 Teresa Rodriguez-Calvo
Teresa Rodriguez-Calvo James D. Johnson
James D. Johnson Lut Overbergh
Lut Overbergh Jessica L. Dunne
Jessica L. Dunne- 1Institute of Diabetes Research, Helmholtz Zentrum Muenchen, German Research Center for Environmental Health, Munich, Germany
- 2Diabetes Research Group, Department of Cellular and Physiological Sciences, Life Sciences Institute, University of British Columbia, Vancouver, BC, Canada
- 3Laboratory Clinical and Experimental Endocrinology, KU Leuven, Leuven, Belgium
- 4Janssen Research and Development, LLC, Raritan, NJ, United States
The mechanisms underlying type 1 diabetes (T1D) pathogenesis remain largely unknown. While autoantibodies to pancreatic beta-cell antigens are often the first biological response and thereby a useful biomarker for identifying individuals in early stages of T1D, their role in T1D pathogenesis is not well understood. Recognition of these antigenic targets by autoreactive T-cells plays a pathological role in T1D development. Recently, several beta-cell neoantigens have been described, indicating that both neoantigens and known T1D antigens escape central or peripheral tolerance. Several questions regarding the mechanisms by which tolerance is broken in T1D remain unanswered. Further delineating the timing and nature of antigenic responses could allow their use as biomarkers to improve staging, as targets for therapeutic intervention, and lead to a better understanding of the mechanisms leading to loss of tolerance. Multiple factors that contribute to cellular stress may result in the generation of beta-cell derived neoepitopes and contribute to autoimmunity. Understanding the cellular mechanisms that induce beta-cells to produce neoantigens has direct implications on development of therapies to intercept T1D disease progression. In this perspective, we will discuss evidence for the role of neoantigens in the pathogenesis of T1D, including antigenic responses and cellular mechanisms. We will additionally discuss the pathways leading to neoepitope formation and the cross talk between the immune system and the beta-cells in this regard. Ultimately, delineating the timing of neoepitope generation in T1D pathogenesis will determine their role as biomarkers as well as therapeutic targets.
The Evolving Understanding of the Pathogenesis of Human T1D
The immune system plays a pivotal role in type 1 diabetes (T1D), as demonstrated by numerous studies and experiments in vitro, ex vivo, in animal models, and in humans (1, 2). However, our understanding of the disease continues to evolve, with a greater recent appreciation for the role of pancreatic beta-cell factors and beta-cells themselves in disease etiology (3, 4). Our knowledge of the immunopathology of T1D is incomplete, partially due to the difficult access to human pancreas samples. This limitation has been partially overcome in recent years thanks to the emergence of several large tissue biobanks like the Network for Pancreatic Organ Donors with Diabetes (nPOD) (5, 6) and the Exeter Archival Diabetes Biobank (EADB) (7), which now permit the investigation of immune cell populations in the human pancreas. The presence of immune cells in the islets, known as insulitis, is a hallmark of T1D (8–11). The importance of CD8+ T-cells in T1D is evident by their abundance in islets that have remaining beta-cells, as well as in those with only a few beta-cells left. CD8+ T-cells are also found in the exocrine portion of the pancreas in individuals with T1D, even when beta-cells are lost (12). While CD4+ T-cells are also present in the islets, they are not considered a major component of the immune infiltrate in established diabetes; this is not unexpected given the more prominent role of CD4+ T-cells in disease initiation (13) rather than in disease progression/amplification. In addition, their potential role in sustaining the effector functions of CD8+ T-cells should not be neglected (14). Both CD4+ and CD8+ T-cell populations decline with beta-cell loss, suggesting that their presence is driven by a beta-cell antigen (11, 12, 15). However, the exact role infiltrating T-cells play in the pathogenesis of the disease remains to be determined both in terms of specificity and function.
Antigen-specific CD8+ T-cells recognizing diverse islet antigens have been detected in the pancreas of individuals with T1D (16). T-cells with single specificity were detected early in the disease process, whereas in long-standing donors, islets usually contained multiple islet-reactive specificities indicating epitope spreading (16). Interestingly, in recent onset cases, different islets harbored different reactivities, which could reflect different stages of the autoimmune process. More recently, a high proportion of preproinsulin (PPI) specific cells have been detected in the islets of donors with T1D (17, 18), confirming previous data obtained from blood samples and highlighting the role of PPI as one of the most prominent antigens in disease pathogenesis (15, 19–22). Attempts to detect neoantigens in situ have not been reported so far but are on the horizon. Characterizing the frequency and localization of neoantigens at different disease stages therefore remains an important goal.
The Expanding Catalogue of Epitopes With Putative Roles in T1D
The exact mechanisms and timing of the antigenic events, the initial loss of tolerance, as well as the role of autoimmunity to both native and modified epitopes in the pathogenesis of T1D remains unknown. Recently, a comprehensive overview of the known T1D epitopes and neoepitopes was published (22). Sixteen CD8+ T-cell conventional epitopes have been identified and five of these are contained within the major known antigens insulin, glutamate decarboxylase (GAD), insulinoma-associated antigen 2 (I-A2), zinc-transporter 8 (ZnT8) and islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP). CD8+ T-cells reactive against epitopes from islet amyloid polypeptide (IAPP), insulin gene enhancer protein (ISL1), urocortin-3 (UCN3) and SLC30A8 (also known as ZnT8) have been also identified in the pancreas (23, 24). Their frequencies were higher in T1D donors compared to non-diabetic donors and their phenotype was predominantly antigen-experienced. In addition, CD8+ T-cell reactivity against other granule proteins such as prohormone convertase 2 (PCSK2), secretogranin 3 (SCG3) and 5 (SCG5) have been recently reported, highlighting the immunogenic potential of beta-cell granule proteins (25). The specificity of islet infiltrating T-cells directly sorted or grown from individual islets isolated from donors with T1D has been also investigated (26). Several CD4+ T-cell clones were obtained, which reacted against proinsulin, GAD65 and chromogranin A (ChgA). CD8+ T-cell clones recognized epitopes from insulin, IA-2 and IGRP. The differential antigenic drivers, including number of targets, diversity of epitopes, environmental triggers, host genetics and even clinical age, may contribute to these varying antigen recognition and disease pathways (27).
Generation of Neoepitopes Through Post-Translational Modifications
In the case of T1D, autoantibodies to native proteins are highly prognostic of future disease but there is little evidence of their pathogenic role (28–30). While loss of tolerance to insulin has long been thought to be involved early in disease pathogenesis, responses to modified proteins may add to disease heterogeneity, in terms of variations in risk and rate of progression to clinical T1D (31). However, clinical evidence has shown that not all cases of T1D start with reactivity to insulin, and reactivities to different non-conventional antigens may be due to different clinical and pathogenic features (32).
