 Bandar Alosaimi1,2
Bandar Alosaimi1,2 Ayman Mubarak3
Ayman Mubarak3 Maaweya E. Hamed3
Maaweya E. Hamed3 Abdullah Z. Almutairi4
Abdullah Z. Almutairi4 Ahmed A. Alrashed5Abdullah AlJuryyan6Mushira Enani7Faris Q. Alenzi8
Ahmed A. Alrashed5Abdullah AlJuryyan6Mushira Enani7Faris Q. Alenzi8 Wael Alturaiki9*
Wael Alturaiki9*- 1Research Center, King Fahad Medical City, Riyadh, Saudi Arabia
- 2College of Medicine, King Fahad Medical City, Riyadh, Saudi Arabia
- 3Department of Botany and Microbiology, College of Science, King Saud University, Riyadh, Saudi Arabia
- 4Laboratory and Blood Bank, King Fahad Hospital, Madina, Saudi Arabia
- 5Pharmaceutical Service Department, Main Hospital, King Fahad Medical City, Riyadh, Saudi Arabia
- 6Pathology and Clinical Laboratory Management, King Fahad Medical City, Riyadh, Saudi Arabia
- 7Medical Specialties Department, King Fahad Medical City, Riyadh, Saudi Arabia
- 8Department of Medical Laboratory Sciences, College of Applied Medical Sciences, Prince Sattam bin Abdulaziz University, Alkharj, Saudi Arabia
- 9Department of Medical Laboratory Sciences, College of Applied Medical Sciences, Majmaah University, Majmaah, Saudi Arabia
COVID-19 severity due to innate immunity dysregulation accounts for prolonged hospitalization, critical complications, and mortality. Severe SARS-CoV-2 infections involve the complement pathway activation for cytokine storm development. Nevertheless, the role of complement in COVID-19 immunopathology, complement‐modulating treatment strategies against COVID-19, and the complement and SARS‐CoV‐2 interaction with clinical disease outcomes remain elusive. This study investigated the potential changes in complement signaling, and the associated inflammatory mediators, in mild-to-critical COVID-19 patients and their clinical outcomes. A total of 53 patients infected with SARS-CoV-2 were enrolled in the study (26 critical and 27 mild cases), and additional 18 healthy control patients were also included. Complement proteins and inflammatory cytokines and chemokines were measured in the sera of patients with COVID-19 as well as healthy controls by specific enzyme-linked immunosorbent assay. C3a, C5a, and factor P (properdin), as well as interleukin (IL)-1β, IL-6, IL-8, tumor necrosis factor (TNF)-α, and IgM antibody levels, were higher in critical COVID-19 patients compared to mild COVID-19 patients. Additionally, compared to the mild COVID-19 patients, factor I and C4-BP levels were significantly decreased in the critical COVID-19 patients. Meanwhile, RANTES levels were significantly higher in the mild patients compared to critical patients. Furthermore, the critical COVID-19 intra-group analysis showed significantly higher C5a, C3a, and factor P levels in the critical COVID-19 non-survival group than in the survival group. Additionally, IL-1β, IL-6, and IL-8 were significantly upregulated in the critical COVID-19 non-survival group compared to the survival group. Finally, C5a, C3a, factor P, and serum IL-1β, IL-6, and IL-8 levels positively correlated with critical COVID-19 in-hospital deaths. These findings highlight the potential prognostic utility of the complement system for predicting COVID-19 severity and mortality while suggesting that complement anaphylatoxins and inflammatory cytokines are potential treatment targets against COVID-19.
Introduction
Sever Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) was reported in China on Dec 31, 2019, after several unexplained cases of pneumonia were reported (1, 2). COVID-19 cases are defined as symptomatic, asymptomatic, and severe disease with mortality rates highest among patients with chronic disease, the elderly, and immunocompromised patients (3). The World Health Organization declared the outbreak a public health emergency of international concern On January 30, 2020 (2). Following community transmission of SARS-CoV-2 in many countries, the WHO declared the COVID-19 outbreak as a pandemic on March 12, 2020. By December 3, 2020, SARS-CoV2 had spread to 206 countries with a total of 63,719,213 laboratory-confirmed cases and 1,482,084 deaths reported around the world (1–4). Severe cases of COVID-19 cannot be predicted early during the onset of symptoms due to the lack of biomarkers or accurate testing for the prediction of COVID-19 disease severity (5, 6). As such, identification of immune parameters as diagnostic biomarkers to predict COVID-19 disease severity might help to accurately select interventional strategies (7–9).
Immune response dysregulation is a primary characteristic of COVID-19 severity, disease evolution and worse clinical outcomes (10). Accumulated evidence suggests that SARS-CoV-2 infections are characterized by dysregulation of innate and adaptive immunity (10–12). Several inflammatory cytokines and chemokines (interleukin (IL)-1β, IL-2, IL-6, IL-7, IL-8, IL-10, granulocyte colony-stimulating factor (G-CSF), granulocyte/macrophage colony-stimulating factor (GM-CSF), interferon gamma-induced protein (IP)-10, monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, interferon (IFN)-γ, tumor necrosis factor (TNF)-α, C-C motif chemokine ligand (CCL)2 and CCL3) as well as C-reactive protein (CRP) are significantly upregulated in severe and critical COVID-19 patients (13, 14). The high inflammatory cytokines and chemokine levels during SARS‐CoV-1, MERS‐CoV, and SARS‐CoV‐2 infections are strongly associated with massive infiltration of immune cells into the lungs and poor disease outcomes (15, 16).
The complement system comprises a proteins network (17). Depending on the type of activation factors, the complement cascade is activated through three pathways: The classical pathway, lectin pathway, and alternative pathway (18–20). Crosstalk between the complement and coagulation systems has been reported to play a crucial role in the vascular endothelial damage and thromboinflammation (21). The complement system is negatively regulated by various complement proteins, including factor I, C1-inhibitor (CI-INH), factor H and C4-binding protein (C4-BP) (11, 21); meanwhile, factor P (properdin) is a positive regulatory complement protein. Several viral infections are associated with complement activation and coagulation dysfunctions (22). Overactivation of pulmonary and systemic complement plays a key role in inflammation, endothelial cell damage, thrombus formation, intravascular coagulation, and multiple organ failure, ultimately leading to death (23, 24). C5a is chemoattractant for neutrophils, monocytes, eosinophils, and T lymphocytes (24). Following infection, complement anaphylatoxins stimulate macrophages to produce TNF-α, IL‐1β, IL‐6, and IL‐8; these mediators promote vascular dysfunction, fibrinolysis, and microvascular thrombosis formation (24). The C5a also plays a major role in inducing higher expression of P‐selectin, intercellular adhesion molecule‐1, fibronectin and fibrinogen. This upregulation of adhesion molecules provokes various cell signaling and pro‐inflammatory pathways (25).
The role of complement in COVID-19 immunopathology and complement‐modulating treatment strategies against COVID-19 has received limited attention with several unanswered questions regarding the interaction between complement, SARS‐CoV‐2, and clinical disease outcomes. In this study, we comprehensively investigated the complement system, pro-inflammatory cytokines/chemokines, and antibody responses of patients with critical and mild COVID-19. The correlation between all examined parameters and COVID-19 in-hospital mortality was also assessed. To the best of our knowledge, this is the first study to investigate the functionality of several complement proteins and complement regulatory factors in the context of critical COVID-19 cases.
Materials and Methods
Patient Selection, Setting, and Sample Collection
This study enrolled 53 patients infected with SARS-CoV-2, as confirmed by RT‐PCR (inclusion criterion). There were 26 critical (intensive care unit [ICU]) and 27 mild COVID-19 cases, and 18 healthy matched control groups. The mild cases were defined based on the absence of symptoms such as shortness of breath and pneumonia and other less severe clinical symptoms (low-grade fever and cough). Blood samples were collected, allowed to clot for 20–35 min at 25°C and subjected to centrifugation for 15 min at 1,000 × g. The serum was immediately assayed, liquated, and stored at ≥ −20°C. Repeated freeze-thaw cycles were avoided. The exclusion criteria were: 1) patients under 14 years of age; 2) patients co-infected with other respiratory pathogens; 3) patients diagnosed with bacteremia and/or viremia caused by any other viruses; 4) immunocompromised patients; 5) patients under treatment with anti-inflammatory and/or anti-complement drugs; 6) patients with preexisting autoimmune diseases; 7) women who were either pregnant or lactating (Table 1). This study was reviewed and approved by the Institutional Review Board of King Fahad Medical City (IRB register number 20‒193) and written informed consent was obtained from all subjects prior to enrollment.
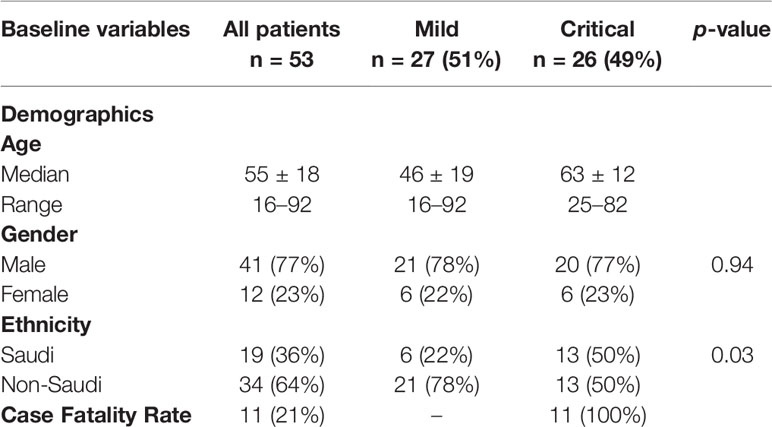
Table 1 Demographics and clinical characteristics of COVID-19 patients.
Clinical Laboratory Investigations
The following blood parameters were examined: CRP, INR, PT, PTT, creatine kinase, Creatine Kinase Myocardial Band, Trop, Hb, platelet, red blood cells, WBC, glucose, ESR, low-density lipoprotein cholesterol, AST, ALT, urea, creatinine, high-density lipoprotein cholesterol, albumin, and total protein.
Measurement of Complement Inflammatory Mediators Anaphylatoxins (C3a/C3b and C5a) and Complement Classical Pathway Component (C1q) and C2 Levels
Serum levels of human complement C3 and C5 fragments, as well as C3b, C1q, and C2 were measured according to the manufacturer’s guidelines using enzyme-linked immunosorbent assay (ELISA) kits (HCA39-K01-Eagle Biosciences, Inc., Columbia, USA; ab193695, Abcam, Cambridge, UK; ab195461, Abcam; ab170246, Abcam; ab154132, Abcam). The concentrations were calculated using standard curves.
Quantification of Serum Complement Regulatory Components (Factors) Levels
Serum concentrations of four complement regulatory factors, P, I, C4-BP, and H were quantified using ELISA kits (ab222864, Abcam; ab195460, Abcam; ab222866, Abcam; HK342, Hycult Biotech, Uden, Netherlands). All ELISA protocols were performed according to the manufacturer’s instructions. The concentrations were calculated using standard curves.
