 Linda Quatrini
Linda Quatrini Biancamaria Ricci
Biancamaria Ricci Cecilia Ciancaglini
Cecilia Ciancaglini Nicola Tumino
Nicola Tumino Lorenzo Moretta
Lorenzo Moretta- Department of Immunology, IRCCS Bambino Gesù Children’s Hospital, Rome, Italy
Through the release of hormones, the neuro-endocrine system regulates the immune system function promoting adaptation of the organism to the external environment and to intrinsic physiological changes. Glucocorticoids (GCs) and sex hormones not only regulate immune responses, but also control the hematopoietic stem cell (HSC) differentiation and subsequent maturation of immune cell subsets. During the development of an organism, this regulation has long-term consequences. Indeed, the effects of GC exposure during the perinatal period become evident in the adulthood. Analogously, in the context of HSC transplantation (HSCT), the immune system development starts de novo from the donor HSCs. In this review, we summarize the effects of GCs and sex hormones on the regulation of HSC, as well as of adaptive and innate immune cells. Moreover, we discuss the short and long-term implications on hematopoiesis of sex steroid ablation and synthetic GC administration upon HSCT.
Introduction
Glucocorticoids (GCs) and the sex hormones estrogens, progesterone and androgens are produced under the control of the hypothalamic-pituitary-adrenal (HPA) and hypothalamic-pituitary-gonadal (HPG) axis, respectively, through a common steroidogenic pathway from cholesterol (1) (Figure 1). GC synthesis occurs in the cortical part of the adrenal gland for both males and females. Androgens are produced in male gonads and, in smaller amounts, in the ovary and adrenal cortex in females. Estrogens are mainly produced from androgens precursors in ovarian granulosa cells and placenta in females, testis in males and non-glandular tissue (fat and bone) in both sexes (2). Progesterone is produced by ovarian granulosa cells, adrenal glands, corpus luteum during the menstrual cycle and placenta (Figure 1) in females and by adrenal glands in both males and females (3).
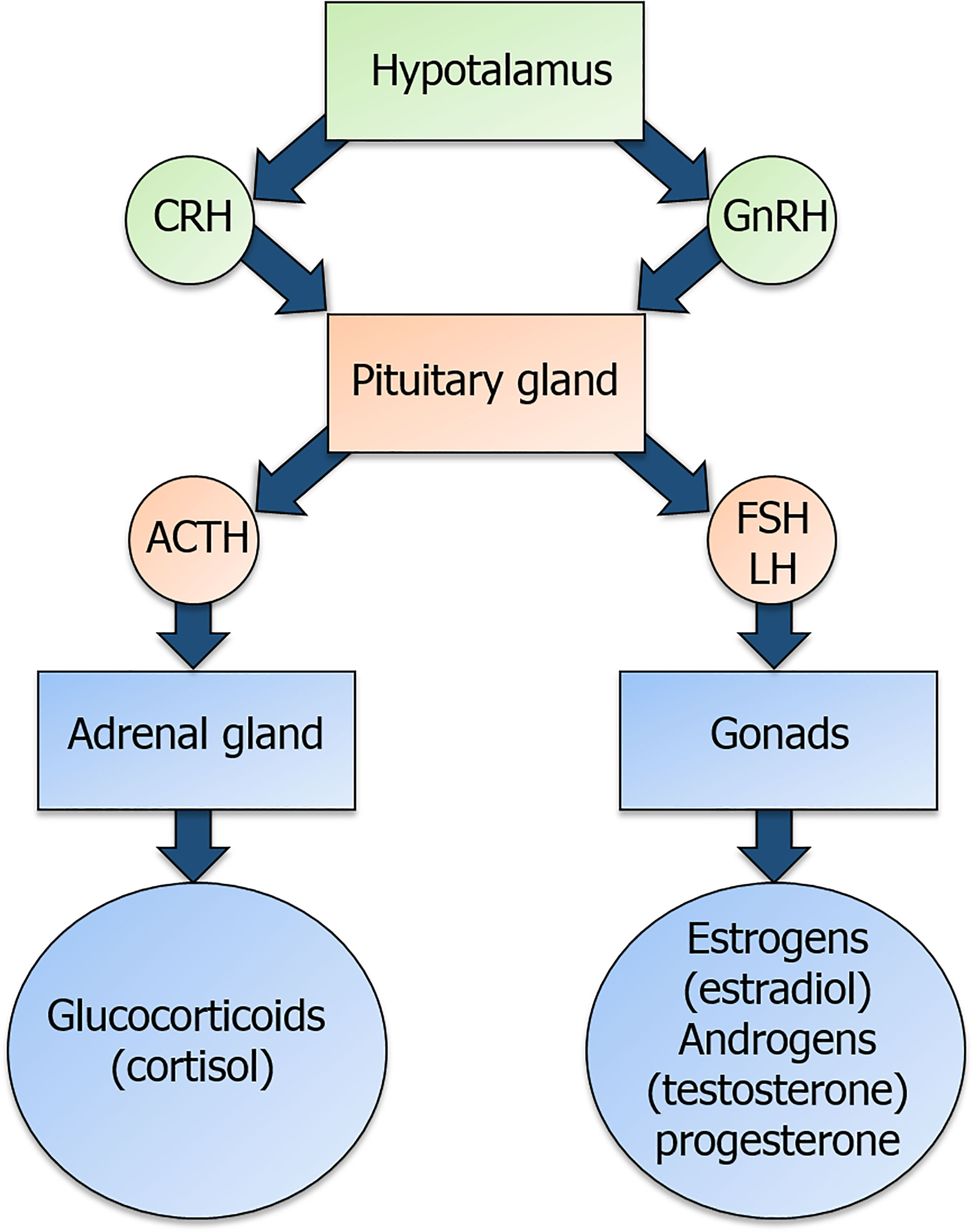
Figure 1 Overview of the hypothalamic-pituitary-adrenal (HPA) axis and of the hypothalamic-pituitary-gonadal (HPG) axis. Corticotropin-releasing hormone (CRH) and gonadotropin-releasing hormone (GnRH) are released by the hypothalamus and act on their receptors in the pituitary gland. In detail, CRH binding to its receptor triggers the production of the adrenocorticotropic-hormone (ACTH), while GnRH binding to its receptor triggers the production of the gonadotropins follicle-stimulating hormone (FSH) and luteinizing hormone (LH). ACTH and gonadotropins are released in the bloodstream and act on the adrenal gland and the gonads, respectively, to induce the secretion of glucocorticoids and sex hormones.
