 Miaoran Xia
Miaoran Xia Bingbing Wang
Bingbing Wang Zihan Wang
Zihan Wang Xulong Zhang
Xulong Zhang Xi Wang
Xi Wang- 1Department of Immunology, School of Basic Medical Sciences, Capital Medical University, Beijing, China
- 2Advanced Innovation Center for Human Brain Protection, Capital Medical University, Beijing, China
- 3Beijing Key Laboratory for Cancer Invasion and Metastasis Research, Capital Medical University, Beijing, China
- 4Department of Oncology, Capital Medical University, Beijing, China
Natural killer (NK) cells are critical innate lymphocytes that can directly kill target cells without prior immunization. NK cell activation is controlled by the balance of multiple germline-encoded activating and inhibitory receptors. NK cells are a heterogeneous and plastic population displaying a broad spectrum of functional states (resting, activating, memory, repressed, and exhausted). In this review, we present an overview of the epigenetic regulation of NK cell-mediated antitumor immunity, including DNA methylation, histone modification, transcription factor changes, and microRNA expression. NK cell-based immunotherapy has been recognized as a promising strategy to treat cancer. Since epigenetic alterations are reversible and druggable, these studies will help identify new ways to enhance NK cell-mediated antitumor cytotoxicity by targeting intrinsic epigenetic regulators alone or in combination with other strategies.
Introduction
Natural killer (NK) cells are potent effector lymphocytes of the innate immune system. They serve as the first line of defense against infected or transformed cells without prior sensitization. Compared with T and B cells, which recognize targets by their antigen-specific cell surface receptors (TCRs/BCRs), NK cell activation is controlled by the balance between activating and inhibitory signals from multiple germline-encoded receptors. These cells patrol for potential target cells that lack major histocompatibility complex class I (MHC I) or overexpress ligands to activate NK cell receptors (NCRs) (1). NK cells are initially recruited to the tumor microenvironment (TME) during the tumor killing process and then are activated by complex signals arising from multiple ligand-receptor interactions. Activated NK cells release cytotoxic granules containing perforin and granzyme B upon forming an immunological synapse with the target cells (2). Perforin forms pores in the membrane of target cells, thus allowing granzymes to enter the cell and initiate cell death (3, 4). NK cells can also induce cell apoptosis through the engagement of Fas ligands (FasL) or tumor necrosis factor-related apoptosis-inducing ligands (TRAIL) with Fas and TRAIL receptors on tumor cells (5, 6). In a process known as antibody-dependent cell cytotoxicity, NK cells recognize opsonized tumor cells via Fc receptors (CD16) and kill them by releasing cytolytic granules. Lysis leads to an increased release of tumor antigens and further primes adaptive immune responses. In addition to direct cytotoxic activity, NK cells can function as central communicators of innate and adaptive immunity in the TME by secreting multiple chemokines (CCL3, CCL4, CCL5, and XCL1), cytokines (IFN-γ, TGF-β, and IL-10), and growth factors (GM-CSF) (7). In this way, these cells communicate with various immune cells within tumor tissues, including monocytes, granulocytes, dendritic cells, T cells, and stromal cells (8).
NK cells play important roles in cancer immunosurveillance, particularly by eliminating early tumors and metastasis (minimal disease). In 1970s, several groups found non-MHC-restricted antitumor activity of NK cells in mice (9–12). Later, the rapid and potent cytotoxicity of NK cells against target cells was also observed in humans (13). Furthermore, an eleven-year follow-up study found that the impaired NK cell killing capacity in the peripheral blood is correlated with tumor incidence and prognosis (14). Compared with the role of T cells in antitumor immunity and adoptive cellular therapy, NK cells have certain advantages and greater potential “off-the-shelf” utility (7). They are as effective as T cells (15, 16) but less toxic because they cause fewer immune-related adverse events. Mature NK cells are effector cells with a broader reactivity to tumors due to their independent recognition of specific receptors and antigen presentation by MHC molecules. Their lytic responses can be triggered within minutes without clone selection and differentiation (1). The “ready-to-go” state is associated with the unique epigenetic features of NK cells, as shown in the following sections.
NK Cell Plasticity
NK cells are a heterogeneous and plastic population. They are classically defined as CD3-CD56+ cells in humans and divided into two major subsets, CD56dimCD16+ and CD56brightCD16low (17–19). CD56dimCD16+ subsets are highly cytotoxic effector cells that are predominantly found in peripheral blood. CD56brightCD16low subsets are recognized as immature NK cells with immune regulation functions through cytokine secretion. They preferentially reside in secondary lymphoid organs, such as lymph nodes. The surface markers of murine NK cells vary depending on the mouse strain. In C57B/6 and SJL mice, NK cells express NK1.1, NKp46, and CD49b (2). For other strains, such as BALB/c, NK cells express CD49b and NKp46 while possessing allelic variants of NK1.1 (2). Tumor necrosis factor receptor superfamily member CD27 and the integrin CD11b are used to mark NK cell differentiation in mice. The most cytotoxic NK cells are recognized as CD27-CD11b+, regulatory NK cells are CD27+CD11b+, and immature NK cells are CD27+CD11b- (20, 21).
NK cells belong to the family of innate lymphoid cells (ILCs). NK cells and ILC1s are grouped into group I innate lymphoid cells (22). ILC1s reside in tissues and function as cytokine secretors. Conventional NK (cNK) cells and ILCs arise from distinct progenitors (23). However, many surface markers initially described on NK cells, such as CD122, NK1.1, and NKp46, can be expressed on ILC1s (24). The mixed phenotype can be explained by imprinting the effects of the tissue microenvironment and cell activation state. Therefore, at present, the definition of NK cells based on their phenotype is essentially at a steady state (24). The majority of human mature NK cells can be identified as CD3-CD127-CD7+CD56+ (or NKp46+)T-bet+Eomes+ lymphocytes, and mature mouse NK cells can be identified as CD3−CD127−NK1.1+ (or NKp46+)T-bet+Eomes+ lymphocytes. There are no markers that can unambiguously distinguish NK cells and ILC1s in human or mouse tissues during infection or inflammation (25).
The conversion between NK cells and ILC1s in the TME was recently described (26). Transforming growth factor-β (TGF-β) in the TME could drive NK cells (CD49a−CD49b+Eomes+) to convert into intermediate ILC1 (intILC1, CD49a+CD49b+Eomes+) populations and ILC1 (CD49a+CD49b−Eomesint) populations. IntILC1s and ILC1s are less cytotoxic and cannot control local tumor growth and metastasis (27). SMAD4, which is a unique common SMAD, acts as a central mediator that facilitates the canonical TGF-β signaling pathway (28). TGF-β induces salivary gland ILC differentiation by suppressing Eomes through a JNK-dependent, Smad4-independent pathway (29). However, Smad4 deficiency does not affect ILC1 differentiation but surprisingly alters the phenotype of cNK cells. Cortez et al. reported that Smad4-deficient NK cells showed features of ILC1s and lost effector functions to control tumor metastasis. Mechanistically, SMAD4 restrained noncanonical TGF-β signaling mediated by the cytokine receptor TGFβR1 in NK cells (30). A subsequent study by Wang et al. showed that selective deletion of Smad4 in NK cells led to impaired NK cell maturation, NK cell homeostasis, and NK cell immune surveillance against melanoma metastases and cytomegalovirus. These changes were associated with a downregulation of granzyme B (Gzmb), Kit, and Prdm1 in Smad4-deficient NK cells and independent of canonical TCF-β signaling (31).
Of note, it has become increasingly clear that various subsets of tissue-resident NK (trNK) cells exist, which differ from cNK cells in their origin, development, and function (32–34). Unlike circulating and widely distributed cNK cells, trNK cells were found to populate multiple tissue sites, including the liver, lung, skin, uterus, salivary gland, adipose tissue, and kidneys (32). trNK cells are distinct from cNK cells in the expression of surface markers and transcription factors. For example, murine liver trNK (LrNK) cells express relatively low levels of NK cell maturation-associated markers, such as CD11b, CD49b (DX5), and Ly49 receptors (35). The development of LrNK is independent of Eomes, while T-bet, Hobit, PLZF, and AhR are more critical for LrNK cell development than cNK cells (34). trNK cells are actively involved in multiple processes, such as antiviral infection, mediating immune tolerance, and promoting fetal growth (34). The accumulation of LrNK cells in hepatocellular carcinoma patients is correlated with poor prognosis (36), suggesting a potential role in tumor development. More comprehensive studies are needed to investigate the role of trNK in antitumor immunity.
Although historically known as innate lymphoid cells, NK cells can also achieve memory characteristics similar to those of adaptive immune cells, such as antigen specificity, longevity, and enhanced recall responses. Memory NK responses were first reported in mouse models of anti-murine cytomegalovirus (MCMV) infection (37) and delayed hypersensitivity reactions to chemical haptens and viral antigens (38, 39). During secondary MCMV infection, memory NK cells bearing the virus-specific Ly49H receptor can rapidly proliferate, degranulate and produce cytokines by recognizing the MCMV-encoded glycoprotein m157 (37). Memory NK cells have also been described in humans expressing NKG2C in HCMV-seropositive individuals (40). Growing evidence suggests that memory-like NK cell responses may occur in response to a broader range of viral, bacterial, and even eukaryotic pathogens (41). The responses of memory-like NK cells against tumors are poorly understood, and two key questions remain to be answered: (1) whether NK cells can acquire memory properties during the antitumor process and (2) whether memory NK cells from infection models can acquire stronger in vivo killing capacity targeting tumor cells.
Compared with cNK cells that live less than ten days (42, 43), memory NK cells can persist for years in some individuals and are important for controlling CMV throughout life (44, 45). Similar to CD8+ T cells, NK cells also exhibit an “exhausted” phenotype in individuals with malignancies or chronic viral infections. This phenotype is represented by a loss of activating receptors (e.g., NKG2D) and increased expression of checkpoint receptors (e.g., NKG2A, TIGIT, PD-1, TIM-3, LAG-3), which severely impair their antitumor function (46). Compared with the “suppression” state, which is reversible after the withdrawal of inhibitory signaling, the “exhaustion” state is not transient and undergoes stable epigenetic changes (47). Antagonistic antibodies (Abs) (e.g., anti-PD-1, anti-TIGIT, and anti-NKG2A monoclonal Abs) can recover NK cell antitumor capacity (46, 48). However, epigenetic intervention should be considered to reactivate exhausted NK cells intrinsically in future studies.
Epigenetic Regulators Modulating NK Cell-Based Antitumor Immunity
Epigenetic alterations are reversible and heritable changes that do not alter DNA sequences, including DNA methylation, posttranslational modifications of histone proteins, changes in transcription factors, and noncoding RNA expression. Despite the deep understanding of NK cell biology, research on epigenetic regulation of NK cell function is just beginning. In this review, we provide an overview of the epigenetic regulators that modulate NK cell-based antitumor immunity, and the findings will hopefully help to identify novel approaches and potential targets for tumor immunotherapy.
