 Theinmozhi Arulraj
Theinmozhi Arulraj Sebastian C. Binder
Sebastian C. Binder Philippe A. Robert
Philippe A. Robert Michael Meyer-Hermann
Michael Meyer-Hermann- 1Department of Systems Immunology, Braunschweig Integrated Centre of Systems Biology, Helmholtz Centre for Infection Research, Braunschweig, Germany
- 2Department of Immunology, University of Oslo, Oslo, Norway
- 3Institute for Biochemistry, Biotechnology and Bioinformatics, Technische Universität Braunschweig, Braunschweig, Germany
Germinal Centres (GCs) are transient structures in secondary lymphoid organs, where affinity maturation of B cells takes place following an infection. While GCs are responsible for protective antibody responses, dysregulated GC reactions are associated with autoimmune disease and B cell lymphoma. Typically, ‘normal’ GCs persist for a limited period of time and eventually undergo shutdown. In this review, we focus on an important but unanswered question – what causes the natural termination of the GC reaction? In murine experiments, lack of antigen, absence or constitutive T cell help leads to premature termination of the GC reaction. Consequently, our present understanding is limited to the idea that GCs are terminated due to a decrease in antigen access or changes in the nature of T cell help. However, there is no direct evidence on which biological signals are primarily responsible for natural termination of GCs and a mechanistic understanding is clearly lacking. We discuss the present understanding of the GC shutdown, from factors impacting GC dynamics to changes in cellular interactions/dynamics during the GC lifetime. We also address potential missing links and remaining questions in GC biology, to facilitate further studies to promote a better understanding of GC shutdown in infection and immune dysregulation.
Introduction
Germinal Centres (GCs) are specialized structures within the secondary lymphoid organs essential for the humoral immune response that form after an infection. B cells in the GCs evolve towards a foreign antigen to progressively improve their affinities (1), by a process referred to as affinity maturation. Affinity maturation is the outcome of somatic hypermutation (SHM) in the genes encoding B cell receptors (BCRs) (2), followed by a selection process aided by follicular dendritic cells (FDCs) and T follicular helper (Tfh) cells within the GCs (3, 4).
A mature GC consists of two distinct compartments: A dark zone (DZ) and a light zone (LZ) (5, 6). The DZ is predominantly filled with actively dividing B cells called centroblasts, mutating with a high rate resulting in BCR affinity changes (2). A network of FDCs and Tfh cells are present in the GC LZ where the selection takes place. Non-dividing B cells (centrocytes) with mutated BCRs are selected in the GC LZ based on the ability to acquire antigen from FDCs and presenting the processed antigen to the Tfh cells that provide survival and proliferative signals (7, 8). Selected centrocytes can either progress to plasma or memory B cells and egress from the GC, or recycle back to the GC DZ and proliferate (9–11). Iterative rounds of such mutation and selection as GC B cells migrate between the two zones (7), result in a stepwise optimization of B cell affinities towards the locally presented antigen, giving rise to affinity-matured memory and plasma cells (Figure 1).
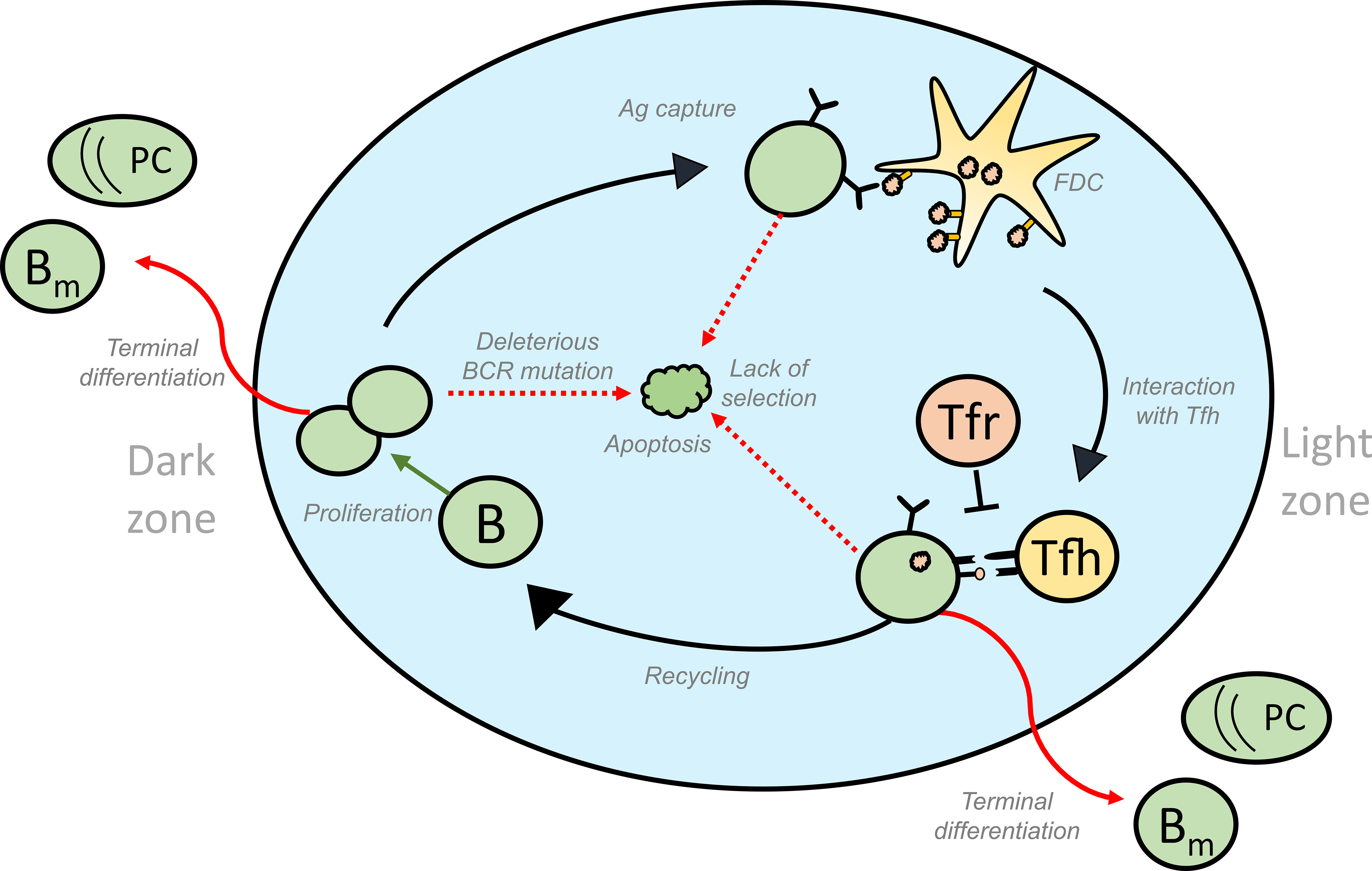
Figure 1 Schematic representation of the different fates of GC B cells that lead to GC volume changes: GC B cells proliferate in the DZ and increase in number. SHM accompanying proliferation might induce deleterious mutations in the BCR gene of some of the B cells, thus activating apoptosis in these GC B cells. Lack of acquisition of antigen, survival signals from FDCs and signals from Tfh cells lead to the apoptosis of GC B cells in the LZ. On the other hand, successful acquisition of these signals could result in the differentiation of the GC B cells into effector cell types such as memory and plasma cells that exit the GC. Alternatively, selected cells can move back to the DZ by a process termed recycling and undergo further rounds of divisions thus contributing to an increase in number of GC B cells. Green and red arrows represent processes that increase or decrease the GC volume, respectively and influence GC shutdown. GC, Germinal Centres; BCR, B cell receptor; FDC, Follicular Dendritic Cells, Tfh, T follicular helper cells, B, GC B cells; Tfr, T follicular regulatory cells; PC, Plasma cell; Bm , Memory B cell; Ag, Antigen.
GCs are highly dynamic structures and the evolution of GC arises as a combination of processes at the cellular level: inflow of new founder B cells (12–14), B cell proliferation, apoptosis, and differentiation to effector cell types. Entry of new founder cells and proliferation scales up the number of GC B cells, while apoptosis and differentiation that leads to GC exit counteracts this effect (11). Their contribution to the GC B cell population is dynamic over time as they are influenced by interactions with FDCs and Tfh cells (15), immune complexes trapped on FDCs (16), soluble signals secreted by Tfh cells (17), presence of T follicular regulatory (Tfr) cells (18) and soluble antibodies from plasma cells (19).
GCs show a typical kinetics with an initial phase of growth followed by a contraction phase. In the initial phase, inflow and clonal expansion predominantly increases the number of GC B cells and the GC expands (20) until it reaches a peak size. During the contraction phase, apoptosis and exit exceed proliferation and the GC begins to shrink (11). This already suggests different phenomenological ways in which GC shutdown can occur (schematically shown in the Figure 1). Hence, shutdown can be brought about by a decrease in recycling or proliferation, or an increase in apoptosis or terminal differentiation over time. Theoretically, in the absence of an explicit signal, shutdown can be influenced by parameters that control GC events like proliferation, differentiation and apoptosis (11). An impact of DZ to LZ phenotype differentiation speed on the GC lifetime has also been predicted (11) and determines whether the GC reaction will terminate or continue growing in the absence of other shutdown signals (21). The GC response in a lymphoid organ lasts for approximately 3 weeks (22, 23) when induced by model antigens but persist longer in response to viral infections (24). However, lifespan of single GCs has not been determined so far. Tracking the same GC over time in vivo is challenging (25), and single measurements might reflect shifted dynamics and are subject to substantial stochastic variation. This hinders the study of single GC dynamics and hence, the experimental analysis of GC shutdown. Consequently, the mechanistic details of natural GC shutdown and which signals induce the shift from expansion to contraction phase are mainly unknown.
In this review, we first briefly discuss the various ways by which B cell fates are regulated in a normal GC. Then, we discuss our present understanding of the biological changes (Figure 2) that could act as a termination signal and promote GC shutdown by inducing changes in the fates of GC B cells. Finally, we also discuss what we can learn from GCs with extended maintenance phase such as chronic GCs induced by viral infections or dysregulated GCs associated with B cell lymphomas (BCLs).
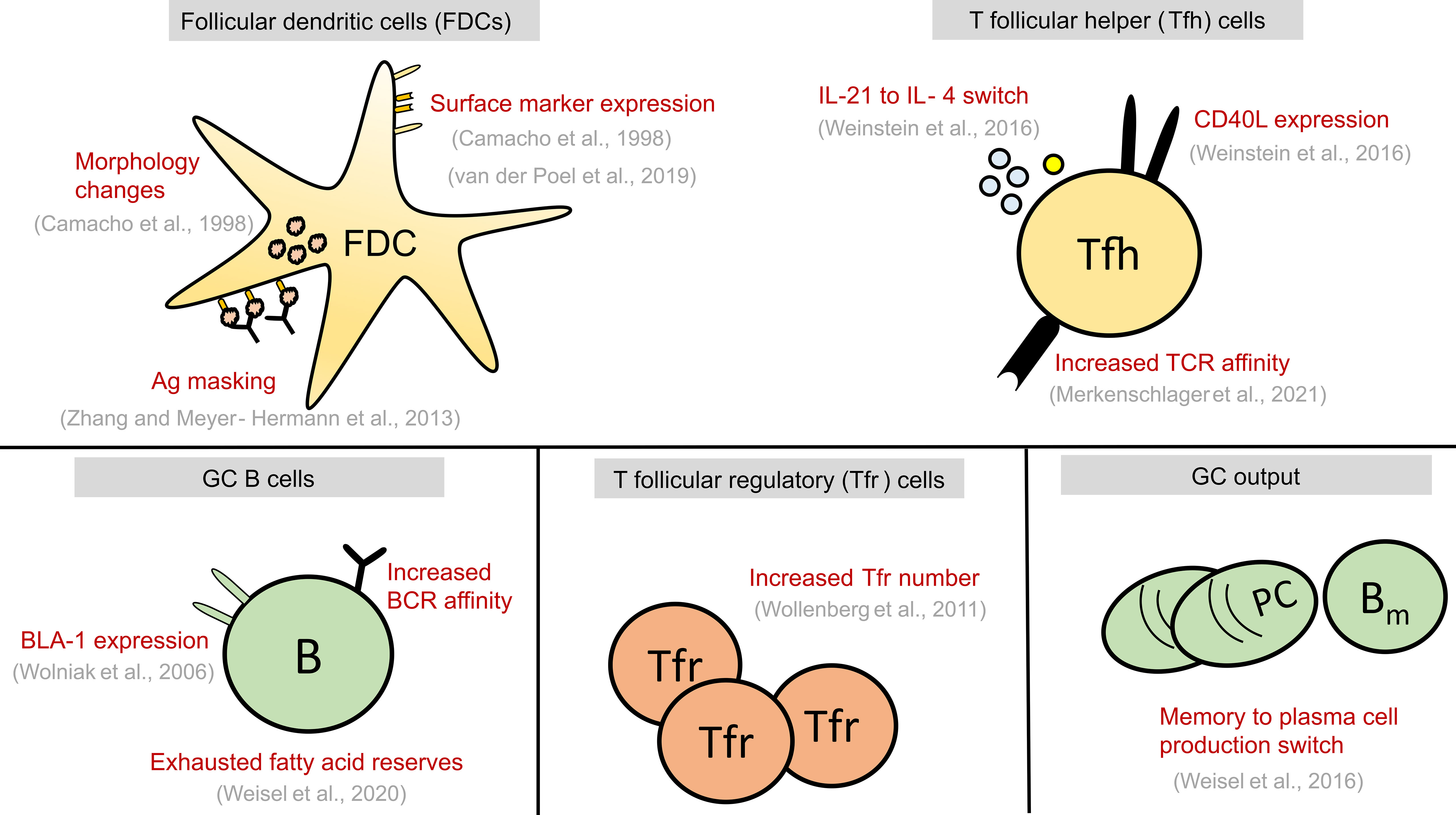
Figure 2 Summary of alterations seen in GC cell types at late stages of GC reaction. These alterations might play a causal role in GC shutdown by influencing the fate decisions of GC B cells shown in Figure 1 or act as a contributing factor to GC shutdown. GC, Germinal centre; FDCs, Follicular dendritic cells; Tfr, T follicular regulatory cells; Tfh, T follicular helper cells, B, GC B cells; TCR, T cell receptor; BCR, B cell receptor; PC, Plasma cell; Bm , Memory B cell; Ag, Antigen.
