 Jasper Iske
Jasper Iske Tomohisa Matsunaga
Tomohisa Matsunaga Hao Zhou1
Hao Zhou1 Stefan G. Tullius
Stefan G. Tullius- 1Division of Transplant Surgery & Transplant Surgery Research Laboratory, Brigham and Women´s Hospital, Harvard Medical School, Boston, MA, United States
- 2Department of Urology, Osaka Medical College, Osaka, Japan
In transplantation, donor and recipients frequently differ in age. Senescent cells accumulate in donor organs with aging and have the potential to promote senescence in adjacent cells when transferred into recipient animals. Characteristically, senescent cells secrete a myriad of pro-inflammatory, soluble molecules as part of their distinct secretory phenotype that have been shown to drive senescence and age-related co-morbidities. Preliminary own data show that the transplantation of old organs limits the physical reserve of recipient animals. Here, we review how organ age may affect transplant recipients and discuss the potential of accelerated aging.
Introduction
Organ transplantation is the treatment of choice for end-stage-organ failure. The supply of organs, however, is limited, resulting in prolonged waiting times with many patients dying or becoming too ill to be transplanted. Aging demographics have incrementally affected the deceased donor population with older donors showing the by far largest proportional increase. Organs from older donors are, at the same time, underutilized, frequently discarded or not even considered (1).
The most obvious strategy that may close the gap between demand and supply may thus be an optimized utilization of older organs from deceased donors (2, 3). Increased donor age, at the same time poses a significant risk for adverse outcomes including more frequent rejections due to an augmented immunogenicity in aging (4, 5). Most relevantly, older organs have shown compromised long-term graft outcomes with inferior graft survival rates in addition to increased rates of chronic allograft dysfunction in kidney, heart and lung transplantation (6).
Senescent cells accumulate with aging and have been identified as critical in driving the immunogenicity of older organs linked to the accumulation of cell-free mitochondrial-DNA that accelerate alloimmune responses (7). Recent evidence also suggests that senescent cells can induce a senescent phenotype in adjacent cells, a potential mechanism on how the engraftment of older organs may facilitate the spread of senescence. Depletion of senescent cells, at the same time, has been shown to ameliorate a wide range of age‐associated disabilities and diseases (8–12).
Here, we introduce potential mechanisms and consequences of prompting bystander senescence and discuss clinically relevant aspects of senescent cell spread when transplanting older organs. Although speculative, age-disparate transplantation may also provide unique opportunities as the transplantation of young organs may contribute to rejuvenation.
Senescent Cells Accelerate Aging
Cellular senescence is characterized as a stable and terminal state of growth arrest based on acquired anti-apoptotic pathways (SCAPS, termed senescent cell anti-apoptotic pathways) that render senescent cells resistant to apoptosis (13). Thus, senescent cells accumulate in many tissues with aging (14–16). Notably, stem cells critical for tissue regeneration have also been shown to undergo senescence associated with the loss of their self-renewal capacity considered as a driver of age-related tissue dysfunction and organismal aging (17–19). Characteristics of senescent cells include distinct transcriptional signatures with upregulated genes causing cell cycle arrest, epi-genetic modifications and a divers non-coding RNA profile (20). Senescence cells also have a compromised mitochondrial function (21) and an altered lysosomal activity with overexpression of endogenous lysosomal beta-galactosidase, serving as the most widely used biomarker to visualize senescent cells, termed senescence-associated β-galactosidase (Sa-β-gal) (22). Of note, high expression of Sa-β-gal is not always associated with cellular senescence as it has also been detected in cells undergoing quiescence (23, 24), which – in contrast to senescence – displays a reversible cell-cycle arrest required for tissue repair and regeneration.
Senescent cells exhibit a distinct, pro-inflammatory secretome consisting of cytokines (IL-6, IL-8, TNF-α), chemokines (CCL2, CCL20) and matrix remodeling enzymes termed the “Senescent Associated Secretory Phenotype” (SASP) (25). The production of SASP is a cardinal feature of senescent cells contributing to age‐related tissue dysfunction, chronic age-associated diseases and organismal aging, impairing tissue homeostasis and impeding neighboring cell function (26).
Cellular senescence can be triggered by oncogenic and DNA‐damaging stressors that induce DNA damage responses, a signaling pathway in which ATM or ATR kinases block cell-cycle progression through stabilization of p53 and transcriptional activation of the cyclin-dependent kinase inhibitors p21 (27). Moreover, the cyclin-dependent kinase inhibitor p16INK4a facilitates cell cycle arrest and can therefore also be used as a senescent cell marker (28). As a consequence of DNA damage, senescent cells exhibit increased frequencies of DNA damage foci containing phosphorylated histone H2A.X that are preferentially located at the telomeres and thus termed telomere associated foci (TAF) (29).
Strikingly, senescent cells are capable of auto-inducing a senescent phenotype in surrounding, non-senescent, bystander cells via gap junction mediated cell–cell contact and processes involving reactive oxygen species (ROS). Thus, continuous exposure of intact fibroblasts to senescent cells resulted in increased numbers of DNA double-strand breaks (DSBs) indicating senescence, which had been inhibited when blocking gap junction-mediated cell–cell contact (30).
