 Marina Saade
Marina Saade Giovanna Araujo de Souza
Giovanna Araujo de Souza Cristoforo Scavone
Cristoforo Scavone Paula Fernanda Kinoshita
Paula Fernanda Kinoshita- Department of Pharmacology, Institute of Biomedical Science, University of São Paulo, São Paulo, Brazil
Inflammation is a response to a lesion in the tissue or infection. This process occurs in a specific manner in the central nervous system and is called neuroinflammation, which is involved in neurodegenerative diseases. GPNMB, an endogenous glycoprotein, has been recently related to inflammation and neuroinflammation. GPNMB is highly expressed in macrophages and microglia, which are cells involved with innate immune response in the periphery and the brain, respectively. Some studies have shown increased levels of GPNMB in pro-inflammatory conditions, such as LPS treatment, and in pathological conditions, such as neurodegenerative diseases and cancer. However, the role of GPNMB in inflammation is still not clear. Even though most studies suggest that GPNMB might have an anti-inflammatory role by promoting inflammation resolution, there is evidence that GPNMB could be pro-inflammatory. In this review, we gather and discuss the published evidence regarding this interaction.
Inflammation
Inflammation is a complex response to microbial infections or tissue damage involving interactions between soluble molecules and cellular effectors to restore homeostasis (1–3). Tissue-resident macrophages and mast cells are relevant to an efficient and rapid immune response that prevents the infection from spreading and/or tissue damage. Those cells can monitor other cells and tissues by sensing any disbalance in homeostasis (3).
Macrophages are phagocytic cells of the innate immune system which are responsible for recognizing an infection, presenting antigens, and also for the production of inflammatory mediators, the removal of apoptotic cells, and the output of growth-factors (1, 4). Macrophages can be polarized in two different subsets that differ in cytokine production, iron, and glucose metabolism (5). The subtype called M1 is activated by pro-inflammatory cytokines or microorganism’s molecules (such as LPS, a lipopolysaccharide and component of gram-negative bacteria membranes), and has killing capacity by the production of reactive oxygen species (ROS), nitric oxide (NO), and inflammatory cytokines (6). M2 macrophages are activated by anti-inflammatory cytokines like interleukins (IL-4, IL-13, IL-10), transforming growth factor-beta (TGF-β), and glucocorticoids. These types of cells establish a healing or growing setting promoting tissue remodeling, angiogenesis, and parasite clearance (7). However, these two macrophages profiles are a simplified version, found in in vitro studies. In vivo, there is a range of functional subsets within M1-M2 extreme profiles, which can be adapted depending on the microenvironment the cells are inserted in (7).
The activation of the immune system starts with the recognition of some patterns known as pathogens-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) by the effector cells of the innate immune system like macrophages, monocytes, and dendritic cells (1, 2). PAMPS are molecules essential for microbial survival. Some examples of PAMPs are nucleic acid, e.g., single-stranded RNA of viruses, plasmatic membrane compounds, or cell walls, e.g., LPS in gram-negative bacteria and b-glucans in fungus (4). DAMPs are endogenous and released due to damage, such as heat-shock proteins, uric acid crystals, serum amyloid A, and nucleic acid fragments (8).
PAMPs and DAMPs are recognized by pattern recognition receptors (PRRs), a set of signaling receptors on transmembrane or intracellular compartments (9). An essential class of PRRs is Toll-like receptors (TLRs), a family of nine transmembrane proteins that activate the expression of transcription factors such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) and interferon regulatory factors (IRFs) (4).
NF-kB is a protein complex expressed constitutively in the cytoplasm that is sequestered by IκB. The translocation of NF-kB to the nucleus is dependent on IκB phosphorylation by IκB kinases (IKK). In pro-inflammatory conditions, the NF-kB dimer activated is composed of p65 (RelA) and p50 subunits, which modulate the transcription of pro-inflammatory genes and the adaptive immune system signaling (2). The production of pro-inflammatory mediators, such as cytokines (tumor necrosis factor (TNF), IL-1, IL-6, and interferon-gamma (IFNγ), chemokines, vasoactive amines (histamine and serotonin), and eicosanoids, recruit effector cells of the acute immune response like monocytes and neutrophils to the site of inflammation, which promotes some damage to the infector agent and may eliminate it (1).
In the early stages of the inflammatory reaction, there is an important loss of tissue-resident macrophages due to cell death. To reestablish this population of cells, two approaches arise recruitment of monocytes and enhancement of tissue macrophage proliferation by their self-renewal ability (7). Although monocytes and macrophages have similar functions during inflammation, they differ from each other in origin. Monocytes originate from the bone marrow, and macrophages from the yolk sac, fetal liver, and bone marrow. The differences also appear in some functions beyond immune response and phagocytosis, since monocytes also patrol and monitor the luminal surface of the endothelium, and macrophages play key roles in tissue development, surveillance, and monitoring of tissue changes, and maintenance of tissue homeostasis (7). They also differ in the localization aspect. Monocytes circulate in the blood, bone marrow, and spleen (7), and macrophages are resident-tissue cells (10, 11).
Different from the innate response, the adaptive response is much more specific and refined not only for the recognition of the antigen, but also for its effector response. The principal cells involved in this response are lymphocytes: T cells and B cells (9).
After the end of the infection, anti-inflammatory cytokines as IL-10 and TGF- β are secreted to promote the resolution of the immune response. The resolution and repair phases are mediated by macrophages (1). The resolution of inflammation is as important as its activation, allowing the cells to return to homeostasis.
Neuroinflammation
The central nervous system (CNS) is an immune-privileged tissue, protected by the blood-brain barrier. Hence, peripheral immune cells do not have access to the brain in physiological circumstances. Besides microglial cells, other macrophages are found in external parts of the CNS, in meninges, choroid plexus, and perivascular spaces. Moreover, other immune cells like dendritic cells, monocytes, and astrocytes can be found in CNS (12). This privileged feature confers a differentiated inflammatory response, which is called neuroinflammation.
Neuroinflammation is present in many disturbances of the CNS, including neurodegenerative diseases, such as Alzheimer’s disease (AD), Amyotrophic lateral sclerosis (ALS), Multiple sclerosis (MS), Parkinson’s disease (PD), and Frontotemporal dementia. Although it is not known if neuroinflammation is the initial cause of neurodegenerative diseases, neuroinflammation promotes a prolonged activation of microglia and astrocytes and is an important factor in disease progression (13, 14). Neuroinflammation also exhibits important roles in the maintenance and progression of diseases, leading to neurotoxicity, neural death, and associated symptoms.
Microglia is a tissue-resident macrophage in the CNS. Similar to peripheral macrophages, microglia plays an important role in environmental surveillance, homeostasis maintenance, and response to any type of disbalance (12, 15). Depending on the type of stimulus secreted by the surrounding cells, different microglia phenotypes are activated, leading to distinct responses. Originally, the microglial activation was classified, the same way as macrophages, in two states: M1 (classically activated) and M2 (alternatively activated). M1 is considered a deleterious and pro-inflammatory state for ROS production and release of inflammatory molecules such as TNF-α, IL-1β, IL-12, whereas M2 is classified as an anti-inflammatory state, that is involved in the resolution of inflammation, the release of trophic factors like TGF-β and brain-derived neurotrophic factor (BDNF) (16, 17). This classification is considered outdated once a variety of microglia phenotypes were found in the brain, and whose activation states are beyond these extreme M1-M2 classifications.
