 Abhimanyu1†
Abhimanyu1† Carlos O. Ontiveros2,3†
Carlos O. Ontiveros2,3† Rosa S. Guerra-Resendez4Tomoki Nishiguchi1Malik Ladki1
Rosa S. Guerra-Resendez4Tomoki Nishiguchi1Malik Ladki1 Isaac B. Hilton4,5,6
Isaac B. Hilton4,5,6 Larry S. Schlesinger2*‡
Larry S. Schlesinger2*‡ Andrew R. DiNardo1*‡
Andrew R. DiNardo1*‡- 1The Global Tuberculosis Program, William T. Shearer Center for Human Immunobiology, Texas Children’s Hospital, Immigrant and Global Health, Baylor College of Medicine, Houston, TX, United States
- 2Host-Pathogen Interactions Program, Texas Biomedical Research Institute, San Antonio, TX, United States
- 3 UT Health San Antonio, San Antonio, TX, United States
- 4Systems, Synthetic, and Physical Biology Graduate Program, Rice University, Houston, TX, United States
- 5Department of Bioengineering, Rice University, Houston, TX, United States
- 6Department of BioSciences, Rice University, Houston, TX, United States
The immune response must balance the pro-inflammatory, cell-mediated cytotoxicity with the anti-inflammatory and wound repair response. Epigenetic mechanisms mediate this balance and limit host immunity from inducing exuberant collateral damage to host tissue after severe and chronic infections. However, following treatment for these infections, including sepsis, pneumonia, hepatitis B, hepatitis C, HIV, tuberculosis (TB) or schistosomiasis, detrimental epigenetic scars persist, and result in long-lasting immune suppression. This is hypothesized to be one of the contributing mechanisms explaining why survivors of infection have increased all-cause mortality and increased rates of unrelated secondary infections. The mechanisms that induce epigenetic-mediated immune suppression have been demonstrated in-vitro and in animal models. Modulation of the AMP-activated protein kinase (AMPK)-mammalian target of rapamycin (mTOR), nuclear factor of activated T cells (NFAT) or nuclear receptor (NR4A) pathways is able to block or reverse the development of detrimental epigenetic scars. Similarly, drugs that directly modify epigenetic enzymes, such as those that inhibit histone deacetylases (HDAC) inhibitors, DNA hypomethylating agents or modifiers of the Nucleosome Remodeling and DNA methylation (NuRD) complex or Polycomb Repressive Complex (PRC) have demonstrated capacity to restore host immunity in the setting of cancer-, LCMV- or murine sepsis-induced epigenetic-mediated immune suppression. A third clinically feasible strategy for reversing detrimental epigenetic scars includes bioengineering approaches to either directly reverse the detrimental epigenetic marks or to modify the epigenetic enzymes or transcription factors that induce detrimental epigenetic scars. Each of these approaches, alone or in combination, have ablated or reversed detrimental epigenetic marks in in-vitro or in animal models; translational studies are now required to evaluate clinical applicability.
Introduction
Epigenetic mechanisms guide gene expression to maintain homeostasis by balancing the nature of expressed and non-expressed genes. This balance can be perturbed either by pathogen- induced epigenetic changes, such as through Rv1998 antigen secreted by Mycobacterium tuberculosis (Mtb) (1) or by chronic and severe stimulation of the immune system as in case of LCMV (2), HCV (3), sepsis (4), Schistosomiasis (5) and TB (6). Long-lasting immune suppression that follows severe or chronic infections increases the risk for secondary infections. This was recognized in 1909 when German researchers noted that TB recurrence occurred after measles (7). In the 1950s, clinicians reported an increased risk for histoplasmosis reactivation among patients recovering from TB (8). Similarly, after surviving sepsis, host immunity remains in a suppressed state that increases the risk for secondary bacterial infections and doubles mortality risk (9, 10). Survivors of pneumonia have increased risk of death with the severity of pneumonia correlated with mortality risk (11). TB survivors also have increased risk of mortality, not only from secondary infections and recurrent TB, but from increased risk of cardiovascular disease and cancer (12, 13). Although epigenetic immune suppression is needed acutely to temper exuberant immunity (14), these immunosuppressive epigenetic marks are long-lived and are thought to be a major contributing factor for increased secondary infections long after resolution of the first insult (12, 15–17). Proper epidemiological studies matched with translational studies need to be conducted, but epigenetic-mediated post-infectious myeloid and lymphoid immune suppression is a suspected explanation for why individuals retain increased mortality risks even after successful treatment for pneumonia, TB or sepsis (11, 13, 15).
After chronic and severe infections, CD4+ T cells are characterized as being anergic and CD8+ cells as being exhausted (18, 19). Functionally, anergic CD4+ T cells fail to recognize and respond to foreign antigen, as measured by decreased antigen-induced cellular proliferation and cytokine production (18). Similarly, CD8+ T cell immune exhaustion is defined by decreased antigen-induced proliferation, cytokine production and an increase in immune checkpoint inhibitors (19). Myeloid cell immune tolerance is defined by a decreased responsiveness, usually measured by decreased phagocytic capacity, killing capacity and cytokine production, e.g., TNF, IL-6 and IL-1β (10, 20–22). Animal models have demonstrated that post-infectious immune suppression is epigenetically mediated and that the detrimental epigenetic marks induced by chronic infections overlap with those induced by cancer (5, 6, 23–30). There are many studies demonstrating how cancer induced epigenetic-mediated immune suppression can be reversed. Herein, we review the growing literature of in-vitro and animal model studies demonstrating how to block or reverse infection induced epigenetic-mediated immune suppression and postulate how these approaches could become clinically relevant to decrease post-infectious morbidity and mortality.
Epigenetic Mechanisms and Gene Expression
Epigenetic mechanisms are one major means of regulating gene expression. This regulation comes from nucleosomal scaffolding of the negatively charged DNA around positively charged proteins, called histones, present as two functional copies apiece of the type H2A, H2B, H3 and H4. Each nucleosome is further condensed in a higher-order structure, the chromatin. Both nucleosome and chromatin can guide accessibility of molecular factors to the DNA, thus resulting in differential gene expression (31). Cells can either circumvent or reinforce these barriers, depending on the context, by dynamically modifying DNA and histones at specific nucleotide or amino acid residues, transiently creating regions of the genome differentially accessible to gene expression machinery. Histones are modified on their free N-terminal tails, or their globular domains that physically interact with the DNA, through chemical modifications including acetylation, methylation, phosphorylation, ubiquitylation, acylation, hydroxylation, glycation, serotonylation, glycosylation, sumoylation and ADP-ribosylation (32). DNA is methylated at cytosine and adenine residues. Epigenetic marks other than acetylation and methylation are not as well studied and are less understood. The gene expression implications of certain epigenetic marks are well established. DNA methylation directly interferes with the binding of DNA and transcription factors, or it can attract proteins that bind specifically to modify DNA, thereby blocking other transcription factors from binding the site (33). Acetylation of histones H3 and H4 relaxes the nucleosome compactness and leads to partial de-condensation of chromatin locally, making the DNA more accessible. Such accessibility is referred to as “permissive” and the loss of accessibility and increased compactness referred to as “restrictive” (34). Histone modifications can be either permissive or restrictive. For example, trimethylation of histone 3 at lysine 4 (H3K4me1, H3K4me3) promotes open chromatin, while trimethylation of histone 3 at lysine 27 (H3K27me3) and at lysine 9 (H3K9me3) promotes restrictive heterochromatin (35).
Signaling Pathways That Induce T Cell Immune Exhaustion
Broadly, T-cell activation involves tightly controlled signaling pathways and cascades, that when perturbed lead to transcription factor (TF) imbalances that then drive epigenetic-mediated gene expression inhibition. Activation of the T-cell receptor (TCR) by MHC-antigen complexes assembles the “TCR signalosome” that results in downstream phosphorylation events and activation of secondary signaling molecules (18, 36–40). Downstream of the TCR, key events include phosphorylation of tyrosine kinases and phospholipase C (PLC)1 (41–43). Activated PLCγ1 cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) to diacylglycerol (DAG) and inositol 1,4,5-triphosphate (IP3), the former being critical for proper activation of activator protein-1 (AP-1) complex, a heterodimer of c-Fos and c-Jun (Figure 1) (44–47). IP3 plays a central role in calcium signaling by releasing intracellular calcium stores from the endoplasmic reticulum (ER) (48) and thereby dephosphorylating NFAT proteins which translocate to the nucleus, and bind with their transcriptional partners, such as AP-1, to activate distinct ranscriptional programs (Figure 1) (49). NFAT/AP-1 transcriptional complexes bind to the promoters of various cytokine genes, including Il2, leading to their active transcription. In the absence of co-stimulation, impaired AP-1 activation results in NFAT homodimerization inducing transcriptional and epigenetic changes that yield anergic and exhausted T cells (Figure 1) (18, 44, 50, 51).
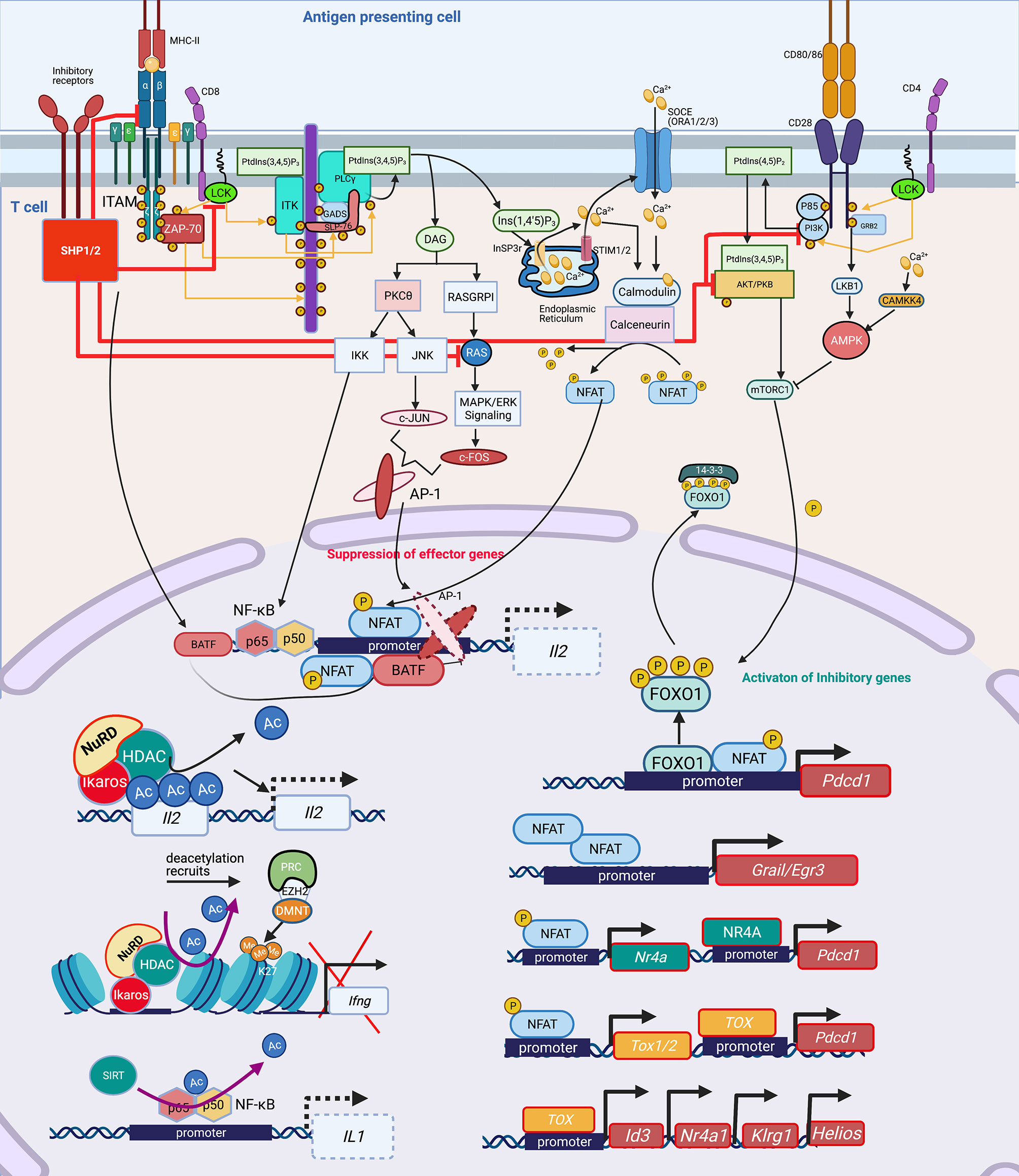
Figure 1 Signaling cascade and transcription factors that mediate epigenetic changes that inhibit host immunity. In T cells, protein kinase Lck and ZAP-70 initiate a signaling cascade that result in activation of PLCγ1 and production of InsP3 (IP3), a second messenger, binding to the InSP3 receptor on the ER leading to release of Ca2+ from the ER. The reduction of Ca2+ activates STIM, which recruits SOCE such as ORAI in the plasma membrane. Opening of ORAI channels in the plasma membrane results in sustained Ca2+ influx and activation of several Ca2+ regulated enzymes such as serine/threonine phosphatase calcineurin, which dephosphorylates NFAT enabling its translocation to the nucleus where it binds to promoters of effector genes including Il2. NFAT requires AP-1 generated through another second messenger DAG activation of PKCΦ and RAS/MAPK/ERK1 pathways. Lck also mediates activation of PI3K activating AKT and mTOR, which govern the phosphorylation of FOXO1. Phosphorylated FOXO1 is transported out of the nucleus and exists in complex with 14-3-3 in the cytoplasm. In exhausted T cells, either through activation of inhibitory receptors such as PD-1, CTL4, a dephosphorylating protein SHP1/2 is activated, which dephosphorylates Lck and ZAP70, suppressing the subsequent signaling cascades. SHP2 inhibits among others RAS, AKT, PI3K and even the TCR-MHCII microcluster, thus weakening or abrogating the effector signals at multiple levels (red inhibition arrows). This leads to widespread change in the cellular TF landscape. SHP1/2 activate BATF3, a TF, due to non-availability of AP-1 to partner with NFAT. Partnerless NFAT, alone leads to transcription of inhibitory genes and receptors including pdcd1, which is also transcribed by increased nuclear retention of unphosphorylated FOXO1 in the nucleus, in absence of a PI3K/AKT/mTOR activation. Unpartnered NFAT transcribes, Nr4A and TOX1/2, which further contribute to inhibitory signaling by increasing transcription of Pdcd1. NFAT homodimer transcribes inhibitory genes Grail3/Erg3. TOX leads to transcription of genes such as Id3, Nr4a1, Klrg1, Helios. Many of these genes and TF lead to epigenetic modifications, which further contribute to exhausted phenotype. TF, Ikaros (Helios family) can directly bind to the Il2 promoter and recruit NuRD, which has HDAC and deacetylates Il2 leading to its transcriptional repression. Deacetylation is usually followed by recruitment of PRC, which through EZH2 can further add to closing of chromatin by adding methylation marks at H3K27, as seen at the Ifng locus. Another, NAD: NADH+ dependent deacetylase, SIRT can directly deacetylate NF-κB to decreases IL1 transcription. Lck, LCK proto-oncogene, Src family tyrosine kinase; ZAP-70, zeta chain of T cell receptor associated protein kinase 70; PLC, Phospholipase C; IP3/InsP3, inositol 1,4,5-trisphosphate; ER, Endoplasmic reticulum; STIM, stromal interaction molecule 1; SOCE, Store-operated calcium entry; ORAI, ORAI calcium release-activated calcium modulator; NFAT, Nuclear factor of activated T-cells; IL, Interleukin; AP-1, Activator protein1; DAG, Di-Acyl Glycerol; PKCΦ, Protein kinase C; MAPK, Mitogen-Activated Protein Kinase; FOXO1, Forkhead box protein O1; 14-3-3, tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein theta (encoded by YWHAQ); PD-1/Pdcd, programmed cell death 1; CTL4, Cytotoxic T-Lymphocyte Associated Protein 4; SHP, Src homology 2 domain-containing tyrosine phosphatase 2; PI3K, phosphatidylinositol 3-kinase; AKT, Protein kinase B; mTOR, mammalian target of rapamycin; MHC, Major Histocompatibility complex; TF, Transcription factors; BATF, Basic Leucine Zipper ATF-Like Transcription Factor; Nr4A, Nuclear Receptor Subfamily 4 Group A Member 1; TOX, Thymocyte Selection Associated High Mobility Group Box; Erg, ETS transcription factor ERG; Id3, Inhibitor Of DNA Binding 3; Klrg1, Killer Cell Lectin Like Receptor G1; NuRD, Nucleosome and DNA Remodeling complex; PRC, Polycomb Repressive Complex; EZH2, Enhancer Of Zeste 2 Polycomb Repressive Complex 2 Subunit; H3K27me3, H3 lysine 27 trimethylation; Ac, Acetylation; Me, Methylation. Created with BioRender.com.