SASP appears to play a critical role in driving bystander senescence. Quantitative proteomics with small molecule screens in transwell two chamber experiments that co-cultured naive human fibroblasts with senescent fibroblasts identified various components of the SASP including TGF-β family ligands, VEGF, CCL2 and CCL20, all capable of inducing paracrine senescence (31). Moreover, culturing naive fibroblasts with conditioned medium derived from senescent fibroblasts demonstrated comparable effects. The senescent phenotype remained detectable 14 days after splitting both cell lines indicating long-term effects (31). A broad range of additional SASP components including IGFBP-7, PAI-1, IL-6 and CXCR2-binding chemokines (such as IL-8 or GROα) have also been shown to drive senescence (32–35).
The spread of senescence has also been confirmed in vivo utilizing transgenic Sos Egfrwa2/+ mice that develop papillomas with a senescent phenotype within their basal and suprabasal layers. Although there were no senescent cells in the tissue close to normal skin, increased frequencies of senescent cells had been detected in surrounding tissue adjacent to senescent papillomas (31).
Can the Engraftment of Old Organs Promote Senescence?
We have previously shown that older donor organs bear increased frequencies of senescent cells (7). Thus, when transplanting an older organ, an increased number of senescent cells is transferred to recipients posing the potential to accelerate senescence. In support of this hypothesis, intraperitoneal transplantation of relatively small numbers of senescent cells into young mice resulted into an augmentation of senescence in visceral adipose tissue associated with a compromised physical capacity (36). In detail, senescent cells from luciferase expressing transgenic mice were intraperitoneally injected and assessed by quantifying SA-βgal+, p16Ink4a+ and TAF+ cells in visceral adipose tissue. By two months, amounts of SA-βgal+ and p16Ink4a+ cells but also luciferase negative TAF+ cells had increased, indicating an augmented number of senescent cells of recipient origin. Consistent with the spread of senescence, distant tissues including the quadriceps muscles displayed an increased frequency of the senescent cell markers such as p16Ink4a, TNF-α, and IL-6 (36). Moreover, autologous transplantation of senescent cells into healthy knee joints promoted the development of an osteoarthritis‐like condition in young mice (37). These observations are consistent with our own preliminary data showing a compromised physical capacity in young mice that had received an old cardiac isograft. Furthermore, when transferring senescent cells into the skeletal muscle of immunocompromised NOD SCID gamma mice, increased numbers of senescent cells and augmented SASP-marker expression including IL‐1α, IL‐1β, IL‐6 and TNF‐α had been detected (38).
Following organ transplantation, significant numbers of passenger leukocytes deriving from the transplanted organ have been shown to disseminate into the recipient tissue (39–42), supporting the concept that senescence may be transferred in organ transplantation (Figure 1).
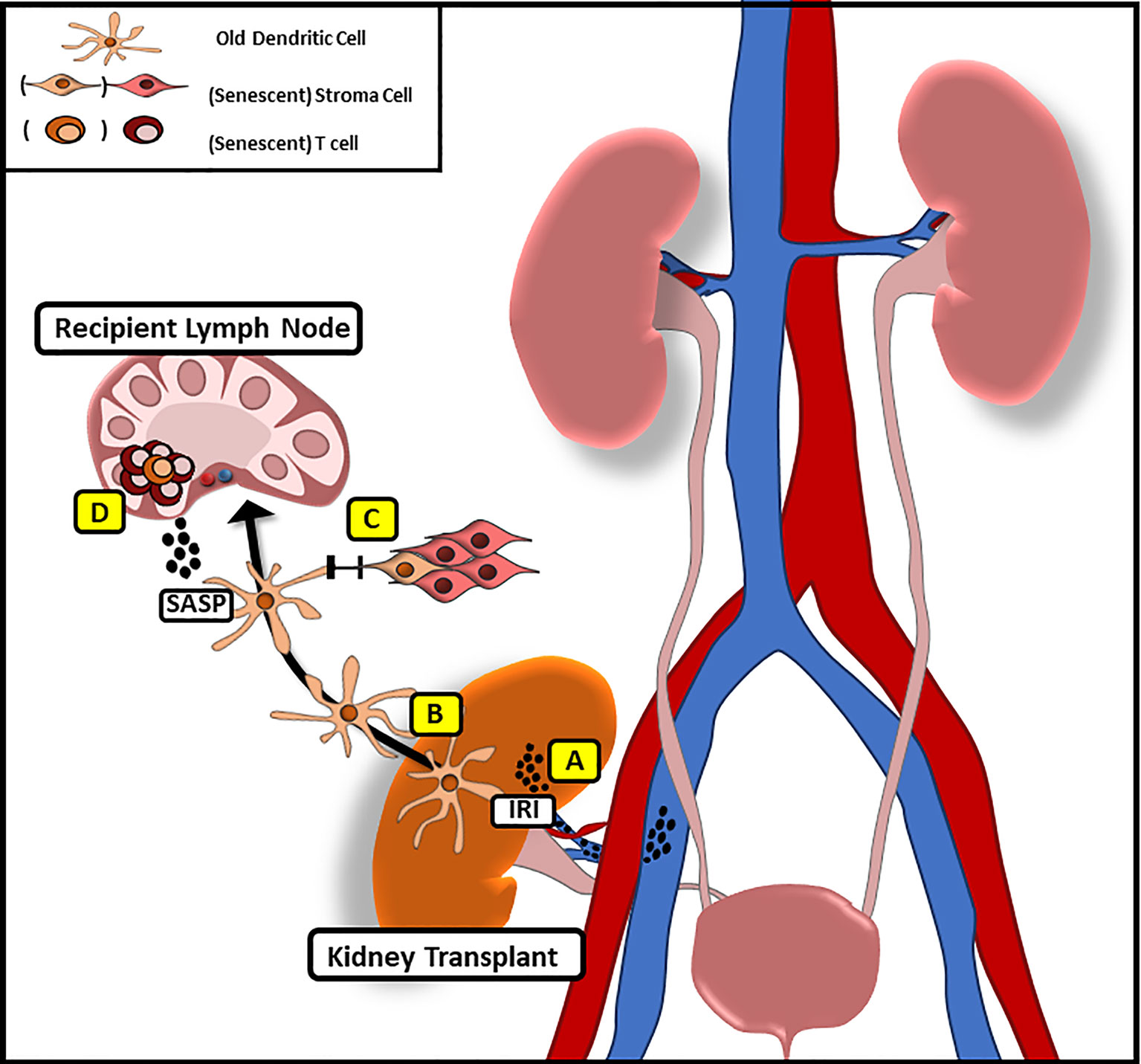