GCs are released into circulation in response to the circadian cycle, are essential regulators of body functions in homeostasis and allow adaptation to the environmental changes. Moreover, due to their potent immune-suppressive and anti-inflammatory effects, synthetic GCs are widely used in clinics to treat acute and chronic inflammation (4). On the other hand, besides regulating reproductive and metabolic body functions throughout the lifespan, sex hormones are also able to regulate immune response and inflammation. Indeed, a key example is given by the predominance of autoimmune diseases in females (5) and by sex dimorphism in antitumor immunity and response to infections (6, 7). In line with this, it was shown that estrogen-dependent protection of women from hepatitis C virus and liver cancer incidence are lost after menopause (8, 9). Age- and sex-related changes in immune response have therefore clinical consequences and should be taken into account because they may affect the efficacy of vaccination and cancer immunotherapy (10).
Because of their lipophilic nature, steroid hormones can cross cell membranes and bind to nuclear receptors belonging to the same superfamily of the ligand-regulated transcription factors (TFs) (11). In particular, they are the GC receptor (GR) and the sex hormone receptors estrogen receptor (ER) ERα and ERβ, progesterone receptor (PR), and androgen receptor (AR). These receptors are characterized by polymorphisms and multiple isoforms, responsible for variations in individual response at both molecular and clinical level. Steroid hormone receptors possess a N-terminal transactivation domain, a conserved central DNA binding domain and a C-terminal ligand-binding domain. In the cytoplasm, they are bound to chaperone proteins (e.g., HSP90). The binding of the ligand frees the receptor from the chaperone, allowing homodimerization, exposure of the nuclear localization sequence, and entry into the nucleus. Once in the nucleus, the ligand–receptor complex can directly bind to DNA sequences termed hormone response elements, and can associate with transcriptional coactivators to facilitate the regulation of target genes. It has been shown that steroid hormones can also mediate rapid “non-genomic” effects by acting directly on cytoplasmatic signal transduction pathways (12, 13). In addition, steroid hormones can alter gene expression through their effects on epigenetic modifications (14–16). For example, GR represses IL-5 transcription by recruiting and interacting with the Histone Deacetylase 1 (HDAC1) (17), and can inhibit p65-mediated transcription by recruiting HDAC2 (18).
Thus, the steroid hormone receptor-driven transcriptional regulation in a given cellular context may be mediated by at least two non-mutually exclusive mechanisms: chromatin remodeling and partnering with a specific repertoire of TFs (19).
Immune cells express receptors for GCs and sex hormones and are therefore regulated by these steroids. However, the response depends on many factors, including the developmental stage, the activation state and the inflammatory signals the cell receives at the same time. In this review, we will summarize the effects of GCs and sex hormones on the development of the immune system and how this regulation affects the immune response in the long term (Table 1).
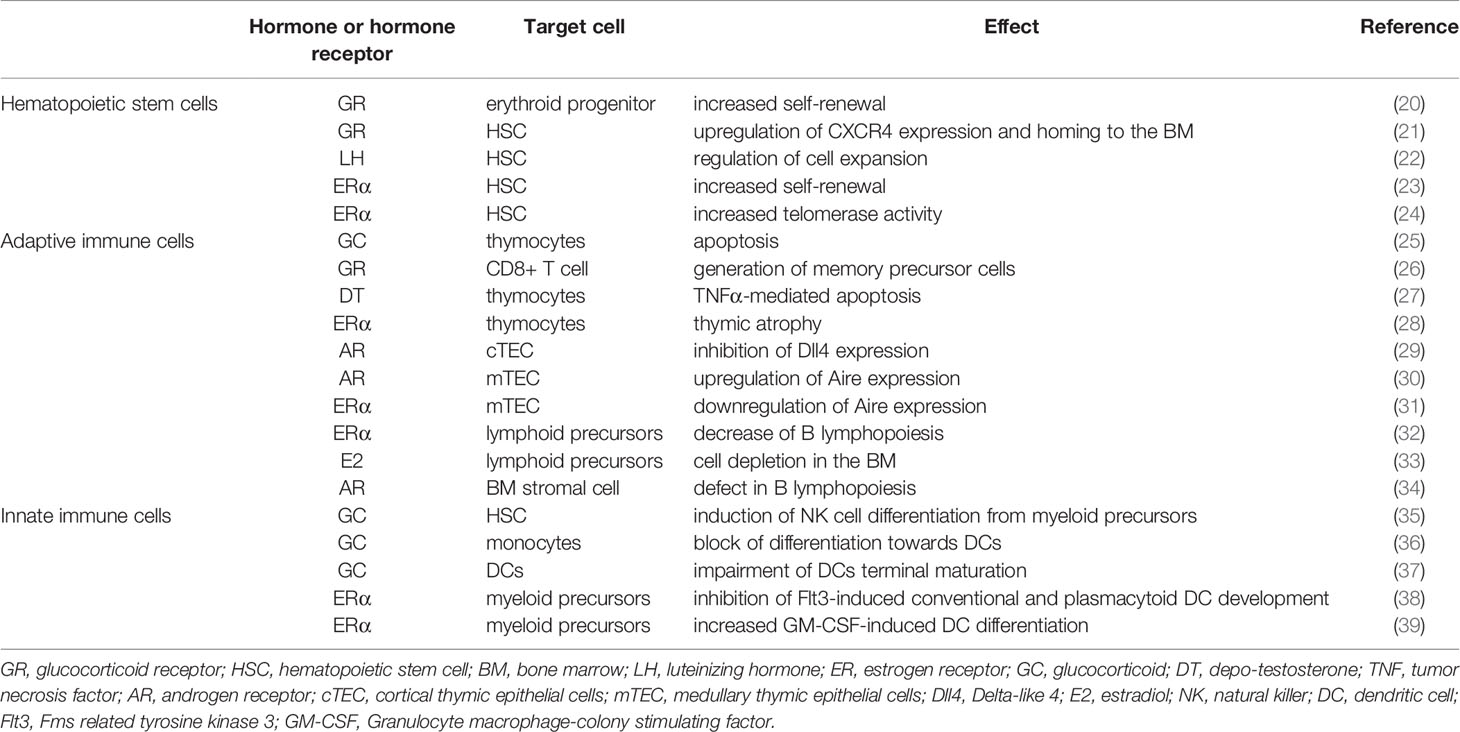
Table 1 Summary of the main effects of GCs and sex hormones on HSC, adaptive and innate immune cells.