Activation of the TCR and CD28 co-stimulatory induces a flux of intracellular Ca2+ and activation of the PI3K-AMPK-mTOR signaling pathway. Activated mTOR engages several downstream effector pathways, including promoting metabolism by activating gene expression of the TFs hypoxia-inducible factor 1α (HIF1α), MYC and sterol regulatory element-binding protein (SREBP). Upregulation of inhibitory receptor signaling recruits Src homology 2 domain-containing tyrosine phosphatase 2 (SHP2) phosphatase which interferes with CD28 costimulatory signaling by blocking PKC-θ (52) and PLCγ1 (53). Increased programmed death protein 1 (PD-1) inhibits AKT and mTOR pathways (54), activates Basic leucine transcription factor (BATF) to repress T-cell proliferation and cytokine secretion in HIV-specific CD8+ T cells (55), and inhibits IL-2 production to limit T-cell proliferation (52). NFAT1 in the absence of AP-1 interaction promotes the expression of Pdcd1 (PD-1 encoding gene) (56). Inactivation of the AKT/mTOR pathway promotes FOXO1 retention in the nucleus to enable continued inhibitory receptor Pdcd1 gene transactivation (57). PD-1 signaling through SHP2 activates AMPK, which is an inhibitor of mTOR signaling (already abrogated by inactivation of PI3K and AKT), leading to downregulation of HIF-1α and MYC, which in turn governs the transcription of the glycolytic enzymes such as GLUT1, thereby decreasing cellular metabolism.
Transcription Factors Driving T Cell Immune Exhaustion
NFAT homodimers play a critical role in induction of the T cell anergy transcriptional program (44). NFAT1 homodimers bind to specific NFAT binding sites on T cell anergy-associated gene promoters. For example, NFAT homodimer consensus binding sites are present in the promoter of Grail, a T cell anergy-associated gene (50). Expression of the early growth response gene 2 (Egr2) and Egr3 is NFAT-dependent, and these TFs are associated with regulation of gene expression of the Casitas B-lineage lymphoma b (Cbl-b) E3 ubiquitin ligase in anergic T cells (58). Downstream of NFAT signaling, the TF Ikaros, binds to the IL2 gene locus, and recruits the NuRD complex, including histone deacetylases (HDACs), thereby facilitating epigenetic remodeling, specifically histone deacetylation, of the Il2 promoter, thus effectively silencing gene expression (59, 60).
Highlighting the critical importance of a balanced NFAT response, a bioengineered constitutively active form of NFAT, termed CA-RIT-NFAT1, closes chromatin conformation inducing epigenetic-mediated immune exhaustion, including decreased microbial killing capacity in CD8+ T cells (56, 61). Constitutively active NFAT1 leads to the enrichment of genes belonging to the nuclear receptor (NR) family of genes, specifically members of the NR4A family. In particular, NR4A2 (NURR1) and NR4A3 (NOR1) exhibit high enrichment upon CA-RIT-NFAT1 expression. NR4A family member genes exhibit greater chromatin accessibility in exhausted tumor infiltrating lymphocytes (62). Using a chimeric antigen receptor T cell (CAR T cell) model, a NR4A triple knockout reversed detrimental chromatin accessibility, and promoted tumor regression and prolonged survival of tumor-bearing mice, thus illustrating the epigenetic and functional relevance of the NR4A family in T cell exhaustion. NR4A is also important for PD-1 and TIM3 expression, markers of T cell exhaustion (62).
NFAT homodimers also induce the thymocyte selection-associated high mobility group box (TOX) proteins which mediate the expression of inhibitory receptors such as PD-1 and TIM3, leading to the T cell exhaustion phenotype. Increased TOX expression occurs in chronic infection models such as LCMV and chronic hepatitis C (HCV) infection (51, 63). Removal of the nuclear localization sequence (NLS) and part of the DNA-binding domain from TOX via deletion of exon 5 resulted in decreased PD-1 expression and impaired generation of the T cell exhaustion phenotype. In addition, TOX exon 5 deletion resulted in differential expression of genes associated with T cell exhaustion such as Id3, Helios (Ikzf2), Nr4a1, Nr4a2, Pdcd1, and Klrg1. Conversely, over-expressing TOX in healthy T cells increases PD-1 expression, demonstrating a role in inducing the T cell exhaustion phenotype (63). Deletion of TOX exon 5 leads to decreased chromatin accessibility of the Pdcd1 gene locus, which encodes PD-1, and increased chromatin accessibility to Tnf (63). Knocking out Tox in CD8+ CAR tumor infiltrating lymphocytes (TILs) increase cytolytic activity further supporting the notion that TOX specifically attenuates CD8+ T cell effector function (Figure 1).
These studies provide evidence for the importance of the NFAT, TOX and NR4A TFs in driving epigenetic-mediated immune exhaustion, and also suggest strategies to alter their activation could be therapeutically pivotal (Figure 1). For example, Cyclosporin A (CsA), a calcineurin inhibitor, inhibits NFAT activation, thereby inhibiting TOX, NR4A1, NR4A2 and NR4A3 and the subsequent detrimental chromatin conformation changes that leads to immune exhaustion (51). Tacrolimus (binding to FK506) inhibits calcineurin by a different mechanism, but similarly decreases NFAT and TOX, blocking the chromatin confirmation changes that upregulate PD-1 and LAG3, thereby preserving capacity to produce TNF and IFN (64–66). Discussed in more detail below, bioengineered upregulation of c-Jun rescues NFAT-AP-1 imbalance, thereby restoring immune function. Therefore, while tacrolimus and CsA are considered immune suppressants, they can prevent detrimental chromatin conformation that leads to immune exhaustion, thereby preserving host immunity. Studies are needed to evaluate if these agents could be of benefit in humans following pneumonia, sepsis or TB.
Signaling Pathways That Induce Myeloid Immune Tolerance
For myeloid cells, the best studied model for immune tolerance is LPS challenge or sepsis. The exact myeloid tolerance mechanism(s) has not yet been elucidated for other infections. Thus, we discuss LPS/sepsis as a central theme for the signaling pathways inducing myeloid tolerance. Following overstimulation, myeloid cells, including monocytes and macrophages, develop tolerance, a state of cell refractoriness defined by an inability to mount an inflammatory response to a secondary stimulation (67). Pathogen- or danger-associated molecular patterns (PAMPs/DAMPs) such as LPS are sensed via pattern recognition receptors (PRR), e.g., toll-like receptors (TLR). LPS is recognized by TLR4, mediating signaling through two distinct adaptor pathways, myeloid differentiation factor 88 (MyD88) and TIR-domain containing adapter-inducing interferon (TRIF). The MyD88 pathway employs interleukin-1 receptor-associated kinase (IRAK)1 and 4 kinases and TNF receptor-associated factor (TRAF) 6 to activate NFκB and AP-1 signaling, promoting transcription of pro-inflammatory cytokines. Activation of TRIF pathway leads to janus kinase (JAK)/signal transducer and activator of transcription (STAT)1 and type I interferon activation and increases the expression of interferon-inducible genes such as TNFA, IFNB, IL1B, IL6, and COX2 (68, 69). PI3K interacts with MYD88 and also influences TLR4 signaling (70). LPS-induced myeloid tolerance involves downregulation of TLR4 expression, decreased recruitment of MyD88 or TRIF to TLR4, decreased activation of IRAK1/4 and diminished canonical NFκB signaling (p65/p50 heterodimer) via formation of inactive p50 homodimers (67, 71), decreased AP-1, reduced expression of TNFA, IL1B, IL6 and IL12B, and increased expression of IL10 and TGFB1 (Figure 2).
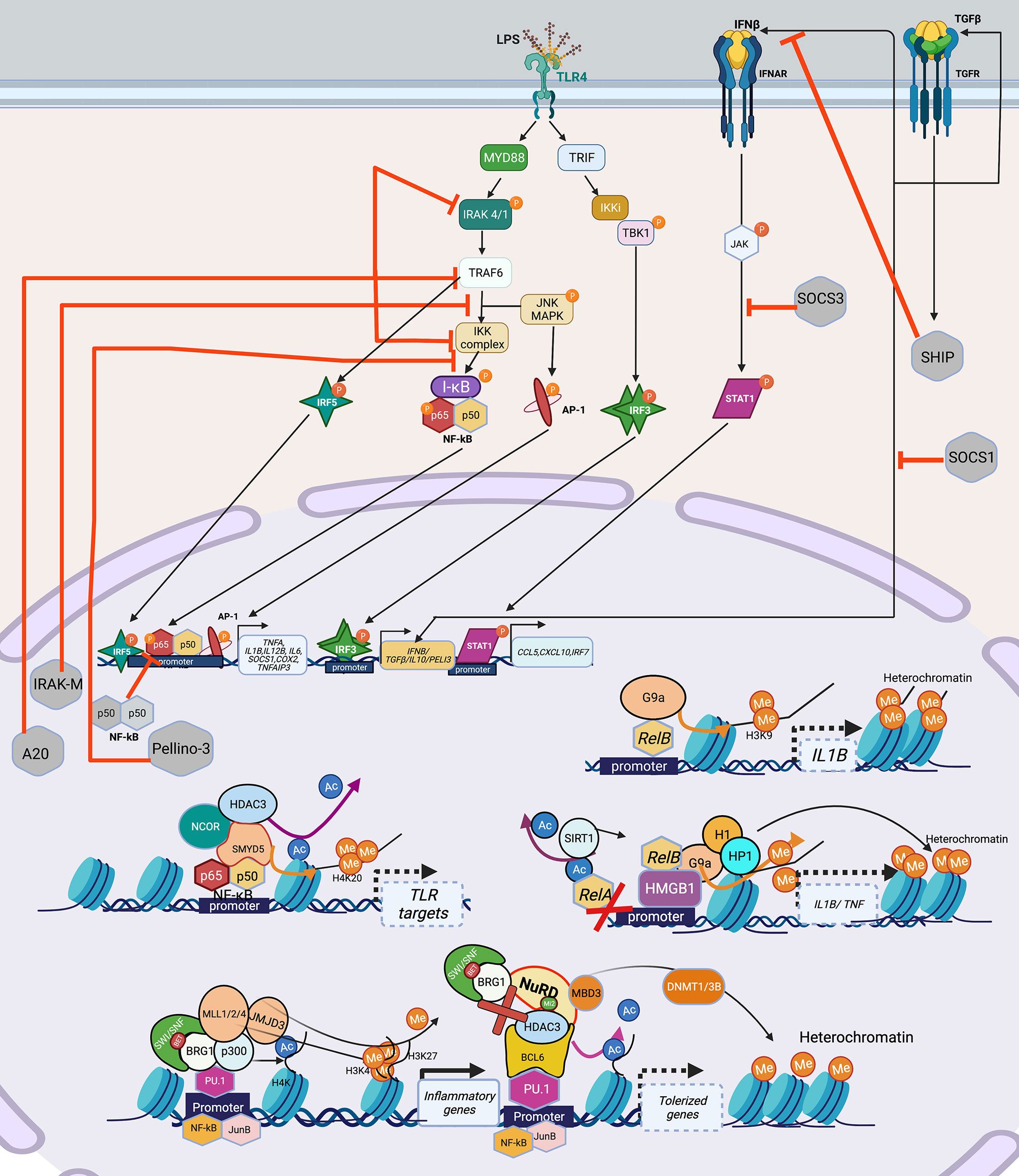
Figure 2 Signaling cascade and transcription factors that mediate epigenetic changes that inhibit host immunity in myeloid cells. TLR4 recognizes LPS, and engages the MyD88-TRIF pathway to induce the TFs: NF-κB, AP-1, IRF3, IRF5 which leads to the induction of pro-inflammatory genes such as TNFA, IL1B, IL6, COX2 etc. IRF3 induces the production of IFNβ and TGFβ, which adds to the IFN signaling and induces STAT1 leading to transcription of CCL5, CXCL10 and IRF7. Overwhelming LPS stimulation as seen in sepsis, leads to lesser production and engagement of TLR4 and its pathway components, with over-inflammation leading to production of inhibitory molecules such as IRAK-M, A20, Pellino-3, SHIP, which inhibit various parts of the LPS-TLR4 signaling cascade, leading to a tolerized phenotype. Epigenetically, multiple mechanisms have been shown to lead to and maintenance of the tolerized phenotype. Guided by TFs such as NF-κB and its isoform RelB, which can recruit HDACs (SIRT included) either alone or in a repressome complex, usually with a chromatin modifier such as SWI/SNF results in deacetylation of histones, followed by addition of repressive methylation (H3K9, H3K3) by DNMT such as SMYD5 (in the NCOR-HDAC3 repressome), or KMT such as G9a bound to HMGB1 (can recruit H1 and HP1) to close the chromatin and suppress gene expression. Lineage TFs such as PU.1 provide good example of this assembly of the SWI/SNF complex containing BRG1 which can recruit HAT (p300) to acetylate H4K, HMT (MLL1/2/3) to add permissive H3K4 and demethylase such as JMJD3 to remove repressive H3K27 to activate inflammatory genes upon LPS stimulation. The same PU.1 when bound to co-repressor BCL6 can induce tolerance by losing the SWI/SNF complex and recruitment of NuRD, which recruits HDAC3 to remove acetylation and induce de novo methylation via DNMT1/3B to close the chromatin and thus shutting down inflammatory gene transcription in tolerance. Created with BioRender.com. TLR4, Toll-like receptor 4; LPS, Bacterial Lipopolysaccharide; MyD88, myeloid differentiation factor 88; TRIF, TIR-domain containing adapter-inducing interferon; TF, Transcription factor; STAT1, signal transducer and activator of transcription; IRAK, interleukin-1 receptor-associated kinase; TRAF6, TNF Receptor Associated Factor 6; SHIP; SH2 domain-containing inositol phosphatase 1; IKK, Ikappa B Kinase; TBK, TANK-binding kinase 1; MAPK, Mitogen-Activated Protein Kinase; I-KB, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor; IRF, Interferon regulatory factors; SWI/SNF, SWItch/Sucrose Non-Fermentable; BRG1, Brahma-related gene-1; HDAC, Histone deacetylase; H1, H1.1 Linker Histone; HP1,Heterochromatin protein-1; HMGB1, High Mobility Group Box 1; DNMT, DNA methyltransferase; MLL, mixed lineage leukemia (lysine methyl transferase); JMJD, Jumonji domain containing protein; BCL6, B-cell lymphoma 6; MBD3, Methyl-CpG Binding Domain Protein 3.