Figure 1 Potential Mechanism of Transferring Senescence Following Solid Organ Transplantation. (A) Following IRI pro-inflammatory factors with similarities to SASP are released that may promote systemic senescence in the recipient. (B) Donor derived, old dendritic cells migrate to recipient lymph nodes following implantation to initiate alloimmune responses through direct antigen presentation. (C) Via gap junction mediated cell–cell contact old DC may promote senescence in recipient stroma cells (D) while inducing a senescent phenotype in recipient T cells through the release of SASP-factors.
Ischemia Reperfusion Injury as a Driver of SASP Promoting Senescence
Ischemia reperfusion injury (IRI) displays an inevitable feature of organ transplantation promoting a sterile inflammation linked to the release of various pro-inflammatory cytokines coinciding with the production of SASP by senescent cells. It appears thus possible that IRI may aid to the promotion of senescence in transplant recipients.
The rapid increase in oxygen demand within the ischemic tissue subsequent to organ reperfusion induces oxidative stress, mitochondrial damage and electrolyte imbalance associated with local inflammation including the release of ROS (43), pro-inflammatory cytokines, in particular TNF-α, IL-1, IL-6 and IL-8 (44, 45) in addition to various proteases (46). Notably, IL-1α expression has been shown to induce an inflammasome mediated SASP activation with the secretion of IL-6 and IL-8 that reinforce senescent growth arrest (31, 47). It has also been demonstrated that ROS induce senescence by promoting mitochondrial dysfunction directly through damaging mitochondrial DNA. Alternatively, ROS may also facilitate senescence in synergy with modifications of the telomerase reverse transcriptase enzyme in addition to the activation of p53 and Ras pathways (48). ROS also inhibit autophagy via p53 activation and the induction of micro-RNAs, effects that further amplify mitochondrial dysfunction (49, 50). Reconciling these cellular effects of IRI on senescence induction, a recent study has confirmed that IRI induces senescence in both cardiomyocytes and interstitial cell populations of murine hearts both within the infarct and in the peri-infarct region of the left ventricular myocardium (51).
Can Senolytic Drugs Inhibit the Spread of Senescence?
Senolytics are a class of drugs that selectively clear senescent cells through inhibiting their SCAPs, thus driving them into apoptosis. According to their targeted SCAP, first generation senolytics can be subdivided in groups including, i), BCL-2 inhibitors such as navitoclax that inhibit the pro-survival pathway BCL-2/BCL-xL, ii), PI3AK/AKT inhibitors including Dasitinib and Quercetin (D & Q) that constrain the synergistic effects of PI3K and Akt inactivating the apoptosis mediating proteins Bad, caspase-9 and FKHRL1 and iii), FOXO regulators such as Foxo4-DRI interfering with the inhibition of p53-mediated apoptosis (52). In addition, cardiac glycosides have been shown to exert senolytics activity through targeting the Na+/K+ Atpase (53, 54) while most recently a BET family protein degrader targeting the non-homologous end joining and autophagy has been delineated as a promising novel senolytic drug (55) (Table 1).
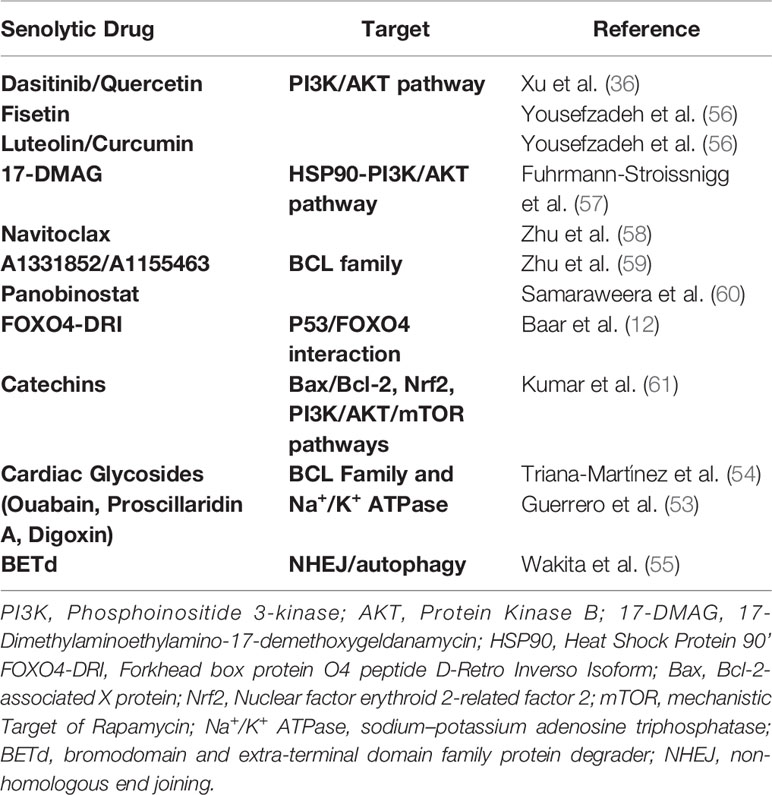
Table 1 Reported senolytic drugs.