After activation, microglia can play different roles including antigen presentation, phagocytosis, secretion of pro- and anti-inflammatory mediators, and production of neurotrophic factors (18). After activation via PRR, different signaling pathways like NF-kB, JAK/STAT, ERK1/2, AP-1, and p68 are responsible for targeting genes to promote the production of cytokines (TNF, IL-6, IL-1β, and IL-18), chemokines (CCL2, CCL3, CCL4, and CCL7), and other molecules like ROS, reactive nitrogen species and glutamate (19). Microglia also produces anti-inflammatory mediators like IL-4, IL-10, IL-13, and TGF-β (12). In this scenario, microglia appears to be the common initial sensor of damage, thus establishing the inflammatory state, and afterward amplification by astrocytes.
In neurodegenerative diseases, a specific type of microglia was found, subsequently classified as disease-associated microglia (DAM) characterized by transcriptional changes driven by apolipoprotein E (APOE) and the triggering receptor expressed on myeloid cells 2 (TREM2) (20). Studies demonstrate that the DAM has a molecular program with upregulation of genes that encode inflammatory molecules such as Itgax, Ccl2, and downregulation of homeostatic genes like Csf1r, Cx3cr1, Hexb, and Trem119 (20). Glycoprotein nonmetastatic melanoma protein B (GPNMB) is also a molecule that has been described to be upregulated in DAM (20, 21).
GPNMB
GPNMB is an endogenous type 1 transmembrane glycoprotein that was first described in 1995 as more highly expressed in low-metastatic melanoma cell lines (22). GPNMB is also known as Osteactivin, DC-HIL (dendritic cell heparan sulfate proteoglycan integrin-dependent ligand), and HGFIN (hematopoietic growth factor inducible neurokinin-1 type). While the name GPNMB is more broadly used, the name Osteoactivin is mostly used in studies with bones, while DC-HIL and HGFIN are commonly used in studies about the immune system and cells. GPNMB is constitutively expressed through most cell types and tissues (23), and its expression can be increased in cancer cells (24).
GPNMB is encoded by the GPNMB gene located at locus 7p15 and has 2 isoforms: one with 572 and the other with 560 amino acids, as a result of alternative splicing (25). After transcription, GPNMB is directed to the cell membrane, in which most of its length is located in the extracellular domain. The extracellular portion of GPNMB has 12 glycosylation sites, a polycystic kidney disease (PKD) domain, and an integrin-recognition (RGD) motif (26). GPNMB can be cleaved by the metalloproteinase ADAM10, releasing a soluble fragment that can bind to many receptors and trigger a cellular response (27).
GPNMB is known to have different functions on different cell types. In bones, GPNMB promotes differentiation of osteoblasts and mineralization of bone matrix (26), fibroblast activation and proliferation (28), maturation of hematopoietic and lymphoid cells (29), and also decreases the activation of lymphocytes T (30). The broad range of GPNMB’s extracellular fragment function is related to its ability to bind to many receptors, such as Na+, K+-ATPase (NKA), CD44, Epidermal Growth Factor Receptor (EGFR), and Vascular Endothelial Growth Factor Receptor (VEGFR) (24) and its ability to interact with integrins, heparin and Syndecan-4 (31, 32).
As illustrated in Figure 1, the interaction between GPNMB and the NKA activates the ERK/MERK and Akt/PI3K pathways (33, 34), which can modulate the activation of NF-κB (35, 36). Previous studies have shown an increase in phosphorylation of ERK and Akt by GPNMB signaling in different models and cell types (33, 34, 37, 38). The Akt residue that appears to be phosphorylated by GPNMB is S473 (34, 38), required for the maximum activation of the kinase (39).
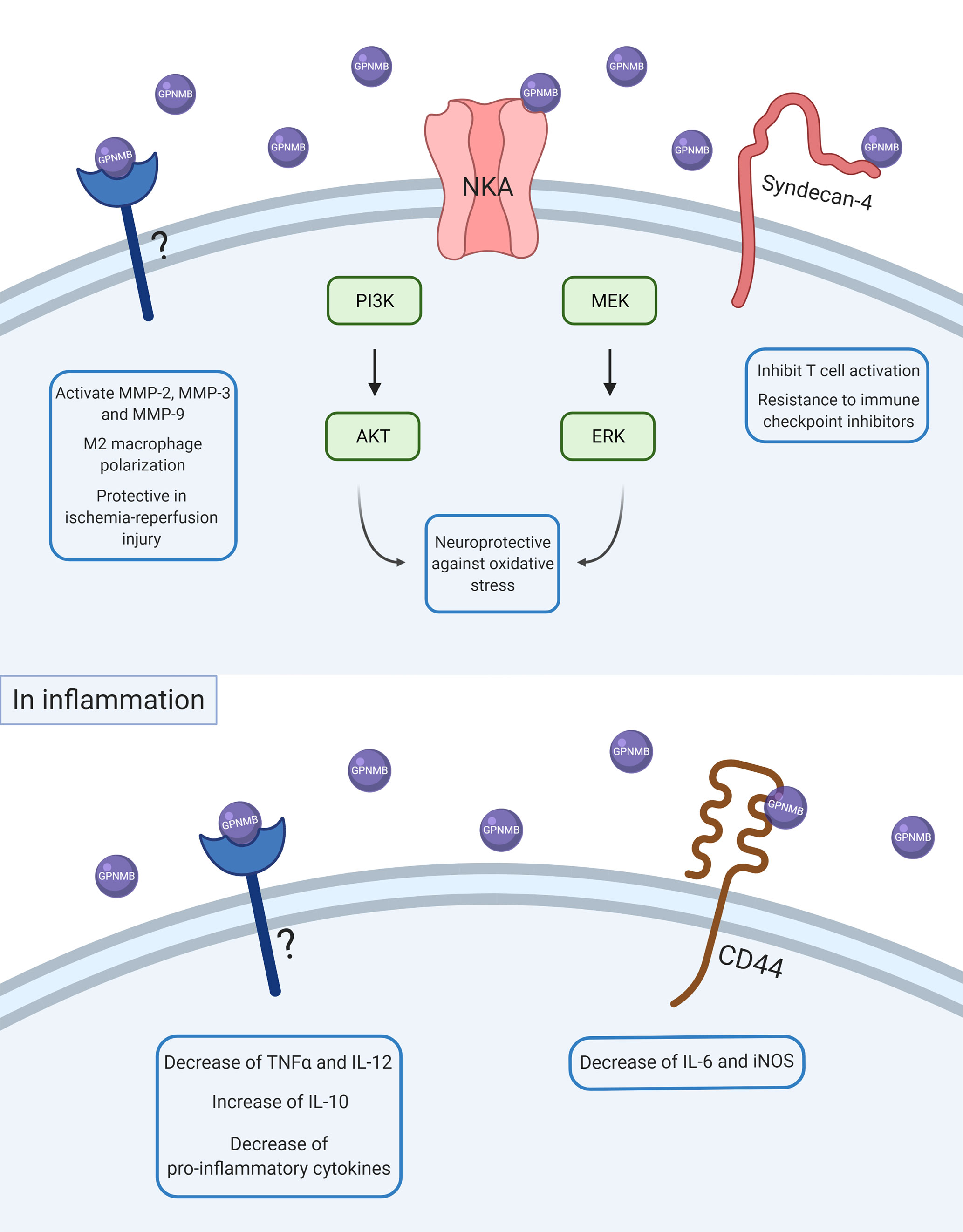
Figure 1 Scheme of GPNMB intracellular signaling in physiological and inflammatory conditions. After ADAM10 cleavage, soluble GPNMB can interact with several receptors, proteins, and other molecules in the cell membrane. Some of these interactions can activate intracellular pathways and lead to changes in other proteins’ expression and consequently in cell response. In basal conditions, GPNMB can interact with NKA and promote neuroprotection against oxidative stress by activation of ERK/MEK and AKT/PI3K pathways. GPNMB can also interact with syndecan-4 and promote inhibition of T cell activation, which is also associated with a resistance of immune checkpoint inhibitors. GPNMB can also promote activation of MMP-2, MMP-3, and MMP-9, M2-macrophage polarization, and a protective role in ischemia-reperfusion injury, but the molecular mechanisms underlying these responses are not known yet. In inflammation, GPNMB seems to have an anti-inflammatory and resolute role by decreasing pro-inflammatory cytokines like TNFα, IL-6, and IL-12 and increasing anti-inflammatory cytokines such as IL-10.
