 Noushin Mossadegh-Keller
Noushin Mossadegh-Keller Gabriel Brisou
Gabriel Brisou Alicia Beyou1
Alicia Beyou1 Bertrand Nadel
Bertrand Nadel Sandrine Roulland
Sandrine Roulland- 1Aix Marseille Univ, CNRS, INSERM, CIML, Marseille, France
- 2Department of Hematology, Institut Paoli-Calmettes, Marseille, France
Lymphomas are cancers deriving from lymphocytes, arising preferentially in secondary lymphoid organs, and represent the 6th cancer worldwide and the most frequent blood cancer. The majority of B cell Non-Hodgkin lymphomas (B-NHL) develop from germinal center (GC) experienced mature B cells. GCs are transient structures that form in lymphoid organs in response to antigen exposure of naive B cells, and where B cell receptor (BCR) affinity maturation occurs to promote B cell differentiation into memory B and plasma cells producing high-affinity antibodies. Genomic instability associated with the somatic hypermutation (SHM) and class-switch recombination (CSR) processes during GC transit enhance susceptibility to malignant transformation. Most B cell differentiation steps in the GC are at the origin of frequent B cell malignant entities, namely Follicular Lymphoma (FL) and GCB diffuse large B cell lymphomas (GCB-DLBCL). Over the past decade, large sequencing efforts have provided a great boost in the identification of candidate oncogenes and tumor suppressors involved in FL and DLBCL oncogenesis. Mouse models have been instrumental to accurately mimic in vivo lymphoma-specific mutations and interrogate their normal function in the GC context and their oncogenic function leading to lymphoma onset. The limited access of biopsies during the initiating steps of the disease, the cellular and (epi)genetic heterogeneity of individual tumors across and within patients linked to perturbed dynamics of GC ecosystems make the development of genetically engineered mouse models crucial to decipher lymphomagenesis and disease progression and eventually to test the effects of novel targeted therapies. In this review, we provide an overview of some of the important genetically engineered mouse models that have been developed to recapitulate lymphoma-associated (epi)genetic alterations of two frequent GC-derived lymphoma entities: FL and GCB-DLCBL and describe how those mouse models have improved our knowledge of the molecular processes supporting GC B cell transformation.
Introduction
The germinal center (GC) is a specialized immune structure localized in secondary lymphoid organs—including lymph nodes, tonsils, and spleen—that forms upon antigenic challenge to support the B cell receptor (BCR) affinity maturation process. In this transient, highly dynamic structures, activated B cells undergo clonal expansion, somatic hypermutation (SHM) of immunoglobulin (Ig) variable genes, selection and eventual differentiation into memory B cells or long-lived plasma cells (PC) (1). The GC is canonically divided into two principal zones: the dark zone (DZ), where B cells undergo clonal expansion and accumulate SHM upon activation-induced-cytidine deaminase (AID) responsible of BCR diversification, and the light zone (LZ), where GC B cell will test their newly acquired mutated Ig for improved affinity to antigen through interaction with immune complex-coated follicular dendritic cells (FDCs) and selection by a limited number of CD4+ T follicular helper cells (TFH) residing in the LZ (2). Within the LZ, B cells can have several fates: (i) a small subset of high-affinity GC B cells, selected in the LZ, will recycle back in the DZ to undergo further cycles of expansion/mutation/selection (3, 4), (ii) some selected LZ B cells can directly exit the GC differentiating into effectors such a memory B cells or plasma cells and (iii) LZ cells with low/no affinity BCRs following SHM due to a lack of antigen engagement and subsequent T cell help die by apoptosis (Figure 1). In the GC LZ, the strength and intensity of the signal received by B cells from TFH cells, which is largely influenced by BCR affinity, mainly determines B cell fate. Recently, the dynamic transcriptional changes characterizing the GC cycle between LZ and DZ have been further refined through single cell gene expression approaches revealing a continuum of cell states between LZ and DZ and highly orchestrated group of molecular programs that co-evolve during the GC response (30–34).
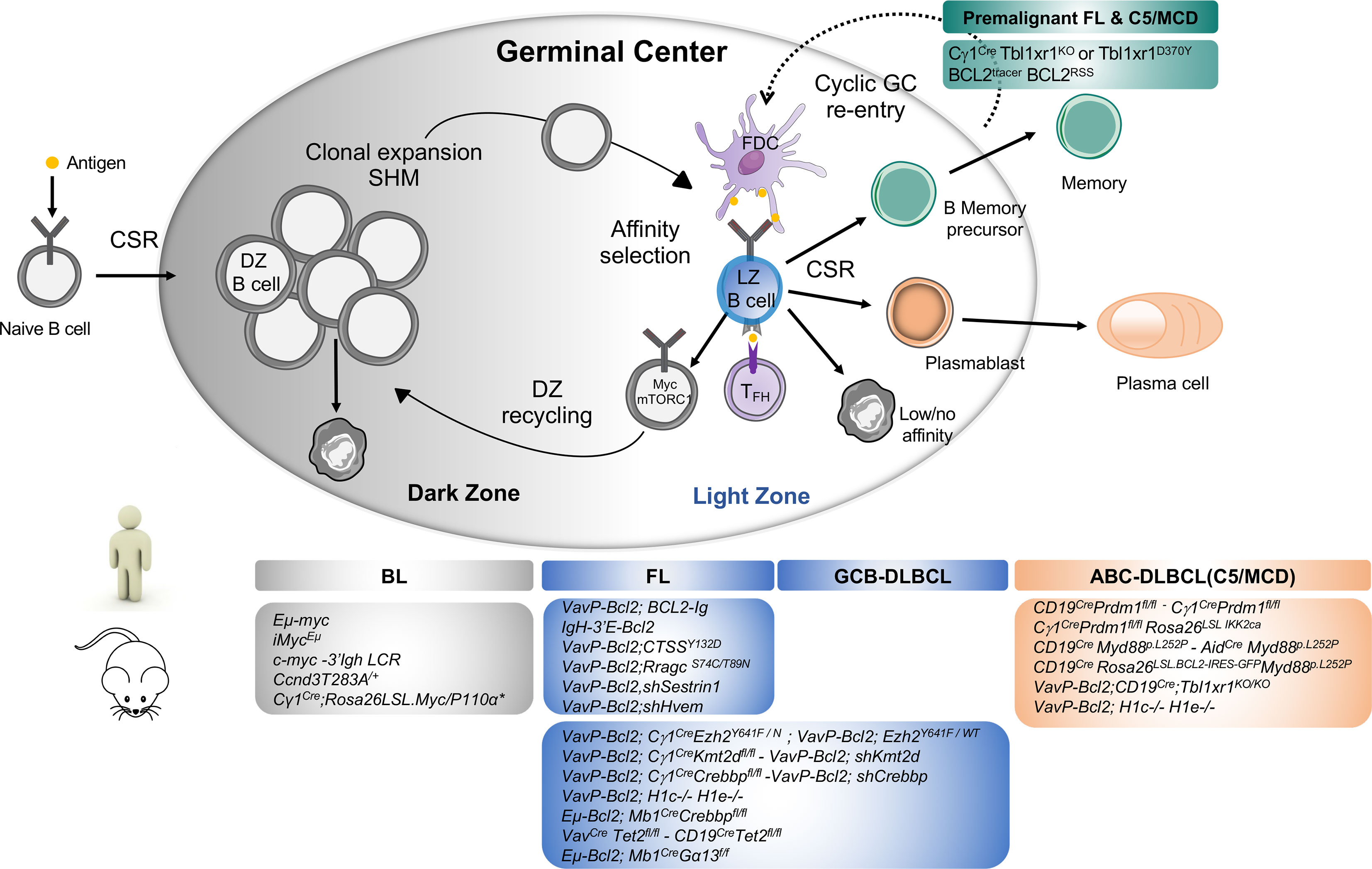
Figure 1 Major mouse models of human B cell lymphomas linked to their putative normal B cell counterpart. Top Panel: Schematic representation of Germinal Center B cells. Activated B cells enter in the GC dark zone (DZ), the site of clonal expansion and somatic hypermutation. Only a subset of DZ B cells will be selected to pass in the GC light zone (LZ) while most DZ B cells undergo apoptosis. GC LZ is the site of affinity-based selection where high-affinity B cells tend to capture more antigen from FDC and receive more T cell help through CD40/CD40L interaction driving their re-entry into the DZ for defined rounds of proliferation and SHM. In the LZ, owing to the failure to receive T cell help and acquisition of damaging BCR mutations, most GC B cells will undergo apoptosis while some LZ B cells that gained productive BCR mutations and enhanced affinity will be selected and terminally differentiate into memory B or plasma cells depending on the strength of T cell help they received [recently reviewed in (5)]. In normal immune response, current models suggest that memory B cells tend to exit early the GC response following low level of T cell help and typically display less SHM and reduced levels of affinity maturation than plasma cells. A limited number of memory B cells can re-enter into secondary GC upon antigenic recall for additional rounds of mutations (6, 7). Cyclic memory cell reactivation of mutated B cells into the GC is however a driving mechanism leading to B cell transformation (8, 9). Bottom panel: The most important genetically-engineered mouse models are linked to the human lymphoma they intend to mimic distinguishing Burkitt Lymphoma (BL) arising from transformation of DZ cells, Follicular Lymphoma and GCB-DLBCL from LZ cells and ABC-DLBCL arising from Activated B/plasmablastic or memory B cells. BL is characterized by Myc translocations between the immunoglobulin heavy or light chain locus. Transgenic mice engineered to dysregulate MYC expression under proximal or distal enhancers Eµ-Myc (10), iMycEµ (11) and c-myc3’LCR (12) led to the development of aggressive lymphomas with Burkitt-like phenotype with high penetrance and short latency in vivo. Conditional overexpression of Myc and PI3K signaling (Cγ1Cre/wt;Rosa26LSL.Myc/P110α*) in the GC cooperate to drive BL-like tumors identifying PI3K pathway activation as a key survival element in Myc-driven lymphomas (13). Modeling Cyclin D3 T283A gain-of-function mutations in B cells—which are recurrent event in DLBCL and sporadic BL (14, 15)—leads to increased DZ proliferation and occasional lymphoproliferative disease in older mice highlighting the need for additional events to exert its oncogenic function (16, 17). ABC-DLBCL are typically characterized by an enhanced activity of the NF-κB survival pathway and the co-occurrence of genomic aberrations in BCR (CD79B), MYD88, TBL1XR1, 18q gains affecting BCL2 and PRDM1 inactivation interfering with normal plasma cell differentiation, all defining features of the C5/MCD genetic subtype (18–20). Single conditional knockout of Prdm1 in B cells or specifically in the GC reveal lymphoma lesions of post-GC origin (Bcl6−, Irf4+) indicative of a preplasmablastic stage in only 20% of animals and with a long latency (18, 21). Constitutive activation of NF-κB pathway with Prdm1 disruption in the GC cooperate to drive DLBCL-like tumor development resembling human ABC-DLBCL (22, 23). Conditional expression in B cells of an oncogenic Myd88L252P allele plus BCL2 overexpression (mimicking BCL2 copy number gains) result in the development of aggressive post-GC lymphomas recapitulating many genotypic, transcriptomic and signaling features of ABC-DLBCL pathogenesis (24, 25) notably the formation of the My-T-BCR (Myd88/TLR9/BCR) supercomplex driving NF-κB mediating survival signals (26) and detection of autoreactive antibodies suggesting a role for self-antigens in driving BCR stimulation as previously proposed in human and mouse models (27, 28). Somatic mutations in TBL1XR1 are enriched in the MCD/C5 genetic subtype (18). Conditional deletion of Tbl1xr1 or expression of TBL1XR1D370Y mutant allele in B cells generates aberrant memory B cells which are more prone to cyclic re-entry into GC reaction thereby providing additional evidence on how skewed GC/Memory B cell dynamics act as a major pathogenic mechanism in lymphoma development (8, 9, 29). Combined with Bcl2 overexpression, Tbl1xr1 mutant mice ultimately give rise to canonical post-GC extranodal ABC-like lymphomas with a proportion of B cells manifesting with a memory B cell phenotype consistent with a putative memory B cell origin of ABC-DLBCL tumors (21).