DNA Methylation
DNA methylation is a heritable epigenetic marker that correlates with gene repression. During the terminal differentiation process, NK cells gradually acquire the ability to produce IFN-γ through demethylation and epigenetic remodeling at the IFNG promoter (Figure 1) (49). DNA methylation has been reported to correlate with the gene expression of a variety of NK cell receptors, including killer Ig-like receptors (KIRs) and natural cytotoxic receptors (NCRs). KIRs are polymorphic groups of molecules, and some are expressed while others are silenced in the same cell. Different KIRs can transmit inhibitory or activating signals to NK cells, and effector function is considered to result from the balance of these contributing signals. The expression repertoire of KIRs is critical for NK killing ability. Moderate demethylation of the inhibitory KIR promoter is essential for normal NK recognition and lysis of abnormal cells. Promoter methylation of KIR genes consistently silences KIR expression (50, 51) and chromatin is condensed in early hemopoietic progenitor cells. During NK cell differentiation and maturation, the chromatin structure opens, and KIR genes sequentially become demethylated and transcribed (Figure 1) (52). Excessive demethylation of the inhibitory KIR promoter represses NK cytolytic function and results in tumor escape. Some studies demonstrated that acute exercise could cause promoter demethylation of the activating NK-cell receptor KIR2DS4 (53) and changed DNA methylation in 33 targets (25 genes) (54). Of the targets, 19 showed decreased methylation and 14 showed increased methylation. Whether these changes lead to functional adaptations needs to be elucidated. In addition, DNA methylation is crucial in maintaining the allele-specific expression of the inhibitory receptor NKG2A. CpGs are methylated in NKG2A-negative stages (hemopoietic stem cells, NK progenitors, and NKG2A-negative NK cells) but hypomethylated specifically in various developmental stages of NKG2A-positive NK cells and NK cell lines (Figure 1) (55). Natural killer group 2 member D (NKG2D) is one of the most crucial activating receptors of NK cells for target recognition. The methylation frequency of the NKG2D promoter can be used as a biomarker for detecting hepatitis B virus-associated hepatocellular carcinoma (HCC). NKG2D promoter methylation in HCC patients was higher than that in chronic hepatitis B patients and healthy controls (56).
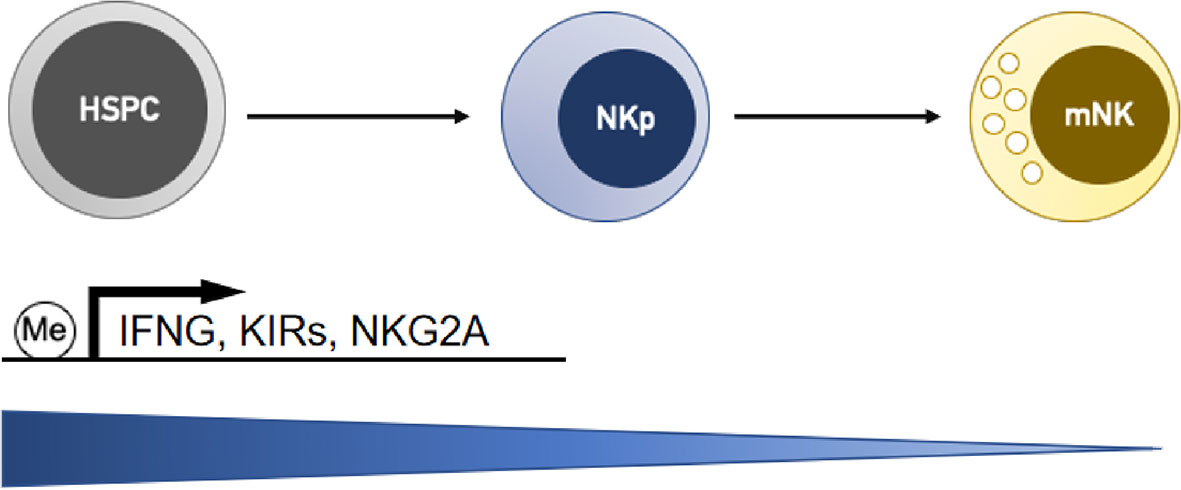
Figure 1 NK cells gradually downregulate DNA methylation levels at the gene promoters of interferon-γ (IFNG) and receptors (KIRs and NKG2A) during the differentiation process, and this activity is correlated with the upregulation of their transcription. HSPC, hemopoietic stem/progenitor cells; NKp, NK cell progenitors; mNK, mature NK cells.
Hypomethylating agents 5-azacytidine (5-aza) and decitabine (Deci) are approved for the treatment of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). However, the direct effect of demethylating treatment on NK cell function remains controversial (Table 1) and should be considered in the application of these drugs. Both 5-aza and Deci can alter the expression of KIRs on NK cells and may thus affect NK reactivity against malignant hematopoietic cells (57–59). Demethylation treatment with 5-aza significantly suppresses the cytolytic activity of the NK-92MI cell line and human polyclonal NK cells, which is related to the overexpression of inhibitory KIRs and impaired granzyme B (GzmB) and perforin (Prf1) release by these cells (57, 58). However, another study reported that systemic treatment with 5-aza leads to an increased proportion of Ki-67+ NK cells expressing multiple KIRs in MDS patients. These proliferating NK cells exhibit increased IFN-γ production and degranulation towards tumor target cells (59). However, Kubler et al. found that low-dose and long-term treatment of humanized NSG mice with 5-aza does not induce common inhibitory KIR expression but instead promotes the differentiation of various NK-cell precursor subsets to enhance the antitumor (pediatric BCP-ALL in vivo) response (60). The different effects could be determined based on the dose, with high doses of the demethylating agents showing cytotoxicity and lower doses mediating DNA hypomethylation. Deci decreases NK cell cytotoxicity at intermediate concentrations and leads to a U-shaped dose-response curve (0-20 μM). In contrast, increased inhibitory KIRs (KIR3DL1, KIR2DL1, KIR2DL2/DL3), decreased NKG2D, and increased NKp44 expression have been induced by Deci treatment in a linear dose-response manner (61). However, another group reported that low-dose Deci (0.2 mg/kg) reduces the antitumor response of NK cells in tumor-bearing mice (75), and Deci has also been shown to increase the cell surface expression of recombinant UL16 binding protein (ULBP) (62) and MHC class I-related molecule B (MICB) (63), the ligands of NKG2D in AML cells, and the NKG2D-dependent sensitivity of these cells to NK-mediated killing in vitro.
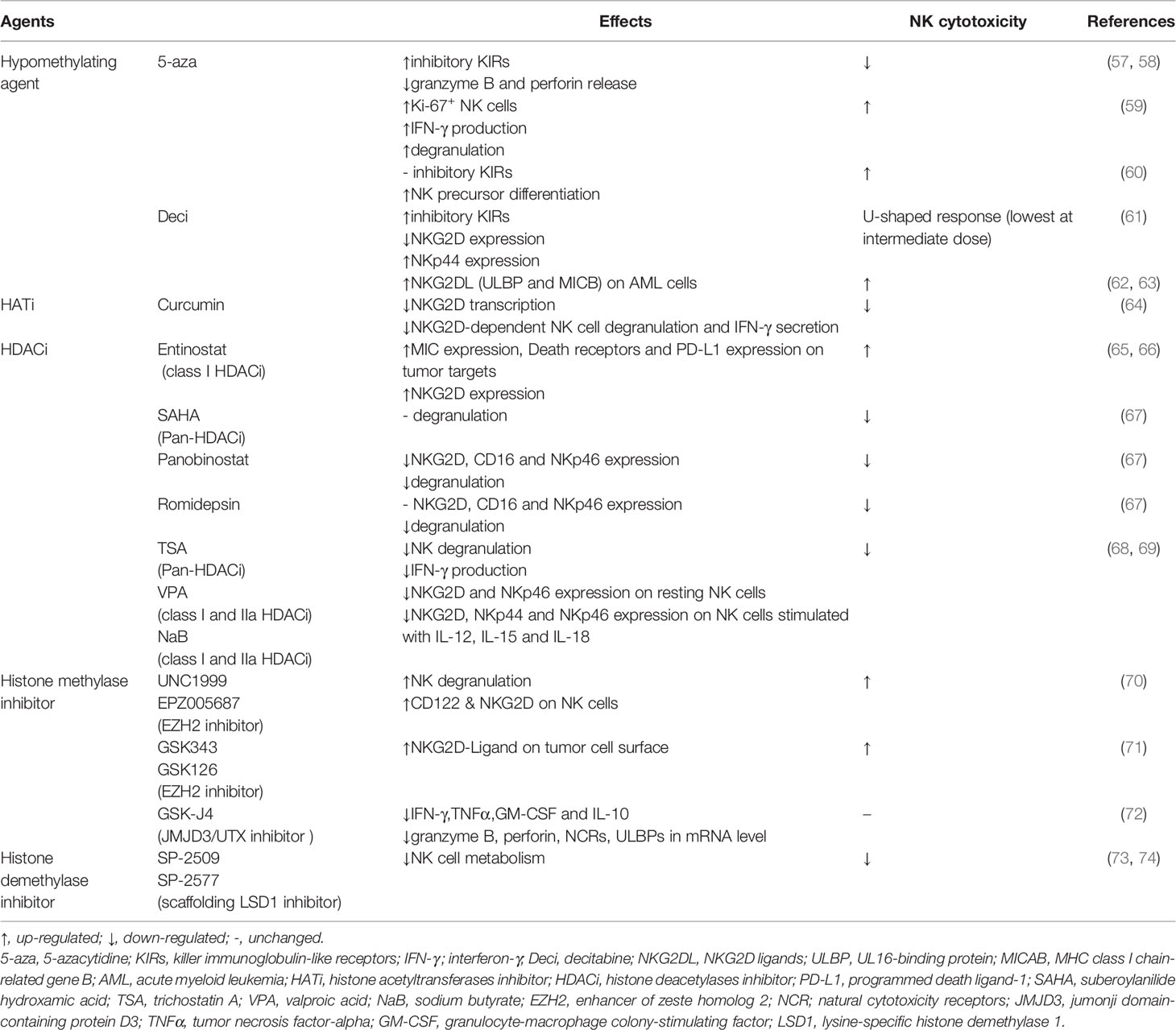
Table 1 Epigenetic drugs targeting DNA methylation and histone modification related to NK antitumor cytotoxicity.
Histone Modification
Histone modifications are associated with the opening or closing state of the chromatin structure, which results in the activation or repression of gene transcription (76). Of particular importance are histone acetylation and methylation. The acetylation of lysine residues on histone 3 (AcH3) and 4 (AcH4) is associated with active transcription (77), while methylation contributes to both active and suppressed states of gene expression. The methylation of histone 3 lysine 9 (H3K9) and H3K27 is inhibitory, whereas the methylation of H3K4, H3K36, and H3K79 is activating (78). The level of histone modification is controlled by the interplay between enzymes: e.g., histone acetyltransferases (HATs) vs. deacetylases (HDACs) (79) and histone methyltransferases vs. demethylases. The dynamic histone modification states determine NK cell activation and effector function in antitumor immunity (80).
Histone Acetylation
Histone acetylation precedes the transcription of many genes (e.g., IFNG and NKG2D) involved in regulating NK cell function (81–83). Chang et al. compared long-range histone hyperacetylation patterns across the Ifng gene region in T cells and NK cells and found that histone acetylation of the Ifng gene depends on stimulation and the transcription factors Stat4 and T-bet in T cells. In contrast, even in resting NK cells, histones along Ifng gene region are already acetylated, and additional proximal domains are hyperacetylated after stimulation of transcription (84). These characteristics may partially explain the quick response of NK cells without prior sensitization. The NKL cell line exhibits high levels of AcH3, AcH4, and H3K4me3 in the NKG2D gene. A significantly high level of AcH3, especially H3K9ac, was observed in the NKG2D gene of NK cells from peripheral blood, while a low level of H3K4me3 was present. Repressive histone modifications (H3K27me3 and H3K9me2) to the NKG2D gene in both NKL and peripheral NK cells were hardly detectable (64).
