 Hazel Ozuna
Hazel Ozuna Silvia M. Uriarte
Silvia M. Uriarte Donald R. Demuth1,2
Donald R. Demuth1,2- 1Department of Microbiology and Immunology, School of Medicine, University of Louisville, Louisville, KY, United States
- 2Department of Oral Immunology and Infectious Diseases, School of Dentistry, University of Louisville, Louisville, KY, United States
Aggregatibacter actinomycetemcomitans is a gram-negative facultative anaerobe and an opportunistic oral pathogen, strongly associated with periodontitis and other inflammatory diseases. Periodontitis is a chronic inflammation of the periodontium resulting from the inflammatory response of the host towards the dysbiotic microbial community present at the gingival crevice. Previously, our group identified catecholamines and iron as the signals that activate the QseBC two-component system in A. actinomycetemcomitans, necessary for the organism to acquire iron as a nutrient to survive in the anaerobic environment. However, the source of catecholamines has not been identified. It has been reported that mouse neutrophils can release catecholamines. In periodontitis, large infiltration of neutrophils is found at the subgingival pocket; hence, we wanted to test the hypothesis that A. actinomycetemcomitans exploits human neutrophils as a source for catecholamines. In the present study, we showed that human neutrophils synthesize, store, and release epinephrine, one of the three main types of catecholamines. Human neutrophil challenge with A. actinomycetemcomitans induced exocytosis of neutrophil granule subtypes: secretory vesicles, specific granules, gelatinase granules, and azurophilic granules. In addition, by selectively inhibiting granule exocytosis, we present the first evidence that epinephrine is stored in azurophilic granules. Using QseC mutants, we showed that the periplasmic domain of the QseC sensor kinase is required for the interaction between A. actinomycetemcomitans and epinephrine. Finally, epinephrine-containing supernatants collected from human neutrophils promoted A. actinomycetemcomitans growth and induced the expression of the qseBC operon under anaerobic conditions. Based on our findings, we propose that A. actinomycetemcomitans promotes azurophilic granule exocytosis by neutrophils as an epinephrine source to promote bacterial survival.
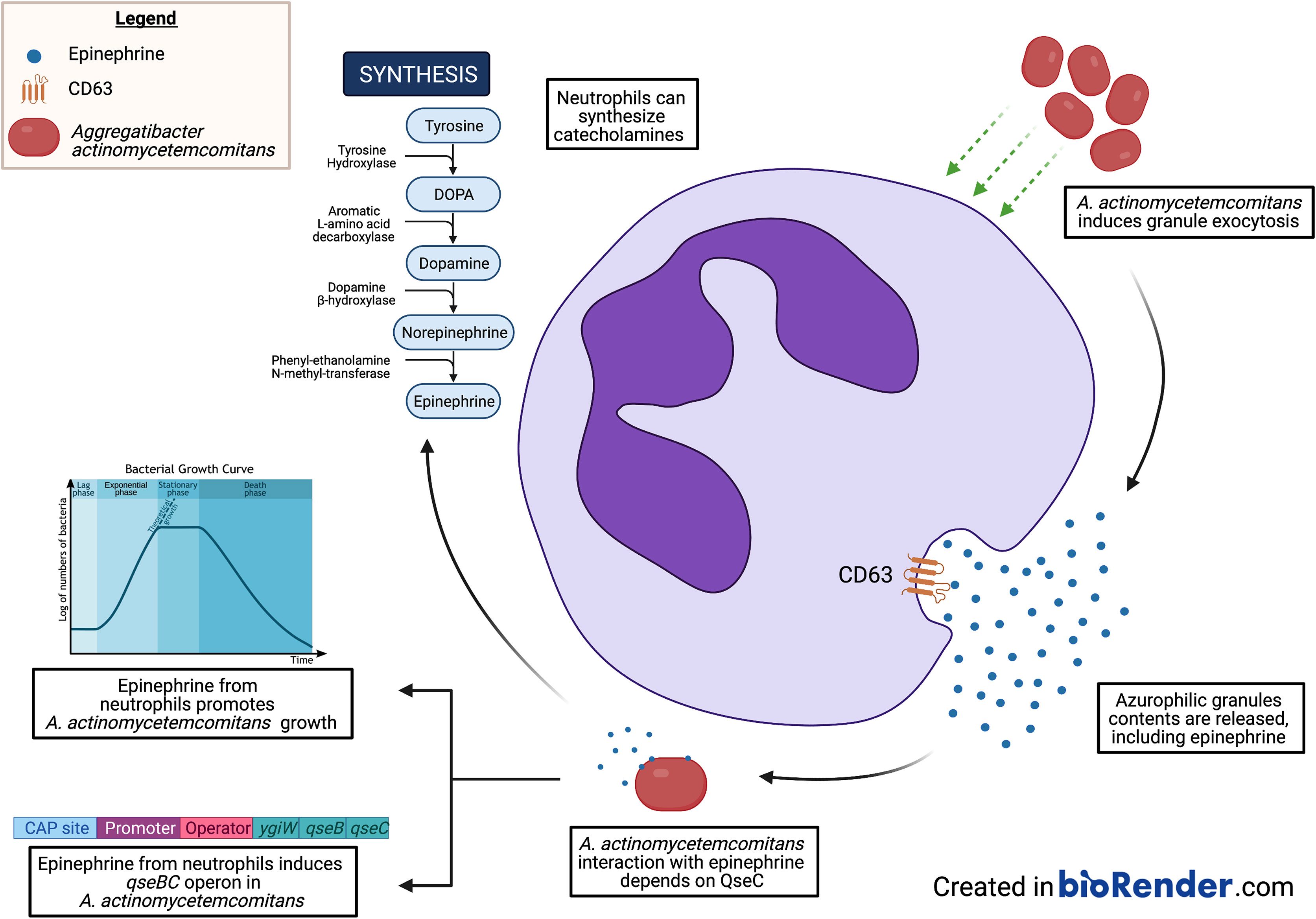
Graphical Abstract
Introduction
In the oral cavity, when host microbe homeostasis is broken, bacterial communities accumulate at the sub-gingival pocket and form biofilms that lead to oral diseases such as periodontitis (1). Periodontitis consists of a chronic inflammation of the periodontium caused by the inflammatory response of the host to plaque biofilm. The disease is characterized by the progressive deepening of the sulcus and loss of attachment between the bone and gingival tissue leading to bone loss. Recurring inflammation of the periodontium has been associated with the initiation, exacerbation, and pathogenesis of a number of other inflammatory diseases (2). Aggregatibacter actinomycetemcomitans (Aa) is a non-motile gram-negative facultative anaerobe of the Pasteurellaceae family (3–5). A. actinomycetemcomitans has been strongly associated with periodontitis and other diseases such as rheumatoid arthritis, cardiovascular diseases, atherosclerosis, urinary tract infections, and brain abscesses (6–10). A. actinomycetemcomitans contributes to tissue inflammation, destruction, and bone resorption by expressing a number of virulence factors such as cytolethal disentin toxin, leukotoxin A of the RTX family of bacterial toxins, and collagenase (11–14).
In the subgingival pocket, Aa must compete for nutrients, such as iron, in order to survive. As part of the host immune response, iron is kept unavailable to bacteria by being sequestered by catecholamines (i.e., epinephrine, norepinephrine, and dopamine) or leukocyte-produced molecules like lactoferrin and transferrin (15, 16). However, some bacteria have evolved to subvert this mechanism by producing iron scavenging molecules, known as siderophores (17–19) or using host-derived catecholamines (15). Catecholamines have been implicated as causative or contributory agents of periodontitis (20–22). Interestingly, Aa does not produce siderophores (23), but expresses the QseBC (quorum sensing in Escherichia coli) two-component system (TCS) that is activated in the presence of both catecholamines and iron (6). The qseBC operon encodes the genes for qseC, a sensor molecule, and qseB, the response regulator (24). Novak et al. (25) demonstrated the requirement of QseC for Aa biofilm growth and virulence. Furthermore, Weigel et al. (6) subsequently showed that Aa growth in a chemically defined media (CDM) supplemented with both epinephrine or norepinephrine and ferrous (Fe2+) or ferric (Fe3+) iron increased qseBC promoter activity and bacterial growth, indicating that both catecholamines and iron are required for the activation of QseBC. Additionally, microarray analysis showed that activation of QseBC induced the expression of genes associated with anaerobic metabolism and respiration and reduced expression of genes involved in iron uptake and transport. However, the source of catecholamines remained to be determined. In periodontitis, neutrophils are found in large numbers at the gingival crevice (26). Murine neutrophils have been shown to release epinephrine or norepinephrine when stimulated with LPS (27). Furthermore, increased levels of the enzymes (tyrosine hydroxylase and dopamine β-hydroxylase) required for the synthesis of catecholamines and their inactivation (catecholamine-O-methyltransferase and monoamine oxidase) enzymes have been observed in murine phagocytes and human lymphocytes (27, 28). Nonetheless, the presence of catecholamines in the host environment has also been shown to promote bacterial iron uptake and growth in media limited conditions (15, 21, 22). Host-derived catecholamines have a higher affinity to iron than antimicrobials like lactoferrin and transferrin (15, 29), serving as excellent pseudo-siderophores (30) to bacteria. Therefore, we proposed that human neutrophils serve as a catecholamine source for Aa to sequester iron, leading to QseBC activation and bacterial growth.
In this study, we demonstrate that human neutrophils have significant levels of tyrosine hydroxylase, monoamine oxidase A, and catecholamine-O-methyltransferase, validating that cells participate in catecholamine metabolism. In addition, we present evidence that human neutrophils store epinephrine in the azurophilic granules and that interaction with Aa induces exocytosis of azurophilic granules and release of epinephrine. Furthermore, we show that treatment with latrunculin A followed by fMLF stimulation, which is known to induce mobilization and content release of azurophilic granules, induces epinephrine release in human neutrophils. The interaction of released epinephrine with Aa activates QseBC and induces Aa growth under anaerobic conditions. These results suggest that neutrophils may serve as an epinephrine source for Aa, to facilitate growth and prime anaerobic metabolism in the subgingival pocket. The findings presented in this article help shed light into the crosstalk that exits between bacteria and the host endocrine system by providing an example of the role that stress hormones play in periodontal disease and potentially other chronic inflammatory diseases.
Methods
Human Neutrophil Isolation and Purification
Recruitment of human donors, blood draws, and use of required materials were done in agreement with guidelines approved by the Institutional Review Board of the University of Louisville. Human neutrophils were isolated from whole blood of healthy donors using plasma-Percoll gradients as previously described (31). When necessary, neutrophils were further purified to obtain >99% pure population. Purification was carried by negative magnetic selection using the Easy Sep Human neutrophil isolation kit (Stemcell Technologies, Vancouver, BC, Canada). Cell purity was assessed by simultaneously staining with FITC-conjugated anti-CD66b (clone G10F5; BioLegend, San Diego, CA, USA) and APC-conjugated anti-CD16 (clone CB16; eBioscience, San Diego, CA, USA) antibodies and determining the percentage of CD66b+CD16+ cells using a BD Celesta flow cytometer (BD Biosciences, San Jose, CA, USA) and FlowJo for analysis (FlowJo, LLC, Ashland, OR, USA). Both pure (>90%–95%) and highly pure (>99%) neutrophils were cultured in RPMI-1640 medium without phenol red (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 2 mM L-glutamine (Sigma-Aldrich, St. Louis, MO, USA) and 5% human serum (Atlanta Biologicals, Flowery Branch, GA, USA).
Bacteria Strains and Media
In this study, we make use of A. actinomycetemcomitans 652 serotype c strain, a low leukotoxic, afimbriated, smooth colony-morphotype variant. Considered an opportunistic pathogen, this strain has been isolated from healthy and in large numbers in periodontitis-positive individuals (32, 33). At sublytic concentrations of leukotoxin A, there is an increase in calcium levels and neutrophil activation leading to oxidative burst and degranulation (34). A. actinomycetemcomitans strains (Supplementary Table 1) were propagated in brain–heart infusion (BHI, Difco, BD Biosciences, Franklin Lakes, NJ, USA) broth supplemented with bacitracin (50 µg/ml) and vancomycin (50 µg/ml), unless indicated otherwise. A. actinomycetemcomitans was grown at 37°C under microaerophilic conditions in a closed tube. The Aa mutant strains (all in Aa 652 background, see Supplementary Table 1) used for epinephrine interaction experiments were previously constructed by Juarez-Rodríguez et al. (24): Aa ΔqseC (non-polar qseC gene deletion mutant, spectinomycin 50 µg/ml), Aa qseCΔp (QseC sensor protein with an in-frame deletion of the periplasmic sensor domain, spectinomycin 50 µg/ml), and Aa ΔqseC-comp (non-polar qseC gene deletion mutant complemented with a single genomic copy of the qseC gene, spectinomycin 50 µg/ml). The Aa strain expressing the plasmid pDJR29, which contains the lacZ gene controlled by the qseBC promoter (35), was used in β-galactosidase assays and was grown under anaerobic conditions in CDM (Supplementary Table 2) (36), supplemented with kanamycin (25 µg/ml). All reagents used in anaerobic experiments were oxygen-depleted.