T dependent humoral response proceeds in several steps triggered by multiple finely orchestrated cellular interactions that affect B cell response through the activation and repression of specific transcriptional programs. Molecular control of this highly dynamic process is complex and involves several transcriptional regulators such as transcription factors and epigenetic regulators that are frequently targeted by somatic mutations driving lymphomagenesis. After antigen encounter and T cell co-stimulation, B cells get activated through BCR, CD40 and toll like receptor (TLR) signalling, inducing NF-κB activation and the expression of genes involved in B cell activation and proliferation driving GC initiation (35, 36). BCL6 is a transcriptional repressor which play a central role in GC initiation and maintenance (37). Its expression is triggered by B–T interaction during the early initiation of the GC response, where it allows B cells to migrate into the center of B cell follicles through the downregulation of EBI2 and S1PR1 and induction of CXCR4. Once GCs are established, BCL6 coordinates the GC response by repressing thousands of genes involved in different cellular processes (T cell mediated B cell activation, BCR/CD40 signaling, apoptosis, DNA damage response, cellular cycle checkpoints,…) (38, 39). In this way, BCL6 allows DZ cells to establish a hyper-proliferative program while tolerating DNA damage caused by SHM without triggering proliferation arrest or apoptosis. In addition, BCL6 prevents signal transduction from several membrane receptors, thus preventing B cells from premature differentiation. BCL6 expression must be repressed to allow B cells to exit the GC. Two signals cooperate to repress BCL6: BCR activation via Ag presented by FDCs and CD40 activation via CD40L expressed by TFH cells (38, 39). The transcription factor cMYC is also essential for GC initiation. Indeed, about 2 h after B cell activation, GC precursors transiently express c-MYC before expressing BCL6 and co-express c-MYC and BCL6 for a short period of time, allowing the initial proliferation phase leading to GC formation. Once the GC is established, c-MYC is then partially repressed by BCL6 (3, 4). Back in the LZ, high-affinity B cells that take up the Ag integrate signals from the BCR and additional signals through several receptors, including CD40, BAFF and TLRs which ultimately activate NF-κB. NF-κB signaling and CD40 costimulation result in cMYC re-expression in selected B cells that return to the DZ for further rounds of cell division (40). On the other hand, this activates IRF4 which, when highly expressed, represses BCL6 expression, thus promoting GC programme silencing and post-GC differentiation (35, 41). Indeed, at high concentration, IRF4 induces BLIMP1 expression which allows plasma cell differentiation (42). Memory B cell differentiation process is less understood but is thought to derive from B cells with low affinity BCR receiving a weak signal from TFH cells, two transcription factors have been involved in this process: BACH2 and more recently HHEX (43, 44).
B cell lymphomas are cancers that develop from the malignant transformation of B lymphocytes at different stages of ontogeny (45). From naive to memory and plasma cells, most differentiation steps are associated with a malignant B cell counterpart defined historically as the cell-of-origin on the basis of histological definitions, phenotype and resemblance of transcriptomic profiles (46). Rapid clonal expansion, genomic instability, tolerance to DNA damage and metabolic reprogramming are physiological GC B cell-specific features that makes them permeable to lymphomagenesis (47, 48). Accordingly, GC B cells are considered at the origin of frequent B cell lymphomas—namely follicular lymphoma (FL), GC B cell-diffuse large B cell lymphoma (GCB-DLBCL), and Burkitt lymphoma (BL) (Figure 1). FL have a follicular growth pattern reminding normal GC architecture, with the presence of TFH cells and FDC stromal cells, carries heavily mutated Ig genes known to occur in the GC primarily and retain a closely related signature to LZ B cells (49, 50). GCB-DLBCL is more transcriptionally reminiscent of LZ B cells while BL is more similar to DZ B cells (46, 51). Of note, single-cell gene expression analyses of mouse (16, 30, 44, 52) and human GC B cells (31–33) have revealed that GC B cell transcriptional states span a continuum from LZ to DZ, and that a large proportion (between 30 and 50%) of GC B cells are in an intermediate state between the two zones. Besides providing important datasets to understand GC transcriptional programs during the normal immune response, these studies offered a more granular survey of human GC B cells states that can serve as references for re-assessing and revisiting the concept of GC B cell lymphoma cell-of-origin (31, 32).
Yet, gene expression profiling only accounts for a portion of GC B cell lymphoma heterogeneity and the advent of next generation sequencing (NGS) has provided a great boost in the identification of genetic alterations (mutations, translocations, copy number alterations…) involved in FL and DLBCL oncogenesis, and has revealed an increased complexity of the lymphoma genetic landscape (14, 18, 19, 53–55). Although GCB-DLBCL and FL represent two clinically and histopathological distinct lymphoma entities, it has become apparent that these two subtypes are far more intricated from a genetic point of view as illustrated by shared multiple recurrent genetic lesions such as BCL2 alterations, mutations in epigenetic regulators (KMT2D, CREBBP, EZH2, EP300…) and immune receptor signaling genes suggesting at least clonal evolution from a similar precursor cell and shared oncogenic pathways (53, 56). Accordingly, the recently defined EZB/C3 genetic subtype is composed by a majority of tumors with a GC B cell gene expression profile and is enriched for the most common genetic abnormalities such as BCL2 translocations and EZH2 mutations.
Among others, mouse models of lymphoid malignancies have advanced our understanding of lymphomagenesis [reviewed in (57–60)] and currently support the biological investigations on the most common putative driver mutations alone or in combination. The limited access to (pre)malignant biopsies during the initiating stage of lymphoma development, the difficulties to recapitulate in in vitro experimental systems the complexity of the GC reaction during an immune response (61), the spatial and (epi)genetic heterogeneity across and within human lymphomas make the development of genetically engineered mice models the most suitable tool i) to characterize the molecular mechanisms by which candidate lymphoma mutations contribute in vivo to lymphomagenesis either alone or in combination and ii) to trace how tumours grow and evolve over time by recapitulating the precise timing at which the genetic lesions happens in human settings iii) to test the effects of targeted pharmacological agents and iv) the synergy between co-occurring genetic alterations. In this review, we will present recent insights on FL and GCB-DLBCL lymphoma mouse models in which genetic alterations targeting the epigenome, immune signaling or metabolic pathways have been accurately recapitulated and for which mechanistic studies yielded new insights on how GC regulatory programs are hijacked by somatic mutations to prevent the resolution of ‘pseudo-tumoral’ GC B cell features and facilitate lymphomagenesis (Table 1). Starting with the founder BCL2 translocation common to the pathogenesis of most FL and GCB-DLBCL, we will focus on how genetically engineered mice (GEM) of (epi)genetic alterations shed new lights on the link between B ‘cell-intrinsic’ lesions and their cell-extrinsic functions to drive lymphoma development by promoting the remodeling of an aberrant immune niche and contributing to immune surveillance mechanisms.
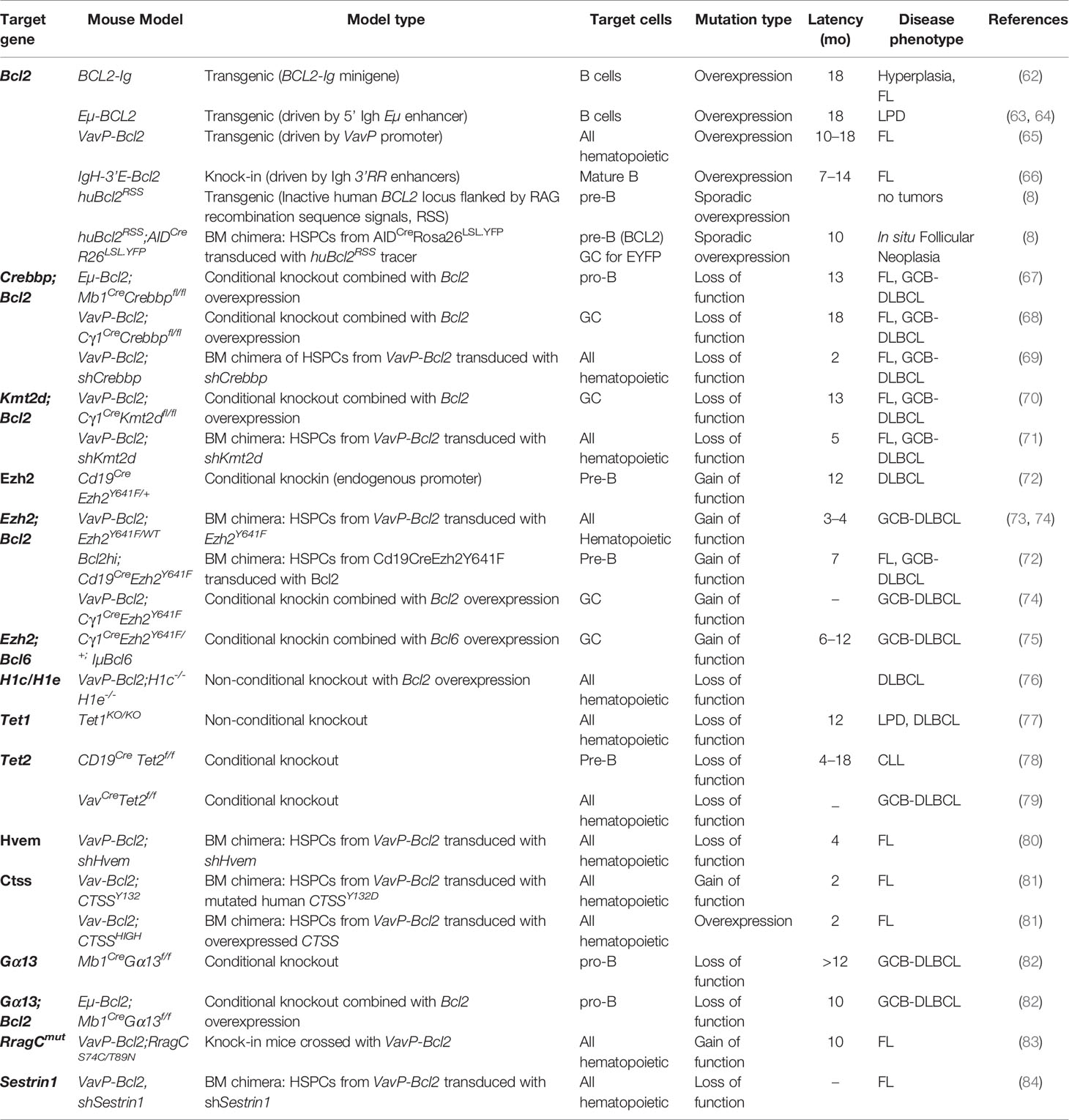
Table 1 Relevant mouse models of follicular lymphoma and germinal center B cell diffuse large B cell lymphomas.
Modeling t(14;18) Translocation and Bcl2 Overexpression
FL represents an attractive model to study the mechanisms by which lymphoid B cells undergo neoplastic initiation and progression in mouse models (50). While FL is considered as a disseminated GC-derived B cell neoplasia, acquisition of the t(14;18) translocation—that lays the BCL2 gene under the transcriptional control of Ig heavy chain (IGH) regulatory regions—constitutes a critical early, likely primary, event in the natural history of the disease occurring in bone marrow (BM) pre B cells during illegitimate V(D)J recombination. It is now well established that t(14;18), although present in 85% of FL and about 30% of GCB-DLBCL patients, is not enough to transform B lymphocytes as t(14;18)-positive circulating B cells are detectable at low frequency (one in a million lymphocytes) in up to 70% of healthy adults who never develop the disease (85), indicating that complementary “hits” must further accumulate, presumably during the later phases of GC B cell maturation (86). Attempts to model the t(14;18) translocation and Bcl2 overexpression have started in the late 80’s with first generation of Bcl2 transgenic mouse models (62).
Eµ-BCL2 and BCL2-Ig
In 1989, to mimic the human BCL2-IGH translocated allele and assess the tumorigenic potential of BCL2 in vivo, Mc Donnell and colleagues developed the first BCL2 transgenic mice bearing a human BCL2-Ig minigene where expression of BCL2 is restricted to B cells (62). The mice developed follicular hyperplasia made of small naive B cells—expressing markers such as B220, IgD, IgM and Igκ—with prolonged survival in vitro. In original studies, after 18 months of age these mice showed high-grade lymphomas although at a low penetrance and interestingly half of those mice harbored c-myc rearrangement. More recent follow-up studies showed that 40% of BCL2-Ig mice develop FL-like tumors expressing GC markers (PNA+BCL6+) by 17–18 months when chronically immunized with sheep red blood cells (SRBC) over 6 months (87). Strasser and colleagues similarly engineered a transgenic strain Eµ-BCL2, where BCL2 is placed under the control of the 5’ IGH enhancer Eµ (63). This model also showed an expansion of small B cells and plasma cells but did not yield tumor development, however an increased incidence of other B-lymphoid neoplasms was observed (64).
VavP-BCL2 and IgH-3’E-Bcl2
The first experimental model that faithfully reproduced the human disease in term of localization, histology, phenotypic and genotypic features involved expression of Bcl2 in all hematopoietic cells. Egle and colleagues generated the VavP-Bcl2 model where the Bcl2 transgene is controlled by Vav gene regulatory sequences, which confer Bcl2 expression in multiple hematopoietic lineages (65). The mice developed, in 15–25% of cases, isotype-switched, somatically mutated Ig and disseminated lymphomas at 10 months of age. In contrast to Eµ-BCL2 transgenic models of the same age, VavP-Bcl2 mice develop spontaneous expansion of PNA+ GC lesions in otherwise ‘healthy’ mice, a premalignant condition which is strongly dependent on CD4 T cell help as in vivo removal of CD4 cells almost abolished GC hyperplasia. In this model, CD4 T cells make a critical input into the exaggerated GC reactions and eventually the onset of FL. A second model developed by the group of Boxer (66) consisted of a IgH-3’E-Bcl2 knock-in mice, where the IgH 3’ enhancer regions was integrated 3’ of Bcl2 locus, thereby mimicking the effects of the long distance IgH 3’ enhancer on Bcl2 expression and limiting Bcl2 expression to mature B cells. In addition to an altered B-cell differentiation and increased B cell numbers in the spleen, lymph node (LN) and BM, these mice recapitulated, between 7- and 14-months, typical histopathological features of GC-experienced FL-like tumors expressing the GC markers PNA and Bcl6 surrounded by FDC networks and CD4+ T cells. Despite its main limitations due to the unspecific Bcl2 deregulated expression in all hematopoietic cells, VavP-Bcl2 is currently one of the most popular models to study in vivo the role of secondary genetic alterations that are frequently found in combination with BCL2 in FL and GCB-DLBCL genetically engineered mouse models.
