 Ruth Pia Duecker
Ruth Pia Duecker Lucia Gronau1
Lucia Gronau1 Patrick C. Baer
Patrick C. Baer Stefan Zielen
Stefan Zielen Ralf Schubert
Ralf Schubert- 1Division for Allergy, Pneumology and Cystic Fibrosis, Department for Children and Adolescence, Goethe-University, Frankfurt am Main, Germany
- 2Division of Nephrology, Department of Internal Medicine III, Goethe-University, Frankfurt am Main, Germany
Hematopoietic stem cell transplantation (HSCT) has been proposed as a promising therapeutic opportunity to improve immunity and prevent hematologic malignancies in Ataxia-telangiectasia (A-T). However, experience in the transplantation strategy for A-T patients is still scarce. The aim of this study was to investigate whether different approaches of HSCT are feasible in regard to graft versus host response and sufficient concerning functional immune reconstitution. Atm-deficient mice were treated with a clinically relevant non-myeloablative host-conditioning regimen and transplanted with CD90.2-depleted, green fluorescent protein (GFP)-expressing, and ataxia telangiectasia mutated (ATM)-competent bone marrow donor cells in a syngeneic, haploidentical or allogeneic setting. Like syngeneic HSCT, haploidentical HSCT, but not allogeneic HSCT extended the lifespan of Atm-deficient mice through the reduction of thymic tumors and normalized T-cell numbers. Donor-derived splenocytes isolated from transplanted Atm-deficient mice filled the gap of cell loss in the naïve T-cell population and raised CD4 cell functionality up to wild-type level. Interestingly, HSCT using heterozygous donor cells let to a significantly improved survival of Atm-deficient mice and increased CD4 cell numbers as well as CD4 cell functionality equivalent to HSCT using with wild-type donor cells. Our data provided evidence that haploidentical HSCT could be a feasible strategy for A-T, possibly even if the donor is heterozygous for ATM. However, this basic research cannot substitute any research in humans.
Introduction
Ataxia-telangiectasia (A-T) is a genomic instability syndrome with progressive neurological deterioration, oculocutaneous telangiectasia, high risk for malignancy, recurrent pulmonary infections, and variable immunodeficiency (1). Immunodeficiency affects numbers as well as functionality of the B and T cell repertoire (2, 3). A-T is based on mutations in the ataxia telangiectasia-mutated (ATM) gene, which is located on chromosome 11q22.3 and regulates complex signaling cascades including DNA damage recognition and repair responses (4–6). Besides lung failure, an elevated cancer susceptibility due to genomic instability and impaired DNA damage responses accounts for morbidity and mortality in A-T (7–10). Occurrence of chromosomal aberrations in T and B lymphocytes are responsible for lymphoid tumors in approximately 10-15% of the patients, making A-T to the disease with the highest malignancy in childhood (11).
Up to date no curative therapy for A-T exist (12). One encouraging approach represents hematopoietic stem cell transplantation (HSCT) to ameliorate the immunodeficiency and prevent the development of leukemia and lymphomas. In Atm-deficient mice, which typically die at the age of 3−6 months, the inhibition of tumorigenesis has been impressively demonstrated (13). Moreover, it has been shown that HSCT restores T-cell (sub)populations in Atm-deficient mice as well as in patients with Ataxia-telangiectasia (14, 15). However, experience in transplantation of A-T patients is scarce and, therefore, using the right conditioning regime and choosing the right donor are of utmost importance. A potential donor such as a HLA-identical sibling is only applicable for a minority of the patients (16). For patients who need a stem cell transplant but do not have an HLA-matched related or unrelated donor, recent medical advances have made the use of a partially matched or haploidentical related donor possible. Thus, haploidentical HSCT increases the chance of finding a donor since everyone has at least one haploidentical relative such as a parent, sibling, or child (17).
The present study aimed to compare different transplantation settings including haploidentical HCST regarding inhibition of tumor development, GvHD and functional immune reestablishment in Atm-deficient mice. In addition, the study also focused on donor derived ATM heterogeneity in regard of haploidentical donors such as biological parents or siblings.
Material and Methods
Animals
In this proof of principle experiments, we used a total of 68 Atm-deficient, 56 wildtype mice and 9 heterozygote mice. For HSCT experiments mice were randomly assigned to the treatment groups and mice where HSCT failed were excluded from the experiments.
The animal studies were performed according to the protocols approved by the German Animal Subjects Committee (Gen. Nr. FK/1034). Atm-deficient mice (Atmtm1Awb; 8 to 10 weeks old), in a 129SvEv background, were housed in IVC cages on a standard light and dark cycle of 12 h with access to food and water ad libitum until harvest. Mice were checked 3 times per week and sacrificed when clinical signs like gradual weight loss indicating emerging thymic lymphomas. Weight was taken twice a week during the observation period.
One hundred percent of our untreated Atm-deficient mice die at the age of 3-6 months due to the development of massive thymic lymphoblastic lymphomas. These tumors grow rapidly causing death from compression of the heart and lungs. As a result, these mice experience significant weight loss (>20%) and signs of disturbed general condition due to an insufficient food and water intake in a very short time and then mice are euthanized to avoid any further animal suffering.
For the HSCT experiments, 11 recipient mice and 9 donor mice (129S6/SvEvGFP+) for the syngeneic transplantation setting, 12 recipient mice and 8 donor mice (F1 generation of C57BL/6J-129S6/SvEv GFP+) for haplo-HSCT, 13 recipients and 8 donor mice (C57BL/6J GFP+) for allo-HSCT, and 7 Atm-deficient mice and 4 heterozygous donor mice for the het-HSCT setting were used.
Mixed Lymphocyte Reaction
For the mixed lymphocyte reaction (MLR), dendritic cells (DC) isolated from bone marrow were obtained from Atm+/+ and Atm-/- mice and were pretreated with Mitomycin C (MMC, 50 µg/mL, Sigma-Aldrich, Steinheim, Germany), preventing the cells to replicate. Spleen cells from syngeneic, haploidentical and allogeneic donor mice were used as responder cells. For the syngeneic stimulation, spleen cells from GFP+ ATM-competent donor mice were used. As haploidentical donor cells spleen cells of the F1-generation of mated 129S6/SvEv-Atm(tm1)Awb/F and C57BL/6Tg(ACTb-EGFP)10sb/J mice were used. Allogeneic splenocytes were taken from RjOrl : Swiss mice (Janvier Labs). The isolation and culture of DCs from progenitor cells of the bone marrow of Atm-deficient and wild-type mice was performed slightly modified accordingly to Lutz et al. (18). Additional detail on the method is provided in an online data supplement. Briefly, femur and tibiae from 6 – 8 weeks old wild-type and Atm-deficient mice were removed and purified from the surrounding muscle tissue by rubbing carefully with kim wipes. Thereafter, intact bones were left in 70% ethanol for 2 - 5 min for disinfection and washed with RPMI1640 (Gibco, Carlsbad, USA). Bones were cut open with scissors and the marrow flushed with culture medium [RPMI1640, 100 U/mL penicillin-streptomycin, 2 mM L-Glutamine, 10% heat-inactivated FBS (Sigma, Steinheim, Germany)] using a syringe with a 0.5 mm diameter needle (25 G). The BM cells were separated by pipetting and passed through a 70 μm cell strainer. The cell strainer was rinsed with PBS three times and bone marrow cells were centrifuged (600 x g, 10 minutes, at room temperature). Subsequently, the cells were taken up in 10 mL culture medium containing 20 ng/mL recombinant mouse granulocyte macrophage-colony stimulating factor (rmGM-CSF, PeproTech, Hamburg, Germany), and cell density was adjusted to 2 x 106 per 9.5 cm2 dish and cultured at 37°C and 5% CO2 in the incubator. After 10 days of culture, the dendritic cells were harvested, labeled with 25 µmol carboxyfluoresceinsuccimidylester (CFSE, Thermo Fisher Scientific, Langenselbold, Germany) and tracked for successful proliferative precursors, according to manufacturer’s protocol, using the FACSVerse flow cytometer (Becton Dickinson, San Jose, CA, USA). The data were evaluated using FACSSuite software.
HSCT in Atm-Deficient Mice
Atm-deficient mice received 0.125 mg/mL anti-CD4 (clone GK1.5) and 0.125 mg/mL anti-CD8 (clone 53 – 6.7) monoclonal antibody (Sigma, Steinheim, Germany) 7 days before bone marrow transplantation (BMT) and a second dose of each antibody in combination with cyclophosphamide (CP, 80 ng/mL, Endoxan, Baxter, Unterschleißheim, Germany) one day before BMT as non-myeloablative conditioning (Figure 2A). Bone marrow donor cells (BMDC) from 129S6/SvEv GFP-transfected wild-type mice (syngeneic) or from mice of the F1 generation of 129S6/SvEv wild-type mice and C57BL/6 mice (haploidentical), or C57BL/6 mice (allogeneic) were depleted using CD90.2 MicroBeads (CD90.2 MicroBeads, Miltenyi Biotec, Bergisch-Gladbach, Germany) according to manufacturer’s protocol to minimize GvH reactions. After conditioning 5 x 106 CD90.2- bone marrow donor cells derived from 129S6/SvEv GFP-transfected wild-type mice were injected intravenously into recipient mice (19, 20).
GvHD Scoring
Some recipient mice began to show clinical signs of GvHD between 4 to 10 days after HSCT. The GvHD scoring system, developed by Cooke et al. were modified and used to quantitate disease progression. Following parameters were scored: fur texture, weight loss, skin integrity, activity, and posture (0 if absent or 1 if present) (21). Mice were scored twice per week and euthanized when weight loss was >20% or at any sign of disturbed general condition.
Bone Marrow T-Cell Depletion
Bone marrow (BM) cells were collected by flushing femurs and tibias from 129S6/SvEv GFP-transfected wild-type mice (syngeneic) or from mice of the F1 generation of 129S6/SvEv wild-type mice and C57BL/6 mice (haploidentical), or C57BL/6 mice (allogeneic) donor mice into MACS buffer (1× PBS, 2 mM EDTA, 0.5% bovine serum albumin). After red blood cell lysis, BM cells were incubated with CD90.2 microbeads for 15 minutes on ice, washed once, and then placed on LS columns (Miltenyi Biotech GmbH, Germany) and magnetically separated using a MiniMACS (Miltenyi Biotech GmbH, Germany). The remaining CD90.2-positive cell fraction was routinely less than 5% of BM cells. Cells were washed and then resuspended in injection medium (DMEM (Gibco, Carlsbad, USA), 10% fetal bovine serum (FBS, Sigma-Aldrich, Steinheim, Germany), 100 U/mL penicillin-streptomycin (Gibco, Carlsbad, USA) prior to transplantation.
Tracking the Migration of GFP+ Donor Cells in Recipient Atm-Deficient Mice After SCT
Two hundred µL of blood was collected by tapping the facial vein of the mice 6, 12 and 24 weeks after HSCT. EDTA containing whole blood samples were first blocked with anti-CD16/32 antibody (Becton Dickinson, Heidelberg, Germany) and then surface stained for anti-CD3-BV421, anti-CD4-PerCp and CD8-PE-Cy7 (BD, Heidelberg, Germany). After staining, erythrocytes were lyzed with FACS lysing solution (BD, Heidelberg, Germany). For analyzing, 10.000 events were acquired using a FACSVerse flow cytometer (BD, Heidelberg, Germany). The data were analyzed using the FACSuite software.
T-cell Activation and Signaling In Vitro
The expression of CD69 following T-cell activation via TCR/CD28 stimulation was evaluated in splenocytes isolated from Atm-deficient mice 12 weeks after syngeneic HSCT and compared to cells from untreated Atm-deficient mice and wild-type mice. Further, the p-Erk intracellular signaling pathway after T-cells activation was investigated in vitro using the BD™ Phosflow Protocol for mouse splenocytes. Additional detail on the method is provided in an online data supplement. Briefly, the splenocytes were stimulated with either purified NA/LE Hamster anti-mouse CD3ϵ ([1 µg/mL], Clone 145-2C11, BD, Heidelberg, Germany) and purified NA/LE Hamster anti-mouse CD28 ([1 µg/mL], Clone 37.51, BD, Heidelberg, Germany) antibody or Concanavalin A (ConA [5 µg/mL], Merck, Darmstadt, Germany). The cells were incubated for 4 h at 37°C. Afterwards, the cells were blocked with CD16/32 (Biolegend, Koblenz, Germany) for 10 min and then surface stained using αCD3-V450 (Clone 17A2, BD, Heidelberg, Germany), αCD4-PerCP-Cy5.5 (Clone RM4-5, BD, Heidelberg, Germany), αCD8-PE (Clone 53-6.7, BD, Heidelberg, Germany), αCD19-APC (Clone 1D3, BD, Heidelberg, Germany), and αCD69-Alexa Fluor 647 (Clone H1.2F3, Biolegend, Koblenz, Germany) for 20 min in the dark. After washing the cells with PBS, they were resuspended in 300 µL PBS and 10,000 events were acquired by flow cytometry.
For intracellular detection of phosphorylated proteins, splenocytes were stimulated in a 37°C water bath via TCR/CD28 for 5 min with αCD3 and αCD28 antibodies. As a positive control served PBu2/Ionomycin which incubated for 15 min. The reaction was stopped by adding cytofix buffer (BD, Heidelberg, Germany) for 10 min at 37°C. After centrifugation, cells were washed with stain buffer (PBS + 2% FBS). Then 1 mL Perm III buffer (BD, Heidelberg, Germany) was added to the cells for 30 min on ice. After 2 washing steps, the cells were resuspended in stain buffer and blocked for 10 min with purified CD16/CD32 (Biolegend, Koblenz, Germany). Then the cells were stained with αCD3-V450, αCD4-PerCP-Cy5.5, and p-Erk-Alexa Fluor 647 (Clone 6B8B69, Biolegend, Koblenz, Germany) for 1 h and measured using a FACSVerse. The data were evaluated using the FACSuite software.
Statistics
Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA). The survival rate was used to generate Kaplan-Meier survival curves which were compared using the log-rank (Mantel-Cox) test. Values are presented as the means (± SEM) and were analyzed using paired or unpaired t-tests or the corresponding Mann–Whitney U-test or a Wilcoxon–Mann–Whitney test. For multiple comparisons, we used Kruskal–Wallis testing. Survival curves comparing the effect of HSCT on survival of Atm-deficient mice were calculated by the Kaplan–Meier method. The following symbols indicate significant P-values: ∗p <0.05, ∗∗p <0.01 and ∗∗∗p <0.001.
Results
T-Cell Proliferation in the MLR
MLR was performed to test T-cell proliferation in a syngeneic, haploidentical and allogeneic setting in vitro. The MLR led to an elevated proliferative response of allogeneic donor T-cells compared to the proliferation of syngeneic and haploidentical donor cells (Figure 1A). The response of syngeneic compared to allogeneic donor T-cells was lower in dendritic cells derived from Atm-deficient mice compared to those from wild-type controls (Atm+/+: 2.99 ± 0.58 fold proliferation, Atm-/-: 1.76 ± 0.27 fold proliferation, p <0.001, Figure 1B).
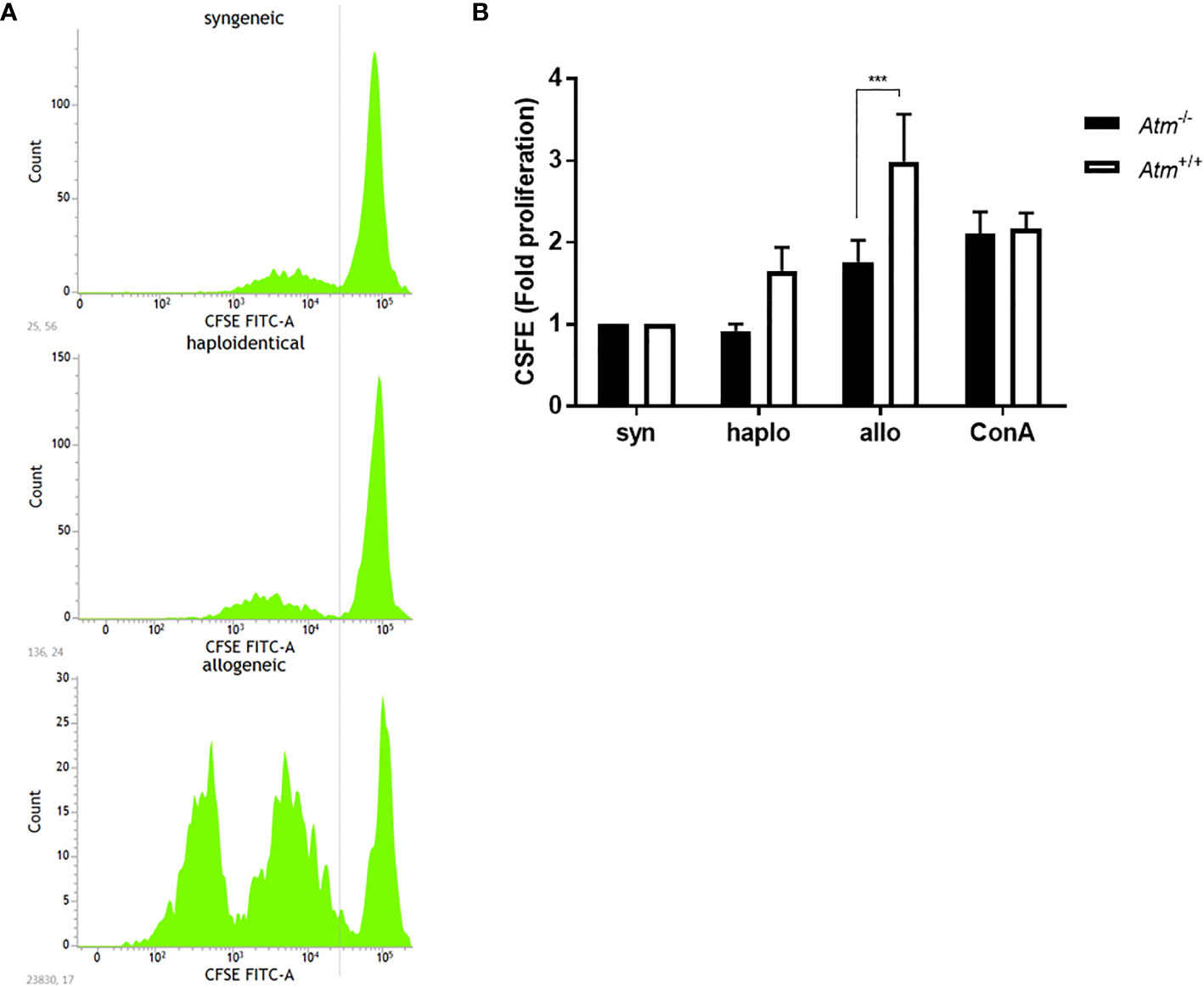
Figure 1 Proliferation of DCs in Atm-/- and Atm+/+ mice following stimulation with syngeneic, haploidentical and allogeneic T-cells in vitro. (A) Representative CFSE intensity histograms showing the mean level of the CFSE expression per generation of T-cells isolated from spleens of syngeneic, haploidentical and allogeneic donor mice. (B) Cell proliferation of T-cells isolated from spleens of syngeneic, haploidentical and allogeneic donor mice and cocultured with DCs isolated from Atm-/- and Atm+/+ mice (n = 8) was followed for 4 days using the CFSE cell proliferation kit. Data were normalized to the syngeneic T-cells and compared to haploidentical and allogeneic T-cell proliferative capacity. Data are presented as mean ± SEM. ***p < 0.001. CFSE, carboxy-fluorescein diacetate succinimidyl ester; DC, dendritic cell.
Survival, Body Weight and GvHD Status After HSCT
To address the different approaches of HSCT, Atm-deficient mice received hematopoietic BMDCs from syngeneic, haploidentical and allogeneic donor mice (Figure 2A). All transplantation settings significantly reduced the mortality rate and inhibited the tumor development in Atm-deficient compared to untreated Atm-deficient mice (Figures 2B, C). The survival rate was higher in the syngeneic (p <0.05) and haploidentical (p =0.058) HSCT compared to allogeneic HSCT.
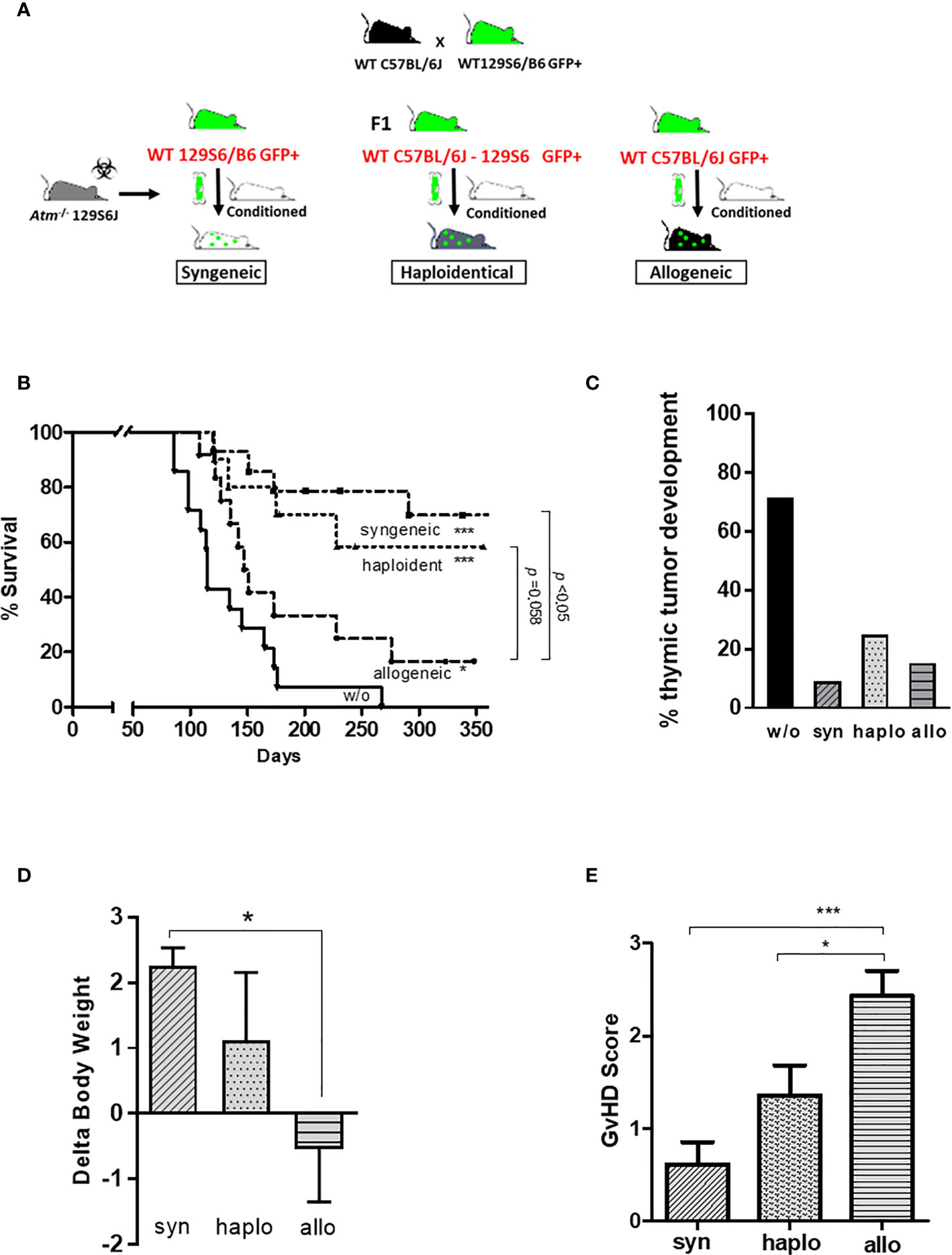
Figure 2 Syngeneic, haploidentical and allogeneic HSCT in Atm-deficient mice. (A) Atm-deficient recipients received a nonmyeloablative conditioning regimen consisting of anti-CD4 mAb and anti-CD8 mAb together with cyclophosphamide. Lin- selected BMDCs derived from 129/SvEv GFP-transgenic WT mice (syngeneic setting) or derived from mice of the F1 generation of 129/SvEv GFP-transgenic and C57BL/6 WT mice (haploidentical setting), or from WT C57BL/6 mice GFP-transgenic (allogeneic setting) was used for HSCT. (B) Survival curves showed as Kaplan-Meier plots derived from Atm-/- mice (w/o; n = 14) before and after syngeneic (n = 11), haploidentical (n = 12) and allogeneic HSCT (n = 13) (* compared to w/o). (C) Percentage of thymic tumor development in untreated Atm-/- (w/o) and syngeneic, haploidentical or syngeneic transplanted Atm-/- mice. (D) Changes in body weight of Atm-/- mice receiving either BMDCs originated from syngeneic, haploidentical or allogeneic donor mice on the day of the transplantation compared to the end of experiments. (E) GvHD score grading performed throughout the observation period after syngeneic, haploidentical and allogeneic HSCT in Atm-/- mice. Data are presented as mean ± SEM. *p < 0.05, ***p < 0.001. BMDC, bone marrow-derived cells; GFP, green fluorescent protein; GvHD, Graft versus Host Disease; HSCT, hematopoietic stem cell transplantation; mAb, monoclonal antibody; WT, wild-type.
Syngeneic and haploidentical HSCT improved the weight gain of Atm-deficient mice compared to the allogeneic transplantation setting in the indicated observation period (Figure 2D; syn: 2.26 ± 0.27 Δbody weight; haplo: 1.12 ± 1.04 Δbody weight; allo: -0.54 ± 0.81 Δbody weight, p <0.05). Atm-deficient mice receiving allogeneic BMDCs revealed a significant increased GvHD score than recipient mice receiving syngeneic and haploidentical BMDCs (syn: 0.62 ± 0.24 GvHD score, haplo: 1.36 ± 0.32 GvHD score, allo: 2.43 ± 0.27 GvHD score, p <0.05 and p <0.001; Figure 2E).
T-Cells and T-Cell Chimerism
In the peripheral blood the percentage of CD3/CD4+ helper T-cells and CD3/CD8+ cytotoxic T−cells was significantly reduced in Atm-deficient mice prior to HSCT (CD4+: Atm+/+ 45.34 ± 2.74%, Atm-/- 33.92 ± 1.46%; CD8+: Atm+/+ 13.98 ± 0.73%, Atm-/- 10.78 ± 0.75%, p <0.01; Figures 3A, B). Syngeneic and haploidentical HSCT increased the percentage of CD4+ helper T-cells significantly compared to untreated Atm-deficient mice (CD3+/CD4+: Atm-/- 33.92 ± 1.46%, Atm-/-syn 40.58 ± 1.13%, Atm-/-haplo 41.22 ± 3.73%, p <0.05; Figure 3A). Interestingly, the percentage if CD4+ helper cells was significantly diminished compared to untreated Atm-deficient mice (CD3+/CD4+: Atm-/- 33.92 ± 1.46%, Atm-/-allo 24.61 ± 3.88%, p <0.05; Figure 3A). Tracking of GFP+ cells revealed an increase in the CD4+ and CD8+ subpopulation 12 months after syngeneic and haploidentical HSCT. In contrast, the percentage of GFP+ T-cells was significantly lower after allogeneic HCST at all measured time points and showed no increase over time (Figures 3C, D).
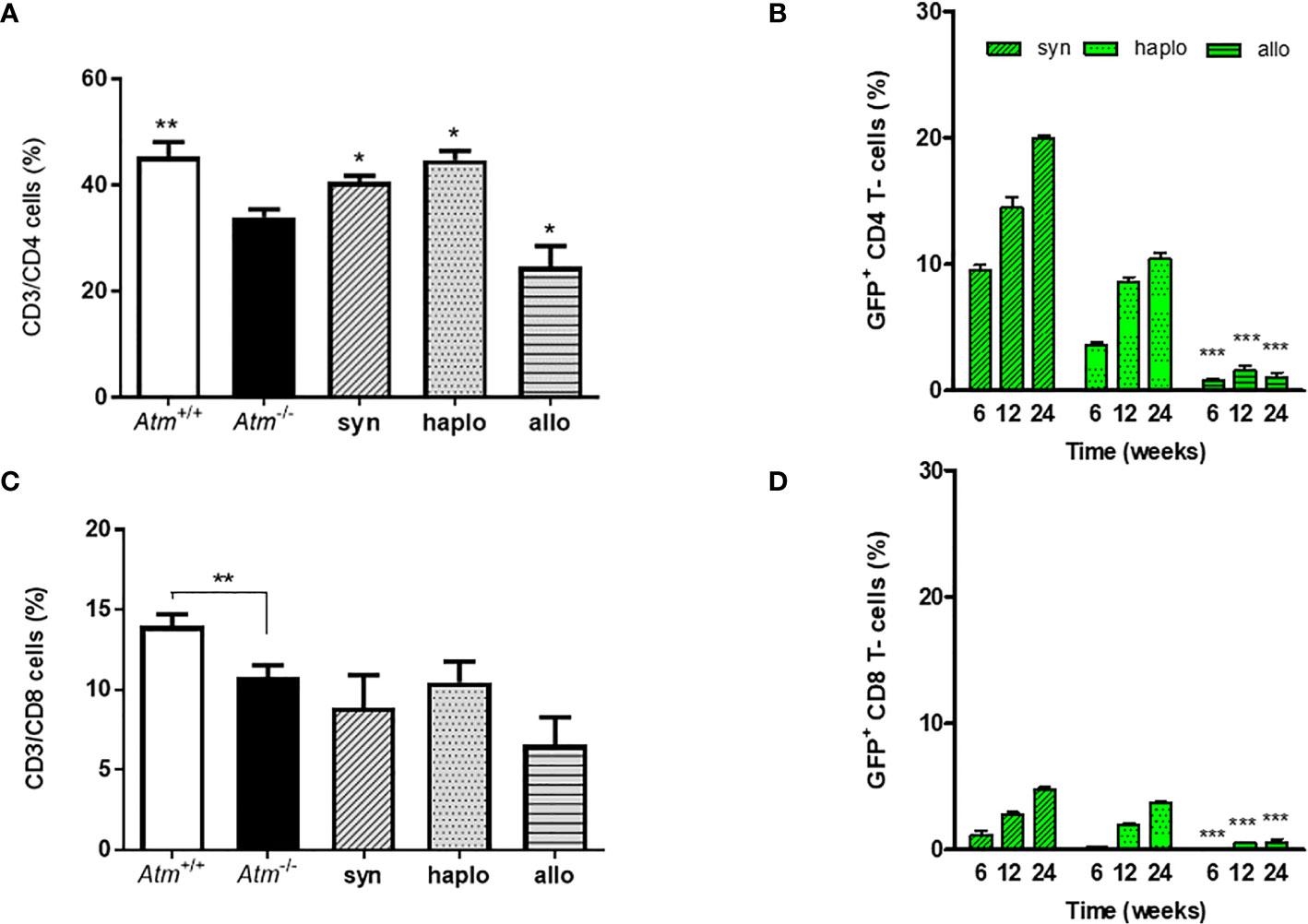
Figure 3 Phenotypic analysis of wild-type and Atm-deficient mice before HSCT and long-term engraftment of GFP expressing donor cells in Atm-deficient mice after syngeneic, haploidentical and allogeneic HSCT in peripheral blood samples. (A, B) Percentage of CD4+ and CD8+ lymphocytes in Atm-/- (n = 8) compared to Atm+/+ mice (n = 8) as well as in syngeneic (n = 4), haploidentical (n = 4) and allogeneic (n = 4) transplanted Atm-/- 24 weeks post transplantation. (C, D) Analysis of the engraftment of GFP+ cells derived from syngeneic, haploidentical and allogeneic donor mice in peripheral blood samples of Atm-/- recipient mice (n = ≥ 5) and surface stained for CD4+ and CD8+ at timepoints 6, 12 and 24 weeks post HSCT (* only compared to syngeneic). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. GFP, green fluorescent protein; HSCT, hematopoietic stem cell transplantation.
T-cell Activation of ATM-Competent Cells in Transplanted Atm-Deficient Mice
CD4 T-cell activation was evaluated by CD69 expression and activation of the MAPK/ERK pathway in isolated splenocytes derived from syngeneic transplanted Atm-deficient mice and compared to those from either wild-type or Atm-deficient mice. Activation via TCR/CD28 (Atm-/-: 3.87 ± 0.39 fold change, Atm-/-trans: 5.8 ± 0.7 fold change, p <0.01) as well as through the TCR cross-linker ConA (Atm-/-: 3.61 ± 0.36 fold change, Atm-/-syn: 6.03 ± 0.61 fold change, p <0.01) resulted in a significant increase in CD69 expression compared to the CD69 expression in untransplanted Atm-deficient mice (Figure 4A).
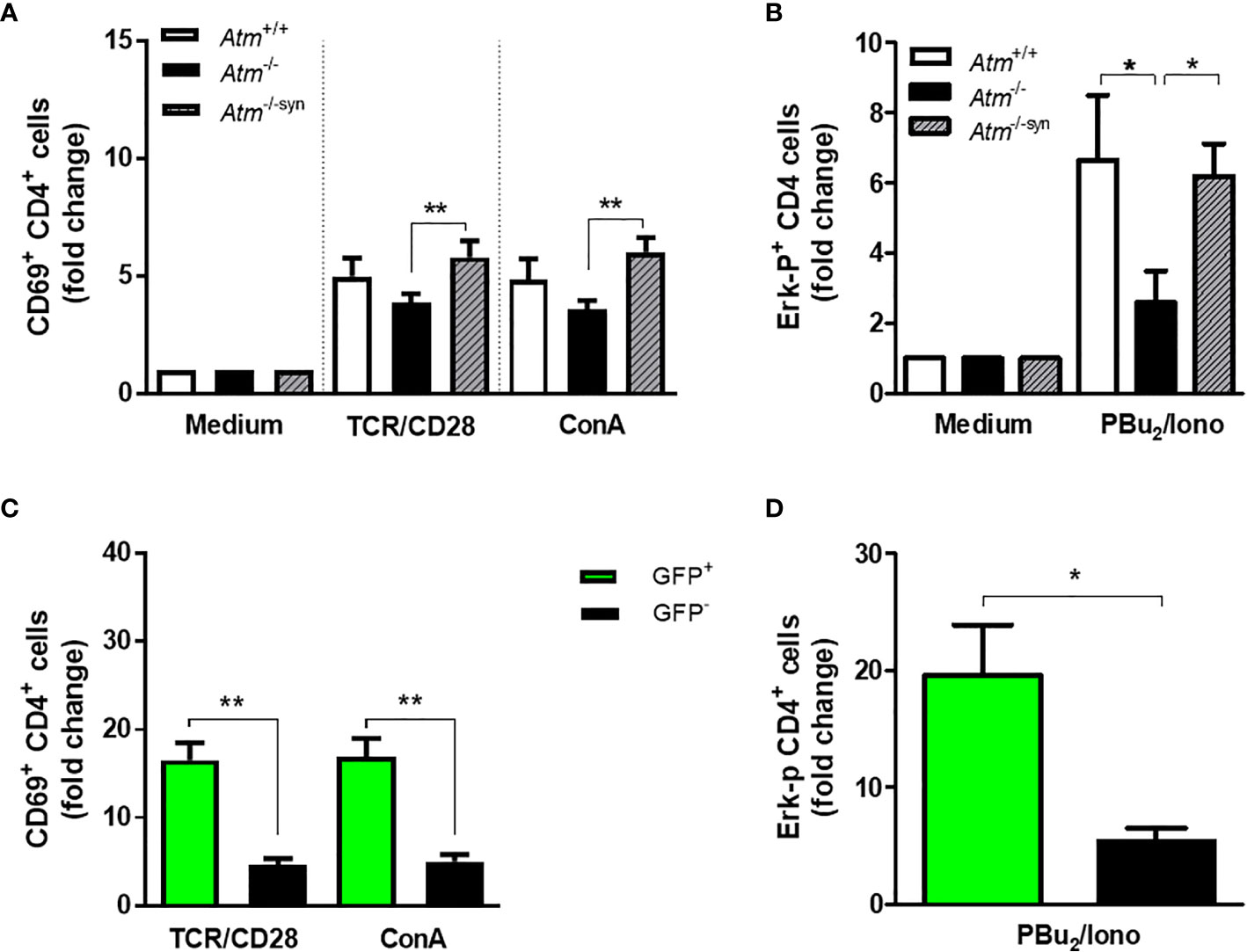
Figure 4 TCR-dependent T-cell activation and Expression of the phosphorylated (p)-Erk protein in Atm-deficient mice after syngeneic HSCT. (A) Flow cytometric analysis of T-cell activation in CD4+ splenocytes from Atm+/+ (n = 7), Atm-/- (n = 7) and syngeneic transplanted Atm-/- mice (n = 9, Atm-/-syn) 12 weeks post HSCT using CD69 as activation marker. The cells were stimulated either via TCR/CD28 or via ConA for 4 h in vitro and the data were normalized to the medium control. (B) Expression of the phosphorylated (p)-Erk protein in stimulated CD4+ splenocytes from Atm+/+ (n = 10), Atm-/- (n = 8) and Atm-/-syn (n = 9) 12 weeks after HSCT measured by flow cytometry. The cells were either stimulated via TCR/CD28 for 5 min and PBu2/Iono for 15 min in vitro. The data were normalized to the unstimulated medium control. (C) T-cell activation in GFP+ and GFP- CD4+ T-cells from Atm-/-syn mice stimulated either directly via αCD3 and αCD28 or nonspecifically by ConA for 4 h 12 weeks after HSCT. (D) Expression of the signaling protein Erk in GFP+ and GFP- CD4+ T-cells from Atm-/-syn stimulated via PBu2/Iono. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01. ConA, Concanavalin A; GFP, green fluorescent protein; HSCT, hematopoietic stem cell transplantation; TCR, T cell receptor.
CD69 expression was significantly higher in ATM-competent (GFP+) splenocytes than in Atm-deficient cells (GFP-) in TCR-stimulated (GFP-: 4.69 ± 0.71 fold change, GFP+: 16.54 ± 1.99 fold change, p <0.01) and ConA-stimulated cells (GFP-: 5.05 ± 0.82 fold change, GFP+: 16.95 ± 2.07 fold change, p <0.01; Figure 4C).
HSCT rescued the ERK-phosphorylation signal of CD4+ splenocytes in Atm-deficient mice (Figure 4B). The stimulation of CD4+ T-cells using PBu2/Ionomycin resulted in a 3.61-fold higher phosphorylation of Erk in transplanted Atm-deficient mice 12 weeks after HSCT compared to cells from untreated Atm-deficient mice (Atm-/-: 2.59 ± 0.9 fold change; Atm-/-syn: 6.2 ± 0.9 fold change; p <0.05; Figure 4B). The GFP+ CD4 T-cell population showed a significantly increased Erk-phosphorylation signal compared to the GFP- CD4+ T-cells (GFP -: 5.38 ± 1.16 fold change; GFP+:19.6 ± 4.32 fold change; p <0.05; Figure 4D).
Activation of Naïve and Memory CD4 T-Cells From Transplanted Atm-Deficient Mice
As previously described by our group, HSCT increases naïve CD4+/CD44dim T-cells rather than memory CD4+/CD44bright cells in Atm-deficient mice (14). To examine whether this fact is also visible on CD4 T-cell activation, CD4+/CD44dim and CD4+/CD44bright T-cell subpopulations derived from the spleen of Atm-deficient mice, wild-type mice and syngeneic transplanted Atm-deficient mice were either activated via TCR or with ConA evaluating CD69 expression using flow cytometry. Syngeneic HSCT significantly increased CD69 expression on CD4+/CD44dim (TCR/CD28: Atm-/-: 7.14 ± 0.71 fold change, Atm-/-syn: 11.59 ± 1.57 fold change, p <0.05; ConA: Atm-/-: 6.26 ± 0.70 fold change, Atm-/-syn: 11.05 ± 1.42 fold change, p <0.05), but only to a small extent on CD4+/CD44bright T cells (Figures 5A, C).
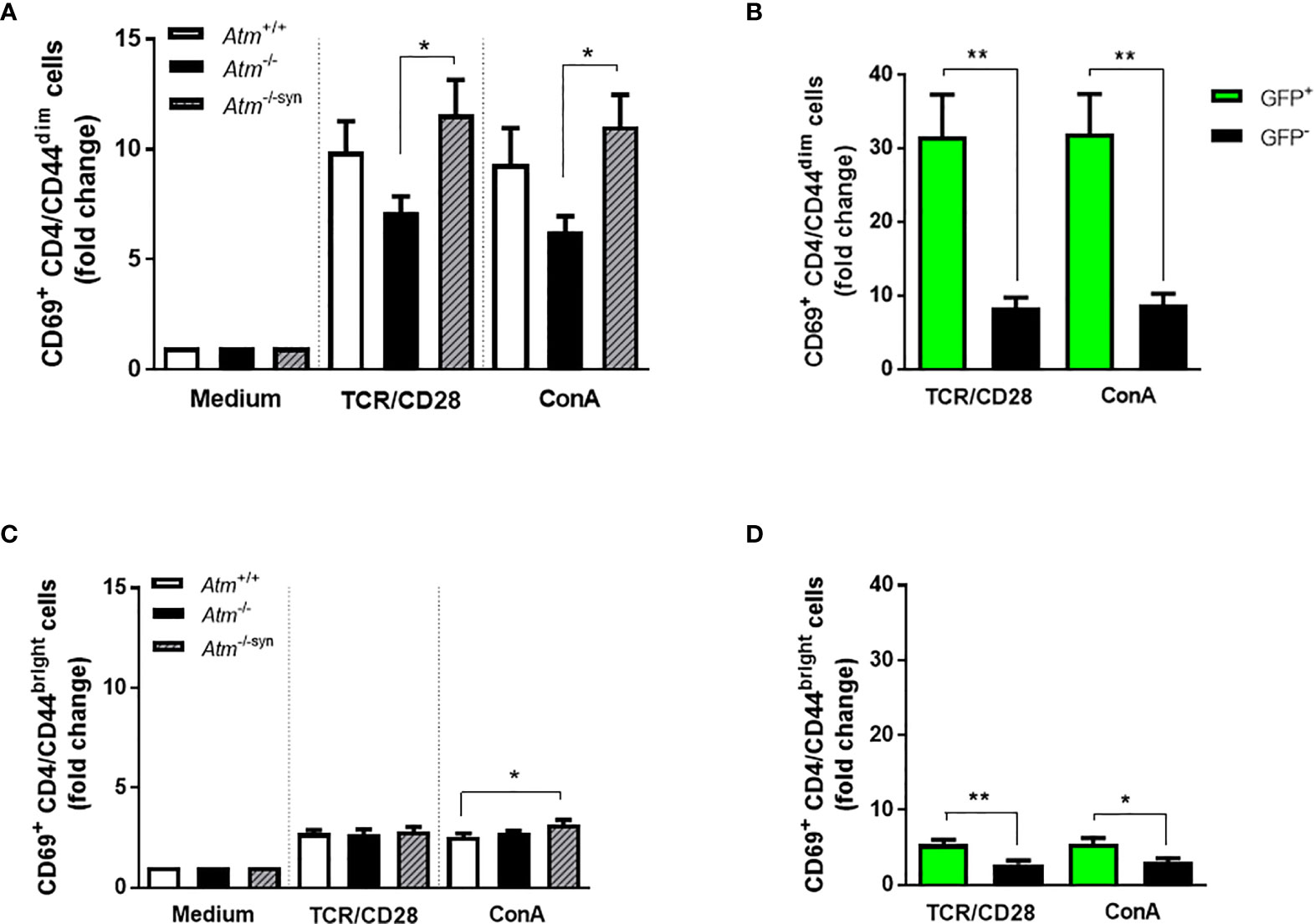
Figure 5 Activation of T-cell subpopulations in splenocytes of Atm-deficient mice in vitro using flow cytometry. (A) Expression of CD69+ in TCR- and ConA-stimulated CD4+/CD44dim splenocytes from untreated Atm+/+, Atm-/- and transplanted Atm-/-syn mice 12 weeks after HSCT (n > 6). The data were normalized to the unstimulated medium control. (B) Expression of CD69+ in GFP+ and GFP- CD4+/CD44dim splenocytes stimulated either via TCR or ConA isolated from Atm-/-syn mice 12 weeks after HSCT (n = 6). (C) T-cell activation in CD4+/CD44bright splenocytes stimulated with either αCD3 and αCD28 or ConA from Atm+/+, Atm-/-, and Atm-/-syn mice (12 weeks after HSCT) (n > 6). The data were normalized to the medium control. (D) Flow cytometric analysis of CD69+ expression GFP+ and GFP- CD4+/CD44bright splenocytes isolated from Atm-/-syn mice either stimulated via TCR or ConA 12 weeks after HSCT (n = 6). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01. ConA, Concanavalin A; GFP, green fluorescent protein; HSCT, hematopoietic stem cell transplantation; TCR, T cell receptor.
In both subpopulations, CD69 activation was significantly increased in GFP+ cells from Atm−deficient mice after syngeneic HSCT compared to the GFP- cell fraction after stimulation via TCR/CD28 (CD4+/CD44dim cells; GFP-: 8.35 ± 1.41 fold change, GFP+: 31.60 ± 5.69 fold change, p <0.01; CD4/CD44bright cells; GFP-: 2.79 ± 0.46 fold change, GFP+: 5.41 ± 0.66 fold change, p <0.01) and after ConA stimulation (CD4/CD44dim cells; GFP-: 8.73 ± 1.55 fold change, GFP+: 31.99 ± 5.36 fold change, p <0.01; CD4/CD44bright cells; GFP-: 3.01 ± 0.53 fold change, GFP+: 5.52 ± 0.76 fold change, p <0.05; Figures 5B, D).
Heterozygous Syngeneic HSCT in Atm-Deficient Mice
This study also examined the effects of grafts from asymptomatic heterozygous donors (Atm+/-) on survival, immunophenotyping and T lymphocyte activation in Atm-deficient mice. Interestingly, the syngeneic HSCT consisting of donors heterozygous for Atm significantly prolonged the lifespan of Atm-deficient mice as well as HSCT with homozygous Atm+/+ donors (Figure 6A). In addition, the HSCT using Atm+/- donors restored the CD4+ T-cell population significantly in Atm-deficient mice 24 weeks after HSCT compared to the T-cell status before HSCT (Atm+/- before: 27.03 ± 0.81%, after Atm+/-: 36.81 ± 2.34%; p <0.01; Figure 6B). The functional analysis of the CD4+ T-cell activation after either direct stimulation via TCR/CD28 or ConA in vitro revealed no differences between splenocytes isolated from heterozygous Atm-mice compared to homozygous Atm-mice (Figure 6C).
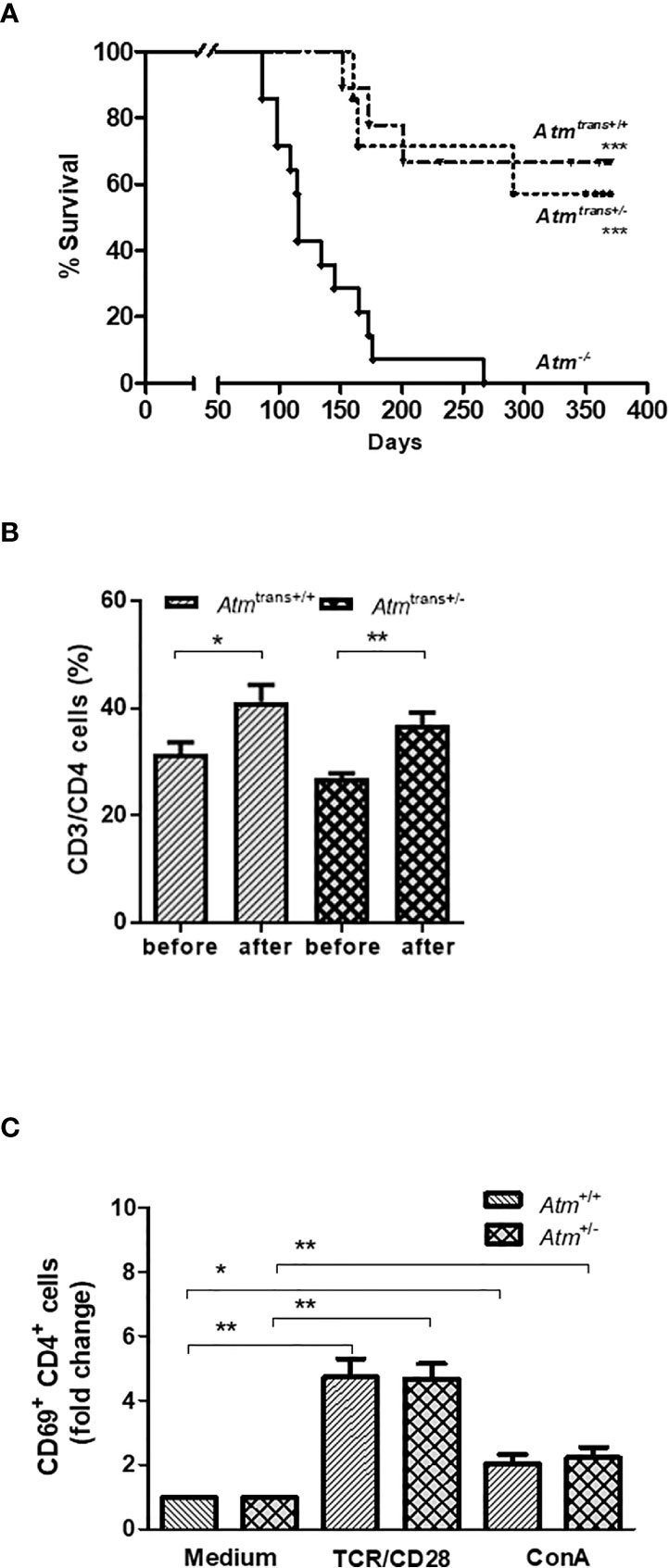
Figure 6 Effects of HSCT with BMDCs isolated from heterozygous (Atm+/-) donors on survival and chimerism in Atm-deficient mice and functional T-cell activation of splenocytes from Atm+/- mice in vitro. (A) Survival curves shown as Kaplan-Meier plot from Atm-/- mice after heterozygous (Atm+/-, n = 7) or homozygous (Atm+/+, n = 9) HSCT and Atm-/- without treatment (n = 14). (B) Phenotypic analysis of CD4+ T cells in peripheral blood taken from Atm-deficient mice before and 24 weeks after receiving BMDCs from Atm+/- or Atm+/+ donors (n ≥ 4) using flow cytometry. (C) Functional analysis of CD4+, CD4+/CD44dim (naïve) and CD4+/CD44bright (memory) CD69 T-cell activation in splenocytes isolated from Atm+/- and Atm+/+ mice stimulated with either TCR/CD28 or ConA for 4 h in vitro (n = 5). The data were normalized to the medium control. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01. ConA, Concanavalin A; GFP, green fluorescent protein; HSCT, hematopoietic stem cell transplantation; TCR, T cell receptor.
Discussion
A-T with its multiple genetic defects results in disruption of DNA repair processes leading to T−cell lymphopenia and impaired antibody production due to a V(D)J recombination defect during lymphocyte maturation (2, 22). This recombination defect is responsible for the immunodeficiency and the high risk of cancer in the immune cell compartment (23). As currently no curative therapy for A-T exists, new treatment strategies are necessary. HSCT, as it is carried out in other genomic instability syndromes as Fanconi anemia or Nijmegen Breakage Syndrome, could be a promising approach, although it is controversially discussed as the survival rate in patients after transplantation is only 25% (24–27). Conditioning is a critical factor in the clinical setting of SCT in A-T. The hypersensitivity to ionizing radiation and radiometric drugs results in a high risk of mortality. Therefore, the selection of a non-myeloablative condition is inevitable before considering SCT in A-T patients. In this regard it is worth to note that potential neurotoxic effects of busulfan-based protocols in patients with underlying neurodegenerative conditions must viewed with caution and should be generally avoided (15, 28, 29). In these patients, the suitability for SCT is controversial and the benefits of SCT are difficult to verify in case reports. The indication should be made cautiously and considered in each individual case. Nevertheless, SCT may be beneficial to improve immunodeficiency and control the risk of lymphoid malignancies and basic research may have the potential to identify some patients as beneficiaries of SCT.
Regarding patient selection, A-T patients with recurrent infections, severe manifestations of immunodeficiency and children with a high risk of lymphoid malignancies are the most likely candidates to benefit from SCT therapy, based on reported data.
According to the literature, HSCT in A-T is not able to ameliorate neurological symptoms, but researchers state cautiously optimistic that the patients exhibit a milder progression of ataxic symptoms and slowed neurologic deterioration (15, 28, 29).
Allogeneic HSCT often results in a severe GvHD, which is the leading complication and cause of non-relapse mortality in the patients (17). Recent clinical progress has been made in the use of transplants from partially matched or haploidentical related donors for patients who are in need of a stem cell transplant but do not have a HLA-matched related or unrelated donor (17, 30). Thus, over the last recent years the use of haploidentical donors has increased as alternative of allo-HSCT since, advantageously haploidentical donors are available for 95% of the patients (17, 31–33). In A-T, haploidentical HSCT has been successfully performed in two patients in Frankfurt who received allo-HSCT from a matched sibling donor (14, 15). However, experience in the transplantation strategy for A-T patients is still scarce (29).
The aim of the present study was to investigate the effects of different HSCT strategies on survival and functional reconstitution of the immune system in Atm-deficient mice. In recent years, we and others were able to show that HSCT significantly rescues the T-cell department and prevents the development of thymic tumors in Atm-deficient recipient mice (13, 14, 19, 20). However, all experiments in Atm-deficient mice were performed in a syngeneic setting. In this study, we compared haploidentical and allogeneic HSCT to the syngeneic setting in these mice. Usually, Atm-deficient mice die from thymic lymphomas at the age of 3-6 months, but however, bone marrow transplantation with ATM-competent cells leads to a stable chimerism and prevents tumorigenesis and prolongs the lifespan of the animals significantly (20). As one might expect, our present study demonstrated that allo-HSCT was associated with an increased death rate compared to the syngeneic setting. In fact, allogeneic transplanted mice showed a reduction in tumor expression, but the mice exhibited significant weight loss (>20%), showed a higher incidence of skin alterations compared to their syngeneic or haploidentical transplanted littermates and exhibited an impaired hematopoiesis due to graft failure (34, 35).
Further, our in vitro MLR experiments revealed that allogeneic-derived ATM-competent T−cells show a lower immunological proliferative activity on Atm-deficient DCs compared to wild-type DCs assuming a low defensive reaction. In contrast, like syngeneic HSCT, haploidentical HSCT significantly extended the lifespan of Atm-deficient mice through the reduction of thymic tumors. Inhibited tumorigenesis and prolonged survival of the Atm-deficient mice in the haploidentical setting is in line with the finding that haploidentical HSCT is a valuable alternative for patients with hematological disorders lacking a HLA-matched unrelated donor (36, 37). Survival was accompanied by an engraftment of donor CD4+ lymphocytes at the level which was reached after syngeneic HSCT (20, 38). Our data show that haploidentical HSCT can fill the gap of the extreme cell loss in the naïve T-cell populations. In addition to the reduction in the numbers of naïve (CD45RA) CD4+ as well as CD8+ T-cell subsets, an intrinsic deficiency in the signal transduction pathway proximal to the PKC has been described in peripheral blood mononuclear cells (PBMCs) from patients with A-T (2). Cellular activation indicated by CD69 expression was reduced compared to controls suggesting an early activation defect in A-T lymphocytes (2). Our analysis provided evidence that syngeneic HSCT can increase T-cell activation and signal transduction to the level of wild-type mice and can rescue the functionality of the T-cell compartment in Atm-deficient mice. Improvement of the T-cell activity was predominantly found in the naïve CD4 cell population (14). However, it must be noted, that the present study did not evaluate cellular and functional T-cell activation in Atm-deficient mice after haplo-HSCT representing a potential weakness of the study design. In addition, beside ex vivo experiments, only in vivo studies can ultimately show the restored functionality of the immune system after transplantation, e.g., by using a lymphocytic choriomeningitis virus (LCMV) transfected murine lung carcinoma cell tumor (39).
Although, outcomes of haploidentical HSCT have improved with remarkable progress within the last years, and HLA-haploidentical donors such as biological parents, biological children, full or half siblings, or collateral related donors might be an acceptable source for donor cells, in A-T, haploidentical donors have a very high probability of heterozygosity for ATM (40). Indeed, since haploidentical donors are carriers of only one ATM allele the outcome of HSCT has not been examined so far. Phenotypically, Atm-heterozygotes mice do not demonstrate reduction in size and body weight compared to wild-type counterparts (41). Cells from heterozygous and wild-type mice showed similar patterns of cell surface marker expression and chromosomal instability (19, 41). Our transplantation experiments let to a significantly improved survival of Atm-deficient mice transplanted with cells from a heterozygous donor equivalent to transplantation with wild-type donor cells. Both transplantation settings significantly increased CD4 cell numbers as well as CD4 cell functionality without any differences between the genotype of the donor cells. However, it is a well-known fact that the results found in murine animal models cannot directly be applied to humans and even that our Atm-deficient mouse model recapitulates the A-T phenotype, only short-term rather than long-term complications after HSCT can be studied. ATM-heterozygous alterations are postulated to be predispose to epithelial neoplasia, in particular breast cancer and a higher rate of lymphoid malignancies are also in the focus of discussion (42). Loss of heterozygosity happens frequently in sporadic tumors of lymphoid origin, and a high prevalence of ATM gene alterations in diverse sporadic lymphoproliferative disorders has been reported (23). In this regard, screening of ATM heterozygosity in potential donors for HSCT in A-T patients has to be considered (42). Our study demonstrates that haploidentical HSCT is a feasible strategy for A-T, even if the donor is heterozygous for ATM regarding restoration of lymphocyte populations and improvement of T-cell functionality. The project delivers substantial data about feasibility, safety, and efficacy of allogeneic and haploidentical HSCT in A-T. However, this basic research cannot substitute any research in humans and further studies are needed to develop a prospective study protocol for HSCT in children with A-T.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
This animal study was reviewed and approved by the German Animal Subjects Committee (Gen. Nr. FK/1034).
Author Contributions
RD and RS conceptualized and designed the study and wrote the manuscript. RD, LG, and PB helped with acquisition of data. RD, LG, PB, RS, and SZ analyzed and interpreted the results. RD, RS, and SZ did the conceptualization, the methodology, and the writing-review and editing of the manuscript. RD, LG, and PB helped with acquisition and validation, formal analysis,visualization, and data curation. RD and RS wrote the original draft. RS and SZ did the supervision and the project administration. RS acquired the funding for the project. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the SPARKS Charity, Action for A-T (Grant Reference 14GOU01, London, England).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We want to thank the SPARKS Charity (London, England) for their support and we thank our colleague Katrin Krug for technical assistance.
References
1. Boder E, Sedgwick RP. Ataxia-Telangiectasia; A Familial Syndrome of Progressive Cerebellar Ataxia, Oculocutaneous Telangiectasia and Frequent Pulmonary Infection. Pediatrics (1958) 21:526–54.
2. Schubert R, Reichenbach J, Zielen S. Deficiencies in CD4+ and CD8+ T Cell Subsets in Ataxia Telangiectasia. Clin Exp Immunol (2002) 129:125–32. doi: 10.1046/j.1365-2249.2002.01830.x
3. Driessen GJ, Ijspeert H, Weemaes CM, Haraldsson Á, Trip M, Warris A, et al. Antibody Deficiency in Patients With Ataxia Telangiectasia is Caused by Disturbed B- and T-Cell Homeostasis and Reduced Immune Repertoire Diversity. J Allergy Clin Immunol (2013) 131:1367–75.e9. doi: 10.1016/j.jaci.2013.01.053
4. Gatti RA, Berkel I, BODER E, Braedt G, Charmley P, Concannon P, et al. Localization of an Ataxia-Telangiectasia Gene to Chromosome 11q22-23. Nature (1988) 336:577–80. doi: 10.1038/336577a0
5. Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A Single Ataxia Telangiectasia Gene With a Product Similar to PI-3 Kinase. Science (1995) 268:1749. doi: 10.1126/science.7792600
6. Lavin MF. Ataxia-Telangiectasia: From a Rare Disorder to a Paradigm for Cell Signalling and Cancer. Nat Rev Mol Cell Biol (2008) 9:759–69. doi: 10.1038/nrm2514
7. Swift M, Morrell D, Massey RB, Chase CL. Incidence of Cancer in 161 Families Affected by Ataxia-Telangiectasia. N Engl J Med (1991) 325:1831–6. doi: 10.1056/NEJM199112263252602
8. Micol R, Ben Slama L, Suarez F, Le Mignot L, Beaute J, Mahlaoui N, et al. Morbidity and Mortality From Ataxia-Telangiectasia Are Associated With ATM Genotype. J Allergy Clin Immunol (2011) 128:382–9.e1. doi: 10.1016/j.jaci.2011.03.052
9. McGrath-Morrow SA, Gower WA, Rothblum-Oviatt C, Brody AS, Langston C, Fan LL, et al. Evaluation and Management of Pulmonary Disease in Ataxia-Telangiectasia. Pediatr Pulmonol (2010) 45:847–59. doi: 10.1002/ppul.21277
10. Shiloh Y, Ziv Y. The ATM Protein Kinase: Regulating the Cellular Response to Genotoxic Stress, and More. Nat Rev Mol Cell Biol (2013) 14:197–210. doi: 10.1038/nrm3546
11. Taylor AM, Metcalfe JA, Thick J, Mak YF. Leukemia and Lymphoma in Ataxia Telangiectasia. Blood (1996) 87:423–38. doi: 10.1182/blood.V87.2.423.bloodjournal872423
12. Di Siena S, Campolo F, Gimmelli R, Di Pietro C, Marazziti D, Dolci S, et al. Atm Reactivation Reverses Ataxia Telangiectasia Phenotypes In Vivo. Cell Death Dis (2018) 9:314. doi: 10.1038/s41419-018-0357-8
13. Bagley J, Cortes ML, Breakefield XO, Iacomini J. Bone Marrow Transplantation Restores Immune System Function and Prevents Lymphoma in Atm-Deficient Mice. Blood (2004) 104:572–8. doi: 10.1182/blood-2003-12-4226
14. Duecker R, Baer PC, Buecker A, Huenecke S, Pfeffermann L-M, Modlich U, et al. Hematopoietic Stem Cell Transplantation Restores Naïve T-Cell Populations in Atm-Deficient Mice and in Preemptively Treated Patients With Ataxia-Telangiectasia. Front Immunol (2019) 10:2785. doi: 10.3389/fimmu.2019.02785
15. Bakhtiar S, Woelke S, Huenecke S, Kieslich M, Taylor AM, Schubert R, et al. Pre-Emptive Allogeneic Hematopoietic Stem Cell Transplantation in Ataxia Telangiectasia. Front Immunol (2018) 9:2495. doi: 10.3389/fimmu.2018.02495
16. Nietfeld JJ, Pasquini MC, Logan BR, Verter F, Horowitz MM. Lifetime Probabilities of Hematopoietic Stem Cell Transplantation in the U.S. Biol Blood Marrow Transplant (2008) 14:316–22. doi: 10.1016/j.bbmt.2007.12.493
17. Montoro J, Sanz J, Sanz GF, Sanz MA. Advances in Haploidentical Stem Cell Transplantation for Hematologic Malignancies. Leuk Lymphoma (2016) 57:1766–75. doi: 10.3109/10428194.2016.1167204
18. Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, et al. An Advanced Culture Method for Generating Large Quantities of Highly Pure Dendritic Cells From Mouse Bone Marrow. J Immunol Methods (1999) 223:77–92. doi: 10.1016/s0022-1759(98)00204-x
19. Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, et al. Atm-Deficient Mice: A Paradigm of Ataxia Telangiectasia. Cell (1996) 86:159–71. doi: 10.1016/s0092-8674(00)80086-0
20. Pietzner J, Baer PC, Duecker RP, Merscher MB, Satzger-Prodinger C, Bechmann I, et al. Bone Marrow Transplantation Improves the Outcome of Atm-Deficient Mice Through the Migration of ATM-Competent Cells. Hum Mol Genet (2013) 22:493–507. doi: 10.1093/hmg/dds448
21. Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J,JR, Crawford JM, et al. An Experimental Model of Idiopathic Pneumonia Syndrome After Bone Marrow Transplantation: I. roles minor H Antigens Endotoxin. Blood (1996) 88:3230–9. doi: 10.1182/blood.V88.8.3230.bloodjournal8883230
22. Vacchio MS, Olaru A, Livak F, Hodes RJ. ATM Deficiency Impairs Thymocyte Maturation Because of Defective Resolution of T Cell Receptor Alpha Locus Coding End Breaks. Proc Natl Acad Sci USA (2007) 104:6323–8. doi: 10.1073/pnas.0611222104
23. Gumy-Pause F, Wacker P, Sappino A-P. ATM Gene and Lymphoid Malignancies. Leukemia (2004) 18:238–42. doi: 10.1038/sj.leu.2403221
24. Slack J, Albert MH, Balashov D, Belohradsky BH, Bertaina A, Bleesing J, et al. Outcome of Hematopoietic Cell Transplantation for DNA Double-Strand Break Repair Disorders. J Allergy Clin Immunol (2018) 141:322–8. doi: 10.1016/j.jaci.2017.02.036
25. Albert MH, Gennery AR, Greil J, Cale CM, Kalwak K, Kondratenko I, et al. Successful SCT for Nijmegen Breakage Syndrome. Bone Marrow Transplant (2010) 45:622–6. doi: 10.1038/bmt.2009.207
26. Ayas M, Al-Seraihi A, El-Solh H, Al-Ahmari A, Khairy A, Aldali A, et al. The Saudi Experience in Fludarabine-Based Conditioning Regimens in Patients With Fanconi Anemia Undergoing Stem Cell Transplantation: Excellent Outcome in Recipients of Matched Related Stem Cells But Not in Recipients of Unrelated Cord Blood Stem Cells. Biol Blood Marrow Transplant (2012) 18:627–32. doi: 10.1016/j.bbmt.2011.08.015
27. Peffault de Latour R, Porcher R, Dalle J-H, Aljurf M, Korthof ET, Svahn J, et al. Allogeneic Hematopoietic Stem Cell Transplantation in Fanconi Anemia: The European Group for Blood and Marrow Transplantation Experience. Blood (2013) 122:4279–86. doi: 10.1182/blood-2013-01-479733
28. Ussowicz M, Wawrzyniak-Dzierżek E, Mielcarek-Siedziuk M, Salamonowicz M, Frączkiewicz J, Rybka B, et al. Allogeneic Stem Cell Transplantation After Fanconi Anemia Conditioning in Children With Ataxia-Telangiectasia Results in Stable T Cell Engraftment and Lack of Infections Despite Mixed Chimerism. Biol Blood Marrow Transplant (2018) 24:2245–9. doi: 10.1016/j.bbmt.2018.07.001
29. Sabino Pinho de Oliveira B, Putti S, Naro F, Pellegrini M. Bone Marrow Transplantation as Therapy for Ataxia-Telangiectasia: A Systematic Review. Cancers (Basel) (2020) 12(11):3207. doi: 10.3390/cancers12113207
30. Piemontese S, Ciceri F, Labopin M, Arcese W, Kyrcz-Krzemien S, Santarone S, et al. A Comparison Between Allogeneic Stem Cell Transplantation From Unmanipulated Haploidentical and Unrelated Donors in Acute Leukemia. J Hematol Oncol (2017) 10:24. doi: 10.1186/s13045-017-0394-2
31. Passweg JR, Baldomero H, Bader P, Bonini C, Duarte RF, Dufour C, et al. Use of Haploidentical Stem Cell Transplantation Continues to Increase: The 2015 European Society for Blood and Marrow Transplant Activity Survey Report. Bone Marrow Transplant (2017) 52:811–7. doi: 10.1038/bmt.2017.34
32. Choo SY. The HLA System: Genetics, Immunology, Clinical Testing, and Clinical Implications. Yonsei Med J (2007) 48:11–23. doi: 10.3349/ymj.2007.48.1.11
33. Fuchs EJ. Haploidentical Transplantation for Hematologic Malignancies: Where do We Stand? Hematol Am Soc Hematol Educ Program (2012) 2012:230–6. doi: 10.1182/asheducation-2012.1.230
34. Cooke KR, Luznik L, Sarantopoulos S, Hakim FT, Jagasia M, Fowler DH, et al. The Biology of Chronic Graft-Versus-Host Disease: A Task Force Report From the National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-Versus-Host Disease. Biol Blood Marrow Transplant (2017) 23:211–34. doi: 10.1016/j.bbmt.2016.09.023
35. Olsson RF, Logan BR, Chaudhury S, Zhu X, Akpek G, Bolwell BJ, et al. Primary Graft Failure After Myeloablative Allogeneic Hematopoietic Cell Transplantation for Hematologic Malignancies. Leukemia (2015) 29:1754–62. doi: 10.1038/leu.2015.75
36. Piguet PF, Grau GE, Allet B, Vassalli P. Tumor Necrosis Factor/Cachectin is an Effector of Skin and Gut Lesions of the Acute Phase of Graft-vs.-Host Disease. J Exp Med (1987) 166:1280–9. doi: 10.1084/jem.166.5.1280
37. Apperley J, Niederwieser D, Huang X, Nagler A, Fuchs E, Szer J, et al. Haploidentical Hematopoietic Stem Cell Transplantation: A Global Overview Comparing Asia, the European Union, and the United States. Biol Blood Marrow Transplant (2016) 22:23–6. doi: 10.1016/j.bbmt.2015.11.001
38. Hill GR, Ferrara JL. The Primacy of the Gastrointestinal Tract as a Target Organ of Acute Graft-Versus-Host Disease: Rationale for the Use of Cytokine Shields in Allogeneic Bone Marrow Transplantation. Blood (2000) 95:2754–9. doi: 10.1182/blood.V95.9.2754.009k25_2754_2759
39. Ghosh S, Carmo M, Calero-Garcia M, Ricciardelli I, Bustamante Ogando JC, Blundell MP, et al. T-Cell Gene Therapy for Perforin Deficiency Corrects Cytotoxicity Defects and Prevents Hemophagocytic Lymphohistiocytosis Manifestations. J Allergy Clin Immunol (2018) 142:904–913.e3. doi: 10.1016/j.jaci.2017.11.050
40. Fabricius WA, Ramanathan M. Review on Haploidentical Hematopoietic Cell Transplantation in Patients With Hematologic Malignancies. Adv Hematol (2016) 2016:5726132. doi: 10.1155/2016/5726132
41. Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, et al. Pleiotropic Defects in Ataxia-Telangiectasia Protein-Deficient Mice. Proc Natl Acad Sci USA (1996) 93:13084–9. doi: 10.1073/pnas.93.23.13084
Keywords: Ataxia-telangiectasia, HSCT, haploidentical, ATM, GvHD, T-lymphocytes
Citation: Duecker RP, Gronau L, Baer PC, Zielen S and Schubert R (2021) Survival and Functional Immune Reconstitution After Haploidentical Stem Cell Transplantation in Atm-Deficient Mice. Front. Immunol. 12:693897. doi: 10.3389/fimmu.2021.693897
Received: 12 April 2021; Accepted: 16 June 2021;
Published: 29 June 2021.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Sujal Ghosh, Heinrich Heine University of Düsseldorf, GermanyJavier Chinen, Baylor College of Medicine, United States
Copyright © 2021 Duecker, Gronau, Baer, Zielen and Schubert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruth Pia Duecker, cnV0aHBpYS5kdWVja2VyQGtndS5kZQ==