Filifactor alocis (F. alocis) ATCC 38596 was cultured in BHI broth supplemented with 5 mg/ml yeast extract, L-cysteine (0.05%), and arginine (0.05%) for 7 days anaerobically at 37°C as previously described (36–38).
A. actinomycetemcomitans Challenge, Epinephrine Detection, and Neutrophil Viability
Human neutrophils (3 × 106 cells/ml) were challenged in suspension with Aa at a multiplicity of infection (MOI) (39) of 50 at 37°C in a shaking water bath for 2 h, 4 h, 8 h, and 24 h, unless indicated otherwise. As a positive control for epinephrine release, neutrophils were treated with latrunculin A (1 μM, Sigma-Aldrich, St. Louis, MO, USA) for 30 min, followed by stimulation with N-formylmethionyl-leucyl-phenylalanine (fMLF, 300 nM, Sigma-Aldrich, St. Louis, MO, USA) for 5 min at 37°C in a shaking water bath. At the end of incubation, samples were centrifuged at 6,000 × g for 30 s and supernatants were collected and supplemented with 100× Halt Protease and Phosphatase inhibitor single-use cocktail (1:10 dilution; Thermo Fisher Scientific, Waltham, MA, USA). The pelleted cells were lysed with ice-cold 1× lysis buffer (10 µl per 1 × 106 of cells; [20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% [vol/vol] Triton X-100, 0.5% [vol/vol] Nonidet P-40, 20 mM NaF, 20 mM NaVO3, 1 mM EDTA, 1 mM EGTA, 5 mM phenylmethylsulfonyl fluoride [PMSF], 2 mM diisopropylfluorophosphate [DFP], 21 μg/ml aprotinin, and 5 μg/ml leupeptin]). Lysates were centrifuged and cell lysate was supplemented with 100× Halt Protease and Phosphatase inhibitor single-use cocktail (1:10 dilution; Thermo Fisher Scientific, Waltham, MA, USA). Epinephrine was measured in supernatants and cell lysate with a commercially available Adrenaline/Epinephrine ELISA kit (Cat. No. E4359, BioVision Inc., Milpitas, CA, USA).
Neutrophil viability when challenged with Aa was determined by Trypan Blue exclusion and cytospin microscopy imaging. For Trypan Blue exclusion, neutrophils were diluted 1:20 in Trypan blue at 0 h, 4 h, 8 h, and 24 h, and live cells were counted using a hemacytometer. For cytospin images, cells were centrifuged at 6,000 × g for 30 s and washed twice with RPMI-1640 (no phenol red, Sigma-Aldrich, St. Louis, MO, USA) to remove bacteria. Neutrophils were resuspended at 1 × 105 cells in 200 µl of RPMI-1640 and 5 µl of human serum was added (Atlanta Biologicals, Flowery Branch, GA, USA). The cell suspension was loaded into the funnel chamber that is assembled on the cytocentrifuge clip, with slide and filter. Cytocentrifuge clip was centrifuged for 5 min at 800 rpm (Shandon Cytospin 3, Thermo Fisher Scientific, Waltham, MA, USA). The microscope slide was removed from the cytocentrifuge clip and fixed and stained using the Hema 3 Protocol staining kit (Thermo Fisher Scientific, Waltham, MA, USA).
Catecholamine Metabolism Enzyme-Linked Immunosorbent Assay (ELISA)
Cell lysates collected from neutrophils challenged with Aa at 2 h, 4 h, 8 h, and 24 h were tested for the presence of enzymes involved in catecholamine metabolism. The levels of tyrosine hydroxylase (Cat. No. NBP3-06920, Novus Biologicals, CO, USA), dopamine β-hydroxylase (Cat. No. NBP2-67945, Novus Biologicals, CO, USA), catechol-o-methyltransferase (Cat. No. OKBB00966, Aviva Systems Biology Corp., San Diego, CA, USA), and monoamine oxidase-A (Cat. No. OKEH02825, Aviva Systems Biology Corp., San Diego, CA, USA) were measured following the indicated protocols supplied by the kit manufacturer.
Neutrophil Granule Exocytosis and Exocytosis Inhibition
Neutrophils (4 × 106 cells/ml) were challenged with fMLF (300 nM, 5 min), latrunculin A (1 μM, 30 min) + fMLF (300 nM, 5 min), or Aa at various time points from 5 min to 24 h at 37°C in a shaking water bath. The exocytosis of secretory vesicles, specific granules, and azurophilic granules was determined by measuring the plasma membrane increase of granule markers using fluorescein isothiocyanate (FITC)-conjugated anti-CD63 (for azurophilic granules, Ancell 215-040, Stillwater, MN, USA), FITC-conjugated anti-CD66b (for specific granules, Biolegend 305104, San Diego, CA, USA), and phycoerythrin (PE)-conjugated anti-CD35 (for secretory vesicles, Biolegend 333406, San Diego, CA, USA) by a FACSCalibur flow cytometer as previously described (31, 40). Following antibody incubation, cells were washed with 0.5% sodium azide (S2002, Sigma, St. Louis, MO, USA) in FTA buffer (211248 BD, Franklin Lakes, NJ, USA) and fixed with 1% paraformaldehyde (PX0055-3, EMD, Darmstadt, Germany). Gelatinase granule exocytosis was determined by measuring the release of matrix metallopeptidase 9 (MMP-9) by ELISA (Cat. No. ab100610, Abcam, Cambridge, MA, USA).
TAT-SNAP23 and TAT-Syntaxin 4 were used to inhibit neutrophil granule exocytosis as previously described by Uriarte et al. (31) and McLeish et al. (40). Briefly, neutrophils (4 × 106 cells/ml) were pretreated with TAT-Syntaxin 4 (0.8 μg/ml), TAT-SNAP23 (0.8 μg/ml), or TAT-control (1 μg/ml) for 15 min, followed by challenge with Aa for 15 min, 2 h, 4 h, and 8 h at 37°C in a shaking water bath. Granule exocytosis was measured by increased plasma membrane expression of CD35 (secretory vesicles), CD66b (specific granules), and CD63 (azurophilic granules) by flow cytometry as described in a previous paragraph. In addition, cell supernatants were collected to measure release of secretory vesicle content by ELISA for albumin (Cat. No. EHALB, Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA), gelatinase content by ELISA for MMP-9, specific granule content by ELISA for lactoferrin (Cat. No. ab108882, Abcam, Cambridge, MA, USA), and azurophilic granule content by ELISA for myeloperoxidase (Cat. No. ab119605, Abcam, Cambridge, MA, USA).
Recombinant Epinephrine Interaction With A. actinomycetemcomitans
Epinephrine interaction with Aa: Increasing colony-forming units (CFUs) of Aa (0.0–4.0 × 108 CFUs) were incubated with 250 pg/ml of recombinant epinephrine (BioVision Inc., Milpitas, CA, USA) for 15 min at 37°C in a shaking water bath. As a control, wells with epinephrine alone were run in parallel. Samples were centrifuged at 6,000 × g for 5 min, and supernatants were collected. Immediately after, epinephrine was measured from supernatants with a commercially available Adrenaline/Epinephrine ELISA kit (Cat. No. E4359, BioVision Inc., Milpitas, CA, USA). For analysis, interpolated values were obtained from recombinant epinephrine standard curve, following manufacturer instructions. The amount of epinephrine associated with Aa was determined by subtracting the interpolated values of bacteria-containing wells from the values obtained for control wells.
Epinephrine interaction with Aa qseC mutant strains: Mutant strains of Aa (ΔqseC, qseC/Δp, and ΔqseC-comp) were incubated at 3.0 × 107 CFUs with increasing concentrations (0–500 pg/ml) of recombinant epinephrine (BioVision Inc., Milpitas, CA, USA) for 15 min at 37°C in a shaking water bath. Wells containing increasing concentrations of recombinant epinephrine serve as control. Afterwards, samples were centrifuged as above, and supernatants were collected. Immediately after, epinephrine was measured and analyzed as described above. The amount of epinephrine associated with Aa was determined by subtracting the interpolated values of Aa-containing wells from the values of epinephrine control.
A. actinomycetemcomitans Growth Kinetics
A. actinomycetemcomitans wild type was inoculated into BHI broth supplemented with bacitracin (50 µg/ml) and vancomycin (50 µg/ml) and grown under microaerophilic conditions at 37°C to an optical density at 600 nm (OD600) of 0.3–0.4. Cells were subcultured in fresh BHI broth at a 1:30 dilution and grown as described above to an OD600 of 0.5–0.6. Cells were washed with CDM (Supplementary Table 2) and inoculated into CDM supplemented as above at a 1:30 dilution. Cultures were supplemented individually or with a combination of 100 µM FeCl2, 50 µM epinephrine, supernatant, or cell lysate collected from neutrophils stimulated with latrunculin A and fMLF (final epinephrine concentration of 30 pg/ml). The OD600 was measured at various time points using a Bio-Rad SmartSpec Plus UV-vis spectrophotometer (Bio-Rad, Hercules, CA, USA).
β-galactosidase Assay
Quantitative evaluation of β-galactosidase activity was determined using permeabilized cells incubated with o-nitrophenyl-b-D-galactopyranoside (ONPG) substrate (Sigma, St Louis, MO, USA) as previously described (41). Briefly, a primary culture of Aa (pDJR29) was grown at 37°C under microaerophilic conditions in a closed tube overnight in BHI broth. The culture was subcultured at a 1:30 dilution in BHI broth and grown as described in the previous paragraph for 24 h. Subsequently, the secondary overnight culture (OD600 of 0.3–0.4) was diluted 1:30 into CDM and grown in an anaerobic chamber for 24 h at 37°C. An aliquot of 0.1 ml was then used to determine the OD600 of the culture at 24 h and triplicate aliquots of 0.1 ml were used to measure β-galactosidase activity.
Statistical Analysis
Unless otherwise noted, statistical experimental conditions and time points were analyzed by a one-way ANOVA, followed by post-hoc Tukey’s multi-comparison test using GraphPad Prism Software (GraphPad PRISM v9, San Diego, CA, USA). Differences were considered significant at p < 0.05.
Results
A. actinomycetemcomitans Challenge Induces Epinephrine Release and Promotes De Novo Catecholamine Synthesis in Human Neutrophils
It has been previously shown that mouse neutrophils stimulated with LPS can release catecholamines (Flier 2007). However, there is as of yet no clear evidence of human neutrophils’ ability to release catecholamines. To determine if human neutrophils release epinephrine, cells were stimulated with fMLF, latrunculin A + fMLF, LPS, or IL-8. Supernatants and cell lysates were collected, and epinephrine levels were determined by ELISA. Unlike what was described in murine neutrophils (27), LPS stimulation did not induce epinephrine release by human neutrophils (Figure 1A). Similarly, stimulation of neutrophils with fMLF or IL-8 failed to induce epinephrine release (Figure 1A). Interestingly, treatment with latrunculin A followed by fMLF induced significant release of epinephrine compared to fMLF alone and basal (Figure 1A). Significant levels of epinephrine were detected in the cell lysates but there was no significant difference between experimental conditions (Figure 1B). Afterward, we examined the ability of Aa to induce epinephrine release by human neutrophils. Cells were challenged with Aa at a MOI of 50 in suspension for 2 h, 4 h, 8 h, and 24 h. Epinephrine levels were measured from supernatants and cell lysates by ELISA. Figure 1C shows that Aa challenge induced significant release of epinephrine compared to basal, with maximum levels reached at 4 h. Epinephrine levels in cell lysates of human neutrophils treated with latrunculin A + fMLF had significantly reduced levels of epinephrine compared to basal (Figure 1D); this correlates the significant increase release of epinephrine observed in supernatants (Figure 1C). Similarly, low epinephrine levels in neutrophil cell lysates were detected after challenged with Aa for 2 h and 4 h, which could be related to the release observed in the supernatants (Figures 1C, D). Interestingly, epinephrine release at 2 h was similar to basal (Figure 1C). A potential explanation for this is that the released epinephrine could be interacting in some way with Aa. We repeated this experiment with highly purified (>99%) human neutrophils (Supplementary Figure 1) and obtained similar results.