Regulation of B Cell Fates in the GCs
As GC kinetics is determined by the balance of apoptosis, proliferation, and differentiation, studies exploring the dynamics of these processes in a normal GC reaction constitute a first approach for a mechanistic understanding of GC shutdown. GC B cell fates are primarily regulated in response to antigen acquisition, soluble cytokines and CD40L signals from Tfh cells. Defects in various B cell intrinsic and extrinsic factors affect normal maintenance and promote GC shutdown (see Table 1). However, there is an intense debate over which biological signals are mainly responsible for the normal GC shutdown. The actual cause might vary depending on the type of GC response such as GCs associated with acute or chronic infection, auto-immunity, tumor or allergy. Here, based on the existing knowledge of GC reactions and examples from Table 1, we discuss the potential mechanisms that might promote a natural GC shutdown (summarized in Figure 2). At first, the different fates of GC B cells relevant to GC shutdown are discussed.
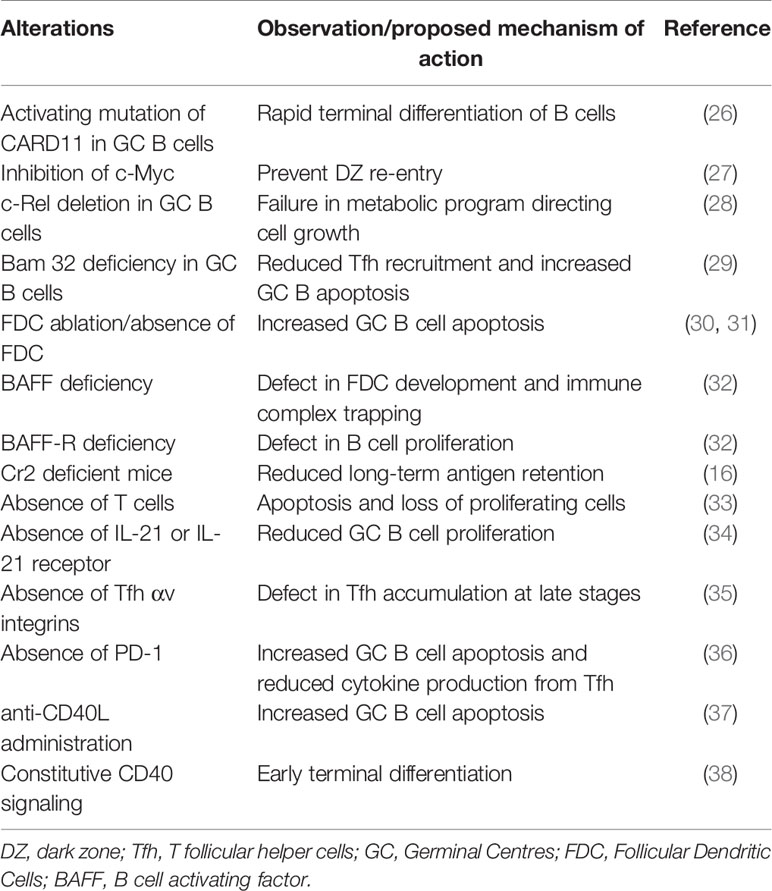
Table 1 Summary of GC alterations resulting in premature termination of GCs and the proposed mechanism of action.
GC B Cell Apoptosis
Extensive apoptosis takes place in both the DZ and LZ compartments of GCs (39). GC B cells spontaneously undergo apoptosis even in the absence of any death signals due to rapidly decaying levels of cFLIP, an inhibitor of apoptosis (40). In addition, FAS activation in B cells leads to apoptosis via the activation of caspases (41). Activation of the Fas-mediated pathways can be prevented by interactions with FDCs or acquisition of CD40 signals due to sustained expression of cFLIP (40). Due to the highly active apoptotic program, the default state of the LZ GC B cells is becoming apoptotic, unless they are positively selected by antigen acquisition and Tfh signals (8, 42).
While GC B cell apoptosis mainly happens due to the absence of survival signals and lack of selection in the LZ, DZ B cells undergo apoptosis primarily due to the deleterious mutations induced in the BCR genes during SHM (39). Stewart et al., found that most B cells with deleterious mutations undergo apoptosis in the DZ and are unable to reach the LZ for selection (43). In addition to Fas, various other molecular players such as BCL-2 have been identified to control B cell apoptosis and are believed to act through distinct mechanisms (44). BCL-2 expression can decrease apoptosis of GC B cells by 10-fold (45) but is insufficient to prevent apoptosis due to deleterious mutations induced in BCR genes (43). Jumaa et al. discovered a mechanism by which BCR expression regulates the metabolism of B cells in a BCR signaling independent manner (46). According to this mechanism, BCR regulates changes in endoplasmic reticulum that in turn controls mitochondrial activity. Thus, the loss of BCR expression leads to altered B cell metabolism (46). Consequently, a similar effect might be expected in GC B cells that cannot express functional BCRs due to deleterious mutations.
Apoptosis is considered an important process that is counteracting the extensive proliferation to maintain the constant size of the GCs during their maintenance phase and in Peyer’s patches (PPs) (39). Consequently, changes in apoptosis are associated with changes in GC size (Table 1). Deficiency in AID (Activation-induced cytidine deaminase), the enzyme responsible for SHM, decreases the loss of B cells due to damaged BCRs and, thus, increases the size of GCs (47). Complete lack of survival signals such as the absence of FDCs (30) or Tfh cells (33) leads to apoptosis of GC B cells and results in GC termination. Aberrations in Fas and BCL-2 are also associated with B-cell lymphoma (48, 49).
GC B Cell Proliferation
A characteristic property of GC B cells is the ability to undergo extensive proliferation. GC founder cells divide (6, 22, 25) and increase the GC volume at initial stages. GC B cells positively selected by the Tfh cells recycle back to the DZ (7) to undergo further rounds of division. The speed of cell cycle progression of selected B cells is regulated by T cell help which shortens the S phase (50). The extent of cell division is found to depend on the amount of antigen captured and presented by the B cells to the Tfh cells (51–53). More specifically, the number of divisions of a selected B cell is determined by the level of c-Myc induced by signals from the Tfh cells (54). Cyclin D3 is important for sustaining proliferative expansion of GC B cells (55–57). Pae et al., discovered that Cyclin D3 controls cell divisions in a dose dependent manner, following transient induction of c-Myc by Tfh signals (58).
Changes in the activity of mTORC1 (59) and in expression of FoxO1 (60, 61) are also involved in the regulation of cell division progression in GC B cells. mTORC1 does not dictate the number of cell divisions but is involved in the cell growth prior to clonal expansion, without which the cell cannot divide (59). Proliferation sustains the number of GC B cells, as GCs collapse rapidly due to defects in proliferation or recycling such as c-Myc ablation (27, 62) or c-Rel deficiency (28).
GC B Cell Terminal Differentiation
Signals from Tfh cells induce the differentiation of certain GC B cells into precursors of output cells, i.e., plasma or memory. These differentiated cells typically exit the GC and decrease the GC volume. It is widely believed that the nature and intensity of Tfh cell signals determine the fate of selected cells although a clear understanding is lacking. Wang et al, discovered that development of memory B cells is dependent on IL-9 produced by Tfh cells (63). Ise et al, discovered a population of LZ B cells that is prone to become plasma cells due to stable interactions with Tfh cells suggesting that differentiation to plasma cells is dependent on the GC B cell-Tfh cell interaction strength (64). However, Kräutler et al. proposed a two-signal based mechanism in which differentiation is initiated by signals delivered to B cells during antigen engagement and Tfh cell signals are only required to complete the process (65). It is also not understood whether the B cells directly differentiate and exit the GC after Tfh selection or recycle and divide further in the DZ before final differentiation and GC exit (52). The latter theory was supported by Radtke and Bannard, who showed that dividing GC B cells in the DZ express low levels of BLIMP-1 and are more sensitized to plasma cell differentiation (66). A faster terminal differentiation can promote faster exit of B cells from GC and leads to early GC termination (38). On the other hand, lack of differentiation and GC exit can lead to excess accumulation of B cells in the GCs (67). In general, it has been accepted that early waves of low affinity cells exiting the GC are enriched in memory cells, while later high affinity cells generate more plasma cells (68–70), suggesting a gradual switch between the decision of memory to plasma cell differentiation (71). CD20 is involved in maintenance of the resting state of Ramos B cell by regulating receptor organization on the surface of the cell and the loss of CD20 induced the differentiation of naïve B cells into plasma cells (72). Similarly, whether changes in the expression or ligation of CD20 is involved in the GC B cell differentiation into plasma cell needs to be investigated. Further work clarifying the signals promoting differentiation or recycling and their dynamics during the GC reaction would help improving knowledge on GC volume changes due to these processes.
Morphological and Functional Changes of Follicular Dendritic Cells
Follicular dendritic cells (FDCs) are necessary for the maintenance of GC reactions as disrupting the FDC network results in premature termination of GCs (30). Similarly, GCs formed in the absence of FDCs are not maintained at later stages (31). FDC disruption results in unavailability of immune complexes carrying the target antigens and lack of cytokines or survival factors such as BAFF (B cell activating factor). Absence of BAFF and BAFF receptor (32) or defect in long term retention of antigen due to the lack of complement receptors (16) result in unsustained GC responses.
FDCs undergo progressive development during the GC response which is observed as changes in morphology and expression of surface markers such as CD23 (6), ICAM-1 (73), VCAM-1 (74) and FcγRIIB receptors (74, 75). Environmental signals have been shown to influence the functional ability of FDCs (76). Cytokines such as IL-2 and IL-10, could also alter the morphology of FDCs by influencing their contractility (77).
Intrinsic defects in aged stromal cells are associated with changes in magnitude and output of GC responses (78). Similarly, such changes in the nature and differentiation state of FDCs over time potentially modulate the interaction of B cells with FDCs (6), but it is unknown if similar changes occurring at late stages trigger GC shutdown. As FDC maintenance requires lymphotoxin signaling delivered by the B cells (79, 80), changes in the differentiation state of FDCs by continuous interactions with GC B cells may promote GC shut down (81) by altering survival signals and/or antigen availability.
Keşmir and De Boer have predicted that limiting antigen rather than Tfh cell help drives the shutdown of GCs as only a few T cells are sufficient to sustain GC reactions (82). A decrease in antigen availability over time would reduce the uptake of antigen by GC B cells and in turn decrease the intensity of Tfh cell signals received. This can terminate the GCs by increasing the fraction of B cells undergoing apoptosis and/or by decreasing the Tfh cell induced proliferation of B cells. As a large amount of antigen is observed in follicles for an extended period of time (83), it was believed that GC shutdown is unlikely to be due to a decrease in antigen availability. However, the antigen access of GC B cells could vary due to the above-described changes in the FDC morphology. A complex organization and non-uniform distribution of immune complexes on the FDC surface has been identified (83, 84). Alterations in FDC morphology leading to a loss of iccosome generation and burial of antigen in the form of pockets could be the reason behind termination of GCs (85). Investigating the organization of immune complexes and receptors on the FDC surface at different stages of the GC reaction might unravel the role of FDCs in GC shutdown.
Heesters et al. found that immune complexes in the FDCs undergo periodic cycling (86, 87), suggesting a mechanism for antigen retention (83, 88, 89). As changes in antigen presentation dynamics might also terminate GCs, it can be speculated that GC termination might be due to modulation of antigen cycling dynamics during the GC reaction. Although changes in the morphology and functional state of FDCs might induce changes in antigen presentation dynamics, there is no experimental evidence that antigen cycling is dynamically modulated in the GC FDCs.
Alternatively, phenotypic changes in FDCs could positively influence GC B cell differentiation rather than limiting antigen access. This mechanism could also potentially terminate GCs by promoting differentiation and exit of GC B cells (81).
Feedback Regulation by Soluble Antibodies
Negative feedback due to soluble antibodies is another mechanism that could decrease the antigen access of GC B cells. Soluble antibodies can suppress or enhance the antibody response, when passively administered with an immunogenic antigen (90). Similarly, maternally derived antibodies can suppress the vaccination response of offspring by influencing the output of GC reactions which can be partially overcome by increasing the dose of antigen (91). Suppression of B cell responses by soluble antibodies can occur due to epitope masking of the administered antigen (19, 92–94).
Further, exogenous antibodies administered are able to enter the GCs, are found deposited on the FDC network and alter GC kinetics and affinity maturation (19). Consequently, soluble antibodies produced from plasma cells might mask the antigen on FDCs and limit available antigen (21, 95). Accordingly, one theory is that the endogenous soluble antibodies terminate the GC by decreasing antigen access over time (19). Mathematical modeling also predicted that high concentration of antibodies can shut down GCs (19, 96). The existence of epitope-specific antibody feedback could shift the B cell response away from immunodominant epitopes by enhancing B cells recognizing other epitopes (93, 97), and generate complex GC dynamics. In the context of secondary stimulation, pre-existing high affinity memory B cells might induce a premature end to the GC reaction due to high antibody feedback derived from differentiation of memory B cells to antibody-forming plasma cells (97). This theory is supported by the finding that reentry of reactivated memory B cells into GCs is limited (98).
It is hard to test the mechanism by which the soluble antibodies influence GC shutdown because lack of soluble antibodies will not only disrupt a potential antigen masking phenomenon but also decrease the clearance of antigen. Both the lack of soluble antibodies (19) and long term persistence of immune complexes (99) are associated with long-lived GCs. However, Bergström et al. showed that the inhibitory effect of passively administered IgG does not correlate with the clearance of antigen from the lymphoid organ and further, inhibition of antibody response was seen in FcRγ knockout mice which lacks the clearance mechanism by antibodies suggesting that suppression is most likely due to antigen masking (92).
On the contrary, early waves of antibodies could support the GC reaction by facilitating the transport of antigen to FDCs in the follicles. Although small antigens can directly enter B cell follicles through conduits, larger antigens are transported by B cells in a complement-dependent manner (100–103) which relies on the formation of immune complexes. This enhancing role of antibodies is likely restricted to early stages of the GC reaction. Whether this mechanism contributes to the maintenance of chronic GCs with persistent antigen due to replicating pathogens needs to be examined.