These studies support the notion that despite a survival advantage conferred to B cells by BCL2, the lymphomagenic process requires additional hits (genetic and/or immunological) for transformation.
Modeling the Early Steps of FL Development: BCL2tracer Model
Although several BCL2-engineered models have provided the initial proof-of-principle that BCL2 ectopic expression leads to FL and high-grade lymphomas, the expression of BCL2 in all B cells (and all T cells for some models) do not represent a true premalignant intermediate stage seen during human lymphomagenesis, where the first t(14;18) event occur in a single B cell in the BM, and is carried on until its ectopic expression in the GC. Instead, pan-B cells BCL2 mouse models generate a polyclonal hyperplasia of naive B cells which is not a known progression step in human FL pathogenesis and as a consequence does not allow studying the early steps of clonal emergence and disease progression. The BCL2tracer mouse model has been engineered to mimic “sporadic” t(14;18) translocation (8). This transgenic model relies on the introduction of potent RAG recombination sites at the vicinity of an inactivated human BCL2 transgenic minilocus. In this construct, recombination allows to turn on ectopic BCL2 expression in only few B cells, and at the appropriate window of B cell development in the BM pre-B cells. The recombination breakpoints provide unique PCR-based clonotypic markers to study early steps of clonal emergence and expansion in mouse blood/tissues at different time points, allowing a precise analysis of clonal progression kinetics. As expected, the emergence of BCL2+ B cells was traced in various tissues at low frequency (1 in 105 to 106) recapitulating the “healthy human t(14;18) carrier” situation. At steady state or after acute immunization with a T-cell dependent antigen, BCL2 alone was not able to drive progression of BCL2-expressing cells into a tumor after 18 months of follow-up, confirming that lymphomagenesis is a stepwise process where premalignant B cells require the accumulation of secondary (epi)genetic alterations to progress into a tumor. However, a chronic immunization protocol with SRBC accelerated genomic instability by allowing BCL2-overexpressing B cells to give rise to memory cells that preferentially underwent iterative rounds of GC entry, allowing multiple rounds of AID-mediated mutagenesis over time to ultimately form premalignant in situ FL structures, the earliest known intermediate preceding human FL. Although this model alone does not form FL lesions, it has been instrumental to propose a revised model of early lymphomagenesis whereby cyclic reactivation of BCL2+ memory B cells within new GC reactions would constitute a major pathogenic mechanism facilitating clonal expansion and accumulation of secondary mutations in FL precursors. It is likely that this reactivation in humans operates over decades before clinical manifestation (that we will never reach during the mouse lifespan) and ultimately contribute to the generation of a heterogeneous population of aberrant memory B cell intermediates resembling clonal FL evolving in asymptomatic patients years before diagnosis. Interestingly, this model also led to identify an “immunological 2nd hit” in FL, departing from the all-genetic, cell-intrinsic concept of lymphomagenesis.
Modeling Epigenetic Alterations
Recurrent mutations affecting histone modifying enzymes are a hallmark of GC-derived lymphomas, more particularly in FL. Within DLBCLs, these mutations are enriched within the GCB-DLBCL subtype, with further enrichment within the newly described EZB/C3 genetic subtype. Inactivating mutations of the H3K4 lysine methyltransferase KMT2D are found in 70 to 90% of FL cases and up to 30% of DLBCL cases representing the most frequently FL mutated genes in these lymphomas after t(14;18) translocations. Approximately 50 to 70% of FLs and ∼25% of DLBCLs carry acquire inactivating mutations in CREBBP, whereas its paralog EP300 is mutated in ∼5% of cases. Activating mutations of the H3K27 histone methyltransferase EZH2, a component of the polycomb repressive complex 2, are found in 10 to 25% of FL cases and 20% of GCB-DLBCL cases (14, 18, 19, 53–55, 88–90). Overall, 95% of FL patients manifest with at least one chromatin modifier gene mutation. Thorough genomic inference analyses of the clonal evolution patterns in sequential pairs of FL at diagnosis vs. relapse/transformation showed that recurrent inactivating mutations in CREBBP and KMT2D represent early events in FL evolution and are likely to be present in the CPC pool supporting a founder role for these events.
CREBBP and EP300 Inactivation
CREBBP and EP300 are highly homologous histone acetyltransferases (HAT) that modify gene expression through H3K27 acetylation at enhancer domains of both histone and non-histone substrates. In 50 to 75% of cases, mutations of CREBBP are missense inactivation of the acetyltransferase catalytic domain, the remaining mutant alleles causing truncation or loss of expression (54, 55, 69, 90). CREBBP and EP300 mutations are usually detected only at one allele and exclusive fashion, which is thought to be explained by the compensatory and redundant role of these enzymes. The precise timing and location where CREBBP or EP300 mutations happens during B cell transformation is still unclear in humans, but there is large agreement from clonal evolution studies and in premalignant FL conditions that CREBBP is an early event (91). Several groups have therefore attempted to investigate the role of hemizygous vs. homozygous Crebbp inactivation on mature B cell differentiation and lymphoma development in vivo using different strategies from early inactivation in all hematopoietic cells, early B-cell specific deletion or GC-specific deletion [Cγ1-Cre (92)], in combination or not with BCL2 transgenic models.
Horton and colleagues studied the consequences of Crebbp deletion in the hematopoietic stem cell compartment thanks to a pIpC-mediated Mx1-Cre recombinase system (93). Those animals developed B cell lymphoproliferative disorders accounting for 29% of all deaths. The B cell lesions localized mostly in the spleen and blood and stained with B220, CD19 and surface IgM indicating their mature B cell origin, however these tumors did not express GC markers. The occurrence of these lymphoproliferative disorders was preceded by the accumulation of lymphoid progenitors characterized by a hyperproliferative state and an altered DNA damage response linked to the loss of p53 activation in the absence of Crebbp. Interestingly, the authors found no enrichment for Bcl6 targets among Crebbp binding sites in their system suggesting different epigenetic changes when Crebbp is deleted in hematopoietic stem cells (HSC). This data suggests that CREBBP mutations acquired early during hematopoiesis may contribute to the emergence of B cell lymphomas although this model might not be relevant for FL or DLBCL progression as the tumors do not exhibit GC features. It also indicates that the timing of mutation acquisition has an important impact on B cell development. In humans, detection of CREBBP mutations in HSCs remains a very rare event (93).
Three others groups investigated the B-cell deletion of Crebbp in combination with BCL2 overexpression to fit with the frequent co-occurrence of the two alterations in human FL and DLBCL (67–69). Jiang et al. recapitulated Crebbp downregulation and Bcl2 overexpression in vivo using a shRNA retroviral infection system of VavP-Bcl2 hematopoietic stem progenitor cells (HSPCs) transplanted into lethally irradiated wild-type recipient mice. They observed an acceleration of lymphoma onset in double-mutant vs. VavP-Bcl2 mice. Lymphoma cells expressed B220, CD19 and IgM and were characterized by somatically mutated Ig locus confirming their GC B cell origin. Importantly, a similar phenotype was observed with a shRNA targeting Ep300. The second model developed by Zhang and colleagues consisted in cohorts of Cγ1-Cre (92) and Cd19-Cre Crebbpflox/flox and Crebbpflox/+ animals where inactivation of Crebbp was induced in GC or developing B cells respectively. After a follow-up of 18 months they did not observe any significant difference between Crebbp mutant mice and their littermate controls concluding that loss of Crebbp alone at early or late stages of B cell differentiation was not sufficient to induce lymphomagenesis. The generation of mice crossing Cγ1-Cre Crebbpflox/flox mice with VavP-Bcl2 transgenic mice led to a significant increase in the incidence of B cell malignancies resembling human FL (92% in double mutant versus 61.5% in VavP-Bcl2 controls). Furthermore, the tumors were characterized by a follicular architecture, were largely of GC origin with Bcl6 expression and presence of mutated Ig genes. In an extension of this study and to investigate in parallel how CREBBP and EP300 contribute to normal GC B cell physiology, Meyer et al. established mouse lines carrying single “floxed” HAT genes that are excised only in activated B cells in the GC (Cγ1-Cre) (94). In accordance with previous studies (95), Meyer et al. confirmed that loss of Crebbp in GC B cells led to increased GC formation while Ep300 loss led to an opposite effect with decreased proportion of GC B cells in immunized mice (94). Transcriptomic analyses of purified GC B cells obtained from these two strains revealed that the set of genes modified by Ep300 loss or Crebbp loss was different with expression of DZ transcripts preferentially repressed in Ep300-deleted GC B cells while LZ genes were preferentially decreased in Crebbp-deleted GC B cells providing a mechanistic explanation for the reduced numbers of GC B cells observed in the GC-deficient Ep300 model. The most interesting phenotype comes from the drastic reduction of GC B cells observed when both HAT genes are deleted from the same GC cells. Indeed, the GC response was completely abrogated indicating that the ability of GC B cells to proliferate and differentiate relies on the combined activity of both acetyltransferases likely due to their overlapping and partially redundant functions. Interestingly, CREBBP-mutated lymphoma B cells maintained this dependency toward EP300 enzymatic activity which identify a unique vulnerability that provide exciting opportunities of targeting single mutant CREBBP or EP300 GC-derived lymphomas.
In a third model, Garcia-Ramirez et al. produced strains of Mb1-Cre Crebbpflox/+ or Crebbpflox/flox mice where Crebbp was deleted at early pro-B cell stage in the BM. Crebbp deletion at early stages of B cell development led to reduced frequencies of B-cell subsets with reduced numbers of total B220+ B cells in BM and spleen. When crossed with the Eµ-BCL2 mouse model, these animals showed higher frequencies of GC B cells in the spleens after SRBC immunization and occasionally develop clonal B cell lymphoma with low penetrance and long latency (13 months). Histology was similar to human FL grade 3 or DLBCL and tumoral cells expressed Pax5 and Bcl6 and displayed SHMs consistent with a GC B cell origin (67).
Overall, these three independent in vivo models confirmed an oncogenic cooperation between CREBBP loss of functions and BCL2 overexpression to promote lymphomagenesis in vivo. Mechanistically, these studies showed that CREBBP, and likely EP300, maintain H3K27 acetylation at certain enhancer that are poised during GC reaction and whose reactivation is required for GC exit and terminal differentiation. This includes genes involved in immune synapse, downstream effectors of BCR and NF-κB or terminal differentiation (Irf4, Nfkb2, Cd40) and antigen presentation, the most notable being MHC class II molecules which are critical for B cell terminal differentiation. In normal GC cells, CREBBP targeted enhancers are direct targets of BCL6 and transiently repressed by BCL6/SMRT/HDAC3 complexes that deacetylate H3K27. Upon selection signals received in the LZ allowing GC exit, these enhancers recover H3K27ac state as CREBBP can directly acetylate BCL6 to inactivate its function by preventing the interaction with co-repressor complexes. By impairing the reactivation of these enhancers leaving unopposed BCL6 oncogenic activity, CREBBP loss of function disrupt the expression of immune synapse genes and their downstream signaling pathways, resulting in accumulation of aberrant GC B cells that fail to properly respond to exit signals from the GC microenvironment thereby promoting lymphoma progression. In human FL, decreased MHC II expression and reduced CD4 and CD8 T cell infiltrations have been described in CREBBP-mutant FL. A similar association between CREBBP inactivation and reduced expression of MHC class II is observed in murine lymphoma models which alters mutant GC cells ability to present antigen to CD4+ cells. Interestingly, CREBBP-mutant lymphomas become dependent to HDAC3, the histone deacetylase opposing the effect of CREBBP, that has been identified as a relevant therapeutic target in these tumors (69). Mondello et al. recently showed that HDAC3-selective inhibitors have a dual effect by reversing CREBBP-mutant aberrant epigenetic programming limiting lymphoma growth inhibition while restoring antitumor immunity, notably antigen presenting-genes (96).
KMT2D Loss of Function
KMT2D is a component of the COMPASS complex involved in transcriptional activation through H3K4 monomethylation of gene enhancers in B cells. The majority of KMT2D mutations are nonsense events leading to a truncated protein lacking the enzymatic SET domain involved in H3K4 methylation resulting in a loss of function (90). To study the consequences of Kmt2d inactivation during B cell differentiation, Zhang and colleagues generated a conditional Kmt2d knock-out model relying on a Cre-Lox system with conditional deletion of B cells early during B cell development (Cd19-Cre Kmt2dflox/flox) or after GC initiation (Cγ1-Cre Kmt2dflox/flox). Early deletion of Kmt2d lead to a higher number and enlarged GC formation while this effect was not seen with late GC deletion and in vitro, Kmt2d-deficient cells displayed a proliferative advantage compared to wild-type cells. However, Kmt2d deficiency in B cells alone upon early or late inactivation was not sufficient to induce FL or DLBCL in vivo. Kmt2d protein was mainly found on putative GC enhancers and global H3K4 methylation levels were diminished in Kmt2d mutant mice. Moreover, although GC-specific deletion was insufficient to initiate malignant transformation, Kmt2dko-VavP-Bcl2 double-mutant mice developed B-cell lymphoproliferative disorders with an incidence of 78% (44% for VavP-Bcl2 alone), with tumors expressing BCL6 and PAX5 consistent with a GC origin and recapitulating a spectrum of histopathological features ranging from early FL to DLBCL (40% early FL, 31% FL and 27% DLBCL) (70).
