 Jasmina Bier
Jasmina Bier Sebastian M. Steiger
Sebastian M. Steiger Holger M. Reichardt
Holger M. Reichardt Fred Lühder
Fred Lühder- 1Institute for Neuroimmunology and Multiple Sclerosis Research, University Medical Center Goettingen, Goettingen, Germany
- 2Institute for Cellular and Molecular Immunology, University Medical Center Goettingen, Goettingen, Germany
Induction of T cell apoptosis constitutes a major mechanism by which therapeutically administered glucocorticoids (GCs) suppress inflammation and associated clinical symptoms, for instance in multiple sclerosis (MS) patients suffering from an acute relapse. The sensitivity of T cells to GC action depends on their maturation and activation status, but the precise effect of antigen-priming in a pathological setting has not been explored. Here we used transgenic and congenic mouse models to compare GC-induced apoptosis between naïve and antigen-specific effector T cells from mice immunized with a myelin peptide. Antigen-primed effector T cells were protected from the pro-apoptotic activity of the synthetic GC dexamethasone in a dose-dependent manner, which resulted in their accumulation relative to naïve T cells in vitro and in vivo. Notably, the differential sensitivity of T cells to GC-induced apoptosis correlated with their expression level of the anti-apoptotic proteins Bcl-2 and Bcl-XL and a loss of the mitochondrial membrane potential. Moreover, accumulation of antigen-primed effector T cells following GC treatment in vitro resulted in an aggravated disease course in an adoptive transfer mouse model of MS in vivo, highlighting the clinical relevance of the observed phenomenon. Collectively, our data indicate that antigen-priming influences the T cells’ sensitivity to therapeutically applied GCs in the context of inflammatory diseases.
Introduction
Glucocorticoids (GCs) have been used as immunosuppressive and anti-inflammatory agents for more than 70 years (1) and have become indispensable for the management of a variety of diseases. In some fields such as dermatology and ophthalmology, GCs are mostly applied in topical form, whereas systemic administration is customary in the treatment of lymphoma and various autoimmune diseases. This also concerns multiple sclerosis (MS) patients suffering from an acute relapse, who commonly receive high-dose intravenous methylprednisolone pulse therapy to ameliorate clinical symptoms (2).
Most of our current knowledge on the mechanisms of GCs in neuroinflammatory diseases stems from the analysis of experimental autoimmune encephalomyelitis (EAE), a frequently used animal model of MS (3). It has turned out that therapeutic GCs mainly act on T cells in this disease whereas GC nanoformulations such as PEGylated liposomes and hybrid nanoparticles rather target myeloid cells (4–6). GCs engage a variety of mechanisms to modulate T-cell function in neuroinflammation. They alter the expression and secretion of cytokines and chemokines, downregulate adhesion molecules, and redirect T cell migration (4, 5, 7, 8). In addition, they induce apoptosis in developing and mature T cells, which shapes the thymocyte repertoire (9) and contributes to the effectiveness of GC therapy (10). In contrast, the role of regulatory T cells (Treg) as targets of GC action remains controversial (4, 11).
Induction of T-cell apoptosis by GCs requires DNA-binding-dependent transcriptional regulation (12), presumably of pro-apoptotic genes such as Bim and PUMA (13), and is mediated via the intrinsic apoptotic pathway (14). The sensitivity of T cells to GC-induced apoptosis depends on their maturation and differentiation stage. While GCs efficiently cause cell death of CD4+CD8+ double-positive (DP) thymocytes, CD4+ and CD8+ single-positive (SP) thymocytes turned out to be highly resistant (15–17). Differential sensitivities to GC-induced apoptosis have also been noticed for peripheral T cell subpopulations such as Th1, Th2 and Th17 cells (18), whereas contradictory results have been obtained for Treg cells (19–21). Experiments in cell culture further indicated that activated T cells are less prone to GC-induced apoptosis than naïve T cells. Such a dichotomy was found for both murine (22, 23) and human T cells (24, 25). However, little information is available about the impact of GCs on antigen-specific effector T cells that have been activated in a pathological context in vivo rather than by artificial treatment with anti-CD3/CD28 antibodies, PMA/ionomycin or ConA in vitro. In order to close this gap, we made use of transgenic mice expressing green or red fluorescent proteins as well as CD45.1/2 congenic mice to distinguish between individual T cell populations, and a protocol to generate antigen-specific effector T cells in vivo by immunizing 2D2 transgenic mice expressing a MOG-specific TCR. Using this approach, we could demonstrate that antigen-primed effector T cells are protected from GC-induced apoptosis in vitro and in vivo, and that this effect is relevant in the context of a model neuroinflammatory disease in mice.
Materials and Methods
Mice
C57BL/6J and B6.SJL-PtprcaPepcb/BoyJ (CD45.1-congenic) mice were originally purchased from Charles River (Sulzfeld, Germany) and bred in the animal facility of the University Medical Center Göttingen. The following mouse strains were also housed in the University Medical Center Göttingen animal facility: 2D2 transgenic mice (26), Act-GFP mice (27), and RFP knock-in mice (28). 2D2 and RFP knock-in mice were intercrossed to obtain mice expressing both the MOG-specific 2D2 TCR transgene and RFP. The mice were kept in individually ventilated cages under specific-pathogen-free conditions and supplied with food and water ad libitum. All animal experiments were performed in accordance with ethical standards of animal welfare and approved by the responsible authorities of Lower Saxony (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit).
Animal Experimentation
Mice were immunized s.c. with 50 µg MOG35-55 in Complete Freund’s Adjuvant (CFA) and injected twice with 200 ng pertussis toxin (PTX) i.p. on day 0 and 2 as described previously (4). The draining lymph nodes were collected on day 12 post immunization in the case of C57BL/6J mice and on day 10 post immunization for 2D2/RFP mice. To investigate GC effects in vivo, 2.5 x 106 restimulated RFP+ antigen-primed T cells and 8-10 x 106 purified GFP+ naïve T cells were injected i.v. into the tail vein of CD45.1-congenic C57BL/6 recipient mice. Three days later, 5 mg/kg or 20 mg/kg Dex (Urbason soluble 32 mg; Sanofi, Paris, France) or PBS as a control were injected i.p. On day 4 after cell transfer, inguinal and mesenteric lymph nodes and spleen were collected, single cell suspensions prepared and analyzed by flow cytometry.
T-Cell Isolation and Cell Culture
CD3+ T cells were isolated from lymph nodes and spleen with the EasySep™ Mouse T Cell Enrichment Kit (Stemcell Technologies, Cologne, Germany) according to the manufacturer’s protocol. Cell purity was routinely determined by flow cytometry and always exceeded 90%. Antigen-specific restimulation of T cells was achieved by seeding cell suspensions from draining lymph nodes of immunized animals in 96-well round bottom plates at 5 x 105 cells/well in 100 µL of restimulation medium consisting of advanced RPMI 1640 (Gibco 51800-035; Thermofisher Scientific, Darmstadt, Germany), 10% fetal calf serum (PAA Laboratories A15-043; Thermofisher Scientific), 1% penicillin/streptomycin (Gibco 150063), 2 mM L-glutamine (Gibco 25030-024), and 0.0002% β-mercaptoethanol. In addition, 25 µg/mL MOG35-55 peptide (wildtype T cells) or 10 µg/mL MOG35-55 peptide (2D2 T cells) provided by the Charité (Berlin, Germany), 25 ng/mL recombinant mouse IL-12 (R&D Systems, 419-ML; Wiesbaden, Germany) and 20 µg/mL anti-IFNγ antibody (XMG1.2, Biolegend, 505847; Uithoorn, The Netherlands) were added. The cell suspension was incubated at 37 °C and 5% CO2 for 48 hrs, after which it was transferred into 96-well flat bottom plates. Dex at concentrations ranging from 10-9 M to 10-6 M or restimulation medium alone as a control were added to the wells, followed by 24 hrs incubation at 37 °C and 5% CO2.