LPS-tolerized myeloid cells are also characterized by negative regulatory molecules IRAK-M, A20, SH2 domain-containing inositol phosphatase 1 (SHIP1) (72), Pellino-3 (73), suppression of tumorigenicity 2 (ST2) (74), suppression of cytokine signaling (SOCS)1 and SOCS3 that inhibit TLR signaling (67, 69) (Figure 2). PI3K pathway, activated in LPS tolerance, also contributes to production of anti-inflammatory cytokines such as sIL-1RA (75) and its inhibition with wortmannin mitigates tolerance and increases TNF production (76). NFκB upregulates HDACs that remove histone acetyl marks and recruit the NuRD complex with the net result of a “repressome” such that euchromatin marks (e.g., histone acetylation) are removed and heterochromatin marks (e.g., DNA methylation and H3K9 and H3K27me) are induced (Figure 2) (20, 77–79). MyD88 activation, via non-coding RNAs, also contributes to decreased chromatin accessibility changes thereby inducing tolerance (80, 81). Acutely, tolerance is beneficial as studies have demonstrated that inhibiting post-sepsis epigenetic-mediated immune suppression too early exacerbates immune pathology (14).
Transcription Factors Driving Myeloid Immune Tolerance
Similar to the situation with immune exhaustion, myeloid cell immune tolerance is mediated by TFs that recruit co-activator and corepressor complexes that modify chromatin accessibility through post-translational modifications (Figure 2). The myeloid lineage defining transcription factor PU.1 facilitates chromatin opening with an increase in H3K4me3 at promoters and H3K4me1 at enhancers. However, in resting macrophages, corepressors such as B-cell lymphoma 6 (BCL-6) associate with PU.1 and recruit HDACs and histone demethylase resulting in repression of many LPS-inducible genes (82). In an analysis of LPS-induced tolerant and non-tolerant genes, NF-κB and MAPK were downregulated in tolerant macrophages (83). NFκB family TF isoform, RelB, mediates epigenetic silencing via facilitating the direct deposition of repressive histone marks by the H3 lysine methyltransferase (KMT) G9a at the IL1B promoter (78). Similarly, binding of high-mobility group box-1 protein (HMGB1) and histone H1 linker at the promoters of TNF and IL1B genes leads to transcription silencing by promoting assembly of RelB, which results in deposition of H3K9me2 mediated by the KMT G9a. Depletion of HMGB1 by siRNA results in dissociation of RelB from the promoter and partially restores TNF transcription (84).
Tolerized myeloid cells exhibit decreased chromatin accessibility due to decreased TLR-induced recruitment of the BRG1-containing SWI/SNF nucleosome remodeling complex and changes in histone acetylation and methylation (83). The NCoR-Hdac3-p50 repressome contains histone deacetylase and SET histone methyltransferases (SMYD5) that result in H3K9/14 deacetylation (77) and H4K20 methylation (85) respectively, both contributing to heterochromatin and repression of tolerizeable genes, thereby inhibiting the expression of genes downstream of TLR4 activation. Genetic disruption of the NcoR-Hdac3 interaction abolishes TLR4 tolerance (83). Interestingly, IFN-γ prevents tolerance by preserving expression of the receptor-interacting protein 140 (RIP140) coactivator and promoting TLR-induced chromatin accessibility upon secondary TLR challenge (86). In contrast, non-tolerized genes maintain an open chromatin state and exhibit more H4 acetylation and maintain H3K4me3 after re-stimulation (83). Interestingly, the NuRD complex acts antagonistically, and in a SWI/SNF-BRG1 dependent manner in LPS stimulated macrophages showing that these complexes exhibit concerted action to guide gene expression in myeloid cells (87) (Figure 2).
In summary, PU.1 facilitates myeloid gene transcription, while tolerance is associated with binding of co-repressor BCL-6 to PU.1, disruption of the NFκB active heterodimer and epigenetic silencing via HMGB1, RelB, NCoR-HDAC3-p50 repressome (Figure 2), increased SMYD5 and G9a methyltransferase and decreased chromatin accessibility due to reduced recruitment of BRG1-NRC. BET inhibitors (that bind to the bromodomain in the BRG1-NRC) such as IBET151, rescue tolerance in a preventative way when administered along with LPS, and not post LPS exposure (88).
Thus, HDAC inhibitors and G9a inhibitors if given after the resolution of acute infection, could potentially mitigate aspects of long-lived myeloid cell tolerance, while BET inhibitors act in a more preventative way (88).
Metabolic Mechanisms Leading to Immune Exhaustion and Tolerance
Upon immune activation via mTOR and NFAT signaling, shifts in cellular metabolism increase glycolysis, the tricarboxylic acid cycle (TCA, also known as the Krebs cycle) and electron transport chain (ETC), not only to meet high energy demands for proliferation and effector function, but also to produce the intermediate metabolites that fuel the biosynthesis of effector protein functions (10, 89–91). While initially beneficial, in severe or chronic infection, these metabolic shifts contribute to epigenetic changes that induce immune suppression (35, 89, 90, 92–95). In both lymphoid and myeloid cells, these metabolic shifts are mediated by the PI3K-Akt-mTOR pathway (96) and if the infection persists, the associated signaling cascades are downregulated and epigenetic mechanisms suppress host immunity, with loss of accessible chromatin that allows for expression of cytokines such as Tnf and Ifng and gain of chromatin accessible regions in inhibitory loci such as Pdcd1 (97, 98), thereby placing cells into an immune suppressed state (97). The described metabolic shifts induce epigenetic changes due to alteration in metabolic precursors required for epigenetic marks. At least three overlapping metabolic-epigenetic rheostats (Figure 3) have been identified that regulate host immunity (99–101).
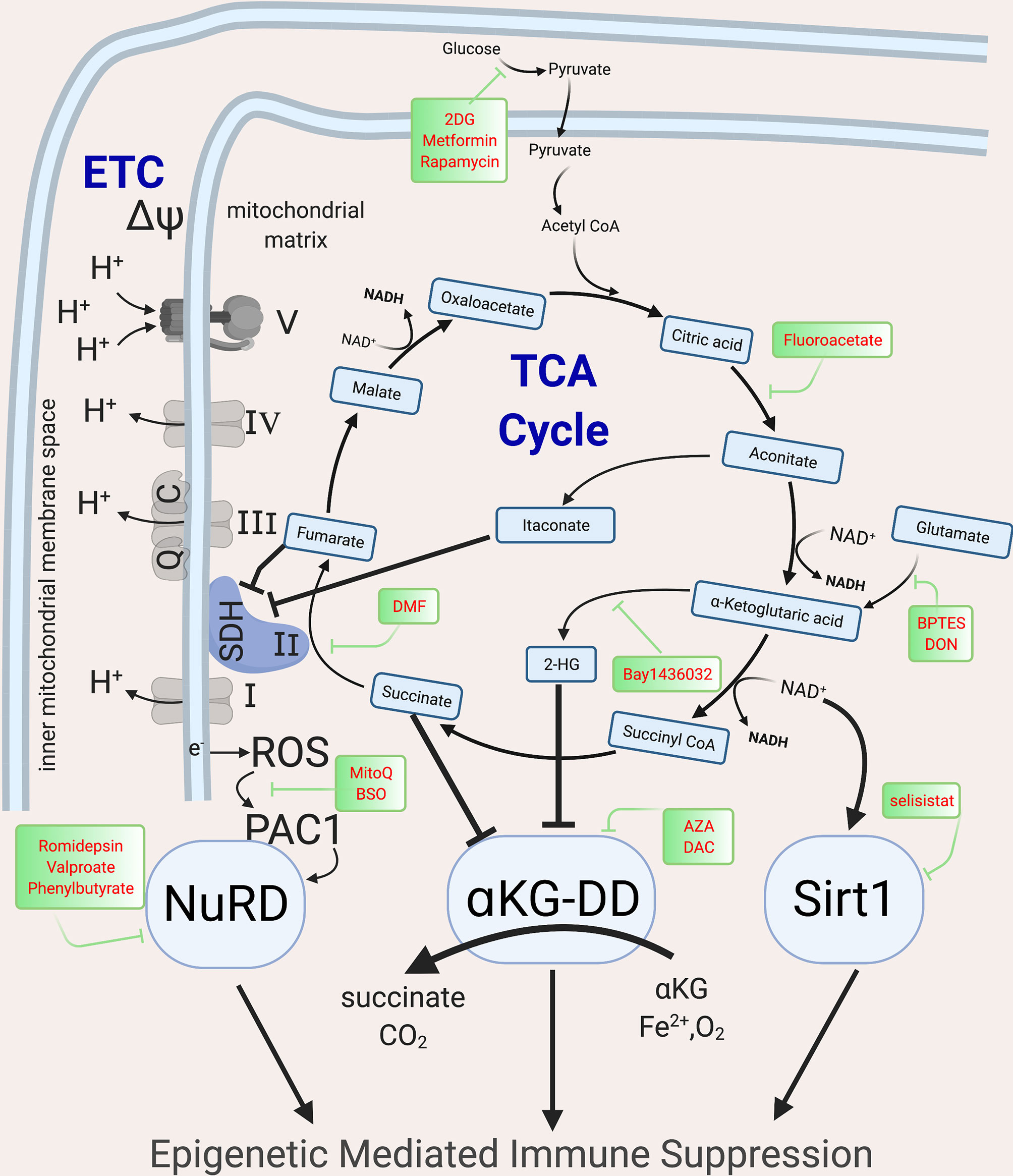
Figure 3 Metabolic intermediates of the TCA cycle guide epigenetic changes that inhibit host immunity. The TCA cycle metabolites act as co-factor for major epigenetic enzymes that shape the epigenomic landscape post infection via three overlapping and redundant major metabolic-epigenetic rheostats (RST). RST1: NAD+: NADH-SIRT: Dependent on the level of NAD+ in the cell, Sirtuins, which are histone deacetylases, can remove acetyl groups and lead to immune suppression; RST2: Succinate-αKG-αKGDD: Dependent on the levels of α−ketoglutarate and succinate (which along with fumarate, malate and Itaconate acts as inhibitors of KGDDs), leads to the activation/inhibition of a family of enzyme dioxygenases which regulate the DNA methylation levels by methylating (via DNMTs) and demethylate (via KDM, JMJD and TET) the DNA; RST3: ROS-PAC1-NuRD: guided by the activation of the ETC, which leads to electron leak and induction of ROS and activation of NuRD, which is multiprotein complex guiding DNA methylation and chromatin accessibility. The main enzymes of the TCA cycle and the ETC are shown, along with the drugs that can be used to block specific enzymes and help with reversing epigenetic mediated Immune suppression. NAD, Nicotinamide adenine dinucleotide; SIRT, Sirtuins; αKGDD, α−ketoglutarate-dependent-dioxygenases; DNMT, DNA methyl transferase; KDM, Histone demethylase; JMJD, Jumonji domain containing protein; ROS, reactive oxygen species; PAC1, Phosphatase of activated cells 1; NuRD, Nucleosome Remodeling and DNA methylation complex; TCA, Tri-carboxylic acid cycle; ETC, Electron transport chain. Created with BioRender.com.
Histone acetylation, an epigenetic mark characteristic of euchromatin is regulated by the availability of nicotinamide adenine dinucleotide (NAD+). High NAD+ levels and NAD+/NADH ratios induce the NAD-dependent deacetylase sirtuins (SIRTs) which deacetylate both histone and non-histone proteins. High dose LPS exposure, via upregulation of IDO1-induced de novo synthesis of NAD+, activates SIRT1, leading to histone deacetylation and gene silencing of proinflammatory genes such as Tnf and Il1b (102, 103). SIRT1 deacetylates p65 lysine of RelA (NF-κB) and nucleosomal H4K16 to terminate NF-κB dependent transcription and remains bound to assembled RelB and recruited transcriptional repressor complex (including heterochromatin linker H1) generating tolerance (102). NAD levels are regulated by CD38 and IDO1. CD38 levels correlate with T cell exhaustion (104, 105) and are elevated in patients with Cytomegalovirus (CMV) (106), Epstein-Barr-virus (EBV) (107), mycobacteria (108) and HIV, and are associated with poor prognosis (109). CD38, an extracellular and intracellular NADase, converts NAD+ molecules to a single cyclic ADP ribose thereby drastically shifting NAD+/NADH ratios, and activating sirtuin-mediated epigenetic mediated immune suppression (104). IDO, another mediator of immune suppression, is the rate limiting enzyme step in de novo NAD synthesis, converting tryptophan to kyneurine. IDO, elevated in sepsis and TB, inhibits host immunity by decreasing nuclear NAD+ concentrations, and initiating sirtuin activation (103, 110). Sirtuins regulate post-infectious immune suppression in both lymphoid and myeloid cells (102, 111), with inhibitors of CD38, IDO1 or SIRT1 able to restore host immunity and prevent mortality in animal models (104, 110, 112).