HAT inhibitor (curcumin) incubation reduced H3K9Ac levels of the NKG2D gene, downregulated NKG2D transcription, and led to a marked reduction in NKG2D-dependent NK cell degranulation and IFN-γ secretion by NKL cells (64). HDAC inhibitors (HDACis) have emerged as novel immunomodulatory drugs and have been reported to affect NK cell cytotoxicity against tumors through both receptor and ligand modulation. The expression of activating ligands for NK cell recognition was increased after HDACi treatment on the cell surfaces of neuroblastoma, melanoma, osteosarcoma, colon, and Merkel cell carcinomas (65, 85). However, different HDAC inhibitors were reported to have varying effects on the NK cell phenotype (Table 1). There are four subclasses of HDACs (HDAC I, II, III, IV). Treatment with a histone deacetylase inhibitor (trichostatin A, TSA) alone was sufficient to induce inhibitory NKG2A receptor expression in mice (55). Entinostat (a class I HDACi) treatment induced NK activation via increased MIC expression in tumor targets as well as enhanced NKG2D expression and ADCC-mediated lysis in primary human NK cells (65, 66). Many HDACis have been reported to negatively regulate the NK antitumor response, including vorinostat (SAHA), panobinostat, romidepsin, TSA, valproic acid (VPA), and sodium butyrate (NaB) (Table 1) (67). They affect NK cell activation through cytokine receptors and activating receptors involved in tumor cell recognition (68, 69). The inhibitory effect on nuclear mobilization of p50 and NK-κB activation caused by HDAC inhibitors also resulted in impaired NK cell activation (82).
Histone Methylation
Li et al. screened 4 upregulated (KMT2C, KDM6B, UTY, and JARID2) and 4 downregulated (ASH1L, PRMT2, KDM2B, and KDM4B) histone methyltransferases/demethylases upon activation of human NK cells by gene expression profiling, which was further confirmed by qPCR and western blot in NK92MI cells. These enzymes were mainly associated with H3K4 methylation and H3K27 methylation, and they only affected limited gene loci instead of the global modification state. Bivalent marks with both H3K4me3 and H3K27me3 determined the “poised” chromatin state of many genes associated with NK activation. This state helps the rapid shift in expression above the baseline during the target recognition process. Treatment with UNC1999 could induce NK cell degranulation. In addition, the expression of IFN-γ and TNF-α is increased after treatment with OG-L002 and MM102 (80).
Histone lysine N-methyltransferase Ezh2 (enhancer of zeste homolog 2) contributes to histone repressive marks H3K27me3. Loss of Ezh2 or inhibition of its enzymatic activity with small molecules in both mouse and human hematopoietic stem and progenitor cells enhanced NK cell expansion and cytotoxicity against tumor cells through upregulation of CD122 and NKG2D (Table 1) (70). The Ezh2 inhibitor EPZ011989 and combination treatment with cisplatin in HT1376 (bladder cancer cell line) xenografts led to increased expression of CD86, MIP-1α, and CD3d at the transcript level as well as CD56 and NCR1 at the protein level, indicating an active state of NK cells (86). Ezh2 was also found to be a transcriptional repressor of NKG2D ligands. Ezh2 inhibition enhanced NK cell eradication of tumor cells in hepatocellular carcinoma (Table 1) (71). Jumonji-type histone H3K27 demethylases (e.g., JMJD3/UTX) have been identified as key regulators of cytokine production in human NK cell subsets. The JMJD3/UTX inhibitor GSK-J4 increased global levels of the repressive H3K27me3 mark around the transcription starting site (TSS) of effector cytokine genes. However, NK cell cytotoxic killing activity against tumor cells was unaffected after treatment with GSK-J4 (Table 1) (72).
Methylation of H3K4 is an activating mark for gene transcription. An H3K4me1-marked latent enhancer at the Ifng locus was essential for NK memory in a systemic endotoxemia model (87). The H3K4me3 demethylase Kdm5a associates with p50 and binds to the suppressor of cytokine signaling 1 (Socs1) promoter region in resting NK cells, thus leading to a repressive chromatin configuration. Kdm5a deficiency impairs the activation of NK cells, leading to decreased IFN-γ production and impaired phosphorylation and nuclear localization of STAT4 (88). LSD1 is a histone demethylase of H3K4me1/2 and H3K9me1/2. Catalytic LSD1 inhibitors blocking demethylase activity are unaffected on NK cells, while scaffolding inhibitors disrupting epigenetic complexes, including LSD1, impair NK cell metabolism and cytotoxicity through depletion of glutathione (Table 1) (73, 74).
Transcription Factors
Transcription factors (TFs) are specific kinds of proteins that can activate or suppress the transcriptional activity of target DNA sequences by specifically recognizing and binding them. Many TFs have been shown to highly modulate the function of human or murine NK cells and affect the eradication of tumor cells (Figure 2A) (89–91). Kwon HJ et al. reported that silencing the expression of the NF-κB p65 subunit caused a significant reduction in the mRNA levels of IFN-γ, TNF-α, MIP-1α/β, GramB, and IκBα induced by NKG2D and 2B4 coengagement (92). The T-box transcription factors T-bet and Eomes are both critical in driving the differentiation and function of NK cells (93). T-bet deficiency impairs the longevity and function of NK cells in inhibiting cancer metastasis, which further precludes the initiation of a potent adaptive response to tumors in mice. Adoptive transfer of wild-type activated NK cells (but not T-bet-/- NK cells) protects T-bet-/- animals after melanoma challenge (94). Aiolos is required for the maturation of CD11b+CD27- NK cells. However, NK cells lacking Aiolos are strongly hyperreactive to various NK cell-mediated tumor models but impaired in controlling viral infection (95). Foxo1 was identified as a negative intrinsic regulator of NK cell homing, late-stage maturation, and effector functions, and it can directly target IFN-γ expression; moreover, Foxo1 deficiency increases the NK cell killing capacity of tumor cells ex vivo and the antimetastatic activity in vivo. Foxo1 suppresses Tbx21 expression through direct binding to its promoter in human NK cells and through association with the promoter via recruitment by Sp1 in murine NK cells (96). Phosphorylation-mediated inactivation of Foxo1 facilitates the activating receptor CD226 regulation of NK cell antitumor responses (97). Krupple-like factor 2 (KLF2) is a key TF responsible for expanding transferred NK cells and prolonging their functionality within the tumor. KLF2 imprints a homeostatic pattern on mature NK cells that allows them to migrate to IL-15-rich microenvironments (98). Cells adapt to hypoxia in solid tumors by upregulating HIF-1α. Inhibition of HIF-1α unleashes the antitumor activity of human tumor-infiltrating NK cells associated with high expression of IFN-γ in an IL-18-dependent manner (99).
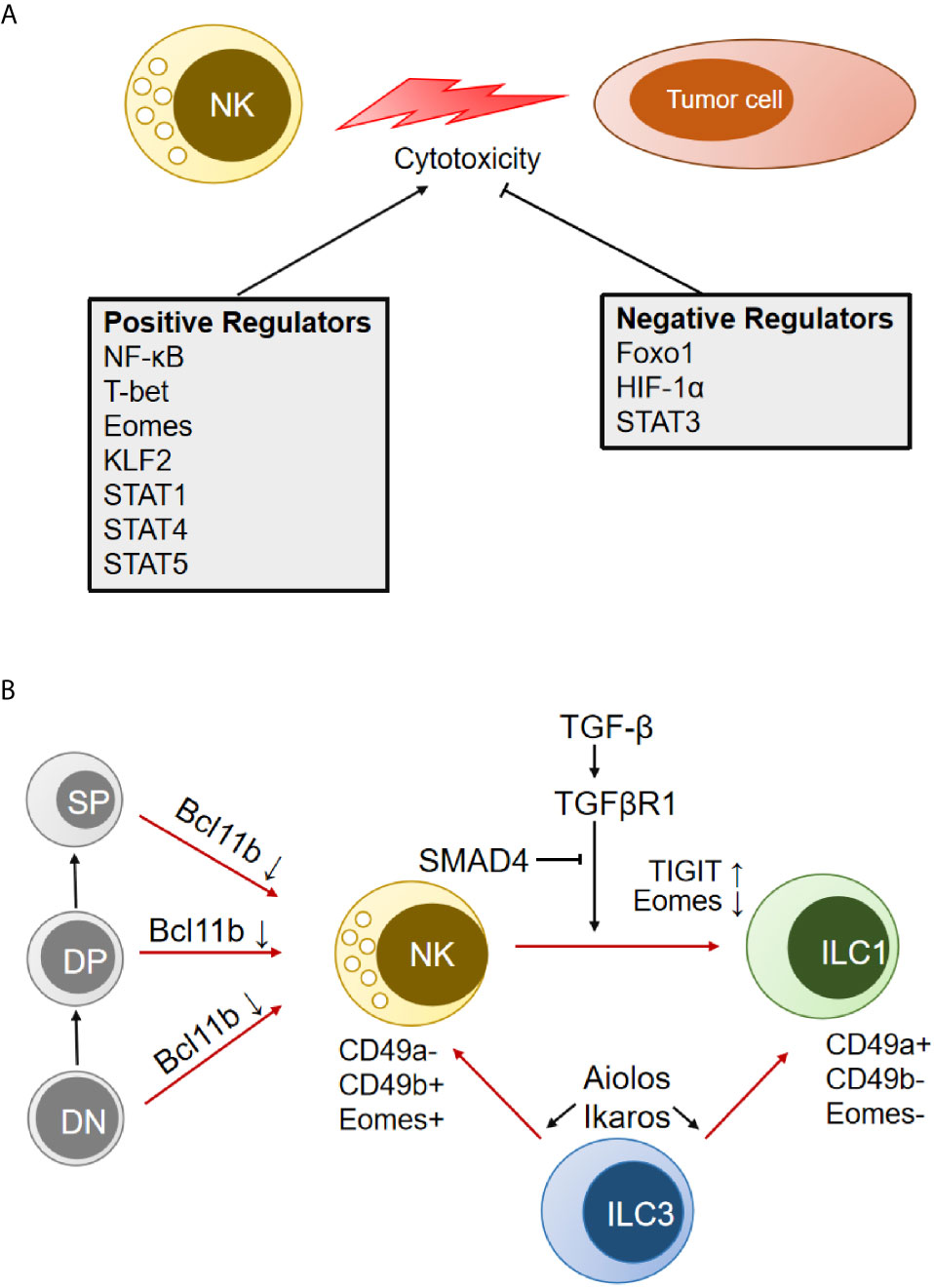
Figure 2 Transcription factors (TFs) that modulate NK cell cytotoxicity and transdifferentiation. (A) TFs that positively and negatively regulate NK antitumor cytotoxicity are indicated separately. (B) Schematic representation of multiple TFs involved in the transdifferentiation between NK cells and other immune cells. DN, double-negative cells in the thymus; DP, double-positive cells in the thymus; SP, single-positive cells in the thymus; ILC, innate lymphoid cells.