PTMs are part of normal physiological processes. However, they can also be formed as a result of an inflammatory assault restricted to inflamed tissues, and as such, be associated with autoimmunity. As neoepitopes are exclusively expressed in the peripheral target tissue, and not present in their modified form in the thymus, their escape from thymic deletion through negative selection in medullary thymic epithelial (mTEC) cells accounts for the lack of central tolerance to PTMs (33, 34). In the context of T1D, several neoantigens generated through PTMs have been described (Figure 1). In some cases, autoantibodies against modified proteins can be both pathogenic and predictors of disease onset (35). Antibodies directed against citrullinated proteins can predate clinical onset of rheumatoid arthritis (RA) by up to two decades (36). The most extensively studied enzymatically mediated PTMs are citrullination, the conversion of arginine into citrulline residues, and deamidation, the conversion of glutamine into glutamic acid residues. These PTMs are mediated by peptidylarginine deiminase (PAD) and tissue transglutaminase (TGM) enzymes, respectively. Citrullinated proteins are preferentially bound by RA-susceptible HLA class II and presented to T-cells, implicating them in disease pathogenesis (37). With similar HLA-susceptible haplotypes shared between RA and T1D, citrullination of multiple proteins has also been implicated in T1D pathogenesis (38, 39). Autoantibodies and circulating and islet infiltrating CD4+ T-cells have been found to react against citrullinated glucose-regulated protein 78 (GRP78) epitopes in T1D individuals (38, 40), following up on earlier observations in NOD mice (41). In addition, circulating CD4+ T-cells reactive against citrullinated GAD65 (39) and islet CD4+ T-cells reactive against citrullinated IAPP have been detected in people with T1D (40). Deamidated peptides, in which glutamine was converted into glutamic acid, were described in both murine T1D (42) and human T1D (39, 43), with several autoantigens identified (39, 42–44).
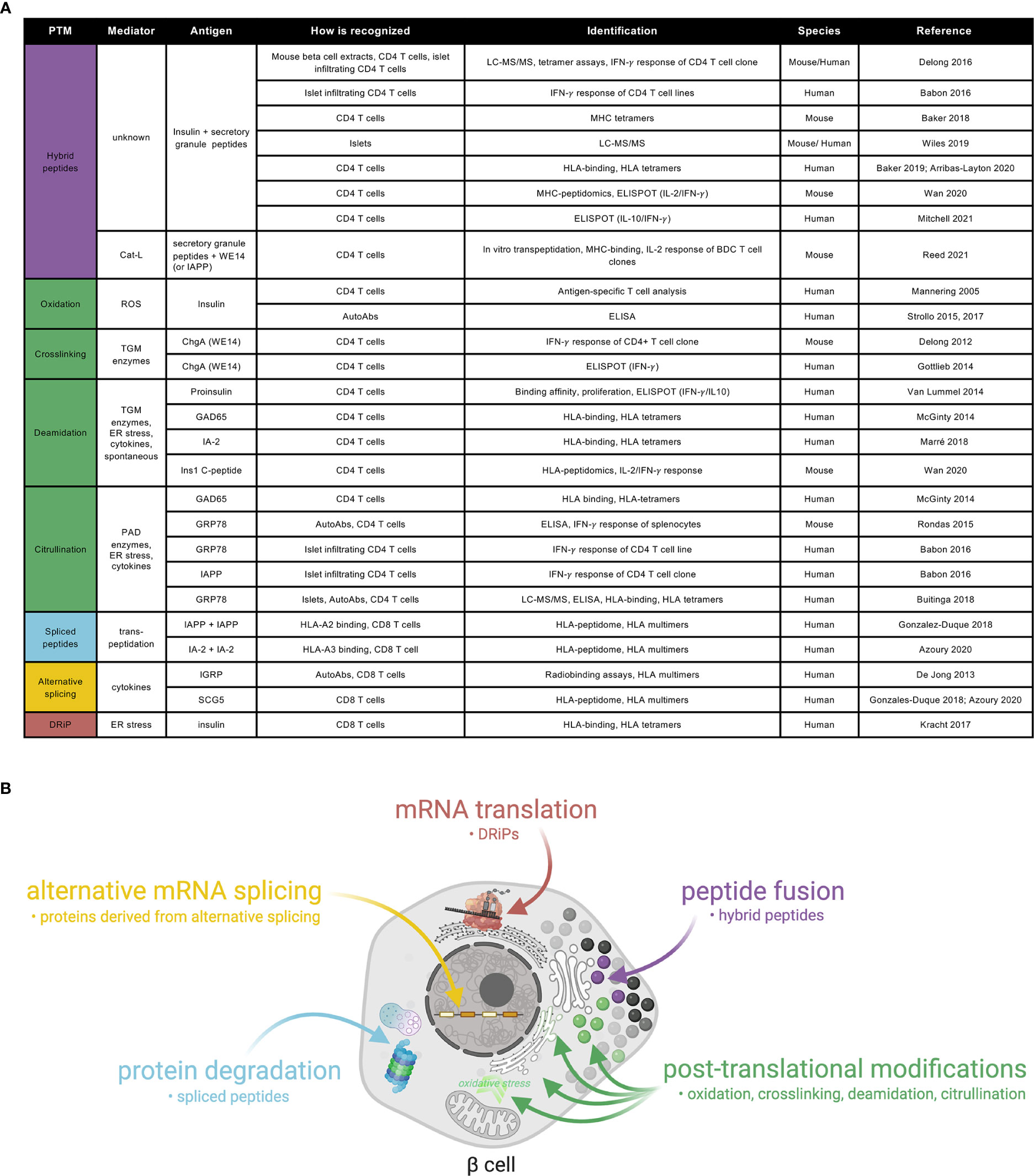
Figure 1 A catalogue of neoepitopes and their production sites/mechanisms in beta-cells. (A) Referenced list of known neoepitopes in type 1 diabetes where the type of PTM, the potential mediator, the antigen and how it is recognized and identified, the species in which it was identified and the reference to the original publication are shown. Cat-L, cathepsin L; ROS, reactive oxygen species; AutoAbs, autoantibodies; TGM, tissue transglutaminase; ChgA, chromogranin A; ER, endoplasmic reticulum; PAD, peptidylarginine deiminase; GAD: glutamate decarboxylase; IA-2: insulinoma-associated antigen 2; ZnT8, zinc-transporter 8; IGRP, islet-specific glucose-6-phosphatase catalytic subunit-related protein; GRP78, glucose-regulated protein 78; IAPP, islet amyloid polypeptide; SCG5, secretogranin 5. (B) Schematic representation of a beta-cell showing the current view on sub-cellular origin of specific classes of neoepitopes, as well as the types of products produced.