Flow Cytometry
All antibodies and reagents for extracellular staining were obtained from BioLegend unless otherwise indicated: anti-CD3ε (145-2C11), anti-CD4 (RM4-5), anti-CD8α (53-6.7), anti-CD19 (6D5), anti-CD44 (IM7), anti-CD45.1 (A20), anti-CD45.2 (104), anti-CD62L (Mel14), AnnexinV, streptavidin, and LIVE/DEAD stain (Thermofisher Scientific). Antibodies and reagents were directly conjugated with FITC, AF647, PE, PerCPCy5.5, PE-Cy7, APC, APC-Cy7, BV421, BV650, BV785 or biotin. Extracellular staining was performed as previously described (29). For intracellular antigen detection, cells were fixed with 4% PFA for 20 min after completion of surface staining, followed by 30 min of incubation with Perm buffer (BD Biosciences, Heidelberg, Germany) and 60 min with an anti-Bcl-2 antibody (clone REA356, conjugated with APC; Miltenyi Biotec, Bergisch Gladbach, Germany), an anti-Bcl-XL antibody (clone 7B2.5, conjugated with biotin; SouthernBiotech, Birmingham, AL, USA) in combination with streptavidin, an anti-Bim antibody (polyclonal, conjugated with AF647, antibodies-online, Aachen, Germany) or an mIgG2b isotype control antibody (clone MPC-11, conjugated with APC or biotin; BioLegend), all diluted in Perm buffer. The mitochondrial membrane potential was measured with the help of the MitoProbe™ TMRM Kit (Thermofisher Scientific) according to the manufacturer’s instructions. All measurements were performed with a Cytoflex® flow cytometry device (Beckmann Coulter, Krefeld, Germany) and analyzed using Cytexpert® (Beckmann) or FlowJo® (Tree Star, Ashland, OR, USA; version 10.7.1.) software.
Transfer EAE
Lymph node cells from immunized C57BL/6J mice were cultured as described above in the absence or presence of 10-6 M or 10-7 M Dex for the last 24 hrs of the culture period. Afterwards, 2.5 x 106 cells were transferred i.p. into C57BL/6J recipient mice (30). The induction of transfer EAE did not require application of PTX. Animals were monitored daily for clinical symptoms by measuring their weight and scoring disease progression as follows: 0 = normal; 1 = reduced tone of tail; 2 = limp tail, impaired righting; 3 = absent righting; 4 = gait ataxia; 5 = mild paraparesis of hindlimbs; 6 = moderate paraparesis; 7 = severe paraparesis or paraplegia; 8 = tetraparesis; 9 = moribund; 10 = death.
Statistical Analysis
All data were analyzed by One-way ANOVA followed by a Newman-Keuls Multiple Comparison test. Analyses were performed with GraphPad Prism software (San Diego, CA, USA). Data are depicted as scatter dots plots with individual data points and a horizontal line representing the mean, or as XY plot with the mean ± SEM. Levels of significance are as follows: n.s., p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001.
Results
Antigen-Priming Protects T Cells From GC-Induced Apoptosis
GC therapy of MS patients and EAE mice induces T cell apoptosis but the differential sensitivity of antigen-primed effector and naïve bystander T cells to the pro-apoptotic activity of GCs has not been explored. To address this issue, we initially established an in vitro model that mimics the situation encountered in the treatment of neuroinflammatory diseases (Figure 1A). Lymph node cells mostly composed of antigen-primed T cells with a MOG-specific TCR were isolated from 2D2/RFP double-transgenic C57BL/6 mice previously immunized with MOG35-55 peptide. These cells were then co-cultured for 3 days in the presence of the same antigen with lymph node cells from GFP transgenic mice containing naïve T cells expressing a natural TCR repertoire. During the last 24 hours of co-culture, Dex was added at concentrations ranging from 10-9 M to 10-6 M to induce apoptosis while leaving T cell differentiation largely unaffected. Flow cytometric analysis revealed that the abundance of naïve GFP+ T cells amongst live cells was gradually diminished with ascending concentrations of Dex, whereas the percentage of antigen-primed RFP+ effector T cells was concomitantly increased (Figure 1B). In contrast, no such effect was observed for other cell types also contained in these lymph node cell preparations. More specifically, the percentages of GFP+ and RFP+ B cells amongst live cells for instance remained unaltered after the addition of Dex (Figure 1B). We conclude that antigen-primed effector T cells are more resistant to GC-induced apoptosis than naïve T cells.
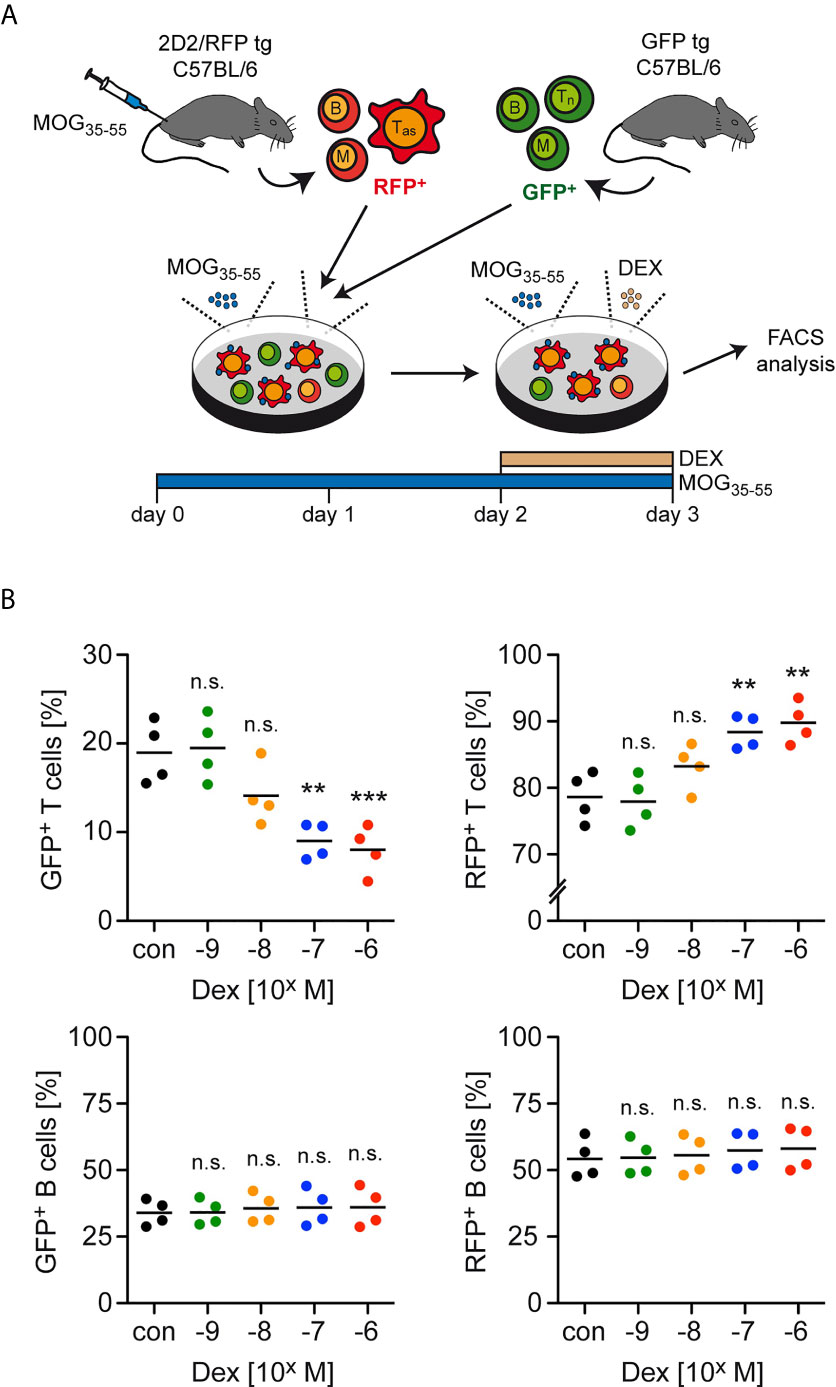
Figure 1 Enrichment of antigen-primed effector T cells after GC treatment in vitro. (A) Setup of the experimental model. (B) Percentages of live/dead– (naïve) GFP+ T cells (upper left panel) as well as live/dead– (antigen-primed) RFP+ T cells (upper right panel) after 3 days of cell culture in the presence of the indicated concentrations of Dex for the last 24 hrs. In control (con) conditions, no Dex was added to the cell culture. Percentages of GFP+ B cells (lower left panel) and RFP+ B cells (lower right panel) were determined in the same cultures. Each dot corresponds to cells originating from one individual mouse, the horizontal line represents the mean. N=4 (biological replicates from individual mice). Statistical analysis was performed by One-way ANOVA followed by a Newman-Keuls Multiple Comparison test. Levels of significance (depicted for the comparison between control and Dex-treated cultures): n.s. = not significant; **p < 0.01; ***p < 0.001. Tas, antigen-specific T cells; Tn, naïve T cells; B, B cells; M, myeloid cells.