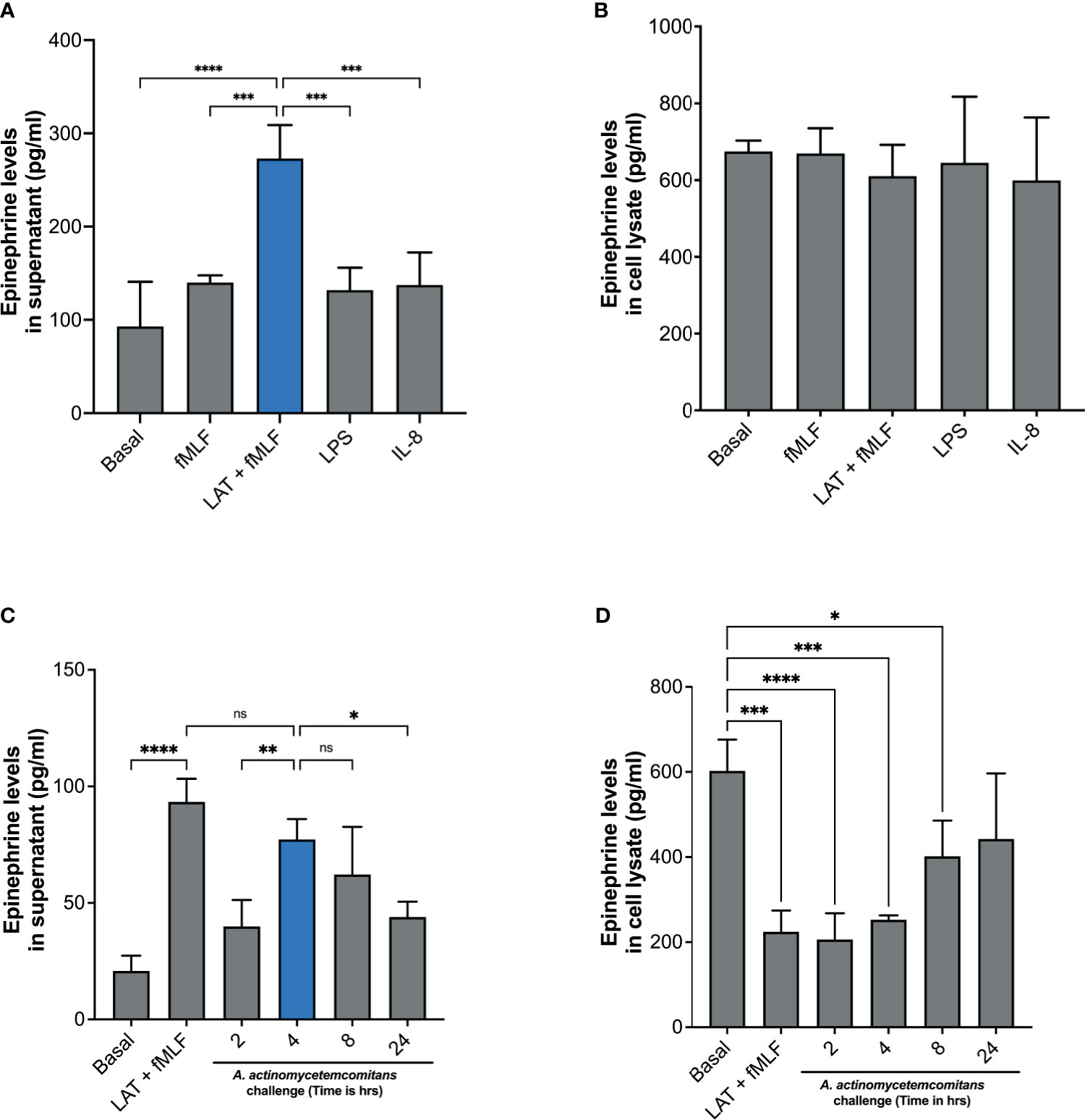
Figure 1 Human neutrophils release epinephrine upon A. actinomycetemcomitans challenge. Human neutrophils (5 × 106 cells/ml) were treated with fMLF, Latrunculin A (LAT) + fMLF, LPS, or IL-8 to induce epinephrine release. Epinephrine content was determined in neutrophil supernatant (A) and cell lysate (B) by ELISA. Human neutrophils (3 × 106 cells/ml) were challenged with A. actinomycetemcomitans (Aa; MOI 50) for 2 h, 4 h, 8 h, and 24 h At the end of each time point, supernatant (C) and cell lysates (D) were collected and epinephrine content was measured by ELISA. Epinephrine concentration is plotted as the mean ± SD of four independent experiments. Statistical differences among experimental conditions and time points were analyzed by one-way ANOVA (A, B) or repeated measures one-way ANOVA (C, D), followed by Tukey’s post-test. ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To confirm that the release of epinephrine is not caused by a cytotoxic effect of Aa on neutrophils, we evaluated the morphology of human neutrophils with cytospin imaging and viability with trypan blue exclusion. Human neutrophil morphology was not altered with Aa challenge (Supplementary Figures 2A, B) and challenge with Aa extended the lifespan of neutrophils (Supplementary Figure 2C) up to 24 h. Taken together, our results demonstrate that Aa stimulates epinephrine release in human neutrophils, depleting storage reservoirs. In addition, the difference in released (Figure 1C) versus stored epinephrine at 2 h (Figure 1D) suggests that epinephrine may be interacting with Aa. The increase of epinephrine levels in cell lysates at later time points of 8 h and 24 h (Figure 1D) suggests that human neutrophils are capable of de novo catecholamine synthesis.
Next, we examined the levels of enzymes involved in catecholamine metabolism. Levels of enzymes involved in catecholamine synthesis—tyrosine hydroxylase and dopamine β-hydroxylase—and levels of enzymes involved in catecholamine inactivation—catechol-o-methyltransferase and monoamine oxidase-A—were measured in cell lysates of human neutrophils exposed to Aa at 2 h, 4 h, 8 h, and 24 h by ELISA. In the absence of Aa, tyrosine hydroxylase levels are significantly low, but after Aa challenge, levels increase significantly with time compared to basal (Figure 2A). In contrast, dopamine β-hydroxylase levels decrease when Aa is present (Figure 2B). The levels of catechol-o-methyltransferase (Figure 2C) and monoamine oxidase-A (Figure 2D) remained comparable to basal, only peaking at 4 h. Catecholamine synthesis enzyme (tyrosine hydroxylase levels) increase at 8 h and 24 h (Figure 2A), whereas inactivation enzymes (Figures 2C, D) decrease at the same time points. These results indicate that Aa challenge induces de novo catecholamine synthesis in human neutrophils.
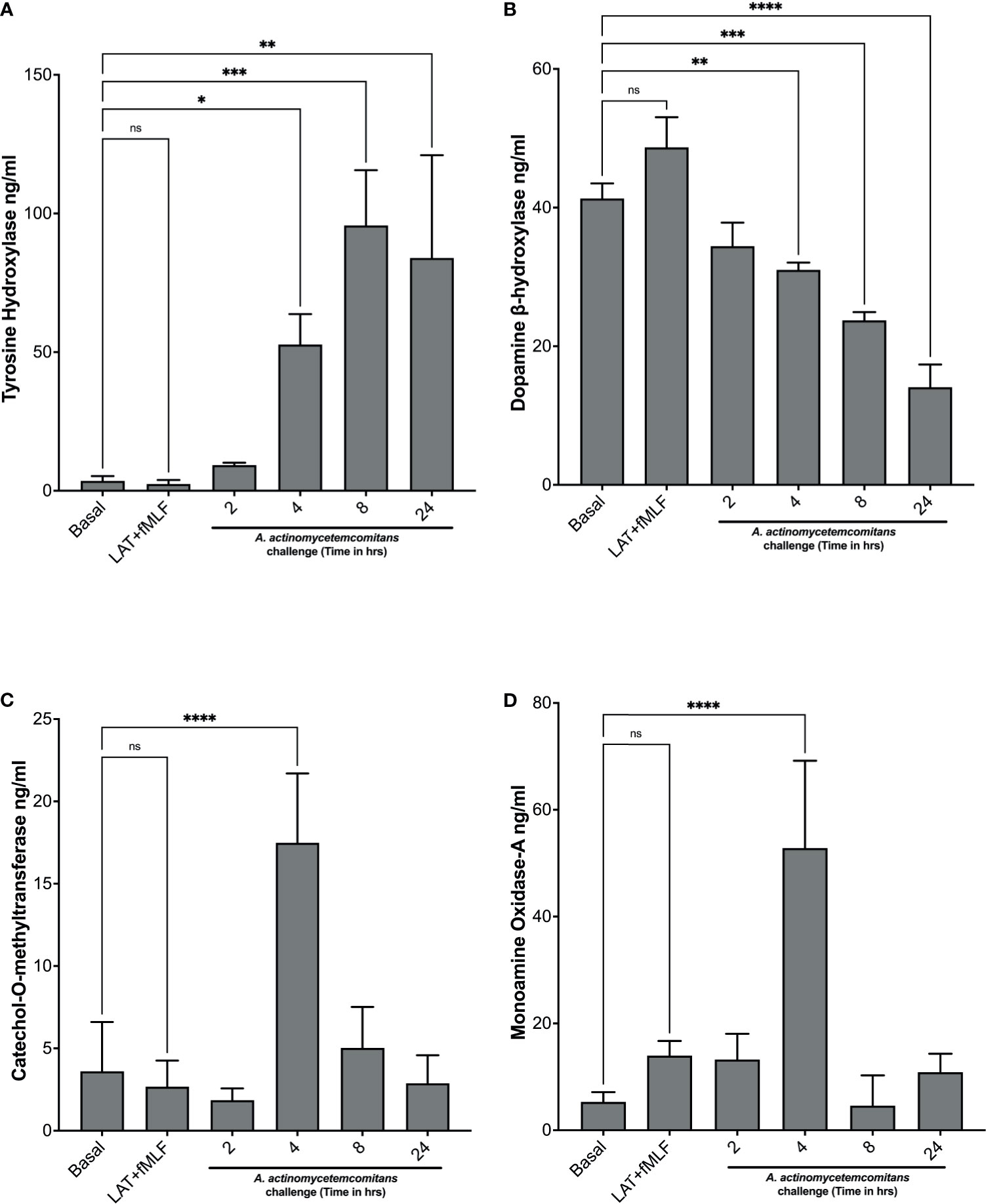
Figure 2 A. actinomycetemcomitans promotes increased levels of enzymes involved in catecholamine metabolism in human neutrophils. Human neutrophils were challenged with A. actinomycetemcomitans (MOI 50) for 2 h, 4 h, 8 h and 24 h; cell lysates were collected, and the levels of tyrosine hydroxylase (A), dopamine β-hydroxylase (B), catechol-O-methyltransferase (C), and monoamine oxidase A (D) were measured by ELISA. Enzyme levels are plotted as the mean ± SD of three independent experiments. Statistical differences among experimental conditions and time points were analyzed by repeated measures one-way ANOVA, followed by Tukey’s post-test. ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
In the infected subgingival pocket, Aa functions in consortium with other bacteria in bacterial communities known as biofilms and Fine et al. showed that A. actinomycetemcomitans positive subjects with bone loss had high levels of the oral pathogen F. alocis (42). In addition, Wang et al. demonstrated that F. alocis accumulation in the oral biofilm was stimulated by the presence of specific strains of Aa (43). To examine if the induction of epinephrine release is specific to Aa, we performed a time course experiment where we investigated the ability of F. alocis to induce epinephrine release in human neutrophils. Human neutrophils were challenged with F. alocis at a MOI of 10 at 1 h, 2 h, and 4 h incubation and epinephrine detection was performed as described above. F. alocis failed to induce significant release of epinephrine (Supplementary Figure 3A). Consistent with this, no significant difference in epinephrine content was detected in cell lysates among experimental conditions (Supplementary Figure 3B). In addition, human neutrophils were challenged with LPS up to 24 h and no significant epinephrine release or difference in epinephrine content was observed (Supplementary Figures 3C, D).
A. actinomycetemcomitans Access Epinephrine by Inducing Exocytosis of Human Neutrophil Granules
In Figure 1, we showed that treatment with latrunculin A followed by stimulation with fMLF, which is known to induce the exocytosis of gelatinase and azurophilic granules, induced epinephrine release by human neutrophils, suggesting that epinephrine may be stored in one or more of the neutrophil granules. To demonstrate this, we first determined the ability of Aa to induce granule exocytosis. Neutrophils were challenged with Aa and granule exocytosis was determined by measuring increase of plasma membrane expression of secretory vesicles, specific granules, and azurophilic granule markers by flow cytometry (see gating strategy and histograms; Supplementary Figure 4) and the extracellular release of MMP-9 by ELISA. Stimulation with Aa induced significant secretory vesicles mobilization by 15 min, as indicated by increased CD35 expression (Figure 3A), which was similar to the exocytosis induced by the positive control fMLF. In addition, gelatinase granule exocytosis, measured by ELISA, was significantly induced after 2 h of Aa challenge (Figure 3B). Specific granule exocytosis, measured by increased CD66b expression, was significantly increased after 4 h of Aa challenge (Figure 3C). Azurophilic granule exocytosis, measured by increased CD63 expression, was significantly increased starting at 8 h post bacterial challenge (Figure 3D). Mobilization of azurophilic granules requires stronger stimulation compared to the other granule subtypes; our results showed that these granules continued to be mobilized by Aa increasingly up to 24 h (Figure 3D). These results demonstrate that Aa challenge induced exocytosis of all four neutrophil granule subtypes.