Oxidative post-translational modifications and cross-linking of proteins are important mechanisms that may contribute to autoreactivity in T1D (Figure 1). TGM-mediated crosslinking of the naturally processed ChgA cleavage product WE14 increased its immunogenicity, eliciting strong CD4 T-cell responses both in NOD mice (45) and T1D patients (46). Oxidation of the insulin A-chain, resulting in a disulfide bond formation between two adjacent cysteines, was shown to be responsible for recognition of the peptide by a T-cell clone isolated from the circulation of a T1D patient (47). More recently, autoantibodies against oxidized insulin were detected in prediabetic children (48) and in recently diagnosed T1D subjects (49).
Hybrid insulin peptides (HIPs) are also potential neoantigens, formed through a covalent cross-linking reaction between the C-terminal carboxylic acid group of proinsulin fragments and the N-terminal amine group of other secretory granule peptides (26). HIPs have been identified by LC-MS/MS in human (50) and mouse islets (51), as well as in the mouse MHC-peptidome (42). An increasing number of studies have shown autoreactive responses against such HIPs, both in NOD mice and in human T1D (26, 52). CD4+ T-cells isolated from pancreatic islets recognized different proinsulin C-peptide fragments fused to IAPP1, IAPP2, neuropeptide-Y or insulin A chain (40, 51). Increased reactivity to several HIPs was shown in peripheral blood of T1D patients (53, 54), Furthermore, in genetically at risk individuals, HIPs were detectable, and were shown to have a predominantly pro-inflammatory profile in those that progressed to developing disease (55), making them interesting candidates for novel biomarkers. Finally, a recent report suggests that transpeptidation of beta-cell antigens, mediated by cathepsin L, generates chimeric epitopes through fusion of secretory granule proteins with WE14, for diabetogenic CD4 T-cells (56). In regard to HLA class I epitopes, spliced peptides, generated in the proteasome through a process referred to as transpeptidation by which two different regions of a protein or of two different proteins are fused (57, 58), have been identified in a human beta-cell line by HLA-peptidomics (21, 25) and were recognized by circulating and pancreas-infiltrating CD8+ T-cells from T1D donors. Furthermore, defective ribosomal products (DRIPs) (24) are also regarded as potential neoepitopes against which CD8+ T-cells were shown to be reactive in human subjects.
Beta-cell Stress as a Contributor to Neoepitope Generation
There are many factors that could theoretically lead to the production of and reactivity to autoantigens in a subset of beta-cells (Figure 1). Beta-cells could produce a modified protein via genetic or epigenetic up-regulation of transcription, or increased translation. Alternatively, errors in mRNA transcription, splicing, or translation processes could generate neoantigens from modified proteins (24, 59), and they could build up as a result of impaired quality control of the modified protein in the endoplasmic reticulum (ER), post-ER compartments, or during proteosomal degradation (Figure 1). Autoantigens may also result from increased expression or activity of PTM modifying enzymes (44). In the case of insulin, the T1D at-risk allele at rs3842753 (60–62) was reported to increase insulin production in a small number of human pancreata (60, 63, 64), a result supported by recent meta-analysis of single-cell RNA sequencing data (65). One might speculate that increased insulin production could both increase neoepitope production and beta-cell vulnerability to ER stress (66). This may be a contributing factor to the increased risk of T1D development observed with childhood obesity (67).
Increasing evidence points to the beta-cell itself as an active player in mediating such processes, thereby participating in its own destruction (3). Beta-cells have a highly developed ER, making it possible for individual cells to react rapidly to changes in metabolic demand and produce enormous amounts of insulin in a short time. On the other hand, this highly specialized secretory task makes the most active beta-cells highly vulnerable to ER stress, which is present to some degree even in normal basal conditions (66). When the demand for protein synthesis and folding overwhelms the capacity of the ER, the cell-autonomous Unfolded Protein Response (UPR) is initiated, aiming to restore ER homeostasis (68). This UPR response is mediated by three UPR ‘sensors’, which are inactive in physiological conditions through their association with the abundant ER chaperone GRP78. With an increased load of unfolded proteins in the ER, GRP78 is released from these UPR sensors, thereby initiating the UPR. When an excessive level of stress is maintained, the UPR fails, and beta-cell apoptosis is triggered (69, 70). ER stress has also been implicated in the generation of neoantigens (71), with increasing recognition of autoreactive T-cell clones specific for deamidated peptides in multiple experimental systems (44, 72). Of importance, such increased immunogenicity was observed when beta-cells were stressed with thapsigargin, but not with tunicamycin, an ER stressor acting through the blocking of glycoprotein synthesis. A similar increase in activity of TGM2 upon inflammatory stress with cytokines has been observed in rodent MIN6 cells and was associated with an increased number of deamidated peptides. Moreover, an increase in non-enzymatically mediated deamidations was observed in this model upon cytokine exposure (73). Finally, insulin DRIP polypeptides increase by Ca-2+ -mediated ER stress, shown by exposure of INS-DRiP-GFP transfected cells to thapsigargin (24).
As to the role of alternative splicing, it has been shown that the beta-cell alternative spliced repertoire is largely affected by the pro-inflammatory cytokines, interleukin-1β (IL1β) and interferon-γ (IFNγ), changing the expression of more than 30 RNA binding proteins, thereby affecting the splicing of more than 3000 genes involved in beta-cell function and survival (3, 59). In addition, PTMs, alternative splicing and first exon usage are induced by interferon-1α (IFNα) (74). The fact that such transcriptional regulation could lead to neoantigen formation was shown for IGRP, with generation of autoantibodies and CD8 T-cells against a pancreas specific IGRP alternative spliced form (75). Recent evidence further showed that the ‘alternative splicing signature’ is also seen in the immunopeptidome of HLA-A2 and HLA-A3 restricted epitopes, leading to the generation of islet-reactive CD8+ T-cells both in T1D patients and healthy subjects (21, 25). All these studies point to a role for ER stress in increasing the prevalence of a variety PTMs where pro-inflammatory cytokines are likely to expand the repertoire of proteins and transcripts generated by beta-cells.
Environmental Factors in the Generation of Neoautoantigens
Viruses and other environmental triggers have been implicated in T1D and may not only contribute to beta-cell stress and production of neoepitopes, but may be responsible for antigen-specific targeting. One such potential pathway is molecular mimicry, where viruses, microbiota or other environmental targets express epitopes similar to those expressed on beta-cells. Cross-reactive T-cells against these epitopes have the potential to eliminate both the environmental stimulus (i.e. viruses) and pancreatic beta-cells (76–78). Cross-reactivity between epitopes present in Coxsackievirus B (CVB) and GAD65 have been reported (79, 80). Furthermore, a dominant epitope present in IA-2 elicited T-cell responses in relatives and shared sequence similarity with a protein of human rotavirus (81). This IA-2 epitope also had some identity and similarity to sequences in Dengue, cytomegalovirus, measles, hepatitis C, and canine distemper viruses, and the bacterium Haemophilus influenzae. Interestingly, two other IA-2 epitopes were similar to amino acid sequences in milk, wheat, and bean proteins (81). More recently, ZnT8-reactive CD8+ T-cell clonotypes were found to cross-recognize a Bacteroides stercoris mimotope (23). Based on this evidence, environmental factors have the potential to elicit autoreactive immune responses. In this scenario, environmental cues could be a requirement or a contributing factor for neoepitope formation in the pancreas.