GCs might also communicate by the exchange of soluble antibodies (19). As newly produced or injected antibodies quickly distribute over the organism, it is likely that antibodies produced by one GC will appear in other GCs. But it is unknown whether the GC shutdown is extrinsically regulated by other GCs.
Progressive Changes in T Follicular Helper Cells
Tfh cells are required to maintain the GC reactions (33). Like FDCs, dynamic changes are also observed in Tfh cells during the course of a GC reaction. Tfh cells switch from a primarily IL-21 to an IL-4 producing state (104). Changes in surface ligand expression such as CD40L are also observed as the GC evolves (104).
Affinity of TCRs could influence the GC lifetime (105) by impacting the GC B cell fate decision. Hence, the repertoire and specificity of Tfh cells might be critical in determining the GC longevity. Recent findings suggested that the Tfh cells undergo selection similar to GC B cells (106). In this process, Tfh cells with high affinities towards the pMHC presented by B cells are selected (106). Implications of such Tfh selection on GC shutdown are presently unknown.
Lack of IL-21 signaling (34) or block of CD40L (37) also result in premature termination of GCs as recycling and proliferation of GC B cells cannot be sustained. On the other hand, constitutive expression of CD40L on B cells mimicking excessive CD40 signaling also resulted in earlier termination of the GCs (38, 107). Understanding this bimodal effect of Tfh cell signals might help interpret the role of Tfh cells in maintenance vs shutdown.
A positive feedback loop of ICOS-ICOSL signaling promotes brief interactions between B and Tfh cells with enhanced surface engagement (108). Pratama et al., have shown that miRNA-146a regulates ICOS-ICOSL signaling in GCs and loss of this miRNA results in excess numbers of Tfh and GC B cells due to increased ICOSL expression on GC B cells (109). As miRNA-146a expression peaks at late stages of GC responses when the Tfh cell response declines (109), this suggests that reduced ICOS-ICOSL signaling might influence the GC dynamics at late stages leading to GC termination.
Absence of PD-1 on Tfh cells also resulted in smaller GCs at late time points due to increased GC B cell apoptosis (36). In summary, these findings suggest that GC shutdown can be altered by changes in Tfh cell signals. However, examining the role of Tfh cells in shutdown faces numerous challenges. B-T interactions in GCs are rather transient (110) compared to B-T interactions at the T-B border zone (111), making it hard to monitor them. As T cell help received by GC B cells is dependent on presented antigen (7), it is possible that underlying changes during shutdown are primarily arising from changes in antigen uptake by B cells as described in previous sections. Moreover, Tfh and GC B cells are known to maintain and influence each other in murine GCs (106, 112). Such intricate mutual dependence makes it hard to distinguish the cause and the effect (105).
Another mode of Tfh cell regulation is inhibition by antigen presentation of plasma cells (113), warranting further studies on maintenance of Tfh cells and the interplay with B cell.
Tfh cells can also migrate between GCs (114), contributing to intercommunication between different GCs. It remains speculative whether GCs regulate the dynamics and shutdown of neighboring GCs by the exchange of Tfh cells.
Suppressive Nature of T Follicular Regulatory Cells
Immune suppressive mechanisms of regulatory T cells (Tregs) play a role in controlling the magnitude of immune responses (115). T follicular regulatory (Tfr) cells, expressing CXCR5, PD-1 and Foxp3 were identified as Treg subset found in GCs (18, 116, 117). Dynamics of Tfr cells suggest that they might play a role in the GC termination. Tfr cells increase in number at the peak of the GC reaction and the Tfh/Tfr ratio decreases during the contraction phase (18, 118).
Studies on the influence of Tfr cell deletion on GC kinetics or GC size reported contradictory results and different subsets of Tfr cells might have different effects (119). These contradictory results could also partly be explained due to differences in experimental approaches and markers used to deplete Tfr cells. Even though CXCR5 is generally targeted to deplete Tfr cells in GCs, it was shown that CXCR5 negative Tfr cells can still access the GCs (120), suggesting a need to better characterize the markers and behavior of Tfr cells in order to specifically deplete them.
However, characterization of Tfr cells remains challenging. For instance, though Tfr and Tfh cells share phenotypic characteristics and are yet distinct (117), unique suppressive T cell subsets which show moderate characteristics of both cell types were found in human tonsils (121). Similarly, in aged mice, IL-10 producing Tfh cells accumulate which is associated with impaired responses (122).
Indeed, in murine Peyer’s patches, Foxp3+ T cells can transform into Foxp3- Tfh cells, showing that Foxp3 expression is plastic (123). Inducing Foxp3 expression is sufficient to convert Tfh to Tfr like cells (124), suggesting that they might cross-differentiate and influence the magnitude of Tfh cell signals over time. Mathematical analysis of the dynamics of naïve T cells and Tregs predicted the contribution of the conversion to pTregs towards maintaining T cell homeostasis and suggested it could be more prominent in lymph nodes than in the spleen (125). Hence, organ specific differences in trans-differentiation of Tfh to Tfr cells and vice versa might be expected and require further investigation.
Several mechanisms have been proposed to explain the influence of Tfr cells on the GC response, which includes a direct inhibitory action on B cells and indirect effects by suppressing Tfh cells (117, 119, 126, 127). Wing et al. and Sage et al. showed that Tfr cells exert their inhibitory action via the inhibitory receptor CTLA-4 (126, 128). In general, CTLA-4 inhibits T cells by outcompeting the CD28 co-stimulatory receptor by binding to B7 ligands (129) on antigen-presenting cells and/or by the trans endocytosis of B7 ligands (130, 131). However, it is not clear whether the inhibitory action of CTLA-4 on GCs depends on trans-endocytosis as the expression levels of B7 ligands on GC B cells in the absence of CTLA-4 (126, 128) showed contradictory results. The role of Tfr cells in GC termination will become easier to probe as future studies improve our understanding of their mechanism of action and influence on GCs.
Potential Role of Tingible Body Macrophages
Phagocytic macrophages called tingible body macrophages (TBMs) present in GCs (132) might play a role in downregulating the GC reaction. Smith et al. observed that TBMs are capable of suppressing IL-2 induction in T cells upon stimulation by B cells in an in vitro culture system through a prostaglandin dependent mechanism (133).
Kinetics of TBMs follow the GC reaction kinetics, and it has been suggested that they play a role in regulating the magnitude of GC reactions (134). However, it is not known whether TBMs modulate the GC dynamics or vice versa. TBMs are capable of endocytosing iccosomes from FDCs (135). Thus, there is a potential inhibitory effect of TBMs on GC by limiting antigen availability which requires further investigations.
Impaired apoptotic cell clearance in the GCs by Mer-deficient TBMs increases the GC B cells at different stages of the GC response (136). Moreover, impaired apoptotic clearance is associated with autoimmune diseases such as systemic lupus erythematosus (137, 138). Even though this rarely studied cell type might not be sufficient for the shutdown, a modulatory effect on GC shutdown could be expected.
Role of Innate Signals
Adjuvants used to enhance the magnitude and longevity of antibody responses have also been shown to alter the kinetics of GC reactions (139). Adjuvants are capable of influencing the differentiation of Tfr (140) and Tfh cells (141) and could alter the Tfh : Tfr ratio even in GC responses towards the same antigen (140). The mechanisms of action of different adjuvants are still being elucidated, but many adjuvants act as ligands for innate immune receptors such as Toll-like receptors (TLRs) (142).
Several studies have demonstrated that TLRs modulate the magnitude and quality of GC responses but it is not known whether only the early development of GCs or also late stages of the GC reaction are affected (143). TLRs enhance the GC response by acting on dendritic cells and/or B cells (143). For instance, TLR9 impacted the GC reaction by acting on both dendritic cells and B cells (144). Action of TLR9 on dendritic cells enhanced the number of Tfh and GC B cells, while TLR9 signaling in B cells was associated with increased ICOS on Tfh cells and a reduced number of Tfr cells (144). FDCs express TLR4, which is upregulated during GC development and loss of TLR4 signaling resulted in a decreased size of GCs (73). Antigen deposition on FDCs has been shown to be enhanced by the TLR ligand-based adjuvant PorB (145).
Kasturi et al. have shown that a combination of TLR4 and 7 ligands can synergistically enhance the antibody response and the persistence of GCs when compared to administering either TLR ligand with antigen (146), suggesting that TLRs can be targeted to modulate the lifetime of GCs. Although TLRs can impact the GC longevity, their role in GC shutdown has not been investigated so far, and would be of importance to understand the relative contribution of different adjuvant strategies to vaccine success.
Metabolic Inhibition
The metabolism of GC B cells has been given minor focus so far, due to technical difficulties in analyzing the GC micro-environment (147, 148). It was acknowledged that GCs are a nutrient-poor and hypoxic environment especially in the LZ (149), possibly required for proper selection of B cells. Weisel et al., found that GC B cells exhibit a rather low glucose uptake and instead use fatty acids as a major source of energy (150). However, subsets of GC B cells such as the positively selected cells might transiently use a higher glycolytic program (150), as mTORC1 activation is associated with increased glucose uptake after DEC205-OVA induced GC B cell interaction with Tfh cells (59). Blockade of glutaminolysis pathways with the DON glutamine analogue leads to GC shrinkage and could be mediated by the Tfh cell sensitivity to glutamine (151). Interestingly, several transcription factors controlling proliferation of centroblasts are also metabolic sensors, such as c-Myc, mTOR, FoxO, suggesting that metabolism can regulate GC dynamics. For instance, mTORC1 controls the anabolic program by regulating the synthesis of lipids, glycolytic flux and by promoting protein synthesis (152), ultimately supporting B cell proliferation. The RNA binding protein PTBP1 that is highly expressed in the positively selected GC B cells, regulates alternative splicing of genes including Pkm (M-type pyruvate kinase), Tyms (Thymidylate synthase), suggesting a post transcriptional control of glycolytic flux and nucleotide synthesis (153).
Metabolic changes have been observed in the GC B cells as the GC matures. Though GC B cells are able to oxidize both endogenous and exogenous fatty acids, they rely more on exogenous fatty acids as the GC progresses, which might be obtained in vivo from dying B cells (150). This is presumably because of an exhausted endogenous supply of fatty acids (150). The significance of such metabolic changes in GC shutdown is unexplored.
In Plasmodium infection, short-lived plasmablasts have been suggested to inhibit GC function by imposing glutamine deprivation (154). This finding suggests that glutamine competition due to the plasma cells in extrafollicular areas might contribute to GC shutdown. As malaria is associated with decreased serum levels of glutamine and anemia, this needs to be shown under physiological metabolic balance in a normal infection.
Son et al., showed that inhibiting the Endoplasmic reticulum associated enzyme, stearoyl-CoA desaturase suppresses Tfh and GC B cell responses (155). Inhibiting this enzyme impaired Tfh cell maintenance and disturbed the balance between Tfh and Tfr cells by enhancing Tfh apoptosis (155). This suggests an impact of metabolic changes on GC responses, but whether such metabolic changes could be responsible for GC shutdown and how such metabolic changes might arise remains unclear.
Dynamic Changes in GC B Cell Characteristics
Studies have revealed characteristic changes in the GC B cells at late stages of GC responses, in addition to the metabolic changes. Very late stage GC B cells were resistant to the antigen depletion (156), suggesting that the maintenance of these GC B cells could be different from the early stages. In addition, a fraction of GC B cells expressing the BLA-1 marker was found to decrease at late stages (157), but its functional significance is unknown. Further, there is a shift in the production of memory cells to plasma cells during the course of the GC reaction (71).
These findings suggest that we do not fully understand the behavior of late GC B cells and there could be undiscovered negative feedback mechanisms acting at late stages which might also play a role in the termination of the GC response. Such mechanisms could be unveiled by studies focused on late stages of GC responses. Similarly, computational modeling suggested that a hypothetical proliferation signal that decays over time would result in reduced number of divisions as GC progresses and leads to GC termination (81). Extensive monitoring of time-dependent changes in GC B cells in parallel with other GC cell types can reveal a great deal of information.
Impact of B Cell Repertoire and GC Seeder Cell Composition
Composition of B cell repertoire is an important determinant of GC B cell recruitment (158). Mathematical modeling has predicted that the affinity of seeder cells influences the efficiency of GC reaction (159). As low affinity and frequency of antigen specific cells in the repertoire can lead to poor vaccination responses, it has been suggested to evaluate these parameters during vaccine development (160). Lifetime of GCs might also be influenced by the B cell repertoire and the founder cell composition of GCs.
In response to a secondary immunization, only limited participation of memory B cells has been observed in recall GCs (98). This finding can be explained by antibody feedback arising due to the differentiation of memory B cells into plasma cells, as the soluble antibodies produced would prevent competitive participation of memory B cells in the GCs (97). Accordingly, recall GCs allow for affinity maturation to a new epitope (97) unlike the concept of “original antigenic sin” (161) and also suggests a role of antibody feedback in the regulation of GC lifetime. As certain subsets of memory cells are capable of participating in GC responses (162), determining factors governing the composition of seeder B cells in primary and recall GC responses might allow for a better understanding of the regulation of GC lifetime by founder cells.
Apart from the composition of seeder cells, activation of naïve B cells and influx of new founder cells can continue after the GC is established (12, 13) and might extend the longevity of ongoing GC responses. Such continued influx of new founder cells might likely contribute to persistence of GCs seen in Peyer’s patches and viral infections.
Hints From GCs With Diverse Kinetics
Perturbation experiments have revealed that defects in a large number of factors can trigger GC shutdown (Table 1). Although such perturbations help infer the role of different factors in maintaining or terminating the GC, they only provide a limited understanding of the primary mechanism of GC shutdown. Studies have shown that the lifetime of GCs varies depending on the nature of the antigen stimuli (24, 163) and other factors such as the organ under consideration (164).
