 Randi Lassiter
Randi Lassiter Todd D. Merchen
Todd D. Merchen Xuexiu Fang2
Xuexiu Fang2 Youli Wang
Youli Wang- 1Department of Surgery, Medical College of Georgia at Augusta University, Augusta, GA, United States
- 2Division of Nephrology, Department of Medicine, Medical College of Georgia at Augusta University, Augusta, GA, United States
Renal tubular epithelial cells (TECs) are the primary targets of ischemia–reperfusion injury (IRI) and rejection by the recipient’s immune response in kidney transplantation (KTx). However, the molecular mechanism of rejection and IRI remains to be identified. Our previous study demonstrated that kynurenine 3-monooxygenase (KMO) and kynureninase were reduced in ischemia–reperfusion procedure and further decreased in rejection allografts among mismatched pig KTx. Herein, we reveal that TEC injury in acutely rejection allografts is associated with alterations of Bcl2 family proteins, reduction of tight junction protein 1 (TJP1), and TEC-specific KMO. Three cytokines, IFNγ, TNFα, and IL1β, reported in our previous investigation were identified as triggers of TEC injury by altering the expression of Bcl2, BID, and TJP1. Allograft rejection and TEC injury were always associated with a dramatic reduction of KMO. 3HK and 3HAA, as direct and downstream products of KMO, effectively protected TEC from injury via increasing expression of Bcl-xL and TJP1. Both 3HK and 3HAA further prevented allograft rejection by inhibiting T cell proliferation and up-regulating aryl hydrocarbon receptor expression. Pig KTx with the administration of DNA nanoparticles (DNP) that induce expression of indoleamine 2,3-dioxygenase (IDO) and KMO to increase 3HK/3HAA showed an improvement of allograft rejection as well as murine skin transplant in IDO knockout mice with the injection of 3HK indicated a dramatic reduction of allograft rejection. Taken together, our data provide strong evidence that reduction of KMO in the graft is a key mediator of allograft rejection and loss. KMO can effectively improve allograft outcome by attenuating allograft rejection and maintaining graft barrier function.
Highlights
Immunosuppressants reduce allograft rejection to prolong allograft survival in kidney transplantation. However, the long-term use of these medications often results in complications that can lead to diseases or even allograft loss. Thus, endogenous immunosuppressive metabolites may represent more ideal medical therapies. Indoleamine 2,3-dioxygenase (IDO) has been reported to prevent allogeneic fetal rejection. To date, neither the molecular mechanism of tolerance induced by IDO nor its downstream enzymes have been clearly defined. This paper demonstrates that kynurenine 3-monooxygenase (KMO), a downstream enzyme of IDO, is the main player in tolerance induced by the kynurenine metabolites, as its direct and downstream products, 3HK and 3HAA, can prevent T cell-induced allograft rejection and protect the tubular epithelial cell from injury caused by the cytokine storm formed in ischemia–reperfusion and early allograft rejection procedure.
Introduction
Kidney transplantation (KTx) is the best treatment for patients with renal failure. Current KTx requires overcoming two major obstacles, including the shortage of available organs for transplant and significant side effects of antirejection medications. Scientists are developing novel organ resources for clinical transplantation. Among all, swine are the most ideal organ donor for clinical transplants due to: (i) the similar size of kidneys and ease of transplantation; (ii) similar renal metabolic function for unrestricted food intake and metabolism; (iii) a variety of swine leukocyte antigens that offer biological diversity and antigen-driven rejection mechanisms similar to humans. Additionally, researchers are also using blastocyst complementation combined with gene editing to increase organs favorable for human transplant (1–3). Although pigs can provide enough organs for transplantation, there is still some distance from the laboratory bench to clinical application. The major barrier is the transmission of infectious microorganisms from pigs to humans (4–9). Although many studies have reported that there are no transmissions of virus in xenotransplantation (10–13), inactivation of endogenous porcine retroviruses using CRISPR-Cas9 technology makes xenotransplantation safer and potentially possible in the future (14, 15).
Rejection of the recipient’s immune system to imported allografts forms the main barrier for successful clinical transplantation and future xenotransplantation (16, 17). Currently, the combination of immunosuppressants to deactivate the recipient’s immune response is used to prevent rejection. These immunosuppressants can temporarily inhibit acute rejection to prolong an allograft’s survival but have less impact on long-term allograft outcomes. The toxicity of immunosuppressants causes complications leading to new diseases that can ultimately lead to graft loss. Endogenous metabolites with antirejection properties may make ideal drugs owing to less toxicity and higher specificity.
Indoleamine 2, 3-dioxygenase (IDO) is the first enzyme in the metabolic pathway from which tryptophan is metabolized to kynurenines. IDO has been shown to prevent allogeneic fetal rejection (18). This antirejection specifically relies on tryptophan depletion and the production of kynurenines (19, 20). IDO downstream enzymes, kynurenine 3-monooxygenase (KMO) and kynureninase produce kynurenine derivatives 3HK (hydroxyl-3 kynurenine) and 3HAA (hydroxyl-3 anthranilic acid), which have been shown to effectively inhibit T cell proliferation (21), thus improving tolerance (22–26). Although induced IDO can increase tolerance in some rodent transplantation models (18, 26–35), our recent study and other scientists’ observations have indicated that IDO, itself, only predicts allograft rejection (36–40). Using our own porcine kidney transplant model, we showed that rejection allografts was associated with down-regulation of KMO and kynureninase and up-regulation of IDO (40).
Tubular epithelial cell (TEC) injury, not glomerular injury, is the primary target of the recipient’s immune system during acute rejection (41, 42). Ischemia–reperfusion, an unavoidable part of the KTx procedure, can also cause ischemia–reperfusion injury (IRI) to TEC. This has been identified through three key signaling pathways (43). Furthermore, a 1-h extension of cold ischemia time increases the risk of rejection by 4% post-transplant (44, 45). Our previous study revealed that alterations of cytokine expression in early allograft rejection were associated with an increase in IDO and a decrease in KMO and other kynurenine metabolic enzymes (40). In the present study, we investigate the influence of cytokine exposure on the gene expression of kynurenine metabolites and cell injury proteins in human primary TEC and allograft rejection. A crucial role of KMO and kynureninase in preventing TEC injury and attenuating allograft rejection will be demonstrated.
Materials and Methods
Animal Study
Studies were performed on 30–40 kg outbred female Yorkshire piglets (Palmetto Research Swine, Reevelville, South Carolina) as we have described previously (40). Briefly, one pair of pigs was operated simultaneously such that the left kidneys were exchanged (allotransplants) or retransplanted (autotransplants). All transplants were ex vivo perfused at 4°C and orthotopically transplanted, followed by right nephrectomy and closure (40). No immunosuppressants were used. All auto- and allotransplanted kidneys and other physiological samples were collected 72 h post-transplantation from the live animals before euthanasia. These studies were approved by Augusta University Institutional Animal Care and Use Committee. Tissues from collected organs were processed for future examination of histology, enzymatic assay, protein expression, and mRNA alterations.
Murine Skin Transplantation
Murine skin transplantation was performed with a modification of the previous protocol (46). Briefly, mouse-ear skin was utilized as a graft onto the back of a recipient mouse. Balbc and C57/B6 mice between 10 and 13 weeks old were used in syngeneic and allogeneic transplants. Rather than suturing, the graft was held in place with a four layer flexible bandage which remained in place in a standard length of time until dressing removal (Fang X, unpublished).
Human Primary Cell Culture
Human cortical renal TEC (HREC, #CC-2554, Lonza) was cultured with a specific medium (#CC-3190, Lonza). TEC expansion, passage, and stocking were performed by following vendor protocols (#CC-5034, Lonza). Cells were seeded in a 1:3 split in six-well plates and grown to 85% confluency with media changes every other day. Then cells were replaced with fresh media and pretreated in the presence or absence of 5–100 μM 3HK (Sigma) or 10–40 μM 3HAA (Sigma) for 16 h. Then cells were challenged with or without cytokine cocktail (15 nM of IFNγ + 6 nM of TNFα + 3 nM of IL1β) for 24 h prior to harvesting. Trizol was utilized for RNA isolation and assays. RIPA buffer was used to lyse cells for Western blot.
Human Peripheral Blood Pan-T Cell Proliferation
Human peripheral blood Pan-T cells (hPBTs, Stemcell Technologies, Canada) were grown in ImmunoCult™-XF T cell expansion medium plus ImmunoCult™-human CD3/CD28/CD2 (25 µl/ml, StemCell Technologies, Canada) and IL-2 (10 ng/ml, StemCell Technologies, Canada) for 4 days. Following this period of expansion/activation, the activated hPBTs were labeled with CellTrace™ Cell Proliferation Kits (#C34557, Invitrogen). Labeled or unlabeled activated hPBTs were grown in proliferation assay medium [PAM, ImmunoCult™-XF T cell expansion medium plus ImmunoCult™-Human CD3/CD28/CD2 (25 µl/ml)] and IL2 (2 ng/ml) in the presence of 3HK. The cells were collected and washed with PBS, and then the cells were fixed with 1% paraformaldehyde in PBS for 20 min at room temperature. The proliferation of T cells was determined with flow cytometry.
Histology and Immunostaining
Freshly dissected kidneys were fixed overnight with 4% paraformaldehyde in PBS (pH 7.2) at 4°C, and kidneys were cut into 4-μm sections. The immunostaining procedure was performed as described previously (40). Immunochemistry was completed using a KMO antibody (Novus Biologicals, Cat.NBP1-86335) and Cyclophilin A (Abcam. Cat. Ab154388) with 1:400 dilutions in 5% donkey serum in standard TBS buffer. The staining was viewed with color developed from horseradish peroxidase complex (Vector Laboratories) and counterstained with hematoxylin.
Quantitative RT-PCR
Quantitative RT-PCR was carried out as previously described (40, 47). Total RNA was isolated from renal tissue and cultured cells with TRIZOL. The RNA was converted to cDNA using SuperScript III First-Strand Synthesis System (Invitrogen). PCR primers producing 100–130 bp amplicon were designed using Primer 3 Input software, the primers for candidate genes were shown in Table 1, and quantitative PCR was performed with SsoAdvanced Universal SYBR Green Kit (Bio-Rad Lab) on a CFX Connect real-time PCR detection system (Bio-Rad Lab). β-actin mRNA was used for normalization.
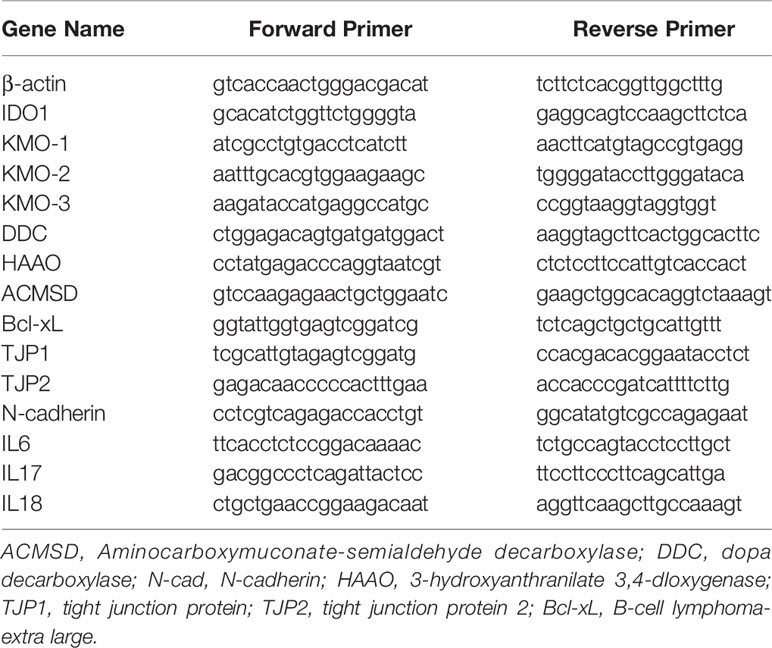
Table 1 Primers used for qPCR.
TUNEL Staining
TUNEL staining was performed with the ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (Millipore S7101) according to the manufacturer’s protocol. TUNEL-positive cells were randomly counted from five different 20× microscopic vision fields for each pig. Total TUNEL-positive cells were quantified from the tissues of four to eight pigs per group.
LDH Assay
3HK toxicity to TEC, tubular endothelial cells (TEndo), and hPBT were assessed with lactate dehydrogenase (LDH) assays using Pierce LDH Cytotoxicity Assay Kit and the chemical compound-mediated cytotoxicity assay protocol (Thermo Scientific). Briefly, TEC, TEndo, and hPBT were grown in their growth media to 85% confluency, while hPBT was grown in the mixture of TEC media and hPBT media in a 2:1 ratio to reduce hPBT medium background in the LDH assay. TEC, TEndo, and hPBT were then treated with different doses of 3HK. LDH assay was performed by following the manufacturer’s instructions.
