 Eleni Stamoula1
Eleni Stamoula1 Spyridon Siafis1
Spyridon Siafis1 Ioannis Dardalas1
Ioannis Dardalas1 Alexandra Ainatzoglou1
Alexandra Ainatzoglou1 Alkis Matsas2
Alkis Matsas2 Theodoros Athanasiadis3
Theodoros Athanasiadis3 Chrysanthi Sardeli1
Chrysanthi Sardeli1 Konstantinos Stamoulas3
Konstantinos Stamoulas3 Georgios Papazisis1*
Georgios Papazisis1*- 1Department of Clinical Pharmacology, School of Medicine, Aristotle University of Thessaloniki, Thessaloniki, Greece
- 2School of Medicine, National and Kapodistrian University of Athens, Athens, Greece
- 3School of Medicine, Aristotle University of Thessaloniki, Thessaloniki, Greece
Background: Increased prevalence of depression has been observed among patients with multiple sclerosis (MS) and correlated with the elevated levels of proinflammatory cytokines and the overall deregulation of monoaminergic neurotransmitters that these patients exhibit. Antidepressants have proved effective not only in treating depression comorbid to MS, but also in alleviating numerous MS symptoms and even minimizing stress-related relapses. Therefore, these agents could prospectively prove beneficial as a complementary MS therapy.
Objective: This review aims at illustrating the underlying mechanisms involved in the beneficial clinical effects of antidepressants observed in MS patients.
Methods: Through a literature search we screened and comparatively assessed papers on the effects of antidepressant use both in vitro and in vivo MS models, taking into account a number of inclusion and exclusion criteria.
Results: In vitro studies indicated that antidepressants promote neural and glial cell viability and differentiation, reduce proinflammatory cytokines and exert neuroprotective activity by eliminating axonal loss. In vivo studies confirmed that antidepressants delayed disease onset and alleviated symptoms in Experimental Autoimmune Encephalomyelitis (EAE), the most prevalent animal model of MS. Further, antidepressant agents suppressed inflammation and restrained demyelination by decreasing immune cell infiltration of the CNS.
Conclusion: Antidepressants were efficient in tackling numerous aspects of disease pathophysiology both in vitro and in vivo models. Given that several antidepressants have already proved effective in clinical trials on MS patients, the inclusion of such agents in the therapeutic arsenal of MS should be seriously considered, following an individualized approach to minimize the adverse events of antidepressants in MS patients.
Introduction
Multiple Sclerosis and Depression
Multiple sclerosis (MS) is the most common demyelinating disease of the central nervous system (CNS), involving inflammatory, neurodegenerative and autoimmune patterns in its pathogenesis (1, 2). Most frequently, the onset of MS is characterized by a clinical course of relapses and remissions (RRMS) present in almost 90% of MS patients (3). Current therapeutic means such as disease modifying therapies (DMTs) are mostly efficient during this stage, as CNS inflammation is still highly prominent and directly implied in the emergence of relapses (4, 5). Along with DMTs, antidepressants are often prescribed to MS patients,as they are quite prone to manifest symptoms of depression and anxiety (6–8). In fact, studies report a 50% lifetime risk of major depression for MS patients (9).
Stress-Related MS Relapses
A significant factor that has been repeatedly held responsible for igniting MS relapses are stressful life events (SLEs) (10, 11). In MS patients, SLEs have proved to spark inflammatory activity by interfering with immune-mediated pathways that regulate autonomic functions, along with the Hypothalamic-Pituitary-Adrenal (HPA) axis (12). Hyper reactivity of the HPA axis is a common finding among MS patients (13). However, chronic stress compromises the ability of endogenous glucocorticoids to regulate inflammation in MS, as it desensitizes immune cells to their regulation by cortisol (12, 14). Resistance to the effects of glucocorticoids has been observed in animals undergoing chronic stress, suggesting that a similar pathway describes the impact of stress on MS patients (15).
Serotonin and MS
Serotoninergic routes are highly responsible for modulating both our autonomic and neuroendocrine reactions to stressful stimuli, as serotonin constitutes a major HPA axis modulator (16, 17). In patients suffering from depression or anxiety, the serotoninergic network is significantly altered by accumulating stress, thereby severely impacting HPA axis function (18). This defect, however, has proved to be reversed upon antidepressant treatment (19, 20). On that premise, antidepressants could constitute a very promising add-on therapy for MS, as elevated bioavailability of serotonin in MS patients may be efficient in reversing the impact of chronic stress on disease progression.
With respects to serotonin or 5-hydroxytriptamine (5-HT), it displays immunomodulatory properties, interfering with T-cell activation, cytokine release from monocytes, and natural killer (NK) cell stimulation (21–25).Multiple pre-clinical studies have unanimously suggested that selective serotonin reuptake inhibitors (SSRIs) promote remission of the clinical signs of experimental autoimmune encephalomyelitis (EAE), the most prevalent animal model of MS, by curbing pro-inflammatory cytokine release (IFN-γ, TNF-a, IL-6, IL-7) and reducing T-cell proliferation (26–29).
In parallel, solid evidence provided by clinical trials has demonstrated that the use of the SSRI escitalopram in women with MS was effective in preventing stress-related relapses (30). To date, long-term impairment remains the inevitable outcome in most MS cases and current drugs fall short of addressing this fervent matter. It has been proved, however, that long-term disability is highly contingent on the build-up of tokens of impairment that remain after the cessation of each relapse (5). Minimizing relapse frequency is of grave importance for achieving a significant delay in the onset of severe impairment and therefore agents like SSRIs that have proved efficient in this field should be seriously considered as a complementary therapeutic option for all MS patients. Given however the individuality of each MS patient and the varying side events exerted by antidepressants, a personalized prescription of these drugs based on the needs of each patient would be highly recommendable (31).
Other Key Neurotransmitters in MS
Accumulating evidence suggests that several motor and non-motor symptoms of MS can be attributed to pathologically reduced levels of key neurotransmitters (32–38). Apart from serotonin (39), studies have detected abnormal fluctuations in the levels of noradrenaline (NE) and γ-aminobutyric acid (GABA) (29, 40) within the CNS of EAE mice. Since agents that increase GABAergic and monoaminergic transmission have been shown to moderate EAE severity (29, 41–43), antidepressants could be deemed as potential therapeutic compounds, capable of suppressing the clinical symptoms and neuropathological characteristics of MS (29, 40, 44).
It is worth noting that these key neurotransmitters display both neuronal and immunomodulatory properties, as 5-HT, NE and GABA not only regulate immune cell function (29, 36–38, 45), but also attenuate EAE severity through anti-inflammatory pathways (29, 41, 45). T cells and macrophages express functional receptors and are capable of synthesizing 5-HT, glutamate, GABA and dopamine (DA) (21, 46, 47). Futher, the alpha and beta 2 adrenergic receptors expressed on the surface of T-cells render them susceptible to regulation by adrenergic transmission (48). Similarly, T-cells and macrophages express functional GABA-A receptors, proving that the maintenance of key neurotransmitters at high concentrations is critical for immunomodulation (29, 49).
Animal Models of MS
As already mentioned, MS is a chronic, autoimmune and demyelinating disease of CNS. While MS is only found in humans, many in vivo models have been developed to better simulate the pathophysiology of this disease. None of the in vivo MS models is perfect; none of these can reproduce the whole range of complex and diverse morphological and functional aspects of this CNS condition. Each one of them has its advantages and disadvantages, all of them have certain limitations. Albeit certain animal models of MS have proved to be valuable tools, mainly in the development of novel MS drugs (50).
According to a review on MS animal models, the experimental autoimmune encephalomyelitis (EAE) model is one of the most representative in vivo MS models as it imitates both the clinical and the pathological characteristics of this condition, followed by the Virus-induced demyelination models (50).
The MS induction on in vivo models could be well categorized into three main classes. These include toxin-induced demyelination models, the virus-induced demyelination model mainly by Theiler’s murine encephalomyelitis virus and the above-mentioned widely used experimental autoimmune encephalomyelitis (EAE) model (50, 51).
Toxin-induced demyelination models are based either on linear inoculation of gliotoxins in the white matter, including ethidium bromide (EtBr) and lysolecithin, or on systemically administered toxins, with cuprizone being the most representative. These models offer duplicability, while the demyelinated area is distinct for further remyelinating studies. Furthermore, ethidium bromide, a toxic intercalating agent, affects both the nucleus DNA and the mitochondrial DNA, but offers well established predictable results, as the magnitude of demyelination is concentration-dependent. Lyso-phosphatidylcholine (lysolecithin) has been used for almost 50 years. Its mechanism of action in the demyelinating process is based on its physicochemical properties, as it can act as a detergent-like agent with selectivity over the myelin-producing cells marking and engaging T and B cells, like activated macrophages. This method can also be implemented in non-human primates, while also the demyelination can be performed in a spatiotemporal manner. On the contrary, this method does not lead to any immune response resembling the one recognized during multiple sclerosis (50).