Experimentally, senolytics have been shown to ameliorate a broad range of age-associated pathologies including diabetes (62), cardiovascular disease (11, 63), Alzheimer disease (64), osteoporosis (8) and cancer (55). Moreover, clinical studies focusing on idiopathic fibrosis, complications of advanced diabetes, osteoarthritis and Alzheimer have been initiated (65). Of note, senolytic drugs such as navitoclax that only target a single SCAP are more likely to exert substantial off-target apoptotic effects on non-senescent cell types including platelets and immune cells while eliminating only a restricted range of senescent cells (58). Thus, efforts have been made to develop novel senolytic drugs that act on multiple SCAPs, increasing the specificity for senescent cells while reducing off-target effects.
Routes of application may play an additional role: site specific delivery of quercetin improved pancreas islet transplant outcome with enhanced glycemic control by delaying cellular senescence in a mouse model of diabetes (66). Moreover, combinatorial treatment with D & Q has been shown to deplete senescent cells in old murine donor organs, thus decreasing ischemia reperfusion derived release of SASP factors which translated into dampened allo-immune responses, prolonging transplant survival (7). The clearance of senescent cells in donor organs may not only exert anti-inflammatory effects but also restrain a potential transfer of senescence underscoring the therapeutic potential of these drugs in organ transplantation. As a proof of concept, administration of D & Q to mice that had received intraperitoneal injections of senescent cells prevented the spread of senescence and resulting physical dysfunction (36). Senolytic treatment of transplant donors may decrease the pool of senescent cells while a subsequent treatment of the recipient may ensure sustainable clearance of senescent cells, preventing a spread of senescence following transplantation. In addition, senomorphic drugs such as ATM kinase inhibitors (67) and Janus kinase inhibitors (68) that attenuate SASP may, in turn, restrain senescence induction through systemic SASP factors released upon ischemia reperfusion injury when transplanting older organs (69). Of relevance, not all senescent cells may promote age-related complications as recent reports have delineated a physiological hemostatic role of a distinct senescent endothelial cell population in liver sinusoids (70).
Envisioning Rejuvenation When Transplanting Young Organs Into Old Recipients
In contrast to the potential of transferring senescence to recipients, it may also be possible that a young donor organ may exert rejuvenating effects when transplanted into an old recipient. We wish to point out that this concept is theoretical at this point, nevertheless worthwhile to speculate on.
Experimental data have shown that young parabiotic animals have the capacity to rejuvenate brain, heart and muscle function of old mice (71–73). Moreover, transferring plasma derived from young into old mice increased neuronal plasticity of the hippocampus and improved cognitive functions providing support for the concept of rejuvenation (74). However, subsequent studies aiming to delineate soluble factors within the blood that may mediate rejuvenation have not yielded clear results (75–77).
Tissue derived factors may be particularly relevant for exerting rejuvenation. Extracellular vesicles (EVs), for instance, containing RNAs, proteins, and lipid components that may be released by transplanted tissue stem cells, have been shown to promote stem cell plasticity and tissue regeneration (78–82). Notably, EVs derived from young mesenchymal stroma cells (MSCs) rejuvenated old endothelial progenitor cells (EPCs) in vitro, while EVs derived from old MSCs lacked this capacity. Furthermore, miRNA-126-loaded EVs have been able to rejuvenate senescent EPCs in vitro (83).
At least in theory, adult stem cells that have been detected in multiple recipient tissues including muscle, liver, myocardium and endothelium following hematopoietic stem cell transplantation (84–86) may support organ function through transdifferentiating into dysfunctional recipient tissue. However, there is no evidence at this time that donor derived MSCs integrated into recipient tissue following solid organ transplantation.
Conclusion
The transplantation of older donor organs is associated with more frequent acute rejection rates and compromised outcomes calling for age-specific treatment approaches that may improve the quality of older organs while reducing immunogenicity. At the same time transplanting an organ from an old donor may pose a risk for young recipients. The clinical relevance and significance of this concept appears obvious and demands therefore a thorough evaluation. The potential of rejuvenation when transplanting young donor organs, in turn, are of theoretical consideration and will also demand a detailed analysis. Critical will therefore be studies that probe either accelerated aging or rejuvenation in relevant pre-clinical transplant models utilizing immunosuppression.
