 Yaoyao Xia
Yaoyao Xia Yikun Li2†
Yikun Li2† Xiaoyan Wu
Xiaoyan Wu Siyuan Chen
Siyuan Chen Xianyong Ma
Xianyong Ma Miao Yu
Miao Yu- 1State Key Laboratory of Livestock and Poultry Breeding, Key Laboratory of Animal Nutrition and Feed Science in South China, Ministry of Agriculture and Rural Affairs, Guangdong Provincial Key Laboratory of Animal Breeding and Nutrition, Guangdong Engineering Technology Research Center of Animal Meat Quality and Safety Control and Evaluation, Institute of Animal Science, Guangdong Academy of Agricultural Sciences, Guangzhou, China
- 2College of Animal Science, South China Agricultural University, Guangzhou, China
- 3Maoming Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Guangzhou, China
Iron fine-tunes innate immune responses, including macrophage inflammation. In this review, we summarize the current understanding about the iron in dictating macrophage polarization. Mechanistically, iron orchestrates macrophage polarization through several aspects, including cellular signaling, cellular metabolism, and epigenetic regulation. Therefore, iron modulates the development and progression of multiple macrophage-associated diseases, such as cancer, atherosclerosis, and liver diseases. Collectively, this review highlights the crucial role of iron for macrophage polarization, and indicates the potential application of iron supplementation as an adjuvant therapy in different inflammatory disorders relative to the balance of macrophage polarization.
Introduction
As an essential microelement, iron is involved in cell proliferation, metabolism and differentiation (1). Generally, recycling iron from red blood cells (RBCs) is essential for maintaining the quantity and function of RBCs. In mammals, approximately 95% of the iron required for physiological processes is dependent on macrophage iron recycling via a series of complex processes (2–7). Of note, the roles of macrophages in regulating iron homeostasis have been well addressed (8, 9).
Reciprocally, iron modulates macrophage fate and function, like the development and differentiation of tissue-resident macrophages (10–18), the function of macrophages in hematopoiesis (19–22), and macrophage-mediated inflammatory responses during pathogen infections (23–28). Noteworthily, in addition to their homeostatic activities, macrophages polarize into disparate phenotypes in response to external signals and stimuli to exert different functions (e.g., regulating the progression of diseases) (29). Numerous regulators, including cellular pathway, intracellular metabolism, and epigenetic regulation contribute to the polarization of macrophages, which have been well summarized elsewhere (30–32). Therefore, in this review, we specifically focus on whether and how iron shapes macrophage polarization.
Regulation of Iron in Macrophage Polarization
It is well accepted that, under normal conditions, iron always binds with ferritin (Ft) in macrophages (33); nevertheless, iron homeostasis and expression of iron-regulated genes strikingly shift during macrophage polarization. For example, in classically-activated macrophages (M1-like, pro-inflammatory phenotype), expression of Hamp (encoding for hepcidin) and FtH/FtL (encoding for ferritin) are highly up-regulated; while FPN (encoding for ferroportin) and IRP1/2 (iron regulatory proteins) are down-regulated (34, 35). Such different expression of Fe-regulated genes in polarized macrophages suggests that iron might be associated with macrophage activation.
Notably, acute iron deprivation decreases expression of activating transcription factor 4 (ATF4) in resting human macrophages, and reduces interleukin (IL)-1β and tumor necrosis factor (TNF)-α expression in human macrophages following LPS treatment (36). Moreover, iron depletion blocks NF-κB activation in rat alveolar macrophage stimulated by LPS plus TNF-α (37). These fascinating findings indicate that iron may be necessary for M1-like macrophage polarization. However, more investigations are needed to support this notion as only few studies focusing on the iron deprivation in modulating macrophage activation (Table 1). Moreover, the current studies seem to be inclined to investigate the effects of iron depletion on M1-like macrophage polarization; therefore, it is interesting to have a comprehensive understanding about iron deprivation on macrophage polarization [e.g., alternatively-activated macrophage polarization (M2-like, anti-inflammatory phenotype)] in the future.
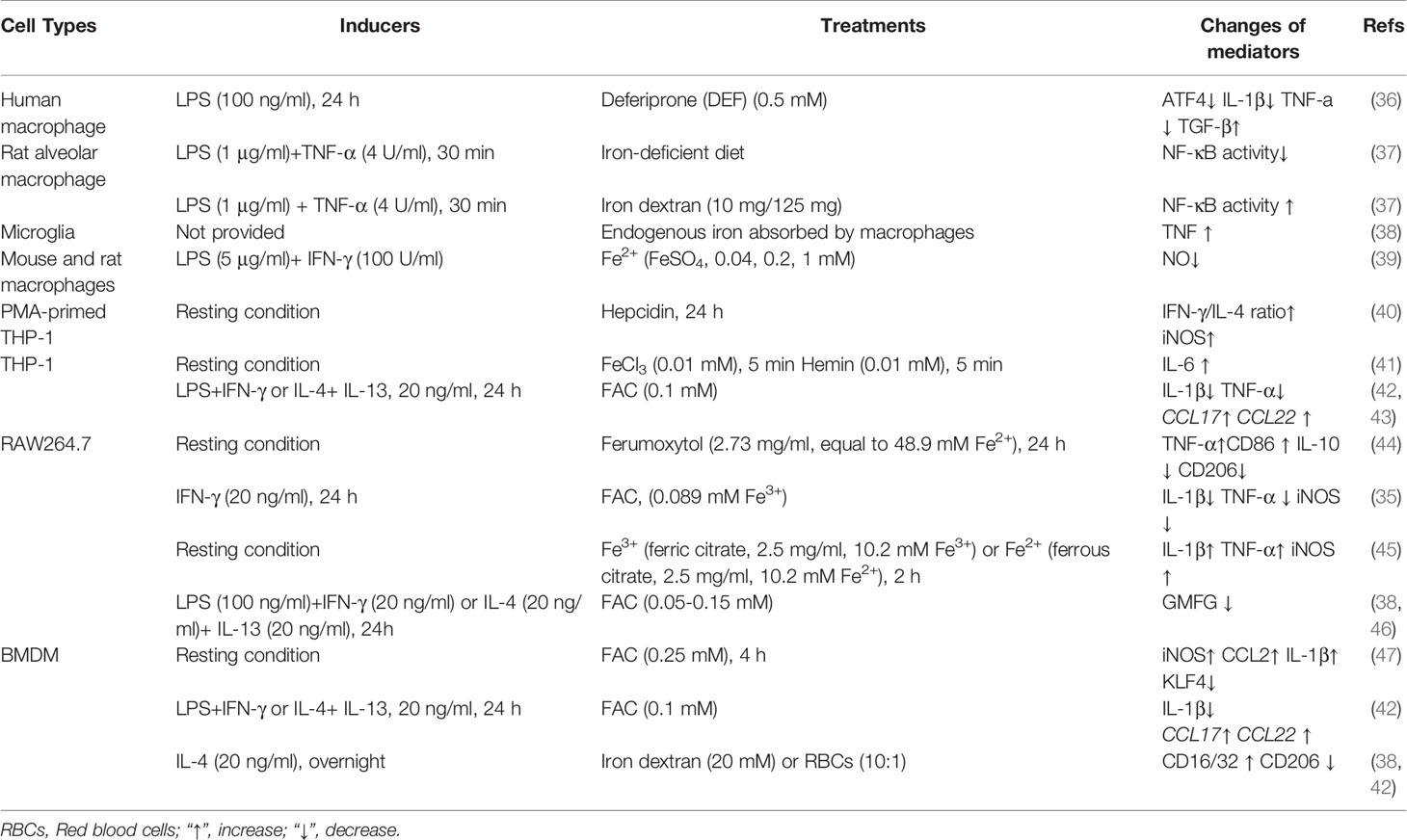
Table 1 Iron affects macrophage phenotypes.
Interestingly, iron supplementation also affects macrophage phenotypes. Increasing endogenous iron induces TNF production in macrophages (38). Besides, exogenous iron supplementation, like ferric ammonium citrate (FAC) (0.25 mM Fe3+), ferric citrate (2.5 mg/ml, equal to 10.2 mM Fe3+), ferrous citrate (2.5 mg/ml, equal to 10.16 mM Fe2+), and Ferumoxytol (Fe3H2O4) (2.73 mg Fe/ml, equal to 48.9 mM Fe2+), promotes the expression of M1 markers (e.g., iNOS, TNF-α, and IL-1β), while lowers the expression of M2 markers (e.g., IL-10 and CD206) in RAW264.7 cells (44, 45), bone marrow-derived macrophages (BMDMs) (47), and THP-1 cells (40, 41). In addition to the in vitro experiments, some in vivo experiments also confirm that iron supplementation enhances pro-inflammatory macrophage polarization (37, 47).
The roles of iron in regulating pro-inflammatory macrophages are not always consistent. In IFN-γ-stimulated RAW264.7 cells, FAC supplementation (25 μg/ml, equal to 0.089 mM Fe3+) lowers the expression of M1 markers (e.g., IL-1β, iNOS, and TNF-α) by blocking signal transducer and activator of transcription (STAT)-1 pathway (35). Moreover, supplementation of Fe2+ (FeSO4, 0~1 mM) reduces LPS/IFN-γ-induced NO synthesis in mouse and rat macrophages (39). The following factors may be conceivably involved in the regulation of the final polarization trends of macrophages: (i) the final iron (Fe2+ and/or Fe3+) concentrations and different processing time (48); (ii) the macrophage types (e.g., RAW264.7 cells, THP-1 cells, and BMDMs); (iii) the various conditions of stimuli/inducers for macrophage polarization; (iv) pretreatment of iron or treatment of iron during macrophage polarization simultaneously. Hence, it is worth conducting experiments to validate the aforementioned speculations, as well as the genders of experimental animals or other factors associating the various results of macrophage polarization status under iron treatment.
In contrast to classically-activated macrophages, alternatively-activated macrophages are dominated by low Ft and high FPN, and the high heme oxygenase (HO)-1 expression promotes the absorption of heme and the excretion of free iron by alternatively-activated macrophages (49). Therefore, iron homeostasis may be also closely related to the functions of M2-like macrophages, which has been validated in models of diabetes (42), spinal cord injuries (SCI) (38), and cancers (44, 45). For example, tumor-associated macrophages (TAMs) are defined as M2-like macrophages that promote the progression of tumor and are associated with poor prognosis (50–52). In tumor-burden BALB/C mice, ferric citrate (2.5 mg per three day, totally 17.5 mg) injection promotes reactive oxygen species (ROS) production and CD86 expression of tumor infiltrating macrophages, and lowers the volume and weight of H22 hepatoma xenograft (45). Likewise, intravenous injection of ferumoxytol (10 mg Fe/kg) to mice profoundly inhibits the function of M2-like macrophages while enhances the function of M1-like macrophages in tumorous tissues (44). Collectively, these findings suggest that iron negatively regulates M2-like macrophage polarization.
In some situations, iron may promote M2-like macrophage polarization. For example, exogenous iron supplementation (FAC, 0.1 mM) and endogenic iron-rich human dermal fibroblasts extracellular matrix (ECM) both enhance THP-1 cells polarizing into M2 phenotype (42). Similar finding is also observed in BMDMs (42). Interestingly, iron status causes different inflammatory response outcomes (44, 53, 54). For example, iron deprivation lowers the expression of M2 markers, while an iron-rich status (under physiological iron loading condition) favors M2-like macrophage polarization and inhibits LPS-induced inflammatory responses, which might be highly associated with the differential regulation of Hamp and FPN in the context of various inflammatory conditions (55). These conflicting results suggest that iron might regulate macrophage polarization by affecting different signal pathways based on different models. Therefore, the underlying mechanisms by which iron shapes the macrophage polarization deserve further exploration.