GCs and Sex Hormones Regulation of HSC Development
Hematopoietic stem cells (HSCs) are multipotent self-renewing units mediating the generation of all blood constituents. An important site for the development of HSC in the embryo is the dorsal aorta, which contains a transient population of blood-producing endothelial cells called hemogenic endothelium (40). Studies on the role of GCs on the early development of immune system in the embryo were done in a zebrafish experimental model (41). It was shown that, upon stress, the central nervous system releases serotonin that, in turn, activates the HPA axis. GC release controls HSC specification promoting hematopoietic stem and progenitor cell (HSPC) formation in the dorsal aorta of embryos (41). These findings suggest that any stress that may be experienced by the embryo during development, such as hypoxia, temperature changes, metabolic or oxidative stress, would promote blood cell formation through activation of the HPA axis.
After birth, steroid hormones control the formation of immune cells by acting on HSCs in the bone marrow (BM). Studies have been performed in mouse models and on ex vivo cultures of human CD34+ HSCs to investigate this matter, especially with the purpose of developing therapeutic strategies to treat hematological diseases (42). For instance, it was suggested that GC treatment may be efficient to treat erythropoietin-resistant anemias because GR signaling regulates erythropoiesis. In particular, it was shown that GR synergizes with peroxisome proliferator-activated receptor α (PPAR-α) to induce self-renewal of erythroid progenitor cells in vivo and increase erythroid cell expansion ex vivo from human CD34+ HSCs (20).
HSC transplantation (HSCT) represents the common therapeutic approach to treat malignant and non-malignant hematological disorders. G-CSF-mediated mobilization of HSC has emerged as the most suitable means to obtain HSCs for transplantation, complemented by plerixafor (a selective CXCR4 antagonist) treatment in “poor-mobilizers” (43, 44). In mice, a neuroendocrine pathway that promotes HSC migration from the BM to the peripheral blood has been identified. It was shown that cholinergic signals through the muscarinic receptor type-1 (Chrm1) in the brain trigger HPA axis activation and GC production. GR signaling in HSC induces an up-regulation of actin-organizing molecules, thus promoting cell migration (45). HSC homing to the BM upon intravenous injection is mediated by coordinated actions of adhesion receptors and the chemokine CXCL12 expressed in the BM microenvironment (46). In a screen of small molecule compounds, GCs were identified as activators of CXCR4 expression in human CD34+ HSC isolated from cord blood (CB). It was shown that short term GC pretreatment is able to induce GR binding to a GC-response element in the CXCR4 promoter and SRC-1-p300 complex recruitment to induce H4K5 and H4K16 histone acetylation, favoring gene transcription. GC-induced CXCR4 up-regulation mediated HSC chemotaxis in response to CXCL12 and homing in the BM. Notably, although GC pretreatment was short, long-term engraftment was observed upon HSC transplantation into primary- and secondary-recipient NSG mice (21).
The regulation of HSC homeostasis by the sex hormones takes place after birth and affects their number and self-renewal capacity at different developmental stages. Luteinizing hormone (LH) is secreted by the pituitary gland at the onset of puberty, promoting the maturation of the reproductive system in both males and females. In post-natal BM, HSCs are a direct target of LH, whose signaling acts like a brake of cell overexpansion, ensuring a proper HSC count for normal hematopoiesis in adulthood (22). HSCs from mice treated with the estrogen estradiol (E2) have increased regenerative capacity after transplantation or irradiation, thus explaining why female immunodeficient recipient mice support reconstitution of the blood system by transplanted human HSCs more efficiently than the male counterpart (47). It was suggested that E2-ERα signaling in HSCs induces the expression of Ern1, which encodes Ire1a, thus activating the Ire1a-Xbp1 pathway of the unfolded protein response and promoting HSC resistance against proteotoxic stress (48). It was shown that Tamoxifen (a selective ER modulator) by signaling through ERα has a minor impact on primitive normal HSCs, but induces apoptosis in malignant HSCs enhancing chemotherapy response in a mouse model of acute myeloid leukemia (49). During pregnancy, ERα signaling in HSCs increases cell division, frequency, and erythropoiesis in the spleen of female mice (23). In particular, E2 promotes increased HSC self-renewal in the bone marrow, while another endogenous ligand for ERα (27-Hydroxycholesterol-27HC) promotes HSC mobilization to the spleen (50). Thus, these two ERα agonists collaborate to favor extramedullary hematopoiesis, required during pregnancy to maintain red blood cell counts despite a rapidly increasing blood volume.
Impaired DNA repair, altered DNA methylation patterns, aberrant metabolism and skewed upregulation of myeloid- (at the expense of lymphoid-) associated genes contribute to altering HSC functions with age (51). Both intrinsic functional changes in the earliest HSCs and extrinsic alterations of the HSC niche contribute to this degeneration (52). Evidence suggests that sex steroids play at least some roles in age-related decline of the immune functions (53) which, indeed, becomes more evident from the onset of puberty, when the levels of these hormones increase (54, 55). A comparison of healthy men and women’s PBMCs revealed a shared epigenomic signature of aging, including declining naïve T cell and increasing monocyte and cytotoxic cell functions. Age-related epigenomic changes first spike around late-thirties with similar timing and magnitude between sexes, whereas the second spike occurs earlier and stronger in men (56). Consistent with a role for sex steroids in the decline of the immune functions, sex steroid ablation (SSA) today represents an attractive therapeutic approach to restore immune competence in immunodeficient individuals (57). It has been shown that SSA has the potential to accelerate the immune recovery in many clinical conditions such as upon autologous and allogeneic HSCT, or after damaging cytoablative treatments like chemotherapy. SSA consists in therapeutically targeting the upstream signaling events such as LH releasing hormone (LHRH) or directly blocking sex steroid receptors (58, 59). This has been achieved clinically for over 35 years because, in addition to its well established role in improving thymopoiesis (see following section), several studies have found that SSA contrasts the age-related decline of early lymphoid progenitors in the BM. SSA does not enhance HSC homing to the BM, but increases their self-renewal and lymphoid differentiation capacities (60). These effects are due to intrinsic changes in HSCs, as well as to an extrinsic effect on the stromal microenvironment. In particular, SSA induces the upregulation of genes implicated in the protection from aging such as Foxo1 in the hematopoietic niche thus increasing its ability to support hematopoiesis (60). Interestingly, while SSA through direct and indirect mechanisms contrasts aging of early lymphoid progenitors, androgens have been shown to increase telomerase activity in primary HSCs (24). For its therapeutic effect on erythropoiesis, the androgen derivative Danazol is now used in the treatment of acquired severe aplastic anemia (61).