Does Neoepitope Generation Lead to Autoimmunity and Beta-Cell Killing in Type 1 Diabetes?
The identification and timing of T-cells reactive against antigens in the pancreas remains a challenging task. Further studies are needed, as illustrated by the apparent lack of correlation between the presence of antigen-specific cells in the periphery and in the pancreas (23). Given that most cells present in the islets have unknown reactivities, it becomes clear that we might be only looking at the tip of the autoreactive iceberg. Models indicate that only 1-2% of antigen-specific cells are enough to achieve effective killing (82). On the other hand, if this is the case, why does it take so long for T-cells to kill their target? It could be that T-cell access to islets is asynchronous due to the expression of different antigens in different islets, at different times. Or perhaps T-cells are not attracted to a given islet unless there is inflammation or a triggering event locally, one that might be able to generate neoantigens. Furthermore, there is little clinical evidence to suggest whether these processes are taking place before disease onset, at a later stage of the disease, or both. A recent longitudinal study on individuals at-risk indicate the presence of neoepitopes in early stages of T1D. Individuals progressing to T1D showed a predominant pro-inflammatory T-cell reactivity against few of the HIPs analyzed (55).
It is clear that individuals carrying certain HLAs are prone to autoreactivity. In this context, it is tempting to speculate that the fragility of beta-cells and their susceptibility to environmental insults and conditions of incremental stress are likely to further unbalance an already compromised genetic system. Considering that neoepitopes are likely to arise before or during disease development, we envision a scenario in which beta-cells themselves actively contribute to neoepitope formation. The upregulation of HLA-I in the islets prior to clinical diagnosis is a good indicator of the potential capacity of beta-cells to present self-epitopes to the immune system, a phenomenon that is not well understood, but that many are actively investigating.
In this regard, and closing the speculative circle, we hypothesize that an environmental insult (i.e. viral infection/s) could induce an anti-viral response and the local production of cytokines (i.e. type I interferons). Anti-viral response molecules can induce the upregulation of HLA-I. At the same time, viruses could induce a translational arrest in beta-cells, hampering insulin production. Also, other forms of environmental stress, such as chemicals, dietary components may cause an initial trigger leading to ER or oxidative stress.
These environmental insults may lead to the formation of a first wave of non-conventional proteins. Impaired clearance of such stressed or dying beta-cells expressing modified proteins will cause activation of APCs and presentation of neoepitopes to CD4+ T-cells, which in turn can trigger several immune responses, including B cell activation with autoantibody production, and the activation of antigen-specific effector T cells that can directly kill beta-cells presenting modified islet peptides. This first cascade of immune activation may in this way cause further beta-cell stress or death, generating an autoreactive loop, with further modification of beta-cell proteins and disease exacerbation. Moreover, epitope spreading to native epitopes may further amplify the immune response (Yang et al, submitted). This proposed model could provide evidence for a role of neoepitopes both in initiation and exacerbation of disease (Figure 2A). It may indeed be that different ways of stress are needed to activate specific types of PTMs, at different times and with unknown duration, during the disease course.
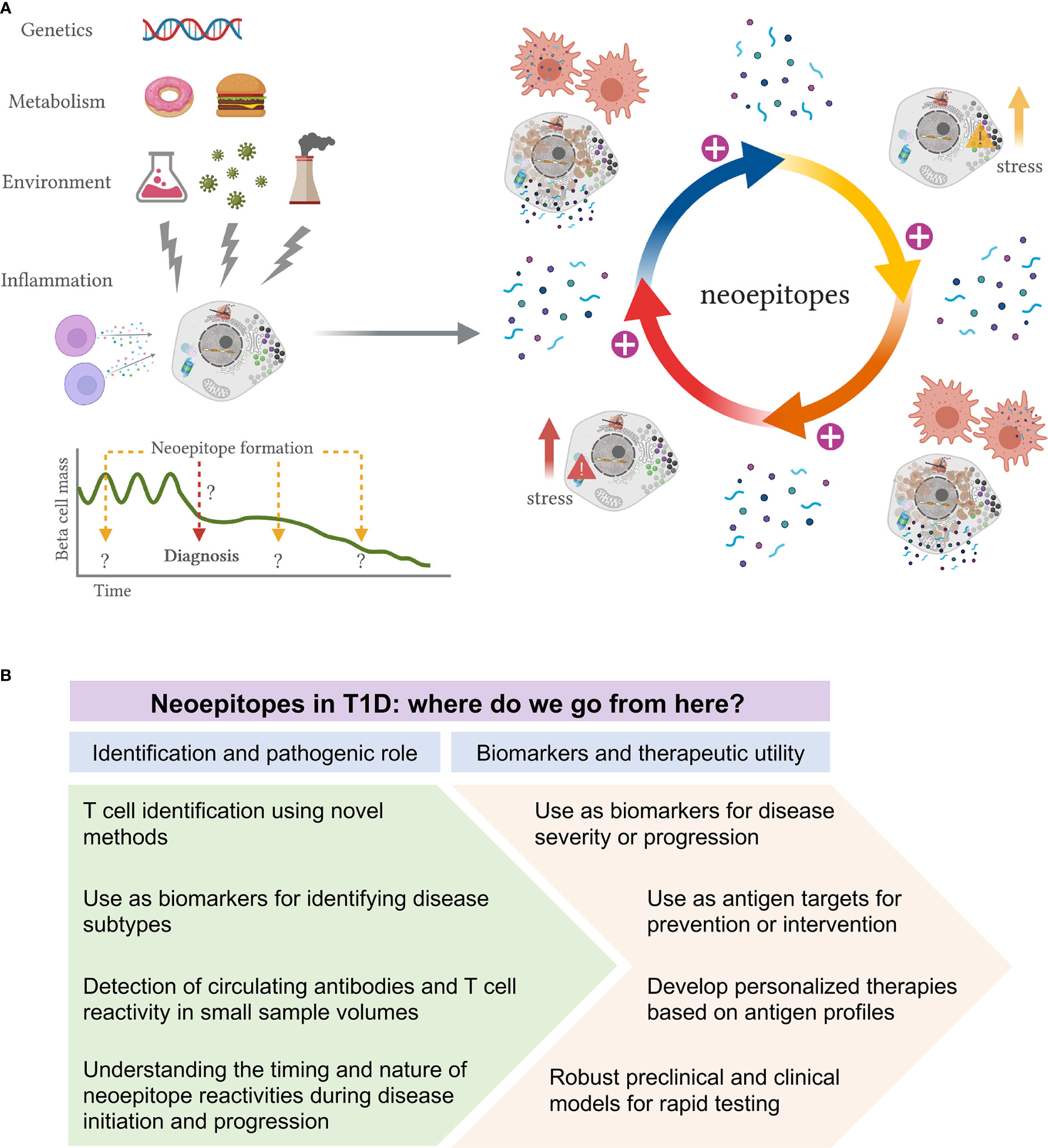
Figure 2 Proposed model of neoepitope generation and roadmap to their identification and utility in T1D. (A) Schematic representation of how neoepitope generation could lead to autoimmunity and beta-cell killing in T1D. Genetic predisposition, metabolism, environmental insults and immune inflammation are some of the factors that may lead to the formation of a first wave of non-conventional proteins. Impaired clearance of such stressed or dying beta-cells expressing modified proteins will cause activation of APCs and presentation of neoepitopes to T-cells, which can trigger several immune responses. This first cascade of immune activation may cause further beta-cell stress or death, generating an autoreactive loop, with further modification of beta-cell proteins and disease exacerbation. This proposed model could provide evidence for a role of neoepitopes both in initiation and exacerbation of disease. It may indeed be that different ways of stress are needed to activate specific types of PTMs, at different times and with unknown duration, during the disease course. (B) Proposed roadmap to identification and utility of neoepitopes in T1D. Multiple neoepitopes have been identified in T1D and yet their precise role in the disease remains ambiguous. We envision two major areas of research: 1) to improve the identification of neoepitopes and to better delineate their role in disease pathogenesis; 2) to evaluate their use as biomarkers and their therapeutic utility.