It has been reported that the signal transducer and activator of transcription (STAT) family (STAT1, STAT3, STAT4, STAT5) positively or negatively regulates NK cell activity (Figure 2A) (100). STAT1 dysfunction in humans and genetic deletion in mice leads to impaired NK cell antitumor cytotoxicity (101). Mutation of the S727 phosphorylation site of STAT1 (Stat1-S727A) increases the expression of perforin and granzyme B and enhances NK cell cytotoxicity in various tumor models, including for melanoma, leukemia, and metastasizing breast cancer. Inhibition of upstream cyclin-dependent kinase 8 (CDK8) may be a therapeutic strategy for stimulating NK cell-mediated tumor surveillance (102). Full-length STAT1α is efficient for NK cell maturation and tumor control in mice, while NK cells from the C-terminally truncated STAT1β isoform show impaired maturation and effector functions (103). STAT-3 regulates all aspects of NK biology, including almost all of the pathways for target cell killing and the reciprocal regulatory interaction between NK cells and other components of the immune system, which has been presented in detail by Nicholas A. Cacalono (104). STAT4 signaling in NK cells could be activated by IL-2 (105) and IL-12 (106), which specifically bind to the human perforin gene and induce activation of NK antitumor activity. Eckelhart et al. found that STAT5fl/fl Ncr1-iCreTag mice show a marked reduction in NK cells in the spleen and lymph nodes and severely impaired NK-dependent antitumor activity (107). There are two homologs of STAT5, STAT5A and STAT5B, which can form homos, heterodimers, and tetramers. It was reported that the loss of STAT5B (but not STAT5A) reduces NK cell numbers and cytotoxicity (108). However, recent studies have shown that STAT5A deficiency is sufficient to compromise NK cell homeostasis, responsiveness, and tumoricidal function (109, 110).
In addition, several TFs have been shown to control the transdifferentiation between NK cells and other immune cells (T cells, ILCs) (Figure 2B). Downregulation of Eomes by TGF-β signaling in the TME could induce the conversion of mouse NK cells to an NK-ILC1 intermediate cell type (intILC1s) and, finally, to ILC1s, which are less cytotoxic and cannot control local tumor growth and metastasis (27). Cortez et al. found that SMAD4 is a negative regulator of NK-ILC1s conversion in a noncanonical TGF-β signaling pathway (30). SMAD4 is the only common SMAD in TGF-β signaling that usually impedes immune cell activation in the tumor microenvironment. Selective deletion of Smad4 in NK cells impairs tumor cell rejection, promotes tumor cell metastases, and impedes NK cell homeostasis and maturation. GzmB was identified as a direct target of a transcriptional complex formed by SMAD4 and JUNB (31). It was also found that ILC3 could transdifferentiate into IFN-γ-producing ILC1 and NK cells by IL-1β plus IL-12 stimulation, which is associated with the upregulation of T-bet and Aiolos. Degradation of Aiolos and Ikaros proteins by lenalidomide inhibits ILC1/NK cell transdifferentiation and ILC1/NK cell function (111). Bcl11b, a zinc finger transcription factor, is essential for the maintenance of T-cell identity. Upon Bcl11b deletion, immature thymic T cells could convert to NK cells and acquire NK cell properties (112, 113). The converted NK cells were called T-to-natural killer (ITNK) cells and exhibited enhanced antitumor activity. They are considered an attractive cell source for cancer immunotherapy (114).
miRNA
MicroRNAs (miRNAs) are small single-stranded noncoding RNAs that target mRNA and promote degradation by binding to the 3’ untranslated region (UTR) (115). miRNAs can modulate gene expression involved in the development, maturation, and effector functions of NK cells (Figure 3) (116).
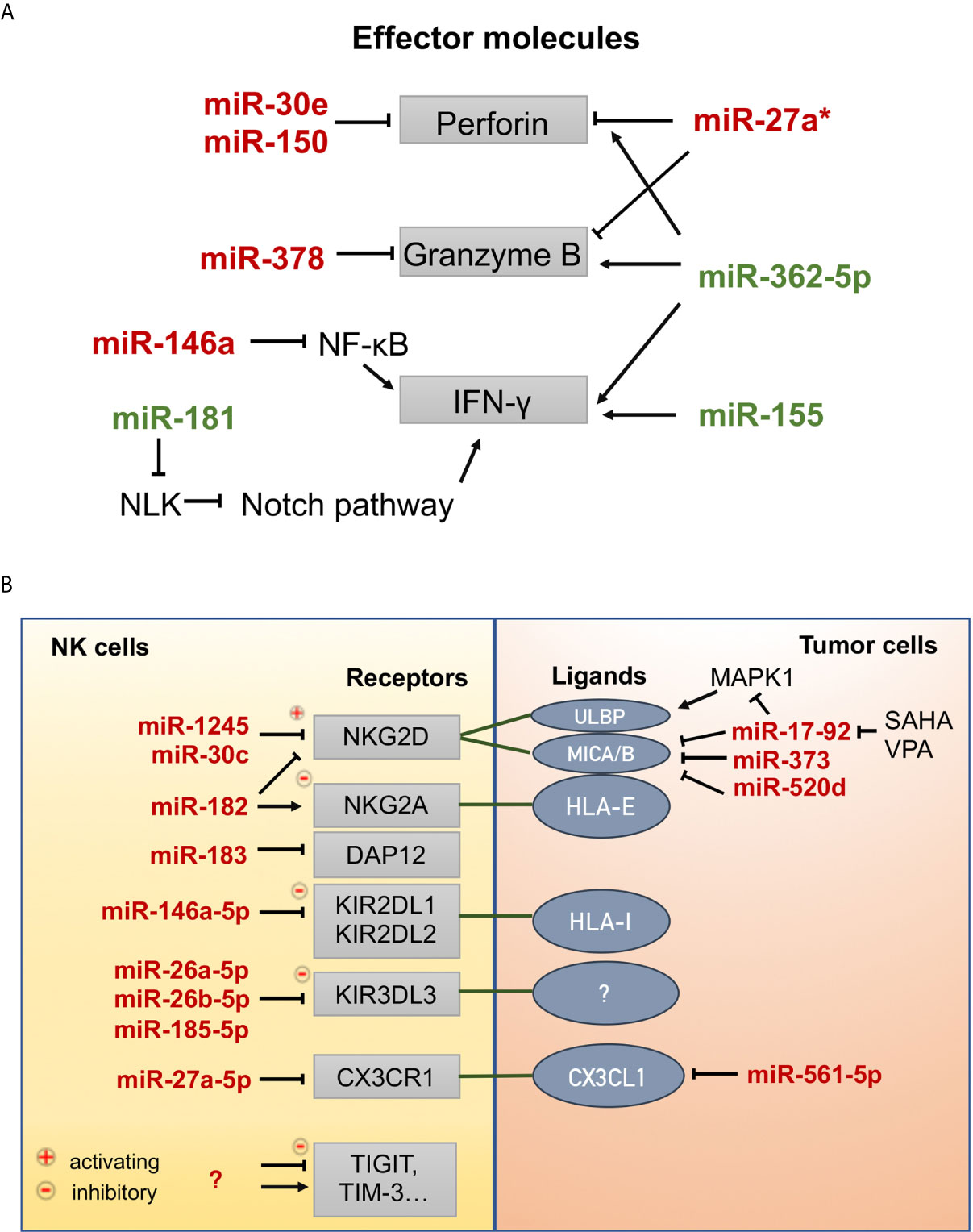
Figure 3 MicroRNAs involved in the effector functions of NK cells. (A) MicroRNAs that positively (green) or negatively (red) regulate the expression of effector molecules (perforin, granzyme B, and interferon-γ). NLK: nemo-like kinase, Notch signaling inhibitor. (B) MicroRNAs that regulate the expression of receptors on NK cells and ligands on tumor cells. CX3CR1, C-X3-C motif chemokine receptor 1; CX3CL1, C-X3-C motif chemokine ligand 1; DAP12, DNAX-activating protein 12 kDa, an exclusive signaling adaptor of many NK cell receptors; HLA-I, human leukocyte antigen, class I; HLA-E, human leukocyte antigen, Class I, E; KIR2DL1, killer cell immunoglobulin-like receptor, two Ig domains and long cytoplasmic tail 1; KIR2DL2, killer cell immunoglobulin-like receptor, two Ig domains and long cytoplasmic tail 2; KIR3DL3, killer cell immunoglobulin-like receptor, three Ig domains and long cytoplasmic tail 3; MICA/B, MHC class I-related molecule A/B; NKG2A, natural-killer group 2 member A; NKG2D, natural-killer group 2 member D; SAHA, suberoylanilide hydroxamic acid (vorinostat), histone deacetylase inhibitor; TIGIT, T cell immunoreceptor with Ig and ITIM domains; TIM-3, T cell immunoglobulin and mucin domain-containing protein 3; ULBP, UL16 binding protein; VPA, valproic acid, histone deacetylase inhibitor.
Prf1 and GzmB are the main effector molecules of NK cells. Prf1 could be targeted by miR-30e (117) and miR-150 (118), GzmB could be targeted by miR-378 (117), while both could be targeted directly by miR-27a* (119) in resting and activated states and indirectly by miR-27a-5p (120) by downregulating the expression of C-X3-C motif chemokine receptor 1 (CX3CR1) under TGF-β1 signaling. Tumor cells upregulate miR-561-5p, which in turn inhibits the production of CX3CL1 and subsequently reduces NK cell recruitment to the tumor (Figure 3A) (121). Wang et al. reported that miR-146a negatively regulates IFN-γ production in human NK cells by targeting the NK-κB signaling pathway (Figure 3A) (122). MiR-146a overexpression significantly suppresses the cytotoxic activity of NK92 cells by targeting STAT1 signal transduction (123). In contrast, miR-181 was found to promote IFN-γ production in primary NK cells in response to cytokine stimulation by targeting nemo-like kinase (NLK), an inhibitor of Notch signaling (124). MiR-362-5p overexpression upregulated Prf1, GzmB, IFN-γ, and CD107a in human NK cells (125). Several reports have shown that miR-155 can enhance NK cell functions by regulating molecules involved in NK cell activation and IFN-γ release (126–128).
Moreover, miRNAs can control the expression of activating and inhibitory receptors on the surface of NK cells or that of their ligands on tumor cells (Figure 3B). Human miR-1245 could downregulate NKG2D on NK cells and, therefore, impair NKG2D-mediated functions of NK cells (129). NKG2D ligands (MICA/B) could also be repressed by miR-20a, miR-93, miR-106b, miR-373, and miR-520d in human cancer cells (HeLa, 293T, DU145, and glioma cells) (130, 131). In breast cancer cells, the miR-17-92 cluster (miR-20a, miR-20b, miR-93, and miR-106b), which could be inhibited by the HDAC inhibitors SAHA and VPA, downregulates the expression of MICA/B by targeting the mRNA 3’-UTR and downregulates ULBP2 by inhibiting the MAPK/ERK signaling pathway (132). The transcription and translation of DNAX-activating protein 12 kDa (DAP12), an exclusive signaling adaptor of many NK cell receptors, could be repressed by human miR-183, thus leading to the abrogation of NK cell antitumor function (133). In contrast, miR-30c-1* (134) promotes NK cell cytotoxicity against hepatoma cells by targeting the transcription factor HMBOX1 and miR-30c (135) could promote the cytotoxicity of NKL cells in vitro by upregulating the expression levels of NKG2D, CD107a, and FasL. Inhibitory receptors (e.g., KIRs, NKG2A, PD-1, TIGIT, TIM-3) function as immune checkpoints associated with NK cell exhaustion and the immune escape of tumor cells. MiR-146a-5p can downregulate the expression of both KIR2DL1 and KIR2DL2 (136). Three miRNAs, miR-26a-5p, miR-26b-5p, and miR-185-5p, were identified as inhibitors of the expression of inhibitory KIR3DL3, whose function has not yet been demonstrated (137). MiR-182 mediates a complex modulation of NKG2D and NKG2A levels at different stages of human hepatocellular carcinoma, resulting in increased Prf1 expression (138). Some miRNAs have been found to target PD-1 [miR-28 (139), miR-138 (140), miR-4717 (141)] and TIM-3 [miR-28 (139)] in T cells and cause T cell exhaustion. Thus, these miRNAs may also play a regulatory role in NK cells; however, experimental evidence has not been presented.