As GPNMB was originally associated with tumorous cells, GPNMB was widely studied in several types of cancer, such as melanoma (40), glioma (41), breast cancer (42), and gastric cancer (43). Although previous work indicated that GPNMB is an anti-metastatic glycoprotein, recent studies question the function of GPNMB in cancer, because GPNMB appears to have a protective role in some types of cancers whereas it may promote metastasis in others (24). In the past years, GPNMB has been studied in breast cancer, whose role has been controversial for years, but most of the evidence suggests that GPNMB is associated with cancer aggressiveness, as it promotes cancer metastasis (42), angiogenesis (27) and immune suppression (40, 44, 45), enhancing tumor migration and invasion. An antibody anti-GPNMB (called CDX-011 or glembatumumab vedotin) has been evaluated in phase I/II trials as a treatment for advanced breast cancer and melanoma (46–48). Clinical trials were discontinued in 2018 after glembatumumab vedotin failed to improve the survival rate and stop tumor progression of women with metastatic triple-negative breast cancers (TNBC) for longer than capecitabine (49).
More recently, the relationship between GPNMB and inflammation has been explored in a few studies, but the function of GPNMB in inflammation is still not clear, as some studies suggest anti-inflammatory action of GPNMB, while others suggest that it acts as a pro-inflammatory mediator (Table 1). In this review, we will gather recent studies about this subject to better understand the relationship between GPNMB and inflammation in different systems.

Table 1 Factors that modulate GPNMB expression.
GPNMB and Neuroinflammation
GPNMB is expressed in all brain regions, but especially in the hippocampal dentate gyrus, choroid plexus, ependyma, periventricular regions, and in layers II and III of the cerebral cortex (50). Besides the brain region, GPNMB is mostly expressed by microglial cells, which are resident brain cells most responsible for mediating inflammatory stimuli and maintaining cerebral homeostasis by phagocytosis, tissue repair, and cytokine release (68).
Bandari and colleagues first described that GPNMB might be related to inflammation. It was shown that GPNMB could interact with Substance P (29), a peptide that facilitates inflammatory response in both the CNS and the Peripheral Nervous System (PNS) by interacting with the receptor NK1. The interaction between substance P and NK1 receptor promotes the activation of mTOR, which regulates inflammation by controlling the balance between IL-10 and IL-12, and the transcription factors AP-1 and NF-κB, which can increase the expression of many chemokines, leading to the promotion of recruitment of immune cells to sites of inflammation (69).
Additionally, there is evidence that pro-inflammatory stimuli could modulate GPNMB expression in the brain. An established model used to induce inflammation and neuroinflammation is the administration of LPS. Studies revealed an increase of pro-inflammatory cytokines released such as IL-1β, TNF-α, and IL-6, and also the activation of microglia in in vitro and in vivo studies after administration of LPS (70, 71). LPS could also disrupt BBB (72). It is important to underline that the route of administration, its dose, and timing are crucial for producing inflammatory changes (73).
Intraperitoneal (i.p.) injection of LPS in rats increases the number of cells expressing GPNMB, especially in the area postrema, which correlates with OX42 expression (50). OX42 is a marker present in both microglia and macrophages. As the area postrema is a region close to ventricular organs, the study suggests that the increase of GPNMB positive cells is due to increased infiltration of macrophages expressing GPNMB in the brain. Even though i.p. injection of LPS resulted in increased GPNMB levels in the brain, the same is still controversial in vitro. Two studies have treated BV-2 cells (microglia cell line) with LPS, but the outcome on mRNA levels of GPNMB was different. One study evidenced increased mRNA levels of Gpnmb after 6h of 10ng/ml or 100ng/ml LPS treatment, and in the 100ng/ml group, Gpnmb mRNA levels remained increased for 12 and 24h. Besides, they have seen an increase in GPNMB protein expression after 12h in both LPS concentrations (51). In the other study, Gpnmb mRNA levels remained the same after 24h treatment with 100ng/ml of LPS, contradicting previous results (21). In one study, BV-2 cells were kept in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), while in the other study, BV-2 were kept in DMEM/F12 media supplemented with 10% FBS.
An increase of GPNMB levels in the brain has also been detected in a diverse number of neurodegenerative diseases (in vitro, in vivo, and patients) such as AD (21), ALS (33, 34) and PD (53). Treatment with β-Amyloid (Aβ), a protein that accumulates in AD and forms aggregates, led to increased levels of Gpnmb mRNA expression. Interestingly, the increase of GPNMB occurs in microglia cells that are surrounding Aβ aggregate plates (21). In humans, it was shown that GPNMB was increased in cerebrospinal fluid (CSF) and post-mortem brains of sporadic AD patients. Sporadic ALS patients also present increased levels of GPNMB in CSF, serum, and lumbar spinal cord tissue (33). Increased levels of GPNMB expression were found in the substantia nigra of PD patients (54). Furthermore, the single nucleotide polymorphism (SNP) rs199347, a known top risk SNP for PD, is located in the GPNMB gene and results in increased GPNMB expression (74). This data supports that GPNMB might be considered a marker for DAM (20, 21).
In addition to being highly expressed, GPNMB seems to have a protective role in neurodegenerative diseases (Table 1). Although the mechanism underlying this protection is not yet well elucidated, it might be related to neuroinflammation. In a cellular ALS model, it has been shown that SOD1G93A inhibits the glycosylation of GPNMB, resulting in increased motoneuron death (33) and treatment with recombinant extracellular fragments of GPNMB (rGPNMB) leads to diminished cell death (33, 34). The changes in cell viability can be due to GPNMB binding to NKA, which, besides maintaining the osmotic equilibrium of the cell, is a receptor known to modulate neuroinflammation (75, 76).
Also in the CNS, GPNMB seems to be protective in cerebral ischemia-reperfusion injury (IRI) (37), which is a condition known to trigger neuroinflammation (77). In this study, a transgenic mice overexpressing GPNMB was generated by the injection of a V5-His-tagged GPNMB cDNA construct containing a CAG hybrid promoter into fertilized mice eggs. Transgenic mice overexpressing GPNMB have resulted in decreased infarct volume after IRI and intraventricular administration of rGPNMB resulted in lessened cerebral infarct damage (37). Increased levels of GPNMB in neurons and astrocytes of both mice and humans were observed during the acute phase of IRI, suggesting that the response to inflammation could be modulated by GPNMB (37). In astrocyte primary culture, it has been evaluated that the administration of rGPNMB, in interaction with the CD44 receptor, leads to a decrease of IL-6 and iNOS levels that were induced by a pro-inflammatory cytokine-mix treatment (54).
Although most of the results suggest that GPNMB has an anti-inflammatory role, there is evidence that neuroinflammation could be dependent on GPNMB (Table 1). The silencing of GPNMB by transfection with GPNMB siRNA in BV-2 cells resulted in decreased levels of pro-inflammatory markers after LPS treatment (51). The increase of iNOS, TNF-α, and IL-1β levels was smaller in the group treated with LPS and GPNMB siRNA in comparison to cells treated with LPS with normal levels of GPNMB (51). Interestingly, phosphorylation of ERK1/2, which is increased with LPS treatment in normal cells, was also diminished in the GPNMB siRNA group treated with LPS. These results led to the conclusion that the induction of neuroinflammation via LPS could be dependent on GPNMB. Another evidence that contradicts previous results is that treatment with the anti-inflammatory cytokine IL-4, but not with a mix of pro-inflammatory cytokines, led to increased levels of GPNMB in astrocytes and mesenchymal cells (38, 54). These contradictory findings highlight that the relationship between GPNMB and neuroinflammation still needs to be more explored and is probably cell-dependent.