In summary, in most cases (concerning nontumorous and/or tumorous tissues), iron affects macrophage phenotypes by promoting classically-activated macrophage functions [especially high iron addition (>0.1 mM Fe3+ and/or >1 mM Fe2+)], while decreasing alternatively-activated macrophage functions (Table 1). It should be noticed that some effects seen are probably the related stress responses rather than iron-driven macrophage polarization (43). The differences in the direction of macrophage polarization caused by iron deficiency, iron supplementation or overload also reflect that the cellular signaling and/or metabolic pathways underlying iron in shaping macrophage polarization are complex and diverse, and the specific mechanisms remain to be explored.
Mechanisms Whereby Iron Mediates Macrophage Polarization
Iron Shapes Macrophage Polarization Through Cellular Signaling Pathways
Within multifarious pathways associating with functions of macrophages, NF-κB functions as a main contributor to regulate macrophage polarization (30, 56). Iron deprivation shapes macrophage polarization through NF-κB involving distinct mechanisms. Interestingly, macrophages isolated from iron deficient rats have reduced Nox activity while iron depletion blocks the activation of NF-κB in LPS+TNF-α-activated macrophages (37), implying the suppression of classically-activated macrophage polarization by iron deprivation (Figure 1A). However, LPS/IFN-γ-polarized macrophages with NADPH oxidase (Nox) 4 deficiency [a member of Nox family (57)] express more Nox2 to enhance p65 nuclear translocation and NF-κB activity to force pro-inflammatory macrophage polarization (58), indicating that there might be another mechanism involving NF-κB for iron deprivation to affect M1-like macrophage polarization (Figure 1B). Here, we speculate that these conflicting results might be associated with the different culture conditions and/or the levels of Nox2; however, the aforementioned process still needs well-designed experimental validation.
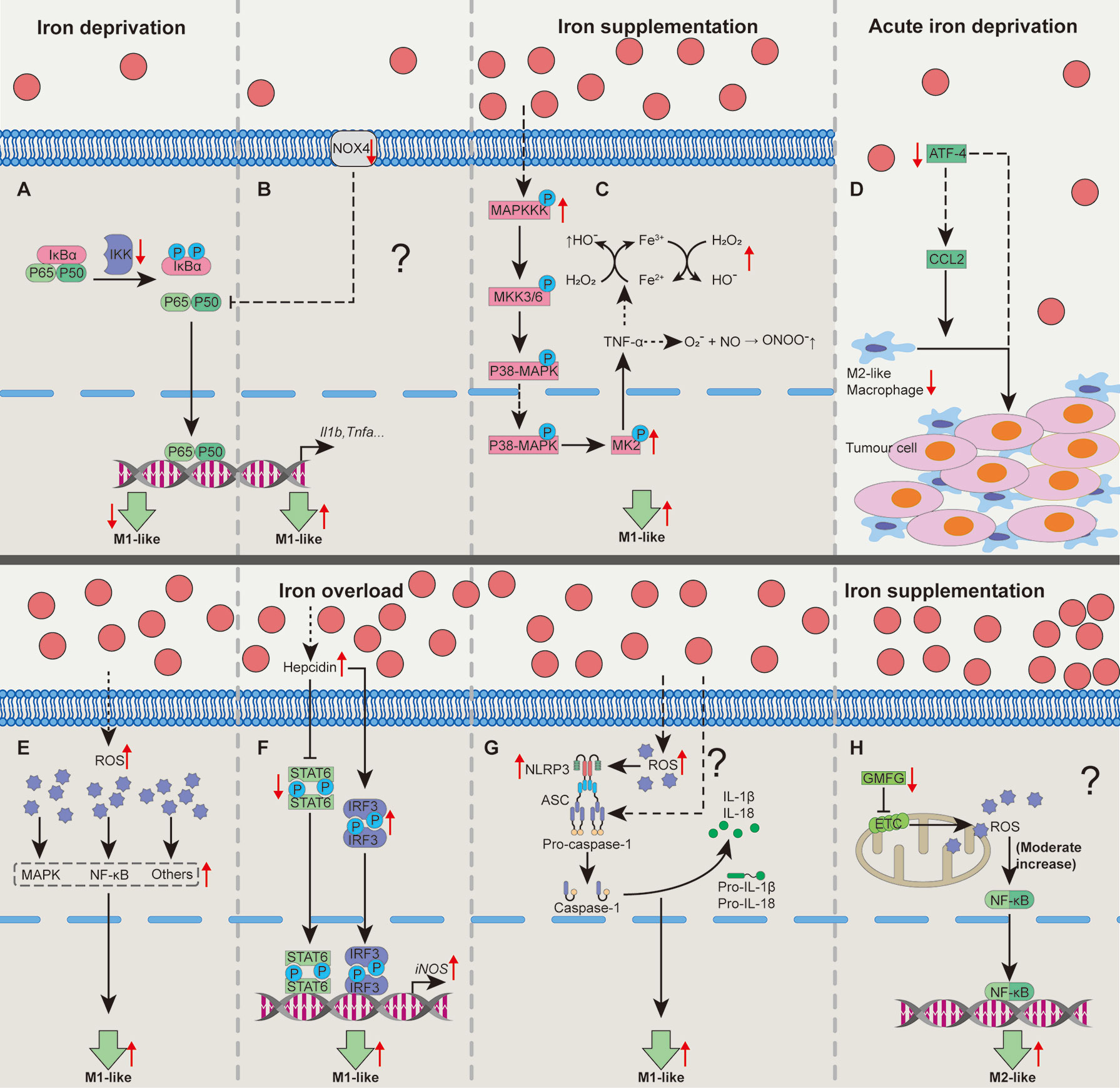
Figure 1 Cellular pathways whereby iron shapes macrophage polarization. Iron deprivation may suppress NF-κB activation to block M1-like macrophage polarization (A). In some special cases, iron deprivation could lower the activity of Nox to promote p65 nuclear translocation and increases NF-κB activity, promoting M1-like macrophage polarization (B). Iron treatment could activate MAPK-p-MK2 pathway to increase TNF-α production to generate a large amount of OH• and ONOO•, promoting M1-like macrophage polarization (C). Acute iron deprivation inhibits ATF4 expression to suppress M2-like macrophage polarization in tumorous tissues (D). Iron treatment may promote M1-like macrophage polarization by inducing the activation of MAPK, NF-κB and other proinflammatory signaling pathways via increasing the production of intracellular ROS (E). Iron overload increases hepcidin expression which can inhibit STAT6 activation while promote IRF3 expression to increase iNOS expression in M1-like macrophages (F). Iron can activate NLRP3 inflammasome to promote M1-like macrophage polarization by the generation of ROS (G). Iron might enhance M2-like macrophage polarization through moderately activating of mtROS cascading with NF-κB pathway by down-regulating GMFG level (H). ATF4, activating transcription factor 4; GMFG, glia maturation factor-γ; IRF3, interferon regulatory factor 3; MAPK, mitogen-activated protein kinase; MK2, kinase 2; NF-κB, nuclear factor κB; NLRP3, NOD‐like receptor; Nox, NADPH oxidase; ROS, reactive oxygen species; STAT6, signal transducer and activator of transcription 6. “↑”, increase; “↓”, decrease. Question mark means that the mechanism needs experimental confirmation.
Iron also shapes macrophage polarization through mitogen-activated protein kinase (MAPK). Iron supplementation or intracellular iron accumulation increases TNF-α expression (38, 44, 54). Mechanistically, iron accumulation enhances expression of MAPK, leading to the phosphorylation of kinase 2 (MK2) to trigger TNF-α production (38, 59). TNF-α continuously stimulates macrophages to produce a large number of hydroxyl radicals (OH•) and peroxynitrite (ONOO•) (54, 60). Thus, iron treatment may promote M1-like macrophage polarization through MAPK-p-MK2 axis (Figure 1C). Lipocalin-2 (Lcn-2) combined with siderophore-bound iron can be absorbed by macrophages (61). Interestingly, Lcn-2 deficiency reduces M1 polarization in BMDMs (62). Mechanistically, Lcn-2 activates ERK1/2 to shape macrophage towards M1-like phenotype (63). However, whether Lcn-2-mediated intracellular iron accumulation causes M1-like macrophage polarization is still lacking the direct evidence.
Other cellular signaling pathways include ATF4, ROS, and NLRP3 inflammasome. ATF4 expression is correlated with the percentage of M2-like macrophages (64, 65). Notably, acute iron deprivation decreases ATF4 (36), indicating that iron deprivation could block M2-like macrophage polarization (Figure 1D). Furthermore, iron treatment may promote M1-like macrophage polarization by increasing the production of intracellular ROS (Figure 1E) (38, 45, 66). Iron overload increases hepcidin expression (45), which inhibits STAT6 activation while promotes interferon regulatory factor (IRF)-3 expression (40), resulting in pro-inflammatory macrophage polarization (Figure 1F). NLRP3 inflammasome (mainly including NLRP3, ASC, Caspase-1, and IL-1β) functions as a crucial platform for M1-like macrophage polarization (30). Of note, iron overload can activate NLRP3 inflammasome through the generation of ROS, and some unrevealed mechanisms (67–69), thereby enhancing M1-like phenotype (Figure 1G).
On the contrary, iron supplementation may also inhibit M1-like polarization by suppressing STAT1 activation (35). Iron supplementation down-regulates actin-regulatory protein glia maturation factor-γ (GMFG), which is negatively associated with the mitochondrial ROS (mtROS) accumulation (46). The moderate increase of mtROS cascading with NF-κB pathway is needed for M2-like macrophage polarization (70); hence, iron supplementation might also shape M2-like macrophage polarization associating with GMFG/mtROS/NF-κB pathway (Figure 1H). Therefore, iron supplementation and/or overload affect macrophage polarization depends on various cellular pathways (Figure 1). And the possible reasons for these different effects of iron in guiding macrophage polarization are largely related to cellular iron status (e.g., marginal or high iron supplementation) and/or heterogeneity of tissue microenvironment.
Iron Shapes Macrophage Polarization Through Cellular Metabolism
The cellular metabolism is highly associated with the functional output of macrophages (31). Unfortunately, there are only several studies concerning the cellular mechanisms whereby iron mediates macrophage polarization. Of note, although acute iron deprivation in human macrophages without pro-inflammatory stimuli (resting macrophages) share the same metabolic switch with pro-inflammatory stimuli (e.g., LPS and/or IFN-γ)-treated macrophages (an induction of glycolysis and fatty acid synthesis, and a repression of OXPHOS), the underlying mechanisms differ (36). Mechanistically, iron deprivation directly inhibits the mRNA expression and protein abundance of respiratory chain Fe-S enzymes (including NDUFS6 and SDHB) and blocks the activity of mitochondrial aconitase to cause citrate accumulation (36), thereby potentiating M1-like phenotype (Figure 2A); nevertheless, LPS-induced M1-like macrophage polarization is limited in iron-deprived human macrophages due to the increase of itaconate:succinate ratio (Figure 2B) (36). Prolyl hydroxylase (PHD), as a key factor in the metabolic pathways, is responsible for M1-like macrophage polarization via post-translational modification of IKKβ and stabilization of HIF‐1α (71–74). Notably, lack of iron inhibits PHD proteins, plausibly explaining how iron deprivation facilitates M1-like macrophage polarization in some specific situation (e.g., acute iron deprivation). Interestingly, iron deprivation also results in reduced transcription of the nuclear DNA-encoded OXPHOS genes [e.g., Ndufa9 and Ndufs7 (complex I), Sdha (complex II), Uqcrc2 (complex III) and Atp5a1 (complex V)] via modulating nuclear epigenome (chiefly decreasing histone acetylation) (75), suggesting that iron depletion may suppress M2-like phenotype (Figure 2C). It is mentionable that the changes in cellular metabolism are likely the outcomes of epigenetic regulation; hence, whether and how iron deprivation facilitates resting and/or pro-inflammatory-activated macrophages towards M1-like phenotype through the decreased acetylation of OXPHOS-related genes still needs experimental validation.