Where Do We Go From Here?
To get a full picture of the neoepitope landscape, more studies should be carried out evaluating such responses in the early stages of T1D and through their progression to T1D to further delineate and describe the importance and timing of neoepitope formation in T1D. Elucidating their role in pathogenesis may also enable their use to improve the current staging paradigm, which is largely based on the presence of autoantibodies to classical islet antigens. Finally, while antigen-specific approaches have shown limited efficacy to date in T1D prevention and treatment, it is feasible that targeting neoepitopes in addition to or instead of classic antigens may provide a better therapeutic benefit.
Understanding how neoantigens are involved in the break of tolerance will lead us towards tolerogenic therapies for T1D. Multiple antigens have been used in T1D trials (83, 84). While no trial achieved its primary outcome, two trials have shown partial successes. First, in a trial where oral insulin was administered to individuals with stage 1 T1D, a pre-defined subgroup had a significantly delayed time to diabetes development (85). Furthermore, in a trial with intranodal injection of GAD65, a predefined subgroup showed preservation of beta-cell function (https://www.diamyd.com/docs/pressClips.aspx?ClipID=3768129). Other groups are exploring combination of antigens and/or antigens plus immunomodulators. Given the limited success of single native antigen in inducing tolerance, the presence of epitope spreading in T1D and the evidence for neoantigen generation and autoreactivity, it is quite feasible that targeting multiple (neo)antigens could aid in promoting antigenic tolerance.
Based on current evidence, we wish to outline two major areas of research to better delineate their exact role, which is a prerequisite for their therapeutic utility (Figure 2B). First, to identify novel epitopes that are most relevant to T1D progression, it will be necessary to develop and use novel detection and analysis methods with improved sensitivity and capacity to identify the nature of the peptides that are presented (86). In turn, being able to assay for T-cell reactivity, even in the periphery, remains a major challenge. The identification of novel epitopes could open opportunities for the characterization of disease subtypes and broaden our understanding of the disease pathogenesis. More efforts on the assessment of circulating antibodies and T-cell reactivity in longitudinal samples are needed to fully understand the timing and nature of neoepitope autoreactivity. Although sample volume continues to be a limitation, especially in studies of pediatric population, we anticipate that new technologies will be able to significantly improve our capacity to detect autoreactive cells in these challenging samples.
It is conceivable that neoepitopes may be used alone or in conjugation with other antigens as biomarkers for disease severity and/or progression, with evident potential to become immune-modifying therapies to induce tolerance in T1D. We could imagine a scenario in which each diabetic patient has its own antigenic profile, which could be used towards personalized medicine. However, more robust preclinical models or in vitro systems to test antigenic candidates should be developed prior to entering clinical studies. In all, neoepitopes possess a yet untapped mechanism to provide better biomarkers for staging progression as well as therapeutic targets, and could be the key to understanding the loss of tolerance in T1D.
Data Availability Statement
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.
Author Contributions
TR-C, JDJ, LO and JLD conceived the concept and co-wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Research in this area in the JJ lab is supported by Diabetes Canada and CIHR. Related research in the LO and TR-C labs is supported by IMI2-JU under grant agreement No 115797 (INNODIA) and No 945268 (INNODIA HARVEST). This Joint Undertaking receives support from the Union’s Horizon 2020 research and innovation program and “EFPIA”, ‘JDRF” and “The Leona M. and Harry B. Helmsley Charitable Trust”. LO lab is supported by JDRF (1-SRA-2019-809-S-B). TR-C is supported by JDRF (5-CDA-2020-949-A-N).
Conflict of Interest
JLD is an employee of Janssen Research and Development, LLC.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the JDRF for its previous coordination of a neoepitopes working group. Figures 1B and 2A were created with BioRender.com.