The second metabolic-epigenetic immune rheostat mechanism is guided by the balance of α-ketoglutarate (α-KG) and succinate. DNA methyltransferases (DNMT), lysine demethylase (KDM), jumonji domain-containing protein D3 (JMJD3) and Ten-eleven translocase (TET), require α-KG (also known as known as 2-oxoglutarate-2OG) as a co-substrate (113). Therefore, these epigenetic enzymes are known as α-KG or 2OG dependent dioxygenases (α-KG-DD) (113). Succinate, the end product of these chemical reactions, acts as a negative feedback loop to inhibit their function (114). In addition to succinate, other late-stage TCA metabolites including fumarate, malate, itaconate and 2-hydroxyglutarate (2HG) inhibit the α-KG-DD epigenetic enzymes (115–120). The importance of TCA metabolites in epigenetic regulation was first described in cancer where mutations in IDH, succinate dehydrogenase (SDH) or fumarate hydratase were found to induce global epigenetic disturbances (121, 122). These mutations lead to TCA metabolite imbalances that drive global DNA and histone hyper-methylation and immune tolerance (123–126). While originally described in cancer, studies in wild type mouse and human healthy T cells demonstrate that, upon immune activation, 2HG is increased via Von Hippel-Lindau (VHL)-HIF1α (120). Initially, 2HG increases T cell IL-2 production, but when 2HG elevations persist, there are global increases in the inhibitory epigenetic mark H3K27me3 with suppression of T cell cytotoxic function (120). Dimethyl fumarate (DMF), an immune suppressive therapy for multiple sclerosis, induces DNA methylation and heterochromatin in monocytes and T cells to suppress exuberant immunity (127, 128).The timing and duration of TCA metabolite shifts need to be further explored as short term shifts induce immune beneficial immunity, while others are immune suppressive (118, 120, 129, 130).
The known metabolic-epigenetic immune rheostats are overlapping and redundant, as demonstrated by the third known metabolic-epigenetic rheostat. SDH is unique in that it is both part of the TCA cycle and the ETC. Upon immune activation, the increase in glycolysis fuels the ETC and when persistent, electrons leak out of the inner mitochondrial space (Figure 3), increasing reactive oxygen species (ROS) in the mitochondrial matrix. This increase in ROS triggers phosphatase of activated cells 1 (PAC1, encoded by DUSP2, dual specificity protein phosphatase 2) and nuclear factor erythroid 2-related factor (NRF2, encoded by NFE2L2) to activate the NuRD complex (131, 132). The NuRD induces histone deacetylation and DNA hypermethylation (via MBD2/3) and is instrumental in limiting exuberant immune pathology in macrophages after sepsis and preventing T cell autoimmunity (133). Mice with tumor-induced immune exhaustion demonstrate elevated mitochondrial ROS that correlates with detrimental epigenetic marks (DNA hypermethylation and closed chromatin conformation) and immune suppression (134). Inhibiting mitochondrial ROS accumulation ablates immune suppression (92, 135), however to date, the direct link via the NuRD has not been demonstrated.
The immune inhibitory effects due to itaconate further demonstrate the overlap of mechanisms by which metabolism acts as an immune rheostat. The direct epigenetic effect of itaconate has not yet been described. Itaconate is produced by diverting cis-aconitate in the TCA cycle by the enzyme cis-aconitate decarboxylase. Itaconate inhibits SDH, leading to succinate accumulation (Figure 3). Therefore, it is presumed but not proven that itaconate induces epigenetic changes akin to succinate. Itaconate acts as a negative feedback to limit exuberant immune pathology, inducing NRF2 nuclear translocation and downregulation of IL1β and IL-6 (93). Upon Mtb infection, mice with knockdown of Irg1 (gene that codes for CAD, the enzyme that converts citrate to itaconate) have increased proinflammatory cytokine production (IL1β, IL-6, IFN-y, IL12) and fatal exuberant pulmonary infiltration of innate immune cells (94, 95).
As noted earlier, most epigenetic marks depend upon metabolic precursors which, when altered, influence the epigenetic landscape. For example, S-adenosylmethionine (SAM)-mediated one-carbon metabolism supplies methyl groups for histone and DNMT. Chronic antigenic stimulation, as occurs in chronic LCMV infection, induces cellular metabolism from glycolysis and glutaminolysis, decreasing amino acid metabolic pathways feeding into the one-carbon metabolism such that reduced threonine, reduces SAM, leading to decreased H3K4me3 levels and impaired cytokine production (136, 137). SAM supplementation increased H4 arginine 31 methylation of STAT1 by Arginine methyl transferase (PRMT1), which is inhibited by HBV and is functionally essential for STAT1 function, improved antiviral effects of IFN-α in HBV infection (138) and HCV (139).
Current knowledge of the metabolic-epigenetic immune rheostat axes indicates functional roles in inhibiting acute exuberant immune pathology at the cost of long-lasting epigenetic marks and long-lasting immune suppression. To date, mechanistic studies have identified inhibitors of glycolysis, glutaminolysis and mTOR as well as transient glucose (140) restriction as possible means to block metabolic-epigenetic immune suppression. However, studies are needed to evaluate the clinical applicability of these mechanisms in severe and chronic infections.
Epigenetic Drugs to Restore Immune Response
Epigenetic drugs have been developed predominantly for cancer therapeutics, however some, such as valproic acid and hydralazine are routinely used as antiepileptics and antihypertensives. Currently approved or in-development epigenetic modifying drugs include DNA hypomethylating agents (HMAs), HDAC inhibitors, lysine methyltransferase inhibitors (targeting EZH2, G9a, DOTL), and BET bromodomain (BRD) inhibitors. Several studies, most in cancer but a growing number in infectious diseases, have demonstrated that epigenetic drugs can reverse epigenetic-mediated immune suppression. Animal models, especially those for sepsis and chronic LCMV, have documented the mechanisms by which epigenetic drugs are able to restore host immunity.
Infection with clone 13 LCMV, the prototypical model for inducing CD8+ T cell immune exhaustion induces global DNA methylation changes associated with immune exhaustion (2, 25, 141). Applying either a conditional knock of DNMT3a or the hypomethylating drug decitabine was able to restore CD8+ T cell effector function (25). Humans with sepsis upregulate DNMT1, DNMT3a and DNMT3b, resulting in global DNA methylation differences, 82.6% of which are suppressive hypermethylated marks (142). In a cecal ligation model of murine sepsis, decitabine restored immune function and decreased mortality (142). Similarly, application of the hypomethylating drug azacytidine or decitabine to cancer cell lines increased interferon responsiveness and antigen presentation (143–145).
DNA methylation changes occur in parallel to other epigenetic modifications to inhibit host immunity. Co-immunoprecipitation studies demonstrated that DNMT associates with EZH2, the catalytic unit of the PRC (146). EZH2 methylates H3K27, inhibiting gene expression (146) and the combination of EZH2 inhibition (RNAi EZH2) and azacytidine can restore gene expression (146). EZH2 acts as an anchor point for multiple epigenetic mechanisms to suppress gene expression (Table 1). In ovarian cancer cell lines, inhibition of EZH2 using DZNep (inhibitor of SAM dependent enzymes), EPZ6438 and GSK126 (selective inhibitor of EZH2) and DNMT (azacytadine) synergistically increase IFN-γ responsiveness, and CXCL9 and CXCL10 expression, while shrinking tumor size (165). In melanoma, prostate cancer, hepatocellular cancer and colon cancer, EZH2-induced epigenetic marks inhibit Th1 polarization and IFN-γ-JAK-STAT signaling, with EZH2 knockdown or pharmacologic inhibition using DZNep or GSK126, restoring IFN-γ-induced gene expression (157–160). In-vitro, EZH2 regulates both Th1 and Th2 polarization, and inhibiting EZH2 genetically or by using EZH2 inhibitor DZNep, results in reduction of the suppressive histone mark H3K27me3, thereby augmenting both Th1 and Th2 polarization and effector cytokine production (166). In exhausted CD8 T cells, GSK126, a specific EZH2 inhibitor, restores CD8 cell cytotoxicity (164). In a CLP model of sepsis, H3K27me3, the repressive epigenetic mark induced by EZH2, persists to inhibit IL12 immunity at least 6 weeks after the initial septic insult (155). In clinical studies of sepsis, EZH2 expression increases proportional to disease severity and correlates with poor clinical outcomes (156). In TB, EZH2 is expressed early (161), with EZH2 inhibition decreasing Ifng and Tnf H3K27me2 and increasing TNF and IFN-γ production (162).
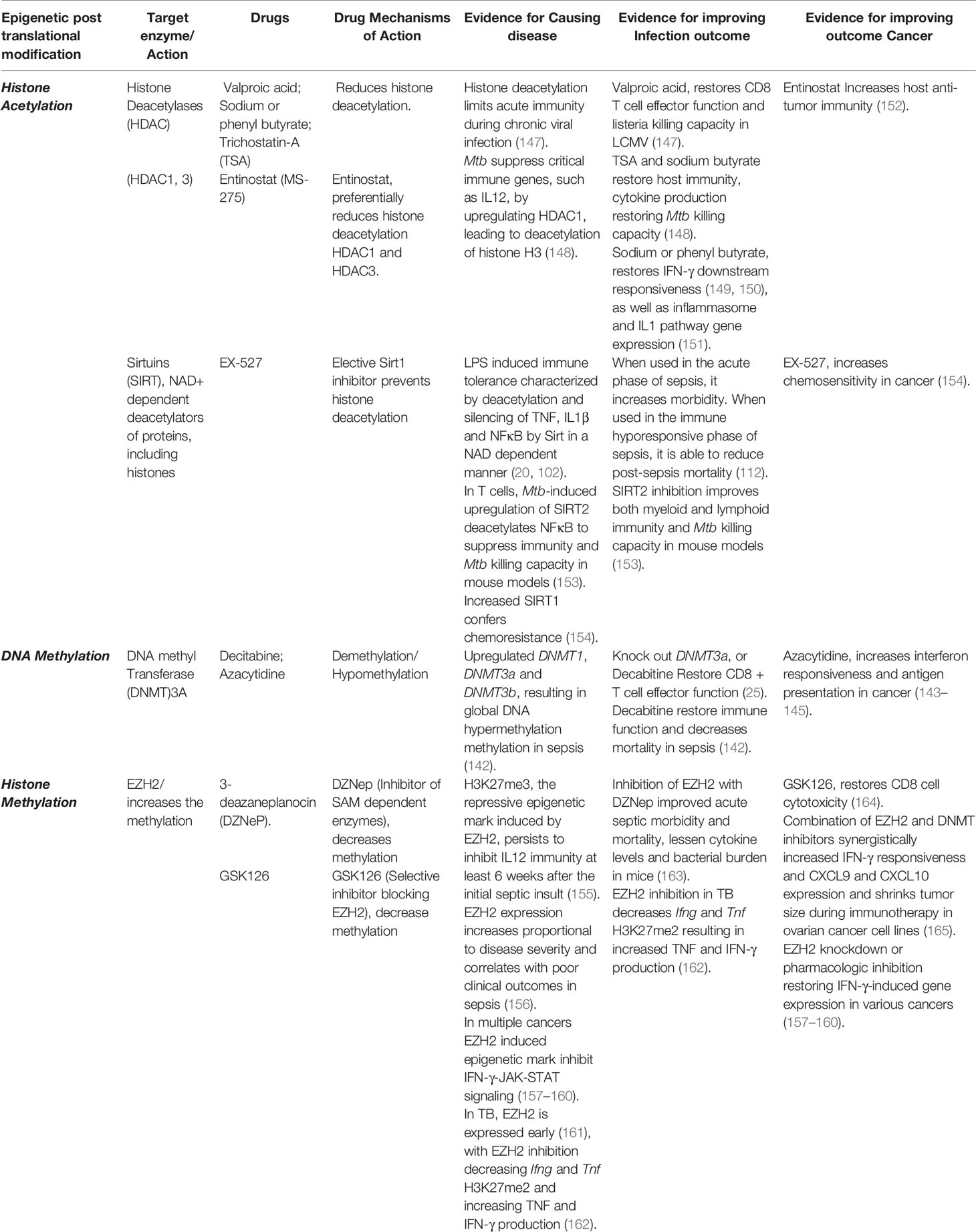
Table 1 Drugs targeting epigenetic enzymes restore Immunity and Reverse Epigenetic-mediated Immune suppression.
The PRC, which includes the HMT EZH2, interacts with DNMTs and also the NuRD complex. Therefore, these three suppressive epigenetic marks, H3K27 deacetylation, H3K27 methylation, and DNA methylation, often occur together, resulting in heterochromatin, thereby silencing gene expression (167). Chronic LCMV infection results in global decreased histone acetylation that limits both LCMV-specific and non-specific CD8 T cell effector function (147). Chronic LCMV infection also non-specifically decreases effector responses to influenza peptides, and decreases salmonella and listeria killing capacity (147, 168). Valproic acid, an HDAC inhibitor, was able to restore LCMV-specific and non-specific CD8 T cell effector function, including non-specific listeria killing capacity (147). Several studies indicate that HDAC inhibitors can restore host immunity when applied to chronic infections. Entinostat (MS-275), an inhibitor of HDAC1 and HDAC3, increases host anti-tumor immunity (152). Considering the long-term increased mortality that persists following a bout of sepsis, it would seem prudent to conduct a clinical trial to evaluate the efficacy of an HDAC inhibitor in reversing long-term sepsis-induced immune suppression (9).
Sirtuins, a class of HDACs, recognize the NAD+: NADH ratio and then deacetylate and silence NFĸB, TNF and IL1b after LPS-induced immune tolerance (20, 102). EX-527, a Sirt1 inhibitor restored myeloid cell IL1B and TNF production when administered after sepsis. Reinforcing the importance of timing, administering EX-527 early during sepsis increases mortality, however if given later during the immune hyporesponsive phase of sepsis, it reduces post-sepsis mortality in mice (112).
TB is the archetypical chronic infection. Macrophages infected with Mtb upregulate HDAC and undergo deacetylation of critical immune genes, such as IL12. Inhibition of HDAC restores immune function including cytokine production and Mtb killing capacity (148). In T cells, Mtb-induced upregulation of SIRT2 deacetylates NFĸB (p65) with SIRT2 inhibition improving both myeloid and lymphoid immunity, and Mtb killing capacity in mouse models (153). Sodium or phenyl butyrate, an HDAC inhibitor, restores IFN-γ downstream responsiveness (149, 150) as well as inflammasome and IL1 pathway gene expression (151). In a clinical trial that did not evaluate epigenetic or immunologic outcomes, the combination of Vitamin D3 and phenylbutyrate did not change time to sputum conversion but did ameliorate TB disease severity (151). Like sepsis, survivors of TB retain detrimental epigenetic scars (6, 22) and have increased all-cause mortality (12, 13). Large clinical trials should evaluate if reversing these detrimental epigenetic marks are able to reverse the post-infectious morbidity and mortality risk due to TB.