Quantification of Serum Chemokine RANTES (CCL5) Levels
Serum RANTES levels were quantified in COVID-19 infection patients (n = 53) and healthy volunteers (n = 18) using a human RANTES ELISA kit (R&D Systems, Minneapolis, USA) following the manufacturer’s protocol. The concentrations were calculated using standard curves, and the results were expressed as pg/mL.
Quantification of the SARS-CoV-2 Antibody
The SARS-CoV-2 IgM antibody was quantified using ELISA kits (AnshLabs, Webster, USA). The SARS-CoV-2 IgM ELISA assay detects antibodies against the spike and nucleocapsid proteins. The assay was performed according to the manufacturer’s protocol. IgM concentration > 12 AU/mL (positive cutoff) was considered positive.
Quantification of Pro-Inflammatory Cytokine and Chemokine Profiles Using ELISArray
The concentrations of the primary 12 human pro-inflammatory cytokines and chemokines (IL1-α, IL1-β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, IL-17A, IFN-γ, TNF-α, and GM-CSF) were measured in the serum of 53 COVID-19 patients and healthy controls, using the multi-analyte ELISArray (Qiagen, Germantown, MD, USA) following the manufacturer’s protocol. The absorbance of the ELISArray was measured at 450 nm and concentrations were calculated using a standard curve. The cytokine or chemokine levels were expressed as pg/mL.
Statistical Analysis
All statistical analyses were performed using GraphPad 5.0 (GraphPad Software, San Diego, CA, USA). Data were assessed using one-way ANOVA followed by Tukey’s multiple comparison test. The correlations between complement proteins, inflammatory cytokines/chemokines, and clinical laboratory blood parameters were tested using the Pearson correlation. Results are presented as mean ± standard deviation unless otherwise specified. A p-value of < 0.05 was considered statistically significant.
Results
Basic Characteristics of COVID-19 Patients
A total of 53 COVID-19 patients (26 critical and 27 mild) were included in this study (41 males and 12 females), with an age ranging from 18 to 92 years and a mean age of 53.2 ± 18.0 years. The mean age was 61.6 ± 11.8 years in the critical group, 45.2 ± 19.2 years in the mild group, and 66.2 ± 6.5 and 58.3 ± 13.6 years in the non-survival and survival patients, respectively. Eleven (42.3%) patients died while 15 recovered (57.7%) in the critical COVID-19 group. All basic characteristics of the enrolled patients are shown in Table 1.
Circulating Complement Activation in Critical and Mild COVID-19 Patients
To determine whether critical COVID-19 patients have high levels of the complement inflammatory mediators anaphylatoxins C3a and C5a, systemic levels of complement (anaphylatoxins) C3a, a product of C3 cleavage, and C5a, a product of C5 cleavage, were measured in samples from patients with confirmed SARS-CoV-2 infection (26 critical and 27 mild), and 17 healthy groups. As shown in Figures 1A, B, both C3a and C5a levels were higher in the COVID group than in the control group. Additionally, severe patients had higher levels of these markers than patients with mild disease (p < 0.0001). The elevated levels of C3a and C5a observed in the critical COVID-19 patients were consistent with decreased levels of complement negative regulatory factor I and C4-BP in the same patients. Overall, these results illustrate the role of complement anaphylatoxins C5a and C3a in pulmonary immunopathology, endothelial damage, vascular leakage, and case fatality in patients with critical COVID-19. The C2 levels were higher in the COVID group than in the control group (Figure 1D). Interestingly, C2 was higher in mild patients compared to severe patients, while there were no significant differences in the C1q levels between these two groups (Figure 1C).
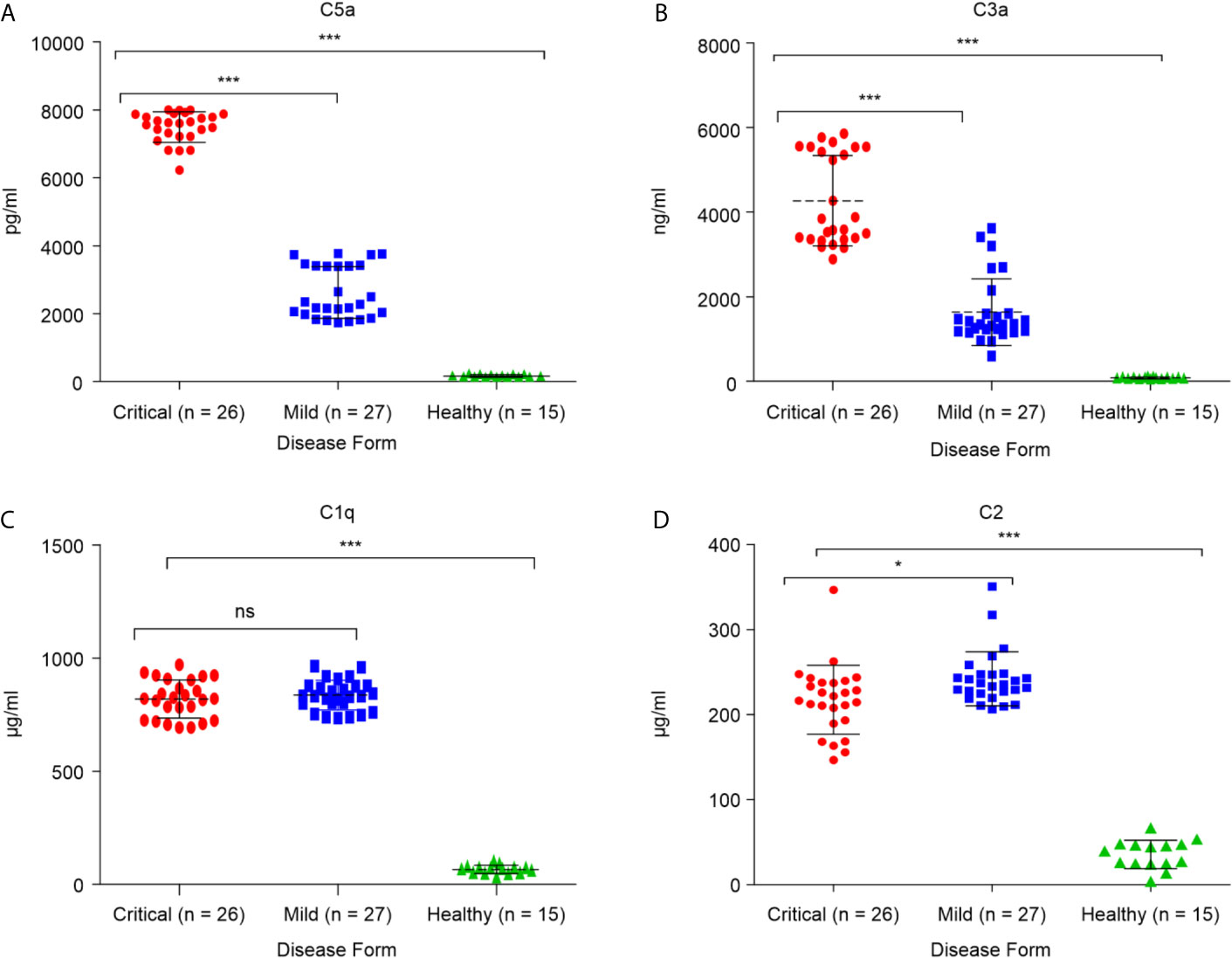
Figure 1 Serum concentrations of the anaphylatoxins in critical and mild COVID-19 patients, and healthy controls. Concentrations of (A) complement fragment C5a; (B) complement fragment C3a; (C) complement fragment C1q; and (D) complement fragment C2 were measured using ELISA. ANOVA and Tukey’s multiple comparison test were used for statistical comparisons. *p < 0.05, ***p < 0.001, and ns p > 0.05.
Critical COVID-19 Condition Related to Abnormal Levels of Complement Regulatory Proteins
Critical COVID-19 Is Associated With Positive Regulation of Complement Activation
These alterations in major complement proteins led us to investigate whether there are changes in regulators of this pathway. The complement regulatory proteins are crucial for controlling complement overactivation to avoid inflammatory pathologies and tissue damage. To determine whether the complement regulatory factors were altered during SARS-CoV-2 infection, four complement regulatory factors, namely factor P, factor I, C4-BP, and factor H, were quantified in critical and mild COVID-19 patients. As shown in Figure 2D, the levels of positive regulatory factor P were significantly higher in the critical COVID-19 patients than in the mild COVID-19 and non-COVID-19 healthy control groups (p < 0.0001), suggesting that the levels of factor P may positively regulate complement activation during the SARS-CoV-2 infection. Furthermore, measurement of factor P in serum might provide evidence for the involvement of the alternative complement pathway since factor P is an important factor in the alternative pathway activation.
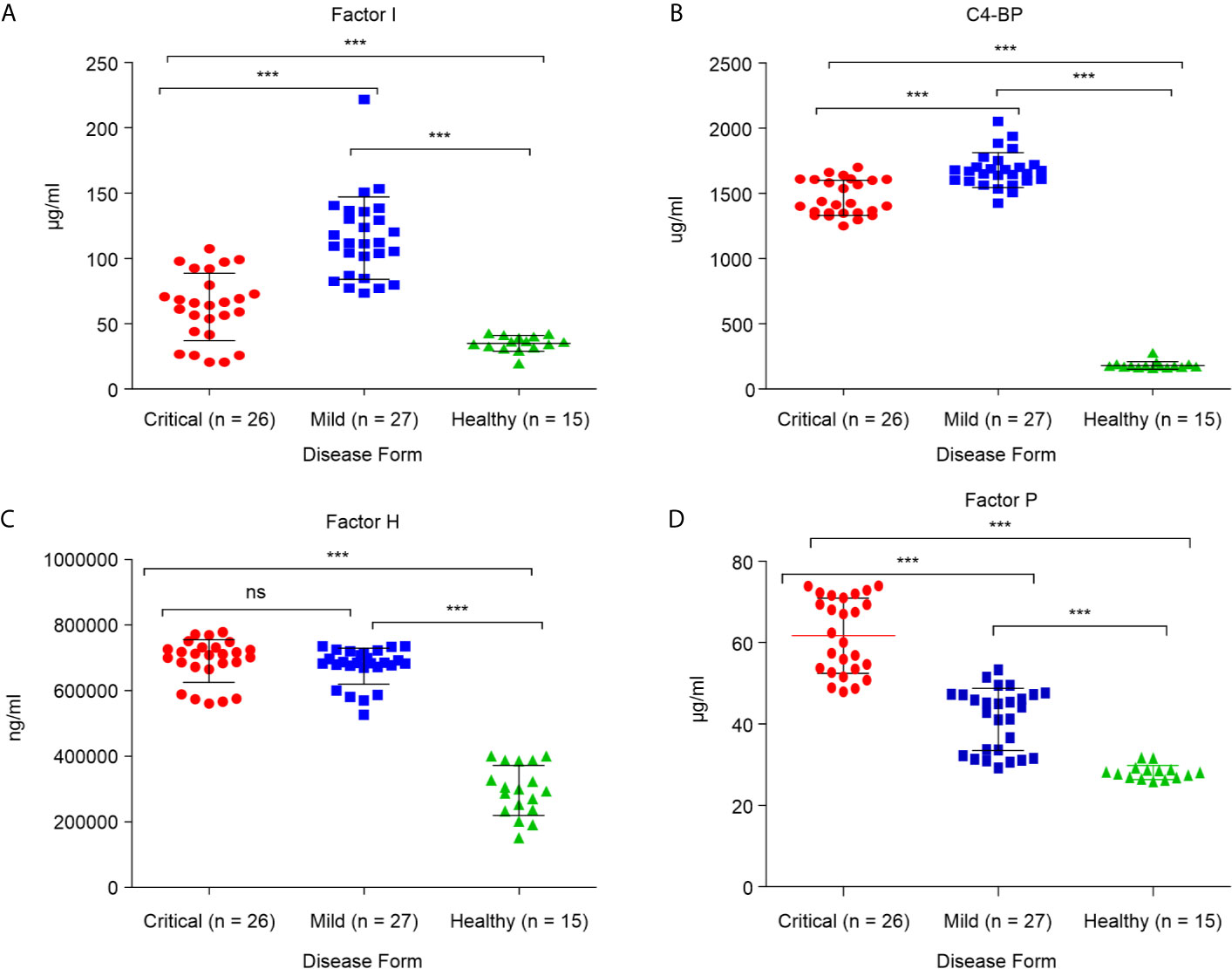
Figure 2 Serum concentrations of complement factors in the critical and mild COVID-19 patients, and healthy controls. Concentrations of (A) factor I; (B) factor C4-BP; (C) factor H; and (D) factor P. ANOVA and Tukey’s multiple comparison test were used for statistical comparisons. ***p < 0.001 and ns p > 0.05.