GCs and Sex Hormones Control the Development of the Adaptive Immune System
The thymus is a critical site for generating a diverse T cell repertoire while maintaining self-tolerance, and both GCs and sex hormones play a critical role in shaping thymic functions. In particular, GCs are crucial in the selection of the appropriate T cell receptor (TCR) self-affinities repertoire in the thymus (62). By dampening the strength of TCR signals, GCs prevent negative selection of T cells that have high affinity for self MHC (63). On the other hand, double positive thymocytes are the most sensitive to GC-induced apoptosis, and indeed stress response inducing GC production (including psychological stress, fasting, injury and infection) causes an acute reduction in thymus size (25, 64, 65). GCs also regulate the subsequent thymus-independent steps of T cell development. A characterization of the epigenetic landscapes of naïve, terminal-effector, memory precursor and memory CD8+ T cells revealed that the expression and binding of specific TFs contribute to the establishment of subset-specific enhancers during differentiation, which control the in vivo response to bacterial infections (26). One of these key TF is Nr3c1, encoding for GR, constitutively expressed during CD8+ T cell differentiation and involved in the generation of memory precursor cells (26). GR has an important role in T cell differentiation not only in physiological, but also in pathological conditions. Indeed, in tumor infiltrating lymphocytes, an increasing gradient of GR expression and signaling from naïve to dysfunctional CD8+ T cells was found, suggesting that endogenous GCs also promote T cell dysfunction and tumor growth (66).
Testosterone can directly induce apoptosis of double positive thymocytes through the upregulation of TNF-α, while estrogens can induce thymic atrophy by eliminating early thymic progenitors and inhibiting the proliferation of β-selected thymocytes (27, 28). They have also an indirect effect on thymopoiesis since the expression of sex steroid receptor is significantly higher on thymic epithelia cells than on thymocytes. Indeed, sex steroids inhibit in cortical thymic epithelial cells (cTECs) the expression of Delta-like 4 (Dll4), a Notch ligand crucial for the commitment and differentiation of T cell progenitors in a dose-dependent manner (29). It was demonstrated that androgen-response elements are present in the promoter of the Dll4 gene and androgen/AR complexes were localized to the Dll4 promoter by chromatin immunoprecipitation (29). Sex hormones also control the expression of Aire (Autoimmune Regulator), the transcription factor responsible for the expression of thousands of tissue-restricted proteins in cells deputed to the presentation of self-antigens to the maturing T lymphocytes. By upregulating Aire expression in medullary TEC (mTEC) by directly binding its promoter, androgens enforce self-tolerance and lead to a more efficient negative selection of self-reactive T cells (30). At the same time, estrogens can decrease Aire expression by epigenetically regulating its methylation (31). Indeed, Aire expression in mice and human thymus is higher in males compared to females, suggesting that increased levels of androgens and decreased levels of estrogens protect males from autoimmunity by promoting Aire expression. All of these molecular mechanisms identified for sex hormones explain why SSA increases thymic cellularity, restores thymic architecture and organization, and enhances thymopoiesis in young and adult animals (58).
Age-related immune system decline is not only restricted to thymic atrophy, but also to an intrinsic defect in the HSC ability to commit to the lymphoid lineage. Therefore, with age there is a significant decline in B lymphopoiesis and humoral immunity, accompanied by a reduced peripheral immune repertoire, leading to increased opportunistic infections and limited recovery following cytoablation after chemo or radiotherapy (67). Estrogens reduce responsiveness to IL-7 causing decreased B lymphopoiesis (32) and deplete lymphoid committed precursors in the BM (33). Androgens affect B lymphopoiesis indirectly, by acting on BM stromal cells (34). Following surgical SSA in mice displaying age- or chemotherapy-induced immunodepletion, a rapid and sustained increase in developing B cells and their upstream lymphoid progenitors was observed. SSA not only increased B cell number, but also enhanced humoral response to challenge by hepatitis B vaccine (68).
GCs and Sex Hormones Regulation of Innate Immune Cells
Innate lymphoid cells (ILCs) differentiate from HSC in the BM, and common ILC precursors continue their development in secondary lymphoid tissues (69). GC treatment of human CB CD34+ HSCs accelerated Natural Killer (NK) cell differentiation, promoted a switch of myeloid precursors towards immature NK cells and induced NK cell cytolytic activity (35). Although a role for the neuroendocrine system in the regulation of helper ILC functions has been shown (70), its contribution to their development has not been investigated so far.
In mice, GC exposure favors myelopoiesis in the bone marrow, suggesting that preservation of granulocytes and their progenitors may be a mechanism to ensure rapid protection of the organism upon stress (71). Neutrophil development and function are modulated also by sex hormones. In particular, healthy young adult women display an activated/mature neutrophil profile characterized by enhanced type I IFN pathway activity, enhanced proinflammatory responses, and distinct immunometabolism compared to young men (72).
Blood monocyte precursors originate in the BM, enter the circulation and are present in the blood until they migrate into tissues where they can differentiate into macrophages and dendritic cells (DC) (73). GCs are long known to act on monocytes influencing their short time mediator release, but there is growing evidence that they are also involved in differentiation processes skewing towards an anti-inflammatory phenotype (74). GCs block the generation of immature DCs from monocytes (36) and impair terminal maturation of already differentiated DCs (37). Similarly, ER signaling inhibits Flt3-induced conventional and plasmacytoid DC development from myeloid precursors (38). On the contrary, it was shown that GM-CSF-induced DC differentiation as Langerhans cells from myeloid progenitors was promoted by adding E2 in vitro and inhibited by ER antagonists and ERα-deficiency (39). Indeed, ERα signaling during this process targets IRF4, a TF critical for Langerhans cell development (75).
Interestingly, mast cell cytokine production seems to be sex dependent. Indeed, in females they mainly produce proinflammatory cytokines, such as TNF and IL-1β, while their activation in males results in a predominant production of IL-33 (76). Although both male and female derived mast cells express AR, testosterone induces IL-33 production only in male-derived cells in vitro (77). These data suggest that, in addition to a direct effect, testosterone may have other effects during development that poise mast cells for IL-33 expression potential, perhaps at an epigenetic level. Therefore, the ability to express a particular array of cytokines may be programmed by prolonged exposure to sex hormones starting early in life and is not solely due to acute sex hormone-receptor interactions.
Conclusions and Future Perspectives: A Role for GCs and Sex Hormones in the Developmental Programming of Immune System?