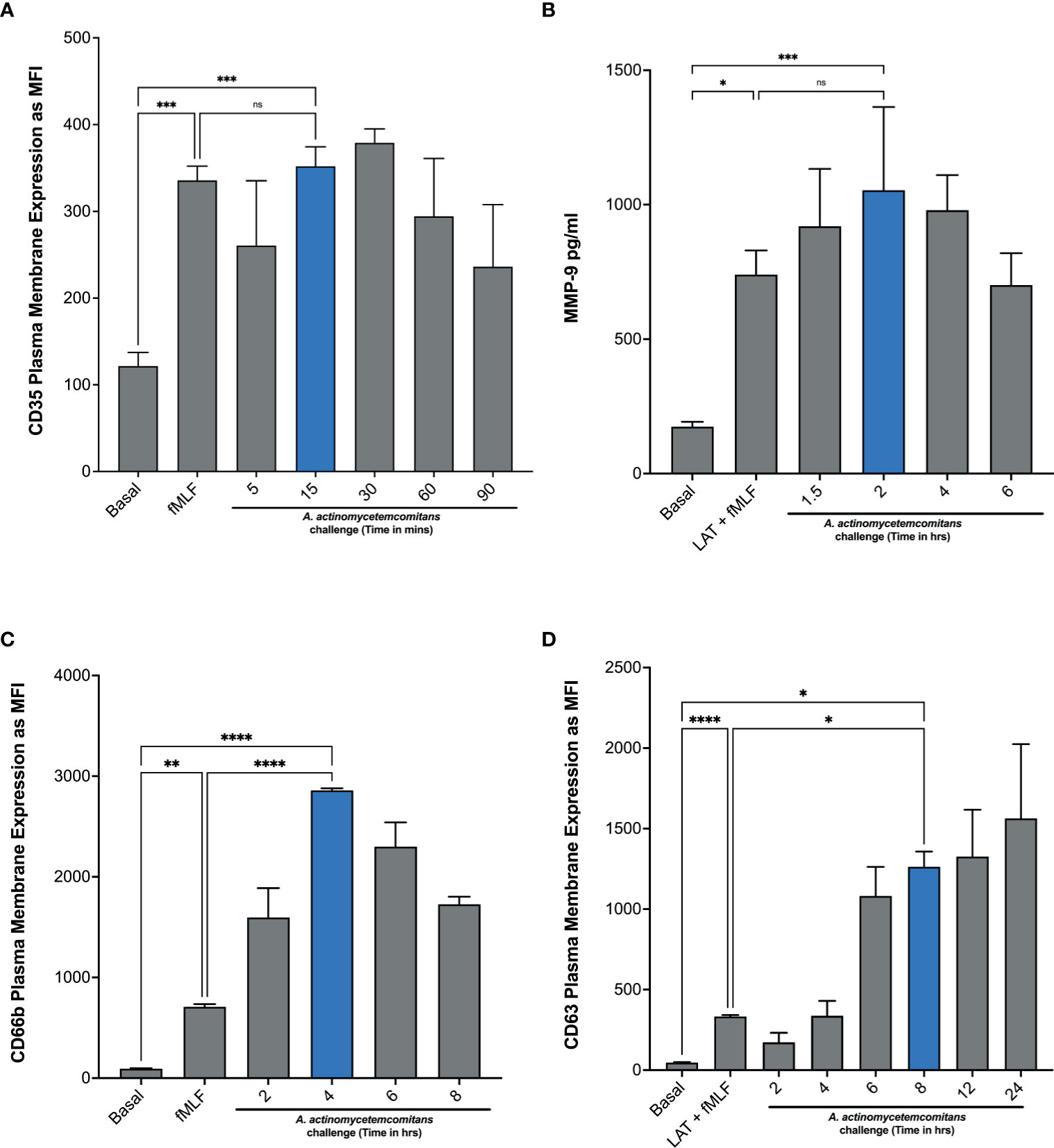
Figure 3 A. actinomycetemcomitans induces exocytosis of all four neutrophil granule subtypes. Induction of neutrophil granule exocytosis was determined by challenging human neutrophils with A. actinomycetemcomitans (MOI 50) for 1.5 h, 6 h, 8 h, and 24 h Increased plasma membrane expression of CD35 (A), CD66b (C), and CD63 (D) was measured to define secretory vesicles, specific granules, and azurophil granule exocytosis, respectively, by flow cytometry. Basal levels and latrunculin (Lat) + fMLF were used as negative and positive control. Granule markers are plotted as the mean channel florescence intensity (MFI) ± SEM of three independent experiments. For gelatinase granule exocytosis, cell supernatants were collected at basal and following each stimulation, and levels of matrix metallopeptidase 9 (MMP-9) were measured by ELISA (B). MMP-9 concentration is plotted as the mean ± SD of three independent experiments. Statistical differences among experimental conditions and time points were analyzed by repeated measures one-way ANOVA, followed by Tukey’s post-test. ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To further characterize if epinephrine release by neutrophils was caused by Aa inducing granule exocytosis, we took advantage of two degranulation inhibitors, TAT-SNAP23, which selectively inhibits secretory vesicles, gelatinase granules, and specific granules (31), and TAT-Syntaxin 4, which blocks exocytosis of all four granule subtypes (40). Human neutrophils were pre-treated with TAT-Syntaxin 4, TAT-SNAP23, or TAT-control peptide fusion proteins, followed by Aa challenge at 15 min, 2 h, 4 h, and 8 h. Granule exocytosis was measured by increased plasma membrane expression of CD35 (secretory vesicles), CD66b (specific granules), and CD63 (azurophilic granules) markers by flow cytometry and the extracellular release of MMP-9 by ELISA. Treatment with TAT-Syntaxin 4 reduced Aa-induced mobilization of secretory vesicles (Figure 4A), gelatinase granules (Figure 4B), specific granules (Figure 4C), and azurophilic granules (Figure 4D). The Aa-induced mobilization of secretory vesicles, gelatinase, and specific granules (Figures 4A–C) was significantly reduced by TAT-SNAP23 pre-treatment. As expected, TAT-SNAP23 failed to inhibit Aa-induced azurophilic granule exocytosis (Figure 4D). Furthermore, cell supernatants were collected at each time point, and granule content release of albumin, lactoferrin, and myeloperoxidase were measured by ELISA. Similar to the results obtained by flow cytometry, challenge of neutrophils with Aa for 15 min induced significant release of albumin, which was significantly reduced by pre-treatment with TAT-SNAP23 and TAT-Syntaxin 4, but not TAT-control (Supplementary Figure 5A). Similarly, Aa challenge for 4 h induced release of lactoferrin, and its release was successfully reduced by TAT-SNAP23 and TAT-Syntaxin 4, but not TAT-control (Supplementary Figure 5B). As expected, neutrophils challenged with Aa for 8 h resulted in significant release of myeloperoxidase, which was inhibited by TAT-Syntaxin 4 but not TAT-SNAP23 or TAT-control (Supplementary Figure 5C).
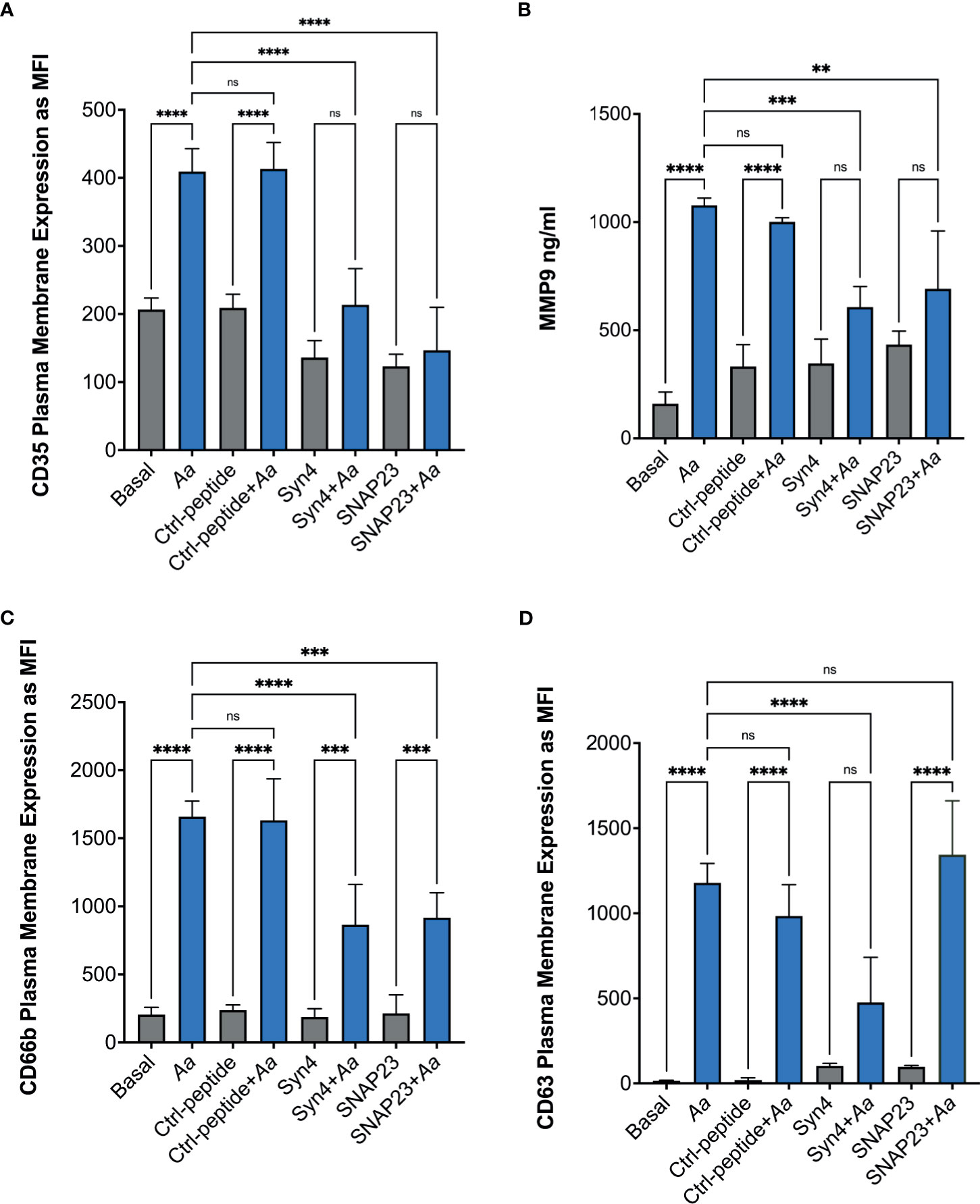
Figure 4 Pre-treatment with TAT fusion proteins prevented A. actinomycetemcomitans-induced neutrophil granule exocytosis. Human neutrophils were unstimulated, challenged with A. actinomycetemcomitans (Aa) at MOI 50, or pre-treated with TAT-Syntaxin 4 (Syn4), TAT-SNAP23 (SNAP23), and TAT-Control peptide (Ctrl-peptide) for 15 min followed by A. actinomycetemcomitans challenge. The peak time point of Aa-induced granule exocytosis was different for each granule subtype. Secretory vesicle peak was at 15 min post-bacterial challenge (A), 2 h for gelatinase granules (B), 4 h for specific granules (C), and 8 h for azurophilic granules (D). Granule markers were measured by flow cytometry and plotted as the mean channel florescence intensity (MFI) ± SEM of four independent experiments (A, C, D). For gelatinase granules, matrix metallopeptidase 9 (MMP-9) was measured from human neutrophil supernatant by ELISA and MMP-9 concentration is plotted as the mean ± SD of four independent experiments (B). Statistical differences among experimental conditions were analyzed by ordinary one-way ANOVA, followed by Tukey’s post-test. ns, not significant, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To further characterize which granule subtype might store epinephrine, supernatants were collected to measure epinephrine levels by ELISA. A. actinomycetemcomitans challenge of human neutrophils at 15 min showed no significant release of epinephrine similar to basal and TAT-control (Supplementary Figure 6A). Moreover, Aa challenge at 2 h induced significant release of epinephrine in human neutrophils; pre-treatment of TAT-Syntaxin 4 was able to inhibit epinephrine release but TAT-SNAP23 failed to inhibit release (Supplementary Figure 6B). On the other hand, only pre-treatment with TAT-Syntaxin 4, but not TAT-SNAP23, blocked Aa-induced epinephrine released after 4 h (Figure 5A) and 8 h (Figure 5B) of bacterial challenge. Since TAT-SNAP23 does not inhibit azurophilic granule exocytosis, these results demonstrate that epinephrine is stored in azurophilic granules. Furthermore, this shows that epinephrine release by neutrophils is linked to granule exocytosis.