Li et al. compared the microbial exposure of germ-free mice at systemic or intestinal mucosa, and found differences in the diversity of the resulting BCR repertoire (165). It would be interesting to test the differences in GC dynamics and shutdown with different routes and sequences of microbial administration. Individual GCs in a lymph node have widely varying rate of loss in clonal diversity (166). Although the exact mechanism behind such differences in clonal evolution of individual GCs was not addressed so far, the observed heterogeneity despite the dynamic exchange of Tfh (114) and potential intercommunication due to soluble antibodies (19), raises the question whether individual GCs have the same lifetime and shutdown mechanisms and whether differences in the pace of clonal evolution are associated with differences in GC population kinetics, maintenance, and shutdown. It may be envisioned that the relative affinity of B cells in a single GC could regulate its dynamics. Although it is unknown whether the mechanism of shutdown is conserved under diverse immunization conditions, comparing the differences in GC response to such diverse conditions including natural viral infections, constitutive GCs seen in Peyer’s patches (Table 2) can provide some hints about the physiological signals that limit the GC lifetime.
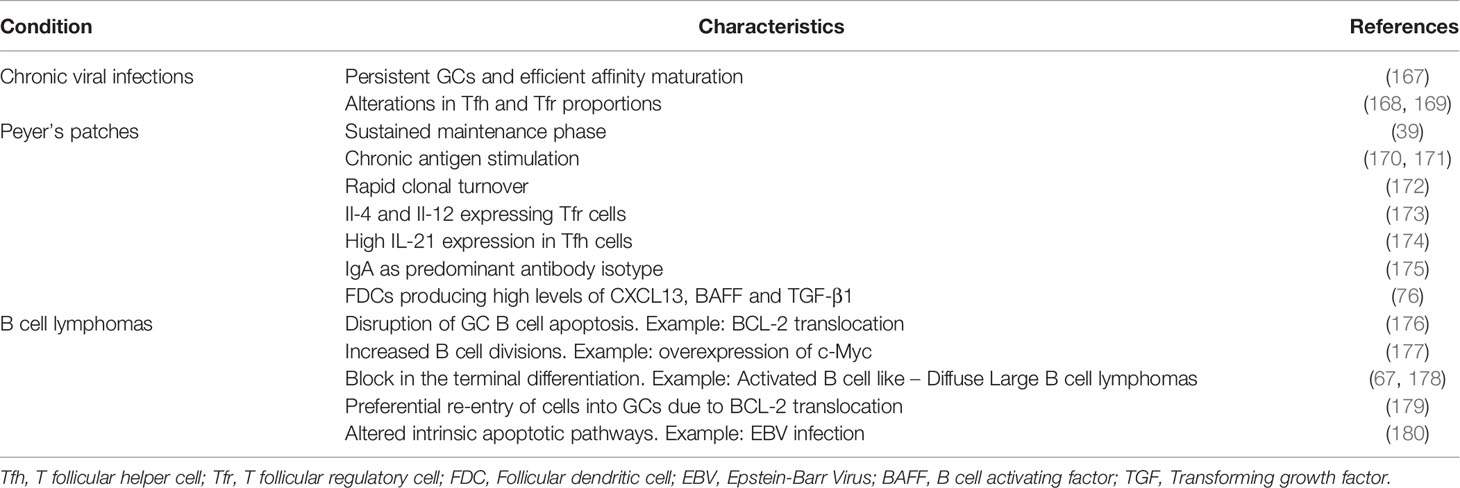
Table 2 Summary of characteristic differences seen in the GCs induced by chronic viral infections, GCs of Peyer’s patches and GC-derived B cell lymphomas when compared to the transient GC responses induced by model protein antigens.
Chronic GCs Due to Viral Infections
Chronic viral infections are capable of inducing long-lived and persistent GCs (181). For instance, after infection with VSV (Vesicular stomatitis virus), GCs were detectable up to 100 days after immunization (24). Chronic viral infections are associated with efficient affinity maturation and production of memory and plasma cells due to persistent GC responses (167). Persistence of GCs and the long-term maintenance of antibody titers is believed to be associated with persistent antigen but there is no direct evidence for this. Also, the resolution of the infection does not always result in GC termination. In the case of influenza virus infection, GCs are formed when the infection is almost resolved but the GCs are still long-lived (182, 183). Sustained GC responses could be due to the persistence of residual viral antigens (184) even after the resolution of infection. In addition to antigen persistence, there are also changes in Tfh and Tfr cell proportions in some viral infections (168, 169). However, the primary reason for the observed longevity of GCs has not been addressed so far.
Constitutive GCs in Peyer’s Patches
Constitutive GC reactions are observed in Peyer’s patches (PPs) of the small intestine, a part of the MALT (Mucosal Associated Lymphoid Tissue). These GCs characteristically have a constant GC volume, which reflects their sustained maintenance phase (39). The key factor that sustains the PP GCs is believed to be the chronic antigen stimulation by intestinal microbiota and antigen encountered in the food (170, 171). In the gut associated GCs, clonal turnover happens at a rapid rate and the rate of selection varies depending on the complexity of microbiota (172). GCs in the PPs also differ from the conventional GCs in lymph nodes and spleen in terms of the Tfh, Tfr cells, FDCs and isotype of antibodies produced (185). Tfr cells in PPs express Il-4 and Il-21 (173) unlike the Tfr cells in conventional GCs. The impact of these differences is not understood. These GCs host a high IL-21 expressing Tfh cell population, which when disrupted reduces the number of GC B cells (174).
In addition to Tfh and Tfr cells, PP FDCs are also considerably different from FDCs in the lymph node in their gene expression profile and produce high levels of CXCL13, BAFF and TGF-β1 due to stimulation by bacterial products and retinoic acid (76). Another major difference is the isotype of antibodies produced; PP GCs predominantly produce IgA antibodies (175) as opposed to IgG in the case of peripheral lymphoid organs. Although, in the latter, class-switch recombination predominantly occurs outside the GCs (186), the isotypic differences of GC B cells and antibodies produced might influence the GC dynamics and is worth investigating.
Microbiota might induce GC formation in a BCR independent manner by the interaction with innate immune receptors (187) and hence, the antigen recognition ability of GC B cells might impact the GC longevity and dynamics. Identifying the key factors promoting the constitutive nature of these GCs would be of importance in enhancing GC response towards vaccination.
Dysregulation in GC-Derived B Cell Lymphomas
GC B cells have a typical gene expression profile to facilitate affinity maturation. Acquisition of antigen and Tfh cell signals dynamically regulate the gene expression profile of GC B cells and promote their transition through different phases of the GC reaction. Intrinsic defects in the GC B cell gene expression programs that influence apoptosis, proliferation and differentiation potentially disrupt normal GC shutdown and trigger lymphomagenesis [reviewed in (188)]. For instance, translocation of BCL-2 can prevent GC B cell apoptosis and increase the number of GC B cells (176). Overexpression of transcription factors such as c-Myc can promote sustained proliferation of GC B cells (177). Further, a block in the differentiation of the GC B cells is seen in ABC-DLBCLs (Activated B cell like – Diffuse Large B cell lymphomas) which result in an accumulation of B cells within the GCs (67, 178). Such intrinsic B cell defects lead to a premalignant state which might undergo malignant transformation when multiple mutations accumulate over time and is facilitated by SHMs in the GCs. In addition, it has been shown that BCL-2 overexpressing cells might preferentially re-enter GCs upon subsequent antigen challenge and can drive lymphomagenesis due to repeated accumulation of diverse mutations (179). There is also evidence suggesting that intrinsic apoptotic pathways of GC B cells are altered by the EBV (Epstein-Barr Virus), a virus associated with Burkitt’s lymphoma facilitating the survival of EBV infected GC B cells (180). Hence, knowledge of the role of B cell genetic defects not only helps to promote an understanding of how the gene expression program of GC B cells dictate normal shutdown but also their dysregulation in lymphomagenesis which could further help to identify targets to block the progression of lymphomas.
Implications in Dissolving Ectopic GCs
In addition to the applications of understanding GC shutdown in designing treatment strategies for GC associated B cell lymphomas, it is also important for disruption of ectopic GCs formed in a certain proportion of patients with autoimmune diseases (189, 190). For instance, ectopic GCs are found in the synovium of Rheumatoid Arthritis (RA) (191, 192), salivary glands of sjögren’s syndrome (193, 194), meninges of multiple sclerosis (195) and thymus of myasthenia gravis patients (196).
Significant correlation between the presence of rheumatoid factors and follicles in the synovium of RA patients (197) and the increase in the affinity of auto antibodies (198) over time suggest that their source could be ectopic GCs although it is not proven (199). These primary follicular structures include FDCs, B cells and T cells organized into typical GC-like structures, which, in contrast to their more static counterparts in secondary lymphoid tissues, were predicted as structures dynamically changing in dependence on chemokines (200) and are thought to promote pathogenesis. They can be disrupted by anti-CD20 antibodies which collapse the B cell follicles (192). It has also been shown that CD8 T cells are important in sustaining the activity of such GCs as disruption of CD8 T cells disrupted GCs (201).
Understanding the natural mechanism and regulation of GC shutdown might help identify efficient ways to target such ectopic GCs. However, as these ectopic GCs might differ from normal GCs, factors in their maintenance apart from the conventional shutdown mechanisms must be determined.
Implications in Vaccine Development
Considering the requirement for effective vaccines against pathogens such as influenza, HIV and more recently, SARS-CoV-2, main focus of several studies is to develop strategies to enhance GC responses by vaccination (202–206). For vaccine development, apart from the choice of antigen and adjuvant, modulation of antigen dynamics has been identified as a strategy to enhance GC responses. Persistent antigen deposition observed during a natural viral infection gave rise to the concept of slow delivery immunization (207). Administering escalating doses of antigen increases immune complex deposition on FDCs and enhances the GC response (204). Similarly, compared to conventional bolus immunization, slow delivery immunization in non-human primates resulted in enhanced neutralizing antibody responses against HIV envelope protein (203). Slow delivery of antigen also enhanced the number of GC B cells, Tfh cells, diversity of GC B cells and antigen retention on FDCs (203).
Longevity of GCs is a key factor that influences the quantity and quality of GC responses. When combined with efficient GC induction, extending the longevity of induced GCs may improve vaccine efficacy. Furthermore, magnitude of recall GC response is limited when compared to the primary GC response (22). Factors influencing the lifetime and magnitude of secondary GCs need to be determined to modulate the GC response to booster immunization. Therefore, a better understanding of the mechanism of GC shutdown in primary and recall GC responses can help identify effective ways to enhance the longevity of GCs and might revolutionize the development of vaccines.
Challenges in Understanding GC Shutdown
Inferring the mechanistic details of GC shutdown from present evidence poses several challenges. Firstly, alterations in GC processes do not always lead to observable changes in GC kinetics and shutdown. For instance, GC kinetics in CD80 deficient mice (208) remains unaltered despite lowered production of plasma cells, as changes in the production of memory cells and apoptosis take over. Also, while TLR9 signaling enhances the magnitude of GC responses on day 14, there is no difference observed at later stages such as on day 21 (144). Hence, changes that influence GC magnitude must be distinguished from those that influence GC kinetics.
Secondly, the dependence of GC progression and kinetics on various factors vary under different conditions. For example, Bam32 deficient mice exhibit a defect in GC maintenance with low doses of SRBC injection but not at high doses (29). Hence, the variation of antigen amount might help disentangle the different mechanisms in their importance for the shutdown. Similarly, GC maintenance was affected by the complete lack of CD40 but was intact with CD40 haploinsufficiency (64). Dependence of GC shutdown on different factors might also vary with the complexity of antigen used for immunization. GC response to complex antigens with different epitopes and Tfh receptor specificities, show GC B cells that have very low affinity or do not bind to the antigen used (209). Such an ability of non-cognate B cells to participate in GC reaction (210) might hide underexplored reasons for peculiar GC dynamics.
Finally, although several factors could alter GC termination, it is experimentally difficult to test which ones play a role in natural termination of GCs. Further, mechanistic understanding of GC shutdown is challenging due to incomplete understanding of how changes in cellular interactions promote molecular changes in GC B cells and how this in turn influences further cellular interactions.
Mathematical Models for Understanding GC Shutdown
Mathematical models as a tool for exploring GCs (211) have contributed substantially to GC research by predicting the recycling of GC B cells (9, 10), limiting nature of Tfh help (95) and antibody feedback (19). Wang et al., developed a stochastic GC model, and predicted that quantity and quality of GC responses can be tuned by the efficiency of T-B interactions (212). De Boer and Perelson developed mathematical models considering selection mechanisms based on density or diversity of pMHC presentation by GC B cells to investigate the evolution of broadly neutralizing antibodies (213). Increasing the breadth of Tfh cell repertoire and magnitude of Tfh responses were found to enhance the selection of broadly reactive GC B cell clones and lead to a quick development of broadly neutralizing antibodies (213).
Due to the technical difficulties in testing the different mechanisms experimentally and non-intuitive behavior of GCs, mathematical modeling studies were more prevalent in exploring the mechanism of shutdown. Predictions of mathematical models have suggested various ways of GC shutdown and the parameters critical for explaining the GC dynamics. Identifying potential mechanisms of shutdown by mathematical models and experimentally testing the predictions might efficiently identify the mechanism of GC shutdown.
Concluding Remarks
Although several mechanisms such as changes in antigen access or nature of Tfh cell signals could promote GC shutdown, identifying the natural cause of GC shutdown remains a challenge. These mechanisms might be inter-related due to complex dependencies in the GCs. A likely response to the GC shutdown mechanism would state that GCs are terminated in a coordinated action of many mechanisms. In this perspective, a partial disruption of one mechanism would not necessarily induce chronic GCs. Nevertheless, several lines of evidence point to antigen availability as a key requirement for GC B cell and Tfh maintenance and suggest a role of antigen limitation. Consequently, a better understanding of antigen availability and accessibility to GC B cells might help solve the long-lasting debate about antigen vs Tfh help as a shutdown signal.
Factors determining the alternative fates of GC B cells are crucial for understanding GC shutdown. Given that loss of function attempts alone might not be informative in the context of GC shutdown, a combination with gain of function mutation studies is necessary. It is important to understand how different factors combined determine the lifetime of GCs and timing of shutdown. In vitro GC development and the approach of synthetic biology might also be powerful in gaining a more detailed understanding. A better characterization of progressive changes happening in GCs, in terms of cellular dynamics, interactions, metabolism, surface markers and other features and the basis of such changes is also important. As GCs develop asynchronously and differ from one another, longitudinal imaging of GCs for sufficiently long periods of time is an important requirement for investigation of GC shutdown. Studies of the late phases of GC reactions and monitoring the GC contraction phase can provide more information on the mechanism of GC shutdown and how different factors coordinate to regulation or pathological dysregulation of the GC contraction.