References
1. Christoffersson G, Rodriguez-Calvo T, von Herrath M. Recent advances in understanding Type 1 Diabetes. F1000Res (2016) 5(F1000 Faculty Rev):110. doi: 10.12688/f1000research.7356.1
2. van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev (2011) 91:79–118. doi: 10.1152/physrev.00003.2010
3. Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PloS Genet (2012) 8:e1002552. doi: 10.1371/journal.pgen.1002552
4. Roep BO, Thomaidou S, van Tienhoven R, Zaldumbide A. Type 1 diabetes mellitus as a disease of the beta-cell (do not blame the immune system?). Nat Rev Endocrinol (2021) 17:150–61. doi: 10.1038/s41574-020-00443-4
5. Campbell-Thompson M, Wasserfall C, Kaddis J, Albanese-O’Neill A, Staeva T, Nierras C, et al. Network for Pancreatic Organ Donors with Diabetes (nPOD): developing a tissue biobank for type 1 diabetes. Diabetes Metab Res Rev (2012) 28:608–17. doi: 10.1002/dmrr.2316
6. Pugliese A, Yang M, Kusmarteva I, Heiple T, Vendrame F, Wasserfall C, et al. The Juvenile Diabetes Research Foundation Network for Pancreatic Organ Donors with Diabetes (nPOD) Program: goals, operational model and emerging findings. Pediatr Diabetes (2014) 15:1–9. doi: 10.1111/pedi.12097
7. Morgan NG, Richardson SJ. Fifty years of pancreatic islet pathology in human type 1 diabetes: insights gained and progress made. Diabetologia (2018) 61:2499–506. doi: 10.1007/s00125-018-4731-y
8. Campbell-Thompson ML, Atkinson MA, Butler AE, Giepmans BN, von Herrath MG, Hyoty H, et al. Re-addressing the 2013 consensus guidelines for the diagnosis of insulitis in human type 1 diabetes: is change necessary? Diabetologia (2017) 60:753–5. doi: 10.1007/s00125-016-4195-x
9. Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, et al. Insulitis and beta-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes (2016) 65:719–31. doi: 10.2337/db15-0779
10. In’t Veld P. Insulitis in human type 1 diabetes: a comparison between patients and animal models. Semin Immunopathol (2014) 36:569–79. doi: 10.1007/s00281-014-0438-4
11. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol (2009) 155:173–81. doi: 10.1111/j.1365-2249.2008.03860.x
12. Rodriguez-Calvo T, Ekwall O, Amirian N, Zapardiel-Gonzalo J, von Herrath MG. Increased immune cell infiltration of the exocrine pancreas: a possible contribution to the pathogenesis of type 1 diabetes. Diabetes (2014) 63:3880–90. doi: 10.2337/db14-0549
13. Unanue ER, Wan X. The Immunoreactive Platform of the Pancreatic Islets Influences the Development of Autoreactivity. Diabetes (2019) 68:1544–51. doi: 10.2337/dbi18-0048
14. Espinosa-Carrasco G, Le Saout C, Fontanaud P, Stratmann T, Mollard P, Schaeffer M, et al. CD4(+) T Helper Cells Play a Key Role in Maintaining Diabetogenic CD8(+) T Cell Function in the Pancreas. Front Immunol (2017) 8:150–161. doi: 10.3389/fimmu.2017.02001
15. Yeo L, Pujol-Autonell I, Baptista R, Eichmann M, Kronenberg-Versteeg D, Heck S, et al. Circulating beta cell-specific CD8(+) T cells restricted by high-risk HLA class I molecules show antigen experience in children with and at risk of type 1 diabetes. Clin Exp Immunol (2020) 199:263–77. doi: 10.1111/cei.13391
16. Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med (2012) 209:51–60. doi: 10.1084/jem.20111187
17. Rodriguez-Calvo T, Krogvold L, Amirian N, Dahl-Jorgensen K, von Herrath M. One in Ten CD8+ Cells in the Pancreas of Living Individuals With Recent Onset Type 1 Diabetes Recognizes the Preproinsulin Epitope PPI15-24. Diabetes (2021) 70(3):752–8. doi: 10.2337/db20-0908
18. Bender C, Rodriguez-Calvo T, Amirian N, Coppieters KT, von Herrath MG. The healthy exocrine pancreas contains preproinsulin-specific CD8 T cells that attack islets in type 1 diabetes. Sci Adv (2020) 6(42):eabc5586. doi: 10.1126/sciadv.abc5586
19. Culina S, Brezar V, Mallone R. Insulin and type 1 diabetes: immune connections. Eur J Endocrinol (2013) 168:R19–31. doi: 10.1530/EJE-12-0693
20. Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature (2005) 435:220–3. doi: 10.1038/nature03523
21. Gonzalez-Duque S, Azoury ME, Colli ML, Afonso G, Turatsinze JV, Nigi L, et al. Conventional and Neo-antigenic Peptides Presented by beta Cells Are Targeted by Circulating Naive CD8+ T Cells in Type 1 Diabetic and Healthy Donors. Cell Metab (2018) 28:946–960 e6. doi: 10.1016/j.cmet.2018.07.007
22. James EA, Mallone R, Kent SC, DiLorenzo TP. T-Cell Epitopes and Neo-epitopes in Type 1 Diabetes: A Comprehensive Update and Reappraisal. Diabetes (2020) 69:1311–35. doi: 10.2337/dbi19-0022
23. Culina S, Lalanne AI, Afonso G, Cerosaletti K, Pinto S, Sebastiani G, et al. Islet-reactive CD8(+) T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci Immunol (2018) 3(20):eaao4013. doi: 10.1126/sciimmunol.aao4013
24. Kracht MJ, van Lummel M, Nikolic T, Joosten AM, Laban S, van der Slik AR, et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med (2017) 23:501–7. doi: 10.1038/nm.4289
25. Azoury ME, Tarayrah M, Afonso G, Pais A, Colli ML, Maillard C, et al. Peptides Derived From Insulin Granule Proteins Are Targeted by CD8(+) T Cells Across MHC Class I Restrictions in Humans and NOD Mice. Diabetes (2020) 69:2678–90. doi: 10.2337/db20-0013
26. Wiles TA, Delong T. HIPs and HIP-reactive T cells. Clin Exp Immunol (2019) 198:306–13. doi: 10.1111/cei.13335
27. Mannering SI, Morris JS, Stone NL, Jensen KP, PM VANE, Harrison LC. CD4+ T cell proliferation in response to GAD and proinsulin in healthy, pre-diabetic, and diabetic donors. Ann N Y Acad Sci (2004) 1037:16–21. doi: 10.1196/annals.1337.003
28. Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA, et al. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care (2015) 38:1964–74. doi: 10.2337/dc15-1419
29. Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA (2013) 309:2473–9. doi: 10.1001/jama.2013.6285
30. Pietropaolo M, Towns R, Eisenbarth GS. Humoral autoimmunity in type 1 diabetes: prediction, significance, and detection of distinct disease subtypes. Cold Spring Harb Perspect Med (2012) 2(10):a012831. doi: 10.1101/cshperspect.a012831
31. Nakayama M. Insulin as a key autoantigen in the development of type 1 diabetes. Diabetes Metab Res Rev (2011) 27:773–7. doi: 10.1002/dmrr.1250
32. Krischer JP, Liu X, Vehik K, Akolkar B, Hagopian WA, Rewers MJ, et al. Predicting Islet Cell Autoimmunity and Type 1 Diabetes: An 8-Year TEDDY Study Progress Report. Diabetes Care (2019) 42:1051–60. doi: 10.2337/dc18-2282
33. Anderson MS, Su MA. Aire and T cell development. Curr Opin Immunol (2011) 23:198–206. doi: 10.1016/j.coi.2010.11.007
34. Doyle HA, Mamula MJ. Autoantigenesis: the evolution of protein modifications in autoimmune disease. Curr Opin Immunol (2012) 24:112–8. doi: 10.1016/j.coi.2011.12.003
35. Watad A, Amital H. ACPAs Are Much More Than Diagnostic Autoantibodies. Rambam Maimonides Med J (2016) 7(4):e0028. doi: 10.5041/RMMJ.10255
36. Jorgensen KT, Wiik A, Pedersen M, Hedegaard CJ, Vestergaard BF, Gislefoss RE, et al. Cytokines, autoantibodies and viral antibodies in premorbid and postdiagnostic sera from patients with rheumatoid arthritis: case-control study nested in a cohort of Norwegian blood donors. Ann Rheum Dis (2008) 67:860–6. doi: 10.1136/ard.2007.073825