Certain other toxins possess analogous demyelinating toxic results but are not in general use. Examples include ionomycin, a calcium ionophore, 6-aminonicotinamide, an antimetabolite of niacin and diphtheria toxin. Antibody-mediated demyelination is also an acknowledged animal model of induced demyelination by galactocerebroside antibodies. Finally, this class of methods included cuprizone, a copper-chelating agent, which has been shown to be toxic for myelin, affecting both white and grey matter leading to oligodendrocyte apoptosis, mitochondrial enzyme malfunction and activation of microglia. Like lysolecithin, cuprizone can also be performed in a spatiotemporal manner while interest is focused on the combined use of cuprizone with other methods of demyelination induction like EAE.
There is growing indication that certain viruses are involved in the pathogenesis of MS, functioning like environmental triggers. The Epstein-Barr virus (EBV) is a typical example that has long been associated with autoimmune conditions including multiple sclerosis despite the exact cause still remains unknown (51). Viruses that have been used in vivo as MS inductors include Theiler’s murine encephalomyelitis virus (TMEV), the canine distemper virus and the mouse hepatitis virus. The former is the most established and serves as a neurotropic viral infection model. TMEV can be separated into two main categories based on the virulence of the viral strains or subgroups and the qualification to induce demyelination. The effects of each viral subgroup extend from severe encephalitis to deadly encephalomyelitis, also being subject to the mouse strains. The most defiant are the BALB/c, C57BL/6J, C57BL/10, and C57/L mouse strains (50). This model can lead to both acute and chronic phase of CNS toxicity, outlined by CNS inflammation and neural apoptosis and affecting the subcortical gray matter, the hippocampus and the basal ganglia.
The most established in vivo model of MS is the EAE model which can mimic a broader spectrum of histopathological and immunological expressions of the disease. EAE can be induced via two different paths, the active immunization with myelin peptides (52)or the passively or adoptively transferred encephalitogenic T cells (53).
Active EAE requires mice, rats, guinea pigs or nonhuman primates, the use of a myelin-related antigen and concomitant injections of pertussis toxin, leading to activated myelin-specific T cells and encephalitogenic lymphocyte–mediated demyelination. Conversely passive EAE is based on the administration of activated, myelin– specific T cells. Passive EAE evolves faster, does not require any adjuvant and showcases better homogeneity, however its main limitation is that the myelin antigen–specific T cells might not have the desired encephalitogenic capacity, when used in vivo (54).
EAE is also affected by the animal strains or species used. The leading option for animals that can accurately imitate the pathophysiology of MS are mice and rats of different strains including Lewis, Dark Agouti (DA) and Brown Norway (BN). Additionally, non-human primates including common marmosets (Callithrix jacchus) and rhesus monkeys (Macaca mulatta), can also be used for in vivo experiments on MS (50).
Therefore, the aim of this review is to provide readers with a useful insight into pre-clinical findings regarding the immunomodulatory effects of antidepressants in in vivo and in vitro models of MS.
Methods
Literature Search
We systematically searched the literature for studies investigating the effects of antidepressants on in vitro and in vivo models of multiple sclerosis. An electronic database literature search was conducted in PubMed, Cochrane and Scopus from inception through 17 April 2021 to provide us with results from in vivo and in vitro studies.
The following keywords were used: for in vivo studies (experimental autoimmune encephalomyelitis OR EAE) AND (MS OR sclerosis) AND antidepressant; for in vitro (In Vitro or cell culture) AND (MS or sclerosis) AND antidepressant. Retrieved articles were imported to EndNote. All articles were independently screened for duplicity and eligibility by author ES and ID.
Inclusion and Exclusion Criteria for In Vitro Papers
The inclusion criteria for in vitro research were the following: i) original research paper, ii) published in English, iii) use of antidepressant drugs/agents, iv) use of antidepressant agents as a monotherapy or combination treatments.
Articles were excluded if: i) the study did not evaluated MS, ii) the pharmacological agent had antidepressant properties but no clinical use as an antidepressant iv) only the abstract was available, v) the research involved patients. In total, our search yielded 271 articles of which 6 were eligible as abstracts. Finally, after the full text of each article was retrieved and all our inclusion criteria were met, 4 articles were included (Figure 1).
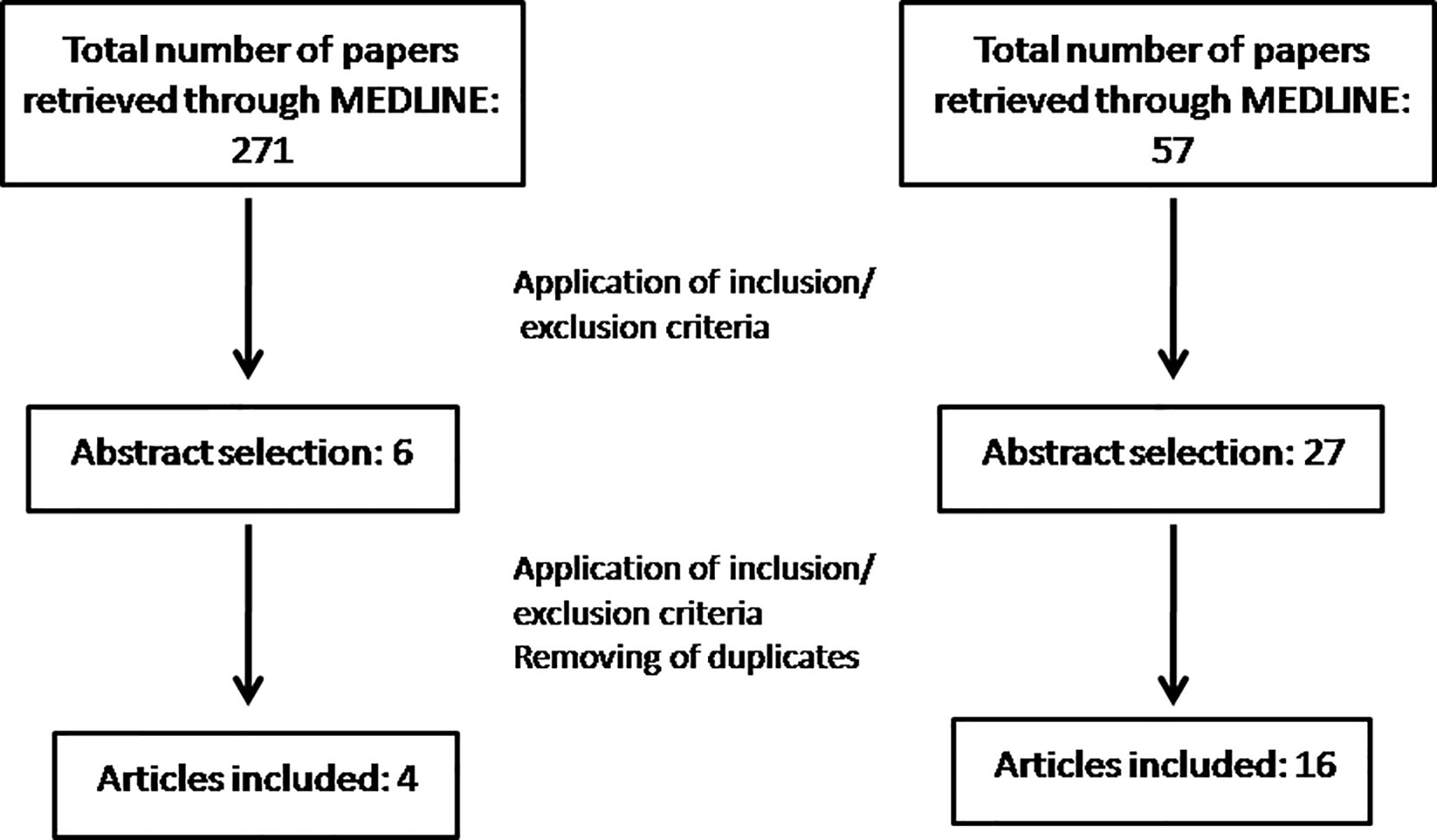
Figure 1 Flow chart of in vitro and in vivo results.