Bioengineering Approaches to Reverse Epigenetic-Mediated Immune Exhaustion & Suppression
Systemic means to reverse immune suppression, such as immune checkpoint inhibitor blockade (e.g., anti-PD-1 and anti-LAG-3), have short and long-term toxicities (169). Newer technology such as the CRISPR/Cas9 system holds promise as a precise and controlled bioengineering tool (170–175) to reverse immune suppression. Typically, the CRISPR/Cas9 system includes a guide RNA (gRNA) complexed with the Cas9 protein to specifically edit a unique genomic address (170). For example, in vitro gene editing of the PD-1 and LAG-3 genes using CRISPR-Cas9 in CAR-T cells has improved their anti-tumor function (169, 176).
A catalytically inactive version of the Cas9 protein called dead, or deactivated, Cas9 (dCas9) (177) repurposes the CRISPR-Cas9 platform for precision edited of the epigenome or gene expression machinery (173–175, 177). A diverse spectrum of epigenetic effectors has been tethered to dCas9 to deliver epigenetic payloads to specific sites across the genome, giving rise to a continually expanding epigenome editing toolkit (173, 178). The Krüppel-associated box (KRAB) is a repressive domain that is a component of several zinc-finger transcription factors (179). A fusion protein between the KRAB domain and dCas9 (dCas9-KRAB) has been shown to promote highly specific gene silencing when targeted to mammalian genes (180) and to distal regulatory elements such as enhancers (181). A version of dCas9-KRAB with a linker for activation of T cells (LAT-dCas9-KRAB) was recently shown to silence the PD-1 gene when targeted to its transcription start site (176).
Targeting the transcriptional start sites and promoters with dCas9 coupled with the de novo methyltransferases DNMT3A and its homolog DNMT3L (dCas9-DNMT3A/3L) has been described to produce widespread DNA methylation of CpG islands at the targeted loci for up to 1200 bp (182). In addition, tethering the catalytic domain of the DNA demethylase TET1 to dCas9 (dCas9-TET1) to promoters previously silenced by engineered transcriptional repressors can generate a stable, long-term reactivation of the silenced gene by demethylation of targeted CpG islands (183). Previous work has also shown that a fusion protein consisting of the catalytic core of the human acetyltransferase p300 and dCas9 (dCas9-p300) can achieve robust genetic transcriptional activation by targeting either promoters, proximal enhancers or distal enhancers (184).
The epigenome editing tools dCas9-TET1 and dCas9-p300 were recently employed to elucidate the epigenetic landscape of the Foxp3 locus, an important transcription factor in T cells. Demethylation of the enhancer region of the Foxp3 locus was achieved in mouse primary T cells, although without strong Foxp3 gene expression. In contrast, targeting dCas9-p300 to the Foxp3 promoter stabilized Foxp3 expression under both normal and inflammatory culture conditions in vitro (185). This technical approach provides new opportunities to revert anomalous post-infectious epigenetic modifications in other immunologically relevant genes using dCas9-based epigenome editing.
Robust targeted transcriptional activation has also been achieved by using CRISPR activation (CRISPRa) tools. dCas9 fused to an engineered tripartie activation domain consisting of VP64, p65 and Rta, (dCas9-VPR) has proven to be a potent synthetic CRISPR/Cas9-based transcriptional activator. For example, dCas9-VPR is able to induce gene activation of some target genes up to 320-fold compared to the original, conventional dCas9-VP64 activator (186).
Another method to increase transcriptional activation is by recruiting several copies of the regulatory proteins at once to the target gene. This can be achieved by fusing dCas9 to the SunTag, an array of a repeated short peptide sequence with strong affinity for a single-chain variable fragment (scFv) antibody fused to the activation domain. The SunTag can recruit up to 24 copies of the antibody-fused protein and has been used to recruit multiple copies of the transcriptional activation domain VP64, increasing gene expression of the targeted locus (187).
RNA aptamers that interact with transcriptional activation domains have been inserted into gRNAs, and these systems have been used to recruit transcriptional regulatory domains via dCas9 (188, 189). For example, the synergistic activation mediator consists of an MS2 bacteriophage coat protein-binding aptamer that is placed in the gRNA loops, which enables a fusion between MS2 p65 and Heat Shock Factor 1 (HSF1) to be successfully recruited to targeted genomic loci (188). Recently the SAM system was used to increase the expression of key endogenous genes related to immunological exhaustion in the context of boosting anticancer immunotherapy. Multiplexed gene activation of Cd70, Cd80, Cd86, Ifnα4, Ifnβ1, and Ifnγ was achieved in mice using a CRISPRa gRNA library improving immunogenicity of the transduced cells and leading to tumor rejection in vivo (190).
Finally, Proteolysis Targeting Chimeras (PROTACs) are small molecules which induce the targeted degradation of a protein by linking it to an E3 ubiquitin ligase. The ubiquitinated protein is then recognized and degraded by the 26S proteasome (191). Recently, Si et al. demonstrated that a hematopoietic progenitor kinase has a key role in T cell exhaustion and could be targeted by employing PROTACs and CRISPR/Cas9 technology. Increased gene expression of the MAP4K1 gene has been correlated with increased T cell exhaustion due to dysregulation of the NFκB signaling pathway. Knocking out this gene using a CRISPR/Cas9 system in CAR-T cells improved their persistence and functionality in vivo. Similarly, developing a small molecule PROTAC that selectively degrades the HPK1 protein encoded by the MAP4K1 gene in CAR-T cells improves their efficacy as well (192).
A major challenge in reducing T cell exhaustion is the enduring epigenetic changes that differ from their normal state (25). The CRISPR/dCas9-based protein fusions to epigenetic writers and erasers are a potential tool to robustly and precisely modulate the epigenome of exhausted T cells, reverting them to their pre-infected functional state.
As previously discussed, T cell function requires balanced AP-1 and NFAT heterodimerization. CAR T cells experience tonic activation that induces characteristic features of exhaustion (193). By manipulating HA-28z CAR T cells to over-express c-Jun, AP-1-NFAT balance was restored, increasing IL-2 production (193). Recent studies have shown that the HDAC SIRT1 functions to deacetylate c-Jun, inactivating it, and thus effectively preventing the formation of the NFAT/AP-1 complexes required to induce Il-2 expression in activated T cells. In this way SIRT1 acts as an epigenetic promoter of immune exhaustion (18, 194). In a follow-up CAR T study, the incorporation of a titratable FK506 binding protein 12 (FKBP) destabilizing domain (DD) emphasized the importance of timing and rest (195). Simply put, this engineered CAR T cell model demonstrated that interrupting tonic T cell activation, either through the titratable FK506 DD or through dasatinib, a tyrosine kinase inhibitor, could block epigenetic-mediated immune exhaustion.
Conclusion
Increasing evidence from in-vitro studies and animal models has demonstrated the signaling pathways, TFs, metabolic intermediates and epigenetic enzymes that remodel chromatin in order to suppress gene expression and limit exuberant immune pathology. Although acutely, this suppression helps regulate an overly exuberant immune response, it makes individuals more susceptible to secondary infections and cancers leading to increased long-term morbidity and mortality. Other fields have harnessed drugs to manipulate epigenetic enzymes, metabolic pathways, TFs and signaling pathways to improve clinical outcomes. Similar studies need to evaluate which strategy limits off-target adverse effects in order to restore host immunity. For example, theoretically, upstream moderation of the three-metabolic-epigenetic-immune rheostats might better block detrimental epigenetic marks than a specific epigenetic modifying drug. Considering the significant long-term mortality that exists after pneumonia, sepsis and TB, translational studies using emerging immunologic approaches and bioengineering tools are needed to evaluate if modulating these pathways improve clinical outcomes.
Author Contributions
All authors made a contribution to the acquisition of the information for the work, critically revised the manuscript for important intellectual content, and gave final approval of the current version to be published. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
AD is supported by NIAID K23 AI141681-02. AD, TN, RG-R, and IH are supported by the John S. Dunn Foundation. LS is supported by NIAID R01AI136831, R21 AI145539, P01AG051428 and OD P51 OD011133. CO is supported by STX- MSTP-NIH T32GM113896. IH is supported by CPRIT RR170030. RG-R is supported by Fulbright-García Robles Scholarship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Yaseen I, Kaur P, Nandicoori VK, Khosla S. Mycobacteria Modulate Host Epigenetic Machinery by Rv1988 Methylation of a Non-Tail Arginine of Histone H3. Nat Commun (2015) 6:8922. doi: 10.1038/ncomms9922
2. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, et al. Chronic Virus Infection Enforces Demethylation of the Locus That Encodes PD-1 in Antigen-Specific CD8(+) T Cells. Immunity (2011) 35(3):400–12. doi: 10.1016/j.immuni.2011.06.015
3. Hamdane N, Juhling F, Crouchet E, El Saghire H, Thumann C, Oudot MA, et al. Hcv-Induced Epigenetic Changes Associated With Liver Cancer Risk Persist After Sustained Virologic Response. Gastroenterology (2019) 1562313-2329(8):e2317. doi: 10.1053/j.gastro.2019.02.038
4. Cross D, Drury R, Hill J, Pollard AJ. Epigenetics in Sepsis: Understanding Its Role in Endothelial Dysfunction, Immunosuppression, and Potential Therapeutics. Front Immunol (2019) 10:1363. doi: 10.3389/fimmu.2019.01363
5. DiNardo AR, Nishiguchi T, Mace EM, Rajapakshe K, Mtetwa G, Kay A, et al. Schistosomiasis Induces Persistent DNA Methylation and Tuberculosis-Specific Immune Changes. J Immunol (2018) 201(1):124–33. doi: 10.4049/jimmunol.1800101
6. DiNardo AR, Rajapakshe K, Nishiguchi T, Grimm SL, Mtetwa G, Dlamini Q, et al. DNA Hypermethylation During Tuberculosis Dampens Host Immune Responsiveness. J Clin Invest (2020) 130(6):3113–23. doi: 10.1172/JCI134622
7. Turk JL. Von Pirquet, Allergy and Infectious Diseases: A Review. J R Soc Med (1987) 80(1):31–3. doi: 10.1177/014107688708000113
8. Furcolow ML, Brasher CA. Chronic Progressive (Cavitary) Histoplasmosis as a Problem in Tuberculosis Sanatoriums. Am Rev Tuberc (1956) 73(5):609–19. doi: 10.1164/artpd.1956.73.5.609
9. Prescott HC, Osterholzer JJ, Langa KM, Angus DC, Iwashyna TJ. Late Mortality After Sepsis: Propensity Matched Cohort Study. BMJ (2016) 353:i2375. doi: 10.1136/bmj.i2375
10. Venet F, Monneret G. Advances in the Understanding and Treatment of Sepsis-Induced Immunosuppression. Nat Rev Nephrol (2018) 14(2):121–37. doi: 10.1038/nrneph.2017.165
11. Sandvall B, Rueda AM, Musher DM. Long-Term Survival Following Pneumococcal Pneumonia. Clin Infect Dis (2013) 56(8):1145–6. doi: 10.1093/cid/cis1207
12. Lee-Rodriguez C, Wada PY, Hung YY, Skarbinski J. Association of Mortality and Years of Potential Life Lost With Active Tuberculosis in the United States. JAMA Netw Open (2020) 3(9):e2014481. doi: 10.1001/jamanetworkopen.2020.14481
13. Romanowski K, Baumann B, Basham CA, Ahmad Khan F, Fox GJ, Johnston JC. Long-Term All-Cause Mortality in People Treated for Tuberculosis: A Systematic Review and Meta-Analysis. Lancet Infect Dis (2019) 19(10):1129–37. doi: 10.1016/S1473-3099(19)30309-3
14. Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is Required to Resolve Inflammation by Recruiting Hdac2 to Specifically Repress IL-6. Nature (2015) 525(7569):389–93. doi: 10.1038/nature15252
15. DiNardo AR, Netea MG, Musher DM. Postinfectious Epigenetic Immune Modifications - A Double-Edged Sword. N Engl J Med (2021) 384(3):261–70. doi: 10.1056/NEJMra2028358
16. Boomer JS, Green JM, Hotchkiss RS. The Changing Immune System in Sepsis: Is Individualized Immuno-Modulatory Therapy the Answer? Virulence (2014) 5(1):45–56. doi: 10.4161/viru.26516
17. Otto GP, Sossdorf M, Claus RA, Rodel J, Menge K, Reinhart K, et al. The Late Phase of Sepsis Is Characterized by an Increased Microbiological Burden and Death Rate. Crit Care (2011) 15(4):R183. doi: 10.1186/cc10332
18. Valdor R, Macian F. Induction and Stability of the Anergic Phenotype in T Cells. Semin Immunol (2013) 25(4):313–20. doi: 10.1016/j.smim.2013.10.010
19. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
