger M, Arbour N, et al. Immunological and Pathological Characterization of Fatal Rebound MS Activity Following Natalizumab Withdrawal. Mult Scler (2017) 23:72–81. doi: 10.1177/1352458516641775
323. Wiendl H, Kieseier B. Multiple Sclerosis: Reprogramming the Immune Repertoire With Alemtuzumab in MS. Nat Rev Neurol (2013) 9:125–6. doi: 10.1038/nrneurol.2013.2
324. Hawker K, O’Connor P, Freedman MS, Calabresi PA, Antel J, Simon J, et al. Rituximab in Patients With Primary Progressive Multiple Sclerosis: Results of a Randomized Double-Blind Placebo-Controlled Multicenter Trial. Ann Neurol (2009) 66:460–71. doi: 10.1002/ana.21867
325. Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab Versus Placebo in Primary Progressive Multiple Sclerosis. N Engl J Med (2017) 376:209–20. doi: 10.1056/NEJMoa1606468
326. Sabatino JJ Jr, Zamvil SS, Hauser SL. B-Cell Therapies in Multiple Sclerosis. Cold Spring Harb Perspect Med (2019) 9(2):a032037. doi: 10.1101/cshperspect.a032037
327. von Essen MR, Ammitzbøll C, Hansen RH, Petersen ERS, McWilliam O, Marquart HV, et al. Proinflammatory CD20+ T Cells in the Pathogenesis of Multiple Sclerosis. Brain (2019) 142:120–32. doi: 10.1093/brain/awy301
328. Pei Y, Wong JHY, Robertson ES. Targeted Therapies for Epstein-Barr Virus-Associated Lymphomas. Cancers (Basel) (2020) 12(9):2565. doi: 10.3390/cancers12092565
329. Petereit HF, Rubbert-Roth A. Rituximab Levels in Cerebrospinal Fluid of Patients With Neurological Autoimmune Disorders. Mult Scler (2009) 15:189–92. doi: 10.1177/1352458508098268
330. Montalban X, Arnold DL, Weber MS, Staikov I, Piasecka-Stryczynska K, Willmer J, et al. Placebo-Controlled Trial of An Oral BTK Inhibitor in Multiple Sclerosis. N Engl J Med (2019) 380:2406–17. doi: 10.1056/NEJMoa1901981
331. Bhargava P, Kim S, Reyes AA, Grenningloh R, Boschert U, Absinta M, et al. Imaging Meningeal Inflammation in CNS Autoimmunity Identifies a Therapeutic Role for BTK Inhibition. Brain (2021) awab045. doi: 10.1093/brain/awab045
332. Kosowicz JG, Lee J, Peiffer B, Guo Z, Chen J, Liao G, et al. Drug Modulators of B Cell Signaling Pathways and Epstein-Barr Virus Lytic Activation. J Virol (2017) 91(16):e00747–17. doi: 10.1128/JVI.00747-17
333. Pender MP, Csurhes PA, Smith C, Douglas NL, Neller MA, Matthews KK, et al. Epstein-Barr Virus-Specific T Cell Therapy for Progressive Multiple Sclerosis. JCI Insight (2018) 3(22):e124714. doi: 10.1172/jci.insight.124714
334. Bar-Or A, Pender MP, Khanna R, Steinman L, Hartung HP, Maniar T, et al. Epstein-Barr Virus in Multiple Sclerosis: Theory and Emerging Immunotherapies. Trends Mol Med (2020) 26:296–310. doi: 10.1016/j.molmed.2021.01.004
335. Endriz J, Ho PP, Steinman L. Time Correlation Between Mononucleosis and Initial Symptoms of MS. Neurol Neuroimmunol Neuroinflamm (2017) 4(3):e308. doi: 10.1212/NXI.0000000000000308
336. Langer-Gould A, Wu J, Lucas R, Smith J, Gonzales E, Amezcua L, et al. Epstein-Barr Virus, Cytomegalovirus, and Multiple Sclerosis Susceptibility: A Multiethnic Study. Neurology (2017) 89:1330–37. doi: 10.1212/WNL.0000000000004412