Using a different experimental system, Ortega-Molina and colleagues also explored cooperation between bcl2 and Kmt2d deletion in B cell lymphomagenesis. Using a retroviral infection system of shRNAs transduction to silence Kmt2d in HSPCs from VavP-Bcl2 donor mice following transplantation into irradiated wild-type mice, an acceleration of the lymphomagenesis process as well as an increase in the incidence of FL-like tumors (from 30 to 60%) was observed in double-mutant mice compared to shRNA control vectors, validating the tumor suppressive role of Kmt2d in B lymphocytes. At preclinical stages of the disease, Ortega Molina et al. showed that after immunization, the number of GC B cells was increased when Kmt2d was suppressed. Moreover, GCs persisted for a longer period than in control mice. Kmt2d loss was associated with a decrease in IgG1 production suggesting a dysfunction of the class switch recombination processes (71). Interestingly, the generation of Kmt2dflox/floxCd19-Cre crossed with a strain overexpressing AID led to the development of aggressive lymphomas resembling DLBCLs and confirms that, independently from BCL2 expression, genetic instability linked to AID overexpression cooperates with Kmt2d loss to promote lymphomagenesis.
Integrative genomic analyses from human samples carrying KMT2D mutations and Kmt2d-mouse FLs showed that genes differentially expressed in Kmt2d-mutated lymphomas were mostly repressed and affected a set of genes involved in terminal differentiation programs and GC exit, such as CD40 and BCR signaling, regulation of apoptosis, control of cell migration and proliferation. KMT2D mutations result in persistent demethylation of enhancers and failure of the respective genes to respond to signals, notably CD40 signaling from TFH. Moreover, absence of Kmt2d affects negatively the expression of major B-cell tumor suppressors such as Tnfaip3, Socs3, Tnfrsf14, Asxl1 or Arid1a. In conclusion, lack of Kmt2d leads to an aberrant repression of key genes normally required for GC exit, favoring an abnormal GC B cell outgrowth and failure to differentiate leading eventually to lymphoma development. Of note, these studies showed once more that the stage and/or the timing of a given (epi)genetic alteration has a strong influence on the transcriptional changes occurring in the GC. The developmental stage at which Kmt2d mutations are introduced in human precursor tumor cells is still unknown but it has been hypothesized that epigenetic reprogramming may require multiple rounds of cell divisions to allow the replacement of modified histones by non-modified histones explaining why Kmt2d inactivation after GC initiation may have a more modest phenotype than early inactivation.
EZH2 Gain of Function
EZH2 is a H3K27 methyltransferase part of the Polycomb Repressive complex-2 (PRC2). Heterozygous gain of function mutations, preferentially affecting the EZH2 SET domain at the Y641 residue and making EZH2 more efficient at H3K27 trimethylation (97), are found in up to 30% of GCB-DLBCL and FL and de facto enriched in EZB/C3 DLBCL subtype (18, 89, 98). Several groups have investigated the functional role of wild-type and mutant EZH2 during GC reaction and B cell lymphomagenesis (73, 75, 99, 100). Using a Cγ1-Cre Ezh2flox/flox strain, Béguelin et al. observed a marked reduction in GC B cells after immunization. They reproduced this phenotype in immunized wild-type mice treated with an EZH2 inhibitor targeting wild-type and mutant EZH2, establishing that EZH2 is required for GC formation (73). Similar observations were made by Caganova et al. underlining that under normal conditions, EZH2 enables GC formation at least in part by suppressing cell-cycle checkpoint genes like CDKN1A, impairing DNA damage responses to support centroblast proliferation and silencing essential plasma cell differentiation genes, particularly Blimp1 and Irf4 (99, 100).
To understand how EZH2Y641 hotspot mutation perturbs GC development and drives lymphomagenesis, Béguelin and colleagues developed two mouse strains conditionally expressing the mutant Ezh2Y641N or Ezh2Y641F in GC B cells upon Cγ1-Cre recombinase (73, 75). Upon immunization, both models caused oversized GCs and displayed increased abundance of H3K27me3 mark at critical GC B cell bivalent promoter leading to permanent silencing of EZH2 target genes. The GC phenotype appears to be mediated through cooperative and mutually interdependent actions of EZH2 together with the transcriptional repressor BCL6 and BCOR repressive complex. These Ezh2 mutant mice did not develop lymphomas. However, early activation (CD19-Cre) of mutant Ezh2 in an independent study led to aggressive DLBCL in about 12 months (72). VavP-Bcl2 HSPCs were transduced with retroviruses expressing Ezh2Y641F, Ezh2WT or an empty vector and transplanted into lethally irradiated recipient mice subjected to SRBC immunization every 4 weeks to induce GC formation. Ezh2Y641F Bcl2+ chimeric mice led to early lymphoma development in 70% of mice at 111 days (vs 20% in mice overexpressing Ezh2WTBcl2+ and none in the Bcl2 control at that stage), characterized by enlarged spleen and liver and resembling morphologically to DLBCL with centroblastic morphology (73, 74). These data demonstrate that EZH2 gain of function mutations accelerate GC B cell lymphomagenesis in cooperation with Bcl2 overexpression, recapitulating features of GCB-DLBCL. Similar results were obtained when transgenic and knock-in Ezh2 strains engineered to express heterozygous mutant Ezh2 in GC B cells were crossed with VavP-Bcl2. Importantly, homozygous expression of mutant Ezh2 phenocopies the Ezh2 knock-out phenotype further attesting the requirement for the maintenance of the wild-type allele for Ezh2 mutant enzymatic activity.
Recent studies questioned whether EZH2 mutation has additional and qualitatively distinct function in lymphomagenesis beyond simply being a more potent version of the wild-type enzyme. Along these lines, Ennishi et al. identified a strong enrichment of EZH2 mutations in human DLBCL cases with loss of MHC-I and MHC-II expression linked to a reduced number of tumor-infiltrating lymphocytes and less T cell cytolytic activity (74). To investigate in detail the consequences of EZH2 mutations on MHC expression and immune microenvironment, the authors relied on two different mouse models. First, they used the Ezh2-mutant model developed by Béguelin et al. where VavP-Bcl2 hematopoietic progenitors are infected with Ezh2-mutant containing retrovirus before re-injection into lethally irradiated recipient mice. Second, they used Cγ1-Cre Ezh2Y641N or Ezh2Y641F mice crossed with VavP-Bcl2 strain which develops DLBCL-like tumors. In both experimental systems, MHC-I and MHC-II expression were significantly reduced in mutant Ezh2 mice compared to wild-type mice, with reduced infiltration of CD3, CD4 and CD8 T cells in the tumor microenvironment, establishing Ezh2 gain-of-function mutation as a driver of MHC downregulation in GC lymphomagenesis, and eventually favoring immune escape. Importantly they also showed that EZH2 epigenetic switch-off of MHC molecules, driven by transcriptional repression of MHC-I/II transactivators, could be reversed with EZH2 inhibitors. The ability to restore MHC expression provides an interesting proof of concept in combining epigenetic reprogramming small molecules with immunotherapeutic approaches.
More recently, Melnick and colleagues further strengthened the concept that Ezh2 mutation initiate GC derived lymphomagenesis by escaping immune effector recognition and inducing a remodeling of the GC immunological niche (101). Using competitive BM chimera to track the cellular dynamics of Ezh2Y641F B cells during GC reaction, they found that Ezh2 mutants manifested a competitive growth advantage in the GC. This competitive advantage led to GC hyperplasia characterized by an increase of a cycling LZ cell population, without maturation blockade, associated with the expansion of FDC network. Using droplet based single cell transcriptomics and advanced histone mass spectrometry technologies, they assessed how Ezh2 mutation affected GC B cell cellular and histone methylation dynamics in an unbiased manner. They found that the epigenetic reprogramming imprinted by Ezh2 mutation through the reinforcement of the repressive program induced by H3K27me3 accumulation led to the abolition of LZ B cells’ dependence toward TFH emanating signals. Indeed, DZ re-entry of Ezh2-mutated centrocytes was clearly diminished and these GC B cell escaped TFH-mediated clonal selection. The ability of Ezh2-mutant GC B cells to proliferate within the LZ was linked to FDC interaction as this ability was impaired when FDC function was abolished through injection of soluble lymphotoxin receptor β. Remarkably, this study revealed how activating mutation of EZH2 induces a premalignant FL-like niche allowing B cells to persist as slowly proliferative centrocytes without TFH help and in a FDC dependent manner.
The above studies provide important insights on an emerging paradigm where one of the most critical function of epigenetic modifier mutations in promoting GC B cell transformation goes beyond the sole reprogramming of the GC epigenome, but instead arise from failure of GC exit signals to restore expression of genes that normally regulate immune signaling pathways and antigen presentation (47). Of therapeutic interest for the targeting of the FL common precursor cells, this remodeling of the immune synapse at least with EZH2 mutations tends to occur early during lymphomagenesis.
Linker Histone Loss of Function
Linker histones (H1) are additional chromatin modifying genes involved in the organization and stabilization of the nucleosome structure, supporting the folding of chromatin into higher-order conformation, and regulating its epigenetic state through the recruitment of histone modifiers. Heterozygous H1 mutations, found in up to 44% of FL and 27% of GCB-DLBCL, are mostly missense events affecting the globular C-terminal domain which led to the loss of protein function with impaired chromatin binding (53). Among them, H1C and H1E are the most common affected H1 isoforms observed in B cell lymphomas (102, 103).
To investigate the functionality of these isoforms in lymphomagenesis, Yusufova and colleagues used the previously described H1c−/− H1e−/− mice model (104). At early time points after chronic SRBC immunization, those animals manifested lymphoproliferative disease with invasiveness of B220+ B cells in extranodal tissues such as liver and lungs. Further analysis using competitive BM chimera revealed a competitive advantage for H1c/H1e-deficient B cells characterized by a specific increase of cycling LZ B cell population expressing Gl7, Fas and cd86 markers whereas no effect was found in other mature and immature B cell populations. Notably, H1 deficiency enables a chromatin decompaction in GC B cells with an enrichment in stem cell genes that become desilenced in H1-deficient GC cells. Given the frequent co-occurrence of H1C and H1E mutated genes with BCL2 overexpression in lymphomas, they crossed H1c−/− H1e−/− mice with VavP-Bcl2 mice. Loss of H1 isoforms caused a more extensive disruption of lymph node architecture with diffuse infiltration of immunoblastic cells, along with an extensive invasion of B220+ B cells and CD3+ T cells in extranodal tissues establishing H1 proteins as bona fide tumor suppressors. RNA sequencing analysis of lymphoma-like tumors in mouse and humans revealed a significant enrichment for stem cell signature and serial transplantations confirms that loss of H1 conferred lymphoma cells with enhanced self-renewal potential. These findings enlighten the contribution of H1 linker deletion in driving malignant transformation where epigenetic marks changes favor a relaxed state chromatin in GC B cells, increasing B cell fitness advantage by allowing self-renewal proprieties and may expose DNA to further AID-mediated additional hits.
TET1 and TET2 Loss
Besides chromatin modifiers genes, a number of studies have demonstrated methylation and disruption of cytosine methylation [5-methylcytosine (5mC)] patterning as another factor linked to the biology of B cell lymphoid malignancies (105). The methyl-cytosine dioxygenase TET2 (ten-eleven translocation 2) missense or truncated mutation is present in 6–12% of GCB-DLBCL (14). TET2 mutations are known to occur early in human HSCs and can be found in individuals with clonal haematopoiesis. Whether early TET2 mutations has a driving role in DLBCL was explored in multiple models. 5mC is well established as an epigenetic mark associated with transcriptional silencing, notably of tumor suppressor genes. TET2 is involved in active DNA demethylation, catalysing the oxidation of 5mC to 5-hydroxymethylcytosine (5hmC). Recently, it has been appreciated that 5hmC also functions as an epigenetic mark, and when linked to gene enhancers, is associated with activation of nearby genes.
Conditional deletion of Tet2 specifically in the B cell compartment with CD19-Cre Tet2fl/fl mice showed B cell transformation mimicking chronic lymphocytic leukemia (78). Programmed deletion of Tet2 in hematopoietic cells (Vav-Cre) or B cells (CD19-Cre) in immunized animals disrupt the ability of GC B cells to undergo CSR and terminal differentiation. Furthermore, conditional deletion of Tet2 at the GC stage results in a preneoplastic GC hyperplasia, blockage of GC exit and PC differentiation evolving in DLBCL-like tumors, confirming its role as a bona fide B cell tumor suppressor (79). Mechanistically, this phenotype is due to the focal loss of 5hmC at enhancers linked to B cell differentiation. Indeed, Tet2−/− GC B cells feature disruption of many enhancers linked to GC exit signaling pathways, antigen presentation, and terminal differentiation genes. This mechanism is conceptually similar to the functions of the histone modifiers in DLBCL which fails to restore the immune synapse. Interestingly, TET2 and CREBBP mutations are mutually exclusive in DLBCL (106), thus a combined mouse model could be engineered to find a potential therapeutic vulnerability in DLBCL.