Perspectives
NK cells play a crucial role in preventing tumor initiation and metastasis. Many studies have illustrated the epigenetic regulatory mechanism of NK cell antitumor cytotoxicity, and they mainly focused on the expression of NK cell receptors and effector molecules, as we reviewed above. Multiple modulators always participate in epigenetic regulation. For example, histone modifications determine the open/closed state of chromatin, which affects the binding of transcription factors to specific regulatory sites. Additional research should focus on the interactions between different epigenetic modulators rather than just studying individual molecules. Recent technological advances have allowed us to gain a deeper understanding of NK cells. For example, single-cell RNA sequencing helps decipher the similarities and differences between humans and mice and between blood and splenic NK cells (142). Very recently, Li et al. applied the transposase accessible chromatin with sequencing (ATAC-seq) technique to define two distinct TF clusters that dynamically regulate NK cell differentiation in a homemade in vitro NK cell differentiation system (143). NK cells are a heterogeneous population that consists of multiple subsets and various states. The tissue site shapes the functional potential of NK cell subsets. Whole transcriptome profiling reveals the site-specific variations of NK cells in the lymph node, lung, blood, bone marrow, and spleen (33). However, the epigenetic features of these subsets are still a mystery.
The “states” (resting, activating, memory, repressed, and exhausted) of NK cells are controlled epigenetically, although insights into the underlying mechanism are very limited. Adaptive NK cells exhibit a unique whole-genome epigenetic signature similar to that of effector memory CD8+ T cells but not conventional NK cells (144). Chronic stimulation (NKG2C Abs with IL-15) could induce exhaustion in primary adaptive NK cells, thereby upregulating the expression of checkpoint receptors LAG-3 and PD-1. These NK cells are dysfunctional when challenged with tumor targets and exhibit a whole genome-DNA methylation profile similar to the epigenetically remodeled profiles of exhausted CD8+ T cells (145). It is reasonable to presume that NK cells are similar to T cells and show susceptibility to exhaustion during the antitumor war. However, there is a lack of consensus on the defining features of NK cell dysfunctional states, such as senescence, suppression, and exhaustion (47). Further consideration is needed to determine the state of NK cells in the antitumor response and how their epigenetic landscape changes during the process.
NK cell-based immunotherapy is an effective supplement to T cell-based therapy. Various approaches have been introduced to activate NK cells in adoptive cell therapy for better clinical outcomes, including generating CAR-NKs and inducing ADCC by mAbs, immune checkpoint blockade, engineered cytokine stimulatory, and so on (146). Even so, NK cell-based therapies are still in the early stages of development. Other than these “extrinsic” strategies, approaches that target “intrinsic” epigenetic regulators should be taken into consideration. Research on the epigenetic control of NK cell functions will provide new evidence for developing drugs and effective cancer prevention approaches. For example, demethylating agents can restore the absence of transcription of NKG2DL associated with high levels of DNA methylation in tumor cells. Some histone modification regulators (e.g., EZH2 and LSD1) have been found to be aberrantly overexpressed in various malignant tumors. Small molecular inhibitors are in clinical or preclinical development. From our perspective, these inhibitors also have potential applications in improving the in vitro expansion of NK cell cytotoxicity. More studies are needed to further elucidate the application of epigenetic drugs in NK cell-based immunotherapy, alone or in combination with other strategies.
Author Contributions
MX and XW conceived and designed the manuscript. MX did literature searching, drafted the manuscript, and drew the figures. BW and ZW did literature searching and drafted several sections. XZ and XW reviewed and revised the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Scientific Research Common Program of Beijing Municipal Commission of Education (KM201910025026 to MX), the Support Project of High-level Teachers in Beijing Municipal Universities in the Period of 13th Five–year Plan (IDHT20190510 to XW and XZ), and the National Natural Science Foundation of China (81972652 to XW).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Lanier LL. NK Cell Recognition. Annu Rev Immunol (2005) 23:225–74. doi: 10.1146/annurev.immunol.23.021704.115526
2. Goh W, Huntington ND. Regulation of Murine Natural Killer Cell Development. Front Immunol (2017) 8:130. doi: 10.3389/fimmu.2017.00130
3. Thiery J, Keefe D, Boulant S, Boucrot E, Walch M, Martinvalet D, et al. Perforin Pores in the Endosomal Membrane Trigger the Release of Endocytosed Granzyme B Into the Cytosol of Target Cells. Nat Immunol (2011) 12(8):770–7. doi: 10.1038/ni.2050
4. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science (2020) 368(6494):eaaz7548. doi: 10.1126/science.aaz7548
5. Arase H, Arase N, Saito T. Fas-Mediated Cytotoxicity by Freshly Isolated Natural Killer Cells. J Exp Med (1995) 181(3):1235–8. doi: 10.1084/jem.181.3.1235
6. Takeda K, Cretney E, Hayakawa Y, Ota T, Akiba H, Ogasawara K, et al. TRAIL Identifies Immature Natural Killer Cells in Newborn Mice and Adult Mouse Liver. Blood (2005) 105(5):2082–9. doi: 10.1182/blood-2004-08-3262
7. Bald T, Krummel MF, Smyth MJ, Barry KC. The NK Cell-Cancer Cycle: Advances and New Challenges in NK Cell-Based Immunotherapies. Nat Immunol (2020) 21(8):835–47. doi: 10.1038/s41590-020-0728-z
8. Bald T, Pedde AM, Corvino D, Bottcher JP. The Role of NK Cell as Central Communicators in Cancer Immunity. Adv Immunol (2020) 147:61–88. doi: 10.1016/bs.ai.2020.06.002
9. Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural Cytotoxic Reactivity of Mouse Lymphoid Cells Against Syngeneic and Allogeneic Tumors. II. Characterization of effector cells. Int J Cancer (1975) 16(2):230–9. doi: 10.1002/ijc.2910160205
10. Herberman RB, Nunn ME, Lavrin DH. Natural Cytotoxic Reactivity of Mouse Lymphoid Cells Against Syngeneic Acid Allogeneic Tumors. I. Distribution of reactivity and specificity. Int J Cancer (1975) 16(2):216–29. doi: 10.1002/ijc.2910160204
11. Kiessling R, Klein E, Pross H, Wigzell H. “Natural” Killer Cells in the Mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol (1975) 5(2):117–21. doi: 10.1002/eji.1830050209
12. Kiessling R, Klein E, Wigzell H. “Natural” Killer Cells in the Mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol (1975) 5(2):112–7. doi: 10.1002/eji.1830050208
13. Pross HF, Baines MG. Spontaneous Human Lymphocyte-Mediated Cytotoxicity Againts Tumour Target Cells. I. The effect of malignant disease. Int J Cancer (1976) 18(5):593–604. doi: 10.1002/ijc.2910180508
14. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural Cytotoxic Activity of Peripheral-Blood Lymphocytes and Cancer Incidence: An 11-Year Follow-Up Study of a General Population. Lancet (2000) 356(9244):1795–9. doi: 10.1016/S0140-6736(00)03231-1
15. Chiang SC, Theorell J, Entesarian M, Meeths M, Mastafa M, Al-Herz W, et al. Comparison of Primary Human Cytotoxic T-cell and Natural Killer Cell Responses Reveal Similar Molecular Requirements for Lytic Granule Exocytosis But Differences in Cytokine Production. Blood (2013) 121(8):1345–56. doi: 10.1182/blood-2012-07-442558
16. Deguine J, Breart B, Lemaitre F, Di Santo JP, Bousso P. Intravital Imaging Reveals Distinct Dynamics for Natural Killer and CD8(+) T Cells During Tumor Regression. Immunity (2010) 33(4):632–44. doi: 10.1016/j.immuni.2010.09.016
17. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity (2017) 47(5):820–33. doi: 10.1016/j.immuni.2017.10.008
18. Bjorkstrom NK, Ljunggren HG, Michaelsson J. Emerging Insights Into Natural Killer Cells in Human Peripheral Tissues. Nat Rev Immunol (2016) 16(5):310–20. doi: 10.1038/nri.2016.34
19. Melsen JE, Lugthart G, Lankester AC, Schilham MW. Human Circulating and Tissue-Resident CD56(Bright) Natural Killer Cell Populations. Front Immunol (2016) 7:262. doi: 10.3389/fimmu.2016.00262
20. Kim S, Iizuka K, Kang HS, Dokun A, French AR, Greco S, et al. In Vivo Developmental Stages in Murine Natural Killer Cell Maturation. Nat Immunol (2002) 3(6):523–8. doi: 10.1038/ni796
21. Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of Mouse NK Cells is a 4-Stage Developmental Program. Blood (2009) 113(22):5488–96. doi: 10.1182/blood-2008-10-187179
22. Colonna M. Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity (2018) 48(6):1104–17. doi: 10.1016/j.immuni.2018.05.013
23. Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, et al. T-Bet and Eomes Instruct the Development of Two Distinct Natural Killer Cell Lineages in the Liver and in the Bone Marrow. J Exp Med (2014) 211(3):563–77. doi: 10.1084/jem.20131560
24. Seillet C, Brossay L, Vivier E. Natural Killers or ILC1s? That is the question. Curr Opin Immunol (2021) 68:48–53. doi: 10.1016/j.coi.2020.08.009
25. Spits H, Bernink JH, Lanier L. NK Cells and Type 1 Innate Lymphoid Cells: Partners in Host Defense. Nat Immunol (2016) 17(7):758–64. doi: 10.1038/ni.3482
26. Bald T, Wagner M, Gao Y, Koyasu S, Smyth MJ. Hide and Seek: Plasticity of Innate Lymphoid Cells in Cancer. Semin Immunol (2019) 41:101273. doi: 10.1016/j.smim.2019.04.001
27. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor Immunoevasion by the Conversion of Effector NK Cells Into Type 1 Innate Lymphoid Cells. Nat Immunol (2017) 18(9):1004–15. doi: 10.1038/ni.3800
28. Massague J. How Cells Read TGF-beta Signals. Nat Rev Mol Cell Biol (2000) 1(3):169–78. doi: 10.1038/35043051
29. Cortez VS, Cervantes-Barragan L, Robinette ML, Bando JK, Wang Y, Geiger TL, et al. Transforming Growth Factor-Beta Signaling Guides the Differentiation of Innate Lymphoid Cells in Salivary Glands. Immunity (2016) 44(5):1127–39. doi: 10.1016/j.immuni.2016.03.007
30. Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, et al. SMAD4 Impedes the Conversion of NK Cells Into ILC1-like Cells by Curtailing non-Canonical TGF-beta Signaling. Nat Immunol (2017) 18(9):995–1003. doi: 10.1038/ni.3809
31. Wang Y, Chu J, Yi P, Dong W, Saultz J, Wang Y, et al. SMAD4 Promotes TGF-beta-independent NK Cell Homeostasis and Maturation and Antitumor Immunity. J Clin Invest (2018) 128(11):5123–36. doi: 10.1172/JCI121227
32. Peng H, Tian Z. Diversity of Tissue-Resident NK Cells. Semin Immunol (2017) 31:3–10. doi: 10.1016/j.smim.2017.07.006
33. Dogra P, Rancan C, Ma W, Toth M, Senda T, Carpenter DJ, et al. Tissue Determinants of Human Nk Cell Development, Function, and Residence. Cell (2020) 180(4):749–63.e13. doi: 10.1016/j.cell.2020.01.022
34. Zhou J, Tian Z, Peng H. Tissue-Resident NK Cells and Other Innate Lymphoid Cells. Adv Immunol (2020) 145:37–53. doi: 10.1016/bs.ai.2019.11.002
35. Sojka DK, Tian Z, Yokoyama WM. Tissue-Resident Natural Killer Cells and Their Potential Diversity. Semin Immunol (2014) 26(2):127–31. doi: 10.1016/j.smim.2014.01.010
36. Sun H, Liu L, Huang Q, Liu H, Huang M, Wang J, et al. Accumulation of Tumor-Infiltrating Cd49a(+) NK Cells Correlates With Poor Prognosis for Human Hepatocellular Carcinoma. Cancer Immunol Res (2019) 7(9):1535–46. doi: 10.1158/2326-6066.CIR-18-0757
37. Sun JC, Beilke JN, Lanier LL. Adaptive Immune Features of Natural Killer Cells. Nature (2009) 457(7229):557–61. doi: 10.1038/nature07665
38. O’Leary JG, Goodarzi M, Drayton DL, von Andrian UH. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol (2006) 7(5):507–16. doi: 10.1038/ni1332
39. Paust S, Gill HS, Wang BZ, Flynn MP, Moseman EA, Senman B, et al. Critical Role for the Chemokine Receptor CXCR6 in NK Cell-Mediated Antigen-Specific Memory of Haptens and Viruses. Nat Immunol (2010) 11(12):1127–35. doi: 10.1038/ni.1953
40. Luetke-Eversloh M, Hammer Q, Durek P, Nordstrom K, Gasparoni G, Pink M, et al. Human Cytomegalovirus Drives Epigenetic Imprinting of the IFNG Locus in NKG2Chi Natural Killer Cells. PloS Pathog (2014) 10(10):e1004441. doi: 10.1371/journal.ppat.1004441
41. Brillantes M, Beaulieu AM. Memory and Memory-Like Nk Cell Responses to Microbial Pathogens. Front Cell Infect Microbiol (2020) 10:102. doi: 10.3389/fcimb.2020.00102
42. Zhang Y, Wallace DL, de Lara CM, Ghattas H, Asquith B, Worth A, et al. In Vivo Kinetics of Human Natural Killer Cells: The Effects of Ageing and Acute and Chronic Viral Infection. Immunology (2007) 121(2):258–65. doi: 10.1111/j.1365-2567.2007.02573.x
43. Wang JW, Howson JM, Ghansah T, Desponts C, Ninos JM, May SL, et al. Influence of SHIP on the NK Repertoire and Allogeneic Bone Marrow Transplantation. Science (2002) 295(5562):2094–7. doi: 10.1126/science.1068438
44. Lopez-Verges S, Milush JM, Schwartz BS, Pando MJ, Jarjoura J, York VA, et al. Expansion of a unique CD57(+)NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci U S A (2011) 108(36):14725–32. doi: 10.1073/pnas.1110900108
45. Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X, et al. Cytomegalovirus Reactivation After Allogeneic Transplantation Promotes a Lasting Increase in Educated NKG2C+ Natural Killer Cells With Potent Function. Blood (2012) 119(11):2665–74. doi: 10.1182/blood-2011-10-386995
46. Zhang C, Liu Y. Targeting NK Cell Checkpoint Receptors or Molecules for Cancer Immunotherapy. Front Immunol (2020) 11:1295. doi: 10.3389/fimmu.2020.01295
47. Merino AM, Kim H, Miller JS, Cichocki F. Unraveling Exhaustion in Adaptive and Conventional NK Cells. J Leukoc Biol (2020) 108(4):1361–8. doi: 10.1002/JLB.4MR0620-091R
48. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK Cells to Immunotherapy Mediated by PD-1/PD-L1 Blockade. J Clin Invest (2018) 128(10):4654–68. doi: 10.1172/JCI99317
49. Luetke-Eversloh M, Cicek BB, Siracusa F, Thom JT, Hamann A, Frischbutter S, et al. NK Cells Gain Higher IFN-gamma Competence During Terminal Differentiation. Eur J Immunol (2014) 44(7):2074–84. doi: 10.1002/eji.201344072
50. Chan HW, Kurago ZB, Stewart CA, Wilson MJ, Martin MP, Mace BE, et al. DNA Methylation Maintains Allele-Specific KIR Gene Expression in Human Natural Killer Cells. J Exp Med (2003) 197(2):245–55. doi: 10.1084/jem.20021127
51. Santourlidis S, Trompeter HI, Weinhold S, Eisermann B, Meyer KL, Wernet P, et al. Crucial Role of DNA Methylation in Determination of Clonally Distributed Killer Cell Ig-like Receptor Expression Patterns in NK Cells. J Immunol (2002) 169(8):4253–61. doi: 10.4049/jimmunol.169.8.4253
52. Santourlidis S, Graffmann N, Christ J, Uhrberg M. Lineage-Specific Transition of Histone Signatures in the Killer Cell Ig-like Receptor Locus From Hematopoietic Progenitor to NK Cells. J Immunol (2008) 180(1):418–25. doi: 10.4049/jimmunol.180.1.418
53. Schenk A, Pulverer W, Koliamitra C, Bauer CJ, Ilic S, Heer R, et al. Acute Exercise Increases the Expression of KIR2DS4 by Promoter Demethylation in NK Cells. Int J sports Med (2019) 40(1):62–70. doi: 10.1055/a-0741-7001
54. Schenk A, Koliamitra C, Bauer CJ, Schier R, Schweiger MR, Bloch W, et al. Impact of Acute Aerobic Exercise on Genome-Wide DNA-Methylation in Natural Killer Cells-A Pilot Study. Genes (Basel) (2019) 10(5):380. doi: 10.3390/genes10050380
55. Rogers SL, Rouhi A, Takei F, Mager DL. A Role for DNA Hypomethylation and Histone Acetylation in Maintaining Allele-Specific Expression of Mouse NKG2A in Developing and Mature NK Cells. J Immunol (2006) 177(1):414–21. doi: 10.4049/jimmunol.177.1.414
56. Zhao NH, Qian Y, Wu CS, Wang JW, Fang Y, Fan XP, et al. Diagnostic Value of NKG2D Promoter Methylation in Hepatitis B Virus-Associated Hepatocellular Carcinoma. Biomark Med (2019) 13(13):1093–105. doi: 10.2217/bmm-2019-0102
57. Gao XN, Lin J, Wang LL, Yu L. Demethylating Treatment Suppresses Natural Killer Cell Cytolytic Activity. Mol Immunol (2009) 46(10):2064–70. doi: 10.1016/j.molimm.2009.02.033
58. Schmiedel BJ, Arelin V, Gruenebach F, Krusch M, Schmidt SM, Salih HR. Azacytidine Impairs NK Cell Reactivity While Decitabine Augments NK Cell Responsiveness Toward Stimulation. Int J Cancer (2011) 128(12):2911–22. doi: 10.1002/ijc.25635
59. Sohlberg E, Pfefferle A, Andersson S, Baumann BC, Hellstrom-Lindberg E, Malmberg KJ. Imprint of 5-Azacytidine on the Natural Killer Cell Repertoire During Systemic Treatment for High-Risk Myelodysplastic Syndrome. Oncotarget (2015) 6(33):34178–90. doi: 10.18632/oncotarget.6213
60. Kubler A, Woiterski J, Witte KE, Buhring HJ, Hartwig UF, Ebinger M, et al. Both Mature KIR+ and Immature KIR- NK Cells Control Pediatric Acute B-cell Precursor Leukemia in NOD. Cg-Prkdcscid IL2rgtmWjl/Sz Mice Blood (2014) 124(26):3914–23. doi: 10.1182/blood-2014-05-572743
61. Kopp LM, Ray A, Denman CJ, Senyukov VS, Somanchi SS, Zhu S, et al. Decitabine has a Biphasic Effect on Natural Killer Cell Viability, Phenotype, and Function Under Proliferative Conditions. Mol Immunol (2013) 54(3-4):296–301. doi: 10.1016/j.molimm.2012.12.012
62. Rohner A, Langenkamp U, Siegler U, Kalberer CP, Wodnar-Filipowicz A. Differentiation-Promoting Drugs Up-Regulate NKG2D Ligand Expression and Enhance the Susceptibility of Acute Myeloid Leukemia Cells to Natural Killer Cell-Mediated Lysis. Leuk Res (2007) 31(10):1393–402. doi: 10.1016/j.leukres.2007.02.020
63. Grimm EA, Mazumder A, Zhang HZ, Rosenberg SA. Lymphokine-Activated Killer Cell Phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J Exp Med (1982) 155(6):1823–41. doi: 10.1084/jem.155.6.1823
64. Fernandez-Sanchez A, Baragano Raneros A, Carvajal Palao R, Sanz AB, Ortiz A, Ortega F, et al. DNA Demethylation and Histone H3K9 Acetylation Determine the Active Transcription of the NKG2D Gene in Human CD8+ T and NK Cells. Epigenetics (2013) 8(1):66–78. doi: 10.4161/epi.23115
65. Hicks KC, Fantini M, Donahue RN, Schwab A, Knudson KM, Tritsch SR, et al. Epigenetic Priming of Both Tumor and NK Cells Augments Antibody-Dependent Cellular Cytotoxicity Elicited by the anti-PD-L1 Antibody Avelumab Against Multiple Carcinoma Cell Types. Oncoimmunology (2018) 7(11):e1466018. doi: 10.1080/2162402X.2018.1466018
66. Zhu S, Denman CJ, Cobanoglu ZS, Kiany S, Lau CC, Gottschalk SM, et al. The Narrow-Spectrum HDAC Inhibitor Entinostat Enhances NKG2D Expression Without NK Cell Toxicity, Leading to Enhanced Recognition of Cancer Cells. Pharm Res (2015) 32(3):779–92. doi: 10.1007/s11095-013-1231-0
67. Pace M, Williams J, Kurioka A, Gerry AB, Jakobsen B, Klenerman P, et al. Histone Deacetylase Inhibitors Enhance Cd4 T Cell Susceptibility to NK Cell Killing But Reduce Nk Cell Function. PloS Pathog (2016) 12(8):e1005782. doi: 10.1371/journal.ppat.1005782
68. Rossi LE, Avila DE, Spallanzani RG, Ziblat A, Fuertes MB, Lapyckyj L, et al. Histone Deacetylase Inhibitors Impair NK Cell Viability and Effector Functions Through Inhibition of Activation and Receptor Expression. J Leukoc Biol (2012) 91(2):321–31. doi: 10.1189/jlb.0711339
69. Shi X, Li M, Cui M, Niu C, Xu J, Zhou L, et al. Epigenetic Suppression of the Antitumor Cytotoxicity of NK Cells by Histone Deacetylase Inhibitor Valproic Acid. Am J Cancer Res (2016) 6(3):600–14.