337. Munger KL, Hongell K, Cortese M, Åivo J, Soilu-Hänninen M, Surcel HM, et al. Epstein-Barr Virus and Multiple Sclerosis Risk in the Finnish Maternity Cohort. Ann Neurol (2019) 86:436–42. doi: 10.1002/ana.25532
338. Hedström AK, Huang J, Michel A, Butt J, Brenner N, Hillert J, et al. High Levels of Epstein-Barr Virus Nuclear Antigen-1-Specific Antibodies and Infectious Mononucleosis Act Both Independently and Synergistically to Increase Multiple Sclerosis Risk. Front Neurol (2020) 10:1368. doi: 10.3389/fneur.2019.01368
339. Biström M, Jons D, Engdahl E, Gustafsson R, Huang J, Brenner N, et al. Epstein-Barr Virus Infection After Adolescence and Human Herpesvirus 6A as Risk Factors for Multiple Sclerosis. Eur. J Neurol (2021) 28:579–86. doi: 10.1111/ene.14597
340. Ricigliano VA, Handel AE, Sandve GK, Annibali V, Ristori G, Mechelli R, et al. EBNA2 Binds to Genomic Intervals Associated With Multiple Sclerosis and Overlaps With Vitamin D Receptor Occupancy. PloS One (2015) 10:e0119605. doi: 10.1371/journal.pone.0119605
341. Harley JB, Chen X, Pujato M, Miller D, Maddox A, Forney C, et al. Transcription Factors Operate Across Disease Loci, With EBNA2 Implicated in Autoimmunity. Nat Genet (2018) 50:699–707. doi: 10.1038/s41588-018-0102-3
342. Afrasiabi A, Parnell GP, Fewings N, Schibeci SD, Basuki MA, Chandramohan R, et al. Evidence From Genome Wide Association Studies Implicates Reduced Control of Epstein-Barr Virus Infection in Multiple Sclerosis Susceptibility. Genome Med (2019) 11(1):26. doi: 10.1186/s13073-019-0640-z
343. Olsson T. Epstein Barr Virus Infection and Immune Defense Related to HLA-DR15: Consequences for Multiple Sclerosis. Eur J Immunol (2020) 51:56–9. doi: 10.1002/eji.202049030
344. Menegatti J, Schub D, Schäfer M, Grässer FA, Ruprecht K. Hla-DRB1*15:01 is a Coreceptor for Epstein-Barr Virus, Linking Genetic and Environmental Risk Factors for Multiple Sclerosis. Eur J Immunol (2021). doi: 10.1002/eji.202149179
345. Lycke J. Trials of Antivirals in the Treatment of Multiple Sclerosis. Acta Neurol Scand (2017) 136(Suppl 201):45–8. doi: 10.1111/ane.12839
346. Drosu NC, Edelman ER, Housman DE. Could Antiretrovirals be Treating EBV in MS? A Case Report. Mult Scler Relat Disord (2018) 22:19–21. doi: 10.1016/j.msard.2018.02.029
347. Cohen JI. Vaccine Development for Epstein-Barr Virus. Adv Exp Med Biol (2018) 1045:477–93. doi: 10.1007/978-981-10-7230-7_22
Keywords: multiple sclerosis, CD8 T cells, B cells, Epstein-Barr virus (EBV), anti-EBV immunity
Citation: Veroni C and Aloisi F (2021) The CD8 T Cell-Epstein-Barr Virus-B Cell Trialogue: A Central Issue in Multiple Sclerosis Pathogenesis. Front. Immunol. 12:665718. doi: 10.3389/fimmu.2021.665718
Received: 08 February 2021; Accepted: 07 June 2021;
Published: 07 July 2021.
Edited by:
Pentti Tienari, University of Helsinki, FinlandReviewed by:
Trygve Holmøy, Akershus University Hospital, NorwayBert A. ‘T Hart, University Medical Center Groningen, Netherlands
Copyright © 2021 Veroni and Aloisi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Aloisi, ZnJhbmNlc2NhLmFsb2lzaUBpc3MuaXQ=