 Ying Li1,2†
Ying Li1,2† Ming Shi
Ming Shi- 1School of Life Science and Technology, Harbin Institute of Technology, Harbin, China
- 2International Research Center for Regenerative Medicine, BOAO International Hospital, Qionghai, China
- 3Department of Infectious and Medicine, Heilongjiang Provincial Hospital, Harbin, China
Inflammasomes are fundamental innate immune mechanisms that promote inflammation and induce an inflammatory form of programmed cell death, pyroptosis. Pyroptotic inflammasome has been reported to be closely associated with tumorigenesis and prognosis of multiple cancers. Emerging studies show that the inflammasome assembly into a higher-order supramolecular complex has been utilized to evaluate the status of the innate immune response. The inflammasomes are now regarded as cellular signaling hubs of the innate immunity that drive the production of inflammatory cytokines and consequent recruitment of immune cells to the tumor sites. Herein, we provided an overview of molecular characteristics and biological properties of canonical and non-canonical inflammasome signaling in cancer immunology and immunotherapy. We also focus on the mechanism of regulating pyroptotic inflammasome in tumor cells, as well as the potential roles of inflammasome-mediated pyroptotic cell death in cancers, to explore the potential diagnostic and therapeutic markers contributing to the prevention and treatment of cancers.
Introduction
Inflammasome, a major class of signalosomes in innate immunity, is a cytosolic multiprotein platform that formed by the oligomerization of a sensor, an adaptor apoptosis-associated speck-like (ASC) and caspases in response to pathogen-associated molecules and cellular stress (1, 2). These inflammasome components are expressed at low levels in normal tissue cells to prevent inappropriate activation, and are primed, activated and assembled through homotypical death domain (DD) interactions (3–6). The DD domains, from caspase recruitment domain (CARD), pyrin domain (PYD), to death effector domain (DED), were found to self-assemble into higher-order helical filaments in inflammasome (1). The higher-order inflammasome complexes carry out intricate signaling and key effector functions in innate immunity and inflammation.
Inflammasome activation induces pyroptosis, a type of programmed cell death. Studies have shown that the gasdermin family members (Gasdermins, GSDMs) play vital roles in inflammasome-induced pyroptosis (7–9). The inflammasome-induced pyroptosis depends on the formation of plasma membrane pores by the pyroptosis effectors GSDMs (7). Emerging studies had indicated that inflammasome activation plays a central role in the tumorigenesis (including immunosuppression, proliferation, angiogenesis and metastasis) and tumor suppression (10). The pyroptosis initiated by inflammasomes induces innate immune responses in cancer tissues, and targeting pyroptosis has exhibited potential anti-tumor capabilities in cancer treatment (8). Thus, targeting the inflammasome and pyroptosis is a promising strategy for cancer immunotherapy. The inflammasome and its related pyroptosis-trigged immune activation in cancer tissues will provide cancer patients with more effective anti-tumor immune responses and a better prognosis (9).
Activation of cell death effector GSDMs also has some connections with a type of cell apoptosis, NETosis, which is related to the formation of inflammasome and noncanonical inflammasome signaling (11, 12). In previous studies, NETosis was believed to associate with the immune defenses, helping to resist various pathogens (13, 14). While emerging studies have shown that noncanonical inflammasome signaling-elicited NETosis also has a positive impact on the tumorigenesis by protecting tumor cells against immune attack and promoting tumor cell metastasis (15–17).
Currently, with an increasing number of studies in innate immunity, the inflammasome assembly into a functional higher-order complex functions as hub platforms for inflammatory cytokine production, and has been considered to utilize in evaluating the activation and regulation status of the innate immune response (2, 18, 19). The inflammasomes are now regarded as cellular signaling hubs of the innate immunity that drive the inflammatory signaling and consequent recruitment of immune cells to the tumor sites, but activation of different inflammasomes may exhibit the exact opposite outcomes in cancers, anti-tumor or pro-tumor effects (20–23). In-depth understanding the functions of these canonical and non-canonical inflammasomes is critical for revealing the molecular mechanisms that govern the innate immune response and inflammatory signaling in cancer Immunotherapy (10, 24).
Canonical and Non-Canonical Inflammasome Signaling in Tumor Immunity
Canonical inflammasomes, assembled by sensor proteins (including pyrin domain containing related protein family (NLRP), absent in melanoma (AIM) 2, interferon-γ inducible factor (IFI) 16, RIG-I, and CARD-domain-containing (NLRC) 4), play key roles in immune surveillance of pathogens infections and danger signals by proteolytically activating caspases 1 and/or 11 (caspase-4/5 in humans) that cleaves interleukin-1β (IL-1β), interleukin-18 (IL-18) and the pore-forming protein gasdermin D (GSDMD), leading to cytokine maturation and pyroptosis (Table 1) (25, 26). Canonical inflammasome-induced pyroptosis is typically marked by the induction of rapid polymerization of the bipartite adapter ASC into large helical filaments with the sensors and caspases to form a single supramolecular ASC punctum (also known as ASC specks), which mediates robust cellular responses and acts as an important hallmark for inflammasome activation (27–29). NAIPs, which could recognize the bacterial ligands, recruit NLRC4 to assembly the NAIP-NLRC4 inflammasome complex, and directly activate the caspase 1 without the adaptor ASC (30–32).

Table 1 Canonical and non-canonical Inflammasomes.
The non-canonical inflammasome activation mediates a caspase 11 (caspase 4/5 in human) dependent innate immune response to the invasion of gram-negative bacteria (33–35). The cytosolic sensor caspase 11 functions as a signal initiator and mediates the recognition of gram-negative bacteria via directly interacting with cytosolic lipopolysaccharide (LPS) and assembling a higher order structure called the non-canonical caspase 11 inflammasome (33). Besides caspase 11, innate immune sensor ZBP1 and the inhibition of kinase TAK1 could regulate the assembly of RIPK1/RIPK3-FADD-caspase-8 cell death complex and induce the Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis) (Table 1) (36, 37). These active caspases cleave and activate GSDMD to promote pyroptosis, and then trigger a secondary activation of the canonical NLRP3 inflammasome for cytokine release (Figure 1) (38, 39).
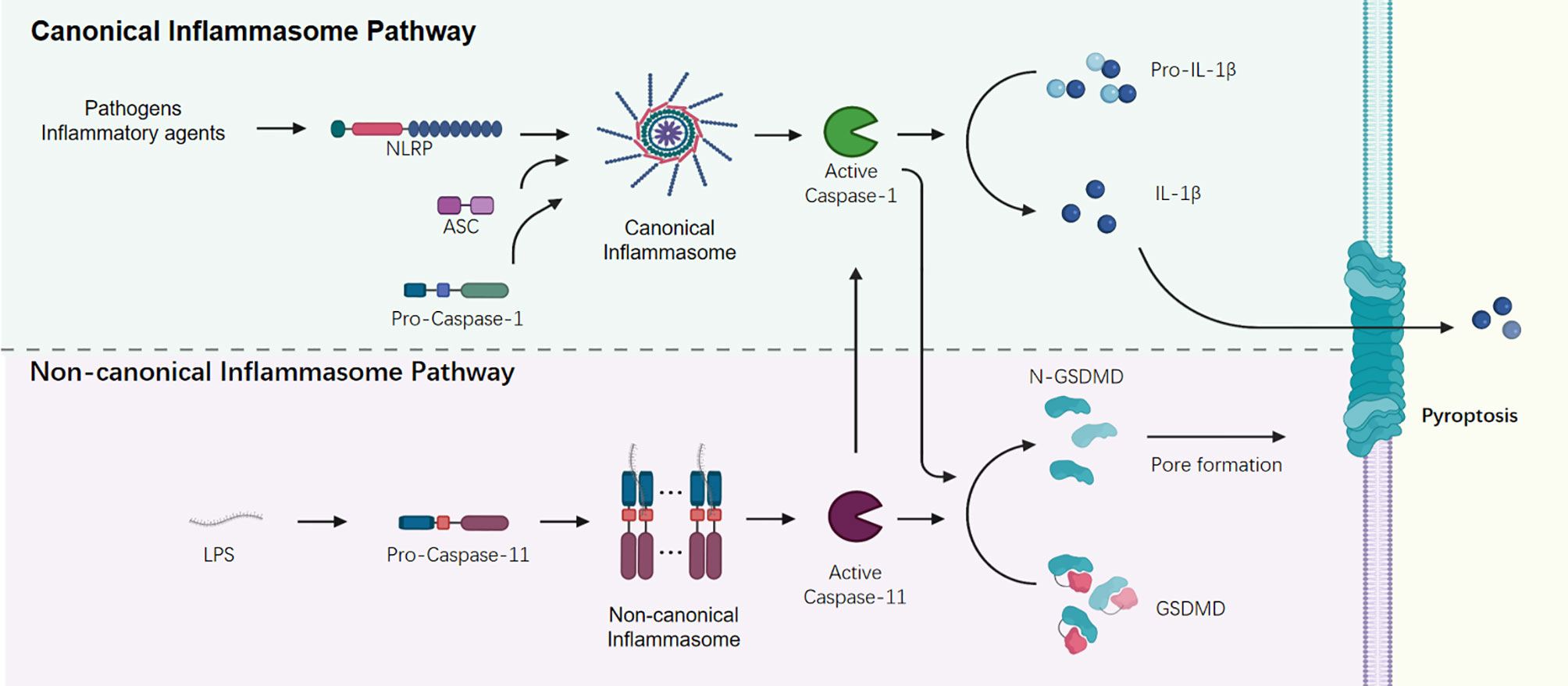
Figure 1 Canonical and non-canonical Inflammasome signaling pathways.
The effects of both canonical and non-canonical Inflammasomes activation-induced pyroptosis may be a double-edged sword on cancers (40). The role of inflammasome activation in promoting tumorigenesis has been previously reviewed by rajendra karki et al., which indicates that inflammasome components could induce cancer cell proliferation, survival and metastasis, and promote cancer cells to evade immune surveillance (10). Except the direct killing of cancer cells and cancer related microenvironmental cells by pyroptosis, the release of inflammasome-dependent cytokines (IL-1β, IL-18, et al.) and other costimulatory molecules (either from the cancer cells or from the cells in the tumor microenvironment) will significantly reshape the cancer immune microenvironment. The composition of cancer immune microenvironment will determine the effect of pyroptosis on cancer. On the one hand, pyroptosis may promote the cancer occurrence by recruiting the immunosuppressive immune cells (such as myeloid-derived suppressor cells, MDSC) (41–43) and inducing chronic inflammation (10, 44, 45); on the other hand, it may also inhibit the cancer occurrence by recruiting the NK and CD8+ T cells to the cancer microenvironment (8).
GSDMs and GSDMs-Dependent Canonical Inflammasome Signaling Modulate Tumor Immunity
The GSDMs, pyroptosis executors, are consists of gasdermin A (GSDMA), gasdermin B (GSDMB), gasdermin C (GSDMC), gasdermin D (GSDMD), gasdermin E (GSDME), and Autosomal Recessive Deafness Type 59 Protein (DFNB59 or PJVK) in homo sapiens, and displayed different tissue expression patterns (46, 47). In 2015, Kayagaki, N. et al. (38), Shi, J. et al. (48) and He, W. T. et al. (49) firstly discovered GSDMD, the executor of pyroptosis, and confirmed that it was cleaved by caspase 1 and caspase 4. Since this discovery, more and more gasdermins were characterized to play vital roles in inflammasome and pyroptosis. Further protein structure analysis of these GSDMs confirmed that by cleaving and releasing their N-terminal domains, these GSDMs can induce cell death by forming large oligomeric pores on cell membrane, disrupting the integrity of cell membrane and releasing the inflammatory mediators (50–54). These functions and mechanisms recently had been reported to relate with cancer therapy, especially the GSDMD and GSDME (8, 55, 56).
GSDMA, GSDMC and PJVK are not detected in most human tissues (both tumor tissues and normal tissues), while GSDMB, GSDMD and GSDME are highly expressed in most human tissues (both tumor tissues and normal tissues), especially GSDMD (47). These also indicate that different GSDMs may perform different functions in cancer development and cancer therapy (Table 2).
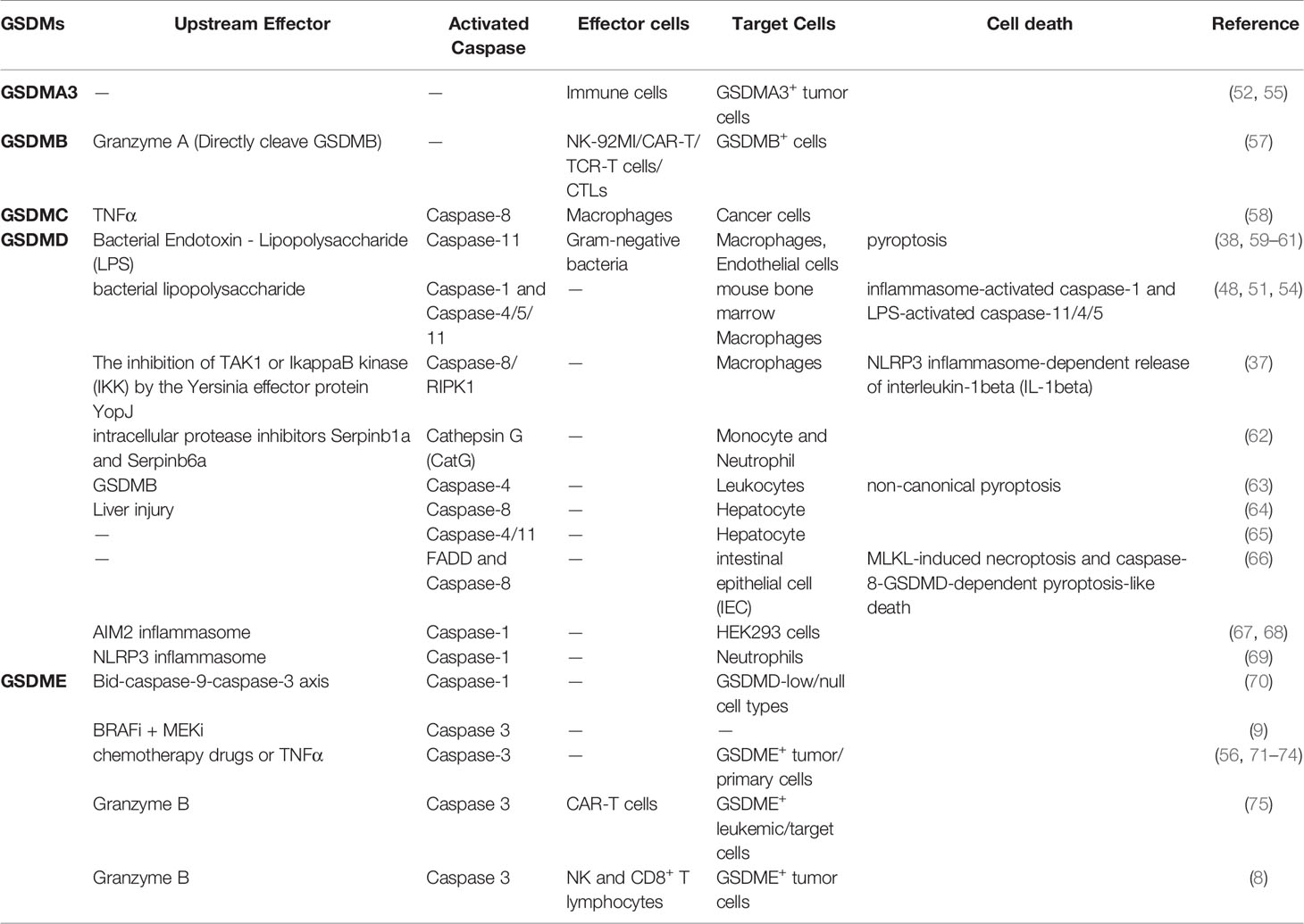
Table 2 Function of GSDMs in anti-tumor immunotherapy.
GSDMA, especially GSDMA3, is expressed in the epidermis and frequently silenced in gastric cancer cell lines (76). Mutations in GSDMA3 with gain-of-function are associated with skin inflammation and hair loss (77). GSDMA3-N domains could form membrane-disrupting pores during pyroptosis (50). Dysregulated GSDMA3 could cause cell necrosis and chronic inflammation (52), and potentially influence the cancer immunotherapy (55).
GSDMB, which has been proven as an independent poor prognostic biomarker in breast cancer, is overexpressed in about 60% of HER2 breast cancers (78). The highly expressed GSDMB significantly promotes cancer cell migration and develops cancer cell resistance to anti-HER2 therapies (79). Knockout GSDMB, or intracellular-delivered anti-GSDMB through nanocapsules could neutralize the effects of GSDMB, reduce the aggressiveness of HER2 breast cancer, and enhance the sensitivity to trastuzumab (80). These results indicate that GSDMB may play a positive role in cancer development. However, Zhou Z et al. found that GSDMB positive cells showed greater sensitivity to granzyme A-mediated cytotoxic lymphocyte killing mechanism, and the upregulation of GSDMB expression in tumor cells might be due to the activation of the interferon gamma signaling (57) (Figure 2B). While the interferon gamma signaling also could trigger the expression of immune checkpoints, such as PD-L1 and PD-L2 (81, 82), which provided the explanation for the poor prognosis of GSDMB-positive tumors. In addition, these studies also indicated that cancer immunotherapy with the immune checkpoint inhibitors might be a better strategy for treating the GSDMB-positive tumors. We are looking forward to see the similar clinical trials for this cancer subtype in the future (83).
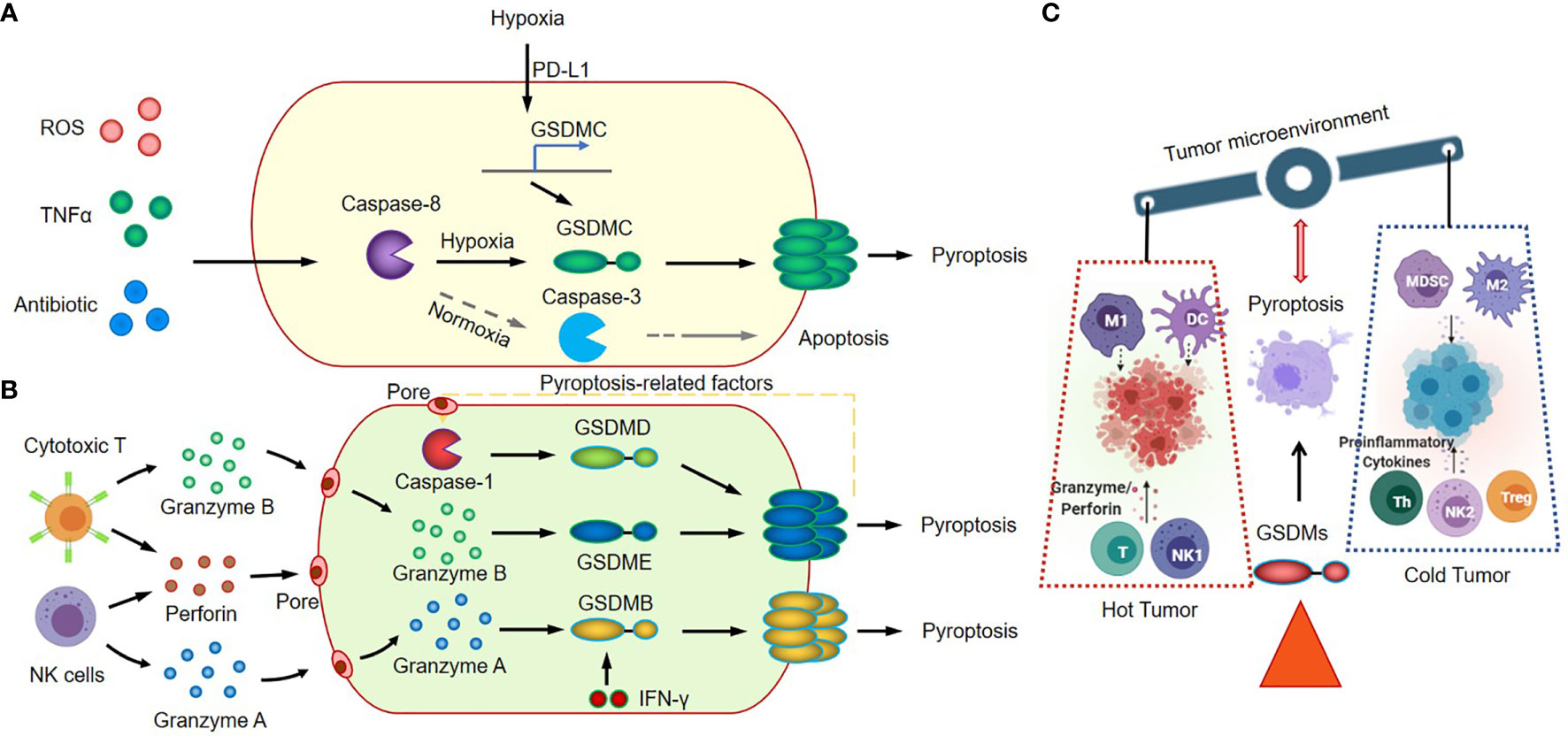
Figure 2 Canonical inflammasome signaling in cancer immunology. (A) GSDMC-dependent inflammasome signaling and pyroptosis pathway. (B) Granzyme A/B-mediated cytotoxic lymphocyte killing mechanism and GSDMB/GSDMD/GSDME induce tumor cell pyroptosis. (C) GSDMs-mediated inflammasome activation and pyroptosis regulate antitumor immunity.
GSDMC, another effector of pyroptosis, correlates with poor survival in cancer patients (84, 85). GSDMC is cleaved by caspase-8 with TNFα treatment, and also can be cleaved by caspase-6 in response to reactive oxygen species (ROS) insult in cancer cells. The elevated expression level of GSDMC, which could be induced by the nuclear translocated PD-L1, is required for the switching TNFα-induced apoptosis to pyroptosis in cancer cells (58). In tumor microenvironment, the tumor-associated macrophages could secret the TNFα, and induce tumor necrosis through the activation of caspase-8, the translocation of PD-L1, and the cleavage of GSDMC (Figure 2A). This GSDMC-dependent inflammasome signaling and pyroptosis pathway will significantly change the tumor microenvironment, promote tumor progression and increase the resistance to chemotherapy, radiotherapy and immunotherapy.
The functions of GSDMD and GSDME are much clearer (48, 51, 54, 59–61). Studies had showed that GSDMD and GSDME-mediated canonical inflammasome signaling and pyroptosis play vital roles in the immune response of cancer tissues through modulating the tumor immune microenvironment. The GSDMD could be cleaved by activated caspase 4/11 (63, 65) and caspase 8 (64, 66), and activated caspase 1 (activated by AIM2 or NLRP3 inflammasome) (67–69). The GSDME could be cleaved by activated caspase 1 and caspase 3, and then trigger the transition of cancer cells from apoptosis to pyroptosis (9, 70, 86). The transition highly relies on the expression level of GSDME in cancer cells (56). The cleavage of GSDMD and GSDME in cancer cells could be induced by various therapeutic strategies, including chemotherapy drugs (56, 71–74, 87), molecular target therapies (62, 88), or immune cell therapy (8, 75). The GSDME is constitutively expressed in many normal tissues, which explains why the chemotherapy drugs could induce direct damage in normal tissues (56). Remarkably, these damages in tumor tissues have a positive function; the damage-induced cleavage of gasdermins, inflammasome activation and subsequent pyroptosis will promote the recruitment and activation of the tumor-infiltrating NK and CD8+ T lymphocytes in tumor sites (8). Thus, GSDMs-mediated inflammasome activation and pyroptosis could turn “cold” tumor into “hot” by modulating tumor immune microenvironment, and consequently regulate antitumor immunity (Figure 2C).
The expression pattern of GSDME is distinguished from GSDMD. The GSDME gene is frequently silenced in cancer cells, and loss of function (LOF) of GSDME by mutations or hypermethylation of promoter region in cancer cells will significantly reduce the anti-tumor innate immune responses (8, 40). Zhang Z et al. reported that the granzyme B released by cytotoxic T cells could cleave GSDME in cancer cells, and the granzyme B/GSDME-mediated pyroptosis suppressed tumor growth through a perforin-dependent T-cell killing mechanism (8). Thus, it will be beneficial to target GSDME or to elevate tumor-derived GSDME expression level in cancer treatment. While the excessive activation of inflammasome-induced pyroptosis in cancer treatment, such as in CAR-T therapy, could also cause serious consequences; The CAR-T cells elicited GSDME–mediated cancer cell pyroptosis and released pyroptosis-related factors. The pyroptosis-related factors activated caspase 1 for GSDMD cleavage in macrophages, and resulted in the release of more cytokines and the subsequent cytokine release syndrome (CRS) (75) (Figure 2B).
Noncanonical Inflammasome Signaling-Elicited NETosis Promotes Tumorigenesis
NETosis, a proinflammatory cell death modality originally identified in neutrophil, provides host defense against extracellular intruders in response to various stimuli. NETosis differs from apoptosis and necrosis, but has some connections with the activation of pyroptosis executor GSDMD (12). Various stimuli promote the release of NE from the neutrophil granules, and NE cleaves and activates GSDMD, leading to nuclear and plasma membrane rupture and neutrophil cell lysis by NETosis (Figure 3A). Exposure of neutrophil to cytosolic LPS also activates the noncanonical inflammasome signaling and triggers GSDMD-dependent NETosis (12, 89). Both Caspase-11 and GSDMD are required for NETosis at multiple stages, including nuclear delobulation, chromatin decondensation, nuclear membrane permeabilization and plasma membrane rupture (89).
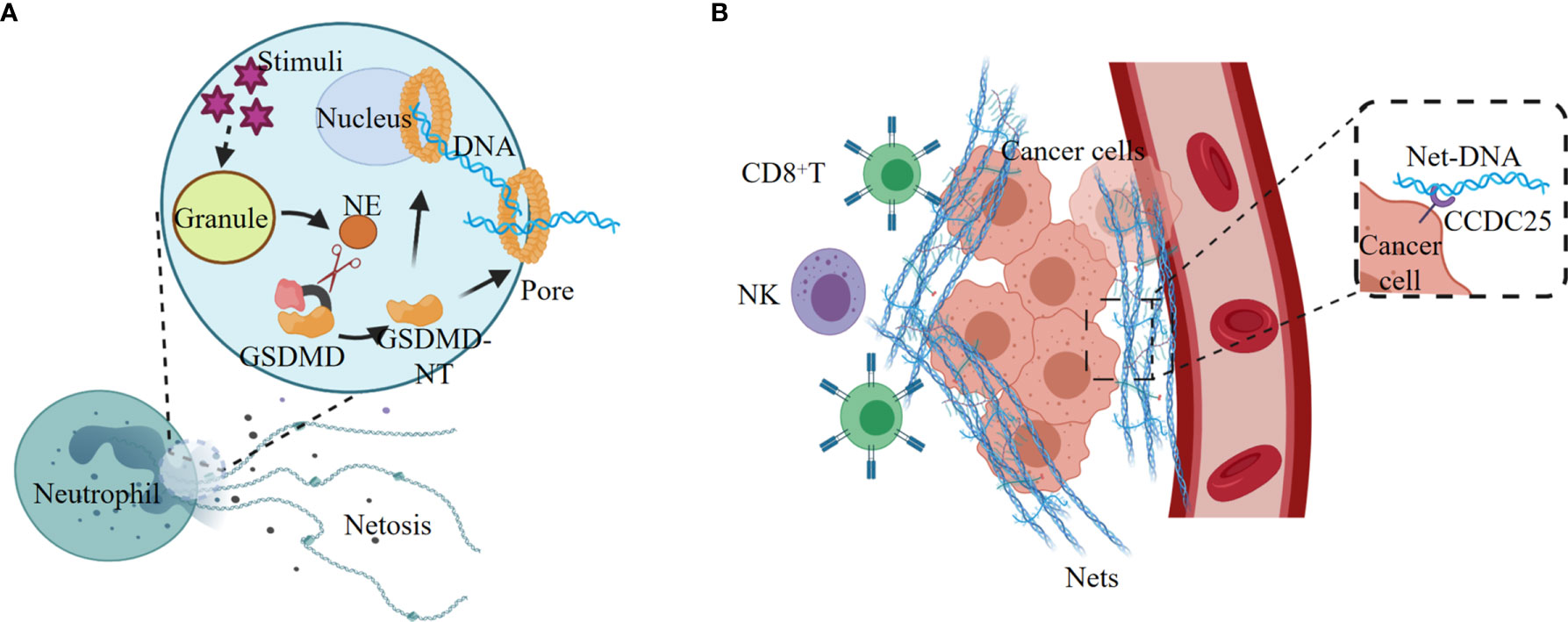
Figure 3 NETosis elicited by noncanonical inflammasome signaling promotes tumorigenesis. (A) Various stimuli promote the release of NE from the granules, and NE cleaves and activates GSDMD, leading to nuclear and plasma membrane rupture and neutrophil cell lysis by NETosis. (B) Tumor-secreted ligands induce extrusion of NETs, and NETs protect tumor cells from CTL and NK cytotoxicity. The extracellular NETs-DNA binds to the transmembrane protein CCDC25 on tumor cells, and thus improve tumor cell migration.
Neutrophil extracellular traps (NETs) are the regulated outcome of NETosis, and the release of NETs is linked with tumorigenesis (90–92). Tumor cells can recruit myeloid cells, mostly neutrophils, by secreting CXCR1 and CXCR2 agonists ELR positive CXCL chemokines, such as CXCL1, CXCL2, and CXCL8 (93). The tumor-derived ELR positive CXCL chemokines are the major mediators of cancer-promoted NETosis and NETs (91). The NETs released from neutrophils are enriched on the tumor surface to form a barrier, which effectively reduces the contact of CD8+ T cells and NK cells with tumor cells, and thus protects tumors from immune cytotoxicity (Figure 3B).
Excessive NETs produced by sustained inflammation contribute to reawakenment of dormant cancer cells (94). The sustained lung inflammation induced by tobacco smoke or LPS instillation recruits and activates neutrophils, and the subsequent NETs formation is greatly induced in the cancer cell dormancy mouse model. The two NETs-associated proteases neutrophil elastase (NE) and matrix metalloproteinase 9 (MMP9) remodel the extracellular matrix (ECM) by hydrolyzing laminin. The proteolytic remodeling laminin induces the proliferation of dormant cancer cells through activating the cell surface ECM receptor integrin α3β1 (94). In mouse models of small bowel tumors, tumor development is related with the accumulation of low-density neutrophils (LDNs). The LDNs aggregation-induced up-regulation of complement 3a receptor (C3aR) and activation of the complement cascade lead to NETosis, coagulation and differentiation of neutrophils into N2 type, which in turn promote tumorigenesis (95).
NETs also play an important role in promoting tumor metastasis. The transmembrane protein CCDC25 is a NET-DNA (the DNA component of NETs) receptor on cancer cells, which activate the integrin‐linked kinase (ILK)/β-parvin pathway by binding to extracellular DNA to enhance the mobility of cells, thereby promoting tumor metastasis (96) (Figure 3B). In tissues of lung and retina, NETosis can induce apoptosis of senescent vascular endothelial cells and promote the formation of new blood vessels that are conducive to tumor growth (97, 98). In addition, NETs can continuously deposit in the lungs, which might be the reason why lung is one of the most common sites of cancer metastasis (99).
Pyroptotic Inflammasome Signaling Acts as a Critical Regulator of Inflammation and TILs Within Tumor Microenvironment
The activation of canonical or non-canonical inflammasome signaling in the cytosolic compartment will lead to pyroptosis (34, 100, 101), which are critical defense mechanisms against endogenous (tissue or cellular injury) or exogenous danger signals (infections, such as microbes) (34, 100–102). The dysregulation of inflammasome activation in cancer development and progression is controversial, due to the inconsistent findings on potential cancer promotion and immunotherapy (103). As the potent contributors to the activation of inflammatory cytokines in cancer tissues, the excessive inflammasome signaling will lead to the cancer progression (10, 104, 105). Thus, inhibition of inflammasome with some certain inhibitors could potentially be used for clinical cancer treatment (106, 107). Meanwhile, in the treatment of cancer, various drugs could induce the activation of inflammasome-related pyroptosis and cause the release of proinflammatory cytokines. Subsequently, the activated inflammatory cytokines could recruit the NK or cytotoxic T cells to the tumor site for killing cancer cells, and eventually delay the tumor progression (108, 109).
Regulation of inflammasome activation might reinforce anti-tumor immunity by boosting the recruitment of TILs (110). Mechanismly, the checkpoint molecule, T cell immunoglobulin and mucin-containing molecule 3 (TIM-3) in DC cells restrains anti-tumor immunity through suppressing inflammasome activation; TIM-3-deficient DCs promote the recruitment of stem-like CD8+ TILs and boost antigen-specific immunity via increasing accumulation of reactive oxygen species resulting in driving inflammasome activation (111). Additionally, the pyroptotic inflammasome-cytokine (IL-18) pathway effectively regulates the NK-cell-mediated tumor attack through promoting the maturation of NK cells and surface expression of the death ligand FasL, which consequently leads to elevate the tumoricidal activity of NK cells (108). Thus, inhibition of inflammasome activation, or downstream effector cytokines might abrogate the protective anti-tumor immunity and expanded TILs.
Recently, emerging insights in cancer immunology indicate that the roles of pyroptotic inflammasomes on tumor immunotherapies may highly rely on the tumor stage and the tumor microenvironment (105). In early-stages of cancer development, the pyroptotic inflammasomes participate in the innate immune response, recruit tumor-infiltrating lymphocytes and promote inflammatory cell death of cancer. The immune cells recruited to tumor sites retain their immunosurveillance properties and anti-tumor immunity prevails (Elimination phase) (112), while the ESCRT-III-mediated plasma membrane repair in pyroptotic cells strongly inhibits pyroptotic cell death (113, 114). A dynamic interplay of pyroptosis with ESCRT-mediated membrane repair in cancer cells occurs in immune equilibrium phase. In addition, cancer cells also develop a series of pyroptosis-resistance strategies to escape immune attack and establish a protumor immune microenvironment (escape phase) (Figure 4A). The levels of pyroptotic inflammasomes regulate the inflammation and TILs within tumor microenvironment, and affect the balance between cancer cell elimination and immune escape (Figure 4B).
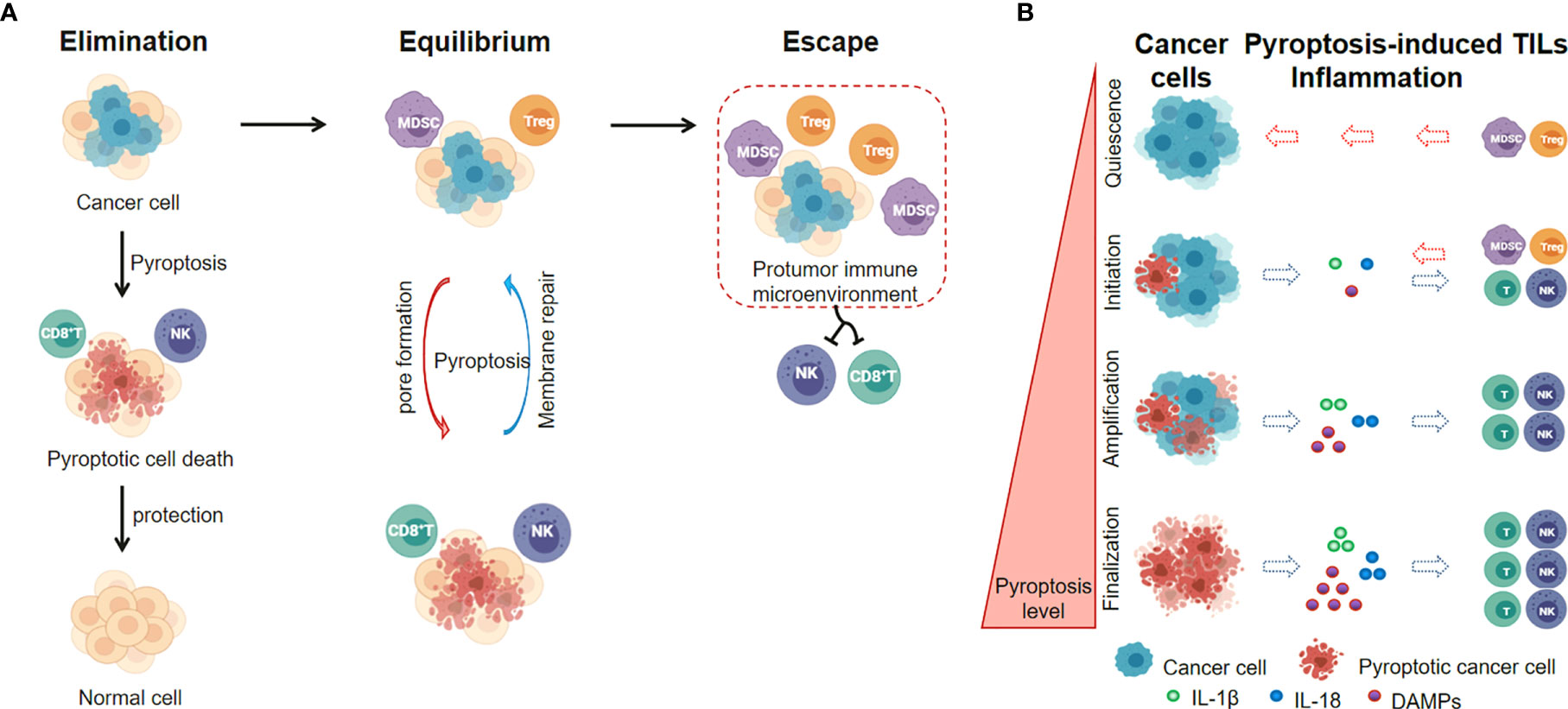
Figure 4 Pyroptotic Inflammasome signaling regulates inflammation and TILs within tumor microenvironment. (A) The level of pyroptotic inflammasomes is associated with the balance between cancer cell elimination and immune escape. (B) Pyroptosis levels in cancer cells affect the inflammation and TILs within tumor microenvironment.
The inflammasomes have been considered as cellular signaling hubs of the innate immunity that drive the production of inflammatory cytokines, promote inflammation and induce pyroptosis in cancer cells. While inflammasome signaling hubs function in innate immune response, the inflammasome activation links with diverse physiological and pathological processes, such as autophagy (2, 115), cellular stress response (116), cell-cycle progression (117). In these processes, inflammasomes activation is tightly regulated by DDX3X-mediated assembly of stress granules, HDAC6-associated autophagosomal degradation, and NEK7-dependent mitotic spindle formation and cytokinesis (2, 115–117).
Conclusion
Inflammasome signaling has shifted the paradigm for the hub platform in innate immune responses. The inflammasomes are considered as cellular signaling hubs of the innate immunity that drive the production of inflammatory cytokines, promote inflammation and induce pyroptosis in cancer cells. The polymerization of pattern recognition receptors, adaptor ASC, and effectors caspases into higher-order supramolecular complexes facilitates signal transduction cascades and proximity-facilitated enzyme activation. In these complexes, pattern recognition receptors (sensor proteins) and adaptor ASC form the center (ASC specks), whereas caspases make up the filaments. The inflammasome activation and assembly into higher-order supramolecular complexes function as inflammasome hub platforms for inflammatory cytokine production, and have been utilized to evaluate the status of the innate immune response.
The pyroptotic inflammasome regulates inflammation, TILs within tumor microenvironment, and the consequent recruitment of immune cells to the tumor sites. But the ESCRT-III-mediated plasma membrane repair in pyroptotic cells strongly inhibits pyroptosis in cancer cells. Thus, a dynamic interplay of pyroptosis with membrane repair in cancer cells occurs in immune equilibrium phase. The level of pyroptotic inflammasomes might related with the balance between cancer cell elimination and immune escape. With the inflammasome examples in cancer immunology presented here, we could see that the regulation of inflammasome level with some certain agonists or antagonists would potentially be used for future clinical cancer treatment.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This work was supported by funding from the National Natural Science Foundation of China (31870891 and 81502781). Figures were created with Biorender.com. We apologize to authors whose work could not be cited due to space limitation.
References
1. Shi M, Zhang P, Vora SM, Wu H. Higher-Order Assemblies in Innate Immune and Inflammatory Signaling: A General Principle in Cell Biology. Curr Opin Cell Biol (2020) 63:194–203. doi: 10.1016/j.ceb.2020.03.002
2. Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, et al. HDAC6 Mediates an Aggresome-Like Mechanism for NLRP3 and Pyrin Inflammasome Activation. Science (2020) 369:eaas8995. doi: 10.1126/science.aas8995
3. Rathinam VAK, Chan FK. Inflammasome, Inflammation, and Tissue Homeostasis. Trends Mol Med (2018) 24:304–18. doi: 10.1016/j.molmed.2018.01.004
4. Poli G, Fabi C, Bellet MM, Costantini C, Nunziangeli L, Romani L, et al. Epigenetic Mechanisms of Inflammasome Regulation. Int J Mol Sci (2020) 21:5758. doi: 10.3390/ijms21165758
5. Cornut M, Bourdonnay E, Henry T. Transcriptional Regulation of Inflammasomes. Int J Mol Sci (2020) 21:8087. doi: 10.3390/ijms21218087
6. Latz E, Xiao TS, Stutz A. Activation and Regulation of the Inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi: 10.1038/nri3452
7. Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol (2017) 27:673–84. doi: 10.1016/j.tcb.2017.05.005
8. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E Suppresses Tumour Growth by Activating Anti-Tumour Immunity. Nature (2020) 579:415–20. doi: 10.1038/s41586-020-2071-9
9. Erkes DA, Cai W, Sanchez IM, Purwin TJ, Rogers C, Field CO, et al. Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov (2020) 10:254–69. doi: 10.1158/2159-8290.CD-19-0672
10. Karki R, Kanneganti TD. Diverging Inflammasome Signals in Tumorigenesis and Potential Targeting. Nat Rev Cancer (2019) 19:197–214. doi: 10.1038/s41568-019-0123-y
11. Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular Mechanisms of NETosis. Annu Rev Cell Dev Biol (2020) 36:191–218. doi: 10.1146/annurev-cellbio-020520-111016
12. Chen KW, Demarco B, Broz P. Beyond Inflammasomes: Emerging Function of Gasdermins During Apoptosis and NETosis. EMBO J (2020) 39:e103397. doi: 10.15252/embj.2019103397
13. Branzk N, Papayannopoulos V. Molecular Mechanisms Regulating NETosis in Infection and Disease. Semin Immunopathol (2013) 35:513–30. doi: 10.1007/s00281-013-0384-6
14. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-Induced NETosis is a Dynamic Process Involving Neutrophil Multitasking. vivo Nat Med (2012) 18:1386–93. doi: 10.1038/nm.2847
15. Demers M, Wong SL, Martinod K, Gallant M, Cabral JE, Wang Y, et al. Priming of Neutrophils Toward NETosis Promotes Tumor Growth. Oncoimmunology (2016) 5:e1134073. doi: 10.1080/2162402X.2015.1134073
16. Arpinati L, Shaul ME, Kaisar-Iluz N, Mali S, Mahroum S, Fridlender ZG. NETosis in Cancer: A Critical Analysis of the Impact of Cancer on Neutrophil Extracellular Trap (NET) Release in Lung Cancer Patients vs. Mice. Cancer Immunol Immunother (2020) 69:199–213. doi: 10.1007/s00262-019-02474-x
17. Papayannopoulos V. Neutrophil Extracellular Traps in Immunity and Disease. Nat Rev Immunol (2018) 18:134–47. doi: 10.1038/nri.2017.105
18. Xia S, Chen Z, Shen C, Fu TM. Higher-Order Assemblies in Immune Signaling: Supramolecular Complexes and Phase Separation. Protein Cell (2021). doi: 10.1007/s13238-021-00839-6
19. Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural Mechanism for NEK7-Licensed Activation of NLRP3 Inflammasome. Nature (2019) 570:338–43. doi: 10.1038/s41586-019-1295-z
20. Prochnicki T, Latz E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab (2017) 26:71–93. doi: 10.1016/j.cmet.2017.06.018
21. Deets KA, Vance RE. Inflammasomes and Adaptive Immune Responses. Nat Immunol (2021) 22:412–22. doi: 10.1038/s41590-021-00869-6
22. Theivanthiran B, Evans KS, DeVito NC, Plebanek M, Sturdivant M, Wachsmuth LP, et al. A Tumor-Intrinsic PD-L1/NLRP3 Inflammasome Signaling Pathway Drives Resistance to Anti-PD-1 Immunotherapy. J Clin Invest (2020) 130:2570–86. doi: 10.1172/JCI133055
23. Gouravani M, Khalili N, Razi S, Keshavarz-Fathi M, Rezaei N. The NLRP3 Inflammasome: A Therapeutic Target for Inflammation-Associated Cancers. Expert Rev Clin Immunol (2020) 16:175–87. doi: 10.1080/1744666X.2020.1713755
24. Minton K. Pyroptosis Heats Tumour Immunity. Nat Rev Immunol (2020) 20:274–5. doi: 10.1038/s41577-020-0297-2
25. Deigendesch N, Zychlinsky A, Meissner F. Copper Regulates the Canonical NLRP3 Inflammasome. J Immunol (2018) 200:1607–17. doi: 10.4049/jimmunol.1700712
26. Monie TP. The Canonical Inflammasome: A Macromolecular Complex Driving Inflammation. Subcell Biochem (2017) 83:43–73. doi: 10.1007/978-3-319-46503-6_2
27. Stutz A, Horvath GL, Monks BG, Latz E. ASC Speck Formation as a Readout for Inflammasome Activation. Methods Mol Biol (2013) 1040:91–101. doi: 10.1007/978-1-62703-523-1_8
28. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The Adaptor ASC has Extracellular and ‘Prionoid’ Activities That Propagate Inflammation. Nat Immunol (2014) 15:727–37. doi: 10.1038/ni.2913