37. James EA, Moustakas AK, Bui J, Papadopoulos GK, Bondinas G, Buckner JH, et al. HLA-DR1001 presents “altered-self” peptides derived from joint-associated proteins by accepting citrulline in three of its binding pockets. Arthritis Rheum (2010) 62:2909–18. doi: 10.1002/art.27594
38. Buitinga M, Callebaut A, Marques Camara Sodre F, Crevecoeur I, Blahnik-Fagan G, Yang ML, et al. Inflammation-Induced Citrullinated Glucose-Regulated Protein 78 Elicits Immune Responses in Human Type 1 Diabetes. Diabetes (2018) 67:2337–48. doi: 10.2337/db18-0295
39. McGinty JW, Chow IT, Greenbaum C, Odegard J, Kwok WW, James EA. Recognition of posttranslationally modified GAD65 epitopes in subjects with type 1 diabetes. Diabetes (2014) 63:3033–40. doi: 10.2337/db13-1952
40. Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med (2016) 22:1482–7. doi: 10.1038/nm.4203
41. Rondas D, Crevecoeur I, D’Hertog W, Ferreira GB, Staes A, Garg AD, et al. Citrullinated glucose-regulated protein 78 is an autoantigen in type 1 diabetes. Diabetes (2015) 64:573–86. doi: 10.2337/db14-0621
42. Wan X, Vomund AN, Peterson OJ, Chervonsky AV, Lichti CF, Unanue ER. The MHC-II peptidome of pancreatic islets identifies key features of autoimmune peptides. Nat Immunol (2020) 21:455–63. doi: 10.1038/s41590-020-0623-7
43. van Lummel M, Duinkerken G, van Veelen PA, de Ru A, Cordfunke R, Zaldumbide A, et al. Posttranslational modification of HLA-DQ binding islet autoantigens in type 1 diabetes. Diabetes (2014) 63:237–47. doi: 10.2337/db12-1214
44. Marre ML, McGinty JW, Chow IT, DeNicola ME, Beck NW, Kent SC, et al. Modifying Enzymes Are Elicited by ER Stress, Generating Epitopes That Are Selectively Recognized by CD4(+) T Cells in Patients With Type 1 Diabetes. Diabetes (2018) 67:1356–68. doi: 10.2337/db17-1166
45. Delong T, Baker RL, He J, Barbour G, Bradley B, Haskins K. Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes (2012) 61:3239–46. doi: 10.2337/db12-0112
46. Gottlieb PA, Delong T, Baker RL, Fitzgerald-Miller L, Wagner R, Cook G, et al. Chromogranin A is a T cell antigen in human type 1 diabetes. J Autoimmun (2014) 50:38–41. doi: 10.1016/j.jaut.2013.10.003
47. Mannering SI, Harrison LC, Williamson NA, Morris JS, Thearle DJ, Jensen KP, et al. The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J Exp Med (2005) 202:1191–7. doi: 10.1084/jem.20051251
48. Strollo R, Vinci C, Napoli N, Pozzilli P, Ludvigsson J, Nissim A. Antibodies to post-translationally modified insulin as a novel biomarker for prediction of type 1 diabetes in children. Diabetologia (2017) 60:1467–74. doi: 10.1007/s00125-017-4296-1
49. Strollo R, Vinci C, Arshad MH, Perrett D, Tiberti C, Chiarelli F, et al. Antibodies to post-translationally modified insulin in type 1 diabetes. Diabetologia (2015) 58:2851–60. doi: 10.1007/s00125-015-3746-x
50. Wiles TA, Delong T, Baker RL, Bradley B, Barbour G, Powell RL, et al. An insulin-IAPP hybrid peptide is an endogenous antigen for CD4 T cells in the non-obese diabetic mouse. J Autoimmun (2017) 78:11–8. doi: 10.1016/j.jaut.2016.10.007
51. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science (2016) 351:711–4. doi: 10.1126/science.aad2791
52. Baker RL, Jamison BL, Wiles TA, Lindsay RS, Barbour G, Bradley B, et al. CD4 T Cells Reactive to Hybrid Insulin Peptides Are Indicators of Disease Activity in the NOD Mouse. Diabetes (2018) 67:1836–46. doi: 10.2337/db18-0200
53. Baker RL, Jamison BL, Haskins K. Hybrid insulin peptides are neo-epitopes for CD4 T cells in autoimmune diabetes. Curr Opin Endocrinol Diabetes Obes (2019) 26:195–200. doi: 10.1097/MED.0000000000000490
54. Arribas-Layton D, Guyer P, Delong T, Dang M, Chow IT, Speake C, et al. Hybrid Insulin Peptides Are Recognized by Human T Cells in the Context of DRB1*04:01. Diabetes (2020) 69:1492–502. doi: 10.2337/db19-0620
55. Mitchell AM, Alkanani AA, McDaniel KA, Pyle L, Waugh K, Steck AK, et al. T-cell responses to hybrid insulin peptides prior to type 1 diabetes development. Proc Natl Acad Sci U S A (2021) 118(6):e2019129118. doi: 10.1073/pnas.2019129118
56. Reed B, Crawford F, Hill RC, Jin N, White J, Krovi SH, et al. Lysosomal cathepsin creates chimeric epitopes for diabetogenic CD4 T cells via transpeptidation. J Exp Med (2021) 218(2):e20192135. doi: 10.1084/jem.20192135
57. Faridi P, Li C, Ramarathinam SH, Vivian JP, Illing PT, Mifsud NA, et al. A subset of HLA-I peptides are not genomically templated: Evidence for cis- and trans-spliced peptide ligands. Sci Immunol (2018) 3(28):eaar3947. doi: 10.1126/sciimmunol.aar3947
58. Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, et al. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science (2016) 354:354–8. doi: 10.1126/science.aaf4384
59. Alvelos MI, Juan-Mateu J, Colli ML, Turatsinze JV, Eizirik DL. When one becomes many-Alternative splicing in beta-cell function and failure. Diabetes Obes Metab (2018) 20:77–87. doi: 10.1111/dom.13388
60. Bennett ST, Lucassen AM, Gough SC, Powell EE, Undlien DE, Pritchard LE, et al. Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat Genet (1995) 9:284–92. doi: 10.1038/ng0395-284
61. Owerbach D, Gabbay KH. Localization of a type I diabetes susceptibility locus to the variable tandem repeat region flanking the insulin gene. Diabetes (1993) 42:1708–14. doi: 10.2337/diabetes.42.12.1708
62. Barratt BJ, Payne F, Lowe CE, Hermann R, Healy BC, Harold D, et al. Remapping the insulin gene/IDDM2 locus in type 1 diabetes. Diabetes (2004) 53:1884–9. doi: 10.2337/diabetes.53.7.1884
63. Vafiadis P, Bennett ST, Colle E, Grabs R, Goodyer CG, Polychronakos C. Imprinted and genotype-specific expression of genes at the IDDM2 locus in pancreas and leucocytes. J Autoimmun (1996) 9:397–403. doi: 10.1006/jaut.1996.0054
64. Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG, et al. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet (1997) 15:289–92. doi: 10.1038/ng0397-289
65. Wang S, Flibotte S, Camunas-Soler J, MacDonald PE, Johnson JD. A new hypothesis for type 1 diabetes risk: The at-risk allele at rs3842753 associates with increased beta cell INS mRNA in a meta-analysis of single cell RNA sequencing data. Can J Diabetes (2021). doi: 10.1016/j.jcjd.2021.03.007
66. Szabat M, Page MM, Panzhinskiy E, Skovso S, Mojibian M, Fernandez-Tajes J, et al. Reduced Insulin Production Relieves Endoplasmic Reticulum Stress and Induces beta Cell Proliferation. Cell Metab (2016) 23:179–93. doi: 10.1016/j.cmet.2015.10.016
67. Nucci AM, Virtanen SM, Cuthbertson D, Ludvigsson J, Einberg U, Huot C, et al. Growth and development of islet autoimmunity and type 1 diabetes in children genetically at risk. Diabetologia (2021) 64:826–35. doi: 10.1007/s00125-020-05358-3
68. Xin Y, Dominguez Gutierrez G, Okamoto H, Kim J, Lee AH, Adler C, et al. Pseudotime Ordering of Single Human beta-Cells Reveals States of Insulin Production and Unfolded Protein Response. Diabetes (2018) 67:1783–94. doi: 10.2337/db18-0365
69. Eizirik DL, Miani M, Cardozo AK. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia (2013) 56:234–41. doi: 10.1007/s00125-012-2762-3
70. Yong J JJ, Han J, Kaufman RJ. Therapeutic opportunities around pancreatic β-cell ER stress in diabetes. Natures Rev Endocrinol (2021) in press.