Early life exposure to environmental cues, particularly during the perinatal period, can have a life-long impact on the organism development and physiology. The biological rationale for this phenomenon is to promote physiologic adaptation to the anticipated environment based on early life experience. A role in this “developmental programming” for early life stress or prenatal treatment with synthetic GCs during sensitive windows of development has been established (78). Excessive or premature exposure to GCs has been associated to long term effects on tissues and organs (hypertension, hyperglycemia, anxiety), as well as to higher risks for atopic diseases, asthma, autoimmune type I diabetes, infectious diseases and decreased adaptive and anti-tumor immune response (79–83). GC-induced programming of the immune system is mediated by effects on HSCs or other persistent progenitors, which endure through the individual’s lifespan (84, 85), rather than upon the short-lived fully differentiated immune cell populations. The long-term consequences of perinatal GC exposure were recently evaluated in mice in a study by putting the synthetic GC dexamethasone in the drinking water from mid-pregnancy to early post-natal period, when hematological and lymphoid organogenesis takes place (86). As a result, the authors observed a diminished CD8+ T cell response in adulthood and impaired control of tumor growth and bacterial infections. Perinatal GC exposure led to HPA axis reprogramming with alterations in its threshold, decreased systemic levels of GCs and persistent changes in the chromatin state of naïve T cells (86). The gene expression program was “imprinted”, as the changes were maintained also upon cell adoptive transfer to new environments and, importantly, the stem cell compartment contributed to compromised T cell responses, as the defect could be observed also in irradiated mice transplanted with the BM of GC-treated mice (86).
These findings suggest that GCs, as well as the other hormones whose receptors function as TFs, may be able to induce an “imprinting” in HSC gene expression program, most likely at the epigenetic level, thus stably affecting HSC developmental trajectories. This kind of regulation that occurs during organism development may be recapitulated upon HSCT when the immune system development from donor-derived HSCs starts de novo. Since SSA and GCs are widely used to improve HSCT outcome and prevent complications such as graft vs host disease, respectively, it would be interesting to gain further insights into the short and long term consequences of these treatments on hematopoiesis. A better understanding of HSC regulation by GCs and sex hormones is extremely important to predict their effects on the immune system development and design therapeutic approaches to improve the long-term reconstitution in HSCT recipients.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants awarded by Associazione Italiana per la Ricerca sul Cancro (AIRC)-Special Program Metastatic disease: the key unmet need in oncology 5X1000 2018 Id. 21147 (LM), AIRC IG 2017 Id. 19920 (LM), and RC-2020 OPBG (LM). LQ has received funding from AIRC and from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no 800924.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Miller WL, Auchus RJ. The Molecular Biology, Biochemistry, and Physiology of Human Steroidogenesis and Its Disorders. Endocrine Rev (2011) 32(1):81–151. doi: 10.1210/er.2010-0013
2. Vermeulen A, Kaufman JM, Goemaere S, van Pottelberg I. Estradiol in Elderly Men. Aging male Off J Int Soc Study Aging Male (2002) 5(2):98–102. doi: 10.1080/tam.5.2.98.102
3. Guennoun R, Labombarda F, Gonzalez Deniselle MC, Liere P, De Nicola AF, Schumacher M. Progesterone and Allopregnanolone in the Central Nervous System: Response to Injury and Implication for Neuroprotection. J Steroid Biochem Mol Biol (2015) 146:48–61. doi: 10.1016/j.jsbmb.2014.09.001
4. Quatrini L, Ugolini S. New Insights Into the Cell- and Tissue-Specificity of Glucocorticoid Actions. Cell Mol Immunol (2020) 18(2):269–78. doi: 10.1038/s41423-020-00526-2
5. Moulton VR. Sex Hormones in Acquired Immunity and Autoimmune Disease. Front Immunol (2018) 9:2279. doi: 10.3389/fimmu.2018.02279
6. Klein SL, Flanagan KL. Sex Differences in Immune Responses. Nat Rev Immunol (2016) 16(10):626–38. doi: 10.1038/nri.2016.90
7. Jaillon S, Berthenet K, Garlanda C. Sexual Dimorphism in Innate Immunity. Clin Rev Allergy Immunol (2019) 56(3):308–21. doi: 10.1007/s12016-017-8648-x
8. Baden R, Rockstroh JK, Buti M. Natural History and Management of Hepatitis C: Does Sex Play a Role? J Infect Dis (2014) 209(Suppl 3):S81–5. doi: 10.1093/infdis/jiu057
9. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender Disparity in Liver Cancer Due to Sex Differences in Dependent IL-6 Production. Science (2007) 317(5834):121–4. doi: 10.1126/science.1140485
10. Gubbels Bupp MR, Potluri T, Fink AL, Klein SL. The Confluence of Sex Hormones and Aging on Immunity. Front Immunol (2018) 9:1269. doi: 10.3389/fimmu.2018.01269
11. Cain DW, Cidlowski JA. Immune Regulation by Glucocorticoids. Nat Rev Immunol (2017) 17(4):233–47. doi: 10.1038/nri.2017.1
12. Kovats S. Estrogen Receptors Regulate Innate Immune Cells and Signaling Pathways. Cell Immunol (2015) 294(2):63–9. doi: 10.1016/j.cellimm.2015.01.018
13. Buttgereit F, Scheffold A. Rapid Glucocorticoid Effects on Immune Cells. Steroids (2002) 67(6):529–34. doi: 10.1016/s0039-128x(01)00171-4
14. Mann M, Cortez V, Vadlamudi RK. Epigenetics of Estrogen Receptor Signaling: Role in Hormonal Cancer Progression and Therapy. Cancers (2011) 3(3):1691–707. doi: 10.3390/cancers3021691
15. Walecki M, Eisel F, Klug J, Baal N, Paradowska-Dogan A, Wahle E, et al. Androgen Receptor Modulates Foxp3 Expression in CD4+CD25+Foxp3+ Regulatory T-Cells. Mol Biol Cell (2015) 26(15):2845–57. doi: 10.1091/mbc.E14-08-1323
16. Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, et al. How Glucocorticoid Receptors Modulate the Activity of Other Transcription Factors: A Scope Beyond Tethering. Mol Cell Endocrinol (2013) 380(1-2):41–54. doi: 10.1016/j.mce.2012.12.014
17. Jee YK, Gilmour J, Kelly A, Bowen H, Richards D, Soh C, et al. Repression of Interleukin-5 Transcription by the Glucocorticoid Receptor Targets GATA3 Signaling and Involves Histone Deacetylase Recruitment. J Biol Chem (2005) 280(24):23243–50. doi: 10.1074/jbc.M503659200