20. Vachharajani V, McCall CE. Epigenetic and Metabolic Programming of Innate Immunity in Sepsis. Innate Immun (2019) 25(5):267–79. doi: 10.1177/1753425919842320
21. Cook JA. Molecular Basis of Endotoxin Tolerance. Ann N Y Acad Sci (1998) 851:426–8. doi: 10.1111/j.1749-6632.1998.tb09020.x
22. Roquilly A, Jacqueline C, Davieau M, Molle A, Sadek A, Fourgeux C, et al. Alveolar Macrophages Are Epigenetically Altered After Inflammation, Leading to Long-Term Lung Immunoparalysis. Nat Immunol (2020). 21(6):636–48. doi: 10.1038/s41590-020-0673-x
23. Di Pietro A, Good-Jacobson KL. Disrupting the Code: Epigenetic Dysregulation of Lymphocyte Function During Infectious Disease and Lymphoma Development. J Immunol (2018) 201(4):1109–18. doi: 10.4049/jimmunol.1800137
24. Tough DF, Tak PP, Tarakhovsky A, Prinjha RK. Epigenetic Drug Discovery: Breaking Through the Immune Barrier. Nat Rev Drug Discovery (2016) 15(12):835–53. doi: 10.1038/nrd.2016.185
25. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell (2017) 170(1):142–57.e119. doi: 10.1016/j.cell.2017.06.007
26. Ahn E, Youngblood B, Lee J, Lee J, Sarkar S, Ahmed R. Demethylation of the PD-1 Promoter Is Imprinted During the Effector Phase of CD8 T Cell Exhaustion. J Virol (2016) 90(19):8934–46. doi: 10.1128/JVI.00798-16
27. Nakayama-Hosoya K, Ishida T, Youngblood B, Nakamura H, Hosoya N, Koga M, et al. Epigenetic Repression of Interleukin 2 Expression in Senescent CD4+ T Cells During Chronic HIV Type 1 Infection. J Infect Dis (2015) 211(1):28–39. doi: 10.1093/infdis/jiu376
28. Youngblood B, Noto A, Porichis F, Akondy RS, Ndhlovu ZM, Austin JW, et al. Cutting Edge: Prolonged Exposure to HIV Reinforces a Poised Epigenetic Program for PD-1 Expression in Virus-Specific CD8 T Cells. J Immunol (2013) 191(2):540–4. doi: 10.4049/jimmunol.1203161
29. Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA Methylation Remodeling Accompanies CD8 T Cell Effector Function. J Immunol (2013) 191(6):3419–29. doi: 10.4049/jimmunol.1301395
30. Youngblood B, Reich NO. The Early Expressed HIV-1 Genes Regulate DNMT1 Expression. Epigenetics (2008) 3(3):149–56. doi: 10.4161/epi.3.3.6372
31. Khorasanizadeh S. The Nucleosome: From Genomic Organization to Genomic Regulation. Cell (2004) 116(2):259–72. doi: 10.1016/s0092-8674(04)00044-3
32. Peterson CL, Laniel MA. Histones and Histone Modifications. Curr Biol (2004) 14(14):R546–551. doi: 10.1016/j.cub.2004.07.007
33. Curradi M, Izzo A, Badaracco G, Landsberger N. Molecular Mechanisms of Gene Silencing Mediated by DNA Methylation. Mol Cell Biol (2002) 22(9):3157–73. doi: 10.1128/mcb.22.9.3157-3173.2002
34. Eberharter A, Becker PB. Histone Acetylation: A Switch Between Repressive and Permissive Chromatin. Second in Review Series on Chromatin Dynamics. EMBO Rep (2002) 3(3):224–9. doi: 10.1093/embo-reports/kvf053
35. Phan AT, Goldrath AW, Glass CK. Metabolic and Epigenetic Coordination of T Cell and Macrophage Immunity. Immunity (2017) 46(5):714–29. doi: 10.1016/j.immuni.2017.04.016
36. Barber EK, Dasgupta JD, Schlossman SF, Trevillyan JM, Rudd CE. The CD4 and CD8 Antigens are Coupled to a Protein-Tyrosine Kinase (p56lck) That Phosphorylates the CD3 Complex. Proc Natl Acad Sci USA (1989) 86(9):3277–81. doi: 10.1073/pnas.86.9.3277
37. Chan AC, Irving BA, Fraser JD, Weiss A. The Zeta Chain is Associated With a Tyrosine Kinase and Upon T-cell Antigen Receptor Stimulation Associates With ZAP-70, a 70-kDa Tyrosine Phosphoprotein. Proc Natl Acad Sci USA (1991) 88(20):9166–70. doi: 10.1073/pnas.88.20.9166
38. Chan AC, Iwashima M, Turck CW, Weiss A. Zap-70: A 70 Kd Protein-Tyrosine Kinase That Associates With the TCR Zeta Chain. Cell (1992) 71(4):649–62. doi: 10.1016/0092-8674(92)90598-7
39. Isakov N, Wange RL, Burgess WH, Watts JD, Aebersold R, Samelson LE. ZAP-70 Binding Specificity to T Cell Receptor Tyrosine-Based Activation Motifs: The Tandem SH2 Domains of ZAP-70 Bind Distinct Tyrosine-Based Activation Motifs With Varying Affinity. J Exp Med (1995) 181(1):375–80. doi: 10.1084/jem.181.1.375
40. Li QJ, Dinner AR, Qi S, Irvine DJ, Huppa JB, Davis MM, et al. CD4 Enhances T Cell Sensitivity to Antigen by Coordinating Lck Accumulation at the Immunological Synapse. Nat Immunol (2004) 5(8):791–9. doi: 10.1038/ni1095
41. Liu SK, Fang N, Koretzky GA, McGlade CJ. The Hematopoietic-Specific Adaptor Protein Gads Functions in T-Cell Signaling Via Interactions With the SLP-76 and LAT Adaptors. Curr Biol (1999) 9(2):67–75. doi: 10.1016/s0960-9822(99)80017-7
42. Zhang W, Sloan-Lancaster J, Kitchen J, Trible RP, Samelson LE. LAT: The ZAP-70 Tyrosine Kinase Substrate That Links T Cell Receptor to Cellular Activation. Cell (1998) 92(1):83–92. doi: 10.1016/s0092-8674(00)80901-0
43. Sommers CL, Rabin RL, Grinberg A, Tsay HC, Farber J, Love PE. A Role for the Tec Family Tyrosine Kinase Txk in T Cell Activation and Thymocyte Selection. J Exp Med (1999) 190(10):1427–38. doi: 10.1084/jem.190.10.1427
44. Baine I, Abe BT, Macian F. Regulation of T-Cell Tolerance by Calcium/NFAT Signaling. Immunol Rev (2009) 231(1):225–40. doi: 10.1111/j.1600-065X.2009.00817.x
45. Jain J, McCaffrey PG, Miner Z, Kerppola TK, Lambert JN, Verdine GL, et al. The T-Cell Transcription Factor NFATp Is a Substrate for Calcineurin and Interacts With Fos and Jun. Nature (1993) 365(6444):352–5. doi: 10.1038/365352a0
46. Jain J, McCaffrey PG, Valge-Archer VE, Rao A. Nuclear Factor of Activated T Cells Contains Fos and Jun. Nature (1992) 356(6372):801–4. doi: 10.1038/356801a0
47. Macian F, Garcia-Rodriguez C, Rao A. Gene Expression Elicited by NFAT in the Presence or Absence of Cooperative Recruitment of Fos and Jun. EMBO J (2000) 19(17):4783–95. doi: 10.1093/emboj/19.17.4783
48. Lewis RS. Calcium Signaling Mechanisms in T Lymphocytes. Annu Rev Immunol (2001) 19:497–521. doi: 10.1146/annurev.immunol.19.1.497
49. Macian F, Lopez-Rodriguez C, Rao A. Partners in Transcription: NFAT and AP-1. Oncogene (2001) 20(19):2476–89. doi: 10.1038/sj.onc.1204386
50. Soto-Nieves N, Puga I, Abe BT, Bandyopadhyay S, Baine I, Rao A, et al. Transcriptional Complexes Formed by NFAT Dimers Regulate the Induction of T Cell Tolerance. J Exp Med (2009) 206(4):867–76. doi: 10.1084/jem.20082731
51. Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 Transcription Factors Cooperate With NR4A Transcription Factors to Impose CD8(+) T Cell Exhaustion. Proc Natl Acad Sci USA (2019) 116(25):12410–5. doi: 10.1073/pnas.1905675116
52. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 Inhibits T-Cell Receptor Induced Phosphorylation of the ZAP70/CD3zeta Signalosome and Downstream Signaling to Pkctheta. FEBS Lett (2004) 574(1-3):37–41. doi: 10.1016/j.febslet.2004.07.083
53. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed Cell Death 1 Forms Negative Costimulatory Microclusters That Directly Inhibit T Cell Receptor Signaling by Recruiting Phosphatase SHP2. J Exp Med (2012) 209(6):1201–17. doi: 10.1084/jem.20112741
54. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol Cell Biol (2005) 25(21):9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
55. Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, et al. Transcriptional Analysis of HIV-specific Cd8+ T Cells Shows That PD-1 Inhibits T Cell Function by Upregulating BATF. Nat Med (2010) 16(10):1147–51. doi: 10.1038/nm.2232
56. Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME, et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8(+) T Cells. Immunity (2015) 42(2):265–78. doi: 10.1016/j.immuni.2015.01.006
57. Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The Transcription Factor FoxO1 Sustains Expression of the Inhibitory Receptor PD-1 and Survival of Antiviral CD8(+) T Cells During Chronic Infection. Immunity (2014) 41(5):802–14. doi: 10.1016/j.immuni.2014.10.013
58. Safford M, Collins S, Lutz MA, Allen A, Huang CT, Kowalski J, et al. Egr-2 and Egr-3 Are Negative Regulators of T Cell Activation. Nat Immunol (2005) 6(5):472–80. doi: 10.1038/ni1193
59. Thomas RM, Chunder N, Chen C, Umetsu SE, Winandy S, Wells AD. Ikaros Enforces the Costimulatory Requirement for IL2 Gene Expression and Is Required for Anergy Induction in CD4+ T Lymphocytes. J Immunol (2007) 179(11):7305–15. doi: 10.4049/jimmunol.179.11.7305
60. Bandyopadhyay S, Dure M, Paroder M, Soto-Nieves N, Puga I, Macian F. Interleukin 2 Gene Transcription Is Regulated by Ikaros-Induced Changes in Histone Acetylation in Anergic T Cells. Blood (2007) 109(7):2878–86. doi: 10.1182/blood-2006-07-037754
61. Scott-Browne JP, Lopez-Moyado IF, Trifari S, Wong V, Chavez L, Rao A, et al. Dynamic Changes in Chromatin Accessibility Occur in CD8(+) T Cells Responding to Viral Infection. Immunity (2016) 45(6):1327–40. doi: 10.1016/j.immuni.2016.10.028
62. Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, et al. NR4A Transcription Factors Limit CAR T Cell Function in Solid Tumours. Nature (2019) 567(7749):530–4. doi: 10.1038/s41586-019-0985-x
63. Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, et al. TOX Reinforces the Phenotype and Longevity of Exhausted T Cells in Chronic Viral Infection. Nature (2019) 571(7764):265–9. doi: 10.1038/s41586-019-1326-9
64. Yao C, Sun HW, Lacey NE, Ji Y, Moseman EA, Shih HY, et al. Single-Cell RNA-seq Reveals TOX as a Key Regulator of CD8(+) T Cell Persistence in Chronic Infection. Nat Immunol (2019) 20(7):890–901. doi: 10.1038/s41590-019-0403-4
65. Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin States Define Tumour-Specific T Cell Dysfunction and Reprogramming. Nature (2017) 545(7655):452–6. doi: 10.1038/nature22367
66. Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. TOX Is a Critical Regulator of Tumour-Specific T Cell Differentiation. Nature (2019) 571(7764):270–4. doi: 10.1038/s41586-019-1324-y
67. Biswas SK, Lopez-Collazo E. Endotoxin Tolerance: New Mechanisms, Molecules and Clinical Significance. Trends Immunol (2009) 30(10):475–87. doi: 10.1016/j.it.2009.07.009
68. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of Adaptor TRIF in the MyD88-Independent Toll-Like Receptor Signaling Pathway. Science (2003) 301(5633):640–3. doi: 10.1126/science.1087262
69. Biswas SK, Tergaonkar V. Myeloid Differentiation Factor 88-Independent Toll-like Receptor Pathway: Sustaining Inflammation or Promoting Tolerance? Int J Biochem Cell Biol (2007) 39(9):1582–92. doi: 10.1016/j.biocel.2007.04.021
70. Laird MH, Rhee SH, Perkins DJ, Medvedev AE, Piao W, Fenton MJ, et al. Tlr4/MyD88/PI3K Interactions Regulate TLR4 Signaling. J Leukoc Biol (2009) 85(6):966–77. doi: 10.1189/jlb.1208763
71. Carmody RJ, Ruan Q, Palmer S, Hilliard B, Chen YH. Negative Regulation of Toll-Like Receptor Signaling by NF-kappaB p50 Ubiquitination Blockade. Science (2007) 317(5838):675–8. doi: 10.1126/science.1142953
72. Xiong Y, Medvedev AE. Induction of Endotoxin Tolerance In Vivo Inhibits Activation of IRAK4 and Increases Negative Regulators IRAK-M, Ship-1, and A20. J Leukoc Biol (2011) 90(6):1141–8. doi: 10.1189/jlb.0611273
73. Murphy MB, Xiong Y, Pattabiraman G, Manavalan TT, Qiu F, Medvedev AE. Pellino-3 Promotes Endotoxin Tolerance and Acts as a Negative Regulator of TLR2 and TLR4 Signaling. J Leukoc Biol (2015) 98(6):963–74. doi: 10.1189/jlb.2VMA0515-229RR
74. Hoogerwerf JJ, Leendertse M, Wieland CW, de Vos AF, de Boer JD, Florquin S, et al. Loss of Suppression of Tumorigenicity 2 (ST2) Gene Reverses Sepsis-Induced Inhibition of Lung Host Defense in Mice. Am J Respir Crit Care Med (2011) 183(7):932–40. doi: 10.1164/rccm.201006-0934OC
75. Learn CA, Boger MS, Li L, McCall CE. The Phosphatidylinositol 3-Kinase Pathway Selectively Controls sIL-1RA Not interleukin-1beta Production in the Septic Leukocytes. J Biol Chem (2001) 276(23):20234–9. doi: 10.1074/jbc.M100316200
76. Bowling WM, Flye MW, Qiu YY, Callery MP. Inhibition of Phosphatidylinositol-3’-Kinase Prevents Induction of Endotoxin Tolerance In Vitro. J Surg Res (1996) 63(1):287–92. doi: 10.1006/jsre.1996.0262
77. Yan Q, Carmody RJ, Qu Z, Ruan Q, Jager J, Mullican SE, et al. Nuclear Factor-Kappab Binding Motifs Specify Toll-like Receptor-Induced Gene Repression Through an Inducible Repressosome. Proc Natl Acad Sci USA (2012) 109(35):14140–5. doi: 10.1073/pnas.1119842109
78. Chen X, El Gazzar M, Yoza BK, McCall CE. The NF-kappaB Factor RelB and Histone H3 Lysine Methyltransferase G9a Directly Interact to Generate Epigenetic Silencing in Endotoxin Tolerance. J Biol Chem (2009) 284(41):27857–65. doi: 10.1074/jbc.M109.000950
79. El Gazzar M, Yoza BK, Chen X, Hu J, Hawkins GA, McCall CE. G9a and HP1 Couple Histone and DNA Methylation to TNFalpha Transcription Silencing During Endotoxin Tolerance. J Biol Chem (2008) 283(47):32198–208. doi: 10.1074/jbc.M803446200
80. Seeley JJ, Ghosh S. Molecular Mechanisms of Innate Memory and Tolerance to LPS. J Leukoc Biol (2017) 101(1):107–19. doi: 10.1189/jlb.3MR0316-118RR