71. Marre ML, James EA, Piganelli JD. beta cell ER stress and the implications for immunogenicity in type 1 diabetes. Front Cell Dev Biol (2015) 3:67. doi: 10.3389/fcell.2015.00067
72. Marre ML, Profozich JL, Coneybeer JT, Geng X, Bertera S, Ford MJ, et al. Inherent ER stress in pancreatic islet beta cells causes self-recognition by autoreactive T cells in type 1 diabetes. J Autoimmun (2016) 72:33–46. doi: 10.1016/j.jaut.2016.04.009
73. Callebaut A, Derua R, Vig S, Delong T, Mathieu C, Overbergh L. Identification of Deamidated Peptides in Cytokine-Exposed MIN6 Cells through LC-MS/MS Using a Shortened Digestion Time and Inspection of MS2 Spectra. J Proteome Res (2021) 20(2):1405–14. doi: 10.1021/acs.jproteome.0c00801
74. Colli ML, Ramos-Rodriguez M, Nakayasu ES, Alvelos MI, Lopes M, Hill JLE, et al. An integrated multi-omics approach identifies the landscape of interferon-alpha-mediated responses of human pancreatic beta cells. Nat Commun (2020) 11:2584. doi: 10.1038/s41467-020-16327-0
75. de Jong VM, Zaldumbide A, van der Slik AR, Persengiev SP, Roep BO, Koeleman BP. Post-transcriptional control of candidate risk genes for type 1 diabetes by rare genetic variants. Genes Immun (2013) 14:58–61. doi: 10.1038/gene.2012.38
76. Coppieters KT, Wiberg A, von Herrath MG. Viral infections and molecular mimicry in type 1 diabetes. APMIS (2012) 120:941–9. doi: 10.1111/apm.12011
77. Dunne JL, Richardson SJ, Atkinson MA, Craig ME, Dahl-Jorgensen K, Flodstrom-Tullberg M, et al. Rationale for enteroviral vaccination and antiviral therapies in human type 1 diabetes. Diabetologia (2019) 62:744–53. doi: 10.1007/s00125-019-4811-7
78. Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest (2017) 127:2881–91. doi: 10.1172/JCI94549
79. Ashton MP, Eugster A, Walther D, Daehling N, Riethausen S, Kuehn D, et al. Incomplete immune response to coxsackie B viruses associates with early autoimmunity against insulin. Sci Rep (2016) 6:32899. doi: 10.1038/srep32899
80. Atkinson MA, Bowman MA, Campbell L, Darrow BL, Kaufman DL, Maclaren NK. Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J Clin Invest (1994) 94:2125–9. doi: 10.1172/JCI117567
81. Honeyman MC, Stone NL, Harrison LC. T-cell epitopes in type 1 diabetes autoantigen tyrosine phosphatase IA-2: potential for mimicry with rotavirus and other environmental agents. Mol Med (1998) 4:231–9. doi: 10.1007/BF03401920
82. Oldstone MB, Edelmann KH, McGavern DB, Cruite JT, Welch MJ. Molecular anatomy and number of antigen specific CD8 T cells required to cause type 1 diabetes. PloS Pathog (2012) 8:e1003044. doi: 10.1371/journal.ppat.1003044
83. Coppieters KT, Harrison LC, von Herrath MG. Trials in type 1 diabetes: Antigen-specific therapies. Clin Immunol (2013) 149:345–55. doi: 10.1016/j.clim.2013.02.002
84. Rodriguez-Fernandez S, Almenara-Fuentes L, Perna-Barrull D, Barneda B, Vives-Pi M. A century later, still fighting back: antigen-specific immunotherapies for type 1 diabetes. Immunol Cell Biol (2021). doi: 10.1111/imcb.12439
85. G. Writing Committee for the Type 1 Diabetes TrialNet Oral Insulin Study, Krischer JP, Schatz DA, Bundy B, Skyler JS, Greenbaum CJ. Effect of Oral Insulin on Prevention of Diabetes in Relatives of Patients With Type 1 Diabetes: A Randomized Clinical Trial. JAMA (2017) 318:1891–902. doi: 10.1001/jama.2017.17070
Keywords: type 1 diabetes, neoepitopes, autoantigens, pathogenesis, beta-cell, biomarker
Citation: Rodriguez-Calvo T, Johnson JD, Overbergh L and Dunne JL (2021) Neoepitopes in Type 1 Diabetes: Etiological Insights, Biomarkers and Therapeutic Targets. Front. Immunol. 12:667989. doi: 10.3389/fimmu.2021.667989
Received: 15 February 2021; Accepted: 29 March 2021;
Published: 19 April 2021.
Edited by:
Arnaud Zaldumbide, Leiden University Medical Center, NetherlandsReviewed by:
Raghavendra G. Mirmira, University of Chicago, United StatesAnne Cooke, University of Cambridge, United Kingdom
Copyright © 2021 Rodriguez-Calvo, Johnson, Overbergh and Dunne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teresa Rodriguez-Calvo, dGVyZXNhLnJvZHJpZ3VlekBoZWxtaG9sdHotbXVlbmNoZW4uZGU=