GPNMB in Peripheral Inflammation
GPNMB is physiologically expressed in most cell types, and pro-inflammatory stimulus can cause its increase in some cell types, especially on macrophages and monocytes (Table 1). Microarray analysis evidenced GPNMB as a gene highly expressed in an inflammatory macrophage population (TEMP) compared with a non-stimulated macrophage population (BMM) or fibroblasts, and the administration of pro-inflammatory stimulus as LPS and IFN-γ led to increased levels of GPNMB in BMM (52).
In the liver of rats, GPNMB is constitutively expressed in peritoneal macrophages and Kupffer cells (resident liver macrophages) rather than in other cell types (57). Liver damage seems to increase GPNMB levels, as the induction of liver damage by CCl4 administration in vivo (57) resulted in higher levels of GPNMB. In human liver samples, GPNMB was not found in basal conditions but GPNMB is expressed in livers presenting hepatitis, cirrhosis, and paracetamol intoxication, which all refer to inflammatory diseases (57, 63). This data suggests that even if the GPNMB expression is different between rats and humans, the increase of GPNMB in a pro-inflammatory scenario is consistent.
GPNMB seems to be overexpressed in macrophages in diseases that have a pro-inflammatory component in different tissues. In colitis, a chronic inflammatory disease in the intestine, infiltrating macrophages in the injured mucosa express GPNMB, and GPNMB expression increases according to the severity of the illness (62). Interestingly, D2 mice, which is a mice strain known to have a premature stop codon mutation in the GPNMB gene, had a more severe condition of colitis, accompanied by increased levels of pro-inflammatory cytokines (62) (Table 2). In end-stage renal disease, a disease in which both inflammatory and mineralization processes are upregulated, patients have upregulation in Gpnmb mRNA levels and protein expression in adhering monocytes, which was sustained and even increased after differentiation to macrophages in vitro (58). In addition, GPNMB is highly expressed in a murine model of IRI in the liver and kidney (55). GPNMB is also induced and secreted in white adipose tissue in obesity (63). Besides being increased in disease, it has been reported in humans that mutations that result in truncation of the GPNMB gene are linked to Autosomal-Recessive Amyloidosis Cutis Dyschromica (ACD), which is a rare condition characterized by generalized hyperpigmentation with small hypopigmented macules on the skin (64). Individuals affected by ACD also present reduced levels of GPNMB in the characteristic skin lesions of this condition (64).
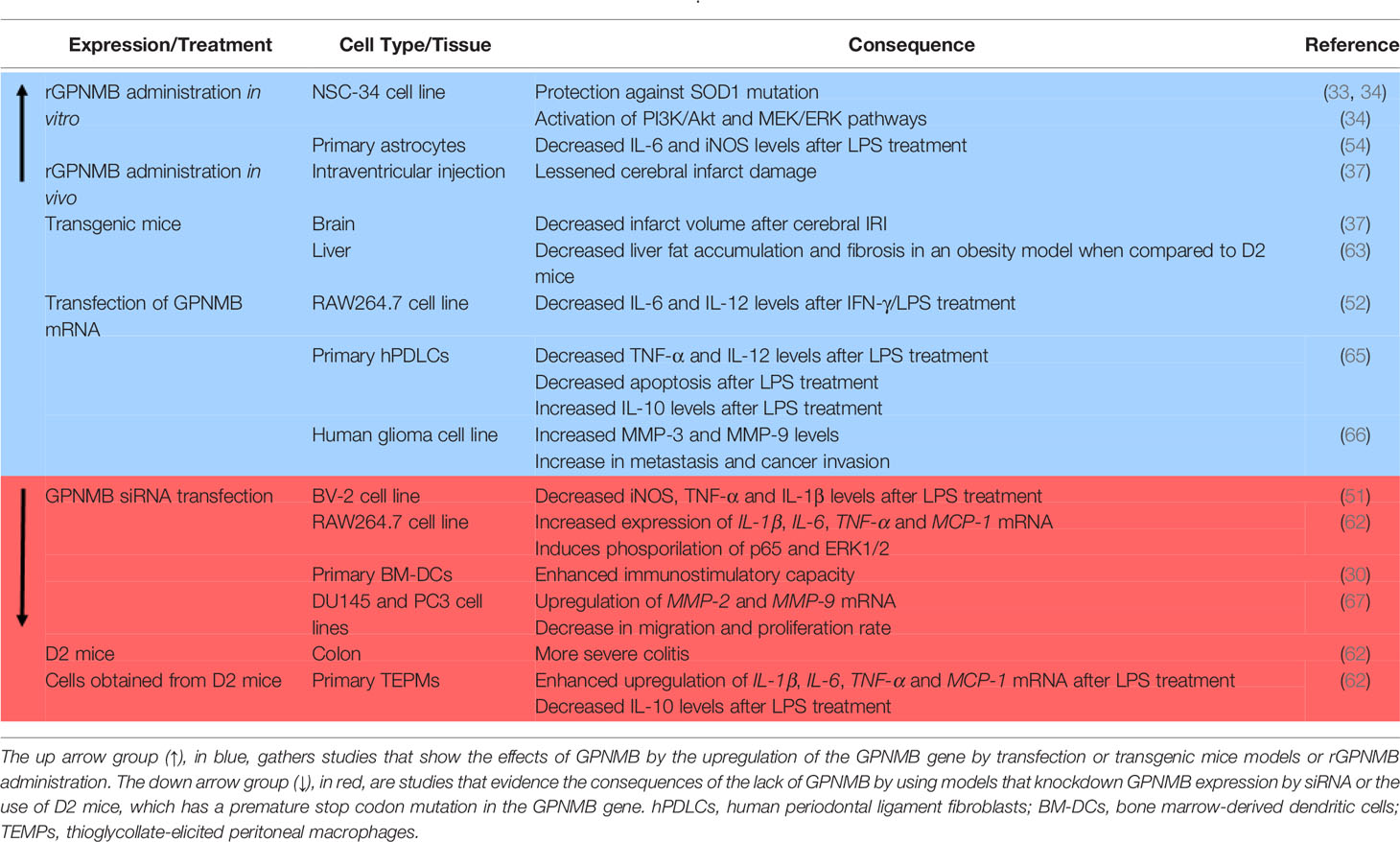
Table 2 Outcome of models that modulate GPNMB expression in vitro or in vivo.
Diseases that are characterized by macrophage dysfunction also seem to increase GPNMB expression (25). Niemann-Pick type C disease is a condition that arises from lysosomal dysfunction in macrophages and presents increased levels of GPNMB in the liver, brain, and spleen in a mouse model of this disease and in the plasma of patients (59). In another disease characterized by lysosomal dysfunction in macrophages, Gaucher disease, GPNMB is increased in the spleen, plasma (60), and CSF (61) of patients. In a genetically modified mice model of type 1 Gaucher Disease, GPNMB levels were also increased, and after treatment, GPNMB levels returned to normal (60).