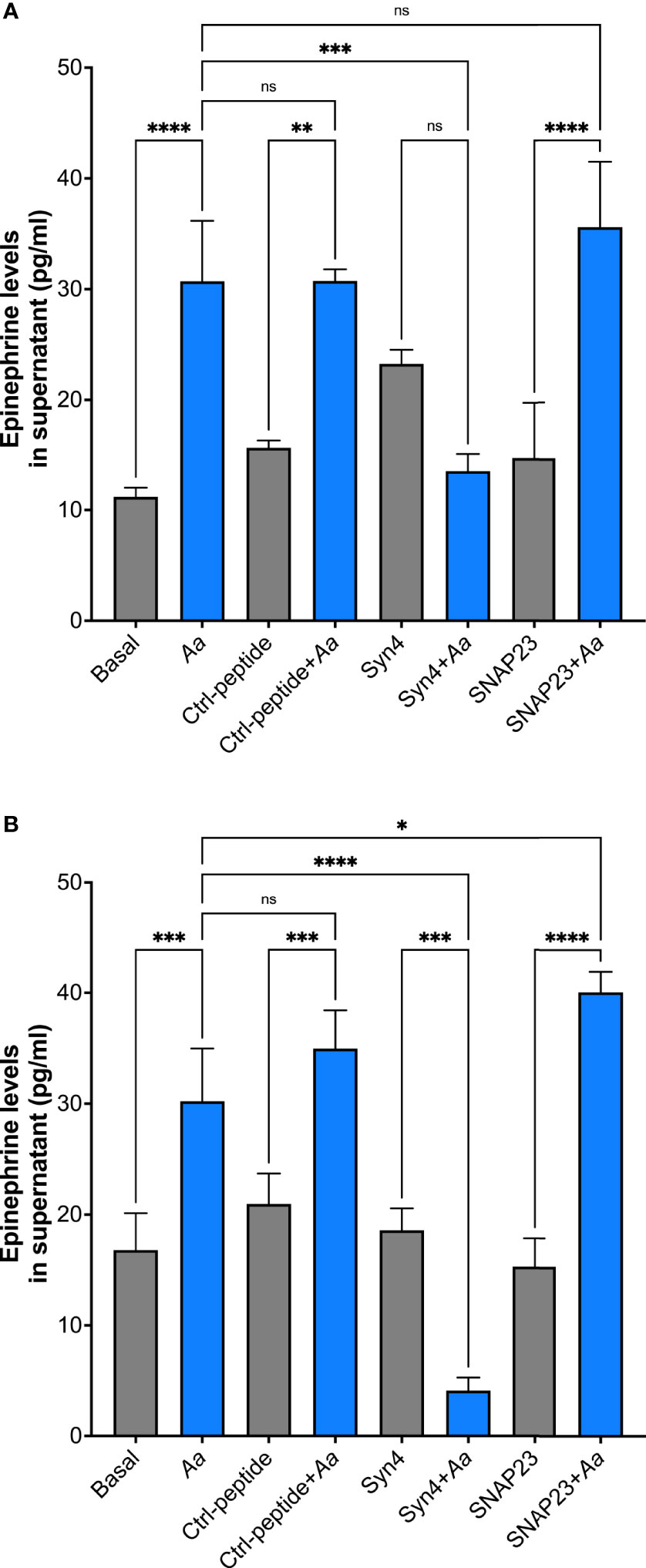
Figure 5 Human neutrophils store epinephrine in azurophilic granules. Epinephrine storage location was determined by pre-treating human neutrophils with TAT-Syntaxin 4 (Syn4), TAT-SNAP23 (SNAP23), or TAT-Ctrl (Ctrl) for 15 min, followed by a 4-h challenge with A. actinomycetemcomitans at MOI 50 (A) or 8 h (B). Epinephrine concentrations are plotted as the mean ± SD of three independent experiments. Statistical differences among experimental conditions were analyzed by ordinary one-way ANOVA, followed by Tukey’s post-test. ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
A. actinomycetemcomitans Interacts With Epinephrine in a QseC-Dependent Manner
To determine if Aa interacts with epinephrine, increasing CFUs of Aa were incubated with a fixed concentration of recombinant epinephrine. Epinephrine interaction with Aa increased with increasing Aa CFUs (Figure 6A). Previous work in our lab demonstrated that QseBC was required for Aa biofilm growth and virulence (25) and that the periplasmic domain of QseC was required for activation of the TCS by catecholamines and iron (6). Therefore, we performed epinephrine dose interaction assays using various Aa strains deficient for QseC. Interaction of epinephrine was diminished by deletion of the QseC sensor (ΔqseC) and in the strain expressing QseC with an in-frame deletion of the periplasmic domain (qseCΔp) (Figure 6B). The ability of epinephrine to interact with Aa was restored when the qseC gene was complemented into the QseC deletion mutant (ΔqseC-comp) (Figure 6B). These results show that Aa interaction with epinephrine requires QseC and is dependent on the QseC periplasmic domain.
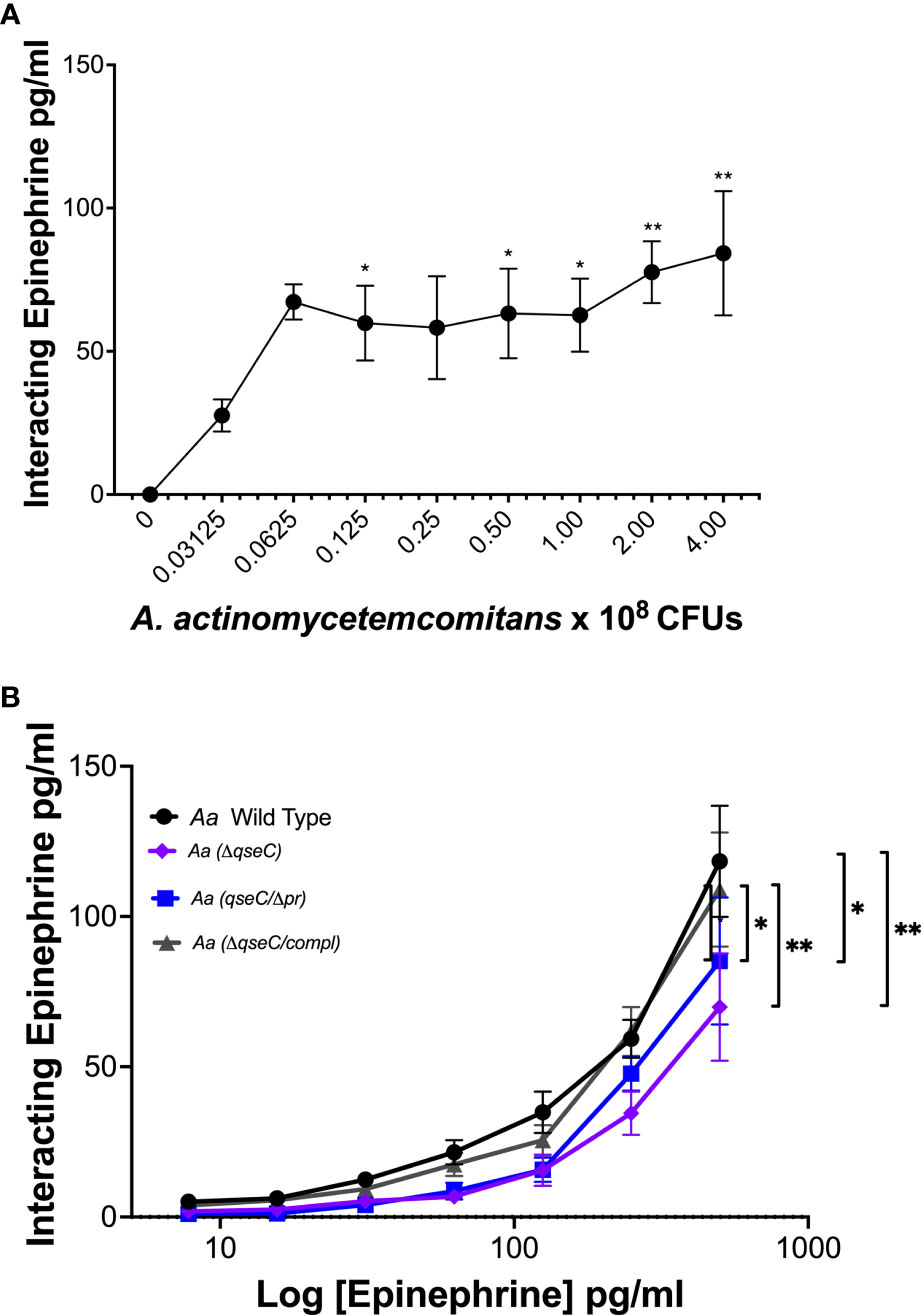
Figure 6 A. actinomycetemcomitans interacts with epinephrine in a QseC-dependent manner. The interaction of epinephrine and A. actinomycetemcomitans was determined by incubation of increasing colony-forming units (CFUs) of A. actinomycetemcomitans with 250 pg/ml of recombinant epinephrine. Associated epinephrine was plotted against increasing CFUs of Aa (A). Requirement of QseC for epinephrine interaction with A. actinomycetemcomitans was determined by incubation of the bacterium with increasing concentrations of recombinant epinephrine (0–500 pg/ml) (B). Epinephrine levels were measured by ELISA and plotted as the mean ± SD of five independent experiments. Statistical differences among experimental conditions were analyzed by ordinary one-way ANOVA (A) or two-way ANOVA (B), followed by Tukey’s post-test. *p < 0.05. **p < 0.01.
Epinephrine From Human Neutrophils Promotes A. actinomycetemcomitans Growth and qseBC Expression
To determine if the epinephrine levels released by human neutrophils could promote bacterial growth, the supernatants and cell lysates were collected from cells stimulated with latrunculin A and fMLF and used to assess Aa growth and expression of the qseBC operon. A. actinomycetemcomitans was grown anaerobically in CDM alone or supplemented individually or in combination with neutrophil supernatant, neutrophil cell lysate, iron (100 μM), or epinephrine (50 μM). Growth in CDM supplemented with neutrophil lysate or epinephrine alone was comparable to the unsupplemented CDM control (Figure 7A). Supplementation with iron, either alone or in combination with epinephrine, resulted in a significant induction of Aa growth (Figure 7A). Furthermore, medium supplemented with neutrophil supernatant alone promoted growth, which was increased when supplemented with iron (Figure 7A). On the other hand, neutrophil cell lysate supplemented with iron promoted growth similar to epinephrine + iron or neutrophil supernatant alone (Figure 7A). Supplementation with epinephrine, neutrophil supernatant, or lysate alone failed to induce expression of QseBC (Figure 7B). QseBC expression was significantly induced by iron and a further significant increase in expression occurred by supplementation with a combination of iron with epinephrine or neutrophil supernatant (Figure 7B). Unexpectedly, supplementation with iron and neutrophil lysate suppressed QseBC expression (Figure 7B). This experiment was performed under aerobic conditions with similar results (data not shown). Here, we show that Aa makes use of neutrophil-derived epinephrine to promote bacterial growth and expression of the QseBC in the anaerobic environment.