Inclusion and Exclusion Criteria for In Vivo Papers
Inclusion criteria for in vivo research were the following: i) original research paper, ii) published in English, iii) use of antidepressant drugs/agents, iv) use of antidepressant agents as a monotherapy or combination treatments, v) use of validated in vivo tests vi) induction of EAE in mice and rats.
Articles were excluded if i) the study did not evaluated MS, ii) no behavioral tests were used, iii) the pharmacological agent had antidepressant properties but no clinical use as an antidepressant iv) only the abstract was available, v) the article was a review or a case report. In total, our search yielded 59 articles of which 27 were eligible as abstracts. Finally, after the full text of each article was retrieved and all our inclusion criteria were met, 16 articles were included (Figure 1).
Results
In Vitro Results
In our research we ended up with 4 studies on antidepressants use, on in vitro models of MS. All studies were performed in in vivo and in vitro models of MS. Cultures involved cells that were either human or rat and mice derived. Among the drugs examined in this review are the tricyclic antidepressants clomipramine, desipramine, imipramine, amitriptyline, the selective serotonin reuptake inhibitors fluvoxamine (55), and the serotonin- norepinephrine reuptake inhibitor (SNRI) drug venlafaxine (38). The antidepressant effects of these drugs on MS models were evaluated using various methods. Real-time PCR, Western blot analysis and ELISA assay were the most widely used techniques, apart from live-cell imaging, immunohistochemistry, immunostaining and immunofluorescence (IF). Ghareghani et al. found that fluvoxamine enhanced cell proliferation, viability and differentiation of astrocytes, oligodendrocytes and embryonic neural stem cells (eNSCs) (55). Venlafaxine reduced the secretion of pro-inflammatory cytokines such as TNF-a, IFN-γ and IL-6, therefore suppressing inflammation in the CNS, while regulating NK cell and T-cell gene expression (38). Tricyclic antidepressant drugs were found to exhibit neuroprotective activity through elimination of neuronal loss. Reduced proliferation of T-cells and activated B-cells was observed, in tandem with suppression of TNF-a secretion (56).
Ghareghani et al. used murine embryonic neural stem cells from Lewis rat embryos to study the effects of fluvoxamine performing MTT assay to assess cell viability, Real-time PCR, Western blot analysis and Immunofluorescence (IF) analyses. Fluvoxamine was found to act through the Notch signaling pathway, enhancing cell proliferation transcription factors at even low concentrations. Astrocyte, oligodendrocyte and neuron differentiation was observed to be upregulated which may be attributed to upregulation of the mRNA expression of Notch1, Hes1 and Ki-67 (55).
In their study Faissner et al. used cell cultures from both human (brain tissues and peripheral blood mononuclear cells) and murine (splenocytes) origin. Neurotoxicity was induced by rotenone, while HORAC assay, Flow cytometry, live cell imaging, Immunocytochemistry and microscopy were performed. The researchers concluded that Clomipramine, Desipramine, Trimipramine, Imipramine and Doxepin all belonging to the tricyclic antidepressant class, exert beneficial effects in the treatment of MS. Prevention of neuronal loss and antioxidative effects were also observed, while T-cell and activated B-cell proliferation, TNF-a production and plasma membrane compromise were all reduced. These findings highlight an overall neuroprotective activity, that is of pivotal importance for a demyelinating autoimmune disease like MS (56).
In Vivo Results
The in vivo results indicated that SSRIs, such as sertraline, fluoxetine and fluvoxamine either delayed disease onset or ameliorated the clinical symptoms in EAE mice. SSRIs mitigated clinical scores and eliminated EAE symptoms, mainly through their actions on immunomodulatory cells. Sertraline-treated mice manifested milder clinical symptoms compared to the untreated EAE group, while sertraline displayed a dose-dependent inhibitory effect on the secretion of the pro-inflammatory cytokines IL-2, TNF-a and INF-γ. Similarly, the reduction of cytokines in mice serum (IL-6, IL-10, TNF-a and INF-γ) was also observed after fluoxetine treatment. Apart from cytokines, fluoxetine also reduced inflammation by directly impacting APC and naïve T-cells. In EAE rats, both fluoxetine (pretreatment/preventive) and fluvoxamine (symptomatic treatment) eliminated clinical symptoms and reduced IFN-γ secretion. Interestingly, fluvoxamine also inhibited the formation of demyelinating plaques, suppressed immune cell infiltration into the CNS and upregulated anti-inflammatory agents. Moreover, in a rat EAE model, duloxetine prevented cold allodynia and showed anti-nociceptive effect on cold hyperalgesia, thus alleviating some clinical signs.
Dose-dependent relief of mechanical allodynia in the bilateral hind paws of EAE mice was also observed after treatment with amitriptyline, a tricyclic antidepressant. In addition, pharmacological intervention with chronic application of amitriptyline in the mild MOG-EAE mice model resulted in a decreased startle reaction and increased hippocampal norepinephrine levels. Another group of researchers (57) utilized the combination treatment or nortriptyline (TCA) and desloratadine (antihistamine) to assess their therapeutic potential on EAE mice. This combination treatment moderated EAE severity by reducing CD4+T cell infiltration in the CNS and suppressing IFN-γ, IL-17 secretion, while boosting anti-inflammatory IL-4 levels. These findings are aligned with other observations supporting that imipramine reduces plasma levels of IL-4 and clomipramine decreases m-RNA expression levels of IFN-γ, TNF-a, IL-17 and chemokine CCL-2. Overproduction of chemokine CCL-5 (also known as RANTES) was mitigated by desipramine, thus restoring glutamate exocytosis and presynaptic cortical defects (57).
In another study, researchers used splenocytes, encephalitogenic T cell clones, primary peritoneal macrophages and brain and spinal cord sections from female mice after the EAE protocol was performed in vivo. They conducted ELISA to determine the cytokine levels in the culture supernatants, while carrying out cell viability assay and real-time PCR after RNA isolation. Venlafaxine an SNRI drug was found to regulate the clinical and histopathological impact of EAE. Pro-inflammatory cytokines such as TNF-a, IFN-γ, IL-6, Ccl5 and IL-12 were downregulated while CNS inflammation was also reduced showcasing a potential efficacy in MS (38). According to Dawson et al, fingolimod inhibits the enzyme acid sphingomyelinase sharing a related mechanism of action with desipramine, a tricyclic antidepressant. The researchers used neural-derived cells and fibroblasts and observed that desipramine suppressed ASMase without inducing significant inhibition of other lysosomal hydrolases (58).
According to Taler et al, antidepressants, especially SSRIs, display an immunomodulatory activity by reducing immune cell viability and attenuating of pro-inflammatory cytokine secretion. In particular, their research demonstrated that treatment of EAE mice with sertraline alleviated the neurological symptoms of MOG-induced chronic EAE (42). In addition, fluoxetine suppresses the adaptive immune response in EAE through the reduction of cytokine release (IL-6, IL-10, TNF-a, IFN-γ) and induction of CD4 T-cell apoptosis (59, 60). Recently, a study indicated that the SNRI venlafaxine suppressed the secretion of the pro-inflammatory agents TNF-a, IFN-γ, IL-2 and chemokines in encephalitogenic T cellclones, splenocytes and macrophages, while increasing BDNF expression (38).
Furthermore, treatment of EAE mice with the SNRI venlafaxine ameliorated EAE symptoms in a dose-dependent manner. Venlafaxine exerted its beneficial effects through suppression or enhancement of mRNA expression of proinflammatory and anti-inflammatory factors, respectively. These proinflammatory factors include IFN-γ, TNF-a, IL-12, chemokine CCL-2, CCL-5. On the contrary, venlafaxine increased mRNA expression of the neurotrophic factor BDNF.
Moreover, phenelzine a MAO inhibitor, has been used as a treatment in established EAE- female C57/BL6 mice. It was observed that phenelzine delayed the onset of clinical signs, reduced impairments, ameliorated locomotor function and demonstrated antinociceptive effects. The aforementioned benefits derive from phenelzine’s ability to normalize the levels of GABA and biogenic amines that have been shown to possess anti-inflammatory properties. In particular, phenelzine increased the levels of 5-HT, NE, DA within the spinal cord, brain and brainstem. Lastly, phenelzine normalized pre-synaptic excitatory synaptic densities in S1 and neuronal morphologies.