It is also not clear whether the mechanism of GC shutdown and their interplay could be variable under different conditions. Main reason leading to variability in the lifetime of GCs induced under different conditions needs to be elucidated. In the context of multiple asynchronous GCs, mechanism of shutdown of individual GCs rather than the overall response needs to be addressed by future studies. Understanding the mechanism and factors regulating GC shutdown would allow for modulation of GC lifetime to improve vaccination responses. Although the primary focus of the review was to motivate studies on understanding natural GC shutdown, it also highlights a need to better understand the diverse ways in which GC shutdown is dysregulated for potential therapeutic developments.
Author Contributions
TA, SB, PR, and MM-H wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
TA was supported by the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement no. 765158. PR was supported by the Human Frontier Science Program (RGP0033/2015).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Gabriel Victora, Michael Reth, Claudia Berek and Julia Jellusova for constructive suggestions and revising the manuscript.
References
1. Berek C, Berger A, Apel M. Maturation of the Immune Response in Germinal Centers. Cell (1991) 67:1121–9. doi: 10.1016/0092-8674(91)90289-B
2. Nossal GJV. The Molecular and Cellular Basis of Affinity Maturation in the Antibody Response. Cell (1992) 68:1–2. doi: 10.1016/0092-8674(92)90198-L
3. MacLennan ICM. Germinal Centers. Annu Rev Immunol (1994) 12:117–39. doi: 10.1146/annurev.iy.12.040194.001001
4. Rajewsky K. Clonal Selection and Learning in the Antibody System. Nature (1996) 381:751–8. doi: 10.1038/381751a0
5. Allen CDC, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal Center Dark and Light Zone Organization Is Mediated by CXCR4 and CXCR5. Nat Immunol (2004) 5:943–52. doi: 10.1038/ni1100
6. Camacho SA, Kosco-Vilbois MH, Berek C. The Dynamic Structure of the Germinal Center. Immunol Today (1998) 19:511–4. doi: 10.1016/S0167-5699(98)01327-9
7. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal Center Dynamics Revealed by Multiphoton Microscopy With a Photoactivatable Fluorescent Reporter. Cell (2010) 143:592–605. doi: 10.1016/j.cell.2010.10.032
8. Lindhout E, de Groot C, Koopman G, Pals ST. Triple Check for Antigen Specificity of B Cells During Germinal Centre Reactions. Immunol Today (1997) 18:573–7. doi: 10.1016/S0167-5699(97)01160-2
9. Kepler TB, Perelson AS. Somatic Hypermutation in B Cells: An Optimal Control Treatment. J Theor Biol (1993) 164:37–64. doi: 10.1006/jtbi.1993.1139
10. Kepler TB, Perelson AS. Cyclic Re-Entry of Germinal Center B Cells and the Efficiency of Affinity Maturation. Immunol Today (1993) 14:412–5. doi: 10.1016/0167-5699(93)90145-B
11. Meyer-Hermann M, Deutsch A, Or-Guil M. Recycling Probability and Dynamical Properties of Germinal Center Reactions. J Theor Biol (2001) 210:265–85. doi: 10.1006/jtbi.2001.2297
12. Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, et al. In Vivo Imaging of Germinal Centres Reveals a Dynamic Open Structure. Nature (2007) 446:83–7. doi: 10.1038/nature05573
13. Schwickert TA, Alabyev B, Manser T, Nussenzweig MC. Germinal Center Reutilization by Newly Activated B Cells. J Exp Med (2009) 206:2907–14. doi: 10.1084/jem.20091225
14. Turner JS, Benet ZL, Grigorova IL. Antigen Acquisition Enables Newly Arriving B Cells to Enter Ongoing Immunization-Induced Germinal Centers. J Immunol (2017) 199:1301–7. doi: 10.4049/jimmunol.1700267
15. Stebegg M, Kumar SD, Silva-Cayetano A, Fonseca VR, Linterman MA, Graca L. Regulation of the Germinal Center Response. Front Immunol (2018) 9:2469. doi: 10.3389/fimmu.2018.02469
16. Gatto D, Pfister T, Jegerlehner A, Martin SW, Kopf M, Bachmann MF. Complement Receptors Regulate Differentiation of Bone Marrow Plasma Cell Precursors Expressing Transcription Factors Blimp-1 and XBP-1. J Exp Med (2005) 201:993–1005. doi: 10.1084/jem.20042239
17. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21 Acts Directly on B Cells to Regulate Bcl-6 Expression and Germinal Center Responses. J Exp Med (2010) 207:353–63. doi: 10.1084/jem.20091738
18. Wollenberg I, Agua-Doce A, Hernández A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the Germinal Center Reaction by Foxp3+ Follicular Regulatory T Cells. J Immunol (2011) 187:4553–60. doi: 10.4049/jimmunol.1101328
19. Zhang Y, Meyer-Hermann M, George LA, Figge MT, Khan M, Goodall M, et al. Germinal Center B Cells Govern Their Own Fate Via Antibody Feedbac. J Exp Med (2013) 210:457–64. doi: 10.1084/jem.20120150
20. Jacob J, Kassir R, Kelsoe G. In Situ Studies of the Primary Immune Response to (4-Hydroxy-3-Nitrophenyl) Acetyl. I. The Architecture and Dynamics of Responding Cell Populations. J Exp Med (1991) 173:1165–75. doi: 10.1084/jem.173.5.1165
21. Iber D, Maini PK. A Mathematical Model for Germinal Centre Kinetics and Affinity Maturation. J Theor Biol (2002) 219:153–75. doi: 10.1006/jtbi.2002.3079
22. Liu Y -J, Zhang J, Lane PJL, Chan E--, Maclennan ICM. Sites of Specific B Cell Activation in Primary and Secondary Responses to T Cell-Dependent and T Cell-Independent Antigens. Eur J Immunol (1991) 21:2951–62. doi: 10.1002/eji.1830211209
23. Hollowood K, Macartney J. Cell Kinetics of the Germinal Center Reaction - a Stathmokinetic Study. Eur J Immunol (1992) 22:261–6. doi: 10.1002/eji.1830220138
24. Bachmann MF, Odermatt B, Hengartner H, Zinkernagel RM. Induction of Long-Lived Germinal Centers Associated With Persisting Antigen After Viral Infection. J Exp Med (1996) 183:2259–69. doi: 10.1084/jem.183.5.2259
25. Firl DJ, Degn SE, Padera T, Carroll MC. Capturing Change in Clonal Composition Amongst Single Mouse Germinal Centers. Elife (2018) 7:e33051. doi: 10.7554/eLife.33051
26. Wray-Dutra MN, Chawla R, Thomas KR, Seymour BJ, Arkatkar T, Sommer KM, et al. Activated CARD11 Accelerates Germinal Center Kinetics, Promoting mTORC1 and Terminal Differentiation. J Exp Med (2018) 215:2445–61. doi: 10.1084/jem.20180230
27. Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, et al. The Proto-Oncogene MYC Is Required for Selection in the Germinal Center and Cyclic Reentry. Nat Immunol (2012) 13:1083–91. doi: 10.1038/ni.2428
28. Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, et al. Germinal Center B Cell Maintenance and Differentiation Are Controlled by Distinct NF-κb Transcription Factor Subunits. J Exp Med (2014) 211:2103–18. doi: 10.1084/jem.20132613
29. Zhang T, Al-Alwan M, Marshall AJ. The Pleckstrin Homology Domain Adaptor Protein Bam32/DAPP1 Is Required for Germinal Center Progression. J Immunol (2010) 184:164–72. doi: 10.4049/jimmunol.0902505
30. Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, et al. Follicular Dendritic Cells Help Establish Follicle Identity and Promote B Cell Retention in Germinal Centers. J Exp Med (2011) 208:2497–510. doi: 10.1084/jem.20111449
31. Boulianne B, Le MX, Ward LA, Meng L, Haddad D, Li C, et al. AID-Expressing Germinal Center B Cells Cluster Normally Within Lymph Node Follicles in the Absence of FDC-M1+ CD35+ Follicular Dendritic Cells But Dissipate Prematurely. J Immunol (2013) 191:4521–30. doi: 10.4049/jimmunol.1300769
32. Rahman ZSM, Rao SP, Kalled SL, Manser T. Normal Induction But Attenuated Progression of Germinal Center Responses in BAFF and BAFF-R Signaling–Deficient Mice. J Exp Med (2003) 198:1157–69. doi: 10.1084/jem.20030495
33. de Vinuesa CG, Cook MC, Ball J, Drew M, Sunners Y, Cascalho M, et al. Germinal Centers Without T Cells. J Exp Med (2000) 191:485–94. doi: 10.1084/jem.191.3.485
34. Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, et al. IL-21 Regulates Germinal Center B Cell Differentiation and Proliferation Through a B Cell–Intrinsic Mechanism. J Exp Med (2010) 207:365–78. doi: 10.1084/jem.20091777
35. Schrock DC, Leddon SA, Hughson A, Miller J, Lacy-Hulbert A, Fowell DJ. Pivotal Role for αv Integrins in Sustained Tfh Support of the Germinal Center Response for Long-Lived Plasma Cell Generation. Proc Natl Acad Sci (2019) 116:4462–70. doi: 10.1073/pnas.1809329116
36. Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. PD-1 Regulates Germinal Center B Cell Survival and the Formation and Affinity of Long-Lived Plasma Cells. Nat Immunol (2010) 11:535–42. doi: 10.1038/ni.1877
37. Han S, Hathcock K, Zheng B, Kepler TB, Hodes R, Kelsoe G. Cellular Interaction in Germinal Centers. Roles of CD40 Ligand and B7-2 in Established Germinal Centers. J Immunol (1995) 155:556–67.
38. Bolduc A, Long E, Stapler D, Cascalho M, Tsubata T, Koni PA, et al. Constitutive CD40L Expression on B Cells Prematurely Terminates Germinal Center Response and Leads to Augmented Plasma Cell Production in T Cell Areas. J Immunol (2010) 185:220 – 230. doi: 10.4049/jimmunol.0901689
39. Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, et al. The Microanatomic Segregation of Selection by Apoptosis in the Germinal Center. Science (2017) 358:0–9. doi: 10.1126/science.aao2602
40. van Eijk M, Medema JP, de Groot C. Cutting Edge: Cellular Fas-Associated Death Domain-Like IL-1-Converting Enzyme-Inhibitory Protein Protects Germinal Center B Cells From Apoptosis During Germinal Center Reactions. J Immunol (2001) 166:6473–6. doi: 10.4049/jimmunol.166.11.6473
41. Krammer PH. Cd95’s Deadly Mission in the Immune System. Nature (2000) 407:789–95. doi: 10.1038/35037728
42. Victora GD, Nussenzweig MC. Germinal Centers. Annu Rev Immunol (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
43. Stewart I, Radtke D, Phillips B, McGowan SJ, Bannard O. Germinal Center B Cells Replace Their Antigen Receptors in Dark Zones and Fail Light Zone Entry When Immunoglobulin Gene Mutations Are Damaging. Immunity (2018) 49:477–89. doi: 10.1016/j.immuni.2018.08.025
44. Strasser A, Harris AW, Huang DC, Krammer PH, Cory S. Bcl-2 and Fas/APO-1 Regulate Distinct Pathways to Lymphocyte Apoptosis. EMBO J (1995) 14:6136–47. doi: 10.1002/j.1460-2075.1995.tb00304.x
45. Smith KGC, Light A, O’Reilly LA, Ang S-M, Strasser A, Tarlinton D. Bcl-2 Transgene Expression Inhibits Apoptosis in the Germinal Center and Reveals Differences in the Selection of Memory B Cells and Bone Marrow Antibody-Forming Cells. J Exp Med (2000) 191:475–84. doi: 10.1084/jem.191.3.475
46. Jumaa H, Caganova M, McAllister EJ, Hoenig L, He X, Saltukoglu D, et al. Immunoglobulin Expression in the Endoplasmic Reticulum Shapes the Metabolic Fitness of B Lymphocytes. Life Sci Alliance (2020) 3:e202000700. doi: 10.26508/lsa.202000700
47. Zaheen A, Boulianne B, Parsa J-Y, Ramachandran S, Gommerman JL, Martin A. AID Constrains Germinal Center Size by Rendering B Cells Susceptible to Apoptosis. Blood (2009) 114:547–54. doi: 10.1182/blood-2009-03-211763
48. Schuetz JM, Johnson NA, Morin RD, Scott DW, Tan K, Ben-Nierah S, et al. BCL2 Mutations in Diffuse Large B-Cell Lymphoma. Leukemia (2012) 26:1383–90. doi: 10.1038/leu.2011.378
49. Razzaghi R, Agarwal S, Kotlov N, Plotnikova O, Nomie K, Huang DW, et al. Compromised Counterselection by FAS Creates an Aggressive Subtype of Germinal Center Lymphoma. J Exp Med (2020) 218(3):e20201173. doi: 10.1084/jem.20201173
50. Gitlin AD, Mayer CT, Oliveira TY, Shulman Z, Jones MJK, Koren A, et al. T Cell Help Controls the Speed of the Cell Cycle in Germinal Center B Cells. Science (2015) 349:643–6. doi: 10.1126/science.aac4919
51. Meyer-Hermann M. Overcoming the Dichotomy of Quantity and Quality in Antibody Responses. J Immunol (2014) 193(11):5414–9. doi: 10.4049/jimmunol.1401828