Critical COVID-19 Cases Are Associated With Decreased Levels of Complement Negative Regulatory Proteins
Measurement of negative regulatory proteins in COVID-19 patients may provide evidence for complement cascade regulation. Quantitation of the complement negative regulatory factors (factor I, C4-BP, and factor H), in critical and mild COVID-19 patients showed that the levels of negative regulatory proteins, factor I and C4-BP, were significantly lower in the critical COVID-19 group than in the mild COVID-19 group (p < 0.0001; Figures 2A, B). These negative regulatory proteins play a critical role in complement system regulation. The low levels of factor I and C4-BP suggest that critical COVID-19 patients have a reduced ability to control and regulate complement activation, suggesting that SARS-CoV-2 somehow suppresses and inhibits the complement negative regulatory proteins during critical COVID-19. In contrast, the level of factor H, which regulates complement by inactivating the C3 convertase and dislodges Bb from the C3bBb complex, did not differ between the critical and mild COVID-19 patients (p > 0.05; Figure 2C).
Critical COVID-19 Induces High Levels of SARS-CoV-2 IgM Antibody
We aimed to determine whether SARS-CoV-2 IgM contributes to COVID-19 severity. Both critical and mild COVID-19 patients showed positive IgM antibody response, with mean values of 2,050.7 AU/mL and 992.2 AU/mL, respectively (positive cutoff > 12 AU/mL). However, the IgM concentration in critical COVID-19 patients was significantly higher than that in patients with mild COVID-19 (p = 0.0021; Figure 3).
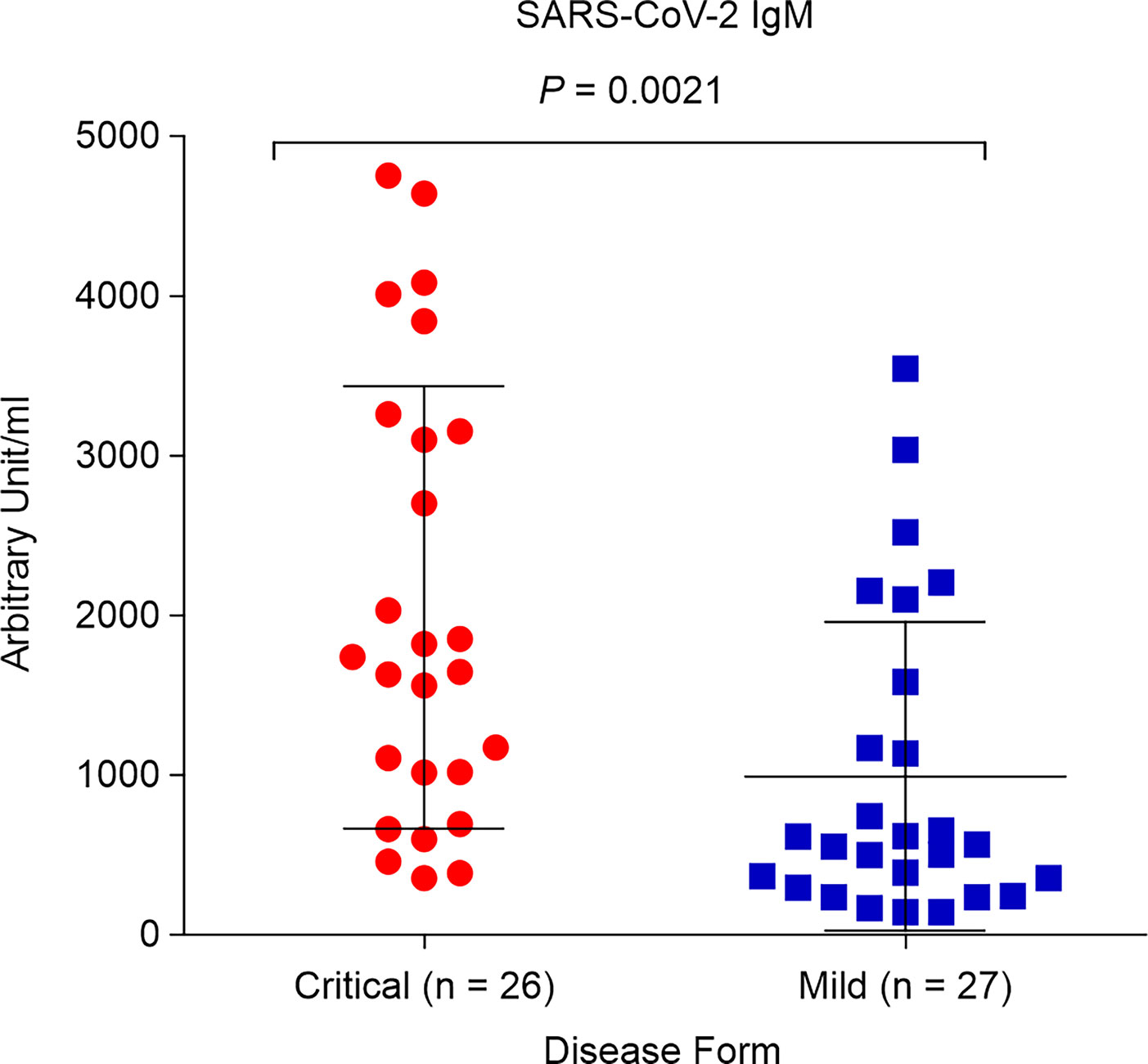
Figure 3 Levels of SARS-CoV-2 immunoglobulin M (IgM) in critical and mild COVID-19 patients (P = 0.002). ELISA assay was used to detect antibodies against spike and nucleocapsid protein.
RANTES (CCL5) Levels Are Increased in Mild COVID-19 Patients
Alterations in the concentration of serum RANTES in the studied patients with mild COVID-19 and non-COVID-19 healthy controls are shown in Figure 4E. The serum RANTES levels were higher in the mild COVID-19 group than in the critical COVID-19 and healthy control groups (p < 0.01, for both). RANTES during mild SARS-CoV-2 infection might play a role in the anti-SARS-CoV-2 immune response.
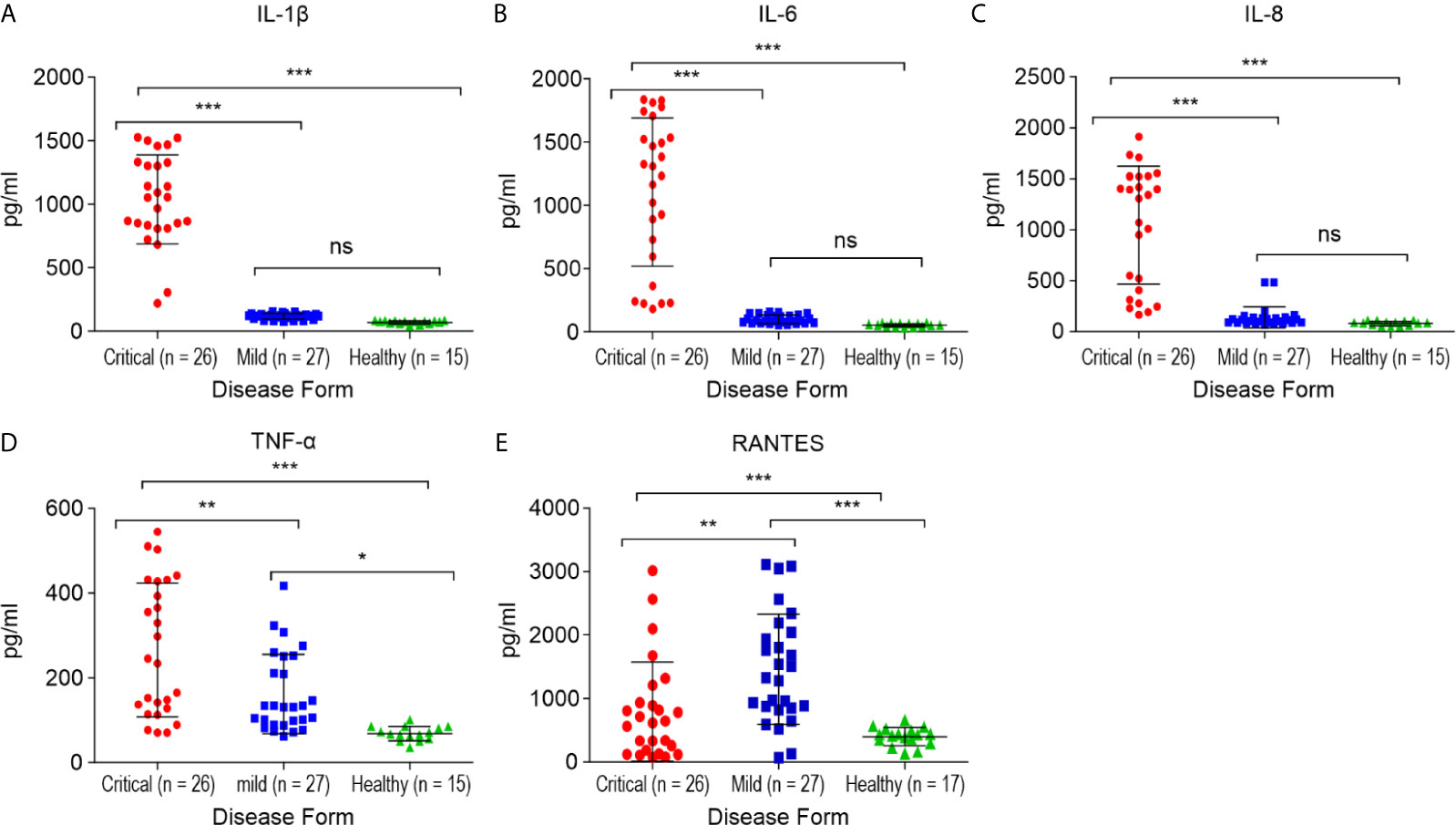
Figure 4 Comparison of the cytokine/chemokine levels between the critical and mild COVID-19 patients, and healthy controls. (A) interleukin 1 β (IL-1β); (B) interleukin 6 (IL-6); (C) interleukin 8 (IL-8); (D) tumor necrosis factor α (TNF-α); (E) RANTES. *p < 0.05, **p < 0.01 and ***p < 0.001 and ns p > 0.05.