Even though pro-inflammatory stimuli lead to increased levels of GPNMB in macrophages and other cell types, not all the cells respond in the same way. In mononuclear phagocytes found in the liver, LPS administration in vitro led to diminished levels of GPNMB (57). On the other hand, LPS treatment in macrophages obtained from D2 mice presented increased expression of pro-inflammatory cytokines and diminished levels of IL-10 when compared to macrophages obtained from mice expressing normal levels of GPNMB (62). Also, RAW264.7 cells (macrophage-like cell lineage) transfected with GPNMB siRNA presented increased levels of pro-inflammatory cytokines and p65 phosphorylation when compared with normal RAW264.7 cells (62), indicating that GPNMB might attenuate pro-inflammatory responses. Contributing to the hypothesis that GPNMB could have an anti-inflammatory role, RAW264.7 cells overexpressing GPNMB showed diminished levels of IL-6 and IL-12 after IFN-γ/LPS treatment when compared to the scramble group (52).
Another proof that GPNMB may contribute to the resolution of inflammation is that GPNMB is highly expressed in M2-polarized macrophages rather than M1 macrophages (38, 56), and GPNMB promotes M2 polarization on macrophages (56), which indicates that GPNMB may promote inflammatory resolution. In a periodontal disease cell model, primary human periodontal ligament cells (hPDLCs) overexpressing GPNMB by lentiviral transfection showed decreased pro-inflammatory cytokines TNF-α and IL-12 and increased anti-inflammatory IL-10 levels after LPS treatment (65). In addition, GPNMB protected hPDLCs from apoptosis driven by LPS administration. Increased levels of GPNMB also appear to attenuate other aspects of the disease, not only the ones associated with inflammation. In nonalcoholic steatosis, transgenic mice containing an aP2 promoter-driven GPNMB overexpression showed decreased fat accumulation and fibrosis in the liver in an obesity model when compared to obese D2 mice (63).
Besides interacting with macrophages and monocytes, GPNMB can have a role in the modulation of T cell activation. Activated T cells present receptors to GPNMB, and GPNMB has the ability to inhibit T cells’ response to primary and secondary activation (30). The ability to bind to T cells seems to be dependent on the PKD domain present in the extracellular domain of GPNMB. Endogenous GPNMB prevents T cells from entering the cell cycle, resulting in the inhibition of its activation and proliferation, while GPNMB knockdown T cells display enhanced capacity for immunostimulation (30). Further studies showed that inhibition of T cell activation by GPNMB is dependent on the syndecan-4 pathway (78) and that GPNMB expression regulates lymphocyte allostimullatory capacity, which means the capability of producing antibodies (45). Several studies agree that GPNMB inhibits T cells, which indicates that GPNMB can weaken the immunological response, which is related to both onset and resolution of inflammation.
GPNMB in Cancer Inflammation
Inflammation is known to predispose the development of cancer and tumor progression (79). Tumor cells can produce cytokines and attract immune cells to the tumor microenvironment, promoting a chronic inflammation at the site of the tumor. The cell types present in the tumor microenvironment may define cancer prognosis and chosen therapeutic approach (80), and the presence of T cells in the tumor microenvironment is required to promote anti-tumoral responses. The ability to inhibit T cell activation may be an important factor that explains the relationship between GPNMB and cancer. Syndecan-4 is an important molecule for the activation of T cells, and GPNMB can bind to syndecan-4, inhibiting T cell activation. A study showed that GPNMB can attenuate the activation of T cells that are activated via syndecan-4, allowing the evasion of melanoma cells for immunological recognition and destruction (40), whereas the blockade of GPNMB by antibodies restores the integrity of T cells, attenuates tumor growth, and increases IFNγ levels in the tumor microenvironment (81). Syndecan-4 is expressed in some activated T cells and presents specific heparan sulfates that bind to GPNMB. This interaction leads to a reduction of pro-inflammatory cytokine secretion and blocks T cells from entering the S phase of the cell cycle (45).
One of the currently available cancer therapies is based on immunotherapy drugs called immune checkpoint inhibitors (ICI). Immune checkpoints are a physiological mechanism that prevents exacerbated immune response and cell death, but this mechanism needs to be blocked in cancer to destroy tumorous cells. ICI acts by blocking checkpoints proteins, amplifying the anti-tumoral response, and allowing T cells to kill cancer cells (82). It seems that higher levels of circulating GPNMB in the blood of patients are associated with resistance to ICI therapy (83). Thus, GPNMB seems to modulate the immune response to cancer through several mechanisms.
Another way that GPNMB might contribute to cancer inflammation is by interacting with molecules involved in tumoral progression. GPNMB seems to activate MMP-3 (51), and MMPs activation is involved with both processes that promote cancer cell migration and invasion and inflammation. Overexpression of GPNMB by retrovirus transfection in glioma cells is accompanied by MMP-3 and MMP-9 increase (66), both increasing metastasis and cancer invasion (66). This phenomenon is also documented in other types of cancer, as GPNMB promotes the invasion and proliferation of prostate cancer cell lines through the activation of MMP-2 and MMP-9, which can be reversed by GPNMB knockdown by siRNA transfection (67). It is important to note that, besides cancer, MMPs are also associated with inflammation signaling (84).
Conclusions
This review shows that GPNMB has a majorly anti-inflammatory role and is important to disease resolution, but there is data that contradicts this tendency. The association of GPNMB with disease resolution is corroborated by the studies that associate GPNMB mutations with a predisposition to Parkinson’s Disease and Autosomal-Recessive Amyloidosis Cutis Dyschromica. However, it is important to notice that GPNMB is associated with cancer progression and metastasis. Regarding inflammation, most of the studies also suggest an anti-inflammatory role of GPNMB, most of them relying on macrophage/microglia activation. As there are also studies suggesting that GPNMB could have a pro-inflammatory role in some cases, more studies should be conducted to clarify this issue. Since GPNMB can interact with different receptors and molecules, the question of whether GPNMB will have a pro- or anti-inflammatory role could depend on which ligand it is interacting with and also the cell type involved.
Author Contributions
Elaborated the figures and wrote the manuscript: MS and GAS. Reviewed topics and concepts: CS. Conceived, reviewed and discussed concepts in the manuscript: PFK. All authors contributed to the article and approved the submitted version.
Funding
MS is supported by a Master fellow grant #2018/07896-3, GAS by a student fellow grant #2019/05970-4, and PFK by a postdoctoral fellow grant #2018/14289-6 from São Paulo Research Foundation (FAPESP). CS is a fellow researcher of the National Council for Scientific and Technological Development (CNPq). This publication was made possible by grants from FAPESP to CS (2016/07427-8); CNPq 405089/2018-0; and CAPES – STINT program 88887.125409/2016-00 (Joint Brazilian-Swedish Research Collaboration) and USP Neuroscience Research Support Centres (NAPNA) to CS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Carolina Fernandez for English Editing. Image 1 was created with BioRender.com.
References
1. Medzhitov R. Origin and Physiological Roles of Inflammation. Nature (2008) 454(7203):428–35. doi: 10.1038/nature07201
2. Barton GM. A Calculated Response: Control of Inflammation by the Innate Immune System. J Clin Invest (2008) 118(2):413–20. doi: 10.1172/JCI34431