The methyl-cytosine dioxygenase Tet1 (ten-eleven translocation 1) is also an important regulator of 5-hydroxymethylcytoine and interestingly transcriptionally silenced in FL. Cimmino and colleagues engineered Tet1-deficient mice where B cell lymphoma development was promoted resulting in a diminished survival compared to wild type mice (77). Tet1-deficient mice exhibited lymphadenopathy and hepato-splenomegaly. Splenic tumors were characterized by a massive infiltration of proliferating lymphocytes disrupting the normal architecture and expressing the GC markers Bcl6 and Irf4 but not the PC marker Cd138. When combined with Bcl2 overexpression, Tet1-deficient B cell lymphomagenesis was accelerated up to 10 weeks post-transplantation. Altogether, deletion of Tet1 and Tet2 in mice induces phenotypically predominant DLBCL tumors supporting a suppressor role in mature B cells.
Modeling Evasion From Immune Surveillance and Dissemination
The influence of the GC microenvironment on B cell development which provides essential signals for the survival, selection and differentiation is well established with several actors residing in the GC LZ such as T follicular helper cells (TFH), follicular dendritic cells (FDC), regulatory T cells (Treg), macrophages and stromal cells (19). FL is the paradigm of a B cell malignancy strongly dependent on direct interaction with a GC-like permissive microenvironment that co-evolves with malignant cell clones as a part of a dynamic interplay (49). Recent studies in both FL patients and genetically-engineered mouse models have started to highlight the link between B ‘cell-intrinsic’ tumor genetic alterations and their cell-extrinsic functions during lymphomagenesis by contributing to escape of immune surveillance mechanisms. We will discuss the latest studies showing how the TNFRSF14 or CTSS frequent alterations in FL modify the TME/malignant B cells crosstalk and contribute to lymphoma development either by affecting antigen processing and hiding from the immune system, or modifying its composition to become tumor-supportive.
Tnfrsf14 Loss in FL
TNFRSF14, the gene encoding the HVEM receptor located on 1p chromosome is one of the most frequent cell surface protein, deleted or mutated in >40% of FL cases and enriched in GCB-DLBCL (14, 18, 19). HVEM is expressed at the surface of B cells as well as other cell types and has multiple ligands including LIGHT or BTLA. Besides its role as a signaling receptor, HVEM can act as ligand and transmit signals into BTLA-expressing cells notably TFH (107). How loss of HVEM contributes to lymphomagenesis has been the focus of two independent and complementary in vivo studies (80, 108). The first study took advantage of the well characterized VavP-Bcl2 model that recapitulates key aspects of the genetics and pathology of human FLs to generate BM chimeras where knockdown of Hvem was mediated by transduction of shHvem into VavP-Bcl2 HSCs followed by reconstitution of irradiated wild-type hosts. Knockdown of Hvem in all hematopoietic system caused a significant acceleration and increased penetrance of lymphoma development compared to VavP-Bcl2 controls with 90% of animals carrying tumors at 100 days. Despite the absence of B-cell specificity of the shRNA transduction strategy, only B cells were enriched with the short-hairpin construct indicating the Hvem knockdown in T cells was unlikely to participate to the lymphomagenic effect. Mechanistically, besides cell-autonomous activation of B cells, Hvem loss and the consequent loss of interaction with Btla triggers the amplification of TFH producing high amounts of TNF and Lymphotoxin, the two non-redundant factors involved in lymphoid stromal cell differentiation and maintenance, and favors lymphoid stromal cell activation including FDC and FRC (Follicular Reticular Cells). This seminal study offers the first demonstration of a functional impact of a B-cell specific genetic alteration on the polarization of a FL-supportive microenvironment (80). Of therapeutic interest, immunotherapeutic delivery of a soluble HVEM receptor, via modified CAR-T cells, inhibited the growth of lymphoma by restoring the BTLA-HVEM interaction highlighting the essential role of the tumor/microenvironment dialog in lymphomagenesis. In an independent study, Mintz et al. reported that in models of Bcl2 overexpression in B cells, Btla deficiency in T cells led to a similar GC B cell outgrowth and accelerated lymphomagenesis than Hvem deficiency in B cells proposing an alternative mechanism by which the Btla-Hvem axis functions as an cell-extrinsic suppressor in lymphomagenesis (108). Using a chimeric mouse system, they identified that during a normal immune response, Hvem restrains B cell proliferation, differentiation and selection by reducing the delivery of signal in trans through the Btla-Hvem axis on TFH cells. Hvem mutation in B cells would lead to a loss of negative signaling in TFH cells and allows Hvem-mutant B cells to receive exaggerated helper signals that promote proliferation and accrual of AID-mediated mutations. Collectively, those data provide important evidence for a cell-extrinsic tumor suppressor role of Hvem. The ways in which increased signaling via CD40 and other T cell–derived helper factors cooperates with Bcl2-overexpression in lymphoma development remain to be fully elucidated.
CTSS Alterations in FL
Cathepsin S (CTSS) is part of a family of cysteine proteases whose role is essential in the regulation of normal immune response through its activity on antigen processing, B cell expansion and communication with CD4+ T cells. By cleaving the CD74 chaperone protein bound on MHC class II molecules, CTSS enzymatic activity results in a smaller peptide CLIP that will be displaced and allow variable antigenic peptides to bond to MHCII and present at the cell surface. Recurrent hotspot mutations and gene amplifications of CTSS have been recently described in 6 and 13% of FL patients respectively (81, 109) and mechanistically, CTSSY132D hotspot mutation promotes activation of the protein and increases its protease activity. To assess how the most common CTSSY132D mutations or CTSS overexpression contribute to accelerate lymphoma development, Dheilly et al. generated a chimera mouse model of FL using the VavP-Bcl2 mice as HSPC donor cells and expressing either mutated human CTSS or overexpressing human CTSS. Both models revealed an oncogenic role of CTSS over-activation with higher penetrance and decreased latency as compared to VavP-Bcl2 tumors alone. Tumors with CTSS alterations were characterized by a remodeled tumor-prone microenvironment with an increased infiltration of CD4+ T cells while limiting CD8+ T cells recruitment. Depletion of CD4+ T cells in VavP-Bcl2/CTSS chimera models confirm that CTSS is essential to support the communication and co-stimulatory signals between tumor B cells and CD4+ cells in the GC context. Conversely, loss of CTSS activity in aggressive mouse lymphoma xenograft restrain lymphoma growth by recruiting and enhancing CD8+ T cells cytotoxic activity while impairing communication with CD4+ TFH cells. These data show for the first time that by altering the processing of antigenic peptides, CTSS mutations or overexpression remodel the immune microenvironment to promote lymphoma growth and implies that targeting a regulator of antigen presentation such as CTSS could modulate the spectrum of processed antigens, promote activation of cytotoxic T cells, enhance tumor immunogenicity and improve response to anti-PD1 immunotherapies.
Disruption of Gα Migration Pathway
Another pathway frequently mutated in GC-derived lymphomas is the GC homing pathway involving S1PR2 and GNA13. The guanine nucleotide binding protein GNA13 (encoding Gα13), is a signaling mediator downstream of transmembrane G-protein-coupled receptors sphingosine-1-phosphate receptor-2 (S1PR2), that confines B cells in the GC and promotes growth regulation by suppressing both Akt and cell migration (110). In about 20% of GCB-DLBCL cases (38% in the EZB subtype), deleterious mutations affect one of the members of the Gα13 homing pathways, namely GNA13, S1PR2 and P2RY8 (another S1P receptor expressed on GC B cells). To model in vivo the impact of GNA13 loss during lymphomagenesis, Muppidi and colleagues utilized a mixed BM chimeric approach, deleting Gα13 in all B-lineage using Mb1Cre;Gα13f/f mice. Deficiency of Gα13 favors the formation of enlarged mesenteric LN with GC B cells expansion associated with a marked disruption of the GC architecture and a loss of GC confinement due to the inability to suppress migration in response to S1P. Occasional transformation in B cell lymphomas displaying a GC-like phenotype (Gl7+CD138-Bcl6+Irf4+IgD-) were observed at 1 year of age (82). One of the critical observations made in Gα13 deficient cells is the loss of confinement which allows egress outside the GC, dissemination and seeding of these tumoral cells at distant sites such as blood and BM. As deficiency in S1PR2 did not phenocopy Gα13 deficiency, the authors searched for additional Gα13 G protein coupled receptor that may be involved in GC B cell regulation and discovered that P2RY8, an orphan receptor also represses GC B cell growth promoting confinement via Gα13 and is mutated in GCB-DLBCL. Cooperation between Bcl2 overexpression and Gna13 loss showed an exacerbated phenotype in double mutant mice leading to greater accumulation of GC B cells in spleen, wider dispersal throughout the follicles and more dissemination in blood and BM suggesting a combinatorial effect of Bcl2 in promoting abnormal B cell survival outside the GC niche. These findings shed new lights on an important mechanism by which disruption of Gα13 signaling exerts dual actions in promoting growth and favoring dissemination of GC B cell in GC-derived lymphomagenesis and offer a biological explanation for factors leading to systematic dissemination of tumoral GC B cells in multiple organs including the BM.
Modeling Dysregulation of GC Metabolism
During T-dependent adaptive immune responses, B cells undergo a quick anabolic shift that sustain the GC proliferative burst (111). Many B-cell lymphomas originating in the GC present an exceptionally high proliferation index. This implies massive metabolic requirements in order to generate sufficient energy and support anabolism for repeated growth and division cycles. Recurrent mutations in components of the nutrient sensing pathway that activates the mechanistic target of rapamycin complex 1 (mTORC1), a driver of cellular anabolism are found in about 25% of FLs (112, 113). In response to growth factors and when there is sufficient intracellular amino acid concentration, mTORC1 promotes protein synthesis (114). Intracellular amino acid concentration is perceived through a protein complex present on lysosome surface comprising RAG GTPases, Ragulator complex, v-ATPase complex (vacuolar H+-adenosine triphosphate ATPase) and SLC38A9 (sodium-coupled amino acid transporter 9). When amino acid concentration is sufficient, the RAG GTPases form heterodimers on lysosome surface allowing the recruitment of mTORC1 and downstream protein synthesis supporting cellular growth (114–116).
RRAGC Activating Mutations
The RRAGC gene encodes for Ras-related GTP-binding protein (RAGC), an essential activator of mTORC1 downstream of the sensing of cellular nutrients. Recurrent point mutations of RRAGC are found in 18% of FLs and are rare in other B cell lymphomas (112, 113). More than half of these events are associated with mutations of certain components of the v-ATPase complex (ATP6V1B2 and ATP6AP1). To characterize the impact of recurrent RRAGC mutations on B cell function and lymphomagenesis, Ortega-Molina et al. used CRISPR/Cas9 genome editing techniques to engineer two independent murine Rragc knock-in mouse strains reproducing hotspot mutations recurrently found in human FL (S75C and T90N mutations) (83). Heterozygous RagCS74C/+ and RagCT89N/+ heterozygous mice showed no obvious phenotype. Lymphoid and myeloid cell populations frequencies were similar in mutant versus wild-type mice in the spleen and BM however RagC knock-in mutations conferred partial insensitivity to nutrient withdrawal. When bred to a VavP-Bcl2 mice, RagC-Bcl2+ double-mutant mice showed exacerbated B cell responses in response to immunization characterized by enlarged GCs, increased plasma cell production without impairment of high-affinity B cell selection and eventually acceleration of lymphoma development. This was observed both in the progeny of VavP-Bcl2-RagCmut mice but also using a BM chimeric system where VavP-Bcl2-RagCmut fetal liver cells were transplanted into lethally irradiated wild-type recipients. Histological analysis of these tumors revealed a follicular growth pattern of Bcl6+ B cells with no difference according to genotype. Bulk RNA sequencing analysis of B220+ cells obtained from RagCmut and RagCWT Bcl2+ FL-like tumors revealed that the mTORC1 signaling signature was enriched in RagCmut FL. Furthermore, when transcriptional profiles from murine and human FL with or without RagC mutations were compared, upregulated genes in murine RagCmut FL were enriched in RRAGC mutated human FL. Interestingly, the mTOR inhibitor rapamycin, given orally to RagCmut and RagCWT Bcl2+ mice during long term, inhibited proliferation selectively in RagCmut FL. Overall, this data shows that RRAGC mutations result in mTORC1 activation regardless of the intracellular amino acid concentration which confers a selective advantage to GC B cells. In addition, the presence of these mutations makes GC B cells less dependent to TFH signals, as blockade of T cell help through anti-CD40L after GC induction makes RagCmut B cells intrinsically resistant to apoptosis despite TFH suppression. Finally, when these mutations are associated with Bcl2 overexpression, the lymphomagenesis process is accelerated in a cell-intrinsic manner where mutant GC B cells with increased fitness could continue to undergo cycles of selection and proliferation favoring further acquisition of additional hits (83).
SESTRIN1 Loss of Function
SESTRIN1 has been identified as a target gene of the recurrent 6q deletion in FLs and it is also epigenetically inactivated by EZH2 gain-of-function (84). SESTRIN1 is a mTORC1 regulator that inhibits cell growth when TP53 is activated in response to DNA damage. Inactivation of SESTRIN1, by del6q or by the EZH2 mutation leads to mTORC1 activation. Therefore, SESTRIN1 loss represents an alternative to RRAGC mutations that maintain mTORC1 activity under nutrient starvation. To recapitulate SESTRIN1 deficiency in vivo, in combination with BCL2 overexpression, Oricchio and colleagues retrovirally transduced VavP-Bcl2 HSPCs with shRNAs targeting Sestrin1. Sestrin1 deficiency led to an acceleration of the Bcl2-mediated lymphoma, establishing his tumor suppressive role in B cells. Morphologically, BCL2/Sestrin1-deficient tumors resembled FL with a follicular architecture, they displayed markers of a GC phenotype (PNA and BCL6), showed evidence of somatic hypermutation, an increased tumor proliferation based on Ki67 staining. Remarkably, pharmacological inhibition of EZH2 promotes SESTRIN1 re-expression and restores its tumor suppressive activity, suggesting the possibility to epigenetically control mTORC1 activity in lymphoma. Interestingly, EZH2/RRAGC gain of function mutations and SESTRIN1 loss are mutually exclusive suggesting that these alterations are involved in mTORC1 activity maintenance allowing tumor cells to escape proliferation inhibition (84) also becoming less dependent to TFH signals as shown in the context of RRAGC and EZH2 mutations (83, 101). In summary, mTORC1 pathway activation, through various mutually exclusive molecular alterations, is clearly involved in B cell lymphomagenesis process and may be associated to a specific sensitivity to mTOR inhibitors.
Conclusions and Perspectives
B cell lymphomas are among the most frequent hematopoietic malignancies and represent a molecularly heterogeneous group of diseases with different therapeutic vulnerabilities (26, 117) and clinical outcomes that largely depend on a complex interplay between a multitude of genomic alterations and heterogeneous tumor microenvironment signatures (118, 119). Sequencing studies allowed the identification of recurrently mutated genes that drive lymphomagenesis and led to refine DLBCL classification that pave the way for personalized therapeutic strategies (14, 19, 118). The latest large-scale genome sequencing studies identified up to seven different molecular subtypes of DLBCL (118) which represent an interesting framework for the development of models mimicking each molecular subtype. Now, recent DLBCL classifications based of TME composition revealed four distinct microenvironment compositions which provide independent contributions to clinical outcome regardless of the GCB/ABC cell-of-origin or the genetic DLBCL subtype classification (119). The challenge of modeling future preclinical mouse models will be to recapitulate both the genomic features of the tumors and the parallel remodeling of the tumor microenvironment. Such tumors likely develop over decades in humans before clinical symptoms appear and rely on a complex combination of hits that have been accumulated in a stepwise and ordered manner leading to a spatially and temporally intra-tumor heterogeneity. Advanced technologies are emerging for a better characterization of the existing and future models and cellular heterogeneity in murine lymphomas can be resolved by measuring gene expression at a single cell resolution, allowing the study of B cells together with their microenvironment. Using those techniques, we and others have started to uncover and compare the underlying transcriptional and functional heterogeneity within FL malignant B cells and normal GC B cells in human and mice (31, 81, 101, 120, 121).
Another important feature of lymphoma biology that need to be properly addressed in mouse models is mimicking the kinetics and cellular context in which genetic hits accumulate overtime sometimes over years. The order of appearance of such hits and how it influences lymphomagenesis remains an area of active research. Mutations in KMT2D, CREBBP or EZH2 represent early events during lymphomagenesis and have been proposed to occur in HSPCs. Mouse models reproducing these alterations do not induce lymphomagenesis on their own but accelerate GC B cell transformation when combined with BCL2 overexpression (68–71). Accordingly, recent genomic data obtained from prediagnostic samples formally demonstrate that BCL2 translocations systematically precede the acquisition of subsequent epigenetic mutations (122).
Murine models developed since several decades have been particularly useful to inform us on the biological mechanisms driving B cell lymphomagenesis, and helping to understand the functional consequences of genetic alterations. As one of the emerging functions of epigenetic modifier genes in GC B cell lymphoma is to favor disease initiation through the reprogramming of the immune niche, mouse models that faithfully recapitulate the complex interactions with the microenvironment will become valuable tools for the testing and development of novel, rationally designed therapeutic approaches (80, 101). In this line, it is remarkable that microenvironment remodeling seems to occur early in the context of EZH2 mutations, suggesting that early epigenetic therapy may be useful to prevent disease progression (105).
GEM models may recapitulate the natural history and histological properties of human tumors. However, they display a limited mutational and immunological complexity compared to the human tumors they are intended to model (119). Novel sophisticated tools for engineering mouse models such as CRISPR-Cas9 gene editing techniques or patient-derived xenografts (especially those mimicking indolent lymphomas such as FL), including in humanized mice, will be useful to fill this gap and efficiently generate preclinical models that reflect the complex genetics of the human tumors (61, 123–125). The generation of transplantable lymphoma cell lines obtained from these GEM can be engineered to develop functional CRISPR screening in vivo; such an approach would also be useful to screen for therapeutic vulnerabilities in an unbiased manner. Now that most of the genetic drivers have been discovered in B cell lymphomas and that we are getting the tools for a more in-depth characterization of their functional consequences both on B cells and its microenvironment, we hope that studies using more complex GEM tumor models will serve to streamline the translation of targeted therapies with novel immunotherapies into the clinics.
Author Contributions
All authors contribute to the writing of the review. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from Fondation pour La Recherche Médicale (GB), Fondation ARC, Institut National du Cancer, Canceropôle PACA and Institut Carnot Calym. We thank all the team members past and present that have contributed to some of the work described in this review.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cyster JG, Allen CDC. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell (2019) 177:524–40. doi: 10.1016/j.cell.2019.03.016
2. Victora GD, Nussenzweig MC. Germinal Centers. Annu Rev Immunol (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
3. Calado DP, Sasaki Y, Godinho SA, Pellerin A, Köchert K, Sleckman BP, et al. The Cell-Cycle Regulator c-Myc Is Essential for the Formation and Maintenance of Germinal Centers. Nat Immunol (2012) 13:1092–100. doi: 10.1038/ni.2418
4. Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, et al. The Proto-Oncogene MYC Is Required for Selection in the Germinal Center and Cyclic Reentry. Nat Immunol (2012) 13:1083–91. doi: 10.1038/ni.2428
5. Laidlaw BJ, Cyster JG. Transcriptional Regulation of Memory B Cell Differentiation. Nat Rev Immunol (2020) 21:1–12. doi: 10.1038/s41577-020-00446-2
6. Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S, et al. Multiple Layers of B Cell Memory With Different Effector Functions. Nat Immunol (2009) 10:1292–9. doi: 10.1038/ni.1814
7. Mesin L, Schiepers A, Ersching J, Barbulescu A, Cavazzoni CB, Angelini A, et al. Restricted Clonality and Limited Germinal Center Reentry Characterize Memory B Cell Reactivation by Boosting. Cell (2020) 180:92–106.e11. doi: 10.1016/j.cell.2019.11.032
8. Sungalee S, Mamessier E, Morgado E, Grégoire E, Brohawn PZ, Morehouse CA, et al. Germinal Center Reentries of BCL2-Overexpressing B Cells Drive Follicular Lymphoma Progression. J Clin Invest (2014) 124:5337–51. doi: 10.1172/JCI72415
9. Venturutti L, Melnick A. The Dangers of Déjà Vu: Memory B-Cells as the Cell-of-Origin of ABC-Dlbcls. Blood (2020) 136:2263–74. doi: 10.1182/blood.2020005857
10. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The C-Myc Oncogene Driven by Immunoglobulin Enhancers Induces Lymphoid Malignancy in Transgenic Mice. Nature (1985) 318:533–8. doi: 10.1038/318533a0
11. Park SS, Kim JS, Tessarollo L, Owens JD, Peng L, Han SS, et al. Insertion of C-Myc Into Igh Induces B-Cell and Plasma-Cell Neoplasms in Mice. Cancer Res (2005) 65:1306–15. doi: 10.1158/0008-5472.CAN-04-0268
12. Truffinet V, Pinaud E, Cogné N, Petit B, Guglielmi L, Cogné M, et al. The 3’ IgH Locus Control Region Is Sufficient to Deregulate a C-Myc Transgene and Promote Mature B Cell Malignancies With a Predominant Burkitt-Like Phenotype. J Immunol (2007) 179:6033–42. doi: 10.4049/jimmunol.179.9.6033
13. Sander S, Calado DP, Srinivasan L, Köchert K, Zhang B, Rosolowski M, et al. Synergy Between Pi3k Signaling and MYC in Burkitt Lymphomagenesis. Cancer Cell (2012) 22:167–79. doi: 10.1016/j.ccr.2012.06.012
14. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell (2017) 171:481–94. doi: 10.1016/j.cell.2017.09.027
15. Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt Lymphoma Pathogenesis and Therapeutic Targets From Structural and Functional Genomics. Nature (2012) 490:116–20. doi: 10.1038/nature11378
16. Pae J, Ersching J, Castro TBR, Schips M, Mesin L, Allon SJ, et al. Cyclin D3 Drives Inertial Cell Cycling in Dark Zone Germinal Center B Cells. J Exp Med (2021) 218:e20201699. doi: 10.1084/jem.20201699
17. Ramezani-Rad P, Chen C, Zhu Z, Rickert RC. Cyclin D3 Governs Clonal Expansion of Dark Zone Germinal Center B Cells. Cell Rep (2020) 33:108403. doi: 10.1016/j.celrep.2020.108403
18. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. New Engl J Med (2018) 378:1396–407. doi: 10.1056/NEJMoa1801445
19. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular Subtypes of Diffuse Large B Cell Lymphoma Are Associated With Distinct Pathogenic Mechanisms and Outcomes. Nat Med (2018) 24:679–90. doi: 10.1038/s41591-018-0016-8
20. Young RM, Phelan JD, Shaffer AL, Wright GW, Huang DW, Schmitz R, et al. Taming the Heterogeneity of Aggressive Lymphomas for Precision Therapy. Annu Rev Cancer Biol (2019) 3:429–55. doi: 10.1146/annurev-cancerbio-030518-055734
21. Venturutti L, Teater M, Zhai A, Chadburn A, Babiker L, Kim D, et al. Tbl1xr1 Mutations Drive Extranodal Lymphoma by Inducing a Pro-Tumorigenic Memory Fate. Cell (2020) 182:297–316.e27. doi: 10.1016/j.cell.2020.05.049
22. Calado DP, Zhang B, Srinivasan L, Sasaki Y, Seagal J, Unitt C, et al. Constitutive Canonical Nf-κb Activation Cooperates With Disruption of BLIMP1 in the Pathogenesis of Activated B Cell-Like Diffuse Large Cell Lymphoma. Cancer Cell (2010) 18:580–9. doi: 10.1016/j.ccr.2010.11.024