Reduced Sensitivity of Antigen-Primed T Cells to Apoptosis Induction
Next, we explored whether the enrichment of antigen-primed effector T cells after Dex treatment was a direct consequence of an altered sensitivity to apoptosis induction. Using the same experimental setup as before, we found that the percentage of AnnexinV+ cells amongst GFP+CD4+ naïve T cells strongly increased in a dose-dependent manner after Dex treatment, whereas the percentage of AnnexinV+ cells amongst RFP+CD4+ effector T cells increased only very moderately (Figure 2A). To further unveil the mechanisms underlying the T cells’ differential sensitivity to GC-induced apoptosis, we investigated the expression of Bcl-2 and Bcl-XL in naïve GFP+CD4+ and antigen-primed RFP+CD4+ T cells. Flow cytometric analysis revealed that antigen-priming increased the levels of both anti-apoptotic proteins compared to the naïve state regardless of whether the cells were treated with Dex or not (Figure 2B). In contrast, the levels of the pro-apoptotic molecule Bim were comparable in both cell types (data not shown). Therefore, the elevated expression of anti-apoptotic proteins presumably explains the observed protection of effector T cells from GC-induced apoptosis.
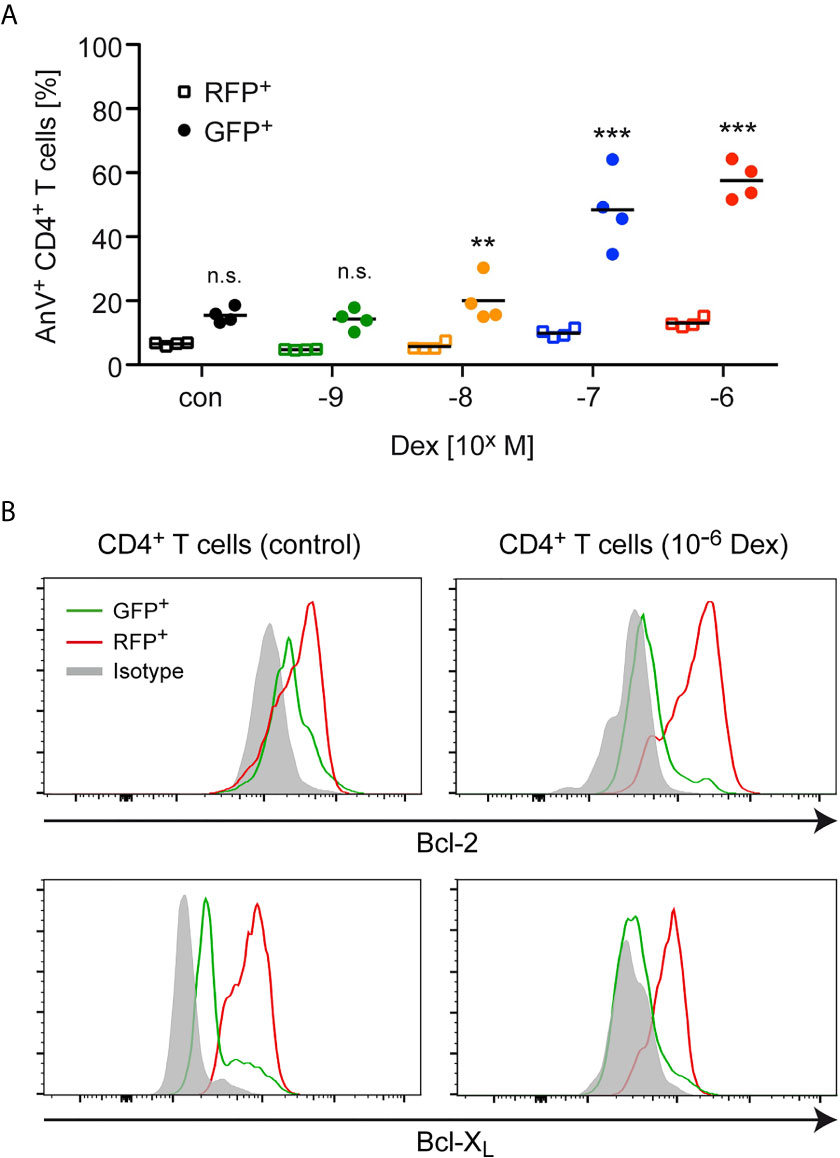
Figure 2 Apoptosis sensitivity of naïve and antigen-primed effector T cells. (A) Percentages of AnnexinV+ (AnV+) cells amongst (naïve) GFP+ and (antigen-primed) RFP+CD4+ T cells after 3 days of cell culture in the presence of the indicated concentrations of Dex for the last 24 hrs. No Dex was added to the cell culture under control (con) conditions. The experimental setup corresponds to the one illustrated in Figure 1A. Open symbols refer to RFP+ cells and filled symbols refer to GFP+ cells. N=4. Statistical analysis was performed by One-way ANOVA followed by a Newman-Keuls Multiple Comparison test. Levels of significance (depicted for each comparison between RFP+ and GFP+ cells): n.s. = not significant; **p < 0.01; ***p < 0.001. (B) Intracellular stainings of Bcl-2 or Bcl-XL in (naïve) GFP+ and (antigen-primed) RFP+CD4+ T cells cultured in the absence (control) or presence of 10-6 M Dex for the last 24 hrs according to the experimental scheme depicted in Figure 1A. Staining with an mIgG2b antibody served as an isotype control. One representative analysis for each condition is depicted as overlayed histograms.
To corroborate our findings, we also measured the mitochondrial membrane potential, as Bcl-2 family members have been reported to exert their apoptosis-modifying activity via this organelle (31). Lymph node cells, mostly composed of antigen-primed T cells with a MOG-specific TCR, were isolated from 2D2 transgenic C57BL/6 mice previously immunized with MOG35-55 peptide. These cells were co-cultured for 3 days in the presence of the same antigen with lymph node cells from CD45.1-congenic mice containing primarily naïve T cells. During the last 24 hours of co-culture, the cells were either treated with 10-6 M Dex or left untreated (Figure 3A). Under control conditions, a more pronounced loss of the mitochondrial membrane potential, as indicated by a reduced staining with TMRM, was observed for naïve CD45.1+CD4+ T cells in comparison to antigen-primed CD45.2+CD4+ T cells (Figure 3B). This difference was even stronger after Dex treatment and correlated well with the expression levels of the anti-apoptotic molecules Bcl-2 and Bcl-XL in these cells. Collectively, our results underscore that naïve T cells are the preferential target of apoptotic cell death after GC treatment.