3. Nathan C. Points of Control in Inflammation. Nature (2002) 420(6917):846–52. doi: 10.1038/nature01320
4. Murphy K, Travers P, Walport M, Janeway C. Janeway’s Immunobiology. 8th. New York: Garland Science (2012).
5. Mills CD. M1 and M2 Macrophages: Oracles of Health and Disease. Crit Rev Immunol (2012) 32(6):463–88. doi: 10.1615/CritRevImmunol.v32.i6.10
6. Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-Macrophage Colony-Stimulating Factor (CSF) and Macrophage Csf-Dependent Macrophage Phenotypes Display Differences in Cytokine Profiles and Transcription Factor Activities: Implications for CSF Blockade in Inflammation. J Immunol (2007) 178(8):5245–52. doi: 10.4049/jimmunol.178.8.5245
7. Italiani P, Boraschi D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front Immunol (2014) 5:514. doi: 10.3389/fimmu.2014.00514
8. Cronkite DA, Strutt TM. The Regulation of Inflammation by Innate and Adaptive Lymphocytes. J Immunol Res (2018) 2018:1–14. doi: 10.1155/2018/1467538
9. Medzhitov R, Janeway CA. Innate Immunity: The Virtues of a Nonclonal System of Recognition. Cell (1997) 91(3):295–8. doi: 10.1016/S0092-8674(00)80412-2
10. Epelman S, Lavine KJ, Randolph GJ. Origin and Function of Tissue Macrophages. Immunity (2014) 41:21–35. doi: 10.1016/j.immuni.2014.06.013
11. Sieweke MH, Allen JE. Beyond Stem Cells: Self-Renewal of Differentiated Macrophages. Science (2013) 342:1242974. doi: 10.1126/science.1242974
12. Voet S, Prinz M, van Loo G. Microglia in Central Nervous System Inflammation and Multiple Sclerosis Pathology. Trends Mol Med (2018) 25(2):112–23. doi: 10.1016/j.molmed.2018.11.005
13. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms Underlying Inflammation in Neurodegeneration. Cell (2010) 140(6):918–34. doi: 10.1016/j.cell.2010.02.016
14. Bachiller S, Jiménez-Ferrer I, Paulus A, Yang Y, Swanberg M, Deierborg T, et al. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front Cell Neurosci (2018) 12:488. doi: 10.3389/fncel.2018.00488
15. Shemer A, Erny D, Jung S, Prinz M. Microglia Plasticity During Health and Disease: An Immunological Perspective. Trends Immunol (2015) 36:614–24. doi: 10.1016/j.it.2015.08.003
16. Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. Microglial and Macrophage Polarization - New Prospects for Brain Repair. Nat Rev Neurol (2015) 11:56–64. doi: 10.1038/nrneurol.2014.207
17. Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol (2016) 53(2):1181–94. doi: 10.1007/s12035-014-9070-5
18. Li Q, Barres BA. Microglia and Macrophages in Brain Homeostasis and Disease. Nat Rev Immunol (2018) 18(4):225–42. doi: 10.1038/nri.2017.125
19. Merson TD, Binder MD, Kilpatrick TJ. Role of Cytokines as Mediators and Regulators of Microglial Activity in Inflammatory Demyelination of the CNS. Neuromol Med (2010) 12(2):99–132. doi: 10.1007/s12017-010-8112-z
20. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, Fatimy REI, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity (2017) 47(3):566–81. doi: 10.1016/j.immuni.2017.08.008
21. Hüttenrauch M, Ogorek I, Klafki H, Otto M, Stadelmann C, Weggen S, et al. Glycoprotein NMB: A Novel Alzheimer’s Disease Associated Marker Expressed in a Subset of Activated Microglia. Acta Neuropathol Commun (2018) 6(1):108. doi: 10.1186/s40478-018-0612-3
22. Weterman MA, Ajubi N, van Dinter IM, Degen WG, van Muijen GN, Ruitter DJ, et al. Nmb, a Novel Gene, is Expressed in Low-Metastatic Human Melanoma Cell Lines and Xenografts. Int J Cancer (1995) 60(1):73–81. doi: 10.1002/ijc.2910600111
23. The Human Protein Atlas. GPNMB. Available at: https://www.proteinatlas.org/ENSG00000136235-GPNMB/tissue (Accessed 08/10/2020). (n.d.).
24. Taya M, Hammes SR. Glycoprotein Non-Metastatic Melanoma Protein B (GPNMB) and Cancer: A Novel Potential Therapeutic Target. Steroids (2018) 133:102–7. doi: 10.1016/j.steroids.2017.10.013
25. van der Lienden MJC, Gaspar P, Boot R, Aerts J, van Eijk M. Glycoprotein non-Metastatic Protein B: An Emerging Biomarker for Lysosomal Dysfunction in Macrophages. Int J Mol Sci (2018) 20(1):66. doi: 10.3390/ijms20010066
26. Abdelmagid SM, Barbe MF, Rico MC, Salihoglu S, Arango-hisijara I, Selim AH, et al. Osteoactivin, an Anabolic Factor That Regulates Osteoblast Differentiation and Function. Exp Cell Res (2008) 314(13):2334–51. doi: 10.1016/j.yexcr.2008.02.006
27. Rose AA, Annis MG, Dong Z, Pepin F, Hallett M, Park M, et al. ADAM10 Releases a Soluble Form of the GPNMB/Osteoactivin Extracellular Domain With Angiogenic Properties. PloS One (2010) 5(8):e12093. doi: 10.1371/journal.pone.0012093
28. Furochi H, Tamura S, Mameoka M, Yamada C, Ogawa T, Hirasaka K, et al. Osteoactivin Fragments Produced by Ectodomain Shedding Induce MMP-3 Expression Via ERK Pathway in Mouse NIH-3T3 Fibroblasts. FEBS Lett (2007) 581(30):5743–50. doi: 10.1016/j.febslet.2007.11.036
29. Bandari PS, Qian J, Yehia G, Joshi DD, Maloof PB, Potian J, et al. Hematopoietic Growth Factor Inducible Neurokinin-1 Type: A Transmembrane Protein That is Similar to Neurokinin 1 Interacts With Substance P. Regul Pept (2003) 111(1–3):169–78. doi: 10.1016/S0167-0115(02)00288-4
30. Chung JS, Sato K, Dougherty II, Cruz PD Jr, Ariizumi K. Dc-HIL is a Negative Regulator of T Lymphocyte Activation. Blood (2007a) 109(10):4320–7. doi: 10.1182/blood-2006-11-053769
31. Shikano S, Bonkobara M, Zukas PK, Ariizumi K. Molecular Cloning of a Dendritic Cell-Associated Transmembrane Protein, DC-HIL, That Promotes RGD-dependent Adhesion of Endothelial Cells Through Recognition of Heparan Sulfate Proteoglycans. J Biol Chem (2001) 276(11):8125–34. doi: 10.1074/jbc.M008539200
32. Maric G, Rose AA, Annis MG, Siegel PM. Glycoprotein non-Metastatic B (Gpnmb): A Metastatic Mediator and Emerging Therapeutic Target in Cancer. Onco Targets Ther (2013) 9(6):839–52. doi: 10.2147/OTT.S44906
33. Tanaka H, Shimazawa M, Kimura M, Takata M, Tsuruma K, Yamada M, et al. The Potential of GPNMB as Novel Neuroprotective Factor in Amyotrophic Lateral Sclerosis. Sci Rep (2012) 2:573. doi: 10.1038/srep00573
