 Daniel Bode
Daniel Bode Alyssa H. Cull
Alyssa H. Cull Juan A. Rubio-Lara
Juan A. Rubio-Lara David G. Kent
David G. Kent- 1Wellcome Medical Research Council (MRC) Cambridge Stem Cell Institute, University of Cambridge, Cambridge, United Kingdom
- 2Department of Haematology, University of Cambridge, Cambridge, United Kingdom
- 3York Biomedical Research Institute, Department of Biology, University of York, York, United Kingdom
Single-cell molecular tools have been developed at an incredible pace over the last five years as sequencing costs continue to drop and numerous molecular assays have been coupled to sequencing readouts. This rapid period of technological development has facilitated the delineation of individual molecular characteristics including the genome, transcriptome, epigenome, and proteome of individual cells, leading to an unprecedented resolution of the molecular networks governing complex biological systems. The immense power of single-cell molecular screens has been particularly highlighted through work in systems where cellular heterogeneity is a key feature, such as stem cell biology, immunology, and tumor cell biology. Single-cell-omics technologies have already contributed to the identification of novel disease biomarkers, cellular subsets, therapeutic targets and diagnostics, many of which would have been undetectable by bulk sequencing approaches. More recently, efforts to integrate single-cell multi-omics with single cell functional output and/or physical location have been challenging but have led to substantial advances. Perhaps most excitingly, there are emerging opportunities to reach beyond the description of static cellular states with recent advances in modulation of cells through CRISPR technology, in particular with the development of base editors which greatly raises the prospect of cell and gene therapies. In this review, we provide a brief overview of emerging single-cell technologies and discuss current developments in integrating single-cell molecular screens and performing single-cell multi-omics for clinical applications. We also discuss how single-cell molecular assays can be usefully combined with functional data to unpick the mechanism of cellular decision-making. Finally, we reflect upon the introduction of spatial transcriptomics and proteomics, its complementary role with single-cell RNA sequencing (scRNA-seq) and potential application in cellular and gene therapy.
Introduction
The crucial role that single-cell approaches play in understanding cell function has been recognised for decades. Early advances in immunology, and particularly hematopoiesis, have demonstrated the power of such approaches for ascribing functional properties to a single cell. Pioneering work by Till and McCulloch uncovered functional heterogeneity of hematopoietic stem cells (HSCs) by performing single cell-derived assays termed colony-forming unit spleen, or CFU-S, assays (1, 2). Similarly, early studies of single multipotent progenitors provided insights into the progenitor cell commitment and the development of mature immune cells, such as T and B lymphocytes (3, 4). Perhaps most transformative was the introduction of fluorescence activated cell sorting (FACS) which enabled the near-ubiquitous adaption of single-cell functional assays in immunology, hematopoiesis, and beyond (5–7).
Efforts to characterize the cellular function of single cells have fuelled an increased desire to understand detailed molecular mechanisms, but the technologies to do so in single cells have lagged substantially. The development of the polymerase chain reaction (PCR) for amplifying DNA ultimately paved the way for the first glimpse into the transcriptome of single cells (8, 9). The initial protocol for the amplification of cDNA using PCR from single macrophages was introduced by Brady et al. (10), where robust exponential amplification was achieved without disturbing the relative abundance of mRNA sequences, enabling the inspection of rare transcripts in a complex single cell-derived cDNA library. In parallel, Eberwine and colleagues developed a linear RNA amplification approach, based on the amplification of antisense RNA using a T7 RNA polymerase (11, 12). By inspecting mRNAs from single pyramidal neurons isolated from rat brains, they provided the first evidence for global molecular heterogeneity between morphologically similar cells (11).
While targeted single-cell PCR-based molecular screens revolutionized molecular biology, the low throughput and hypothesis-driven nature prevented unbiased exploratory screening. In 1991, Fodor and colleagues developed a novel photolithography-based approach for efficient synthesis of complex oligonucleotides on the microscale (13). This pioneering work would lead to the development of microarray technology where several years later, Schena et al. first applied this method for monitoring gene expression, examining the expression of 45 Arabidopsis genes from total mRNA (14). The following decade saw a rapid expansion of the technology, resulting in genome-wide genomic, transcriptomic and epigenetic screening using microarrays [reviewed elsewhere: (15–18)]. This ultimately enabled microarray analysis at single cell level (19), leading to insights into the molecular pathways governing cell fate (20, 21).
Microarrays, a hybridisation-based approach, assayed the known transcriptome and was therefore unsuitable for unbiased detection of novel transcripts. In 1977, Sanger and colleagues published the first genome to be sequenced (22) and soon after early generation sequencing methods began to rapidly develop (23). However, these approaches were extremely costly and time consuming (23). This opened up space for next generation sequencing (NGS) to lead to a revolution in molecular profiling, enabling low-cost, high-throughput and highly parallelised sequencing of nucleic acids. To date, a wide variety of NGS platforms have been developed [reviewed in (24, 25)] and in all cases, sheared DNA is bound to adapter sequences which are immobilised within flow cells, facilitating the synthesis of complementary DNA fragments for subsequent amplification (26). By using fluorophore-labelled nucleotides and simultaneous fluorescence readouts across the entire flow cell, the respective sequences can be determined and ultimately mapped against the reference genome (24, 27, 28). NGS for routine DNA and RNA sequencing provides multiple advantages over microarray technology, including reduced background noise, an increased dynamic range and the detection of novel transcripts (25, 29, 30).
For these reasons, NGS was rapidly adapted to a variety of model systems, including the inspection of rare cell types at single cell resolution (31–36). Tang et al. pioneered the first protocol for single-cell RNA sequencing (scRNA-seq) in single mouse blastomeres with improved performance compared to microarray-based single-cell protocols (36). Following this there has been an explosion of single-cell molecular technologies, enabling unbiased screening of the transcriptome (37, 38), genome (39, 40), DNA methylation (41), chromatin accessibility (42) and spatial resolution of gene expression (43). While these methods provide comprehensive snapshots of molecular states, their integration with cellular phenotype and function is less common and remains vital to the inspection of tissue complexity, disease progression, therapeutic intervention, and beyond. To achieve this goal, pioneering work to integrate omics protocols led to the development of several multimodal technologies. These include simultaneous screening of I) cell surface proteins and mRNA (44, 45), II) DNA methylation and mRNA (46), III) perturbations and mRNA (47), IV) DNA and mRNA (48), V) lineage tracing and mRNA, and VI) cellular function and mRNA (44, 49, 50).
Single-cell technologies have thus provided insight into a wide-range of disease mechanisms, especially in illnesses with significant heterogeneity (51), leading to a long list of potential new therapeutic options. In recent years, the fields of cellular and gene therapy have been steadily evolving for treatment of some monogenic diseases (gene therapy) and B cell leukemias (cell therapy) in particular (52, 53). However, to enable further improvements and applications to other more complex disease types such as autoimmune type 1 diabetes, key aspects such as characterizing target tissues, identifying novel targets in heterogeneous diseases and assessing efficacy of therapeutic interventions all require deeper interrogation. Recent advances in single-cell technologies are ideally positioned to address a number of these unmet needs (51).
In this review, we outline a wide range of recent technologies for screening the genome, epigenome, transcriptome and proteome of single cells and the multimodal integration of these platforms. We focus on the integration of functional cellular phenotypes with molecular profiles and emphasise the use of single-cell technologies in gene and cell therapies.
A Golden Age for Gene Therapy - Recent Successes in Treating Monogenic Disorders
In its simplest form, gene therapy aims to cure a patient’s disease by introducing a normal or corrected copy of a gene into target cells. In 1972, Friedmann and Roblin first proposed the concept of gene therapy as a treatment for inherited genetic defects that largely affected children, many of whom experienced severe, life-threatening symptoms (54). Initially, HSC transplantation represented the primary curative option for many of these disorders, but the availability of matched sibling donors and the risk of severe graft-versus-host disease were barriers for many patients (55). To circumvent these issues, the first gene therapy clinical trials used patient-derived differentiated (T lymphocytes) or immature (hematopoietic stem and progenitor cells, HSPCs) cells that were engineered ex vivo to express a disease-correcting transgene (56, 57). Pioneering studies in the late 1990s and early 2000s initially reported successful treatment of adenosine deaminase-deficient severe combined immunodeficiency (ADA-SCID) and other hematological disorders (56–59); however, these successes were soon overshadowed by reports of patients who experienced significant adverse events including the development of treatment-related leukemias and severe immune reactions (60–65). Many of these unanticipated biological effects were later directly linked to the viral vectors used for transgene delivery (66, 67). Consequently, research efforts became focused on improving the safety of viral vectors (68–70) and monitoring for pre-leukemic mutations became a standard feature of treatment follow-up (71–74).
Following these improvements, a number of clinical trials have demonstrated the long-term benefits achieved in individuals with various primary immunodeficiencies and monogenic blood disorders who have received gene therapy treatments (75–84). The follow-up data being reported for these patients mainly focus on disease-relevant parameters such as blood counts and overall clinical symptoms. As a result, numerous questions related to the gene therapy process still remain (Figure 1). For example, which HSPC populations are readily transduced during drug product creation and how does this impact outcomes? Do gene corrected terminally differentiated cells have any advantage over their non-transduced counterparts? These types of questions can best be answered using single-cell technologies. Another area of active research involves the development of in vivo non-viral delivery systems. These strategies include the use of nanoparticles, aptamers/oligonucleotides and extracellular vesicles to deliver transgenes or siRNAs/shRNAs (85–90). While in vivo treatments circumvent issues related to the isolation and manipulation of target cells, they have the potential to induce expression of transgenes or siRNAs/shRNAs in cell types that are not relevant to curing disease. High resolution single-cell transcriptomic and proteomic data will be vital in dissecting how these new treatments affect cell populations receiving the correcting vector. These types of information, especially at the level of preclinical studies, will greatly aid in the development of these technologies.
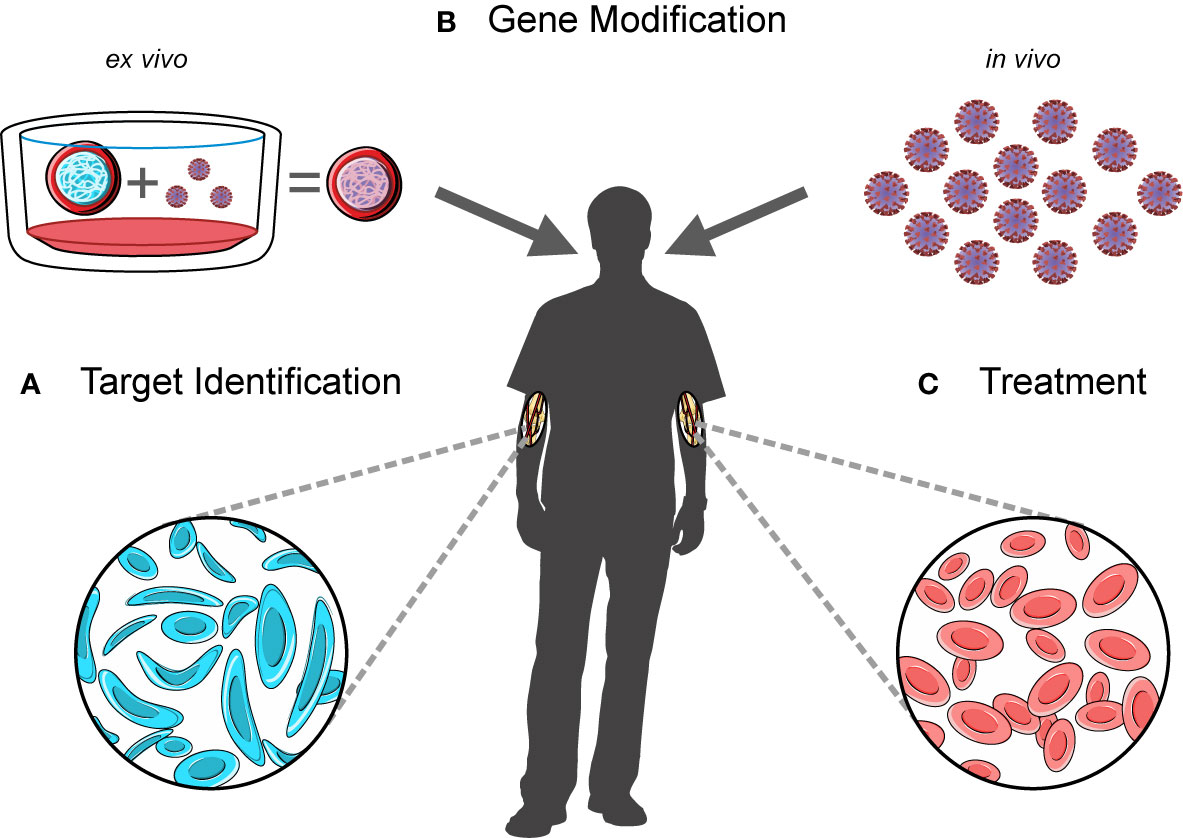
Figure 1 A workflow for developing and administering gene therapy. Novel gene therapy approaches involve (A) the identification of therapeutic targets, (B) an ex vivo gene modification step to create a transduced drug product (left) or the production of an in vivo product (right), and (C) the infusion of these products into patients following myeloablative conditioning.
Moving beyond monogenic disorders, multi-target approaches may be useful in treating complex acquired diseases, such as cancers or autoimmune diseases like type 1 diabetes. Large-scale bulk pan-cancer genomics studies have suggested that tumors harbour an average of 4-5 driver mutations (91–94). While this represents an opportunity for the simultaneous manipulation of multiple drivers, the efficacy of this approach in individual patients depends on the specific combinations of these mutations within tumor cell subpopulations. As most genetic profiling of tumors is done using bulk sequencing, the resolution of major/minor clones and subclones becomes very difficult without the use of single-cell approaches. If individual cancers could be profiled to such high resolution, gene therapy strategies could be imagined to target genes essential to cancer cell survival (95–98) or disrupt processes such as angiogenesis that facilitate tumor growth (99–102). Combination therapies may also prove to be highly effective in some contexts (103, 104).
Type 1 diabetes is an autoimmune disease driven by loss of T cell tolerance resulting in islet autoimmunity. During disease development, insulin-producing β-cells in the pancreas are abnormally targeted by infiltrating immune cells (105). For monogenic disorders such as immune dysregulation polyendocrinopathy enteropathy X-linked syndrome where patients are at a much higher risk of developing secondary type 1 diabetes, gene therapy treatment could offer a potential cure (106). However, the genetic drivers of primary type 1 diabetes are complex and may act at the level of β-cells themselves and/or various T cell populations (105). Preclinical studies exploring the use of gene therapy to treat type 1 diabetes have clearly demonstrated the need for treatments that function on two levels - one to create or maintain functional insulin-secreting β-cells and another to protect these cells from autoimmune responses (107–110). Regardless of disease context, the overall diversity of cellular interactions driving human disease presents many challenges to the development of successful treatments. Single-cell studies can address questions pertaining to cell type interactions, disease-specific immunity, clonal dynamics of gene corrected cells and therapy-escape mechanisms, moving gene therapy forward to the next level.
Cell Therapy as a Promising Treatment for More Complex Diseases
While gene therapy has revolutionized the treatment of primary immunodeficiencies and monogenic disorders, other strategies may be required to treat more complex diseases. Currently, the primary standard of care for many cancers is chemotherapy, radiation therapy or, in the case of solid tumors, surgery. Immune-based treatments including cell therapy and immune checkpoint inhibitors are now being developed, already showing promise in treating refractory or relapsed patient cohorts. Cell therapy strategies involving chimeric antigen receptor (CAR) T cells have been particularly successful in the treatment of B-cell malignancies (111–113). In brief, these therapies use autologous lymphocytes with synthetically engineered antigen receptors to target tumor-specific antigens (114), thereby harnessing the immune system to trigger anti-tumor immunity (Figure 2). Pioneering work by several groups led to the first successful application of this technology in the treatment of B-cell malignancies (111–113), with the first therapy approved by the US-FDA in 2017 for use in B-cell acute lymphoblastic leukemia and diffuse large B-cell lymphoma (115).
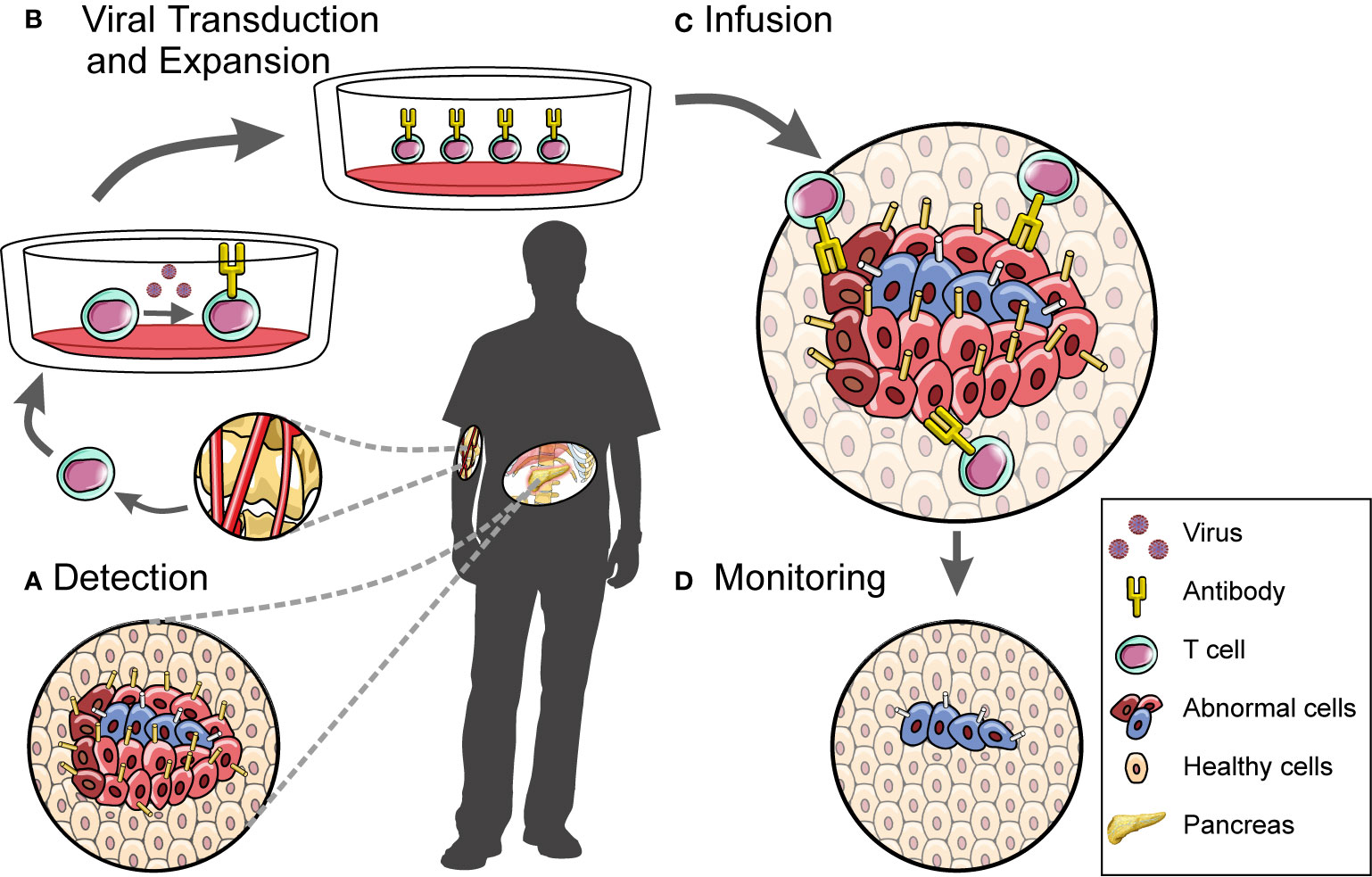
Figure 2 A workflow for developing and administering cell therapy. CAR T cell-based therapies involve (A) the discovery of disease-associated antigens which can then be used to target the cytotoxic effects of engineered CAR T cells, (B) the isolation and manipulation of patient-derived T cell populations, (C) the infusion of these cells into patients, and (D) downstream monitoring of disease.
Although stable remission is reportedly achieved in approximately 40-60% of patients with these B-cell malignancies (116), a number of significant barriers to increasing treatment efficacy have been identified. CAR T cell persistence and expansion has been shown to be variable between patients. Researchers have suggested that the use of less differentiated T cell subsets or T cells with an altered genetic background (for example, TET2 disruption) during the manufacturing phase may improve outcomes (115, 117–123). However, a better understanding of the key molecular drivers of T cell expansion and persistence is required to inform future efforts to tailor the production of CAR T cells. Single-cell technologies can be used here to dissect these processes at the molecular level. In addition to increasing the overall performance of CAR T cells, another key aspect required to improve therapeutic outcomes is to control immune responses not directly mediated by CAR T cells (111–113, 124). In order to minimise these responses, a more thorough understanding of immune cell interactions must first be developed. In this context, single-cell approaches will provide the resolution required to dissect these complex systems. On a different level, selective pressures applied by anti-CD19 CAR T cells may also lead to antigen escape and lineage switching as 10-25% of patients go on to develop a CD19- cancer (125). While groups reported acquired CD19 loss-of-function mutations (126) and abnormal splicing events leading to loss of CD19 expression (127, 128), the specific origin of CD19- cancer cells was not clear. A recent paper using single-cell techniques provides evidence that in at least some patients, treatment-resistant CD19- cancer cells exist prior to treatment (129), underscoring the vital role of single-cell approaches pinpointing the mechanisms by which cancer cells escape treatment and informing strategies targeting refractory disease.
On the other hand, there has been relatively limited success seen in CAR T cell treatments outside of B cell malignancies, despite the development of therapeutics targeting multiple antigens simultaneously or sequentially [reviewed in (130–132)]. In solid cancers, tumor-specific antigens (TSAs) first need to be comprehensively profiled to allow for selection of appropriate candidate TSAs (133) which is especially important when dealing with heterogeneous tumors. Understanding the consequences of on-target/off-tumor effects is also essential to creating safe and effective therapies as evidenced by recent reports of adverse events experienced by patients in two separate cell therapy clinical trials (134, 135). Even once promising TSAs have been selected and tested in both animal models and early phase clinical trials, a number of other tumor-specific factors will likely interfere with the effectiveness of this treatment strategy. For example, immunosuppressive mechanisms that dampen T cell anti-tumor responses may also impact CAR T cell function. Combination therapies or further disruptions to create CAR T cells that are resistant to these immune evasion pathways may therefore become essential (136, 137). Other CAR immune cell populations such as B cells, natural killer (NK) cells and macrophages may also be useful in treating certain diseases (138–140).
In the context of diabetes, both CAR T cell and regulatory T cell (Tregs)-based treatments are currently being developed (141–146). Under normal conditions, Tregs mediate immune tolerance by expressing anti-inflammatory cytokines and dampening the inflammatory or cytotoxic responses of other types of T lymphocytes (147). While patients with type 1 diabetes have similar frequencies of Tregs compared to control individuals, it has been shown that these Tregs have reduced immunosuppressive capacity (148–150). Adoptive Treg transfers from healthy donors into patients have shown promise in preclinical models for a number of different diseases driven by immune dysregulation including type 1 diabetes (145, 151–156). However, a thorough understanding of the heterogeneous cell types that facilitate disease initiation and progression will be crucial to optimizing these treatment regimens.
Using Single-Cell Approaches to Refine Treatment And Inform the Development of Novel Therapeutics
Although great strides have been made in gene and cell therapy, applications to a wider range of diseases requires more information. Key aspects, such as characterizing target tissues, identifying novel targets in heterogeneous diseases and assessing efficacy of therapeutic interventions require deeper interrogation and single-cell approaches are well-positioned to provide this information.
While a number of groups have begun to use single-cell approaches to dissect various aspects of CAR T cell-based therapy (129, 157, 158), the gene therapy field has not explored this to the same extent. That said, a handful of studies have used bulk sequencing approaches to examine post-transplantation clonal dynamics in a small number of patients (159–161). Biasco and colleagues used this approach to estimate transduced HSPC population size and describe the contributions of HSPC subpopulations to various stages of hematopoietic reconstitution (159, 160). Most recently, Six and colleagues addressed questions pertaining to clonal selection following gene therapy in WAS, sickle cell disease (SCD) or beta-thalassemia patients and found no indications of clonal skewing caused by insertional mutagenesis (161). While all three of these studies provide important insights into human hematopoiesis, the reliance on bulk sequencing approaches to map viral integration sites means that several key questions remain unanswerable. For example, these methods do not allow unedited cells or low abundance clones to be tracked or the effects of multiple integration sites to be assessed. Furthermore, relationships between transduced and non-transduced cells cannot be assayed. These details can only be examined using strategies that analyse single cells and their clonal progeny (162).
In contrast, studies employing single-cell technologies have already begun to deconstruct the fundamental biology behind anti-CD19 CAR T cell therapeutic outcomes. Shieh et al. used single-cell transcriptomics to identify gene signatures associated with good treatment outcomes for patients with B cell malignancies, providing insights relevant to the optimisation of CAR T cell production (157). Deng et al. used a similar approach to discover transcriptional signatures connected to both complete and poor treatment responses (158). This study also identified a novel, transcriptionally distinct cell population found specifically in the infusion products of patients who went on to develop high-grade immune effector cell-associated neurotoxicity syndrome (158). This finding demonstrates the value of single-cell approaches in generating essential information that can then be fed back into clinical practice. Another recent publication applying single-cell technologies reported that the disease-driving clone observed in one patient’s relapsed B cell acute lymphoblastic leukemia existed prior to anti-CD19 CAR T treatment (129). Taken together, these studies clearly illustrate how single cell-based datasets can provide clinically relevant insights into various aspects of the cell therapy process (Figure 2).
For every stage of the gene and cell therapy process, a number of important questions remain unanswered (Table 1). Ultimately, single-cell approaches will be instrumental both in informing our understanding of human disease and in developing the effective therapeutics required to treat them. Data generated using these methods has the potential to better inform our understanding of the numerous complex factors influencing treatment outcomes. The generation of novel targets and delivery methods for heterogeneous diseases relies on a high level of detail and the ability to map cell-cell interactions, especially for disorders with a strong immune component.
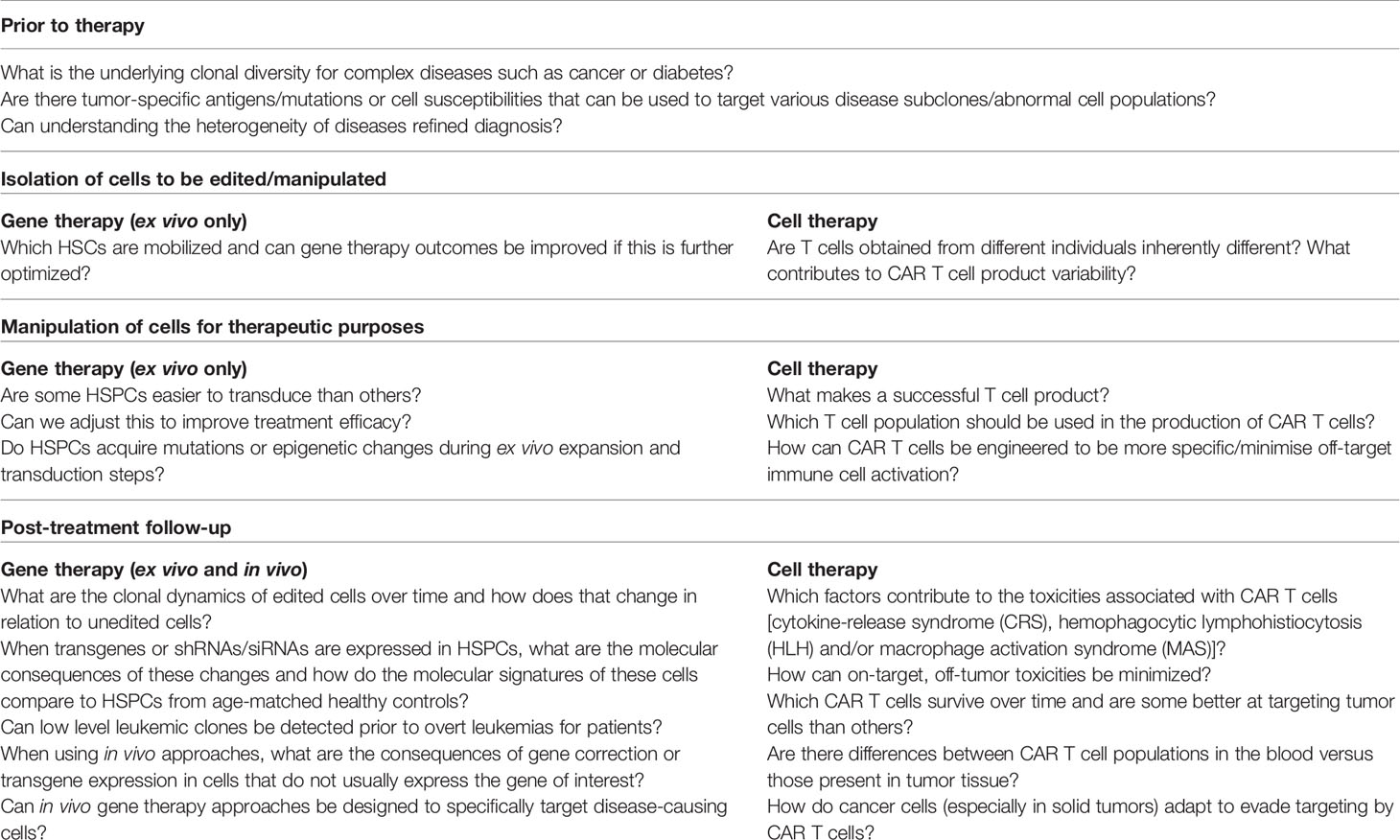
Table 1 Unmet needs and addressable questions in gene and cell therapy.
Single-Cell Multi-Omics Platforms and Their Prospect in Gene and Cell Therapy
A wide array of screening platforms have been developed to interrogate molecular states at the single cell level to give insight into tumor heterogeneity and clonal evolution of complex tissues. Here, we describe a selection of the most widely used omics tools and discuss their application in gene or cell therapy, including their potential role in addressing future clinical challenges.
Genome
The first protocol for DNA sequencing at the single cell level, termed single nucleus sequencing (SNS), was described by Navin and colleagues (40). Comparable and reproducible detection levels of copy number variations were observed in single cell and bulk (106) samples. By sequencing the genomes of 100 single monogenomic breast tumor cells and the associated liver metastatic tissue, the authors also observed substantial clonal heterogeneity (40). After FACS of single nuclei and whole genome amplification (WGA), each nucleus is sequenced in an individual flow lane. The requirement of full sequencing lanes for single nuclei limited the throughput of such experiments and consequently, several groups introduced barcoding technologies to permit multiplexing of single cells in a single sequencing lane (163–167). To address this challenge, Amini et al. developed a combinatorial barcoding approach, first using Tn5 transposome-mediated labelling followed by PCR-based indexing to yield nearly 10,000 unique barcodes (165). In turn, Vitak et al. demonstrated the efficacy of a single-cell combinatorial indexed sequencing (SCI-seq) platform by acquiring >1500 single cell genomes from a primary pancreatic ductal adenocarcinoma sample (39). To date, a multitude of single-cell sequencing platforms rely on these barcoding principles (168, 169). However, only ~32% of sequenced cells had sufficient coverage for copy-number variation (CNV) detection (39). To address this issue and avoid amplification biases of exponential WGA, Chen and colleagues developed a linear amplification protocol, significantly reducing the required resolution for CNV calling and this was further complemented by experimental and computational approaches to improve the detection of single nucleotide variants (170, 171).
Despite experimental drawbacks related to coverage, single-cell whole genome sequencing (scWGS) has enabled an unprecedented insight into clonal dynamics during tumorigenesis and normal hematopoiesis (162, 172). One notable example includes a temporal study of single human B lymphocytes that explored the evolution of mutational signatures and age-related accumulation of oncogenic mutations (173), only achievable through scWGS.
While bulk WGS studies can infer which disease-causing mutations co-occur based on average variant allele frequencies, there is the potential to group populations of cells that in reality are part of distinct clonal entities. scWGS provides a more precise overview of clonal subpopulations while also capturing information that can be used to pinpoint mutation co-occurrence and order of acquisition (174–178). This approach has been used to profile mutant clones in diseases such as childhood acute lymphoblastic leukemia, childhood T cell acute lymphoblastic leukemia and adult acute myeloid leukemia (179–181). Rare cancer cell populations missed in bulk WGS may also be detected in scWGS assays, as demonstrated by Xu and colleagues (181). Capturing this heterogeneity is essential to understanding how clones with certain mutational profiles impact disease evolution and response to treatment.
Once gene corrected cells have been infused into a patient receiving gene therapy, it is important to track the clonal evolution of these corrected cells. scWGS could be used to track these dynamics as well as answer questions surrounding whether treatment-related mutations are acquired in cells during the gene therapy process. While this method is particularly effective at identifying copy number variants and aneuploidy, technical challenges exist such as low read coverage and sequencing depth. This may significantly hamper efforts to profile single nucleotide changes in gene corrected cells. For HSPCs, bulk WGS of single cell-derived clonal cultures or colonies has bypassed these obstacles (182); however, this approach is not feasible for cell types where ex vivo expansion is not possible. Provided that technical challenges are overcome, scWGS represents a promising avenue to explore clonal dynamics. However, the cost for sufficient whole genome coverage in bulk and scWGS currently remains a major barrier for routine adoption.
Following cell therapy treatments, scWGS can be used to assess mutation profiles at the single cell level for highly heterogeneous tumors during the follow-up stage. This information would be particularly helpful in determining why certain patients experience disease relapse, allowing for the identification of specific clones that are either highly susceptible or resistant to CAR T cell cytotoxicity. Additionally, building a more comprehensive understanding of tumor cell clonal dynamics will be key to dissecting out subpopulations that could then be profiled with the aim of identifying new TSAs. This type of approach can be applied to any group of diseases where complex mutation profiles are expected to impact the effectiveness of treatment.
Immune receptor repertoire analysis facilitates the interrogation of clonal dynamics of the adaptive immune response and thus provides a crucial tool for immunotherapy (183). In particular, the development of VDJ-sequencing and single-cell T cell receptor (TCR) sequencing enabled robust profiling of the output of VDJ recombination, using targeted PCR and NGS (184, 185). A multitude of studies outlined the efficacy of TCR sequencing for immune cell profiling in cancer patients to help stratify patient cohorts for immunotherapy, identify the T cell repertoire in the tumour microenvironment and determine the response to PD-1 therapy (186–188). Intriguingly, computational tools have also been developed to enable retrospective VDJ profiling from global single cell sequencing data, thus negating the need for separate immune receptor profiling (157). Nevertheless, limited availability of patient tissue samples and peripheral blood can prevent identification of rare clones and sequential PCR amplification increases risk of amplification biases (189).
Epigenome
The epigenome plays a crucial role in determining cell identity and function with chromatin organization playing a critical role in modulating gene expression and other regulatory functions (190). Chromatin accessibility is governed by the core epigenetic mechanisms of DNA methylation and post-translational modifications of histones (191). Thus, being able to screen DNA methylation, chromatin accessibility and histone modification at single cell resolution can provide crucial insight into tissue heterogeneity.
To identify open chromatin regions and characterize regulatory elements, Buenrostro and colleagues pioneered the assay for transposase-accessible chromatin using sequencing (ATAC-seq) protocol (192). In brief, this protocol leveraged the previously described hyperactive Tn5 transposase to simultaneously fragment open chromatin regions and introduce sequencing adaptors for subsequent library synthesis (164, 192, 193). While the original ATAC-seq protocol required 500-50,000 cells, the adaptation to inspect single cells soon followed. Buenrostro et al. used the Fluidigm microfluidic platform, allowing single cell capture and downstream processing of hundreds or thousands of single cells (42). Since its inception, others have developed approaches to increase the throughput of scATAC-seq to tens, or even hundreds of thousands of cells (194, 195). Illustrating its power, Sapathy et al. generated scATAC-seq profiles for over 60,000 primary human bone marrow and peripheral blood mononuclear cells (PBMC) (194). Here, the authors identified cell-type specific cis-elements, key transcription factor (TF) activity across a broad range of hematopoietic populations and gene activity, using aggregate accessibility of multiple cis-elements for a single gene. Most intriguingly, such high density of single cell clusters permits the inference of complex differentiation trajectories. Using the well-characterized development of B cells, the authors were able to reconstruct the differentiation pathway, characterize cis-elements of each cell type, and identify active TF programs along the entire differentiation trajectory. Unsurprisingly, scATAC-seq enabled a previously unseen insight into tumor evolution, such as the role of naïve cell types in driving tumorigenesis (194, 196, 197).
DNA methylation of cytosine residues (5mC) plays a crucial role in epigenetic regulation, including the modulation of cis-regulatory elements (198). In particular, DNA methylation has been implicated in gene silencing to regulate transcriptional activity during development and altering transcription factor binding (199, 200). The development of bisulphite sequencing (BS-seq) enabled unbiased, genome-wide inspection of the DNA methylome (201). To enable BS-seq at single cell resolution (scBS-seq), pioneering work by Smallwood et al. adapted the existing post-bisulfite adapter tagging protocol to derive quantitative DNA methylation signatures at up to 50% of CpG islands (202–204). Smallwood et al. and others have extensively applied scBS-seq to interrogate mouse gastrulation, human implantation, embryonic stem cells and alternative splicing at single cell resolution (203, 205–207).
The clinically-relevant utility of scATAC-seq in building a comprehensive understanding of the tumor microenvironment has been clearly shown by Sapathy et al. (194) where chromatin accessibility was mapped for more than 37,000 cells from five sets of serial basal cell carcinoma tumor biopsies. Pre- and post-PD-1 inhibitor treated samples were profiled and cell types formed clearly defined clusters, with tumor cells and non-tumor populations clustering away from one another (194). One major strength of this method is the ability to assess chromatin accessibility at specific cis-elements in disease-associated loci across multiple cell types. This allows for the annotation of tumor-specific, immune cell population-specific or stromal-cell specific active cis-elements. Aside from describing active and inactive chromosomal regions for various cell populations, scATAC-seq can also be combined with individual lentiviral integration site mapping, enabling researchers to examine where these sites fall in relation to open chromosome regions (208). This type of information can be useful in assessing whether integration of viral components in or near specific genes can be connected to robust expansion or in vivo persistence of CAR T cells (208). The same approaches could be used to assess how viral integration in certain chromosomal regions affects outcomes in gene therapy. These studies clearly demonstrate how this approach permits comparison of diverse cell populations that directly impact both the disease microenvironment and response to treatment.
In some diseases, therapeutic benefits may be attained through the de-repression of epigenetically silenced genes. One such example involves triggering the expression of fetal gamma-globin (HbF) to correct the pathophysiological defects associated with SCD (80, 209). One preclinical study aiming to identify a novel treatment for Fragile X syndrome used a directed DNA demethylation tool to remove methylation marks in the FMR1 promoter region, leading to increased FMR1 expression (210). Newly developed CRISPR/Cas9-mediated demethylation and methylation tools allow for the manipulation of the methylome (211–214). In order for these strategies to be developed into viable treatments, techniques such as scBS-seq will be required to ensure that targeting is specific and that it does not lead to outgrowth of modified cells.
Recent evidence suggests that changes in CAR T cell global methylation status may have some bearing on treatment efficacy. One study found enhanced proliferation and persistence of a dominant CAR T clone with biallelic disruption of the TET2 gene, which encodes a demethylating enzyme (121). Another study provided evidence that decitabine treatment-mediated epigenetic reprogramming of CAR T cells led to enhanced cytotoxicity and persistence (215). scBS-seq profiling of CAR T cells in a variety of patient samples has the potential to identify novel mechanisms that play a role in determining overall treatment response.
Single-cell epigenomic screening, such as scATAC-seq and scBS-seq, can provide crucial insights into the disease microenvironment, tumor-infiltrating lymphocytes or epigenetic disruption in disease. However, the rapid technological advances in single cell epigenomics posed a new challenge – the computational analysis of large data volumes. In addition, high background noise levels, low sequencing depth and limited capture rates of single-cell epigenetic screens restricts the analytical scope of pipelines developed for bulk sequencing protocols (216). Hence, current analytical strategies leverage a pseudo-bulk approach. First, single cells are aggregated for peak calling, then individuals cells are inspected for identified pseudo-bulk peaks (217). More recently, comprehensive tools have been developed to integrate dimensionality reduction, peak calling, identification of variable peaks, motif analysis, prediction of gene association and differentiation trajectories into single pipelines (218, 219).
Transcriptome
Single-cell RNA sequencing (scRNA-seq) is arguably the most widely applied and established single-cell molecular screening platform. Consequently, a multitude of novel scRNA-seq protocols and adaptations have been developed [extensively reviewed elsewhere: (220, 221)]. Amongst these, two major groups have emerged, primarily differing in sequence coverage to either profile full-length transcripts or sequence the 3’ or 5’ ends of captured transcripts. Picelli and colleagues pioneered Smart-seq2 for full-length transcriptomic profiling of hundreds of cells (38). Alternatively, platforms for 3’ mRNA profiling, such as Drop-seq (37) and more recently Chromium (10X Genomics) (222), utilise droplet-based microfluidic devices and unique molecular identifiers for massively high-throughput single-cell screens. This technological advance allowed profiling of tens or hundreds of thousands of cells at significantly reduced sequencing costs per cell compared to full-length profiling protocols. These high throughput techniques enable deep molecular profiling of complex tissues and are particularly beneficial for the identification of rare cell types. In contrast, full-length profiling protocols are not compatible with droplet-based approaches, thus reducing the throughput by 10- to 1000-fold at increased sequencing cost per cell (221). However, Smart-seq2 provides deeper sequencing coverage, resulting in the detection of a larger number of genes with fewer sequencing dropouts (223, 224), allowing much more robust conclusions about transcript co-expression in single cells. Increased sequencing depth also provides increased detection of low-abundance transcripts. Perhaps most useful, full-length transcript profiling also permits the detection of alternative splicing and novel transcripts (221). Taken together, both sequencing platforms provide a diverse toolbox to cover a broad range of biological questions, but it is imperative to choose the right tool for the biological question being addressed.
Multiple studies have demonstrated the utility of scRNA-seq in describing cell-cell interactions, discovering unique disease-associated cell populations, identifying minimal residual disease following treatment and even distinguishing host- versus donor-derived cells following transplantation (222, 225–228). These types of applications can easily be used to address a number of currently unanswered questions relating to all phases of the gene therapy process (Table 1). As a lower-cost alternative to WGS, scRNA-seq can be used to identify single nucleotide variants (SNVs) and splice variants in gene corrected cells (221, 229). Given that scRNA-seq is also particularly powerful in separating heterogeneous groups of cells (225), these datasets can be very useful in identifying genes and pathways relevant to the function of abnormal cell types that participate in the establishment of diseases such as diabetes (230, 231). In turn, this information can be employed to develop new therapeutic avenues.
Similar to its applications in gene therapy, scRNA-seq can also be used to dissect basic biological processes such as T cell development (232), aspects of which may inform the optimization of CAR T cell therapies. As discussed above, a number of studies profiling anti-CD19 CAR T cell populations before and after infusion into patients have been able to draw clinically relevant conclusions about transcriptional profiles that mark CAR T cells associated with both good and poor clinical outcomes (158, 232). scRNA-seq studies can also be used to examine interactions occurring within the tumor microenvironment between various endogenous immune cell types and CAR T cells (233).
Proteome
The eukaryotic proteome provides the greatest molecular complexity within the genotype-phenotype paradigm. With the addition of post-translational modification, the number of functionally distinct proteins considerably exceeds the ~20,000 identified protein-coding genes (234). In addition to the complexity of the proteome, the absence of protein amplification tools has limited our ability to perform unbiased proteomic screens. Traditional hypothesis-driven approaches, such as high-resolution microscopy, flow cytometry and immunohistochemistry, have enabled protein quantification at single cell resolution (235); however, these techniques are limited by the number of screened proteins, cell throughput, and the need to know the target a priori. These limitations are partly addressed by mass cytometry, a high-throughput quantitative screen for up to 60 proteins using currently available protocols and a theoretical capacity of up to 120 proteins (236). The principle of mass cytometry, or cytometry by time-of-flight (CyTOF), was based on the core concept of covalent conjugation of multiple individual antibodies with unique heavy metal reporter isotopes with district ion masses (237). In brief, single cells, labelled with a complex set of reporter-conjugated antibodies, are vaporised by inductively coupled plasma to release reporter ions for analysis by time-of-flight mass spectrometry (238–240). Unique ion mass sizes permit deconvolution and ultimately the quantitative comparison of labelled proteins on individual cells.
Pioneering work by Palii and colleagues utilised CyTOF to determine the role of lineage-specific transcription factors (LS-TF) in hematopoietic lineage specification (241). By performing a temporal screen during erythropoiesis, the authors demonstrated that multipotent progenitor populations undergo gradual LS-TF changes to commit to single lineages at the single cell level. Furthermore, CyTOF has been widely applied in immune cell profiling, biomarker discovery and treatment response studies (236, 242, 243). Such findings demonstrate the power of single-cell approaches to decipher complex molecular interactions, which would otherwise be masked in bulk studies.
As previously mentioned, one of the potential risks of virus-based gene therapy is the development of an immune response targeting the delivery vehicle. A major strength of CyTOF is its ability to profile multiple cell types simultaneously, allowing researchers to create snapshots of proteins being expressed both on the cell surface and intracellularly (244, 245). With the aim of determining whether healthy donor PBMCs were reactive to viral vector components used in many gene therapy clinical trials, Kuranda et al. simultaneously profiled cytokine secretion, immune cell activation, and T cell exhaustion using CyTOF (246). Different immune cell responses were observed, some of which correlated with whether or not the donor had previously been exposed to the virus originally used to develop clinical viral vectors. These findings indicate that it may be possible to predict which patients will go on to develop vector immunogenicity (246). This type of approach can also be applied to the monitoring of immune cell interactions following CAR T cell infusion.
While CyTOF was originally developed for the screening of suspension cells, Giesen et al. pioneered imaging mass cytometry (IMC) to introduce spatially resolved mass cytometry of ~30 proteins (247). Giesen and colleagues elegantly combined traditional immunohistochemistry with laser ablation and mass cytometry, thus enabling mass cytometric screening across tissue sections with subcellular resolution. Two concurrent studies utilised IMC for screening islets and the immune cell compartment of type 1 diabetes patients at single-cell resolution (248, 249). The authors demonstrated the alterations in islet topology during disease progression and the role of T lymphocytes in β-cell destruction.
As outlined above, high-throughput single-cell phenotyping plays a crucial role in gene and cell therapy. CyTOF and other flow cytometry-based technologies, such as full spectrum flow cytometry (FSFC) and Chipcytometry, enable phenotyping of dozens of distinct cell types (250, 251). In brief, Chipcytometry utilises microfluidics to enable iterative inspection of cell surface markers, while FSFC relies on full spectral acquisition to enable parallel screening of dozens of cell surface markers (250, 251). Near limitless throughput and high capture efficiency paired with the ability to distinguish rare cell populations provides a powerful tool for immunophenotyping. Indeed, FSFC has been successfully applied to identify therapy-mediated alterations in peripheral blood mononucleocyte profiles of head and neck squamous cell carcinoma patients (252).
Despite these advances, the high cell throughput and complexity of acquired CyTOF data provides a significant computational challenge and remains a key focus area for technical development [comprehensively reviewed elsewhere: (253)]. Recent technological advances in mass spectrometry and upstream sample processing have also raised the prospect of unbiased proteomic screens. Separate work by the Slavov and Mann groups have shown a capacity to capture ~3000 and ~800 proteins per cell, respectively (254–256). At present, however, the technology is prohibitive for routine application and will require substantial development to become a powerful tool in the near future.
Multimodal Sequencing of Complex Tissues
The development of single-cell uni-modal sequencing platforms to independently interrogate the genome, epigenome, transcriptome or proteome has raised the prospect of screening multiple components simultaneously (multimodal profiling).
Numerous approaches for separating genomic DNA and mRNA from the same single cell have been proposed [various approaches extensively reviewed elsewhere: (163)]. Amongst these, the elegant G&T-seq protocol, pioneered by Macaulay et al., separates mRNA from genomic DNA by using magnetic beads and biotinylated oligo(dT) primers against poly-A tails of mRNA molecules (Figure 3 and Table 2) (48). The full-length transcript profiling in G&T-seq assays provides a powerful tool for identifying alternatively spliced transcripts, fusion transcripts and expression of single nucleotide variants (SNVs) (269). The ability to associate such information with DNA copy number and structural variants at the single cell level allows unprecedented insight into the relationship of the genotype and its gene expression profiles. Nevertheless, manual separation of DNA and mRNA during the G&T-seq protocol increases sample handling, thereby limiting the throughput to hundreds of cells (269) which is further compounded by the high sequencing costs to ensure sufficient genome coverage.
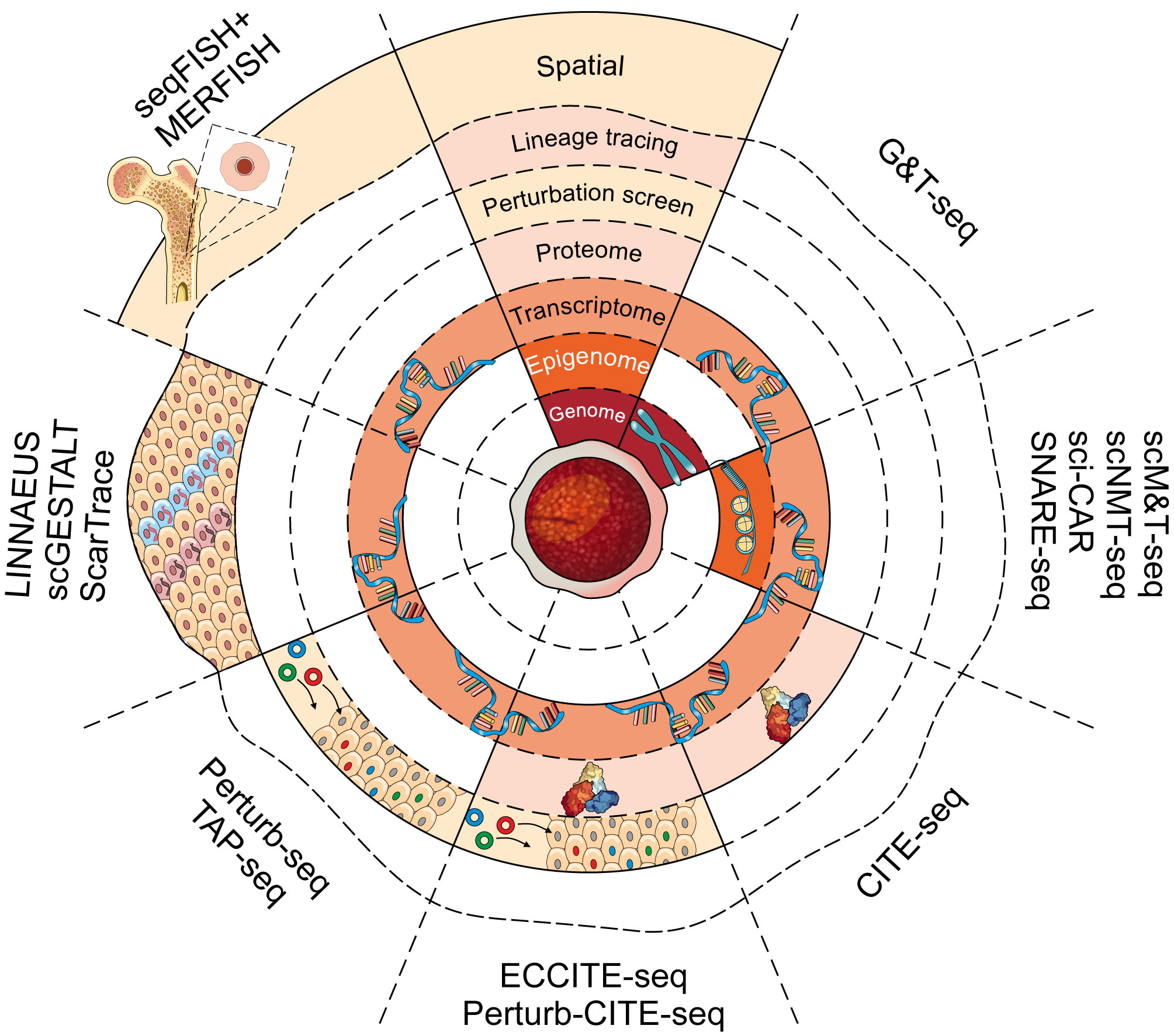
Figure 3 Single-cell multimodal platforms and their uses. A number of recently developed technologies can be used to assess the genomic, transcriptomic, epigenomic and proteomic landscape of a single cell. Each layer of the concentric circle represents a different molecular dimension that can be assessed using each method (from inside to outside: genome, epigenome, transcriptome, proteome, genetic perturbation, lineage tracing, spatial transcriptome). Method names are indicated along the periphery.

Table 2 Multimodal single-cell tools.
Whole genome sequencing (WGS) approaches provide a crucial tool for characterizing genomic abnormalities in primary tumors (270). Zhu et al. recently applied G&T-seq to a subset of lymphovascular invasive cells, isolated from a breast cancer patient (271), describing the relationship between RNA and CNV clones and outlining multiple functionally distinct clones and their role in metastatic dynamics. This illustrates the power of G&T-seq to uniquely integrate genomic abnormalities with transcriptional consequences, potentially of substantial utility in deciphering tumor heterogeneity and intra-tumoral clonal dynamics post CAR T therapy.
Existing epigenetic single-cell assays have also been adapted to enable multimodal approaches (Figure 3 and Table 2). For example, Angermueller et al. adapted the existing principles of G&T-seq by introducing a bisulfite treatment step which allowed DNA methylation profiles and gene expression to be obtained from the same cell (scM&T-seq) (46). A more recent adaptation to the scM&T-seq protocol introduced chromatin accessibility as the third dimension for simultaneous single-cell nucleosome, methylation and transcription sequencing (scNMT-seq) (257). Here, a methyltransferase is used to label accessible DNA prior to scBS-seq. Such labelling permits downstream computational deconvolution of DNA methylation and chromatin accessibility profiles (272). To date, scM&T-seq and scNMT-seq have provided intriguing insight into stem cell biology and mouse gastrulation. For instance, pioneering work by Argelaguet and colleagues described the role of epigenetic priming at lineage-specific enhancers during lineage commitment (205). A second pioneering study revealed that changes in DNA methylation drive increasing transcriptional heterogeneity during stem cell ageing (273). These studies demonstrate the impact of a multi-modal scNMT-seq for characterising the role of the epigenome in complex tissues and biological processes, including the underlying cellular heterogeneity.
Taking into account the role of DNA methylation in driving autoimmune defects, age-related diseases and tumorigenesis (274, 275), scNMT-seq can provide a powerful and versatile tool for uncovering novel therapeutic avenues. These principles can also be applied for assessing the extent to which normal tissue function can be restored following corrective gene therapies. Similarly, multimodal epigenetic and gene expression profiling can provide a valuable tool for characterizing the tumor microenvironment and its interaction with CAR T cells to increase therapeutic efficacy. However, the relatively low-throughput of scNMT-seq can limit the coverage of large, complex tissues.
To determine the impact of cis- and trans-regulatory elements on gene expression profiles, collecting chromatin accessibility and gene expression profiles from the same cell are of paramount importance. Cao et al. pioneered sci-CAR to simultaneously perform nuclear scRNA-seq and scATAC-seq (258) by adapting previously established principles of single-cell combinatorial indexing to barcode mRNA and open chromatin regions from single nuclei extracts. Shortly thereafter, Chen and colleagues developed SNARE-seq for performing simultaneous gene expression and chromatin accessibility profiling (259). In contrast to sci-CAR, SNARE-seq utilized the high-throughput Drop-seq platform to incorporate single nuclei and adapter-coated beads. Upon nuclei lysis within each droplet, released nuclear RNA and chromatin fragments bind to the uniquely barcoded beads allowing connectivity of ATAC-seq and RNA-seq profiles of individual cells. Furthermore, SNARE-seq enabled significantly improved capture of chromatin fragments and improved the transcript sequencing depth (259). That said, the potential of SNARE-seq is partially restricted by the complexity of downstream data analysis and this prompted the development of integrated analysis pipelines, such as Signac (218) and the Chromium Single-Cell Multiome ATAC + Gene Expression platform. The simplification of the sample preparation process and analysis pipelines will be required to facilitate the wider adoption of multi-modal epigenetic and gene expression screening.
A vast array of computational tools has been developed for the analysis of unimodal single cell data. For instance, advances in dimensionality reduction, clustering and algorithms for identifying marker genes, constructing lineage trajectories and batch correction contributed greatly to current widespread access to scRNA-seq analysis tools (163). The assembly and curation of key tools into unified analysis pipelines, such as Seurat, SCRAN or SCANPY, has enabled bench-trained scientists to independently analyse scRNA-seq data (276–279). Datasets from multimodal analysis with distinct cellular dimensions inherently do not share common features (280), making data integration across distinct modalities from the same cell a profound and novel computational challenge. To integrate multiple modalities collected from the same cells into a single reference describing cell identities, Hao et al. developed a Weighted Nearest Neighbour (WNN) framework (281). In brief, WNN utilises nearest neighbour analysis and computes modality weights to derive a single landscape, reflecting the similarities of all modalities. The increased adoption of single-cell multimodal screens provides another computational challenge - the integration of multimodal data across distinct experiments, platforms and batches. While multiple strategies to integrate and batch-correct unimodal scRNA-seq datasets have been proposed (278, 282), their applicability to multimodal datasets is limited. To overcome this limitation, Stuart and colleagues adapted canonical correlation analysis and L2 normalisation to derive anchors for data integration (283). To enable integration in a variety of experimental settings, several anchoring methods have been proposed [reviewed in (284)]. Nevertheless, the rapidly expanding landscape of novel multimodal screening technologies continues to require bespoke analytical approaches and recent developments in multimodal data analysis are expansively reviewed elsewhere (163, 280).
Overall, technological advances have resulted in an unprecedented proliferation of novel single-cell molecular assays. Intriguingly, the capability of incorporating such approaches to acquire multiple elements from single cells has allowed the interrogation of the direct relationship of multiple molecular dimensions. Such extensive single-cell profiling is particularly beneficial for application in future cell therapies where the interrogation of tumor infiltrating lymphocytes and tumor microenvironments will provide a crucial component for target discovery and monitoring of therapeutic efficacy. Due to the heterogeneous nature and shifting clonal dynamics of malignant tissues, single-cell approaches are of paramount importance for the development of effective cell therapies.
Multimodal Single-Cell Approaches Integrating Functional and Molecular Data
Simultaneously acquiring functional and molecular readouts from the same cells have historically represented an experimental challenge, as omics profiling tools typically result in destruction of the target cell. This is particularly challenging when the functional state of a cell is determined by a retrospective assay, thereby making its prospective isolation and molecular characterization impossible. Hence, most technical developments that combine functional and molecular multimodal approaches have focused on capturing cellular function prior to a destructive single-cell assay.
Transcriptome and the Cell Surface Proteome
One of the first applications of multimodal omics technologies arose from the desire to connect cell surface phenotypes with gene expression profiles. Several well-characterized biological systems, particularly immune cell subtypes and hematopoiesis, have benefited from in-depth characterization of cell surface markers for a variety of functionally distinct cellular populations (285). As a result, quantitative phenotypic information of selected cell surface markers can permit inference of cellular function. Fluorescence-activated cell sorting (FACS) in combination with index sorting allows simultaneous recording of cell surface protein levels prior to deposition in lysis buffer for downstream destructive molecular assay, such as the Smart-seq2 protocol for gene expression profiling (38). The application of such approaches has allowed the linkage of stem cell function with global molecular profile for the first time and provided numerous insights into our understanding of transcriptional heterogeneity throughout hematopoiesis (44, 285–287).
Strategies involving index sorting and downstream scRNA-seq are particularly powerful when combined with functional outcome analyses. Wilson et al. and others have shown how these methods can be applied to understanding the heterogeneity inherent to many normal tissues and identifying features that differentiate normal and disease-causing cell types (44, 287–292). These methods would be particularly useful in linking T cell function to distinct gene expression profiles, allowing for the identification of subpopulations of cells that are associated with specific clinical outcomes.
Nevertheless, isolation strategies of functional cell types frequently do not achieve homogeneity and contaminating cells cannot be fully excluded from destructive molecular assays. This is in contrast to selective single-cell functional assays that can distinguish truly functional cells from contaminants, meaning that cellular heterogeneity is often the first to be identified (i.e., they drop out of the assay and do not generate a confusing data point) (293). Furthermore, cell isolation by FACS requires prior knowledge of distinct cell types, thereby precluding the discovery of novel cell types. In addition, index-sorting FACS-based approaches are not compatible with droplet-based high-throughput sequencing platforms. To overcome these limitations, Stoeckius et al. pioneered CITE-seq (cellular indexing of transcriptomes and epitopes by sequencing, Figure 3 and Table 2) (45). Here, antibodies against cell surface proteins of interest are labelled using unique oligonucleotide barcodes. Antibody-labelled cells are subjected to the Drop-seq protocol, encapsulating single cells in droplets containing beads to introduce unique cellular barcodes to mRNA and the antibody-derived tags (ADTs). Subsequently, ADT counts are used to quantify antibody-bound cell surface proteins and provide a link to the corresponding single-cell gene expression profiles. Consistent surface proteome quantification and resolution were achieved compared to traditional flow cytometry approaches, while providing a theoretically unlimited scope for antibody multiplexing (45).
The application of CITE-seq in tumor microenvironment biology has been noted previously (294, 295). Praktiknjo et al. screened healthy and tumor-bearing mouse salivary glands, including the immune compartment of the tissue (295). By performing CITE-seq, the authors were able to construct a comprehensive gene expression atlas and simultaneously recorded a comprehensive set of 63 immune-specific cell surface proteins. Most notably, they derived a comprehensive cell atlas of the tumor microenvironment, using gene expression profiles and quantification of cell surface proteins, underscoring the utility of CITE-seq in the discovery of novel tumor-specific cell surface antigens for cell therapy. By linking surface protein quantification with gene expression profiling at single cell resolution, CITE-seq can identify novel antigens associated with specific clones within heterogeneous cancer tissues, ultimately raising the prospect of a broader spectrum of effective cell therapies. The efficacy of multimodal single-cell screens, such as CITE-seq has been particularly evident throughout the scientific response to the COVID-19 outbreak. Combined efforts to screen >780,000 single PBMCs from COVID-19 patients and healthy donors using CITE-seq revealed the immune response to COVID-19 infections and its role in disease pathology (296). Such studies provide a prominent example how single-cell multiomics can provide rapid insight into previously unknown diseases and help inform the development of effective therapeutics.
Perturbation Screens
Large-scale perturbation screens have previously provided unprecedented insights into gene functions and their role in complex biological mechanisms (297). The advent of CRISPR/Cas9 has revolutionized our ability to conduct high-throughput perturbation screening and multiple groups have now developed multimodal single-cell perturbation screens, combining CRISPR technology with scRNA-seq (47, 298–301). In Perturb-seq (Figure 3 and Table 2), a pool of barcoded single-guide RNAs (sgRNAs) is constructed against a set of 24 transcription factors and transduced cells are subjected to high-throughput droplet-based sequencing, whereby unique cell barcodes are also introduced. The dual barcoding approach allows connection of single-cell gene expression profiles with a respective perturbation. Such single-cell CRISPR screens and their ability to interrogate transcriptional consequences of perturbations provided a novel method to assess the functional effectors of complex biological mechanism and tissues (301, 302). Of note, Jin et al. demonstrated the application of Perturb-seq in an in vivo setting (303). To interrogate the underlying molecular mechanisms driving autism, the authors introduced a guide RNA pool against risk genes to the forebrain of a developing embryo in utero. The progeny of perturbed cells was then collected at P7 for downstream scRNA-seq analysis, providing key insights into the molecular mechanisms of neocortical cell types.
Perturb-seq can be very useful in trying to understand larger pathways that integrate multiple signals. For example, Adamson et al. used Perturb-seq to understand how activation of the unfolded protein response (UPR) differed between individual cells (301). This type of data has the potential to disentangle larger signaling networks, all of which is important for understanding complex processes such as immune responses.
Despite the demonstrated efficacy, application of Perturb-seq is limited by the sequencing depth of high-throughput approaches. Acquired data is subject to significant background noise and low-abundant transcripts are frequently missed (47, 298). Furthermore, the multiplicity problem of combining multiplexed perturbations with single-cell gene expression profiles poses a computational challenge. Schraivogel proposed an intriguing adaptation, termed targeted Perturb-seq (TAP-seq) (262). By performing targeted amplification of a selected set of genes prior to sequencing, the cost and analytical complexity could be significantly reduced. This approach provides a powerful tool for screening cellular pathways with defined genetic biomarkers. In the context of cell therapy, TAP-seq could thus provide a cost-effective tool for identifying underlying molecular mechanisms of immune cell evasion of CAR T therapy.
There have been a wide variety of additional approaches to integrate single-cell perturbation screens with the surface proteome of the same cell. Most notably, Mimitou et al. proposed ECCITE-seq (260) and Frangieh et al. described Perturb-CITE-seq (261). In brief, Mimitou et al., adapted the existing CITE-seq protocol by introducing addition oligonucleotides against unique sgRNA identifiers to cellular barcoding beads. Thus, sgRNA, transcripts, antibody-oligonucleotides and up to 2 other parameters can be recorded for individual cells (260). More recently, Frangieh et al. proposed Perturb-CITE-seq to provide a scalable solution for Perturb-seq with simultaneous screening of cell surface proteins (261). Here, the authors demonstrated the benefits of Perturb-CITE-seq by identifying molecular pathways driving immune evasion of a melanoma cell line against primary tumor infiltrating lymphocytes (261). Overall, the ability to connect gene expression profiles and the cell surface proteome from single cells under perturbation provides a comprehensive characterisation of complex molecular systems. As demonstrated by Frangieh et al., such technologies can help identify and characterize immune evasion drivers and ultimately reveal novel targets that might lead to enhanced therapeutic potency of immunotherapies.
Clonal Tracking and Lineage Tracing
Recent work by Lee-Six et al. outlined the application of whole genome sequencing (WGS) approaches to establish the clonal dynamics of human HSPCs (182). The authors isolated single HSPCs from a healthy donor and were able to retrospectively reconstruct the phylogenetic tree of single cell-derived colonies, based on a broad set of shared or unique acquired somatic mutations. By simultaneously screening mature cells isolated from peripheral blood samples of the same individual, Lee-Six et al. were able to infer the progeny and extended relatedness of stem cell clones. Using this approach in a 59 year old human, the authors could map all the way back to the most recent common ancestor for blood and buccal epithelium, observed an early expansion of the stem cell compartment and confirmed hematopoietic activity of a large number of diverse HSC clones estimated to be between 50,000 and 200,000 actively contributing HSCs (162, 182).
This technique could be powerfully applied to gain insight into the clonal dynamics of HSCs used in gene therapy. Careful patient monitoring must be undertaken to ensure therapeutic efficacy and restoration of normal tissue function. As multipotent cells provide the most common target for gene therapies, gene corrections can significantly impact the clonal dynamics of the target tissue. Intriguingly, previous efforts to track therapeutic efficacy of corrective therapies large depended on monitoring progeny cells, their homeostatic function and particularly the proportion of target cells expressing the desired gene edit (159, 304). However, such approaches do not provide sufficient resolution to fully characterize clonal dynamics of corrected cell types and their impact on homeostatic tissue function. WGS of single cell-derived colonies allows to monitor naturally occurring somatic mutations in multipotent cells and their progeny to establish their relationship and infer clonal dynamics of single cells (162). When applied to a pool of edited cell and mature cell progeny post-gene therapy, such approaches can provide a direct insight into therapeutic efficacy and long-term tissue health.
In contrast, upfront labelling of target cells followed by temporal tracking of their progeny can reveal patterns of clonal evolution. Here, the advent of routine and cost-effective sequencing also revolutionised lineage tracing, providing a compelling alternative to traditional imaging-based approaches. In the context of diabetes, lineage tracing has been used to track the various cell types which originate from pancreatic progenitor cell populations (305–307) and identify cell types that are able to transdifferentiate into insulin-secreting cells (110, 308, 309). High-throughput screening at single cell resolution and integration into multimodal approaches greatly expand the scope of lineage tracing (310). While fluorescent tags limit the capacity of parallel barcoding, DNA sequence complexity provides a scalable barcoding approach. In principle, unique DNA barcodes are first introduced into a large population of target cells. Subsequently, amplification of the unique set of DNA barcodes in cell progeny can be used to compute lineage phylogenies (311, 312). A prominent barcoding approach relies on CRISPR/Cas9-mediated dynamic lineage tracing. Here, CRISPR/Cas9-mediated double-stranded breaks are introduced at defined genomic loci (313). The resulting insertions and deletions (indels) create unique cellular barcodes, which evolve over time. By sequencing such regions, the mutational patterns can be used to establish phylogeny and clonal evolution. Multiple groups have independently pioneered such CRISPR/Cas9-based lineage tracing approaches, which predominantly differentiate in the number of loci used to store lineage barcodes (263, 265, 314–318). Of note, using genome editing of synthetic target arrays for lineage tracing (GESTALT), McKenna et al. were able to trace and reconstruct early developmental pathways in a whole organism.
Dynamic lineage tracing protocols outlined above have been integrated in multimodal screens to link cellular progeny to their respective gene expression profiles, including single-cell GESTALT (scGESTALT), linear tracing by nuclease-activated editing of ubiquitous sequences (LINNAEUS) and ScarTrace (Figure 3 and Table 2) (263–265). Raj et al. integrated the underlying principles of GESTALT with scRNA-seq to simultaneously acquire lineage information and gene expression profiles of the same cell (264). Instead of targeted sequencing of genomic DNA, scGESTALT relies on sequencing of expressed transgenes, which encode the unique cellular barcode. The use of droplet-based high-throughput gene expression thus provides cell type information, otherwise lost in previous lineage tracing protocols. Intriguingly, the LINNAEUS and ScarTrace protocols introduce barcodes in fluorescent transgenes to allow monitoring of successful integration of cellular barcodes. Thus, providing a crucial quality control mechanism prior to performing computational- and capital-intense sequencing (263, 265).
While prospective lineage tracing is not possible in humans, the use of these techniques in preclinical studies has the potential to unlock cellular relationships that are relevant to understanding cell origins in normal and diseased tissues. Furthermore, lineage tracing may also be used to link immature immune cell types to their immunologically active terminally differentiated counterparts. This could feed into refinements of CAR T cell production protocols for example, allowing for the selection of specific populations with maximal effector function (117).
Nevertheless, these multimodal lineage tracing technologies are currently in their infancy and a variety of experimental and computational limitations require attention. Shallow sequencing depth of high-throughput approaches can prevent barcode detection and CRISPR/Cas9-induced cell toxicity has recently been described, thus potentially disrupt the effective construction of phylogeny or distort separation of cell types (310, 319, 320). Furthermore, Spanjaard et al. noted the probability of double scarring, whereby a subset of non-homologous end joining-mediated errors have a higher probability of occurring (263). Thus, if not excluded, high-frequency scars can result in false inference of lineage relationship. To address the issue of barcode duplications and noise, Zafar et al. recently proposed a novel analytical pipeline for improved lineage tree reconstruction and integration of separate single-cell lineage tracing experiments (49). While these advances are promising, further computational innovation will be of paramount importance for the adoption of single-cell lineage tracing in gene and cell therapy developments.
Introducing Spatial Resolution in Gene and Cell Therapy
Single-cell sequencing technologies and their multimodal integration continue to push the boundaries of understanding the mechanisms governing complex tissue organization. However, such single-cell screening protocols are largely based on removing the cells and destroying them, typically discarding any spatial information of the underlying tissue from which they were extracted. The crucial role of cellular location and spatial gene expression throughout early embryogenesis has been widely recognized (321). Similarly, cellular location in heterogeneous tumors and the surrounding tumor microenvironment are vital to cell function (322). Therefore, resolving spatial dimensions and linking these with gene expression profiles to infer gene function and cell identity can help us understand disease pathology and complex tissue function. Here, we discuss selected technological developments in spatial transcriptomics and their prospect in the development of novel cell and gene therapies [spatial omics protocols are comprehensively described elsewhere: (321, 323)].
The development of fluorescence in situ hybridisation (FISH) techniques first enabled the detection of DNA and RNA molecules in structurally preserved, fixed tissue sections (43, 324, 325). Oligonucleotides, complementary to a target nucleotide sequence, are labelled with single or multiple fluorophores. In turn, fluorescently labelled oligos bound to a target region can be observed using optical microscopy. Ultimately, the principles of FISH facilitated quantitative detection of mRNA at subcellular resolution (43, 324, 326). Here, the authors constructed a library, consisting of short single fluorophore-labelled oligos, against a single mRNA target to estimate the number of mRNA molecules in a single cell, screening up to 3 mRNA sequences in parallel.
To enable high-throughput spatial transcriptomic screening, Lubeck et al. first established the principles of sequential FISH (seqFISH), providing a strategy with theoretically whole transcriptome coverage (327, 328). In brief, multiple single fluorophore-labelled probes are used for mRNA labelling during a single hybridization round. By stripping probes and performing multiple rounds of hybridisation, the number of unique barcoding increases exponentially. Shah et al. demonstrated the efficacy of seqFISH for screening hundreds of genes at sub-cellular resolution, providing a novel insight into the spatial organisation and transcriptional heterogeneity of the mouse brain (329). The recent introduction of an additional fluorophore to sequential hybridisation allowed further scaling of seqFISH (seqFISH+) (Figure 3 and Table 2) (266). This strategy avoids optical crowding by effectively diluting mRNA molecules into separate images. The result was a robust protocol for screening 10,000 genes in spatially resolved tissues, spanning thousands of cells (266). Here, the use of confocal microscopy for the seqFISH+ protocol provides a key advantage to facilitate wider adaption. A recent study by Lohoff et al. applied seqFISH to construct spatially resolved gene expression profiles for mouse organogenesis using a computational framework for the integration of spatially-resolved gene expression maps with scRNA-seq profiles of cell types in early mouse development (330, 331). In parallel, Chen et al. pioneered a multiplexed error-robust FISH (MERFISH) approach which combined error-corrected barcoded probes and sequential imaging to perform a multiplexed screen of hundreds of genes (Figure 3 and Table 2) (267). Further MERFISH developments, such as the use of expansion microscopy, enabled quantification of thousands of genes in hundreds spatially resolved cells at a detection efficiency of ~80% (268). This high capture efficiency is a major advantage of MERFISH.
While the methods outlined above drove innovation in spatial transcriptomics, their relative infancy is accompanied by experimental and computational complexity, which currently provides a barrier to wide-spread adoption. Several commercially available platforms have been established to provide a standardised experimental framework. The Visium platform utilised NGS for deriving spatially resolved gene expression profiles (323, 332). Here, a tissue section of interest is deposited onto a slide, coated with uniquely barcoded arrays (barcode spacing permits 55um resolution). Following barcoding of captured mRNA molecules, cDNA libraries are subjected to high-throughput NGS and spatial deconvolution based on the unique barcoding. However, the current barcode spacing prevents interrogation of neighbouring cells. Here, in situ analysis can provide a complementary approach, allowing interrogation of a defined set of mRNA targets at spatial singe cell resolution (333–335).
Collectively, spatial transcriptomics technologies are currently in the developmental and early adaption phase. As a result, several key limitations persist. For instance, the tissue-dependent optimisation and sequential hybridisation rounds require significant experimental time, while the use of customised equipment also impacts implementation. However, increasing throughput and the desire to reach whole-transcriptome coverage will greatly increase imaging time and data complexity, making the most prominent limiting factor the development of robust analytical tools. To overcome the computational barrier, recent advances aim to address key unmet needs in data analysis and its scalability (336, 337).
Despite these challenges, several major advances have already been made using spatial transcriptomics, including studies in tumor heterogeneity and transcriptional changes in the microenvironment. In one study, Berglund et al. constructed a comprehensive spatial map of tumor and healthy prostate tissue biopsies from a prostate adenocarcinoma patient (322). The authors uncovered significant transcriptional differences between the tumor core and its periphery. Intriguingly, thorough interrogation of stromal and immune cell types, surrounding the primary tumor, facilitated the identification of heterogeneous gene expression networks in the tumor microenvironment (322). Spatial transcriptomics has also been applied for mapping the localisation of Cxcl12-abundant reticular cells in the bone marrow niche and for the characterisation of stromal cell heterogeneity in tumor microenvironments (338, 339). These and other studies demonstrate that the potential of spatial transcriptomics in deciphering tumor architecture, heterogeneity and microenvironments has been widely recognised. Beside its role in therapeutic discovery and disease pathology, spatially resolved gene expression profiles can become of paramount importance for monitoring therapeutic outcomes of cell therapies and identify evasion mechanisms in response to cell therapies. In addition, spatial characterisation post CAR T cell therapy could provide an insight into the impact of off-target effects on the function of proximal tissues. Similarly, spatial transcriptomics could aid in long-term monitoring of patients undergoing corrective gene therapies.
Concluding Remarks
The past decade has produced an abundance of novel single-cell molecular tools, facilitating the unbiased screening of a wide array of molecular dimensions at unprecedented resolution. Unimodal sequencing technologies have proved particularly impactful in the first wave of wide-scale adoption, but more approaches have been focused on combining such techniques into multimodal screens to allow simultaneous capture of multiple molecular dimensions from the same cell. These technologies have allowed researchers to unpick the molecular mechanisms driving disease pathology at a scale not previously considered possible. Tissue and disease heterogeneity, previously masked in bulk sequencing approaches, are now routinely being explored at single cell resolution.
Techniques such as scRNA-seq have been widely adopted due to the production of robust experimental protocols and increasing consensus surrounding the computational approaches for quality control and data analysis. On the other hand, multimodal screens have not yet enjoyed similar uptake due to their reliance on high sequencing costs, advanced integrative computational tools and technical expertise. However, just as moving to single cells was a technical hurdle of 10 years ago, the research benefits derived from novel multimodal screening platforms will push the limits of discovery and accelerate technical development and method standardization. The next few years should see these technical and computational approaches streamlined to create reproducible protocols and standardised analytical pipelines to facilitate rapid adoption rates, as has occurred for scRNA-seq historically.
Concomitant with the technical challenges and need for standardization, the increased accessibility of single-cell technologies has exponentially increased the amount of data generated during these studies. This provides a unique opportunity to leverage the power of these studies by integrating datasets but also makes for substantial computing and processing challenges. Batch correction and data integration across experiments and different sequencing platforms are areas that will require particular attention and novel computational approaches for handling and analysing increasing amounts of data will be of paramount importance. Ultimately, the continuous technical improvements and aggregation of data could provide the foundation for a fully characterized reference atlas of the human body at single cell resolution. The drive towards such a resource is evident in the recently announced efforts to establish a common coordinate framework (CCF) for data collection and integration (340). In line with that, initiatives such as the Human Biomolecular Atlas Program and the CCF aim to provide a publicly available tool to help researchers map data from diseased states onto healthy single-cell datasets and provide a reference for the entire scientific community (340, 341).
A number of recent studies have clearly demonstrated the utility of these approaches in (1) understanding complex biological processes such as cell fate determination and immune response, (2) dissecting tissue and disease heterogeneity, and (3) stimulating innovative research aimed at developing novel therapeutics (342–344). Within the next decade, it is anticipated that an increasing number of patients across many disease types will be treated with gene and cell therapy. Using samples obtained from these growing patient cohorts, single-cell technologies will undoubtedly be used to answer essential questions related to the relationships between disease-causing cells, normal or corrected cell types, tumor-targeting lymphocytes such as CAR T cells, and endogenous immune populations. For autoimmune diseases such as type 1 diabetes where the risk of relapse is relatively high due to immunogenicity, this level of detail will be essential to finding new ways to increase treatment efficacy. Additionally, due to the relatively recent wider application of these therapeutics, only a limited number of gene or cell therapy clinical trial patients have been monitored for more than 10 years following treatment initiation (65, 84, 345–347). Depending on the stability of edited cells and the influence of other comorbidities, detailed studies using single-cell approaches may also become relevant during long-term follow up. As patients enter the later decades of life, the intersection of age-related and treatment-related abnormalities may present unique clinical challenges. Further refinements and innovations to single-cell profiling technologies have the potential to unlock and disentangle relationships between key drivers of disease phenotypes, leading to wider delivery of authentic personalised medicine.
Author Contributions
DB and AC wrote and compiled the review. JR-L designed and created the figures. DK supervised the work and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The DK laboratory is supported by an ERC Starting Grant (ERC-2016-STG–715371), an MRC-AMED joint award (MR/V005502/1), and the Bill and Melinda Gates Foundation (INV-002189). DB was supported by a Wellcome PhD Studentship and AC by the Bill and Melinda Gates Foundation (INV-002189).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Tim Lohoff for the valuable discussions and advice on current development in multi-omics and spatial transcriptomics.
References
1. Till JE, McCulloch EA. A Direct Measurement of the Radiation Sensitivity of Normal Mouse Bone Marrow Cells. Radiat Res (1961) 14:213. doi: 10.2307/3570892
2. Becker AJ, McCulloch EA, Till JE. Cytological Demonstration of the Clonal Nature of Spleen Colonies Derived From Transplanted Mouse Marrow Cells. Nature (1963) 197:452–4. doi: 10.1038/197452a0
3. Kingston R, Jenkinson EJ, Owen JJT. A Single Stem Cell can Recolonize an Embryonic Thymus, Producing Phenotypically Distinct T-Cell Populations. Nature (1985) 317:811–3. doi: 10.1038/317811a0
4. Pike BL, Vaux DL, Clark-Lewis I, Schrader JW, Nossal GJ. Proliferation and Differentiation of Single Hapten-Specific B Lymphocytes Is Promoted by T-Cell Factor(s) Distinct From T-Cell Growth Factor. Proc Natl Acad Sci USA (1982) 79:6350–4. doi: 10.1073/pnas.79.20.6350
5. Spangrude G, Heimfeld S, Weissman I. Purification and Characterization of Mouse Hematopoietic Stem Cells. Science (80-) (1988) 241(4861):58–62. doi: 10.1126/science.2898810
6. Itoh Y, Germain RN. Single Cell Analysis Reveals Regulated Hierarchical T Cell Antigen Receptor Signaling Thresholds and Intraclonal Heterogeneity for Individual Cytokine Responses of CD4+ T Cells. J Exp Med (1997) 186:757–66. doi: 10.1084/jem.186.5.757
7. Osawa M, Hanada K, Hamada H, Nakauchi H. Long-Term Lymphohematopoietic Reconstitution by a Single CD34-low/negative Hematopoietic Stem Cell. Science (1996) 273:242–5. doi: 10.1126/science.273.5272.242
8. Mullis KB, Faloona FA. Specific Synthesis of DNA In Vitro Via a Polymerase-Catalyzed Chain Reaction. Methods Enzymol (1987) 155:335–50. doi: 10.1016/0076-6879(87)55023-6
9. Scharf SJ, Horn GT, Erlich HA. Direct Cloning and Sequence Analysis of Enzymatically Amplified Genomic Sequences. Science (80-) (1986) 233:1076–8. doi: 10.1126/science.3461561
10. Brady G, Barbara M, Iscove NN. Representative In Vitro cDNA Amplification From Individual Hemopoietic Cells and Colonies. Methods Mol Cell Biol (1990) 2:17–25.
11. Eberwine J, Yeh H, Miyashiro K, Cao Y, Nair S, Finnell R, et al. Analysis of Gene Expression in Single Live Neurons. Proc Natl Acad Sci USA (1992) 89:3010–4. doi: 10.1073/pnas.89.7.3010
12. Van Gelder RN, Von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA Synthesized From Limited Quantities of Heterogeneous cDNA. Proc Natl Acad Sci USA (1990) 87:1663–7. doi: 10.1073/pnas.87.5.1663
13. Fodor SPA, Read JL, Pirrung MC, Stryer L, Lu AT, Solas D. Light-Directed, Spatially Addressable Parallel Chemical Synthesis. Science (80-) (1991) 251:767–73. doi: 10.1126/science.1990438
14. Schena M, Shalon D, Davis RW, Brown PO. Quantitative Monitoring of Gene Expression Patterns With a Complementary DNA Microarray. Science (80-) (1995) 270:467–70. doi: 10.1126/science.270.5235.467
15. Gresham D, Dunham MJ, Botstein D. Comparing Whole Genomes Using DNA Microarrays. Nat Rev Genet (2008) 9:291–302. doi: 10.1038/nrg2335
16. Shendure J, Mitra RD, Varma C, Church GM. Advanced Sequencing Technologies: Methods and Goals. Nat Rev Genet (2004) 5:335–44. doi: 10.1038/nrg1325
17. Fan JB, Chee MS, Gunderson KL. Highly Parallel Genomic Assays. Nat Rev Genet (2006) 7:632–44. doi: 10.1038/nrg1901
19. Kurimoto K, Yabuta Y, Ohinata Y, Ono Y, Uno KD, Yamada RG, et al. An Improved Single-Cell cDNA Amplification Method for Efficient High-Density Oligonucleotide Microarray Analysis. Nucleic Acids Res (2006) 34(5):e42. doi: 10.1093/nar/gkl050
20. Jensen KB, Watt FM. Single-Cell Expression Profiling of Human Epidermal Stem and Transit-Amplifying Cells: Lrig1 Is a Regular of Stem Cell Quiescence. Proc Natl Acad Sci USA (2006) 103:11958–63. doi: 10.1073/pnas.0601886103
21. Xie D, Chen CC, Ptaszek LM, Xiao S, Cao X, Fang F, et al. Rewirable Gene Regulatory Networks in the Preimplantation Embryonic Development of Three Mammalian Species. Genome Res (2010) 20:804–15. doi: 10.1101/gr.100594.109
22. Sanger F, Coulson AR, Friedmann T, Air GM, Barrell BG, Brown NL, et al. The Nucleotide Sequence of Bacteriophage φx174. J Mol Biol (1978) 125(2):225–46. doi: 10.1016/0022-2836(78)90346-7
23. Heather JM, Chain B. The Sequence of Sequencers: The History of Sequencing DNA. Genomics (2016) 107(1):1–8. doi: 10.1016/j.ygeno.2015.11.003
24. Goodwin S, McPherson JD, McCombie WR. Coming of Age: Ten Years of Next-Generation Sequencing Technologies. Nat Rev Genet (2016) 17:333–51. doi: 10.1038/nrg.2016.49
25. Wang Z, Gerstein M, Snyder M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat Rev Genet (2009) 10:57–63. doi: 10.1038/nrg2484
26. Fedurco M, Romieu A, Williams S, Lawrence I, Turcatti G. BTA, a Novel Reagent for DNA Attachment on Glass and Efficient Generation of Solid-Phase Amplified DNA Colonies. Nucleic Acids Res (2006) 34:e22–2. doi: 10.1093/nar/gnj023
27. Guo J, Xu N, Li Z, Zhang S, Wu J, Dae HK, et al. Four-Color DNA Sequencing With 3′-O-Modified Nucleotide Reversible Terminators and Chemically Cleavable Fluorescent Dideoxynucleotides. Proc Natl Acad Sci USA (2008) 105:9145–50. doi: 10.1073/pnas.0804023105
28. Ju J, Kim DH, Bi L, Meng Q, Bai X, Li Z, et al. Four-Color DNA Sequencing by Synthesis Using Cleavable Fluorescent Nucleotide Reversible Terminators. Proc Natl Acad Sci USA (2006) 103:19635–40. doi: 10.1073/pnas.0609513103
29. Wilhelm BT, Landry JR. RNA-Seq-Quantitative Measurement of Expression Through Massively Parallel RNA-Sequencing. Methods (2009) 48:249–57. doi: 10.1016/j.ymeth.2009.03.016
30. Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and Microarray in Transcriptome Profiling of Activated T Cells. PloS One (2014) 9(1):e78644. doi: 10.1371/journal.pone.0078644
31. Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, et al. The Transcriptional Landscape of the Yeast Genome Defined by RNA Sequencing. Science (80-) (2008) 320:1344–9. doi: 10.1126/science.1158441
32. Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, et al. Dynamic Repertoire of a Eukaryotic Transcriptome Surveyed at Single-Nucleotide Resolution. Nature (2008) 453:1239–43. doi: 10.1038/nature07002
33. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and Quantifying Mammalian Transcriptomes by RNA-Seq. Nat Methods (2008) 5:621–8. doi: 10.1038/nmeth.1226
34. Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, et al. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell (2008) 133:523–36. doi: 10.1016/j.cell.2008.03.029
35. Cloonan N, Forrest ARR, Kolle G, Gardiner BBA, Faulkner GJ, Brown MK, et al. Stem Cell Transcriptome Profiling Via Massive-Scale mRNA Sequencing. Nat Methods (2008) 5:613–9. doi: 10.1038/nmeth.1223
36. Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. mRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat Methods (2009) 6:377–82. doi: 10.1038/nmeth.1315
37. Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly Parallel Genome-Wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell (2015) 161:1202–14. doi: 10.1016/j.cell.2015.05.002
38. Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-Seq2 for Sensitive Full-Length Transcriptome Profiling in Single Cells. Nat Methods (2013) 10:1096–100. doi: 10.1038/nmeth.2639
39. Vitak SA, Torkenczy KA, Rosenkrantz JL, Fields AJ, Christiansen L, Wong MH, et al. Sequencing Thousands of Single-Cell Genomes With Combinatorial Indexing. Nat Methods (2017) 14:302–8. doi: 10.1038/nmeth.4154
40. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour Evolution Inferred by Single-Cell Sequencing. Nature (2011) 472:90–5. doi: 10.1038/nature09807
41. Luo C, Keown CL, Kurihara L, Zhou J, He Y, Li J, et al. Single-Cell Methylomes Identify Neuronal Subtypes and Regulatory Elements in Mammalian Cortex. Science (80-) (2017) 357:600–4. doi: 10.1126/science.aan3351
42. Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. Single-Cell Chromatin Accessibility Reveals Principles of Regulatory Variation. Nature (2015) 523:486–90. doi: 10.1038/nature14590
43. Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging Individual mRNA Molecules Using Multiple Singly Labeled Probes. Nat Methods (2008) 5:877–9. doi: 10.1038/nmeth.1253
44. Wilson NK, Kent DG, Buettner F, Shehata M, Macaulay IC, Calero-Nieto FJ, et al. Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity Within Stem Cell Populations. Cell Stem Cell (2015) 16:712–24. doi: 10.1016/j.stem.2015.04.004
45. Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous Epitope and Transcriptome Measurement in Single Cells. Nat Methods (2017) 14:865–8. doi: 10.1038/nmeth.4380
46. Angermueller C, Clark SJ, Lee HJ, Macaulay IC, Teng MJ, Hu TX, et al. Parallel Single-Cell Sequencing Links Transcriptional and Epigenetic Heterogeneity. Nat Methods (2016) 13:229–32. doi: 10.1038/nmeth.3728
47. Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, et al. Perturb-Seq: Dissecting Molecular Circuits With Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell (2016) 167:1853–66.e17. doi: 10.1016/j.cell.2016.11.038
48. Macaulay IC, Haerty W, Kumar P, Li YI, Hu TX, Teng MJ, et al. G&T-Seq: Parallel Sequencing of Single-Cell Genomes and Transcriptomes. Nat Methods (2015) 12:519–22. doi: 10.1038/nmeth.3370
49. Zafar H, Lin C, Bar-Joseph Z. Single-Cell Lineage Tracing by Integrating CRISPR-Cas9 Mutations With Transcriptomic Data. Nat Commun (2020) 11:1–14. doi: 10.1038/s41467-020-16821-5
50. Shepherd MS, Li J, Wilson NK, Oedekoven CA, Li J, Belmonte M, et al. Single-Cell Approaches Identify the Molecular Network Driving Malignant Hematopoietic Stem Cell Self-Renewal. Blood (2018) 132(8):791–803. doi: 10.1182/blood-2017-12-821066
51. Cha J, Lee I. Single-Cell Network Biology for Resolving Cellular Heterogeneity in Human Diseases. Exp Mol Med (2020) 52:1798–808. doi: 10.1038/s12276-020-00528-0
52. Charrot S, Hallam S. CAR-T Cells: Future Perspectives. HemaSphere (2019) 3(2):e188. doi: 10.1097/HS9.0000000000000188
53. Naldini L. Gene Therapy Returns to Centre Stage. Nature (2015) 526:351–60. doi: 10.1038/nature15818
54. Friedmann T, Roblin R. Gene Therapy for Human Genetic Disease? Science (80-) (1972) 175:949–55. doi: 10.1126/science.175.4025.949
55. Slatter MA, Gennery AR. Hematopoietic Cell Transplantation in Primary Immunodeficiency – Conventional and Emerging Indications. Expert Rev Clin Immunol (2018) 14(2):103–14. doi: 10.1080/1744666X.2018.1424627
56. Bordignon C, Notarangelo LD, Nobili N, Ferrari G, Casorati G, Panina P, et al. Gene Therapy in Peripheral Blood Lymphocytes and Bone Marrow for ADA- Immunodeficient Patients. Science (80-) (1995) 270(5235):470–5. doi: 10.1126/science.270.5235.470
57. Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, et al. T Lymphocyte-Directed Gene Therapy for ADA- SCID: Initial Trial Results After 4 Years. Science (80-) (1995) 270(5235):475–80. doi: 10.1126/science.270.5235.475
58. Kang EM, Choi U, Theobald N, Linton G, Long Priel DA, Kuhns D, et al. Retrovirus Gene Therapy for X-linked Chronic Granulomatous Disease can Achieve Stable Long-Term Correction of Oxidase Activity in Peripheral Blood Neutrophils. Blood (2010) 115(4):783–91. doi: 10.1182/blood-2009-05-222760
59. Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Díez IA, Dewey RA, et al. Stem-Cell Gene Therapy for the Wiskott–Aldrich Syndrome. N Engl J Med (2010) 363:1918–27. doi: 10.1056/NEJMoa1003548
60. Nienhuis AW, Dunbar CE, Sorrentino BP. Genotoxicity of Retroviral Integration In Hematopoietic Cells. Mol Ther (2006) 13(6):1031–49. doi: 10.1016/j.ymthe.2006.03.001
61. Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JEJ, et al. CD8+ T-cell Responses to Adeno-Associated Virus Capsid in Humans. Nat Med (2007) 13:419–22. doi: 10.1038/nm1549
62. Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional Mutagenesis Combined With Acquired Somatic Mutations Causes Leukemogenesis Following Gene Therapy of SCID-X1 Patients. J Clin Invest (2008) 118:3143–50. doi: 10.1172/JCI35798
63. Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional Oncogenesis in 4 Patients After Retrovirus-Mediated Gene Therapy of SCID-X1. J Clin Invest (2008) 118:3132–42. doi: 10.1172/JCI35700
64. Rogers GL, Martino AT, Zolotukhin I, Ertl HC, Herzog RW. Role of the Vector Genome and Underlying Factor IX Mutation in Immune Responses to AAV Gene Therapy for Hemophilia B. J Transl Med (2014) 12:25. doi: 10.1186/1479-5876-12-25
65. Braun CJ, Boztug K, Paruzynski A, Witzel M, Schwarzer A, Rothe M, et al. Gene Therapy for Wiskott-Aldrich Syndrome–Long-Term Efficacy and Genotoxicity. Sci Transl Med (2014) 6(227):227ra33. doi: 10.1126/scitranslmed.3007280
66. Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, et al. A Serious Adverse Event After Successful Gene Therapy for X-Linked Severe Combined Immunodeficiency. N Engl J Med (2003) 348:255–6. doi: 10.1056/NEJM200301163480314
67. Montini E, Cesana D, Schmidt M, Sanvito F, Bartholomae CC, Ranzani M, et al. The Genotoxic Potential of Retroviral Vectors Is Strongly Modulated by Vector Design and Integration Site Selection in a Mouse Model of HSC Gene Therapy. J Clin Invest (2009) 119(4):964–75. doi: 10.1172/JCI37630
68. Yu SF, von Ruden T, Kantoff PW, Garber C, Seiberg M, Ruther U, et al. Self-Inactivating Retroviral Vectors Designed for Transfer of Whole Genes Into Mammalian Cells. Proc Natl Acad Sci (1986) 83(10):3194–8. doi: 10.1073/pnas.83.10.3194
69. Asokan A, Schaffer DV, Jude Samulski R. The AAV Vector Toolkit: Poised at the Clinical Crossroads. Mol Ther (2012) 20(4):P699–708. doi: 10.1038/mt.2011.287
70. Wang D, Tai PWL, Gao G. Adeno-Associated Virus Vector as a Platform for Gene Therapy Delivery. Nat Rev Drug Discov (2019) 18:358–78. doi: 10.1038/s41573-019-0012-9
71. Paruzynski A, Arens A, Gabriel R, Bartholomae CC, Scholz S, Wang W, et al. Genome-Wide High-Throughput Integrome Analyses by nrLAM-PCR and Next-Generation Sequencing. Nat Protoc (2010) 5:1379–95. doi: 10.1038/nprot.2010.87
72. Giordano FA, Sorg UR, Appelt J-U, Lachmann N, Bleier S, Roeder I, et al. Clonal Inventory Screens Uncover Monoclonality Following Serial Transplantation ofMgmtP140K -Transduced Stem Cells and Dose-Intense Chemotherapy. Hum Gene Ther (2011) 22:697–710. doi: 10.1089/hum.2010.088
73. Giordano FA, Appelt J-U, Link B, Gerdes S, Lehrer C, Scholz S, et al. High-Throughput Monitoring of Integration Site Clonality in Preclinical and Clinical Gene Therapy Studies. Mol Ther Methods Clin Dev (2015) 2:14061. doi: 10.1038/mtm.2014.61
74. Beard BC, Adair JE, Trobridge GD, Kiem H-P. High-Throughput Genomic Mapping of Vector Integration Sites in Gene Therapy Studies. Methods Mol Biol (2014) 1185:321–44. doi: 10.1007/978-1-4939-1133-2_22
75. George LA, Sullivan SK, Giermasz A, Rasko JEJ, Samelson-Jones BJ, Ducore J, et al. Hemophilia B Gene Therapy With a High-Specific-Activity Factor IX Variant. N Engl J Med (2017) 377:2215–27. doi: 10.1056/nejmoa1708538
76. Pasi KJ, Rangarajan S, Mitchell N, Lester W, Symington E, Madan B, et al. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N Engl J Med (2020) 382:29–40. doi: 10.1056/NEJMoa1908490
77. Rangarajan S, Walsh L, Lester W, Perry D, Madan B, Laffan M, et al. AAV5–Factor VIII Gene Transfer in Severe Hemophilia A. N Engl J Med (2017) 377:2519–30. doi: 10.1056/NEJMoa1708483
78. Ferrua F, Cicalese MP, Galimberti S, Giannelli S, Dionisio F, Barzaghi F, et al. Lentiviral Haemopoietic Stem/Progenitor Cell Gene Therapy for Treatment of Wiskott-Aldrich Syndrome: Interim Results of a Non-Randomised, Open-Label, Phase 1/2 Clinical Study. Lancet Haematol (2019) 6(5):E239–53. doi: 10.1016/S2352-3026(19)30021-3
79. Kohn DB, Booth C, Kang EM, Pai SY, Shaw KL, Santilli G, et al. Lentiviral Gene Therapy for X-Linked Chronic Granulomatous Disease. Nat Med (2020) 26:200–6. doi: 10.1038/s41591-019-0735-5
80. Esrick EB, Lehmann LE, Biffi A, Achebe M, Brendel C, Ciuculescu MF, et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N Engl J Med (2021) 384:205–15. doi: 10.1056/nejmoa2029392
81. Ribeil J-A, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, et al. Gene Therapy in a Patient With Sickle Cell Disease. N Engl J Med (2017) 376:848–55. doi: 10.1056/NEJMoa1609677
82. Mamcarz E, Zhou S, Lockey T, Abdelsamed H, Cross SJ, Kang G, et al. Lentiviral Gene Therapy Combined With Low-Dose Busulfan in Infants With SCID-X1. N Engl J Med (2019) 380:1525–34. doi: 10.1056/NEJMoa1815408
83. Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Zhang F, Adams S, et al. Immunodeficiency: Hematopoietic Stem Cell Gene Therapy for Adenosine Deaminase-Deficient Severe Combined Immunodeficiency Leads to Long-Term Immunological Recovery and Metabolic Correction. Sci Transl Med (2011) 3(97):97ra80. doi: 10.1126/scitranslmed.3002716
84. Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, et al. Efficacy of Gene Therapy for X-Linked Severe Combined Immunodeficiency. N Engl J Med (2010) 363:355–64. doi: 10.1056/nejmoa1000164
85. Chen J, Guo Z, Tian H, Chen X. Production and Clinical Development of Nanoparticles for Gene Delivery. Mol Ther Methods Clin Dev (2016) 3:16023. doi: 10.1038/mtm.2016.23
86. Yang S, Li H, Xu L, Deng Z, Han W, Liu Y, et al. Oligonucleotide Aptamer-Mediated Precision Therapy of Hematological Malignancies. Mol Ther Nucleic Acids (2018) 13:164–75. doi: 10.1016/j.omtn.2018.08.023
87. Sung YK, Kim SW. Recent Advances in the Development of Gene Delivery Systems. Biomater Res (2019) 23:8. doi: 10.1186/s40824-019-0156-z
88. Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, Goodspeed K, Gray SJ, Kay CN, et al. Current Clinical Applications of In Vivo Gene Therapy With AAVs. Mol Ther (2021) 29:464–88. doi: 10.1016/j.ymthe.2020.12.007
89. Mandal H, Katiyar SS, Swami R, Kushwah V, Katare PB, Kumar Meka A, et al. ϵ-Poly-L-Lysine/Plasmid DNA Nanoplexes for Efficient Gene Delivery In Vivo. Int J Pharm (2018) 542:142–52. doi: 10.1016/j.ijpharm.2018.03.021
90. Murphy DE, de Jong OG, Brouwer M, Wood MJ, Lavieu G, Schiffelers RM, et al. Extracellular Vesicle-Based Therapeutics: Natural Versus Engineered Targeting and Trafficking. Exp Mol Med (2019) 51:1–12. doi: 10.1038/s12276-019-0223-5
91. Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, et al. DNA Sequencing of a Cytogenetically Normal Acute Myeloid Leukaemia Genome. Nature (2008) 456:66–72. doi: 10.1038/nature07485
92. Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, et al. A Comprehensive Catalogue of Somatic Mutations From a Human Cancer Genome. Nature (2010) 463:191–6. doi: 10.1038/nature08658
93. Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, et al. A Small-Cell Lung Cancer Genome With Complex Signatures of Tobacco Exposure. Nature (2010) 463:184–90. doi: 10.1038/nature08629
94. Campbell PJ, Getz G, Korbel JO, Stuart JM, Jennings JL, Stein LD, et al. Pan-Cancer Analysis of Whole Genomes. Nature (2020) 578:82–93. doi: 10.1038/s41586-020-1969-6
95. Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, et al. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions With Oncogenic Ras. Cell (2017) 168(5):P890–903. doi: 10.1016/j.cell.2017.01.013
96. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi Screen Identifies Brd4 as a Therapeutic Target in Acute Myeloid Leukaemia. Nature (2011) 478:524–8. doi: 10.1038/nature10334
97. Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of Essential Genes for Cancer Immunotherapy. Nature (2017) 548:537–42. doi: 10.1038/nature23477
98. Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational Correction of Copy Number Effect Improves Specificity of CRISPR–Cas9 Essentiality Screens in Cancer Cells. Nat Genet (2017) 49:1779–84. doi: 10.1038/ng.3984
99. Sun E, Han R, Lu B. Gene Therapy of Renal Cancer Using Recombinant Adeno−Associated Virus Encoding Human Endostatin. Oncol Lett (2018) 16(3):2789–96. doi: 10.3892/ol.2018.9036
100. Shen Z, Yao C, Wang Z, Yue L, Fang Z, Yao H, et al. Vastatin, an Endogenous Antiangiogenesis Polypeptide That Is Lost in Hepatocellular Carcinoma, Effectively Inhibits Tumor Metastasis. Mol Ther (2016) 24(8):P1358–68. doi: 10.1038/mt.2016.56
101. Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A Phase I Clinical Trial of Adoptive T Cell Therapy Using IL-12 Secreting MUC-16ecto Directed Chimeric Antigen Receptors for Recurrent Ovarian Cancer. J Transl Med (2015) 13:102. doi: 10.1186/s12967-015-0460-x
102. Daud A, Takamura K, Diep T, Heller R, Pierce RH. Long-Term Overall Survival From a Phase I Trial Using Intratumoral Plasmid Interleukin-12 With Electroporation in Patients With Melanoma. J Transl Med (2015) 13:O3. doi: 10.1186/1479-5876-13-S1-O3
103. Xia Y, Du Z, Wang X, Li X. Treatment of Uterine Sarcoma With rAd-p53 (Gendicine) Followed by Chemotherapy: Clinical Study of TP53 Gene Therapy. Hum Gene Ther (2018) 29(2):242–50. doi: 10.1089/hum.2017.206
104. Chakraborty C, Sharma AR, Sharma G, Sarkar BK, Lee S-S. The Novel Strategies for Next-Generation Cancer Treatment: miRNA Combined With Chemotherapeutic Agents for the Treatment of Cancer. Oncotarget (2018) 9:10164–74. doi: 10.18632/oncotarget.24309
105. Roep BO, Thomaidou S, van Tienhoven R, Zaldumbide A. Type 1 Diabetes Mellitus as a Disease of the β-Cell (Do Not Blame the Immune System?). Nat Rev Endocrinol (2021) 17:150–61. doi: 10.1038/s41574-020-00443-4
106. Goodwin M, Lee E, Lakshmanan U, Shipp S, Froessl L, Barzaghi F, et al. CRISPR-Based Gene Editing Enables FOXP3 Gene Repair in IPEX Patient Cells. Sci Adv (2020) 6(19):eaaz0571. doi: 10.1126/sciadv.aaz0571
107. Xia F, Cao H, Du J, Liu X, Liu Y, Xiang M. Reg3g Overexpression Promotes β Cell Regeneration and Induces Immune Tolerance in Nonobese-Diabetic Mouse Model. J Leukoc Biol (2016) 99:1131–40. doi: 10.1189/jlb.3a0815-371rrr
108. Mallol C, Casana E, Jimenez V, Casellas A, Haurigot V, Jambrina C, et al. AAV-Mediated Pancreatic Overexpression of Igf1 Counteracts Progression to Autoimmune Diabetes in Mice. Mol Metab (2017) 6(7):664–80. doi: 10.1016/j.molmet.2017.05.007
109. Manzoor F, Johnson MC, Li C, Samulski RJ, Wang B, Tisch R. β-Cell-Specific IL-35 Therapy Suppresses Ongoing Autoimmune Diabetes in NOD Mice. Eur J Immunol (2017) 47:144–54. doi: 10.1002/eji.201646493
110. Xiao X, Guo P, Shiota C, Zhang T, Coudriet GM, Fischbach S, et al. Endogenous Reprogramming of Alpha Cells Into Beta Cells, Induced by Viral Gene Therapy, Reverses Autoimmune Diabetes. Cell Stem Cell (2018) 22:78–90.e4. doi: 10.1016/j.stem.2017.11.020
111. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med (2017) 377:2545–54. doi: 10.1056/nejmoa1708566
112. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
113. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
114. Sadelain M, Rivière I, Riddell S. Therapeutic T Cell Engineering. Nature (2017) 545:423–31. doi: 10.1038/nature22395
115. Rafiq S, Hackett CS, Brentjens RJ. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat Rev Clin Oncol (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
116. Larson RC, Maus MV. Recent Advances and Discoveries in the Mechanisms and Functions of CAR T Cells. Nat Rev Cancer (2021) 21:145–61. doi: 10.1038/s41568-020-00323-z
117. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric Antigen Receptor-Modified T Cells Derived From Defined CD8+ and CD4+ Subsets Confer Superior Antitumor Reactivity In Vivo. Leukemia (2016) 30:492–500. doi: 10.1038/leu.2015.247
118. Das RK, Vernau L, Grupp SA, Barrett DM. Naïve T-Cell Deficits at Diagnosis and After Chemotherapy Impair Cell Therapy Potential in Pediatric Cancers. Cancer Discov (2019) 9(4):492–9. doi: 10.1158/2159-8290.CD-18-1314
119. Finney OC, Brakke H, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al. CD19 CAR T Cell Product and Disease Attributes Predict Leukemia Remission Durability. J Clin Invest (2019) 129(5):2123–32. doi: 10.1172/JCI125423
120. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat Med (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
121. Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 Promotes the Therapeutic Efficacy of CD19-Targeted T Cells. Nature (2018) 558:307–12. doi: 10.1038/s41586-018-0178-z
122. Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, et al. Genome-Wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell (2018) 175(7):1958–71.e15. doi: 10.1016/j.cell.2018.10.024
123. Nobles CL, Sherrill-Mix S, Everett JK, Reddy S, Fraietta JA, Porter DL, et al. CD19-Targeting CAR T Cell Immunotherapy Outcomes Correlate With Genomic Modification by Vector Integration. J Clin Invest (2019) 130(2):673–85. doi: 10.1172/JCI130144
124. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric Antigen Receptor T-Cell Therapy — Assessment and Management of Toxicities. Nat Rev Clin Oncol (2018) 15(1):47–62. doi: 10.1038/nrclinonc.2017.148
125. Majzner RG, Mackall CL. Tumor Antigen Escape From CAR T-Cell Therapy. Cancer Discov (2018) 8(10):1219–26. doi: 10.1158/2159-8290.CD-18-0442
126. Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, et al. Genetic Mechanisms of Target Antigen Loss in CAR19 Therapy of Acute Lymphoblastic Leukemia. Nat Med (2018) 24(10):1504–6. doi: 10.1038/s41591-018-0146-z
127. Asnani M, Hayer KE, Naqvi AS, Zheng S, Yang SY, Oldridge D, et al. Retention of CD19 Intron 2 Contributes to CART-19 Resistance in Leukemias With Subclonal Frameshift Mutations in CD19. Leukemia (2020) 34(4):1202–7. doi: 10.1038/s41375-019-0580-z
128. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of Acquired Mutations and Alternative Splicing ofCD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov (2015) 5(12):1282–95. doi: 10.1158/2159-8290.CD-15-1020
129. Rabilloud T, Potier D, Pankaew S, Nozais M, Loosveld M, Payet-Bornet D. Single-Cell Profiling Identifies Pre-Existing CD19-Negative Subclones in a B-ALL Patient With CD19-Negative Relapse After CAR-T Therapy. Nat Commun (2021) 12(1):865. doi: 10.1038/s41467-021-21168-6
130. Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol Ther (2020) 28(11):2320–39. doi: 10.1016/j.ymthe.2020.09.015
131. Mahadeo KM, Khazal SJ, Abdel-Azim H, Fitzgerald JC, Taraseviciute A, Bollard CM, et al. Management Guidelines for Paediatric Patients Receiving Chimeric Antigen Receptor T Cell Therapy. Nat Rev Clin Oncol (2019) 16(1):45–63. doi: 10.1038/s41571-018-0075-2
132. Pan J, Niu Q, Deng B, Liu S, Wu T, Gao Z, et al. CD22 CAR T-Cell Therapy in Refractory or Relapsed B Acute Lymphoblastic Leukemia. Leukemia (2019) 33(12):2854–66. doi: 10.1038/s41375-019-0488-7
133. Zamora AE, Crawford JC, Thomas PG. Hitting the Target: How T Cells Detect and Eliminate Tumors. J Immunol (2018) 200(2):392–9. doi: 10.4049/jimmunol.1701413
134. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol Ther (2010) 18(4):843–51. doi: 10.1038/mt.2010.24
135. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor–Transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J Immunother (2019) 42(4):126–35. doi: 10.1097/CJI.0000000000000260
136. Yoon D, Osborn M, Tolar J, Kim C. Incorporation of Immune Checkpoint Blockade Into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int J Mol Sci (2018) 19(2):340. doi: 10.3390/ijms19020340
137. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Anti-Tumor Efficacy of Human Chimeric Antigen Receptor T Cells. Sci Rep (2017) 7(1):737. doi: 10.1038/s41598-017-00462-8
138. Pesch T, Bonati L, Kelton W, Parola C, Ehling RA, Csepregi L, et al. Molecular Design, Optimization, and Genomic Integration of Chimeric B Cell Receptors in Murine B Cells. Front Immunol (2019) 10:2630. doi: 10.3389/fimmu.2019.02630
139. Shimasaki N, Jain A, Campana D. NK Cells for Cancer Immunotherapy. Nat Rev Drug Discov (2020) 19(3):200–18. doi: 10.1038/s41573-019-0052-1
140. Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res (2021) 81(5):1201–8. doi: 10.1158/0008-5472.CAN-20-2990
141. Kobayashi S, Thelin MA, Parrish HL, Deshpande NR, Lee MS, Karimzadeh A, et al. A Biomimetic Five-Module Chimeric Antigen Receptor (5M CAR) Designed to Target and Eliminate Antigen-Specific T Cells. Proc Natl Acad Sci (2020) 117(46):28950–9. doi: 10.1073/pnas.2012495117
142. Tenspolde M, Zimmermann K, Weber LC, Hapke M, Lieber M, Dywicki J, et al. Regulatory T Cells Engineered With a Novel Insulin-Specific Chimeric Antigen Receptor as a Candidate Immunotherapy for Type 1 Diabetes. J Autoimmun (2019) 103:102289. doi: 10.1016/j.jaut.2019.05.017
143. Zhang L, Sosinowski T, Cox AR, Cepeda JR, Sekhar NS, Hartig SM, et al. Chimeric Antigen Receptor (CAR) T Cells Targeting a Pathogenic MHC Class II:peptide Complex Modulate the Progression of Autoimmune Diabetes. J Autoimmun (2019) 96:50–8. doi: 10.1016/j.jaut.2018.08.004
144. Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of Type 1 Diabetes With CD4+CD25highCD127-Regulatory T Cells Prolongs Survival of Pancreatic Islets — Results of One Year Follow-Up. Clin Immunol (2014) 153(1):23–30. doi: 10.1016/j.clim.2014.03.016
145. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 Diabetes Immunotherapy Using Polyclonal Regulatory T Cells. Sci Transl Med (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
146. Gliwiński M, Iwaszkiewicz-Grześ D, Wołoszyn-Durkiewicz A, Tarnowska M, Żalińska M, Hennig M, et al. Proinsulin-Specific T Regulatory Cells may Control Immune Responses in Type 1 Diabetes: Implications for Adoptive Therapy. BMJ Open Diabetes Res Care (2020) 8(1):e000873. doi: 10.1136/bmjdrc-2019-000873
147. Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. Regulatory T Cells: How do They Suppress Immune Responses? Int Immunol (2009) 21:1105–11. doi: 10.1093/intimm/dxp095
148. Lindley S, Dayan CM, Bishop A, Roep BO, Peatman M, Tree TIM. Defective Suppressor Function in CD4+CD25+ T-Cells From Patients With Type 1 Diabetes. Diabetes (2005) 54:92–9. doi: 10.2337/diabetes.54.1.92
149. Tree TIM, Lawson J, Edwards H, Skowera A, Arif S, Roep BO, et al. Naturally Arising Human CD4 T-Cells That Recognize Islet Autoantigens and Secrete Interleukin-10 Regulate Proinflammatory T-Cell Responses Via Linked Suppression. Diabetes (2010) 59:1451–60. doi: 10.2337/db09-0503
150. Buckner JH. Mechanisms of Impaired Regulation by CD4+ CD25+ FOXP3+ Regulatory T Cells in Human Autoimmune Diseases. Nat Rev Immunol (2010) 10:849–59. doi: 10.1038/nri2889
151. Cohen JL, Trenado A, Vasey D, Klatzmann D, Salomon BL. CD4+CD25+ Immunoregulatory T Cells: New Therapeutics for Graft-Versus-Host Disease. J Exp Med (2002) 196:401–6. doi: 10.1084/jem.20020090
152. Taylor PA, Lees CJ, Blazar BR. The Infusion of Ex Vivo Activated and Expanded CD4 +CD25 + Immune Regulatory Cells Inhibits Graft-Versus-Host Disease Lethality. Blood (2002) 99:3493–9. doi: 10.1182/blood.V99.10.3493
153. Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T Cells, Expanded With Dendritic Cells Presenting a Single Autoantigenic Peptide, Suppress Autoimmune Diabetes. J Exp Med (2004) 199:1467–77. doi: 10.1084/jem.20040180
154. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In Vitro-Expanded Antigen-Specific Regulatory T Cells Suppress Autoimmune Diabetes. J Exp Med (2004) 199:1455–65. doi: 10.1084/jem.20040139
155. Esensten JH, Muller YD, Bluestone JA, Tang Q. Regulatory T-cell Therapy for Autoimmune and Autoinflammatory Diseases: The Next Frontier. J Allergy Clin Immunol (2018) 142:1710–8. doi: 10.1016/j.jaci.2018.10.015
156. Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front Immunol (2019) 10:43. doi: 10.3389/fimmu.2019.00043
157. Sheih A, Voillet V, Hanafi LA, DeBerg HA, Yajima M, Hawkins R, et al. Clonal Kinetics and Single-Cell Transcriptional Profiling of CAR-T Cells in Patients Undergoing CD19 CAR-T Immunotherapy. Nat Commun (2020) 11:1–13. doi: 10.1038/s41467-019-13880-1
158. Deng Q, Han G, Puebla-Osorio N, Ma MCJ, Strati P, Chasen B, et al. Characteristics of Anti-CD19 Car T Cell Infusion Products Associated With Efficacy and Toxicity in Patients With Large B Cell Lymphomas. Nat Med (2020) 26:1878–87. doi: 10.1038/s41591-020-1061-7
159. Biasco L, Pellin D, Scala S, Dionisio F, Basso-Ricci L, Leonardelli L, et al. In Vivo Tracking of Human Hematopoiesis Reveals Patterns of Clonal Dynamics During Early and Steady-State Reconstitution Phases. Cell Stem Cell (2016) 19:107–19. doi: 10.1016/j.stem.2016.04.016
160. Scala S, Basso-Ricci L, Dionisio F, Pellin D, Giannelli S, Salerio FA, et al. Dynamics of Genetically Engineered Hematopoietic Stem and Progenitor Cells After Autologous Transplantation in Humans. Nat Med (2018) 24(11):1683–90. doi: 10.1038/s41591-018-0195-3
161. Six E, Guilloux A, Denis A, Lecoules A, Magnani A, Vilette R, et al. Clonal Tracking in Gene Therapy Patients Reveals a Diversity of Human Hematopoietic Differentiation Programs. Blood (2020) 135(15):1219–31. doi: 10.1182/blood.2019002350
162. Lee-Six H, Kent DG. Tracking Hematopoietic Stem Cells and Their Progeny Using Whole-Genome Sequencing. Exp Hematol (2020) 83:12–24. doi: 10.1016/j.exphem.2020.01.004
163. Lee J, Hyeon DY, Hwang D. Single-Cell Multiomics: Technologies and Data Analysis Methods. Exp Mol Med (2020) 52:1428–42. doi: 10.1038/s12276-020-0420-2
164. Adey A, Morrison HG, Asan, Xun X, Kitzman JO, Turner EH, et al. Rapid, Low-Input, Low-Bias Construction of Shotgun Fragment Libraries by High-Density In Vitro Transposition. Genome Biol (2010) 11:R119. doi: 10.1186/gb-2010-11-12-r119
165. Amini S, Pushkarev D, Christiansen L, Kostem E, Royce T, Turk C, et al. Haplotype-Resolved Whole-Genome Sequencing by Contiguity-Preserving Transposition and Combinatorial Indexing. Nat Genet (2014) 46:1343–9. doi: 10.1038/ng.3119
166. Adey A, Kitzman JO, Burton JN, Daza R, Kumar A, Christiansen L, et al. In Vitro, Long-Range Sequence Information for De Novo Genome Assembly Via Transposase Contiguity. Genome Res (2014) 24:2041–9. doi: 10.1101/gr.178319.114
167. Baslan T, Kendall J, Rodgers L, Cox H, Riggs M, Stepansky A, et al. Genome-Wide Copy Number Analysis of Single Cells. Nat Protoc (2012) 7:1024–41. doi: 10.1038/nprot.2012.039
168. Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, et al. Comprehensive Single-Cell Transcriptional Profiling of a Multicellular Organism. Science (80-) (2017) 357:661–7. doi: 10.1126/science.aam8940
169. Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, et al. Single-Cell Profiling of the Developing Mouse Brain and Spinal Cord With Split-Pool Barcoding. Science (80-) (2018) 360:176–82. doi: 10.1126/science.aam8999
170. Dong X, Zhang L, Milholland B, Lee M, Maslov AY, Wang T, et al. Accurate Identification of Single-Nucleotide Variants in Whole-Genome-Amplified Single Cells. Nat Methods (2017) 14:491–3. doi: 10.1038/nmeth.4227
171. Bae T, Tomasini L, Mariani J, Zhou B, Roychowdhury T, Franjic D, et al. Different Mutational Rates and Mechanisms in Human Cells at Pregastrulation and Neurogenesis. Science (80-) (2018) 359:550–5. doi: 10.1126/science.aan8690
172. Lawson DA, Kessenbrock K, Davis RT, Pervolarakis N, Werb Z. Tumour Heterogeneity and Metastasis at Single-Cell Resolution. Nat Cell Biol (2018) 20:1349–60. doi: 10.1038/s41556-018-0236-7
173. Zhang L, Dong X, Lee M, Maslov AY, Wang T, Vijg J. Single-Cell Whole-Genome Sequencing Reveals the Functional Landscape of Somatic Mutations in B Lymphocytes Across the Human Lifespan. Proc Natl Acad Sci USA (2019) 116:9014–9. doi: 10.1073/pnas.1902510116
174. Kent DG, Green AR. Order Matters: The Order of Somatic Mutations Influences Cancer Evolution. Cold Spring Harb Perspect Med (2017) 7(4):a027060. doi: 10.1101/cshperspect.a027060
175. Ortmann CA, Kent DG, Nangalia J, Silber Y, Wedge DC, Grinfeld J, et al. Effect of Mutation Order on Myeloproliferative Neoplasms. N Engl J Med (2015) 372:601–12. doi: 10.1056/nejmoa1412098
176. Rodriguez-Meira A, Buck G, Clark SA, Povinelli BJ, Alcolea V, Louka E, et al. Unravelling Intratumoral Heterogeneity Through High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. Mol Cell (2019) 73:1292–1305.e8. doi: 10.1016/j.molcel.2019.01.009
177. Anderson K, Lutz C, Van Delft FW, Bateman CM, Guo Y, Colman SM, et al. Genetic Variegation of Clonal Architecture and Propagating Cells in Leukaemia. Nature (2011) 469:356–61. doi: 10.1038/nature09650
178. Notta F, Mullighan CG, Wang JCY, Poeppl A, Doulatov S, Phillips LA, et al. Evolution of Human BCR-ABL1 Lymphoblastic Leukaemia-Initiating Cells. Nature (2011) 469:362–7. doi: 10.1038/nature09733
179. Gawad C, Koh W, Quake SR. Dissecting the Clonal Origins of Childhood Acute Lymphoblastic Leukemia by Single-Cell Genomics. Proc Natl Acad Sci USA (2014) 111:17947–52. doi: 10.1073/pnas.1420822111
180. Albertí-Servera L, Demeyer S, Govaerts I, Swings T, De Bie J, Gielen O, et al. Single-Cell DNA Amplicon Sequencing Reveals Clonal Heterogeneity and Evolution in T-Cell Acute Lymphoblastic Leukemia. Blood (2021) 137:801–11. doi: 10.1182/blood.2020006996
181. Xu L, Durruthy-Durruthy R, Eastburn DJ, Pellegrino M, Shah O, Meyer E, et al. Clonal Evolution and Changes in Two AML Patients Detected With A Novel Single-Cell DNA Sequencing Platform. Sci Rep (2019) 9(1):11119. doi: 10.1038/s41598-019-47297-z
182. Lee-Six H, Øbro NF, Shepherd MS, Grossmann S, Dawson K, Belmonte M, et al. Population Dynamics of Normal Human Blood Inferred From Somatic Mutations. Nature (2018) 561:473–8. doi: 10.1038/s41586-018-0497-0
183. Barennes P, Quiniou V, Shugay M, Egorov ES, Davydov AN, Chudakov DM, et al. Benchmarking of T Cell Receptor Repertoire Profiling Methods Reveals Large Systematic Biases. Nat Biotechnol (2021) 39:236–45. doi: 10.1038/s41587-020-0656-3
184. Chovanec P, Bolland DJ, Matheson LS, Wood AL, Krueger F, Andrews S, et al. Unbiased Quantification of Immunoglobulin Diversity at the DNA Level With VDJ-Seq. Nat Protoc (2018) 13:1232–52. doi: 10.1038/nprot.2018.021
185. De Simone M, Rossetti G, Pagani M. Single Cell T Cell Receptor Sequencing: Techniques and Future Challenges. Front Immunol (2018) 9:1638. doi: 10.3389/fimmu.2018.01638
186. Hogan SA, Courtier A, Cheng PF, Jaberg-Bentele NF, Goldinger SM, Manuel M, et al. Peripheral Blood TCR Repertoire Profiling may Facilitate Patient Stratification for Immunotherapy Against Melanoma. Cancer Immunol Res (2019) 7:77–85. doi: 10.1158/2326-6066.CIR-18-0136
187. Jin Y-B, Luo W, Zhang G-Y, Lin K-R, Cui J-H, Chen X-P, et al. TCR Repertoire Profiling of Tumors, Adjacent Normal Tissues, and Peripheral Blood Predicts Survival in Nasopharyngeal Carcinoma. Cancer Immunol Immunother (2018) 67:1719–30. doi: 10.1007/s00262-018-2237-6
188. Wieland A, Kamphorst AO, Adsay NV, Masor JJ, Sarmiento J, Nasti TH, et al. T Cell Receptor Sequencing of Activated CD8 T Cells in the Blood Identifies Tumor-Infiltrating Clones That Expand After PD-1 Therapy and Radiation in a Melanoma Patient. Cancer Immunol Immunother (2018) 67:1767–76. doi: 10.1007/s00262-018-2228-7
189. Ichinohe T, Miyama T, Kawase T, Honjo Y, Kitaura K, Sato H, et al. Next-Generation Immune Repertoire Sequencing as a Clue to Elucidate the Landscape of Immune Modulation by Host-Gut Microbiome Interactions. Front Immunol (2018) 9:668. doi: 10.3389/fimmu.2018.00668
190. Holliday R. Epigenetics: A Historical Overview. Epigenetics (2006) 1:76–80. doi: 10.4161/epi.1.2.2762
191. Cazaly E, Saad J, Wang W, Heckman C, Ollikainen M, Tang J. Making Sense of the Epigenome Using Data Integration Approaches. Front Pharmacol (2019) 10:126. doi: 10.3389/fphar.2019.00126
192. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of Native Chromatin for Fast and Sensitive Epigenomic Profiling of Open Chromatin, DNA-Binding Proteins and Nucleosome Position. Nat Methods (2013) 10:1213–8. doi: 10.1038/nmeth.2688
193. Goryshin IY, Reznikoff WS. Tn5 In Vitro Transposition. J Biol Chem (1998) 273:7367–74. doi: 10.1074/jbc.273.13.7367
194. Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, et al. Massively Parallel Single-Cell Chromatin Landscapes of Human Immune Cell Development and Intratumoral T Cell Exhaustion. Nat Biotechnol (2019) 37:925–36. doi: 10.1038/s41587-019-0206-z
195. Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, et al. Multiplex Single-Cell Profiling of Chromatin Accessibility by Combinatorial Cellular Indexing. Science (80-) (2015) 348:910–4. doi: 10.1126/science.aab1601
196. Guilhamon P, Chesnelong C, Kushida MM, Nikolic A, Singhal D, Macleod G, et al. Single-Cell Chromatin Accessibility Profiling of Glioblastoma Identifies an Invasive Cancer Stem Cell Population Associated With Lower Survival. Elife (2021) 10:1–20. doi: 10.7554/ELIFE.64090
197. Litzenburger UM, Buenrostro JD, Wu B, Shen Y, Sheffield NC, Kathiria A, et al. Single-Cell Epigenomic Variability Reveals Functional Cancer Heterogeneity. Genome Biol (2017) 18(1):15. doi: 10.1186/s13059-016-1133-7
198. Greenberg MVC, Bourc’his D. The Diverse Roles of DNA Methylation in Mammalian Development and Disease. Nat Rev Mol Cell Biol (2019) 20:590–607. doi: 10.1038/s41580-019-0159-6
199. Smith ZD, Meissner A. DNA Methylation: Roles in Mammalian Development. Nat Rev Genet (2013) 14:204–20. doi: 10.1038/nrg3354
200. Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, et al. Impact of Cytosine Methylation on DNA Binding Specificities of Human Transcription Factors. Science (80-) (2017) 356(6337):eaaj2239. doi: 10.1126/science.aaj2239
201. Bock C, Tomazou EM, Brinkman AB, Müller F, Simmer F, Gu H, et al. Quantitative Comparison of Genome-Wide DNA Methylation Mapping Technologies. Nat Biotechnol (2010) 28:1106–14. doi: 10.1038/nbt.1681
202. Miura F, Enomoto Y, Dairiki R, Ito T. Amplification-Free Whole-Genome Bisulfite Sequencing by Post-Bisulfite Adaptor Tagging. Nucleic Acids Res (2012) 40:e136–6. doi: 10.1093/nar/gks454
203. Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, et al. Single-Cell Genome-Wide Bisulfite Sequencing for Assessing Epigenetic Heterogeneity. Nat Methods (2014) 11:817–20. doi: 10.1038/nmeth.3035
204. Clark SJ, Smallwood SA, Lee HJ, Krueger F, Reik W, Kelsey G. Genome-Wide Base-Resolution Mapping of DNA Methylation in Single Cells Using Single-Cell Bisulfite Sequencing (scBS-Seq). Nat Protoc (2017) 12:534–47. doi: 10.1038/nprot.2016.187
205. Argelaguet R, Clark SJ, Mohammed H, Stapel LC, Krueger C, Kapourani CA, et al. Multi-Omics Profiling of Mouse Gastrulation at Single-Cell Resolution. Nature (2019) 576:487–91. doi: 10.1038/s41586-019-1825-8
206. Linker SM, Urban L, Clark SJ, Chhatriwala M, Amatya S, McCarthy DJ, et al. Combined Single-Cell Profiling of Expression and DNA Methylation Reveals Splicing Regulation and Heterogeneity. Genome Biol (2019) 20:30. doi: 10.1186/s13059-019-1644-0
207. Zhou F, Wang R, Yuan P, Ren Y, Mao Y, Li R, et al. Reconstituting the Transcriptome and DNA Methylome Landscapes of Human Implantation. Nature (2019) 572:660–4. doi: 10.1038/s41586-019-1500-0
208. Wang W, Fasolino M, Cattau B, Goldman N, Kong W, Frederick MA, et al. Joint Profiling of Chromatin Accessibility and CAR-T Integration Site Analysis at Population and Single-Cell Levels. Proc Natl Acad Sci USA (2020) 117:5442–52. doi: 10.1073/pnas.1919259117
209. Frangoul H, Altshuler D, Cappellini MD, Chen Y-S, Domm J, Eustace BK, et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med (2021) 384:252–60. doi: 10.1056/nejmoa2031054
210. Liu XS, Wu H, Krzisch M, Wu X, Graef J, Muffat J, et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell (2018) 172:979–92.e6. doi: 10.1016/j.cell.2018.01.012
211. Xu X, Tao Y, Gao X, Zhang L, Li X, Zou W, et al. A CRISPR-based Approach for Targeted DNA Demethylation. Cell Discov (2016) 2:16009. doi: 10.1038/celldisc.2016.9
212. Vojta A, Dobrinic P, Tadic V, Bockor L, Korac P, Julg B, et al. Repurposing the CRISPR-Cas9 System for Targeted DNA Methylation. Nucleic Acids Res (2016) 44:5615–28. doi: 10.1093/nar/gkw159
213. Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, et al. Editing DNA Methylation in the Mammalian Genome. Cell (2016) 167:233–47.e17. doi: 10.1016/j.cell.2016.08.056
214. Choudhury SR, Cui Y, Lubecka K, Stefanska B, Irudayaraj J. CRISPR-dCas9 Mediated TET1 Targeting for Selective DNA Demethylation at BRCA1 Promoter. Oncotarget (2016) 7:46545–56. doi: 10.18632/oncotarget.10234
215. Wang Y, Tong C, Dai H, Wu Z, Han X, Guo Y, et al. Low-Dose Decitabine Priming Endows CAR T Cells With Enhanced and Persistent Antitumour Potential Via Epigenetic Reprogramming. Nat Commun (2021) 12(1):409. doi: 10.1038/s41467-020-20696-x
216. Kelsey G, Stegle O, Reik W. Single-Cell Epigenomics: Recording the Past and Predicting the Future. Science (80-) (2017) 358:69–75. doi: 10.1126/science.aan6826
217. Zamanighomi M, Lin Z, Daley T, Chen X, Duren Z, Schep A, et al. Unsupervised Clustering and Epigenetic Classification of Single Cells. Nat Commun (2018) 9(1):2410. doi: 10.1038/s41467-018-04629-3
218. Stuart T, Srivastava A, Lareau C, Satija R. Multimodal Single-Cell Chromatin Analysis With Signac. bioRxiv (2020). doi: 10.1101/2020.11.09.373613 2020.11.09.373613.
219. Fang R, Preissl S, Li Y, Hou X, Lucero J, Wang X, et al. Comprehensive Analysis of Single Cell ATAC-seq Data With SnapATAC. Nat Commun (2021) 12:1–15. doi: 10.1038/s41467-021-21583-9
220. Hwang B, Lee JH, Bang D. Single-Cell RNA Sequencing Technologies and Bioinformatics Pipelines. Exp Mol Med (2018) 50:96. doi: 10.1038/s12276-018-0071-8
221. Chen G, Ning B, Shi T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front Genet (2019) 10:317. doi: 10.3389/fgene.2019.00317
222. Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively Parallel Digital Transcriptional Profiling of Single Cells. Nat Commun (2017) 8:1–12. doi: 10.1038/ncomms14049
223. Wang X, He Y, Zhang Q, Ren X, Zhang Z. Direct Comparative Analyses of 10X Genomics Chromium and Smart-Seq2. Genomics Proteomics Bioinformatics (2021). doi: 10.1016/j.gpb.2020.02.005
224. Ziegenhain C, Vieth B, Parekh S, Reinius B, Guillaumet-Adkins A, Smets M, et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol Cell (2017) 65:631–643.e4. doi: 10.1016/j.molcel.2017.01.023
225. Ochocka N, Segit P, Walentynowicz KA, Wojnicki K, Cyranowski S, Swatler J, et al. Single-Cell RNA Sequencing Reveals Functional Heterogeneity of Glioma-Associated Brain Macrophages. Nat Commun (2021) 12(1):1151. doi: 10.1038/s41467-021-21407-w
226. Aissa AF, Islam ABMMK, Ariss MM, Go CC, Rader AE, Conrardy RD, et al. Single-Cell Transcriptional Changes Associated With Drug Tolerance and Response to Combination Therapies in Cancer. Nat Commun (2021) 12(1):1628. doi: 10.1038/s41467-021-21884-z
227. Zhou S, Huang Y-E, Liu H, Zhou X, Yuan M, Hou F, et al. Single-Cell RNA-Seq Dissects the Intratumoral Heterogeneity of Triple-Negative Breast Cancer Based on Gene Regulatory Networks. Mol Ther Nucleic Acids (2021) 23:682–90. doi: 10.1016/j.omtn.2020.12.018
228. Hua P, Roy N, de la Fuente J, Wang G, Thongjuea S, Clark K, et al. Single-Cell Analysis of Bone Marrow–Derived CD341 Cells From Children With Sickle Cell Disease and Thalassemia. Blood (2019) 134:2111–5. doi: 10.1182/blood.2019002301
229. Liu S, Zhou B, Wu L, Sun Y, Chen J, Liu S. Single-Cell Differential Splicing Analysis Reveals High Heterogeneity of Liver Tumor-Infiltrating T Cells. Sci Rep (2021) 11(1):5325. doi: 10.1038/s41598-021-84693-w
230. Xin Y, Kim J, Okamoto H, Ni M, Wei Y, Adler C, et al. RNA Sequencing of Single Human Islet Cells Reveals Type 2 Diabetes Genes. Cell Metab (2016) 24:608–15. doi: 10.1016/j.cmet.2016.08.018
231. Segerstolpe Å, Palasantza A, Eliasson P, Andersson EM, Andréasson AC, Sun X, et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab (2016) 24:593–607. doi: 10.1016/j.cmet.2016.08.020
232. Zhou W, Yui MA, Williams BA, Yun J, Wold BJ, Cai L, et al. Single-Cell Analysis Reveals Regulatory Gene Expression Dynamics Leading to Lineage Commitment in Early T Cell Development. Cell Syst (2019) 9:321–37.e9. doi: 10.1016/j.cels.2019.09.008
233. Boulch M, Cazaux M, Loe-Mie Y, Thibaut R, Corre B, Lemaître F, et al. A Cross-Talk Between CAR T Cell Subsets and the Tumor Microenvironment Is Essential for Sustained Cytotoxic Activity. Sci Immunol (2021) 6(57):eabd4344. doi: 10.1126/sciimmunol.abd4344
234. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-Based Map of the Human Proteome. Science (80-) (2015) 347:1260419–1260419. doi: 10.1126/science.1260419
235. Labib M, Kelley SO. Single-Cell Analysis Targeting the Proteome. Nat Rev Chem (2020) 4:143–58. doi: 10.1038/s41570-020-0162-7
236. Hartmann FJ, Bendall SC. Immune Monitoring Using Mass Cytometry and Related High-Dimensional Imaging Approaches. Nat Rev Rheumatol (2020) 16:87–99. doi: 10.1038/s41584-019-0338-z
237. Lou X, Zhang G, Herrera I, Kinach R, Ornatsky O, Baranov V, et al. Polymer-Based Elemental Tags for Sensitive Bioassays. Angew Chem Int Ed (2007) 46:6111–4. doi: 10.1002/anie.200700796
238. Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, et al. Mass Cytometry: Technique for Real Time Single Cell Multitarget Immunoassay Based on Inductively Coupled Plasma Time-of-Flight Mass Spectrometry. Anal Chem (2009) 81:6813–22. doi: 10.1021/ac901049w
239. Bendall SC, Simonds EF, Qiu P, Amir EAD, Krutzik PO, Finck R, et al. Single-Cell Mass Cytometry of Differential Immune and Drug Responses Across a Human Hematopoietic Continuum. Science (80-) (2011) 332:687–96. doi: 10.1126/science.1198704
240. Ornatsky O, Bandura D, Baranov V, Nitz M, Winnik MA, Tanner S. Highly Multiparametric Analysis by Mass Cytometry. J Immunol Methods (2010) 361:1–20. doi: 10.1016/j.jim.2010.07.002
241. Palii CG, Cheng Q, Gillespie MA, Morrissey E, Higgs DR. Single-Cell Proteomics Reveal That Quantitative Changes in Co-Expressed Lineage-Specific Transcription Factors Determine Cell Fate. Cell Stem Cell (2019) 24:812–20. doi: 10.1016/j.stem.2019.02.006
242. Rubin SJS, Bai L, Haileselassie Y, Garay G, Yun C, Becker L, et al. Mass Cytometry Reveals Systemic and Local Immune Signatures That Distinguish Inflammatory Bowel Diseases. Nat Commun (2019) 10:1–14. doi: 10.1038/s41467-019-10387-7
243. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically Expanded Peripheral T Helper Cell Subset Drives B Cells in Rheumatoid Arthritis. Nature (2017) 542:110–4. doi: 10.1038/nature20810
244. Grandi FC, Baskar R, Smeriglio P, Murkherjee S, Indelli PF, Amanatullah DF, et al. Single-Cell Mass Cytometry Reveals Cross-Talk Between Inflammation-Dampening and Inflammation-Amplifying Cells in Osteoarthritic Cartilage. Sci Adv (2020) 6(11):eaay5352. doi: 10.1126/sciadv.aay5352
245. Levine JH, Simonds EF, Bendall SC, Davis KL, Amir EAD, Tadmor MD, et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells That Correlate With Prognosis. Cell (2015) 162:184–97. doi: 10.1016/j.cell.2015.05.047
246. Kuranda K, Jean-Alphonse P, Leborgne C, Hardet R, Collaud F, Marmier S, et al. Exposure to Wild-Type AAV Drives Distinct Capsid Immunity Profiles in Humans. J Clin Invest (2018) 128:5267–79. doi: 10.1172/JCI122372
247. Giesen C, Wang HAO, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, et al. Highly Multiplexed Imaging of Tumor Tissues With Subcellular Resolution by Mass Cytometry. Nat Methods (2014) 11:417–22. doi: 10.1038/nmeth.2869
248. Damond N, Engler S, Zanotelli VRT, Schapiro D, Wasserfall CH, Kusmartseva I, et al. A Map of Human Type 1 Diabetes Progression by Imaging Mass Cytometry. Cell Metab (2019) 29:755–68.e5. doi: 10.1016/j.cmet.2018.11.014
249. Wang YJ, Traum D, Schug J, Gao L, Liu C, Atkinson MA, et al. Multiplexed In Situ Imaging Mass Cytometry Analysis of the Human Endocrine Pancreas and Immune System in Type 1 Diabetes. Cell Metab (2019) 29:769–83.e4. doi: 10.1016/j.cmet.2019.01.003
250. Bonilla DL, Reinin G, Chua E. Full Spectrum Flow Cytometry as a Powerful Technology for Cancer Immunotherapy Research. Front Mol Biosci (2021) 7:612801. doi: 10.3389/fmolb.2020.612801
251. Hümmert MW, Alvermann S, Gingele S, Gross CC, Wiendl H, Mirenska A, et al. Immunophenotyping of Cerebrospinal Fluid Cells by Chipcytometry. J Neuroinflamm (2018) 15:1–11. doi: 10.1186/s12974-018-1176-7
252. Bauman JE, Ohr J, Gooding WE, Ferris RL, Duvvuri U, Kim S, et al. Phase I Study of Ficlatuzumab and Cetuximab in Cetuximab-Resistant, Recurrent/Metastatic Head and Neck Cancer. Cancers (Basel) (2020) 12:1–17. doi: 10.3390/cancers12061537
253. Palit S, Heuser C, De Almeida GP, Theis FJ, Zielinski CE. Meeting the Challenges of High-Dimensional Single-Cell Data Analysis in Immunology. Front Immunol (2019) 10:1515. doi: 10.3389/fimmu.2019.01515
254. Specht H, Emmott E, Petelski AA, Huffman RG, Perlman DH, Serra M, et al. Single-Cell Proteomic and Transcriptomic Analysis of Macrophage Heterogeneity Using SCoPE2. Genome Biol (2021) 22:50. doi: 10.1186/s13059-021-02267-5
255. Budnik B, Levy E, Harmange G, Slavov N. SCoPE-MS: Mass Spectrometry of Single Mammalian Cells Quantifies Proteome Heterogeneity During Cell Differentiation. Genome Biol (2018) 19:161. doi: 10.1186/s13059-018-1547-5
256. Brunner AD, Thielert M, Vasilopoulou C, Ammar C, Coscia F, Mund A, et al. Ultra-High Sensitivity Mass Spectrometry Quantifies Single-Cell Proteome Changes Upon Perturbation. bioRxiv (2020). doi: 10.1101/2020.12.22.423933 2020.12.22.423933.
257. Clark SJ, Argelaguet R, Kapourani CA, Stubbs TM, Lee HJ, Alda-Catalinas C, et al. ScNMT-seq Enables Joint Profiling of Chromatin Accessibility DNA Methylation and Transcription in Single Cells E. Nat Commun (2018) 9:1–9. doi: 10.1038/s41467-018-03149-4
258. Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, et al. Joint Profiling of Chromatin Accessibility and Gene Expression in Thousands of Single Cells. Science (80-) (2018) 361:1380–5. doi: 10.1126/science.aau0730
259. Chen S, Lake BB, Zhang K. High-Throughput Sequencing of the Transcriptome and Chromatin Accessibility in the Same Cell. Nat Biotechnol (2019) 37:1452–7. doi: 10.1038/s41587-019-0290-0
260. Mimitou EP, Cheng A, Montalbano A, Hao S, Stoeckius M, Legut M, et al. Multiplexed Detection of Proteins, Transcriptomes, Clonotypes and CRISPR Perturbations in Single Cells. Nat Methods (2019) 16:409–12. doi: 10.1038/s41592-019-0392-0
261. Frangieh CJ, Melms JC, Thakore PI, Geiger-Schuller KR, Ho P, Luoma AM, et al. Multimodal Pooled Perturb-CITE-seq Screens in Patient Models Define Mechanisms of Cancer Immune Evasion. Nat Genet (2021) 53:332–41. doi: 10.1038/s41588-021-00779-1
262. Schraivogel D, Gschwind AR, Milbank JH, Leonce DR, Jakob P, Mathur L, et al. Targeted Perturb-seq Enables Genome-Scale Genetic Screens in Single Cells. Nat Methods (2020) 17:629–35. doi: 10.1038/s41592-020-0837-5
263. Spanjaard B, Hu B, Mitic N, Olivares-Chauvet P, Janjuha S, Ninov N, et al. Simultaneous Lineage Tracing and Cell-Type Identification Using CrIsPr-Cas9-Induced Genetic Scars. Nat Biotechnol (2018) 36:469–73. doi: 10.1038/nbt.4124
264. Raj B, Wagner DE, McKenna A, Pandey S, Klein AM, Shendure J, et al. Simultaneous Single-Cell Profiling of Lineages and Cell Types in the Vertebrate Brain. Nat Biotechnol (2018) 36:442–50. doi: 10.1038/nbt.4103
265. Alemany A, Florescu M, Baron CS, Peterson-Maduro J, Van Oudenaarden A. Whole-Organism Clone Tracing Using Single-Cell Sequencing. Nature (2018) 556:108–12. doi: 10.1038/nature25969
266. Eng CHL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, et al. Transcriptome-Scale Super-Resolved Imaging in Tissues by RNA SeqFISH+. Nature (2019) 568:235–9. doi: 10.1038/s41586-019-1049-y
267. Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. Spatially Resolved, Highly Multiplexed RNA Profiling in Single Cells. Science (80-) (2015) 348:aaa6090–aaa6090. doi: 10.1126/science.aaa6090
268. Xia C, Fan J, Emanuel G, Hao J, Zhuang X. Spatial Transcriptome Profiling by MERFISH Reveals Subcellular RNA Compartmentalization and Cell Cycle-Dependent Gene Expression. Proc Natl Acad Sci USA (2019) 116:19490–9. doi: 10.1073/pnas.1912459116
269. Macaulay IC, Teng MJ, Haerty W, Kumar P, Ponting CP, Voet T. Separation and Parallel Sequencing of the Genomes and Transcriptomes of Single Cells Using G&T-Seq. Nat Protoc (2016) 11:2081–103. doi: 10.1038/nprot.2016.138
270. Nakagawa H, Fujita M. Whole Genome Sequencing Analysis for Cancer Genomics and Precision Medicine. Cancer Sci (2018) 109:513–22. doi: 10.1111/cas.13505
271. Zhu Z, Wang W, Lin F, Jordan T, Li G, Silverman S, et al. Genome Profiles of Lymphovascular Breast Cancer Cells Reveal Multiple Clonally Differentiated Outcomes With Multi-Regional LCM and G&T-Seq. bioRxiv (2019) 807156. doi: 10.1101/807156
272. Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, Jones PA. Genome-Wide Mapping of Nucleosome Positioning and DNA Methylation Within Individual DNA Molecules. Genome Res (2012) 22:2497–506. doi: 10.1101/gr.143008.112
273. Hernando-Herraez I, Evano B, Stubbs T, Commere PH, Jan Bonder M, Clark S, et al. Ageing Affects DNA Methylation Drift and Transcriptional Cell-to-Cell Variability in Mouse Muscle Stem Cells. Nat Commun (2019) 10:1–11. doi: 10.1038/s41467-019-12293-4
274. Jin Z, Liu Y. DNA Methylation in Human Diseases. Genes Dis (2018) 5:1–8. doi: 10.1016/j.gendis.2018.01.002
275. Salameh Y, Bejaoui Y, El Hajj N. DNA Methylation Biomarkers in Aging and Age-Related Diseases. Front Genet (2020) 11:171. doi: 10.3389/fgene.2020.00171
276. Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial Reconstruction of Single-Cell Gene Expression Data. Nat Biotechnol (2015) 33:495–502. doi: 10.1038/nbt.3192
277. Wolf FA, Angerer P, Theis FJ. Scanpy: Large-Scale Single-Cell Gene Expression Data Analysis. Genome Biol (2018) 19:15. doi: 10.1186/s13059-017-1382-0
278. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating Single-Cell Transcriptomic Data Across Different Conditions, Technologies, and Species. Nat Biotechnol (2018) 36:411–20. doi: 10.1038/nbt.4096
279. Lun ATL, McCarthy DJ, Marioni JC. A Step-by-Step Workflow for Low-Level Analysis of Single-Cell RNA-Seq Data With Bioconductor. F1000Research (2016) 5:2122. doi: 10.12688/f1000research.9501.2
280. Stuart T, Satija R. Integrative Single-Cell Analysis. Nat Rev Genet (2019) 20:257–72. doi: 10.1038/s41576-019-0093-7
281. Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, et al. Integrated Analysis of Multimodal Single-Cell Data. bioRxiv (2020). doi: 10.1101/2020.10.12.335331 2020.10.12.335331.
282. Haghverdi L, Lun ATL, Morgan MD, Marioni JC. Batch Effects in Single-Cell RNA-Sequencing Data Are Corrected by Matching Mutual Nearest Neighbors. Nat Biotechnol (2018) 36:421–7. doi: 10.1038/nbt.4091
283. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive Integration of Single-Cell Data. Cell (2019) 177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031
284. Argelaguet R, Cuomo ASE, Stegle O, Marioni JC. Computational Principles and Challenges in Single-Cell Data Integration. Nat Biotechnol (2021). doi: 10.1038/s41587-021-00895-7
285. Laurenti E, Göttgens B. From Haematopoietic Stem Cells to Complex Differentiation Landscapes. Nature (2018) 553:418–26. doi: 10.1038/nature25022
286. Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell (2015) 163:1663–77. doi: 10.1016/j.cell.2015.11.013
287. Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, et al. A Single-Cell Resolution Map of Mouse Hematopoietic Stem and Progenitor Cell Differentiation. Blood (2016) 128(8):e20–31. doi: 10.1182/blood-2016-05-716480
288. Giustacchini A, Thongjuea S, Barkas N, Woll PS, Povinelli BJ, Booth CAG, et al. Single-Cell Transcriptomics Uncovers Distinct Molecular Signatures of Stem Cells in Chronic Myeloid Leukemia. Nat Med (2017) 23:692–702. doi: 10.1038/nm.4336
289. Karamitros D, Stoilova B, Aboukhalil Z, Hamey F, Reinisch A, Samitsch M, et al. Single-Cell Analysis Reveals the Continuum of Human Lympho-Myeloid Progenitor Cells. Nat Immunol (2018) 19:85–97. doi: 10.1038/s41590-017-0001-2
290. Schulte R, Wilson NK, Prick JCM, Cossetti C, Maj MK, Gottgens B, et al. Index Sorting Resolves Heterogeneous Murine Hematopoietic Stem Cell Populations. Exp Hematol (2015) 43:803–11. doi: 10.1016/j.exphem.2015.05.006
291. Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment Within Human Melanoma. Cell (2019) 176:775–789.e18. doi: 10.1016/j.cell.2018.11.043
292. Bhagwat N, Dulmage K, Pletcher CH, Wang L, DeMuth W, Sen M, et al. An Integrated Flow Cytometry-Based Platform for Isolation and Molecular Characterization of Circulating Tumor Single Cells and Clusters. Sci Rep (2018) 8(1):5035. doi: 10.1038/s41598-018-23217-5
293. Shepherd MS, Kent DG. Emerging Single-Cell Tools Are Primed to Reveal Functional and Molecular Heterogeneity in Malignant Hematopoietic Stem Cells. Curr Opin Hematol (2019) 26:214–21. doi: 10.1097/MOH.0000000000000512
294. de Vries NL, Mahfouz A, Koning F, de Miranda NFCC. Unraveling the Complexity of the Cancer Microenvironment With Multidimensional Genomic and Cytometric Technologies. Front Oncol (2020) 10:1254. doi: 10.3389/fonc.2020.01254
295. Praktiknjo SD, Obermayer B, Zhu Q, Fang L, Liu H, Quinn H, et al. Tracing Tumorigenesis in a Solid Tumor Model at Single-Cell Resolution. Nat Commun (2020) 11:1–12. doi: 10.1038/s41467-020-14777-0
296. Stephenson E, Reynolds G, Botting RA, Calero-Nieto FJ, Morgan MD, Tuong ZK, et al. Single-Cell Multi-Omics Analysis of the Immune Response in COVID-19. Nat Med (2021) 27:904–16. doi: 10.1038/s41591-021-01329-2
297. Liberali P, Snijder B, Pelkmans L. Single-Cell and Multivariate Approaches in Genetic Perturbation Screens. Nat Rev Genet (2015) 16:18–32. doi: 10.1038/nrg3768
298. Xie S, Duan J, Li B, Zhou P, Hon GC. Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol Cell (2017) 66:285–299.e5. doi: 10.1016/j.molcel.2017.03.007
299. Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, et al. Pooled CRISPR Screening With Single-Cell Transcriptome Readout. Nat Methods (2017) 14:297–301. doi: 10.1038/nmeth.4177
300. Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, et al. Dissecting Immune Circuits by Linking Crispr-Pooled Screens With Single-Cell RNA-Seq. Cell (2016) 167:1883–96.e15. doi: 10.1016/j.cell.2016.11.039
301. Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell (2016) 167:1867–82.e21. doi: 10.1016/j.cell.2016.11.048
302. Giladi A, Paul F, Herzog Y, Lubling Y, Weiner A, Yofe I, et al. Single-Cell Characterization of Haematopoietic Progenitors and Their Trajectories in Homeostasis and Perturbed Haematopoiesis. Nat Cell Biol (2018) 20:836–46. doi: 10.1038/s41556-018-0121-4
303. Jin X, Simmons SK, Guo A, Shetty AS, Ko M, Nguyen L, et al. In Vivo Perturb-Seq Reveals Neuronal and Glial Abnormalities Associated With Autism Risk Genes. Science (80-) (2020) 370(6520):eaaz6063. doi: 10.1126/science.aaz6063
304. Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, et al. Hematopoietic Stem Cell Gene Therapy With a Lentiviral Vector in X-linked Adrenoleukodystrophy. Science (80-) (2009) 326:818–23. doi: 10.1126/science.1171242
305. Wang D, Wang J, Bai L, Pan H, Feng H, Clevers H, et al. Long-Term Expansion of Pancreatic Islet Organoids From Resident Procr+ Progenitors. Cell (2020) 180:1198–211.e19. doi: 10.1016/j.cell.2020.02.048
306. Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, Billestrup N, et al. The Ectopic Expression of Pax4 in the Mouse Pancreas Converts Progenitor Cells Into α and Subsequently β Cells. Cell (2009) 138:449–62. doi: 10.1016/j.cell.2009.05.035
307. Gu G, Dubauskaite J, Melton DA. Direct Evidence for the Pancreatic Lineage: NGN3+ Cells Are Islet Progenitors and Are Distinct From Duct Progenitors. Development (2002) 129:2447–57. doi: 10.1242/dev.129.10.2447
308. Minami K, Okuno M, Miyawaki K, Okumachi A, Ishizaki K, Oyama K, et al. Lineage Tracing and Characterization of Insulin-Secreting Cells Generated From Adult Pancreatic Acinar Cells. Proc Natl Acad Sci USA (2005) 102:15116–21. doi: 10.1073/pnas.0507567102
309. Piran R, Lee SH, Li CR, Charbono A, Bradley LM, Levine F. Pharmacological Induction of Pancreatic Islet Cell Transdifferentiation: Relevance to Type I Diabetes. Cell Death Dis (2014) 5(7):e1357. doi: 10.1038/cddis.2014.311
310. Wagner DE, Klein AM. Lineage Tracing Meets Single-Cell Omics: Opportunities and Challenges. Nat Rev Genet (2020) 21:410–27. doi: 10.1038/s41576-020-0223-2
311. Kester L, van Oudenaarden A. Single-Cell Transcriptomics Meets Lineage Tracing. Cell Stem Cell (2018) 23:166–79. doi: 10.1016/j.stem.2018.04.014
312. McKenna A, Gagnon JA. Recording Development With Single Cell Dynamic Lineage Tracing. Dev (2019) 146(12):dev169730. doi: 10.1242/dev.169730
313. McKenna A, Findlay GM, Gagnon JA, Horwitz MS, Schier AF, Shendure J. Whole-Organism Lineage Tracing by Combinatorial and Cumulative Genome Editing. Science (80-) (2016) 353(6298):aaf7907. doi: 10.1126/science.aaf7907
314. Wagner DE, Weinreb C, Collins ZM, Briggs JA, Megason SG, Klein AM. Single-Cell Mapping of Gene Expression Landscapes and Lineage in the Zebrafish Embryo. Science (80-) (2018) 360:981–7. doi: 10.1126/science.aar4362
315. Weinreb C, Rodriguez-Fraticelli A, Camargo FD, Klein AM. Lineage Tracing on Transcriptional Landscapes Links State to Fate During Differentiation. Science (80-) (2020) 367(6479):eaaw3381. doi: 10.1126/science.aaw3381
316. Kalhor R, Kalhor K, Mejia L, Leeper K, Graveline A, Mali P, et al. Developmental Barcoding of Whole Mouse Via Homing CRISPR. Science (80-) (2018) 361(6405):eaat9804. doi: 10.1126/science.aat9804
317. Kalhor R, Mali P, Church GM. Rapidly Evolving Homing CRISPR Barcodes. Nat Methods (2017) 14:195–200. doi: 10.1038/nmeth.4108
318. Chan MM, Smith ZD, Grosswendt S, Kretzmer H, Norman TM, Adamson B, et al. Molecular Recording of Mammalian Embryogenesis. Nature (2019) 570:77–82. doi: 10.1038/s41586-019-1184-5
319. Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 Genome Editing Induces a P53-Mediated DNA Damage Response. Nat Med (2018) 24:927–30. doi: 10.1038/s41591-018-0049-z
320. Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, et al. P53 Inhibits CRISPR-Cas9 Engineering in Human Pluripotent Stem Cells. Nat Med (2018) 24:939–46. doi: 10.1038/s41591-018-0050-6
321. Waylen LN, Nim HT, Martelotto LG, Ramialison M. From Whole-Mount to Single-Cell Spatial Assessment of Gene Expression in 3D. Commun Biol (2020) 3:1–11. doi: 10.1038/s42003-020-01341-1
322. Berglund E, Maaskola J, Schultz N, Friedrich S, Marklund M, Bergenstråhle J, et al. Spatial Maps of Prostate Cancer Transcriptomes Reveal an Unexplored Landscape of Heterogeneity. Nat Commun (2018) 9:1–13. doi: 10.1038/s41467-018-04724-5
323. Asp M, Bergenstråhle J, Lundeberg J. Spatially Resolved Transcriptomes—Next Generation Tools for Tissue Exploration. BioEssays (2020) 42:1900221. doi: 10.1002/bies.201900221
324. Femino AM, Fay FS, Fogarty K, Singer RH. Visualization of Single RNA Transcripts in Situ. Science (80-) (1998) 280:585–90. doi: 10.1126/science.280.5363.585
325. Bauman JGJ, Wiegant J, Borst P, van Duijn P. A New Method for Fluorescence Microscopical Localization of Specific DNA Sequences by in Situ Hybridization of Fluorochrome-Labelled RNA. Exp Cell Res (1980) 128:485–90. doi: 10.1016/0014-4827(80)90087-7
326. Lyubimova A, Itzkovitz S, Junker JP, Fan ZP, Wu X, Van Oudenaarden A. Single-Molecule mRNA Detection and Counting in Mammalian Tissue. Nat Protoc (2013) 8:1743–58. doi: 10.1038/nprot.2013.109
327. Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L. Single-Cell in Situ RNA Profiling by Sequential Hybridization. Nat Methods (2014) 11:360–1. doi: 10.1038/nmeth.2892
328. Lubeck E, Cai L. Single-Cell Systems Biology by Super-Resolution Imaging and Combinatorial Labeling. Nat Methods (2012) 9:743–8. doi: 10.1038/nmeth.2069
329. Shah S, Lubeck E, Zhou W, Cai L. In Situ Transcription Profiling of Single Cells Reveals Spatial Organization of Cells in the Mouse Hippocampus. Neuron (2016) 92:342–57. doi: 10.1016/j.neuron.2016.10.001
330. Lohoff T, Ghazanfar S, Missarova A, Koulena N, Pierson N, Griffiths JA, et al. Highly Multiplexed Spatially Resolved Gene Expression Profiling of Mouse Organogenesis. bioRxiv (2020) 24(7):939–46. doi: 10.1101/2020.11.20.391896 2020.11.20.391896.
331. Moncada R, Barkley D, Wagner F, Chiodin M, Devlin JC, Baron M, et al. Integrating Microarray-Based Spatial Transcriptomics and Single-Cell RNA-Seq Reveals Tissue Architecture in Pancreatic Ductal Adenocarcinomas. Nat Biotechnol (2020) 38:333–42. doi: 10.1038/s41587-019-0392-8
332. Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and Analysis of Gene Expression in Tissue Sections by Spatial Transcriptomics. Science (80-) (2016) 353:78–82. doi: 10.1126/science.aaf2403
333. Chen WT, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell (2020) 182:976–91.e19. doi: 10.1016/j.cell.2020.06.038
334. Carow B, Hauling T, Qian X, Kramnik I, Nilsson M, Rottenberg ME. Spatial and Temporal Localization of Immune Transcripts Defines Hallmarks and Diversity in the Tuberculosis Granuloma. Nat Commun (2019) 10:1–15. doi: 10.1038/s41467-019-09816-4
335. Asp M, Giacomello S, Larsson L, Wu C, Fürth D, Qian X, et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell (2019) 179:1647–60.e19. doi: 10.1016/j.cell.2019.11.025
336. Dries R, Zhu Q, Dong R, Eng CHL, Li H, Liu K, et al. Giotto: A Toolbox for Integrative Analysis and Visualization of Spatial Expression Data. Genome Biol (2021) 22:78. doi: 10.1186/s13059-021-02286-2
337. Palla G, Spitzer H, Klein M, Fischer D, Christina A, Benedikt Kuemmerle L, et al. Squidpy: A Scalable Framework for Spatial Single Cell 2 Analysis. bioRxiv (2021). doi: 10.1101/2021.02.19.431994 2021.02.19.431994.
338. Grauel AL, Nguyen B, Ruddy D, Laszewski T, Schwartz S, Chang J, et al. TGFβ-Blockade Uncovers Stromal Plasticity in Tumors by Revealing the Existence of a Subset of Interferon-Licensed Fibroblasts. Nat Commun (2020) 11:1–17. doi: 10.1038/s41467-020-19920-5
339. Baccin C, Al-Sabah J, Velten L, Helbling PM, Grünschläger F, Hernández-Malmierca P, et al. Combined Single-Cell and Spatial Transcriptomics Reveal the Molecular, Cellular and Spatial Bone Marrow Niche Organization. Nat Cell Biol (2020) 22:38–48. doi: 10.1038/s41556-019-0439-6
340. Rood JE, Stuart T, Ghazanfar S, Biancalani T, Fisher E, Butler A, et al. Toward a Common Coordinate Framework for the Human Body. Cell (2019) 179:1455–67. doi: 10.1016/j.cell.2019.11.019
341. Snyder MP, Lin S, Posgai A, Atkinson M, Regev A, Rood J, et al. The Human Body at Cellular Resolution: The NIH Human Biomolecular Atlas Program. Nature (2019) 574:187–92. doi: 10.1038/s41586-019-1629-x
342. Pijuan-Sala B, Griffiths JA, Guibentif C, Hiscock TW, Jawaid W, Calero-Nieto FJ, et al. A Single-Cell Molecular Map of Mouse Gastrulation and Early Organogenesis. Nature (2019) 566:490–5. doi: 10.1038/s41586-019-0933-9
343. Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Domínguez Á, et al. The Bone Marrow Microenvironment at Single-Cell Resolution. Nature (2019) 569:222–8. doi: 10.1038/s41586-019-1104-8
344. Peng J, Sun BF, Chen CY, Zhou JY, Chen YS, Chen H, et al. Single-Cell RNA-Seq Highlights Intra-Tumoral Heterogeneity and Malignant Progression in Pancreatic Ductal Adenocarcinoma. Cell Res (2019) 29:725–38. doi: 10.1038/s41422-019-0195-y
345. Candotti F, Shaw KL, Muul L, Carbonaro D, Sokolic R, Choi C, et al. Gene Therapy for Adenosine Deaminase-Deficient Severe Combined Immune Deficiency: Clinical Comparison of Retroviral Vectors and Treatment Plans. Blood (2012) 120:3635–46. doi: 10.1182/blood-2012-02-400937
346. Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Adams S, Howe SJ, et al. Immunodeficiency: Long-term Persistence of a Polyclonal T Cell Repertoire After Gene Therapy for X-Linked Severe Combined Immunodeficiency. Sci Transl Med (2011) 3(97):97ra79. doi: 10.1126/scitranslmed.300271
Keywords: cell therapy, gene therapy, single-cell sequencing, scRNA-seq, multimodal omics, multiomics, CAR T cell therapy, disease heterogeneity
Citation: Bode D, Cull AH, Rubio-Lara JA and Kent DG (2021) Exploiting Single-Cell Tools in Gene and Cell Therapy. Front. Immunol. 12:702636. doi: 10.3389/fimmu.2021.702636
Received: 29 April 2021; Accepted: 28 June 2021;
Published: 12 July 2021.
Edited by:
F. Susan Wong, Cardiff University, United KingdomReviewed by:
Georgia Fousteri, San Raffaele Hospital (IRCCS), ItalyGurudutt Pendyala, University of Nebraska Medical Center, United States
Stephan Schlickeiser, Charité - Universitätsmedizin Berlin, Germany
Copyright © 2021 Bode, Cull, Rubio-Lara and Kent. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David G. Kent, ZGF2aWQua2VudEB5b3JrLmFjLnVr