Critical COVID-19 Patients Exhibit High Levels of IL-1β, IL-6, TNF-α, and Neutrophil Chemoattractant Chemokine IL-8 (CXCL8)
The serum samples from critical COVID-19, mild COVID-19, and healthy controls were collected to quantify 12 pro-inflammatory cytokines and chemokines. The levels of IL1-β, IL-6, IL-8 (p < 0.0001, for each), and TNF-α (p < 0.01) were significantly higher in the critical COVID-19 patients than in the mild COVID-19 and non-COVID-19 healthy controls (Figure 4).
Critical COVID-19 Non-Survival Patients Have Elevated Levels of Complement Components and Pro-Inflammatory Cytokines/Chemokines
It was found that 11/26 (42%) critical COVID-19 patients died during hospitalization. The C5a (7,770.3 ± 191; p =0.0047), C3a (5,019.2 ± 920.4; p = 0.0008), and factor P (68.8 ± 6.7; p = 0.0002) concentrations were significantly higher in the critical COVID-19 non-survival group than in the critical COVID-19 survival group (Figure 5). Additionally, the concentrations of IL-1β (1,236.5 ± 398.1; p = 0.0105), IL-6 (1,408.9 ± 490.3; p = 0.0198), and IL-8 (1,443.4 ± 195.8; p = 0.0012) were significantly upregulated in the critical COVID-19 non-survival group compared to the critical COVID-19 survival group (Figure 5).
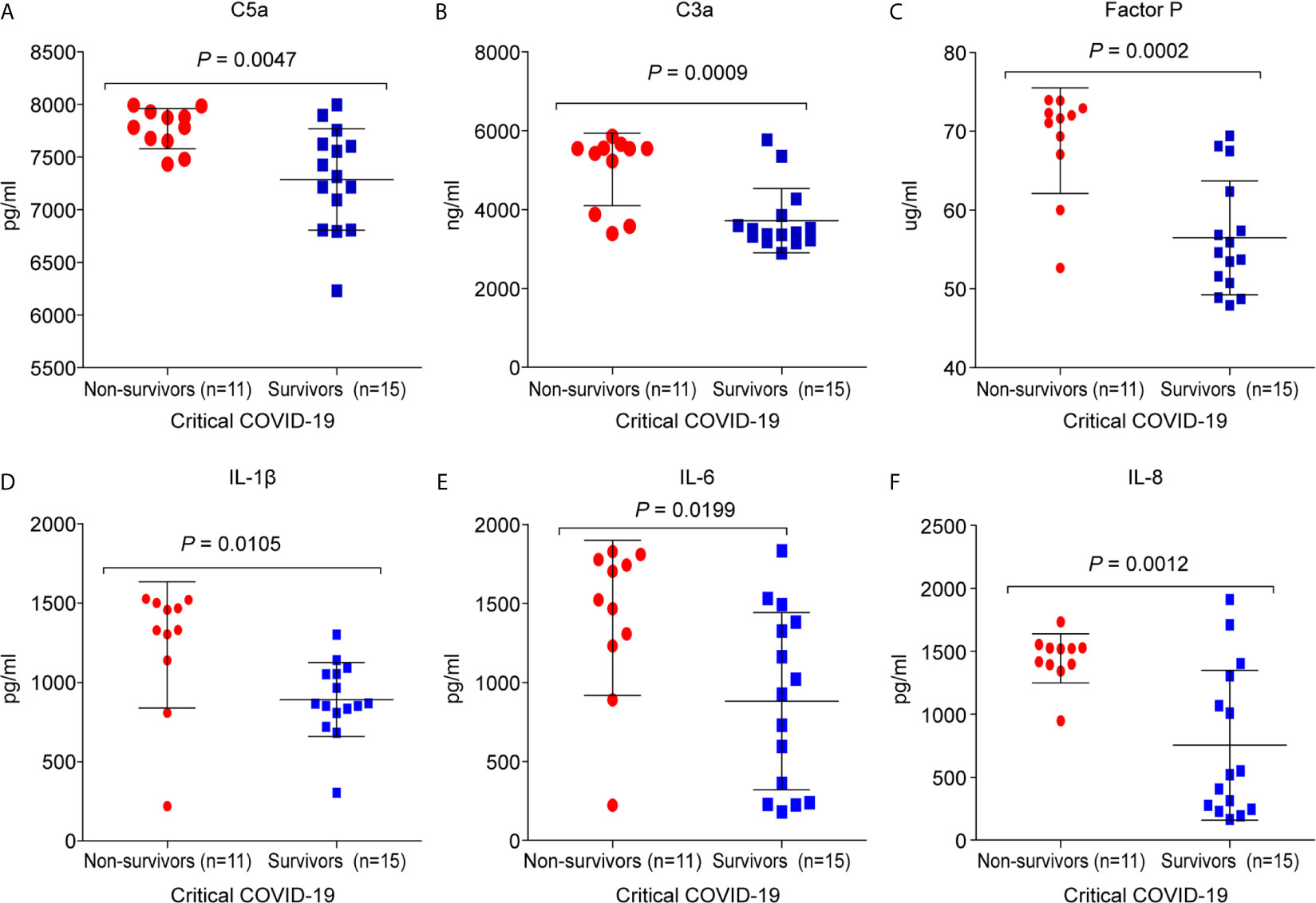
Figure 5 Comparison of the complement proteins and cytokine/chemokine levels between critical COVID-19 patients who survived and those who did not survive. The serum samples were collected from the day of admission or day 14 during the intensive care unit (ICU)stay. (A) complement fragment C5a (P = 0.005); (B) complement fragment C3a (P < 0.001); (C) factor P (P < 0.001); (D) interleukin 1 β (IL-1β) (P = 0.01); (E) interleukin 6 (IL-6) (P = 0.02); (F) interleukin 8 (IL-8) (P = 0.001).
Association Between Circulating Levels of Complement Proteins and Pro-Inflammatory Cytokines/Chemokines With Critical COVID-19 Patients In-Hospital Mortality
Pearson’s correlation analysis showed that serum levels of C5a, C3a, and factor P strongly correlated with critical COVID-19 in-hospital death (r = 0.5366; r = 0.6138; r = 0.6716, respectively; Figures 6A–C). Likewise, serum IL-1β, IL-6, and IL-8 levels were positively correlated with critical COVID-19 in-hospital death (r = 0.4929; r = 0.4539; r = 0.6002, respectively; Figures 6D–F). Our results indicated that the overactivation of the complement system and higher levels of pro-inflammatory cytokines/chemokines in critical COVID-19 patients were significantly correlated with critical COVID-19 in-hospital mortality.
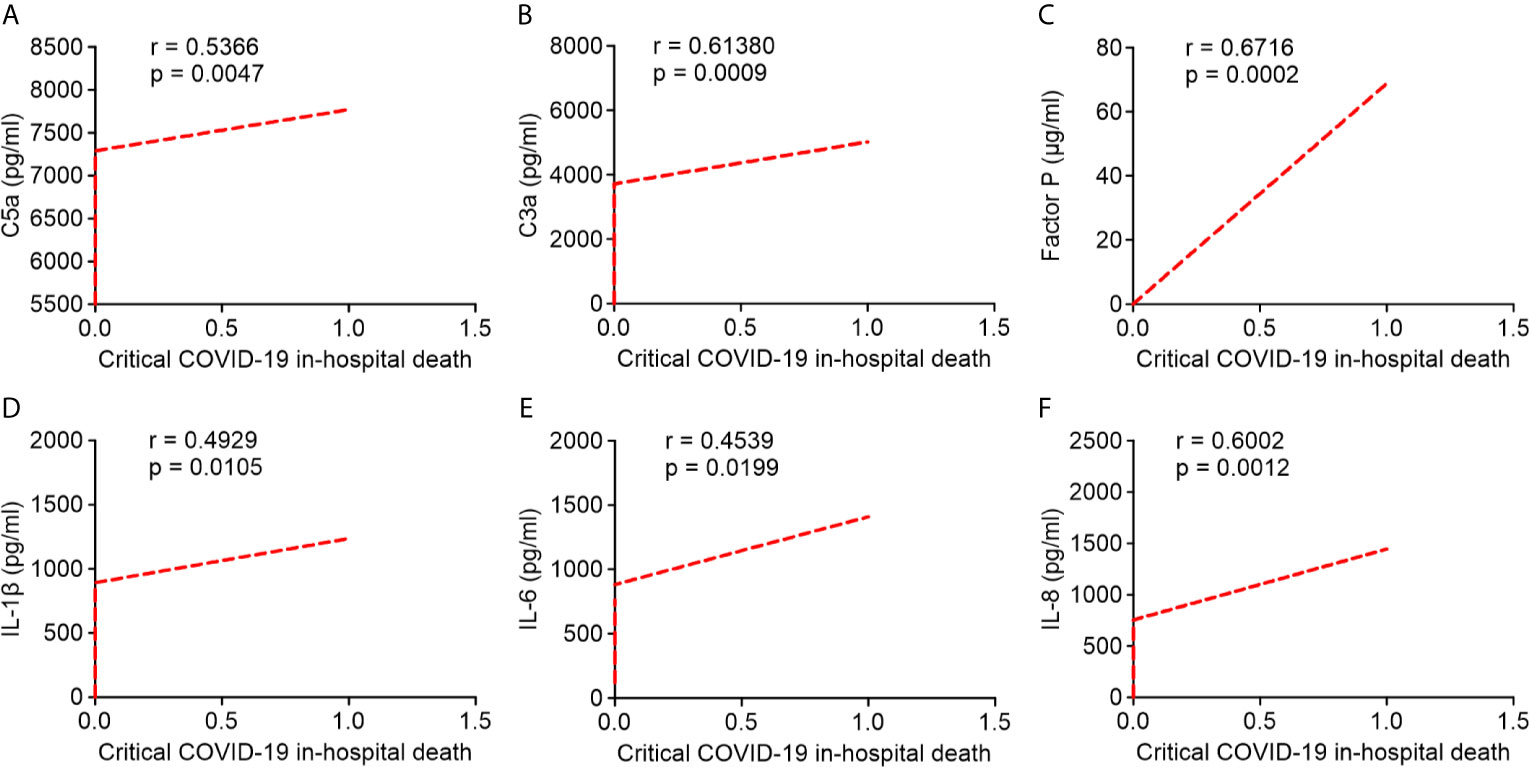
Figure 6 Correlation between serum complement components and inflammatory cytokine/chemokine levels in critical COVID-19 patients who died in hospital. (A) complement fragment C5a (P = 0.005); (B) complement fragment C3a (P < 0.001); (C) factor P (P < 0.001); (D) interleukin 1 β (IL-1β) (P = 0.01); (E) interleukin 6 (IL-6) (P = 0.02); (F) interleukin 8 (IL-8) (P = 0.001).