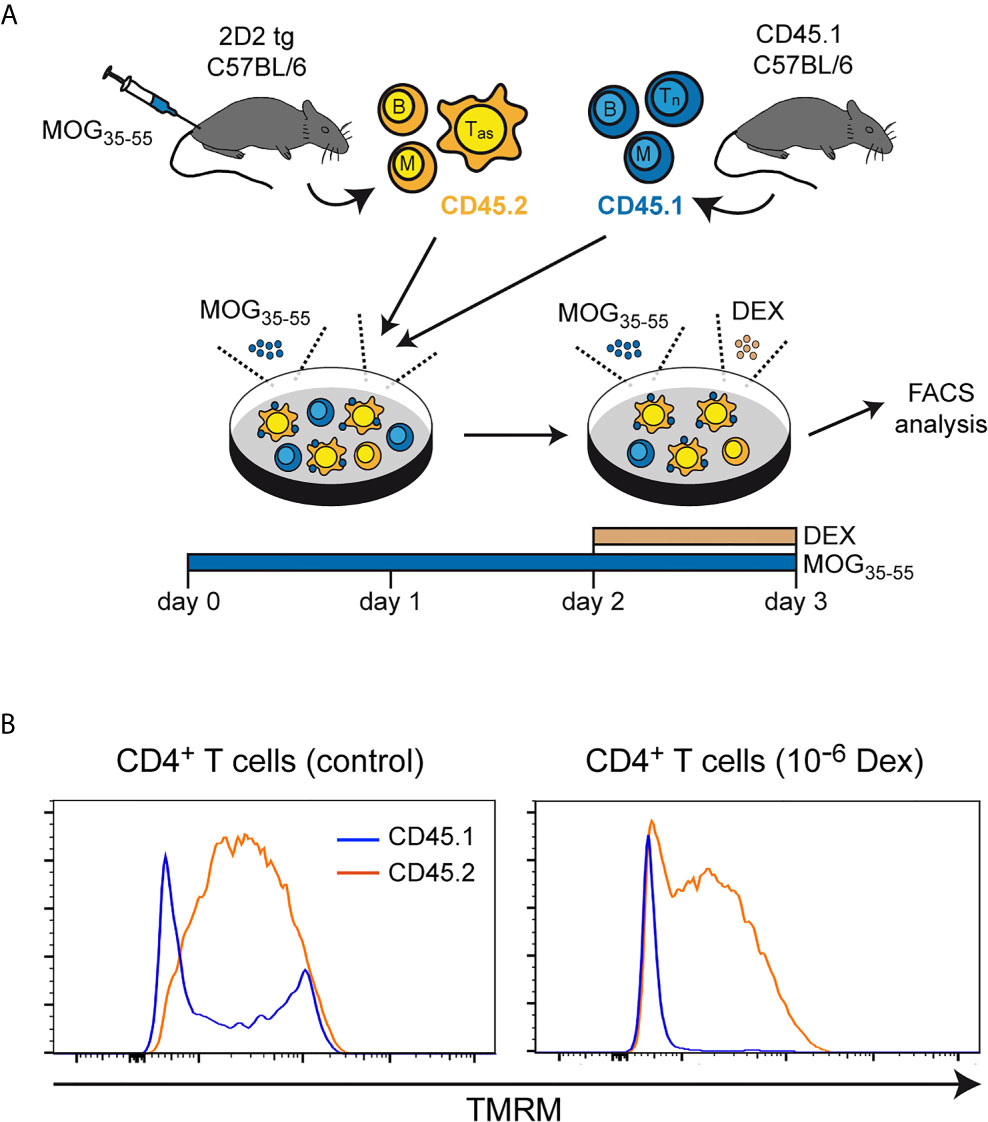
Figure 3 Mitochondrial membrane potential in naïve and antigen-primed effector T cells. (A) Setup of the experimental model. (B) Intracellular stainings of (naïve) CD45.1+ and (antigen-primed) CD45.2+CD4+ T cells with tetramethylrhodamine methylester (TMRM) after culturing the cells in the absence (control) or presence of 10-6 M Dex for the last 24 hrs. One representative analysis is depicted for each condition as overlayed histograms. Tas, antigen-specific T cells; Tn, naïve T cells; B, B cells; M, myeloid cells.
Enrichment of Antigen-Primed T Cells Coincides With Their Activation State
In the last step, we investigated late activation/memory markers as an alternative strategy to distinguish between naïve and effector T cells. Here, we used total lymph node cells from C57BL/6 wildtype mice previously immunized with MOG35-55 and restimulated them for 3 days in cell culture. During the last 24 hours, Dex was added at ascending concentrations ranging from 10-9 to 10-6 M (Figure 4A). In agreement with our earlier findings, the abundance of naïve CD44lowCD62L+ cells amongst live CD4+ T cells decreased from approximately 60% to 20% with increasing concentrations of Dex, while CD4+ T cells with an activated phenotype (CD44highCD62L–) were concomitantly enriched amongst live cells (Figure 4B). In summary, our data suggest that upregulation of anti-apoptotic proteins after activation specifically protects effector T cells from GC-induced apoptosis.
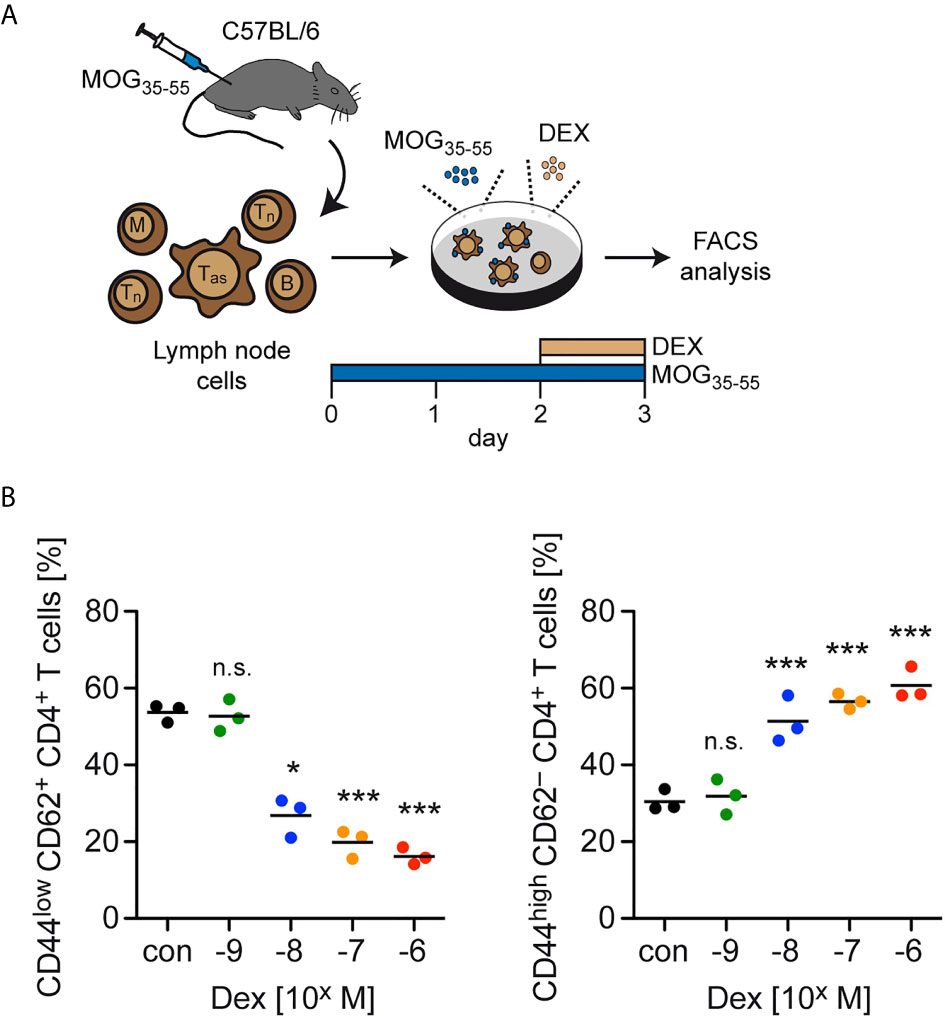
Figure 4 Enrichment of T cells with an activation/memory phenotype after GC treatment in vitro. (A) Setup of the experimental model. (B) Percentages of live/dead– (naïve) CD44lowCD62L+ CD4+ T cells (left panel) and live/dead– (activated) CD44highCD62L– CD4+ T cells (right panel) after 3 days of cell culture in the absence (con) or presence of the indicated concentrations of Dex for the last 24 hrs. Each dot corresponds to cells from one individual mouse, the horizontal line represents the mean. N=3 (biological replicates from individual mice). Statistical analysis was performed by One-way ANOVA followed by a Newman-Keuls Multiple Comparison test. Levels of significance (depicted for the comparison between control and Dex-treated cultures): n.s., not significant; *p < 0.05; ***p < 0.001. Tas, antigen-specific T cells; Tn, naïve T cells; B, B cells; M, myeloid cells.