52. Meyer-Hermann M, Mohr E, Pelletier N, Zhang Y, Victora GD, Toellner K-M. A Theory of Germinal Center B Cell Selection, Division, and Exit. Cell Rep (2012) 2:162–74. doi: 10.1016/j.celrep.2012.05.010
53. Gitlin AD, Shulman Z, Nussenzweig MC. Clonal Selection in the Germinal Centre by Regulated Proliferation and Hypermutation. Nature (2014) 509:637–40. doi: 10.1038/nature13300
54. Finkin S, Hartweger H, Oliveira TY, Kara EE, Nussenzweig MC. Protein Amounts of the MYC Transcription Factor Determine Germinal Center B Cell Division Capacity. Immunity (2019) 51:324–36.e5. doi: 10.1016/j.immuni.2019.06.013
55. Ramezani-Rad P, Chen C, Zhu Z, Rickert RC. Cyclin D3 Governs Clonal Expansion of Dark Zone Germinal Center B Cells. Cell Rep (2020) 33(7):108403. doi: 10.1016/j.celrep.2020.108403
56. Peled JU, Yu JJ, Venkatesh J, Bi E, Ding BB, Krupski-Downs M, et al. Requirement for Cyclin D3 in Germinal Center Formation and Function. Cell Res (2010) 20:631–46. doi: 10.1038/cr.2010.55
57. Cato MH, Chintalapati SK, Yau IW, Omori SA, Rickert RC. Cyclin D3 Is Selectively Required for Proliferative Expansion of Germinal Center B Cells. Mol Cell Biol (2011) 31:127 – 137. doi: 10.1128/MCB.00650-10
58. Pae J, Ersching J, Castro TBR, Schips M, Mesin L, Allon SJ, et al. Cyclin D3 Drives Inertial Cell Cycling in Dark Zone Germinal Center B Cells. J Exp Med (2020) 218(4):e20201699. doi: 10.1084/jem.20201699
59. Ersching J, Efeyan A, Mesin L, Jacobsen JT, Pasqual G, Grabiner BC, et al. Germinal Center Selection and Affinity Maturation Require Dynamic Regulation of mTORC1 Kinase. Immunity (2017) 46:1045–58. doi: 10.1016/j.immuni.2017.06.005
60. Inoue T, Shinnakasu R, Ise W, Kawai C, Egawa T, Kurosaki T. The Transcription Factor Foxo1 Controls Germinal Center B Cell Proliferation in Response to T Cell Help. J Exp Med (2017) 214:1181–98. doi: 10.1084/jem.20161263
61. Luo W, Weisel F, Shlomchik MJ. B Cell Receptor and CD40 Signaling Are Rewired for Synergistic Induction of the c-Myc Transcription Factor in Germinal Center B Cells. Immunity (2018) 48:313–26.e5. doi: 10.1016/j.immuni.2018.01.008
62. Calado DP, Sasaki Y, Godinho SA, Pellerin A, Köchert K, Sleckman BP, et al. The Cell-Cycle Regulator C-Myc Is Essential for the Formation and Maintenance of Germinal Centers. Nat Immunol (2012) 13:1092–100. doi: 10.1038/ni.2418
63. Wang Y, Shi J, Yan J, Xiao Z, Hou X, Lu P, et al. Germinal-Center Development of Memory B Cells Driven by IL-9 From Follicular Helper T Cells. Nat Immunol (2017) 18:921–30. doi: 10.1038/ni.3788
64. Ise W, Fujii K, Shiroguchi K, Ito A, Kometani K, Takeda K, et al. T Follicular Helper Cell-Germinal Center B Cell Interaction Strength Regulates Entry Into Plasma Cell or Recycling Germinal Center Cell Fate. Immunity (2018) 48:702–15. doi: 10.1016/j.immuni.2018.03.027
65. Kräutler NJ, Suan D, Butt D, Bourne K, Hermes JR, Chan TD, et al. Differentiation of Germinal Center B Cells Into Plasma Cells Is Initiated by High-Affinity Antigen and Completed by Tfh Cells. J Exp Med (2017) 214:1259–67. doi: 10.1084/jem.20161533
66. Radtke D, Bannard O. Expression of the Plasma Cell Transcriptional Regulator Blimp-1 by Dark Zone Germinal Center B Cells During Periods of Proliferation. Front Immunol (2019) 9:3106. doi: 10.3389/fimmu.2018.03106
67. Mandelbaum J, Bhagat G, Tang H, Mo T, Brahmachary M, Shen Q, et al. BLIMP1 Is a Tumor Suppressor Gene Frequently Disrupted in Activated B Cell-Like Diffuse Large B Cell Lymphoma. Cancer Cell (2010) 18:568–79. doi: 10.1016/j.ccr.2010.10.030
68. Viant C, Weymar GHJ, Escolano A, Chen S, Hartweger H, Cipolla M, et al. Antibody Affinity Shapes the Choice Between Memory and Germinal Center B Cell Fates. Cell (2020) 183(5):1298–311. doi: 10.1016/j.cell.2020.09.063
69. Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, et al. Regulated Selection of Germinal-Center Cells Into the Memory B Cell Compartment. Nat Immunol (2016) 17:861–9. doi: 10.1038/ni.3460
70. Smith KGC, Light A, Nossal GJV, Tarlinton DM. The Extent of Affinity Maturation Differs Between the Memory and Antibody-Forming Cell Compartments in the Primary Immune Response. EMBO J (1997) 16:2996–3006. doi: 10.1093/emboj/16.11.2996
71. Weisel FJ, Zuccarino-Catania GV, Chikina M, Shlomchik MJ. A Temporal Switch in the Germinal Center Determines Differential Output of Memory B and Plasma Cells. Immunity (2016) 44:116–30. doi: 10.1016/j.immuni.2015.12.004
72. Kläsener K, Jellusova J, Andrieux G, Salzer U, Böhler C, Steiner SN, et al. CD20 as a Gatekeeper of the Resting State of Human B Cells. Proc Natl Acad Sci (2021) 118:e2021342118. doi: 10.1073/pnas.2021342118
73. Garin A, Meyer-Hermann M, Contie M, Figge MT, Buatois V, Gunzer M, et al. Toll-Like Receptor 4 Signaling by Follicular Dendritic Cells Is Pivotal for Germinal Center Onset and Affinity Maturation. Immunity (2010) 33:84–95. doi: 10.1016/j.immuni.2010.07.005
74. El Shikh ME, El Sayed R, Szakal AK, Tew JG. Follicular Dendritic Cell (FDC)-Fcγriib Engagement via Immune Complexes Induces the Activated FDC Phenotype Associated With Secondary Follicle Development. Eur J Immunol (2006) 36:2715–24. doi: 10.1002/eji.200636122
75. van der Poel CE, Bajic G, Macaulay CW, van den Broek T, Ellson CD, Bouma G, et al. Follicular Dendritic Cells Modulate Germinal Center B Cell Diversity Through Fcγriib. Cell Rep (2019) 29:2745–55. doi: 10.1016/j.celrep.2019.10.086
76. Suzuki K, Maruya M, Kawamoto S, Sitnik K, Kitamura H, Agace WW, et al. The Sensing of Environmental Stimuli by Follicular Dendritic Cells Promotes Immunoglobulin A Generation in the Gut. Immunity (2010) 33:71–83. doi: 10.1016/j.immuni.2010.07.003
77. Muñoz-Fernández R, Prados A, Tirado-González I, Martín F, Abadía AC, Olivares EG. Contractile Activity of Human Follicular Dendritic Cells. Immunol Cell Biol (2014) 92:851–9. doi: 10.1038/icb.2014.61
78. Denton AE, Silva-Cayetano A, Dooley J, Hill DL, Carr EJ, Robert PA, et al. Intrinsic Defects in Lymph Node Stromal Cells Underpin Poor Germinal Center Responses During Aging. bioRxiv (2020) 2020:5. doi: 10.1101/2020.05.07.082255
79. Gonzalez M, Mackay F, Browning JL, Kosco-Vilbois MH, Noelle RJ. The Sequential Role of Lymphotoxin and B Cells in the Development of Splenic Follicles. J Exp Med (1998) 187:997–1007. doi: 10.1084/jem.187.7.997
80. Endres R, Alimzhanov MB, Plitz T, Fütterer A, Kosco-Vilbois MH, Nedospasov SA, et al. Mature Follicular Dendritic Cell Networks Depend on Expression of Lymphotoxin β Receptor by Radioresistant Stromal Cells and of Lymphotoxin β and Tumor Necrosis Factor by B Cells. J Exp Med (1999) 189:159–68. doi: 10.1084/jem.189.1.159
81. Moreira JS, Faro J. Modelling Two Possible Mechanisms for the Regulation of the Germinal Center Dynamics. J Immunol (2006) 177:3705–10. doi: 10.4049/jimmunol.177.6.3705
82. Keşmir C, De Boer RJ. A Mathematical Model on Germinal Center Kinetics and Termination. J Immunol (1999) 163:2463–9.
83. Mandel TE, Phipps RP, Abbot AP, Tew JG. Long-Term Antigen Retention by Dendritic Cells in the Popliteal Lymph Node of Immunized Mice. Immunology (1981) 43:353.
84. Radoux D, Heinen E, Kinet-Denoël C, Tihange E, Simar L. Precise Localization of Antigens on Follicular Dendritic Cells. Cell Tissue Res (1984) 235:267–74. doi: 10.1007/BF00217850
85. Kosco-Vilbois MH, Zentgraf H, Gerdes J, Bonnefoy J-Y. To ‘B’or Not to ‘B’a Germinal Center? Immunol Today (1997) 18:225–30. doi: 10.1016/S0167-5699(97)01048-7
86. Heesters BA, Chatterjee P, Kim Y-A, Gonzalez SF, Kuligowski MP, Kirchhausen T, et al. Endocytosis and Recycling of Immune Complexes by Follicular Dendritic Cells Enhances B Cell Antigen Binding and Activation. Immunity (2013) 38:1164–75. doi: 10.1016/j.immuni.2013.02.023
87. Heesters BA, Lindqvist M, Vagefi PA, Scully EP, Schildberg FA, Altfeld M, et al. Follicular Dendritic Cells Retain Infectious HIV in Cycling Endosomes. PloS Pathog (2015) 11(12):e1005285. doi: 10.1371/journal.ppat.1005285
88. Tew JG, Mandel TE. Prolonged Antigen Half-Life in the Lymphoid Follicles of Specifically Immunized Mice. Immunology (1979) 37:69.
89. Kosco-Vilbois MH. Are Follicular Dendritic Cells Really Good for Nothing? Nat Rev Immunol (2003) 3:764–9. doi: 10.1038/nri1179
90. Heyman B. Regulation of Antibody Responses via Antibodies, Complement, and Fc Receptors. Annu Rev Immunol (2000) 18:709–37. doi: 10.1146/annurev.immunol.18.1.709
91. Vono M, Eberhardt CS, Auderset F, Mastelic-Gavillet B, Lemeille S, Christensen D, et al. Maternal Antibodies Inhibit Neonatal and Infant Responses to Vaccination by Shaping the Early-Life B Cell Repertoire Within Germinal Centers. Cell Rep (2019) 28:1773–84.e5. doi: 10.1016/j.celrep.2019.07.047
92. Bergström JJE, Xu H, Heyman B. Epitope-Specific Suppression of IgG Responses by Passively Administered Specific IgG: Evidence of Epitope Masking. Front Immunol (2017) 8:238. doi: 10.3389/fimmu.2017.00238
93. Forsell MNE, Kvastad L, Sedimbi SK, Andersson J, Karlsson MCI. Regulation of Subunit-Specific Germinal Center B Cell Responses to the HIV-1 Envelope Glycoproteins by Antibody-Mediated Feedback. Front Immunol (2017) 8:738. doi: 10.3389/fimmu.2017.00738
94. Xu H, Zhang L, Heyman B. IgG-Mediated Immune Suppression in Mice Is Epitope Specific Except During High Epitope Density Conditions. Sci Rep (2018) 8:1–10. doi: 10.1038/s41598-018-33087-6
95. Meyer-Hermann ME, Maini PK, Iber D. An Analysis of B Cell Selection Mechanisms in Germinal Centers. Math Med Biol A J IMA (2006) 23:255–77. doi: 10.1093/imammb/dql012
96. Arulraj T, Binder SC, Robert PA, Meyer-Hermann M. Synchronous Germinal Center Onset Impacts the Efficiency of Antibody Responses. Front Immunol (2019) 10:2116. doi: 10.3389/fimmu.2019.02116
97. Meyer-Hermann M. Injection of Antibodies Against Immunodominant Epitopes Tunes Germinal Centers to Generate Broadly Neutralizing Antibodies. Cell Rep (2019) 29:1066–73. doi: 10.1016/j.celrep.2019.09.058
98. Mesin L, Schiepers A, Ersching J, Barbulescu A, Cavazzoni CB, Angelini A, et al. Restricted Clonality and Limited Germinal Center Reentry Characterize Memory B Cell Reactivation by Boosting. Cell (2020) 180:92–106. doi: 10.1016/j.cell.2019.11.032
99. Nordström E, Abedi-Valugerdi M, Möller E. Longevity of Immune Complexes and Abnormal Germinal Centre Formation in NZB Mice. Scand J Immunol (2000) 52:477–82. doi: 10.1046/j.1365-3083.2000.00800.x
100. Roozendaal R, Mempel TR, Pitcher LA, Gonzalez SF, Verschoor A, Mebius RE, et al. Conduits Mediate Transport of Low-Molecular-Weight Antigen to Lymph Node Follicles. Immunity (2009) 30:264–76. doi: 10.1016/j.immuni.2008.12.014
101. Phan TG, Grigorova I, Okada T, Cyster JG. Subcapsular Encounter and Complement-Dependent Transport of Immune Complexes by Lymph Node B Cells. Nat Immunol (2007) 8:992–1000. doi: 10.1038/ni1494
102. Heesters BA, Myers RC, Carroll MC. Follicular Dendritic Cells: Dynamic Antigen Libraries. Nat Rev Immunol (2014) 14:495–504. doi: 10.1038/nri3689
103. Batista FD, Harwood NE. The Who, How and Where of Antigen Presentation to B Cells. Nat Rev Immunol (2009) 9:15–27. doi: 10.1038/nri2454
104. Weinstein JS, Herman EI, Lainez B, Licona-Limón P, Esplugues E, Flavell R, et al. T FH Cells Progressively Differentiate to Regulate the Germinal Center Response. Nat Immunol (2016) 17:1197–205. doi: 10.1038/ni.3554
105. Jain RW, Parham KA, Tesfagiorgis Y, Craig HC, Romanchik E, Kerfoot SM. Autoreactive, Low-Affinity T Cells Preferentially Drive Differentiation of Short-Lived Memory B Cells at the Expense of Germinal Center Maintenance. Cell Rep (2018) 25:3342–55. doi: 10.1016/j.celrep.2018.11.070