Pro-Inflammatory Cytokine/Chemokine Profiles in the Serum of Critical COVID-19 Patients and Their Correlation With Serum Concentrations of Complement Proteins
We further investigated any potential correlation between pro-inflammatory cytokines/chemokines and anaphylatoxins and showed that the levels of C3a positively correlated with the levels of IL-6 (r = 0.3886, P = 0.0498) and IL-1β (r = 0.3517, but P = 0.0781; Table 2). Additionally, C5a levels mildly, correlated with the levels of IL-1β (r = 0.2979, but P = 0.1394) and IL-6 (r = 0.2862, but P = 0.1564), however these results were not significant (Table 2). In addition, serum factor P levels positively correlated with the levels of C3a (r = 0.6766, P = 0.0001). We also observed a positive correlation between C5a and C3a levels (r = -0.5672, P = 0.05; Table 2).

Table 2 Correlation between serum complement proteins and inflammatory. cytokines/chemokines.
Discussion
In this study, we sought to determine the level of complement factors and pro-inflammatory cytokines/chemokines in the critical and mild patient responses to SARS-CoV-2. This study offers the first insights into the protein expression profile of the complement factors during SARS-CoV-2 infection. Different markers of human serum obtained from patients with COVID-19 and healthy controls were analyzed. The concentration of complement inflammatory mediators (C3a and C5a), responsible for attracting phagocytic cells to infection sites, were higher in critical COVID-19 patients than in the mild and healthy groups. However, lower levels of complement regulators were associated with the critical groups. We also analyzed a panel of 12 inflammatory cytokines/chemokines, including IL1-α, IL1-β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, IL-17A, IFN-γ, TNF-α, and GM-CSF, in the sera obtained from critical and mild patients diagnosed with SARS-CoV-2 infection. Among these cytokines, serum levels of IL1-β, IL-6, IL-8, and TNF-α were found to be significantly increased in the critical COVID-19 patients relative to mild patients. In addition, the RANTES serum levels were significantly elevated in mild COVID-19 patients relative to the critical cases.
The complement system is an important component of the innate immune response to pathogens. Activation of complements by viral infections induces acute and chronic inflammation, as well as intravascular coagulation and cell damage. Consequently, organ failure and death occur (23, 26). Indeed, complement activation has been implicated in the pathogenesis of MERS-CoV and SAR-CoV infections, which is similar to the current pandemic SARS-CoV-2 infection (27).
C5a and C3a are potent mediators that induce inflammatory reactions. C5a triggers the recruitment of neutrophils and monocytes followed by their accumulation and activation. In addition, it initiates mast cells activation and degradation while increasing the induction of cytokines and vascular permeability (28, 29). This observation was evident following SARS-CoV infection (30) as well as demonstrated in vitro and in vivo following pathogenic H5N1 influenza virus (31). Our work has demonstrated that the concentrations of the pro-inflammatory mediators, anaphaylatoxin C3a and C5a, were significantly increased in critical COVID-19 patients relative to mild and non-infected groups. In turn, these anaphylatoxins likely enhanced the release of pro-inflammatory cytokines and chemokines (IL-1, IL-8, IL-6 and TNF-a) from phagocytic cells and T cells, as previously reviewed (19). This indicates that the amount of C3 is this group is higher than that of healthy volunteers. Additionally, the non-survival group demonstrated a higher concentration of C3a, C5a, and factor P in comparison to the critical survival group. A significant correlation was observed between serum complement components and inflammatory cytokines/chemokines levels in patients with critical COVID-19 in-hospital mortality.
A recent study on SARS-CoV infections has shown that C3 activation exacerbates acute respiratory distress syndrome (ARDS) which is associated with SARS-CoV. Low lung infiltration of neutrophils, inflammatory monocytes, and decreased concentration of cytokines and chemokines in the lungs and serum have also been reported (32). A deficiency of C3 in these patients led to low production of C3a and C3b, a skewed immune response toward Th2 and induction of regulatory T cell development from CD4+ cells (33, 34).
Another study (35) has described the increased serum levels of C5a in critical cases of COVID-19 patients, which is consistent with our findings. A study conducted in Italy showed high induction levels of C5a and C5b-C9 in the plasma of a COVID-19 infected group (36). These molecules were observed in the serum and lung tissue of mice infected with MERS-CoV (37). However, in severe COVID-19 patients, only the deposition of C5b-9 was observed and was significantly elevated in pulmonary microvasculature (22). Patients hospitalized with COVID-19, who were admitted to ICU, had elevated levels of C3b on red blood cells peaking on day 7, relative to healthy non-infected COVID-19 donors (38). Collectively, MERS-CoV, SARS-CoV, and SARS-CoV-2 over-activate the complement system and contributes to dysregulation of the host immune response. High serum levels of complement protein C5a have also been reported in non-survivor H5N1 patients compared with survivors (24). Animal studies have further demonstrated that complement C3 knockout mice are protected from lung inflammation and respiratory failure as well as inflammatory cytokines (32). Furthermore, a MERS-CoV mouse model treated with a C5a receptor (C5aR, CD88) blocker reduced lung inflammation and decreased viral replication in the lungs (39).
Furthermore, other proteins mediators of complement, such as C3, C4, and C5b-9, as well as inflammatory cells were found to be significantly increased in alveolar epithelial cells of the non-survivors infected with COVID-19. A probable inhibitory effect was noticed in this study when patients were treated with anti-C5a monoclonal antibody (35), which may be a potential strategy against SARS-CoV-2 infection. In addition, strong deposition of C5b-9 in the kidney tissue of patients infected with SARS-CoV-2 has recently been reported, which caused tissue damage (40).
To the best of our knowledge, this is the first study to report significantly elevated levels of other complement components such as factor P, C1q, C2, and C3 in COVID-19 patients in comparison to healthy non-infected individuals. Moreover, factor P was higher in the critical group than in the mildly affected group. However, no difference was observed between the critical and mildly affected groups in terms of C1q. These mediators have important roles in enhancing complement activation during SARS-CoV-2 infection. Thus, COVID-19 may trigger activation of complement via different pathways, especially the classical and alternative pathways, causing an accumulation of opsonin molecules, such as C3b, in COVID-19 patients. Another study showed that SARS-CoV, MERS-CoV, and SARS-CoV-2 can activate the lectin pathway through interactions between N-protein and MASP-2 (35). Hence, we can conclude that 1) the lower C2 and C1q level in critical patients is believed to complement the depletion during activation; 2) regulatory mediators are upregulated; and 3) the time of sample collection may have impacted the results, as levels were increased during the early stage and gradually decreased during infection.
Several regulatory molecules, including C1-INH, C4-BP, decay-accelerating factor (DAF/CD55), factor H, and factor I, control complement activation (11, 19). This is the first report regarding SARS-CoV-2 infection, showing that both critical and mild cases of COVID-19 patients have elevated levels of factor I, factor H, and C4-BP compared to those in non-COVID-19 healthy individuals (Figure 3). However, the critical groups had a lower proportion of these regulatory proteins, except for factor H, in their serum compared to mild cases (Figures 3A–C). This might be attributed to the high concentration of anaphylatoxins, C3a, and C5a, among the critical groups resulting from complement activation.
The first published autopsy study of severe COVID-19 infection showed an association between activation of the alternative pathway and lectin pathway cascades and microvascular injury and thrombosis, suggesting complement-mediated thrombotic microvascular injury syndrome through alternative and lectin pathway activation (22). Accumulated evidence demonstrated an association between coagulopathy (pulmonary embolism or venous, arterial, and/or microvascular thrombosis) and SARS-CoV-2 infection (41). In fact, systemic microthrombi and multi-organ injury following SARS‐CoV-1, MERS‐CoV, SARS‐CoV‐2, H1N1, H5N1, and H7N9 influenza infections have been described (24, 42). Patients with SARS-CoV-2 infection are characterized by overactivation of the complement pathways (17, 43). Meanwhile, data from COVID-19 patients showed that inflammation and respiratory failure are associated with systemic complement activation (44). A recent study confirmed that severe COVID-19 cases have a higher level of circulating C5a and sC5b-9, signifying C5a blockade as a potential treatment strategy to control and reduce disease severity (36, 44). Numerous randomized controlled trials reported increased survival in severe cases of COVID-19 patients treated with anti-C5 therapy (27, 36, 45). Collectively, these results confirm that inhibition of an over-activated complement response significantly reduces COVID-19 disease severity.
We have also demonstrated a significant correlation between complement concentration and coagulation in patients infected with SARS-CoV-2, compared to mild cases and healthy controls. Moreover, patients with high complement proteins showed increased platelet counts. Several studies have shown that COVID-19 patients with excessive coagulant factors are more susceptible to increased thrombosis and deteriorating clinical outcomes (21). Thus, the lack of C3 in mouse models demonstrated a decrease in apparent defect of thrombus and activation of platelets (46). However, after treatment with different anticoagulants (Clexane & heparins), the level was decreased. The levels were significantly decreased in the same group that received Clexane but not heparin. Moreover, critical patients treated with hydroxyl chloroquine and antiviral drugs (ritonavir and lopinavir) showed inhibition of coagulation with no thrombosis, although antiviral drugs (remdesivir, favipiravir) alone had no significant effect.
The levels of cytokines and chemokines in patients with COVID-19 remains controversial (47). Moreover, ICU patients have elevated plasma levels of IL-2, IL-7, IL-10, GSCF, IP10, MCP1, MIP1A, and TNF-α compared to non-ICU patients (13). However, one study has reported no significant differences in serum levels of TNF-α, IL-1, IL-8, and IL-10 in mild, severe, and critical ICU patients (48). Similarly, we did not observe differences in IL-10 levels between critical and mild COVID-19 patients.