70. Yin J, Leavenworth JW, Li Y, Luo Q, Xie H, Liu X, et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proc Natl Acad Sci U S A (2015) 112(52):15988–93. doi: 10.1073/pnas.1521740112
71. Bugide S, Green MR, Wajapeyee N. Inhibition of Enhancer of Zeste Homolog 2 (EZH2) Induces Natural Killer Cell-Mediated Eradication of Hepatocellular Carcinoma Cells. Proc Natl Acad Sci (2018) 115(15):E3509–18. doi: 10.1073/pnas.1802691115
72. Cribbs A, Hookway ES, Wells G, Lindow M, Obad S, Oerum H, et al. Inhibition of Histone H3K27 Demethylases Selectively Modulates Inflammatory Phenotypes of Natural Killer Cells. J Biol Chem (2018) 293(7):2422–37. doi: 10.1074/jbc.RA117.000698
73. Bailey CP, Figueroa M, Gangadharan A, Yang Y, Romero MM, Kennis BA, et al. Pharmacologic Inhibition of Lysine-Specific Demethylase 1 as a Therapeutic and Immune-Sensitization Strategy in Pediatric High-Grade Glioma. Neuro Oncol (2020) 22(9):1302–14. doi: 10.1093/neuonc/noaa058
74. Bailey CP, Figueroa M, Gangadharan A, Lee DA, Chandra J. Scaffolding LSD1 Inhibitors Impair Nk Cell Metabolism and Cytotoxic Function Through Depletion of Glutathione. Front Immunol (2020) 11:2196. doi: 10.3389/fimmu.2020.02196
75. Triozzi PL, Aldrich W, Achberger S, Ponnazhagan S, Alcazar O, Saunthararajah Y. Differential Effects of Low-Dose Decitabine on Immune Effector and Suppressor Responses in Melanoma-Bearing Mice. Cancer Immunol Immunother (2012) 61(9):1441–50. doi: 10.1007/s00262-012-1204-x
76. Bannister AJ, Kouzarides T. Regulation of Chromatin by Histone Modifications. Cell Res (2011) 21(3):381–95. doi: 10.1038/cr.2011.22
77. Zhang Y, Sun Z, Jia J, Du T, Zhang N, Tang Y, et al. Overview of Histone Modification. Adv Exp Med Biol (2021) 1283:1–16. doi: 10.1007/978-981-15-8104-5_1
78. Black JC, Van Rechem C, Whetstine JR. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol Cell (2012) 48(4):491–507. doi: 10.1016/j.molcel.2012.11.006
79. Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, et al. Genome-Wide Mapping of HATs and HDACs Reveals Distinct Functions in Active and Inactive Genes. Cell (2009) 138(5):1019–31. doi: 10.1016/j.cell.2009.06.049
80. Li Y, Wang J, Yin J, Liu X, Yu M, Li T, et al. Chromatin State Dynamics During NK Cell Activation. Oncotarget (2017) 8(26):41854–65. doi: 10.18632/oncotarget.16688
81. Ni L, Wang L, Yao C, Ni Z, Liu F, Gong C, et al. The Histone Deacetylase Inhibitor Valproic Acid Inhibits NKG2D Expression in Natural Killer Cells Through Suppression of STAT3 and HDAC3. Sci Rep (2017) 7:45266. doi: 10.1038/srep45266
82. Ogbomo H, Michaelis M, Kreuter J, Doerr HW, Cinatl J Jr. Histone Deacetylase Inhibitors Suppress Natural Killer Cell Cytolytic Activity. FEBS Lett (2007) 581(7):1317–22. doi: 10.1016/j.febslet.2007.02.045
83. Schenk A, Bloch W, Zimmer P. Natural Killer Cells–an Epigenetic Perspective of Development and Regulation. Int J Mol Sci (2016) 17(3):326. doi: 10.3390/ijms17030326
84. Chang S, Aune TM. Histone Hyperacetylated Domains Across the Ifng Gene Region in Natural Killer Cells and T Cells. Proc Natl Acad Sci U S A (2005) 102(47):17095–100. doi: 10.1073/pnas.0502129102
85. Jennings VA, Scott GB, Rose AMS, Scott KJ, Migneco G, Keller B, et al. Potentiating Oncolytic Virus-Induced Immune-Mediated Tumor Cell Killing Using Histone Deacetylase Inhibition. Mol Ther (2019) 27(6):1139–52. doi: 10.1016/j.ymthe.2019.04.008
86. Ramakrishnan S, Granger V, Rak M, Hu Q, Attwood K, Aquila L, et al. Inhibition of EZH2 Induces NK Cell-Mediated Differentiation and Death in Muscle-Invasive Bladder Cancer. Cell Death Differ (2019) 26(10):2100–14. doi: 10.1038/s41418-019-0278-9
87. Rasid O, Chevalier C, Camarasa TM-N, Fitting C, Cavaillon J-M, Hamon MA. H3k4me1 Supports Memory-Like NK Cells Induced by Systemic Inflammation. Cell Rep (2019) 29(12):3933–45.e3. doi: 10.1016/j.celrep.2019.11.043
88. Zhao D, Zhang Q, Liu Y, Li X, Zhao K, Ding Y, et al. H3k4me3 Demethylase Kdm5a Is Required for NK Cell Activation by Associating With p50 to Suppress Socs1. Cell Rep (2016) 15(2):288–99. doi: 10.1016/j.celrep.2016.03.035
89. Held W, Jeevan-Raj B, Charmoy M. Transcriptional Regulation of Murine Natural Killer Cell Development, Differentiation and Maturation. Cell Mol Life Sci (2018) 75(18):3371–9. doi: 10.1007/s00018-018-2865-1
90. Bugide S, Janostiak R, Wajapeyee N. Epigenetic Mechanisms Dictating Eradication of Cancer by Natural Killer Cells. Trends Cancer (2018) 4(8):553–66. doi: 10.1016/j.trecan.2018.06.004
91. Kee BL, Morman RE, Sun M. Transcriptional Regulation of Natural Killer Cell Development and Maturation. Adv Immunol (2020) 146:1–28. doi: 10.1016/bs.ai.2020.01.001
92. Kwon HJ, Choi GE, Ryu S, Kwon SJ, Kim SC, Booth C, et al. Stepwise Phosphorylation of p65 Promotes NF-kappaB Activation and NK Cell Responses During Target Cell Recognition. Nat Commun (2016) 7:11686. doi: 10.1038/ncomms11686
93. Zhang J, Marotel M, Fauteux-Daniel S, Mathieu AL, Viel S, Marcais A, et al. T-Bet and Eomes Govern Differentiation and Function of Mouse and Human NK Cells and ILC1. Eur J Immunol (2018) 48(5):738–50. doi: 10.1002/eji.201747299
94. Werneck MB, Lugo-Villarino G, Hwang ES, Cantor H, Glimcher LH. T-bet plays a key role in NK-mediated control of melanoma metastatic disease. J Immunol (2008) 180(12):8004–10. doi: 10.4049/jimmunol.180.12.8004
95. Holmes ML, Huntington ND, Thong RP, Brady J, Hayakawa Y, Andoniou CE, et al. Peripheral natural killer cell maturation depends on the transcription factor Aiolos. EMBO J (2014) 33(22):2721–34. doi: 10.15252/embj.201487900
96. Deng Y, Kerdiles Y, Chu J, Yuan S, Wang Y, Chen X, et al. Transcription Factor Foxo1 is a Negative Regulator of Natural Killer Cell Maturation and Function. Immunity (2015) 42(3):457–70. doi: 10.1016/j.immuni.2015.02.006
97. Du X, de Almeida P, Manieri N, de Almeida Nagata D, Wu TD, Harden Bowles K, et al. CD226 regulates natural killer cell antitumor responses via phosphorylation-mediated inactivation of transcription factor FOXO1. Proc Natl Acad Sci U S A (2018) 115(50):E11731–40. doi: 10.1073/pnas.1814052115
98. Rabacal W, Pabbisetty SK, Hoek KL, Cendron D, Guo Y, Maseda D, et al. Transcription factor KLF2 regulates homeostatic NK cell proliferation and survival. Proc Natl Acad Sci U S A (2016) 113(19):5370–5. doi: 10.1073/pnas.1521491113
99. Ni J, Wang X, Stojanovic A, Zhang Q, Wincher M, Buhler L, et al. Single-Cell RNA Sequencing of Tumor-Infiltrating Nk Cells Reveals That Inhibition of Transcription Factor HIF-1alpha Unleashes NK Cell Activity. Immunity (2020) 52(6):1075–87 e8. doi: 10.1016/j.immuni.2020.05.001
100. Gotthardt D, Sexl V. Stats in NK-Cells: The Good, the Bad, and the Ugly. Front Immunol (2016) 7:694. doi: 10.3389/fimmu.2016.00694
101. Vargas-Hernandez A, Forbes LR. JAK/STAT Proteins and Their Biological Impact on NK Cell Development and Function. Mol Immunol (2019) 115:21–30. doi: 10.1016/j.molimm.2018.12.005
102. Putz EM, Gotthardt D, Hoermann G, Csiszar A, Wirth S, Berger A, et al. CDK8-Mediated STAT1-S727 Phosphorylation Restrains NK Cell Cytotoxicity and Tumor Surveillance. Cell Rep (2013) 4(3):437–44. doi: 10.1016/j.celrep.2013.07.012
103. Meissl K, Simonovic N, Amenitsch L, Witalisz-Siepracka A, Klein K, Lassnig C, et al. Stat1 Isoforms Differentially Regulate Nk Cell Maturation and Anti-tumor Activity. Front Immunol (2020) 11:2189. doi: 10.3389/fimmu.2020.02189
104. Cacalano NA. Regulation of Natural Killer Cell Function by STAT3. Front Immunol (2016) 7:128. doi: 10.3389/fimmu.2016.00128
105. Wang KS, Ritz J, Frank DA. IL-2 Induces STAT4 Activation in Primary NK Cells and NK Cell Lines, But Not in T Cells. J Immunol (1999) 162(1):299–304.