81. Seeley JJ, Baker RG, Mohamed G, Bruns T, Hayden MS, Deshmukh SD, et al. Induction of Innate Immune Memory Via microRNA Targeting of Chromatin Remodelling Factors. Nature (2018) 559(7712):114–9. doi: 10.1038/s41586-018-0253-5
82. Barish GD, Yu RT, Karunasiri M, Ocampo CB, Dixon J, Benner C, et al. Bcl-6 and NF-kappaB Cistromes Mediate Opposing Regulation of the Innate Immune Response. Genes Dev (2010) 24(24):2760–5. doi: 10.1101/gad.1998010
83. Foster SL, Hargreaves DC, Medzhitov R. Gene-Specific Control of Inflammation by TLR-induced Chromatin Modifications. Nature (2007) 447(7147):972–8. doi: 10.1038/nature05836
84. El Gazzar M, Yoza BK, Chen X, Garcia BA, Young NL, McCall CE. Chromatin-Specific Remodeling by HMGB1 and Linker Histone H1 Silences Proinflammatory Genes During Endotoxin Tolerance. Mol Cell Biol (2009) 29(7):1959–71. doi: 10.1128/MCB.01862-08
85. Stender JD, Pascual G, Liu W, Kaikkonen MU, Do K, Spann NJ, et al. Control of Proinflammatory Gene Programs by Regulated Trimethylation and Demethylation of Histone H4K20. Mol Cell (2012) 48(1):28–38. doi: 10.1016/j.molcel.2012.07.020
86. Chen J, Ivashkiv LB. IFN-Gamma Abrogates Endotoxin Tolerance by Facilitating Toll-like Receptor-Induced Chromatin Remodeling. Proc Natl Acad Sci USA (2010) 107(45):19438–43. doi: 10.1073/pnas.1007816107
87. Ramirez-Carrozzi VR, Nazarian AA, Li CC, Gore SL, Sridharan R, Imbalzano AN, et al. Selective and Antagonistic Functions of SWI/SNF and Mi-2beta Nucleosome Remodeling Complexes During an Inflammatory Response. Genes Dev (2006) 20(3):282–96. doi: 10.1101/gad.1383206
88. Novakovic B, Habibi E, Wang SY, Arts RJW, Davar R, Megchelenbrink W, et al. Beta-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell (2016) 1671354-1368(5):e1314. doi: 10.1016/j.cell.2016.09.034
89. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell (2017) 169(4):570–86. doi: 10.1016/j.cell.2017.04.004
90. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity (2016) 45(2):358–73. doi: 10.1016/j.immuni.2016.07.008
91. Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol (1927) 8(6):519–30. doi: 10.1085/jgp.8.6.519
92. Russell SL, Lamprecht DA, Mandizvo T, Jones TT, Naidoo V, Addicott KW, et al. Compromised Metabolic Reprogramming Is an Early Indicator of CD8(+) T Cell Dysfunction During Chronic Mycobacterium Tuberculosis Infection. Cell Rep (2019) 29(11):3564–79.e3565. doi: 10.1016/j.celrep.2019.11.034
93. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an Anti-Inflammatory Metabolite That Activates Nrf2 Via Alkylation of KEAP1. Nature (2018) 556(7699):113–7. doi: 10.1038/nature25986
94. Nair S, Huynh JP, Lampropoulou V, Loginicheva E, Esaulova E, Gounder AP, et al. Irg1 Expression in Myeloid Cells Prevents Immunopathology During M. Tuberculosis Infection. J Exp Med (2018) 215(4):1035–45. doi: 10.1084/jem.20180118
95. Hoffmann E, Machelart A, Belhaouane I, Deboosere N, Pauwels A-M, Saint-André J-P, et al. IRG1 Controls Immunometabolic Host Response and Restricts Intracellular Mycobacterium Tuberculosis Infection. bioRxiv (2019), 761551. doi: 10.1101/761551
96. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell (2013) 153(6):1239–51. doi: 10.1016/j.cell.2013.05.016
97. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic Stability of Exhausted T Cells Limits Durability of Reinvigoration by PD-1 Blockade. Science (2016) 354(6316):1160–5. doi: 10.1126/science.aaf2807
98. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The Epigenetic Landscape of T Cell Exhaustion. Science (2016) 354(6316):1165–9. doi: 10.1126/science.aae0491
99. Donohoe DR, Bultman SJ. Metaboloepigenetics: Interrelationships Between Energy Metabolism and Epigenetic Control of Gene Expression. J Cell Physiol (2012) 227(9):3169–77. doi: 10.1002/jcp.24054
100. Annunziata I, van de Vlekkert D, Wolf E, Finkelstein D, Neale G, Machado E, et al. MYC Competes With MiT/TFE in Regulating Lysosomal Biogenesis and Autophagy Through an Epigenetic Rheostat. Nat Commun (2019) 10(1):3623. doi: 10.1038/s41467-019-11568-0
101. Matilainen O, Quiros PM, Auwerx J. Mitochondria and Epigenetics - Crosstalk in Homeostasis and Stress. Trends Cell Biol (2017) 27(6):453–63. doi: 10.1016/j.tcb.2017.02.004
102. Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, McCall CE. NAD+-Dependent SIRT1 Deacetylase Participates in Epigenetic Reprogramming During Endotoxin Tolerance. J Biol Chem (2011) 286(11):9856–64. doi: 10.1074/jbc.M110.196790
103. Zhang J, Tao J, Ling Y, Li F, Zhu X, Xu L, et al. Switch of NAD Salvage to De Novo Biosynthesis Sustains SIRT1-RelB-Dependent Inflammatory Tolerance. Front Immunol (2019) 10:2358. doi: 10.3389/fimmu.2019.02358
104. Katsuyama E, Suarez-Fueyo A, Bradley SJ, Mizui M, Marin AV, Mulki L, et al. The CD38/NAD/SIRTUIN1/EZH2 Axis Mitigates Cytotoxic Cd8 T Cell Function and Identifies Patients With SLE Prone to Infections. Cell Rep (2020) 30112-123(1):e114. doi: 10.1016/j.celrep.2019.12.014
105. Horenstein AL, Bracci C, Morandi F, Malavasi F. CD38 in Adenosinergic Pathways and Metabolic Re-Programming in Human Multiple Myeloma Cells: In-tandem Insights From Basic Science to Therapy. Front Immunol (2019) 10:760. doi: 10.3389/fimmu.2019.00760
106. Booiman T, Wit FW, Girigorie AF, Maurer I, De Francesco D, Sabin CA, et al. Terminal Differentiation of T Cells is Strongly Associated With CMV Infection and Increased in HIV-positive Individuals on ART and Lifestyle Matched Controls. PloS One (2017) 12(8):e0183357. doi: 10.1371/journal.pone.0183357
107. Zidovec Lepej S, Vince A, Dakovic Rode O, Remenar A, Jeren T. Increased Numbers of CD38 Molecules on Bright CD8+ T Lymphocytes in Infectious Mononucleosis Caused by Epstein-Barr Virus Infection. Clin Exp Immunol (2003) 133(3):384–90. doi: 10.1046/j.1365-2249.2003.02219.x
108. Adekambi T, Ibegbu CC, Cagle S, Kalokhe AS, Wang YF, Hu Y, et al. Biomarkers on Patient T Cells Diagnose Active Tuberculosis and Monitor Treatment Response. J Clin Invest (2015) 125(5):1827–38. doi: 10.1172/JCI77990
109. Dentone C, Fenoglio D, Schenone E, Cenderello G, Prinapori R, Signori A, et al. Increased CD38 Expression on T Lymphocytes as a Marker of HIV Dissemination Into the Central Nervous System. HIV Clin Trials (2015) 16(5):190–6. doi: 10.1179/1945577115Y.0000000005
110. Gautam US, Foreman TW, Bucsan AN, Veatch AV, Alvarez X, Adekambi T, et al. In Vivo Inhibition of Tryptophan Catabolism Reorganizes the Tuberculoma and Augments Immune-Mediated Control of Mycobacterium Tuberculosis. Proc Natl Acad Sci USA (2018) 115(1):E62–71. doi: 10.1073/pnas.1711373114
111. Kuroda S, Yamazaki M, Abe M, Sakimura K, Takayanagi H, Iwai Y. Basic Leucine Zipper Transcription Factor, ATF-like (BATF) Regulates Epigenetically and Energetically Effector CD8 T-Cell Differentiation Via Sirt1 Expression. Proc Natl Acad Sci USA (2011) 108(36):14885–9. doi: 10.1073/pnas.1105133108
112. Vachharajani VT, Liu T, Brown CM, Wang X, Buechler NL, Wells JD, et al. SIRT1 Inhibition During the Hypoinflammatory Phenotype of Sepsis Enhances Immunity and Improves Outcome. J Leukoc Biol (2014) 96(5):785–96. doi: 10.1189/jlb.3MA0114-034RR
113. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-Hydroxyglutarate is a Competitive Inhibitor of Alpha-Ketoglutarate-Dependent Dioxygenases. Cancer Cell (2011) 19(1):17–30. doi: 10.1016/j.ccr.2010.12.014
114. Chandel NS. Navigating Metabolism. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press (2015).
115. Ntranos A, Ntranos V, Bonnefil V, Liu J, Kim-Schulze S, He Y, et al. Fumarates Target the Metabolic-Epigenetic Interplay of Brain-Homing T Cells in Multiple Sclerosis. Brain (2019) 142(3):647–61. doi: 10.1093/brain/awy344
116. Laukka T, Mariani CJ, Ihantola T, Cao JZ, Hokkanen J, Kaelin WG Jr, et al. Fumarate and Succinate Regulate Expression of Hypoxia-Inducible Genes Via TET Enzymes. J Biol Chem (2016) 291(8):4256–65. doi: 10.1074/jbc.M115.688762
117. Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, et al. Dimethyl Fumarate Targets GAPDH and Aerobic Glycolysis to Modulate Immunity. Science (2018) 360(6387):449–53. doi: 10.1126/science.aan4665
118. Dominguez-Andres J, Novakovic B, Li Y, Scicluna BP, Gresnigt MS, Arts RJW, et al. The Itaconate Pathway Is a Central Regulatory Node Linking Innate Immune Tolerance and Trained Immunity. Cell Metab (2019) 29211-220(1):e215. doi: 10.1016/j.cmet.2018.09.003
119. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of alpha-KG-Dependent Histone and DNA Demethylases by Fumarate and Succinate That Are Accumulated in Mutations of FH and SDH Tumor Suppressors. Genes Dev (2012) 26(12):1326–38. doi: 10.1101/gad.191056.112
120. Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, et al. S-2-Hydroxyglutarate Regulates CD8(+) T-Lymphocyte Fate. Nature (2016) 540(7632):236–41. doi: 10.1038/nature20165
121. Martinez-Reyes I, Chandel NS. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat Commun (2020) 11(1):102. doi: 10.1038/s41467-019-13668-3
122. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell (2010) 18(6):553–67. doi: 10.1016/j.ccr.2010.11.015
123. Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, et al. Suppression of Antitumor T Cell Immunity by the Oncometabolite (R)-2-Hydroxyglutarate. Nat Med (2018) 24(8):1192–203. doi: 10.1038/s41591-018-0095-6
124. DiNardo CD, Jabbour E, Ravandi F, Takahashi K, Daver N, Routbort M, et al. IDH1 and IDH2 Mutations in Myelodysplastic Syndromes and Role in Disease Progression. Leukemia (2016) 30(4):980–4. doi: 10.1038/leu.2015.211
125. Glass JL, Hassane D, Wouters BJ, Kunimoto H, Avellino R, Garrett-Bakelman FE, et al. Epigenetic Identity in AML Depends on Disruption of Nonpromoter Regulatory Elements and Is Affected by Antagonistic Effects of Mutations in Epigenetic Modifiers. Cancer Discovery (2017) 7(8):868–83. doi: 10.1158/2159-8290.CD-16-1032
126. Zhang X, Rao A, Sette P, Deibert C, Pomerantz A, Kim WJ, et al. IDH Mutant Gliomas Escape Natural Killer Cell Immune Surveillance by Downregulation of NKG2D Ligand Expression. Neuro Oncol (2016) 18(10):1402–12. doi: 10.1093/neuonc/now061
127. Carlstrom KE, Ewing E, Granqvist M, Gyllenberg A, Aeinehband S, Enoksson SL, et al. Therapeutic Efficacy of Dimethyl Fumarate in Relapsing-Remitting Multiple Sclerosis Associates With ROS Pathway in Monocytes. Nat Commun (2019) 10(1):3081. doi: 10.1038/s41467-019-11139-3
128. Luckel C, Picard F, Raifer H, Campos Carrascosa L, Guralnik A, Zhang Y, et al. Il-17(+) Cd8(+) T Cell Suppression by Dimethyl Fumarate Associates With Clinical Response in Multiple Sclerosis. Nat Commun (2019) 10(1):5722. doi: 10.1038/s41467-019-13731-z
129. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated Aerobic Glycolysis as Metabolic Basis for Trained Immunity. Science (2014) 345(6204):1250684. doi: 10.1126/science.1250684
130. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab (2016) 24(6):807–19. doi: 10.1016/j.cmet.2016.10.008
131. Bracken AP, Brien GL, Verrijzer CP. Dangerous Liaisons: Interplay Between SWI/SNF, NuRD, and Polycomb in Chromatin Regulation and Cancer. Genes Dev (2019) 33(15-16):936–59. doi: 10.1101/gad.326066.119
132. Dan L, Liu L, Sun Y, Song J, Yin Q, Zhang G, et al. The Phosphatase PAC1 Acts as a T Cell Suppressor and Attenuates Host Antitumor Immunity. Nat Immunol (2020) 21(3):287–97. doi: 10.1038/s41590-019-0577-9
133. Hultqvist M, Olofsson P, Holmberg J, Backstrom BT, Tordsson J, Holmdahl R. Enhanced Autoimmunity, Arthritis, and Encephalomyelitis in Mice With a Reduced Oxidative Burst Due to a Mutation in the Ncf1 Gene. Proc Natl Acad Sci USA (2004) 101(34):12646–51. doi: 10.1073/pnas.0403831101
134. Yu YR, Imrichova H, Wang H, Chao T, Xiao Z, Gao M, et al. Disturbed Mitochondrial Dynamics in CD8(+) Tils Reinforce T Cell Exhaustion. Nat Immunol (2020) 21(12):1540–51. doi: 10.1038/s41590-020-0793-3
135. Fisicaro P, Barili V, Montanini B, Acerbi G, Ferracin M, Guerrieri F, et al. Targeting Mitochondrial Dysfunction can Restore Antiviral Activity of Exhausted HBV-Specific Cd8 T Cells in Chronic Hepatitis B. Nat Med (2017) 23(3):327–36. doi: 10.1038/nm.4275
136. Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, et al. Influence of Threonine Metabolism on S-Adenosylmethionine and Histone Methylation. Science (2013) 339(6116):222–6. doi: 10.1126/science.1226603
137. Roy DG, Chen J, Mamane V, Ma EH, Muhire BM, Sheldon RD, et al. Methionine Metabolism Shapes T Helper Cell Responses Through Regulation of Epigenetic Reprogramming. Cell Metab (2020) 31(2):250–266 e259. doi: 10.1016/j.cmet.2020.01.006
138. Li J, Chen F, Zheng M, Zhu H, Zhao D, Liu W, et al. Inhibition of STAT1 Methylation Is Involved in the Resistance of Hepatitis B Virus to Interferon Alpha. Antiviral Res (2010) 85(3):463–9. doi: 10.1016/j.antiviral.2009.10.011