34. Ono Y, Tsuruma K, Takata M, Shimazawa M, Hara H. Glycoprotein Nonmetastatic Melanoma Protein B Extracellular Fragment Shows Neuroprotective Effects and Activates the PI3K/Akt and MEK/ERK Pathways Via the Na +/K +-Atpase. Sci Rep (2016) 6(1):23241. doi: 10.1038/srep23241
35. Bai D, Ueno L, Vogt PK. Akt-Mediated Regulation of Nfκb and the Essentialness of Nfκb for the Oncogenicity of PI3K and Akt. Int J Cancer (2009) 125(12):2863–70. doi: 10.1002/ijc.24748
36. Maeng YS, Min JK, Kim JH, Yamagishi A, Mochizuki N, Kwon JY, et al. ERK is an Anti-Inflammatory Signal That Suppresses Expression of NF-kappaB-dependent Inflammatory Genes by Inhibiting IKK Activity in Endothelial Cells. Cell Signal (2006) 18(7):994–1005. doi: 10.1016/j.cellsig.2005.08.007
37. Nakano Y, Suzuki Y, Takagi T, Kitashoji A, Ono Y, Tsuruma K, et al. Glycoprotein Nonmetastatic Melanoma Protein B (GPNMB) as a Novel Neuroprotective Factor in Cerebral Ischemia-Reperfusion Injury. Neuroscience (2014) 277:123–31. doi: 10.1016/j.neuroscience.2014.06.065
38. Yu B, Sondag GR, Malcuit C, Kim MH, Safadi FF. Macrophage-Associated Osteoactivin/GPNMB Mediates Mesenchymal Stem Cell Survival, Proliferation, and Migration Via a CD44-Dependent Mechanism. J Cell Biochem (2016) 117(7):1511–21. doi: 10.1002/jcb.25394
39. Linton MF, Moslehi JJ, Babaev VR. Akt Signaling in Macrophage Polarization, Survival, and Atherosclerosis. Int J Mol Sci (2019) 20(11):2703. doi: 10.3390/ijms20112703
40. Tomihari M, Chung JS, Akiyoshi H, Cruz PD Jr, Ariizumi K. DC-HIL/Glycoprotein Nmb Promotes Growth of Melanoma in Mice by Inhibiting the Activation of Tumor-Reactive T Cells. Cancer Res (2010) 70(14):5778–87. doi: 10.1158/0008-5472.CAN-09-2538
41. Bao G, Wang N, Li R, Xu G, Liu P, He B. Glycoprotein non-Metastatic Melanoma Protein B Promotes Glioma Motility and Angiogenesis Through the Wnt/β-Catenin Signaling Pathway. Exp Biol Med (2016) 241(17):1968–76. doi: 10.1177/1535370216654224
42. Rose AA, Siegel PM. Osteoactivin/HGFIN: Is it a Tumor Suppressor or Mediator of Metastasis in Breast Cancer? Breast Cancer Res (2007) 9(6):403. doi: 10.1186/bcr1791
43. Ren F, Zhao Q, Liu B, Sun X, Tang Y, Huang H, et al. Transcriptome analysis reveals GPNMB as a potential therapeutic target for gastric cancer. J Cell Physiol (2020) 235(3):2738–52. doi: 10.1002/jcp.29177
44. Schwarzbich MA, Gutknecht M, Salih J, Salih HR, Brossart P, Rittig SM. The Immune Inhibitory Receptor Osteoactivin is Upregulated in Monocyte-Derived Dendritic Cells by BCR-ABL Tyrosine Kinase Inhibitors. Cancer Immunol Immunother (2012) 61(2):193–202. doi: 10.1007/s00262-011-1096-1
45. Chung JS, Bonkobara M, Tomihari M, Cruz PD Jr, Ariizumi K. The DC-HIL/syndecan-4 Pathway Inhibits Human Allogeneic T-cell Responses. Eur J Immunol (2009) 39(4):965–74. doi: 10.1002/eji.200838990
46. Ott PA, Hamid O, Pavlick AC, Kluger H, Kim KB, Boasberg PD, et al. Phase I/II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Patients With Advanced Melanoma. J Clin Oncol (2014) 32(32):3659–66. doi: 10.1200/JCO.2013.54.8115
47. Bendell J, Saleh M, Rose AAN, Siegel PM, Hart L, Sirpal S, et al. Phase I/II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Patients With Locally Advanced or Metastatic Breast Cancer. J Clin Oncol (2014) 32(32):3619–25. doi: 10.1200/JCO.2013.52.5683
48. Yardley DA, Weaver R, Melisko ME, Saleh MN, Arena FP, Forero A, et al. EMERGE: A Randomized Phase II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Advanced Glycoprotein NMB-expressing Breast Cancer. J Clin Oncol (2015) 33(14):1609–19. doi: 10.1200/JCO.2014.56.2959
49. U.S. National Library of Medicine. Clinicaltrials.Gov Identifier Nct01997333, Study of Glembatumumab Vedotin (Cdx-011) in Patients With Metastatic, Gpnmb Over-Expressing, Triple Negative Breast Cancer (Metric) (2018). Available at: https://clinicaltrials.gov/ct2/show/study/NCT01997333 (Accessed January 12, 2021).
50. Huang JJ, Ma WJ, Yokoyama S. Expression and Immunolocalization of Gpnmb, a Glioma-Associated Glycoprotein, in Normal and Inflamed Central Nervous Systems of Adult Rats. Brain Behav (2012) 2(2):85–96. doi: 10.1002/brb3.39
51. Shi F, Duan S, Cui J, Yan X, Li H, Wang Y, et al. Induction of Matrix Metalloproteinase-3 (Mmp-3) Expression in the Microglia by Lipopolysaccharide (LPS) Via Upregulation of Glycoprotein Nonmetastatic Melanoma B (Gpnmb) Expression. J Mol Neurosci (2014) 54(2):234–42. doi: 10.1007/s12031-014-0280-0
52. Ripoll VM, Irvine KM, Ravasi T, Sweet MJ, Hume DA. Gpnmb is Induced in Macrophages by IFN-gamma and Lipopolysaccharide and Acts as a Feedback Regulator of Proinflammatory Responses. J Immunol (2007) 178(10):6557–66. doi: 10.4049/jimmunol.178.10.6557
53. Moloney EB, Moskites A, Ferrari EJ, Isacson I, Hallett PJ. The Glycoprotein GPNMB is Selectively Elevated in the Substantia Nigra of Parkinson’s Disease Patients and Increases After Lysosomal Stress. Neurobiol Dis (2018) 120:1–11. doi: 10.1016/j.nbd.2018.08.013
54. Neal ML, Boyle AM, Budge KM, Safadi FF, Richardson R. The Glycoprotein GPNMB Attenuates Astrocyte Inflammatory Responses Through the CD44 Receptor. J Neuroinflammation (2018) 15(1):73. doi: 10.1186/s12974-018-1100-1
55. Michalinos A, Tsaroucha AK, Lambropoulou M, Schizas D, Valsami G, Kostomitsopoulos N, et al. Glycoprotein non-Metastatic Melanoma B Expression After Hepatic Ischemia Reperfusion and the Effect of Silibinin. Transl Gastroenterol Hepatol (2020) 5:7. doi: 10.21037/tgh.2019.11.01
56. Zhou L, Zhuo H, Ouyang H, Liu Y, Yuan F, Sun L, et al. Glycoprotein non-Metastatic Melanoma Protein B (Gpnmb) is Highly Expressed in Macrophages of Acute Injured Kidney and Promotes M2 Macrophages Polarization. Cell Immunol (2017) 316:53–60. doi: 10.1016/j.cellimm.2017.03.006
57. Haralanova-Ilieva B, Ramadori G, Armbrust T. Expression of Osteoactivin in Rat and Human Liver and Isolated Rat Liver Cells. J Hepatol (2005) 42(4):565–72. doi: 10.1016/j.jhep.2004.12.021
58. Pahl MV, Vaziri ND, Yuan J, Adler SG. Upregulation of Monocyte/Macrophage HGFIN (Gpnmb/Osteoactivin) Expression in End-Stage Renal Disease. Clin J Am Soc Nephrol (2010) 5(1):56–61. doi: 10.2215/CJN.03390509
59. Marques ARA, Gabriel TL, Aten J, van Roomen CPAA, Ottenhoff R, Claessen N, et al. Gpnmb is a Potential Marker for the Visceral Pathology in Niemann-Pick Type C Disease. PloS One (2016) 11(1):e0147208. doi: 10.1371/journal.pone.0147208