18. Ito K, Barnes PJ, Adcock IM. Glucocorticoid Receptor Recruitment of Histone Deacetylase 2 Inhibits Interleukin-1Beta-induced Histone H4 Acetylation on Lysines 8 and 12. Mol Cell Biol (2000) 20(18):6891–903. doi: 10.1128/mcb.20.18.6891-6903.2000
19. Weikum ER, Knuesel MT, Ortlund EA, Yamamoto KR. Glucocorticoid Receptor Control of Transcription: Precision and Plasticity Via Allostery. Nat Rev Mol Cell Biol (2017) 18(3):159–74. doi: 10.1038/nrm.2016.152
20. Lee HY, Gao X, Barrasa MI, Li H, Elmes RR, Peters LL, et al. PPAR-Alpha and Glucocorticoid Receptor Synergize to Promote Erythroid Progenitor Self-Renewal. Nature (2015) 522(7557):474–7. doi: 10.1038/nature14326
21. Guo B, Huang X, Cooper S, Broxmeyer HE. Glucocorticoid Hormone-Induced Chromatin Remodeling Enhances Human Hematopoietic Stem Cell Homing and Engraftment. Nat Med (2017) 23(4):424–8. doi: 10.1038/nm.4298
22. Peng YJ, Yu H, Hao X, Dong W, Yin X, Lin M, et al. Luteinizing Hormone Signaling Restricts Hematopoietic Stem Cell Expansion During Puberty. EMBO J (2018) 37(17):e98984. doi: 10.15252/embj.201898984
23. Nakada D, Oguro H, Levi BP, Ryan N, Kitano A, Saitoh Y, et al. Oestrogen Increases Haematopoietic Stem-Cell Self-Renewal in Females and During Pregnancy. Nature (2014) 505(7484):555–8. doi: 10.1038/nature12932
24. Calado RT, Yewdell WT, Wilkerson KL, Regal JA, Kajigaya S, Stratakis CA, et al. Sex Hormones, Acting on the TERT Gene, Increase Telomerase Activity in Human Primary Hematopoietic Cells. Blood (2009) 114(11):2236–43. doi: 10.1182/blood-2008-09-178871
25. Tarcic N, Ovadia H, Weiss DW, Weidenfeld J. Restraint Stress-Induced Thymic Involution and Cell Apoptosis Are Dependent on Endogenous Glucocorticoids. J Neuroimmunol (1998) 82(1):40–6. doi: 10.1016/S0165-5728(97)00186-0
26. Yu B, Zhang K, Milner JJ, Toma C, Chen R, Scott-Browne JP, et al. Epigenetic Landscapes Reveal Transcription Factors That Regulate CD8(+) T Cell Differentiation. Nat Immunol (2017) 18(5):573–82. doi: 10.1038/ni.3706
27. Guevara Patino JA, Marino MW, Ivanov VN, Nikolich-Zugich J. Sex Steroids Induce Apoptosis of CD8+CD4+ Double-Positive Thymocytes Via TNF-Alpha. Eur J Immunol (2000) 30(9):2586–92. doi: 10.1002/1521-4141(200009)30:9<2586::AID-IMMU2586>3.0.CO;2-L
28. Zoller AL, Kersh GJ. Estrogen Induces Thymic Atrophy by Eliminating Early Thymic Progenitors and Inhibiting Proliferation of Beta-Selected Thymocytes. J Immunol (2006) 176(12):7371–8. doi: 10.4049/jimmunol.176.12.7371
29. Velardi E, Tsai JJ, Holland AM, Wertheimer T, Yu VW, Zakrzewski JL, et al. Sex Steroid Blockade Enhances Thymopoiesis by Modulating Notch Signaling. J Exp Med (2014) 211(12):2341–9. doi: 10.1084/jem.20131289
30. Zhu ML, Bakhru P, Conley B, Nelson JS, Free M, Martin A, et al. Sex Bias in CNS Autoimmune Disease Mediated by Androgen Control of Autoimmune Regulator. Nat Commun (2016) 7:11350. doi: 10.1038/ncomms11350
31. Dragin N, Bismuth J, Cizeron-Clairac G, Biferi MG, Berthault C, Serraf A, et al. Estrogen-Mediated Downregulation of AIRE Influences Sexual Dimorphism in Autoimmune Diseases. J Clin Invest (2016) 126(4):1525–37. doi: 10.1172/JCI81894
32. Medina KL, Kincade PW. Pregnancy-Related Steroids are Potential Negative Regulators of B Lymphopoiesis. Proc Natl Acad Sci USA (1994) 91(12):5382–6. doi: 10.1073/pnas.91.12.5382
33. Medina KL, Garrett KP, Thompson LF, Rossi MI, Payne KJ, Kincade PW. Identification of Very Early Lymphoid Precursors in Bone Marrow and Their Regulation by Estrogen. Nat Immunol (2001) 2(8):718–24. doi: 10.1038/90659
34. Olsen NJ, Gu X, Kovacs WJ. Bone Marrow Stromal Cells Mediate Androgenic Suppression of B Lymphocyte Development. J Clin Invest (2001) 108(11):1697–704. doi: 10.1172/JCI13183
35. Vitale C, Cottalasso F, Montaldo E, Moretta L, Mingari MC. Methylprednisolone Induces Preferential and Rapid Differentiation of CD34+ Cord Blood Precursors Toward NK Cells. Int Immunol (2008) 20(4):565–75. doi: 10.1093/intimm/dxn014
36. Woltman AM, de Fijter JW, Kamerling SW, Paul LC, Daha MR, van Kooten C. The Effect of Calcineurin Inhibitors and Corticosteroids on the Differentiation of Human Dendritic Cells. Eur J Immunol (2000) 30(7):1807–12. doi: 10.1002/1521-4141(200007)30:7<1807::AID-IMMU1807>3.0.CO;2-N
37. Piemonti L, Monti P, Allavena P, Sironi M, Soldini L, Leone BE, et al. Glucocorticoids Affect Human Dendritic Cell Differentiation and Maturation. J Immunol (1999) 162(11):6473–81. doi: 10.1093/intimm/11.9.1519
38. Carreras E, Turner S, Paharkova-Vatchkova V, Mao A, Dascher C, Kovats S. Estradiol Acts Directly on Bone Marrow Myeloid Progenitors to Differentially Regulate GM-CSF or Flt3 Ligand-Mediated Dendritic Cell Differentiation. J Immunol (2008) 180(2):727–38. doi: 10.4049/jimmunol.180.2.727
39. Paharkova-Vatchkova V, Maldonado R, Kovats S. Estrogen Preferentially Promotes the Differentiation of CD11c+ CD11b(Intermediate) Dendritic Cells From Bone Marrow Precursors. J Immunol (2004) 172(3):1426–36. doi: 10.4049/jimmunol.172.3.1426
40. Tober J, Maijenburg MW, Speck NA. Taking the Leap: Runx1 in the Formation of Blood From Endothelium. Curr Topics Dev Biol (2016) 118:113–62. doi: 10.1016/bs.ctdb.2016.01.008
41. Kwan W, Cortes M, Frost I, Esain V, Theodore LN, Liu SY, et al. The Central Nervous System Regulates Embryonic Hspc Production Via Stress-Responsive Glucocorticoid Receptor Signaling. Cell Stem Cell (2016) 19(3):370–82. doi: 10.1016/j.stem.2016.06.004
42. Guo B, Huang X, Broxmeyer HE. Enhancing Human Cord Blood Hematopoietic Stem Cell Engraftment by Targeting Nuclear Hormone Receptors. Curr Opin Hematol (2018) 25(4):245–52. doi: 10.1097/MOH.0000000000000429
43. Levesque JP, Winkler IG. Mobilization of Hematopoietic Stem Cells: State of the Art. Curr Opin Organ Transplant (2008) 13(1):53–8. doi: 10.1097/MOT.0b013e3282f42473
44. To LB, Levesque JP, Herbert KE. How I Treat Patients Who Mobilize Hematopoietic Stem Cells Poorly. Blood (2011) 118(17):4530–40. doi: 10.1182/blood-2011-06-318220
45. Pierce H, Zhang D, Magnon C, Lucas D, Christin JR, Huggins M, et al. Cholinergic Signals From the CNS Regulate G-CSF-Mediated Hsc Mobilization From Bone Marrow Via a Glucocorticoid Signaling Relay. Cell Stem Cell (2017) 20(5):648–58.e4. doi: 10.1016/j.stem.2017.01.002
46. Magnon C, Frenette PS. Hematopoietic Stem Cell Trafficking. In: Stembook. Cambridge (MA): Harvard Stem Cell Institute (2008).