139. Feld JJ, Modi AA, El-Diwany R, Rotman Y, Thomas E, Ahlenstiel G, et al. S-Adenosyl Methionine Improves Early Viral Responses and Interferon-Stimulated Gene Induction in Hepatitis C Nonresponders. Gastroenterology (2011) 140(3):830–9. doi: 10.1053/j.gastro.2010.09.010
140. Klein Geltink RI, Edwards-Hicks J, Apostolova P, O’Sullivan D, Sanin DE, Patterson AE, et al. Metabolic Conditioning of CD8(+) Effector T Cells for Adoptive Cell Therapy. Nat Metab (2020) 2(8):703–16. doi: 10.1038/s42255-020-0256-z
141. Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, et al. Effector CD8 T Cells Dedifferentiate Into Long-Lived Memory Cells. Nature (2017) 552(7685):404–9. doi: 10.1038/nature25144
142. Cao L, Zhu T, Lang X, Jia S, Yang Y, Zhu C, et al. Inhibiting DNA Methylation Improves Survival in Severe Sepsis by Regulating NF-KappaB Pathway. Front Immunol (2020) 11:1360. doi: 10.3389/fimmu.2020.01360
143. Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, et al. Immune Regulation by Low Doses of the DNA Methyltransferase Inhibitor 5-Azacitidine in Common Human Epithelial Cancers. Oncotarget (2014) 5(3):587–98. doi: 10.18632/oncotarget.1782
144. Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, et al. Alterations of Immune Response of Non-Small Cell Lung Cancer With Azacytidine. Oncotarget (2013) 4(11):2067–79. doi: 10.18632/oncotarget.1542
145. Mikyskova R, Indrova M, Vlkova V, Bieblova J, Simova J, Parackova Z, et al. DNA Demethylating Agent 5-Azacytidine Inhibits Myeloid-Derived Suppressor Cells Induced by Tumor Growth and Cyclophosphamide Treatment. J Leukoc Biol (2014) 95(5):743–53. doi: 10.1189/jlb.0813435
146. Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb Group Protein EZH2 Directly Controls DNA Methylation. Nature (2006) 439(7078):871–4. doi: 10.1038/nature04431
147. Zhang F, Zhou X, DiSpirito JR, Wang C, Wang Y, Shen H. Epigenetic Manipulation Restores Functions of Defective CD8(+) T Cells From Chronic Viral Infection. Mol Ther (2014) 22(9):1698–706. doi: 10.1038/mt.2014.91
148. Chandran A, Antony C, Jose L, Mundayoor S, Natarajan K, Kumar RA. Mycobacterium Tuberculosis Infection Induces HDAC1-Mediated Suppression of IL-12B Gene Expression in Macrophages. Front Cell Infect Microbiol (2015) 5:90. doi: 10.3389/fcimb.2015.00090
149. Pennini ME, Pai RK, Schultz DC, Boom WH, Harding CV. Mycobacterium Tuberculosis 19-kDa Lipoprotein Inhibits IFN-gamma-Induced Chromatin Remodeling of MHC2TA by TLR2 and MAPK Signaling. J Immunol (2006) 176(7):4323–30. doi: 10.4049/jimmunol.176.7.4323
150. Wang Y, Curry HM, Zwilling BS, Lafuse WP. Mycobacteria Inhibition of IFN-Gamma Induced HLA-DR Gene Expression by Up-Regulating Histone Deacetylation at the Promoter Region in Human THP-1 Monocytic Cells. J Immunol (2005) 174(9):5687–94. doi: 10.4049/jimmunol.174.9.5687
151. Coussens AK, Wilkinson RJ, Martineau AR. Phenylbutyrate Is Bacteriostatic Against Mycobacterium Tuberculosis and Regulates the Macrophage Response to Infection, Synergistically With 25-Hydroxy-Vitamin D3. PloS Pathog (2015) 11(7):e1005007. doi: 10.1371/journal.ppat.1005007
152. Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, Cruz KA, et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers Into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating Mdscs. Cancer Immunol Res (2018) 6(12):1561–77. doi: 10.1158/2326-6066.CIR-18-0070
153. Bhaskar A, Kumar S, Khan MZ, Singh A, Dwivedi VP, Nandicoori VK. Host Sirtuin 2 as an Immunotherapeutic Target Against Tuberculosis. Elife (2020) 9. doi: 10.7554/eLife.55415
154. Yousafzai NA, Zhou Q, Xu W, Shi Q, Xu J, Feng L, et al. SIRT1 Deacetylated and Stabilized XRCC1 to Promote Chemoresistance in Lung Cancer. Cell Death Dis (2019) 10(5):363. doi: 10.1038/s41419-019-1592-3
155. Wen H, Dou Y, Hogaboam CM, Kunkel SL. Epigenetic Regulation of Dendritic Cell-Derived Interleukin-12 Facilitates Immunosuppression After a Severe Innate Immune Response. Blood (2008) 111(4):1797–804. doi: 10.1182/blood-2007-08-106443
156. Zhao D, Li Z, Liu X, Liu N, Bao X, Sun H, et al. Lymphocyte Expression of EZH2 Is Associated With Mortality and Secondary Infectious Complications in Sepsis. Int Immunopharmacol (2020) 89(Pt B):107042. doi: 10.1016/j.intimp.2020.107042
157. Wee ZN, Li Z, Lee PL, Lee ST, Lim YP, Yu Q. EZH2-Mediated Inactivation of IFN-gamma-JAK-STAT1 Signaling Is an Effective Therapeutic Target in MYC-driven Prostate Cancer. Cell Rep (2014) 8(1):204–16. doi: 10.1016/j.celrep.2014.05.045
158. Xiao G, Jin LL, Liu CQ, Wang YC, Meng YM, Zhou ZG, et al. EZH2 Negatively Regulates PD-L1 Expression in Hepatocellular Carcinoma. J Immunother Cancer (2019) 7(1):300. doi: 10.1186/s40425-019-0784-9
159. Tiffen JC, Gallagher SJ, Tseng HY, Filipp FV, Fazekas de St. Groth B, Hersey P. EZH2 as a Mediator of Treatment Resistance in Melanoma. Pigment Cell Melanoma Res (2016) 29(5):500–7. doi: 10.1111/pcmr.12481
160. Koyanagi M, Baguet A, Martens J, Margueron R, Jenuwein T, Bix M. EZH2 and Histone 3 Trimethyl Lysine 27 Associated With IL4 and IL13 Gene Silencing in Th1 Cells. J Biol Chem (2005) 280(36):31470–7. doi: 10.1074/jbc.M504766200
161. Yaqubi M, Mohammadnia A, Fallahi H. Transcription Factor Regulatory Network for Early Lung Immune Response to Tuberculosis in Mice. Mol Med Rep (2015) 12(2):2865–71. doi: 10.3892/mmr.2015.3721
162. Wang Y, Zhong H, Xie X, Chen CY, Huang D, Shen L, et al. Long Noncoding RNA Derived From CD244 Signaling Epigenetically Controls CD8+ T-Cell Immune Responses in Tuberculosis Infection. Proc Natl Acad Sci USA (2015) 112(29):E3883–3892. doi: 10.1073/pnas.1501662112
163. Zhang Q, Sun H, Zhuang S, Liu N, Bao X, Liu X, et al. Novel Pharmacological Inhibition of EZH2 Attenuates Septic Shock by Altering Innate Inflammatory Responses to Sepsis. Int Immunopharmacol (2019) 76:105899. doi: 10.1016/j.intimp.2019.105899
164. Barili V, Fisicaro P, Montanini B, Acerbi G, Filippi A, Forleo G, et al. Targeting p53 and Histone Methyltransferases Restores Exhausted CD8+ T Cells in HCV Infection. Nat Commun (2020) 11(1):604. doi: 10.1038/s41467-019-14137-7
165. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic Silencing of TH1-Type Chemokines Shapes Tumour Immunity and Immunotherapy. Nature (2015) 527(7577):249–53. doi: 10.1038/nature15520
166. Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, et al. The Polycomb Protein Ezh2 Regulates Differentiation and Plasticity of CD4(+) T Helper Type 1 and Type 2 Cells. Immunity (2013) 39(5):819–32. doi: 10.1016/j.immuni.2013.09.012
167. Reynolds N, Salmon-Divon M, Dvinge H, Hynes-Allen A, Balasooriya G, Leaford D, et al. NuRD-mediated Deacetylation of H3K27 Facilitates Recruitment of Polycomb Repressive Complex 2 to Direct Gene Repression. EMBO J (2012) 31(3):593–605. doi: 10.1038/emboj.2011.431
168. Mbanwi AN, Wang C, Geddes K, Philpott DJ, Watts TH. Irreversible Splenic Atrophy Following Chronic LCMV Infection Is Associated With Compromised Immunity in Mice. Eur J Immunol (2017) 47(1):94–106. doi: 10.1002/eji.201646666
169. Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 Mediated LAG-3 Disruption in CAR-T Cells. Front Med (2017) 11(4):554–62. doi: 10.1007/s11684-017-0543-6
170. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science (2012) 337(6096):816–21. doi: 10.1126/science.1225829
171. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science (2013) 339(6121):819–23. doi: 10.1126/science.1231143
172. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-Guided Human Genome Engineering Via Cas9. Science (2013) 339(6121):823–6. doi: 10.1126/science.1232033
173. Thakore PI, Black JB, Hilton IB, Gersbach CA. Editing the Epigenome: Technologies for Programmable Transcription and Epigenetic Modulation. Nat Methods (2016) 13(2):127–37. doi: 10.1038/nmeth.3733
174. Hilton IB, Gersbach CA. Enabling Functional Genomics With Genome Engineering. Genome Res (2015) 25(10):1442–55. doi: 10.1101/gr.190124.115
175. Pickar-Oliver A, Gersbach CA. The Next Generation of CRISPR-Cas Technologies and Applications. Nat Rev Mol Cell Biol (2019) 20(8):490–507. doi: 10.1038/s41580-019-0131-5
176. Li L, Gao Y, Srivastava R, Wang W, Xiong Q, Fang Z, et al. Lentiviral Delivery of Combinatorial CAR/CRISPRi Circuit Into Human Primary T Cells Is Enhanced by TBK1/IKKvarepsilon Complex Inhibitor BX795. J Transl Med (2020) 18(1):363. doi: 10.1186/s12967-020-02526-2
177. Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell (2013) 152(5):1173–83. doi: 10.1016/j.cell.2013.02.022
178. Li J, Mahata B, Escobar M, Goell J, Wang K, Khemka P, et al. Programmable Human Histone Phosphorylation and Gene Activation Using a CRISPR/Cas9-Based Chromatin Kinase. Nat Commun (2021) 12(1):896. doi: 10.1038/s41467-021-21188-2
179. Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The Human Transcription Factors. Cell (2018) 172(4):650–65. doi: 10.1016/j.cell.2018.01.029
180. Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell (2013) 154(2):442–51. doi: 10.1016/j.cell.2013.06.044
181. Thakore PI, D’Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, et al. Highly Specific Epigenome Editing by CRISPR-Cas9 Repressors for Silencing of Distal Regulatory Elements. Nat Methods (2015) 12(12):1143–9. doi: 10.1038/nmeth.3630
182. Stepper P, Kungulovski G, Jurkowska RZ, Chandra T, Krueger F, Reinhardt R, et al. Efficient Targeted DNA Methylation With Chimeric dCas9-Dnmt3a-Dnmt3L Methyltransferase. Nucleic Acids Res (2017) 45(4):1703–13. doi: 10.1093/nar/gkw1112
183. Amabile A, Migliara A, Capasso P, Biffi M, Cittaro D, Naldini L, et al. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell (2016) 167(1):219–32.e214. doi: 10.1016/j.cell.2016.09.006
184. Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, et al. Epigenome Editing by a CRISPR-Cas9-Based Acetyltransferase Activates Genes From Promoters and Enhancers. Nat Biotechnol (2015) 33(5):510–7. doi: 10.1038/nbt.3199
185. Okada M, Kanamori M, Someya K, Nakatsukasa H, Yoshimura A. Stabilization of Foxp3 Expression by CRISPR-dCas9-Based Epigenome Editing in Mouse Primary T Cells. Epigenet Chromatin (2017) 10:24. doi: 10.1186/s13072-017-0129-1
186. Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, Iyer E,PR, et al. Highly Efficient Cas9-Mediated Transcriptional Programming. Nat Methods (2015) 12(4):326–8. doi: 10.1038/nmeth.3312
187. Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell (2014) 159(3):635–46. doi: 10.1016/j.cell.2014.09.039
188. Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, et al. Genome-Scale Transcriptional Activation by an Engineered CRISPR-Cas9 Complex. Nature (2015) 517(7536):583–8. doi: 10.1038/nature14136
189. Zalatan JG, Lee ME, Almeida R, Gilbert LA, Whitehead EH, La Russa M, et al. Engineering Complex Synthetic Transcriptional Programs With CRISPR RNA Scaffolds. Cell (2015) 160(1-2):339–50. doi: 10.1016/j.cell.2014.11.052
190. Wang G, Chow RD, Bai Z, Zhu L, Errami Y, Dai X, et al. Multiplexed Activation of Endogenous Genes by CRISPRa Elicits Potent Antitumor Immunity. Nat Immunol (2019) 20(11):1494–505. doi: 10.1038/s41590-019-0500-4
191. Sun X, Gao H, Yang Y, He M, Wu Y, Song Y, et al. Protacs: Great Opportunities for Academia and Industry. Signal Transduct Target Ther (2019) 4:64. doi: 10.1038/s41392-019-0101-6
192. Si J, Shi X, Sun S, Zou B, Li Y, An D, et al. Hematopoietic Progenitor Kinase1 (Hpk1) Mediates T Cell Dysfunction and Is a Druggable Target for T Cell-Based Immunotherapies. Cancer Cell (2020) 38(4):551–66.e511. doi: 10.1016/j.ccell.2020.08.001
193. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. c-Jun Overexpression in CAR T Cells Induces Exhaustion Resistance. Nature (2019) 576(7786):293–300. doi: 10.1038/s41586-019-1805-z
194. Zhang J, Lee SM, Shannon S, Gao B, Chen W, Chen A, et al. The Type III Histone Deacetylase Sirt1 Is Essential for Maintenance of T Cell Tolerance in Mice. J Clin Invest (2009) 119(10):3048–58. doi: 10.1172/JCI38902
Keywords: tolerance, immune exhaustion, epigenetics, chronic infections, bioengineering
Citation: Abhimanyu, Ontiveros CO, Guerra-Resendez RS, Nishiguchi T, Ladki M, Hilton IB, Schlesinger LS and DiNardo AR (2021) Reversing Post-Infectious Epigenetic-Mediated Immune Suppression. Front. Immunol. 12:688132. doi: 10.3389/fimmu.2021.688132
Received: 30 March 2021; Accepted: 17 May 2021;
Published: 07 June 2021.
Edited by:
Veron Ramsuran, University of KwaZulu-Natal, South AfricaReviewed by:
Aleksander M. Grabiec, Jagiellonian University, PolandKrishnendu Mukherjee, University Hospital Münster, Germany
Copyright © 2021 Abhimanyu, Ontiveros, Guerra-Resendez, Nishiguchi, Ladki, Hilton, Schlesinger and DiNardo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew R. DiNardo, QW5kcmV3LmRpbmFyZG9AYmNtLmVkdQ==; Larry S. Schlesinger, TFNjaGxlc2luZ2VyQHR4YmlvbWVkLm9yZw==
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship