 Maria J. E. Visser
Maria J. E. Visser Gareth Tarr
Gareth Tarr Etheresia Pretorius
Etheresia Pretorius- 1Department of Physiological Sciences, Faculty of Science, Stellenbosch University, Stellenbosch, South Africa
- 2Division of Rheumatology, Institute of Orthopaedics and Rheumatology, Winelands Mediclinic Orthopaedic Hospital, Stellenbosch, South Africa
Psoriasis (PsO) is a common T cell-mediated inflammatory disorder of the skin with an estimated prevalence of 2%. The condition manifests most commonly as erythematous plaques covered with scales. The aetiology of PsO is multifactorial and disease initiation involves interactions between environmental factors, susceptibility genes, and innate and adaptive immune responses. The underlying pathology is mainly driven by interleukin-17. In addition, various inflammatory mediators from specific T helper (TH) cell subsets, namely TH1, TH17, and TH22, are overexpressed in cutaneous lesions and may also be detected in the peripheral blood of psoriatic patients. Moreover, these individuals are also at greater risk, compared to the general population, of developing multiple comorbid conditions. Cardiovascular disease (CVD) has been recognised as a prominent comorbidity of PsO. A potential mechanism contributing to this association may be the presence of a hypercoagulable state in these individuals. Inflammation and coagulation are closely related. The presence of chronic, low-grade systemic inflammation may promote thrombosis – one of the major determinants of CVD. A pro-inflammatory milieu may induce the expression of tissue factor, augment platelet activity, and perturb the vascular endothelium. Altogether, these changes will result in a prothrombotic state. In this review, we describe the aetiology of PsO, as well as the pathophysiology of the condition. We also consider its relationship to CVD. Given the systemic inflammatory nature of PsO, we evaluate the potential contribution of prominent inflammatory mediators (implicated in PsO pathogenesis) to establishing a prothrombotic state in psoriatic patients.
Introduction
Psoriasis (PsO) is a chronic immune-mediated inflammatory disease of the skin, often associated with multiple comorbidities, affecting approximately 2% of the global population (1). Of those affected, 30% may develop an inflammatory arthritis (psoriatic arthritis). The reported prevalence of PsO in childhood may be up to 1.37% (2–4), while the estimated prevalence in adults ranges from 0.51% to 11.43% (3, 5, 6). Research suggests that disease development involves a complex interplay between genetic predisposition, environmental stimuli, and disordered innate and adaptive immune responses. Lesions may assume a variety of clinical forms (7), with plaque PsO being the most common disease variant. Typically, the condition manifests as well-circumscribed, erythematous papules and/or plaques covered with scales. In addition to its physical symptoms, PsO also imposes a significant psychosocial burden that may lead to anxiety, depression, and in severe cases, suicidality (8). The physical and psychological impact of PsO may significantly influence a patients’ quality of life (9), as well as contribute to an increased risk of mortality in severe forms of the disease (10).
PsO is considered to be a T cell-mediated disease and the corresponding cytokine profile of psoriatic lesions indicates important roles for interferon (IFN)-α, interleukin (IL)-22, the IL-23/IL-17 axis, and tumour necrosis factor (TNF) in psoriatic pathology (11–13). In addition to localised cutaneous inflammation, these molecules have also been detected in the systemic circulation of psoriatic patients – increasing the risk of comorbidities (14–16). Considering these observations, a paradigm shift has occurred from viewing PsO as merely ‘skin-deep’ to viewing it as a systemic inflammatory condition that can affect various extracutaneous tissues (17). Cardiovascular disease (CVD) is a notable comorbidity in patients suffering from PsO. An increased risk of major cardiovascular events, as well as an increased CVD mortality in severe forms of the disease, has been reported in these individuals (18–22). The relationship between PsO and CVD is widely acknowledged, however, the mechanisms responsible remain uncertain.
Hypercoagulability is a potential mechanistic link accounting for the association between CVD and PsO. It is becoming increasingly clear that inflammation and coagulation are interrelated processes (23, 24). A hypercoagulable state may develop as a result of an imbalance in haemostatic and inflammatory activity mediated by pro-inflammatory cytokines. These molecules may promote the initiation of coagulation, inhibit endogenous anticoagulant systems, and impair fibrinolytic activity (24, 25). The vascular endothelium also plays a central role in clot formation, as it is located at the nexus of inflammation and coagulation. Under normal physiological conditions, the resting endothelium displays anticoagulant and anti-inflammatory effects (26, 27). This homeostasis is disrupted in inflammatory conditions as a result of pro-inflammatory cytokines perturbing the vessel wall, resulting in a transition to an activated state that favours coagulation (28).
This review will briefly discuss genetic and environmental risk factors associated with the development of PsO, as well as the pathogenesis of the condition. In addition, the literature describing the systemic inflammatory nature of PsO and its subsequent relationship to CVD will also be considered. Finally, the potential contribution of prominent inflammatory mediators in PsO, to promote a prothrombotic state, is also appraised.
The Aetiology of Psoriasis
Genetic Risk Factors
The important role of a genetic component in the molecular pathogenesis of PsO has been supported by various family-based studies (29), including twin studies. An increased disease concordance rate has been reported in monozygotic twins, compared to dizygotic twins (30–32). Despite this disease concordance, the incidence never reaches 100% – suggesting a role for environmental factors in addition to genetic susceptibility in PsO development. The current view is that the mode of inheritance for PsO is multifactorial. Classic genome-wide linkage analysis has identified at least nine different chromosomal regions, termed psoriasis susceptibility (PSORS1–PSORS9) loci, which have shown statistically significant associations with PsO (33). Multiple studies have validated PSORS1 as the most important genomic region in PsO predisposition and that it may account for up to 35% of disease heritability (34). PSORS1 is located on human chromosome 6p21.3 within the region of the major histocompatibility complex (MHC). Human leukocyte antigen (HLA)-Cw*0602 has been accepted as the most likely PSORS1 disease allele (35). The HLA-C gene encodes a class 1 MHC protein and participates in the priming of cluster of differentiation (CD) 8+ T cell immune responses. More than 80 PsO susceptibility loci have been identified in genome-wide association studies (GWASs), conducted mainly on European and Asian populations (36). In 2007, in the first GWAS for PsO, two genes (IL12B and IL23R) were identified that were associated with PsO risk (37). Specific pathways implicated in PsO pathogenesis that have been identified through GWASs include interferon signalling, the IL-23 pathway, the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) cascade and the regulation of T cell responses (38–42).
Environmental Risk Factors
Environmental triggers play a critical role in the onset and development of PsO in genetically predisposed individuals through the interaction with genes and the induction of epigenetic modifications (43). Various environmental exposures have been associated with the initiation and/or exacerbation of psoriatic lesions. Stressful life events are well known to contribute to the initiation and aggravation of PsO. A potential explanation for stress-induced PsO onset or flares may be dysregulation of hypothalamic–pituitary–adrenal axis activity (44, 45). Obesity is another significant risk factor for PsO and body mass index has been demonstrated to correlate with disease severity (46). Potential mediators of this association may include adipokines, such as leptin and resistin, which possess pro-inflammatory actions (47). Furthermore, epidemiological studies have suggested an association between smoking and the development of PsO, based on the increased incidence of PsO reported among current and former smokers compared to non-smokers (48–50). Smoking may induce PsO by augmenting the existing systemic oxidative stress, interacting with immune cells, and altering gene expression (51, 52). Bacterial infections, specifically by Staphylococcus aureus and Streptococcus pyogenes, are recognised triggers of PsO. β-haemolytic streptococci have been isolated more frequently from throat swabs from PsO patients, compared to controls (53), and enterotoxins from Staphylococcus aureus have been linked to more severe PsO (54). Dysbiosis of both the gut and skin microbiome has become a recurrent theme in psoriatic individuals (55). Gut dysbiosis, characterised by a decrease in microbes with anti-inflammatory and immunomodulatory properties (56, 57), seems to be prevalent in these individuals (58, 59). An altered skin microbiome has also been associated with the condition (60, 61).
Pathogenesis of Psoriasis
Environmental factors (e.g., stress, obesity, smoking, infection, and dysbiosis) or physical trauma (the Koebner phenomenon) may perturb keratinocytes, resulting in the release of self-deoxyribonucleic acid (DNA) or self-ribonucleic acid (RNA) (62, 63). In turn, these self-nucleic acids may form a complex with the endogenous antimicrobial peptide (AMP), cathelicidin/LL37 (63). DNA-LL37 complexes activate plasmacytoid dendritic cells (pDCs), via Toll-like receptor (TLR) 9 signalling, which secrete high levels of IFN-α (64). In a similar fashion, RNA-LL37 complexes activate pDCs in a TLR7-dependent manner (62). Consequently, conventional dendritic cells (cDCs), which function as professional antigen-presenting cells (APCs), are activated by IFN-α. RNA-LL37 complexes also possess the capacity to activate cDCs directly, leading to the production of IL-6 and TNF by these cells. Once activated, cDCs migrate to secondary lymphoid organs and secrete IL-12 and IL-23 (65), which induce the differentiation of naïve CD4+ T cells into TH1 (66) and TH17 (67) cells, respectively. This process provides a potential mechanism for the initiation of the inflammatory sequelae in PsO and highlights the central role of DCs to establish a link between the innate and adaptive branches of the immune system.
TH1 cells express cytokines such as IFN-γ, IL-2, and TNF (66, 68). IFN-γ induces the expression of several chemokines and cytokines in the skin and also promotes the accumulation and infiltration of inflammatory cells (69, 70). TNF has been identified as a key regulatory molecule in the cytokine network of PsO. In addition to TH1 cells, a distinct subset of DCs, namely TNF- and inducible nitric oxide synthase-producing DCs, also release large amounts of TNF (71). It has been suggested that TNF production mediates the proliferation of resident T cells in the development of psoriatic lesions (72). In addition, TNF regulates the expression of cell adhesion molecules, which mediate the extravasation of leukocytes, on endothelial cells and keratinocytes in psoriatic skin (73). TNF also stimulate keratinocytes to induce the expression of pro-inflammatory cytokines such as IL-6, IL-8, and TNF itself, through the activation of NF-κB (74). Furthermore, TNF may synergise with IL-17 to enhance the expression of key inflammatory genes in keratinocytes (75).
TH17 cytokines such as IL-17A, IL-17F, and IL-22 (76, 77) are key pathogenic effectors in PsO. Therefore, PsO has been regarded as a TH17-mediated disease. However, this paradigm is shifting towards understanding PsO as an IL-17-driven disease (78). Various other cellular sources of IL-17 exist, including CD8+ T cells (comprising mucosa-associated invariant T cells and conventional T cells) (79), dermal γδ T cells (80), group 3 innate lymphoid cells (81), mast cells, and neutrophils (82). IL-17A has been shown to govern the expression of signature PsO genes in keratinocytes (83). IL-17 acts on keratinocytes to induce the expression of AMPs, such as β-defensin 2, and neutrophil chemoattractants, namely C-X-C motif chemokine ligand (CXCL)1, CXCL3, CXCL5, CXCL6, and CXCL8 (70). Specifically, IL-17F promotes neutrophil accumulation in the dermis by stimulating the release of IL-8 by keratinocytes (84). TH17 cytokines (IL-17A, IL-22, and TNF) also cause keratinocytes to produce C-C motif chemokine ligand (CCL) 20 and its receptor, C-C motif chemokine receptor (CCR) 6, thereby facilitating the recruitment and infiltration of TH17 cells (85). IL-17A-induced keratinocyte production of IL-19 has been shown to upregulate the production of antimicrobial proteins, namely S100 calcium-binding protein (S100)A7, S100A8, and S100A9. IL-19 and IL-17A may also interact synergistically to enhance the keratinocyte response. These observations seem to suggest IL-19 as a potentially novel component of the IL-23/IL-17 axis (86), which plays a crucial role in the development of psoriatic inflammation (87). Apart from its role in TH17 differentiation and expansion, IL-23 has been demonstrated to induce dermal inflammation and epidermal hyperplasia – mediated through the combined effects of IL-17A (88) and IL-22 (77).
IL-22 is co-expressed with IL-17A and IL-17F by activated TH17 cells (76, 77) as well as TH22 cells (89). IL-22 does not affect immune cells (90) and primarily targets epithelial cells, mediating innate immune responses and contributing to wound healing (91). This cytokine has a dual nature, exhibiting both anti- and pro-inflammatory properties (92). IL-22 induces the overexpression of AMPs, such as β-defensin 2, and the S100 protein family (76, 93, 94). In addition, IL-22 has been reported to downregulate the expression of genes involved in the regulation of keratinocyte differentiation, resulting in acanthosis (94–96). As a pro-inflammatory cytokine, IL-22 stimulates keratinocyte production of chemokines (CCL2, CCL20, CXCL5, and CXCL8) (93, 94). These chemotactic agents will promote the infiltration of monocytes/macrophages (97), neutrophils (98), and T cells (99) at sites of cutaneous inflammation. A graphical representation of key cytokine circuits in the immunopathogenesis of PsO is provided in Figure 1.
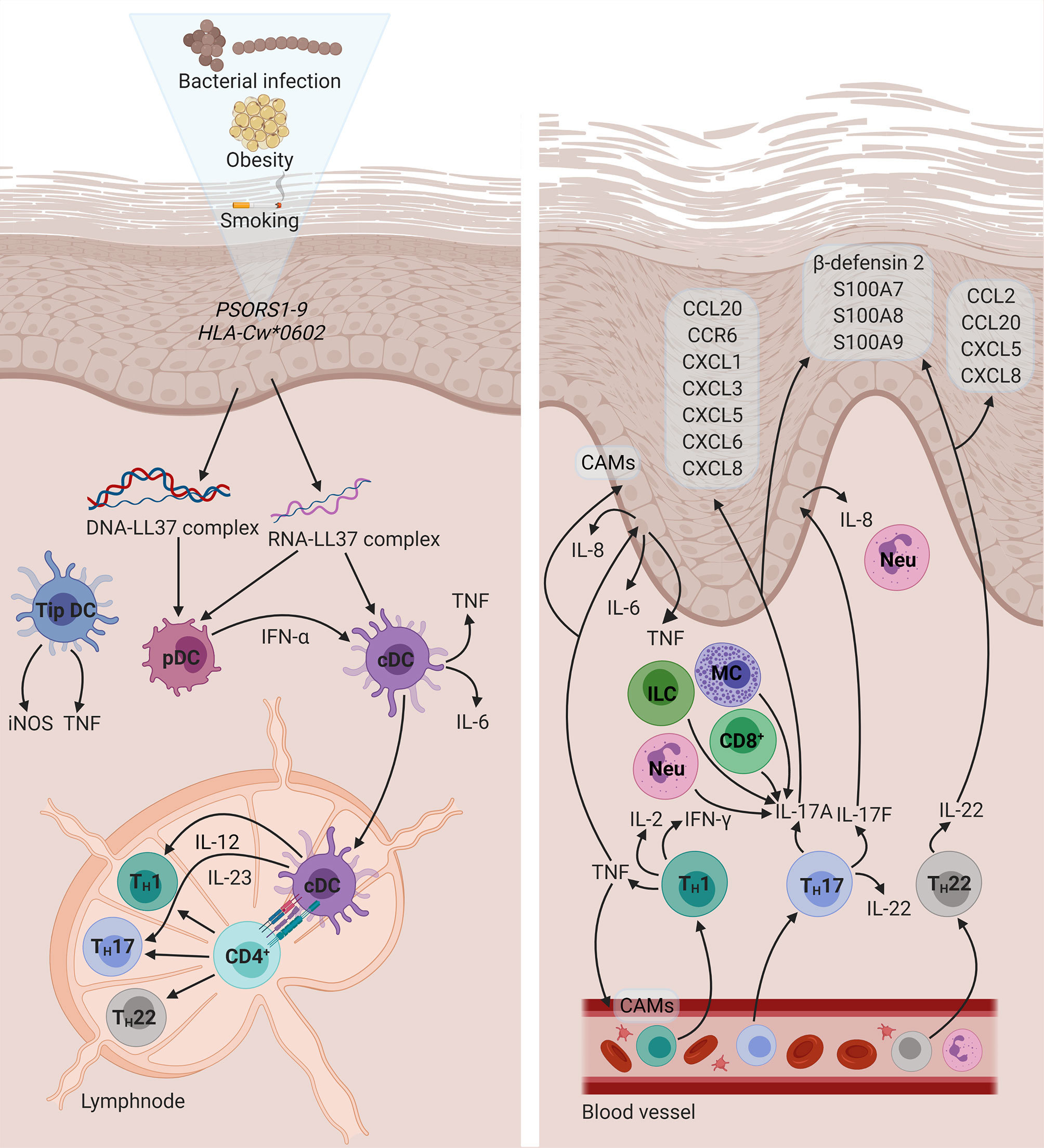
Figure 1 The immunopathogenesis of PsO. A combination of genetic and environmental factors activates pDCs. In turn, cDCs activate naïve CD 4+ T cells through the presentation of an unknown antigen. Subsequently, activated CD4+ T cells differentiate into TH1, TH17, and TH22 cells, which migrate to the dermis and give rise to a psoriatic plaque. Diagram created with BioRender.com. CAM, cell adhesion molecule; CCL, C-C motif chemokine ligand; CCR, C-C motif chemokine receptor; CD, cluster of differentiation; cDC, conventional dendritic cell; CXCL, C-X-C motif chemokine ligand; DNA, deoxyribonucleic acid; HLA, human leukocyte antigen; IFN, interferon; IL, interleukin; ILC, innate lymphoid cell; iNOS, inducible nitric oxide synthase; MC, mast cell; Neu, neutrophil; pDC, plasmacytoid dendritic cell; PSORS: psoriasis susceptibility; RNA, ribonucleic acid; S100, S100 calcium-binding protein; TH, T helper; Tip DC, tumour necrosis factor- and inducible nitric oxide synthase-producing dendritic cell; TNF, tumour necrosis factor.
Other cytokines that have been implicated in psoriatic inflammation include IL-9, IL-19, IL-20, IL-24, IL-33, and IL-36. IL-9 has been suggested to contribute to PsO pathogenesis via its pro-angiogenic activity and induction of IL-17 production (100). In addition, members of the IL-20 subfamily of cytokines (IL-19, IL-20, and IL-24) have been shown to affect keratinocyte proliferation and differentiation and to induce the expression of various PsO-related molecules (101). Indeed, suppression of these cytokines resulted in alleviation of epidermal hyperplasia in psoriatic patients (102). Furthermore, IL-33 was recently demonstrated to act on keratinocytes in an autocrine manner, thereby perpetuating the psoriatic inflammatory response (103). Finally, IL-36 has been implicated in keratinocyte-specific pathways that mediate dermal inflammation in PsO (104, 105).
Treatment of Psoriasis
When determining the appropriate treatment regimen for a PsO patient, various factors should be taken into consideration, such as disease severity the presence of psoriatic arthritis, comorbidities, and the impact on the patient’s quality of life. The severity of PsO may be determined by the percentage of the total body surface area (BSA) that is involved, with <3% BSA considered mild, 3–10% BSA considered moderate, and >10% BSA considered severe. Mild to moderate PsO is treated with topical agents including corticosteroids, calcineurin inhibitors, vitamin D3 analogues, keratolytic agents, anthralin, retinoids, and coal tar preparations (106). For moderate to severe PsO, phototherapy and systemic treatments are prescribed (107). Oral systemic therapies, such as methotrexate, apremilast, cyclosporine, and acitretin, possess anti-inflammatory and immunomodulatory properties (108). The advent of biologic systemic therapies has drastically changed the treatment of PsO. Currently, there are four classes of biologicals available, namely TNF inhibitors, IL-12/23 inhibitors, IL-17 inhibitors, and IL-23 inhibitors (109). These agents exert their effects by targeting prominent cytokines involved in the pathogenesis of PsO. When compared to traditional systemic agents these therapies have better safety profiles and may also be more efficacious (110).
The Systemic Inflammatory Nature of Psoriasis
PsO was initially primarily regarded as a hyperkeratotic disorder solely confined to affected skin areas. However, the systemic inflammatory nature of the condition has become increasingly apparent in recent years (111, 112). Various pro-inflammatory products are overexpressed in psoriatic skin lesions. These mediators also seem to be released into the systemic circulation of psoriatic patients and may reflect disease severity (113). Peripheral inflammation is evident by the abnormal expression of a host of inflammatory molecules in the blood of these individuals (113–116). Moreover, inflammation may also be detected at extracutaneous sites. In a pilot study by Mehta et al. (117), 18F-fluorodeoxyglucose emission tomography-computed tomography was utilised to localise and quantify inflammatory activity in individuals with moderate to severe PsO. The authors detected systemic inflammation in the skin, joints, liver, and vasculature, with significantly greater aortic and hepatic inflammation in psoriatic patients compared to age- and gender-matched controls (117). Furthermore, individuals with PsO have an increased risk of developing multiple comorbid diseases (presented in Table 1), which serves as further evidence for the presence of systemic inflammation in the condition. It has been proposed that the chronic course of PsO, as well as common inflammatory molecules and/or pathways, may act as the driving forces of the development and/or worsening of these extracutaneous manifestations (15, 111).

Table 1 Comorbidities associated with PsO.
Besides systemic comorbid conditions, individuals with PsO may also develop metabolic abnormalities. An increased prevalence of metabolic syndrome (MetS) – a constellation of dyslipidaemia, hypertension, insulin resistance, and visceral obesity – has been reported in psoriatic patients compared to healthy controls (132, 133, 138–140). However, the directionality of these associations remains unknown. It is possible that PsO may initiate inflammatory pathways that drive the development of metabolic disturbances. Inflamed adipose tissue produces adipocytokines (141) that may worsen existing PsO (142). We note that metabolic dysfunction may also contribute to an increased cardiovascular burden in psoriatic individuals. This is however, beyond the scope of this paper. There are several excellent review articles in which the association between MetS and PsO are discussed (143–145).
The Association Between Psoriasis and Cardiovascular Disease
In recent years, CVD has emerged as a particularly prominent comorbidity of PsO. Several epidemiological studies have reported an increased risk of CVD in psoriatic individuals, compared to the general population. Furthermore, recent guidelines for the primary prevention of CVD, by the American College of Cardiology and the American Heart Association, indicated PsO as a risk-enhancing factor for the development of atherosclerotic CVD (146).
In a landmark study by Gelfand and colleagues (20), patients with PsO were reported to have a higher incidence of myocardial infarction (MI). Patients with mild and severe PsO had an incidence of 4.04 (95% confidence interval [CI]: 3.88–4.21) and 5.13 (95% CI: 4.22–6.17) per 1000 person-years, compared to an incidence of 3.58 in healthy control subjects (95% CI: 3.52–3.65) (20). In addition, traditional CVD risk factors such as hyperlipidaemia, hypertension, and smoking were more prevalent among psoriatic individuals. PsO remained an independent risk factor for CVD, after adjusting for established CVD risk factors. Similar associations were noted in studies assessing the risk of stroke (19) and venous thromboembolism (VTE) (18) in psoriatic individuals. Gelfand et al. (19) reported an increased risk of stroke in mild (hazard ratio [HR] 1.06; 95% CI: 1.0–1.1) and severe (HR 1.43; 95% CI: 1.1–1.9) PsO, after adjusting for major risk factors (19). Furthermore, Ahlehoff and colleagues (18) reported higher incidence rates of VTE in psoriatic individuals (1.92 and 3.20 per 1000 person-years for mild and severe PsO) compared to healthy controls (1.29 per 1000 person-years) (18). In a meta-analysis by Gaeta and colleagues (147), it was shown that PsO confers an excess risk of 24% for the development of CVD (147). It has also been reported that severe disease activity contributes substantially to increased mortality due to CVD (148–150). Mehta et al. (21) identified severe PsO as an independent risk factor for CVD deaths (HR 1.57; 95% CI: 1.26–1.96), after adjusting for traditional CVD risk factors (21). Lastly, in a study by Abuabara and colleagues (149), it was determined that CVD was responsible for the highest absolute (61.9 deaths per 1000 person-years) and excess (3.1 deaths per 1000 person-years) risk in PsO patients (149).
The link between PsO and CVD may potentially be explained by the chronic course of the disease and the associated systemic inflammation. A considerable body of literature supports the notion that chronic low-grade systemic inflammation is a central theme in the pathogenesis and propagation of CVD (151–154). Moreover, elevated levels of C-reactive protein (CRP) – a sensitive marker of systemic inflammation – has been suggested as a predictor of future CVD events (155). Two landmark trials, namely the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) study (156), and the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) (157), also provided evidence for a prominent role of systemic inflammation in the etiopathogenesis of CVD. In the following section, we will elaborate on the interplay between inflammation and coagulation as well as how an imbalance in these activities may promote the development of CVD – specifically via thrombosis.
Psoriatic Inflammation and Coagulopathy
Inflammation and coagulation are interdependent processes, demonstrated by the dynamic crosstalk between these systems. Under normal physiological conditions, these systems function as protective mechanisms and are tightly regulated. However, dysregulation may result in chronic, systemic inflammation and/or thrombotic complications. In response to invading pathogens or tissue damage, inflammation ensues to eliminate the original insult and to promote wound healing and tissue repair. However, if the inflammatory process is not duly resolved; acute inflammation may transition to chronic, systemic inflammation. In turn, the sustained activation of the coagulation cascade, driven primarily by pro-inflammatory cytokines, may follow.
A particularly prominent molecule in the context of inflammation-induced coagulation is tissue factor (TF) (25). The tenase complex, comprising TF and factor VIIa, activates the extrinsic coagulation pathway, culminating in the generation of thrombin. In the final steps of coagulation, thrombin catalyses the conversion of soluble fibrinogen to insoluble fibrin. Pro-inflammatory cytokines may upregulate the expression of TF on endothelial cells and monocytes (158). In addition, inflammation favours the suppression of natural anticoagulant mechanisms, namely the antithrombin pathway, the protein C pathway, and tissue factor pathway inhibitor (TFPI) (24, 25). Finally, fibrinolytic activity may also decrease due to a continuous increase in plasminogen activator inhibitor (PAI)-1 levels stimulated by pro-inflammatory molecules (25). Thus, chronic systemic inflammation may alter the haemostatic balance to favour a prothrombotic state.
In turn, coagulation may also modulate and perpetuate the inflammatory response. Coagulation proteases may bind to protease-activated receptors on the activated endothelium, inducing the synthesis and expression of cell adhesion molecules (159, 160). These molecules play a pivotal role in the extravasation of leukocytes to sites of inflammation. Furthermore, activated coagulation factors may also elicit an inflammatory response by interacting with immune cells to induce the production of cytokines (161, 162). Thrombocytes or platelets are also increasingly recognised for their ability to mediate and regulate inflammation. Activated platelets release their granular content, which comprises a plethora of procoagulant and pro-inflammatory molecules. Platelets are also implicated in the recruitment of leukocytes and the regulation of vascular permeability (163).
From these observations, it is evident that inflammation and coagulation should not be viewed as separate entities and that an imbalance in these activities may culminate in the development of prothrombotic conditions. Thrombosis is the most common pathology underlying the three major cardiovascular conditions, namely ischaemic heart disease, ischaemic stroke, and VTE (164–166). Arterial thrombosis, which ensues after the rupture of an atherosclerotic plaque or damage to the vessel wall, may give rise to MI or stroke. Platelets play a central role in the formation of an arterial thrombus (167). In contrast, the pathomechanism of venous thrombosis, associated with deep vein thrombosis (DVT) and pulmonary embolism, is less clear. Virchow’s triad describes three factors, namely hypercoagulability, endothelial dysfunction, and altered blood flow, which may predispose an individual to the development of venous thrombosis (168).
As discussed in the previous sections, PsO is characterised by chronic, systemic inflammation and accompanied by an increased risk for CVD. A potential mechanism may be the development of a prothrombotic state, via the action of multiple pro-inflammatory cytokines, due to psoriatic pathology. The most prominent cytokines that could play a role in the development of a prothrombotic state - IL-6, IL-17, and TNF - will be discussed below. We focus specifically on the effects of these cytokines on platelets and the endothelium. In Figures 2 and 3 schematic representations are provided of the signalling pathways that could be initiated in platelets and endothelial cells, respectively, upon stimulation with these cytokines.
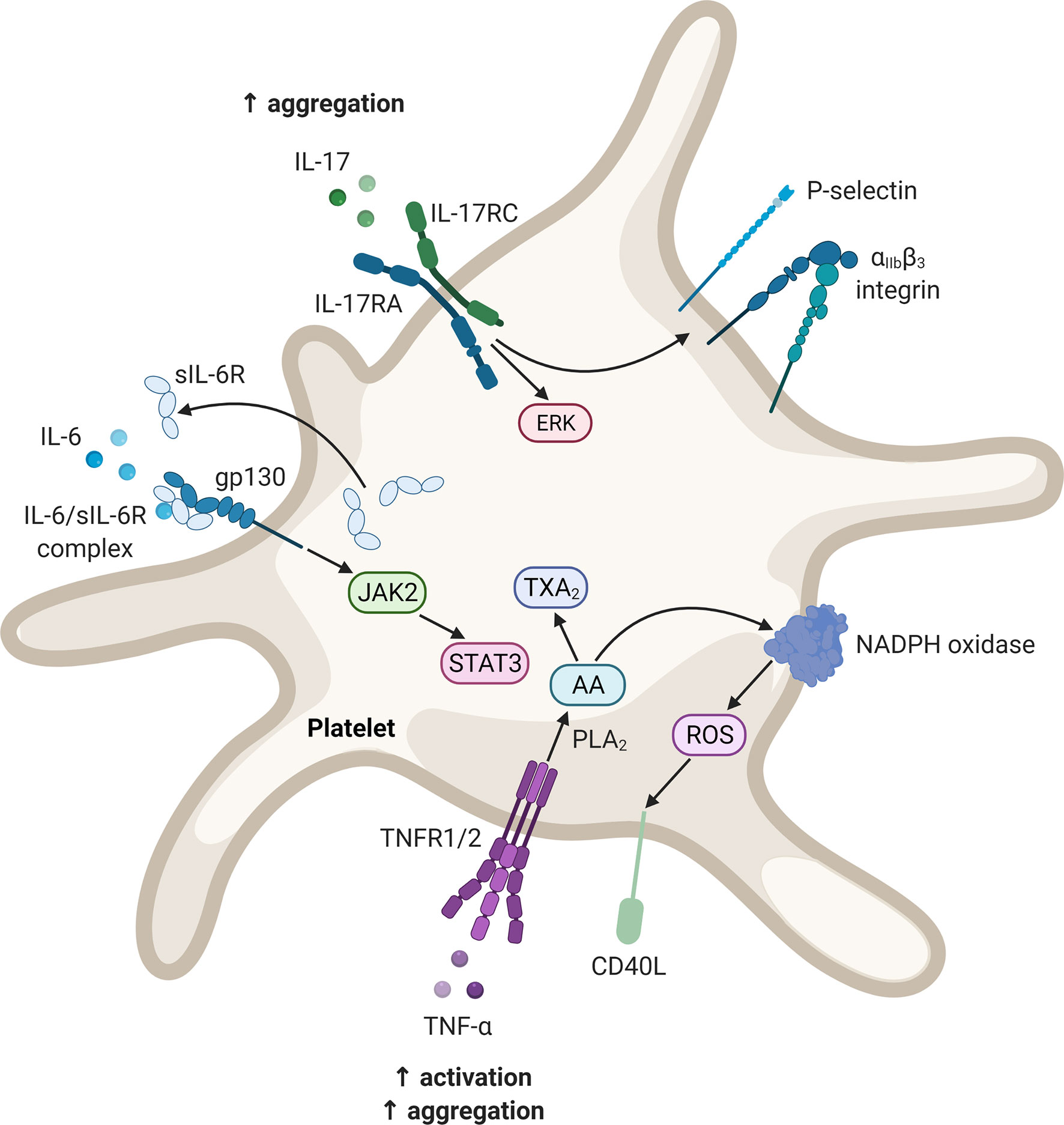
Figure 2 Potential signalling pathways in platelets upon stimulation with IL-6, IL-17, and TNF. These mediators may contribute to platelet activation as well as enhancing the platelet response to agonist-induced platelet activation. Diagram created with BioRender.com. AA, arachidonic acid; CD40L, CD40 ligand; ERK, extracellular signal-regulated kinase; gp, glycoprotein; IL, interleukin; IL-17RA, interleukin-17 receptor A; IL-17RC, interleukin-17 receptor C; JAK, Janus kinase; NADPH, nicotinamide adenine dinucleotide phosphate; PLA2, phospholipase A2; ROS, reactive oxygen species; sIL-6R, soluble interleukin-6 receptor; STAT, signal transducer and activator of transcription; TXA2, thromboxane A2; TNF, tumour necrosis factor; TNFR, tumour necrosis factor receptor.
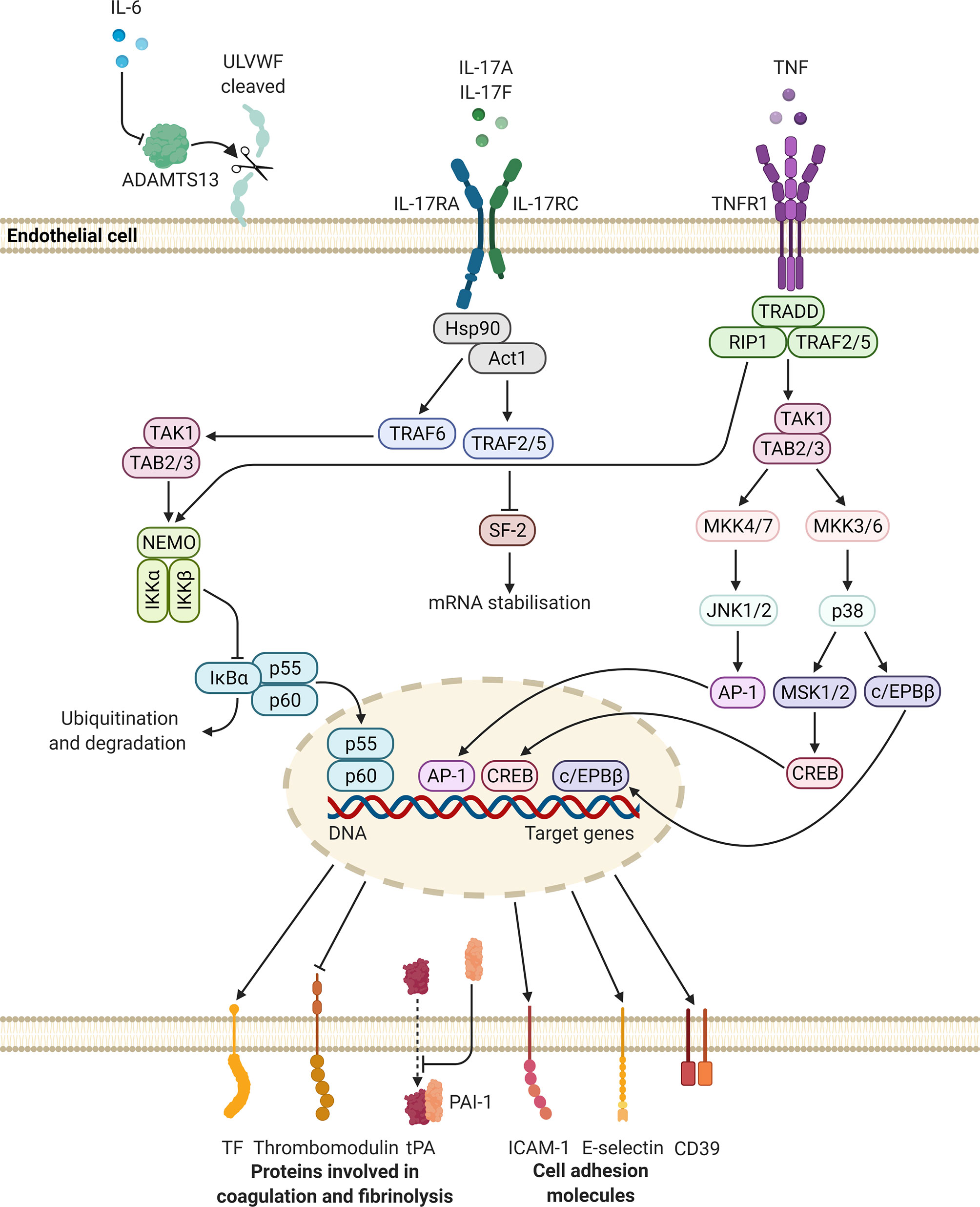
Figure 3 Potential intracellular signalling pathways initiated in endothelial cells, upon stimulation with IL-6, IL-17, and TNF. These mediators may upregulate the synthesis and release of molecules that promote platelet adhesion and coagulation, while suppressing the expression of proteins involved in fibrinolysis. Altogether these changes may lead to a prothrombotic state. Diagram created with BioRender.com. ADAMTS13, a disintegrin-like and metalloprotease with thrombospondin type 1 motif, 13; AP, activator protein; c/EPB, CCAAT/enhancer-binding protein; CD, cluster of differentiation; CREB, cAMP response element binding protein; DNA, deoxyribonucleic acid; Hsp, heat shock protein; ICAM-1, intercellular adhesion molecule-1; IKBα, NF-kappa-B inhibitor alpha; IKK, inhibitor of nuclear factor kappa-B kinase; IL, interleukin; IL-17RA, interleukin-17 receptor A; IL-17RC, interleukin-17 receptor C; JNK, c-Jun N-terminal kinase; MKK, mitogen-activated protein kinase kinase; MSK, mitogen- and stress-activated protein kinase; NEMO, NF-kappa-B essential modulator; PAI-1, plasminogen activator inhibitor-1; SF, splicing factor; TF, tissue factor; TNF, tumour necrosis factor; TNFR, tumour necrosis factor receptor; tPA, tissue-type plasminogen activator; TRADD, tumour necrosis factor receptor 1-associated death domain protein; TRAF, tumour necrosis factor receptor-associated factor; RIP, receptor-interacting protein kinase; ULVWF, ultra-large von Willebrand factor.
Interleukin-6
IL-6 is a pleiotropic cytokine involved in several physiological processes including the acute-phase response and antibody production (169). The effects of IL-6 are mediated via two receptors, namely the IL-6 receptor (IL-6R) and the signal-transducing subunit, glycoprotein (gp) 130 (169). In PsO, IL-6, combined with IL-23, contributes to the differentiation of TH17 cells (12). The overexpression of this cytokine has been detected in psoriatic skin lesions (170) as well as in the circulation of these individuals (113, 171, 172). In addition, IL-6 signalling has been suggested to diminish regulatory T cell activity in PsO, thereby allowing the expansion of effector T cells (170). In the context of CVD, IL-6 has emerged as a pivotal mediator of thrombotic disease. Raised IL-6 levels have been found to be related to recurrent venous thrombosis (173), with detectable levels of IL-6 associated with a two-fold increase in the risk of venous thrombosis (174). The potential involvement of IL-6 in the development of adverse cardiovascular events is further supported by findings of a sub-study of the CANTOS trial. In this study, baseline IL-6 levels were associated with an increased risk of cardiovascular events. More importantly, it was demonstrated for the first time that lowering IL-6 levels – via the inhibition of IL-1β by canakinumab – resulted in a 15% reduction of atherothrombotic events (MI and stroke) (175).
During inflammation, IL-6 acts on hepatocytes to induce the synthesis of acute-phase proteins such as CRP, fibrinogen, and serum amyloid A (SAA) (176). Both CRP and SAA have demonstrated the ability to promote coagulation via the induction of TF synthesis in endothelial cells and suppression of the TFPI pathway (177, 178). With regard to fibrinogen, it has been suggested that a causal relationship exists between hyperfibrinogenaemia and thrombosis (179).
Although platelets do not express IL-6R, they have been shown to express gp130 (180, 181). Upon thrombin-induced platelet activation, the soluble form of IL-6R is secreted which can form a complex with IL-6 (180, 181). This complex may activate gp130, in a process termed trans-signalling, resulting in the activation of signal transducer and activator of transcription (STAT) 3 (181). However, IL-6 trans-signalling does not seem to affect platelet activation and/or aggregation. Nevertheless, IL-6 may promote platelet adhesion and aggregation indirectly. IL-6 inhibits the activity of a disintegrin-like and metalloprotease with thrombospondin type 1 motif, 13 (ADAMTS13) (182). ADAMTS13 is responsible for the cleavage of ultra-large von Willebrand factor multimers released by the endothelium. In the ultra-large form, these multimers are hyperreactive and may interact with platelets to induce adhesion and aggregation.
Interleukin-17
IL-17 plays a pivotal role in the innate immune response, particularly in host defence against microbial invasion (183). The IL-17 family consists of six members, namely IL-17A to IL-17F, and signalling is mediated through the IL-17 receptor (IL-17R) family that comprises five members, IL-17RA to IL-17RE (184). This pro-inflammatory cytokine has been implicated in the pathogenesis of diverse autoimmune and inflammatory diseases, including PsO (185). Indeed, IL-17 is regarded as the principal driver of psoriatic inflammation. The overexpression of this cytokine has been detected in lesional psoriatic skin, when compared to nonlesional skin (186, 187). In addition, serum levels of IL-17A are elevated in psoriatic patients (188) and may also reflect disease severity (186). With respect to cardiovascular pathology, conflicting results have been reported on the role of IL-17. Some studies have found a protective effect of IL-17 (189), while others have reported a pro-atherogenic role for IL-17 (190). Nevertheless, prothrombotic effects of IL-17 have been demonstrated. In a murine model of DVT, IL-17A was found to promote thrombus formation by enhancing platelet aggregation and neutrophil infiltration of thrombi (191). Moreover, it has been shown that targeted blocking of IL-12/23 and IL-17 resulted in improved skin phenotype and lengthened clotting times to occlusive thrombus formation in a murine model of psoriatic disease (192).
Platelets have been demonstrated to express a functional IL-17 receptor, namely IL-17RA (193). Exposing activated platelets (induced by adenosine diphosphate) to IL-17 augments platelet aggregation (193–195). This action is suggested to be mediated by the opening of the mitochondrial permeability transition pore (196) and the phosphorylation of extracellular signal-regulated kinase-2 (194, 196). Furthermore, stimulation with IL-17 causes an increased expression and accelerated externalisation of P-selectin and exposure of the αIIbβ3 integrin (193, 194, 196). Both of the aforementioned molecules facilitate platelet aggregation and are used as markers of platelet activation.
IL-17 may also alter the endothelium towards an activated state. IL-17RA is constitutively expressed in endothelial cells (197). It has been reported that IL-17 and TNF act synergistically to activate the endothelium, resulting in the synthesis of inflammatory mediators, and the expression of cell adhesion molecules (E-selectin and intercellular adhesion molecule-1) and TF by these cells (195). Anticoagulant activity by the endothelium also becomes diminished, as thrombomodulin expression is downregulated (195). It should be noted that IL-17 alone is not a strong inducer of inflammatory activity, however, IL-17 in concert with TNF potently induce pro-inflammatory gene expression. This effect is thought to be mediated by the ability of IL-17 to stabilise messenger RNA (198, 199). Finally, the vascular expression of CD39, an inhibitor of platelet aggregation, is also downregulated by IL-17A (195, 200).
Tumour Necrosis Factor
TNF is a potent inflammatory cytokine, orchestrating various processes such as inflammation, cell differentiation, and apoptosis (201). The effects of TNF are mediated by two receptors, namely TNF receptor (TNFR) 1 and TNFR2 (202). In PsO, this cytokine amplifies the inflammatory response via the generation of reactive oxygen species and inducing the expression of cytokines and cell adhesion molecules (73, 203). Increased TNF activity has been detected in involved skin from psoriatic individuals (204) and TNF levels were also elevated in the serum of these individuals (113, 205). In relation to cardiovascular diseases, increased concentrations of TNF have been associated with recurrent coronary events (206), and venous thrombosis (174). In addition, TNF levels have been suggested to be an independent predictor of cardiovascular events such as MI, stroke, and CVD mortality (207). Regarding the prothrombotic properties of TNF, discrepant results have been reported. In a murine model of atherothrombosis, potent antithrombotic effects of TNF were reported (208). In contrast, another study found that treatment with this cytokine resulted in accelerated thrombus formation (209).
It has been suggested that TNF may promote the (hyper)activation of platelets by interacting with TNFR1 and TNFR2, which is expressed on the platelet membrane (210). TNF may also facilitate platelet activation by stimulating the arachidonic acid pathway (210). Furthermore, CD40 ligand (CD40L) expression by activated platelets may be induced by TNF via an arachidonic acid-dependent oxidative stress mechanism (211, 212). In turn, CD40L enhances platelet activation and aggregation, as well as thrombus formation (213). TNF also induces the release of large von Willebrand factor multimers from endothelial cells, which propagates platelet thrombus formation (182). Nevertheless, the ability of TNF to stimulate platelet activation directly remains contested, with some studies reporting no effect (209).
TNF may alter the properties of the endothelium by inducing the synthesis of procoagulant molecules and suppressing natural anticoagulant mechanisms. Engagement of TNF with TNFR1 results in the expression of TF on the surface of endothelial cells and the subsequent deposition of fibrin (209, 214–216). In addition, the production of platelet-activating factor (PAF), by the endothelium, is also induced by TNF (217, 218). PAF induces platelet aggregation (219) and functions as a potent inflammatory mediator (220). The activity of the anticoagulant molecule, activated protein C, is dependent on the presence of functional endothelial thrombomodulin, which has been shown to be downregulated by TNF (216). Mechanistically, this inhibitory effect is mediated by downregulating the transcription of the thrombomodulin gene (209, 221). Finally, TNF inhibits fibrinolysis by induction of the PAI-1 gene (209) through the activity of NF-κB (222) and by decreasing the release of tPA (223).
Concluding Remarks
PsO is an immune-mediated inflammatory disorder of the skin, characterised by the overexpression of TH1-, TH17-, and TH22-derived inflammatory cytokines. These mediators are upregulated in the lesional skin of psoriatic individuals and may also be released into the circulatory system of these patients. The systemic inflammatory nature of the condition is reflected by a multitude of dysregulated inflammatory molecules, which may give rise to various comorbidities. PsO has been identified as an independent risk factor for the development of CVD (Figure 4). The chronic subclinical systemic inflammation associated with the condition may predispose psoriatic individuals to thrombosis – a major determinant of CVD. Inflammatory cytokines may contribute to the development of a prothrombotic state, via the induction of TF, platelet activation and/or enhancing the platelet response, and endothelial dysfunction.
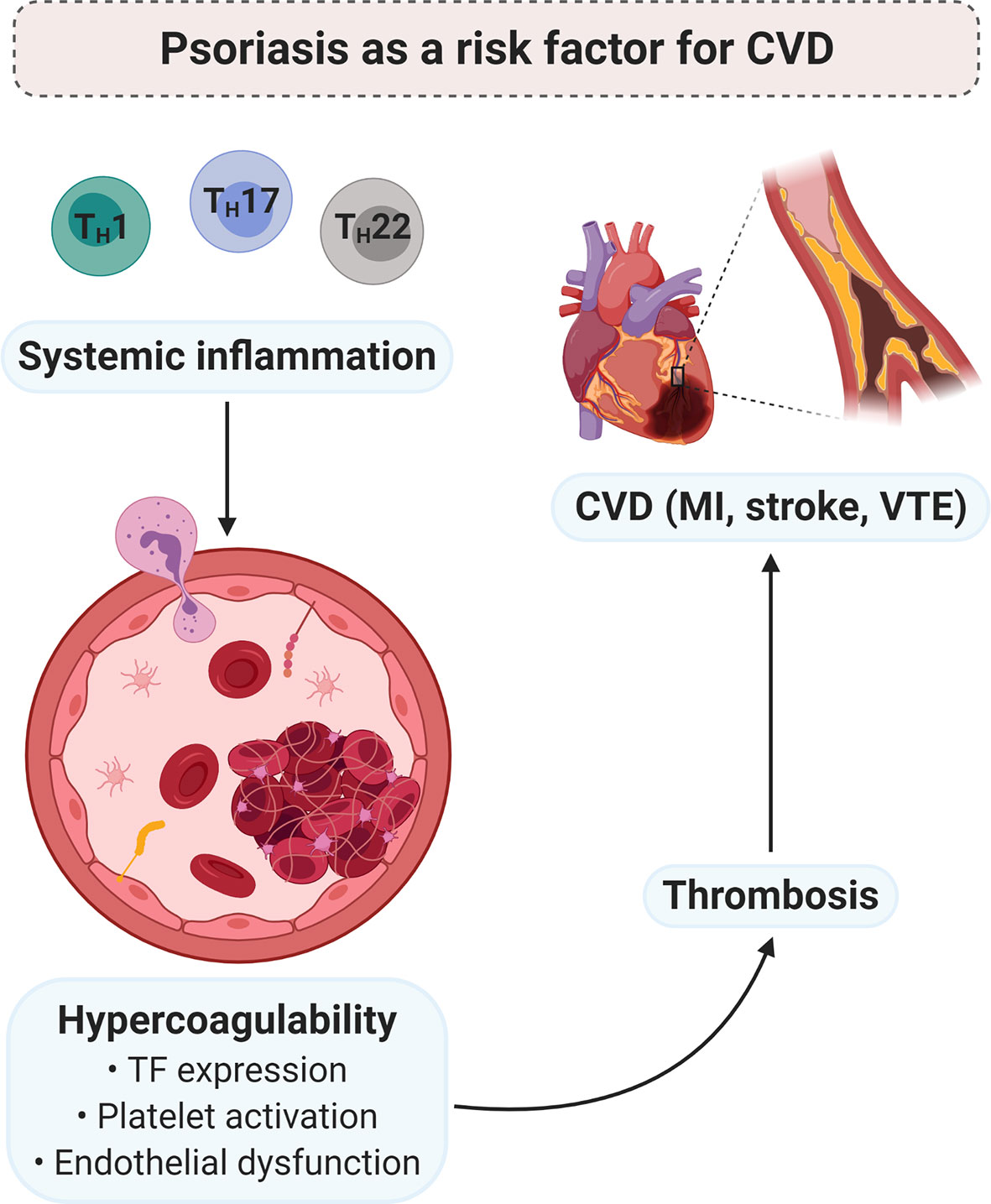
Figure 4 PsO as a risk factor for CVD. Chronic low-grade systemic inflammation, due to psoriatic pathology, may contribute to the development of a hypercoagulable state. Pro-inflammatory mediators induce the expression of TF, enhance platelet activation and/or the platelet response, and endothelial dysfunction. In turn, hypercoagulability predisposes thrombosis, which is the major underlying pathology of CVD. Diagram created with BioRender.com. CVD, cardiovascular disease; MI, myocardial infarction; TF, tissue factor; TH, T helper; VTE, venous thromboembolism.
The exact mechanisms underlying the association between PsO and CVD remains elusive. However, it is imperative that both physicians and patients must be aware of the potential cardiovascular risk that PsO may pose. Therefore, effective management of the condition should not only aim to ameliorate cutaneous inflammation but also systemic inflammation, in order to prevent secondary comorbidities such as CVD. Finally, as pointed out in a recent review by Aksentijevich and colleagues (224), PsO presents a unique opportunity as a human model for the study of chronic systemic inflammation in the development of CVD (224). Treatment of psoriatic patients with targeted biological therapies may shed light on the contribution of specific cytokines to cardiovascular morbidity, and provide novel treatment targets.
Author Contributions
MV: writing of paper, and preparation of figures. GT: editing of paper. EP: study leader, corresponding author, editing of paper, funding. All authors contributed to the article and approved the submitted version.
Funding
This work is based on the research supported in part by the National Research Foundation (NRF) of South Africa (Grant Numbers: 117473 and 132825). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Nestle FO, Kaplan DH, Barker J. Psoriasis. New Engl J Med (2009) 361(5):496–509. doi: 10.1056/NEJMra0804595
2. Chen GY, Cheng YW, Wang CY, Hsu TJ, Hsu MM, Yang PT, et al. Prevalence of Skin Diseases Among Schoolchildren in Magong, Penghu, Taiwan: A Community-Based Clinical Survey. J Formosan Med Assoc = Taiwan yi zhi (2008) 107(1):21–9. doi: 10.1016/s0929-6646(08)60004-2
3. Michalek IM, Loring B, John SM. A Systematic Review of Worldwide Epidemiology of Psoriasis. J Eur Acad Dermatol Venereol: JEADV (2017) 31(2):205–12. doi: 10.1111/jdv.13854
4. Yang YC, Cheng YW, Lai CS, Chen W. Prevalence of Childhood Acne, Ephelides, Warts, Atopic Dermatitis, Psoriasis, Alopecia Areata and Keloid in Kaohsiung County, Taiwan: A Community-Based Clinical Survey. J Eur Acad Dermatol Venereol: JEADV (2007) 21(5):643–9. doi: 10.1111/j.1468-3083.2006.02036.x
5. Danielsen K, Olsen AO, Wilsgaard T, Furberg AS. Is the Prevalence of Psoriasis Increasing? A 30-Year Follow-Up of a Population-Based Cohort. Br J Dermatol (2013) 168(6):1303–10. doi: 10.1111/bjd.12230
6. Takeshita J, Gelfand JM, Li P, Pinto L, Yu X, Rao P, et al. Psoriasis in the US Medicare Population: Prevalence, Treatment, and Factors Associated With Biologic Use. J Invest Dermatol (2015) 135(12):2955–63. doi: 10.1038/jid.2015.296
7. Naldi L, Gambini D. The Clinical Spectrum of Psoriasis. Clinics Dermatol (2007) 25(6):510–8. doi: 10.1016/j.clindermatol.2007.08.003
8. Kurd SK, Troxel AB, Crits-Christoph P, Gelfand JM. The Risk of Depression, Anxiety, and Suicidality in Patients With Psoriasis: A Population-Based Cohort Study. Arch Dermatol (2010) 146(8):891–5. doi: 10.1001/archdermatol.2010.186
9. Armstrong AW, Schupp C, Wu J, Bebo B. Quality of Life and Work Productivity Impairment Among Psoriasis Patients: Findings From the National Psoriasis Foundation Survey Data 2003-2011. PloS One (2012) 7(12):e52935. doi: 10.1371/journal.pone.0052935
10. Gelfand JM, Troxel AB, Lewis JD, Kurd SK, Shin DB, Wang X, et al. The Risk of Mortality in Patients With Psoriasis: Results From a Population-Based Study. JAMA Dermatol (2007) 143(12):1493–9. doi: 10.1001/archderm.143.12.1493
11. Hao J-Q. Targeting Interleukin-22 in Psoriasis. Inflammation (2014) 37(1):94–9. doi: 10.1007/s10753-013-9715-y
12. Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 Axis in the Immunopathogenesis of Psoriasis. J Invest Dermatol (2009) 129(6):1339–50. doi: 10.1038/jid.2009.59
13. Grine L, Dejager L, Libert C, Vandenbroucke RE. An Inflammatory Triangle in Psoriasis: TNF, Type I IFNs and IL-17. Cytokine Growth Factor Rev (2015) 26(1):25–33. doi: 10.1016/j.cytogfr.2014.10.009
14. Oliveira M, Rocha B, Duarte GV. Psoriasis: Classical and Emerging Comorbidities. Bras Dermatol (2015) 90(1):9–20. doi: 10.1590/abd1806-4841.20153038
15. Reich K. The Concept of Psoriasis as a Systemic Inflammation: Implications for Disease Management. J Eur Acad Dermatol Venereol: JEADV (2012) 26 Suppl 2:3–11. doi: 10.1111/j.1468-3083.2011.04410.x
16. Takeshita J, Grewal S, Langan SM, Mehta NN, Ogdie A, Van Voorhees AS, et al. Psoriasis and Comorbid Diseases: Epidemiology. J Am Acad Dermatol (2017) 76(3):377–90. doi: 10.1016/j.jaad.2016.07.064
17. Davidovici BB, Sattar N, Prinz J, Puig L, Emery P, Barker JN, et al. Psoriasis and Systemic Inflammatory Diseases: Potential Mechanistic Links Between Skin Disease and Co-Morbid Conditions. J Invest Dermatol (2010) 130(7):1785–96. doi: 10.1038/jid.2010.103
18. Ahlehoff O, Gislason GH, Lindhardsen J, Charlot MG, Jorgensen CH, Olesen JB, et al. Psoriasis Carries an Increased Risk of Venous Thromboembolism: A Danish Nationwide Cohort Study. PloS One (2011) 6(3):e18125. doi: 10.1371/journal.pone.0018125
19. Gelfand JM, Dommasch ED, Shin DB, Azfar RS, Kurd SK, Wang X, et al. The Risk of Stroke in Patients With Psoriasis. J Invest Dermatol (2009) 129(10):2411–8. doi: 10.1038/jid.2009.112
20. Gelfand JM, Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of Myocardial Infarction in Patients With Psoriasis. JAMA (2006) 296(14):1735–41. doi: 10.1001/jama.296.14.1735
21. Mehta NN, Azfar RS, Shin DB, Neimann AL, Troxel AB, Gelfand JM. Patients With Severe Psoriasis Are at Increased Risk of Cardiovascular Mortality: Cohort Study Using the General Practice Research Database. Eur Heart J (2010) 31(8):1000–6. doi: 10.1093/eurheartj/ehp567
22. Manolis AA, Manolis TA, Melita H, Manolis AS. Psoriasis and Cardiovascular Disease: The Elusive Link. Int Rev Immunol (2019) 38(1):33–54. doi: 10.1080/08830185.2018.1539084
23. Foley JH, Conway EM. Cross Talk Pathways Between Coagulation and Inflammation. Circ Res (2016) 118(9):1392–408. doi: 10.1161/CIRCRESAHA.116.306853
24. Esmon CT. The Interactions Between Inflammation and Coagulation. Br J Haematol (2005) 131(4):417–30. doi: 10.1111/j.1365-2141.2005.05753.x
25. Levi M, van der Poll T. Two-Way Interactions Between Inflammation and Coagulation. Trends Cardiovasc Med (2005) 15(7):254–9. doi: 10.1016/j.tcm.2005.07.004
26. Abeyama K, Stern DM, Ito Y, Kawahara K, Yoshimoto Y, Tanaka M, et al. The N-Terminal Domain of Thrombomodulin Sequesters High-Mobility Group-B1 Protein, a Novel Antiinflammatory Mechanism. J Clin Invest (2005) 115(5):1267–74. doi: 10.1172/jci22782
27. Iwaki T, Cruz DT, Martin JA, Castellino FJ. A Cardioprotective Role for the Endothelial Protein C Receptor in Lipopolysaccharide-Induced Endotoxemia in the Mouse. Blood (2005) 105(6):2364–71. doi: 10.1182/blood-2004-06-2456
28. Bevilacqua MP, Pober JS, Majeau GR, Fiers W, Cotran RS, Gimbrone MA Jr. Recombinant Tumor Necrosis Factor Induces Procoagulant Activity in Cultured Human Vascular Endothelium: Characterization and Comparison With the Actions of Interleukin 1. Proc Natl Acad Sci USA (1986) 83(12):4533–7. doi: 10.1073/pnas.83.12.4533
29. Abele DC, Dobson RL, Graham JB. Heredity and Psoriasis: Study of a Large Family. JAMA Dermatol (1963) 88(1):38–47. doi: 10.1001/archderm.1963.01590190044005
30. Duffy DL, Spelman LS, Martin NG. Psoriasis in Australian Twins. J Am Acad Dermatol (1993) 29(3):428–34. doi: 10.1016/0190-9622(93)70206-9
31. Grjibovski AM, Olsen AO, Magnus P, Harris JR. Psoriasis in Norwegian Twins: Contribution of Genetic and Environmental Effects. J Eur Acad Dermatol Venereol (2007) 21(10):1337–43. doi: 10.1111/j.1468-3083.2007.02268.x
32. Farber EM, Nall ML, Watson W. Natural History of Psoriasis in 61 Twin Pairs. Arch Dermatol (1974) 109(2):207–11. doi: 10.1001/archderm.109.2.207
33. Duffin KC, Chandran V, Gladman DD, Krueger GG, Elder JT, Rahman P. Genetics of Psoriasis and Psoriatic Arthritis: Update and Future Direction. J Rheumatol (2008) 35(7):1449–53.
34. Trembath RC, Clough RL, Rosbotham JL, Jones AB, Camp RD, Frodsham A, et al. Identification of a Major Susceptibility Locus on Chromosome 6p and Evidence for Further Disease Loci Revealed by a Two Stage Genome-Wide Search in Psoriasis. Hum Mol Genet (1997) 6(5):813–20. doi: 10.1093/hmg/6.5.813
35. Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NVC, Jenisch S, et al. Sequence and Haplotype Analysis Supports HLA-C as the Psoriasis Susceptibility 1 Gene. Am J Hum Genet (2006) 78(5):827–51. doi: 10.1086/503821
36. Hwang ST, Nijsten T, Elder JT. Recent Highlights in Psoriasis Research. J Invest Dermatol (2017) 137(3):550–6. doi: 10.1016/j.jid.2016.11.007
37. Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, et al. A Large-Scale Genetic Association Study Confirms IL12B and Leads to the Identification of IL23R as Psoriasis-Risk Genes. Am J Hum Genet (2007) 80(2):273–90. doi: 10.1086/511051
38. Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-Wide Scan Reveals Association of Psoriasis With IL-23 and NF-kappaB Pathways. Nat Genet (2009) 41(2):199–204. doi: 10.1038/ng.311
39. Tsoi LC, Stuart PE, Tian C, Gudjonsson JE, Das S, Zawistowski M, et al. Large Scale Meta-Analysis Characterizes Genetic Architecture for Common Psoriasis Associated Variants. Nat Commun (2017) 8(1):15382. doi: 10.1038/ncomms15382
40. Strange A, Capon F, Spencer CC, Knight J, Weale ME, Allen MH, et al. A Genome-Wide Association Study Identifies New Psoriasis Susceptibility Loci and an Interaction Between HLA-C and ERAP1. Nat Genet (2010) 42(11):985–90. doi: 10.1038/ng.694
41. Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 New Psoriasis Susceptibility Loci Highlights the Role of Innate Immunity. Nat Genet (2012) 44(12):1341–8. doi: 10.1038/ng.2467
42. Sun LD, Cheng H, Wang ZX, Zhang AP, Wang PG, Xu JH, et al. Association Analyses Identify Six New Psoriasis Susceptibility Loci in the Chinese Population. Nat Genet (2010) 42(11):1005–9. doi: 10.1038/ng.690
43. Zeng J, Luo S, Huang Y, Lu Q. Critical Role of Environmental Factors in the Pathogenesis of Psoriasis. J Dermatol (2017) 44(8):863–72. doi: 10.1111/1346-8138.13806
44. Evers AWM, Verhoeven EWM, Kraaimaat FW, Jong EMGJD, Brouwer SJMD, Schalkwijk J, et al. How Stress Gets Under the Skin: Cortisol and Stress Reactivity in Psoriasis. Br J Dermatol (2010) 163(5):986–91. doi: 10.1111/j.1365-2133.2010.09984.x
45. Richards HL, Ray DW, Kirby B, Mason D, Plant D, Main CJ, et al. Response of the Hypothalamic–Pituitary–Adrenal Axis to Psychological Stress in Patients With Psoriasis. Br J Dermatol (2005) 153(6):1114–20. doi: 10.1111/j.1365-2133.2005.06817.x
46. Duarte GV, Oliveira M, Cardoso TM, Follador I, Silva TS, Cavalheiro CMA, et al. Association Between Obesity Measured by Different Parameters and Severity of Psoriasis. Int J Dermatol (2013) 52(2):177–81. doi: 10.1111/j.1365-4632.2011.05270.x
47. Johnston A, Arnadottir S, Gudjonsson JE, Aphale A, Sigmarsdottir AA, Gunnarsson SI, et al. Obesity in Psoriasis: Leptin and Resistin as Mediators of Cutaneous Inflammation. Br J Dermatol (2008) 159(2):342–50. doi: 10.1111/j.1365-2133.2008.08655.x
48. Armstrong AW, Harskamp CT, Dhillon JS, Armstrong EJ. Psoriasis and Smoking: A Systematic Review and Meta-Analysis. Br J Dermatol (2014) 170(2):304–14. doi: 10.1111/bjd.12670
49. Li W, Han J, Choi HK, Qureshi AA. Smoking and Risk of Incident Psoriasis Among Women and Men in the United States: A Combined Analysis. Am J Epidemiol (2012) 175(5):402–13. doi: 10.1093/aje/kwr325
50. Naldi L, Chatenoud L, Linder D, Belloni Fortina A, Peserico A, Virgili AR, et al. Cigarette Smoking, Body Mass Index, and Stressful Life Events as Risk Factors for Psoriasis: Results From an Italian Case–Control Study. J Invest Dermatol (2005) 125(1):61–7. doi: 10.1111/j.0022-202X.2005.23681.x
51. Attwa E, Swelam E. Relationship Between Smoking-Induced Oxidative Stress and the Clinical Severity of Psoriasis. J Eur Acad Dermatol Venereol (2011) 25(7):782–7. doi: 10.1111/j.1468-3083.2010.03860.x
52. Armstrong AW, Armstrong EJ, Fuller EN, Sockolov ME, Voyles SV. Smoking and Pathogenesis of Psoriasis: A Review of Oxidative, Inflammatory and Genetic Mechanisms. Br J Dermatol (2011) 165(6):1162–8. doi: 10.1111/j.1365-2133.2011.10526.x
53. Guðjónsson JE, Thorarinsson AM, Sigurgeirsson B, Kristinsson KG, Valdimarsson H. Streptococcal Throat Infections and Exacerbation of Chronic Plaque Psoriasis: A Prospective Study. Br J Dermatol (2003) 149(3):530–4. doi: 10.1046/j.1365-2133.2003.05552.x
54. Tomi NS, Kränke B, Aberer E. Staphylococcal Toxins in Patients With Psoriasis, Atopic Dermatitis, and Erythroderma, and in Healthy Control Subjects. J Am Acad Dermatol (2005) 53(1):67–72. doi: 10.1016/j.jaad.2005.02.034
55. Visser MJE, Kell DB, Pretorius E. Bacterial Dysbiosis and Translocation in Psoriasis Vulgaris. Front Cell Infect Microbiol (2019) 9:7. doi: 10.3389/fcimb.2019.00007
56. Mosca A, Leclerc M, Hugot JP. Gut Microbiota Diversity and Human Diseases: Should We Reintroduce Key Predators in Our Ecosystem? Front Microbiol (2016) 7:455. doi: 10.3389/fmicb.2016.00455
57. Hiippala K, Jouhten H, Ronkainen A, Hartikainen A, Kainulainen V, Jalanka J, et al. The Potential of Gut Commensals in Reinforcing Intestinal Barrier Function and Alleviating Inflammation. Nutrients (2018) 10(8):1–23. doi: 10.3390/nu10080988
58. Codoñer FM, Ramírez-Bosca A, Climent E, Carrión-Gutierrez M, Guerrero M, Pérez-Orquín JM, et al. Gut Microbial Composition in Patients With Psoriasis. Sci Rep (2018) 8(1):3812. doi: 10.1038/s41598-018-22125-y
59. Eppinga H, Sperna Weiland CJ, Thio HB, van der Woude CJ, Nijsten TEC, Peppelenbosch MP, et al. Similar Depletion of Protective Faecalibacterium Prausnitzii in Psoriasis and Inflammatory Bowel Disease, But Not in Hidradenitis Suppurativa. J Crohn’s Colitis (2016) 10(9):1067–75. doi: 10.1093/ecco-jcc/jjw070
60. Alekseyenko AV, Perez-Perez GI, De Souza A, Strober B, Gao Z, Bihan M, et al. Community Differentiation of the Cutaneous Microbiota in Psoriasis. Microbiome (2013) 1(1):31. doi: 10.1186/2049-2618-1-31
61. Gao Z, Tseng CH, Strober BE, Pei Z, Blaser MJ. Substantial Alterations of the Cutaneous Bacterial Biota in Psoriatic Lesions. PloS One (2008) 3(7):e2719. doi: 10.1371/journal.pone.0002719
62. Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, et al. Self-RNA-Antimicrobial Peptide Complexes Activate Human Dendritic Cells Through TLR7 and TLR8. J Exp Med (2009) 206(9):1983–94. doi: 10.1084/jem.20090480
63. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang Y-H, Homey B, et al. Plasmacytoid Dendritic Cells Sense Self-DNA Coupled With Antimicrobial Peptide. Nature (2007) 449(7162):564–9. doi: 10.1038/nature06116
64. Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, et al. Plasmacytoid Predendritic Cells Initiate Psoriasis Through Interferon-Alpha Production. J Exp Med (2005) 202(1):135–43. doi: 10.1084/jem.20050500
65. Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel P19 Protein Engages IL-12p40 to Form a Cytokine, IL-23, With Biological Activities Similar as Well as Distinct From IL-12. Immunity (2000) 13(5):715–25. doi: 10.1016/s1074-7613(00)00070-4
66. Nestle FO, Turka LA, Nickoloff BJ. Characterization of Dermal Dendritic Cells in Psoriasis. Autostimulation of T Lymphocytes and Induction of Th1 Type Cytokines. J Clin Invest (1994) 94(1):202–9. doi: 10.1172/jci117308
67. Zaba LC, Fuentes-Duculan J, Eungdamrong NJ, Abello MV, Novitskaya I, Pierson KC, et al. Psoriasis Is Characterized by Accumulation of Immunostimulatory and Th1/Th17 Cell-Polarizing Myeloid Dendritic Cells. J Invest Dermatol (2009) 129(1):79–88. doi: 10.1038/jid.2008.194
68. Austin LM, Ozawa M, Kikuchi T, Walters IB, Krueger JG. The Majority of Epidermal T Cells in Psoriasis Vulgaris Lesions can Produce Type 1 Cytokines, Interferon-Gamma, Interleukin-2, and Tumor Necrosis Factor-Alpha, Defining TC1 (Cytotoxic T Lymphocyte) and TH1 Effector Populations: A Type 1 Differentiation Bias Is Also Measured in Circulating Blood T Cells in Psoriatic Patients. J Invest Dermatol (1999) 113(5):752–9. doi: 10.1046/j.1523-1747.1999.00749.x
69. Johnson-Huang LM, Suárez-Fariñas M, Pierson KC, Fuentes-Duculan J, Cueto I, Lentini T, et al. A Single Intradermal Injection of IFN-γ Induces an Inflammatory State in Both non-Lesional Psoriatic and Healthy Skin. J Invest Dermatol (2012) 132(4):1177–87. doi: 10.1038/jid.2011.458
70. Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suarez-Farinas M, Cardinale I, et al. Th17 Cytokines Interleukin (IL)-17 and IL-22 Modulate Distinct Inflammatory and Keratinocyte-Response Pathways. Br J Dermatol (2008) 159(5):1092–102. doi: 10.1111/j.1365-2133.2008.08769.x
71. Lowes MA, Chamian F, Abello MV, Fuentes-Duculan J, Lin SL, Nussbaum R, et al. Increase in TNF-Alpha and Inducible Nitric Oxide Synthase-Expressing Dendritic Cells in Psoriasis and Reduction With Efalizumab (Anti-CD11a). Proc Natl Acad Sci USA (2005) 102(52):19057–62. doi: 10.1073/pnas.0509736102
72. Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, Nestle FO. Spontaneous Development of Psoriasis in a New Animal Model Shows an Essential Role for Resident T Cells and Tumor Necrosis Factor-Alpha. J Exp Med (2004) 199(5):731–6. doi: 10.1084/jem.20031482
73. Terajima S, Higaki M, Igarashi Y, Nogita T, Kawashima M. An Important Role of Tumor Necrosis Factor-Alpha in the Induction of Adhesion Molecules in Psoriasis. Arch Dermatol Res (1998) 290(5):246–52. doi: 10.1007/s004030050299
74. Young CN, Koepke JI, Terlecky LJ, Borkin MS, Boyd SL, Terlecky SR. Reactive Oxygen Species in Tumor Necrosis Factor-Alpha-Activated Primary Human Keratinocytes: Implications for Psoriasis and Inflammatory Skin Disease. J Invest Dermatol (2008) 128(11):2606–14. doi: 10.1038/jid.2008.122
75. Chiricozzi A, Guttman-Yassky E, Suárez-Fariñas M, Nograles KE, Tian S, Cardinale I, et al. Integrative Responses to IL-17 and TNF-α in Human Keratinocytes Account for Key Inflammatory Pathogenic Circuits in Psoriasis. J Invest Dermatol (2011) 131(3):677–87. doi: 10.1038/jid.2010.340
76. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 Are Coexpressed by Th17 Cells and Cooperatively Enhance Expression of Antimicrobial Peptides. J Exp Med (2006) 203(10):2271–9. doi: 10.1084/jem.20061308
77. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 Cytokine, Mediates IL-23-Induced Dermal Inflammation and Acanthosis. Nature (2007) 445(7128):648–51. doi: 10.1038/nature05505
78. Brembilla NC, Senra L, Boehncke WH. The IL-17 Family of Cytokines in Psoriasis: IL-17A and Beyond. Front Immunol (2018) 9:1682. doi: 10.3389/fimmu.2018.01682
79. Teunissen MBM, Yeremenko NG, Baeten DLP, Chielie S, Spuls PI, de Rie MA, et al. The IL-17A-Producing CD8+ T-Cell Population in Psoriatic Lesional Skin Comprises Mucosa-Associated Invariant T Cells and Conventional T Cells. J Invest Dermatol (2014) 134(12):2898–907. doi: 10.1038/jid.2014.261
80. Cai Y, Shen X, Ding C, Qi C, Li K, Li X, et al. Pivotal Role of Dermal IL-17-Producing γδ T Cells in Skin Inflammation. Immunity (2011) 35(4):596–610. doi: 10.1016/j.immuni.2011.08.001
81. Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of Innate Lymphoid Cells in Human Skin and Blood Demonstrates Increase of NKp44+ ILC3 in Psoriasis. J Invest Dermatol (2014) 134(4):984–91. doi: 10.1038/jid.2013.477
82. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast Cells and Neutrophils Release IL-17 Through Extracellular Trap Formation in Psoriasis. J Immunol (2011) 187(1):490–500. doi: 10.4049/jimmunol.1100123
83. Muromoto R, Hirao T, Tawa K, Hirashima K, Kon S, Kitai Y, et al. IL-17A Plays a Central Role in the Expression of Psoriasis Signature Genes Through the Induction of IkappaB-Zeta in Keratinocytes. Int Immunol (2016) 28(9):443–52. doi: 10.1093/intimm/dxw011
84. Watanabe H, Kawaguchi M, Fujishima S, Ogura M, Matsukura S, Takeuchi H, et al. Functional Characterization of IL-17F as a Selective Neutrophil Attractant in Psoriasis. J Invest Dermatol (2009) 129(3):650–6. doi: 10.1038/jid.2008.294
85. Harper EG, Guo C, Rizzo H, Lillis JV, Kurtz SE, Skorcheva I, et al. Th17 Cytokines Stimulate CCL20 Expression in Keratinocytes In Vitro and In Vivo: Implications for Psoriasis Pathogenesis. J Invest Dermatol (2009) 129(9):2175–83. doi: 10.1038/jid.2009.65
86. Witte E, Kokolakis G, Witte K, Philipp S, Doecke WD, Babel N, et al. IL-19 Is a Component of the Pathogenetic IL-23/IL-17 Cascade in Psoriasis. J Invest Dermatol (2014) 134(11):2757–67. doi: 10.1038/jid.2014.308
87. van der Fits L, Mourits S, Voerman JSA, Kant M, Boon L, Laman JD, et al. Imiquimod-Induced Psoriasis-Like Skin Inflammation in Mice Is Mediated via the IL-23/IL-17 Axis. J Immunol (2009) 182(9):5836. doi: 10.4049/jimmunol.0802999
88. Rizzo HL, Kagami S, Phillips KG, Kurtz SE, Jacques SL, Blauvelt A. IL-23-Mediated Psoriasis-Like Epidermal Hyperplasia Is Dependent on IL-17a. J Immunol (2011) 186(3):1495–502. doi: 10.4049/jimmunol.1001001
89. Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, et al. Th22 Cells Represent a Distinct Human T Cell Subset Involved in Epidermal Immunity and Remodeling. J Clin Invest (2009) 119(12):3573–85. doi: 10.1172/jci40202
90. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 Increases the Innate Immunity of Tissues. Immunity (2004) 21(2):241–54. doi: 10.1016/j.immuni.2004.07.007
91. Zenewicz LA, Flavell RA. Recent Advances in IL-22 Biology. Int Immunol (2011) 23(3):159–63. doi: 10.1093/intimm/dxr001
92. Alabbas SY, Begun J, Florin TH, Oancea I. The Role of IL-22 in the Resolution of Sterile and Nonsterile Inflammation. Clin Transl Immunol (2018) 7(4):e1017–e. doi: 10.1002/cti2.1017
93. Tohyama M, Shirakata Y, Hanakawa Y, Dai X, Shiraishi K, Murakami M, et al. Bcl-3 Induced by IL-22 via STAT3 Activation Acts as a Potentiator of Psoriasis-Related Gene Expression in Epidermal Keratinocytes. Eur J Immunol (2018) 48(1):168–79. doi: 10.1002/eji.201747017
94. Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 Inhibits Epidermal Differentiation and Induces Proinflammatory Gene Expression and Migration of Human Keratinocytes. J Immunol (2005) 174(6):3695–702. doi: 10.4049/jimmunol.174.6.3695
95. Wolk K, Haugen H, Xu W, Witte E, Waggie K, Anderson M, et al. IL-22 and IL-20 Are Key Mediators of the Epidermal Alterations in Psoriasis While IL-17 and IFN-Gamma Are Not. J Mol Med (Berlin Germany) (2009) 87:523–36. doi: 10.1007/s00109-009-0457-0
96. Wolk K, Witte E, Wallace E, Docke WD, Kunz S, Asadullah K, et al. IL-22 Regulates the Expression of Genes Responsible for Antimicrobial Defense, Cellular Differentiation, and Mobility in Keratinocytes: A Potential Role in Psoriasis. Eur J Immunol (2006) 36(5):1309–23. doi: 10.1002/eji.200535503
97. Vestergaard C, Just H, Baumgartner Nielsen J, Thestrup-Pedersen K, Deleuran M. Expression of CCR2 on Monocytes and Macrophages in Chronically Inflamed Skin in Atopic Dermatitis and Psoriasis. Acta Derm Venereol (2004) 84(5):353–8. doi: 10.1080/00015550410034444
98. Gillitzer R, Ritter U, Spandau U, Goebeler M, Bröcker E-B. Differential Expression of GRO-α and IL-8 mRNA Psoriasis: A Model for Neutrophil Migration and Accumulation In Vivo. J Invest Dermatol (1996) 107(5):778–82. doi: 10.1111/1523-1747.ep12371803
99. Homey B, Dieu-Nosjean MC, Wiesenborn A, Massacrier C, Pin JJ, Oldham E, et al. Up-Regulation of Macrophage Inflammatory Protein-3 Alpha/CCL20 and CC Chemokine Receptor 6 in Psoriasis. J Immunol (2000) 164(12):6621–32. doi: 10.4049/jimmunol.164.12.6621
100. Singh TP, Schön MP, Wallbrecht K, Gruber-Wackernagel A, Wang X-J, Wolf P. Involvement of IL-9 in Th17-Associated Inflammation and Angiogenesis of Psoriasis. PloS One (2013) 8(1):e51752. doi: 10.1371/journal.pone.0051752
101. Sa SM, Valdez PA, Wu J, Jung K, Zhong F, Hall L, et al. The Effects of IL-20 Subfamily Cytokines on Reconstituted Human Epidermis Suggest Potential Roles in Cutaneous Innate Defense and Pathogenic Adaptive Immunity in Psoriasis. J Immunol (2007) 178(4):2229–40. doi: 10.4049/jimmunol.178.4.2229
102. Wang F, Smith N, Maier L, Xia W, Hammerberg C, Chubb H, et al. Etanercept Suppresses Regenerative Hyperplasia in Psoriasis by Acutely Downregulating Epidermal Expression of Interleukin (IL)-19, IL-20 and IL-24. Br J Dermatol (2012) 167(1):92–102. doi: 10.1111/j.1365-2133.2012.10961.x
103. Zeng F, Chen H, Chen L, Mao J, Cai S, Xiao Y, et al. An Autocrine Circuit of IL-33 in Keratinocytes Is Involved in the Progression of Psoriasis. J Invest Dermatol (2021) 141(3):596–606. doi: 10.1016/j.jid.2020.07.027
104. Hernández-Santana YE, Leon G, St Leger D, Fallon PG, Walsh PT. Keratinocyte Interleukin-36 Receptor Expression Orchestrates Psoriasiform Inflammation in Mice. Life Sci Alliance (2020) 3(4):e201900586. doi: 10.26508/lsa.201900586
105. Goldstein JD, Bassoy EY, Caruso A, Palomo J, Rodriguez E, Lemeille S, et al. IL-36 Signaling in Keratinocytes Controls Early IL-23 Production in Psoriasis-Like Dermatitis. Life Sci Alliance (2020) 3(6):e202000688. doi: 10.26508/lsa.202000688
106. Elmets CA, Korman NJ, Prater EF, Wong EB, Rupani RN, Kivelevitch D, et al. Joint AAD-NPF Guidelines of Care for the Management and Treatment of Psoriasis With Topical Therapy and Alternative Medicine Modalities for Psoriasis Severity Measures. J Am Acad Dermatol (2021) 84(2):432–70. doi: 10.1016/j.jaad.2020.07.087
107. Armstrong AW, Read C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. Jama (2020) 323(19):1945–60. doi: 10.1001/jama.2020.4006
108. Menter A, Gelfand JM, Connor C, Armstrong AW, Cordoro KM, Davis DMR, et al. Joint American Academy of Dermatology-National Psoriasis Foundation Guidelines of Care for the Management of Psoriasis With Systemic Nonbiologic Therapies. J Am Acad Dermatol (2020) 82(6):1445–86. doi: 10.1016/j.jaad.2020.02.044
109. Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker JNWN. Psoriasis. Lancet (2021) 397(10281):1301–15. doi: 10.1016/S0140-6736(20)32549-6
110. Kaushik SB, Lebwohl MG. Review of Safety and Efficacy of Approved Systemic Psoriasis Therapies. Int J Dermatol (2019) 58(6):649–58. doi: 10.1111/ijd.14246
111. Grozdev I, Korman N, Tsankov N. Psoriasis as a Systemic Disease. Clinics Dermatol (2014) 32(3):343–50. doi: 10.1016/j.clindermatol.2013.11.001
112. Korman NJ. Management of Psoriasis as a Systemic Disease: What Is the Evidence? Br J Dermatol (2020) 182(4):840–8. doi: 10.1111/bjd.18245
113. Arican O, Aral M, Sasmaz S, Ciragil P. Serum Levels of TNF-Alpha, IFN-Gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in Patients With Active Psoriasis and Correlation With Disease Severity. Mediators Inflammation (2005) 2005(5):273–9. doi: 10.1155/mi.2005.273
114. Bai F, Zheng W, Dong Y, Wang J, Garstka MA, Li R, et al. Serum Levels of Adipokines and Cytokines in Psoriasis Patients: A Systematic Review and Meta-Analysis. Oncotarget (2018) 9(1):1266–78. doi: 10.18632/oncotarget.22260
115. Cataldi C, Mari NL, Lozovoy MAB, Martins LMM, Reiche EMV, Maes M, et al. Proinflammatory and Anti-Inflammatory Cytokine Profiles in Psoriasis: Use as Laboratory Biomarkers and Disease Predictors. Inflammation Res: Off J Eur Histamine Res Soc [et al] (2019) 68(7):557–67. doi: 10.1007/s00011-019-01238-8
116. Rashmi R, Rao KS, Basavaraj KH. A Comprehensive Review of Biomarkers in Psoriasis. Clin Exp Dermatol (2009) 34(6):658–63. doi: 10.1111/j.1365-2230.2009.03410.x
117. Mehta NN, Yu Y, Saboury B, Foroughi N, Krishnamoorthy P, Raper A, et al. Systemic and Vascular Inflammation in Patients With Moderate to Severe Psoriasis as Measured by [18F]-Fluorodeoxyglucose Positron Emission Tomography-Computed Tomography (FDG-PET/CT): A Pilot Study. Arch Dermatol (2011) 147(9):1031–9. doi: 10.1001/archdermatol.2011.119
118. Chi C-C, Wang J, Chen Y-F, Wang S-H, Chen F-L, Tung T-H. Risk of Incident Chronic Kidney Disease and End-Stage Renal Disease in Patients With Psoriasis: A Nationwide Population-Based Cohort Study. J Dermatol Sci (2015) 78(3):232–8. doi: 10.1016/j.jdermsci.2015.03.012
119. Chiu HY, Huang HL, Li CH, Yin YJ, Chen HA, Hsu ST, et al. Increased Risk of Glomerulonephritis and Chronic Kidney Disease in Relation to the Severity of Psoriasis, Concomitant Medication, and Comorbidity: A Nationwide Population-Based Cohort Study. Br J Dermatol (2015) 173(1):146–54. doi: 10.1111/bjd.13599
120. Wan J, Wang S, Haynes K, Denburg MR, Shin DB, Gelfand JM. Risk of Moderate to Advanced Kidney Disease in Patients With Psoriasis: Population Based Cohort Study. BMJ: Br Med J (2013) 347:f5961. doi: 10.1136/bmj.f5961
121. Dowlatshahi EA, Wakkee M, Arends LR, Nijsten T. The Prevalence and Odds of Depressive Symptoms and Clinical Depression in Psoriasis Patients: A Systematic Review and Meta-Analysis. J Invest Dermatol (2014) 134(6):1542–51. doi: 10.1038/jid.2013.508
122. Olivier C, Robert PD, Daihung DO, UrbÀ G, Catalin MP, Hywel W, et al. The Risk of Depression, Anxiety, and Suicidality in Patients With Psoriasis: A Population-Based Cohort Study. Arch Dermatol (2010) 146(8):891–5. doi: 10.1001/archdermatol.2010.186
123. Khalid U, Hansen PR, Gislason GH, Lindhardsen J, Kristensen SL, Winther SA, et al. Psoriasis and New-Onset Diabetes: A Danish Nationwide Cohort Study. Diabetes Care (2013) 36(8):2402–7. doi: 10.2337/dc12-2330
124. Armstrong AW, Harskamp CT, Armstrong EJ. Psoriasis and the Risk of Diabetes Mellitus: A Systematic Review and Meta-Analysis. JAMA Dermatol (2013) 149(1):84–91. doi: 10.1001/2013.jamadermatol.406
125. Cohen AD, Dreiher J, Shapiro Y, Vidavsky L, Vardy DA, Davidovici B, et al. Psoriasis and Diabetes: A Population-Based Cross-Sectional Study. J Eur Acad Dermatol Venereol (2008) 22(5):585–9. doi: 10.1111/j.1468-3083.2008.02636.x
126. Cohen AD, Dreiher J, Birkenfeld S. Psoriasis Associated With Ulcerative Colitis and Crohn’s Disease. J Eur Acad Dermatol Venereol: JEADV (2009) 23(5):561–5. doi: 10.1111/j.1468-3083.2008.03031.x
127. Egeberg A, Mallbris L, Warren RB, Bachelez H, Gislason GH, Hansen PR, et al. Association Between Psoriasis and Inflammatory Bowel Disease: A Danish Nationwide Cohort Study. Br J Dermatol (2016) 175(3):487–92. doi: 10.1111/bjd.14528
128. Li WQ, Han JL, Chan AT, Qureshi AA. Psoriasis, Psoriatic Arthritis and Increased Risk of Incident Crohn’s Disease in US Women. Ann Rheumatic Dis (2013) 72(7):1200–5. doi: 10.1136/annrheumdis-2012-202143
129. Brauchli YB, Jick SS, Miret M, Meier CR. Psoriasis and Risk of Incident Cancer: An Inception Cohort Study With a Nested Case-Control Analysis. J Invest Dermatol (2009) 129(11):2604–12. doi: 10.1038/jid.2009.113
130. Egeberg A, Thyssen JP, Gislason GH, Skov L. Skin Cancer in Patients With Psoriasis. J Eur Acad Dermatol Venereol: JEADV (2016) 30(8):1349–53. doi: 10.1111/jdv.13619
131. Pouplard C, Brenaut E, Horreau C, Barnetche T, Misery L, Richard MA, et al. Risk of Cancer in Psoriasis: A Systematic Review and Meta-Analysis of Epidemiological Studies. J Eur Acad Dermatol Venereol: JEADV (2013) 27 Suppl 3:36–46. doi: 10.1111/jdv.12165
132. Langan SM, Seminara NM, Shin DB, Troxel AB, Kimmel SE, Mehta NN, et al. Prevalence of Metabolic Syndrome in Patients With Psoriasis: A Population-Based Study in the United Kingdom. J Invest Dermatol (2012) 132(3, Part 1):556–62. doi: 10.1038/jid.2011.365
133. Sommer DM, Jenisch S, Suchan M, Christophers E, Weichenthal M. Increased Prevalence of the Metabolic Syndrome in Patients With Moderate to Severe Psoriasis. Arch Dermatol Res (2006) 298(7):321. doi: 10.1007/s00403-006-0703-z
134. Love TJ, Qureshi AA, Karlson EW, Gelfand JM, Choi HK. Prevalence of the Metabolic Syndrome in Psoriasis: Results From the National Health and Nutrition Examination Survey, 2003-2006. Arch Dermatol (2011) 147(4):419–24. doi: 10.1001/archdermatol.2010.370
135. Gisondi P, Targher G, Zoppini G, Girolomoni G. Non-Alcoholic Fatty Liver Disease in Patients With Chronic Plaque Psoriasis. J Hepatol (2009) 51(4):758–64. doi: 10.1016/j.jhep.2009.04.020
136. Candia R, Ruiz A, Torres-Robles R, Chávez-Tapia N, Méndez-Sánchez N, Arrese M. Risk of non-Alcoholic Fatty Liver Disease in Patients With Psoriasis: A Systematic Review and Meta-Analysis. J Eur Acad Dermatol Venereol: JEADV (2015) 29(4):656–62. doi: 10.1111/jdv.12847
137. Roberts KK, Cochet AE, Lamb PB, Brown PJ, Battafarano DF, Brunt EM, et al. The Prevalence of NAFLD and NASH Among Patients With Psoriasis in a Tertiary Care Dermatology and Rheumatology Clinic. Aliment Pharmacol Ther (2015) 41(3):293–300. doi: 10.1111/apt.13042
138. Cohen AD, Gilutz H, Henkin Y, Zahger D, Shapiro J, Bonneh DY, et al. Psoriasis and the Metabolic Syndrome. Acta Derm Venereol (2007) 87(6):506–9. doi: 10.2340/00015555-0297
139. Cohen AD, Sherf M, Vidavsky L, Vardy DA, Shapiro J, Meyerovitch J. Association Between Psoriasis and the Metabolic Syndrome. A Cross-Sectional Study. Dermatology (2008) 216(2):152–5. doi: 10.1159/000111512
140. Tobin A-M, Veale DJ, Fitzgerald O, Rogers S, Collins P, O’Shea D, et al. Cardiovascular Disease and Risk Factors in Patients With Psoriasis and Psoriatic Arthritis. J Rheumatol (2010) 37:1386–94. doi: 10.3899/jrheum.090822 jrheum.090822.
141. Fantuzzi G. Adipose Tissue, Adipokines, and Inflammation. J Allergy Clin Immunol (2005) 115(5):911–9. doi: 10.1016/j.jaci.2005.02.023
142. Gerdes S, Rostami-Yazdi M, Mrowietz U. Adipokines and Psoriasis. Exp Dermatol (2011) 20(2):81–7. doi: 10.1111/j.1600-0625.2010.01210.x
143. Gisondi P, Fostini AC, Fossà I, Girolomoni G, Targher G. Psoriasis and the Metabolic Syndrome. Clin Dermatol (2018) 36(1):21–8. doi: 10.1016/j.clindermatol.2017.09.005
144. Takahashi H, Iizuka H. Psoriasis and Metabolic Syndrome. J Dermatol (2012) 39(3):212–8. doi: 10.1111/j.1346-8138.2011.01408.x
145. Azfar RS, Gelfand JM. Psoriasis and Metabolic Disease: Epidemiology and Pathophysiology. Curr Opin Rheumatol (2008) 20(4):416–22. doi: 10.1097/BOR.0b013e3283031c99
146. Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation (2019) 140(11):e596–646. doi: 10.1161/cir.0000000000000678
147. Gaeta M, Castelvecchio S, Ricci C, Pigatto P, Pellissero G, Cappato R. Role of Psoriasis as Independent Predictor of Cardiovascular Disease: A Meta-Regression Analysis. Int J Cardiol (2013) 168(3):2282–8. doi: 10.1016/j.ijcard.2013.01.197
148. Mallbris L, Akre O, Granath F, Yin L, Lindelof B, Ekbom A, et al. Increased Risk for Cardiovascular Mortality in Psoriasis Inpatients But Not in Outpatients. Eur J Epidemiol (2004) 19(3):225–30. doi: 10.1023/b:ejep.0000020447.59150.f9
149. Abuabara K, Azfar RS, Shin DB, Neimann AL, Troxel AB, Gelfand JM. Cause-Specific Mortality in Patients With Severe Psoriasis: A Population-Based Cohort Study in the U.K. Br J Dermatol (2010) 163(3):586–92. doi: 10.1111/j.1365-2133.2010.09941.x
150. Armstrong EJ, Harskamp CT, Armstrong AW. Psoriasis and Major Adverse Cardiovascular Events: A Systematic Review and Meta-Analysis of Observational Studies. J Am Heart Assoc (2013) 2(2):e000062. doi: 10.1161/jaha.113.000062
151. Golia E, Limongelli G, Natale F, Fimiani F, Maddaloni V, Pariggiano I, et al. Inflammation and Cardiovascular Disease: From Pathogenesis to Therapeutic Target. Curr Atheroscl Rep (2014) 16(9):435. doi: 10.1007/s11883-014-0435-z
152. Kofler S, Nickel T, Weis M. Role of Cytokines in Cardiovascular Diseases: A Focus on Endothelial Responses to Inflammation. Clin Sci (2005) 108(3):205–13. doi: 10.1042/cs20040174
153. Demetz G, Ott I. The Interface Between Inflammation and Coagulation in Cardiovascular Disease. Int J Inflammation (2012) 2012:860301. doi: 10.1155/2012/860301
154. Nagareddy P, Smyth SS. Inflammation and Thrombosis in Cardiovascular Disease. Curr Opin Hematol (2013) 20(5):457–63. doi: 10.1097/MOH.0b013e328364219d
155. Marcovina SM, Crea F, Davignon J, Kaski JC, Koenig W, Landmesser U, et al. Biochemical and Bioimaging Markers for Risk Assessment and Diagnosis in Major Cardiovascular Diseases: A Road to Integration of Complementary Diagnostic Tools. J Internal Med (2007) 261(3):214–34. doi: 10.1111/j.1365-2796.2006.01734.x
156. Ridker PM, Danielson E, Fonseca FAH, Genest J, Gotto AM, Kastelein JJP, et al. Rosuvastatin to Prevent Vascular Events in Men and Women With Elevated C-Reactive Protein. New Engl J Med (2008) 359(21):2195–207. doi: 10.1056/NEJMoa0807646
157. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory Therapy With Canakinumab for Atherosclerotic Disease. New Engl J Med (2017) 377(12):1119–31. doi: 10.1056/NEJMoa1707914
158. Szotowski B, Antoniak S, Poller W, Schultheiss HP, Rauch U. Procoagulant Soluble Tissue Factor Is Released From Endothelial Cells in Response to Inflammatory Cytokines. Circ Res (2005) 96(12):1233–9. doi: 10.1161/01.RES.0000171805.24799.fa
159. Camerer E, Huang W, Coughlin SR. Tissue Factor- and Factor X-Dependent Activation of Protease-Activated Receptor 2 by Factor VIIa. Proc Natl Acad Sci USA (2000) 97(10):5255–60. doi: 10.1073/pnas.97.10.5255
160. Camerer E, Kataoka H, Kahn M, Lease K, Coughlin SR. Genetic Evidence That Protease-Activated Receptors Mediate Factor Xa Signaling in Endothelial Cells. J Biol Chem (2002) 277(18):16081–7. doi: 10.1074/jbc.M108555200
161. Vorlova S, Koch M, Manthey HD, Cochain C, Busch M, Chaudhari SM, et al. Coagulation Factor XII Induces Pro-Inflammatory Cytokine Responses in Macrophages and Promotes Atherosclerosis in Mice. Thromb Haemostasis (2017) 117(1):176–87. doi: 10.1160/th16-06-0466
162. de Jonge E, Friederich PW, Vlasuk GP, Rote WE, Vroom MB, Levi M, et al. Activation of Coagulation by Administration of Recombinant Factor VIIa Elicits Interleukin 6 (IL-6) and IL-8 Release in Healthy Human Subjects. Clin Diagn Lab Immunol (2003) 10(3):495–7. doi: 10.1128/cdli.10.3.495-497.2003
163. Gros A, Ollivier V, Ho-Tin-Noé B. Platelets in Inflammation: Regulation of Leukocyte Activities and Vascular Repair. Front Immunol (2014) 5:678. doi: 10.3389/fimmu.2014.00678
164. Raskob GE, Angchaisuksiri P, Blanco AN, Buller H, Gallus A, Hunt BJ, et al. Thrombosis: A Major Contributor to Global Disease Burden. Arterioscler Thromb Vasc Biol (2014) 34(11):2363–71. doi: 10.1161/atvbaha.114.304488
165. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and Regional Mortality From 235 Causes of Death for 20 Age Groups in 1990 and 2010: A Systematic Analysis for the Global Burden of Disease Study 2010. Lancet (2012) 380(9859):2095–128. doi: 10.1016/S0140-6736(12)61728-0
166. Blann AD, Lip GY. Venous Thromboembolism. BMJ (Clinical Res ed) (2006) 332(7535):215–9. doi: 10.1136/bmj.332.7535.215
167. Koupenova M, Kehrel BE, Corkrey HA, Freedman JE. Thrombosis and Platelets: An Update. Eur Heart J (2017) 38(11):785–91. doi: 10.1093/eurheartj/ehw550
168. Mackman N. New Insights Into the Mechanisms of Venous Thrombosis. J Clin Invest (2012) 122(7):2331–6. doi: 10.1172/jci60229
169. Tanaka T, Narazaki M, Kishimoto T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb Perspect Biol (2014) 6(10):a016295–a. doi: 10.1101/cshperspect.a016295
170. Goodman WA, Levine AD, Massari JV, Sugiyama H, McCormick TS, Cooper KD. IL-6 Signaling in Psoriasis Prevents Immune Suppression by Regulatory T Cells. J Immunol (2009) 183(5):3170–6. doi: 10.4049/jimmunol.0803721
171. Oliveira P, Cardoso PRG, Lima E, Pereira MC, Duarte ALBP, Pitta I, et al. IL-17a, IL-22, IL-6, and IL-21 Serum Levels in Plaque-Type Psoriasis in Brazilian Patients. Mediators Inflammation (2015) 2015:819149. doi: 10.1155/2015/819149
172. Neuner P, Urbanski A, Trautinger F, Möller A, Kirnbauer R, Kapp A, et al. Increased IL-6 Production by Monocytes and Keratinocytes in Patients With Psoriasis. J Invest Dermatol (1991) 97(1):27–33. doi: 10.1111/1523-1747.ep12477880
173. van Aken BE, den Heijer M, Bos GM, van Deventer SJ, Reitsma PH. Recurrent Venous Thrombosis and Markers of Inflammation. Thromb Haemostasis (2000) 83(4):536–9. doi: 10.1055/s-0037-1613858
174. Reitsma PH, Rosendaal FR. Activation of Innate Immunity in Patients With Venous Thrombosis: The Leiden Thrombophilia Study. J Thromb Haemost (2004) 2(4):619–22. doi: 10.1111/j.1538-7836.2004.00689.x
175. Ridker PM, Libby P, MacFadyen JG, Thuren T, Ballantyne C, Fonseca F, et al. Modulation of the Interleukin-6 Signalling Pathway and Incidence Rates of Atherosclerotic Events and All-Cause Mortality: Analyses From the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Eur Heart J (2018) 39(38):3499–507. doi: 10.1093/eurheartj/ehy310
176. Heinrich PC, Castell JV, Andus T. Interleukin-6 and the Acute Phase Response. Biochem J (1990) 265(3):621–36. doi: 10.1042/bj2650621
177. Chen Y, Wang J, Yao Y, Yuan W, Kong M, Lin Y, et al. CRP Regulates the Expression and Activity of Tissue Factor as Well as Tissue Factor Pathway Inhibitor via NF-kappaB and ERK 1/2 MAPK Pathway. FEBS Lett (2009) 583(17):2811–8. doi: 10.1016/j.febslet.2009.07.037
178. Zhao Y, Zhou S, Heng CK. Impact of Serum Amyloid A on Tissue Factor and Tissue Factor Pathway Inhibitor Expression and Activity in Endothelial Cells. Arterioscler Thromb Vasc Biol (2007) 27(7):1645–50. doi: 10.1161/atvbaha.106.137455
179. Machlus KR, Cardenas JC, Church FC, Wolberg AS. Causal Relationship Between Hyperfibrinogenemia, Thrombosis, and Resistance to Thrombolysis in Mice. Blood (2011) 117(18):4953–63. doi: 10.1182/blood-2010-11-316885
180. Marta RF, Goette NP, Lev PR, Chazarreta CD, Pirola CJ, Molinas FC. Normal Platelets Possess the Soluble Form of IL-6 Receptor. Cytokine (2005) 29(1):13–7. doi: 10.1016/j.cyto.2004.09.003
181. Marino M, Scuderi F, Ponte E, Maiuri MT, De Cristofaro R, Provenzano C, et al. Novel Path to IL-6 Trans-Signaling Through Thrombin-Induced Soluble IL-6 Receptor Release by Platelets. J Biol Regul Homeost Agents (2013) 27(3):841–52.
182. Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of Inflammatory Cytokines on the Release and Cleavage of the Endothelial Cell-Derived Ultralarge Von Willebrand Factor Multimers Under Flow. Blood (2004) 104(1):100–6. doi: 10.1182/blood-2004-01-0107
183. Jin W, Dong C. IL-17 Cytokines in Immunity and Inflammation. Emerg Microbes Infect (2013) 2(1):1–5. doi: 10.1038/emi.2013.58
184. Kolls JK, Lindén A. Interleukin-17 Family Members and Inflammation. Immunity (2004) 21(4):467–76. doi: 10.1016/j.immuni.2004.08.018
185. Onishi RM, Gaffen SL. Interleukin-17 and its Target Genes: Mechanisms of Interleukin-17 Function in Disease. Immunology (2010) 129(3):311–21. doi: 10.1111/j.1365-2567.2009.03240.x
186. Kolbinger F, Loesche C, Valentin MA, Jiang X, Cheng Y, Jarvis P, et al. β-Defensin 2 Is a Responsive Biomarker of IL-17A-Driven Skin Pathology in Patients With Psoriasis. J Allergy Clin Immunol (2017) 139(3):923–32. doi: 10.1016/j.jaci.2016.06.038
187. Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K. Characterization of the Interleukin-17 Isoforms and Receptors in Lesional Psoriatic Skin. Br J Dermatol (2009) 160(2):319–24. doi: 10.1111/j.1365-2133.2008.08902.x
188. de Oliveira PS, Cardoso PR, Lima EV, Pereira MC, Duarte AL, Pitta Ida R, et al. IL-17a, IL-22, IL-6, and IL-21 Serum Levels in Plaque-Type Psoriasis in Brazilian Patients. Mediators Inflammation (2015) 2015:819149. doi: 10.1155/2015/819149
189. Simon T, Taleb S, Danchin N, Laurans L, Rousseau B, Cattan S, et al. Circulating Levels of Interleukin-17 and Cardiovascular Outcomes in Patients With Acute Myocardial Infarction. Eur Heart J (2013) 34(8):570–7. doi: 10.1093/eurheartj/ehs263
190. Erbel C, Chen L, Bea F, Wangler S, Celik S, Lasitschka F, et al. Inhibition of IL-17a Attenuates Atherosclerotic Lesion Development in ApoE-Deficient Mice. J Immunol (2009) 183(12):8167–75. doi: 10.4049/jimmunol.0901126
191. Ding P, Zhang S, Yu M, Feng Y, Long Q, Yang H, et al. IL-17A Promotes the Formation of Deep Vein Thrombosis in a Mouse Model. Int Immunopharmacol (2018) 57:132–8. doi: 10.1016/j.intimp.2018.02.006
192. Li Y, Golden JB, Camhi MI, Zhang X, Fritz Y, Diaconu D, et al. Protection From Psoriasis-Related Thrombosis After Inhibition of IL-23 or IL-17a. J Invest Dermatol (2018) 138(2):310–5. doi: 10.1016/j.jid.2017.09.021
193. Maione F, Cicala C, Liverani E, Mascolo N, Perretti M, D’Acquisto F. IL-17A Increases ADP-Induced Platelet Aggregation. Biochem Biophys Res Commun (2011) 408(4):658–62. doi: 10.1016/j.bbrc.2011.04.080
194. Zhang S, Yuan J, Yu M, Fan H, Guo ZQ, Yang R, et al. IL-17A Facilitates Platelet Function Through the ERK2 Signaling Pathway in Patients With Acute Coronary Syndrome. PloS One (2012) 7(7):e40641. doi: 10.1371/journal.pone.0040641
195. Hot A, Lenief V, Miossec P. Combination of IL-17 and TNFalpha Induces a Pro-Inflammatory, Pro-Coagulant and Pro-Thrombotic Phenotype in Human Endothelial Cells. Ann Rheumatic Dis (2012) 71(5):768–76. doi: 10.1136/annrheumdis-2011-200468
196. Yuan J, Ding PW, Yu M, Zhang SS, Long Q, Cheng X, et al. IL-17 Induces MPTP Opening Through ERK2 and P53 Signaling Pathway in Human Platelets. J Huazhong Univ Sci Technol Med Sci = Hua zhong ke ji da xue xue bao Yi xue Ying wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban (2015) 35(5):679–83. doi: 10.1007/s11596-015-1489-z
197. Zhu F, Wang Q, Guo C, Wang X, Cao X, Shi Y, et al. IL-17 Induces Apoptosis of Vascular Endothelial Cells: A Potential Mechanism for Human Acute Coronary Syndrome. Clin Immunol (Orlando Fla) (2011) 141(2):152–60. doi: 10.1016/j.clim.2011.07.003
198. Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 Enhances Chemokine Gene Expression Through mRNA Stabilization. J Immunol (2007) 179(6):4135–41. doi: 10.4049/jimmunol.179.6.4135
199. Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T. Treatment With IL-17 Prolongs the Half-Life of Chemokine CXCL1 mRNA via the Adaptor TRAF5 and the Splicing-Regulatory Factor SF2 (ASF). Nat Immunol (2011) 12(9):853–60. doi: 10.1038/ni.2081
200. Maione F, Parisi A, Caiazzo E, Morello S, D’Acquisto F, Mascolo N, et al. Interleukin-17a Exacerbates Ferric Chloride-Induced Arterial Thrombosis in Rat Carotid Artery. Int J Inflammation (2014) 2014:247503. doi: 10.1155/2014/247503
201. Zelová H, Hošek J. TNF-α Signalling and Inflammation: Interactions Between Old Acquaintances. Inflammation Res (2013) 62(7):641–51. doi: 10.1007/s00011-013-0633-0
202. Baud V, Karin M. Signal Transduction by Tumor Necrosis Factor and its Relatives. Trends Cell Biol (2001) 11(9):372–7. doi: 10.1016/s0962-8924(01)02064-5
203. Bonifati C, Ameglio F. Cytokines in Psoriasis. Int J Dermatol (1999) 38(4):241–51. doi: 10.1046/j.1365-4362.1999.00622.x
204. Ettehadi P, Greaves MW, Wallach D, Aderka D, Camp RD. Elevated Tumour Necrosis Factor-Alpha (TNF-Alpha) Biological Activity in Psoriatic Skin Lesions. Clin Exp Immunol (1994) 96(1):146–51. doi: 10.1111/j.1365-2249.1994.tb06244.x
205. Kyriakou A, Patsatsi A, Vyzantiadis T-A, Sotiriadis D. Serum Levels of TNF-α, IL-12/23p40, and IL-17 in Plaque Psoriasis and Their Correlation With Disease Severity. J Immunol Res (2014) 2014:467541. doi: 10.1155/2014/467541
206. Ridker PM, Rifai N, Pfeffer M, Sacks F, Lepage S, Braunwald E. Elevation of Tumor Necrosis Factor-Alpha and Increased Risk of Recurrent Coronary Events After Myocardial Infarction. Circulation (2000) 101(18):2149–53. doi: 10.1161/01.cir.101.18.2149
207. Kablak-Ziembicka A, Przewlocki T, Sokołowski A, Tracz W, Podolec P. Carotid Intima-Media Thickness, Hs-CRP and TNF-α Are Independently Associated With Cardiovascular Event Risk in Patients With Atherosclerotic Occlusive Disease. Atherosclerosis (2011) 214(1):185–90. doi: 10.1016/j.atherosclerosis.2010.10.017
208. Cambien B, Bergmeier W, Saffaripour S, Mitchell HA, Wagner DD. Antithrombotic Activity of TNF-Alpha. J Clin Invest (2003) 112(10):1589–96. doi: 10.1172/jci19284
209. Pircher J, Merkle M, Wörnle M, Ribeiro A, Czermak T, Stampnik Y, et al. Prothrombotic Effects of Tumor Necrosis Factor Alpha In Vivo Are Amplified by the Absence of TNF-Alpha Receptor Subtype 1 and Require TNF-Alpha Receptor Subtype 2. Arthritis Res Ther (2012) 14(5):R225–R. doi: 10.1186/ar4064
210. Pignatelli P, De Biase L, Lenti L, Tocci G, Brunelli A, Cangemi R, et al. Tumor Necrosis Factor-Alpha as Trigger of Platelet Activation in Patients With Heart Failure. Blood (2005) 106(6):1992–4. doi: 10.1182/blood-2005-03-1247
211. Pignatelli P, Cangemi R, Celestini A, Carnevale R, Polimeni L, Martini A, et al. Tumour Necrosis Factor Alpha Upregulates Platelet CD40L in Patients With Heart Failure. Cardiovasc Res (2008) 78(3):515–22. doi: 10.1093/cvr/cvn040
212. Pignatelli P, Sanguigni V, Lenti L, Ferro D, Finocchi A, Rossi P, et al. Gp91phox-Dependent Expression of Platelet CD40 Ligand. Circulation (2004) 110(10):1326–9. doi: 10.1161/01.CIR.0000134963.77201.55
213. Yacoub D, Hachem A, Theoret JF, Gillis MA, Mourad W, Merhi Y. Enhanced Levels of Soluble CD40 Ligand Exacerbate Platelet Aggregation and Thrombus Formation Through a CD40-Dependent Tumor Necrosis Factor Receptor-Associated Factor-2/Rac1/p38 Mitogen-Activated Protein Kinase Signaling Pathway. Arterioscler Thromb Vasc Biol (2010) 30(12):2424–33. doi: 10.1161/atvbaha.110.216143
214. Conway EM, Bach R, Rosenberg RD, Konigsberg WH. Tumor Necrosis Factor Enhances Expression of Tissue Factor mRNA in Endothelial Cells. Thromb Res (1989) 53(3):231–41. doi: 10.1016/0049-3848(89)90098-4
215. Kirchhofer D, Tschopp TB, Hadváry P, Baumgartner HR. Endothelial Cells Stimulated With Tumor Necrosis Factor-Alpha Express Varying Amounts of Tissue Factor Resulting in Inhomogenous Fibrin Deposition in a Native Blood Flow System. Effects of Thrombin Inhibitors. J Clin Invest (1994) 93(5):2073–83. doi: 10.1172/JCI117202
216. Nawroth PP, Stern DM. Modulation of Endothelial Cell Hemostatic Properties by Tumor Necrosis Factor. J Exp Med (1986) 163(3):740–5. doi: 10.1084/jem.163.3.740
217. Bussolino F, Camussi G, Baglioni C. Synthesis and Release of Platelet-Activating Factor by Human Vascular Endothelial Cells Treated With Tumor Necrosis Factor or Interleukin 1 Alpha. J Biol Chem (1988) 263(24):11856–61. doi: 10.1016/S0021-9258(18)37865-7
218. Camussi G, Bussolino F, Salvidio G, Baglioni C. Tumor Necrosis Factor/Cachectin Stimulates Peritoneal Macrophages, Polymorphonuclear Neutrophils, and Vascular Endothelial Cells to Synthesize and Release Platelet-Activating Factor. J Exp Med (1987) 166(5):1390–404. doi: 10.1084/jem.166.5.1390
219. Chignard M, Le Couedic JP, Tence M, Vargaftig BB, Benveniste J. The Role of Platelet-Activating Factor in Platelet Aggregation. Nature (1979) 279(5716):799–800. doi: 10.1038/279799a0
220. Camussi G, Tetta C, Baglioni C. The Role of Platelet-Activating Factor in Inflammation. Clin Immunol Immunopathol (1990) 57(3):331–8. doi: 10.1016/0090-1229(90)90108-3
221. Conway EM, Rosenberg RD. Tumor Necrosis Factor Suppresses Transcription of the Thrombomodulin Gene in Endothelial Cells. Mol Cell Biol (1988) 8(12):5588–92. doi: 10.1128/mcb.8.12.5588
222. Hou B, Eren M, Painter CA, Covington JW, Dixon JD, Schoenhard JA, et al. Tumor Necrosis Factor Alpha Activates the Human Plasminogen Activator Inhibitor-1 Gene Through a Distal Nuclear Factor kappaB Site. J Biol Chem (2004) 279(18):18127–36. doi: 10.1074/jbc.M310438200
223. Schleef RR, Bevilacqua MP, Sawdey M, Gimbrone MA Jr., Loskutoff DJ. Cytokine Activation of Vascular Endothelium. Effects on Tissue-Type Plasminogen Activator and Type 1 Plasminogen Activator Inhibitor. J Biol Chem (1988) 263(12):5797–803. doi: 10.1016/S0021-9258(18)60636-2
Keywords: cardiovascular disease, endothelium, hypercoagulability, platelets, psoriasis, systemic inflammation
Citation: Visser MJE, Tarr G and Pretorius E (2021) Thrombosis in Psoriasis: Cutaneous Cytokine Production as a Potential Driving Force of Haemostatic Dysregulation and Subsequent Cardiovascular Risk. Front. Immunol. 12:688861. doi: 10.3389/fimmu.2021.688861
Received: 31 March 2021; Accepted: 06 July 2021;
Published: 16 July 2021.
Edited by:
Beate E. Kehrel, University Hospital Münster, GermanyReviewed by:
Paola Di Meglio, King’s College London, United KingdomEva Reali, University of Milano-Bicocca, Italy
Copyright © 2021 Visser, Tarr and Pretorius. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Etheresia Pretorius, cmVzaWFwQHN1bi5hYy56YQ==; orcid.org/0000-0002-9108-2384