60. Kramer G, Wegdam W, Donker-Koopman W, Ottenhoff R, Gaspar P, Verhoek M, et al. Elevation of Glycoprotein Nonmetastatic Melanoma Protein B in Type 1 Gaucher Disease Patients and Mouse Models. FEBS Open Bio (2016) 6(9):902–13. doi: 10.1002/2211-5463.12078
61. Zigdon H, Savidor A, Levin Y, Meshcheriakova A, Schiffmann R, Futerman AH. Identification of a Biomarker in Cerebrospinal Fluid for Neuronopathic Forms of Gaucher Disease. PloS One (2015) 10(3):e0120194. doi: 10.1371/journal.pone.0120194
62. Sasaki F, Kumagai K, Uto H, Takami Y, Kure T, Tabu K, et al. Expression of Glycoprotein Nonmetastatic Melanoma Protein B in Macrophages Infiltrating Injured Mucosa is Associated With the Severity of Experimental Colitis in Mice. Mol Med Rep (2015) 12(5):7503–11. doi: 10.3892/mmr.2015.4408
63. Katayama A, Nakatsuka A, Eguchi J, Murakami K, Teshigawara S, Kanzaki M, et al. Beneficial Impact of Gpnmb and its Significance as a Biomarker in Nonalcoholic Steatohepatitis. Sci Rep (2015) 5:16920. doi: 10.1038/srep16920
64. Yang CF, Lin SP, Chiang CP, Wu YH, H’ng WS, Chang CP, et al. Loss of GPNMB Causes Autosomal-Recessive Amyloidosis Cutis Dyschromica in Humans. Am J Hum Genet (2018) 102(2):219–32. doi: 10.1016/j.ajhg.2017.12.012
65. Song R, Lin L. Glycoprotein Nonmetastatic Melanoma Protein B (Gpnmb) Ameliorates the Inflammatory Response in Periodontal Disease. Inflammation (2019) 42(4):1170–8. doi: 10.1007/s10753-019-00977-4
66. Rich JN, Shi Q, Hjelmeland M, Cummings TJ, Kuan CT, Bigner DD, et al. Bone-Related Genes Expressed in Advanced Malignancies Induce Invasion and Metastasis in a Genetically Defined Human Cancer Model. J Biol Chem (2003) 278(18):15951–7. doi: 10.1074/jbc.M211498200
67. Fiorentini C, Bodei S, Bedussi F, Fragni M, Bonini SA, Simeone C, et al. GPNMB/OA Protein Increases the Invasiveness of Human Metastatic Prostate Cancer Cell Lines DU145 and PC3 Through MMP-2 and MMP-9 Activity. Exp Cell Res (2014) 323(1):100–11. doi: 10.1016/j.yexcr.2014.02.025
68. Graeber MB, Li W, Rodriguez ML. Role of Microglia in CNS Inflammation. FESB Lett (2011) 585(23):3798–805. doi: 10.1016/j.febslet.2011.08.033
69. Mashaghi A, Marmalidou A, Tehrani M, Grace PM, Pothoulakis C, Dana R. Neuropeptide Substance P and the Immune Response. Cell Mol Life Sci (2016) 73(22):4249–64. doi: 10.1007/s00018-016-2293-z
70. Layé S, Parnet P, Goujon E, Dantzer R. Peripheral Administration of Lipopolysaccharide Induces the Expression of Cytokine Transcripts in the Brain and Pituitary of Mice. Brain Research. Mol Brain Res (1994) 27(1):157–62. doi: 10.1016/0169-328x(94)90197-x
71. Hagan P, Poole S, Bristow AF. Endotoxin-Stimulated Production of Rat Hypothalamic Interleukin-1 Beta In Vivo and In Vitro, Measured by Specific Immunoradiometric Assay. J Mol Endocrinol (1993) 11(1):31–6. doi: 10.1677/jme.0.0110031
72. Banks WA, Gray AM, Michelle A, Erickson MA, Salameh TS, Damodarasamy M, et al. Lipopolysaccharide-Induced Blood-Brain Barrier Disruption: Roles of Cyclooxygenase, Oxidative Stress, Neuroinflammation, and Elements of the Neurovascular Unit. J Neuroinflamm (2015) 12:223. doi: 10.1186/s12974-015-0434-1
73. Lopes PC. LPS and Neuroinflammation: A Matter of Timing. Inflammopharmacology (2016) 24(5):291–3. doi: 10.1007/s10787-016-0283-2
74. Murthy MN, Blauwendraat C, Guelfi S, Hardy J, Lewis PA, Trabzuni D. Increased Brain Expression of GPNMB is Associated With Genome Wide Significant Risk for Parkinson’s Disease on Chromosome 7p15.3. Neurogenetics (2017) 18:121–33. doi: 10.1007/s10048-017-0514-8
75. Kinoshita PF, Yshii LM, Orellana AMM, Paixão AG, Vasconcelos AR, Lima LS, et al. Alpha 2 Na +,K +-ATPase Silencing Induces Loss of Inflammatory Response and Ouabain Protection in Glial Cells. Sci Rep (2017) 7(1):4894. doi: 10.1038/s41598-017-05075-9
76. Orellana AM, Kinoshita PF, Leite JA, Kawamoto EM, Scavone C. Cardiotonic Steroids as Modulators of Neuroinflammation. Front Endocrinol (2016) 7:10. doi: 10.3389/fendo.2016.00010
77. Wu L, Xiong X, Wu X, Ye Y, Jian Z, Zhi Z, et al. Targeting Oxidative Stress and Inflammation to Prevent Ischemia-Reperfusion Injury. Front Mol Neurosci (2020) 13:28. doi: 10.3389/fnmol.2020.00028
78. Chung JS, Dougherty I, Cruz PD Jr, Ariizumi K. Syndecan-4 Mediates the Coinhibitory Function of DC-HIL on T Cell Activation. J Immunol (2007b) 179(9):5778–84. doi: 10.4049/jimmunol.179.9.5778
79. Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity (2019) 51(1):27–41. doi: 10.1016/j.immuni.2019.06.025
80. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat Med (2018) 24(5):541–50. doi: 10.1038/s41591-018-0014-x
81. Kobayashi M, Chung JS, Beg M, Arriaga Y, Verma U, Courtney K, et al. Blocking Monocytic Myeloid-Derived Suppressor Cell Function Via Anti-DC-HIL/GPNMB Antibody Restores the In Vitro Integrity of T Cells From Cancer Patients. Clin Cancer Res (2019) 25(2):828–38. doi: 10.1158/1078-0432.CCR-18-0330
82. Lee L, Gupta M, Sahasranaman S. Immune Checkpoint Inhibitors: An Introduction to the Next-Generation Cancer Immunotherapy. J Clin Pharmacol (2016) 56(2):157–69. doi: 10.1002/jcph.591
83. Chung JS, Ramani V, Kobayashi M, Fattah F, Popat V, Zhang S, et al. Dc-Hil/Gpnmb Is a Negative Regulator of Tumor Response to Immune Checkpoint Inhibitors. Clin Cancer Res (2020) 26(6):1449–59. doi: 10.1158/1078-0432
Keywords: GPNMB, inflammation, neuroinflammation, macrophages, cancer, protection
Citation: Saade M, Araujo de Souza G, Scavone C and Kinoshita PF (2021) The Role of GPNMB in Inflammation. Front. Immunol. 12:674739. doi: 10.3389/fimmu.2021.674739
Received: 01 March 2021; Accepted: 23 April 2021;
Published: 12 May 2021.
Edited by:
James Harris, Monash University, AustraliaReviewed by:
Michelle Coleman, Seattle Children's Research Institute, United StatesJames Vince, Walter and Eliza Hall Institute of Medical Research, Australia
Copyright © 2021 Saade, Araujo de Souza, Scavone and Kinoshita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paula Fernanda Kinoshita, cGF1bGEuZi5raW5vc2hpdGFAZ21haWwuY29t