Elevated serum levels of pro-inflammatory cytokines, including IL1-β, IL6, IL-12, IFN-γ, IP10, and MCP1 are associated with lung inflammation and damage in patients with SARS (49). Additionally, MERS-CoV infection has been reported to induce high levels of pro-inflammatory cytokines, including TNF-α, IFN-γ, IL-15, and IL-17 (50). Consistent with previous findings (51, 52), we also observed that serum levels of IL1-β, IL-6, IL-8, and TNF-α, but not IL1-α, IFN-γ, IL-2, IL-4, IL-10, IL-12, or IL-17A were significantly elevated in ICU patients compared to non-ICU patients, suggesting a possible role for T-helper-1 cell responses during SARS-CoV-2 infection. Moreover, the increased levels of Th1 cytokines, including IL-1β, IL-6, and TNF-α, may be associated with the cytokine storm that is associated with disease severity. Nonetheless, SARS-CoV-2 infection was also shown to induce increased levels of Th-2 cytokines, such as IL-4 and IL-10, which can suppress inflammation (49).
Previous studies have reported that TNF-α exerts strong antiviral activity against swine, avian, and human influenza viruses (53). These cytokines are involved in the regulation of inflammatory processes and infectious diseases (54). It has further been reported that TNF-α serum levels are increased in COVID-19 patients with higher levels detected in severe cases (13, 51, 52). However, normal levels of TNF-α have been reported in severe COVID-19 patients (55). Previous studies have also reported that overproduction of TNF-α is associated with poor disease outcomes in MERS-CoV and SARS-CoV (50, 56), while administration of anti-TNF-α antibody (certolizumab) may have positive effects on COVID-19 patients (57). Additionally, although elevated levels of TNF-α and IFN-γ contribute to lung damage and high case fatality in COVID-19 patients, combination therapy of anti-TNF-α and anti-IFN-γ neutralizing antibodies effectively reduces inflammatory cell death, tissue damage, and mortality (58)
IL-1 is actively involved in inflammatory response to infection (59). Meanwhile, SARS-CoV-2 has been shown to affect the activation and maturation of IL-1β, which can sequentially activate IL-6 and TNF-α (60–62). IL-1β also contributes to the cytokine storm induced by SARS-CoV-2 (63). In fact, most COVID-19 patients with severe illness have increased levels of IL-1β, which is associated with ARDS (64); therefore, inhibition of IL-1β can mitigate development of the cytokine storm that causes death in COVID-19 patients (65). Additionally, IL-1 and IL-18 are secreted following inflammasome activation and have critical roles in the regulation of innate and adaptive immune responses (66). Severe SARS-CoV-2 infection engages inflammasome activation and pyroptosis associated with elevated levels of IL-1ß. However, therapy targeting IL-1ß has demonstrated beneficial outcomes in the prevention of SARS-CoV-2-induced cell death (67). Similarly, early blockade of the IL-1 receptor was found to be a promising therapeutic strategy against hyperinflammation, development of the cytokine storm, and respiratory failure in COVID-19 patients (68).
Furthermore, IL-6 levels are elevated in patients with COVID-19 and are associated with a poor prognosis (48, 69, 70), and are further increased in severe COVID-19 associated symptoms compared to mild patients, suggesting the importance of this cytokine as a marker to monitor disease progression and development (52, 55). Additionally, the IL-6 levels were higher in patients who died from COVID-19 than in the recovered individuals (71). Recently, it has been reported that increased levels of IL-6 enhance the inflammatory process and contribute to cytokine storm thereby worsening prognosis (72); however, targeting IL-6 receptors (IL-6r) with a specific monoclonal antibody (tocilizumab) has been shown to mediate an effective therapeutic option in COVID-19 patients who are at risk of developing cytokine storms (69).
In addition, IL-8 is a strong pro-inflammatory cytokine that plays an essential role in the activation and recruitment of neutrophil cells during inflammation (73); neutrophilia is more frequently observed in severe COVID-19 patients than in the mildly affected patients (14), suggesting that IL-8 participates in the pathophysiology of COVID-19. Similarly, in our study, high IL-8 levels were associated with increased numbers of neutrophils, particularly in the ICU patients relative to mild patients and healthy normal controls. IL-8 also contributes to the occurrence of ARDS and results in a cytokine storm linked to death in patients infected with SARS-CoV or MERS-CoV (74, 75). It has been established that pre-treatment with an anti-CXCL8 antibody prevents the development of severe lung injury (76); thus, targeting this cytokine or its receptor may offer an effective treatment option for COVID-19 patients.
RANTES (CCL5) is a strong leucocyte chemoattractant that can activate and induce migration of several immune cells, including T cells, natural killer cells, dendritic cells, monocytes, basophils, and eosinophils, to the site of inflammation (77, 78). It has been reported that, at the early stage of SARS-CoV-2 infection, increased serum levels of CCL5 were observed in patients with mild COVID-19 symptoms compared to severe patients, suggesting that CCL5 may protect against viral infection, where virus-specific CD8+ T cells respond to the removal of the virus before lung inflammation occurs (79). In addition, it has been shown that mild cases, not severe COVID-19 patients, are characterized by high expression of clonally expanded CD8+ T cells in the bronchoalveolar fluid, which suggests the potential mechanisms underlying pathogenesis and recovery in mild patients (80). Moreover, in agreement with these findings, we found that serum levels of RANTES were significantly elevated in mild patients and critical ICU patients compared to healthy normal controls. However, RANTES levels were significantly increased in the mildly affected patients compared to the critical ICU patients (81). Taken together, these results suggest that targeting RANTES, at least in the early stages of viral infection, may enhance viral clearance and prevent the spread of the virus to the lung and other parts of the body. In addition, this finding highlights the fact that RANTES might be important in antiviral responses in patients mildly affected with COVID-19 but also contributes to cytokine storm and mortality in severe cases.
The detection of antibodies during SARS-CoV-2 provides insights into the disease diagnosis and clinical course of COVID-19 infection. However, recent studies have indicated that COVID-19 patients with high levels of antibody responses, including the total antibodies, were associated with COVID-19 disease severity (9, 82). Likewise, increased B cell activation and proliferation in severe COVID-19 patients is associated with adverse outcomes (83, 84).
Additionally, a higher antibody response during several viral infections, including COVID-19 and MERS-CoV, was associated with antibody-dependent enhancement of immunopathological and robust inflammatory response (85, 86). In this study, high levels of IgM were detected in critical COVID-19 patients. Our findings suggest that high levels of SARS-CoV-2 IgM response could act as an early biomarker to differentiate between critical and mild COVID-19 patients. Furthermore, this result provides evidence of the possible antibody-dependent enhancement phenomenon during SARS-CoV-2 infection. In fact, markedly-increased humoral immunity has been linked to antibody-dependent enhancement, which was observed during SARS-CoV-1 infection (87). Therefore, we believe that high levels of IgM may play a critical role in the immunopathology and severity of COVID-19. In any case, further studies are required to confirm this finding.
The limitations of the current study can be summarized as follows: 1) Assessment of T-helper-1 and T-helper-2 cytokine expression in the sera of infected patients may not reflect the host response against SARS-CoV-2 in the airways. 2) COVID-19 patients were not tracked, and serum samples were collected once and, therefore, it is difficult to determine the kinetics of cytokine levels. Furthermore, future studies should examine the expression of cytokines in bronchoalveolar fluid samples from COVID-19 patients to achieve a better understanding of the host immune response to SARS-CoV-2 infection. In addition, conducting multiple sampling time points may help to determine the peak levels for each cytokine.
In conclusion, we showed an increase in the serum levels of cytokines, including IL1-β, IL-6, IL-8, and TNF-α in ICU patients relative to mild COVID-19, while RANTES serum levels were increased in the mild, not ICU patients. In addition, SARS-CoV-2 infection contributes to the potent activation of complement mediators including C5a, C3a, factor P, factor I, factor C4-BP, and factor H. Despite the advantage of complement activation and protection against COVID-19, there is a strong association in the pathogenesis of SARS-CoV-2, especially in tissue inflammation and coagulation. Thus, this outcome might have contributed to tissue damage and dysfunction, whether in circulation or other organs. There is an urgent need to identify an effective therapeutic strategy against COVID-19 by targeting and suppressing excessive mediators of complements in controlling SARS-CoV-2 infection, as well as pro-inflammatory cytokines (IL1-β, IL-6, and TNF-α) and chemokines (IL-8 and RANTES) that might be used as predictive markers to evaluate disease severity and, thus, can be targeted as treatment options in COVID-19 disease.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by The Institutional Review Board of King Fahad Medical City (IRB register number 20‒193). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
WA contributed to study design, planning, and supervision. WA and BA secured funding. MH and BA performed the experiments and data analysis. FA, AZA, and AAA conducted sample collection. AA and ME contributed to clinical data collection and manuscript revision. WA, BA, AM, and MH wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Targeted Research Grant Program of King Abdulaziz City for Science and Technology (grant number: 5-20-01-675-0002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the Research Center at the King Fahad Medical City for their continued support and the Regional Lab in Medina for their valuable contribution to sample collection. The authors extend their sincere gratitude toward the Science and Technology Unit (STU), Vice Rector for Graduate Studies and Scientific Research, Majmaah University for supporting the study. We would like to thank Editage (www.editage.com) for English language editing.
References
1. Perlman S. Another Decade, Another Coronavirus. N Engl J Med (2020) 382(8):760–2. doi: 10.1056/NEJMe2001126
2. World Health Organization. Coronavirus Disease 2019 (COVID-19). Available at: https://apps.who.int/iris/handle/10665/331219 (Accessed February 22, 2020).