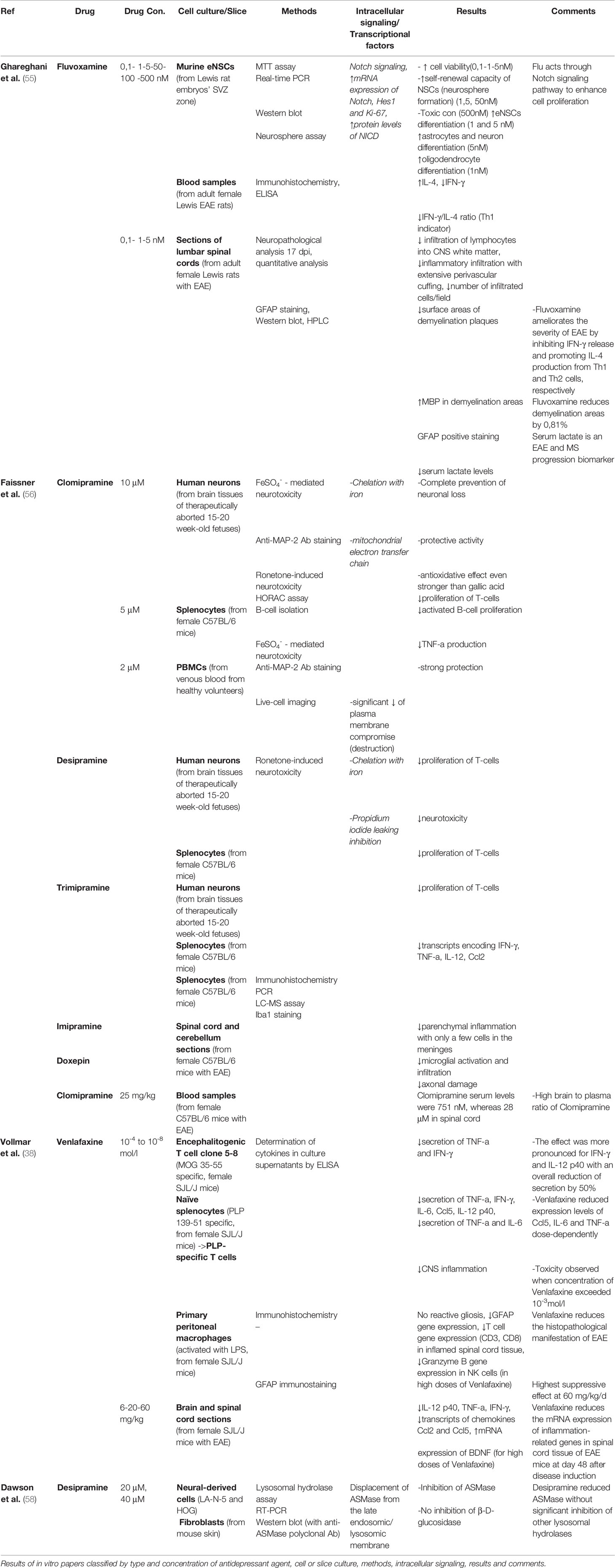
Table 1 Comparative assessment of in vitro studies on the effects of antidepressants in cell and slice cultures.
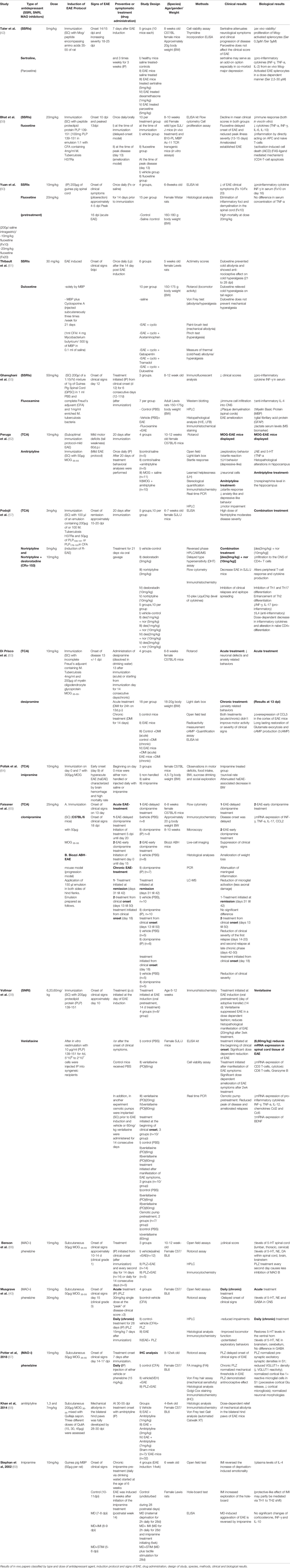
Table 2 Comparative assessment of in vivo studies on the effects of antidepressants on disease scores and progression.
Discussion
Among MS patients, depression constitutes a highly frequent comorbidity, as studies indicate a 25% prevalence of depression in MS (6, 70). This trend severely affects the quality of life perceived by MS patients, as following disability, depression is the second most impactful factor determining the health-related quality of life (71). Moreover, depression can compromise patient adherence to DMTs, further affecting MS prognosis (72, 73). Although to date, about 86% of depressive MS patients receive antidepressant therapy, depressive symptoms often remain, pointing towards an underdosage or poor matching of these drugs to each patient (74).
Findings encompassed in this review have documented the efficacy of antidepressants in promoting oligodendrocyte maturation and proliferation (55). In MS patients, demyelination is often accompanied by compensatory remyelinating activity, an effect that is principally mediated by oligodendrocyte maturation (75). Therefore, agents like antidepressants or phosphodiesterase inhibitors (76) that stimulate the differentiation of oligodendrocyte precursor cells (OPCs) into mature oligodendrocytes also boost remyelination, thus exerting a neuroprotective effect. This effect can also be indirectly attained through suppression of cytokines that curb Oligodendrogenesis.
The regulation of T cell proliferation and stimulation by antidepressants reported in some studies of this review (38, 56)is of great significance, as these aspects are directly involved in MS pathogenesis. Myelin-reactive T cells are present in MS patients and held accountable for igniting demyelination, therefore the suppression of their activation, proliferation and migration constitute a very salutary property displayed by antidepressants. Lately, the role of B cells in MS has also been described as crucial, involving actions like the orchestration of effector T cell activity through antigen presentation and priming, as well as the secretion of proinflammatory cytokines (77, 78), rendering them principally responsible for the formation of a proinflammatory milieu in the CNS (79).
Studies included in this review also reported the suppression of proinflammatory cytokines induced by antidepressants. Along with several established proinflammatory cytokines such as IL-2, IL-6, IL-12, IL-17, TNFa and IFNγ, antidepressants were also found to reduce serum levels of anti-inflammatory cytokines IL-4 and IL-10, though there has been some evidence supporting some of their immunostimulatory properties (80, 81).
Although MS is considered a Th1 autoimmune disease in which prevails a CD4+ immune response, CD8+ T cells seem to play a pivotal role in the pathogenesis of major depressive disorder (MDD). Clinical studies revealed that CD8+ T cells are increased in MS patients with depression compared to those without, being traceable in their serum during active phases (82). According to other studies, however, CD4+ T cells also seem to be augmented during MDDs in MS (83).
In a clinical scope, antidepressants have proved to be efficient not only in tackling depression comorbid to MS (84, 85), but also even in minimizing stress-related relapses, as shown by the clinical trials of escitalopram on female MS patients (30). Therefore, the use of antidepressants is not only a consolation therapy to improve the quality of life in MS, but also has the potential to significantly modify the course of the disease. Other antidepressants such as vortioxetine combine their antidepressant properties with an enhancing effect on patients’ cognition (86–88). This constitutes a very significant aspect, as about half of MS patients are estimated to manifest cognitive impairment (89). This agent however has neither yet undergone clinical trials on MS patients nor is its efficacy on cognitive enhancement unanimously accepted (90).
Regarding antidepressant use in MS, several adverse events of these drugs could potentially overlap some of the existing deficits that are to be found in MS patients, therefore exacerbating them. To draw an example, SSRIs are known to cause sexual dysfunction, a state that might be already prominent in MS patients, even reaching 85% in female MS patients (91). Therefore, given the heterogeneity of the clinical course of MS in each individual patient, a personalized and patient-oriented approach is necessary to ensure both safety and efficacy in the use of antidepressants in MS (31, 92).
Antidepressants, however, also have the capacity to alleviate numerous MS symptoms. Bupropion can benefit MS patients suffering from chronic fatigue, as this drug has been clinically shown to improve the fatigue severity scale when tested on a patient with MS (93, 94). Fatigue accounts for one of the most prevalent symptoms among MS patients, severely impacting their experienced quality of life. However, the multifactorial and diverse nature of this symptom impedes its management, calling for personalized treatments (95). Therefore, although randomized-controlled trials (RCTs) with numerous participants are required to secure this observation, the identification of a soothing effect of antidepressants on fatigue would constitute a highly significant discovery.
With respect to neuropathic pain, the SNRI duloxetine has been proved to adequately treat this distressing symptom prevalent in more than 25% of MS patients (96), as signified in a double-blind RCT (97). This drug has already received FDA approval for the treatment of peripheral neuropathy in diabetic patients, therefore its inclusion in MS therapy would not be far-fetched. Venlafaxine has also demonstrated some promising qualities regarding neuropathic pain (98), while also tackling the issue of migraines. Although the prevalence of migraines in MS remains unclarified, the importance of their treatment has been repeatedly stressed, as this comorbidity has been correlated with a more symptomatic clinical course of MS (99). Finally, duloxetine has been clinically documented to relieve stress urinary incontinence (100–102), without having yet been tested on MS patients that exhibit this symptom. However, on MS patients suffering from overactive bladder syndrome, a precursor of urinary incontinence, duloxetine was found to be efficient (103).
Taken together, this evidence suggests that antidepressants have proved to be highly effective not only in treating depression in MS patients (85), but also in alleviating numerous distressing symptoms that these patients exhibit (31). Nonetheless, apart from relieving MS comorbidities, antidepressants have even proved to alter disease course and delay progression by curbing stress-related relapses that form a significant pharmacological target in RRMS (30). This clinical background further intensifies the importance of our findings, as basic research studies incorporated in this review unanimously attested to the benefits of antidepressants in MS, both in vitro and in the EAE animal model. Regarding in vivo MS models, one of the limitations of this review is that it examined only the EAE animal model, which however constitutes the most prevalent and representative animal model currently used in MS research.
However, clinical trials on the matter remain scarce and inconclusive due to the relatively confined number of participants and the uniqueness of each trial, rendering their comparison futile (31). Therefore, clinical testing of antidepressant agents in MS should be further intensified to provide us with reliable assumptions, as existing evidence remains promising.
Conclusion
All things considered, antidepressants have proved effective both in alleviating EAE, an animal model of MS and in vitro, displaying salutary immunomodulatory and anti-inflammatory properties. Clinical studies have also verified the efficacy and safety profile of antidepressants in MS. However, this field warrants further research that would elucidate the underlying mechanisms of action of these agents in MS and highlight their eligibility as a complementary MS therapy.
Author Contributions
ES: manuscript writing, editing, acquisition of data. ID, SS, AA, AM, TA, KS: Analysis and interpretation of data. CS: manuscript editing. GP: manuscript writing, review of the final manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hagemeier K, Brück W, Kuhlmann T. Multiple Sclerosis - Remyelination Failure as a Cause of Disease Progression. Histol Histopathol (2012) 27(3):277–87. doi: 10.14670/HH-27.277
2. Compston A, Coles A. Multiple Sclerosis. Lancet (2008) 372(9648):1502–17. doi: 10.1016/S0140-6736(08)61620-7
3. Tsang BKT, Macdonell R. Multiple Sclerosis: Diagnosis, Management and Prognosis. Aust Fam Phys (2011) 40(12):948–55.
4. Aktas O, Kieseier B, Hartung HP. Neuroprotection, Regeneration and Immunomodulation: Broadening the Therapeutic Repertoire in Multiple Sclerosis. Trends Neurosci (2010) 33(3):140–52. doi: 10.1016/j.tins.2009.12.002
5. Dutta R, Trapp BD. Mechanisms of Neuronal Dysfunction and Degeneration in Multiple Sclerosis [Internet]. Prog Neurobiol Prog Neurobiol (2011) 93:1–12. doi: 10.1016/j.pneurobio.2010.09.005
6. Patten SB, Beck CA, Williams JVA, Barbui C, Metz LM. Major Depression in Multiple Sclerosis: A Population-Based Perspective [Internet]. Neurol Lippincott Williams Wilkins (2003) 61:1524–7. doi: 10.1212/01.WNL.0000095964.34294.B4
7. Mohr DC, Hart SL, Fonareva I, Tasch ES. Treatment of Depression for Patients With Multiple Sclerosis in Neurology Clinics. Mult Scler (2006) 12(2):204–8. doi: 10.1191/135248506ms1265oa
8. Vattakatuchery JJ, Rickards H, Cavanna AE. Pathogenic Mechanisms of Depression in Multiple Sclerosis. J Neuropsychiatr Clin Neurosci (2011) 23(3):261–76. doi: 10.1176/jnp.23.3.jnp261
9. Chwastiak L, Ehde DM, Gibbons LE, Sullivan M, Bowen JD, Kraft GH. Depressive Symptoms and Severity of Illness in Multiple Sclerosis: Epidemiologic Study of a Large Community Sample. Am J Psychiatry (2002) 159(11):1862–8. doi: 10.1176/appi.ajp.159.11.1862
10. Brown RF, Tennant CC, Dunn SM, Pollard JD. A Review of Stress-Relapse Interactions in Multiple Sclerosis: Important Features and Stress-Mediating and -Moderating Variables [Internet]. Mult Scler (2005) 11:477–84. doi: 10.1191/1352458505ms1170oa
11. Mitsonis CI, Potagas C, Zervas I, Sfagos K. The Effects of Stressful Life Events on the Course of Multiple Sclerosis: A Review [Internet]. Int J Neurosci (2009) 119:315–35. doi: 10.1080/00207450802480192
12. Mohr DC, Pelletier D. A Temporal Framework for Understanding the Effects of Stressful Life Events on Inflammation in Patients With Multiple Sclerosis [Internet]. Brain Behav Immun (2006) 20:27–36. doi: 10.1016/j.bbi.2005.03.011
13. Heesen C, Gold SM, Huitinga I, Reul JMHM. Stress and Hypothalamic-Pituitary-Adrenal Axis Function in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis-a Review [Internet]. Psychoneuroendocrinology (2007) 32:604–18. doi: 10.1016/j.psyneuen.2007.05.002
14. Bruno A, Dolcetti E, Rizzo FR, Fresegna D, Musella A, Gentile A, et al. Inflammation-Associated Synaptic Alterations as Shared Threads in Depression and Multiple Sclerosis [Internet]. Front Media S A (2020) 14:169. doi: 10.3389/fncel.2020.00169
15. Avitsur R, Stark JL, Sheridan JF. Social Stress Induces Glucocorticoid Resistance in Subordinate Animals. Horm Behav (2001) 39(4):247–57. doi: 10.1006/hbeh.2001.1653
16. Lowry CA. Functional Subsets of Serotonergic Neurones: Implications for Control of the Hypothalamic-Pituitary-Adrenal Axis. J Neuroendocr (2002) 14(11):911–23. doi: 10.1046/j.1365-2826.2002.00861.x
17. Melnikov M, Sviridova A, Rogovskii V, Oleskin A, Boziki M, Bakirtzis C, et al. Serotoninergic System Targeting in Multiple Sclerosis: The Prospective for Pathogenetic Therapy. Mult Scler Relat Disord (2021) 51:102888. doi: 10.1016/j.msard.2021.102888