23. Mandelbaum J, Bhagat G, Tang H, Mo T, Brahmachary M, Shen Q, et al. Blimp1 Is a Tumor Suppressor Gene Frequently Disrupted in Activated B Cell-Like Diffuse Large B Cell Lymphoma. Cancer Cell (2010) 18:568–79. doi: 10.1016/j.ccr.2010.10.030
24. Knittel G, Liedgens P, Korovkina D, Seeger JM, Al-Baldawi Y, Al-Maarri M, et al. B-Cell-Specific Conditional Expression of Myd88p.L252P Leads to the Development of Diffuse Large B-Cell Lymphoma in Mice. Blood (2016) 127:2732–41. doi: 10.1182/blood-2015-11-684183
25. Flümann R, Rehkämper T, Nieper P, Pfeiffer P, Holzem A, Klein S, et al. An Autochthonous Mouse Model of Myd88 - and BCL2 -Driven Diffuse Large B-Cell Lymphoma Reveals Actionable Molecular Vulnerabilities. Blood Cancer Discov (2021) 2:70–91. doi: 10.1158/2643-3230.BCD-19-0059
26. Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, et al. A Multiprotein Supercomplex Controlling Oncogenic Signalling in Lymphoma. Nature (2018) 560:387–91. doi: 10.1038/s41586-018-0290-0
27. Young RM, Wu T, Schmitz R, Dawood M, Xiao W, Phelan JD, et al. Survival of Human Lymphoma Cells Requires B-Cell Receptor Engagement by Self-Antigens. Proc Natl Acad Sci USA (2015) 112:13447–54. doi: 10.1073/pnas.1514944112
28. Perez-Chacon G, Adrados M, Vallejo-Cremades MT, Lefebvre S, Reed JC, Zapata JM. Dysregulated TRAF3 and BCL2 Expression Promotes Multiple Classes of Mature Non-Hodgkin B Cell Lymphoma in Mice. Front Immunol (2018) 9:3114. doi: 10.3389/fimmu.2018.03114
29. Milpied P, Nadel B, Roulland S. Premalignant Cell Dynamics in Indolent B-Cell Malignancies. Curr Opin Hematol (2015) 22:388–96. doi: 10.1097/MOH.0000000000000159
30. McHeyzer-Williams LJ, Milpied PJ, Okitsu SL, McHeyzer-Williams MG. Class-Switched Memory B Cells Remodel Bcrs Within Secondary Germinal Centers. Nat Immunol (2015) 16:296–305. doi: 10.1038/ni.3095
31. Milpied P, Cervera-Marzal I, Mollichella M-L, Tesson B, Brisou G, Traverse-Glehen A, et al. Human Germinal Center Transcriptional Programs Are De-Synchronized in B Cell Lymphoma. Nat Immunol (2018) 19:1013–24. doi: 10.1038/s41590-018-0181-4
32. Holmes AB, Corinaldesi C, Shen Q, Kumar R, Compagno N, Wang Z, et al. Single-Cell Analysis of Germinal-Center B Cells Informs on Lymphoma Cell of Origin and Outcome. J Exp Med (2020) 217:e20200483. doi: 10.1084/jem.20200483
33. King HW, Orban N, Riches JC, Clear AJ, Warnes G, Teichmann SA, et al. Single-Cell Analysis of Human B Cell Maturation Predicts How Antibody Class Switching Shapes Selection Dynamics. Sci Immunol (2021) 6:eabe6291. doi: 10.1126/sciimmunol.abe6291
34. Kennedy DE, Okoreeh MK, Maienschein-Cline M, Ai J, Veselits M, McLean KC, et al. Novel Specialized Cell State and Spatial Compartments Within the Germinal Center. Nat Immunol (2020) 21:1–11. doi: 10.1038/s41590-020-0660-2
35. Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, et al. Transcriptional Regulation of Germinal Center B and Plasma Cell Fates by Dynamical Control of IRF4. Immunity (2013) 38:918–29. doi: 10.1016/j.immuni.2013.04.009
36. De Silva NS, Klein U. Dynamics of B Cells in Germinal Centres. Nat Rev Immunol (2015) 15:137–48. doi: 10.1038/nri3804
37. Basso K, Dalla-Favera R. Roles of BCL6 in Normal and Transformed Germinal Center B Cells. Immunol Rev (2012) 247:172–83. doi: 10.1111/j.1600-065X.2012.01112.x
38. Basso K, Dalla-Favera R. Bcl6: Master Regulator of the Germinal Center Reaction and Key Oncogene in B Cell Lymphomagenesis. Adv Immunol (2010) 105:193–210. doi: 10.1016/S0065-2776(10)05007-8
39. Hatzi K, Melnick A. Breaking Bad in the Germinal Center: How Deregulation of BCL6 Contributes to Lymphomagenesis. Trends Mol Med (2014) 20:343–52. doi: 10.1016/j.molmed.2014.03.001
40. Luo W, Weisel F, Shlomchik MJ. B Cell Receptor and CD40 Signaling Are Rewired for Synergistic Induction of the C-Myc Transcription Factor in Germinal Center B Cells. Immunity (2018) 48:313–26.e5. doi: 10.1016/j.immuni.2018.01.008
41. Saito M, Gao J, Basso K, Kitagawa Y, Smith PM, Bhagat G, et al. A Signaling Pathway Mediating Downregulation of BCL6 in Germinal Center B Cells Is Blocked by BCL6 Gene Alterations in B Cell Lymphoma. Cancer Cell (2007) 12:280–92. doi: 10.1016/j.ccr.2007.08.011
42. Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription Factor Irf4 Controls Plasma Cell Differentiation and Class-Switch Recombination. Nat Immunol (2006) 7:773–82. doi: 10.1038/ni1357
43. Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, et al. Regulated Selection of Germinal-Center Cells Into the Memory B Cell Compartment. Nat Immunol (2016) 17:861–9. doi: 10.1038/ni.3460
44. Laidlaw BJ, Duan L, Xu Y, Vazquez SE, Cyster JG. The Transcription Factor Hhex Cooperates With the Corepressor Tle3 to Promote Memory B Cell Development. Nat Immunol (2020) 21:1–12. doi: 10.1038/s41590-020-0713-6
45. Basso K, Dalla-Favera R. Germinal Centres and B Cell Lymphomagenesis. Nat Rev Immunol (2015) 15:172–84. doi: 10.1038/nri3814
46. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct Types of Diffuse Large B-Cell Lymphoma Identified by Gene Expression Profiling. Nature (2000) 403:503–11. doi: 10.1038/35000501
47. Mlynarczyk C, Fontán L, Melnick A. Germinal Center-Derived Lymphomas: The Darkest Side of Humoral Immunity. Immunol Rev (2019) 288:214–39. doi: 10.1111/imr.12755
48. Pasqualucci L. Molecular Pathogenesis of Germinal Center-Derived B Cell Lymphomas. Immunol Rev (2019) 288:240–61. doi: 10.1111/imr.12745
49. Verdière L, Mourcin F, Tarte K. Microenvironment Signaling Driving Lymphomagenesis. Curr Opin Hematol (2018) 25:335–45. doi: 10.1097/MOH.0000000000000440
50. Carbone A, Roulland S, Gloghini A, Younes A, von Keudell G, López-Guillermo A, et al. Follicular Lymphoma. Nat Rev Dis Primers (2019) 5:83. doi: 10.1038/s41572-019-0132-x
51. Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, Nussenzweig MC. Identification of Human Germinal Center Light and Dark Zone Cells and Their Relationship to Human B-Cell Lymphomas. Blood (2012) 120:2240–8. doi: 10.1182/blood-2012-03-415380
52. Pikor NB, Mörbe U, Lütge M, Gil-Cruz C, Perez-Shibayama C, Novkovic M, et al. Remodeling of Light and Dark Zone Follicular Dendritic Cells Governs Germinal Center Responses. Nat Immunol (2020) 21:1–11. doi: 10.1038/s41590-020-0672-y
53. Okosun J, Bödör C, Wang J, Araf S, Yang C-Y, Pan C, et al. Integrated Genomic Analysis Identifies Recurrent Mutations and Evolution Patterns Driving the Initiation and Progression of Follicular Lymphoma. Nat Genet (2014) 46:176–81. doi: 10.1038/ng.2856
54. Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating Mutations of Acetyltransferase Genes in B-Cell Lymphoma. Nature (2011) 471:189–95. doi: 10.1038/nature09730
55. Green MR, Kihira S, Liu CL, Nair RV, Salari R, Gentles AJ, et al. Mutations in Early Follicular Lymphoma Progenitors Are Associated With Suppressed Antigen Presentation. Proc Natl Acad Sci USA (2015) 112:E1116–25. doi: 10.1073/pnas.1501199112
56. Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of Follicular Lymphoma Transformation. Cell Rep (2014) 6:130–40. doi: 10.1016/j.celrep.2013.12.027
57. Ramezani-Rad P, Rickert RC. Murine Models of Germinal Center Derived-Lymphomas. Curr Opin Immunol (2017) 45:31–6. doi: 10.1016/j.coi.2016.12.002
58. Pasqualucci L, Klein U. Mouse Models in the Study of Mature B-Cell Malignancies. Cold Spring Harb Perspect Med (2021) 11:a034827. doi: 10.1101/cshperspect.a034827
59. Donnou S, Galand C, Touitou V, Sautès-Fridman C, Fabry Z, Fisson S. Murine Models of B-Cell Lymphomas: Promising Tools for Designing Cancer Therapies. Adv Hematol (2012) 2012:701704. doi: 10.1155/2012/701704
60. Flümann R, Nieper P, Reinhardt HC, Knittel G. New Murine Models of Aggressive Lymphoma. Leukemia Lymphoma (2020) 61:788–98. doi: 10.1080/10428194.2019.1691200
61. Purwada A, Shah SB, Béguelin W, August A, Melnick AM, Singh A. Ex Vivo Synthetic Immune Tissues With T Cell Signals for Differentiating Antigen-Specific, High Affinity Germinal Center B Cells. Biomaterials (2019) 198:27–36. doi: 10.1016/j.biomaterials.2018.06.034
62. McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP, et al. Bcl-2-Immunoglobulin Transgenic Mice Demonstrate Extended B Cell Survival and Follicular Lymphoproliferation. Cell (1989) 57:79–88. doi: 10.1016/0092-8674(89)90174-8
63. Strasser A, Whittingham S, Vaux DL, Bath ML, Adams JM, Cory S, et al. Enforced BCL2 Expression in B-Lymphoid Cells Prolongs Antibody Responses and Elicits Autoimmune Disease. Proc Natl Acad Sci USA (1991) 88:8661–5. doi: 10.1073/pnas.88.19.8661
64. Strasser A, Harris AW, Cory S. E Mu-Bcl-2 Transgene Facilitates Spontaneous Transformation of Early Pre-B and Immunoglobulin-Secreting Cells But Not T Cells. Oncogene (1993) 8:1–9.
65. Egle A, Harris AW, Bath ML, O’Reilly L, Cory S. Vavp-Bcl2 Transgenic Mice Develop Follicular Lymphoma Preceded by Germinal Center Hyperplasia. Blood (2004) 103:2276–83. doi: 10.1182/blood-2003-07-2469
66. Xiang H, Noonan EJ, Wang J, Duan H, Ma L, Michie S, et al. The Immunoglobulin Heavy Chain Gene 3’ Enhancers Induce Bcl2 Deregulation and Lymphomagenesis in Murine B Cells. Leukemia (2011) 25:1484–93. doi: 10.1038/leu.2011.115
67. García-Ramírez I, Tadros S, González-Herrero I, Martín-Lorenzo A, Rodríguez-Hernández G, Moore D, et al. Crebbp Loss Cooperates With Bcl2 Overexpression to Promote Lymphoma in Mice. Blood (2017) 129:2645–56. doi: 10.1182/blood-2016-08-733469
68. Zhang J, Vlasevska S, Wells VA, Nataraj S, Holmes AB, Duval R, et al. The CREBBP Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B-Cell Lymphoma. Cancer Discov (2017) 7:322–37. doi: 10.1158/2159-8290.CD-16-1417
69. Jiang Y, Ortega-Molina A, Geng H, Ying H-Y, Hatzi K, Parsa S, et al. Crebbp Inactivation Promotes the Development of HDAC3-Dependent Lymphomas. Cancer Discov (2017) 7:38–53. doi: 10.1158/2159-8290.CD-16-0975
70. Zhang J, Dominguez-Sola D, Hussein S, Lee J-E, Holmes AB, Bansal M, et al. Disruption of KMT2D Perturbs Germinal Center B Cell Development and Promotes Lymphomagenesis. Nat Med (2015) 21:1190–8. doi: 10.1038/nm.3940
71. Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, et al. The Histone Lysine Methyltransferase Kmt2d Sustains a Gene Expression Program That Represses B Cell Lymphoma Development. Nat Med (2015) 21:1199–208. doi: 10.1038/nm.3943
72. Souroullas GP, Jeck WR, Parker JS, Simon JM, Liu J-Y, Paulk J, et al. An Oncogenic Ezh2 Mutation Induces Tumors Through Global Redistribution of Histone 3 Lysine 27 Trimethylation. Nat Med (2016) 22:632–40. doi: 10.1038/nm.4092
73. Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 Is Required for Germinal Center Formation and Somatic Ezh2 Mutations Promote Lymphoid Transformation. Cancer Cell (2013) 23:677–92. doi: 10.1016/j.ccr.2013.04.011
74. Ennishi D, Takata K, Béguelin W, Duns G, Mottok A, Farinha P, et al. Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov (2019) 9:546–63. doi: 10.1158/2159-8290.CD-18-1090
75. Béguelin W, Teater M, Gearhart MD, Calvo Fernández MT, Goldstein RL, Cárdenas MG, et al. EZH2 and BCL6 Cooperate to Assemble Cbx8-Bcor Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell (2016) 30:197–213. doi: 10.1016/j.ccell.2016.07.006
76. Yusufova N, Kloetgen A, Teater M, Osunsade A, Camarillo JM, Chin CR, et al. Histone H1 Loss Drives Lymphoma by Disrupting 3d Chromatin Architecture. Nature (2020) 22:1–7. doi: 10.1038/s41586-020-3017-y
77. Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu Y, et al. TET1 Is a Tumor Suppressor of Hematopoietic Malignancy. Nat Immunol (2015) 16:653–62. doi: 10.1038/ni.3148
78. Mouly E, Ghamlouch H, Della-Valle V, Scourzic L, Quivoron C, Roos-Weil D, et al. B-Cell Tumor Development in Tet2-Deficient Mice. Blood Adv (2018) 2:703–14. doi: 10.1182/bloodadvances.2017014118
79. Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Béguelin W, Fontán L, et al. Tet2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-Cell Lymphomagenesis. Cancer Discov (2018) 8:1632–53. doi: 10.1158/2159-8290.CD-18-0657
80. Boice M, Salloum D, Mourcin F, Sanghvi V, Amin R, Oricchio E, et al. Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified Car-T Cells. Cell (2016) 167:405–18.e13. doi: 10.1016/j.cell.2016.08.032
81. Dheilly E, Battistello E, Katanayeva N, Sungalee S, Michaux J, Duns G, et al. Cathepsin S Regulates Antigen Processing and T Cell Activity in Non-Hodgkin Lymphoma. Cancer Cell (2020) 37:674–89. doi: 10.1016/j.ccell.2020.03.016
82. Muppidi JR, Schmitz R, Green JA, Xiao W, Larsen AB, Braun SE, et al. Loss of Signalling Via Gα13 in Germinal Centre B-Cell-Derived Lymphoma. Nature (2014) 516:254–8. doi: 10.1038/nature13765
83. Ortega-Molina A, Deleyto-Seldas N, Carreras J, Sanz A, Lebrero-Fernández C, Menéndez C, et al. Oncogenic Rag Gtpase Signalling Enhances B Cell Activation and Drives Follicular Lymphoma Sensitive to Pharmacological Inhibition of Mtor. Nat Metab (2019) 1:775–89. doi: 10.1038/s42255-019-0098-8
84. Oricchio E, Katanayeva N, Donaldson MC, Sungalee S, Pasion JP, Béguelin W, et al. Genetic and Epigenetic Inactivation of SESTRIN1 Controls mTORC1 and Response to EZH2 Inhibition in Follicular Lymphoma. Sci Transl Med (2017) 9:eaak9969. doi: 10.1126/scitranslmed.aak9969
85. Roulland S, Navarro J-M, Grenot P, Milili M, Agopian J, Montpellier B, et al. Follicular Lymphoma-Like B Cells in Healthy Individuals: A Novel Intermediate Step in Early Lymphomagenesis. J Exp Med (2006) 203:2425–31. doi: 10.1084/jem.20061292
86. Roulland S, Faroudi M, Mamessier E, Sungalee S, Salles G, Nadel B. Chapter 1 - Early Steps of Follicular Lymphoma Pathogenesis. In: FW Alt, editor. Advances in Immunology. Academic Press (2011). 111:1–46. doi: 10.1016/B978-0-12-385991-4.00001-5
87. Brescia P, Schneider C, Holmes AB, Shen Q, Hussein S, Pasqualucci L, et al. Mef2b Instructs Germinal Center Development and Acts as an Oncogene in B Cell Lymphomagenesis. Cancer Cell (2018) 34:453–65. doi: 10.1016/j.ccell.2018.08.006
88. Kridel R, Chan FC, Mottok A, Boyle M, Farinha P, Tan K, et al. Histological Transformation and Progression in Follicular Lymphoma: A Clonal Evolution Study. PloS Med (2016) 13:e1002197. doi: 10.1371/journal.pmed.1002197
89. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic Mutations Altering EZH2 (Tyr641) in Follicular and Diffuse Large B-Cell Lymphomas of Germinal-Center Origin. Nat Genet (2010) 42:181–5. doi: 10.1038/ng.518
90. Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent Mutation of Histone-Modifying Genes in Non-Hodgkin Lymphoma. Nature (2011) 476:298–303. doi: 10.1038/nature10351
91. Schmidt J, Ramis-Zaldivar JE, Bonzheim I, Steinhilber J, Müller I, Haake A, et al. Crebbp Gene Mutations Are Frequently Detected in In Situ Follicular Neoplasia. Blood (2018) 132:2687–90. doi: 10.1182/blood-2018-03-837039
92. Casola S, Cattoretti G, Uyttersprot N, Koralov SB, Seagal J, Segal J, et al. Tracking Germinal Center B Cells Expressing Germ-Line Immunoglobulin Gamma1 Transcripts by Conditional Gene Targeting. Proc Natl Acad Sci USA (2006) 103:7396–401. doi: 10.1073/pnas.0602353103
93. Horton SJ, Giotopoulos G, Yun H, Vohra S, Sheppard O, Bashford-Rogers R, et al. Early Loss of Crebbp Confers Malignant Stem Cell Properties on Lymphoid Progenitors. Nat Cell Biol (2017) 19:1093–104. doi: 10.1038/ncb3597
94. Meyer SN, Scuoppo C, Vlasevska S, Bal E, Holmes AB, Holloman M, et al. Unique and Shared Epigenetic Programs of the CREBBP and EP300 Acetyltransferases in Germinal Center B Cells Reveal Targetable Dependencies in Lymphoma. Immunity (2019) 51:S1074761319303322. doi: 10.1016/j.immuni.2019.08.006
95. Hashwah H, Schmid CA, Kasser S, Bertram K, Stelling A, Manz MG, et al. Inactivation of CREBBP Expands the Germinal Center B Cell Compartment, Down-Regulates MHCII Expression and Promotes Dlbcl Growth. Proc Natl Acad Sci USA (2017) 114:9701–6. doi: 10.1073/pnas.1619555114
96. Mondello P, Tadros S, Teater M, Fontan L, Chang AY, Jain N, et al. Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma. Cancer Discov (2020) 10:CD–19-0116. doi: 10.1158/2159-8290.CD-19-0116
97. McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, et al. Mutation of A677 in Histone Methyltransferase EZH2 in Human B-Cell Lymphoma Promotes Hypertrimethylation of Histone H3 on Lysine 27 (H3k27). Proc Natl Acad Sci USA (2012) 109:2989–94. doi: 10.1073/pnas.1116418109
98. Bödör C, Grossmann V, Popov N, Okosun J, O’Riain C, Tan K, et al. Ezh2 Mutations Are Frequent and Represent an Early Event in Follicular Lymphoma. Blood (2013) 122:3165–8. doi: 10.1182/blood-2013-04-496893
99. Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain P-L, et al. Germinal Center Dysregulation by Histone Methyltransferase EZH2 Promotes Lymphomagenesis. J Clin Invest (2013) 123:5009–22. doi: 10.1172/JCI70626
100. Béguelin W, Rivas MA, Calvo Fernández MT, Teater M, Purwada A, Redmond D, et al. Ezh2 Enables Germinal Centre Formation Through Epigenetic Silencing of CDKN1A and an Rb-E2F1 Feedback Loop. Nat Commun (2017) 8:877. doi: 10.1038/s41467-017-01029-x
101. Béguelin W, Teater M, Meydan C, Hoehn KB, Phillip JM, Soshnev AA, et al. Mutant EZH2 Induces a Pre-Malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell (2020) 37:655–73.e11. doi: 10.1016/j.ccell.2020.04.004
102. Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S, et al. Mutations in Linker Histone Genes Hist1h1 B, C, D, and E; Oct2 (Pou2f2); IRF8; and ARID1A Underlying the Pathogenesis of Follicular Lymphoma. Blood (2014) 123:1487–98. doi: 10.1182/blood-2013-05-500264
103. Yusufova N, Kloetgen A, Teater M, Osunsade A, Camarillo JM, Chin CR, et al. Histone H1 Loss Drives Lymphoma by Disrupting 3d Chromatin Architecture. Nature (2021) 589:299–305. doi: 10.1038/s41586-020-3017-y
104. Fan Y, Nikitina T, Morin-Kensicki EM, Zhao J, Magnuson TR, Woodcock CL, et al. H1 Linker Histones Are Essential for Mouse Development and Affect Nucleosome Spacing In Vivo. Mol Cell Biol (2003) 23:4559–72. doi: 10.1128/mcb.23.13.4559-4572.2003
105. Duy C, Béguelin W, Melnick A. Epigenetic Mechanisms in Leukemias and Lymphomas. Cold Spring Harb Perspect Med (2020) 10:a034959. doi: 10.1101/cshperspect.a034959
106. Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. Tet2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event During Human Lymphomagenesis. Cancer Cell (2011) 20:25–38. doi: 10.1016/j.ccr.2011.06.003
107. Mintz MA, Cyster JG. T Follicular Helper Cells in Germinal Center B Cell Selection and Lymphomagenesis. Immunol Rev (2020) 296:48–61. doi: 10.1111/imr.12860
108. Mintz MA, Felce JH, Chou MY, Mayya V, Xu Y, Shui J-W, et al. The HVEM-BTLA Axis Restrains T Cell Help to Germinal Center B Cells and Functions as a Cell-Extrinsic Suppressor in Lymphomagenesis. Immunity (2019) 51:S1074761319302420. doi: 10.1016/j.immuni.2019.05.022
109. Bararia D, Hildebrand JA, Stolz S, Haebe S, Alig S, Trevisani CP, et al. Cathepsin S Alterations Induce a Tumor-Promoting Immune Microenvironment in Follicular Lymphoma. Cell Rep (2020) 3:107522. doi: 10.1016/j.celrep.2020.107522
110. Lu E, Cyster JG. G-Protein Coupled Receptors and Ligands That Organize Humoral Immune Responses. Immunol Rev (2019) 289:158–72. doi: 10.1111/imr.12743
111. Ersching J, Efeyan A, Mesin L, Jacobsen JT, Pasqual G, Grabiner BC, et al. Germinal Center Selection and Affinity Maturation Require Dynamic Regulation of mTORC1 Kinase. Immunity (2017) 46:1045–58.e6. doi: 10.1016/j.immuni.2017.06.005
112. Okosun J, Wolfson RL, Wang J, Araf S, Wilkins L, Castellano BM, et al. Recurrent mTORC1-Activating Rragc Mutations in Follicular Lymphoma. Nat Genet (2016) 48:183–8. doi: 10.1038/ng.3473
113. Ying ZX, Jin M, Peterson LF, Bernard D, Saiya-Cork K, Yildiz M, et al. Recurrent Mutations in the MTOR Regulator RRAGC in Follicular Lymphoma. Clin Cancer Res (2016) 22:5383–93. doi: 10.1158/1078-0432.CCR-16-0609
114. Zoncu R, Efeyan A, Sabatini DM. Mtor: From Growth Signal Integration to Cancer, Diabetes and Ageing. Nat Rev Mol Cell Biol (2011) 12:21–35. doi: 10.1038/nrm3025
115. Rebsamen M, Pochini L, Stasyk T, de Araújo MEG, Galluccio M, Kandasamy RK, et al. SLC38A9 Is a Component of the Lysosomal Amino Acid Sensing Machinery That Controls Mtorc1. Nature (2015) 519:477–81. doi: 10.1038/nature14107
116. Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag Complex Targets mTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell (2010) 141:290–303. doi: 10.1016/j.cell.2010.02.024
117. Young RM, Phelan JD, Wilson WH, Staudt LM. Pathogenic B-Cell Receptor Signaling in Lymphoid Malignancies: New Insights to Improve Treatment. Immunol Rev (2019) 291:190–213. doi: 10.1111/imr.12792
118. Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma With Therapeutic Implications. Cancer Cell (2020) 37:551–68. doi: 10.1016/j.ccell.2020.03.015
119. Kotlov N, Bagaev A, Revuelta MV, Phillip JM, Cacciapuoti MT, Antysheva Z, et al. Clinical and Biological Subtypes of B-Cell Lymphoma Revealed by Microenvironmental Signatures. Cancer Discov (2021) 11:candisc.0839.2020. doi: 10.1158/2159-8290.CD-20-0839
120. Andor N, Simonds EF, Czerwinski DK, Chen J, Grimes SM, Wood-Bouwens C, et al. Single-Cell RNA-Seq of Follicular Lymphoma Reveals Malignant B-Cell Types and Coexpression of T-Cell Immune Checkpoints. Blood (2019) 133:1119–29. doi: 10.1182/blood-2018-08-862292
121. Brisou G, Zala M, Gil L, Pagano G, Bru A, Cervera-Marzal I, et al. Desynchronization of the Germinal Center Dynamics and Remodeling of the Tumor Microenvironment Characterize Kmt2d-Driven Lymphomagenesis. Blood (2018) 132:670–0. doi: 10.1182/blood-2018-99-116617
122. Schroers-Martin J. Recurrent Crebbp Mutations in Follicular Lymphoma Appear Localized to the Committed B-Cell Lineage. In (ASH). Available at: https://ash.confex.com/ash/2020/webprogram/Paper142761.html (Accessed March 21, 2021).
123. Townsend EC, Murakami MA, Christodoulou A, Christie AL, Köster J, DeSouza TA, et al. The Public Repository of Xenografts Enables Discovery and Randomized Phase II-Like Trials in Mice. Cancer Cell (2016) 29:574–86. doi: 10.1016/j.ccell.2016.03.008
124. Tothova Z, Krill-Burger JM, Popova KD, Landers CC, Sievers QL, Yudovich D, et al. Multiplex CRISPR/Cas9-Based Genome Editing in Human Hematopoietic Stem Cells Models Clonal Hematopoiesis and Myeloid Neoplasia. Cell Stem Cell (2017) 21:547–55. doi: 10.1016/j.stem.2017.07.015
Keywords: germinal center (GC), follicular lymphoma (FL), diffuse large B cell lymphoma (DLBCL), genetically engineered mouse (GEMs), epigenetic modifier mutations
Citation: Mossadegh-Keller N, Brisou G, Beyou A, Nadel B and Roulland S (2021) Human B Lymphomas Reveal Their Secrets Through Genetic Mouse Models. Front. Immunol. 12:683597. doi: 10.3389/fimmu.2021.683597
Received: 21 March 2021; Accepted: 12 May 2021;
Published: 16 July 2021.
Edited by:
Juan M. Zapata, Instituto de Investigaciones Biomédicas Alberto Sols, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Parham Ramezani-Rad, Sanford Burnham Prebys Medical Discovery Institute, United StatesMiguel Gallardo, Spanish National Cancer Research Center (CNIO), Spain
Copyright © 2021 Mossadegh-Keller, Brisou, Beyou, Nadel and Roulland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandrine Roulland, cm91bGxhbmRAY2ltbC51bml2LW1ycy5mcg==
†These authors have contributed equally to this work