47. Notta F, Doulatov S, Dick JE. Engraftment of Human Hematopoietic Stem Cells Is More Efficient in Female NOD/SCID/IL-2Rgc-Null Recipients. Blood (2010) 115(18):3704–7. doi: 10.1182/blood-2009-10-249326
48. Chapple RH, Hu T, Tseng YJ, Liu L, Kitano A, Luu V, et al. Eralpha Promotes Murine Hematopoietic Regeneration Through the Ire1alpha-mediated Unfolded Protein Response. eLife (2018) 7:e31159. doi: 10.7554/eLife.31159
49. Sanchez-Aguilera A, Arranz L, Martin-Perez D, Garcia-Garcia A, Stavropoulou V, Kubovcakova L, et al. Estrogen Signaling Selectively Induces Apoptosis of Hematopoietic Progenitors and Myeloid Neoplasms Without Harming Steady-State Hematopoiesis. Cell Stem Cell (2014) 15(6):791–804. doi: 10.1016/j.stem.2014.11.002
50. Oguro H, McDonald JG, Zhao Z, Umetani M, Shaul PW, Morrison SJ. 27-Hydroxycholesterol Induces Hematopoietic Stem Cell Mobilization and Extramedullary Hematopoiesis During Pregnancy. J Clin Invest (2017) 127(9):3392–401. doi: 10.1172/JCI94027
51. Geiger H, de Haan G, Florian MC. The Ageing Haematopoietic Stem Cell Compartment. Nat Rev Immunol (2013) 13(5):376–89. doi: 10.1038/nri3433
52. Woolthuis CM, de Haan G, Huls G. Aging of Hematopoietic Stem Cells: Intrinsic Changes or Micro-Environmental Effects? Curr Opin Immunol (2011) 23(4):512–7. doi: 10.1016/j.coi.2011.05.006
53. Chinn IK, Blackburn CC, Manley NR, Sempowski GD. Changes in Primary Lymphoid Organs With Aging. Semin Immunol (2012) 24(5):309–20. doi: 10.1016/j.smim.2012.04.005
54. Medina KL, Strasser A, Kincade PW. Estrogen Influences the Differentiation, Proliferation, and Survival of Early Lineage Precursors. Blood (2000) 95(6):2059–67. doi: 10.1182/blood.V95.6.2059
55. Smithson G, Couse JF, Lubahn DB, Korach KS, Kincade PW. The Role of Estrogen Receptors and Androgen Receptors in Sex Steroid Regulation of B Lymphopoiesis. J Immunol (1998) 161(1):27–34.
56. Marquez EJ, Chung CH, Marches R, Rossi RJ, Nehar-Belaid D, Eroglu A, et al. Sexual-Dimorphism in Human Immune System Aging. Nat Commun (2020) 11(1):751. doi: 10.1038/s41467-020-14396-9
57. Velardi E, Dudakov JA, van den Brink MR. Sex Steroid Ablation: An Immunoregenerative Strategy for Immunocompromised Patients. Bone Marrow Transplant (2015) 50 Suppl 2:S77–81. doi: 10.1038/bmt.2015.101
58. Goldberg GL, King CG, Nejat RA, Suh DY, Smith OM, Bretz JC, et al. Luteinizing Hormone-Releasing Hormone Enhances T Cell Recovery Following Allogeneic Bone Marrow Transplantation. J Immunol (2009) 182(9):5846–54. doi: 10.4049/jimmunol.0801458
59. Lai KP, Lai JJ, Chang P, Altuwaijri S, Hsu JW, Chuang KH, et al. Targeting Thymic Epithelia AR Enhances T-Cell Reconstitution and Bone Marrow Transplant Grafting Efficacy. Mol Endocrinol (2013) 27(1):25–37. doi: 10.1210/me.2012-1244
60. Khong DM, Dudakov JA, Hammett MV, Jurblum MI, Khong SM, Goldberg GL, et al. Enhanced Hematopoietic Stem Cell Function Mediates Immune Regeneration Following Sex Steroid Blockade. Stem Cell Rep (2015) 4(3):445–58. doi: 10.1016/j.stemcr.2015.01.018
61. Jaime-Perez JC, Colunga-Pedraza PR, Gomez-Ramirez CD, Gutierrez-Aguirre CH, Cantu-Rodriguez OG, Tarin-Arzaga LC, et al. Danazol as First-Line Therapy for Aplastic Anemia. Ann Hematol (2011) 90(5):523–7. doi: 10.1007/s00277-011-1163-x
62. Brown MA, Su MA. An Inconvenient Variable: Sex Hormones and Their Impact on T Cell Responses. J Immunol (2019) 202(7):1927–33. doi: 10.4049/jimmunol.1801403
63. Taves MD, Ashwell JD. Glucocorticoids in T Cell Development, Differentiation and Function. Nat Rev Immunol (2020) 21(4):233–43. doi: 10.1038/s41577-020-00464-0
64. Dominguez-Gerpe L, Rey-Mendez M. Time-Course of the Murine Lymphoid Tissue Involution During and Following Stressor Exposure. Life Sci (1997) 61(10):1019–27. doi: 10.1016/s0024-3205(97)00606-1
65. Ruzek MC, Pearce BD, Miller AH, Biron CA. Endogenous Glucocorticoids Protect Against Cytokine-Mediated Lethality During Viral Infection. J Immunol (1999) 162(6):3527–33.