106. Yamamoto K, Shibata F, Miyasaka N, Miura O. The Human Perforin Gene is a Direct Target of STAT4 Activated by IL-12 in NK Cells. Biochem Biophys Res Commun (2002) 297(5):1245–52. doi: 10.1016/S0006-291X(02)02378-1
107. Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, et al. A Novel Ncr1-Cre Mouse Reveals the Essential Role of STAT5 for NK-cell Survival and Development. Blood (2011) 117(5):1565–73. doi: 10.1182/blood-2010-06-291633
108. Imada K, Bloom ET, Nakajima H, Horvath-Arcidiacono JA, Udy GB, Davey HW, et al. Stat5b is Essential for Natural Killer Cell-Mediated Proliferation and Cytolytic Activity. J Exp Med (1998) 188(11):2067–74. doi: 10.1084/jem.188.11.2067
109. Chehboun S, Leiva-Torres GA, Charbonneau B, Eveleigh R, Bourque G, Vidal SM. A Point Mutation in the Linker Domain of Mouse STAT5A is Associated With Impaired NK-cell Regulation. Genes Immun (2020) 21(2):136–41. doi: 10.1038/s41435-019-0088-6
110. Villarino AV, Sciume G, Davis FP, Iwata S, Zitti B, Robinson GW, et al. Subset- and Tissue-Defined STAT5 Thresholds Control Homeostasis and Function of Innate Lymphoid Cells. J Exp Med (2017) 214(10):2999–3014. doi: 10.1084/jem.20150907
111. Mazzurana L, Forkel M, Rao A, Van Acker A, Kokkinou E, Ichiya T, et al. Suppression of Aiolos and Ikaros Expression by Lenalidomide Reduces Human ILC3-ILC1/NK Cell Transdifferentiation. Eur J Immunol (2019) 49(9):1344–55. doi: 10.1002/eji.201848075
112. Li L, Leid M, Rothenberg EV. An Early T Cell Lineage Commitment Checkpoint Dependent on the Transcription Factor Bcl11b. Science (2010) 329(5987):89–93. doi: 10.1126/science.1188989
113. Li P, Burke S, Wang J, Chen X, Ortiz M, Lee SC, et al. Reprogramming of T Cells to Natural Killer-Like Cells Upon Bcl11b Deletion. Science (2010) 329(5987):85–9. doi: 10.1126/science.1188063
114. Liu P, Li P, Burke S. Critical Roles of Bcl11b in T-cell Development and Maintenance of T-cell Identity. Immunol Rev (2010) 238(1):138–49. doi: 10.1111/j.1600-065X.2010.00953.x
115. Lu TX, Rothenberg ME. Microrna. J Allergy Clin Immunol (2018) 141(4):1202–7. doi: 10.1016/j.jaci.2017.08.034
116. Pesce S, Greppi M, Ferretti E, Obino V, Carlomagno S, Rutigliani M, et al. miRNAs in NK Cell-Based Immune Responses and Cancer Immunotherapy. Front Cell Dev Biol (2020) 8:119. doi: 10.3389/fcell.2020.00119
117. Wang P, Gu Y, Zhang Q, Han Y, Hou J, Lin L, et al. Identification of Resting and Type I IFN-Activated Human NK Cell miRNomes Reveals microRNA-378 and microRNA-30e as Negative Regulators of NK Cell Cytotoxicity. J Immunol (2012) 189(1):211–21. doi: 10.4049/jimmunol.1200609
118. Kim N, Kim M, Yun S, Doh J, Greenberg PD, Kim TD, et al. MicroRNA-150 Regulates the Cytotoxicity of Natural Killers by Targeting Perforin-1. J Allergy Clin Immunol (2014) 134(1):195–203. doi: 10.1016/j.jaci.2014.02.018
119. Kim TD, Lee SU, Yun S, Sun HN, Lee SH, Kim JW, et al. Human microRNA-27a* Targets Prf1 and GzmB Expression to Regulate NK-cell Cytotoxicity. Blood (2011) 118(20):5476–86. doi: 10.1182/blood-2011-04-347526
120. Regis S, Caliendo F, Dondero A, Casu B, Romano F, Loiacono F, et al. TGF-Beta1 Downregulates the Expression of CX3CR1 by Inducing miR-27a-5p in Primary Human NK Cells. Front Immunol (2017) 8:868. doi: 10.3389/fimmu.2017.00868
121. Chen EB, Zhou ZJ, Xiao K, Zhu GQ, Yang Y, Wang B, et al. The Mir-561-5p/CX3CL1 Signaling Axis Regulates Pulmonary Metastasis in Hepatocellular Carcinoma Involving Cx3cr1(+) Natural Killer Cells Infiltration. Theranostics (2019) 9(16):4779–94. doi: 10.7150/thno.32543
122. Wang H, Zhang Y, Wu X, Wang Y, Cui H, Li X, et al. Regulation of Human Natural Killer Cell IFN-gamma Production by MicroRNA-146a Via Targeting the NF-kappaB Signaling Pathway. Front Immunol (2018) 9:293. doi: 10.3389/fimmu.2018.00293
123. Xu D, Han Q, Hou Z, Zhang C, Zhang J. miR-146a Negatively Regulates NK Cell Functions Via STAT1 Signaling. Cell Mol Immunol (2017) 14(8):712–20. doi: 10.1038/cmi.2015.113
124. Cichocki F, Felices M, McCullar V, Presnell SR, Al-Attar A, Lutz CT, et al. Cutting Edge: microRNA-181 Promotes Human NK Cell Development by Regulating Notch Signaling. J Immunol (2011) 187(12):6171–5. doi: 10.4049/jimmunol.1100835
125. Ni F, Guo C, Sun R, Fu B, Yang Y, Wu L, et al. MicroRNA Transcriptomes of Distinct Human NK Cell Populations Identify miR-362-5p as an Essential Regulator of NK Cell Function. Sci Rep (2015) 5:9993. doi: 10.1038/srep09993
126. Trotta R, Chen L, Ciarlariello D, Josyula S, Mao C, Costinean S, et al. miR-155 Regulates IFN-gamma Production in Natural Killer Cells. Blood (2012) 119(15):3478–85. doi: 10.1182/blood-2011-12-398099
127. Sullivan RP, Fogel LA, Leong JW, Schneider SE, Wong R, Romee R, et al. MicroRNA-155 Tunes Both the Threshold and Extent of NK Cell Activation Via Targeting of Multiple Signaling Pathways. J Immunol (2013) 191(12):5904–13. doi: 10.4049/jimmunol.1301950
128. Cheng YQ, Ren JP, Zhao J, Wang JM, Zhou Y, Li GY, et al. MicroRNA-155 Regulates Interferon-Gamma Production in Natural Killer Cells Via Tim-3 Signalling in Chronic Hepatitis C Virus Infection. Immunology (2015) 145(4):485–97. doi: 10.1111/imm.12463
129. Espinoza JL, Takami A, Yoshioka K, Nakata K, Sato T, Kasahara Y, et al. Human microRNA-1245 Down-Regulates the NKG2D Receptor in Natural Killer Cells and Impairs NKG2D-mediated Functions. Haematologica (2012) 97(9):1295–303. doi: 10.3324/haematol.2011.058529
130. Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, et al. Human microRNAs Regulate Stress-Induced Immune Responses Mediated by the Receptor NKG2D. Nat Immunol (2008) 9(9):1065–73. doi: 10.1038/ni.1642
131. Codo P, Weller M, Meister G, Szabo E, Steinle A, Wolter M, et al. MicroRNA-mediated Down-Regulation of NKG2D Ligands Contributes to Glioma Immune Escape. Oncotarget (2014) 5(17):7651–62. doi: 10.18632/oncotarget.2287
132. Shen J, Pan J, Du C, Si W, Yao M, Xu L, et al. Silencing NKG2D Ligand-Targeting miRNAs Enhances Natural Killer Cell-Mediated Cytotoxicity in Breast Cancer. Cell Death Dis (2017) 8(4):e2740. doi: 10.1038/cddis.2017.158
133. Donatelli SS, Zhou JM, Gilvary DL, Eksioglu EA, Chen X, Cress WD, et al. TGF-beta-inducible microRNA-183 silences tumor-associated natural killer cells. Proc Natl Acad Sci U S A (2014) 111(11):4203–8. doi: 10.1073/pnas.1319269111
134. Gong J, Liu R, Zhuang R, Zhang Y, Fang L, Xu Z, et al. miR-30c-1* Promotes Natural Killer Cell Cytotoxicity Against Human Hepatoma Cells by Targeting the Transcription Factor HMBOX1. Cancer Sci (2012) 103(4):645–52. doi: 10.1111/j.1349-7006.2012.02207.x
135. Ma Y, Gong J, Liu Y, Guo W, Jin B, Wang X, et al. MicroRNA-30c Promotes Natural Killer Cell Cytotoxicity Via Up-Regulating the Expression Level of NKG2D. Life Sci (2016) 151:174–81. doi: 10.1016/j.lfs.2016.03.012
136. Pesce S, Squillario M, Greppi M, Loiacono F, Moretta L, Moretta A, et al. New Mirna Signature Heralds Human Nk Cell Subsets At Different Maturation Steps: Involvement of miR-146a-5p in the Regulation of KIR Expression. Front Immunol (2018) 9:2360. doi: 10.3389/fimmu.2018.02360
137. Nutalai R, Gaudieri S, Jumnainsong A, Leelayuwat C. Regulation of KIR3DL3 Expression Via Mirna. Genes (Basel) (2019) 10(8):603. doi: 10.3390/genes10080603
138. Abdelrahman MM, Fawzy IO, Bassiouni AA, Gomaa AI, Esmat G, Waked I, et al. Enhancing NK Cell Cytotoxicity by miR-182 in Hepatocellular Carcinoma. Hum Immunol (2016) 77(8):667–73. doi: 10.1016/j.humimm.2016.04.020
139. Li Q, Johnston N, Zheng X, Wang H, Zhang X, Gao D, et al. miR-28 Modulates Exhaustive Differentiation of T Cells Through Silencing Programmed Cell Death-1 and Regulating Cytokine Secretion. Oncotarget (2016) 7(33):53735–50. doi: 10.18632/oncotarget.10731
140. Wei J, Nduom EK, Kong LY, Hashimoto Y, Xu S, Gabrusiewicz K, et al. MiR-138 Exerts Anti-Glioma Efficacy by Targeting Immune Checkpoints. Neuro Oncol (2016) 18(5):639–48. doi: 10.1093/neuonc/nov292
141. Zhang G, Li N, Li Z, Zhu Q, Li F, Yang C, et al. microRNA-4717 Differentially Interacts With its Polymorphic Target in the PD1 3’ Untranslated Region: A Mechanism for Regulating PD-1 Expression and Function in HBV-associated Liver Diseases. Oncotarget (2015) 6(22):18933–44. doi: 10.18632/oncotarget.3662
142. Crinier A, Milpied P, Escaliere B, Piperoglou C, Galluso J, Balsamo A, et al. High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved Nk Cell Subsets in Humans and Mice. Immunity (2018) 49(5):971–86.e5. doi: 10.1016/j.immuni.2018.09.009
143. Li K, Wu Y, Li Y, Yu Q, Tian Z, Wei H, et al. Landscape and Dynamics of the Transcriptional Regulatory Network During Natural Killer Cell Differentiation. Genomics Proteomics Bioinf (2020). doi: 10.1016/j.gpb.2020.12.003
144. Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, et al. Cytomegalovirus Infection Drives Adaptive Epigenetic Diversification of NK Cells With Altered Signaling and Effector Function. Immunity (2015) 42(3):443–56. doi: 10.1016/j.immuni.2015.02.008
145. Merino A, Zhang B, Dougherty P, Luo X, Wang J, Blazar BR, et al. Chronic Stimulation Drives Human NK Cell Dysfunction and Epigenetic Reprograming. J Clin Invest (2019) 129(9):3770–85. doi: 10.1172/JCI125916
Keywords: natural killer (NK) cells, epigenetics, DNA methylation, histone modification, transcription factor, microRNA, antitumor immunity
Citation: Xia M, Wang B, Wang Z, Zhang X and Wang X (2021) Epigenetic Regulation of NK Cell-Mediated Antitumor Immunity. Front. Immunol. 12:672328. doi: 10.3389/fimmu.2021.672328
Received: 25 February 2021; Accepted: 19 April 2021;
Published: 04 May 2021.
Edited by:
Dianwen Ju, Fudan University, ChinaReviewed by:
Hui Peng, University of Science and Technology of China, ChinaJianhua Yu, City of Hope National Medical Center, United States
Copyright © 2021 Xia, Wang, Wang, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi Wang, eGl3YW5nQGNjbXUuZWR1LmNu; Xulong Zhang, emh4bHdsQGNjbXUuZWR1LmNu