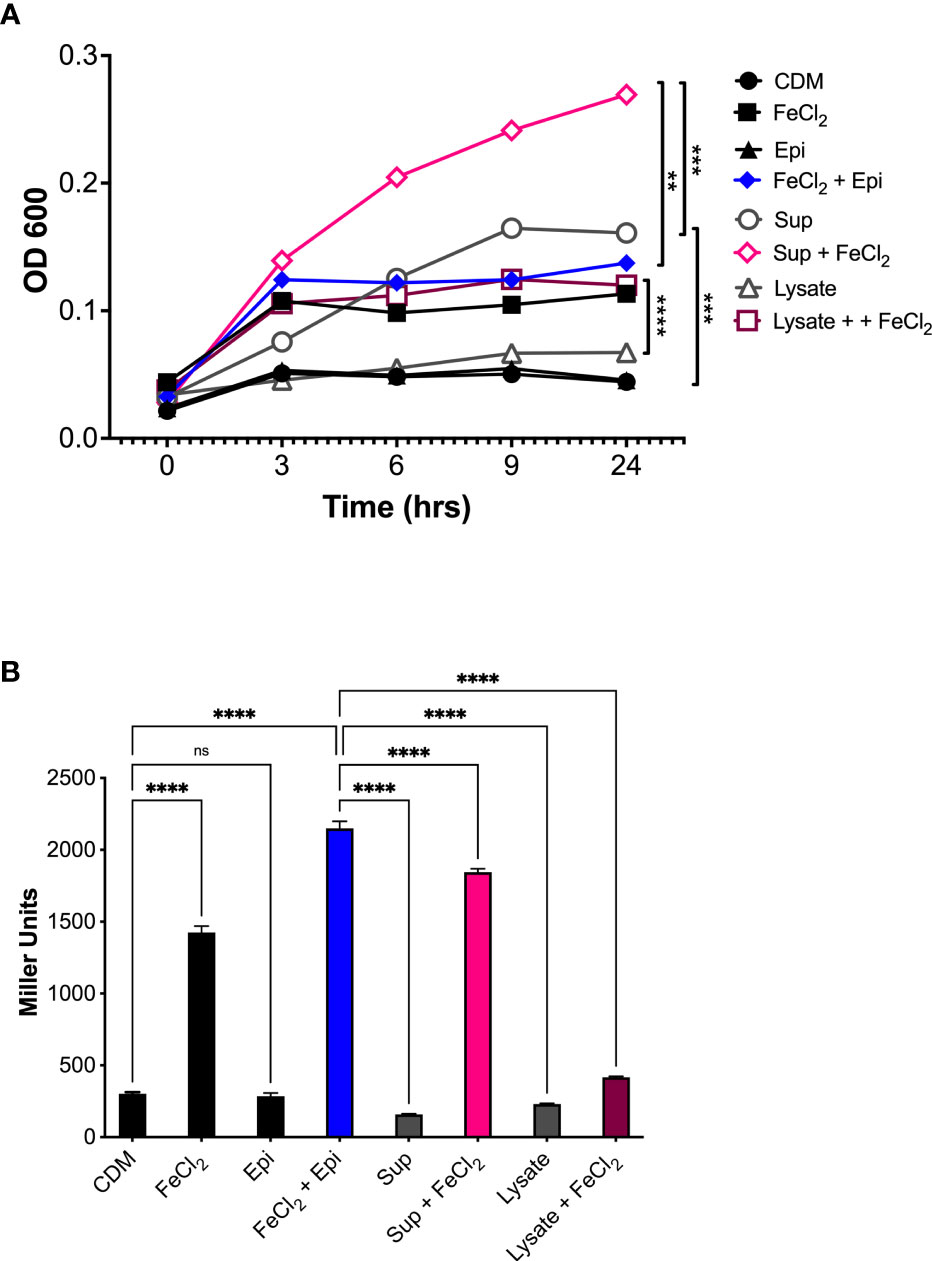
Figure 7 Host-derived epinephrine promotes A. actinomycetemcomitans growth and qseBC expression. The supernatants and lysates from human neutrophils stimulated with latrunculin A and fMLF were collected and used as supplement for chemically defined media (CDM). A. actinomycetemcomitans was grown under anaerobic conditions in CDM alone or supplemented with human neutrophil supernatant (Sup), cell lysate (Lysate), iron (FeCl2, 100 μM), or Epinephrine (Epi, 50 μM). At designated times, growth was measured by determining the optical density at 600 nm (OD600) (A). A. actinomycetemcomitans strain (that harbors the qseBC promoter-lacZ reporter plasmid pDJR29) was grown under anaerobic conditions in CDM alone or supplemented with human neutrophil supernatant (Sup), cell lysate (Lysate), iron (FeCl2, 100 μM), or Epinephrine (Epi, 50 μM). Expression of the qseBC operon was determined by measuring β-galactosidase activity after 24 h (B). The OD600 measurements for growth were plotted as the mean ± SEM of three independent experiments (A). The expression of the qseBC operon is presented in miller units as the mean ± SD of three independent experiments (B). Statistical differences among experimental conditions were analyzed by two-way ANOVA (A) or ordinary one-way ANOVA (B), followed by Tukey’s post-test. ns, not significant, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
The newly emerging field of microbial endocrinology (16, 44) studies the communication or inter-kingdom signaling (45) that has evolved between microorganisms and their hosts. The study of inter-kingdom signaling includes the study of hormonal communication, where microorganisms respond to the host neurohormones as environmental cues to regulate the expression of genes necessary for virulence and survival (44, 46–49). There are reports of host–pathogen crosstalk involving Aa biofilm sequestering and taking up IL-1β (50). Additionally, it has been shown previously that Aa expresses a cytokine binding receptor, BilRI, that allows it to benefit from cytokine release to promote biofilm formation and bacterial growth (51, 52). Previous work by our group demonstrated that the presence of catecholamines and iron in vitro promoted Aa growth and regulated the expression of genes necessary for virulence and survival in the anaerobic environment (6). However, those studies did not determine if similar effects occurred in vivo. Based on work by others, it is known that there is a large infiltration of neutrophils at the subgingival pocket during periodontitis (26, 53, 54) and murine neutrophils have been shown to release and synthesize catecholamines when stimulated with LPS (27). Therefore, we considered human neutrophils as a potential source of catecholamines for Aa. In our ex vivo studies, we showed that Aa induces epinephrine release in human neutrophils depleting stored catecholamine levels and indirectly inducing catecholamine synthesis. As a novel finding, we identified azurophilic granules as the storage location of epinephrine in human neutrophils and that Aa gains access to epinephrine by inducing granule exocytosis. Finally, we proved that host-derived epinephrine promoted Aa growth and QseBC expression under anaerobic conditions.
Marino et al. (28) detected catecholamines in human peripheral blood mononuclear cell medium, but the report failed to determine if these catecholamines were released and, if so, what stimuli induced release. Likewise, catecholamine release has been shown in other immune cells such as rat and human lymphocytes (55, 56). Although there is extensive work done in murine neutrophils, there was no report of human neutrophils releasing catecholamines and identified stimuli. Here, we present for the first time that human neutrophils release epinephrine when stimulated with latrunculin A + fMLF and when challenged with Aa, but not by F. alocis, which occupies the same oral niche as Aa (see below). These observations support previous findings by Parantainen et al. (39), the first to identify endogenous catecholamines in human neutrophils. However, at the time, the source of these catecholamines was not determined or if neutrophils participated in catecholamine synthesis. Later, further evidence of catecholamines present intracellularly in human neutrophils was found and enzymes involved in catecholamine metabolism were detected by high-performance liquid chromatography (HPLC) (57). In our experiments, we observed that the levels of epinephrine in cell lysates increased after 8 h and 24 h of bacterial challenge, suggesting de novo catecholamine synthesis by human neutrophils. This was confirmed by the presence of significant levels of tyrosine hydroxylase levels in the presence of Aa. In contrast, dopamine β-hydroxylase levels decreased when Aa was present. Tyrosine hydroxylase catalyzes the first and rate-limiting step of the catecholamine synthesis pathway, explaining larger amounts of this enzyme. A potential reason for this is that dopamine β-hydroxylase catalyzes dopamine into norepinephrine, and not all dopamine will be catalyzed, explaining the decrease in enzyme levels. We also found significant levels at 4 h of the catecholamine inactivating enzymes catechol-o-methyltransferase and monoamine oxidase-A. This correlates with the increased epinephrine release induced by Aa at 4 h; as epinephrine is released, demand for degradation increases. Similarly, as stored epinephrine is depleted, there is demand for catecholamine synthesis at later time points, and inactivation enzymes decrease at this time. A potential limitation of our study is that granule exocytosis experiments were done under aerobic conditions and not under low oxygen or anaerobic conditions. The periodontal pocket is considered a hypoxic environment, with decreasing oxygen levels as the depth of the periodontal pocket increases, becoming increasingly hypoxic during infection due to activation of the NADPH oxidase complex (58, 59). Therefore, it is likely that neutrophils are exposed to hypoxic conditions in vivo when recruited to the gingival pocket. Although we did not perform experiments with both Aa and neutrophils under anaerobic conditions, preliminary work in our lab with neutrophils challenged with F. alocis showed no difference in the lifespan of neutrophils grown aerobically or anaerobically. Additionally, untreated neutrophils under anaerobic conditions resulted in enhanced viability compared to cells grown aerobically. Though we infer that human neutrophils will undergo granule exocytosis under hypoxic conditions, this remains to be tested in the future.
We demonstrated that Aa induces the mobilization of secretory vesicles, gelatinase granules, specific granules, and azurophilic granules in human neutrophils. These findings align with previous observations that identified enhanced surface expression of CD63 and CD66b in oral neutrophils, characteristic of neutrophils that are undergoing active granule exocytosis (60, 61). Johansson et al. (62) showed that purified leukotoxin A induces release of granule contents from human neutrophils, and the authors observed increased expression of CD63 and CD66b along with the release of elastase and lactoferrin. In our ex vivo model, we obtained similar observations by challenging human neutrophils with a low leukotoxic strain of Aa compared to high concentrations of purified leukotoxin. Furthermore, through selective inhibition of granule exocytosis, we identified azurophilic granules as the storage location for epinephrine in human neutrophils. To the best of our knowledge, this observation serves as the first evidence of the storage location of epinephrine in human neutrophils. Human neutrophils are recruited out of circulation by interaction with neutrophil-specific adhesion molecules on the blood vessel wall. This interaction in addition to chemoattractant gradient such as IL-8 promotes the mobilization of secretory vesicles. Once neutrophils cross through the epithelium, they continue to receive signals such as increasing IL-8 gradient potentially released by gingiva epithelial cells and formyl peptides from invading bacteria, resulting in mobilization of gelatinase granules. The primed neutrophil arrives at the infected subgingival pocket where there are overwhelming inflammatory signals that may induce the release of specific and azurophilic granules. Alternatively, these granules can fuse with vacuoles containing phagocytized bacteria, forming a phagolysosome (63–65). Thus, Aa may induce the release of azurophilic granules at the subgingival pocket to gain access to epinephrine and in turn the release of potent antimicrobial protein contents of these granules may contribute to tissue damage and disease progression.
When human neutrophils were challenged with Aa for 2 h, we expected to see a high release of epinephrine, due to depleted epinephrine levels in cell lysate. A plausible explanation to the difference in levels of epinephrine released versus stored at 2 h is that some of the epinephrine released could be interacting with Aa and therefore no longer in the supernatant. Epinephrine interaction with Aa increased with increasing Aa CFUs. Furthermore, Aa interaction with epinephrine was found to require QseC, in a QseC periplasmic domain-dependent manner. This supports previous findings made by our group that identified catecholamines (norepinephrine and epinephrine) as the signal that is sensed by QseC and that the periplasmic domain of QseC is required for qseBC operon expression in the presence of catecholamines and iron (6). It also strengthens the importance of QseC in recognizing catecholamines for growth and virulence of Aa (25). The exact details of the interaction of Aa with epinephrine remains unclear, but the discussed results make us conclude that the QseC periplasmic domain is required. As for a receptor for epinephrine, when Aa is grown in the presence of catecholamines and iron, the expression of the enterobactin operon, which contains a gene for FepA, remains unchanged (6). In E. coli, FepA is an outer membrane protein that serves as a receptor for the ferric enterobactin siderophore (66, 67). The uptake of this siderophore into the bacterial cells is TonB-dependent (67, 68). We performed different experiments to determine if FepA was required for Aa interaction with epinephrine using a ΔfepA mutant Aa strain and we found no difference in interaction compared with the wild-type strain (data not shown). Moreover, as an alternative for iron entry into the bacterial cells, there are the ferrous iron transport system (Feo) and periplasmic-binding protein-dependent transport (PBT) (69–72). These are predominant iron transport systems at low oxygen conditions when ferrous iron is stable and is more prevalent than ferric iron (17). In the case the ferric iron is predominant, it will be chelated by ferric iron reductases and then uptake is mediated by ferrous permeases (73, 74). Rhodes et al. (19) demonstrated that Aa makes use of inorganic iron to grow under chelated conditions and proposed that Aa may acquire iron through the expression of systems which function independently of an outer membrane receptor or TonB-dependent transport. The elucidation of which TonB and receptor independent iron transport system is used by Aa will require further investigation.
Under anaerobic conditions, we showed that Aa growth in limited media is promoted in the presence of neutrophil supernatant or cell lysate supplemented with iron. Likewise, supplementation with neutrophil supernatant and iron induced expression of QseBC. Interestingly, neutrophil cell lysate alone or supplemented with iron QseBC expression was suppressed. Human neutrophil lysates contain all internal contents of neutrophils, including components such as calprotectin. Calprotectin is a metal binding protein, abundant in neutrophil cytoplasm (75, 76). Another protein that is found in specific granules is the neutrophil gelatinase-associated lipocalin. This protein scavenges iron from siderophores, and it has been shown to remove iron from catecholamines as well (77). These proteins can be sequestering the supplemented iron competing with epinephrine present in lysate, making it unavailable to Aa and consequentially suppressing the induction of the QseBC.
In Aa-positive subjects, F. alocis was detected at high levels (42) and F. alocis accumulation in the oral biofilm has been shown to be stimulated by the presence of specific strains of Aa (43). Based on our experiments, we determined that F. alocis does not induce release of epinephrine in human neutrophils. This result goes in hand with previous findings that F. alocis does not induce the exocytosis of azurophilic granules (38, 78). In the case that F. alocis is found to be catecholamine-responsive, it will suggest that it may be benefiting from the ability of Aa to induce epinephrine release. However, at present, there is no report of F. alocis being influenced in some way by catecholamines. A. actinomycetemcomitans has also been found to be associated to Fusobacterium nucleatum, whose growth is increased by hormones like epinephrine and norepinephrine (21); Lactobacillus spp. expresses transporter systems for uptake of catecholamines (79) and other catecholamine-responsive species such as Prevotella spp. and Leptotrichia (16, 80, 81). Some of these bacteria have been shown to lack the ability to produce siderophores as is the case of Aa (82, 83). Therefore, it is likely that these bacteria work as a team to thrive in the subgingival pocket, where the role of Aa may be to induce catecholamine (i.e., epinephrine) release by the infiltrating human neutrophils.
In conclusion, our ex vivo experiments demonstrated that human neutrophils store and release epinephrine upon stimulation with latrunctin A + fMLF. We show that Aa can induce epinephrine release in human neutrophils, consequently depleting epinephrine storage and promoting catecholamine metabolism. Furthermore, one of the most important contributions of this study was the finding of azurophilic granules as the storage location of epinephrine in human neutrophils and that Aa gains access to it by inducing granule exocytosis. Additionally, we found that epinephrine interaction with Aa depends on QseC expression and requires the QseC periplasmic domain. Finally, human-neutrophil-derived epinephrine supplemented with iron promoted Aa growth and QseBC expression under anaerobic conditions. The presented findings supply evidence towards the growing field of microbial endocrinology by contributing to the understanding of the crosstalk between bacteria and the host endocrine system. Furthermore, it expands the knowledge of the role of stress hormones in periodontal disease and potentially other chronic inflammatory diseases.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board of the University of Louisville. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
HO performed the experiments and participated in the study design, data analysis and interpretation, and drafting of the manuscript. SU and DD equally participated in the study design, data interpretation, critical revision of the manuscript, and study supervision. DD obtained funding for the study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Integrated Programs in Biomedical Sciences Graduate Fellowship at the University of Louisville School of Medicine, by the National Institute of Dental and Craniofacial Research (NIDCR) grants RO1DE014605 (DD/SU) and R01DE024509 (SU), and by the Ruth L. Kirschstein National Research Service Award by the NIDCR F31DE027585 (HO).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors want to thank Terri Manning for neutrophil isolation. For the preparation and testing of the TAT fusion proteins, we thank Shweta Tandon. For the creation of the QseC mutants, we want to thank Asención Torres-Escobar and María Dolores Juárez-Rodríguez.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.707096/full#supplementary-material
References
1. Hajishengallis G. Periodontitis: From Microbial Immune Subversion to Systemic Inflammation. Nat Rev Immunol (2015) 15:30–44. doi: 10.1038/nri3785
2. Konkel JE, O’Boyle C, Krishnan S. Distal Consequences of Oral Inflammation. Front Immunol (2019) 10:1403. doi: 10.3389/fimmu.2019.01403
3. Kesic L, Petrovic M, Obradovic R, Pejcic A. The Importance of Aggregatibacter actinomycetemcomitans in the Etiology of Periodontal Disease. Acta Med Medianae (2009) 48(3):35–7.
4. Raja M, Ummer F, Dhivakar CP. Aggregatibacter actinomycetemcomitans - A Tooth Killer? J Clin Diagn Res (2014) 8:ZE13–16. doi: 10.7860/JCDR/2014/9845.4766
5. Gholizadeh P, Pormohammad A, Eslami H, Shokouhi B, Fakhrzadeh V, Kafil HS. Oral Pathogenesis of Aggregatibacter actinomycetemcomitans. Microb Pathog (2017) 113:303–11. doi: 10.1016/j.micpath.2017.11.001
6. Weigel WA, Demuth DR, Torres-Escobar A, Juarez-Rodriguez MD. Aggregatibacter actinomycetemcomitans QseBC Is Activated by Catecholamines and Iron and Regulates Genes Encoding Proteins Associated With Anaerobic Respiration and Metabolism. Mol Oral Microbiol (2015) 30:384–98. doi: 10.1111/omi.12101
7. Rahamat-Langendoen JC, van Vonderen MG, Engstrom LJ, Manson WL, van Winkelhoff AJ, Mooi-Kokenberg E. Brain Abscess Associated With Aggregatibacter actinomycetemcomitans: Case Report and Review of Literature. J Clin Periodontol (2011) 38:702–6. doi: 10.1111/j.1600-051X.2011.01737.x
8. Townsend TR. Urinary Tract Infection Due to Actinobacillus actinomycetemcomitans. JAMA: J Am Med Assoc (1969) 210:558. doi: 10.1001/jama.1969.03160290110041
9. Yew HS, Chambers ST, Roberts SA, Holland D, Julian KA, Raymond NJ, et al. Association Between HACEK Bacteraemia and Endocarditis. J Med Microbiol (2014) 63:892–5. doi: 10.1099/jmm.0.070060-0
10. Zhang T, Kurita-Ochiai T, Hashizume T, Du Y, Oguchi S, Yamamoto M. Aggregatibacter actinomycetemcomitans Accelerates Atherosclerosis With an Increase in Atherogenic Factors in Spontaneously Hyperlipidemic Mice. FEMS Immunol Med Microbiol (2010) 59:143–51. doi: 10.1111/j.1574-695X.2010.00674.x
11. Robertson PB, Lantz M, Marucha PT, Kornman KS, Trummel CL, Holt SC. Collagenolytic Activity Associated With Bacteroides Species and Actinobacillus actinomycetemcomitans. J Periodontal Res (1982) 17:275–83. doi: 10.1111/j.1600-0765.1982.tb01154.x
12. Jinadasa RN, Bloom SE, Weiss RS, Duhamel GE. Cytolethal Distending Toxin: A Conserved Bacterial Genotoxin That Blocks Cell Cycle Progression, Leading to Apoptosis of a Broad Range of Mammalian Cell Lineages. Microbiology (2011) 157:1851–75. doi: 10.1099/mic.0.049536-0
13. Brogan JM, Lally ET, Poulsen K, Kilian M, Demuth DR. Regulation of Actinobacillus Actinomycetemcomitans Leukotoxin Expression: Analysis of the Promoter Regions of Leukotoxic and Minimally Leukotoxic Strains. Infect Immun (1994) 62:501–8. doi: 10.1128/iai.62.2.501-508.1994
14. Hritz M, Fisher E, Demuth DR. Differential Regulation of the Leukotoxin Operon in Highly Leukotoxic and Minimally Leukotoxic Strains of Actinobacillus actinomycetemcomitans. Infect Immun (1996) 64:2724–9. doi: 10.1128/iai.64.7.2724-2729.1996
15. Freestone PP, Lyte M, Neal CP, Maggs AF, Haigh RD, Williams PH. The Mammalian Neuroendocrine Hormone Norepinephrine Supplies Iron for Bacterial Growth in the Presence of Transferrin or Lactoferrin. J Bacteriol (2000) 182:6091–8. doi: 10.1128/jb.182.21.6091-6098.2000
16. Sandrini S, Aldriwesh M, Alruways M, Freestone P. Microbial Endocrinology: Host-Bacteria Communication Within the Gut Microbiome. J Endocrinol (2015) 225:R21–34. doi: 10.1530/JOE-14-0615
17. Andrews SC, Robinson AK, Rodríguez-Quiñones F. Bacterial Iron Homeostasis. FEMS Microbiol Rev (2003) 27:215–37. doi: 10.1016/S0168-6445(03)00055-X
18. Chatterjee A, O’Brian MR. Rapid Evolution of a Bacterial Iron Acquisition System. Mol Microbiol (2018) 108:90–100. doi: 10.1111/mmi.13918
19. Rhodes ER, Menke S, Shoemaker C, Tomaras AP, McGillivary G, Actis LA. Iron Acquisition in the Dental Pathogen Actinobacillus actinomycetemcomitans: What Does It Use as a Source and How Does It Get This Essential Metal? Biometals (2007) 20:365. doi: 10.1007/s10534-006-9058-3
20. Graziano TS, Closs P, Poppi T, Franco GC, Cortelli JR, Groppo FC, et al. Catecholamines Promote the Expression of Virulence and Oxidative Stress Genes in Porphyromonas gingivalis. J Periodontal Res (2014) 49:660–9. doi: 10.1111/jre.12148
21. Jentsch HF, Marz D, Kruger M. The Effects of Stress Hormones on Growth of Selected Periodontitis Related Bacteria. Anaerobe (2013) 24:49–54. doi: 10.1016/j.anaerobe.2013.09.001
22. Roberts A, Matthews JB, Socransky SS, Freestone PPE, Williams PH, Chapple ILC. Stress and the Periodontal Diseases: Effects of Catecholamines on the Growth of Periodontal Bacteria In Vitro. Oral Microbiol Immunol (2002) 17:296–303. doi: 10.1034/j.1399-302x.2002.170506.x
23. Winston JL CC, Neiders ME, Dyer DW. Membrane Protein Expression by Actinobacillus actinomycetemcomitans in Response to Iron Availability. J Dent Res (1993) 72:1366–73. doi: 10.1177/00220345930720100501
24. Juarez-Rodriguez MD, Torres-Escobar A, Demuth DR. ygiW and qseBC are Co-Expressed in Aggregatibacter actinomycetemcomitans and Regulate Biofilm Growth. Microbiology (2013) 159:989–1001. doi: 10.1099/mic.0.066183-0
25. Novak EA, Shao H, Daep CA, Demuth DR. Autoinducer-2 and QseC Control Biofilm Formation and In Vivo Virulence of Aggregatibacter actinomycetemcomitans. Infect Immun (2010) 78:2919–26. doi: 10.1128/IAI.01376-09
26. Delima AJ, Van Dyke TE. Origin and Function of the Cellular Components in Gingival Crevice Fluid. Periodontol 2000 (2003) 31:55–76. doi: 10.1034/j.1600-0757.2003.03105.x
27. Flierl MA, Rittirsch D, Nadeau BA, Chen AJ, Vidya Sarma J, Zetoune FS, et al. Phagocyte-Derived Catecholamines Enhance Acute Inflammatory Injury. Nature (2007) 449:721–5. doi: 10.1038/nature06185
28. Marino F, Cosentino M, Bombelli R, Ferrari M, Lecchini S, Frigo G, et al. Endogenous Catecholamine Synthesis, Metabolism, Storage, and Uptake in Human Peripheral Blood Mononuclear Cells. Exp Hematol (1999) 27:489–95. doi: 10.1016/s0301-472x(98)00057-5
29. Sandrini SM, Shergill R, Woodward J, Muralikuttan R, Haigh RD, Lyte M, et al. Elucidation of the Mechanism by Which Catecholamine Stress Hormones Liberate Iron From the Innate Immune Defense Proteins Transferrin and Lactoferrin. J Bacteriol (2010) 192:587–94. doi: 10.1128/JB.01028-09
30. Verbrugghe E, Boyen F, Gaastra W, Bekhuis E, Leyman B, Van Parys A, et al. The Complex Interplay Between Stress and Bacterial Infections in Animals. Veterinary Microbiol (2012) 155:115–27. doi: 10.1016/j.vetmic.2011.09.012
31. Uriarte SM, Rane MJ, Luerman GC, Barati MT, Ward RA, Nauseef WM, et al. Granule Exocytosis Contributes to Priming and Activation of the Human Neutrophil Respiratory Burst. J Immunol (2011) 187:391–400. doi: 10.4049/jimmunol.1003112
32. Fine DH, Markowitz K, Furgang D, Fairlie K, Ferrandiz J, Nasri C, et al. Aggregatibacter Actinomycetemcomitans and its Relationship to Initiation of Localized Aggressive Periodontitis: Longitudinal Cohort Study of Initially Healthy Adolescents. J Clin Microbiol (2007) 45:3859–69. doi: 10.1128/JCM.00653-07
33. Tan KS, Woo CH, Ong G, Song KP. Prevalence of Actinobacillus actinomycetemcomitans in an Ethnic Adult Chinese Population. J Clin Periodontol (2001) 28:886–90. doi: 10.1034/j.1600-051x.2001.028009886.x
34. Czuprynski CJ, Noel EJ, Ortiz-Carranza O, Srikumaran S. Activation of Bovine Neutrophils by Partially Purified Pasteurella Haemolytica Leukotoxin. Infect Immun (1991) 59:3126–33. doi: 10.1128/iai.59.9.3126-3133.1991
35. Juarez-Rodriguez MD, Torres-Escobar A, Demuth DR. Construction of New Cloning, lacZ Reporter and Scarless-Markerless Suicide Vectors for Genetic Studies in Aggregatibacter actinomycetemcomitans. Plasmid (2013) 69:211–22. doi: 10.1016/j.plasmid.2013.01.002
36. Socransky SS, Dzink JL, Smith CM. Chemically Defined Medium for Oral Microorganisms. J Clin Microbiol (1985) 22:303–5. doi: 10.1128/JCM.22.2.303-305.1985
37. Armstrong CL, Klaes CK, Vashishta A, Lamont RJ, Uriarte SM. Filifactor alocis Manipulates Human Neutrophils Affecting Their Ability to Release Neutrophil Extracellular Traps Induced by PMA. Innate Immun (2018) 24:210–20. doi: 10.1177/1753425918767507
38. Armstrong CL, Miralda I, Neff AC, Tian S, Vashishta A, Perez L, et al. Filifactor alocis Promotes Neutrophil Degranulation and Chemotactic Activity. Infect Immun (2016) 84:3423–33. doi: 10.1128/iai.00496-16
39. Parantainen J, Alanko J, Moilanen E, Metsa-Ketela T, Asmawi MZ, Vapaatalo H. Catecholamines Inhibit Leukotriene Formation and Decrease Leukotriene/Prostaglandin Ratio. Biochem Pharmacol (1990) 40:961–6. doi: 10.1016/0006-2952(90)90480-9
40. McLeish KR, Uriarte SM, Tandon S, Creed TM, Le J, Ward RA. Exocytosis of Neutrophil Granule Subsets and Activation of Prolyl Isomerase 1 Are Required for Respiratory Burst Priming. J Innate Immun (2013) 5:277–89. doi: 10.1159/000345992
41. Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory (1972).
42. Fine DH, Markowitz K, Fairlie K, Tischio-Bereski D, Ferrendiz J, Furgang D, et al. A Consortium of Aggregatibacter actinomycetemcomitans, Streptococcus parasanguinis, and Filifactor alocis Is Present in Sites Prior to Bone Loss in a Longitudinal Study of Localized Aggressive Periodontitis. J Clin Microbiol (2013) 51:2850–61. doi: 10.1128/JCM.00729-13
43. Wang Q, Wright CJ, Dingming H, Uriarte SM, Lamont RJ. Oral Community Interactions of Filifactor alocis In Vitro. PloS One (2013) 8:e76271. doi: 10.1371/journal.pone.0076271
44. Freestone PP, Sandrini SM, Haigh RD, Lyte M. Microbial Endocrinology: How Stress Influences Susceptibility to Infection. Trends Microbiol (2008) 16:55–64. doi: 10.1016/j.tim.2007.11.005
45. Hughes DT, Sperandio V. Inter-Kingdom Signalling: Communication Between Bacteria and Their Hosts. Nat Rev Microbiol (2008) 6:111–20. doi: 10.1038/nrmicro1836
46. Freestone P. Communication Between Bacteria and Their Hosts. Scientifica (Cairo) (2013) 2013:361073. doi: 10.1155/2013/361073
47. Lyte M. Microbial Endocrinology and Infectious Disease in the 21st Century. Trends Microbiol (2004) 12:14–20. doi: 10.1016/j.tim.2003.11.004
48. Lyte M, Freestone P. Microbial Endocrinology Comes of Age. Microbe (2009) 4:169–76. doi: 10.1128/microbe.4.169.1
49. Lyte M, Freestone PP eds. Microbial Endocrinology: Interkingdom Signaling in Infectious Disease and Health. New York: Springer (2010).
50. Paino A, Ahlstrand T, Nuutila J, Navickaite I, Lahti M, Tuominen H, et al. Identification of a Novel Bacterial Outer Membrane Interleukin-1β-Binding Protein From Aggregatibacter actinomycetemcomitans. PloS One (2013) 8:e70509. doi: 10.1371/journal.pone.0070509
51. Ahlstrand T, Torittu A, Elovaara H, Valimaa H, Pollanen MT, Kasvandik S, et al. Interactions Between the Aggregatibacter actinomycetemcomitans Secretin HofQ and Host Cytokines Indicate a Link Between Natural Competence and Interleukin-8 Uptake. Virulence (2018) 9:1205–23. doi: 10.1080/21505594.2018.1499378
52. Ahlstrand T, Tuominen H, Beklen A, Torittu A, Oscarsson J, Sormunen R, et al. A Novel Intrinsically Disordered Outer Membrane Lipoprotein of Aggregatibacter actinomycetemcomitans Binds Various Cytokines and Plays a Role in Biofilm Response to Interleukin-1β and Interleukin-8. Virulence (2017) 8:115–34. doi: 10.1080/21505594.2016.1216294
53. Hajishengallis G, Chavakis T, Hajishengallis E, Lambris JD. Neutrophil Homeostasis and Inflammation: Novel Paradigms From Studying Periodontitis. J Leukocyte Biol (2015) 98:539–48. doi: 10.1189/jlb.3VMR1014-468R
54. Sima C, Glogauer M. Neutrophil Dysfunction and Host Susceptibility to Periodontal Inflammation: Current State of Knowledge. Curr Oral Health Rep (2014) 1:95–103. doi: 10.1007/s40496-014-0015-x
55. Jiang JL, Peng YP, Qiu YH, Wang JJ. Effect of Endogenous Catecholamines on Apoptosis of Con A-Activated Lymphocytes of Rats. J Neuroimmunol (2007) 192:79–88. doi: 10.1016/j.jneuroim.2007.09.012
56. Musso NR, Brenci S, Setti M, Indiveri F, Lotti G. Catecholamine Content and In Vitro Catecholamine Synthesis in Peripheral Human Lymphocytes. J Clin Endocrinol Metab (1996) 81:3553–7. doi: 10.1210/jcem.81.10.8855800
57. Cosentino M, Marino F, Bombelli R, Ferrari M, Lecchini S, Frigo G. Endogenous Catecholamine Synthesis, Metabolism, Storage and Uptake in Human Neutrophils. Life Sci (1999) 64:975–81. doi: 10.1016/s0024-3205(99)00023-5
58. Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, et al. Transmigrating Neutrophils Shape the Mucosal Microenvironment Through Localized Oxygen Depletion to Influence Resolution of Inflammation. Immunity (2014) 40:66–77. doi: 10.1016/j.immuni.2013.11.020
59. Loesche WJ, Gusberti F, Mettraux G, Higgins T, Syed S. Relationship Between Oxygen Tension and Subgingival Bacterial Flora in Untreated Human Periodontal Pockets. Infect Immun (1983) 42:659–67. doi: 10.1128/iai.42.2.659-667.1983
60. Lakschevitz FS, Hassanpour S, Rubin A, Fine N, Sun C, Glogauer M. Identification of Neutrophil Surface Marker Changes in Health and Inflammation Using High-Throughput Screening Flow Cytometry. Exp Cell Res (2016) 342:200–9. doi: 10.1016/j.yexcr.2016.03.007
61. Uriarte SM, Edmisson JS, Jimenez-Flores E. Human Neutrophils and Oral Microbiota: A Constant Tug-of-War Between a Harmonious and a Discordant Coexistence. Immunol Rev (2016) 273:282–98. doi: 10.1111/imr.12451
62. Johansson A, Claesson R, Hanstrom L, Sandstrom G, Kalfas S. Polymorphonuclear Leukocyte Degranulation Induced by Leukotoxin From Actinobacillus actinomycetemcomitans. J Periodontal Res (2000) 35:85–92. doi: 10.1034/j.1600-0765.2000.035002085.x
63. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil Function: From Mechanisms to Disease. Annu Rev Immunol (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
64. Lacy P. Mechanisms of Degranulation in Neutrophils. Allergy Asthma Clin Immunol (2006) 2:98–108. doi: 10.1186/1710-1492-2-3-98
65. Sheshachalam A, Srivastava N, Mitchell T, Lacy P, Eitzen G. Granule Protein Processing and Regulated Secretion in Neutrophils. Front Immunol (2014) 5:448. doi: 10.3389/fimmu.2014.00448
66. Armstrong SK, Francis CL, McIntosh MA. Molecular Analysis of the Escherichia Coli Ferric Enterobactin Receptor FepA. J Biol Chem (1990) 265:14536–43. doi: 10.1016/S0021-9258(18)77336-5
67. Liu J, Rutz JM, Feix JB, Klebba PE. Permeability Properties of a Large Gated Channel Within the Ferric Enterobactin Receptor, FepA. Proc Natl Acad Sci (1993) 90:10653–7. doi: 10.1073/pnas.90.22.10653
68. Skare JT, Ahmer BM, Seachord CL, Darveau RP, Postle K. Energy Transduction Between Membranes. TonB, A Cytoplasmic Membrane Protein, can be Chemically Cross-Linked In Vivo to the Outer Membrane Receptor FepA. J Biol Chem (1993) 268:16302–8. doi: 10.1016/S0021-9258(19)85421-2
69. Clarke TE, Rohrbach MR, Tari LW, Vogel HJ, Koster W. Ferric Hydroxamate Binding Protein FhuD From Escherichia Coli: Mutants in Conserved and Non-Conserved Regions. Biometals (2002) 15:121–31. doi: 10.1023/a:1015249530156
70. Higgins CF, Gallagher HG, Hyde SC, Mimmack ML, Pearce SR. Periplasmic Binding Protein-Dependent Transport Systems: The Membrane-Associated Components. Philos Trans R Soc London B Biol Sci (1990) 326:353–65. doi: 10.1098/rstb.1990.0017
71. Lau CKY, Krewulak KD, Vogel HJ. Bacterial Ferrous Iron Transport: The Feo System. FEMS Microbiol Rev (2016) 40:273–98. doi: 10.1093/femsre/fuv049
72. Wyckoff EE, Mey AR, Leimbach A, Fisher CF, Payne SM. Characterization of Ferric and Ferrous Iron Transport Systems in Vibrio cholerae. J Bacteriol (2006) 188:6515–23. doi: 10.1128/JB.00626-06
73. Kosman DJ. Iron Metabolism in Aerobes: Managing Ferric Iron Hydrolysis and Ferrous Iron Autoxidation. Coord Chem Rev (2013) 257:210–7. doi: 10.1016/j.ccr.2012.06.030
74. Sheldon JR, Laakso HA, Heinrichs DE. Iron Acquisition Strategies of Bacterial Pathogens. Microbiol Spectr (2016) 4(2):VMBF-0010-2015. doi: 10.1128/microbiolspec.VMBF-0010-2015
75. Nakashige TG, Zhang B, Krebs C, Nolan EM. Human Calprotectin Is an Iron-Sequestering Host-Defense Protein. Nat Chem Biol (2015) 11:765–71. doi: 10.1038/nchembio.1891
76. Voganatsi A, Panyutich A, Miyasaki KT, Murthy RK. Mechanism of Extracellular Release of Human Neutrophil Calprotectin Complex. J Leukocyte Biol (2001) 70:130–4. doi: 10.1189/jlb.70.1.130
77. Miethke M, Skerra A. Neutrophil Gelatinase-Associated Lipocalin Expresses Antimicrobial Activity by Interfering With L-Norepinephrine-Mediated Bacterial Iron Acquisition. Antimicrob Agents Chemother (2010) 54:1580–9. doi: 10.1128/AAC.01158-09
78. Edmisson JS, Tian S, Armstrong CL, Vashishta A, Klaes CK, Miralda I, et al. Filifactor alocis Modulates Human Neutrophil Antimicrobial Functional Responses. Cell Microbiol (2018) 20:e12829. doi: 10.1111/cmi.12829
79. Lyte M, Villageliú DN, Crooker BA, Brown DR. Symposium Review: Microbial Endocrinology—Why the Integration of Microbes, Epithelial Cells, and Neurochemical Signals in the Digestive Tract Matters to Ruminant Health. J Dairy Sci (2018) 101:5619–28. doi: 10.3168/jds.2017-13589
80. Boyanova L. Stress Hormone Epinephrine (Adrenaline) and Norepinephrine (Noradrenaline) Effects on the Anaerobic Bacteria. Anaerobe (2017) 44:13–9. doi: 10.1016/j.anaerobe.2017.01.003
81. Velusamy SK, Sampathkumar V, Ramasubbu N, Paster BJ, Fine DH. Aggregatibacter actinomycetemcomitans Colonization and Persistence in a Primate Model. Proc Natl Acad Sci USA (2019) 116:22307–13. doi: 10.1073/pnas.1905238116
82. Miyashita M, Oishi S, Kiso A, Kikuchi Y, Ueda O, Hirai K, et al. Hemoglobin Binding Activity and Hemoglobin-Binding Protein of Prevotella Nigrescens. Eur J Med Res (2010) 15:314. doi: 10.1186/2047-783X-15-7-314
Keywords: Aggregatibacter actinomycetemcomitans, human neutrophils, catecholamines, epinephrine, granule exocytosis, QseBC system, periodontitis
Citation: Ozuna H, Uriarte SM and Demuth DR (2021) The Hunger Games: Aggregatibacter actinomycetemcomitans Exploits Human Neutrophils As an Epinephrine Source for Survival. Front. Immunol. 12:707096. doi: 10.3389/fimmu.2021.707096
Received: 08 May 2021; Accepted: 21 July 2021;
Published: 12 August 2021.
Edited by:
Nicolas Dutzan, University of Chile, ChileReviewed by:
Anders Johansson, Umeå University, SwedenTomoki Maekawa, Niigata University, Japan
Josefine Hirschfeld, University of Birmingham, United Kingdom
Copyright © 2021 Ozuna, Uriarte and Demuth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia M. Uriarte, c2lsdmlhLnVyaWFydGVAbG91aXN2aWxsZS5lZHU=