3. Jaimes JA, André NM, Chappie JS, Millet JK, Whittaker GR. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically-Sensitive Activation Loop. J Mol Biol (2020) 432(10):3309–25. doi: 10.1016/j.jmb.2020.04.009
4. Guo Y-R, Cao Q-D, Hong Z-S, Tan Y-Y, Chen S-D, Jin H-J, et al. The Origin, Transmission and Clinical Therapies on Coronavirus Disease 2019 (COVID-19) Outbreak – an Update on the Status. Mil Med Res (2020) 7(1):11. doi: 10.1186/s40779-020-00240-0
5. Jiang A-M, Shi X, Liu N, Gao H, Ren M-D, Zheng X-Q, et al. Nosocomial Infections Due to Multidrug-Resistant Bacteria in Cancer Patients: A Six-Year Retrospective Study of an Oncology Center in Western China. BMC Infect Dis (2020) 20(1):452. doi: 10.1186/s12879-020-05181-6
6. Poston JT, Patel BK, Davis AM. Management of Critically Ill Adults With Covid-19. JAMA (2020) 323(18):1839–41. doi: 10.1001/jama.2020.4914
7. Chen X, Ling J, Mo P, Zhang Y, Jiang Q, Ma Z, et al. Restoration of Leukomonocyte Counts Is Associated With Viral Clearance in COVID-19 Hospitalized Patients. medRxiv (2020). doi: 10.1101/2020.03.03.20030437
8. Vázquez Y, González L, Noguera L, González PA, Riedel CA, Bertrand P, et al. Cytokines in the Respiratory Airway as Biomarkers of Severity and Prognosis for Respiratory Syncytial Virus Infection: An Update. Front Immunol (2019) 10:1154. doi: 10.3389/fimmu.2019.01154
9. Zhang B, Zhou X, Zhu C, Song Y, Feng F, Qiu Y, et al. Immune Phenotyping Based on the Neutrophil-to-Lymphocyte Ratio and IgG Level Predicts Disease Severity and Outcome for Patients With COVID-19. [Original Research]. Front Mol Biosci (2020) 7:157. doi: 10.3389/fmolb.2020.00157
10. Abou-Ismail MY, Diamond A, Kapoor S, Arafah Y, Nayak L. The Hypercoagulable State in COVID-19: Incidence, Pathophysiology, and Management. Thromb Res (2020) 194:101–15. doi: 10.1016/j.thromres.2020.06.029
11. Afshar-Kharghan V. The Role of the Complement System in Cancer. J Clin Investig (2017) 127(3):780–9. doi: 10.1172/JCI90962
12. Del Valle DM, Kim-Schulze S, Huang H-H, Beckmann ND, Nirenberg S, Wang B, et al. An Inflammatory Cytokine Signature Predicts COVID-19 Severity and Survival. Nat Med (2020) 26(10):1636–43. doi: 10.1038/s41591-020-1051-9
13. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/s0140-6736(20)30183-5
14. Liu J, Li S, Liu J, Liang B, Wang X, Wang H, et al. Longitudinal Characteristics of Lymphocyte Responses and Cytokine Profiles in the Peripheral Blood of SARS-CoV-2 Infected Patients. EBioMedicine (2020) 55:102763. doi: 10.1016/j.ebiom.2020.102763
15. Alosaimi B, Hamed ME, Naeem A, Alsharef AA, AlQahtani SY, AlDosari KM, et al. Mers-CoV Infection Is Associated With Downregulation of Genes Encoding Th1 and Th2 Cytokines/Chemokines and Elevated Inflammatory Innate Immune Response in the Lower Respiratory Tract. Cytokine (2020) 126:154895. doi: 10.1016/j.cyto.2019.154895
16. Reghunathan R, Jayapal M, Hsu L-Y, Chng H-H, Tai D, Leung BP, et al. Expression Profile of Immune Response Genes in Patients With Severe Acute Respiratory Syndrome. BMC Immunol (2005) 6(1):2. doi: 10.1186/1471-2172-6-2
17. Marchetti M. Covid-19-Driven Endothelial Damage: Complement, HIF-1, and ABL2 Are Potential Pathways of Damage and Targets for Cure. Ann Hematol (2020) 99(8):1701–07. doi: 10.1007/s00277-020-04138-8
18. Cserhalmi M, Papp A, Brandus B, Uzonyi B, Józsi M. Regulation of Regulators: Role of the Complement Factor H-related Proteins. Semin Immunol (2019) 45:101341. doi: 10.1016/j.smim.2019.101341
19. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement System Part II: Role in Immunity. [Review]. Front Immunol (2015) 6:257. doi: 10.3389/fimmu.2015.00257
20. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The Renaissance of Complement Therapeutics. Nat Rev Nephrol (2018) 14(1):26–47. doi: 10.1038/nrneph.2017.156
21. Fletcher-Sandersjöö A, Bellander B-M. Is COVID-19 Associated Thrombosis Caused by Overactivation of the Complement Cascade? A Literature Review. Thromb Res (2020) 194:36–41. doi: 10.1016/j.thromres.2020.06.027
22. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Trans Res: J Lab Clin Med (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
23. Noris M, Benigni A, Remuzzi G. The Case of Complement Activation in COVID-19 Multiorgan Impact. Kidney Int (2020) 98(2):314–22. doi: 10.1016/j.kint.2020.05.013
24. Wang R, Xiao H, Guo R, Li Y, Shen B. The Role of C5a in Acute Lung Injury Induced by Highly Pathogenic Viral Infections. Emerg Microbes Infect (2015) 4(5):e28–8. doi: 10.1038/emi.2015.28
25. Ohta R, Torii Y, Imai M, Kimura H, Okada N, Ito Y. Serum Concentrations of Complement Anaphylatoxins and Proinflammatory Mediators in Patients With 2009 H1N1 Influenza. Microbiol Immunol (2011) 55(3):191–8. doi: 10.1111/j.1348-0421.2011.00309.x
26. Noris M, Remuzzi G. Overview of Complement Activation and Regulation. Semin Nephrol (2013) 33(6):479–92. doi: 10.1016/j.semnephrol.2013.08.001
27. Stahel PF, Barnum SR. Complement Inhibition in Coronavirus Disease (Covid)-19: A Neglected Therapeutic Option. [Opinion]. Front Immunol (2020) 11:1661. doi: 10.3389/fimmu.2020.01661
28. Huber-Lang M, Sarma VJ, Lu KT, McGuire SR, Padgaonkar VA, Guo RF, et al. Role of C5a in Multiorgan Failure During Sepsis. J Immunol (2001) 166(2):1193–99. doi: 10.4049/jimmunol.166.2.1193
29. Woodruff TM, Nandakumar KS, Tedesco F. Inhibiting the C5-C5a Receptor Axis. Mol Immunol (2011) 48(14):1631–42. doi: 10.1016/j.molimm.2011.04.014
30. Franks TJ, Chong PY, Chui P, Galvin JR, Lourens RM, Reid AH, et al. Lung Pathology of Severe Acute Respiratory Syndrome (SARS): A Study of 8 Autopsy Cases From Singapore. Hum Pathol (2003) 34(8):743–48. doi: 10.1016/s0046-8177(03)00367-8
31. Hu Y, Jin Y, Han D, Zhang G, Cao S, Xie J, et al. Mast Cell-Induced Lung Injury in Mice Infected With H5N1 Influenza Virus. J Virol (2012) 86(6):3347–56. doi: 10.1128/jvi.06053-11
32. Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, et al. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio (2018) 9(5):e01753-18. doi: 10.1128/mBio.01753-18
33. Ghannam A, Pernollet M, Fauquert JL, Monnier N, Ponard D, Villiers MB, et al. Human C3 Deficiency Associated With Impairments in Dendritic Cell Differentiation, Memory B Cells, and Regulatory T Cells. J Immunol (2008) 181(7):5158–66. doi: 10.4049/jimmunol.181.7.5158
34. Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of Signaling Into CD4+ Cells Via C3aR and C5aR Enables Autoinductive TGF-β1 Signaling and Induction of Foxp3+ Regulatory T Cells. Nat Immunol (2013) 14(2):162–71. doi: 10.1038/ni.2499
35. Gao T, Hu M, Zhang X, Li H, Zhu L, Liu H, et al. Highly Pathogenic Coronavirus N Protein Aggravates Lung Injury by MASP-2-Mediated Complement Over-Activation. medRxiv (2020). doi: 10.1101/2020.03.29.20041962
36. Cugno M, Meroni PL, Gualtierotti R, Griffini S, Grovetti E, Torri A, et al. Complement Activation in Patients With COVID-19: A Novel Therapeutic Target. J Allergy Clin Immunol (2020) 146(1):215–17. doi: 10.1016/j.jaci.2020.05.006
37. Jiang Y, Zhao G, Song N, Li P, Chen Y, Guo Y, et al. Blockade of the C5a–C5aR Axis Alleviates Lung Damage in hDPP4-Transgenic Mice Infected With MERS-Cov. Emerg Microbes Infect (2018b) 7(1):1–12. doi: 10.1038/s41426-018-0063-8
38. Lam LM, Murphy SJ, Kuri-Cervantes L, Weisman AR, Ittner CAG, Reilly JP, et al. Erythrocytes Reveal Complement Activation in Patients With COVID-19. medRxiv (2020). doi: 10.1101/2020.05.20.20104398
39. Jiang Y, Zhao G, Song N, Li P, Chen Y, Guo Y, et al. Blockade of the C5a-C5aR Axis Alleviates Lung Damage in hDPP4-Transgenic Mice Infected With MERS-Cov. Emerg Microbes Infect (2018a) 7(1):77–7. doi: 10.1038/s41426-018-0063-8
40. Diao B, Wang C, Wang R, Feng Z, Tan Y, Wang H, et al. Human Kidney Is a Target for Novel Severe Acute Respiratory Syndrome Coronavirus 2 (Sars-CoV-2) Infection. medRxiv (2020). doi: 10.1101/2020.03.04.20031120
41. Kipshidze N, Dangas G, White CJ, Kipshidze N, Siddiqui F, Lattimer CR, et al. Viral Coagulopathy in Patients With COVID-19: Treatment and Care. Clin Appl Thromb Hemost (2020) 26:1076029620936776. doi: 10.1177/1076029620936776
42. Chauhan AJ, Wiffen LJ, Brown TP. Covid-19: A Collision of Complement, Coagulation and Inflammatory Pathways. J Thromb Haemost (2020). doi: 10.1111/jth.14981
43. Lo MW, Kemper C, Woodruff TM. Covid-19: Complement, Coagulation, and Collateral Damage. J Immunol (2020) 205(6):1488–95. doi: 10.4049/jimmunol.2000644
44. Yu J, Yuan X, Chen H, Chaturvedi S, Braunstein EM, Brodsky RA. Direct Activation of the Alternative Complement Pathway by SARS-CoV-2 Spike Proteins Is Blocked by Factor D Inhibition. Blood (2020) 136(18):2080–89. doi: 10.1182/blood.2020008248
45. Diurno F, Numis FG, Porta G, Cirillo F, Maddaluno S, Ragozzino A, et al. Eculizumab Treatment in Patients With COVID-19: Preliminary Results From Real Life ASL Napoli 2 Nord Experience. Eur Rev Med Pharmacol Sci (2020) 24(7):4040–7. doi: 10.26355/eurrev_202004_20875
46. Subramaniam S, Jurk K, Hobohm L, Jäckel S, Saffarzadeh M, Schwierczek K, et al. Distinct Contributions of Complement Factors to Platelet Activation and Fibrin Formation in Venous Thrombus Development. Blood (2017) 129(16):2291–302. doi: 10.1182/blood-2016-11-749879
47. Costela-Ruiz VJ, Illescas-Montes R, Puerta-Puerta JM, Ruiz C, Melguizo-Rodríguez L. Sars-CoV-2 Infection: The Role of Cytokines in COVID-19 Disease. Cytokine Growth Factor Rev (2020) 54:62–75. doi: 10.1016/j.cytogfr.2020.06.001