GC Treatment of Mice Preferentially Induces Apoptosis in Naïve T Cells
As a first step to confirm our results in vivo, lymph node cells containing antigen-primed effector T cells expressing a MOG-specific TCR were isolated from 2D2/RFP double-transgenic C57BL/6 mice previously immunized with MOG35-55 peptide and restimulated in cell culture with the same antigen. These RFP+ effector T cells were then transferred into CD45.1-congenic C57BL/6 mice together with purified GFP+ naïve T cells, and 3 days later the mice were injected with 5 or 20 mg/kg Dex or PBS as a control. CD45.1-congenic mice were used as recipients to unequivocally allow to distinguish donor T cells from endogenous cells. After another 24 hours, the ratio between GFP+ and RFP+ T cells was determined in secondary lymphoid organs by flow cytometric analysis (Figure 5A). Similar to our in vitro findings, a dose-dependent accumulation of RFP+ antigen-primed effector T cells compared to GFP+ naïve T cells was observed in lymph nodes and spleen (Figures 5B, C). Moreover, also in this in vivo setting, a higher expression level of Bcl-2 was found in antigen-primed RFP+CD4+ T cells than in naïve GFP+CD4+ T cells regardless of whether or not the cells were treated with Dex (Figure 5D). Our data collectively provide evidence that the sensitivity of antigen-primed effector T cells to GC-induced apoptosis in mice is diminished in vivo as well.
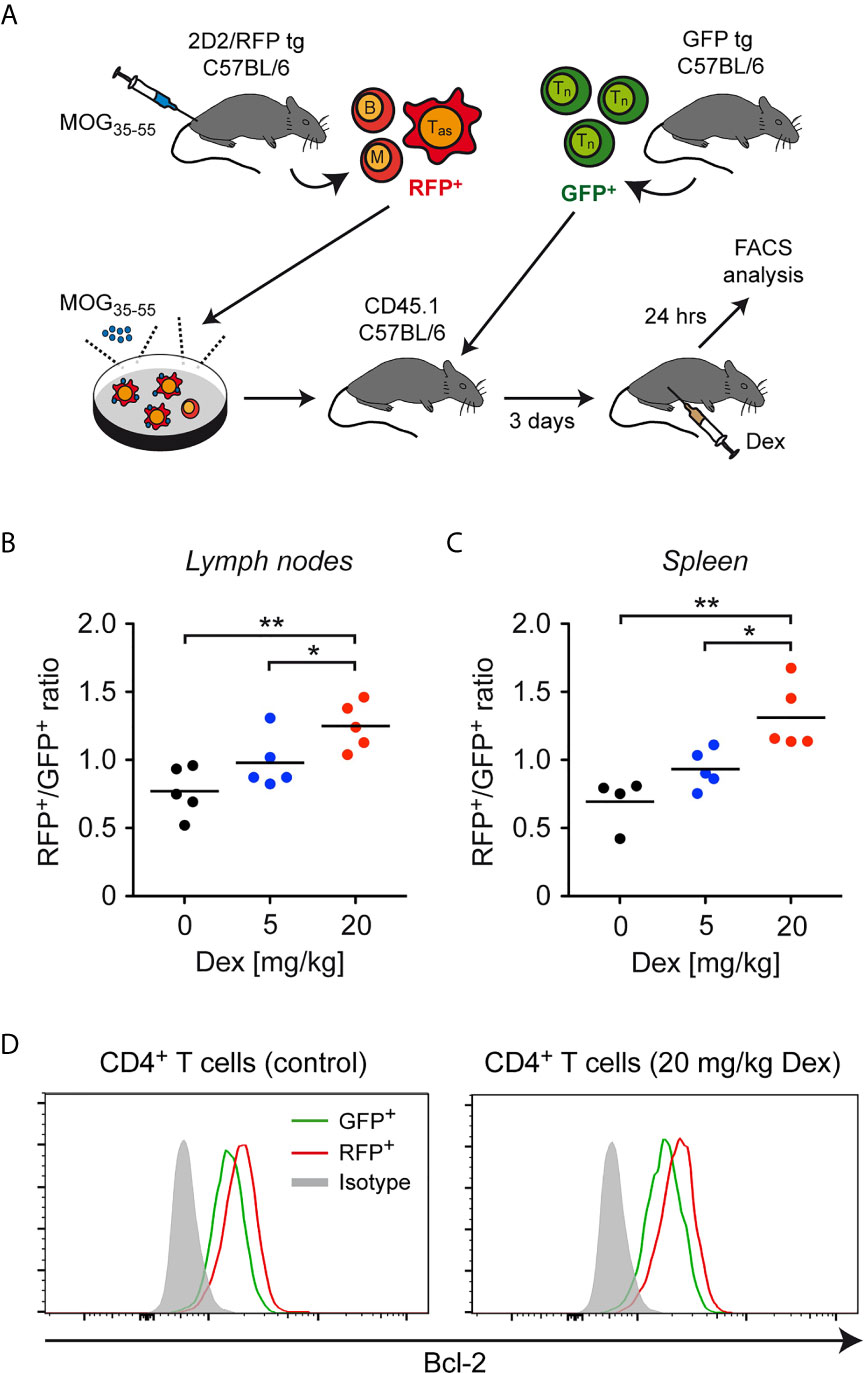
Figure 5 Enrichment of antigen-primed effector T cells after GC treatment in vivo. (A) Setup of the experimental model. (B, C) Weighted ratio of live/dead– CD45.2+ (antigen-primed) RFP+ T cells and (naïve) GFP+ T cells from lymph nodes (B) and spleen (C) on day 4 after transfer and 24 hrs after application of the indicated doses of Dex (5 mg/kg or 20 mg/kg) in vivo. Control mice (0 mg/kg Dex) received PBS. Data are depicted as the mean +/- SEM. N=4-5 (biological replicates from individual mice). Statistical analysis was performed by One-way ANOVA followed by a Newman-Keuls Multiple Comparison test. Levels of significance (depicted for the comparison between control and Dex-treated cultures): *p < 0.05; **p < 0.01. (D) Intracellular stainings of Bcl-2 in (naïve) GFP+ and (antigen-primed) RFP+CD4+ T cells isolated from mice treated with 20 mg/kg Dex or left untreated as outlined in panel (A). Staining with an mIgG2b antibody served as an isotype control. One representative analysis for each condition is depicted as overlayed histograms. Tas, antigen-specific T cells; Tn, naïve T cells; B, B cells; M, myeloid cells.
GC-Treatment of T Cells Aggravates Adoptive Transfer EAE in Mice
Finally, we asked whether the accumulation of antigen-primed effector T cells after in vitro treatment with Dex had any consequences in an adoptive transfer EAE model. To address this issue, C57Bl/6 wildtype mice were immunized with MOG35-55 peptide and 12 days later the lymph nodes containing both antigen-primed effector and naïve T cells were isolated and restimulated in vitro with the same antigen for 3 days. During the last 24 hours, Dex was added at two different concentrations (10-7 M and 10-6 M), or the cells were left untreated as a control. Eventually, 2.5 x 106 live cells from each condition were transferred into naïve C57Bl/6 wildtype mice and the disease course was followed over 3 weeks (Figure 6A). Mice receiving T cells from cell cultures treated with Dex showed a significantly aggravated disease course dependent on the employed Dex concentration compared to mice receiving control T cells (Figure 6B). This negative effect was also reflected by a higher incidence of EAE [10-6 M Dex: 91% (10/11), 10-7 M Dex: 85% (11/13), control: 75% (9/12)], an earlier onset of the disease (Figure 6C), and an increase of the cumulative disease score (Figure 6D). In summary, our findings suggest that protection of antigen-primed effector T cells from GC-induced apoptosis is relevant for the pathogenesis of T cell-dependent inflammatory diseases in mice and presumably MS in humans as well.
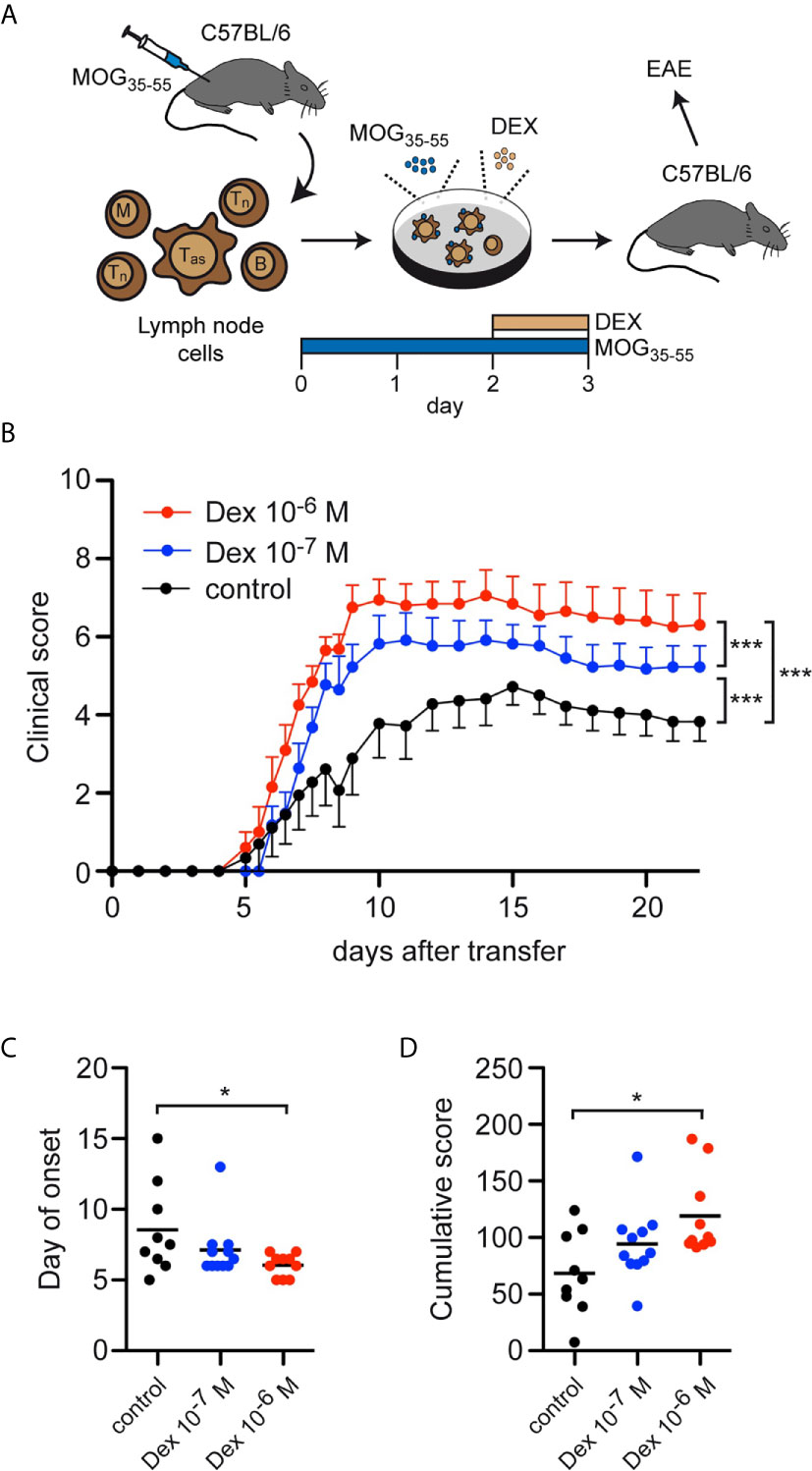
Figure 6 Enrichment of antigen-primed effector T cells after GC treatment in vitro aggravates clinical symptoms in an adoptive transfer EAE model. (A) Setup of the experimental model. (B-D) EAE development after transfer of 2.5 x 106 T cells under control conditions (black) or after treatment with 10-7 M (blue) or 10-6 M (red) Dex for the last 24 hrs of the cell culture period in vitro. Clinical score of all mice that became sick, depicted as the mean +/- SEM (B), the day of disease onset (C), and the cumulative clinical score (D) are depicted. N = 11-13. Statistical analysis was performed by One-way ANOVA followed by a Newman-Keuls Multiple Comparison test. Levels of significance: *p < 0.05; ***p < 0.001.
Discussion
Induction of T-cell apoptosis was one of the first modes of action identified for GCs and is still believed to be highly relevant for their clinical effectiveness in the treatment of various diseases. It was soon noticed that profound differences exist between individual developmental stages, cellular subsets and differentiation states with regard to apoptosis sensitivity. DP thymocytes are presumably the most sensitive type of immune cell in this respect (16, 17), but differences were also found for mature T-cell subpopulations such as Th1, Th2 and Th17 cells (21, 32). Furthermore, it was observed that T-cell activation conferred protection against GC-induced apoptosis, which was linked to the cell cycle entry into the G1/G2/M phase (33, 34). Along these lines, T cells from burn-injured mice were shown to have a reduced sensitivity to GC-induced apoptosis, which correlated with their enhanced activation status (23). In contrast, very little is known concerning GC-induced apoptosis of antigen-primed effector T cells in the pathological setting of inflammatory diseases. It is noteworthy that a stimulation with anti-CD3/CD28 antibodies, PMA/ionomycin or ConA, as done in most published studies, mimics T cell activation only partially. In natural conditions, activation of T cells is achieved by interaction of an MHC:peptide complex on antigen-presenting cells with the TCR, and engagement of CD28 by B7 molecules in the context of an immune microenvironment composed of other leukocytes, cytokines and growth factors. Since these diverse influences not only impact the fate of effector T cells but presumably determine their sensitivity to GC-induced apoptosis, too, the latter process should be preferably investigated in more sophisticated experimental setups.
To uncover whether antigen-primed effector T cells show a different sensitivity towards GC-induced apoptosis compared to naïve T cells, we developed a co-culture system in which both cell types could be distinguished based on their expression of a red and green fluorescent protein or the expression of the congenic markers CD45.1 and 2, respectively. T cell priming in vivo was achieved by immunization of 2D2-transgenic mice expressing a TCR specific for the pathologically relevant myelin antigen MOG35-55 followed by restimulation in cell culture. With this setup we ensured that T cell activation occurred in a microenvironment that mimics the situation encountered during an autoimmune response. Importantly, only antigen-specific T cells but neither B cells isolated from the same immunized mice nor naïve T cells were protected from GC-induced cell death. This effect could be assigned to a reduced induction of apoptosis based on the staining of T cell subtypes with AnnexinV and their differential loss of the mitochondrial membrane potential. While these results collectively confirmed that antigen-priming alters apoptosis induction by GCs, this effect was not only clear in vitro but also observed in vivo. In fact, we could demonstrate that the protective effect of antigen-priming with regard to GC-induced apoptosis was dose-dependent in corresponding concentration ranges in vitro (10-8 and 10-7 M Dex) and in vivo (5 and 20 mg/kg Dex). This observation indicates that the mechanism in cell culture and living mice is very similar.
Several mechanisms are being discussed as possible explanations for the diminished sensitivity of activated T cells to GC-induced apoptosis in vitro, including a critical role of cytokines as part of the immune microenvironment. This concerns IL-2, IL-4, IL-10 and IL-12 acting on individual T-cell subsets (24, 35), and downstream pathways involving IκBα (36) and PI3K/AKT (37). Importantly, cytokine signaling has been shown to upregulate the anti-apoptotic proteins Bcl-2 and Bcl-XL in activated T cells, which seems to be an important mechanism of their reduced sensitivity to apoptosis induction (38). Intriguingly, Bcl-2 in T cells was found to be only induced after stimulation of total splenocyte preparations with ConA but not after activation of purified T cells with PMA/ionomycin. In contrast, Bcl-XL was induced under both conditions (39). These findings support the notion that the immune microenvironment dictates if anti-apoptotic proteins become elevated during T cell activation or not. In our study, we could show that both proteins were expressed at higher levels in antigen-primed effector T cells compared to naïve T cells, regardless of GC treatment, a finding which reflects the natural context of an autoimmune response in vivo. Noteworthy, the previous observation that T cells obtained from Bcl-2-transgenic mice are resistant to GC-induced apoptosis is also in line with our current finding that an increased expression of this anti-apoptotic protein in antigen-primed effector T cells is linked to a protection against cell death by GCs (8). Hence, we consider it fair to conclude that T cells, which were naturally activated in the context of an immune response, become resistant against GC-induced apoptosis through induction of Bcl-2 and Bcl-XL.
The biological consequences of the observed GC-resistance of antigen-primed T cells have not been studied in physiologically relevant mouse models up to now. Adoptive transfer EAE mimics many hallmarks of human MS and hence makes it possible to test the in vivo relevance of the altered balance between naïve and antigen-primed T cells after Dex treatment in vitro (30). Importantly, the MOG-specific T cells analyzed in this study are able to induce clinical symptoms after transfer into C57BL/6 mice and therefore resemble pathogenic T cells found in MS patients. Our results unveiled that the enrichment of antigen-primed effector T cells after incubation with Dex enhanced disease development after adoptive transfer into mice. These data further indicate that our model is very sensitive to the input of antigen-primed effector T cells and that already a slight increase in their relative number suffices to induce a statistically significant worsening of the disease course.
In summary, our study extents previous findings concerning GC-induced apoptosis of activated T cells to the analysis of pathologically relevant antigen-primed effector T cells in a setting which is relevant for neuroinflammatory diseases. The data have several practical implications. Antigen-primed effector T cells can be enriched in cell culture with the help of GCs, which can be exploited for the analysis of autoimmune diseases but also in approaches to treat cancer where pure preparations of such cells are required. Furthermore, the observation that preventive treatment of EAE with methylprednisolone paradoxically worsened the disease outcome can now be explained by an unintended enrichment of effector T cells prior to disease induction (40). Although GCs undoubtedly remain a powerful tool to interfere with inflammatory conditions including MS, our results highlight the importance of critically considering whether their application might result in an enrichment of effector T cells during an ongoing immune response and therefore might even prove harmful.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit.
Author Contributions
JB: performed and analyzed experiments. SS: performed and analyzed experiments. HR: designed the project, analyzed experiments, and wrote the manuscript. FL: designed the project, analyzed experiments, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Deutsche Forschungsgemeinschaft (RE 1631/17-1 and FL 377/4-1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Birgit Curdt and Martina Weig for their expert technical assistance, Leonard Hammermann and Judith Strauß for help with the adoptive transfer EAE model, and Catherine Ludwig for language editing.
References
1. Hench P. Effects of Cortisone in the Rheumatic Diseases. Lancet (1950) 2:483–4. doi: 10.1016/S0140-6736(50)91461-9
2. Milligan NM, Newcombe R, Compston DA. A Double-Blind Controlled Trial of High Dose Methylprednisolone in Patients With Multiple Sclerosis: 1. Clinical Effects. J Neurol Neurosurg Psychiatry (1987) 50:511–6. doi: 10.1136/jnnp.50.5.511
3. Gold R, Linington C, Lassmann H. Understanding Pathogenesis and Therapy of Multiple Sclerosis Via Animal Models: 70 Years of Merits and Culprits in Experimental Autoimmune Encephalomyelitis Research. Brain (2006) 129:1953–71. doi: 10.1093/brain/awl075
4. Wüst S, van den Brandt J, Tischner D, Kleiman A, Tuckermann JP, Gold R, et al. Peripheral T Cells are the Therapeutic Targets of Glucocorticoids in Experimental Autoimmune Encephalomyelitis. J Immunol (2008) 180:8434–43. doi: 10.4049/jimmunol.180.12.8434
5. Schweingruber N, Haine A, Tiede K, Karabinskaya A, van den Brandt J, Wüst S, et al. Liposomal Encapsulation of Glucocorticoids Alters Their Mode of Action in the Treatment of Experimental Autoimmune Encephalomyelitis. J Immunol (2011) 187:4310–8. doi: 10.4049/jimmunol.1101604
6. Montes-Cobos E, Ring S, Fischer HJ, Heck J, Strauss J, Schwaninger M, et al. Targeted Delivery of Glucocorticoids to Macrophages in a Mouse Model of Multiple Sclerosis Using Inorganic-Organic Hybrid Nanoparticles. J Control Release (2017) 245:157–69. doi: 10.1016/j.jconrel.2016.12.003
7. Wüst S, Tischner D, John M, Tuckermann JP, Menzfeld C, Hanisch UK, et al. Therapeutic and Adverse Effects of a non-Steroidal Glucocorticoid Receptor Ligand in a Mouse Model of Multiple Sclerosis. PloS One (2009) 4:e8202. doi: 10.1371/journal.pone.0008202
8. Schweingruber N, Fischer HJ, Fischer L, van den Brandt J, Karabinskaya A, Labi V, et al. Chemokine-Mediated Redirection of T Cells Constitutes a Critical Mechanism of Glucocorticoid Therapy in Autoimmune CNS Responses. Acta Neuropathol (2014) 127:713–29. doi: 10.1007/s00401-014-1248-4
9. Mittelstadt PR, Monteiro JP, Ashwell JD. Thymocyte Responsiveness to Endogenous Glucocorticoids is Required for Immunological Fitness. J Clin Invest (2012) 122:2384–94. doi: 10.1172/JCI63067
10. Schmidt J, Gold R, Schonrock L, Zettl UK, Hartung HP, Toyka KV. T-Cell Apoptosis in Situ in Experimental Autoimmune Encephalomyelitis Following Methylprednisolone Pulse Therapy. Brain (2000) 123( Pt 7):1431–41. doi: 10.1093/brain/123.7.1431
11. Kim D, Nguyen QT, Lee J, Lee SH, Janocha A, Kim S, et al. Anti-Inflammatory Roles of Glucocorticoids are Mediated by Foxp3(+) Regulatory T Cells Via a miR-342-Dependent Mechanism. Immunity (2020) 53:581–96.e5. doi: 10.1016/j.immuni.2020.07.002
12. Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, et al. DNA Binding of the Glucocorticoid Receptor is Not Essential for Survival. Cell (1998) 93:531–41. doi: 10.1016/S0092-8674(00)81183-6
13. Erlacher M, Michalak EM, Kelly PN, Labi V, Niederegger H, Coultas L, et al. BH3-Only Proteins Puma and Bim are Rate-Limiting for Gamma-Radiation- and Glucocorticoid-Induced Apoptosis of Lymphoid Cells In Vivo. Blood (2005) 106:4131–8. doi: 10.1182/blood-2005-04-1595
14. Wang D, Müller N, McPherson KG, Reichardt HM. Glucocorticoids Engage Different Signal Transduction Pathways to Induce Apoptosis in Thymocytes and Mature T Cells. J Immunol (2006) 176:1695–702. doi: 10.4049/jimmunol.176.3.1695
15. Wiegers GJ, Knoflach M, Bock G, Niederegger H, Dietrich H, Falus A, et al. CD4(+)CD8(+)TCR(Low) Thymocytes Express Low Levels of Glucocorticoid Receptors While Being Sensitive to Glucocorticoid-Induced Apoptosis. Eur J Immunol (2001) 31:2293–301. doi: 10.1002/1521-4141(200108)31:8<2293::AID-IMMU2293>3.0.CO;2-I
16. Berki T, Palinkas L, Boldizsar F, Nemeth P. Glucocorticoid (GC) Sensitivity and GC Receptor Expression Differ in Thymocyte Subpopulations. Int Immunol (2002) 14:463–9. doi: 10.1093/intimm/14.5.463
17. van den Brandt J, Wang D, Reichardt HM. Resistance of Single-Positive Thymocytes to Glucocorticoid-Induced Apoptosis is Mediated by CD28 Signaling. Mol Endocrinol (2004) 18:687–95. doi: 10.1210/me.2003-0390
18. Banuelos J, Lu NZ. A Gradient of Glucocorticoid Sensitivity Among Helper T Cell Cytokines. Cytokine Growth Factor Rev (2016) 31:27–35. doi: 10.1016/j.cytogfr.2016.05.002
19. Chen X, Murakami T, Oppenheim JJ, Howard OM. Differential Response of Murine CD4+CD25+ and CD4+CD25- T Cells to Dexamethasone-Induced Cell Death. Eur J Immunol (2004) 34:859–69. doi: 10.1002/eji.200324506
20. Sbiera S, Dexneit T, Reichardt SD, Michel KD, van den Brandt J, Schmull S, et al. Influence of Short-Term Glucocorticoid Therapy on Regulatory T Cells In Vivo. PloS One (2011) 6:e24345. doi: 10.1371/journal.pone.0024345
21. Prenek L, Litvai T, Balazs N, Kugyelka R, Boldizsar F, Najbauer J, et al. Regulatory T Cells are Less Sensitive to Glucocorticoid Hormone Induced Apoptosis Than CD4(+) T Cells. Apoptosis (2020) 25:715–29. doi: 10.1007/s10495-020-01629-x
22. Zacharchuk CM, Mercep M, Chakraborti PK, Simons SS Jr., Ashwell JD. Programmed T Lymphocyte Death. Cell Activation- and Steroid-Induced Pathways are Mutually Antagonistic. J Immunol (1990) 145:4037–45.
23. D’Elia M, Patenaude J, Dupras C, Bernier J. T Cells From Burn-Injured Mice Demonstrate a Loss of Sensitivity to Glucocorticoids. Am J Physiol Endocrinol Metab (2010) 299:E299–307. doi: 10.1152/ajpendo.00084.2010
24. Brunetti M, Martelli N, Colasante A, Piantelli M, Musiani P, Aiello FB. Spontaneous and Glucocorticoid-Induced Apoptosis in Human Mature T Lymphocytes. Blood (1995) 86:4199–205. doi: 10.1182/blood.V86.11.4199.bloodjournal86114199
25. Strauss G, Osen W, Debatin KM. Induction of Apoptosis and Modulation of Activation and Effector Function in T Cells by Immunosuppressive Drugs. Clin Exp Immunol (2002) 128:255–66. doi: 10.1046/j.1365-2249.2002.01777.x
26. Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin Oligodendrocyte Glycoprotein-Specific T Cell Receptor Transgenic Mice Develop Spontaneous Autoimmune Optic Neuritis. J Exp Med (2003) 197:1073–81. doi: 10.1084/jem.20021603
27. Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green Mice’ as a Source of Ubiquitous Green Cells. FEBS Lett (1997) 407:313–9. doi: 10.1016/S0014-5793(97)00313-X
28. Luche H, Weber O, Nageswara Rao T, Blum C, Fehling HJ. Faithful Activation of an Extra-Bright Red Fluorescent Protein in “Knock-in” Cre-reporter Mice Ideally Suited for Lineage Tracing Studies. Eur J Immunol (2007) 37:43–53. doi: 10.1002/eji.200636745
29. van den Brandt J, Voss K, Schott M, Hünig T, Wolfe MS, Reichardt HM. Inhibition of Notch Signaling Biases Rat Thymocyte Development Towards the NK Cell Lineage. Eur J Immunol (2004) 34:1405–13. doi: 10.1002/eji.200324735
30. Flach AC, Litke T, Strauss J, Haberl M, Gomez CC, Reindl M, et al. Autoantibody-Boosted T-cell Reactivation in the Target Organ Triggers Manifestation of Autoimmune CNS Disease. Proc Natl Acad Sci U S A (2016) 113:3323–8. doi: 10.1073/pnas.1519608113
31. Rasmussen ML, Gama V. A Connection in Life and Death: The BCL-2 Family Coordinates Mitochondrial Network Dynamics and Stem Cell Fate. Int Rev Cell Mol Biol (2020) 353:255–84. doi: 10.1016/bs.ircmb.2019.12.005
32. Banuelos J, Shin S, Cao Y, Bochner BS, Morales-Nebreda L, Budinger GR, et al. BCL-2 Protects Human and Mouse Th17 Cells From Glucocorticoid-Induced Apoptosis. Allergy (2016) 71:640–50. doi: 10.1111/all.12840
33. Walker PR, Kwast-Welfeld J, Gourdeau H, Leblanc J, Neugebauer W, Sikorska M. Relationship Between Apoptosis and the Cell Cycle in Lymphocytes: Roles of Protein Kinase C, Tyrosine Phosphorylation, and AP1. Exp Cell Res (1993) 207:142–51. doi: 10.1006/excr.1993.1173
34. Tuosto L, Cundari E, Gilardini Montani MS, Piccolella E. Analysis of Susceptibility of Mature Human T Lymphocytes to Dexamethasone-Induced Apoptosis. Eur J Immunol (1994) 24:1061–5. doi: 10.1002/eji.1830240508
35. Zubiaga AM, Munoz E, Huber BT. IL-4 and IL-2 Selectively Rescue Th Cell Subsets From Glucocorticoid-Induced Apoptosis. J Immunol (1992) 149:107–12.
36. Xie H, Seward RJ, Huber BT. Cytokine Rescue From Glucocorticoid Induced Apoptosis in T Cells is Mediated Through Inhibition of Ikappabalpha. Mol Immunol (1997) 34:987–94. doi: 10.1016/S0161-5890(97)00128-4
37. Kawabe K, Lindsay D, Braitch M, Fahey AJ, Showe L, Constantinescu CS. IL-12 Inhibits Glucocorticoid-Induced T Cell Apoptosis by Inducing GMEB1 and Activating PI3K/Akt Pathway. Immunobiology (2012) 217:118–23. doi: 10.1016/j.imbio.2011.07.018
38. Akbar AN, Borthwick NJ, Wickremasinghe RG, Panayoitidis P, Pilling D, Bofill M, et al. Interleukin-2 Receptor Common Gamma-Chain Signaling Cytokines Regulate Activated T Cell Apoptosis in Response to Growth Factor Withdrawal: Selective Induction of Anti-Apoptotic (bcl-2, bcl-xL) But Not Pro-Apoptotic (Bax, bcl-xS) Gene Expression. Eur J Immunol (1996) 26:294–9. doi: 10.1002/eji.1830260204
39. Broome HE, Dargan CM, Krajewski S, Reed JC. Expression of Bcl-2, Bcl-X, and Bax After T Cell Activation and IL-2 Withdrawal. J Immunol (1995) 155:2311–7.
Keywords: glucocorticoids, apoptosis, T cells, antigen-priming, adoptive transfer EAE
Citation: Bier J, Steiger SM, Reichardt HM and Lühder F (2021) Protection of Antigen-Primed Effector T Cells From Glucocorticoid-Induced Apoptosis in Cell Culture and in a Mouse Model of Multiple Sclerosis. Front. Immunol. 12:671258. doi: 10.3389/fimmu.2021.671258
Received: 23 February 2021; Accepted: 28 May 2021;
Published: 10 June 2021.
Edited by:
Oxana Bereshchenko, University of Perugia, ItalyReviewed by:
Ulrike Baschant, University Hospital Carl Gustav Carus, GermanySabine Vettorazzi, University of Ulm, Germany
Copyright © 2021 Bier, Steiger, Reichardt and Lühder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Holger M. Reichardt, aHJlaWNoYXJkdEBtZWQudW5pLWdvZXR0aW5nZW4uZGU=; Fred Lühder, ZnJlZC5sdWVoZGVyQG1lZC51bmktZ29ldHRpbmdlbi5kZQ==
†Present address: Jasmina Bier, Institute of Neuroimmunology and Multiple Sclerosis, University Medical Center Hamburg, Hamburg, Germany
‡These authors have contributed equally to this work