66. Acharya N, Madi A, Zhang H, Klapholz M, Escobar G, Dulberg S, et al. Endogenous Glucocorticoid Signaling Regulates Cd8(+) T Cell Differentiation and Development of Dysfunction in the Tumor Microenvironment. Immunity (2020) 53(3):658–71.e6. doi: 10.1016/j.immuni.2020.08.005
67. Frasca D, Diaz A, Romero M, Landin AM, Blomberg BB. Age Effects on B Cells and Humoral Immunity in Humans. Ageing Res Rev (2011) 10(3):330–5. doi: 10.1016/j.arr.2010.08.004
68. Dudakov JA, Goldberg GL, Reiseger JJ, Chidgey AP, Boyd RL. Withdrawal of Sex Steroids Reverses Age- and Chemotherapy-Related Defects in Bone Marrow Lymphopoiesis. J Immunol (2009) 182(10):6247–60. doi: 10.4049/jimmunol.0802446
69. Quatrini L, Tumino N, Moretta F, Besi F, Vacca P, Moretta L. Helper Innate Lymphoid Cells in Allogenic Hematopoietic Stem Cell Transplantation and Graft Versus Host Disease. Front Immunol (2020) 11:582098. doi: 10.3389/fimmu.2020.582098
70. Quatrini L, Vivier E, Ugolini S. Neuroendocrine Regulation of Innate Lymphoid Cells. Immunological Rev (2018) 286(1):120–36. doi: 10.1111/imr.12707
71. Trottier MD, Newsted MM, King LE, Fraker PJ. Natural Glucocorticoids Induce Expansion of All Developmental Stages of Murine Bone Marrow Granulocytes Without Inhibiting Function. Proc Natl Acad Sci USA (2008) 105(6):2028–33. doi: 10.1073/pnas.0712003105
72. Gupta S, Nakabo S, Blanco LP, O’Neil LJ, Wigerblad G, Goel RR, et al. Sex Differences in Neutrophil Biology Modulate Response to Type I Interferons and Immunometabolism. Proc Natl Acad Sci USA (2020) 117(28):16481–91. doi: 10.1073/pnas.2003603117
73. Ginhoux F, Guilliams M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity (2016) 44(3):439–49. doi: 10.1016/j.immuni.2016.02.024
74. Yona S, Gordon S. Inflammation: Glucocorticoids Turn the Monocyte Switch. Immunol Cell Biol (2007) 85(2):81–2. doi: 10.1038/sj.icb.7100034
75. Carreras E, Turner S, Frank MB, Knowlton N, Osban J, Centola M, et al. Estrogen Receptor Signaling Promotes Dendritic Cell Differentiation by Increasing Expression of the Transcription Factor IRF4. Blood (2010) 115(2):238–46. doi: 10.1182/blood-2009-08-236935
76. Christy AL, Walker ME, Hessner MJ, Brown MA. Mast Cell Activation and Neutrophil Recruitment Promotes Early and Robust Inflammation in the Meninges in EAE. J Autoimmun (2013) 42:50–61. doi: 10.1016/j.jaut.2012.11.003
77. Russi AE, Ebel ME, Yang Y, Brown MA. Male-Specific IL-33 Expression Regulates Sex-Dimorphic EAE Susceptibility. Proc Natl Acad Sci USA (2018) 115(7):E1520–E9. doi: 10.1073/pnas.1710401115
78. Solano ME, Holmes MC, Mittelstadt PR, Chapman KE, Tolosa E. Antenatal Endogenous and Exogenous Glucocorticoids and Their Impact on Immune Ontogeny and Long-Term Immunity. Semin Immunopathol (2016) 38(6):739–63. doi: 10.1007/s00281-016-0575-z
79. Beijers R, Jansen J, Riksen-Walraven M, de Weerth C. Maternal Prenatal Anxiety and Stress Predict Infant Illnesses and Health Complaints. Pediatrics (2010) 126(2):e401–9. doi: 10.1542/peds.2009-3226
80. Nielsen NM, Hansen AV, Simonsen J, Hviid A. Prenatal Stress and Risk of Infectious Diseases in Offspring. Am J Epidemiol (2011) 173(9):990–7. doi: 10.1093/aje/kwq492
81. Henriksen RE, Thuen F. Marital Quality and Stress in Pregnancy Predict the Risk of Infectious Disease in the Offspring: The Norwegian Mother and Child Cohort Study. PloS One (2015) 10(9):e0137304. doi: 10.1371/journal.pone.0137304
82. O’Connor TG, Winter MA, Hunn J, Carnahan J, Pressman EK, Glover V, et al. Prenatal Maternal Anxiety Predicts Reduced Adaptive Immunity in Infants. Brain Behavior Immun (2013) 32:21–8. doi: 10.1016/j.bbi.2013.02.002
83. Witek Janusek L, Tell D, Albuquerque K, Mathews HL. Childhood Adversity Increases Vulnerability for Behavioral Symptoms and Immune Dysregulation in Women With Breast Cancer. Brain Behavior Immun (2013) 30 Suppl:S149–62. doi: 10.1016/j.bbi.2012.05.014
84. Mikkola HK, Orkin SH. The Journey of Developing Hematopoietic Stem Cells. Development (2006) 133(19):3733–44. doi: 10.1242/dev.02568
85. Moisiadis VG, Matthews SG. Glucocorticoids and Fetal Programming Part 2: Mechanisms. Nat Rev Endocrinol (2014) 10(7):403–11. doi: 10.1038/nrendo.2014.74
Keywords: glucocorticoids, sex hormones, hematopoietic stem cell transplantation, hematopoietic stem and progenitor cell, immune system development
Citation: Quatrini L, Ricci B, Ciancaglini C, Tumino N and Moretta L (2021) Regulation of the Immune System Development by Glucocorticoids and Sex Hormones. Front. Immunol. 12:672853. doi: 10.3389/fimmu.2021.672853
Received: 26 February 2021; Accepted: 08 June 2021;
Published: 23 June 2021.
Edited by:
Chander Raman, University of Alabama at Birmingham, United StatesReviewed by:
Michele Ardolino, University of Ottawa, CanadaCristina Tecchio, University of Verona, Italy
Christoph Baerwald, Leipzig University, Germany
Copyright © 2021 Quatrini, Ricci, Ciancaglini, Tumino and Moretta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linda Quatrini, bGluZGEucXVhdHJpbmlAb3BiZy5uZXQ=