106. Merkenschlager J, Finkin S, Ramos V, Kraft J, Cipolla M, Nowosad CR, et al. Dynamic Regulation of TFH Selection During the Germinal Centre Reaction. Nature (2021) 591:458–63. doi: 10.1038/s41586-021-03187-x
107. Kishi Y, Aiba Y, Higuchi T, Furukawa K, Tokuhisa T, Takemori T, et al. Augmented Antibody Response With Premature Germinal Center Regression in CD40L Transgenic Mice. J Immunol (2010) 185:211–9. doi: 10.4049/jimmunol.0901694
108. Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, et al. T–B-Cell Entanglement and ICOSL-Driven Feed-Forward Regulation of Germinal Centre Reaction. Nature (2015) 517:214–8. doi: 10.1038/nature13803
109. Pratama A, Srivastava M, Williams NJ, Papa I, Lee SK, Dinh XT, et al. MicroRNA-146a Regulates ICOS–ICOSL Signalling to Limit Accumulation of T Follicular Helper Cells and Germinal Centres. Nat Commun (2015) 6:6436. doi: 10.1038/ncomms7436
110. Allen CDC, Okada T, Tang HL, Cyster JG. Imaging of Germinal Center Selection Events During Affinity Maturation. Science (2007) 315:528 – 531. doi: 10.1126/science.1136736
111. Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, et al. Antigen-Engaged B Cells Undergo Chemotaxis Toward the T Zone and Form Motile Conjugates With Helper T Cells. PloS Biol (2005) 3:e150. doi: 10.1371/journal.pbio.0030150
112. Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, et al. Persistent Antigen and Germinal Center B Cells Sustain T Follicular Helper Cell Responses and Phenotype. Immunity (2013) 38:596–605. doi: 10.1016/j.immuni.2012.11.020
113. Pelletier N, McHeyzer-Williams LJ, Wong KA, Urich E, Fazilleau N, McHeyzer-Williams MG. Plasma Cells Negatively Regulate the Follicular Helper T Cell Program. Nat Immunol (2010) 11:1110–8. doi: 10.1038/ni.1954
114. Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, et al. T Follicular Helper Cell Dynamics in Germinal Centers. Science (2013) 341:673–7. doi: 10.1126/science.1241680
115. Corthay A. How do Regulatory T Cells Work? Scand J Immunol (2009) 70:326–36. doi: 10.1111/j.1365-3083.2009.02308.x
116. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular Regulatory T Cells Expressing Foxp3 and Bcl-6 Suppress Germinal Center Reactions. Nat Med (2011) 17:983–8. doi: 10.1038/nm.2426
117. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ Follicular Regulatory T Cells Control the Germinal Center Response. Nat Med (2011) 17:975–82. doi: 10.1038/nm.2425
118. Wallin EF. T Follicular Regulatory Cells and Antibody Responses in Transplantation. Transplantation (2018) 102:1614. doi: 10.1097/TP.0000000000002224
119. Wing JB, Tekgüç M, Sakaguchi S. Control of Germinal Center Responses by T-Follicular Regulatory Cells. Front Immunol (2018) 9:1910. doi: 10.3389/fimmu.2018.01910
120. Vanderleyden I, Fra-Bido SC, Innocentin S, Stebegg M, Okkenhaug H, Evans-Bailey N, et al. Follicular Regulatory T Cells Can Access the Germinal Center Independently of CXCR5. Cell Rep (2020) 30:611–9. doi: 10.1016/j.celrep.2019.12.076
121. Cañete PF, Sweet RA, Gonzalez-Figueroa P, Papa I, Ohkura N, Bolton H, et al. Regulatory Roles of IL-10–Producing Human Follicular T Cells. J Exp Med (2019) 216:1843–56. doi: 10.1084/jem.20190493
122. Almanan M, Raynor J, Ogunsulire I, Malyshkina A, Mukherjee S, Hummel SA, et al. IL-10–Producing Tfh Cells Accumulate With Age and Link Inflammation With Age-Related Immune Suppression. Sci Adv (2020) 6:eabb0806. doi: 10.1126/sciadv.abb0806
123. Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, et al. Preferential Generation of Follicular B Helper T Cells From Foxp3+ T Cells in Gut Peyer’s Patches. Science (2009) 323:1488–92. doi: 10.1126/science.1169152
124. Hou S, Clement RL, Diallo A, Blazar BR, Rudensky AY, Sharpe AH, et al. FoxP3 and Ezh2 Regulate Tfr Cell Suppressive Function and Transcriptional Program. J Exp Med (2019) 216:605–20. doi: 10.1084/jem.20181134
125. Milanez-Almeida P, Meyer-Hermann M, Toker A, Khailaie S, Huehn J. Foxp3+ Regulatory T-Cell Homeostasis Quantitatively Differs in Murine Peripheral Lymph Nodes and Spleen. Eur J Immunol (2015) 45:153–66. doi: 10.1002/eji.201444480
126. Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The Coinhibitory Receptor CTLA-4 Controls B Cell Responses by Modulating T Follicular Helper, T Follicular Regulatory, and T Regulatory Cells. Immunity (2014) 41:1026–39. doi: 10.1016/j.immuni.2014.12.005
127. Ritvo P-GG, Churlaud G, Quiniou V, Florez L, Brimaud F, Fourcade G, et al. Tfr Cells Lack IL-2rα But Express Decoy IL-1R2 and IL-1Ra and Suppress the IL-1–Dependent Activation of Tfh Cells. Sci Immunol (2017) 2:eaan0368. doi: 10.1126/sciimmunol.aan0368
128. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T Cells Control Antigen-Specific Expansion of Tfh Cell Number and Humoral Immune Responses Via Coreceptor CTLA-4. Immunity (2014) 41:1013–25. doi: 10.1016/j.immuni.2014.12.006
129. Krummel MF, Allison JP. CD28 and CTLA-4 Have Opposing Effects on the Response of T Cells to Stimulation. J Exp Med (1995) 182:459–65. doi: 10.1084/jem.182.2.459
130. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science (2011) 332:600–3. doi: 10.1126/science.1202947
131. Khailaie S, Rowshanravan B, Robert PA, Waters E, Halliday N, Herrera JDB, et al. Characterization of CTLA4 Trafficking and Implications for Its Function. Biophys J (2018) 115:1330–43. doi: 10.1016/j.bpj.2018.08.020
132. Swartzendruber DC, Congdon CC. Electron Microscope Observations on Tingible Body Macrophages in Mouse Spleen. J Cell Biol (1963) 19:641–6. doi: 10.1083/jcb.19.3.641
133. Smith JP, Burton GF, Tew JG, Szakal AK. Tinigible Body Macrophages in Regulation of Germinal Center Reactions. Dev Immunol (1998) 6:38923. doi: 10.1155/1998/38923
134. Smith JP, Lister AM, Tew JG, Szakal AK. Kinetics of the Tingible Body Macrophage Response in Mouse Germinal Center Development and Its Depression With Age. Anat Rec (1991) 229:511–20. doi: 10.1002/ar.1092290412
135. Szakal AK, Kosco MH, Tew JG. A Novel In Vivo Follicular Dendritic Cell-Dependent Iccosome-Mediated Mechanism for Delivery of Antigen to Antigen-Processing Cells. J Immunol (1988) 140:341–53.
136. Rahman ZSM, Shao W-H, Khan TN, Zhen Y, Cohen PL. Impaired Apoptotic Cell Clearance in the Germinal Center by Mer-Deficient Tingible Body Macrophages Leads to Enhanced Antibody-Forming Cell and Germinal Center Responses. J Immunol (2010) 185:5859–68. doi: 10.4049/jimmunol.1001187
137. Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, et al. Impaired Uptake of Apoptotic Cells Into Tingible Body Macrophages in Germinal Centers of Patients With Systemic Lupus Erythematosus. Arthritis Rheum (2002) 46:191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K
138. Gaipl US, Munoz LE, Grossmayer G, Lauber K, Franz S, Sarter K, et al. Clearance Deficiency and Systemic Lupus Erythematosus (SLE). J Autoimmun (2007) 28:114–21. doi: 10.1016/j.jaut.2007.02.005
139. Pedersen GK, Wørzner K, Andersen P, Christensen D. Vaccine Adjuvants Differentially Affect Kinetics of Antibody and Germinal Center Responses. Front Immunol (2020) 11:579761. doi: 10.3389/fimmu.2020.579761
140. Aloulou M, Carr EJ, Gador M, Bignon A, Liblau RS, Fazilleau N, et al. Follicular Regulatory T Cells can be Specific for the Immunizing Antigen and Derive From Naive T Cells. Nat Commun (2016) 7:10579. doi: 10.1038/ncomms10579
141. Hill DL, Pierson W, Bolland DJ, Mkindi C, Carr EJ, Wang J, et al. The Adjuvant GLA-SE Promotes Human Tfh Cell Expansion and Emergence of Public Tcrβ Clonotypes. J Exp Med (2019) 216:1857–73. doi: 10.1084/jem.20190301
142. Reed SG, Orr MT, Fox CB. Key Roles of Adjuvants in Modern Vaccines. Nat Med (2013) 19:1597–608. doi: 10.1038/nm.3409
143. DeFranco AL, Rookhuizen DC, Hou B. Contribution of Toll-Like Receptor Signaling to Germinal Center Antibody Responses. Immunol Rev (2012) 247:64–72. doi: 10.1111/j.1600-065X.2012.01115.x
144. Rookhuizen DC, DeFranco AL. Toll-Like Receptor 9 Signaling Acts on Multiple Elements of the Germinal Center to Enhance Antibody Responses. Proc Natl Acad Sci (2014) 111:E3224–33. doi: 10.1073/pnas.1323985111
145. Lisk C, Yuen R, Kuniholm J, Antos D, Reiser ML, Wetzler LM. Toll-Like Receptor Ligand Based Adjuvant, PorB, Increases Antigen Deposition on Germinal Center Follicular Dendritic Cells While Enhancing the Follicular Dendritic Cells Network. Front Immunol (2020) 11:1254. doi: 10.3389/fimmu.2020.01254
146. Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, et al. Programming the Magnitude and Persistence of Antibody Responses With Innate Immunity. Nature (2011) 470:543–7. doi: 10.1038/nature09737
147. Jellusova J. The Role of Metabolic Checkpoint Regulators in B Cell Survival and Transformation. Immunol Rev (2020) 295:39–53. doi: 10.1111/imr.12855
148. Choi S-C, Morel L. Immune Metabolism Regulation of the Germinal Center Response. Exp Mol Med (2020) 52:348–55. doi: 10.1038/s12276-020-0392-2
149. Cho SH, Raybuck AL, Stengel K, Wei M, Beck TC, Volanakis E, et al. Germinal Centre Hypoxia and Regulation of Antibody Qualities by a Hypoxia Response System. Nature (2016) 537:234–8. doi: 10.1038/nature19334
150. Weisel FJ, Mullett SJ, Elsner RA, Menk AV, Trivedi N, Luo W, et al. Germinal Center B Cells Selectively Oxidize Fatty Acids for Energy While Conducting Minimal Glycolysis. Nat Immunol (2020) 21:331–42. doi: 10.1038/s41590-020-0598-4
151. Choi S-C, Titov AA, Abboud G, Seay HR, Brusko TM, Roopenian DC, et al. Inhibition of Glucose Metabolism Selectively Targets Autoreactive Follicular Helper T Cells. Nat Commun (2018) 9:4369. doi: 10.1038/s41467-018-06686-0
152. Laplante M, Sabatini DM. mTOR Signaling in Growth Control and Disease. Cell (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
153. Monzón-Casanova E, Screen M, Díaz-Muñoz MD, Coulson RMR, Bell SE, Lamers G, et al. The RNA-Binding Protein PTBP1 Is Necessary for B Cell Selection in Germinal Centers. Nat Immunol (2018) 19:267–78. doi: 10.1038/s41590-017-0035-5
154. Vijay R, Guthmiller JJ, Sturtz AJ, Surette FA, Rogers KJ, Sompallae RR, et al. Infection-Induced Plasmablasts Are a Nutrient Sink That Impairs Humoral Immunity to Malaria. Nat Immunol (2020) 21:790–801. doi: 10.1038/s41590-020-0678-5
155. Min Son Y, Cheon IS, Goplen NP, Dent AL, Sun J. Inhibition of Stearoyl-CoA Desaturase Suppresses Follicular Help T and Germinal Center B Cell Responses. bioRxiv (2019) 601021. doi: 10.1101/601021
156. Gatto D, Martin SW, Bessa J, Pellicioli E, Saudan P, Hinton HJ, et al. Regulation of Memory Antibody Levels: The Role of Persisting Antigen Versus Plasma Cell Life Span. J Immunol (2007) 178:67–76. doi: 10.4049/jimmunol.178.1.67
157. Wolniak KL, Noelle RJ, Waldschmidt TJ. Characterization of (4-Hydroxy-3-Nitrophenyl) Acetyl (NP)-Specific Germinal Center B Cells and Antigen-Binding B220– Cells After Primary NP Challenge in Mice. J Immunol (2006) 177:2072–9. doi: 10.4049/jimmunol.177.4.2072
158. Abbott RK, Lee JH, Menis S, Skog P, Rossi M, Ota T, et al. Precursor Frequency and Affinity Determine B Cell Competitive Fitness in Germinal Centers, Tested With Germline-Targeting HIV Vaccine Immunogens. Immunity (2018) 48:133–146.e6. doi: 10.1016/j.immuni.2017.11.023
159. Meyer-Hermann M, Beyer T. The Type of Seeder Cells Determines the Efficiency of Germinal Center Reactions. Bull Math Biol (2004) 66:125–41. doi: 10.1016/j.bulm.2003.08.003
160. Havenar-Daughton C, Abbott RK, Schief WR, Crotty S. When Designing Vaccines, Consider the Starting Material: The Human B Cell Repertoire. Curr Opin Immunol (2018) 53:209–16. doi: 10.1016/j.coi.2018.08.002
162. Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S, et al. Multiple Layers of B Cell Memory With Different Effector Functions. Nat Immunol (2009) 10:1292–9. doi: 10.1038/ni.1814