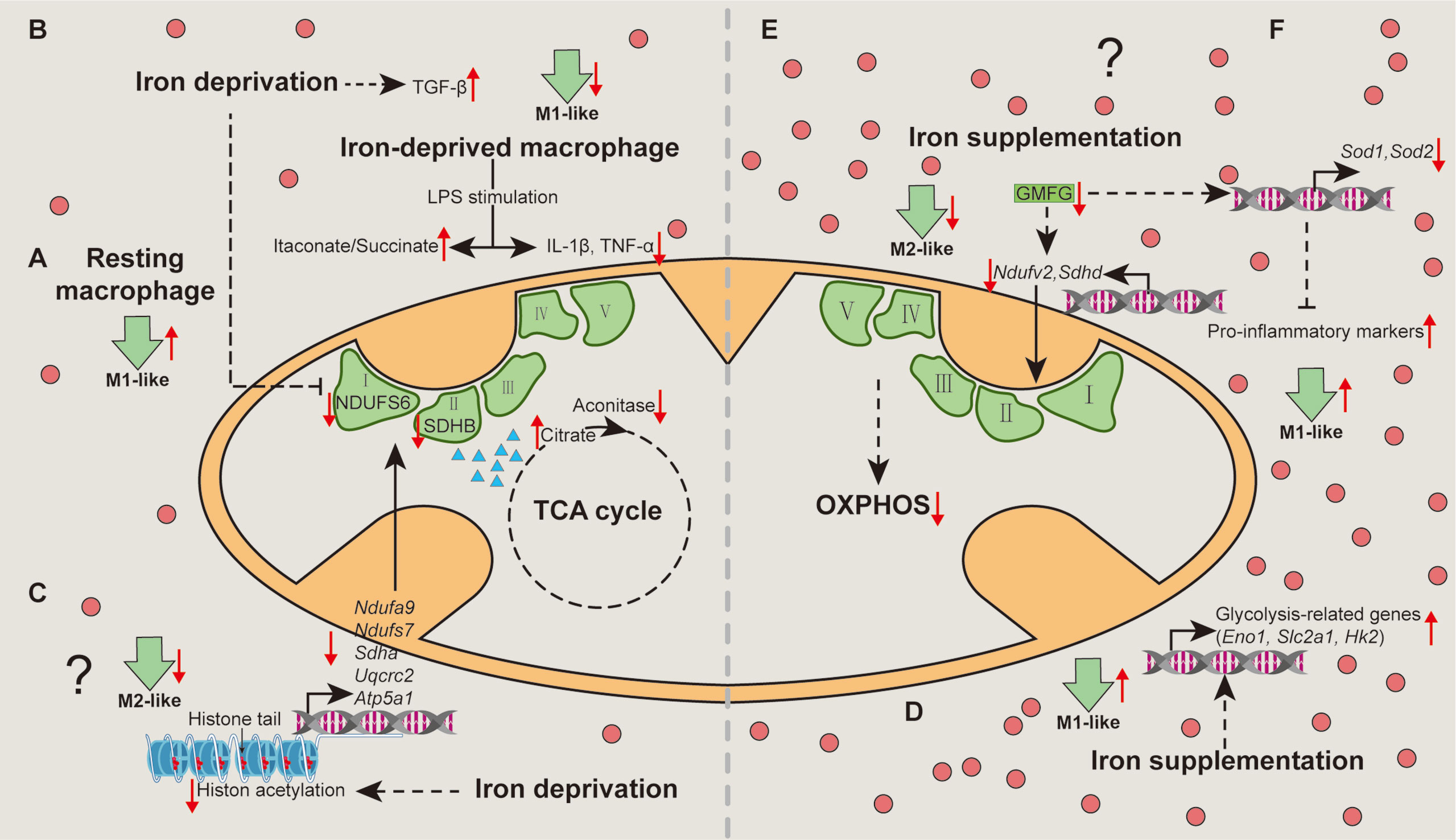
Figure 2 Mechanisms associated with metabolic pathways whereby iron shapes macrophage polarization. Iron deprivation directly inhibits the expression of NDUFS6 and SDHB and blocks the activity of mitochondrial aconitase to cause citrate accumulation, thereby switching resting macrophages towards M1-like macrophages (A). Iron deprivation limits M1 polarization by increasing itaconate:succinate ratio through altering metabolic fluxes and inducing TGF-β signaling pathway (B). Specially, iron deprivation decreases histone acetylation to reduce transcription of the nuclear DNA-encoded OXPHOS genes, suppressing M2-like phenotype (C). Iron-load increases the expressions of glycolysis-related genes to promote glycolysis in macrophages, enhancing M1-like macrophage polarization (D). Iron might restrain M2-like macrophage, while favors M1-like macrophage polarization via reducing the expression of NDUFV2 and SDHD (E), and SOD1 and SOD2 through down-regulating GMFG (F), respectively. OXPHOS, oxidative phosphorylation; TGF-β, transforming growth factor-β. “↑”, increase; “↓”, decrease. Question mark means that the mechanism needs experimental confirmation.
As for iron supplementation, it has been demonstrated that iron-load promotes glycolysis in macrophages, manifested by increased expression of glycolysis-related genes, such as Eno1, Slc2a1, and Hk2 (76), indicating that iron promotes M1-like macrophage polarization through glycolysis (Figure 2D). Considering that iron-load down-regulates GMFG (46), and that blockade of GMFG reduces the expression of NDUFV2 (complex І), SDHD (complex II), and SOD (77), iron might restrain M2-like macrophage polarization via GMFG-OXPHOS pathway (Figure 2E) and/or favor M1-like macrophage polarization through GMFG-SOD signaling (Figure 2F). Overall, iron mainly shapes activated macrophages polarize into pro-inflammatory phenotype via targeting cellular metabolism (Figure 2).
Iron Shapes Macrophage Polarization Through Epigenetic Regulation/Modification
Generally, epigenetic regulation/modification includes small RNA, DNA modification (methylation), histone modification, and chromatin remodeling. Epigenetic regulation/modification causes chromosome-bound, heritable changes to gene expression that are not dependent on changes to DNA sequence (78), thus becoming a key platform for macrophage reprogramming (79–81). In this section, we summarize how iron shapes macrophage phenotypes through the interactions between iron and several epigenetic regulators (Figure 3).
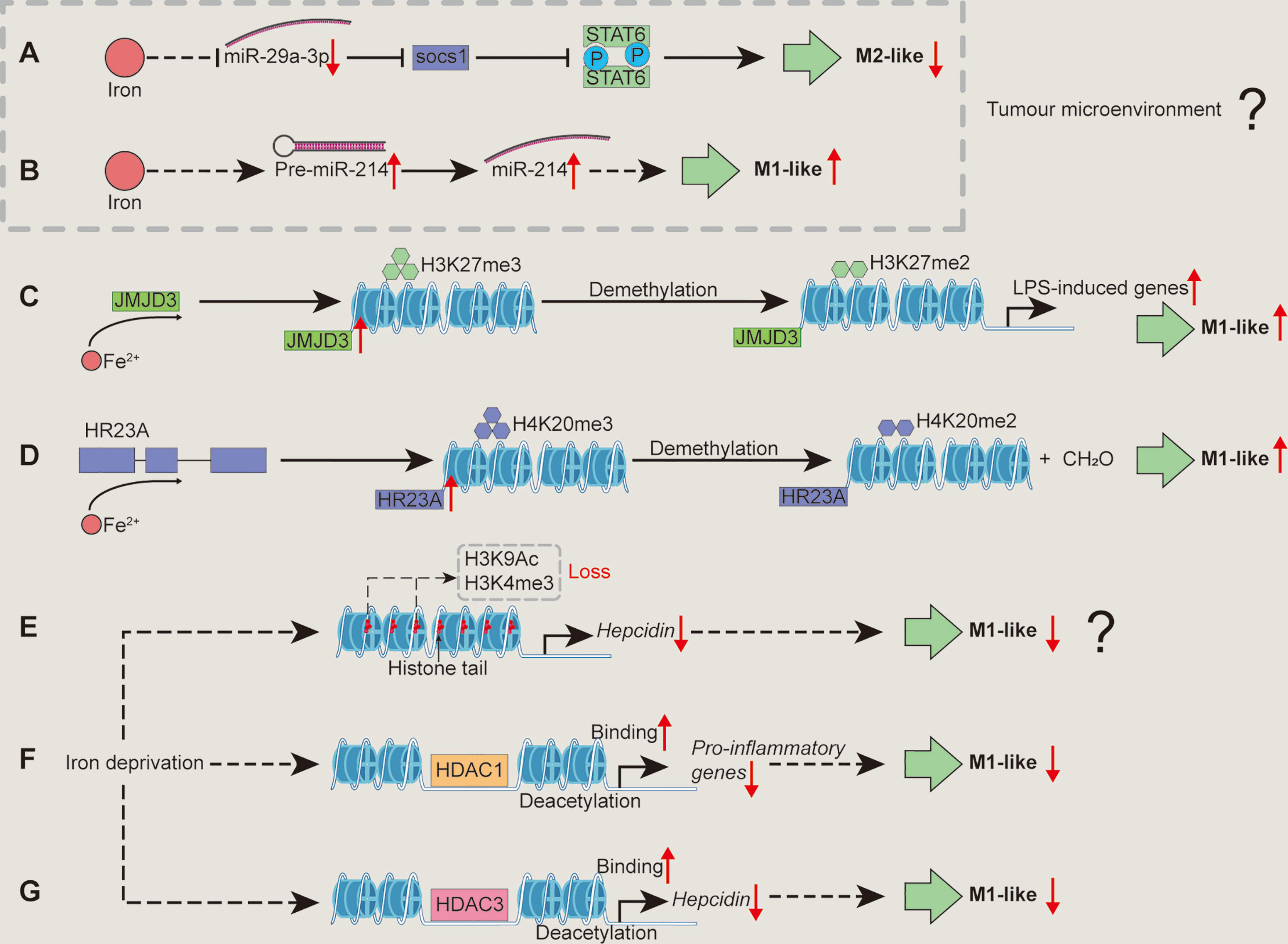
Figure 3 Mechanisms associated with epigenetic regulation/modification whereby iron shapes macrophage polarization. Iron supplementation may reduce miR-29a expression to block the activation of SOCS1/STAT6 signal pathway, inhibiting TAM polarization (A). Iron could promote M1-like macrophage polarization by increasing miR-214 expression (B). Iron could increase the enzyme activity of JMJD3 to demethylate H3K27me3, relieving the transcriptional silencing of LPS-induced genes and facilitating M1-like macrophage polarization (C). Iron might promote macrophages toward pro-inflammatory phenotype through demethylating H4K20me3 by enhancing the enzyme activity of HR23A (D). Iron deficiency might cause the loss of H3K9Ac and H3K4me3 to reduce hepcidin expression, inhibiting M1-like macrophage polarization (E). Iron deficiency inhibits M1 activation (e.g., pro-inflammatory responses) possibly through increasing HDAC1 binding to the related genes (F). Iron deficiency inhibits M1-like phenotype by lowering hepcidin expression via enhancing HDAC3 binds chromatin at the hepcidin locus (G). H3K27me3, histone 3 lysine 27 trimethylation; H4K20me3, histone 4 lysine 20 trimethylation; HDAC, histone deacetylase; SOCS1, suppressor of cytokine signaling 1. “↑”, increase; “↓”, decrease. Question mark means that the mechanism needs experimental confirmation.
MiRNAs
MicroRNAs (miRNAs) are a series of non-coding RNA involved in regulating gene expression. With the establishment of miRNAs expression profiles associated with the polarized human and mouse macrophages, it is not surprising that miRNAs function in controlling macrophage polarization (82). The expression of miR-141-3p or miR-152 is highly up-regulated in pro-inflammatory macrophages; while the expression of miR-124 is increased in anti-inflammatory macrophages (82). MiR141/200c could inhibit adenosine 5’-monophosphate (AMP)-activated protein kinase (AMPK) phosphorylation to block the activation of peroxisome proliferators-activated receptor (PPAR)-α, leading to an impairment of fatty acid β-oxidation in macrophages (83). Therefore, miR-141 favors M1-like phenotype (83). Interestingly, the anti-inflammatory effects of miR152 appear to be inconsistent with their up-regulation in M1-like macrophages. MiR-152 directly targets to Kruppel-like factor 5 (KLF5) to inhibit M1-like macrophage polarization (84). MiR-124 favors M2-like phenotype by inhibiting LPS-induced STAT3 phosphorylation in LPS-exposed RAW264.7 cells (85).
Of note, some miRNAs associating with macrophage polarization are supposed to regulate the expression of iron-regulated genes. For example, miR-124 modulates the stability of TFR1 mRNA by targeting iron-responsive element (IRE)-B (86); miR-152 negatively regulates TFR1 at post-transcriptional level (87); miR-141-3p regulates TFR1 mRNA degradation by directly targeting 3’-untranslated region (UTR) of TFR1 (86) or not (88, 89). Insofar, it is not surprising that iron shapes macrophage polarization through miRNAs.
MiR-29a-3p enhances the polarization of M2-like macrophages by activating suppressor of cytokine signaling (SOCS)-1/STAT6 signal pathway (90). Considering iron negatively regulates miR-29a (91), iron supplementation may reduce miR-29a expression to inhibit M2-like macrophage polarization (Figure 3A). Moreover, miR-214 is related to M1 polarization (92–94) and iron saccharate promotes miR-214 transcription (95); thus, it is likely that iron could promote M1-like macrophage polarization by increasing miR-214 expression (Figure 3B). However, these observations about iron-mediated the expression of miRNAs are achieved by TAM; therefore, whether iron shapes the polarization of conventional macrophages via miRNAs needs further investigations.
DNA Methylation
DNA methylation is associated with the transcriptional inhibition via DNA methyltransferases (DNMTs, including DNMT1, DNMT2, DNMT3A, and DNMT3B) (96). Up to now, DNA methylation in modulating M1 activation is still in its infancy. Only DNMT1 has been found to inhibit the expression of SOCS1 (which functions as a negative regulator of cytokine signals, including STAT1) through hypermethylation, thereby enhancing M1-like macrophage polarization (97). Interestingly, different concentrations of exogenous iron (Fe3+, 0.0625~0.25 mM) directly decrease the enzyme activity of purified DNMT in vitro (98). In addition, as mentioned above, FAC supplementation (~0.089 mM Fe3+) suppresses M1-like macrophage polarization by blocking STAT1 pathway in IFN-γ-stimulated RAW264.7 cells (35); thus, it remains to know whether in some special cases, iron blocks STAT1 pathway through regulating DNA methylation.
Histone Modification
Histone 4 lysine 20 trimethylation (H4K20me3), H3K9me3, and H3K27me3 restrain inflammatory cytokine gene transcription (e.g., IL-12 p40) in the absence of TLR signaling to keep macrophages in a “poised” state (32). Notably, these epigenetic “brakes” related to transcriptional silencing could be released by using demethylases in macrophages followed by inflammatory stimulation (e.g., TLR ligands) (32). Indeed, these modifiers involved in the regulation of macrophage polarization are also inextricably linked to iron. LPS-triggered NF-κB activation promotes JMJD3 (specially demethylates H3K27me3) binding to the promoter regions of LPS-induced genes in macrophages (99, 100). Notably, the enzyme activities of JumonjiC histone demethylases (including JMJD3) depend on 2-oxoglutarate and iron (II); thus, it is likely that iron could increase the enzyme activity of JMJD3 to demethylate H3K27me3, relieving the transcriptional silencing of LPS-induced genes and facilitating M1-like macrophage polarization (Figure 3C). Moreover, HR23A is a histone H4K20 demethylase that specifically demethylates H4K20me1/2/3 to produce formaldehyde, and iron (II) is required as cofactor for the enzyme activity of HR23A (101). Herein, we envision that iron might promote macrophages towards pro-inflammatory phenotype through demethylating H4K20me3 by enhancing the enzyme activity of HR23A (Figure 3D). Moreover, it has been shown that ten-eleven translocation family proteins (Tets) [which are iron (II) and α-ketoglutarate (α-KG)-dependent demethylases (102–104)] promote (by Tet1) (105) or restrain (through Tet2) (106, 107) pro-inflammatory responses in macrophages through unique mechanisms; thus, iron might also shape M1-like macrophage polarization through regulating Tet activity (Figure not shown).
Indeed, after removal of “negative histone markers” (which could inhibit gene expression, e.g., H4K20me3, H3K9me3, and H3K27me3) of inflammatory genes, “positive histone markers” (which could promote gene expression, including H3K4me3, H3K9Ac, and H3K27Ac) increase, that could be enriched in the promoter region of Cxcl10 (M1 marker gene) (32). Notably, iron deficiency suppresses hepcidin expression involving reversible loss of H3K9Ac and H3K4me3 at the hepcidin locus (108). Given that hepcidin supplementation promotes M1-like polarization (40), we speculate that iron deficiency might cause the loss of H3K9Ac and H3K4me3 to reduce hepcidin expression, inhibiting M1-like macrophage polarization (Figure 3E). Nevertheless, the above hypothesis still requires the convincing experimental evidence.
It should be noticed that HDACs highly regulate M1 activation; and LPS stimulation affect almost all classes of HDACs, thereby controlling the expression of inflammatory genes. For instance, HDAC 2/3/6/9 positively regulate M1 polarization, whereas HDAC 1/4/5 have repressive effects (32, 109). It has been found that the initial decrease in HDAC1 is responsible for IL-8 induction during L. pneumophila infection (110). Fetal iron deficiency increases HDAC1 binding to its target gene promoter (111). Therefore, iron deficiency inhibits M1 activation (e.g., pro-inflammatory responses) possibly through increasing HDAC1 binding to the related genes (Figure 3F). Besides, HDAC3 depletion leads to the loss of basic expression of IFN-β in macrophages, which affects subsequent STAT1-dependent gene expression and thus inhibits inflammatory activation (109). Intriguingly, during iron deficiency, RGFP966 (the HDAC3 inhibitor) counteracts hepcidin suppression (108). As mentioned above, hepcidin could promote M1-like macrophage polarization (40); thus, it is possible that iron deficiency inhibits M1-like phenotype by lowering hepcidin expression via enhancing HDAC3 binds chromatin at the hepcidin locus (Figure 3G). Besides, HDAC1 reduces expression of hepcidin by interacting with SMAD4 rather than deacetylation of SMAD4 or histone-H3 on the hepcidin promoter (112); therefore, it is interesting to further investigate whether iron deficiency directs pro-inflammatory macrophage polarization that is independent of the effect of deacetylation of HDAC1.
Sirtuin 2 (SIRT2) depletion results in a decrease in cellular iron levels; mechanistically, SIRT2 deacetylates nuclear factor erythroid-derived 2-related factor 2 (NRF2) on K506 and K508 to reduce FPN1 expression, leading to the decreased cellular iron export (113). After TNF-α stimulation, Sirt2-/- mouse embryonic fibroblasts (MEFs) cells become highly acetylated, resulting in increased expression of NF-κB target genes, including Mpa2l, Cxcl5, Ip10, and Il6 (114). Reciprocally, whether iron status shapes M1 polarization as an outcome of the effect of SIRT2/NRF2 is still unknown. Importantly, a large number of iron deprivation-related histone acetylation and methylation modifications have been found in iron chelator-treated myoblasts cells (75), thus it would be very interesting to explore whether these epigenetic modifications could participate in regulating macrophage polarization in the context of iron deprivation.
Indeed, chromatin remodeling occurs through the ATP-dependent SWI/SNF complex during M1 activation (115), and SWI/SNF directly represses iron transport-related genes (116); therefore, it is of great interest to investigate whether iron shapes M1 polarization involving chromatin remodeling in the future.
Iron in Macrophage-Associated Diseases
Notably, defects in macrophage phenotypic switch, such as the prolonged activation of a pro-inflammatory property in macrophages, are associated with various macrophage-associated diseases (117). Thus, the significant role of iron in dictating macrophage polarization suggests that iron has potential for affecting the progression of macrophage-associated diseases.
Nonalcoholic Fatty Liver Disease
Liver is the main organ for systemic iron regulation and storage, and the increase of iron in the liver may cause oxidative damage, lipid peroxidation, and even cell death (118). Nonalcoholic fatty liver disease (NAFLD) is one of the most common chronic liver diseases, including steatosis, nonalcoholic steatohepatitis (NASH), cirrhosis, and hepatocellular carcinoma (119).
In patients with NAFLD, iron deposits in different liver cells [including reticuloendothelial system (RES)]. Of note, iron deposits in RES could aggravate clinical symptoms of patients (120) due to the accumulation of inflammation-induced iron in Kupffer cells via IL-6/hepcidin pathway (121, 122). Simultaneously, the accumulation of iron in RES also promotes the production of ROS, inducing lipid peroxidation, apoptosis and/or necrosis of Kupffer cells (123). Similarly, in high-fat-induced NASH model, dietary iron supplementation induces iron accumulation in sinusoidal macrophages as well as high level of TNF-α in liver, aggravating disease symptoms (124). However, in thioacetamide-induced cirrhosis model, after fed with high-iron diet, although macrophages are recruited in the liver sinusoid, the number of macrophages infiltration within the hepatic lesions is reduced, thereby the hepatocellular injury could be attenuated, which is probably due to iron alters the apoptotic pathway (125).
Atherosclerosis
Atherosclerosis is a common vascular disease with the formation of plaques or lesions in the arterial wall (126). Macrophages are the main inflammatory cells in atherosclerotic plaques. During atherogenesis, circulating monocyte-derived macrophages could be recruited to vascular endothelium and form lipid-laden foam cells (one of the main characteristics of atherosclerosis) (127). Indeed, according to the “iron hypothesis”, overloaded iron generates a large amount of ROS and promotes the lipid-peroxidation, inducing fatty deposits and inflammatory cell infiltration (128). During atherogenesis, high levels of iron in circulation promotes the recruitment of monocyte-derived macrophages to vascular endothelium, and excessive iron stimulates M1-like macrophage polarization in plaques, thereby facilitating the formation of foam cells (129–131). Hepcidin level is supposed to be a major determinant of the level of iron retained within macrophages (20, 132). Of note, impaired clearance of iron from plaque macrophages caused by high inflammation-induced levels of hepcidin might be associated with atherogenesis (133). Thus, it may be possible to prevent atherosclerosis by reducing iron retention in the plaque macrophages through decreasing hepcidin concentration (134, 135).
Cancer/Tumor
TAMs are M2-like macrophages related to the progression and poor prognosis of tumor (136–138). Iron-containing M2 macrophage conditioned medium contributes to the growth of human renal cell carcinoma cells (RCC10) or mouse NIH-3T3 cells; and chelation of iron by desferrioxamine (DFO) supplementation reduces cancer cell proliferation (139). Moreover, co-culture of TAM and melanoma cells exhibit coexpression of HO-1 and CD163, suggesting that tumor cells-derived M2 macrophages promote heme metabolism (140). These studies indicate that M2 macrophages metabolize iron and delivery iron to tumor cells. Indeed, iron chelation therapy for cancer is used clinically. In addition to DFO, there are several other drugs (like deferiprone, deferasirox, and curcumin) are applied for slowing down cancer progression (141). Interestingly, except for iron chelation-based cancer therapy, iron supplementation (e.g., iron oxide nanoparticles) also shows highly anti-tumor potential. For example, ferumoxytol inhibits tumor growth and metastases that are closely associated with M1 polarization and cancer cell apoptosis induced by Fenton reaction (44).
Collectively, these interesting findings suggest the various roles of iron in macrophage-associated diseases through altering macrophage responses. Together with the other macrophage-associated diseases summarized in Table 2, the specific pathways transmitting the effects of iron on these diseases need to be fully understood.

Table 2 Iron in other macrophage-associated diseases.
Concluding Remarks
Iron is an essential metal that fine-tunes a series of physiological functions, including macrophage-dependent responses. In this review, we summarize how iron shapes macrophage polarization. Mechanistically, iron modulates macrophage polarization via cellular signaling pathways (e.g., MAPK, NF-κB, and STATs), cellular metabolism (mainly targeting OXPHOS), and epigenetic regulation (e.g., miRNAs, DNMT1, JMJD3, H3K4me3, H4K20me3, and HDAC1/3). Other pathways discussed in this review seem to additionally contribute; however, these require a broader experimental confirmation. It has been well documented that α-KG produced by glutaminolysis is responsible for JMJD3-dependent demethylation of H3K27 on the promoter of M2‐specific marker genes (149). Thus, it would be very interesting to study whether iron shapes macrophage polarization (M2 polarization in particular) through the correlation of epigenetic and metabolic reprogramming. As we mentioned above, iron may have various roles in regulating macrophage polarization based on different types of macrophages, therefore, it is also meaningful to conduct comparative studies on the effects of iron on polarization of macrophage with different origins (embryonic origin compared with monocyte derivation), tissues (e.g., osteoclasts from bone and alveolar macrophage from lung), or even species (murine and human).
Mitophagy activation or inhibition fine-tunes macrophage polarization (150). Interestingly, iron affects mitophagy through diverse pathways (including mtROS and MAPK) (151, 152). However, the exact mechanisms whereby iron targets mitophagy to switch macrophage polarization should still be revealed with some reservation. Furthermore, mitochondrial dynamics (fission and fusion) is intertwined with macrophage polarization. It has been demonstrated that iron chelation selectively increases mitochondrial fusion protein (Mfn2) expression in neuronal HT22 cells (153), while iron overload induces dephosphorylation of dynamin-related protein (Drp)-1 S637 loci and activation of Drp1 in hippocampal HT-22 neuronal cells (154) as well as promotes mitochondrial fragmentation in mesenchymal stromal cells (MSC) by activating the AMPK/mitochondrial fission factor (MFF)/Drp1 pathway (155). Nevertheless, a broader basis concerning cell types is needed to allow generalizations and robust conclusions on iron in directing macrophage polarization. More recently, it has found that endogenous and/or exogenous iron-induced iron overload can trigger macrophage ferroptosis (156, 157). Macrophage under hypoxia condition undergoes ferroptosis (158, 159), thus whether iron shapes macrophage polarization (chiefly M1 polarization) through affecting ferroptosis should also be taken into consideration. More importantly, it is urgent to dig out the intrinsic relationship between ferroptosis, macrophage phenotype, and cancer progression (160).
Author Contributions
YX designed the review article. YX, YL, and XW wrote the review article. YX, XM, and MY revised the review article. YL, XW, QZ, and SC helped with designing figures and finding relevant literatures. YX, XM, and MY approved the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (31902200), Special fund for scientific innovation strategy-construction of high level Academy of Agriculture Science (R2017PY-JG001, R2020YJ-YB2002), and Agricultural competitive industry discipline team building project of Guangdong Academy of Agricultural Sciences (202118TD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Our profound admiration and respect go to researchers in this field and in our laboratories, for their dedication and hard work. We apologize to scientists whose work is in this field if their papers are not cited owing to space limitations.
References
1. Kosman DJ. Redox Cycling in Iron Uptake, Efflux, and Trafficking. J Biol Chem (2010) 285(35):26729–35. doi: 10.1074/jbc.R110.113217
2. Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell (2010) 142(1):24–38. doi: 10.1016/j.cell.2010.06.028
3. Korolnek T, Hamza I. Macrophages and Iron Trafficking At the Birth and Death of Red Cells. Blood (2015) 125(19):2893–7. doi: 10.1182/blood-2014-12-567776
4. Low PS, Waugh SM, Zinke K, Drenckhahn D. The Role of Hemoglobin Denaturation and Band 3 Clustering in Red Blood Cell Aging. Sci (New York N.Y.) (1985) 227(4686):531–3. doi: 10.1126/science.2578228
5. Willekens FLA, Werre JM, Groenen-Dop YAM, Roerdinkholder-Stoelwinder B, de Pauw B, Bosman G. Erythrocyte Vesiculation: A Self-Protective Mechanism? Br J Haematol (2008) 141(4):549–56. doi: 10.1111/j.1365-2141.2008.07055.x
6. White C, Yuan XJ, Schmidt PJ, Bresciani E, Samuel TK, Campagna D, et al. Hrg1 Is Essential for Heme Transport From the Phagolysosome of Macrophages During Erythrophagocytosis. Cell Metab (2013) 17(2):261–70. doi: 10.1016/j.cmet.2013.01.005
7. Soe-Lin S, Apte SS, Andriopoulos B, Andrews MC, Schranzhofer M, Kahawita T, et al. Nramp1 Promotes Efficient Macrophage Recycling of Iron Following Erythrophagocytosis In Vivo. Proc Natl Acad Sci USA (2009) 106(14):5960–5. doi: 10.1073/pnas.0900808106
8. Soares MP, Hamza I. Macrophages and Iron Metabolism. Immunity (2016) 44(3):492–504. doi: 10.1016/j.immuni.2016.02.016
9. Ganz T. Macrophages and Systemic Iron Homeostasis. J Innate Immun (2012) 4(5-6):446–53. doi: 10.1159/000336423
10. Ginhoux F, Guilliams M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity (2016) 44(3):439–49. doi: 10.1016/j.immuni.2016.02.024
11. Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, Rouault TA. Dysfunction of the Heme Recycling System in Heme Oxygenase 1-Deficient Mice: Effects on Macrophage Viability and Tissue Iron Distribution. Blood (2010) 116(26):6054–62. doi: 10.1182/blood-2010-03-272138
12. Haldar M, Kohyama M, So AY-L, Wumesh KC, Wu X, Briseno CG, et al. Heme-Mediated SPI-C Induction Promotes Monocyte Differentiation Into Iron-Recycling Macrophages. Cell (2014) 156(6):1223–34. doi: 10.1016/j.cell.2014.01.069
13. Hume DA. Differentiation and Heterogeneity in the Mononuclear Phagocyte System. Mucosal Immunol (2008) 1(6):432–41. doi: 10.1038/mi.2008.36
14. Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, et al. Local Macrophage Proliferation, Rather Than Recruitment From the Blood, is a Signature of T(H)2 Inflammation. Science (2011) 332(6035):1284–8. doi: 10.1126/science.1204351
15. Xie WJ, Lorenz S, Dolder S, Hofstetter W. Extracellular Iron is a Modulator of the Differentiation of Osteoclast Lineage Cells. Calcified Tissue Int (2016) 98(3):275–83. doi: 10.1007/s00223-015-0087-1
16. Zhao GY, Di DH, Wang B, Huang X, Xu YJ. Effects of Mouse Hepcidin 1 Treatment on Osteoclast Differentiation and Intracellular Iron Concentration. Inflammation (2015) 38(2):718–27. doi: 10.1007/s10753-014-9982-2
17. Baschant U, Rauner MT, Balaian E, Weidner H, Roetto A, Platzbecker U, et al. Wnt5a is a Key Target for the Pro-Osteogenic Effects of Iron Chelation on Osteoblast Progenitors. Haematologica (2016) 101(12):1499–507. doi: 10.3324/haematol.2016.144808
18. Winn NC, Volk KM, Hasty AH. Regulation of Tissue Iron Homeostasis: The Macrophage “Ferrostat”. JCI Insight (2020) 5(2):14. doi: 10.1172/jci.insight.132964
19. Muckenthaler MU, Rivella S, Hentze MW, Galy B. A Red Carpet for Iron Metabolism. Cell (2017) 168(3):344–61. doi: 10.1016/j.cell.2016.12.034
20. Nemeth E, Ganz T. Regulation of Iron Metabolism by Hepcidin. Annu Rev Nutr (2006) 26:323–42. doi: 10.1146/annurev.nutr.26.061505.111303
21. Leimberg MJ, Prus E, Konijn AM, Fibach E. Macrophages Function as a Ferritin Iron Source for Cultured Human Erythroid Precursors. J Cell Biochem (2008) 103(4):1211–8. doi: 10.1002/jcb.21499
22. Rhodes MM, Kopsombut P, Bondurant MC, Price JO, Koury MJ. Adherence to Macrophages in Erythroblastic Islands Enhances Erythroblast Proliferation and Increases Erythrocyte Production by a Different Mechanism Than Erythropoietin. Blood (2008) 111(3):1700–8. doi: 10.1182/blood-2007-06-098178
23. Collins HL, Kaufmann SHE, Schaible UE. Iron Chelation Via Deferoxamine Exacerbates Experimental Salmonellosis Via Inhibition of the Nicotinamide Adenine Dinucleotide Phosphate Oxidase-Dependent Respiratory Burst. J Immunol (2002) 168(7):3458–63. doi: 10.4049/jimmunol.168.7.3458
24. Mair SM, Nairz M, Bellmann-Weiler R, Muehlbacher T, Schroll A, Theurl I, et al. Nifedipine Affects the Course of Salmonella Enterica Serovar Typhimurium Infection by Modulating Macrophage Iron Homeostasis. J Infect Dis (2011) 204(5):685–94. doi: 10.1093/infdis/jir395
25. Nairz M, Theurl I, Ludwiczek S, Theurl M, Mair SM, Fritsche G, et al. The Co-Ordinated Regulation of Iron Homeostasis in Murine Macrophages Limits the Availability of Iron for Intracellular Salmonella Typhimurium. Cell Microbiol (2007) 9(9):2126–40. doi: 10.1111/j.1462-5822.2007.00942.x
26. Paradkar PN, De Domenico I, Durchfort N, Zohn I, Kaplan J, Ward DM. Iron Depletion Limits Intracellular Bacterial Growth in Macrophages. Blood (2008) 112(3):866–74. doi: 10.1182/blood-2007-12-126854
27. Bellmann-Weiler R, Martinz V, Kurz K, Engl S, Feistritzer C, Fuchs D, et al. Divergent Modulation of Chlamydia Pneumoniae Infection Cycle in Human Monocytic and Endothelial Cells by Iron, Tryptophan Availability and Interferon Gamma. Immunobiology (2010) 215(9-10):842–8. doi: 10.1016/j.imbio.2010.05.021
28. Isanaka S, Mugusi F, Urassa W, Willett WC, Bosch RJ, Villamor E, et al. Iron Deficiency and Anemia Predict Mortality in Patients With Tuberculosis. J Nutr (2012) 142(2):350–7. doi: 10.3945/jn.111.144287
29. Osborn O, Olefsky JM. The Cellular and Signaling Networks Linking the Immune System and Metabolism in Disease. Nat Med (2012) 18(3):363–74. doi: 10.1038/nm.2627
30. Xia YY, Chen SY, Zeng SJ, Zhao YY, Zhu CR, Deng BC, et al. Melatonin in Macrophage Biology: Current Understanding and Future Perspectives. J Pineal Res (2019) 66(2):21. doi: 10.1111/jpi.12547
31. O’Neill LA, Kishton RJ, Rathmell J. A Guide to Immunometabolism for Immunologists. Nat Rev Immunol (2016) 16(9):553–65. doi: 10.1038/nri.2016.70
32. Chen S, Yang J, Wei Y, Wei X. Epigenetic Regulation of Macrophages: From Homeostasis Maintenance to Host Defense. Cell Mol Immunol (2020) 17(1):36–49. doi: 10.1038/s41423-019-0315-0
33. Kraml P. The Role of Iron in the Pathogenesis of Atherosclerosis. Physiol Res (2017) 66(Suppl 1):S55–67. doi: 10.33549/physiolres.933589
34. Mesquita G, Silva T, Gomes AC, Oliveira PF, Alves MG, Fernandes R, et al. H-Ferritin is Essential for Macrophages’ Capacity to Store or Detoxify Exogenously Added Iron. Sci Rep (2020) 10(1):15. doi: 10.1038/s41598-020-59898-0
35. Gan ZS, Wang QQ, Li JH, Wang XL, Wang YZ, Du HH. Iron Reduces M1 Macrophage Polarization in RAW264.7 Macrophages Associated With Inhibition of STAT1. Mediators Inflammation (2017) 2017:8570818. doi: 10.1155/2017/8570818
36. Pereira M, Chen T-D, Buang N, Olona A, Ko J-H, Prendecki M, et al. Acute Iron Deprivation Reprograms Human Macrophage Metabolism and Reduces Inflammation In Vivo. Cell Rep (2019) 28(2):498–511.e5. doi: 10.1016/j.celrep.2019.06.039
37. Ward RJ, Wilmet S, Legssyer R, Leroy D, Toussaint L, Crichton RR, et al. Effects of Marginal Iron Overload on Iron Homeostasis and Immune Function in Alveolar Macrophages Isolated From Pregnant and Normal Rats. Biometals (2009) 22(2):211–23. doi: 10.1007/s10534-008-9155-6
38. Kroner A, Greenhalgh AD, Zarruk JG, Passos Dos Santos R, Gaestel M, David S. TNF and Increased Intracellular Iron Alter Macrophage Polarization to a Detrimental M1 Phenotype in the Injured Spinal Cord. Neuron (2014) 83(5):1098–116. doi: 10.1016/j.neuron.2014.07.027
39. Harhaji L, Vuckovic O, Miljkovic D, Stosic-Grujicic S, Trajkovic V. Iron Down-Regulates Macrophage Anti-Tumour Activity by Blocking Nitric Oxide Production. Clin Exp Immunol (2004) 137(1):109–16. doi: 10.1111/j.1365-2249.2004.02515.x
40. Liu E, Li Z, Zhang Y, Chen K. Hepcidin Induces M1 Macrophage Polarization in Monocytes or THP-1 Derived Macrophages. Iran J Immunol (2019) 16(3):190–9. doi: 10.22034/IJI.2019.80270
41. Dufrusine B, Di Francesco A, Oddi S, Scipioni L, Angelucci CB, D’Addario C, et al. Iron-Dependent Trafficking of 5-Lipoxygenase and Impact on Human Macrophage Activation. Front Immunol (2019) 10:1347. doi: 10.3389/fimmu.2019.01347
42. Wilkinson HN, Roberts ER, Stafford AR, Banyard KL, Matteucci P, Mace KA, et al. Tissue Iron Promotes Wound Repair Via M2 Macrophage Polarization and the Chemokine (C-C Motif) Ligands 17 and 22. Am J Pathol (2019) 189(11):2196–208. doi: 10.1016/j.ajpath.2019.07.015
43. Polati R, Castagna A, Bossi AM, Alberio T, De Domenico I, Kaplan J, et al. Murine Macrophages Response to Iron. J Proteomics (2012) 76:10–27. doi: 10.1016/j.jprot.2012.07.018
44. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, et al. Iron Oxide Nanoparticles Inhibit Tumour Growth by Inducing Pro-Inflammatory Macrophage Polarization in Tumour Tissues. Nat Nanotechnol (2016) 11(11):986–94. doi: 10.1038/nnano.2016.168
45. Zhou Y, Que KT, Zhang Z, Yi ZJ, Zhao PX, You Y, et al. Iron Overloaded Polarizes Macrophage to Proinflammation Phenotype Through ROS/acetyl-p53 Pathway. Cancer Med (2018) 7(8):4012–22. doi: 10.1002/cam4.1670
46. Aerbajinai W, Ghosh MC, Liu J, Kumkhaek C, Zhu J, Chin K, et al. Glia Maturation Factor-γ Regulates Murine Macrophage Iron Metabolism and M2 Polarization Through Mitochondrial ROS. Blood Adv (2019) 3(8):1211–25. doi: 10.1182/bloodadvances.2018026070
47. Handa P, Thomas S, Morgan-Stevenson V, Maliken BD, Gochanour E, Boukhar S, et al. Iron Alters Macrophage Polarization Status and Leads to Steatohepatitis and Fibrogenesis. J Leukoc Biol (2019) 105(5):1015–26. doi: 10.1002/JLB.3A0318-108R
48. Kao JK, Wang SC, Ho LW, Huang SW, Lee CH, Lee MS, et al. M2-Like Polarization of THP-1 Monocyte-Derived Macrophages Under Chronic Iron Overload. Ann Hematol (2020) 99(3):431–41. doi: 10.1007/s00277-020-03916-8
49. Cairo G, Recalcati S, Mantovani A, Locati M. Iron Trafficking and Metabolism in Macrophages: Contribution to the Polarized Phenotype. Trends Immunol (2011) 32(6):241–7. doi: 10.1016/j.it.2011.03.007
50. Sumitomo R, Hirai T, Fujita M, Murakami H, Otake Y, Huang CL. M2 Tumor-Associated Macrophages Promote Tumor Progression in non-Small-Cell Lung Cancer. Exp Ther Med (2019) 18(6):4490–8. doi: 10.3892/etm.2019.8068
51. Lee C, Jeong H, Bae Y, Shin K, Kang S, Kim H, et al. Targeting of M2-like Tumor-Associated Macrophages With a Melittin-Based Pro-Apoptotic Peptide. J Immunother Cancer (2019) 7(1):147. doi: 10.1186/s40425-019-0610-4
52. Yamaguchi T, Fushida S, Yamamoto Y, Tsukada T, Kinoshita J, Oyama K, et al. Tumor-Associated Macrophages of the M2 Phenotype Contribute to Progression in Gastric Cancer With Peritoneal Dissemination. Gastric Cancer (2016) 19(4):1052–65. doi: 10.1007/s10120-015-0579-8
53. Hoeft K, Bloch DB, Graw JA, Malhotra R, Ichinose F, Bagchi A. Iron Loading Exaggerates the Inflammatory Response to the Toll-like Receptor 4 Ligand Lipopolysaccharide by Altering Mitochondrial Homeostasis. Anesthesiology (2017) 127(1):121–35. doi: 10.1097/aln.0000000000001653
54. Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, et al. An Unrestrained Proinflammatory M1 Macrophage Population Induced by Iron Impairs Wound Healing in Humans and Mice. J Clin Invest (2011) 121(3):985–97. doi: 10.1172/jci44490
55. Agoro R, Taleb M, Quesniaux VFJ, Mura C. Cell Iron Status Influences Macrophage Polarization. PloS One (2018) 13(5):e0196921. doi: 10.1371/journal.pone.0196921
56. Locati M, Curtale G, Mantovani A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu Rev Pathol (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
57. Bedard K, Krause KH. The NOX Family of ROS-generating NADPH Oxidases: Physiology and Pathophysiology. Physiol Rev (2007) 87(1):245–313. doi: 10.1152/physrev.00044.2005
58. Helfinger V, Palfi K, Weigert A, Schröder K. The NADPH Oxidase Nox4 Controls Macrophage Polarization in an Nfκb-Dependent Manner. Oxid Med Cell Longev (2019) 2019:3264858. doi: 10.1155/2019/3264858
59. Ghasemlou N, Lopez-Vales R, Lachance C, Thuraisingam T, Gaestel M, Radzioch D, et al. Mitogen-Activated Protein Kinase-Activated Protein Kinase 2 (MK2) Contributes to Secondary Damage After Spinal Cord Injury. J Neurosci (2010) 30(41):13750–9. doi: 10.1523/jneurosci.2998-10.2010
60. Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: Biochemistry, Pathophysiology and Development of Therapeutics. Nat Rev Drug Discovery (2007) 6(8):662–80. doi: 10.1038/nrd2222
61. Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, et al. Lipocalin 2 Mediates an Innate Immune Response to Bacterial Infection by Sequestrating Iron. Nature (2004) 432(7019):917–21. doi: 10.1038/nature03104
62. Wang Q, Li S, Tang X, Liang L, Wang F, Du H. Lipocalin 2 Protects Against Escherichia Coli Infection by Modulating Neutrophil and Macrophage Function. Front Immunol (2019) 10:2594. doi: 10.3389/fimmu.2019.02594
63. Ye D, Yang K, Zang S, Lin Z, Chau HT, Wang Y, et al. Lipocalin-2 Mediates non-Alcoholic Steatohepatitis by Promoting Neutrophil-Macrophage Crosstalk Via the Induction of CXCR2. J Hepatol (2016) 65(5):988–97. doi: 10.1016/j.jhep.2016.05.041
64. Xia HF, Zhu JY, Wang JN, Ren JG, Cai Y, Wang FQ, et al. Association of ATF4 Expression With Tissue Hypoxia and M2 Macrophage Infiltration in Infantile Hemangioma. J Histochem Cytochem (2017) 65(5):285–94. doi: 10.1369/0022155417694872
65. Liu B, Chen P, Xi D, Zhu H, Gao Y. ATF4 Regulates CCL2 Expression to Promote Endometrial Cancer Growth by Controlling Macrophage Infiltration. Exp Cell Res (2017) 360(2):105–12. doi: 10.1016/j.yexcr.2017.08.031
66. Rendra E, Riabov V, Mossel DM, Sevastyanova T, Harmsen MC, Kzhyshkowska J. Reactive Oxygen Species (ROS) in Macrophage Activation and Function in Diabetes. Immunobiology (2019) 224(2):242–53. doi: 10.1016/j.imbio.2018.11.010
67. Gelfand BD, Wright CB, Kim Y, Yasuma T, Yasuma R, Li S, et al. Iron Toxicity in the Retina Requires Alu RNA and the NLRP3 Inflammasome. Cell Rep (2015) 11(11):1686–93. doi: 10.1016/j.celrep.2015.05.023
68. Nakamura K, Kawakami T, Yamamoto N, Tomizawa M, Fujiwara T, Ishii T, et al. Activation of the NLRP3 Inflammasome by Cellular Labile Iron. Exp Hematol (2016) 44(2):116–24. doi: 10.1016/j.exphem.2015.11.002
69. Erdei J, Tóth A, Balogh E, Nyakundi BB, Bányai E, Ryffel B, et al. Induction of NLRP3 Inflammasome Activation by Heme in Human Endothelial Cells. Oxid Med Cell Longev (2018) 2018:4310816. doi: 10.1155/2018/4310816
70. Formentini L, Santacatterina F, Núñez de Arenas C, Stamatakis K, López-Martínez D, Logan A, et al. Mitochondrial ROS Production Protects the Intestine From Inflammation Through Functional M2 Macrophage Polarization. Cell Rep (2017) 19(6):1202–13. doi: 10.1016/j.celrep.2017.04.036
71. Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, et al. Prolyl Hydroxylase-1 Negatively Regulates IkappaB Kinase-Beta, Giving Insight Into Hypoxia-Induced NFkappaB Activity. Proc Natl Acad Sci USA (2006) 103(48):18154–9. doi: 10.1073/pnas.0602235103
72. Takeda Y, Costa S, Delamarre E, Roncal C, Leite de Oliveira R, Squadrito ML, et al. Macrophage Skewing by Phd2 Haplodeficiency Prevents Ischaemia by Inducing Arteriogenesis. Nature (2011) 479(7371):122–6. doi: 10.1038/nature10507
73. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an Inflammatory Signal That Induces IL-1β Through HIF-1α. Nature (2013) 496(7444):238–42. doi: 10.1038/nature11986
74. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell (2016) 167(2):457–470.e13. doi: 10.1016/j.cell.2016.08.064
75. Rensvold JW, Krautkramer KA, Dowell JA, Denu JM, Pagliarini DJ. Iron Deprivation Induces Transcriptional Regulation of Mitochondrial Biogenesis. J Biol Chem (2016) 291(40):20827–37. doi: 10.1074/jbc.M116.727701
76. Hu X, Cai X, Ma R, Fu W, Zhang C, Du X. Iron-Load Exacerbates the Severity of Atherosclerosis Via Inducing Inflammation and Enhancing the Glycolysis in Macrophages. J Cell Physiol (2019) 234(10):18792–800. doi: 10.1002/jcp.28518
77. Perriotte-Olson C, Adi N, Manickam DS, Westwood RA, Desouza CV, Natarajan G, et al. Nanoformulated Copper/Zinc Superoxide Dismutase Reduces Adipose Inflammation in Obesity. Obes (Silver Spring) (2016) 24(1):148–56. doi: 10.1002/oby.21348
78. Deans C, Maggert KA. What do You Mean, “Epigenetic”? Genetics (2015) 199(4):887–96. doi: 10.1534/genetics.114.173492
79. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic Programming of Monocyte-to-Macrophage Differentiation and Trained Innate Immunity. Science (2014) 345(6204):1251086. doi: 10.1126/science.1251086
80. Phan AT, Goldrath AW, Glass CK. Metabolic and Epigenetic Coordination of T Cell and Macrophage Immunity. Immunity (2017) 46(5):714–29. doi: 10.1016/j.immuni.2017.04.016
81. Ivashkiv LB. Epigenetic Regulation of Macrophage Polarization and Function. Trends Immunol (2013) 34(5):216–23. doi: 10.1016/j.it.2012.11.001
82. Essandoh K, Li Y, Huo J, Fan GC. Mirna-Mediated Macrophage Polarization and its Potential Role in the Regulation of Inflammatory Response. Shock (2016) 46(2):122–31. doi: 10.1097/SHK.0000000000000604
83. Tran M, Lee SM, Shin DJ, Wang L. Loss of miR-141/200c Ameliorates Hepatic Steatosis and Inflammation by Reprogramming Multiple Signaling Pathways in NASH. JCI Insight (2017) 2(21):e96094. doi: 10.1172/jci.insight.96094
84. Wang W, Zhang Y, Wang L, Li J, Li Y, Yang X, et al. mircroRNA-152 Prevents the Malignant Progression of Atherosclerosis Via Down-Regulation of KLF5. BioMed Pharmacother (2019) 109:2409–14. doi: 10.1016/j.biopha.2018.08.014
85. Sun Y, Li Q, Gui H, Xu DP, Yang YL, Su DF, et al. MicroRNA-124 Mediates the Cholinergic Anti-Inflammatory Action Through Inhibiting the Production of Pro-Inflammatory Cytokines. Cell Res (2013) 23(11):1270–83. doi: 10.1038/cr.2013.116
86. Miyazawa M, Bogdan AR, Hashimoto K, Tsuji Y. Regulation of Transferrin Receptor-1 mRNA by the Interplay Between IRE-binding Proteins and miR-7/miR-141 in the 3’-IRE Stem-Loops. Rna (2018) 24(4):468–79. doi: 10.1261/rna.063941.117
87. Kindrat I, Tryndyak V, de Conti A, Shpyleva S, Mudalige TK, Kobets T, et al. MicroRNA-152-mediated Dysregulation of Hepatic Transferrin Receptor 1 in Liver Carcinogenesis. Oncotarget (2016) 7(2):1276–87. doi: 10.18632/oncotarget.6004
88. Corral VM, Schultz ER, Connell GJ. Neither miR-7-5p Nor miR-141-3p is a Major Mediator of Iron-Responsive Transferrin Receptor-1 mRNA Degradation. Rna (2019) 25(11):1407–15. doi: 10.1261/rna.072371.119
89. Miyazawa M, Bogdan AR, Hashimoto K, Tsuji Y. Iron-Induced Transferrin Receptor-1 mRNA Destabilization: A Response to “Neither miR-7-5p Nor miR-141-3p is a Major Mediator of Iron-Responsive Transferrin Receptor-1 mRNA Degradation”. Rna (2019) 25(11):1416–20. doi: 10.1261/rna.073270.119
90. Cai J, Qiao B, Gao N, Lin N, He W. Oral Squamous Cell Carcinoma-Derived Exosomes Promote M2 Subtype Macrophage Polarization Mediated by Exosome-Enclosed Mir-29a-3p. Am J Physiol Cell Physiol (2019) 316(5):C731–40. doi: 10.1152/ajpcell.00366.2018
91. Zhang L, Ye Y, Tu H, Hildebrandt MA, Zhao L, Heymach JV, et al. MicroRNA-related Genetic Variants in Iron Regulatory Genes, Dietary Iron Intake, microRNAs and Lung Cancer Risk. Ann Oncol (2017) 28(5):1124–9. doi: 10.1093/annonc/mdx046
92. Gao Y, Fang P, Li WJ, Zhang J, Wang GP, Jiang DF, et al. Lncrna NEAT1 Sponges miR-214 to Regulate M2 Macrophage Polarization by Regulation of B7-H3 in Multiple Myeloma. Mol Immunol (2020) 117:20–8. doi: 10.1016/j.molimm.2019.10.026
93. Lu S, Gao Y, Huang X, Wang X. Cantharidin Exerts Anti-Hepatocellular Carcinoma by miR-214 Modulating Macrophage Polarization. Int J Biol Sci (2014) 10(4):415–25. doi: 10.7150/ijbs.8002
94. Li XF, Wang Y, Zheng DD, Xu HX, Wang T, Pan M, et al. M1 Macrophages Promote Aortic Valve Calcification Mediated by microRNA-214/TWIST1 Pathway in Valvular Interstitial Cells. Am J Transl Res (2016) 8(12):5773–83.
95. Okazaki Y, Chew SH, Nagai H, Yamashita Y, Ohara H, Jiang L, et al. Overexpression of miR-199/214 is a Distinctive Feature of Iron-Induced and Asbestos-Induced Sarcomatoid Mesothelioma in Rats. Cancer Sci (2020) 111(6):2016–27. doi: 10.1111/cas.14405
96. Holliday R, Pugh JE. DNA Modification Mechanisms and Gene Activity During Development. Science (1975) 187(4173):226–32. doi: 10.1126/science.1111098
97. Cheng C, Huang C, Ma TT, Bian EB, He Y, Zhang L, et al. SOCS1 Hypermethylation Mediated by DNMT1 is Associated With Lipopolysaccharide-Induced Inflammatory Cytokines in Macrophages. Toxicol Lett (2014) 225(3):488–97. doi: 10.1016/j.toxlet.2013.12.023
98. Ye Q, Trivedi M, Zhang Y, Böhlke M, Alsulimani H, Chang J, et al. Brain Iron Loading Impairs DNA Methylation and Alters GABAergic Function in Mice. FASEB J (2019) 33(2):2460–71. doi: 10.1096/fj.201801116RR
99. De Santa F, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L, et al. Jmjd3 Contributes to the Control of Gene Expression in LPS-activated Macrophages. EMBO J (2009) 28(21):3341–52. doi: 10.1038/emboj.2009.271
100. Chen S, Ma J, Wu F, Xiong LJ, Ma H, Xu W, et al. The Histone H3 Lys 27 Demethylase JMJD3 Regulates Gene Expression by Impacting Transcriptional Elongation. Genes Dev (2012) 26(12):1364–75. doi: 10.1101/gad.186056.111
101. Cao X, Chen Y, Wu B, Wang X, Xue H, Yu L, et al. Histone H4K20 Demethylation by Two hHR23 Proteins. Cell Rep (2020) 30(12):4152–64.e6. doi: 10.1016/j.celrep.2020.03.001
102. Ciesielski P, Jóźwiak P, XXXA. Krześlak: [TET Proteins and Epigenetic Modifications in Cancers]. Postepy Hig Med Dosw (Online) (2015) 69:1371–83. doi: 10.5604/17322693.1186346
103. Tamanaha E, Guan S, Marks K, Saleh L. Distributive Processing by the Iron(II)/α-Ketoglutarate-Dependent Catalytic Domains of the TET Enzymes Is Consistent With Epigenetic Roles for Oxidized 5-Methylcytosine Bases. J Am Chem Soc (2016) 138(30):9345–8. doi: 10.1021/jacs.6b03243
104. Zhao B, Yang Y, Wang X, Chong Z, Yin R, Song SH, et al. Redox-Active Quinones Induces Genome-Wide DNA Methylation Changes by an Iron-Mediated and Tet-dependent Mechanism. Nucleic Acids Res (2014) 42(3):1593–605. doi: 10.1093/nar/gkt1090
105. Huang Y, Tian C, Li Q, Xu Q. Tet1 Knockdown Inhibits Porphyromonas Gingivalis LPS/IFN-γ-Induced M1 Macrophage Polarization Through the NF-κb Pathway in THP-1 Cells. Int J Mol Sci (2019) 20(8):2023. doi: 10.3390/ijms20082023
106. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal Hematopoiesis Associated With TET2 Deficiency Accelerates Atherosclerosis Development in Mice. Science (2017) 355(6327):842–7. doi: 10.1126/science.aag1381
107. Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is Required to Resolve Inflammation by Recruiting Hdac2 to Specifically Repress IL-6. Nature (2015) 525(7569):389–93. doi: 10.1038/nature15252
108. Pasricha SR, Lim PJ, Duarte TL, Casu C, Oosterhuis D, Mleczko-Sanecka K, et al. Hepcidin is Regulated by Promoter-Associated Histone Acetylation and HDAC3. Nat Commun (2017) 8(1):403. doi: 10.1038/s41467-017-00500-z
109. Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, et al. Requirement for the Histone Deacetylase Hdac3 for the Inflammatory Gene Expression Program in Macrophages. Proc Natl Acad Sci USA (2012) 109(42):E2865–74. doi: 10.1073/pnas.1121131109
110. Schmeck B, Lorenz J, N’Guessan P D, Opitz B, van Laak V, Zahlten J, et al. Histone Acetylation and Flagellin are Essential for Legionella Pneumophila-Induced Cytokine Expression. J Immunol (2008) 181(2):940–7. doi: 10.4049/jimmunol.181.2.940
111. Tran PV, Kennedy BC, Lien YC, Simmons RA, Georgieff MK. Fetal Iron Deficiency Induces Chromatin Remodeling At the Bdnf Locus in Adult Rat Hippocampus. Am J Physiol Regul Integr Comp Physiol (2015) 308(4):R276–82. doi: 10.1152/ajpregu.00429.2014
112. Yin X, Wu Q, Monga J, Xie E, Wang H, Wang S, et al. Hdac1 Governs Iron Homeostasis Independent of Histone Deacetylation in Iron-Overload Murine Models. Antioxid Redox Signal (2018) 28(13):1224–37. doi: 10.1089/ars.2017.7161
113. Yang X, Park SH, Chang HC, Shapiro JS, Vassilopoulos A, Sawicki KT, et al. Sirtuin 2 Regulates Cellular Iron Homeostasis Via Deacetylation of Transcription Factor NRF2. J Clin Invest (2017) 127(4):1505–16. doi: 10.1172/jci88574
114. Rothgiesser KM, Erener S, Waibel S, Lüscher B, Hottiger MO. Correction: SIRT2 Regulates NF-κb-Dependent Gene Expression Through Deacetylation of P65 Lys310 (Doi:10.1242/Jcs.073783). J Cell Sci (2019) 132(8):jcs232801. doi: 10.1242/jcs.232801
115. Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W, et al. The SWI/SNF Chromatin Remodelling Complex is Required for Maintenance of Lineage Specific Enhancers. Nat Commun (2017) 8:14648. doi: 10.1038/ncomms14648
116. Monahan BJ, Villén J, Marguerat S, Bähler J, Gygi SP, Winston F. Fission Yeast SWI/SNF and RSC Complexes Show Compositional and Functional Differences From Budding Yeast. Nat Struct Mol Biol (2008) 15(8):873–80. doi: 10.1038/nsmb.1452
117. Xia Y, He F, Wu X, Tan B, Chen S, Liao Y, et al. GABA Transporter Sustains IL-1β Production in Macrophages. Sci Adv (2021) 7(15):eabe9274. doi: 10.1126/sciadv.abe9274
118. Nelson JE, Wilson L, Brunt EM, Yeh MM, Kleiner DE, Unalp-Arida A, et al. Relationship Between the Pattern of Hepatic Iron Deposition and Histological Severity in Nonalcoholic Fatty Liver Disease. Hepatology (2011) 53(2):448–57. doi: 10.1002/hep.24038
119. Rinella ME. Nonalcoholic Fatty Liver Disease A Systematic Review. Jama-Journal Am Med Assoc (2015) 313(22):2263–73. doi: 10.1001/jama.2015.5370
120. Blonder N, Yamada G, Wilson L, Van Natta M, Tonascia J, Handa P, et al. Relationship Between the Pattern of Hepatic Iron Deposition and Histological Severity in Nonalcoholic Fatty Liver Disease. Hepatology (2018) 68:949A–A. doi: 10.1002/hep.24038
121. Nelson JE, Brunt EM, Kowdley KV, Nonalcoholic Steatohepatitis C. Lower Serum Hepcidin and Greater Parenchymal Iron in Nonalcoholic Fatty Liver Disease Patients With C282Y HFE Mutations. Hepatology (2012) 56(5):1730–40. doi: 10.1002/hep.25856
122. Otogawa K, Kinoshita K, Fujii H, Sakabe M, Shiga R, Nakatani K, et al. Erythrophagocytosis by Liver Macrophages (Kupffer Cells) Promotes Oxidative Stress, Inflammation, and Fibrosis in a Rabbit Model of Steatohepatitis - Implications for the Pathogenesis of Human Nonalcoholic Steatohepatitis. Am J Pathol (2007) 170(3):967–80. doi: 10.2353/ajpath.2007.060441
123. Maliken BD, Nelson JE, Klintworth HM, Beauchamp M, Yeh MM, Kowdley KV. Hepatic Reticuloendothelial System Cell Iron Deposition is Associated With Increased Apoptosis in Nonalcoholic Fatty Liver Disease. Hepatology (2013) 57(5):1806–13. doi: 10.1002/hep.26238
124. Atarashi M, Izawa T, Miyagi R, Ohji S, Hashimoto A, Kuwamura M, et al. Dietary Iron Supplementation Alters Hepatic Inflammation in a Rat Model of Nonalcoholic Steatohepatitis. Nutrients (2018) 10(2):14. doi: 10.3390/nu10020175
125. Atarashi M, Izawa T, Mori M, Inai Y, Kuwamura M, Yamate J. Dietary Iron Overload Abrogates Chemically-Induced Liver Cirrhosis in Rats. Nutrients (2018) 10(10):1400. doi: 10.3390/nu10101400
126. Libby P, Buring JE, Badimon L, Hansson CK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers (2019) 5(1):57. doi: 10.1038/s41572-019-0116-x
127. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in Atherosclerosis: A Dynamic Balance. Nat Rev Immunol (2013) 13(10):709–21. doi: 10.1038/nri3520
128. Ramakrishna G, Rooke TW, Cooper LT. Iron and Peripheral Arterial Disease: Revisiting the Iron Hypothesis in a Different Light. Vasc Med (2003) 8(3):203–10. doi: 10.1191/1358863x03vm493ra
129. Glass CK, Witztum JL. Atherosclerosis: The Road Ahead. Cell (2001) 104(4):503–16. doi: 10.1016/s0092-8674(01)00238-0
130. Gordon S. Alternative Activation of Macrophages. Nat Rev Immunol (2003) 3(1):23–35. doi: 10.1038/nri978
131. Vinchi F, Muckenthaler MU, Da Silva MC, Balla G, Balla J, Jeney V. Atherogenesis and Iron: From Epidemiology to Cellular Level. Front Pharmacol (2014) 5:94. doi: 10.3389/fphar.2014.00094
132. Ganz T, Nemeth E. Iron Imports. IV. Hepcidin and Regulation of Body Iron Metabolism. Am J Physiol Gastrointest Liver Physiol (2006) 290(2):G199–203. doi: 10.1152/ajpgi.00412.2005
133. Sullivan JL. Macrophage Iron, Hepcidin, and Atherosclerotic Plaque Stability. Exp Biol Med (Maywood) (2007) 232(8):1014–20. doi: 10.3181/0703-mr-54
134. Sullivan JL. Iron in Arterial Plaque: A Modifiable Risk Factor for Atherosclerosis. Biochim Et Biophys Acta-General Subj (2009) 1790(7):718–23. doi: 10.1016/j.bbagen.2008.06.005
135. Chan KH, Ng MKC, Stocker R. Haem Oxygenase-I and Cardiovascular Disease: Mechanisms and Therapeutic Potential. Clin Sci (2011) 120(11-12):493–504. doi: 10.1042/cs20100508
136. Condeelis J, Pollard JW. Macrophages: Obligate Partners for Tumor Cell Migration, Invasion, and Metastasis. Cell (2006) 124(2):263–6. doi: 10.1016/j.cell.2006.01.007
137. Bingle L, Brown NJ, Lewis CE. The Role of Tumour-Associated Macrophages in Tumour Progression: Implications for New Anticancer Therapies. J Pathol (2002) 196(3):254–65. doi: 10.1002/path.1027
138. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-Related Inflammation. Nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
139. Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, et al. Differential Regulation of Iron Homeostasis During Human Macrophage Polarized Activation. Eur J Immunol (2010) 40(3):824–35. doi: 10.1002/eji.200939889
140. Sierra-Filardi E, Vega MA, Sanchez-Mateos P, Corbi AL, Puig-Kroger A. Heme Oxygenase-1 Expression in M-CSF-polarized M2 Macrophages Contributes to LPS-induced IL-10 Release. Immunobiology (2010) 215(9-10):788–95. doi: 10.1016/j.imbio.2010.05.020
141. Torti SV, Torti FM. Iron and Cancer: More Ore to be Mined. Nat Rev Cancer (2013) 13(5):342–55. doi: 10.1038/nrc3495
142. Zohn IE, De Domenico I, Pollock A, Ward DM, Goodman JF, Liang X, et al. The Flatiron Mutation in Mouse Ferroportin Acts as a Dominant Negative to Cause Ferroportin Disease. Blood (2007) 109(10):4174–80. doi: 10.1182/blood-2007-01-066068
143. Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, et al. The Iron Exporter Ferroportin/Slc40a1 is Essential for Iron Homeostasis. Cell Metab (2005) 1(3):191–200. doi: 10.1016/j.cmet.2005.01.003
144. Hagemeier J, Ramanathan M, Schweser F, Dwyer MG, Lin F, Bergsland N, et al. Iron-Related Gene Variants and Brain Iron in Multiple Sclerosis and Healthy Individuals. Neuroimage-Clinical (2018) 17:530–40. doi: 10.1016/j.nicl.2017.11.003
145. Mallucci G, Peruzzotti-Jametti L, Bernstock JD, Pluchino S. The Role of Immune Cells, Glia and Neurons in White and Gray Matter Pathology in Multiple Sclerosis. Prog Neurobiol (2015) 127:1–22. doi: 10.1016/j.pneurobio.2015.02.003
146. Haider L, Simeonidou C, Steinberger G, Hametner S, Grigoriadis N, Deretzi G, et al. Multiple Sclerosis Deep Grey Matter: The Relation Between Demyelination, Neurodegeneration, Inflammation and Iron. J Neurol Neurosurg Psychiatry (2014) 85(12):1386–95. doi: 10.1136/jnnp-2014-307712
148. Zumbrennen-Bullough K, Babitt JL. The Iron Cycle in Chronic Kidney Disease (CKD): From Genetics and Experimental Models to CKD Patients. Nephrol Dialysis Transplant (2014) 29(2):263–73. doi: 10.1093/ndt/gft443
149. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. α-Ketoglutarate Orchestrates Macrophage Activation Through Metabolic and Epigenetic Reprogramming. Nat Immunol (2017) 18(9):985–94. doi: 10.1038/ni.3796
150. Zhao Y, Guo Y, Jiang Y, Zhu X, Liu Y, Zhang X. Mitophagy Regulates Macrophage Phenotype in Diabetic Nephropathy Rats. Biochem Biophys Res Commun (2017) 494(1-2):42–50. doi: 10.1016/j.bbrc.2017.10.088
151. Allen GF, Toth R, James J, Ganley IG. Loss of Iron Triggers PINK1/Parkin-independent Mitophagy. EMBO Rep (2013) 14(12):1127–35. doi: 10.1038/embor.2013.168
152. Zhang J, Hu W, Ding C, Yao G, Zhao H, Wu S. Deferoxamine Inhibits Iron-Uptake Stimulated Osteoclast Differentiation by Suppressing Electron Transport Chain and MAPKs Signaling. Toxicol Lett (2019) 313:50–9. doi: 10.1016/j.toxlet.2019.06.007
153. Chen XL, Zhang GP, Guo SL, Ding JQ, Lin JJ, Yang Q, et al. Mfn2-Mediated Preservation of Mitochondrial Function Contributes to the Protective Effects of BHAPI in Response to Ischemia. J Mol Neurosci (2017) 63(3-4):267–74. doi: 10.1007/s12031-017-0976-z
154. Park J, Lee DG, Kim B, Park SJ, Kim JH, Lee SR, et al. Iron Overload Triggers Mitochondrial Fragmentation Via Calcineurin-Sensitive Signals in HT-22 Hippocampal Neuron Cells. Toxicology (2015) 337:39–46. doi: 10.1016/j.tox.2015.08.009
155. Zheng Q, Zhao Y, Guo J, Zhao S, Fei C, Xiao C, et al. Iron Overload Promotes Mitochondrial Fragmentation in Mesenchymal Stromal Cells From Myelodysplastic Syndrome Patients Through Activation of the AMPK/MFF/Drp1 Pathway. Cell Death Dis (2018) 9(5):515. doi: 10.1038/s41419-018-0552-7
156. Zhang X, Xing X, Liu H, Feng J, Tian M, Chang S, et al. Ionizing Radiation Induces Ferroptosis in Granulocyte-Macrophage Hematopoietic Progenitor Cells of Murine Bone Marrow. Int J Radiat Biol (2020) 96(5):584–95. doi: 10.1080/09553002.2020.1708993
157. Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of Ferroptosis in Murine Models of Hemochromatosis. Hepatology (2017) 66(2):449–65. doi: 10.1002/hep.29117
158. Fuhrmann DC, Mondorf A, Beifuß J, Jung M, Brüne B. Hypoxia Inhibits Ferritinophagy, Increases Mitochondrial Ferritin, and Protects From Ferroptosis. Redox Biol (2020) 36:101670. doi: 10.1016/j.redox.2020.101670
159. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox Lipid Reprogramming Commands Susceptibility of Macrophages and Microglia to Ferroptotic Death. Nat Chem Biol (2020) 16(3):278–90. doi: 10.1038/s41589-019-0462-8
Keywords: iron, inflammation, macrophage, metabolism, epigenetics
Citation: Xia Y, Li Y, Wu X, Zhang Q, Chen S, Ma X and Yu M (2021) Ironing Out the Details: How Iron Orchestrates Macrophage Polarization. Front. Immunol. 12:669566. doi: 10.3389/fimmu.2021.669566
Received: 19 February 2021; Accepted: 26 April 2021;
Published: 12 May 2021.
Edited by:
Pinyi Lu, Frederick National Laboratory for Cancer Research (NIH), United StatesReviewed by:
Rafiou Agoro, Indiana University, United StatesLikui Feng, The Rockefeller University, United States
Copyright © 2021 Xia, Li, Wu, Zhang, Chen, Ma and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xianyong Ma, bWF4aWFueW9uZ0BnZGFhcy5jbg==; Miao Yu, eXVtaWFvQGdkYWFzLmNu
†These authors have contributed equally to this work