Abstract
Rapid recruitment of neutrophils to an inflamed site is one of the hallmarks of an effective host defense mechanism. The main pathway through which this happens is by the innate immune response. Neutrophils, which play an important part in innate immune defense, migrate into lungs through the modulation actions of chemokines to execute a variety of pro-inflammatory functions. Despite the importance of chemokines in host immunity, little has been discussed on their roles in host immunity. A holistic understanding of neutrophil recruitment, pattern recognition pathways, the roles of chemokines and the pathophysiological roles of neutrophils in host immunity may allow for new approaches in the treatment of infectious and inflammatory disease of the lung. Herein, this review aims at highlighting some of the developments in lung neutrophil-immunity by focusing on the functions and roles of CXC/CC chemokines and pattern recognition receptors in neutrophil immunity during pulmonary inflammations. The pathophysiological roles of neutrophils in COVID-19 and thromboembolism have also been summarized. We finally summarized various neutrophil biomarkers that can be utilized as prognostic molecules in pulmonary inflammations and discussed various neutrophil-targeted therapies for neutrophil-driven pulmonary inflammatory diseases.
Introduction
Infections of the lower respiratory tract accounts for about 35% of all deaths accruing from infectious diseases, resulting in a yearly death rate of about 4 million patients (1). They increase the worldwide disease burden than most infectious diseases such as HIV infection and malaria (1, 2). The effectiveness of a host defense mechanism against infections of the lung is very crucial and this is basically dependent on the quick clearance of the disease-causing agent from the airways. The main pathway through which this happens is by the innate immune response (3, 4). Therefore, any inabilities in the host innate immune response can lead to heavy microbial colonization which can compromise the integrity of the lung parenchyma. The exact mechanism of immune activation during pulmonary inflammation and infection remains unclear. However, some studies have iterated that the host, through resident and non-resident immune cells and receptors such as pattern recognition receptors (PRR), recognizes and destroys these organisms. One of the key players in innate immunity is neutrophils and their recruitment to the site of inflammation. Because of their ability to enter into organs and tissues to execute important host defense mechanisms, neutrophils are usually refer to as all-terrain vehicle of the innate immune system. Neutrophils form the first line of host defense mechanism because of how quick they are recruited when the lung is challenged with a microbial infection or other particles. These functions of neutrophils are highly regulated by signals received from their repertoire of PRRs, and this allow neutrophils, whose recruitment are modulated by chemokines to sense both pattern-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) at the site of inflammation. Robust recruitment of neutrophils to an inflamed site is the hallmark of all injuries and acute microbial infections (5, 6). This robust recruitment is made possible through neutrophil concentration gradients across the epithelium, extracellular matrix (ECM) and endothelium (7, 8). Neutrophils are guided to the sites of action by chemokines expressed by resident cells. It has been confirmed through animal and clinical studies that, CXC and CC chemokines play important roles in innate immunity by recruiting and activating neutrophils during microbial infections and injuries of the lungs. During some inflammatory diseases, the levels of these chemokines increase and vary according to the stage of the disease (9–11). Inappropriate neutrophil recruitment, or when neutrophil activation is impaired, it can lead to lung inflammations. Also, when the recruitment is not controlled or when the activation is not sustained, this could lead to collateral tissue damage and disease.
Despite the importance of chemokines and pattern recognition receptors in host immunity, little has been discussed on their roles in neutrophil-induced immunity. Herein, this review aims at highlighting some of the developments in lung neutrophil immunity and focuses on the functions of (1) CXC chemokines, (2) CC chemokines, (3) pattern recognition receptors and (4) integrins in neutrophil immunity during lung infections and inflammations. The pathophysiological role of neutrophils in COVID-19 and thromboembolism have also been summarized. Finally, we discussed various neutrophilic biomarkers, and various neutrophil-targeted therapies for neutrophil-driven pulmonary inflammatory diseases.
Neutrophils
In humans, neutrophils form the largest proportion of leukocytes that circulate in the blood and form the major component in organs such as the lungs. They play a major role in innate immunity despite being described as having terminal differentiation and being characterized with a short lifespan after leaving the hematopoietic organ. Their main distinguishing characteristic is the removal of debris and pathogens through phagocytosis but can also play important roles in immune functions. Aside the direct phagocytosis of bacteria (12) and fungi (13), neutrophils, through the process of NETosis reduce the spread of microbes by releasing neutrophil extracellular traps (NETs) (14). Although neutrophils destroy pathogenic agents, they also have the ability to significantly modulate the functions of other immune cells. The recruitment of neutrophils into the lung usually occurs in the small capillaries of the alveolar network (15, 16). Neutrophils change their shapes in order to move across the capillary bed because of the small nature of the lung capillaries (15). In addition, the velocity of blood flow within the lungs’ capillary network is low (17). The velocity of blood flow and the change in shape of neutrophils account for the increase in the time required by neutrophils to transit during physiological conditions in the pulmonary microvasculature and this has accounted for the name ‘marginated pool of neutrophils’ (15) (Figure 1A). The extracellular secretion of oxidases and proteases (e.g. Myeloperoxidase and neutrophil elastase) following the mobilization of granule to the surface of a cell is a trademark of neutrophil activities within an inflamed airway. This mechanism modulates the upregulation of primary and secondary granule markers (CD63 and CD66b, respectively) on the surface of airway neutrophils resulting in high proteolytic and oxidative activities. Further findings have shown that airway neutrophils play a role in the regulation of adaptive immune system, demonstrating the multifaceted significance of neutrophil plasticity (19). For instance, in cystic fibrosis airway neutrophils, a significant immunosuppressive feature has been detected. This feature is immunosuppressive because it causes the downregulation of T-cell through the activation of arginase I (20). When neutrophils are activated in cystic fibrosis airways, they don’t only impact on T cells but also play important role in the lymphatic compartment by showcasing its antigen-presenting cell capabilities (e.g., expression of CD80, CD86, and MHC II). One feature of immature neutrophils is the expression of CXCR4 and is highly upregulated when airway neutrophils are activated. This could be responsible for their acquired ability to migrate from inflammatory tissues into lymphatic vessels (21–23). The transiting of neutrophils to lymph nodes has been related to the proliferation of T-cell, suggesting that neutrophils are also involved in the active regulation of the adaptive immune response (23, 24). Intracellular pathogens can use this lymphatic neutrophils as a “Trojan horse” to spread within the body (25, 26).
Figure 1

Schematic representation of how neutrophils are recruited to the lung. (A) Neutrophils change their shapes in order to move across the capillary bed because of the small nature of the lung capillaries. The increase in time required by neutrophils to transit in the lungs accounts for the name ‘marginated pool of neutrophils’. Neutrophil stiffening by cytoskeleton rearrangement after stimulation participates in neutrophil recruitment into the lung. (B) 12-HETE, a lipid mediator made by 12/15-LO in lung macrophages regulates the balance of chemokine-chemokine receptors and increases vascular permeability and neutrophil recruitment in the lungs. (C) The increase in chemokine concentration gradient in the alveolar region coupled with the presentation of chemokines by glycosaminoglycans, modulates the recruitment of neutrophils to this region of the lung (18).
Integrin-Mediated Neutrophil Interaction With Endothelia and Epithelial Cells
Until recently, adhesion of neutrophils to the endothelium was known to involve three phases: rolling which is mediated by selectin, chemokine-triggered activation of neutrophils, and firm arrest of neutrophils initiated by integrins (27, 28). However, studies have shown that this process is a rather complex event made up of additional phases such as tethering, slow rolling, modulation of adhesion strength, intraluminal crawling, and transcellular or paracellular migration (27, 29). Currently, it is believed that the first stage in neutrophil and other leukocyte adhesion to the endothelium is neutrophil capture (or tethering), which is mediated by interactions between L, E, and P-selectins, as well as P-selectin glycoprotein ligand (PSGL1) and α4β1 (VLA4) integrin. Leukocytes, inflamed endothelium and platelets, and inflamed endothelium are known to respectively express L-selectin, P-selectin and E-selectin. Also, endothelium and some leukocytes are known to express PSGL1. Subsequently, an interaction between selectins, PSGL-1 and other glycosylated ligands mediate the rolling of neutrophils on the endothelium. More importantly, adhesions which are mediated by L-selectin and P-selectin require shear stress (30, 31). This phase is ensued by a selectin-triggered signaling phase (“slow rolling”), followed by a firm capture of neutrophils on the endothelial surface, a step that involves β1- and β2-integrins and their respective binding partners.
Integrins are αβ heterodimeric, transmembrane proteins that mediate both cell-substrate and cell-cell interactions with a diverse group of ligands (32). Integrins have numerous functions, one of which is to mediate cell migration. Integrins are made up of several α subfamily (α2β1, α3β1, α4β1, α5β1, α6β1 and α9β1) which are usually expressed and upregulated on neutrophils (33, 34). Integrins, including those of the β1 and β2 subfamilies are known to be expressed by human neutrophils. Proteins of the extracellular matrix (ECM) binds to the β1 family integrin upon recognition of the amino acid sequences Arg-Gly-Asp (RGD) and Leu-Asp-Val (LDV) (35). The β2-integrins consist of a common β-chain (CD18) and a variable α-chain (CD11a, b, c, or d). Interactions between CD11a/CD18 (LFA-1), α4β1 (VLA-4), α4β7 and some intercellular adhesion molecules such as ICAM-1, VCAM1, and MADCAM1 mediate neutrophil arrest. A series of outside-in and inside-out intracellular signaling pathways are then activated, resulting in the strengthening of adhesion followed by the spreading of neutrophils. Conformational changes in the structure of inserted (I) domain of the αL subunit of LFA-149 enhance firm neutrophils adhesion under shear flow (36, 37). Neutrophils then crawl along endothelial cells (“intraluminal crawling”) by a process involving interactions between CD11b/CD18 (αmβ2 or Mac-1) and ICAM-1. Neutrophils, through a paracellular or a transcellular route, finally transmigrate across the endothelium. In a murine experiment, integrin α4β1 was found to be involved in neutrophil adhesion during pneumonia caused by Streptococcus pneumoniae (38). Also, in a study by Ulyanova et al., it was opined that integrin α4β1 plays a key role in adhesion and migration during lung inflammation, and mediates integrin β2-independent neutrophil accumulation (39). However, the expression of integrin α4β1 on neutrophils in early stage acute respiratory distress syndrome (ARDS), was found to be downregulated (40). During lung infection, it has been reported that extravasation of neutrophils to the site of inflammation is aided by integrin α9β1 (33). In a study involving older people with aspiration pneumonia, it was found that integrin α9β1 and CD11b expression levels on circulating neutrophils were increased (41).
Neutrophil Degranulation
Pro- and anti-inflammatory substances are the major components of neutrophil granules and these can be released to destroy pathogenic organisms. This process is termed as degranulation (42). Neutrophils play important roles by releasing granules that aid in the killing of invading pathogens. Primary (azurophilic), secondary (specific) and tertiary (gelatinase) granules, as well as secretory vesicles are the four types of granules found in neutrophils. Each of these granules has a different protein content (43). They have distinct functions and are released sequentially to cell surface or to the microbe-containing phagolysosome by exocytosis in response to various signals. Following initial contact of neutrophils and endothelial cells, secretory vesicles are released via exocytosis which results in the expression of some key surface membrane proteins leading to the rolling of neutrophils through the endothelial monolayer in blood vessels. This then initiates extravasation at the infection site (43). The release of a secretory vesicles is respectively followed by the release of tertiary, primary and secondary granules (16). The release of the contents of the primary and secondary granules into the phagolysosome, or degranulation into surrounding tissues, initiates a series of antimicrobial activities. Antimicrobial enzymes and peptides such as serine proteases and defensins, as well as myeloperoxidase, which converts H2O2 to antiseptics hypobromous acid, hypochlorous acid and hypoiodous acid are all constituents of the primary granules (44). Lactoferrin, lipocalin, lysozyme, LL37 and matrix metalloproteinases are among the overlapping proteins contained in secondary and tertiary granules (44). The actions of these molecules within the granules are required for effective pulmonary immunity without causing significant tissue damage (45).
Neutrophil Extracellular Traps
Until recently, neutrophils were mainly known to utilize both extracellular killing by exocytosis and intracellular killing by phagocytosis to recognize and kill pathogenic organisms. Recently, the release of extracellular “traps” or complexes created by cationic effectors (including neutrophil myeloperoxidase and elastase), histones, and decondensed nuclear DNA (e.g., after histone citrullination), have been identified as the third effector mechanism utilized by neutrophils. The release of neutrophil extracellular traps (NETs) (Figure 2), through a process call “NETosis”, is believed to immobilize pathogens and probably destroy them while also precipitating neutrophil death (47, 48). Although NETs have been linked to a variety of diseases, including viral, fungal, and bacterial infectious diseases as well as other autoimmune disorders, their functions in chronic and acute inflammation is still not fully elucidated (47, 49). Studies focusing on NETs have demonstrated both advantageous and harmful impacts of these systems in the context of airway diseases (50). NET can increase the killing efficiency and reduce the burden caused by pathogens because of its ability to spread out and trap these pathogens. A lot of organisms have developed a mechanism to evade these destructive impacts of the NET system and this can result in the accumulation of host DNA, histones, neutrophil elastase, and myeloperoxidase thereby causing direct or indirect cell toxicity and subsequent lung injury (51–53). This can also lead to obstruction in the airways because the presence of these proteins and enzymes may result in an increased mucus viscosity (54–56).
Figure 2
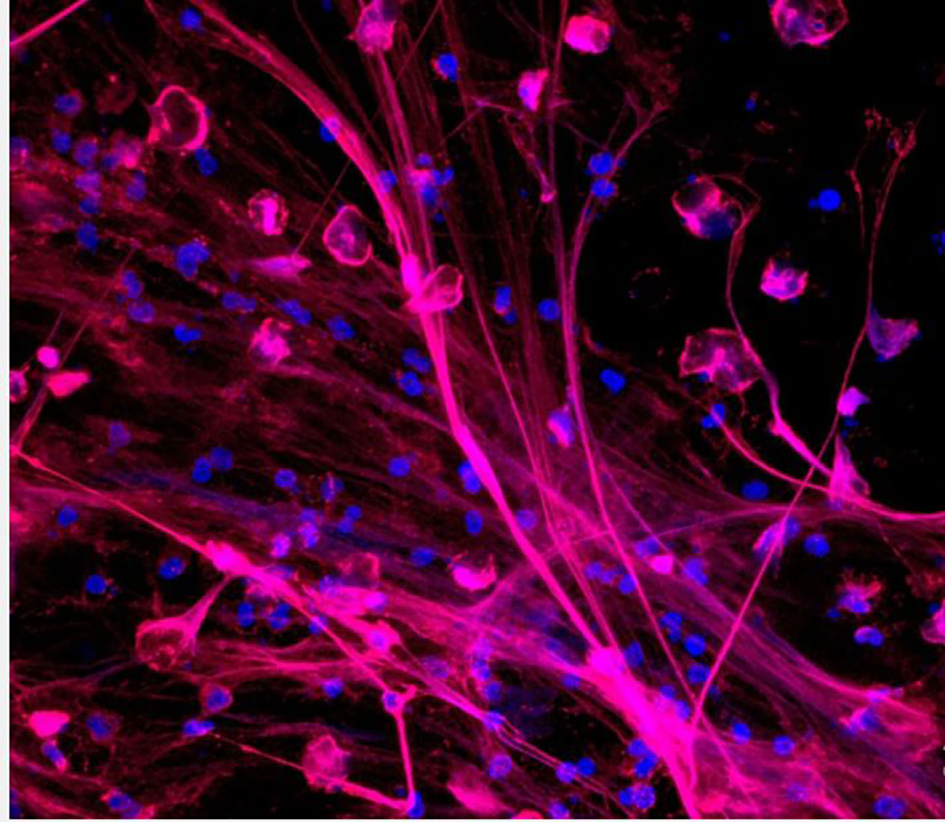
Neutrophils releasing neutrophil extracellular traps (NETs). NETs are stained to visualize neutrophil myeloperoxidase (red) and DNA (blue) (46).
Regulatory Activities of Neutrophils During Pulmonary Inflammation and Infection
The most common lung infectious disease which is characterized by inflammation in the interstitial lung, alveolar and the airways is pneumonia. Pneumonia development hinges on the interplay between mucosal immunity and mucosal colonization by the etiological agent (57). During pneumonia, there is recruitment of neutrophils into the lung. Neutrophils can act in unison with other immune cells to regulate infections caused by pathogens (58). In recent times, evidence have been documented on the roles of neutrophils in pneumonia pathogenesis. According to Onishi et al. (59), the level of neutrophil in bronchoalveolar lavage fluid (BALF) was greater in the relapse category of organizing pneumonia patients. On the other hand, community-acquired pneumonia patients had lower levels of neutrophils in peripheral blood (60). During acute lung injury (ALI), chemokines present on the inflamed pulmonary endothelial cells play important roles in the recruitment of neutrophils into the lung (61). Intracellular signaling cascade is initiated through the binding of chemokines to their neutrophil receptors which leads to integrin activation and cytoskeleton rearrangement. This process is essential for the recruitment of neutrophils (61). As a response to lung injury, resident pulmonary macrophages produce and transmit chemokines, and also server as a major source of pro-inflammatory mediators, such as IL-1, tumor necrosis factor- (TNF-α), 12-hydroxyeicosatetraenoic acid (12-HETE), and interleukin (IL)-8 (CXCL8) (62, 63). During inflammation, the lung macrophages/monocytes express 12/15-lipoxygenase (12/15-LO). The 12-HETE lipid mediator, a product of 12/15-LO, has been involved in the regulation of vascular permeability and recruitment of neutrophils into the lungs during lipopolysaccharide-induced injury in the lungs (Figure 1B) (63, 64). The 12/15-LO is known to be essential for the mobilization of neutrophil into the lung’s intra-alveolar and interstitial regions by hematopoietic cells. However, trafficking of neutrophils to the lung’s microvasculature is controlled by non-hematopoietic 12/15-LO (64). This was confirmed when the vascular permeability was drastically reduced in both 12/15-LO-deficient and 12/15-LO-blocked WT mice during an ALI-induced experiment. A CXCR2-dependent mechanism has also been shown to mediate vascular permeability (Figure 1B) (63, 64). A concentration gradient of chemokine within the intravascular, interstitial and the alveolar space has been observed during the development of ALI, with the maximum accumulation been observed in the alveolar area (Figure 1C) (65, 66). The migration of neutrophils into the alveolar area is stimulated by this gradient. Distinct chemokines presented by glycosaminoglycans get stuck to endothelial cells (67, 68). CXCL8’s monomer-dimer equilibrium is vital for the attachment of CXCL8 to glycosaminoglycans which influences its capacity to mobilize neutrophils (69). CXCR2 receptors on neutrophils have been found to be among the key significant chemokine receptors which are active in lung neutrophil mobilization during ALI animal models (Figure 1C) (70–73).
One of the hallmark of acute respiratory distress syndrome (ARDS) is the infiltration of neutrophils to the inflamed lung (74). Endothelial cells stimulate and arrest circulating neutrophils in patients with ARDS (75). The formation of neutrophil extracellular traps (NETs), oxidative stress and the release of proteases usually occurs when there is activation of neutrophil during ARDS. During ARDS, selectin sequesters neutrophils, leading to an “inside-out” activation of CD11a/CD18, which then binds to intercellular adhesion molecules (ICAMs) of the endothelium (76). During ARDS, neutrophils help to repair the damaged lung tissue by releasing MMP-9 and activating the Wnt/b-catenin pathway (77). The role of neutrophil recruitment in ARDS is complicated, and more research is required. Some researchers have shed light on the pathogenic function of neutrophils in inflammatory ARDS (78). Higher levels of neutrophils have been observed in patients presenting with ARDS and this can serve as a predictor of poor prognostic outcome (79). As a result, it has been projected that strategies to reduce neutrophils in lung tissue, including the reduction of neutrophil recruitment and the activation of its immune functions, would reduce lung injury. Elevated levels of neutrophils in pulmonary tissues contribute to the pathogenesis of ARDS, while decreasing levels reduce the generation of cytotoxic mediators (80). The assembly and activation of reactive oxygen species (ROS)-producing nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex (NOX2) by neutrophils can contribute to the progression of ARDS (81). NOX2, which is found on membranes, converts oxygen to superoxide anion, which is then released to the outside of the cells. The highly reactive superoxide anion spontaneously dismutates into a more stable hydrogen peroxide (H2O2). H2O2 can pass through the cell membrane and disseminate to the extracellular or intracellular environment. H2O2 is used by the enzyme myeloperoxidase to generate hydroxyl radicals, hypochlorous acid, and other reactive products (82). Since ROS are harmful to pulmonary tissues, it is desirable to reduce ROS production in order to reduce lung inflammatory injury (83).
Pattern Recognition Receptors in Pulmonary Infections and Inflammation
The identification of pathogens during infection is one of the most significant roles of the innate immune system and this recognition is driven by cell-surface pattern recognition receptors (PRRs) (84). Intracellular and extracellular PRRs ligation can mediate chemokine/cytokine expression and also induce neutrophil recruitment into the lungs during lung inflammations. PRRs specifically recognize unique molecular patterns found on the surfaces of microbes and this leads to a series of upstream and downstream events. This eventually causes the migration of neutrophils to the lungs followed by the influx of monocyte/macrophage to the site of infection (57). The first host cells that encounter antigens of microorganisms during infection are the airway epithelial cells, dendritic cells, and alveolar macrophages. These cells trigger proinflammatory or anti-inflammatory downstream immune responses. PRRs are present in soluble forms like mannan-binding lectin (MBL) and in the form of transmembraneous or intracellular molecules that directly mediate cellular immune responses. The major families of airway epithelial PPRs include protease-activated receptors (PAR), Toll-like receptors (TLRs), Nod-like receptors (NLRs), C-type lectin receptors, RIG-I-like receptors (RLRs), and the bitter- and sweet-taste receptors (Table 1). These PRRs initiate a cascade of downstream signal transduction pathways which leads to the recognition of PAMPs and DAMPs in response to pulmonary infections (Figure 3). PAMPs are highly conserved structures presented by several groups of microorganisms. PAMPs may include bacterial lipopolysaccharide (LPS), peptidoglycan (PGN) or lipoteichoic acid (LTA).
Table 1
| Ligand (adaptors in parentheses) | PRRs | Ligand (origin in parentheses) | Localization | Function in neutrophil | Signals | Response | Ref |
|---|---|---|---|---|---|---|---|
| TLRs (TRAM, Trif, Mal, MyD88) | TLR1 | Triacyl lipopeptides (bacterial lipoprotein) Di-/triacyl lipopeptides | Cell surface | Migration, Inhibit apoptosis, NET formation, Respiratory burst, Degranulation, Phagocytosis | IRFs NF-κB MAPKs | Pro–IL-1, pro–IL-18 Antiviral proteins Chemokines Cytokines | (85, 86) |
| TLR2 | Multiple lipoproteins, Lipoteichoic acid, Zymosan (fungi) | Cell surface | Formation of heterophilic dimers with TLR1 and TLR6, Respiratory burst, Neutrophil migration, Degranulation, apoptotic regulator, Phagocytosis | (87, 88) | |||
| TLR4 | LPS (Gram-negative bacteria) | Cell surface | Recognition of LPS together with myeloid differentiation factor 2, Respiratory burst, NET formation, Neutrophil migration, Phagocytosis, Inhibit apoptosis, Degranulation | (89–91) | |||
| TLR5 | Flagellin (flagellated bacteria) | Cell surface | Activation of lung epithelial cells to induce inflammatory cytokine, NET formation, Inhibit apoptosis, Phagocytosis, Degranulation | (92, 93) | |||
| TLR6 | Triacyl lipopeptides (bacterial lipoprotein) | Cell surface | Formation of heterophilic dimers with TLR2, Respiratory burst, Inhibition of apoptosis, Phagocytosis, NET formation, Degranulation, | (94) | |||
| TLR8 | ssRNA (viruses); small antiviral compounds | Endosome | Recognition of synthetic compound imidazoquinoline, Neutrophil migration, NET formation, Respiratory burst, Degranulation, Inhibition of apoptosis, Phagocytosis | (90, 91) | |||
| TLR9 | Unmethylated CpG DNA | Endosome | Degranulation, Phagocytosis, Respiratory burst, Inhibit apoptosis, NET formation, Migration, Proinflammatory cytokines | (95, 96) | |||
| TLR10 | Unknown | No neutrophil function described | (94, 97) | ||||
| NLRs (MyD88) | NLRC4 (IPAF) | Bacterial flagellin and other components of the bacterial secretion apparatus | Cytoplasm | Trigger the secretion of IL-1β from neutrophils. | NF-κB Caspase-1 | IL-1, IL-18 | (98, 99) |
| NOD1 | Peptidoglycan (Gram-negative bacteria) | Cytoplasm | Recognition of intracellular bacterial cell products, Phagocytosis, Bacterial killing, neutrophil migration | (100, 101) | |||
| NOD2 | Peptidoglycan (Gram-positive bacteria) | Cytoplasm | Recognition of intracellular bacterial cell products, Phagocytosis, neutrophil migration, Bacterial killing, Degranulation | (100, 102) | |||
| NOD5/NLRX1 | dsRNA (viruses) | In response to TLR2 ligands, NLRX1 induces the production of neutrophil ROS. | (103) | ||||
| NLRP1 | Muramyl dipeptide moiety of PGN (bacterial cell wall); Anthrax lethal toxin (Bacillus anthracis) | No neutrophil function described | (98, 104) | ||||
| NLRP3 | PAMPs, virulence factor, DAMPs | Endosome | Response to multiple stimuli via forming a NALP3 inflammasome and secreting IL-1β, caspase-1 activation | (98, 100) | |||
| NLRP6 | Unknown | It negatively regulates TLR-induced canonical NF-kB and MAPK pathways in murine | (105, 106) | ||||
| NLRP12 | It mediates the secretion of IL-1β induced by inflammasomes in neutrophils. | (107) |
Summary of some pattern recognition receptors and their functions in innate immunity.
Figure 3
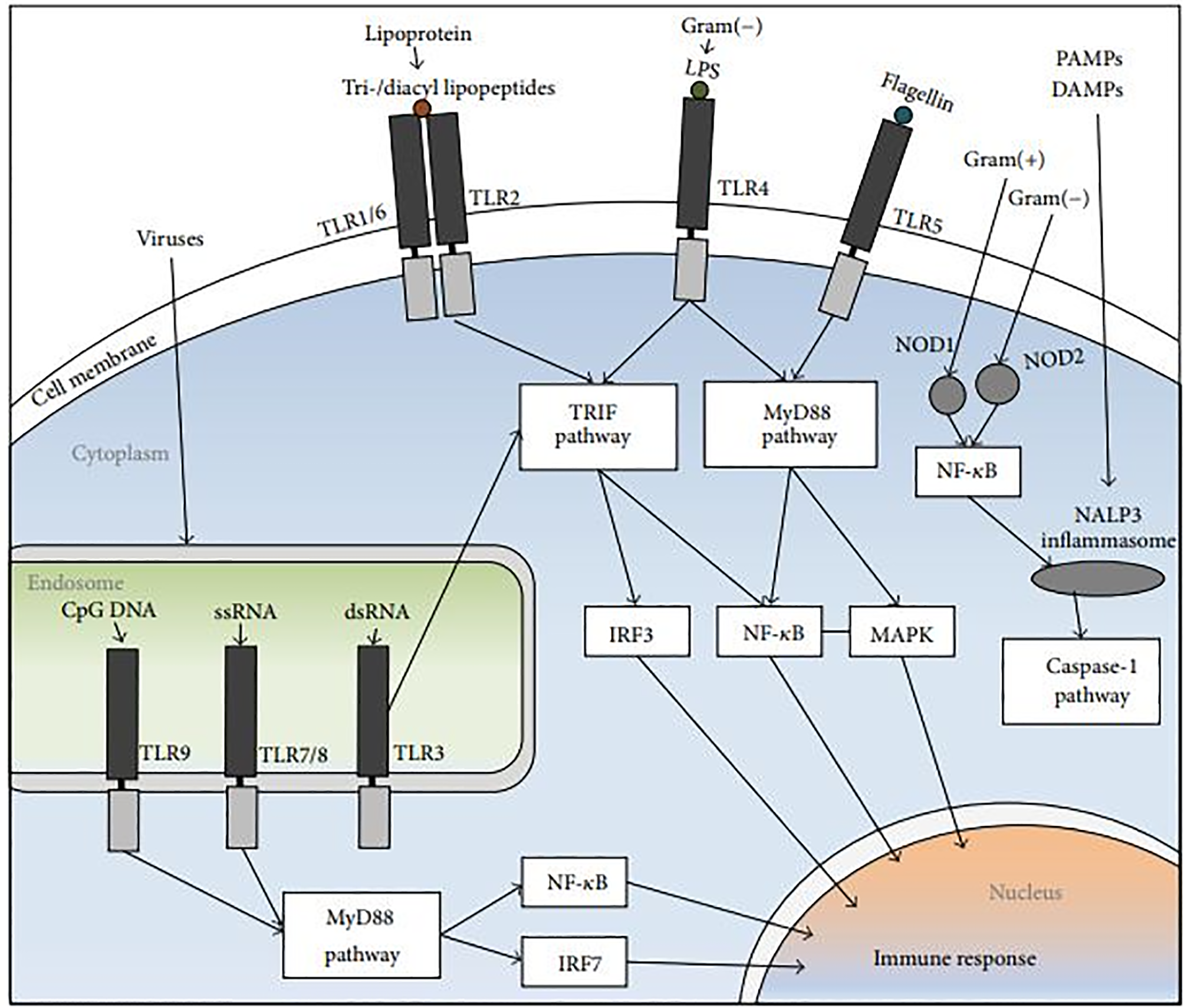
The NLR and TLR signal-transduction pathways. The PRRs identify DAMPs and PAMPs. Endosomal TLR3, and TLR4, TLR1/6+TLR2 heterodimers stimulate the TRIF pathway, followed by NF-κB and IRF induction. The MyD88 pathway is activated by TLR5 and endosomal TLR9 and TLR7, followed by IRF7, NF-κB and MAPK activation. NOD2 and NOD1 which are cytoplasmic PRRs, induce the recruitment of NALP3 inflammasome, trigger the release of NF-κB, and activate the caspase-1 pathway (108).
In order to ascertain the role of NOD-receptors in COPD, Barton et al. studied the expression of NOD1 in some alveolar epithelial type II cells, airway epithelial cells, endothelial cells as well as alveolar macrophages (109). They found that patients with chronic bronchitis had decreased expression of NOD1 in their lung tissues (109). DAMP-activated inflammasome may also have a contributing role in the pathophysiology of COPD. Conditions such as infections, hypercapnia, inhaled toxic agents, focal hypoperfusion, oxidative stress, tissue acidification, necrotic cell death and hypoxia may induce damaged lung tissues to release DAMPs (e.g., uric acid, ATP), leading to the activation of NLRP3 inflammasome. NLRP3 activation protects the host from infections caused by several pneumonia-causing bacteria such as C. pneumoniae, S. pneumoniae, K. pneumoniae, S. aureus, and L. pneumophila (110–112). Aged mice had decreased NLRP3 expression and function, which made them more susceptible to pneumonia, ALI, and death (113). Due to a diminished expression and function of NLRP3 in the lungs of an aging population, their susceptibility to secondary pneumonia caused by S. pneumoniae was enhanced (114). NLRP3 also increases the incidence and mortality rate of ALI (115). Decreased expression of NLRP3 also inhibited the onset of severe necrotic pneumonia caused by S. aureus by enhancing bacteria clearance (116). When NLRP3 is activated by α-hemolysin during S. aureus-induced pneumonia, it causes necrotizing pneumonia or necrotic lung injury that is independent of IL-1β signaling (116, 117). S. aureus-induced pneumonia does not only stimulate NLRP3 inflammasome but also stimulates NLRC4 inflammasome which induces necroptosis by inhibiting IL-17A-induced neutrophil trafficking to the lungs and the production of IL-18 (118). NLRC4 deficiency promotes neutrophil infiltration in the lungs, reduces necroptosis, improves pathogen clearance, and improves host survival. Thus, NLRC4 deficiency in both hematopoietic and non-hematopoietic cells protects the host from S. aureus-induced pneumonia (118). Activation of NLRC4 stimulates the production of IL-1 β, IL-17A, and neutrophil chemoattractants in the lung, which prove beneficial to the host during pneumonia caused by Gram-negative bacteria such as K. pneumoniae and P. aeruginosa (119). NLRC4 activation, on the other hand, causes inflammatory lung injury, increases lung bacterial burden, and causes necroptosis during P. aeruginosa-induced pneumonia (120). Furthermore, following S. aureus infections, NLRC4 suppressed IL-17A-dependent neutrophil accumulation by triggering necroptosis and IL-18 activation in the lungs (118). NLRP6-/- mice were more resistant to pulmonary infection caused by S. aureus than their wild-type counterparts, as they recorded improved survival rates and increased bacterial clearance in the lungs (121).
TLR2, TLR4, and NLRP3 expressed by mRNA in patients with COPD were significantly increased in neutrophils during acute exacerbation compared to stable disease. However, TLR9 expression by mRNA did not differ significantly between stable disease and exacerbation. Increased expression of TLR2, TLR4, and NLRP3 on neutrophils could make peripheral blood neutrophils more sensitive to DAMPs generated during COPD exacerbations. This means that, elevated TLR2 and TLR4 expression in neutrophils, along with higher DAMP levels, may contribute to DAMP-induced neutrophilic airway inflammation during COPD exacerbation (122). TLR2 and TLR4 are not receptors for only DAMPs but also pathogen-associated molecular patterns (PAMPs). Thus, PAMPs may play a role in the inflammatory response during airway infection-associated exacerbations. ATP, a recently classified DAMP, is known to activate NLRP3 and has been shown to be elevated in BAL fluid of COPD patients (123). Pro-inflammatory cytokines IL-1 and IL-18 are released when NLRP3 is activated on neutrophils, and this has been linked to the development of COPD (123, 124). Exposure to cigarette smoke extract is known to increase TLR4 expression in bronchial and nasal epithelial cells during pulmonary inflammation (125, 126).
Airway epithelial cells recognize different pathogens during pneumonia due to the expression of various PRRs. These PRRs can be either extracellular TLRs (TLR1, TLR2, TLR4, TLR5, and TLR6), intracellular TLRs (TLR3, TLR7, TLR8, and TLR9) or NLRs inflammasome (127–129). In response to TLR activation during K. pneumoniae-induced pneumonia, TRIF signaling pathway can provide some antibacterial defense by inducing interferon (IFN)-x03B3 in the lungs (85). Due to the attenuation of neutrophil sequestration and the production of MIP-2, TNF-, IL-6, and LIX, Toll/IL-1R Domain-Containing Adaptor Protein (TIRAP), has been reported to play a critical role in pneumonia caused by K. pneumoniae (130). During a P. aeruginosa-induced pneumonia, it was evident that TIRAP is not required for neutrophil infiltration, LIX production, and bacterial clearance (130). Also, TLR2-induced MyD88 activation was not required for the clearance of S. aureus during pneumonia. However, TLR2-induced MyD88 activation is known to trigger an important inflammatory immune response and it is very critical for the clearance of P. aeruginosa-induced pneumonia (131). When compared to airway neutrophils from healthy subjects, a large fraction of neutrophils isolated from the BALF of patients suffering from chronic airway inflammation had upregulated levels of TLR2, TLR4, TLR5, and TLR9, as seen in CF and non-CF-bronchiectasis. These changes are associated with neutrophil respiratory burst activity, and is concomitant with de novo protein synthesis, granule exocytosis, and later induction of apoptosis (21, 96, 132). For mucosal intrinsic defense activity against non-pathogenic E. coli, S. enterica, S. pneumoniae, and P. aeruginosa, TLR5 has been confirmed to play such critical role in a rodent model (133, 134). TLR9-deficient mice are unable to produce Th1 effector cells, resulting in a higher bacterial load in the lungs (135). Thus, TLR9 plays a detrimental role in pneumonia caused by both P. aeruginosa pneumonia and methicillin-resistant S. aureus (136).
Roles of Chemokines in Lung Neutrophil Immunity
Chemokines have a low molecular weight that ranges from 7 to 15kDa and are the largest family among small cytokines. Together with their receptors, chemokines are able to regulate the migration and residence of all immune cells. Although some chemokines are considered pro-inflammatory because they are influenced by immune reactions, others are considered homeostatic and operate to regulate the migration of cells during tissue growth and repair. Chemokines are very important because of their specific physiological role, i.e. they induce the recruitment of specific subset of leukocytes (137, 138). Through subtraction hybridization process, several chemokines were originally recognized as early response genes that are stimulated by growth factors. The assumption was that, chemokines, based on this property, acted as nuclear factors and took part in the proliferation of cells. However, based on complete amino acid sequencing, chemokines were confirmed as secretory proteins. CXC chemokines: CXCL1-8 and CXCL12, and CC chemokines: CCL2, CCL17 (TARC), CCL18 (PARC), and CCL20, are the most important chemokines involve in the recruitment of neutrophils into the airways (Table 2) (151). Different cytokines produced by local airway cells (Thymic stromal lymphopoietin and IL-33, IL-25, IL-23, IL-17, IL-10, IL-1-β, IL-1-alpha), are those that transmit the relevant biological impact of chemokines.
Table 2
| Chemo-attractants | Receptors | Ref | |||
|---|---|---|---|---|---|
| Systemic name | Name in Human | Name in Murine | Human neutrophils | Murine neutrophils | |
| CXCL1 | GROα | KC | CXCR2 | CXCR2 | (139, 140) |
| CXCL2 | GROβ | MIP-2 | CXCR2 | CXCR2 | (141) |
| CXCL5 | ENA-78 | LIX | CXCR2 | CXCR2 | (142, 143) |
| CXCL6 | GCP-2 | NA | CXCR1/CXCR2 | NA | (144) |
| CXCL8 | IL-8 | NA | CXCR1/CXCR2 | CXCR2 | (65, 145) |
| CXCL12 | SDF-1α | SDF-1α | CXCR4 | CXCR4 | (146) |
| CCL3 | MlP-lα | MlP-lα | NA | CCR1 | (147, 148) |
| CCL5 | RANTES | RANTES | NA | CCR1 | (140, 149) |
| CCL7 | MCP-3 | MARC | NA | CCR1 | (150) |
Main murine and human chemo-attractants and their receptors expressed in neutrophils during pulmonary infection and inflammation.
NA, not applicable.
Both CXC and CC chemokines play important roles in the stimulation of neutrophil chemotaxis. Under normal conditions, CXCL1 levels are negligible, however, they increase substantially during active infections in the lungs. According to Paudel et al., Cxcl1-/- mice showed impaired neutrophil recruitment and poor bacteria elimination from their BALF and lungs when challenged with a S. pneumoniae (152). In a pneumococcal infection of the lungs, CXCL1 was known to regulate neutrophil recruitment through a CD62L- and CD49d-dependent process. Results from Batra et al. showed that CXCL1 is an important chemokine for neutrophil influx and the production of Leukotriene B4 (LTB4) in the lungs during K. pneumoniae infection and is also very important for ROS regeneration in the lungs (153). Their analysis also validated the significance of CXCL1 in NADPH oxidase expression and the formation of NO and free radicals of oxygen in neutrophils after K. pneumoniae infections in the lungs. Studies have shown that CXCL1 is essential in NF-kB activation within the lungs after K. pneumoniae infection (Figure 4). NF-kB protects the lungs by avoiding excessive injury and inflammation during pneumococcal and E. Coli-induced pneumonia (155, 156). NF-kB has again been shown to be important in antibacterial host defense (157, 158) and hence reiterating the importance of CXCL1 in neutrophil recruitment (154). After infection with an influenza A virus (IAV), CXCL1, CXCL2, and neutrophils were found in lung tissues and airways of neonatal mice (159, 160). Furthermore, non-endothelial cells, non-epithelial cells (ATII cells) and lung stromal cells were shown to be important for the induction of Cxcl1 in a MyD88/TRIF signaling dependent manner during infection with a respiratory syncytial virus (RSV) (161). The precise source of neutrophil chemoattractants during viral infection is an important research topic for future targeted neutrophilic inflammation therapies. During viral infections, many neutrophil chemoattractants such as CXCL1, CXCL2, and IL-17 (161, 162) are produced in the lungs and airways, which cause neutrophil trafficking into the lungs of mice and ferrets (161, 163). By the activation of CXCR2 on neutrophils and their interaction with GAGs, CXCL2/3 chemokines can orchestrate the recruitment of neutrophils to the lungs during pulmonary infections. By binding to its receptor (CXCR2), CXCL5 chemokine, also known as lipopolysaccharide-induced chemokine (LIX), plays a significant role in the trafficking of neutrophils to the lungs during infection and inflammation (57). According to Gibbs et al., clock-controlled-CXCL5 mediates circadian variation and activates the rhythmic recruitment of neutrophils to the lung during pulmonary infection (164). CXCL8, also known as IL-8, is one of the most effective chemo-attractants (165), which can bind to the G protein–coupled receptors CXCR1 and CXCR2 on neutrophils (166). One of its numerous functions is to guide neutrophils through the tissue matrix until they reach the inflammation site. By using different signaling mechanisms, CXCL8 induces specific intracellular signaling cascades that result in rapid neutrophil recruitment (167–170). CXCL8 influences the movement of neutrophils across the endothelium (171), pulmonary epithelium (172), and fibroblasts (173). CXCL-12 is expressed in the lungs by cells such as the endothelial and epithelial cells (146). Both the ligand (CXCL12) and its receptor (CXCR4), play an important role in neutrophil influx to the lungs during infection (174–176). The signaling pathway of CXCL12/CXCR4 plays a crucial role in modulating neutrophil action in ALI not only by enhancing chemotaxis but by preventing cell death. CXCR4 inhibition reduces the trans-endothelial and trans-epithelial trafficking of neutrophils in pulmonary inflammation (175). Following a tissue damage, there was an increase in the local production of CXCL12; an important chemokine in the reparative cascade, which resulted in the guidance and recruitment of stem cells to the lungs (173). Although some studies have iterated the role of CXCR4 in the augmentation of pulmonary fibrosis (177), others have also reported on their role in neo-alveolarization (178). In a recent study, it was also discovered that CXCR4hi neutrophils are more likely to induce NETs, which increase the uptake of house dust mite by inflammatory dendritic cells, thereby increasing the risk of allergic asthma (179). The overexpression of neutrophil chemoattractants such as CXCL1, CXCL2, CXCL3, CXCL5, IL-8 (CXCL8), and CCL20 in the lungs of COVID-19 patients suggest that these cells can express neutrophil chemokines after SARS-CoV-2 infection (180). Chemokines also play important roles in metastasis and apoptosis. It has been shown that CXCL1 promotes cell migration. Because of this, Guo et al., performed a cell-proliferation inhibitory experiment by using Jinrong granule. Their results showed that Jinrong granule could inhibit the ability of CXCL-1 to promote the migration and proliferation of breast cancer cells and it could also reverse the promoting effect of CXCL-1 on breast cancer through the CXCL-1- CLCR2/CCL20 pathway (181). By using this analogy, therapies can be designed to target the cell migratory and proliferation potential of CXCL-1 during pulmonary infections and inflammation.
Figure 4
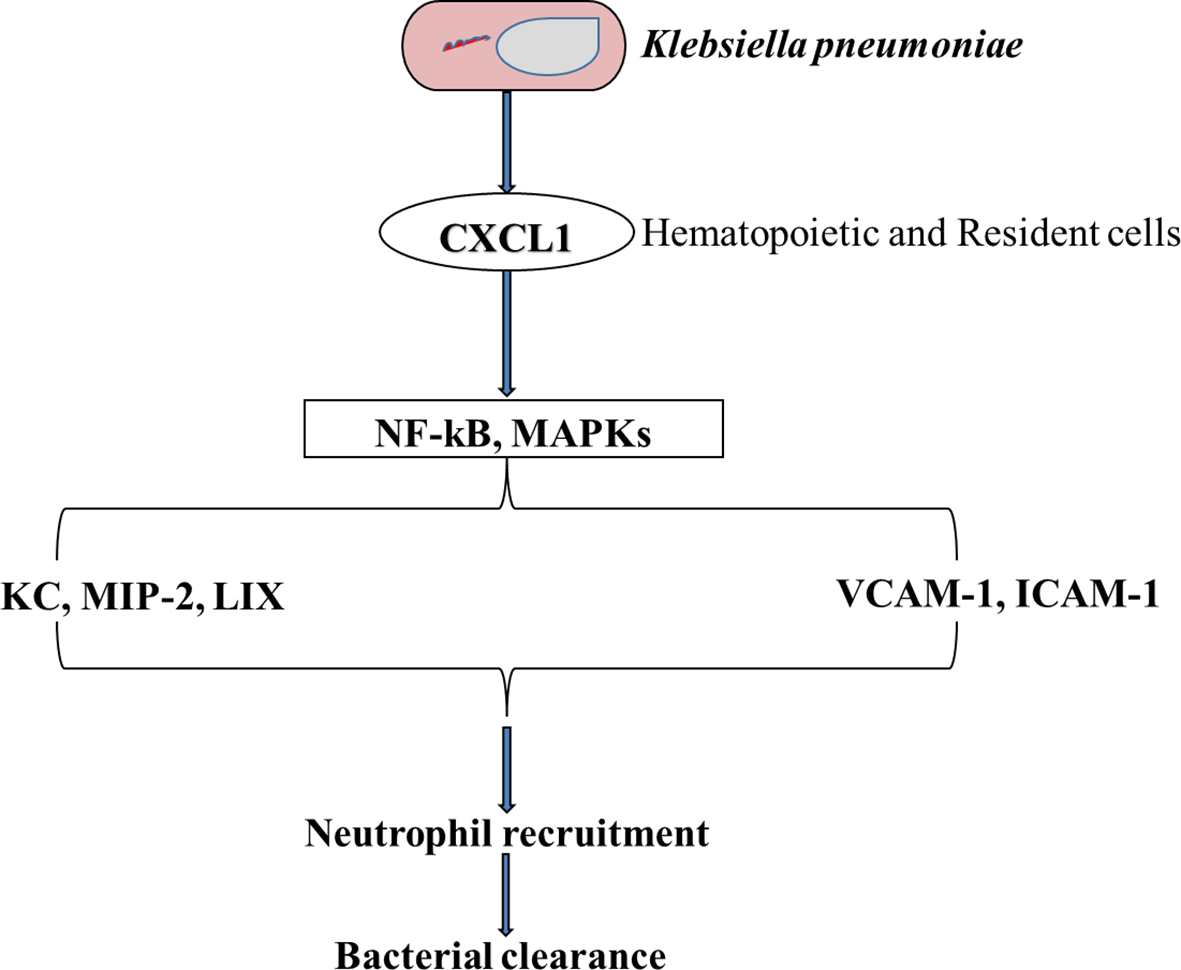
A Schematic representation of the importance of CXCL1 in neutrophil influx when the lung is challenged with K. pneumoniae. CXCL1 influences the activation of both NF-kB and MAPK which leads to an increase in MIP-2 and LIX chemokines, and other adhesion molecules such as VCAM-1 and ICAM-1. This cascade of events leads to an influx of the lungs with neutrophils followed by the clearance of the bacteria (154).
Some CC chemokines are classified as inflammatory (CCL2, CCL3, CCL4, CCL5, CCL11 and CCL13) while others are classified as homeostatic (CCL18, CCL19, CCL21, CCL25 and CCL27). However, some are considered as having both homeostatic and inflammatory function (CCL14, CCL15, CCL16 and CCL23) (182). Under normal conditions, there is no expression of CC chemokine receptors (CRs) (183), and these receptors do not respond to their CC chemokine ligand even upon stimulations. However, it has been shown that under inflammatory conditions in the lungs, neutrophils after their migration, expand their CR expression repertoire (21, 184). Functions of neutrophils which include chemotaxis, phagocytosis and respiratory bust are usually altered when there is an induction of CC chemokines and their receptors (CRs) (21, 184). The expression of these receptors are modulated by pro-inflammatory cytokines including IFN-γ, TNF-α, and GM-CSF. Macrophage inflammatory protein 1 (MIP-1/CCL3), which is one of the important members of the CC chemokine family, mediates the development of neutrophils and regulates their recruitment to the lungs during infection. MIP-1 has been reported to be chemotactic for neutrophils (185). In a study by Bonville et al., it was confirmed that neutrophil recruitment to the lung parenchyma in response to heterologous CCL3 expression in the respiratory epithelium, is directly dependent on IFNγ signaling (186). During a S. pneumoniae-induced inflammation, CCL2 chemokine binds to its receptor (CCR2), which triggers a PI3Kγ-dependent downstream signaling cascade, resulting in neutrophil immigration into the lung (187, 188). Also, CCL2 and CCL7 chemokines are known to work synergistically with CXCL8 to drive neutrophil trafficking to the lungs during acute respiratory distress syndrome (ARDS) (150). Grommes et al., indicated that the recruitment of neutrophils during an LPS-, acid-, and sepsis-induced ALI, is made possible because of the release of CCL5-CXCL4 heterodimer from platelets, and any disruption of this heterodimer decreases the amount of neutrophils recruited to the lungs (189). CCR1, CCR2, CCR3, CCR5, CXCR3, and CXCR4 were upregulated by a large fraction of neutrophils isolated from the BALF of patients suffering from chronic airway inflammation, as seen in CF, COPD, and asthma (190).
Neutrophil Oxidative Burst
Oxidative burst is a critical antimicrobial mechanism of neutrophils and is mediated by nicotinamide adenine dinucleotide phosphate (NADP) oxidase (191). Despite having the ability to protect against pulmonary infection, neutrophils, if left uncontrolled, can cause pathogenic effects through a variety of functions (192). Reduced NADP (NADPH2), thiocyanate, ergothioneine, thiosulfate, reduced glutathione (GSH), reduced nicotinamide adenine dinucleotide (NADH2), azide, Tapazole, thiourea, cyanide, cysteine, and tyrosine are the main components of the myeloperoxidase system, which performs this antibacterial activity. Genetic mutations in the NADPH oxidase subunit, gp91 (also referred to as NOX2), are associated with chronic recurrent and life-threatening microbial infections. When neutrophils are stimulated by microbes or by integrin-dependent adhesion to the ECM, they release reactive oxygen intermediates (ROIs). In mice, both the Vav family of Rho GTPase guanine nucleotide exchange factors (GEFs) and phospholipase C–γ2 (PLC-γ2) have been shown to be critical mediators of adhesion-dependent ROI production by neutrophils. Vav is critical for neutrophil-dependent host defense against S. aureus- and P. aeruginosa-induced hospital-acquired pneumonia (193). Compared to healthy controls, PMBCs and erythrocytes isolated from patients with tuberculosis had a significantly decreased levels of GSH (194). However, elevated levels of GSH have been shown to improve the inhibition capacity of T-cells against the growth of M. tuberculosis during pulmonary inflammation (195).
During oxidative burst, neutrophils produce reactive oxygen specie (ROS) which has been shown to induce necrosis cause by M. tuberculosis (196). Rapid assessment of neutrophil oxidative burst capacity has been proposed as an effective way to identify patients at risk of excessive immune responses during pulmonary inflammation (197). Therefore, the correlation between GSH and/or NADPH2 levels in TB patients and neutrophils oxidative burst capacity can provide host-targeted therapies. While some authors have reported an increased ROS production by blood PMN in cystic fibrosis (198, 199), others reported that the production of ROS is dependent on the pathogenic agent (200) or the detection method employed to measure the respiratory burst activity (201). Montemurro et al. (202) have demonstrated that blood neutrophils of CF patients had higher ROS release than their control counterparts. Lung ischemia-reperfusion injury has been linked to the production ROS and oxidative burst (203).
Pathophysiological Role of Neutrophils, NETs in COVID-19
Numerous studies have suggested that the recruitment of neutrophils to the lungs is linked to disease severity during viral infection. During an RSV-induced severe bronchiolitis, neutrophils accounted for nearly >90% of the BAL cell composition, confirming the role of neutrophils in disease pathogenesis (204, 205). Elevated levels of neutrophils and their markers in the lungs have also been observed in both rhinovirus and hMPV-infected children and in severe cases of influenza and SARS-CoV-2 infection (206, 207). Elevation in neutrophil level is one common phenomena observe during severe respiratory viral infections, and it is reasonable to postulate that their recruitment to the lungs and subsequent activation can exacerbate tissue pathology and cause disease. The current global pandemic, COVID-19, is a multisystem inflammatory disease caused by the SARS-CoV-2 virus. The innate immune response has been widely linked to COVID-19 immunopathogenesis. After reaching the alveoli, SARS-CoV-2 activates alveolar macrophages, which induces innate immune responses. A complement cascade is then activated by the viral particles through the lectin pathway. C3a and C5a are complementary peptides which are generated after the activation of the complement system. This then stimulate the migration of neutrophils to the site of infection. SARS-CoV-2 S-protein induces the release of proteins such as epithelial membrane protein 2 (Emp2) by the lung epithelial cells. The Emp2 of alveolar epithelial type 1 cells upregulate neutrophil migration. COVID-19 pathogenesis has been linked to neutrophil infiltration into the lungs. However, the numerous functions of neutrophils which include its interaction with other immune cell population, virus internalization and killing, cytokines release, degranulation, oxidative burst, and the production of neutrophil extracellular traps (NETs), helps to improve antiviral defenses (208, 209). Degranulation and the activation of neutrophils are the highly activated processes in SARS infection (210).
Neutrophilia has been identified as one of the markers linked with poor prognosis and severe respiratory symptoms in COVID-19 patients (211–213). According to Wang et al., neutrophilia is associated with lung injury in patients with severe COVID-19 (214). During autopsies of COVID-19 victims, neutrophilic mucositis was found in the lungs, indicating that the entire LRT was inflamed (215, 216). By using a Myeloperoxidase (MPO), Neutrophil Elastase (NE) and a Citrullinated Histone H3 (citH3) staining method, neutrophil infiltration via neutrophilic plugs was detected in patients with COVID-19 (217). Similarly, elevated levels of neutrophils have been found in peripheral blood of both severe and non-surviving COVID-19 patients (218, 219). According to a research by Parackova et al., neutrophils enhance the stimulation of Th17 in patients with COVID-19, and these cell population have been implicated in immune-mediated injury (220). It has been revealed that the infiltration of lungs with immature and/or dysfunctional neutrophils, as defined by the expression of CD11b, CD16, CD24, CD34, and CD38 and the infiltration with recently activated neutrophils which is characterized by the expression of CD64, RANK, RANKL and reduced CD62L, have been implicated as the causes of imbalance immune response during severe COVID-19 cases (220, 221). Severe COVID-19 pathophysiology is also characterized by altered neutrophil quantity, phenotype, and neutrophil functioning. Increased numbers of neutrophils have been reported in the nasopharyngeal epithelium (222) and later in the more distant regions of the lung following SARS-CoV-2 infection (223). An increase in the number of neutrophils has also been detected as a characteristic feature in the blood of COVID-19 patients (224–226) and markers of neutrophil activation are an important feature of blood transcriptomes in severe cases (227, 228). Increased levels of CXCL2 and CXCL8 have been shown through Transcriptional analysis of peripheral blood mononuclear cells and BALF from COVID-19 patients, to contribute to the recruitment of neutrophils to the lung, which then exacerbate the inflammatory response (206). Moreover, activated neutrophils express properdin, factor B, and C3, thus driving complement activation (229), a marker of severe COVID-19 disease (230, 231).
Infiltration is not the only mechanism by which neutrophils cause pathology in COVID-19 patients. Indeed, several inflammatory conditions including thrombosis, sepsis, and respiratory failure have all been linked to the pathological effects of NETs (232–234). A disproportionate release of virus-induced NET have been reported in COVID-19 patients and this is linked to the pathogenesis of this disease. The release of NETs by neutrophils has been linked to organ damage and death in COVID-19 patients (215). Another recent study found that markers of NET release (myeloperoxidase-DNA and citrullinated histone H3) in the sera of COVID-19 patients potently triggered NETosis in control neutrophils (235). COVID-19 patients have elevated levels of IL-6, which is a likely driver of NETosis. Similar in other inflammatory diseases, IL-6 stimulates the release of NETs throughout the body of COVID-19 patients (236, 237). Virus-damaged epithelial cells (56, 238), activated endothelial cells (239), activated platelets (240, 241), and inflammatory cytokines like IL-1β are all triggers of NETosis (242, 243). Higher levels of NETs have been seen in COVID-19 patients (235, 244, 245), and an increase in plasma NETs has been linked to increased COVID-19 severity (245), lung damage and microvascular thrombosis (244). Uncontrolled NETs can potentially trigger thrombosis.
Pathological Role of Neutrophils in Thromboembolism
Rather than platelets, neutrophils can play the lead role in thrombotic complications associated with COVID-19 (Figure 5). NETs have been demonstrated to exert thrombogenic activity in several inflammatory diseases by expressing functionally active tissue factor (TF). Thrombin-antithrombin (TAT) activity which indicates the activation of a TF/thrombin axis has been strongly linked with the levels of myeloperoxidase (MPO)/DNA complexes. NET release has also been identified as a key contributor to neutrophil-related thromboinflammation, providing the scaffold for platelet entrapment and activation. An in vitro and ex vivo models have been used to demonstrate the role of NETs in neutrophil-related thromboinflammation (247, 248). Leppkes et al. concluded that the development of NETs inside the microvessels of patients suffering from COVID-19 is associated with the severity of the disease. Rapid vessel occlusion caused by the intravascular development of NETs with platelet aggregation results in organ damage (249). In a study by Nicolai et al., it was noted that the kidney, lung, and heart of patients with COVID-19 had inflammatory microvascular thrombi which contained NETs and platelets (250). Thrombotic complications contribute to morbidity and mortality in severe COVID-19 (251, 252). In COVID-19 patients, abnormal coagulation parameters and elevated levels of proinflammatory cytokines are correlated with disease severity, poor prognosis, and incidence of venous thromboembolism. Thrombosis affects circulation in both the veins and the arteries of patients with COVID-19, leading to deep vein thrombosis, acute coronary syndrome, pulmonary embolism, stroke and microvascular thrombosis (253, 254). NET-remnants, such as citrullinated H3, circulating cell-free DNA, or MPO-DNA complexes have been found in abundance in the blood of COVID-19 patients (244, 249). Furthermore, patients with severe illness had elevated levels of both neutrophil activation markers and neutrophil-platelet aggregates (250, 255). Importantly, tissue factor is abundant in NETs from COVID-19 patients (TF). The release of thrombogenic NETs decorated with TF have been associated with activation of the complement system (256). Vascular injury is one of the outcome of excessive formation of NETs (257). Excessive NET can leads to the formation of autoantibodies that determine the appearance of various forms of autoimmune vasculitis (258). Immunothrombosis associated with NETs release has been shown via a histopathological study, to be linked to organ damage in severe COVID-19 (259). Occlusion of small pulmonary vessels caused by aggregated NETs was found in lungs during autopsies from victims of COVID-19-related ARDS (244). In a K18-hACE2 transgenic mice infected with SARS-CoV-2, neutrophils were seen infiltrating the alveolar and interstitial areas, which resulted in severe pulmonary pathology (162). Aggregated NETs may clog microvessels and this contribute to poor outcomes in COVID-19. DNAses prevent vascular occlusions which are caused by non-canonical NET-driven thrombosis during a steady-state condition (260). Thisfinding suggests that NET-dissolving mediators in patients can be impaired or elevated (261). In a quest to understand the role of neutrophil-lymphocyte in SARS-CoV-2 infection, Nicholai and his team compared histopathological lung specimens of COVID-19 with that of a viral pneumonia caused by H1N1 or seasonal influenza virus. Their findings highlighted neutrophil-driven immunothrombosis as a key element of severe COVID-19, as immunothrombotic vessel occlusion and NETosis were strongly elevated compared to influenza pneumonia (262).
Figure 5
Neutrophil rather than platelet activation are associated with thrombotic complications in COVID-19 patients (246).
Neutrophilia and Neutrophils in Lung Destruction and Resolution
Increased pulmonary vasculature permeability, accumulation of neutrophils in alveoli and disruption of the alveolar epithelium are all characteristic features of acute inflammatory response (263). Alteration in alveolar function i.e. leakage of plasma and interstitial fluid into airspace, is linked to damaged alveolar epithelial cells caused by transmigration of neutrophils from the alveolar capillaries to the airspace. Excessive neutrophils can cause tissue damage by increasing inflammatory response and by directly releasing toxic effectors. At high concentrations, many neutrophil effector mediators can cause tissue damage. For instance, although NE plays a role in digesting extracellular matrix (264) and induces mucus production (helps in pathogen clearance), when produced in excess, it can contribute to airway pathology because mucus plugs can obstruct the airways (265). The combination of mucus production and NET release can damage tissues and impair lung function (56, 266). Increase in blood neutrophil levels can predict severe respiratory damage and is an early stage marker for SARS-CoV-2 infection (267, 268). Thus, cytokines release and respiratory failure may be as a result of neutrophilia and excessive NETs. Excessive NETs can directly kill epithelial and endothelial cells (191, 269), damage the epithelium in pulmonary fungal infection (270) and cause severe injury to the endothelium during acute lung injury (271).
However, some researchers have opined that transmigration of neutrophils can occur without any destruction to major barriers and that, neutrophil accumulation can lead to the repair and regeneration of lung epithelium. The ability of accumulated neutrophils to repair tissues is partly due to the clearance of epithelial debris from the damaged sites which creates a clean matrix for regeneration of the epithelium (272). Furthermore, neutrophils can induce a repair response by activating the proliferation of lung epithelial cell (273) and by secreting pro-resolution products such as Annexin A1 (274). Neutrophil transmigration through an elastase-mediated cleavage of E-cadherin has been shown to activate the β-catenin signaling in alveolar type II epithelial cells in mice treated with keratinocyte chemokine or intratracheal LPS (275). In an acid-induced acute lung injury model, neutrophilia was essential for the proliferation of type II pneumocytes, which are necessary in regenerating alveolar epithelium. Neutrophilia, according to proteomic analysis, promote multiple regenerative pathways, including MMP9, MMP2, and FGF1 (276).
Diagnostic Neutrophilic Biomarkers of Pulmonary Inflammations
Biomarkers have been defined as “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention”. Many neutrophilic biomarkers associated with pulmonary inflammation have been studied (Table 3). The large number of biomarkers studied reflect the complex pathophysiology of pulmonary inflammations and the heterogeneity of the host response. This portion of the review summarizes some of these molecular biomarkers.
Table 3
| Type of Pulmonary inflammation | Biomarkers | Tissue/Detection level | Ref | Type of Pulmonary inflammation | Biomarkers | Tissue/Detection level | Ref |
|---|---|---|---|---|---|---|---|
| ARDS | Von-Willebrand Factor | Plasma/ 351% ± 265% | (277) | Pneumonia | CRP (mg/L) | fingerstick blood/ 60 (18-134) | (278) |
| SP-D (ng/ml) | Plasma/ 275 (80 – 462) | (279) | PCT (ng/ml) | Serum/ 3.64 ± 12.32 | (280) | ||
| LDH (IU/L) | Serum/ 274 ± 104 | (281) | sTREM-1 (pg/ml) | Serum/ 183.9 (119.8-232.1) | (282) | ||
| TNF-a (pg/mL) | Plasma/ 7.5 (3.8–13.4) | (283) | ProADM (nmol/L) | Venous blood/ 2.341 (1.188-4.226) | (284) | ||
| IL-6 (pg/mL) | Plasma/ 240 (139-498) | (283) | IL-6 (pg/mL) | Serum/ 242.2(92.33-473.97) | (285) | ||
| IL-10 (pg/mL) | Plasma/ 77 (31-169) | (283) | Angiopoietin-2 (ng/ml) | Serum/ 5.92 (3.48–9.99) | (286) | ||
| Protein C | Pulmonary edema fluid and plasma/ 37% ± 14% | (287) | Presepsin (pg/mL) | Plasma/ 1734 (1014-3128) | (288) | ||
| Plasminogen Activator Inhibitor (ng/ml) | alveolar fluid and plasma/ 2687 ± 1498 | (289) | Calprotectin (mg/L) | Serum/ 7.43 (4.60, 10.33) | (290) | ||
| CC16 (ng/ml) | Plasma/ 14.3 (9.0 - 19.0) | (279) | FGF21 (pg/mL) | Serum/ 456.5(181.2−1127.9) | (291) | ||
| KL-6 (U/l) | Plasma/ 477 (287-636) | (279) | NETs (U/mL) | BALF/ 223 (40.6–766) | (292) | ||
| sRAGE (ng/ml) | Plasma/ 1932 (960-4267) | (293) | PTX3 (ng/ml) | BALF/ ≥1 | (294) | ||
| sE-selectin (ng/mL) | Serum/ 53.0 ± 17.8 | (281) | TNF-α (pg/mL) | Plasma/ 44 (37–62) | (295) | ||
| CRP (mg/dL) | Serum/ 18.6 ± 11.0 | (281) | D-dimer (mcg/L) | Plasma/ 2080 (1050–3410) | (296) | ||
| COVID-19 | (MPO)-DNA (AU) | Serum/ 1.31 ± 0.18 | (297) | ||||
| NE (ng/mL) | Plasma/ 144 (84–248) | (298) | |||||
| NLR | Serum/ 8.78 (5.76-25.10) | (299) | |||||
| TAT (µg/mL) | Plasma/ 7.30 (4.50-12.2) | (300) | |||||
| VWF | Plasma/ 306% (200-421) | (300) | |||||
| ADAMTS13 | Plasma/ 47.3% (25.8-66.1) | (300) | |||||
| PAP (ng/mL) | Plasma/ 984 (648-2377) | (300) | |||||
| CRP (μg/L) | Serum/ 81.49 (19.40-107.42) | (299) |
Neutrophilic biomarkers of pulmonary inflammations.
ADAMTS13: a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; CRP, C-reactive protein; CC16, Clara cell secretory protein; FGF21, fibroblast growth factor 21; IL-6, interleukin-6; KL-6, Krebs von den Lungen-6; LDH, lactate dehydrogenase; MR-proADM, midregional-proadrenomedullin; (MPO)-DNA, myeloperoxidase (MPO)-DNA; NE, neutrophil elastase; NLR, neutrophil-to-lymphocyte ratio; NETs, neutrophil extracellular traps; PAP, plasmin-antiplasmin complex; PCT, procalcitonin; PTX3, pentraxin 3; sRAGE, soluble receptor of advanced glycation end products; SP-D, surfactant protein; sE-selectin, soluble endothelia-selectin; sTNF, soluble tumor necrosis factor receptors TNF-α: tumour necrosis factor alpha; TAT, thrombin-antithrombin complex.
Alpha-1 antitrypsin and CD16b (AAT : CD16b) protein complex released by primed neutrophils has been found to be significantly elevated in sera of patients with CF, making it a potential biomarker to diagnose exacerbated cystic fibrosis. The expression of these neutrophil priming-associated biomarkers in peripheral blood can be used to expound the inflammatory process in CF (301). Adenosine monophosphate and purines adenosine triphosphate have also been identified as potential neutrophilic biomarkers of pulmonary inflammation in CF. In chronic obstructive pulmonary disease, research have shown that, the development of systemic inflammation (302–305), exacerbations and other lung functioning parameters (306, 307) are positively linked with the levels of fibrinogen, C-reactive protein, and IL-6. COPD exacerbation has been linked to elevated levels of neutrophil gelatinase associated lipocalin (NGAL), osteoprorotegerin and soluble TNF receptor-1 (sTNFR-1) (308, 309). Additionally, vascular endothelial growth factor (VEGF) and MPO were predicting factors in determining the severity of lung function impairment and dyspnea (310). There have been numerous studies reporting decreased concentrations of Clara cell protein (CC-16) in the blood of people suffering from COPD, supporting the notion that this protein may be an important biomarker in the prediction of bronchial epithelial cell dysfunction (311–315). Compared to their control counterparts, people with COPD had elevated levels of NGAL, heparin-binding EGF-like growth factor (HB-EGF), extracellular newly identified RAGE-binding protein (EN-RAGE; also known as S100A12), MPO, fibrinogen and transforming growth factor alpha (TGF-α). Conversely, COPD patients had lower levels of soluble receptor for advanced glycation end products (sRAGE) than the control group (316). Active neutrophil elastase (NE), a serine proteinase secreted by neutrophils in response to inflammation and pathogen invasion is elevated during exacerbations of COPD and may be a viable biomarker for distinguishing a bacterial exacerbation in patients with COPD (317). A number of biomarkers have been used as predictive biomarkers for pneumonia. These include copeptin, CRP, proadrenomedullin (proADM), and procalcitonin (PCT) (318–322). Calprotectin, PCT, plasma pentraxin 3 (PTX3) and presepsin have been used as promising acute-phase molecular predictive markers for community-acquired pneumonia (323). von Willebrand factor (VWF), adhesion molecules (such as E-selectin, L-selectin, intercellular adhesion molecule [ICAM], and vascular cell adhesion protein-1), thrombomodulin (TM), protein C, and plasminogen activator inhibitor-1 (PAI-1) are also known as important markers of endothelial activation and injury in acute respiratory distress syndrome (ARDS). A number of coagulation biomarkers including protein C, thrombomodulin and PAI-1 have been shown to be abnormal in ALI (324). The epithelial mucin protein, Kerbs von den Lungren-6 (KL-6), has also been studied as a potential biomarker. KL-6 is unregulated when type II pneumocytes become injured (325).
Inflammation and immunity play a critical role in many chronic diseases. Neutrophil-lymphocyte ratio (NLR) is a biomarker that reflects the balance between acute and chronic inflammation (neutrophil count) as well as adaptive immunity (lymphocyte count). It can be computed as the ratio between neutrophils and lymphocytes in peripheral blood. NLR, as determined by a recent meta-analysis (326), appeared to be a predictive biomarker for acute exacerbations in patients with COPD. In patients with COVID-19 infection, NLR was shown to have a good predictive value on disease severity and mortality (327). Data from recent studies suggest that NLR is an important predictor of mortality among patients with the novel Coronavirus disease (79). Numerous studies have implicated NLR in the development of COPD. In an examination of acute episode of COPD, Lee et al. reported that the NLR in the acute episode was significantly higher than that in the stable period and in the healthy control group. However, the NLR significantly decreased in the recovery stage patients with acute exacerbations (328). According to Taylan et al, NLR gradually increased with the severity of COPD, suggesting that NLR can be used as an early biomarker of COPD (329). The results of these studies confirm that NLR has great value in the assessment of COPD severity and acute exacerbations. Although not related to pulmonary inflammation, Huang et al., through a research to explore the applicative value of preoperative NLR combined with serum carcinoembryonic antigen (CEA), carbohydrate antigen (CA) 19-9, CA 125 and CA 72-4 levels, proved that NLR can reflect the inflammatory and immune status in gastric cancer patients and that, these combinations are closely related to clinical pTNM stage in gastric cancer (330).
Clinical Trials Targeting Neutrophils in Neutrophil-Driven Pulmonary Inflammatory Diseases
All around the world, clinical trials are being conducted with the aim of targeting neutrophils in order to improve the conditions of patients with pulmonary infections and inflammations. Once a clear definition of the roles of neutrophils in pulmonary infections is established, researchers will be able to design effective treatments to target neutrophil dysregulation and development. Clinical trials of various promising pharmacological modulators of neutrophil recruitment/activation and NETosis are currently in Phase II and III. These include agents targeting NADPH oxidase, colony-stimulating factors and receptors, as well as CXCR2. In humans, CXCR2 appears to be the predominant neutrophil chemokine receptor despite having overlapping functions with CXCR1. CXCR2 has been implicated in several functions, including neutrophil egress from the bone marrow (331) and the activation of other cell types. CXCR1/CXCR2 have been a target for a number of pharmacological studies. For instance, patients with cystic fibrosis who were treated with a CXCR2 antagonist, SB-656933, had a decreased level of sputum inflammatory biomarkers (332). Another CXCR2 inhibitor, navarixin (MK-7123), improved pulmonary function in COPD, and the blockage of CXCR2 in patients suffering from moderate neutrophilic asthma, reduced the accumulation of neutrophil in the lungs (333). AZD5069, a selective CXCR2 antagonist, has been ineffective in controlling acute exacerbations in severe cases of refractory asthma, in spite of its ability to decrease neutrophil numbers in sputum (334) (Table 4). A phase II clinical trial of QBM076, another CXCR2 antagonist, was terminated due to elevated liver transaminase levels in patients with COPD (Table 4). In a murine experiment to inhibit CXCR2, an orally active CXCR2-antagonist molecule was used. It was confirmed that the molecule reduced inflammation caused by neutrophils, reduced infiltration of neutrophils to the lungs and decreased the level of enzymes that cause tissue damage (335). Danirixin, another CXCR2 antagonist, has also shown positive effects on respiratory symptoms and health status (336). Targeting the CXCL12-CXCR4 axis may also provide an effective treatment option for COPD. Plerixafor was found to reduce lung damage in mice with emphysema caused by cigarette smoke exposure (337). Although there are potential adverse effects in antagonizing the CXCL12-CXCR4 axis, it still provides a promising strategies in the treatment of pulmonary infections and inflammations (Figure 6). A summary of some selected clinical trials based on therapeutic strategies that specifically target neutrophils are provided in Table 4.
Table 4
| Target | Name of drug | Company/sponsor | Indication | Phase | Identifier | Comments |
|---|---|---|---|---|---|---|
| CXCR2 | AZD5069 | AstraZeneca | Asthma | II | NCT01704495 | Completed in 2014; the frequency of severe exacerbation in patients with severe asthma was not reduced |
| QBM076 | Novartis Pharmaceuticals | COPD | II | NCT01972776 | Part 1 was completed; part 2 was terminated (for safety reasons) in 2015 | |
| AZD5069 | AstraZeneca | Asthma | I | NCT01890148 | Completed in 2014. The inhibitor decreased sputum neutrophil numbers | |
| NETs | Fostamatinib | NHLBI | COVID-19 | II | NCT04579393 | Ongoing |
| Eculizumab | Hudson Medical | COVID-19 | II | NCT04346797 | Ongoing | |
| AIR DNase™ | Protalix | Cystic fibrosis | II | NCT02722122 | Status unknown | |
| PDE4 | Roflumilast | QuantumLeap Healthcare Collaborative | COVID-19 | II | NCT04488081 | Ongoing |
| NE | Alvelestat (AZD9668) | Mereo BioPharma | COPD | II | NCT03636347 | Ongoing |
| Lonodelestat (POL6014) | Santhera Pharmaceuticals | Cystic fibrosis | II | NCT03748199 | Status unknown | |
| Elafin | Peking University Third Hospital | ARDS | I | NCT02944279 | Completed in 2014 | |
| CXCR2 | Danirixin (GSK1325756) | GlaxoSmithKline | COPD | II | NCT03034967 | Completed in 2018 |
| PDE4 | Ensifentrine (RPL554) | Verona Pharma plc | COVID-19 | II | NCT04527471 | Ongoing |
| IL-6, IFNs, NETs | Baricitinib | Hospital of Prato | COVID-19 | II/III | NCT04320277 | Not yet recruiting |
Selected clinical trials targeting neutrophils in neutrophil-driven pulmonary inflammatory diseases.
Figure 6
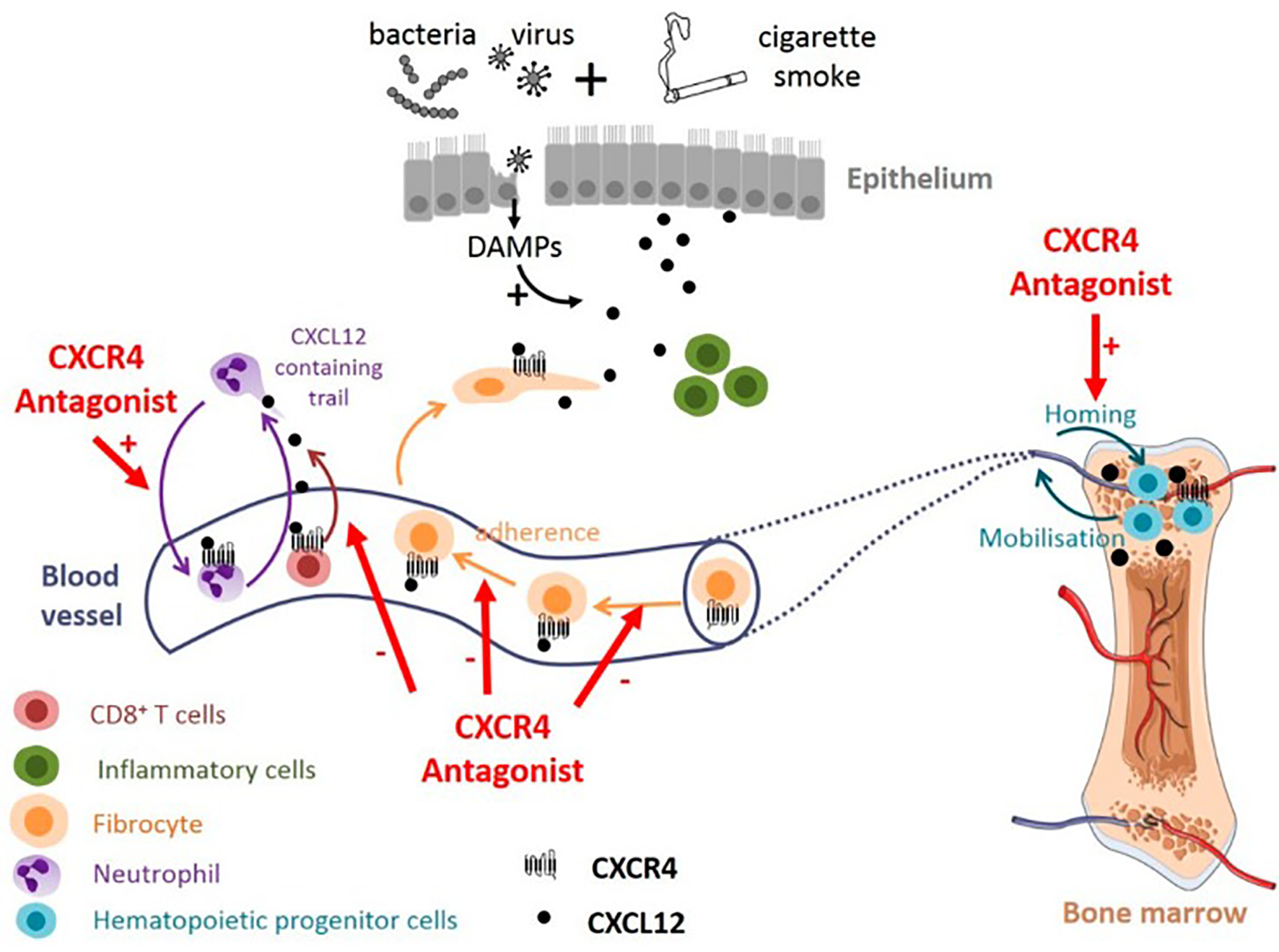
Potential effect of targeting CXCL12/CXCR4 axis in COPD. CXCR4 antagonists could promote neutrophil demargination from the lungs and inhibit T-cells and fibrocytes recruitment into bronchial tissue. CXCR4 antagonists may also contribute to maintaining the pool of hematopoeitic progenitor cells in the bone marrow, available for tissue repair (338).
Concluding Remarks
Neutrophils, which destroy pathogenic agents, have the ability to significantly modulate the functions of other immune cells and their recruitment into the lung. They play a major role in innate immunity during pulmonary inflammation. It is becoming increasingly clear that all of these processes are highly regulated by the signals they receive from their repertoire of PRRs which allow neutrophils, whose recruitment are modulated by chemokines, to sense PAMPs and DAMPs at the sites of inflammation. A holistic understanding of neutrophil pattern recognition pathways and the roles of CXC and CC chemokines in host immunity may allow for new approaches in the treatment of infectious and inflammatory disease of the lungs. In this review, we have demonstrated the roles and importance of PRRs, CXC and CC chemokines in host immunity during lung pathogenesis. We have herein summarize some of the signal transduction pathways through which neutrophils are recruited and guided to the lungs. The pathophysiological role of neutrophils in COVID-19 and thromboembolism have also been summarized. Finally, we discussed various neutrophilic biomarkers, and neutrophil-targeted therapies for neutrophil-driven pulmonary inflammatory diseases.
Funding
This work is supported by the National Natural Science Foundation of China (No. 81973099).
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
CYE conceptualized, structured and drafted the manuscript; EKD, CA, LD, SH, SL, SYA, EN, TS, XLZ, HL, ZX and FF contributed to literature search and writing of the manuscript. YW and XJZ contributed to the conceptualization, fund sourcing and supervision of the work. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
MizgerdJP. Lung Infection—A Public Health Priority. PloS Med (2006) 3(2):e76. doi: 10.1371/journal.pmed.0030076
2
MizgerdJP. Acute Lower Respiratory Tract Infection. N Engl J Med (2008) 358:716–27. doi: 10.1056/NEJMra074111
3
MartinTRFrevertCW. Innate Immunity in the Lungs. Proc Am Thorac Society (2005) 2(5):403–11. doi: 10.1513/pats.200508-090JS
4
ZhangPSummerWRBagbyGJNelsonS. Innate Immunity and Pulmonary Host Defense. Immunol Rev (2000) 173:39–51. doi: 10.1034/j.1600-065X.2000.917306.x
5
PhillipsonMKubesP. The Healing Power of Neutrophils. Trends Immunol (2019) 40:635–47. doi: 10.1016/j.it.2019.05.001
6
MayadasTNCullereXLowellCA. The Multifaceted Functions of Neutrophils. Annu Rev Pathol (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
7
GriffithJWSokolCLLusterAD. Chemokines and Chemokine Receptors: Positioning Cells for Host Defense and Immunity. Annu Rev Immunol (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
8
ColditzIGSchneiderMAPruensterMRotA. Chemokines at Large: In-Vivo Mechanisms of Their Transport, Presentation and Clearance. Thromb Haemost (2007) 97:688–93. doi: 10.1160/TH07-02-0105
9
JinLBatraSDoudaDNPalaniyarNJeyaseelanS. CXCL1 Contributes to Host Defense in Polymicrobial Sepsis via Modulating T Cell and Neutrophil Functions. J Immunol (2014) 193:3549–58. doi: 10.4049/jimmunol.1401138
10
MetkarSKimKSSilverJGoyertSM. Differential Expression of CD14-Dependent and Independent Pathways for Chemokine Induction Regulates Neutrophil Trafficking in Infection. J Leukoc Biol (2012) 92:389–96. doi: 10.1189/jlb.0112011
11
CraciunFLSchullerERRemickDG. Early Enhanced Local Neutrophil Recruitment in Peritonitis-Induced Sepsis Improves Bacterial Clearance and Survival. J Immunol (2010) 185:6930–8. doi: 10.4049/jimmunol.1002300
12
SegalAW. How Neutrophils Kill Microbes. Annu Rev Immunol (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
13
GazendamRPvan de GeerARoosDvan den BergTKKuijpersTW. How Neutrophils Kill Fungi. Immunol Rev (2016) 273(1):299–311. doi: 10.1111/imr.12454
14
JenneCNWongCHZempFJMcDonaldBRahmanMMForsythPAet al. Neutrophils Recruited to Sites of Infection Protect From Virus Challenge by Releasing Neutrophil Extracellular Traps. Cell Host Microbe (2013) 13(2):169–80. doi: 10.1016/j.chom.2013.01.005
15
DoerschukCM. Leukocyte Trafficking in Alveoli and Airway Passages. Respir Res (2000) 1(3):1–5. doi: 10.1186/rr24
16
GrommesJSoehnleinO. Contribution of Neutrophils to Acute Lung Injury. Mol Med (2011) 17(3):293–307. doi: 10.2119/molmed.2010.00138
17
KueblerWMGoetzAE. The Marginated Pool. Eur Surg Res (2002) 34(1-2):92–100. doi: 10.1159/000048894
18
RossaintJZarbockA. Tissue-Specific Neutrophil Recruitment Into the Lung, Liver, and Kidney. J Innate Immun (2013) 5(4):348–57. doi: 10.1159/000345943
19
ScapiniPMariniOTecchioCCassatellaMA. Human Neutrophils in the Saga of Cellular Heterogeneity: Insights and Open Questions. Immunol Rev (2016) 273(1):48–60. doi: 10.1111/imr.12448
20
IngersollSALavalJForrestOAPreiningerMBrownMRArafatDet al. Mature Cystic Fibrosis Airway Neutrophils Suppress T Cell Function: Evidence for a Role of Arginase 1 But Not Programmed Death-Ligand 1. J Immunol (2015) 194(11):5520–8. doi: 10.4049/jimmunol.1500312
21
HartlDKrauss-EtschmannSKollerBHordijkPLKuijpersTWHoffmannFet al. Infiltrated Neutrophils Acquire Novel Chemokine Receptor Expression and Chemokine Responsiveness in Chronic Inflammatory Lung Diseases. J Immunol (2008) 181(11):8053–67. doi: 10.4049/jimmunol.181.11.8053
22
TirouvanziamRGernezYConradCKMossRBSchrijverIDunnCEet al. Profound Functional and Signaling Changes in Viable Inflammatory Neutrophils Homing to Cystic Fibrosis Airways. Proc Natl Acad Sci (2008) 105(11):4335–9. doi: 10.1073/pnas.0712386105
23
HamptonHRBaileyJTomuraMBrinkRChtanovaT. Microbe-Dependent Lymphatic Migration of Neutrophils Modulates Lymphocyte Proliferation in Lymph Nodes. Nat Commun (2015) 6(1):1–1. doi: 10.1038/ncomms8139
24
DuffyDPerrinHAbadieVBenhabilesNBoissonnasALiardCet al. Neutrophils Transport Antigen From the Dermis to the Bone Marrow, Initiating a Source of Memory CD8+ T Cells. Immunity (2012) 37(5):917–29. doi: 10.1016/j.immuni.2012.07.015
25
AbadieVBadellEDouillardPEnsergueixDLeenenPJTanguyMet al. Neutrophils Rapidly Migrate via Lymphatics After Mycobacterium Bovis BCG Intradermal Vaccination and Shuttle Live Bacilli to the Draining Lymph Nodes. Blood (2005) 106(5):1843–50. doi: 10.1182/blood-2005-03-1281
26
CoombesJLCharsarBAHanSJHalkiasJChanSWKoshyAAet al. Motile Invaded Neutrophils in the Small Intestine of Toxoplasma Gondii-Infected Mice Reveal a Potential Mechanism for Parasite Spread. Proc Natl Acad Sci (2013) 110(21):E1913–22. doi: 10.1073/pnas.1220272110
27
LeyKLaudannaCCybulskyMINoursharghS. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat Rev Immunol (2007) 7:678–89. doi: 10.1038/nri2156
28
RoseDMAlonRGinsbergMH. Integrin Modulation and Signaling in Leukocyte Adhesion and Migration. Immunol Rev (2007) 218:126–34. doi: 10.1111/j.1600-065X.2007.00536.x
29
ZarbockALeyK. Mechanisms and Consequences of Neutrophil Interaction With the Endothelium. Am J Pathol (2008) 172:1–7. doi: 10.2353/ajpath.2008.070502
30
JungUBullardDCTedderTFLeyK. Velocity Differences Between L- and P-Selectin-Dependent Neutrophil Rolling in Venules of Mouse Cremaster Muscle In Vivo. Am J Physiol Heart Circ Physiol (1996) 271:H2740–7. doi: 10.1152/ajpheart.1996.271.6.H2740
31
YagoTZarnitsynaVIKlopockiAGMcEverRPZhuC. Transport Governs Flow-Enhanced Cell Tethering Through L-Selectin at Threshold Shear. Biophys J (2007) 92:330–42. doi: 10.1529/biophysj.106.090969
32
TaookaYMaedaAHiyamaKIshiokaSYamakidoM. Effects of Neutrophil Elastase Inhibitor on Bleomycin-Induced Pulmonary Fibrosis in Mice. Am J Respir Crit Care Med (1997) 156:260–5. doi: 10.1164/ajrccm.156.1.9612077
33
TaookaYChenJYednockTSheppardD. The Integrin α9β1 Mediates Adhesion to Activated Endothelial Cells and Transendothelial Neutrophil Migration Through Interaction With Vascular Cell Adhesion Molecule-1. J Cell Biol (1999) 145:413–20. doi: 10.1083/jcb.145.2.413
34
ShangTYednockTIssekutzAC. α9β1 Integrin is Expressed on Human Neutrophils and Contributes to Neutrophil Migration Through Human Lung and Synovial Fibroblast Barriers. J Leukoc Biol (1999) 66:809–16. doi: 10.1002/jlb.66.5.809
35
HumphriesJDByronAHumphriesMJ. Integrin Ligands at a Glance. J Cell Sci (2006) 119:3901–3. doi: 10.1242/jcs.03098
36
SalasAShimaokaMChenSCarmanCVSpringerT. Transition From Rolling to Firm Adhesion is Regulated by the Conformation of the I Domain of the Integrin Lymphocyte Function-Associated Antigen-1. J Biol Chem (2002) 277:50255–62. doi: 10.1074/jbc.M209822200
37
ConstantinGMajeedMGiagulliCPiccioLKimJYButcherECet al. Chemokines Trigger Immediate β2 Integrin Affinity and Mobility Changes: Differential Regulation and Roles in Lymphocyte Arrest Under Flow. Immunity (2000) 13:759–69. doi: 10.1016/S1074-7613(00)00074-1
38
KadiogluADe FilippoAKBangertMFernandesVERichardsLJonesKet al. The Integrins Mac-1 and α4β1 Perform Crucial Roles in Neutrophil and T Cell Recruitment to Lungs During Streptococcus Pneumoniae Infection. J Immunol (2011) 186:5907–15. doi: 10.4049/jimmunol.1001533
39
UlyanovaTPriestleyGVBanerjeeERThalia PapayannopoulouT. Unique and Redundant Roles of α4 and β2 Integrins in Kinetics of Recruitment of Lymphoid Versus Myeloid Cell Subsets to the Inflamed Peritoneum Revealed by Studies of Genetically Deficient Mice. Exp Hematol (2007) 35:1256–65. doi: 10.1016/j.exphem.2007.04.015
40
PreiraPForelJMRobertPNègrePBiarnes-PelicotMXeridatFet al. The Leukocyte-Stiffening Property of Plasma in Early Acute Respiratory Distress Syndrome (ARDS) Revealed by a Microfluidic Single-Cell Study: The Role of Cytokines and Protection With Antibodies. Crit Care (2016) 20:8. doi: 10.1186/s13054-015-1157-5
41
TaookaYOheMChenLSutaniAHigashiYIsobeT. Increased Expression Levels of Integrin α9β1 and CD11b on Circulating Neutrophils and Elevated Serum IL-17A in Elderly Aspiration Pneumonia. Respiration (2013) 86:367–75. doi: 10.1159/000345390
42
MollinedoF. Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol (2019) 40:228e42. doi: 10.1016/j.it.2019.01.006
43
BordonJAlibertiSFernandez-BotranRUriarteSMRaneMJDuvvuriPet al. Understanding the Roles of Cytokines and Neutrophil Activity and Neutrophil Apoptosis in the Protective Versus Deleterious Inflammatory Response in Pneumonia. Int J Infect Dis (2013) 17:e76–83. doi: 10.1016/j.ijid.2012.06.006
44
NathanC. Neutrophils and Immunity: Challenges and Opportunities. Nat Publ Group (2006) 6:173–82. doi: 10.1038/nri1785
45
MizgerdJP. Respiratory Infection and the Impact of Pulmonary Immunity on Lung Health and Disease. Am J Respir Crit Care Med (2012) 186:824–9. doi: 10.1164/rccm.201206-1063PP
46
ZdenekH. Could NET-Wielding Neutrophils be Driving Respiratory Distress and Death in COVID-19 Patients? (2020). Available at: https://www.uab.edu/medicine/pathology/news-events/archive/690-could-net-wielding-neutrophils-be-driving-respiratory-distress-and-death-in-covid-19-patients.
47
BrinkmannVZychlinskyA. Beneficial Suicide: Why Neutrophils Die to Make NETs. Nat Rev Microbiol (2007) 5(8):577–82. doi: 10.1038/nrmicro1710
48
BrinkmannVReichardUGoosmannCFaulerBUhlemannYWeissDSet al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
49
KrugerPSaffarzadehMWeberANRieberNRadsakMvon BernuthHet al. Neutrophils: Between Host Defence, Immune Modulation, and Tissue Injury. PloS Pathog (2015) 11(3):e1004651. doi: 10.1371/journal.ppat.1004651
50
CortjensBvan WoenselJBBemRA. Neutrophil Extracellular Traps in Respiratory Disease: Guided Anti-Microbial Traps or Toxic Webs? Paediatric Respir Rev (2017) 21:54–61. doi: 10.1016/j.prrv.2016.03.007
51
ChengOZPalaniyarN. NET Balancing: A Problem in Inflammatory Lung Diseases. Front Immunol (2013) 4:1. doi: 10.3389/fimmu.2013.00001
52
YippBGKubesP. NETosis: How Vital is it? Blood (2013) 122(16):2784–94. doi: 10.1182/blood-2013-04-457671
53
SørensenOEBorregaardN. Neutrophil Extracellular Traps—The Dark Side of Neutrophils. J Clin Invest (2016) 126(5):1612–20. doi: 10.1172/JCI84538
54
Grabcanovic-MusijaFObermayerAStoiberWKrautgartnerWDSteinbacherPWinterbergNet al. Neutrophil Extracellular Trap (NET) Formation Characterises Stable and Exacerbated COPD and Correlates With Airflow Limitation. Respir Res (2015) 16(1):1–2. doi: 10.1186/s12931-015-0221-7
55
WrightTKGibsonPGSimpsonJLMcDonaldVMWoodLGBainesKJ. Neutrophil Extracellular Traps Are Associated With Inflammation in Chronic Airway Disease. Respirology (2016) 21(3):467–75. doi: 10.1111/resp.12730
56
CortjensBde BoerOJde JongRAntonisAFSabogal PiñerosYSLutterRet al. Neutrophil Extracellular Traps Cause Airway Obstruction During Respiratory Syncytial Virus Disease. J Pathol (2016) 238(3):401–11. doi: 10.1002/path.4660
57
BalamayooranGBatraSFesslerMBHappelKIJeyaseelanS. Mechanisms of Neutrophil Accumulation in the Lungs Against Bacteria. Am J Respir Cell Mol Biol (2010) 43(1):5–16. doi: 10.1165/rcmb.2009-0047TR
58
ParkBParkGKimJLimSALeeKM. Innate Immunity Against Legionella Pneumophila During Pulmonary Infections in Mice. Arch Pharmacal Res (2017) 40(2):131–45. doi: 10.1007/s12272-016-0859-9
59
OnishiYKawamuraTNakaharaYKagamiRSasakiSTakahashiSet al. Factors Associated With the Relapse of Cryptogenic and Secondary Organizing Pneumonia. Respir Invest (2017) 55(1):10–5. doi: 10.1016/j.resinv.2016.09.001
60
PatamatamkulSKlungboonkrongVPraisarntiPJirakiatK. A Case-Control Study of Community-Acquired Acinetobacter Baumannii Pneumonia and Melioidosis Pneumonia in Northeast Thailand: An Emerging Fatal Disease With Unique Clinical Features. Diagn Microbiol Infect Disease (2017) 87(1):79–86. doi: 10.1016/j.diagmicrobio.2016.10.014
61
BhatiaMZemansRLJeyaseelanS. Role of Chemokines in the Pathogenesis of Acute Lung Injury. Am J Respir Cell Mol Biol (2012) 46(5):566–72. doi: 10.1165/rcmb.2011-0392TR
62
WangJNikradMPTravantyEAZhouBPhangTGaoBet al. Innate Immune Response of Human Alveolar Macrophages During Influenza A Infection. PloS One (2012) 7(3):e29879. doi: 10.1371/journal.pone.0029879
63
ZarbockADiStasiMRSmithESandersJMKronkeGHarryBLet al. Improved Survival and Reduced Vascular Permeability by Eliminating or Blocking 12/15-Lipoxygenase in Mouse Models of Acute Lung Injury (ALI). J Immunol (2009) 183(7):4715–22. doi: 10.4049/jimmunol.0802592
64
RossaintJNadlerJLLeyKZarbockA. Eliminating or Blocking 12/15-Lipoxygenase Reduces Neutrophil Recruitment in Mouse Models of Acute Lung Injury. Crit Care (2012) 16(5):1–5. doi: 10.1186/cc11518
65
MillerEJCohenABMatthayMA. Increased Interleukin-8 Concentrations in the Pulmonary Edema Fluid of Patients With Acute Respiratory Distress Syndrome From Sepsis. Crit Care Med (1996) 24(9):1448–54. doi: 10.1097/00003246-199609000-00004
66
QuintonLJNelsonSZhangPBoéDMHappelKIPanWet al. Selective Transport of Cytokine-Induced Neutrophil Chemoattractant From the Lung to the Blood Facilitates Pulmonary Neutrophil Recruitment. Am J Physiol Lung Cell Mol Physiol (2004) 286(3):L465–72. doi: 10.1152/ajplung.00153.2003
67
SchmidtEPYangYJanssenWJGandjevaAPerezMJBarthelLet al. The Pulmonary Endothelial Glycocalyx Regulates Neutrophil Adhesion and Lung Injury During Experimental Sepsis. Nat Med (2012) 18(8):1217–23. doi: 10.1038/nm.2843
68
HandelTMJohnsonZCrownSELauEKSweeneyMProudfootAE. Regulation of Protein Function by Glycosaminoglycans—As Exemplified by Chemokines. Annu Rev Biochem (2005) 74:385–410. doi: 10.1146/annurev.biochem.72.121801.161747
69
DasSTRajagopalanLGuerrero-PlataASaiJRichmondAGarofaloRPet al. Monomeric and Dimeric CXCL8 Are Both Essential for In Vivo Neutrophil Recruitment. PloS One (2010) 5(7):e11754. doi: 10.1371/journal.pone.0011754
70
BelperioJAKeaneMPLynchJPStrieterRM. The Role of Cytokines During the Pathogenesis of Ventilator-Associated and Ventilator-Induced Lung Injury. In: Seminars in Respiratory and Critical Care Medicine, vol. 27(04). 333 Seventh Avenue, New York, NY 10001, USA: Thieme Medical Publishers, Inc. (2006). p. 350–64.
71
StrieterRMKeaneMPBurdickMDSakkourAMurrayLABelperioJA. The Role of CXCR2/CXCR2 Ligands in Acute Lung Injury. Curr Drug Targets Inflamm Allergy (2005) 4(3):299–303. doi: 10.2174/1568010054022178
72
StadtmannAZarbockA. CXCR2: From Bench to Bedside. Front Immunol (2012) 3. doi: 10.3389/fimmu.2012.00263
73
ZarbockAAllegrettiMLeyK. Therapeutic Inhibition of CXCR2 by Reparixin Attenuates Acute Lung Injury in Mice. Br J Pharmacol (2008) 155(3):357–64. doi: 10.1038/bjp.2008.270
74
ZemansRLMatthayMA. What Drives Neutrophils to the Alveoli in ARDS? Thorax (2017) 72:1e3. doi: 10.1136/thoraxjnl-2016-209170
75
ZindelJKubesP. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu Rev Pathol (2020) 15:493e518. doi: 10.1146/annurev-pathmechdis-012419-032847
76
LamFWDaQGuilloryBCruzMA. Recombinant Human Vimentin Binds to P-Selectin and Blocks Neutrophil Capture and Rolling on Platelets and Endothelium. J Immunol (2018) 200:1718e26. doi: 10.4049/jimmunol.1700784
77
Blazquez-PrietoJLopez-AlonsoIAmado-RodriguezLHuidobroCGonzalez-LopezAKueblerWMet al. Impaired Lung Repair During Neutropenia can be Reverted by Matrix Metalloproteinase-9. Thorax (2018) 73:321e30. doi: 10.1136/thoraxjnl-2017-210105
78
PolverinoERosales-MayorEDaleGEDembowskyKTorresA. The Role of Neutrophil Elastase Inhibitors in Lung Diseases. Chest (2017) 152:249e62. doi: 10.1016/j.chest.2017.03.056
79
LiuYDuXChenJJinYPengLWangHHXet al. Neutrophil-to-Lymphocyte Ratio as an Independent Risk Factor for Mortality in Hospitalized Patients With COVID-19. J Infect (2020) 81:e6e12. doi: 10.1016/j.jinf.2020.04.002
80
ChenCYTsaiYFHuangWJChangSHHwangTL. Propofol Inhibits Endogenous Formyl Peptide-Induced Neutrophil Activation and Alleviates Lung Injury. Free Radic Biol Med (2018) 129:372e82. doi: 10.1016/j.freeradbiomed.2018.09.048
81
NguyenGTGreenERMecsasJ. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front Cell Infect Microbiol (2017) 7:373. doi: 10.3389/fcimb.2017.00373
82
PakOSydykovAKosanovicDSchermulyRTDietrichASchroderKet al. Lung Ischaemia-Reperfusion Injury: The Role of Reactive Oxygen Species. Adv Exp Med Biol (2017) 967:195e225. doi: 10.1007/978-3-319-63245-2_12
83
WagnerJStrosingKMSpassovSGLinZEngelstaedterHTackeSet al. Sevoflurane Posttreatment Prevents Oxidative and Inflammatory Injury in Ventilator-Induced Lung Injury. PloS One (2018) 13:e0192896. doi: 10.1371/journal.pone.0192896
84
KumarHKawaiTAkiraS. Pathogen Recognition by the Innate Immune System. Int Rev Immunol (2011) 30(1):16–34. doi: 10.3109/08830185.2010.529976
85
WildeILotzSEngelmannDStarkeAZandbergenGVSolbachWet al. Direct Stimulatory Effects of the TLR2/6 Ligand Bacterial Lipopeptide MALP-2 on Neutrophil Granulocytes. Med Microbiol Immunol (2007) 196:61–71. doi: 10.1007/s00430-006-0027-9
86
MarcosVNussbaumCVitkovLHectorAWiedenbauerERoosDet al. Delayed But Functional Neutrophil Extracellular Trap Formation in Neonates. Blood (2009) 114:4908–4911 author reply 4911– 4902. doi: 10.1182/blood-2009-09-242388
87
TsudaYTakahashiHKobayashiMHanafusaTHerndonDNSuzukiF. Three Different Neutrophil Subsets Exhibited in Mice With Different Susceptibilities to Infection by Methicillin-Resistant Staphylococcus Aureus. Immunity (2004) 21:215–26. doi: 10.1016/j.immuni.2004.07.006
88
SabroeIPrinceLRJonesECHorsburghMJFosterSJVogelSNet al. Selective Roles for Toll-Like Receptor (TLR)2 and TLR4 in the Regulation of Neutrophil Activation and Life Span. J Immunol (2003) 170:5268–75. doi: 10.4049/jimmunol.170.10.5268
89
van BruggenRDrewniakAToolATJansenMvan HoudtMGeisslerJet al. Toll-Like Receptor Responses in IRAK-4- Deficient Neutrophils. J Innate Immun (2010) 2:280–7. doi: 10.1159/000268288
90
BercotBKannengiesserCOudinCGrandchampBSanson-le PorsMJMoulySet al. First Description of NOD2 Variant Associated With Defective Neutrophil Responses in a Woman With Granulomatous Mastitis Related to Corynebacteria. J Clin Microbiol (2009) 47:3034–7. doi: 10.1128/JCM.00561-09
91
RadsakMPSalihHRRammenseeHGSchildH. Triggering Receptor Expressed on Myeloid Cells-1 in Neutrophil Inflammatory Responses: Differential Regulation of Activation and Survival. J Immunol (2004) 172:4956–63. doi: 10.4049/jimmunol.172.8.4956
92
ZhangXMajlessiLDeriaudELeclercCLo-ManR. Coactivation of Syk Kinase and MyD88 Adaptor Protein Pathways by Bacteria Promotes Regulatory Properties of Neutrophils. Immunity (2009) 31:761–71. doi: 10.1016/j.immuni.2009.09.016
93
SalamoneGVPetraccaYFuxman BassJIRumboMNahmodKAGabelloniMLet al. Flagellin Delays Spontaneous Human Neutrophil Apoptosis. Lab Invest (2010) 90:1049–59. doi: 10.1038/labinvest.2010.77
94
HayashiFMeansTKLusterAD. Toll-Like Receptors Stimulate Human Neutrophil Function. Blood (2003) 102:2660–9. doi: 10.1182/blood-2003-04-1078
95
HoarauCGérardBLescanneEHenryDFrançoisSLacapèreJJet al. TLR9 Activation Induces Normal Neutrophil Responses in a Child With IRAK-4 Deficiency: Involvement of the Direct PI3K Pathway. J Immunol (2007) 179:4754–65. doi: 10.4049/jimmunol.179.7.4754
96
KollerBKapplerMLatzinPGaggarASchreinerMTakyarSet al. TLR Expression on Neutrophils at the Pulmonary Site of Infection: TLR1/TLR2-Mediated Up-Regulation of TLR5 Expression in Cystic Fibrosis Lung Disease. J Immunol (2008) 181:2753–63. doi: 10.4049/jimmunol.181.4.2753
97
JankeMPothJWimmenauerVGieseTCochCBarchetWet al. Selective and Direct Activation of Human Neutrophils But Not Eosinophils by Toll-Like Receptor 8. J Allergy Clin Immunol (2009) 123:1026–33. doi: 10.1016/j.jaci.2009.02.015
98
MankanAKDauTJenneDHornungV. The NLRP3/ASC/Caspase-1 Axis Regulates IL-1beta Processing in Neutrophils. Eur J Immunol (2012) 42:710–5. doi: 10.1002/eji.201141921
99
KarmakarMSunYHiseAGRietschAPearlmanE. Cutting Edge: IL-1beta Processing During Pseudomonas Aeruginosa Infection is Mediated by Neutrophil Serine Proteases and Is Independent of NLRC4 and Caspase-1. J Immunol (2012) 189:4231–5. doi: 10.4049/jimmunol.1201447
100
EkmanAKCardellLO. The Expression and Function of Nod-Like Receptors in Neutrophils. Immunology (2010) 130:55–63. doi: 10.1111/j.1365-2567.2009.03212.x
101
ClarkeTBDavisKMLysenkoESZhouAYYuYWeiserJN. Recognition of Peptidoglycan From the Microbiota by Nod1 Enhances Systemic Innate Immunity. Nat Med (2010) 16:228–31. doi: 10.1038/nm.2087
102
TheivanthiranBBatraSBalamayooranGCaiSKobayashiKFlavellRAet al. NOD2 Signaling Contributes to Host Defense in the Lungs Against Escherichia Coli Infection. Infect Immun (2012) 80:2558–69. doi: 10.1128/IAI.06230-11
103
AllenICMooreCBSchneiderMLeiYDavisBKScullMAet al. NLRX1 Protein Attenuates Inflammatory Responses to Infection by Interfering With the RIG-I-MAVS and TRAF6-NF-kappaB Signaling Pathways. Immunity (2011) 34:854–65. doi: 10.1016/j.immuni.2011.03.026
104
KummerJABroekhuizenREverettHAgostiniLKuijkLMartinonFet al. Inflammasome Components NALP 1 and 3 Show Distinct But Separate Expression Profiles in Human Tissues Suggesting a Site-Specific Role in the Inflammatory Response. J Histochem Cytochem (2007) 55:443–52. doi: 10.1369/jhc.6A7101.2006
105
GrenierJMWangLManjiGAHuangWJAl-GarawiAKellyRet al. Functional Screening of Five PYPAF Family Members Identifies PYPAF5 as a Novel Regulator of NF-kappaB and Caspase-1. FEBS Lett (2002) 530:73–8. doi: 10.1016/S0014-5793(02)03416-6
106
AnandPKMalireddiRKLukensJRVogelPBertinJLamkanfiMet al. NLRP6 Negatively Regulates Innate Immunity and Host Defence Against Bacterial Pathogens. Nature (2012) 488:389–93. doi: 10.1038/nature11250
107
VladimerGIWengDPaquetteSWVanajaSKRathinamVAAuneMHet al. The NLRP12 Inflammasome Recognizes Yersinia Pestis. Immunity (2012) 37:96–107. doi: 10.1016/j.immuni.2012.07.006
108
JangJHShinHWLeeJMLeeHWKimECParkSH. An Overview of Pathogen Recognition Receptors for Innate Immunity in Dental Pulp. Mediators Inflamm (2015) 2015:794143. doi: 10.1155/2015/794143
109
BartonJLBergTDidonLNordM. The Pattern Recognition Receptor Nod1 Activates CCAAT/enhancer Binding Protein Beta Signalling in Lung Epithelial Cells. Eur Respir J (2007) 30(2):214–22. doi: 10.1183/09031936.00143906
110
ChaputCSanderLESuttorpNOpitzB. NOD-Like Receptors in Lung Diseases. Front Immunol (2013) 4:393. doi: 10.3389/fimmu.2013.00393
111
de NardoDde NardoCMLatzE. New Insights Into Mechanisms Controlling the NLRP3 Inflammasome and Its Role in Lung Disease. Am J Pathol (2014) 184:42–54. doi: 10.1016/j.ajpath.2013.09.007
112
LeissingerMKulkarniRZemansRLDowneyGPJeyaseelanS. Investigating the Role of Nucleotide-Binding Oligomerization Domain-Like Receptors in Bacterial Lung Infection. Am J Respir Crit Care Med (2014) 189:1461–8. doi: 10.1164/rccm.201311-2103PP
113
Stout-DelgadoHWVaughanSEShiraliACJaramilloRJHarrodKS. Impaired NLRP3 Inflammasome Function in Elderly Mice During Influenza Infection Is Rescued by Treatment With Nigericin. J Immunol (2012) 188:2815–24. doi: 10.4049/jimmunol.1103051
114
ChoSJPlatakiMMitzelDLowryGRooneyKStout-DelgadoH. Decreased NLRP3 Inflammasome Expression in Aged Lung may Contribute to Increased Susceptibility to Secondary Streptococcus Pneumoniae Infection. Exp Gerontol (2018) 105:40–6. doi: 10.1016/j.exger.2017.11.010
115
van LieshoutMHPde VosAFDessingMCde PortoAPNAde BoerOJde BeerRet al. ASC and NLRP3 Impair Host Defense During Lethal Pneumonia Caused by Serotype 3 Streptococcus Pneumoniae in Mice. Eur J Immunol (2018) 48:66–79. doi: 10.1002/eji.201646554
116
KebaierCChamberlandRRAllenICGaoXBrogliePMHallJDet al. Staphylococcus Aureus Alpha-Hemolysin Mediates Virulence in a Murine Model of Severe Pneumonia Through Activation of the NLRP3 Inflammasome. J Infect Dis (2012) 205:807–17. doi: 10.1093/infdis/jir846
117
CravenRRGaoXAllenICGrisDBubeck WardenburgJMcElvania-TekippeEet al. Staphylococcus Aureus Alpha-Hemolysin Activates the NLRP3-Inflammasome in Human and Mouse Monocytic Cells. PloS One (2009) 4:e7446. doi: 10.1371/journal.pone.0007446
118
PaudelSGhimireLJinLBaralPCaiSJeyaseelanS. NLRC4 Suppresses IL-17A-Mediated Neutrophil-Dependent Host Defense Through Upregulation of IL-18 and Induction of Necroptosis During Gram-Positive Pneumonia. Mucosal Immunol (2019) 12:247–57. doi: 10.1038/s41385-018-0088-2
119
CaiSBatraSWakamatsuNPacherPJeyaseelanS. NLRC4 Inflammasome-Mediated Production of Il-1β Modulates Mucosal Immunity in the Lung Against Gram-Negative Bacterial Infection. J Immunol (2012) 188:5623–35. doi: 10.4049/jimmunol.1200195
120
CohenTSPrinceAS. Activation of Inflammasome Signaling Mediates Pathology of Acute P. Aeruginosa Pneumonia. J Clin Invest (2013) 123:1630–7. doi: 10.1172/JCI66142
121
GhimireLPaudelSJinLBaralPCaiSJeyaseelanS. NLRP6 Negatively Regulates Pulmonary Host Defense in Gram-Positive Bacterial Infection Through Modulating Neutrophil Recruitment and Function. PloS Pathog (2018) 14:e1007308. doi: 10.1371/journal.ppat.1007308
122
PouwelsSDvan GeffenWHJonkerMRKerstjensHAMNawijnMCHeijinkIH. Increased Neutrophil Expression of Pattern Recognition Receptors During COPD Exacerbations. Respirology (2016) 22(2):401–4. doi: 10.1111/resp.12912
123
PouwelsSDHeijinkIHten HackenNHTVandenabeelePKryskoDVNawijnMCet al. DAMPs Activating Innate and Adaptive Immune Responses in COPD. Mucosal Immunol (2014) 7:215–26. doi: 10.1038/mi.2013.77
124
YangWNiHWangHGuH. NLRP3 Inflammasome is Essential for the Development of Chronic Obstructive Pulmonary Disease. Int J Clin Exp Pathol (2015) 8:13209–16.
125
BondìMLFerraroMDi VincenzoSGerbinoSCavallaroGGiammonaGet al. Effects in Cigarette Smoke Stimulated Bronchial Epithelial Cells of a Corticosteroid Entrapped Into Nanostructured Lipid Carriers. J Nanobiotechnol (2014) 12:46. doi: 10.1186/s12951-014-0046-4
126
PaceEFerraroMDi VincenzoSGerbinoSBrunoALanataLet al. Oxidative Stress and Innate Immunity Responses in Cigarette Smoke Stimulated Nasal Epithelial Cells. Toxicol In Vitro (2014) 28:292–9. doi: 10.1016/j.tiv.2013.11.004
127
Leiva-JuárezMMKollsJKEvansSE. Lung Epithelial Cells: Therapeutically Inducible Effectors of Antimicrobial Defense. Mucosal Immunol (2018) 11:21–34. doi: 10.1038/mi.2017.71
128
GayNJSymmonsMFGangloffMBryantCE. Assembly and Localization of Toll-Like Receptor Signalling Complexes. Nat Rev Immunol (2014) 14:546–58. doi: 10.1038/nri3713
129
LeeSSuhGYRyterSWChoiAM. Regulation and Function of the Nucleotide Binding Domain Leucine-Rich Repeat-Containing Receptor, Pyrin Domain-Containing-3 Inflammasome in Lung Disease. Am J Respir Cell Mol Biol (2016) 54:151–60. doi: 10.1165/rcmb.2015-0231TR
130
JeyaseelanSYoungSKYamamotoMArndtPGAkiraSKollsJKet al. Toll/IL-1R Domain-Containing Adaptor Protein (TIRAP) Is a Critical Mediator of Antibacterial Defense in the Lung Against Klebsiella Pneumoniae But Not Pseudomonas Aeruginosa. J Immunol (2006) 177:538–47. doi: 10.4049/jimmunol.177.1.538
131
SkerrettSJLiggittHDHajjarAMWilsonCB. Cutting Edge: Myeloid Differentiation Factor 88 is Essential for Pulmonary Host Defense Against Pseudomonas Aeruginosa But Not Staphylococcus Aureus. J Immunol (2004) 172:3377–81. doi: 10.4049/jimmunol.172.6.3377
132
KollerBBalsRRoosDKortingHCGrieseMHartlD. Innate Immune Receptors on Neutrophils and Their Role in Chronic Lung Disease. Eur J Clin Invest (2009) 39:535–47. doi: 10.1111/j.1365-2362.2009.02145.x
133
MorrisAELiggittHDHawnTRSkerrettSJ. Role of Toll-Like Receptor 5 in the Innate Immune Response to Acute P. Aeruginosa Pneumonia. Am J Physiol Lung Cell Mol Physiol (2009) 297(6):L1112. doi: 10.1152/ajplung.00155.2009
134
PorteRFougeronDMuñoz-WolfNTabareauJSirardJ-C. A Toll-Like Receptor 5 Agonist Improves the Efficacy of Antibiotics in Treatment of Primary and Influenza Virus-Associated Pneumococcal Mouse Infections. Antimicrob Agents Chemother (2015) 59(10):6064. doi: 10.1128/AAC.01210-15
135
BengoecheaJASa PessoaJ. Klebsiella Pneumoniaeinfection Biology: Living to Counteract Host Defences. FEMS Microbiol Rev (2018) 43(2):123–44. doi: 10.1093/femsre/fuy043
136
BenmohamedFMedinaMWuYZMaschalidiSTouquiL. Toll-Like Receptor 9 Deficiency Protects Mice Against Pseudomonas Aeruginosa Lung Infection. J Cystic Fibrosis (2013) 12(3):e90466. doi: 10.1371/journal.pone.0090466
137
OppenheimJJZachariaeCOMukaidaNMatsushimaK. Properties of the Novel Proinflammatory Supergene “Intercrine” Cytokine Family. Annu Rev Immunol (1991) 9(1):617–48. doi: 10.1146/annurev.iy.09.040191.003153
138
RazEMahabaleshwarH. Chemokine Signaling in Embryonic Cell Migration: A Fisheye View. Development (2009) 136(8):1223–9. doi: 10.1242/dev.022418
139
De FilippoKHendersonRBLaschingerMHoggN. Neutrophil Chemokines KC and Macrophage-Inflammatory Protein-2 are Newly Synthesized by Tissue Macrophages Using Distinct TLR Signaling Pathways. J Immunol (2008) 80:4308–15. doi: 10.4049/jimmunol.180.6.4308
140
CallDRNemzekJAEbongSJBolgosGRNewcombDEWollenbergGKet al. Differential Local and Systemic Regulation of the Murine Chemokines KC and MIP2. Shock (2001) 15:278–84. doi: 10.1097/00024382-200115040-00005
141
De FilippoKDudeckAHasenbergMNyeEvan RooijenNHartmannKet al. Mast Cell and Macrophage Chemokines CXCL1/CXCL2 Control the Early Stage of Neutrophil Recruitment During Tissue Inflammation. Blood (2013) 121:4930–7. doi: 10.1182/blood-2013-02-486217
142
JeyaseelanSManzerRYoungSKYamamotoMAkiraSMasonRJet al. Induction of CXCL5 During Inflammation in the Rodent Lung Involves Activation of Alveolar Epithelium. Am J Respir Cell Mol Biol (2005) 32:531–9. doi: 10.1165/rcmb.2005-0063OC
143
JeyaseelanSChuHWYoungSKWorthenGS. Transcriptional Profiling of Lipopolysaccharide-Induced Acute Lung Injury. Infect Immun (2004) 72:7247–56. doi: 10.1128/IAI.72.12.7247-7256.2004
144
OpitzBvan LaakVEitelJSuttorpN. Innate Immune Recognition in Infectious and Noninfectious Diseases of the Lung. Am J Respir Crit Care Med (2010) 181(12):1294–309. doi: 10.1164/rccm.200909-1427SO
145
KurdowskaANobleJMGrantISRobertsonCRHaslettCDonnellySC. Anti-Interleukin-8 Autoantibodies in Patients at Risk for Acute Respiratory Distress Syndrome. Crit Care Med (2002) 30:2335–7. doi: 10.1097/00003246-200210000-00024
146
PettyJMSueblinvongVLenoxCCJonesCCCosgroveGPCoolCDet al. Pulmonary Stromal-Derived Factor-1 Expression and Effect on Neutrophil Recruitment During Acute Lung Injury. J Immunol (2007) 178(12):8148–57. doi: 10.4049/jimmunol.178.12.8148
147
RollinsBJMorrisonEDStilesCD. Cloning and Expression of JE, a Gene Inducible by Platelet-Derived Growth Factor and Whose Product has Cytokinelike Properties. Proc Natl Acad Sci USA (1988) 85(11):3738–42. doi: 10.1073/pnas.85.11.3738
148
OpdenakkerGFroyenGFitenPProostPVan DammeJ. Human Monocyte Chemotactic Protein-3 (MCP-3): Molecular Cloning of the cDNA and Comparison With Other Chemokines. Biochem Biophys Res Commun (1993) 191(2):535–42. doi: 10.1006/bbrc.1993.1251
149
BroxmeyerHESherryBLuLCooperSCarowCWolpeSDet al. Myelopoietic Enhancing Effects of Murine Macrophage Inflammatory Proteins 1 and 2 on Colony Formation In Vitro by Murine and Human Bone Marrow Granulocyte/ Macrophage Progenitor Cells. J Exp Med (1989) 170(5):1583–94. doi: 10.1084/jem.170.5.1583
150
PoteyPMRossiAGLucasCDDorwardDA. Neutrophils in the Initiation and Resolution of Acute Pulmonary Inflammation: Understanding Biological Function and Therapeutic Potential. J Pathol (2019) 247(5):672–85. doi: 10.1002/path.5221
151
SadikCDKimNDLusterAD. Neutrophils Cascading Their Way to Inflammation. Trends Immunol (2011) 32(10):452–60. doi: 10.1016/j.it.2011.06.008
152
PaudelSBaralPGhimireLBergeronSJinLDeCorteJAet al. CXCL1 Regulates Neutrophil Homeostasis in Pneumonia-Derived Sepsis Caused by Streptococcus Pneumoniae Serotype 3. Blood (2019) 133(12):1335–45. doi: 10.1182/blood-2018-10-878082
153
BatraSCaiSBalamayooranGJeyaseelanS. Intrapulmonary Administration of Leukotriene B4 Augments Neutrophil Accumulation and Responses in the Lung to Klebsiella Infection in CXCL1 Knockout Mice. J Immunol (2012) 188(7):3458–68. doi: 10.4049/jimmunol.1101985
154
CaiSBatraSLiraSAKollsJKJeyaseelanS. CXCL1 Regulates Pulmonary Host Defense to Klebsiella Infection via CXCL2, CXCL5, NF-κb, and MAPKs. J Immunol (2010) 185(10):6214–25. doi: 10.4049/jimmunol.0903843
155
MizgerdJPLupaMMKoganMSWarrenHBKobzikLTopulosGP. Nuclear Factor-κb P50 Limits Inflammation and Prevents Lung Injury During Escherichia Coli Pneumonia. Am J Respir Crit Care Med (2003) 168(7):810–7. doi: 10.1164/rccm.200303-412OC
156
QuintonLJJonesMRSimmsBTKoganMSRobsonBESkerrettSJet al. Functions and Regulation of NF-κb RelA During Pneumococcal Pneumonia. J Immunol (2007) 178(3):1896–903. doi: 10.4049/jimmunol.178.3.1896
157
PuneetPMoochhalaSBhatiaM. Chemokines in Acute Respiratory Distress Syndrome. Am J Physiol Lung Cell Mol Physiol (2005) 288(1):L3–15. doi: 10.1152/ajplung.00405.2003
158
RemickDGGreenLBNewcombDEGargSJBolgosGLCallDR. CXC Chemokine Redundancy Ensures Local Neutrophil Recruitment During Acute Inflammation. Am J Pathol (2001) 159(3):1149–57. doi: 10.1016/S0002-9440(10)61791-9
159
MakrisSJohanssonC. R848 or Influenza Virus can Induce Potent Innate Immune Responses in the Lungs of Neonatal Mice. Mucosal Immunol (2020) 10:5–10. doi: 10.1038/s41385-020-0314-6
160
LinesJLHoskinsSHollifieldMCauleyLSGarvyBA. The Migration of T Cells in Response to Influenza Virus is Altered in Neonatal Mice. J Immunol (2010) 185:2980–8. doi: 10.4049/jimmunol.0903075
161
KirsebomFCMKausarFNurievRMakrisSJohanssonC. Neutrophil Recruitment and Activation are Differentially Dependent on MyD88/TRIF and MAVS Signaling During RSV Infection. Mucosal Immunol (2019) 12:1244–55. doi: 10.1038/s41385-019-0190-0
162
WinklerESBaileyALKafaiNMNairSMcCuneBTYuJet al. SARS-CoV-2 Infection of Human ACE2-Transgenic Mice Causes Severe Lung Inflammation and Impaired Function. Nat Immunol (2020) 21:1327–35. doi: 10.1038/s41590-020-0778-2
163
PeiróTPatelDFAktharSGregoryLGPyleCJHarkerJAet al. Neutrophils Drive Alveolar Macrophage IL-1β Release During Respiratory Viral Infection. Thorax (2017) 73:546–56. doi: 10.1136/thoraxjnl-2017-210010
164
GibbsJInceLMatthewsLMeiJBellTYangNet al. An Epithelial Circadian Clock Controls Pulmonary Inflammation and Glucocorticoid Action. Nat Med (2014) 20(8):919–26. doi: 10.1038/nm.3599
165
HoffmannEDittrich-BreiholzOHoltmannHKrachtM. Multiple Control of Interleukin-8 Gene Expression. J Leukocyte Biol (2002) 72(5):847–55. doi: 10.1189/jlb.72.5.847
166
SarmientoJShumateCSuetomiKRavindranAVillegasLRajarathnamKet al. Diverging Mechanisms of Activation of Chemokine Receptors Revealed by Novel Chemokine Agonists. PloS One (2011) 6(12):e27967. doi: 10.1371/journal.pone.0027967
167
ViolaALusterAD. Chemokines and Their Receptors: Drug Targets in Immunity and Inflammation. Annu Rev Pharmacol Toxicol (2008) 48:171–97. doi: 10.1146/annurev.pharmtox.48.121806.154841
168
NasserMWRaghuwanshiSKGrantDJJalaVRRajarathnamKRichardsonRM. Differential Activation and Regulation of CXCR1 and CXCR2 by CXCL8 Monomer and Dimer. J Immunol (2009) 183(5):3425–32. doi: 10.4049/jimmunol.0900305
169
RichardsonRMMarjoramRJBarakLSSnydermanR. Role of the Cytoplasmic Tails of CXCR1 and CXCR2 in Mediating Leukocyte Migration, Activation, and Regulation. J Immunol (2003) 170(6):2904–11. doi: 10.4049/jimmunol.170.6.2904
170
RaghuwanshiSKSuYSinghVHaynesKRichmondARichardsonRM. The Chemokine Receptors CXCR1 and CXCR2 Couple to Distinct G Protein-Coupled Receptor Kinases to Mediate and Regulate Leukocyte Functions. J Immunol (2012) 189(6):2824–32. doi: 10.4049/jimmunol.1201114
171
HuberARKunkelSLToddRFWeissSJ. Regulation of Transendothelial Neutrophil Migration by Endogenous Interleukin-8. Science (1991) 254(5028):99–102. doi: 10.1126/science.1718038
172
MulFPZuurbierAEJanssenHCalafatJvan WeteringSHiemstraPSet al. Sequential Migration of Neutrophils Across Monolayers of Endothelial and Epithelial Cells. J Leukocyte Biol (2000) 68(4):529–37. doi: 10.1189/jlb.68.4.529
173
BurnsARSimonSIKukielkaGLRowenJLLuHMendozaLHet al. Chemotactic Factors Stimulate CD18-Dependent Canine Neutrophil Adherence and Motility on Lung Fibroblasts. J Immunol (1996) 156(9):3389–401.
174
IslesHMHermanKRobertsonALLoynesCAPrinceLRElksPMet al. The CXCL12/CXCR4 Signalling Axis Retains Neutrophils at Inflammatory Sites in Zebrafish. BioRxiv (2019) 1:626978. doi: 10.3389/fimmu.2019.01784
175
KonradFMMeichssnerNBuryANgamsriKCReutershanJ. Inhibition of SDF-1 Receptors CXCR4 and CXCR7 Attenuates Acute Pulmonary Inflammation via the Adenosine A 2B-Receptor on Blood Cells. Cell Death Dis (2017) 8(5):e2832–. doi: 10.1038/cddis.2016.482
176
TulottaCStefanescuCChenQTorracaVMeijerAHSnaar-JagalskaBE. CXCR4 Signaling Regulates Metastatic Onset by Controlling Neutrophil Motility and Response to Malignant Cells. Sci Rep (2019) 9(1):2399. doi: 10.1038/s41598-019-38643-2
177
XuJMoraAShimHStecenkoABrighamKLRojasM. Role of the SDF-1/CXCR4 Axis in the Pathogenesis of Lung Injury and Fibrosis. Am J Respir Cell Mol Biol (2007) 37(3):291–9. doi: 10.1165/rcmb.2006-0187OC
178
RafiiSCaoZLisRSiemposIIChavezDShidoKet al. Platelet-Derived SDF-1 Primes the Pulmonary Capillary Vascular Niche to Drive Lung Alveolar Regeneration. Nat Cell Biol (2015) 17(2):123–36. doi: 10.1038/ncb3096
179
RadermeckerCSabatelCVanwingeCRuscittiCMaréchalPPerinFet al. Locally Instructed CXCR4hi Neutrophils Trigger Environment-Driven Allergic Asthma Through the Release of Neutrophil Extracellular Traps. Nat Immunol (2019) 20:1444–55. doi: 10.1038/s41590-019-0496-9
180
DidangelosA. COVID-19 Hyperinflammation: What About Neutrophils? mSphere (2020) 5:e00367–20. doi: 10.1128/mSphere.00367-20
181
GuoLSituHLWangZYLinYChenQJJ. Mechanism of Jinrong Granule in Inhibiting the Invasion of Breast Cancer Cells by the CXCL-1-CXCR2/CCL20 Pathway. Biol Regul Homeost Agents (2020) 34(3):969–76. doi: 10.23812/19-512-A-38
182
LeYZhouYIribarrenPWangJ. Chemokines and Chemokine Receptors: Their Manifold Roles in Homeostasis and Disease. Cell Mol Immunol (2004) 1(2):95–104.
183
SallustoFMackayCRLanzavecchiaA. The Role of Chemokine Receptors in Primary, Effector, and Memory Immune Responses. An Rev Immunol (2000) 18(1):593–620. doi: 10.1146/annurev.immunol.18.1.593
184
ChouRCKimNDSadikCDSeungELanYByrneMHet al. Lipid-Cytokine-Chemokine Cascade Drives Neutrophil Recruitment in a Murine Model of Inflammatory Arthritis. Immunity (2010) 33(2):266–78. doi: 10.1016/j.immuni.2010.07.018
185
SherryBTekamp-OlsonPGallegosCBauerDDavatelisGWolpeSDet al. Resolution of the Two Components of Macrophage Inflammatory Protein 1, and Cloning and Characterization of One of Those Components, Macrophage Inflammatory Protein 1 Beta. J Exp Med (1988) 168(6):2251–9. doi: 10.1084/jem.168.6.2251
186
BonvilleCAPercopoCMDyerKDGaoJPrussinCFosterBet al. Interferon-Gamma Coordinates CCL3-Mediated Neutrophil Recruitment In Vivo. BMC Immunol (2009) 10(1):1–3. doi: 10.1186/1471-2172-10-14
187
WinterCTautKSrivastavaMLängerFMackMBrilesDEet al. Lung-Specific Overexpression of CC Chemokine Ligand (CCL) 2 Enhances the Host Defense to Streptococcus Pneumoniae Infection in Mice: Role of the CCL2-CCR2 Axis. J Immunol (2007) 178(9):5828–38. doi: 10.4049/jimmunol.178.9.5828
188
MausUABackiMWinterCSrivastavaMSchwarzMKR̈ckleTet al. Importance of Phosphoinositide 3-Kinase γ in the Host Defense Against Pneumococcal Infection. Am J Respir Crit Care Med (2007) 175(9):958–66. doi: 10.1164/rccm.200610-1533OC
189
GrommesJAlardJEDrechslerMWanthaSMörgelinMKueblerWMet al. Disruption of Platelet-Derived Chemokine Heteromers Prevents Neutrophil Extravasation in Acute Lung Injury. Am J Respir Crit Care Med (2012) 185(6):628–36. doi: 10.1164/rccm.201108-1533OC
190
HartlDTirouvanziamRLavalJGreeneC,MHabielDSharmaLet al. Innate Immunity of the Lung: From Basic Mechanisms to Translational Medicine. J Innate Immun (2018) 10:487–501. doi: 10.1159/000487057
191
BabiorBM. Oxidants From Phagocytes: Agents of Defense and Destruction. Blood (1984) 64:959–66. doi: 10.1182/blood.V64.5.959.959
192
VillanuevaEYalavarthiSBerthierCCHodginJBKhandpurRLinAMet al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J Immunol (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
193
GrahamDBRobertsonCMBautistaJMascarenhasFDiacovoMJMontgrainVet al. Neutrophil-Mediated Oxidative Burst and Host Defense Are Controlled by a Vav-Plcγ2 Signaling Axis in Mice. J Clin Invest (2007) 117(11):3445–52. doi: 10.1172/JCI32729
194
VenketaramanVMillmanASalmanMSwaminathanSGoetzMLardizabalAet al. Glutathione Levels and Immune Responses in Tuberculosis Patients. Microb Pathog (2008) 44:255–61. doi: 10.1016/j.micpath.2007.09.002
195
GuerraCMorrisDSipinAKungSFranklinMGrayDet al. Glutathione and Adaptive Immune Responses Against Mycobacterium Tuberculosis Infection in Healthy and HIV Infected Individuals. PloS One (2011) 6:e28378. doi: 10.1371/journal.pone.0028378
196
DallengaTRepnikUCorleisBEichJReimerRGriffithsGWet al. Tuberculosis-Induced Necrosis of Infected Neutrophils Promotes Bacterial Growth Following Phagocytosis by Macrophages. Cell Host Microbe (2017) 22:519–30.e3. doi: 10.1016/j.chom.2017.09.003
197
VernonPJSchaubLJDalleluccaJJPusateriAESheppardFR. Rapid Detection of Neutrophil Oxidative Burst Capacity Is Predictive of Whole Blood Cytokine Responses. PloS One (2015) 10:1–13. doi: 10.1371/journal.pone.0146105
198
VaismanNKerasinEHahnTTrifonSVoetHTabachnikE. Increased Neutrophil Chemiluminescence Production in Patients With Cystic Fibrosis. Metabolism (1994) 43:719–22. doi: 10.1016/0026-0495(94)90120-1
199
McKeonDJCadwalladerKAIdrisSCowburnASPasteurMCBarkerHet al. Cystic Fibrosis Neutrophils Have Normal Intrinsic Reactive Oxygen Species Generation. Eur Respir J (2010) 35:1264–72. doi: 10.1183/09031936.00089709
200
FruhwirthMRuedlCEllemunterHBockGWolfH. Flow-Cytometric Evaluation of Oxidative Burst in Phagocytic Cells of Children With Cystic Fibrosis. Int Arch Allergy Immunol (1998) 117:270–5. doi: 10.1159/000024022
201
Witko-SarsatVAllenRCPaulaisMNguyenATBessouGLenoirGet al. Disturbed Myeloperoxidase-Dependent Activity of Neutrophils in Cystic Fibrosis Homozygotes and Heterozygotes, and Its Correction by Amiloride. J Immunol (1996) 157:2728–35.
202
MontemurroPMariggiòMABarbutiGCassanoAVincentiASerioGet al. Increase in Interleukin-8 Production From Circulating Neutrophils Upon Antibiotic Therapy in Cystic Fibrosis Patients. J Cyst Fibros (2012) 11:518–24. doi: 10.1016/j.jcf.2012.04.010
203
ZimmermanBJGrangerDN. Mechanisms of Reperfusion Injury. Am J Med Sci (1994) 307(4):284–92. doi: 10.1097/00000441-199404000-00009
204
McNamaraPSRitsonPSelbyAHartCASmythRL. Bronchoalveolar Lavage Cellularity in Infants With Severe Respiratory Syncytial Virus Bronchiolitis. Arch Dis Child (2003) 88:922–6. doi: 10.1136/adc.88.10.922
205
EverardMLSwarbrickAWrighthamMMcIntyreJDunkleyCJamesPDet al. Analysis of Cells Obtained by Bronchial Lavage of Infants With Respiratory Syncytial Virus Infection. Arch Dis Child (1994) 71:428–32. doi: 10.1136/adc.71.5.428
206
XiongYLiuYCaoLWangDGuoMJiangAet al. Transcriptomic Characteristics of Bronchoalveolar Lavage Fluid and Peripheral Blood Mononuclear Cells in COVID-19 Patients. Emerg Microbes Infect (2020) 9:761–70. doi: 10.1080/22221751.2020.1747363
207
LucasCWongPKleinJCastroTBRSilvaJSundaramMet al. Longitudinal Analyses Reveal Immunological Misfiring in Severe COVID-19. Nature (2020) 584:463–9. doi: 10.1038/s41586-020-2588-y
208
GalaniIEAndreakosE. Neutrophils in Viral Infections: Current Concepts and Caveats. J Leukoc Biol (2015) 98:557–64. doi: 10.1189/jlb.4vmr1114-555r
209
NaumenkoVTurkMJenneCNKimSJ. Neutrophils in Viral Infection. Cell Tissue Res (2018) 371:505–16. doi: 10.1007/s00441-017-2763-0
210
HemmatNDerakhshaniABannazadeh BaghiHSilvestrisNBaradaranBDe SummaS. Neutrophils, Crucial, or Harmful Immune Cells Involved in Coronavirus Infection: A Bioinformatics Study. Front Genet (2020) 11:641. doi: 10.3389/fgene.2020.00641
211
WangJJiangMChenXMontanerLJ. Cytokine Storm and Leukocyte Changes in Mild Versus Severe SARS-CoV-2 Infection: Review of 3939 COVID-19 Patients in China and Emerging Pathogenesis and Therapy Concepts. J Leukoc Biol (2020) 108:17–41. doi: 10.1002/JLB.3COVR0520-272R
212
SinghKMittalSGollapudiSButzmannAKumarJOhgamiRS. A Meta-Analysis of SARS-CoV-2 Patients Identifies the Combinatorial Significance of D-Dimer, C-Reactive Protein, Lymphocyte, and Neutrophil Values as a Predictor of Disease Severity. Int J Lab Hematol (2020) 43(2):324–8. doi: 10.1111/ijlh.13354
213
GuanJWeiXQinSLiuXJiangYChenYet al. Continuous Tracking of COVID-19 Patients’ Immune Status. Int Immunopharmacol (2020) 89:107034. doi: 10.1016/j.intimp.2020.107034
214
WangJLiQYinYZhangYCaoYLinXet al. Excessive Neutrophils and Neutrophil Extracellular Traps in COVID-19. Front Immunol (2020) 11:2063. doi: 10.3389/fimmu.2020.02063
215
BarnesBJAdroverJMBaxter-StoltzfusABorczukACools-LartigueJCrawfordJMet al. Targeting Potential Drivers of COVID-19: Neutrophil Extracellular Traps. J Exp Med (2020) 217:e20200652. doi: 10.1084/jem.20200652
216
YaoXHLiTYHeZCPingYFLiuHWYuSCet al. A Pathological Report of Three COVID-19 Cases by Minimally Invasive Autopsies. Zhonghua Bing Li Xue Za Zhi = Chin J Pathol (2020) 49:E009. doi: 10.3760/cma.j.cn112151-20200312-00193
217
SchurinkBRoosERadonicTBarbeEBoumanCSCde BoerHHet al. Viral Presence and Immunopathology in Patients With Lethal COVID-19: A Prospective Autopsy Cohort Study. Lancet Microbe (2020) 1:e290–9. doi: 10.1016/S2666-5247(20)30144-0
218
WangDHuBHuC. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
219
DuRHLiangLRYangCQWangWCaoTZLiMet al. Predictors of Mortality for Patients With COVID-19 Pneumonia Caused by SARS-CoV-2: A Prospective Cohort Study. Eur Respir J (2020) 55:e2000524. doi: 10.1183/13993003.00524-2020
220
ParackovaZZentsovaIBloomfieldMVrabcovaPSmetanovaJKlocperkAet al. Disharmonic Inflammatory Signatures in COVID-19: Augmented Neutrophils’ But Impaired Monocytes’ and Dendritic Cells’ Responsiveness. Cells (2020) 9:2206. doi: 10.3390/cells9102206
221
Schulte-SchreppingJReuschNPaclikDBaßlerKSchlickeiserSZhangBet al. Severe COVID-19 is Marked by a Dysregulated Myeloid Cell Compartment. Cell (2020) 182(6):1419–40. doi: 10.1016/j.cell.2020.08.001
222
ChuaRLLukassenSTrumpSHennigBPWendischDPottFet al. COVID-19 Severity Correlates With Airway Epithelium-Immune Cell Interactions Identified by Single-Cell Analysis. Nat Biotechnol (2020) 38:970–9. doi: 10.1038/s41587-020-0602-4
223
LiaoMLiuYYuanJWenYXuGZhaoJet al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With COVID-19. Nat Med (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
224
GuanW-JNiZ-YHuYLiangW-HOuC-QHeJ-Xet al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
225
Giamarellos-BourboulisEJNeteaMGRovinaNAkinosoglouKAntoniadouAAntonakosNet al. Complex Immune Dysregulation in COVID-19 Patients With Severe Respiratory Failure. Cell Host Microbe (2020) 27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009
226
QinCZhouLHuZZhangSYangSTaoYet al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis (2020) 71:762–8. doi: 10.1093/cid/ciaa248
227
AschenbrennerACMouktaroudiMKrämerBOestreichMAntonakosNNuesch-GermanoMet al. Disease Severity-Specific Neutrophil Signatures in Blood Transcriptomes Stratify COVID-19 Patients. Genome Med (2021) 13:7. doi: 10.1186/s13073-020-00823-5
228
MeizlishMLPineABBishaiJDGoshuaGNadelmannERSimonovMet al. A Neutrophil Activation Signature Predicts Critical Illness and Mortality in COVID-19. Blood Adv (2021) 5(5):1164–77. doi: 10.1182/bloodadvances.2020003568
229
de BontCMBoelensWCPruijnGJM. NETosis, Complement, and Coagulation: A Triangular Relationship. Cell Mol Immunol (2019) 16:19–27. doi: 10.1038/s41423-018-0024-0
230
GaoTHuMZhangXLiHZhuLLiuHet al. Highly Pathogenic Coronavirus N Protein Aggravates Lung Injury by MASP-2-Mediated Complement Over-Activation. medRxiv (2020) 2020.03.29:20041962. doi: 10.1101/2020.03.29.20041962
231
LlitjosJ-FLeclercMChochoisCMonsallierJ-MRamakersMAuvrayMet al. High Incidence of Venous Thromboembolic Events in Anticoagulated Severe COVID-19 Patients. J Thromb Haemost JTH (2020) 18:1743–6. doi: 10.1111/jth.14869
232
AliRAGandhiAAMengHYalavarthiSVreedeAPEstesSKet al. Adenosine Receptor Agonism Protects Against NETosis and Thrombosis in Antiphospholipid Syndrome. Nat Commun (2019) 10:1916. doi: 10.1038/s41467-019-09801-x
233
GrailerJJCanningBAKalbitzMHaggadoneMDDhondRMAndjelkovicAVet al. Critical Role for the NLRP3 Inflammasome During Acute Lung Injury. J Immunol (2014) 192:5974. doi: 10.4049/jimmunol.1400368
234
XuJZhangXPelayoRMonestierMAmmolloCTSemeraroFet al. Extracellular Histones Are Major Mediators of Death in Sepsis. Nat Med (2009) 15:1318–21. doi: 10.1038/nm.2053
235
ZuoYYalavarthiSShiHGockmanKZuoMMadisonJAet al. Neutrophil Extracellular Traps in COVID-19. JCI Insight (2020) 5(11):e138999. doi: 10.1172/jci.insight.138999
236
MehtaPMcAuleyDFBrownMSanchezETattersallRSMansonJJ. COVID-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet (2020) 395:1033–4. doi: 10.1016/s0140-6736(20)30628-0
237
MerzaMHartmanHRahmanMHwaizRZhangERenströmEet al. Neutrophil Extracellular Traps Induce Trypsin Activation, Inflammation, and Tissue Damage in Mice With Severe Acute Pancreatitis. Gastroenterology (2015) 149:1920–31.e8. doi: 10.1053/j.gastro.2015.08.026
238
NarasarajuTYangESamyRPNgHHPohWPLiewA-Aet al. Excessive Neutrophils and Neutrophil Extracellular Traps Contribute to Acute Lung Injury of Influenza Pneumonitis. Am J Pathol (2011) 179:199–210. doi: 10.1016/j.ajpath.2011.03.013
239
GuptaAKJoshiMBPhilippovaMErnePHaslerPHahnSet al. Activated Endothelial Cells Induce Neutrophil Extracellular Traps and Are Susceptible to NETosis-Mediated Cell Death. S Lett (2010) 584:3193–7. doi: 10.1016/j.febslet.2010.06.006
240
ClarkSRMaACTavenerSAMcDonaldBGoodarziZKellyMMet al. Platelet TLR4 Activates Neutrophil Extracellular Traps to Ensnare Bacteria in Tic Blood. Nat Med (2007) 13:463–9. doi: 10.1038/nm1565
241
McDonaldBDavisRPKimS-JTseMEsmonCTKolaczkowskaEet al. Platelets and Neutrophil Extracellular Traps Collaborate to Promote Intravascular Coagulation During Sis in Mice. Blood (2017) 129:1357–67. doi: 10.1182/blood-2016-09-741298
242
Meher AkshayaKSpinosaMDavisJPPopeNLaubachVESuGet al. Novel Role of IL (Interleukin)-1β in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler Thromb Vasc Biol (2018) 38:843–53. doi: 10.1161/ATVBAHA.117.309897
243
SilPWicklumHSurellCRadaB. Macrophage-Derived IL-1β Enhances Monosodium Urate Crystal-Triggered NET Formation. Inflamm Res (2017) 66:227–37. doi: 10.1007/s00011-016-1008-0
244
MiddletonEAHeXYDenormeFCampbellRANgDSalvatoreSPet al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood (2020) 136(10):1169–79. doi: 10.1182/blood.2020007008
245
VerasFPPontelliMCSilvaCMToller-KawahisaJEde LimaMNascimentoDCet al. SARS-CoV-2–Triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J Exp Med (2020) 217(12):e20201129. doi: 10.1084/jem.20201129
246
NIAID/Flickr Photos. Available at: https://www.flickr.com/photos/tags/niaid/ (Accessed August 10th 2021).
247
BrillAFuchsTASavchenkoASThomasGMMartinodKDe MeyerSFet al. Neutrophil Extracellular Traps Promote Deep Vein Thrombosis in Mice. J Thromb Haemost (2012) 10:136–44. doi: 10.1111/j.1538-7836.2011.04544.x
248
KambasKMarkiewskiMMPneumatikosIARafailSSTheodorouVKonstantonisDet al. C5a and TNF-Alpha Up-Regulate the Expression of Tissue Factor in Intra-Alveolar Neutrophils of Patients With the Acute Respiratory Distress Syndrome. J Immunol (2008) 180:7368–75. doi: 10.4049/jimmunol.180.11.7368
249
LeppkesMKnopfJNaschbergerELindemannASinghJHerrmannIet al. Vascular Occlusion by Neutrophil Extracellular Traps in COVID-19. EBioMedicine (2020) 58:102925. doi: 10.1016/j.ebiom.2020.102925
250
NicolaiLLeunigABrambsSKaiserRWeinbergerTWeigandMet al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation (2020) 142:1176–89. doi: 10.1161/CIRCULATIONAHA.120.048488
251
LeviMThachilJIbaTLevyJH. Coagulation Abnormalities and Thrombosis in Patients With COVID-19. Lancet Haematol (2020) 7:e438–40. doi: 10.1016/S2352-3026(20)30145-9
252
SeitzRGurtlerLSchrammW. Thromboinflammation in COVID-19: Can Alpha2 -Macroglobulin Help to Control the Fire? J Thromb Haemost (2021) 19:351–4. doi: 10.1111/jth.15190
253
GuptaAMadhavanMVSehgalKNairNMahajanSSehrawatTSet al. Extrapulmonary Manifestations of COVID-19. Nat Med (2020) 26:1017–32. doi: 10.1038/s41591-020-0968-3
254
PramitasuriTILaksmidewiAPutraIBKDalimarthaFA. Neutrophil Extracellular Traps in Coronavirus Disease-19-Associated Ischemic Stroke: A Novel Avenue in Neuroscience. Exp Neurobiol (2021) 30:1–12. doi: 10.5607/en20048
255
PetitoEFalcinelliEPalianiUCesariEVaudoGSebastianoMet al. Association of Neutrophil Activation, More Than Platelet Activation, With Thrombotic Complications in Coronavirus Disease 2019. J Infect Dis (2021) 223:933–44. doi: 10.1093/infdis/jiaa756
256
SkendrosPMitsiosAChrysanthopoulouAMastellosDCMetallidisSRafailidisPet al. Complement and Tissue Factor-Enriched Neutrophil Extracellular Traps Are Key Drivers in COVID19 Immunothrombosis. J Clin Investig (2020) 130:6151–7. doi: 10.1172/JCI141374
257
Carmona-RiveraCZhaoWYalavarthiSKaplanMJ. Neutrophil Extracellular Traps Induce Endothelial Dysfunction in Systemic Lupus Erythematosus Through the Activation of Matrix Metalloproteinase-2. Ann Rheum Dis (2015) 74:1417–24. doi: 10.1136/annrheumdis-2013-204837
258
O’SullivanKMHoldsworthSR. Neutrophil Extracellular Traps: A Potential Therapeutic Target in MPO-ANCA Associated Vasculitis? Front Immunol (2021) 12:635188. doi: 10.3389/fimmu.2021.635188
259
MorrisGBortolasciCCPuriBKOliveLMarxWO’NeilAet al. Preventing the Development of Severe COVID-19 by Modifying Immunothrombosis. Life Sci (2021) 264:118617. doi: 10.1016/j.lfs.2020.118617
260
Jimenez-AlcazarMRangaswamyCPandaRBitterlingJSimsekYJLongATet al. Host DNases Prevent Vascular Occlusion by Neutrophil Extracellular Traps. Science (2017) 358:1202–6. doi: 10.1126/science.aam8897
261
GustineJNJonesD. Immunopathology of Hyperinflammation in COVID-19. Am J Pathol (2021) 191:4–17. doi: 10.1016/j.ajpath.2020.08.009
262
NicolaiLLeunigABrambsSKaiserRJoppichMHoffknechtMLet al. Vascular Neutrophilic Inflammation and Immunothrombosis Distinguish Severe COVID-19 From Influenza Pneumonia. J Thromb Haemost (2021) 19:574–81. doi: 10.1111/jth.15179
263
Gonzalez-LopezAAlbaicetaGM. Repair After Acute Lung Injury: Molecular Mechanisms and Therapeutic Opportunities. Crit Care (2012) 16:209. doi: 10.1186/cc11224
264
JasperAEMcIverWJSapeyEWaltonGM. Understanding the Role of Neutrophils in Chronic Inflammatory Airway Disease. F1000Research (2019) 8(F1000 Faculty Rev):557. doi: 10.12688/f1000research.18411.1
265
GehrigSDuerrJWeitnauerMWagnerCJGraeberSYSchatternyJet al. Lack of Neutrophil Elastase Reduces Inflammation, Mucus Hypersecretion, and Emphysema, But Not Mucus Obstruction, in Mice With Cystic Fibrosis–Like Lung Disease. Am J Respir Crit Care Med (2014) 189(9):1082–92. doi: 10.1164/rccm.201311-1932OC
266
PortoBNSteinRT. Neutrophil Extracellular Traps in Pulmonary Diseases: Too Much of a Good Thing? Front Immunol (2016) 7:311. doi: 10.3389/fimmu.2016.00311
267
ZhangBZhouXZhuCFengFQiuYFengJet al. Immune Phenotyping Based on Neutrophil-to-Lymphocyte Ratio and IgG Predicts Disease Severity and Outcome for Patients With COVID-19. Front Mol Biosci (2020) 7:157. doi: 10.3389/fmolb.2020.00157
268
SongC-YXuJHeJ-QLuY-Q. COVID-19 Early Warning Score: A Multi-Parameter Screening Tool to Identify Highly Suspected Patients. medRxiv (2020) 2020.03.05:20031906. doi: 10.1101/2020.03.05.20031906
269
SaffarzadehMJuenemannCQueisserMALochnitGBarretoGGaluskaSPet al. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PloS One (2012) 7:e32366. doi: 10.1371/journal.pone.0032366
270
BranzkNLubojemskaAHardisonSEWangQGutierrezMGBrownGDet al. Neutrophils Sense Microbe Size and Selectively Release Neutrophil Extracellular Traps in Response to Large Pathogens. Nat Immunol (2014) 15:1017–25. doi: 10.1038/ni.2987
271
ThomasGMCarboCCurtisBRMartinodKMazoIBSchatzbergDet al. Extracellular DNA Traps Are Associated With the Pathogenesis of TRALI in Humans and Mice. Blood (2012) 119:6335–43. doi: 10.1182/blood-2012-01-405183
272
HydeDMMillerLAMcDonaldRJStovallMYWongVPinkertonKEet al. Neutrophils Enhance Clearance of Necrotic Epithelial Cells in Ozone-Induced Lung Injury in Rhesus Monkeys. Am J Phys (1999) 277:L1190–8. doi: 10.1152/ajplung.1999.277.6.L1190
273
WangJ. Neutrophils in Tissue Injury and Repair. Cell Tissue Res (2018) 371(3):531–9. doi: 10.1007/s00441-017-2785-7
274
JonesHRRobbCTPerrettiMRossiAG. The Role of Neutrophils in Inflammation Resolution. Semin Immunol (2016) 28(2):137–45. doi: 10.1016/j.smim.2016.03.007
275
ZemansRLBrionesNCampbellMMcClendonJYoungSKSuzukiTet al. Neutrophil Transmigration Triggers Repair of the Lung Epithelium via Beta-Catenin Signaling. Proc Natl Acad Sci U S A (2011) 108:15990–5. doi: 10.1073/pnas.1110144108
276
ParisAJLiuYMeiJDaiNGuoLSpruceLAet al. Neutrophils Promote Alveolar Epithelial Regeneration by Enhancing Type II Pneumocyte Proliferation in a Model of Acid-Induced Acute Lung Injury. Am J Physiol Lung Cell Mol Physiol (2016) 311:L1062–75. doi: 10.1152/ajplung.00327.2016
277
WareLBEisnerMDThompsonBTParsonsPEMatthayMA. Significance of Von Willebrand Factor in Septic and Nonseptic Patients With Acute Lung Injury. Am J Respir Crit Care Med (2004) 170(7):766–72. doi: 10.1164/rccm.200310-1434OC
278
FlandersSASteinJShochatGSellersKHollandMMaselliJet al. Performance of a Bedside C-Reactive Protein Test in the Diagnosis of Community-Acquired Pneumonia in Adults With Acute Cough. Am J Med (2004) 116:529–35. doi: 10.1016/j.amjmed.2003.11.023
279
DetermannRMRoyakkersAAHaitsmaJJZhangHSlutskyASRanieriVMet al. Plasma Levels of Surfactant Protein D and KL-6 for Evaluation of Lung Injury in Critically Ill Mechanically Ventilated Patients. BMC Pulm Med (2010) 10:6. doi: 10.1186/1471-2466-10-6
280
Keramat FGhasemi BasirHRAbdoliEShafiee AghdamAPoorolajalJ. Association of Serum Procalcitonin and C-Reactive Protein Levels With CURB-65 Criteria Among Patients With Community-Acquired Pneumonia. Int J Gen Med (2018) 11:217–23. doi: 10.2147/IJGM.S165190
281
OsakaDShibataYKanouchiKNishiwakiMKimuraTKishiHet al. Soluble Endothelial Selectin in Acute Lung Injury Complicated by Severe Pneumonia. Int J Med Sci (2011) 8:302–8. doi: 10.7150/ijms.8.302
282
ZhaoXXuLYangZSunBWangYLiGet al. Significance of sTREM-1 in Early Prediction of Ventilator-Associated Pneumonia in Neonates: A Single-Center, Prospective, Observational Study. BMC Infect Dis (2020) 20(1):1–8. doi: 10.1186/s12879-020-05196-z
283
FremontRDKoyamaTCalfeeCSWuWDossettLABossertFRet al. Acute Lung Injury in Patients With Traumatic Injuries: Utility of a Panel of Biomarkers for Diagnosis and Pathogenesis. J Trauma (2010) 68:1121–7. doi: 10.1097/TA.0b013e3181c40728
284
BelloSLasierraABMincholéEFandosSRuizMAVeraEet al. Prognostic Power of Proadrenomedullin in Community-Acquired Pneumonia Is Independent of Aetiology. Eur Respir J (2012) 39(5):1144–55. doi: 10.1183/09031936.00080411
285
KhattabAAEl-MekkawyMSShehataAMWhdanNA. Clinical Study of Serum Interleukin-6 in Children With Community-Acquired Pneumonia. Egyptian Pediatr Assoc Gazette (2018) 66(2):43–8. doi: 10.1016/j.epag.2018.03.003
286
GutbierBNeuhaußAKReppeKEhrlerCSantelAKaufmannJet al. Prognostic and Pathogenic Role of Angiopoietin-1 and-2 in Pneumonia. Am J Respir Crit Care Med (2018) 198(2):220–31. doi: 10.1164/rccm.201708-1733OC
287
MatthayMAWareLB. Plasma Protein C Levels in Patients With Acute Lung Injury: Prognostic Significance. Crit Care Med (2004) 32(5):S229–32. doi: 10.1097/01.CCM.0000126121.56990.D3
288
KloucheKCristolJPDevinJGillesVKusterNLarcherRet al. Diagnostic and Prognostic Value of Soluble CD14 Subtype (Presepsin) for Sepsis and Community-Acquired Pneumonia in ICU Patients. Ann Intensive Care (2016) 6:59. doi: 10.1186/s13613-016-0160-6
289
El SolhAABhoraMPinedaLAquilinaAAbbetessaLBerbaryE. Alveolar Plasminogen Activator Inhibitor-1 Predicts ARDS in Aspiration Pneumonitis. Intensive Care Med (2005) 32(1):110–5. doi: doi: 10.1007/s00134-005-2847-2
290
TitovaEVLøfbladLÅsbergAHenriksenAH. Calprotectin as a Biomarker of Pneumonia in Patients Hospitalized With AECOPD. Eur Respir J (2019) 54(suppl 63):PA2895. doi: 10.1183/13993003.congress-2019.PA2895
291
EbrahimiFWolffenbuttelCBlumCABaumgartnerCMuellerBSchuetzPet al. Fibroblast Growth Factor 21 Predicts Outcome in Community-Acquired Pneumonia: Secondary Analysis of Two Randomised Controlled Trials. Eur Respir J (2019) 53(2):1800973. doi: 10.1183/13993003.00973-2018
292
MikacenicCMooreRDmyterkoVWestTEAltemeierWALilesWCet al. Neutrophil Extracellular Traps (NETs) Are Increased in the Alveolar Spaces of Patients With Ventilator-Associated Pneumonia. Crit Care (2018) 22(1):1–8. doi: 10.1186/s13054-018-2290-8
293
JonesTKFengRKerchbergerVEReillyJPAndersonBJShashatyMGet al. Plasma sRAGE Acts as a Genetically Regulated Causal Intermediate in Sepsis-Associated Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med (2020) 201(1):47–56. doi: 10.1164/rccm.201810-2033OC
294
MauriTCoppadoroABombinoMBellaniGZambelliVFornariCet al. Alveolar Pentraxin 3 as an Early Marker of Microbiologically Confirmed Pneumonia: A Threshold-Finding Prospective Observational Study. Crit Care (2014) 18(5):1–0. doi: 10.1186/s13054-014-0562-5
295
RamírezPFerrerMMartíVReyesSMartínezRMenéndezRet al. Inflammatory Biomarkers and Prediction for Intensive Care Unit Admission in Severe Community-Acquired Pneumonia. Crit Care Med (2011) 39(10):2211–7. doi: 10.1097/CCM.0b013e3182257445
296
Cerda-MancillasMCSantiago-GermánDAndrade-BravoBPedraza-OlivaresFValenzo-HernándezFLeaños-MirandaAet al. D-Dimer as a Biomarker of Severity and Adverse Outcomes in Patients With Community Acquired Pneumonia. Arch Med Res (2020) 51(5):429–35. doi: 10.1016/j.arcmed.2020.04.014
297
GuéantJLFromonotJGuéant-RodriguezRMLacolleyPGuieuRRegnaultV. Blood Myeloperoxidase-DNA, a Biomarker of Early Response to SARS-CoV-2 Infection? Allergy (2021) 76(3):892–6. doi: 10.1111/all.14533
298
NgHHavervallSRosellAAguileraKParvKVon MeijenfeldtFAet al. Circulating Markers of Neutrophil Extracellular Traps Are of Prognostic Value in Patients With COVID-19. Arteriosc Thromb Vasc Biol (2021) 41(2):988–94. doi: 10.1161/ATVBAHA.120.315267
299
ImranMMAhmadUUsmanUAliMShaukatAGulN. Neutrophil/lymphocyte Ratio—A Marker of COVID-19 Pneumonia Severity. Int J Clin Pract (2021) 75(4):e13698. doi: 10.1111/ijcp.13698
300
BlasiAvon MeijenfeldtFAAdelmeijerJCalvoAIbañezCPerdomoJet al. In Vitro Hypercoagulability and Ongoing In Vivo Activation of Coagulation and Fibrinolysis in COVID-19 Patients on Anticoagulation. J Thromb Haemostasis (2020) 18(10):2646–53. doi: 10.1111/jth.15043
301
ReevesEPBerginDAFitzgeraldSHayesEKeenanJHenryMet al. A Novel Neutrophil Derived Inflammatory Biomarker of Pulmonary Exacerbation in Cystic Fibrosis. J Cystic Fibrosis (2012) 11(2):100–7. doi: 10.1016/j.jcf.2011.09.010
302
Pinto-PlataVMMüllerovaHTosoJFFeudjo-TepieMSorianoJBVesseyRSet al. C-Reactive Protein in Patients With COPD, Control Smokers and Non-Smokers. Thorax (2006) 61:23–8. doi: 10.1136/thx.2005.042200
303
Pinto-PlataVTosoJLeeKParkDBilelloJMullerovaHet al. Profiling Serum Biomarkers in Patients With COPD: Associations With Clinical Parameters. Thorax (2007) 62:595–601. doi: 10.1136/thx.2006.064428
304
YanbaevaDGDentenerMASpruitMAHouwing-DuistermaatJJKotzDPassosVLet al. IL6 and CRP Haplotypes are Associated With COPD Risk and Systemic Inflammation: A Case-Control Study. BMC Med Genet (2009) 10:23. doi: 10.1186/1471-2350-10-23
305
GanWQManSFSenthilselvanASinDD. Association Between Chronic Obstructive Pulmonary Disease and Systemic Inflammation: A Systematic Review and a Meta-Analysis. Thorax (2004) 59:574–80. doi: 10.1136/thx.2003.019588
306
HurstJRDonaldsonGCPereraWRWilkinsonTMBilelloJAHaganGWet al. Use of Plasma Biomarkers at Exacerbation of Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2006) 174:867–74. doi: 10.1164/rccm.200604-506OC
307
DickensJAMillerBEEdwardsLDSilvermanEKLomasDATal-SingerRet al. COPD Association and Repeatability of Blood Biomarkers in the ECLIPSE Cohort. Respir Res (2011) 12:146. doi: 10.1186/1465-9921-12-146
308
EaganTMUelandTWagnerPDHardieJAMollnesTEDamåsJKet al. Systemic Inflammatory Markers in COPD: Results From the Bergen COPD Cohort Study. Eur Respir J (2010) 35:540–8. doi: 10.1183/09031936.00088209
309
EaganTMDamåsJKUelandTVoll-AanerudMMollnesTEHardieJAet al. Neutrophil Gelatinase-Associated Lipocalin: A Biomarker in COPD. Chest (2010) 138:888–95. doi: 10.1378/chest.09-2718
310
AaronSDVandemheenKLRamsayTZhangCAvnurZNikolchevaTet al. Multi Analyte Profiling and Variability of Inflammatory Markers in Blood and Induced Sputum in Patients With Stable COPD. Respir Res (2010) 11:41. doi: 10.1186/1465-9921-11-41
311
BernardAMarchandiseFXDepelchinSLauwerysRSibilleY. Clara Cell Protein in Serum and Bronchoalveolar Lavage. Eur Respir J (1992) 5:1231–8.
312
ShijuboNItohYYamaguchiTShibuyaYMoritaYHirasawaMet al. Serum and BAL Clara Cell 10 kDa Protein (CC10) Levels and CC10-Positive Bronchiolar Cells Are Decreased in Smokers. Eur Respir J (1997) 10:1108–14. doi: 10.1183/09031936.97.10051108
313
BraidoFRiccioAMGuerraLGamaleroCZolezziATarantiniFet al. Clara Cell 16 Protein in COPD Sputum: A Marker of Small Airways Damage? Respir Med (2007) 101:2119–24. doi: 10.1016/j.rmed.2007.05.023
314
LomasDASilvermanEKEdwardsLDMillerBECoxsonHOTal-SingerR. Evaluation of Serum CC-16 as a Biomarker for COPD in the ECLIPSE Cohort. Thorax (2008) 63:1058–63. doi: 10.1136/thx.2008.102574
315
SinDDMillerBEDuvoixAManSFZhangXSilvermanEKet al. Serum PARC/CCL-18 Concentrations and Health Outcomes in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2011) 183:1187–92. doi: 10.1164/rccm.201008-1220OC
316
CockayneDAChengDTWaschkiBSridharSRavindranPHiltonHet al. Systemic Biomarkers of Neutrophilic Inflammation, Tissue Injury and Repair in COPD Patients With Differing Levels of Disease Severity. PloS One (2012) 7(6):e38629. doi: 10.1371/journal.pone.0038629
317
ThulbornSJMistryVBrightlingCEMoffittKLRibeiroDBafadhelM. Neutrophil Elastase as a Biomarker for Bacterial Infection in COPD. Respir Res (2019) 20:170. doi: 10.1186/s12931-019-1145-4
318
SchuetzPSuter-WidmerIChaudriAChrist-CrainMZimmerliWMuellerBet al. Prognostic Value of Procalcitonin in Community-Acquired Pneumonia. Eur Respir J (2011) 37:384–92. doi: 10.1183/09031936.00035610
319
HuangDTAngusDCKellumJAPughNAWeissfeldLAStruckJet al. Midregional Proadrenomedullin as a Prognostic Tool in Community-Acquired Pneumonia. Chest (2009) 136:823–31. doi: 10.1378/chest.08-1981
320
KolditzMHalankMSchulte-HubbertBBergmannSAlbrechtSHöffkenG. Copeptin Predicts Clinical Deterioration and Persistent Instability in Community-Acquired Pneumonia. Respir Med (2012) 106:1320–8. doi: 10.1016/j.rmed.2012.06.008
321
KrügerSEwigSGiersdorfSHartmannOSuttorpNWelteT. Cardiovascular and Inflammatory Biomarkers to Predict Short- and Long-Term Survival in Community-Acquired Pneumonia: Results From the German Competence Network, CAPNETZ. Am J Respir Crit Care Med (2010) 182:1426–34. doi: 10.1164/rccm.201003-0415OC
322
CavallazziREl-KershKAbu-AtherahESinghSLokeYKWiemkenTet al. Midregional Proadrenomedullin for Prognosis in Community-Acquired Pneumonia: A Systematic Review. Respir Med (2014) 108:1569–80. doi: 10.1016/j.rmed.2014.09.018
323
SiljanWWHolterJCMichelsenAENymoSHLauritzenTOppenKet al. Inflammatory Biomarkers Are Associated With Aetiology and Predict Outcomes in Community-Acquired Pneumonia: Results of a 5-Year Follow-Up Cohort Study. ERJ Open Res (2019) 5(1):00014–2019. doi: 10.1183/23120541.00014-2019
324
WareLBMatthayMAParsonsPEThompsonBTJanuzziJLEisnerMD. Pathogenetic and Prognostic Significance of Altered Coagulation and Fibrinolysis in Acute Lung Injury/Acute Respiratory Distress Syndrome. Crit Care Med (2007) 35(8):1821–8. doi: 10.1097/01.CCM.0000275386.95968.5F
325
IshizakaAMatsudaTAlbertineKHKohHTasakaSHasegawaNet al. Elevation of KL-6, a Lung Epithelial Cell Marker in Plasma and Epithelial Lining Fluid in Acute Respiratory Distress Syndrome. Am J Physiol Lung Cell Mol Physiol (2004) 286:L1088–94. doi: 10.1152/ajplung.00420.2002
326
YeZAiXLiaoZYouCChengY. The Prognostic Values of Neutrophil to Lymphocyte Ratio for Outcomes in Chronic Obstructive Pulmonary Disease. Medicine (2019) 98:e16371. doi: 10.1097/MD.0000000000016371
327
LiXLiuCMaoZXiaoMWangLQiSet al. Predictive Values of Neutrophil-to-Lymphocyte Ratio on Disease Severity and Mortality in COVID-19 Patients: A Systematic Review and Meta-Analysis. Crit Care (2020) 24:647. doi: 10.1186/s13054-020-03374-8
328
LeeSJLeeHRLeeTWJuSLimSGoSIet al. Usefulness of Neutrophil to Lymphocyte Ratio in Patients With Chronic Obstructive Pulmonary Disease: A Prospective Observational Study. Korean J Intern Med (2016) 31:891–8. doi: 10.3904/kjim.2015.084
329
TaylanMDemirMKayaHSelimoglu SenHAbakayOCarkanatAİet al. Alterations of the Neutrophil-Lymphocyte Ratio During the Period of Stable and Acute Exacerbation of Chronic Obstructive Pulmonary Disease Patients. Clin Respir J (2017) 11:311–7. doi: 10.1111/crj.12336
330
HuangCXiaoLLuoHLZhuZMJ. Preoperative Neutrophil-to-Lymphocyte Ratio Combined With Serum CEA, CA19-9, CA125 and CA72-4 Levels in the Clinical Pathological Staging of Gastric Cancer-Based on Propensity Score Matching. Biol Regul Homeost Agents (2020) 34(3):1111–6. doi: 10.23812/19-458-L-46
331
MartinCBurdonPCBridgerGGutierrez-RamosJCWilliamsTJRankinSM. Chemokines Acting via CXCR2 and CXCR4 Control the Release of Neutrophils From the Bone Marrow and Their Return Following Senescence. Immunity (2003) 19(4):583–93. doi: 10.1016/S1074-7613(03)00263-2
332
MossRBMistrySJKonstanMWPilewskiJMKeremETal-SingerRet al. Safety and Early Treatment Effects of the CXCR2 Antagonist SB-656933 in Patients With Cystic Fibrosis. J Cystic Fibrosis (2013) 12(3):241–8. doi: 10.1016/j.jcf.2012.08.016
333
RennardSIDaleDCDonohueJFKanniessFMagnussenHSutherlandERet al. CXCR2 Antagonist MK-7123. A Phase 2 Proof-of-Concept Trial for Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2015) 191(9):1001–11. doi: 10.1164/rccm.201405-0992OC
334
O'ByrnePMMetevHPuuMRichterKKeenCUddinMet al. Efficacy and Safety of a CXCR2 Antagonist, AZD5069, in Patients With Uncontrolled Persistent Asthma: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Respir Med (2016) 4(10):797–806. doi: 10.1016/S2213-2600(16)30227-2
335
ThatcherTHMcHughNAEganRWChapmanRWHeyJATurnerCKet al. Role of CXCR2 in Cigarette Smoke-Induced Lung Inflammation. Am J Physiol Lung Cell Mol Physiol (2005) 289(2):L322–8, 289. doi: 10.1152/ajplung.00039.2005
336
LazaarALMillerBETabbererMYonchukJLeidyNAmberyCet al. Effect of the CXCR2 Antagonist Danirixin on Symptoms and Health Status in COPD. Eur Respir J (2018) 52:1801020. doi: 10.1183/13993003.01020-2018
337
BarwinskaDOueiniHPoirierCAlbrechtMEBogatchevaNVJusticeMJet al. AMD3100 Ameliorates Cigarette Smoke-Induced Emphysema-Like Manifestations in Mice. Am J Physiol Lung Cell Mol Physiol (2018) 315:L382–6. doi: 10.1152/ajplung.00185.2018
338
HenrotPPrevelRBergerPDupinI. Chemokines in COPD: From Implication to Therapeutic Use. Int J Mol Sci (2019) 20(11):2785. doi: 10.3390/ijms20112785
Summary
Keywords
lung, innate immune response, neutrophils, chemokines, pattern recognition receptors, biomarkers
Citation
Effah CY, Drokow EK, Agboyibor C, Ding L, He S, Liu S, Akorli SY, Nuamah E, Sun T, Zhou X, Liu H, Xu Z, Feng F, Wu Y and Zhang X (2021) Neutrophil-Dependent Immunity During Pulmonary Infections and Inflammations. Front. Immunol. 12:689866. doi: 10.3389/fimmu.2021.689866
Received
01 April 2021
Accepted
23 September 2021
Published
19 October 2021
Volume
12 - 2021
Edited by
Jean Sylvia Marshall, Dalhousie University, Canada
Reviewed by
Madhumita Chatterjee, Tübingen University Hospital, Germany; Mohib Uddin, AstraZeneca, Sweden; Kymberly Mae Gowdy, The Ohio State University, United States; Pio Conti, University of Studies G. d’Annunzio Chieti and Pescara, Italy
Updates
Copyright
© 2021 Effah, Drokow, Agboyibor, Ding, He, Liu, Akorli, Nuamah, Sun, Zhou, Liu, Xu, Feng, Wu and Zhang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongjun Wu, wuyongjun@zzu.edu.cn; Xiaoju Zhang, 15837101166@163.com
This article was submitted to Molecular Innate Immunity, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.