163. Bergen MJ, Pan C-H, Greer CE, Legg HS, Polo JM, Griffin DE. Comparison of the Immune Responses Induced by Chimeric Alphavirus-Vectored and Formalin-Inactivated Alum-Precipitated Measles Vaccines in Mice. PloS One (2010) 5:e10297. doi: 10.1371/journal.pone.0010297
164. Adachi Y, Onodera T, Yamada Y, Daio R, Tsuiji M, Inoue T, et al. Distinct Germinal Center Selection at Local Sites Shapes Memory B Cell Response to Viral Escape. J Exp Med (2015) 212:1709–23. doi: 10.1084/jem.20142284
165. Li H, Limenitakis JP, Greiff V, Yilmaz B, Schären O, Urbaniak C, et al. Mucosal or Systemic Microbiota Exposures Shape the B Cell Repertoire. Nature (2020) 584:274–8. doi: 10.1038/s41586-020-2564-6
166. Tas JMJ, Mesin L, Pasqual G, Targ S, Jacobsen JT, Mano YM, et al. Visualizing Antibody Affinity Maturation in Germinal Centers. Science (2016) 351:1048–54. doi: 10.1126/science.aad3439
167. Fallet B, Hao Y, Florova M, Cornille K, de los Aires AV, Girelli Zubani G, et al. Chronic Viral Infection Promotes Efficient Germinal Center B Cell Responses. Cell Rep (2020) 30:1013–26.e7. doi: 10.1016/j.celrep.2019.12.023
168. León B, Bradley JE, Lund FE, Randall TD, Ballesteros-Tato A. FoxP3+ Regulatory T Cells Promote Influenza-Specific Tfh Responses by Controlling IL-2 Availability. Nat Commun (2014) 5:3495. doi: 10.1038/ncomms4495
169. Greczmiel U, Oxenius A. The Janus Face of Follicular T Helper Cells in Chronic Viral Infections. Front Immunol (2018) 9:1162. doi: 10.3389/fimmu.2018.01162
170. Hara S, Sasaki T, Satoh-Takayama N, Kanaya T, Kato T, Takikawa Y, et al. Dietary Antigens Induce Germinal Center Responses in Peyer’s Patches and Antigen-Specific IgA Production. Front Immunol (2019) 10:2432. doi: 10.3389/fimmu.2019.02432
171. Reboldi A, Cyster JG. Peyer’s Patches: Organizing B-Cell Responses at the Intestinal Frontier. Immunol Rev (2016) 271:230–45. doi: 10.1111/imr.12400
172. Nowosad CR, Mesin L, Castro TBR, Wichmann C, Donaldson GP, Araki T, et al. Tunable Dynamics of B Cell Selection in Gut Germinal Centres. Nature (2020) 588(7837):321–6. doi: 10.1038/s41586-020-2865-9
173. Georgiev H, Ravens I, Papadogianni G, Halle S, Malissen B, Loots GG, et al. Shared and Unique Features Distinguishing Follicular T Helper and Regulatory Cells of Peripheral Lymph Node and Peyer’s Patches. Front Immunol (2018) 9:714. doi: 10.3389/fimmu.2018.00714
174. Jones L, Ho WQ, Ying S, Ramakrishna L, Srinivasan KG, Yurieva M, et al. A Subpopulation of High IL-21-Producing CD4+ T Cells in Peyer’s Patches Is Induced by the Microbiota and Regulates Germinal Centers. Sci Rep (2016) 6:30784. doi: 10.1038/srep30784
175. Pabst O. New Concepts in the Generation and Functions of IgA. Nat Rev Immunol (2012) 12:821–32. doi: 10.1038/nri3322
176. Saito M, Novak U, Piovan E, Basso K, Sumazin P, Schneider C, et al. BCL6 Suppression of BCL2 via Miz1 and Its Disruption in Diffuse Large B Cell Lymphoma. Proc Natl Acad Sci (2009) 106:11294–9. doi: 10.1073/pnas.0903854106
177. Scheller H, Tobollik S, Kutzera A, Eder M, Unterlehberg J, Pfeil I, et al. C-Myc Overexpression Promotes a Germinal Center-Like Program in Burkitt’s Lymphoma. Oncogene (2010) 29:888–97. doi: 10.1038/onc.2009.377
178. Xia Y, Xu-Monette ZY, Tzankov A, Li X, Manyam GC, Murty V, et al. Loss of PRDM1/BLIMP-1 Function Contributes to Poor Prognosis of Activated B-Cell-Like Diffuse Large B-Cell Lymphoma. Leukemia (2017) 31:625–36. doi: 10.1038/leu.2016.243
179. Sungalee S, Mamessier E, Morgado E, Grégoire E, Brohawn PZ, Morehouse CA, et al. Germinal Center Reentries of BCL2-Overexpressing B Cells Drive Follicular Lymphoma Progression. J Clin Invest (2014) 124:5337–51. doi: 10.1172/JCI72415
180. Mancao C, Altmann M, Jungnickel B, Hammerschmidt W. Rescue of “Crippled” Germinal Center B Cells From Apoptosis by Epstein-Barr Virus. Blood (2005) 106:4339–44. doi: 10.1182/blood-2005-06-2341
181. Kräutler NJ, Yermanos A, Pedrioli A, Welten SPM, Lorgé D, Greczmiel U, et al. Quantitative and Qualitative Analysis of Humoral Immunity Reveals Continued and Personalized Evolution in Chronic Viral Infection. Cell Rep (2020) 30:997–1012.e6. doi: 10.1016/j.celrep.2019.12.088
182. Kavaler J, Caton AJ, Staudt LM, Gerhard W. A B Cell Population That Dominates the Primary Response to Influenza Virus Hemagglutinin Does Not Participate in the Memory Response. Eur J Immunol (1991) 21:2687–95. doi: 10.1002/eji.1830211107
183. Lam JH, Baumgarth N. The Multifaceted B Cell Response to Influenza Virus. J Immunol (2019) 202:351 – 359. doi: 10.4049/jimmunol.1801208
184. Zammit DJ, Turner DL, Klonowski KD, Lefrançois L, Cauley LS. Residual Antigen Presentation After Influenza Virus Infection Affects CD8 T Cell Activation and Migration. Immunity (2006) 24:439–49. doi: 10.1016/j.immuni.2006.01.015
185. Kato LM, Kawamoto S, Maruya M, Fagarasan S. Gut TFH and IgA: Key Players for Regulation of Bacterial Communities and Immune Homeostasis. Immunol Cell Biol (2014) 92:49–56. doi: 10.1038/icb.2013.54
186. Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity (2019) 51:337–50.e7. doi: 10.1016/j.immuni.2019.07.001
187. Casola S, Otipoby KL, Alimzhanov M, Humme S, Uyttersprot N, Kutok JL, et al. B Cell Receptor Signal Strength Determines B Cell Fate. Nat Immunol (2004) 5:317–27. doi: 10.1038/ni1036
188. Mlynarczyk C, Fontán L, Melnick A. Germinal Center-Derived Lymphomas: The Darkest Side of Humoral Immunity. Immunol Rev (2019) 288:214–39. doi: 10.1111/imr.12755
189. Aloisi F, Pujol-Borrell R. Lymphoid Neogenesis in Chronic Inflammatory Diseases. Nat Rev Immunol (2006) 6:205–17. doi: 10.1038/nri1786
190. Domeier PP, Schell SL, Rahman ZSM. Spontaneous Germinal Centers and Autoimmunity. Autoimmunity (2017) 50:4–18. doi: 10.1080/08916934.2017.1280671
191. Schröder AE, Greiner A, Seyfert C, Berek C. Differentiation of B Cells in the Nonlymphoid Tissue of the Synovial Membrane of Patients With Rheumatoid Arthritis. Proc Natl Acad Sci (1996) 93:221 – 225. doi: 10.1073/pnas.93.1.221
192. Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O’Fallon WM, et al. Lymphoid Neogenesis in Rheumatoid Synovitis. J Immunol (2001) 167:1072–80. doi: 10.4049/jimmunol.167.2.1072
193. Aziz KE, McCluskey PJ, Wakefield D. Characterisation of Follicular Dendritic Cells in Labial Salivary Glands of Patients With Primary Sjogren Syndrome: Comparison With Tonsillar Lymphoid Follicles. Ann Rheum Dis (1997) 56:140–3. doi: 10.1136/ard.56.2.140
194. Salomonsson S, Jonsson MV, Skarstein K, Brokstad KA, Hjelmström P, Wahren-Herlenius M, et al. Cellular Basis of Ectopic Germinal Center Formation and Autoantibody Production in the Target Organ of Patients With Sjögren’s Syndrome. Arthritis Rheum (2003) 48:3187–201. doi: 10.1002/art.11311
195. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of Ectopic B-Cell Follicles With Germinal Centers in the Meninges of Patients With Secondary Progressive Multiple Sclerosis. Brain Pathol (2004) 14:164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x
196. Levinson AI, Wheatley LM. The Thymus and the Pathogenesis of Myasthenia Gravis. Clin Immunol Immunopathol (1996) 78:1–5. doi: 10.1006/clin.1996.0001
197. Randen I, Mellbye OJ, Førre Ø, Natvig JB. The Identification of Germinal Centres and Follicular Dendritic Cell Networks in Rheumatoid Synovial Tissue. Scand J Immunol (1995) 41:481–6. doi: 10.1111/j.1365-3083.1995.tb03596.x
198. Elliott SE, Kongpachith S, Lingampalli N, Adamska JZ, Cannon BJ, Mao R, et al. Affinity Maturation Drives Epitope Spreading and Generation of Proinflammatory Anti–Citrullinated Protein Antibodies in Rheumatoid Arthritis. Arthritis Rheumatol (2018) 70:1946–58. doi: 10.1002/art.40587
199. Holmdahl R, Malmström V, Burkhardt H. Autoimmune Priming, Tissue Attack and Chronic Inflammation — The Three Stages of Rheumatoid Arthritis. Eur J Immunol (2014) 44:1593–9. doi: 10.1002/eji.201444486
200. Beyer T, Meyer-Hermann M. Cell Transmembrane Receptors Determine Tissue Pattern Stability. Phys Rev Lett (2008) 101:148102. doi: 10.1103/PhysRevLett.101.148102
201. Kang YM, Zhang X, Wagner UG, Yang H, Beckenbaugh RD, Kurtin PJ, et al. CD8 T Cells Are Required for the Formation of Ectopic Germinal Centers in Rheumatoid Synovitis. J Exp Med (2002) 195:1325–36. doi: 10.1084/jem.20011565
202. Andersen TK, Huszthy PC, Gopalakrishnan RP, Jacobsen JT, Fauskanger M, Tveita AA, et al. Enhanced Germinal Center Reaction by Targeting Vaccine Antigen to Major Histocompatibility Complex Class II Molecules. NPJ Vaccines (2019) 4:9. doi: 10.1038/s41541-019-0101-0
203. Cirelli KM, Carnathan DG, Nogal B, Martin JT, Rodriguez OL, Upadhyay AA, et al. Slow Delivery Immunization Enhances HIV Neutralizing Antibody and Germinal Center Responses Via Modulation Immunodominance. Cell (2019) 177:1153–71.e28. doi: 10.1016/j.cell.2019.04.012
204. Tam HH, Melo MB, Kang M, Pelet JM, Ruda VM, Foley MH, et al. Sustained Antigen Availability During Germinal Center Initiation Enhances Antibody Responses to Vaccination. Proc Natl Acad Sci (2016) 113:E6639–48. doi: 10.1073/pnas.1606050113
205. Havenar-Daughton C, Carnathan DG, Boopathy AV, Upadhyay AA, Murrell B, Reiss SM, et al. Rapid Germinal Center and Antibody Responses in Non-Human Primates After a Single Nanoparticle Vaccine Immunization. Cell Rep (2019) 29:1756–66.e8. doi: 10.1016/j.celrep.2019.10.008
206. Lederer K, Castaño D, Gómez Atria D, Oguin TH, Wang S, Manzoni TB, et al. SARS-CoV-2 mRNA Vaccines Foster Potent Antigen-Specific Germinal Center Responses Associated With Neutralizing Antibody Generation. Immunity (2020) 53:1281–95.e5. doi: 10.1016/j.immuni.2020.11.009
207. Cirelli KM, Crotty S. Germinal Center Enhancement by Extended Antigen Availability. Curr Opin Immunol (2017) 47:64–9. doi: 10.1016/j.coi.2017.06.008
208. Good-Jacobson KL, Song E, Anderson S, Sharpe AH, Shlomchik MJ. CD80 Expression on B Cells Regulates Murine T Follicular Helper Development, Germinal Center B Cell Survival, and Plasma Cell Generation. J Immunol (2012) 188:4217–25. doi: 10.4049/jimmunol.1102885
209. Kuraoka M, Schmidt AG, Nojima T, Feng F, Watanabe A, Kitamura D, et al. Complex Antigens Drive Permissive Clonal Selection in Germinal Centers. Immunity (2016) 44:542–52. doi: 10.1016/j.immuni.2016.02.010
210. Silver J, Zuo T, Chaudhary N, Kumari R, Tong P, Giguere S, et al. Stochasticity Enables BCR-Independent Germinal Center Initiation and Antibody Affinity Maturation. J Exp Med (2017) 215:77–90. doi: 10.1084/jem.20171022
211. Meyer-Hermann M, Figge MT, Toellner K-M. Germinal Centres Seen Through the Mathematical Eye: B-Cell Models on the Catwalk. Trends Immunol (2009) 30:157–64. doi: 10.1016/j.it.2009.01.005
212. Wang P, Shih C, Qi H, Lan Y. A Stochastic Model of the Germinal Center Integrating Local Antigen Competition, Individualistic T–B Interactions, and B Cell Receptor Signaling. J Immunol (2016) 197:1169–82. doi: 10.4049/jimmunol.1600411
Keywords: germinal centre shutdown, vaccination, chronic germinal centres, B cell lymphoma, ectopic germinal centres, antibody responses
Citation: Arulraj T, Binder SC, Robert PA and Meyer-Hermann M (2021) Germinal Centre Shutdown. Front. Immunol. 12:705240. doi: 10.3389/fimmu.2021.705240
Received: 04 May 2021; Accepted: 24 June 2021;
Published: 07 July 2021.
Edited by:
Stamatis-Nick C Liossis, General University Hospital of Patras, GreeceReviewed by:
Constantina A Bounia, General University Hospital of Patras, GreeceElena Solomou, University of Patras, Greece
Copyright © 2021 Arulraj, Binder, Robert and Meyer-Hermann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Meyer-Hermann, bW1oQHRoZW9yZXRpY2FsLWJpb2xvZ3kuZGU=