Western Blot
Renal tissue and cells were lysed in RIPA buffer containing the cocktail of proteinase inhibitors (Roche) and phosphatase inhibitors. Protein concentrations were determined using a bicinchoninic acid protein assay reagent kit (Pierce). Proteins were separated by SDS-PAGE, and Western blotting was carried out as previously described (40). The following antibodies were used as probes for Western blotting: β-actin (Sigma, A5316); KMO (NBP1-86335), kynureninase (AF4887-SP), alpha-smooth muscle actin (NB300-978SS), snail (NBP2-27184SS), E-cadherin (NBP2-19051SS), tight junction protein 1 (NBP1-85047) were from Novus Biologicals; Bax (sc-7480), Bcl-xL (sc-8392), BID (sc-373939), caspase 3 (sc-7272), caspase 8 (sc-56070), VDR (sc-1008), AhR (sc-133088), pNFκB65-S536 (sc-136548), NFκB65 (sc-8008), ERK-Th202/Ty204 (sc-136521), MHCI (sc-55582), ERK (sc-514302) were from Santa Cruz Biotechnology. Akt-S473 (9271S), Akt (9272S), pPTEN-S380 (9551), and PTEN (9552), MHCII (LGII-612.14), and Bcl2 (4223T) were from Cell Signaling; IDO1 (M256) was obtained from CalBioreagents. Secondary antibodies were horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (Pierce Biotechnology), and signals were detected using SuperSignal West Pico or Dura Extended Duration Substrate (Pierce). β-actin was used for loading normalization. The relative amount of each protein was quantified using NIH ImageJ software.
DNA Nanoparticles
DNA nanoparticles (DNP) were prepared as previously described (48). Briefly, 21 µg of pCpG free–Lacz DNA (InvivoGen, CA) was mixed with 200 µl of 10% of D-(+)-glucose solution at room temperature. Then 7 µl of 150 mM polyethylenimine (PEI, Polysciences Inc, PA) was mixed with another 200 µl of 10% of D-(+)-glucose solution in room temperature. The DNA–glucose solution was then added to PEI–glucose solution under gentle vortex. The mixture of PEI–DNA was kept at room temperature for 10–15 min to form DNA complex. Twenty-one micrograms of DNA in 0.4 ml 10% glucose solution was intravenously injected into 20 g mice or ex vivo perfused into 20 g of pig kidney. For the kidneys larger than 20 g, the ratio of DNA micrograms per PEI in microliters was maintained at 3:1. Thus the amount of DNA, PEI, and 10% glucose solution increased proportionately in accordance with the increase in kidney weight.
Statistical Analyses
Data were presented as means ± SEM. Statistical comparisons were carried out using unpaired t-test or one-way ANOVA as appropriate, with p <0.05 considered as statistically significant
Results
Pathology and Gene Expression of Grafts in Pig Kidney Transplantation
Many factors influence the outcome of allografts in KTx. These factors include the results of ischemia–reperfusion which accompanies the surgical procedure and rejection by the recipient’s immune system. Allograft injury is always the first phenomenon in early stage post-transplant before allograft failure and loss. A number of observations have indicated that tubular epithelial cells are the primary targets of IRI and rejection (41–43, 49). Therefore, the alterations of TEC gene profile and cell phenotype as well as TEC death change cell structure and physiological function leading to allograft failure. We first explored cell death patterns in autograft and allograft in pig kidney transplantation. A huge number of cells with positive staining for necrotic biomarker cyclophilin A are seen in allograft rejection within infiltrated recipient circulatory cells and graft’s interstitial structural and tubular cells. No necrotic cells were found in autograft (Figure 1A). Interestingly, both H&E staining and TUNEL staining for apoptotic cells indicated specific tubular injury and increasing apoptotic cells in allograft tubules. Apoptotic cells are increased four-fold in autografts and increased 16-fold in allografts (Figures 1A, C).
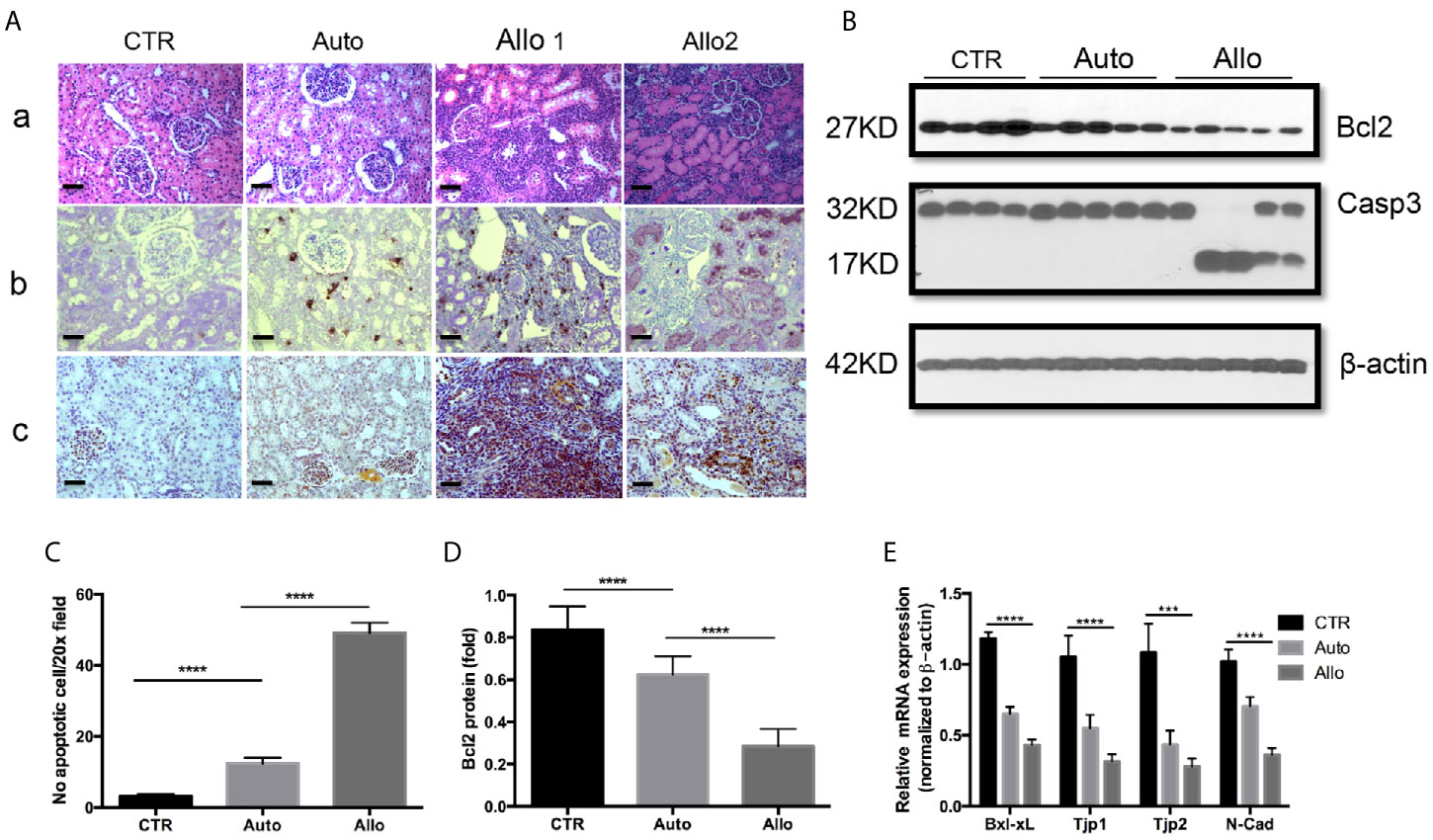
Figure 1 Histology, pathology, and gene expression of kidney tissues. (A): (a) Histology of a representative control right kidney (CTR), an autotransplant kidney (auto), and 2 Banff level III rejection allografts (allo). N-18 for the CTR group, n = 5 for the auto group, and n = 18 for the allo group. (b) Apoptotic cells of TUNEL positive staining on representative CTR, auto, and allograft. (c) Necrotic cells of cyclophilin A (CypA) positive staining on representative CTR, auto, and allograft. (B): Gene expression of anti-apoptotic protein Bcl2 and caspase 3 in representative CTR, auto, and allograft. (C) The quantitative average number of apoptotic cells on the 20× microscope view field from random five pictures on each animal tissue. n = 4 for the CTR group, n = 5 for the autotransplant group, n = 7 for the allotransplant group. (D) Quantitative analysis of Bcl2 protein expression in representative graft. (E) Quantitative PCR of Bcl-xL, tight junction proteins 1 and 2, and N-cadherin mRNA in grafts. ***P < 0.001, ****P < 0.0001, one-way ANOVA, auto versus CTR, allo versus auto. The scale bar is 50 µm.
Transplantation procedures were performed on 52 pigs. Among them, 20 pigs underwent allotransplant without any drug treatment. Five of the twenty pigs (two of which were twins) experienced allograft rejection rated IIA on the Banff scale, while six pigs experienced allograft rejection below IIA on the Banff scale. Pigs with allograft rejection rated IIA and lower had normal physiological parameters (40, 50). Nine of the twenty pigs had allograft rejection rated above Banff scale IIA and abnormal physiological function (Figure 1A). We picked one pig allograft with Banff scale IIA to group with allografts rated higher on the Banff scale to investigate the expression of apoptotic markers Bcl2 and caspase 3 (Casp 3).
Bcl2 family proteins play an important role in cell apoptosis. Western blot showed that anti-apoptotic Bcl2 is moderately reduced in the autograft and dramatically decreased in allograft. Bcl2 reduction is further associated with the activation of Casp 3 (Figures 1B, D). Allografts with Banff II had normal physiological function which was only shown on reduction of Bcl2 but no activation of Casp 3 (Figure 1B). Bcl-xL is another anti-apoptotic protein in the Bcl2 family (51). Quantitative real-time PCR indicated that Bcl-xL was greatly decreased in allograft rejection (Figure 1E). In addition to cell death, epithelial–mesenchymal transition (EMT) is another phenotype of epithelial cell injury which results in changes in epithelial cell physiological function. Western blot indicated that the biomarker of EMT, N-cadherin (N-cad), was reduced in allograft rejection and was associated with the reduction of tight junction protein 1 (TJP1) and tight junction protein 2 (TJP2) expression (Figure 1E).
Rejection and Biological Function of Allograft Were Associated With Expression of KMO and Its Downstream Genes in Kynurenine Metabolism
Our recent article in pig KTx demonstrated the dramatic increase of IDO and a decrease of KMO in allograft rejection (40). KMO is a mitochondrial outer membrane protein. Immunohistochemistry (IHC) of KMO on human kidney tissues from antibody providers indicated that KMO was specifically located in tubular epithelial cells. We further confirmed that high expression of KMO was observed in pig kidney tubular cells. However, KMO expression was reduced in autografts and was almost silenced in allograft rejection (Figure 2A). Recipient cells that infiltrate the allograft also have a high expression of KMO (arrows in Figure 2A). The ratio of KMO mRNA in allograft rejection to average KMO mRNA from 19 normal grafts was positively associated with allograft biological function measured by creatinine (Figure 2B) and was negatively associated with the grade of allograft rejection on the Banff scale (Figure 2C). However, the ratio of KMO mRNA in allograft rejection to average KMO mRNA from normal grafts was not associated with the BUN level (Figure 2D). The expression of more genes in the kynurenine metabolic pathway was analyzed with quantitative PCR. The results indicated that 3-hydroxyanthranilic 3,4 dioxygenase (HAAO), aminocarboxymuconate-semialdehyde decarboxylase (ACMSD), and dopa decarboxylase (DDC) were dramatically reduced in autografts and further decreased in allograft rejection (Figures 2E–G).
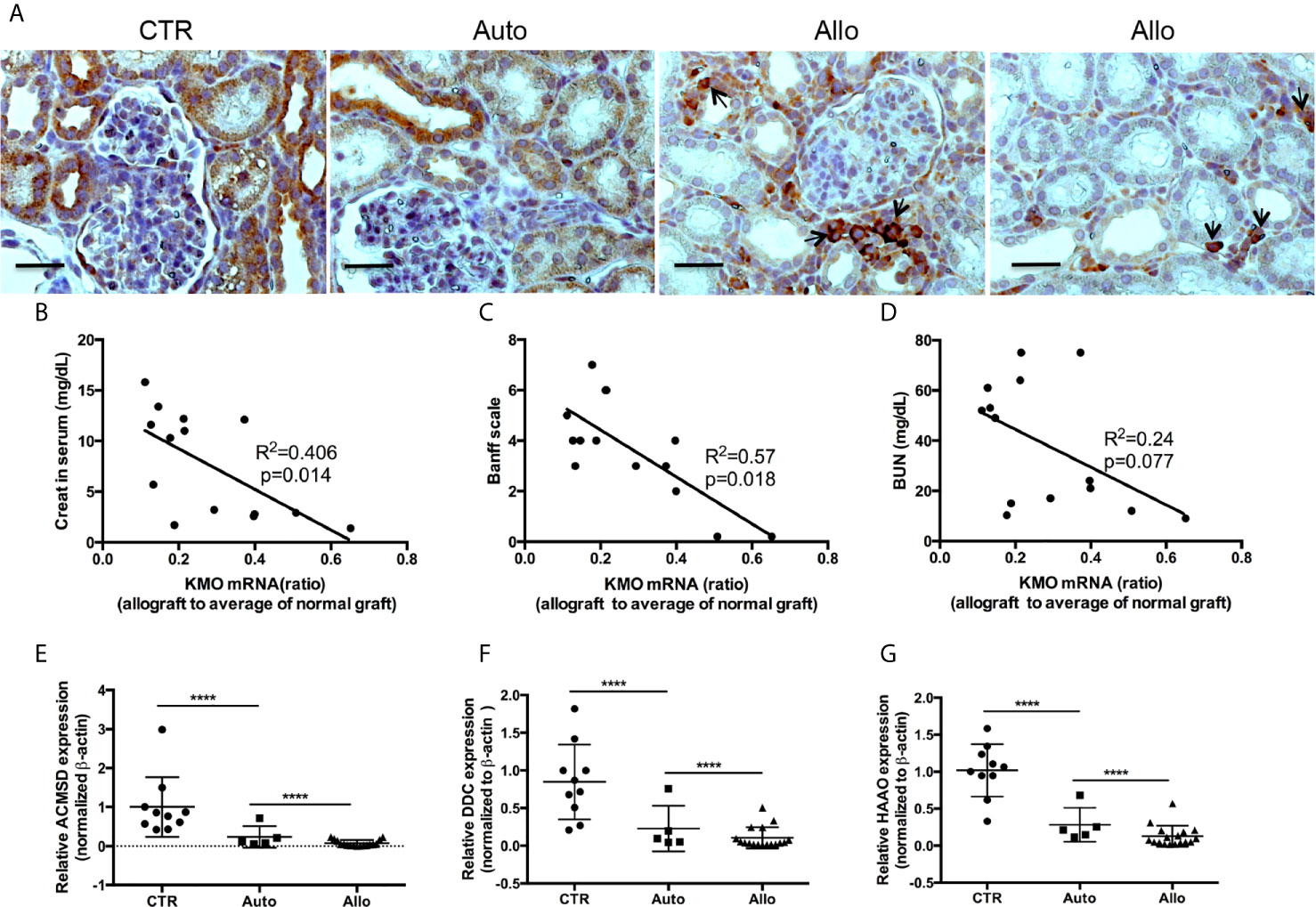
Figure 2 The expression of KMO and other kynurenine metabolic enzymes in grafts. KMO mRNA in grafts were determined by real-time PCR via using three pairs of PCR primers which amplify the 5 terminal (161–265 bp), middle (493–600 bp), and 3 terminal (1,239–1,349 bp) specific sequence of KMO gene; the real-time PCR from three pairs of primers showed almost the same result. β-actin mRNA for normalization, an average of KMO mRNA was calculated from the results of real-time PCR from 19 normal pig kidneys. The ratio of KMO mRNA in allograft rejection to the average of KMO mRNA in normal kidneys is KMO mRNA fold alterations. (A) IHC of KMO on graft tissue section. (B–D) Correlation of KMO mRNA ratio in allograft rejection with serum creatinine, allograft Banff scale, and BUN in recipients. (E–G) Quantitative analysis of HAAQ, DDC, and ACMSD in grafts. ****P < 0.0001, one-way ANOVA, Auto versus CTR, Allo versus Auto. The scale bar is 500 µm.
Cytokine Storm From IRI and Rejection Induced Gene Expression and Signaling Alterations on TEC
Our previous study showed that many cytokines were over-produced in autografts and allografts (40). IL17 is associated with rejection (52–55). IL17 and IL6 are associated with rejection injury (56–58), while IL18 induces local cytokines and chemokine expression in the allograft (59, 60). Quantitative PCR analysis showed that IL17 expression did not change in autografts but is increased 3.03-fold in the allografts. IL18 increased 1.38-fold in autografts and increased 2.34-fold in the allografts. IL6 increased 2.11-fold in the autografts, but decreased a little in allograft rejection (data not shown). Furthermore, allograft rejection in pig KTx had increased expression of IFNγ, TNFα, and IL1β (40). Therefore, allografts are exposed to many altered cytokines or chemokines from ischemia–reperfusion and early rejection procedure.
Next, we used TEC to investigate their injury caused by cytokine storm using a cytokine cocktail consisting of IFNγ, TNFα and IL1β. Western blot indicated TEC challenged with cytokine cocktail showed increased IDO, KY, and MHC I & II. We also found both E-cadherin (E-cad) and α-smooth muscle actin (SMA) increased. KMO, AhR, and TJP1 were greatly decreased. Snail expression was reduced (Figure 3). We also found the expression of Bax was unchanged while BID was greatly induced, and Bcl2 and Bcl-xL were dramatically reduced. Both total Casp 3 and caspase 8 (Casp 8) were increased (Figure 4). Alterations of Bcl2 family members’ expression and activation of caspases causing epithelial cell apoptosis in allograft rejection confirmed cell apoptosis in allograft rejection in vivo (Figure 1B).
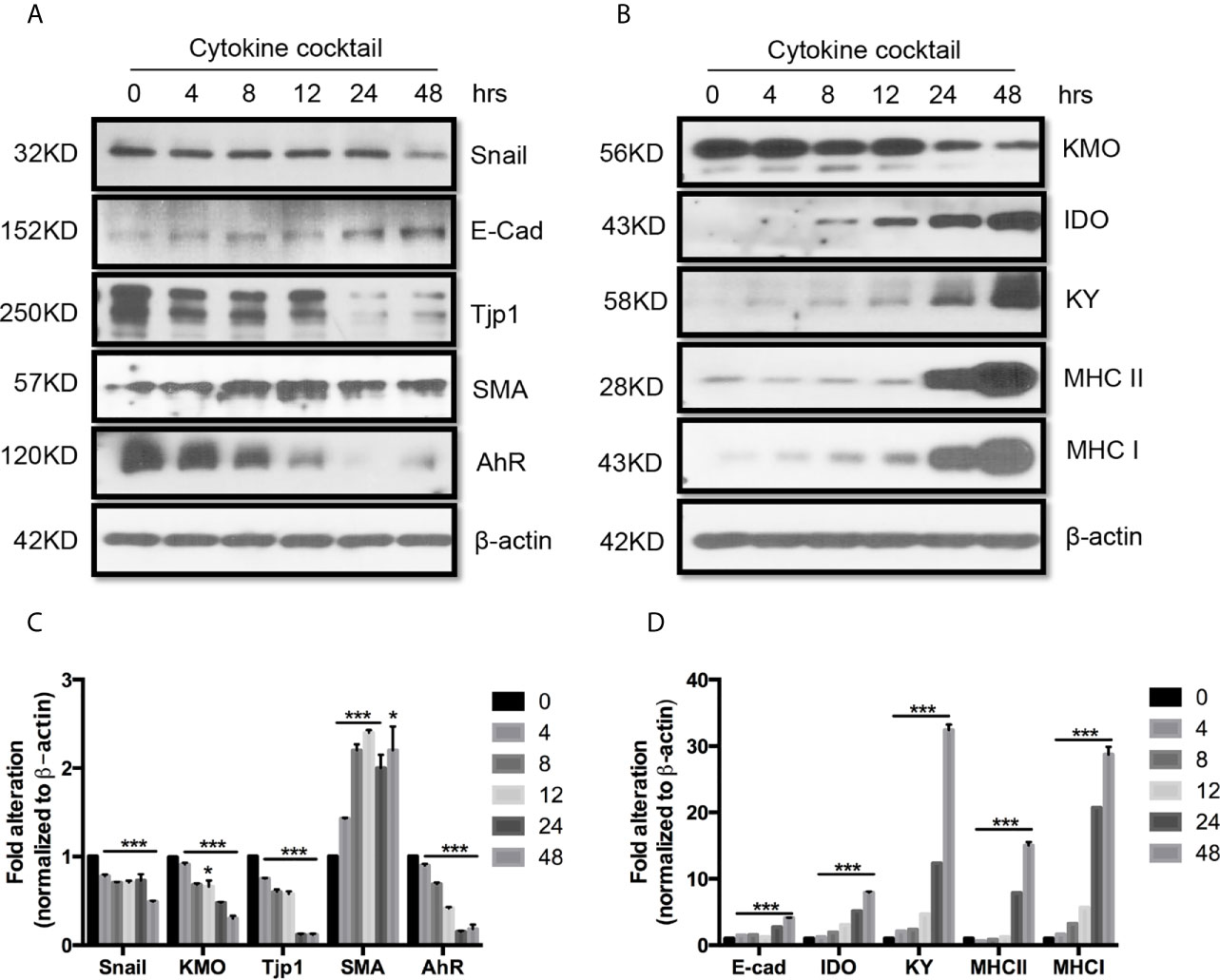
Figure 3 Western blot analysis of function protein expression on primary human renal cortical epithelial cells challenged with cytokines. Primary human renal tubular epithelial cells are challenged with cytokine cocktail (15nM IFNγ, 6nM TNFα, and 3nM IL1β). (A, B) Expression of Snail, E-Cad, TJP1, SMA. AhR, IDO, KMO, KY, MHCI & II. (C, D) Densitometric quantitation of protein in (A, B). All Western blots are representative of at least three independent experiments. *P < 0.05, ***P < 0.0001 versus to cells without treatment, analysis is multiple t-tests, pairwise comparison of individual time-point to control indicated same p-value.
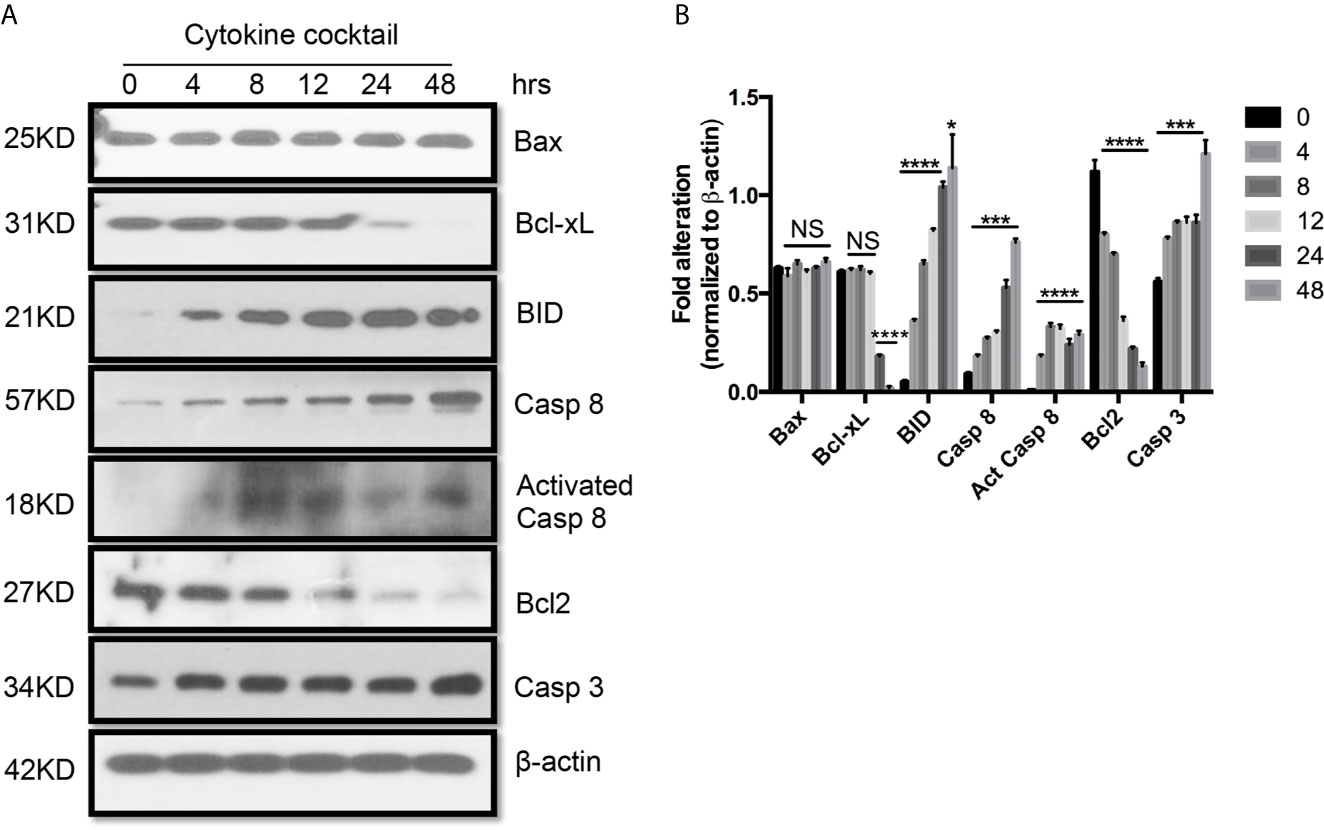
Figure 4 Western blot analysis of apoptotic protein expression on primary human renal cortical epithelial cells challenged with cytokines. (A) The expression of Bcl2, Bcl-xL, BID; Bax caspase 3 and 8 (Casp 3 and 8), activated caspase 8 (Act Casp 8) in cells treated with cytokine cocktail. (B) Densitometric quantitation of proteins in (A). All Western blots are representative of three independent experiments. NS, no significant, *P < 0.05, ***P < 0.0001, ****P < 0.00001 versus cells without treatment; analysis is multiple unpaired t-tests. Pairwise comparison of individual time-point to control indicated the same p-value.
Western blot also indicated that multiple signaling pathways including AktS473, pNFκB65, ErkTh202/Ty204, and PTenS38 in TEC challenged with cytokine cocktail were activated in different time-points (Figure 5). These activated pathways showed the complex biological responses in allograft rejection. Indeed, three signaling pathways can be activated in TEC just from ischemia–reperfusion injury only (43). It is expected that allograft rejection in KTx will activate much more complex biological procedures.
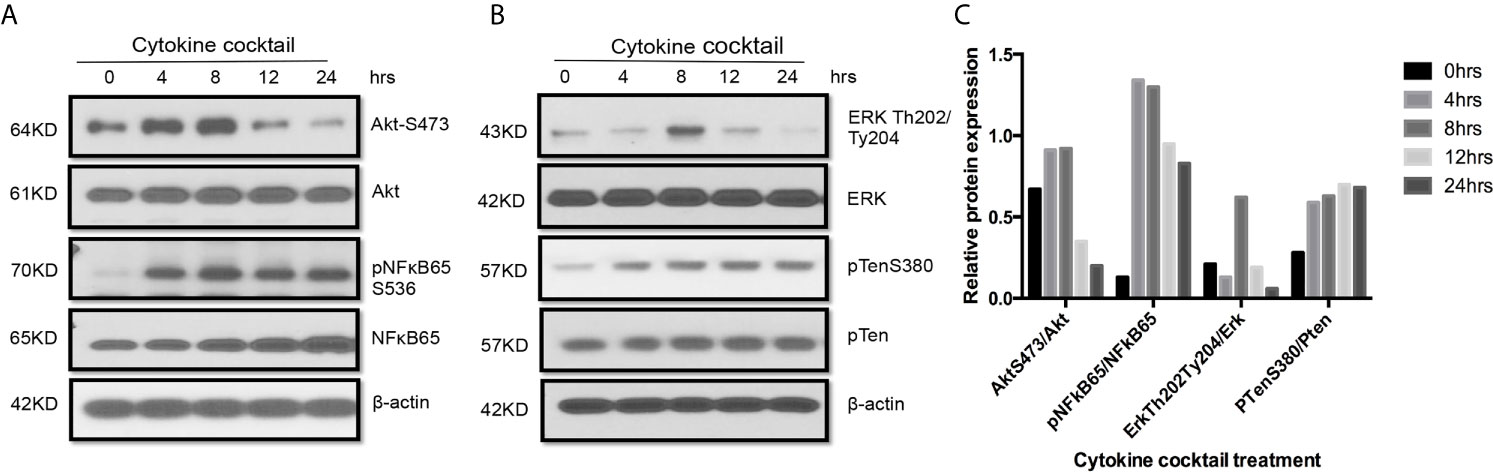
Figure 5 Activation of multiple signaling pathways in TEC challenged by cytokines. Primary human renal TECs were treated with the cytokine cocktail. (A, B) Western blot of PI3K–Akt pathway, NFkb pathway, and Erk signaling pathway in cells treated with cytokine cocktail. (C) Densitometric quantitation of AktS473, pNFkB65, ErkTh202/Ty204, and pTenS380. All Western blots are representative of two independent experiments.
The 3HK and 3HAA From KMO and Its Downstream Partners in Kynurenine Metabolism Effectively Protected TEC From Cytokines-Induced Injury and Inhibited T Cell Proliferation
KMO was down-regulated in autografts and was almost completely silenced in allograft rejection (40). KY was also decreased in autografts and further reduced in allograft rejection, negatively affecting the production of 3HK and 3HAA. This contrasts with greatly increased IDO expression in allograft rejection. Upon exposing TEC to the cytokine cocktail in the presence of 3HK and 3HAA, Western blot showed that both 3HK and 3HAA caused up-regulation of Bcl-xL and TJP1 expression (Figures 6A–C). 3HAA, but not 3HK, reversed cell tolerance protein AhR expression (Figure 6A). These findings suggest that both compounds have a different specific function, and both play crucial roles in protecting TEC from injury.
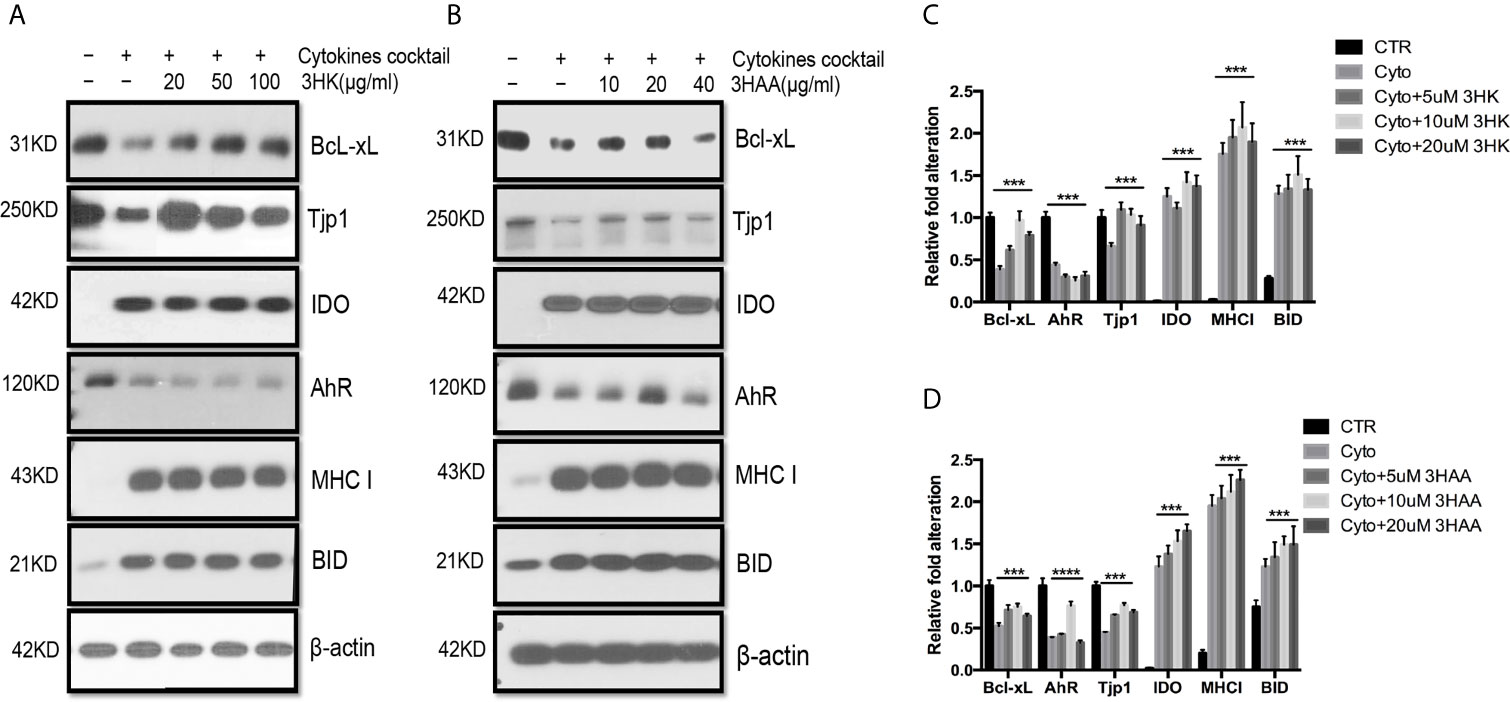
Figure 6 Protective role of 3HK and 3HAA in TEC stimulated with cytokines. Primary human renal TEC pre-incubated with different doses of 3HK or 3HAA overnight; the cells are challenged with the cytokine cocktail (15 nM IFNγ, 6 nM TNFα, and 3 nM IL1β) for 24 h; protein expression was assayed with Western blot: (A, B) 3HK and 3HAA cannot reverse IDO, MHC I, BID expression induced by cytokines. 3HK and 3HAA effectively restore Bcl-xL and Tjp1 expression. 3HAA shows its different function in up-regulation of AhR expression in TEC in inflammatory conditions. (C) Qualification of protein expression in TEC challenged with cytokine cocktail in the presence of 3HK. ***P < 0.0001 versus cell treated with Cyto. (D) Qualification of protein expression in TEC challenged with cytokine cocktail in the presence of 3HAA. ***P < 0.0001, ****P < 0.00001 versus cell treated with Cyto. Pairwise comparison of individual dose to control indicated same p-value. All Western blots are representative of three independent experiments; analysis is multiple one-way ANOVA.
Flow cytometry analysis shown that cytokine cocktail dramatically increased T cell proliferation (Figures 7A, B), whereas 3HK and 3HAA effectively inhibited T cell proliferation at 100 and 10uM (Figures 7C–F), which confirmed previous observations (24, 25, 61). 3HK is toxic to neurons and pancreatic tissue in high doses (62–64). We performed an LDH assay to address the toxicity of 3HK on TEC, TEndo, and hPBT. We found that 3HK had a protective role on TEC and TEndo at the concentration below 100 µM and is toxic to hPBT even at the concentration of 10 µM (Figure 7G).
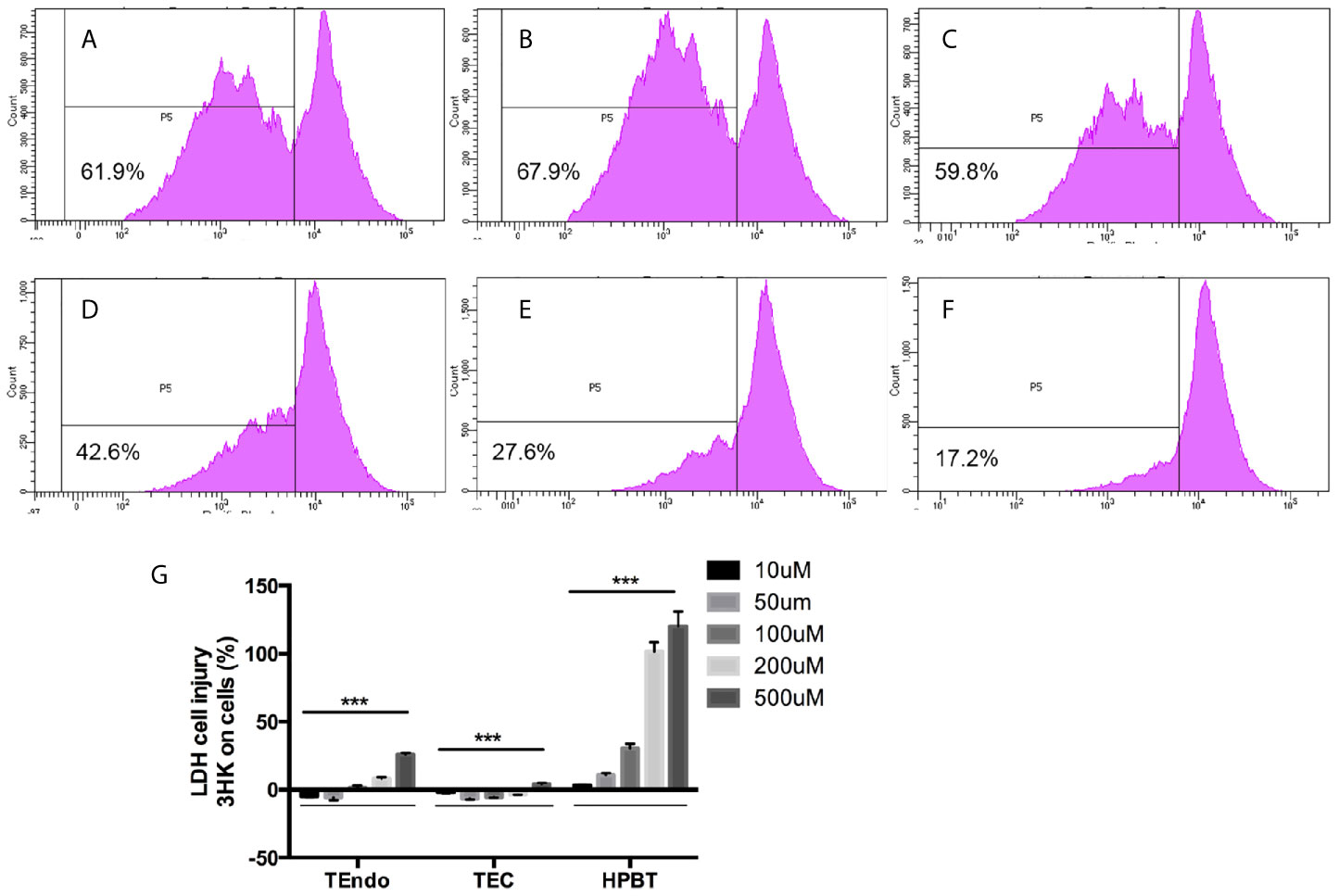
Figure 7 Effects of 3HK and 3HAA on hPBT proliferation and 3HK toxicity on TEndo, TEC, and hPBT. hPBT cell proliferation in (A) normal medium, (B) cytokine cocktail; (C) cytokine cocktail plus 3HK (50 µg/ml); (D) cytokine cocktail plus 3HK (100 µg/ml); (E) cytokine cocktail plus 3HAA (10 µg/ml) and (F) cytokine cocktail plus 3HAA (20 µg/ml); the data of flow cytometry is representative of four independent experiments. (G) Cytotoxicity of 3HK on Endo, TEC, and hPBT. The assay is representative of four independent experiments. ***P < 0.001 versus cells without treatment; analysis is the unpaired t-tests.
DNP-Induced KMO Expression or Increasing 3HK Specifically Reduced Allograft Rejection and Saved Graft Biological Function
Induced or transgene IDO has been reported to attenuate allograft rejection in rodent transplantation model (27, 30, 31, 33, 34, 65), while tolerance was found to be dependent on kynurenines (36–38). However, IDO was greatly induced in allograft rejection in pig kidney transplantation (40). Thus, we subjected the skin transplant from the ear of mice with balb/c background to the trunk of mice and IDO knockout mice with C57black/6 background. Free CpG-Lacz DNA nanoparticles (DNPs) were reported to induce IDO and mediate Treg-relevant tolerance (48, 66). We performed DNP injection to skin transplanted mice for 13 days post-transplant. DNP effectively attenuated skin allograft rejection in wild-type C57black/6 mice but did not reduce skin allograft rejection on IDO knockout C57Black/6 mice (Figure 8). Meanwhile 3HK injection effectively reduced allograft rejection on IDO knockout C57Black/6 mice (Figure 8) and wild-type C57black/6 mice (data not shown).
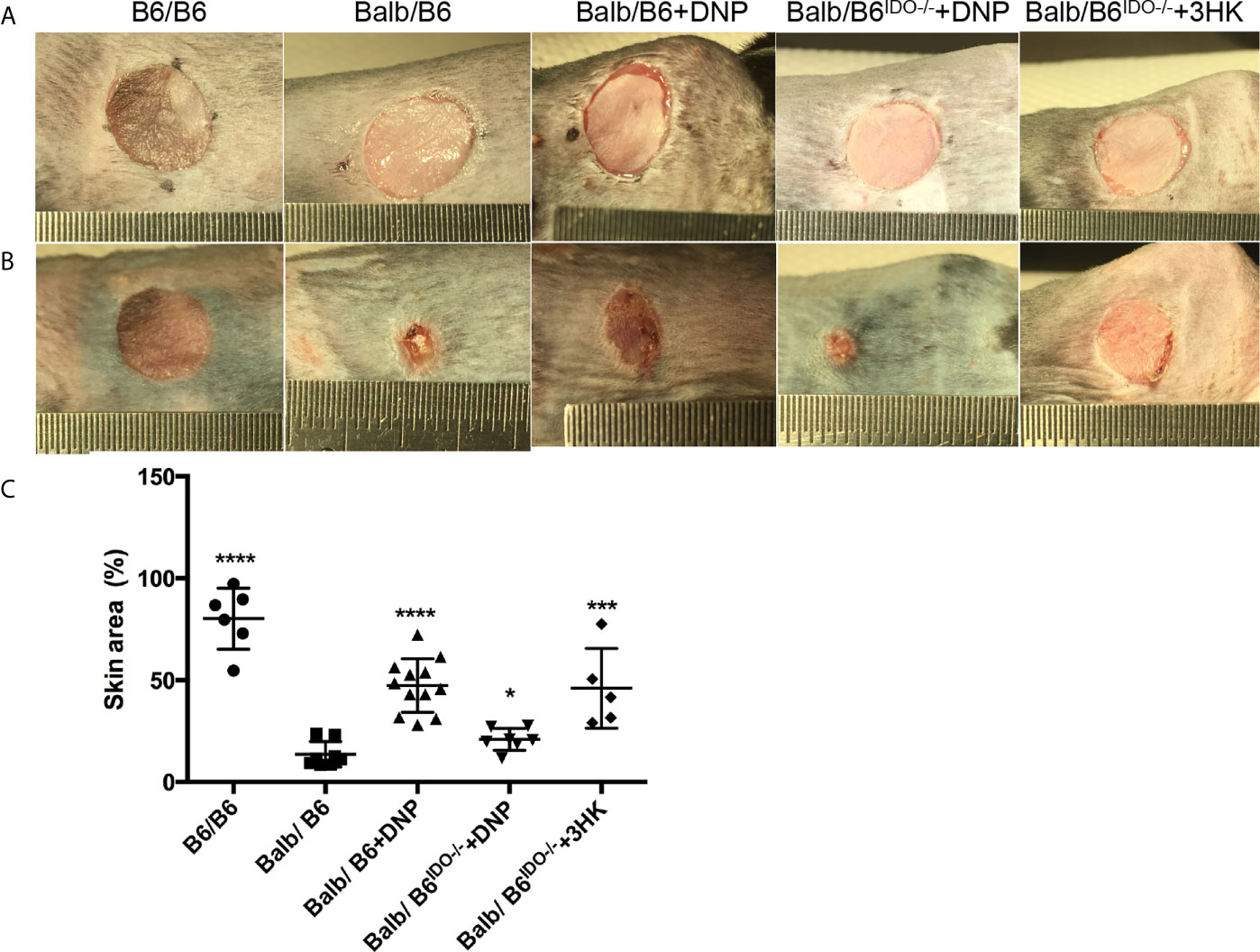
Figure 8 Tolerance effect of kynurenine and its metabolic derivatives in skin graft rejection. (A) Eight-week-old B6 or IDO knockout (B6IDO−/−, IDO-KO) recipients were transplanted with skin from donor Balb/c (Balbc) mice and treated with DNP or 3HK (i.p. injection) on day 0 (the day before operation). Each panel shows the appearance of donor ear skin tissues immediately following grafting onto the recipient on day 1 (operation day). (B) Skin grafts on day 14 from the same mice as (A). (C) Graft remaining area in percent on day 14 compared to day 1 from different treatments. *P < 0.05, ***P < 0.001, ****P < 0.0001 versus Balb/B6; analysis is multiple one-way ANOVA.
We further investigated whether DNP treatment attenuated allograft rejection in a pig kidney transplantation model. Ex vivo perfusion of allografts with DNP pretransplantation resulted in a 2.4-fold increase in IDO mRNA (Figure 9A), a 2.7-fold increase in IDO protein, and a 2.45-fold increase in IDO activity (data not shown). In association with increased IDO, KMO mRNA was also increased 2.5-fold (Figure 9B). The allograft treated with DNP showed less rejection (Figure 9C). IHC of KMO also indicated that allografts pretreated with DNP had less cell infiltration, and TEC retained high expression of KMO in graft tubules (Figure 9D).
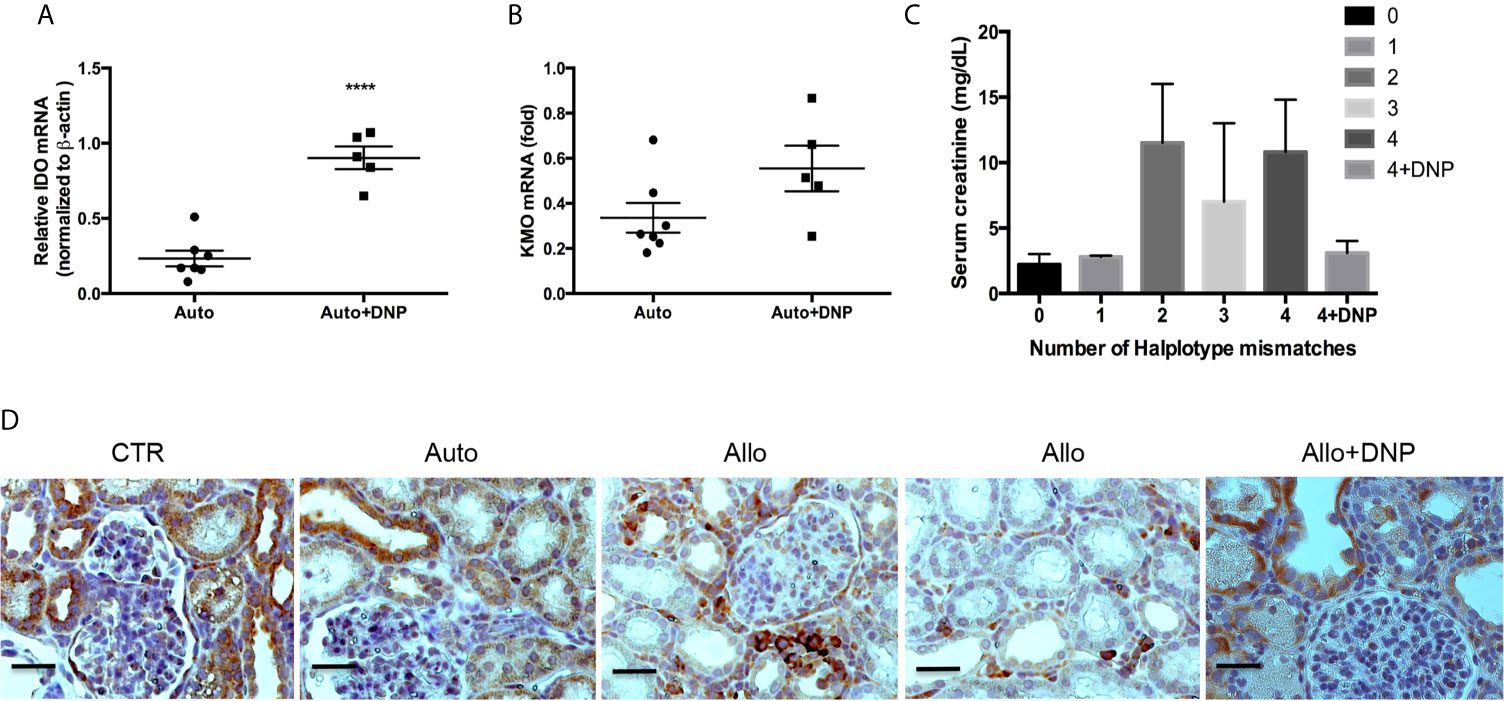
Figure 9 Reduction of allograft rejection by DNP-induced KMO upregulation. Grafts were ex vivo perfused with DNP, transplanted, and harvested at 72hours post-transplantation. (A) DNP-induced IDO expression (B) DNP-induced KMO expression, (C) Effect of DNP treatment on renal function (serum creatinine) from mismatched transplants. (D) Representative KMO IHC expression from normal graft right kidney as control (CTR), and autograft (Auto), mismatched allograft (Allo), and mismatched allograft treated with DNP (Allo+DNP). ****p < 0.0001, analysis is multiple unpaired t-tests. The scale bar is 500mm.
Discussion
Rejection of renal allograft and acquired complications of long-term immunosuppressant medication aimed at attenuating rejection by the recipient’s immune system are the major causes of subsequent allograft loss. Therefore, investigation of the molecular mechanism of rejection and treatment of rejection has been the foremost subject of interest since the establishment of allogeneic transplantation. Targeted improvements in immunosuppressive therapy could have a substantial impact on patients’ long-term survival. Although immunosuppressants have already improved in terms of specificity and lower toxicity secondary to a better understanding of molecular mechanism and pathology of rejection, the renal complex structure and complicated cell biological function in glomeruli and tubules, especially the cells in different tubular segments, have a different biological function, which restricts the dose and usage of antirejection drugs. In multiple organs or many types of cells in one organ, the drugs may favor some specific cells but injure other cells. Therefore, the overdose or long-term usage of antirejection medication results in a series of complications. Indeed, current clinical use of antirejection drugs in KTx including glucocorticoids, cyclophosphamide, chlorambucil, azathioprine, mycophenolate salts, cyclosporine, and tacrolimus can cause toxicity to allograft and other organs (67–73). Thus immunosuppressants with low toxicity and high efficiency are in high demand. The ideal drugs are metabolites preventing rejection of the transplanted organ while also providing normal physiological function, effectively optimizing the recipient’s immune response with diminished toxicity.
IDO has been reported to prevent allogeneic fetal rejection by depletion of tryptophan or increase the ratio of kynurenine to tryptophan (18). Studies have indicated that induced or transgene IDO can attenuate allograft rejection in rodent transplant models (18, 26–35). Given these findings, we planned to improve the outcome of allograft rejection in pig mismatch kidney transplantation via inducing IDO expression. We found that IDO activity increased from 10 to 100-fold without a clear correlation with allograft function. Some allografts with high IDO activity had normal physiological function, while some allografts with high IDO activity had an abnormal function other allografts with low IDO activity lost function. Interestingly, allografts with normal function had low IDO activity and relatively high KMO expression. KMO expression had an inverse association with creatinine concentration in the serum of the recipient holding allograft rejection. This led us to hypothesize that KMO may be the main mediator preventing rejection in IDO-initiating tryptophan to kynurenine metabolites. Indeed, a 2.5 to 5.2-fold kynurenine increase and a 6.6 to 58.3-fold 3HAA increase have been found in cord blood (74). As the intermediate between kynurenine and 3HAA in the kynurenine metabolic pathway, 3HK should play an important role in preventing fetus rejection.
TECs are the primary targets in IRI and acute allograft rejection (43, 75), and they exist in recipient urine as a biomarker of acute rejection in the early stage of KTx (76). However, the molecular mechanism of TEC injury and allograft rejection has not been fully described. We and other scientists found that many increased cytokines in the early allograft rejection stage were associated with TEC injury (40, 77–82). These secreted cytokines should play the important role in inducing TEC injury, which leads to graft failure from epithelial to mesenchymal transition (EMT) (83) or cell death including necrosis and apoptosis (76, 84, 85).
Since a number of cytokines have been found to induce TEC injury, and TEC injury is relevant to rejection (86–88), scientists investigate their relationship in order to provide effective prevention and treatment of allograft rejection. Altered gene expression in the ischemia–reperfusion process may induce reversible injury for allograft organ and tissues or may cause an irreversible chain of injuries which lead to rejection. In this study, three cytokines in low-dose induced TEC injury via activation of many signaling pathways; injured cells had an abnormal physiological function, altered cell phenotype, and finally resulting in cell death. These cytokines possibly induced dramatic accumulation of MHC I & II on TEC cell surface to trigger direct and indirect immune response by recipient’s immune response. Additionally, the three cytokines almost silenced KMO expression resulting in the reduction of 3HK, which also inevitably affected the tandem enzymatic reaction of kynureninase together with KMO to produce 3HAA. Although 3HAA can also be produced by the combination of kynureninase and anthranilic acid 3-hydroxylase to biologically avoid KMO involvement in producing 3HAA, a previous study revealed 3HK is a stronger inhibitor of T cell proliferation than 3HAA in physiological condition (21). In addition, 3HK has other important biological functions as the metabolic precursor of kynurenine aminotransferase to produce important biological product xanthurenic acid and downstream metabolites. Our current study also revealed that 3HK is a stronger protector of the epithelial cell barrier (Figure 6A). Thus, KMO showed its importance in kynurenine metabolism and kynurenines’ immune regulation.
The novel findings here indicated 3HK and 3HAA prevented TEC from injury via increasing Bcl-xL to prevent TEC apoptosis and increasing the expression of tight junction protein 1 to maintain the epithelial cell barrier intact. These findings suggest that reduction of KMO in the ischemia–reperfusion process is responsible for TEC and allograft injury in the early stage of KTx.
Half a century ago, experimental observation on murine skin transplant indicated that graft perfusion with allogeneic RNA attenuated allograft rejection to prolong allograft survival (89). It was further discovered that allograft perfused with any RNA and DNA had a better outcome (90). DNA-nanoparticles (DNPs) increase tolerance by IDO-induced Tregs (48). The free-CpG LacZ DNP administration through blood circulation can improve allograft survival in an IDO dependent manner (48, 66). 3HK and 3HAA effectively reduced rejection through inhibition of T cell proliferation, which suggested that kynurenine derivatives are the real players in preventing rejection. IDO initiates the production of kynurenine, which is further metabolized by downstream enzymes KMO and kynureninase to produce 3HK and 3HAA respectively; both kynurenine derivatives strongly prevent allograft rejection.
Ex vivo perfusion of graft pre-transplant with DNP induced IDO and KMO in grafts in a concerted regulation action similar to the molecular regulation of IDO and KMO in the brain (91). KMO was dramatically down-regulated in allograft rejection and TEC in cytokine pool (40); the administration of DNP with ex vivo perfusion increased KMO expression associated with reduction of allograft rejection. A previous study revealed increasing KMO may be unfavorable in the brain and pancreas (62–64, 91), but the real risk of pancreatitis from KMO requires further investigation. KMO likely has dual functions that may or may not favor some specific cells (92). Although the reduction of KMO has recently been reported to induce abnormal kidney function (93), the beneficial functions of KMO on other tissues or organs may have been overlooked. The administration of 3HK to the recipient of skin transplant can effectively improve the outcome of allograft on the IDO knockout model; this clearly defines the tolerogenic role of KMO.
When pig grafts were ex vivo perfused with DNP, we found that pig grafts are very sensitive to lipid toxicity; the ratio of lipid to DNA is very crucial for attenuation of allograft rejection and improvement of allograft function. Some liposomes and nanomedicine can induce complement activation-related pseudoallergy on pig models (94). The ideal nano-therapeutic should be carefully tested to remove lipid toxicity on drug treatment.
The combination of IFNγ–TNFα–IL1β causing TEC injury with increased MHC I and II provides the potential molecular mechanism of rejection on TEC and allograft during transplantation. The expression and regulation as well as location of MHC I and II induced by cytokines should be further investigated in the future, which may decipher the molecular mechanism of allograft rejection from ischemia–reperfusion and early stage of rejection procedure in organ transplantation. In current study, KMO served as a crucial mediator in linking ischemia procedure to rejection as evidenced by its dramatic down-regulation in ischemia–reperfusion resulting in low 3HK and 3HAA production and ultimately leading to loss of TEC self-protection. This allowed disruption of endogenous metabolites in TEC in preventing T cell proliferation with the final consequence of rejection.
The formation, prevention, and outcome of allograft rejection in KTx is a very complex subject, which relies on many factors such as ischemia–reperfusion procedure and operation procedure, instinctive genetic match, and side effect of long-term medication. The early disruption of gene expression will trigger a reaction chain to result in graft rejection and loss. The current study demonstrated that down-regulation of KMO caused by ischemia and early stage of rejection is a key mediator to cause allograft rejection due to reduction of 3HK and 3HAA production. 3HK and 3HAA strongly prevent allogeneic T cell proliferation and effectively protect graft barrier function via increasing Bcl-xl and TJP1 expression. In addition, an effective elimination of cytokine storm caused in IRI procedure or blocking cytokine-induced reduction of KMO, and moderate supplementation of 3HK and 3HAA after transplantation can provide graft better outcome.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Augusta University Institutional Animal Care and Use Committee.
Author Contributions
YW designed the research. YW, RL, and XF performed the research. XF performed murine skin transplantation. TM performed all pig kidney transplantation. YW analyzed the data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by grants from the Carlos and Marquerite Mason Charitable Trust and Reserve Research grant fund #S-2444 from Dialysis Clinic. Inc.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Nagashima H, Matsunari H. Growing Human Organs in Pigs-A Dream or Reality? Theriogenology (2016) 86:422–6. doi: 10.1016/j.theriogenology.2016.04.056
2. Crane AT, Aravalli RN, Asakura A, Grande AW, Krishna VD, Carlson DF, et al. Interspecies Organogenesis for Human Transplantation. Cell Transplant (2019) 28:1091–105. doi: 10.1177/0963689719845351
3. Kobayashi E. Challenges for Production of Human Transplantable Organ Grafts. Cell Med (2017) 9:9–14. doi: 10.3727/215517916X693113
4. Blusch JH, Roos C, Nitschko H. A Polymerase Chain Reaction-Based Protocol for the Detection of Transmission of Pig Endogenous Retroviruses in Pig to Human Xenotransplantation. Transplantation (2000) 69:2167–72. doi: 10.1097/00007890-200005270-00036
5. Heo Y, Cho Y, Oh KB, Park KH, Cho H, Choi H, et al. Detection of Pig Cells Harboring Porcine Endogenous Retroviruses in Non-Human Primate Bladder After Renal Xenotransplantation. Viruses (2019) 11. doi: 10.3390/v11090801
6. Brewer L, Brown C, Murtaugh MP, Njenga MK. Transmission of Porcine Encephalomyocarditis Virus (EMCV) to Mice by Transplanting EMCV-Infected Pig Tissues. Xenotransplantation (2003) 10:569–76. doi: 10.1034/j.1399-3089.2003.00058.x
7. Kimsa MC, Strzalka-Mrozik B, Kimsa MW, Gola J, Nicholson P, Lopata K, et al. Porcine Endogenous Retroviruses in Xenotransplantation–Molecular Aspects. Viruses (2014) 6:2062–83. doi: 10.3390/v6052062
8. Denner J. Recent Progress in Xenotransplantation, With Emphasis on Virological Safety. Ann Transplant (2016) 21:717–27. doi: 10.12659/AOT.900531
9. Fiebig U, Abicht JM, Mayr T, Langin M, Bahr A, Guethoff S, et al. Distribution of Porcine Cytomegalovirus in Infected Donor Pigs and in Baboon Recipients of Pig Heart Transplantation. Viruses (2018) 10. doi: 10.3390/v10020066
10. Martin U, Tacke SJ, Simon AR, Schroder C, Wiebe K, Lapin B, et al. Absence of PERV Specific Humoral Immune Response in Baboons After Transplantation of Porcine Cells or Organs. Transplant Int Off J Eur Soc Organ Transplant (2002) 15:361–8. doi: 10.1111/j.1432-2277.2002.tb00179.x
11. Denner J, Specke V, Karlas A, Chodnevskaja I, Meyer T, Moskalenko V, et al. No Transmission of Porcine Endogenous Retroviruses (Pervs) in a Long-Term Pig to Rat Xenotransplantation Model and No Infection of Immunosuppressed Rats. Ann Transplant (2008) 13:20–31.
12. Moscoso I, Hermida-Prieto M, Manez R, Lopez-Pelaez E, Centeno A, Diaz TM, et al. Lack of Cross-Species Transmission of Porcine Endogenous Retrovirus in Pig-to-Baboon Xenotransplantation With Sustained Depletion of Anti-Alphagal Antibodies. Transplantation (2005) 79:777–82. doi: 10.1097/01.TP.0000152662.55720.83
13. Fishman JA, Sachs DH, Yamada K, Wilkinson RA. Absence of Interaction Between Porcine Endogenous Retrovirus and Porcine Cytomegalovirus in Pig-to-Baboon Renal Xenotransplantation In Vivo. Xenotransplantation (2018) 25:e12395. doi: 10.1111/xen.12395
14. Niu D, Wei HJ, Lin L, George H, Wang T, Lee IH, et al. Inactivation of Porcine Endogenous Retrovirus in Pigs Using CRISPR-Cas9. Science (2017) 357:1303–7. doi: 10.1126/science.aan4187
15. Yang L, Guell M, Niu D, George H, Lesha E, Grishin D, et al. Genome-Wide Inactivation of Porcine Endogenous Retroviruses (Pervs). Science (2015) 350:1101–4. doi: 10.1126/science.aad1191
16. Cooper DKC, Iwase H, Wang L, Yamamoto T, Li Q, Li J, et al. Bringing Home the Bacon: Update on The State of Kidney Xenotransplantation. Blood Purif (2018) 45:254–9. doi: 10.1159/000485163
17. Cooper DKC, Pierson RN 3rd, Hering BJ, Mohiuddin MM, Fishman JA, Denner J, et al. Regulation of Clinical Xenotransplantation-Time for a Reappraisal. Transplantation (2017) 101:1766–9. doi: 10.1097/TP.0000000000001683
18. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of Allogeneic Fetal Rejection by Tryptophan Catabolism. Science (1998) 281:1191–3. doi: 10.1126/science.281.5380.1191
19. Jia L, Tian P, Ding C. Immunoregulatory Effects of Indoleamine 2, 3-Dioxygenase in Transplantation. Transplant Immunol (2009) 21:18–22. doi: 10.1016/j.trim.2009.01.004
20. Mbongue JC, Nicholas DA, Torrez TW, Kim NS, Firek AF, Langridge WH. The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity. Vaccines (2015) 3:703–29. doi: 10.3390/vaccines3030703
21. Dai X, Zhu BT. Suppression of T-Cell Response and Prolongation of Allograft Survival in a Rat Model by Tryptophan Catabolites. Eur J Pharmacol (2009) 606:225–32. doi: 10.1016/j.ejphar.2008.12.053
22. Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-Derived Catabolites Are Responsible for Inhibition of T and Natural Killer Cell Proliferation Induced by Indoleamine 2,3-Dioxygenase. J Exp Med (2002) 196:459–68. doi: 10.1084/jem.20020121
23. Fallarino F, Grohmann U, Vacca C, Orabona C, Spreca A, Fioretti MC, et al. T Cell Apoptosis by Kynurenines. Adv Exp Med Biol (2003) 527:183–90. doi: 10.1007/978-1-4615-0135-0_21
24. Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T Cell Apoptosis by Tryptophan Catabolism. Cell Death Differ (2002) 9:1069–77. doi: 10.1038/sj.cdd.4401073
25. Bauer TM, Jiga LP, Chuang JJ, Randazzo M, Opelz G, Terness P. Studying the Immunosuppressive Role of Indoleamine 2,3-Dioxygenase: Tryptophan Metabolites Suppress Rat Allogeneic T-Cell Responses In Vitro and In Vivo. Transplant Int Off J Eur Soc Organ Transplant (2005) 18:95–100. doi: 10.1111/j.1432-2277.2004.00031.x
26. Sucher R, Fischler K, Oberhuber R, Kronberger I, Margreiter C, Ollinger R, et al. IDO and Regulatory T Cell Support Are Critical for Cytotoxic T Lymphocyte-Associated Ag-4 Ig-Mediated Long-Term Solid Organ Allograft Survival. J Immunol (2012) 188:37–46. doi: 10.4049/jimmunol.1002777
27. Vavrincova-Yaghi D, Deelman LE, van Goor H, Seelen MA, Vavrinec P, Kema IP, et al. Local Gene Therapy With Indoleamine 2,3-Dioxygenase Protects Against Development of Transplant Vasculopathy in Chronic Kidney Transplant Dysfunction. Gene Ther (2016) 23:797–806. doi: 10.1038/gt.2016.59
28. Na N, Luo Y, Zhao D, Yang S, Hong L, Li H, et al. Prolongation of Kidney Allograft Survival Regulated by Indoleamine 2, 3-Dioxygenase in Immature Dendritic Cells Generated From Recipient Type Bone Marrow Progenitors. Mol Immunol (2016) 79:22–31. doi: 10.1016/j.molimm.2016.09.005
29. Li C, Liu T, Zhao N, Zhu L, Wang P, Dai X. Dendritic Cells Transfected With Indoleamine 2,3-Dioxygenase Gene Suppressed Acute Rejection of Cardiac Allograft. Int Immunopharmacol (2016) 36:31–8. doi: 10.1016/j.intimp.2016.03.048
30. Ebrahimi A, Kardar GA, Teimoori-Toolabi L, Ghanbari H, Sadroddiny E. Inducible Expression of Indoleamine 2,3-Dioxygenase Attenuates Acute Rejection of Tissue-Engineered Lung Allografts in Rats. Gene (2016) 576:412–20. doi: 10.1016/j.gene.2015.10.054
31. He Y, Zhou S, Liu H, Shen B, Zhao H, Peng K, et al. Indoleamine 2, 3-Dioxgenase Transfected Mesenchymal Stem Cells Induce Kidney Allograft Tolerance by Increasing the Production and Function of Regulatory T Cells. Transplantation (2015) 99:1829–38. doi: 10.1097/TP.0000000000000856
32. Zoso A, Mazza EM, Bicciato S, Mandruzzato S, Bronte V, Serafini P, et al. Human Fibrocytic Myeloid-Derived Suppressor Cells Express IDO and Promote Tolerance Via Treg-cell Expansion. Eur J Immunol (2014) 44:3307–19. doi: 10.1002/eji.201444522
33. Vavrincova-Yaghi D, Deelman LE, Goor H, Seelen M, Kema IP, Smit-van Oosten A, et al. Gene Therapy With Adenovirus-Delivered Indoleamine 2,3-Dioxygenase Improves Renal Function and Morphology Following Allogeneic Kidney Transplantation in Rat. J Gene Med (2011) 13:373–81. doi: 10.1002/jgm.1584
34. Yu G, Dai H, Chen J, Duan L, Gong M, Liu L, et al. Gene Delivery of Indoleamine 2,3-Dioxygenase Prolongs Cardiac Allograft Survival by Shaping the Types of T-Cell Responses. J Gene Med (2008) 10:754–61. doi: 10.1002/jgm.1201
35. Mulley WR, Li YQ, Wee JL, Dodge N, Christiansen D, Simeonovic C, et al. Local Expression of IDO, Either Alone or in Combination With CD40Ig, IL10 or CTLA4Ig, Inhibits Indirect Xenorejection Responses. Xenotransplantation (2008) 15:174–83. doi: 10.1111/j.1399-3089.2008.00472.x
36. Belladonna ML, Grohmann U, Guidetti P, Volpi C, Bianchi R, Fioretti MC, et al. Kynurenine Pathway Enzymes in Dendritic Cells Initiate Tolerogenesis in the Absence of Functional IDO. J Immunol (2006) 177:130–7. doi: 10.4049/jimmunol.177.1.130
37. Laurence JM, Wang C, Park ET, Buchanan A, Clouston A, Allen RD, et al. Blocking Indoleamine Dioxygenase Activity Early After Rat Liver Transplantation Prevents Long-Term Survival But Does Not Cause Acute Rejection. Transplantation (2008) 85:1357–61. doi: 10.1097/TP.0b013e31816fc27f
38. Lahdou I, Sadeghi M, Daniel V, Schenk M, Renner F, Weimer R, et al. Increased Pretransplantation Plasma Kynurenine Levels Do Not Protect From But Predict Acute Kidney Allograft Rejection. Hum Immunol (2010) 71:1067–72. doi: 10.1016/j.humimm.2010.08.013
39. Vavrincova-Yaghi D, Seelen MA, Kema IP, Deelman LE, van der Heuvel MC, Breukelman H, et al. Early Posttransplant Tryptophan Metabolism Predicts Long-term Outcome of Human Kidney Transplantation. Transplantation (2015) 99:e97–104. doi: 10.1097/TP.0000000000000603
40. Wang Y, Merchen TD, Fang X, Lassiter R, Ho CS, Jajosky R, et al. Regulation of Indoleamine 2,3 Dioxygenase and Its Role in a Porcine Model of Acute Kidney Allograft Rejection. J Invest Med Off Publ Am Fed Clin Res (2018) 66:1109–17. doi: 10.1136/jim-2018-000742
41. Busch GJ, Galvanek EG, Reynolds ES Jr. Human Renal Allografts. Anal Lesions Long-Term Survivors Hum Pathol (1971) 2:253–98. doi: 10.1016/S0046-8177(71)80037-0
42. Valenzuela R, Hamway SA, Deodhar SD, Braun WE, Banowsky LH, Magnusson MO, et al. Histologic, Ultrastructural, and Immunomicroscopic Findings in 96 One Hour Human Renal Allograft Biopsy Specimens. Immunol Clin Significance Hum Pathol (1980) 11:187–95. doi: 10.1016/S0046-8177(80)80143-2
43. Smith SF, Hosgood SA, Nicholson ML. Ischemia-Reperfusion Injury in Renal Transplantation: 3 Key Signaling Pathways in Tubular Epithelial Cells. Kidney Int (2019) 95:50–6. doi: 10.1016/j.kint.2018.10.009
44. Mikhalski D, Wissing KM, Ghisdal L, Broeders N, Touly M, Hoang AD, et al. Cold Ischemia Is a Major Determinant of Acute Rejection and Renal Graft Survival in the Modern Era of Immunosuppression. Transplantation (2008) 85:S3–9. doi: 10.1097/TP.0b013e318169c29e
45. Debout A, Foucher Y, Trebern-Launay K, Legendre C, Kreis H, Mourad G, et al. Each Additional Hour of Cold Ischemia Time Significantly Increases the Risk of Graft Failure and Mortality Following Renal Transplantation. Kidney Int (2015) 87:343–9. doi: 10.1038/ki.2014.304
46. Cheng C-H, Lee C-F, Fryer M, Furtmuller GJ, Oh B, Powell JD, et al. Murine Full-Thickness Skin Transplantation. J Vis Exp (2017) 119:e55105. doi: 10.3791/55105
47. Wang Y, Zhou J, Minto AW, Hack BK, Alexander JJ, Haas M, et al. Altered Vitamin D Metabolism in Type II Diabetic Mouse Glomeruli May Provide Protection From Diabetic Nephropathy. Kidney Int (2006) 70:882–91. doi: 10.1038/sj.ki.5001624
48. Huang L, Li L, Lemos H, Chandler PR, Pacholczyk G, Baban B, et al. Cutting Edge: DNA Sensing Via the STING Adaptor in Myeloid Dendritic Cells Induces Potent Tolerogenic Responses. J Immunol (2013) 191:3509–13. doi: 10.4049/jimmunol.1301419
49. Robertson H, Kirby JA. Post-Transplant Renal Tubulitis: The Recruitment, Differentiation and Persistence of Intra-Epithelial T Cells. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surgeons (2003) 3:3–10. doi: 10.1034/j.1600-6143.2003.30102.x
50. Lassiter R, Wang Y, Fang X, Winn M, Ghaffari A, Ho CS, et al. A Model of Acute Renal Allograft Rejection in Outbred Yorkshire Piglets. Transplant Immunol (2017) 42:40–6. doi: 10.1016/j.trim.2017.05.001
51. Wang Y, Cao R, Liu D, Chervin A, Yuan J, An J, et al. Oligomerization of BH4-truncated Bcl-X(L) in Solution. Biochem Biophys Res Commun (2007) 361:1006–11. doi: 10.1016/j.bbrc.2007.07.122
52. Kwan T, Chadban SJ, Ma J, Bao S, Alexander SI. Il-17 Deficiency Attenuates Allograft Injury and Prolongs Survival in a Murine Model of Fully MHC-mismatched Renal Allograft Transplantation. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surgeons (2015) 15:1555–67. doi: 10.1111/ajt.13140
53. Chung BH, Kim KW, Kim BM, Doh KC, Cho ML, Yang CW. Increase of Th17 Cell Phenotype in Kidney Transplant Recipients With Chronic Allograft Dysfunction. PloS One (2015) 10:e0145258. doi: 10.1371/journal.pone.0145258
54. Heidt S, Segundo DS, Chadha R, Wood KJ. The Impact of Th17 Cells on Transplant Rejection and the Induction of Tolerance. Curr Opin Organ Transplant (2010) 15:456–61. doi: 10.1097/MOT.0b013e32833b9bfb
55. Chung BH, Oh HJ, Piao SG, Hwang HS, Sun IO, Choi SR, et al. Clinical Significance of the Ratio Between FOXP3 Positive Regulatory T Cell and Interleukin-17 Secreting Cell in Renal Allograft Biopsies With Acute T-Cell-mediated Rejection. Immunology (2012) 136:344–51. doi: 10.1111/j.1365-2567.2012.03588.x
56. Millan O, Rafael-Valdivia L, San Segundo D, Boix F, Castro-Panete MJ, Lopez-Hoyos M, et al. Should IFN-Gamma, IL-17 and IL-2 Be Considered Predictive Biomarkers of Acute Rejection in Liver and Kidney Transplant? Results Multicentric Study Clin Immunol (2014) 154:141–54. doi: 10.1016/j.clim.2014.07.007
57. Uehara M, Solhjou Z, Banouni N, Kasinath V, Xiaqun Y, Dai L, et al. Ischemia Augments Alloimmune Injury Through IL-6-Driven CD4(+) Alloreactivity. Sci Rep (2018) 8:2461. doi: 10.1038/s41598-018-20858-4
58. Jordan SC, Choi J, Kim I, Wu G, Toyoda M, Shin B. Interleukin-6, A Cytokine Critical to Mediation of Inflammation, Autoimmunity and Allograft Rejection: Therapeutic Implications of IL-6 Receptor Blockade. Transplantation (2017) 101:32–44. doi: 10.1097/TP.0000000000001452
59. Liu C, Chen J, Liu B, Yuan S, Shou D, Wen L, et al. Role of IL-18 in Transplant Biology. Eur Cytokine Netw (2018) 29:48–51. doi: 10.1684/ecn.2018.0410
60. Wyburn K, Wu H, Chen G, Yin J, Eris J, Chadban S. Interleukin-18 Affects Local Cytokine Expression But Does Not Impact on the Development of Kidney Allograft Rejection. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surgeons (2006) 6:2612–21. doi: 10.1111/j.1600-6143.2006.01536.x
61. Zaher SS, Germain C, Fu H, Larkin DF, George AJ. 3-Hydroxykynurenine Suppresses CD4+ T-Cell Proliferation, Induces T-Regulatory-Cell Development, and Prolongs Corneal Allograft Survival. Invest Ophthalmol Visual Sci (2011) 52:2640–8. doi: 10.1167/iovs.10-5793
62. Parrott JM, O’Connor JC. Kynurenine 3-Monooxygenase: An Influential Mediator of Neuropathology. Front Psychiatry (2015) 6:116. doi: 10.3389/fpsyt.2015.00116
63. Skouras C, Zheng X, Binnie M, Homer NZ, Murray TB, Robertson D, et al. Increased Levels of 3-Hydroxykynurenine Parallel Disease Severity in Human Acute Pancreatitis. Sci Rep (2016) 6:33951. doi: 10.1038/srep33951
64. Mole DJ, Webster SP, Uings I, Zheng X, Binnie M, Wilson K, et al. Kynurenine-3-monooxygenase Inhibition Prevents Multiple Organ Failure in Rodent Models of Acute Pancreatitis. Nat Med (2016) 22:202–9. doi: 10.1038/nm.4020
65. Beutelspacher SC, Tan PH, McClure MO, Larkin DF, Lechler RI, George AJ. Expression of Indoleamine 2,3-Dioxygenase (IDO) by Endothelial Cells: Implications for the Control of Alloresponses. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surgeons (2006) 6:1320–30. doi: 10.1111/j.1600-6143.2006.01324.x
66. Lemos H, Huang L, Chandler PR, Mohamed E, Souza GR, Li L, et al. Activation of the STING Adaptor Attenuates Experimental Autoimmune Encephalitis. J Immunol (2014) 192:5571–8. doi: 10.4049/jimmunol.1303258
67. Freitas MC, Uchida Y, Lassman C, Danovitch GM, Busuttil RW, Kupiec-Weglinski JW. Type I Interferon Pathway Mediates Renal Ischemia/Reperfusion Injury. Transplantation (2011) 92:131–8. doi: 10.1097/TP.0b013e318220586e
68. Zuidwijk K, de Fijter JW, Mallat MJ, Eikmans M, van Groningen MC, Goemaere NN, et al. Increased Influx of Myeloid Dendritic Cells During Acute Rejection is Associated With Interstitial Fibrosis and Tubular Atrophy and Predicts Poor Outcome. Kidney Int (2012) 81:64–75. doi: 10.1038/ki.2011.289
69. Coombes JD, Mreich E, Liddle C, Rangan GK. Rapamycin Worsens Renal Function and Intratubular Cast Formation in Protein Overload Nephropathy. Kidney Int (2005) 68:2599–607. doi: 10.1111/j.1523-1755.2005.00732.x
70. Ponticelli C, Glassock RJ. Prevention of Complications From Use of Conventional Immunosuppressants: A Critical Review. J Nephrol (2019) 32:851–70. doi: 10.1007/s40620-019-00602-5
71. Wiseman AC. Immunosuppressive Medications. Clin J Am Soc Nephrol CJASN (2016) 11:332–43. doi: 10.2215/CJN.08570814
72. Lizotti Ciliao H, Batista de Oliveira Camargo-Godoy R, Mazzaron Barcelos GR, Zanuto A, Daher Alvares Delfino V, de Syllos Colus IM. Long-Term Genotoxic Effects of Immunosuppressive Drugs on Lymphocytes of Kidney Transplant Recipients. Mutat Res Genet Toxicol Environ Mutagen (2016) 806:47–52. doi: 10.1016/j.mrgentox.2016.07.001
73. Oyouni AAA, Saggu S, Tousson E, Rehman H. Immunosuppressant Drug Tacrolimus Induced Mitochondrial Nephrotoxicity, Modified PCNA and Bcl-2 Expression Attenuated by Ocimum Basilicum L. @ in CD1 Mice. Toxicol Rep (2018) 5:687–94. doi: 10.1016/j.toxrep.2018.06.003
74. Badawy AA. Tryptophan Metabolism, Disposition and Utilization in Pregnancy. Biosci Rep (2015) 35. doi: 10.1042/BSR20150197
75. Ting YT, Coates PT, Walker RJ, McLellan AD. Urinary Tubular Biomarkers as Potential Early Predictors of Renal Allograft Rejection. Nephrology (2012) 17:11–6. doi: 10.1111/j.1440-1797.2011.01536.x
76. Oberbauer R, Rohrmoser M, Regele H, Muhlbacher F, Mayer G. Apoptosis of Tubular Epithelial Cells in Donor Kidney Biopsies Predicts Early Renal Allograft Function. J Am Soc Nephrol JASN (1999) 10:2006–13. doi: 10.1681/ASN.V1092006
77. Lin Y, Kirby JA, Browell DA, Morley AR, Shenton BK, Proud G, et al. Renal Allograft Rejection: Expression and Function of VCAM-1 on Tubular Epithelial Cells. Clin Exp Immunol (1993) 92:145–51. doi: 10.1111/j.1365-2249.1993.tb05961.x
78. De Serres SA, Mfarrej BG, Grafals M, Riella LV, Magee CN, Yeung MY, et al. Derivation and Validation of a Cytokine-Based Assay to Screen for Acute Rejection in Renal Transplant Recipients. Clin J Am Soc Nephrol CJASN (2012) 7:1018–25. doi: 10.2215/CJN.11051011
79. Zhao Y, Cooper DKC, Wang H, Chen P, He C, Cai Z, et al. Potential Pathological Role of Pro-Inflammatory Cytokines (IL-6, TNF-alpha, and IL-17) in Xenotransplantation. Xenotransplantation (2019) 26:e12502. doi: 10.1111/xen.12502
80. Anders HJ. Of Inflammasomes and Alarmins: Il-1beta and IL-1alpha in Kidney Disease. J Am Soc Nephrol JASN (2016) 27:2564–75. doi: 10.1681/ASN.2016020177
81. Woltman AM, de Haij S, Boonstra JG, Gobin SJ, Daha MR, van Kooten C. Interleukin-17 and CD40-Ligand Synergistically Enhance Cytokine and Chemokine Production by Renal Epithelial Cells. J Am Soc Nephrol JASN (2000) 11:2044–55. doi: 10.1681/ASN.V11112044
82. Loverre A, Tataranni T, Castellano G, Divella C, Battaglia M, Ditonno P, et al. IL-17 Expression by Tubular Epithelial Cells in Renal Transplant Recipients With Acute Antibody-Mediated Rejection. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surgeons (2011) 11:1248–59. doi: 10.1111/j.1600-6143.2011.03529.x
83. Hertig A, Verine J, Mougenot B, Jouanneau C, Ouali N, Sebe P, et al. Risk Factors for Early Epithelial to Mesenchymal Transition in Renal Grafts. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surgeons (2006) 6:2937–46. doi: 10.1111/j.1600-6143.2006.01559.x
84. Du C, Jiang J, Guan Q, Yin Z, Masterson M, Parbtani A, et al. Renal Tubular Epithelial Cell Self-Injury Through Fas/Fas Ligand Interaction Promotes Renal Allograft Injury. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surgeons (2004) 4:1583–94. doi: 10.1111/j.1600-6143.2004.00552.x
85. Salahudeen AK, Joshi M, Jenkins JK. Apoptosis Versus Necrosis During Cold Storage and Rewarming of Human Renal Proximal Tubular Cells. Transplantation (2001) 72:798–804. doi: 10.1097/00007890-200109150-00010
86. Nguan CY, Du C. Renal Tubular Epithelial Cells as Immunoregulatory Cells in Renal Allograft Rejection. Transplant Rev (2009) 23:129–38. doi: 10.1016/j.trre.2009.02.003
87. Samsonov D, Geehan C, Woda CB, Briscoe DM. Differential Activation of Human T Cells to Allogeneic Endothelial Cells, Epithelial Cells and Fibroblasts In Vitro. Transplant Res (2012) 1:4. doi: 10.1186/2047-1440-1-4
88. Demmers MW, Korevaar SS, Roemeling-van Rhijn M, van den Bosch TP, Hoogduijn MJ, Betjes MG, et al. Human Renal Tubular Epithelial Cells Suppress Alloreactive T Cell Proliferation. Clin Exp Immunol (2015) 179:509–19. doi: 10.1111/cei.12469
89. Guttmann RD, Kraus ED, Dolan MF. Rejection of Isogeneic Murine Skin Grafts Following Exposure to Allogeneic Ribonucleic Acid. Nature (1964) 203:196–8. doi: 10.1038/203196a0
90. Billingham RE. The Feasibility of Altering the Immunogenicity of Grafts. J Invest Dermatol (1976) 67:149–59. doi: 10.1111/1523-1747.ep12513006
91. Campbell BM, Charych E, Lee AW, Moller T. Kynurenines in CNS Disease: Regulation by Inflammatory Cytokines. Front Neurosci (2014) 8:12. doi: 10.3389/fnins.2014.00012
92. Colin-Gonzalez AL, Maldonado PD, Santamaria A. 3-Hydroxykynurenine: An Intriguing Molecule Exerting Dual Actions in the Central Nervous System. Neurotoxicology (2013) 34:189–204. doi: 10.1016/j.neuro.2012.11.007
93. Korstanje R, Deutsch K, Bolanos-Palmieri P, Hanke N, Schroder P, Staggs L, et al. Loss of Kynurenine 3-Mono-Oxygenase Causes Proteinuria. J Am Soc Nephrol JASN (2016) 27:3271–7. doi: 10.1681/ASN.2015070835
Keywords: kidney transplantation, kynurenine 3-monooxygenase, allograft rejection, kynurenine metabolism, immunosuppresant
Citation: Lassiter R, Merchen TD, Fang X and Wang Y (2021) Protective Role of Kynurenine 3-Monooxygenase in Allograft Rejection and Tubular Injury in Kidney Transplantation. Front. Immunol. 12:671025. doi: 10.3389/fimmu.2021.671025
Received: 22 February 2021; Accepted: 11 June 2021;
Published: 07 July 2021.
Edited by:
Lei Huang, Newcastle University, United KingdomReviewed by:
Henrique De Paula Lemos, Newcastle University, United KingdomChristian Lücht, Charité – Universitätsmedizin Berlin, Germany
Yanjun Liu, Southern Medical University, China
Copyright © 2021 Lassiter, Merchen, Fang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youli Wang, eXdhbmcyQGF1Z3VzdGEuZWR1
†Present address: Randi Lassiter, Nicklaus Children’s Health System, Miami, FL, United States
Todd D. Merchen, Transplant Institute at Research Medical Center, Kansas City, MO, United States