18. Chaouloff F. Serotonin, Stress and Corticoids. J Psychopharmacol (2000) 14(2):139–51. doi: 10.1177/026988110001400203
19. Nikisch G, Mathé AA, Czernik A, Thiele J, Bohner J, Eap CB, et al. Long-Term Citalopram Administration Reduces Responsiveness of HPA Axis in Patients With Major Depression: Relationship With S-citalopram Concentrations in Plasma and Cerebrospinal Fluid (CSF) and Clinical Response. Psychopharmacol (Berl) (2005) 181(4):751–60. doi: 10.1007/s00213-005-0034-3
20. Schüle C. Neuroendocrinological Mechanisms of Actions of Antidepressant Drugs. J Neuroendocr (2007) 19(3):213–26. doi: 10.1111/j.1365-2826.2006.01516.x
21. Yin J, Albert RH, Tretiakova AP, Jameson BA. 5-HT(1B) Receptors Play a Prominent Role in the Proliferation of T-Lymphocytes. J Neuroimmunol (2006) 181(1–2):68–81. doi: 10.1016/j.jneuroim.2006.08.004
22. Dürk T, Panther E, Müller T, Sorichter S, Ferrari D, Pizzirani C, et al. 5-Hydroxytryptamine Modulates Cytokine and Chemokine Production in LPS-primed Human Monocytes Via Stimulation of Different 5-HTR Subtypes. Int Immunol (2005) 17(5):599–606. doi: 10.1093/intimm/dxh242
23. León-Ponte M, Ahern GP, O’Connell PJ. Serotonin Provides an Accessory Signal to Enhance T-cell Activation by Signaling Through the 5-HT7 Receptor. Blood (2007) 109(8):3139–46. doi: 10.1182/blood-2006-10-052787
24. Mössner R, Lesch K-P. Role of Serotonin in the Immune System and in Neuroimmune Interactions. Brain Behav Immun (1998) 12(4):249–71. doi: 10.1006/brbi.1998.0532
25. O’Connell PJ, Wang X, Leon-Ponte M, Griffiths C, Pingle SC, Ahern GP. A Novel Form of Immune Signaling Revealed by Transmission of the Inflammatory Mediator Serotonin Between Dendritic Cells and T Cells. Blood (2006) 107(3):1010–7. doi: 10.1182/blood-2005-07-2903
26. Mohr DC, Goodkin DE, Islar J, Hauser SL, Genain CP. Treatment of Depression is Associated With Suppression of Nonspecific and Antigen-Specific T(H)1 Responses in Multiple Sclerosis. Arch Neurol (2001) 58(7):1081–6. doi: 10.1001/archneur.58.7.1081
27. Diamond M, Kelly JP, Connor TJ. Antidepressants Suppress Production of the Th1 Cytokine Interferon-Gamma, Independent of Monoamine Transporter Blockade. Eur Neuropsychopharmacol J Eur Coll Neuropsychopharmacol (2006) 16(7):481–90. doi: 10.1016/j.euroneuro.2005.11.011
28. Mostert JP, Admiraal-Behloul F, Hoogduin JM, Luyendijk J, Heersema DJ, van Buchem MA, et al. Effects of Fluoxetine on Disease Activity in Relapsing Multiple Sclerosis: A Double-Blind, Placebo-Controlled, Exploratory Study. J Neurol Neurosurg Psychiatry (2008) 79(9):1027–31. doi: 10.1136/jnnp.2007.139345
29. Bhat R, Axtell R, Mitra A, Miranda M, Lock C, Tsien RW, et al. Inhibitory Role for GABA in Autoimmune Inflammation. Proc Natl Acad Sci USA (2010) 107(6):2580–5. doi: 10.1073/pnas.0915139107
30. Mitsonis CI, Zervas IM, Potagas CM, Mitropoulos PA, Dimopoulos NP, Sfagos CA, et al. Effects of Escitalopram on Stress-Related Relapses in Women With Multiple Sclerosis: An Open-Label, Randomized, Controlled, One-Year Follow-Up Study. Eur Neuropsychopharmacol (2010) 20(2):123–31. doi: 10.1016/j.euroneuro.2009.10.004
31. Nathoo N, Mackie A. Treating Depression in Multiple Sclerosis With Antidepressants: A Brief Review of Clinical Trials and Exploration of Clinical Symptoms to Guide Treatment Decisions [Internet]. Multiple Sclerosis Related Disord (2017) 18:177–80. Elsevier B.V. doi: 10.1016/j.msard.2017.10.004
32. Barkhatova VP, Zavalishin IA, Askarova LS, Shavratskii VK, Demina EG. Changes in Neurotransmitters in Multiple Sclerosis. Neurosci Behav Physiol (1998) 28(4):341–4. doi: 10.1007/BF02464784
33. Stover JF, Pleines UE, Morganti-Kossmann MC, Kossmann T, Lowitzsch K, Kempski OS. Neurotransmitters in Cerebrospinal Fluid Reflect Pathological Activity. Eur J Clin Invest (1997) 27(12):1038–43. doi: 10.1046/j.1365-2362.1997.2250774.x
34. White SR, Bhatnagar RK, Bardo MT. Norepinephrine Depletion in the Spinal Cord Gray Matter of Rats With Experimental Allergic Encephalomyelitis. J Neurochem (1983) 40(6):1771–3. doi: 10.1111/j.1471-4159.1983.tb08156.x
35. Froh M, Thurman RG, Wheeler MD. Molecular Evidence for a Glycine-Gated Chloride Channel in Macrophages and Leukocytes. Am J Physiol Gastrointest Liver Physiol (2002) 283(4):G856–63. doi: 10.1152/ajpgi.00503.2001
36. Hofstetter HH, Mössner R, Lesch KP, Linker RA, Toyka KV, Gold R. Absence of Reuptake of Serotonin Influences Susceptibility to Clinical Autoimmune Disease and Neuroantigen-Specific Interferon-Gamma Production in Mouse EAE. Clin Exp Immunol (2005) 142(1):39–44. doi: 10.1111/j.1365-2249.2005.02901.x
37. Lee M, Schwab C, McGeer PL. Astrocytes are GABAergic Cells That Modulate Microglial Activity. Glia (2011) 59(1):152–65. doi: 10.1002/glia.21087
38. Vollmar P, Nessler S, Kalluri SR, Hartung H-P, Hemmer B. The Antidepressant Venlafaxine Ameliorates Murine Experimental Autoimmune Encephalomyelitis by Suppression of Pro-Inflammatory Cytokines. Int J Neuropsychopharmacol (2009) 12(4):525–36. doi: 10.1017/S1461145708009425
39. Hodo TW, de Aquino MTP, Shimamoto A, Shanker A. Critical Neurotransmitters in the Neuroimmune Network [Internet]. Front Immunol (2020) 11:1869. Frontiers Media S.A. doi: 10.3389/fimmu.2020.01869
40. Musgrave T, Tenorio G, Rauw G, Baker GB, Kerr BJ. Tissue Concentration Changes of Amino Acids and Biogenic Amines in the Central Nervous System of Mice With Experimental Autoimmune Encephalomyelitis (EAE). Neurochem Int (2011) 59(1):28–38. doi: 10.1016/j.neuint.2011.03.020
41. Simonini MV, Polak PE, Sharp A, McGuire S, Galea E, Feinstein DL. Increasing CNS Noradrenaline Reduces EAE Severity. J Neuroimmune Pharmacol Off J Soc NeuroImmune Pharmacol (2010) 5(2):252–9. doi: 10.1007/s11481-009-9182-2
42. Taler M, Gil-Ad I, Korob I, Weizman A. The Immunomodulatory Effect of the Antidepressant Sertraline in an Experimental Autoimmune Encephalomyelitis Mouse Model of Multiple Sclerosis. Neuroimmunomodulation (2011) 18(2):117–22. doi: 10.1159/000321634
43. Vansant G, Trauger RJ, Cameron A, Vendemelio M, Kreitschitz S, Carlo AT, et al. Propofol Hemisuccinate Suppression of Experimental Autoimmune Encephalomyelitis. Autoimmunity (2007) 40(3):180–6. doi: 10.1080/08916930701204467
44. Baker GB, Sowa B, Todd KG. Amine Oxidases and Their Inhibitors: What can They Tell Us About Neuroprotection and the Development of Drugs for Neuropsychiatric Disorders? J Psychiatry Neurosci (2007) 32(5):313–5.
45. Levite M. Neurotransmitters Activate T-cells and Elicit Crucial Functions Via Neurotransmitter Receptors. Curr Opin Pharmacol (2008) 8(4):460–71. doi: 10.1016/j.coph.2008.05.001
46. Besser MJ, Ganor Y, Levite M. Dopamine by Itself Activates Either D2, D3 or D1/D5 Dopaminergic Receptors in Normal Human T-cells and Triggers the Selective Secretion of Either IL-10, Tnfalpha or Both. J Neuroimmunol (2005) 169(1–2):161–71. doi: 10.1016/j.jneuroim.2005.07.013
47. Sarchielli P, Di Filippo M, Candeliere A, Chiasserini D, Mattioni A, Tenaglia S, et al. Expression of Ionotropic Glutamate Receptor GLUR3 and Effects of Glutamate on MBP- and MOG-specific Lymphocyte Activation and Chemotactic Migration in Multiple Sclerosis Patients. J Neuroimmunol (2007) 188(1–2):146–58. doi: 10.1016/j.jneuroim.2007.05.021
48. McAlees JW, Smith LT, Erbe RS, Jarjoura D, Ponzio NM, Sanders VM. Epigenetic Regulation of beta2-adrenergic Receptor Expression in T(H)1 and T(H)2 Cells. Brain Behav Immun (2011) 25(3):408–15. doi: 10.1016/j.bbi.2010.10.019
49. Mendu SK, Bhandage A, Jin Z, Birnir B. Different Subtypes of GABA-A Receptors Are Expressed in Human, Mouse and Rat T Lymphocytes. PloS One (2012) 7(8):e42959. doi: 10.1371/journal.pone.0042959
50. Bjelobaba I, Begovic-Kupresanin V, Pekovic S, Lavrnja I. Animal Models of Multiple Sclerosis: Focus on Experimental Autoimmune Encephalomyelitis. J Neurosci Res (2018) 96:1021–42. John Wiley and Sons Inc. doi: 10.1002/jnr.24224
51. Dalgleish AG. Viruses and Multiple Sclerosis. Acta Neurol Scand Suppl (1997) 169:8–15. doi: 10.1111/j.1600-0404.1997.tb08144.x
52. Stromnes IM, Goverman JM. Active Induction of Experimental Allergic Encephalomyelitis. Nat Protoc (2006) 1(4):1810–9. doi: 10.1038/nprot.2006.285
53. Stromnes IM, Goverman JM. Passive Induction of Experimental Allergic Encephalomyelitis. Nat Protoc (2006) 1(4):1952–60. doi: 10.1038/nprot.2006.284
54. Robinson AP, Harp CT, Noronha A, Miller SD. The Experimental Autoimmune Encephalomyelitis (EAE) Model of MS. Utility for Understanding Disease Pathophysiology and Treatment. Handb Clin Neurol (2014) 122:173–89. doi: 10.1016/B978-0-444-52001-2.00008-X
55. Ghareghani M, Zibara K, Sadeghi H, Dokoohaki S, Sadeghi H, Aryanpour R, et al. Fluvoxamine Stimulates Oligodendrogenesis of Cultured Neural Stem Cells and Attenuates Inflammation and Demyelination in an Animal Model of Multiple Sclerosis. Sci Rep (2017) 7(1):4923. doi: 10.1038/s41598-017-04968-z
56. Faissner S, Mishra M, Kaushik DK, Wang J, Fan Y, Silva C, et al. Systematic Screening of Generic Drugs for Progressive Multiple Sclerosis Identifies Clomipramine as a Promising Therapeutic. Nat Commun (2017) 8(1):1–15. doi: 10.1038/s41467-017-02119-6
57. Podojil JR, Padval MV, Miller SD. Combination Treatment of Mice With CRx-153 (Nortriptyline and Desloratadine) Decreases the Severity of Experimental Autoimmune Encephalomyelitis. Cell Immunol (2011) 270(2):237–50. doi: 10.1016/j.cellimm.2011.05.015
58. Dawson G, Qin J. Gilenya (FTY720) Inhibits Acid Sphingomyelinase by a Mechanism Similar to Tricyclic Antidepressants. Biochem Biophys Res Commun (2011) Jan404(1):321–3. doi: 10.1016/j.bbrc.2010.11.115
59. Bhat R, Mahapatra S, Axtell RC, Steinman L. Amelioration of Ongoing Experimental Autoimmune Encephalomyelitis With Fluoxetine. J Neuroimmunol (2017) 313:77–81. doi: 10.1016/j.jneuroim.2017.10.012
60. Yuan X, Qiu G, Liu X, Liu S, Wu Y, Wang X, et al. Fluoxetine Promotes Remission in Acute Experimental Autoimmune Encephalomyelitis in Rats. Neuroimmunomodulation (2012) 19(4):201–8. doi: 10.1159/000334095
61. Thibault K, Calvino B, Pezet S. Characterisation of Sensory Abnormalities Observed in an Animal Model of Multiple Sclerosis: A Behavioural and Pharmacological Study. Eur J Pain (2011) 15(3):231.e1–231.e16. doi: 10.1016/j.ejpain.2010.07.010
62. Peruga I, Hartwig S, Thöne J, Hovemann B, Gold R, Juckel G, et al. Inflammation Modulates Anxiety in an Animal Model of Multiple Sclerosis. Behav Brain Res (2011) 220(1):20–9. doi: 10.1016/j.bbr.2011.01.018
63. Di Prisco S, Merega E, Lanfranco M, Casazza S, Uccelli A, Pittaluga A. Acute Desipramine Restores Presynaptic Cortical Defects in Murine Experimental Autoimmune Encephalomyelitis by Suppressing Central CCL5 Overproduction. Br J Pharmacol (2014) 171(9):2457–67. doi: 10.1111/bph.12631
64. Pollak Y, Orion E, Goshen I, Ovadia H, Yirmiya R. Experimental Autoimmune Encephalomyelitis-Associated Behavioral Syndrome as a Model of “Depression Due to Multiple Sclerosis”. Brain Behav Immun (2002) 16(5):533–43. doi: 10.1016/S0889-1591(02)00010-7
65. Faissner A, Kruse J, Klaus K, Schachner M. Binding of the J 1 Adhesion Molecules to Extracellular Matrix Constituents. J Neurochem (1990) 54(3):1004–15. doi: 10.1111/j.1471-4159.1990.tb02350.x
66. Benson CA, Wong G, Tenorio G, Baker GB, Kerr BJ. The MAO Inhibitor Phenelzine can Improve Functional Outcomes in Mice With Established Clinical Signs in Experimental Autoimmune Encephalomyelitis (EAE). Behav Brain Res (2013) 252:302–11. doi: 10.1016/j.bbr.2013.06.019
67. Potter LE, Paylor JW, Suh JS, Tenorio G, Caliaperumal J, Colbourne F, et al. Altered Excitatory-Inhibitory Balance Within Somatosensory Cortex is Associated With Enhanced Plasticity and Pain Sensitivity in a Mouse Model of Multiple Sclerosis. J Neuroinflamm (2016) 13(1):142. doi: 10.1186/s12974-016-0609-4
68. Khan N, Woodruff TM, Smith MT. Establishment and Characterization of an Optimized Mouse Model of Multiple Sclerosis-Induced Neuropathic Pain Using Behavioral, Pharmacologic, Histologic and Immunohistochemical Methods. Pharmacol Biochem Behav (2014) 126:13–27. doi: 10.1016/j.pbb.2014.09.003
69. Stephan M, Straub RH, Breivik T, Pabst R, von Hörsten S. Postnatal Maternal Deprivation Aggravates Experimental Autoimmune Encephalomyelitis in Adult Lewis Rats: Reversal by Chronic Imipramine Treatment. Int J Dev Neurosci (2002) 20(2):125–32. doi: 10.1016/S0736-5748(02)00007-2
70. Marrie RA, Reingold S, Cohen J, Stuve O, Trojano M, Sorensen PS, et al. The Incidence and Prevalence of Psychiatric Disorders in Multiple Sclerosis: A Systematic Review. Multiple Sclerosis J (2015) 21:305-17. SAGE Publications Ltd. doi: 10.1177/1352458514564487
71. Berrigan LI, Fisk JD, Patten SB, Tremlett H, Wolfson C, Warren S, et al. Health-Related Quality of Life in Multiple Sclerosis: Direct and Indirect Effects of Comorbidity. Neurology (2016) 86(15):1417–24. doi: 10.1212/WNL.0000000000002564
72. Treadaway K, Cutter G, Salter A, Lynch S, Simsarian J, Corboy J, et al. Factors That Influence Adherence With Disease-Modifying Therapy in MS. J Neurol (2009) 256(4):568–76. doi: 10.1007/s00415-009-0096-y
73. Tarrants M, Oleen-Burkey M, Castelli-Haley J, Lage MJ. The Impact of Comorbid Depression on Adherence to Therapy for Multiple Sclerosis. Mult Scler Int (2011) 2011:1–10. doi: 10.1155/2011/271321
74. Raissi A, Bulloch AGM, Fiest KM, McDonald K, Jetté N, Patten SB. Exploration of Undertreatment and Patterns of Treatment of Depression in Multiple Sclerosis. Int J MS Care (2015) 17(6):292–300. doi: 10.7224/1537-2073.2014-084
75. Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating Oligodendrocytes in Chronic Lesions of Multiple Sclerosis. N Engl J Med (2002) 346(3):165–73. doi: 10.1056/NEJMoa010994
76. Ainatzoglou A, Stamoula E, Dardalas I, Siafis S, Papazisis G. The Effects of PDE Inhibitors on Multiple Sclerosis: A Review of In Vitro and In Vivo Models. Curr Pharm Des (2021) 27. doi: 10.2174/1381612827666210303142356
77. Kinzel S, Weber MS. B Cell-Directed Therapeutics in Multiple Sclerosis: Rationale and Clinical Evidence. CNS Drugs (2016) 30:1137–48. Springer International Publishing. doi: 10.1007/s40263-016-0396-6
78. Lehmann Horn K, Kronsbein HC, Weber MS. Targeting B Cells in the Treatment of Multiple Sclerosis: Recent Advances and Remaining Challenges. Ther Adv Neurol Disord (2013) 6:161–73. doi: 10.1177/1756285612474333
79. Harp CT, Ireland S, Davis LS, Remington G, Cassidy B, Cravens PD, et al. Memory B Cells From a Subset of Treatment-Naïve Relapsing-Remitting Multiple Sclerosis Patients Elicit CD4+ T-Cell Proliferation and IFN-γ Production in Response to Myelin Basic Protein and Myelin Oligodendrocyte Glycoprotein. Eur J Immunol (2010) 40(10):2942–56. doi: 10.1002/eji.201040516
80. Lauw FN, Pajkrt D, Hack CE, Kurimoto M, van Deventer SJH, van der Poll T. Proinflammatory Effects of IL-10 During Human Endotoxemia. J Immunol (2000) 165(5):2783–9. doi: 10.4049/jimmunol.165.5.2783
81. Van Kampen C, Gauldie J, Collins SM. Proinflammatory Properties of IL-4 in the Intestinal Microenvironment. Am J Physiol - Gastrointest Liver Physiol (2005) 288(1 51-1):111–7. doi: 10.1152/ajpgi.00014.2004
82. Kaskow BJ, Baecher-Allan C. Effector T Cells in Multiple Sclerosis. Cold Spring Harb Perspect Med (2018) 8(4):a029025. doi: 10.1101/cshperspect.a029025
83. Morris G, Berk M, Puri BK. A Comparison of Neuroimaging Abnormalities in Multiple Sclerosis, Major Depression and Chronic Fatigue Syndrome (Myalgic Encephalomyelitis): Is There a Common Cause? Mol Neurobiol (2018) 55:3592–609. Humana Press Inc. doi: 10.1007/s12035-017-0598-z
84. Patten SB. Antidepressant Treatment for Major Depression in Multiple Sclerosis. Int J MS Care (2009) 11(4):174–9. doi: 10.7224/1537-2073-11.4.174
85. Fiest KM, Walker JR, Bernstein CN, Graff LA, Zarychanski R, Abou-Setta AM, et al. Systematic Review and Meta-Analysis of Interventions for Depression and Anxiety in Persons With Multiple Sclerosis. Multiple Sclerosis Related Disord (2016) 5:12–26. Elsevier B.V. doi: 10.1016/j.msard.2015.10.004
86. Frampton JE. Vortioxetine: A Review in Cognitive Dysfunction in Depression. Drugs (2016) 76(17):1675–82. doi: 10.1007/s40265-016-0655-3
87. McIntyre RS, Lophaven S, Olsen CK. A Randomized, Double-Blind, Placebo-Controlled Study of Vortioxetine on Cognitive Function in Depressed Adults. Int J Neuropsychopharmacol (2014) 17(10):1557–67. doi: 10.1017/S1461145714000546
88. Mahableshwarkar AR, Zajecka J, Jacobson W, Chen Y, Keefe RSE. A Randomized, Placebo-Controlled, Active-Reference, Double-Blind, Flexible-Dose Study of the Efficacy of Vortioxetine on Cognitive Function in Major Depressive Disorder. Neuropsychopharmacology (2015) 40(8):2025–37. doi: 10.1038/npp.2015.52
89. DeSousa EA, Albert RH, Kalman B. Cognitive Impairments in Multiple Sclerosis: A Review. Am J Alzheimer’s Dis other Dementias (2002) 17:23–9. Sage PublicationsSage CA: Thousand Oaks, CA. doi: 10.1177/153331750201700104
90. Zuckerman H, Pan Z, Park C, Brietzke E, Musial N, Shariq AS, et al. Recognition and Treatment of Cognitive Dysfunction in Major Depressive Disorder. Front Psychiatry (2018) 9:655. doi: 10.3389/fpsyt.2018.00655
91. Cordeau D, Courtois F. Sexual Disorders in Women With MS: Assessment and Management. Ann Phys Rehabil Med (2014) Jul 157(5):337–47. doi: 10.1016/j.rehab.2014.05.008
92. Jarvis SE, Mackie A, Metz LM. Symptomatic Therapy for Multiple Sclerosis. Future Med (2013) doi: 10.2217/ebo.12.461
93. Siniscalchi A, Gallelli L, Tolotta GA, Loiacono D, De Sarro G. Open, Uncontrolled, Nonrandomized, 9-Month, Off-Label Use of Bupropion to Treat Fatigue in a Single Patient With Multiple Sclerosis. Clin Ther (2010) 32(12):2030–4. doi: 10.1016/j.clinthera.2010.10.012
94. Solaro C, Gamberini G, Masuccio FG. Depression in Multiple Sclerosis: Epidemiology, Aetiology, Diagnosis and Treatment. CNS Drugs (2018) 32:117–33. Springer International Publishing. doi: 10.1007/s40263-018-0489-5
95. Manjaly ZM, Harrison NA, Critchley HD, Do CT, Stefanics G, Wenderoth N, et al. Pathophysiological and Cognitive Mechanisms of Fatigue in Multiple Sclerosis. J Neurol Neurosurg Psychiatry (2019) 90:642–51. BMJ Publishing Group. doi: 10.1136/jnnp-2018-320050
96. Foley PL, Vesterinen HM, Laird BJ, Sena ES, Colvin LA, Chandran S, et al. Prevalence and Natural History of Pain in Adults With Multiple Sclerosis: Systematic Review and Meta-Analysis. Pain (2013) 154:632–42. Elsevier B.V. doi: 10.1016/j.pain.2012.12.002
97. Vollmer TL, Robinson MJ, Risser RC, Malcolm SK. A Randomized, Double-Blind, Placebo-Controlled Trial of Duloxetine for the Treatment of Pain in Patients With Multiple Sclerosis. Pain Pract (2014) 14(8):732–44. doi: 10.1111/papr.12127
98. Aiyer R, Barkin R, Medicine AB-P. Treatment of Neuropathic Pain With Venlafaxine: A Systematic Review. Pain Med (2017) 1;18(10):1999–2012. doi: 10.1093/pm/pnw261
99. Kister I, Caminero AB, Monteith TS, Soliman A, Bacon TE, Bacon JH, et al. Migraine is Comorbid With Multiple Sclerosis and Associated With a More Symptomatic MS Course. J Headache Pain (2010) 11(5):417–25. doi: 10.1007/s10194-010-0237-9
100. Dmochowski RR, Miklos JR, Norton PA, Zinner NR, Yalcin I, Bump RC. Duloxetine Versus Placebo for the Treatment of North American Women With Stress Urinary Incontinence. J Urol (2003) 170(4 I):1259–63. doi: 10.1097/01.ju.0000080708.87092.cc
101. Millard RJ, Moore K, Rencken R, Yalcin I, Bump RC. Duloxetine vs Placebo in the Treatment of Stress Urinary Incontinence: A Four-Continent Randomized Clinical Trial. BJU Int (2004) 93(3):311–8. doi: 10.1111/j.1464-410X.2004.04607.x
102. Van Kerrebroeck P, Abrams P, Lange R, Slack M, Wyndaele JJ, Yalcin I, et al. Duloxetine Versus Placebo in the Treatment of European and Canadian Women With Stress Urinary Incontinence. BJOG Int J Obstet Gynaecol (2004) 111(3):249–57. doi: 10.1111/j.1471-0528.2004.00067.x
Keywords: MS, antidepressants, EAE, neurotransmitters, in vivo, in vitro, immunomodulation
Citation: Stamoula E, Siafis S, Dardalas I, Ainatzoglou A, Matsas A, Athanasiadis T, Sardeli C, Stamoulas K and Papazisis G (2021) Antidepressants on Multiple Sclerosis: A Review of In Vitro and In Vivo Models. Front. Immunol. 12:677879. doi: 10.3389/fimmu.2021.677879
Received: 08 March 2021; Accepted: 06 May 2021;
Published: 20 May 2021.
Edited by:
Sharon Glynn Lynch, University of Kansas Medical Center, United StatesReviewed by:
Andreia Barroso, Brigham and Women’s Hospital and Harvard Medical School, United StatesUlises Gomez-Pinedo, Instituto de Investigación Sanitaria del Hospital Clínico San Carlos, Spain
Copyright © 2021 Stamoula, Siafis, Dardalas, Ainatzoglou, Matsas, Athanasiadis, Sardeli, Stamoulas and Papazisis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Georgios Papazisis, cGFwYXppc2dAYXV0aC5ncg==; orcid.org/0000-0003-1641-9095