48. Chen L, Liu HG, Liu W, Liu J, Liu K, Shang J, et al. Analysis of Clinical Features of 29 Patients With 2019 Novel Coronavirus Pneumonia. Zhonghua Jie He He Hu Xi Za Zhi (2020) 43(0):E005. doi: 10.3760/cma.j.issn.1001-0939.2020.0005
49. Wong CK, Lam CW, Wu AK, Ip WK, Lee NL, Chan IH, et al. Plasma Inflammatory Cytokines and Chemokines in Severe Acute Respiratory Syndrome. Clin Exp Immunol (2004) 136(1):95–103. doi: 10.1111/j.1365-2249.2004.02415.x
50. Mahallawi WH, Khabour OF, Zhang Q, Makhdoum HM, Suliman BA. Mers-CoV Infection in Humans Is Associated With a Pro-Inflammatory Th1 and Th17 Cytokine Profile. Cytokine (2018) 104:8–13. doi: 10.1016/j.cyto.2018.01.025
51. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and Immunological Features of Severe and Moderate Coronavirus Disease 2019. J Clin Invest (2020) 130(5):2620–29. doi: 10.1172/jci137244
52. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (Covid-19) in Wuhan, China. Clin Infect Dis (2020) 71(15):762–68. doi: 10.1093/cid/ciaa248
53. Seo SH, Webster RG. Tumor Necrosis Factor Alpha Exerts Powerful Anti-Influenza Virus Effects in Lung Epithelial Cells. J Virol (2002) 76(3):1071–76. doi: 10.1128/jvi.76.3.1071-1076.2002
54. Pasquereau S, Kumar A, Herbein G. Targeting TNF and TNF Receptor Pathway in HIV-1 Infection: From Immune Activation to Viral Reservoirs. Viruses (2017) 9(4):64. doi: 10.3390/v9040064
55. Wan S, Yi Q, Fan S, Lv J, Zhang X, Guo L, et al. Characteristics of Lymphocyte Subsets and Cytokines in Peripheral Blood of 123 Hospitalized Patients With 2019 Novel Coronavirus Pneumonia (NCP). medRxiv (2020). doi: 10.1101/2020.02.10.20021832
56. Law HK, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W, et al. Chemokine Up-Regulation in SARS-Coronavirus-Infected, Monocyte-Derived Human Dendritic Cells. Blood (2005) 106(7):2366–74. doi: 10.1182/blood-2004-10-4166
57. Zhang R, Wang X, Ni L, Di X, Ma B, Niu S, et al. Covid-19: Melatonin as a Potential Adjuvant Treatment. Life Sci (2020) 250:117583. doi: 10.1016/j.lfs.2020.117583
58. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell (2021) 184(1):149–68 e117. doi: 10.1016/j.cell.2020.11.025
59. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and Chemokines: At the Crossroads of Cell Signalling and Inflammatory Disease. Biochim Biophys Acta (2014) 1843(11):2563–82. doi: 10.1016/j.bbamcr.2014.05.014
60. DeDiego ML, Nieto-Torres JL, Regla-Nava JA, Jimenez-Guardeño JM, Fernandez-Delgado R, Fett C, et al. Inhibition of NF-κb-Mediated Inflammation in Severe Acute Respiratory Syndrome Coronavirus-Infected Mice Increases Survival. J Virol (2014) 88(2):913–24. doi: 10.1128/jvi.02576-13
61. Nieto-Torres JL, DeDiego ML, Verdiá-Báguena C, Jimenez-Guardeño JM, Regla-Nava JA, Fernandez-Delgado R, et al. Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis. PloS Pathog (2014) 10(5):e1004077. doi: 10.1371/journal.ppat.1004077
62. Siu KL, Yuen KS, Castaño-Rodriguez C, Ye ZW, Yeung ML, Fung SY, et al. Severe Acute Respiratory Syndrome Coronavirus ORF3a Protein Activates the NLRP3 Inflammasome by Promoting TRAF3-Dependent Ubiquitination of ASC. FASEB J (2019) 33(8):8865–77. doi: 10.1096/fj.201802418R
63. Conti P, Ronconi G, Caraffa A, Gallenga CE, Ross R, Frydas I, et al. Induction of Pro-Inflammatory Cytokines (IL-1 and IL-6) and Lung Inflammation by Coronavirus-19 (Covi-19 or SARS-CoV-2): Anti-Inflammatory Strategies. J Biol Regul Homeost Agents (2020) 34(2):327–31. doi: 10.23812/conti-e
64. Zhang W, Zhao Y, Zhang F, Wang Q, Li T, Liu Z, et al. The Use of Anti-Inflammatory Drugs in the Treatment of People With Severe Coronavirus Disease 2019 (COVID-19): The Perspectives of Clinical Immunologists From China. Clin Immunol (2020) 214:108393. doi: 10.1016/j.clim.2020.108393
65. Kritas SK, Ronconi G, Caraffa A, Gallenga CE, Ross R, Conti P. Mast Cells Contribute to Coronavirus-Induced Inflammation: New Anti-Inflammatory Strategy. J Biol Regul Homeost Agents (2020) 34(1):9–14. doi: 10.23812/20-Editorial-Kritas
66. Zalinger ZB, Elliott R, Weiss SR. Role of the Inflammasome-Related Cytokines IL-1 and IL-18 During Infection With Murine Coronavirus. J Neurovirol (2017) 23(6):845–54. doi: 10.1007/s13365-017-0574-4
67. Ferreira AC, Soares VC, de Azevedo-Quintanilha IG, Dias S, Fintelman-Rodrigues N, Sacramento CQ, et al. SARS-CoV-2 Engages Inflammasome and Pyroptosis in Human Primary Monocytes. Cell Death Discov (2021) 7(1):43. doi: 10.1038/s41420-021-00428-w
68. Cauchois R, Koubi M, Delarbre D, Manet C, Carvelli J, Blasco VB, et al. Early IL-1 Receptor Blockade in Severe Inflammatory Respiratory Failure Complicating COVID-19. Proc Natl Acad Sci USA (2020) 117(32):18951–3. doi: 10.1073/pnas.2009017117
69. Luo P, Liu Y, Qiu L, Liu X, Liu D, Li J. Tocilizumab Treatment in COVID-19: A Single Center Experience. J Med Virol (2020) 92(7):814–18. doi: 10.1002/jmv.25801
70. Wang Z, Yang B, Li Q, Wen L, Zhang R. Clinical Features of 69 Cases With Coronavirus Disease 2019 in Wuhan, China. Clin Infect Dis (2020) 71(15):769–77. doi: 10.1093/cid/ciaa272
71. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients With COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet (2020) 395(10229):1054–62. doi: 10.1016/s0140-6736(20)30566-3
72. Zhou Y, Fu B, Zheng X, Wang D, Zhao C, qi Y, et al. Aberrant Pathogenic GM-CSF+T Cells and Inflammatory CD14+CD16+ Monocytes in Severe Pulmonary Syndrome Patients of a New Coronavirus. bioRxiv (2020). doi: 10.1101/2020.02.12.945576
73. Baggiolini M, Walz A, Kunkel S. Neutrophil-Activating Peptide-1/Interleukin 8, a Novel Cytokine That Activates Neutrophils. J Clin Invest (1989) 84(4):1045–49. doi: 10.1172/JCI114265
74. Drosten C, Seilmaier M, Corman VM, Hartmann W, Scheible G, Sack S, et al. Clinical Features and Virological Analysis of a Case of Middle East Respiratory Syndrome Coronavirus Infection. Lancet Infect Dis (2013) 13(9):745–51. doi: 10.1016/s1473-3099(13)70154-3
75. Lew TW, Kwek TK, Tai D, Earnest A, Loo S, Singh K, et al. Acute Respiratory Distress Syndrome in Critically Ill Patients With Severe Acute Respiratory Syndrome. JAMA (2003) 290(3):374–80. doi: 10.1001/jama.290.3.374
76. Folkesson HG, Matthay MA, Hébert CA, Broaddus VC. Acid Aspiration-Induced Lung Injury in Rabbits Is Mediated by Interleukin-8-Dependent Mechanisms. J Clin Invest (1995) 96(1):107–16. doi: 10.1172/jci118009
77. Alturaiki WH. Evaluation of C-C Chemokine Ligand 5 (Ccl5) Chemokine, Interleukin 5 (IL-5) Cytokine, and Eosinophil Counts as Potential Biomarkers in Saudi Patients With Chronic Asthma During Sandstorms. Cureus (2020) 12(4):e7809. doi: 10.7759/cureus.7809
78. Marques RE, Guabiraba R, Russo RC, Teixeira MM. Targeting CCL5 in Inflammation. Expert Opin Ther Tar (2013) 17(12):1439–60. doi: 10.1517/14728222.2013.837886
79. Zhao Y, Qin L, Zhang P, Li K, Liang L, Sun J, et al. Longitudinal COVID-19 Profiling Associates IL-1RA and IL-10 With Disease Severity and RANTES With Mild Disease. JCI Insight (2020) 5(13):e139834. doi: 10.1172/jci.insight.139834
80. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With COVID-19. Nat Med (2020) 26(6):842–44. doi: 10.1038/s41591-020-0901-9
81. Thevarajan I, Nguyen THO, Koutsakos M, Druce J, Caly L, van de Sandt CE, et al. Breadth of Concomitant Immune Responses Prior to Patient Recovery: A Case Report of Non-Severe COVID-19. Nat Med (2020) 26(4):453–55. doi: 10.1038/s41591-020-0819-2
82. Zhao J, Yuan Q, Wang H, Liu W, Liao X, Su Y, et al. Antibody Responses to SARS-CoV-2 in Patients With Novel Coronavirus Disease 2019. Clin Infect Dis (2020) 71(16):2027–34. doi: 10.1093/cid/ciaa344
83. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (Covid-19). [Original Research]. Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
84. Yang L, Gou J, Gao J, Huang L, Zhu Z, Ji S, et al. Immune Characteristics of Severe and Critical COVID-19 Patients. Signal Transduct Target Ther (2020) 5(1):179. doi: 10.1038/s41392-020-00296-3
85. Chang FY, Chen HC, Chen PJ, Ho MS, Hsieh SL, Lin JC, et al. Immunologic Aspects of Characteristics, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19). J BioMed Sci (2020) 27(1):72. doi: 10.1186/s12929-020-00663-w
86. Murphy BR, Whitehead SS. Immune Response to Dengue Virus and Prospects for a Vaccine. Annu Rev Immunol (2011) 29:587–619. doi: 10.1146/annurev-immunol-031210-101315
Keywords: COVID-19, SARS‐CoV‐2, complement anaphylatoxins, inflammatory cytokines, in-hospital mortality, prognosis
Citation: Alosaimi B, Mubarak A, Hamed ME, Almutairi AZ, Alrashed AA, AlJuryyan A, Enani M, Alenzi FQ and Alturaiki W (2021) Complement Anaphylatoxins and Inflammatory Cytokines as Prognostic Markers for COVID-19 Severity and In-Hospital Mortality. Front. Immunol. 12:668725. doi: 10.3389/fimmu.2021.668725
Received: 17 February 2021; Accepted: 28 May 2021;
Published: 01 July 2021.
Edited by:
Tengchuan Jin, University of Science and Technology of China, ChinaReviewed by:
Rksubbarao Rksubbarao, St. Jude Children’s Research Hospital, United StatesMisbah Abbas, University of Science and Technology of China, China
Copyright © 2021 Alosaimi, Mubarak, Hamed, Almutairi, Alrashed, AlJuryyan, Enani, Alenzi and Alturaiki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wael Alturaiki, dy5hbHR1cmFpa2lAbXUuZWR1LnNh