Optimizing the use of available organs for transplant including those from older donors continues to be of critical importance to close the gap between demand and supply in organ transplantation. Utilizing senolytic drugs that selectively deplete senescent cells may constitute a potential approach to improve the outcomes of older organs while restricting the spread of senescence.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Messina M, Diena D, Dellepiane S, Guzzo G, Lo Sardo L, Fop F, et al. Long-Term Outcomes and Discard Rate of Kidneys by Decade of Extended Criteria Donor Age. Clin J Am Soc Nephrol (2017) 12:323–31. doi: 10.2215/CJN.06550616
2. Saidi RF, Hejazii Kenari SK. Challenges of Organ Shortage for Transplantation: Solutions and Opportunities. Int J Organ Transplant Med (2014) 5:87–96.
3. Moffatt E, Collins KA, Jares L. Improving the Supply and Quality of Deceased-Donor Organs for Transplantation. N Engl J Med (2018) 379:692–3. doi: 10.1056/NEJMc1808003
4. Tullius SG, Milford E. Kidney Allocation and the Aging Immune Response. N Engl J Med (2011) 364:1369–70. doi: 10.1056/NEJMc1103007
5. Tullius SG, Tran H, Guleria I, Malek SK, Tilney NL, Milford E. The Combination of Donor and Recipient Age is Critical in Determining Host Immunoresponsiveness and Renal Transplant Outcome. Ann Surg (2010) 252:662–74. doi: 10.1097/SLA.0b013e3181f65c7d
6. Dayoub JC, Cortese F, Anžič A, Grum T, de Magalhães JP. The Effects of Donor Age on Organ Transplants: A Review and Implications for Aging Research. Exp Gerontol (2018) 110:230–40. doi: 10.1016/j.exger.2018.06.019
7. Iske J, Seyda M, Heinbokel T, Maenosono R, Minami K, Nian Y, et al. Senolytics Prevent mt-DNA-Induced Inflammation and Promote the Survival of Aged Organs Following Transplantation. Nat Commun (2020) 11:4289. doi: 10.1038/s41467-020-18039-x
8. Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, et al. Targeting Cellular Senescence Prevents Age-Related Bone Loss in Mice. Nat Med (2017) 23:1072–9. doi: 10.1038/nm.4385
9. Moncsek A, Al-Suraih MS, Trussoni CE, O’Hara SP, Splinter PL, Zuber C, et al. Targeting Senescent Cholangiocytes and Activated Fibroblasts With B-cell Lymphoma-Extra Large Inhibitors Ameliorates Fibrosis in Multidrug Resistance 2 Gene Knockout (Mdr2(-/-)) Mice. Hepatology (2018) 67:247–59. doi: 10.1002/hep.29464
10. Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, et al. Targeting Senescent Cells Enhances Adipogenesis and Metabolic Function in Old Age. Elife (2015) 4:e12997. doi: 10.7554/eLife.12997
11. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic Senolytic Treatment Alleviates Established Vasomotor Dysfunction in Aged or Atherosclerotic Mice. Aging Cell (2016) 15:973–7. doi: 10.1111/acel.12458
12. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell (2017) 169:132–47.e116. doi: 10.1016/j.cell.2017.02.031
13. Soto-Gamez A, Quax WJ, Demaria M. Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions. J Mol Biol (2019) 431:2629–43. doi: 10.1016/j.jmb.2019.05.036
14. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally Occurring p16(Ink4a)-positive Cells Shorten Healthy Lifespan. Nature (2016) 530:184–9. doi: 10.1038/nature16932
15. Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, et al. Ink4a/Arf Expression is a Biomarker of Aging. J Clin Invest (2004) 114:1299–307. doi: 10.1172/JCI22475
16. Yamakoshi K, Takahashi A, Hirota F, Nakayama R, Ishimaru N, Kubo Y, et al. Real-Time In Vivo Imaging of p16Ink4a Reveals Cross Talk With P53. J Cell Biol (2009) 186:393–407. doi: 10.1083/jcb.200904105
17. Sousa-Victor P, Gutarra S, García-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, et al. Geriatric Muscle Stem Cells Switch Reversible Quiescence Into Senescence. Nature (2014) 506:316–21. doi: 10.1038/nature13013
18. Stenderup K, Justesen J, Clausen C, Kassem M. Aging is Associated With Decreased Maximal Life Span and Accelerated Senescence of Bone Marrow Stromal Cells. Bone (2003) 33:919–26. doi: 10.1016/j.bone.2003.07.005
19. Dong Y, Lian X, Xu Y, Hu H, Chang C, Zhang H, et al. Hematopoietic Stem/Progenitor Cell Senescence is Associated With Altered Expression Profiles of Cellular Memory-Involved Gene. Biosci Rep (2018) 38. doi: 10.1042/BSR20171589
20. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular Senescence: Defining a Path Forward. Cell (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
21. Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine (2017) 21:7–13. doi: 10.1016/j.ebiom.2017.03.020
22. Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. Senescence-Associated Beta-Galactosidase is Lysosomal Beta-Galactosidase. Aging Cell (2006) 5:187–95. doi: 10.1111/j.1474-9726.2006.00199.x
23. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin In Vivo. Proc Natl Acad Sci USA (1995) 92:9363–7. doi: 10.1073/pnas.92.20.9363
24. Imai Y, Takahashi A, Hanyu A, Hori S, Sato S, Naka K, et al. Crosstalk Between the Rb Pathway and AKT Signaling Forms a Quiescence-Senescence Switch. Cell Rep (2014) 7:194–207. doi: 10.1016/j.celrep.2014.03.006
25. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PloS Biol (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
26. Kirkland JL, Tchkonia T. Cellular Senescence: A Translational Perspective. EBioMedicine (2017) 21:21–8. doi: 10.1016/j.ebiom.2017.04.013
27. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-Induced Senescence is a DNA Damage Response Triggered by DNA Hyper-Replication. Nature (2006) 444:638–42. doi: 10.1038/nature05327
28. Rayess H, Wang MB, Srivatsan ES. Cellular Senescence and Tumor Suppressor Gene P16. Int J Cancer (2012) 130:1715–25. doi: 10.1002/ijc.27316
29. Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, et al. Telomeres are Favoured Targets of a Persistent DNA Damage Response in Ageing and Stress-Induced Senescence. Nat Commun (2012) 3:708. doi: 10.1038/ncomms1708
30. Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. A Senescent Cell Bystander Effect: Senescence-Induced Senescence. Aging Cell (2012) 11:345–9. doi: 10.1111/j.1474-9726.2012.00795.x
31. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat Cell Biol (2013) 15:978–90. doi: 10.1038/ncb2784
32. Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine Signaling Via the CXCR2 Receptor Reinforces Senescence. Cell (2008) 133:1006–18. doi: 10.1016/j.cell.2008.03.038
33. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell (2008) 133:1019–31. doi: 10.1016/j.cell.2008.03.039
34. Kortlever RM, Higgins PJ, Bernards R. Plasminogen Activator Inhibitor-1 is a Critical Downstream Target of p53 in the Induction of Replicative Senescence. Nat Cell Biol (2006) 8:877–84. doi: 10.1038/ncb1448
35. Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF Induces Senescence and Apoptosis Through Pathways Mediated by the Secreted Protein IGFBP7. Cell (2008) 132:363–74. doi: 10.1016/j.cell.2007.12.032
36. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics Improve Physical Function and Increase Lifespan in Old Age. Nat Med (2018) 24:1246–56. doi: 10.1038/s41591-018-0092-9
37. Xu M, Bradley EW, Weivoda MM, Hwang SM, Pirtskhalava T, Decklever T, et al. Transplanted Senescent Cells Induce an Osteoarthritis-Like Condition in Mice. J Gerontol A Biol Sci Med Sci (2017) 72:780–5. doi: 10.1093/gerona/glw154
38. da Silva PFL, Ogrodnik M, Kucheryavenko O, Glibert J, Miwa S, Cameron K, et al. The Bystander Effect Contributes to the Accumulation of Senescent Cells In Vivo. Aging Cell (2019) 18:e12848. doi: 10.1111/acel.12848
39. Richter N, Raddatz G, Graeter T, Schäfers HJ, Schlitt HJ. Allogeneic Lymphocyte Chimerism After Clinical Lung Transplantation. Transpl Immunol (1995) 3:74–80. doi: 10.1016/0966-3274(95)80010-7
40. Monaco AP. Chimerism in Organ Transplantation: Conflicting Experiments and Clinical Observations. Transplantation (2003) 75:13s–6s. doi: 10.1097/01.TP.0000067945.90241.F4
41. Starzl TE, Demetris AJ, Trucco M, Murase N, Ricordi C, Ildstad S, et al. Cell Migration and Chimerism After Whole-Organ Transplantation: The Basis of Graft Acceptance. Hepatology (1993) 17:1127–52. doi: 10.1002/hep.1840170629
42. Starzl TE, Demetris AJ, Trucco M, Ramos H, Zeevi A, Rudert WA, et al. Systemic Chimerism in Human Female Recipients of Male Livers. Lancet (1992) 340:876–7. doi: 10.1016/0140-6736(92)93286-V
43. Fisher AB, Dodia C, Tan ZT, Ayene I, Eckenhoff RG. Oxygen-Dependent Lipid Peroxidation During Lung Ischemia. J Clin Invest (1991) 88:674–9. doi: 10.1172/JCI115352
44. Krishnadasan B, Naidu BV, Byrne K, Fraga C, Verrier ED, Mulligan MS. The Role of Proinflammatory Cytokines in Lung Ischemia-Reperfusion Injury. J Thorac Cardiovasc Surg (2003) 125:261–72. doi: 10.1067/mtc.2003.16
45. Naidu BV, Krishnadasan B, Farivar AS, Woolley SM, Thomas R, Van Rooijen N, et al. Early Activation of the Alveolar Macrophage is Critical to the Development of Lung Ischemia-Reperfusion Injury. J Thorac Cardiovasc Surg (2003) 126:200–7. doi: 10.1016/S0022-5223(03)00390-8
46. Yano M, Omoto Y, Yamakawa Y, Nakashima Y, Kiriyama M, Saito Y, et al. Increased Matrix Metalloproteinase 9 Activity and mRNA Expression in Lung Ischemia-Reperfusion Injury. J Heart Lung Transpl (2001) 20:679–86. doi: 10.1016/S1053-2498(01)00250-9
47. Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell Surface-Bound IL-1alpha is an Upstream Regulator of the Senescence-Associated IL-6/IL-8 Cytokine Network. Proc Natl Acad Sci USA (2009) 106:17031–6. doi: 10.1073/pnas.0905299106
48. Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, et al. Feedback Between p21 and Reactive Oxygen Production is Necessary for Cell Senescence. Mol Syst Biol (2010) 6:347. doi: 10.1038/msb.2010.5
49. Lawless C, Jurk D, Gillespie CS, Shanley D, Saretzki G, von Zglinicki T, et al. A Stochastic Step Model of Replicative Senescence Explains ROS Production Rate in Ageing Cell Populations. PloS One (2012) 7:e32117. doi: 10.1371/journal.pone.0032117
50. Lauri A, Pompilio G, Capogrossi MC. The Mitochondrial Genome in Aging and Senescence. Ageing Res Rev (2014) 18:1–15. doi: 10.1016/j.arr.2014.07.001
51. Dookun E, Walaszczyk A, Redgrave R, Palmowski P, Tual-Chalot S, Suwana A, et al. Clearance of Senescent Cells During Cardiac Ischemia-Reperfusion Injury Improves Recovery. Aging Cell (2020) 19:e13249. doi: 10.1111/acel.13249
52. Zhu M, Meng P, Ling X, Zhou L. Advancements in Therapeutic Drugs Targeting of Senescence. Ther Adv Chronic Dis (2020) 11:2040622320964125. doi: 10.1177/2040622320964125
53. Guerrero A, Herranz N, Sun B, Wagner V, Gallage S, Guiho R, et al. Cardiac Glycosides are Broad-Spectrum Senolytics. Nat Metab (2019) 1:1074–88. doi: 10.1038/s42255-019-0122-z
54. Triana-Martínez F, Picallos-Rabina P, Da Silva-Álvarez S, Pietrocola F, Llanos S, Rodilla V, et al. Identification and Characterization of Cardiac Glycosides as Senolytic Compounds. Nat Commun (2019) 10:4731. doi: 10.1038/s41467-019-12888-x
55. Wakita M, Takahashi A, Sano O, Loo TM, Imai Y, Narukawa M, et al. A BET Family Protein Degrader Provokes Senolysis by Targeting NHEJ and Autophagy in Senescent Cells. Nat Commun (2020) 11:1935. doi: 10.1038/s41467-020-15719-6
56. Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, et al. Fisetin is a Senotherapeutic That Extends Health and Lifespan. EBioMedicine (2018) 36:18–28. doi: 10.1016/j.ebiom.2018.09.015
57. Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, et al. Identification of HSP90 Inhibitors as a Novel Class of Senolytics. Nat Commun (2017) 8:422. doi: 10.1038/s41467-017-00314-z
58. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, et al. Identification of a Novel Senolytic Agent, Navitoclax, Targeting the Bcl-2 Family of Anti-Apoptotic Factors. Aging Cell (2016) 15:428–35. doi: 10.1111/acel.12445
59. Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, et al. New Agents That Target Senescent Cells: The Flavone, Fisetin, and the BCL-X(L) Inhibitors, A1331852 and A1155463. Aging (Albany NY) (2017) 9:955–63. doi: 10.18632/aging.101202
60. Samaraweera L, Adomako A, Rodriguez-Gabin A, McDaid HM. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci Rep (2017) 7:1900. doi: 10.1038/s41598-017-01964-1
61. Kumar R, Sharma A, Kumari A, Gulati A, Padwad Y, Sharma R. Epigallocatechin Gallate Suppresses Premature Senescence of Preadipocytes by Inhibition of PI3K/Akt/mTOR Pathway and Induces Senescent Cell Death by Regulation of Bax/Bcl-2 Pathway. Biogerontology (2019) 20:171–89. doi: 10.1007/s10522-018-9785-1
62. Aguayo-Mazzucato C, Andle J, Lee TB Jr, Midha A, Talemal L, Chipashvili V, et al. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab (2019) 30:129–42.e124. doi: 10.1016/j.cmet.2019.05.006
63. Lewis-McDougall FC, Ruchaya PJ, Domenjo-Vila E, Shin Teoh T, Prata L, Cottle BJ, et al. Aged-Senescent Cells Contribute to Impaired Heart Regeneration. Aging Cell (2019) 18:e12931. doi: 10.1111/acel.12931
64. Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, et al. Senolytic Therapy Alleviates Aβ-Associated Oligodendrocyte Progenitor Cell Senescence and Cognitive Deficits in an Alzheimer’s Disease Model. Nat Neurosci (2019) 22:719–28. doi: 10.1038/s41593-019-0372-9
65. Kirkland JL, Tchkonia T. Senolytic Drugs: From Discovery to Translation. J Intern Med (2020) 288:518–36. doi: 10.1111/joim.13141
66. Pathak S, Regmi S, Nguyen TT, Gupta B, Gautam M, Yong CS, et al. Polymeric Microsphere-Facilitated Site-Specific Delivery of Quercetin Prevents Senescence of Pancreatic Islets In Vivo and Improves Transplantation Outcomes in Mouse Model of Diabetes. Acta Biomater (2018) 75:287–99. doi: 10.1016/j.actbio.2018.06.006
67. Kang HT, Park JT, Choi K, Kim Y, Choi HJC, Jung CW, et al. Chemical Screening Identifies ATM as a Target for Alleviating Senescence. Nat Chem Biol (2017) 13:616–23. doi: 10.1038/nchembio.2342
68. Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. JAK Inhibition Alleviates the Cellular Senescence-Associated Secretory Phenotype and Frailty in Old Age. Proc Natl Acad Sci USA (2015) 112:E6301–6310. doi: 10.1073/pnas.1515386112
69. Birch J, Gil J. Senescence and the SASP: Many Therapeutic Avenues. Genes Dev (2020) 34:1565–76. doi: 10.1101/gad.343129.120
70. Grosse L, Wagner N, Emelyanov A, Molina C, Lacas-Gervais S, Wagner KD, et al. Defined P16(High) Senescent Cell Types are Indispensable for Mouse Healthspan. Cell Metab (2020) 32:87–99.e86. doi: 10.1016/j.cmet.2020.05.002
71. Baht GS, Silkstone D, Vi L, Nadesan P, Amani Y, Whetstone H, et al. Exposure to a Youthful Circulaton Rejuvenates Bone Repair Through Modulation of β-Catenin. Nat Commun (2015) 6:7131. doi: 10.1038/ncomms8761
72. Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR, et al. Vascular and Neurogenic Rejuvenation of the Aging Mouse Brain by Young Systemic Factors. Science (2014) 344:630–4. doi: 10.1126/science.1251141
73. Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of Aged Progenitor Cells by Exposure to a Young Systemic Environment. Nature (2005) 433:760–4. doi: 10.1038/nature03260
74. Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, et al. Young Blood Reverses Age-Related Impairments in Cognitive Function and Synaptic Plasticity in Mice. Nat Med (2014) 20:659–63. doi: 10.1038/nm.3569
75. Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, et al. Restoring Systemic GDF11 Levels Reverses Age-Related Dysfunction in Mouse Skeletal Muscle. Science (2014) 344:649–52. doi: 10.1126/science.1251152
76. Liu W, Zhou L, Zhou C, Zhang S, Jing J, Xie L, et al. GDF11 Decreases Bone Mass by Stimulating Osteoclastogenesis and Inhibiting Osteoblast Differentiation. Nat Commun (2016) 7:12794. doi: 10.1038/ncomms12794
77. Egerman MA, Cadena SM, Gilbert JA, Meyer A, Nelson HN, Swalley SE, et al. Gdf11 Increases With Age and Inhibits Skeletal Muscle Regeneration. Cell Metab (2015) 22:164–74. doi: 10.1016/j.cmet.2015.05.010
78. Takasugi M. Emerging Roles of Extracellular Vesicles in Cellular Senescence and Aging. Aging Cell (2018) 17. doi: 10.1111/acel.12734
79. Vader P, Breakefield XO, Wood MJ. Extracellular Vesicles: Emerging Targets for Cancer Therapy. Trends Mol Med (2014) 20:385–93. doi: 10.1016/j.molmed.2014.03.002
80. Lonati C, Bassani GA, Brambilla D, Leonardi P, Carlin A, Maggioni M, et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles Improve the Molecular Phenotype of Isolated Rat Lungs During Ischemia/Reperfusion Injury. J Heart Lung Transpl (2019) 38:1306–16. doi: 10.1016/j.healun.2019.08.016
81. Harting MT, Srivastava AK, Zhaorigetu S, Bair H, Prabhakara KS, Toledano Furman NE, et al. Inflammation-Stimulated Mesenchymal Stromal Cell-Derived Extracellular Vesicles Attenuate Inflammation. Stem Cells (2018) 36:79–90. doi: 10.1002/stem.2730
82. McBride JD, Rodriguez-Menocal L, Guzman W, Candanedo A, Garcia-Contreras M, Badiavas EV. Bone Marrow Mesenchymal Stem Cell-Derived CD63(+) Exosomes Transport Wnt3a Exteriorly and Enhance Dermal Fibroblast Proliferation, Migration, and Angiogenesis In Vitro. Stem Cells Dev (2017) 26:1384–98. doi: 10.1089/scd.2017.0087
83. Wang L, Wei J, Da Fonseca Ferreira A, Wang H, Zhang L, Zhang Q, et al. Rejuvenation of Senescent Endothelial Progenitor Cells by Extracellular Vesicles Derived From Mesenchymal Stromal Cells. JACC Basic Transl Sci (2020) 5:1127–41. doi: 10.1016/j.jacbts.2020.08.005
84. Lagasse E, Connors H, Al-Dhalimy M, Reitsma M, Dohse M, Osborne L, et al. Purified Hematopoietic Stem Cells can Differentiate Into Hepatocytes In Vivo. Nat Med (2000) 6:1229–34. doi: 10.1038/81326
85. Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, et al. Bone Marrow Cells Regenerate Infarcted Myocardium. Nature (2001) 410:701–5. doi: 10.1038/35070587
Keywords: immunosenescence and inflammaging, immunosenescence, immune aging, senescent cell, passenger leukocytes, senescent associated secretory phenotype
Citation: Iske J, Matsunaga T, Zhou H and Tullius SG (2021) Donor and Recipient Age-Mismatches: The Potential of Transferring Senescence. Front. Immunol. 12:671479. doi: 10.3389/fimmu.2021.671479
Received: 23 February 2021; Accepted: 08 April 2021;
Published: 28 April 2021.
Edited by:
Martin Johannes Hoogduijn, Erasmus University Rotterdam, NetherlandsReviewed by:
Kotb Abdelmohsen, National Institute on Aging (NIH), United StatesNaoko Ohtani, Osaka City University, Japan
Copyright © 2021 Iske, Matsunaga, Zhou and Tullius. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan G. Tullius, c3R1bGxpdXNAYndoLmhhcnZhcmQuZWR1