29. Kesavardhana S, Kanneganti TD. Mechanisms Governing Inflammasome Activation, Assembly and Pyroptosis Induction. Int Immunol (2017) 29:201–10. doi: 10.1093/intimm/dxx018
30. Diebolder CA, Halff EF, Koster AJ, Huizinga EG, Koning RI. Cryoelectron Tomography of the NAIP5/NLRC4 Inflammasome: Implications for NLR Activation. Structure (2015) 23:2349–57. doi: 10.1016/j.str.2015.10.001
31. von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, et al. Rapid Induction of Inflammatory Lipid Mediators by the Inflammasome In Vivo. Nature (2012) 490:107–11. doi: 10.1038/nature11351
32. Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, et al. Cryo-EM Structure of the Activated NAIP2-NLRC4 Inflammasome Reveals Nucleated Polymerization. Science (2015) 350:404–9. doi: 10.1126/science.aac5789
33. Yi YS. Caspase-11 non-Canonical Inflammasome: A Critical Sensor of Intracellular Lipopolysaccharide in Macrophage-Mediated Inflammatory Responses. Immunology (2017) 152:207–17. doi: 10.1111/imm.12787
34. Downs KP, Nguyen H, Dorfleutner A, Stehlik C. An Overview of the non-Canonical Inflammasome. Mol Aspects Med (2020) 76:100924. doi: 10.1016/j.mam.2020.100924
35. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory Caspases are Innate Immune Receptors for Intracellular LPS. Nature (2014) 514:187–92. doi: 10.1038/nature13683
36. Malireddi RKS, Kesavardhana S, Kanneganti TD. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-Optosis). Front Cell Infect Microbiol (2019) 9:406. doi: 10.3389/fcimb.2019.00406
37. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen Blockade of TAK1 Triggers Caspase-8-Dependent Cleavage of Gasdermin D and Cell Death. Science (2018) 362:1064–9. doi: 10.1126/science.aau2818
38. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature (2015) 526:666–71. doi: 10.1038/nature15541
39. Yang J, Zhao Y, Shao F. Non-Canonical Activation of Inflammatory Caspases by Cytosolic LPS in Innate Immunity. Curr Opin Immunol (2015) 32:78–83. doi: 10.1016/j.coi.2015.01.007
40. Xia X, Wang X, Cheng Z, Qin W, Lei L, Jiang J, et al. The Role of Pyroptosis in Cancer: Pro-Cancer or Pro-”Host”? Cell Death Dis (2019) 10:650. doi: 10.1038/s41419-019-1883-8
41. Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, et al. CD11b+/Gr-1+ Immature Myeloid Cells Mediate Suppression of T Cells in Mice Bearing Tumors of IL-1beta-Secreting Cells. J Immunol (2005) 175:8200–8. doi: 10.4049/jimmunol.175.12.8200
42. Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced Inflammation in the Tumor Microenvironment Delays the Accumulation of Myeloid-Derived Suppressor Cells and Limits Tumor Progression. Cancer Res (2007) 67:10019–26. doi: 10.1158/0008-5472.CAN-07-2354
43. van Deventer HW, Burgents JE, Wu QP, Woodford RM, Brickey WJ, Allen IC, et al. The Inflammasome Component NLRP3 Impairs Antitumor Vaccine by Enhancing the Accumulation of Tumor-Associated Myeloid-Derived Suppressor Cells. Cancer Res (2010) 70:10161–9. doi: 10.1158/0008-5472.CAN-10-1921
44. Ahechu P, Zozaya G, Marti P, Hernandez-Lizoain JL, Baixauli J, Unamuno X, et al. NLRP3 Inflammasome: A Possible Link Between Obesity-Associated Low-Grade Chronic Inflammation and Colorectal Cancer Development. Front Immunol (2018) 9:2918. doi: 10.3389/fimmu.2018.02918
45. Karki R, Man SM, Kanneganti TD. Inflammasomes and Cancer. Cancer Immunol Res (2017) 5:94–9. doi: 10.1158/2326-6066.CIR-16-0269
46. Feng S, Fox D, Man SM. Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death. J Mol Biol (2018) 430:3068–80. doi: 10.1016/j.jmb.2018.07.002
47. Broz P, Pelegrin P, Shao F. The Gasdermins, a Protein Family Executing Cell Death and Inflammation. Nat Rev Immunol (2020) 20:143–57. doi: 10.1038/s41577-019-0228-2
48. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
49. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an Executor of Pyroptosis and Required for Interleukin-1beta Secretion. Cell Res (2015) 25:1285–98. doi: 10.1038/cr.2015.139
50. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-Forming Activity and Structural Autoinhibition of the Gasdermin Family. Nature (2016) 535:111–6. doi: 10.1038/nature18590
51. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature (2016) 535:153–8. doi: 10.1038/nature18629
52. Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo-EM Structure of the Gasdermin A3 Membrane Pore. Nature (2018) 557:62–7. doi: 10.1038/s41586-018-0058-6
53. Yue E, Tuguzbaeva G, Chen X, Qin Y, Li A, Sun X, et al. Anthocyanin is Involved in the Activation of Pyroptosis in Oral Squamous Cell Carcinoma. Phytomedicine (2019) 56:286–94. doi: 10.1016/j.phymed.2018.09.223
54. Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, et al. Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell (2020) 180:941–55.e20. doi: 10.1016/j.cell.2020.02.002
55. Wang Q, Wang Y, Ding J, Wang C, Zhou X, Gao W, et al. A Bioorthogonal System Reveals Antitumour Immune Function of Pyroptosis. Nature (2020) 579:421–6. doi: 10.1038/s41586-020-2079-1
56. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy Drugs Induce Pyroptosis Through Caspase-3 Cleavage of a Gasdermin. Nature (2017) 547:99–103. doi: 10.1038/nature22393
57. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A From Cytotoxic Lymphocytes Cleaves GSDMB to Trigger Pyroptosis in Target Cells. Science (2020) 368(6494):eaaz7548. doi: 10.1126/science.aaz7548
58. Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-Mediated Gasdermin C Expression Switches Apoptosis to Pyroptosis in Cancer Cells and Facilitates Tumour Necrosis. Nat Cell Biol (2020) 22:1264–75. doi: 10.1038/s41556-020-0575-z
59. Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD Membrane Pore Formation Constitutes the Mechanism of Pyroptotic Cell Death. EMBO J (2016) 35:1766–78. doi: 10.15252/embj.201694696
60. Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, et al. Bacterial Endotoxin Activates the Coagulation Cascade Through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity (2019) 51:983–96.e6. doi: 10.1016/j.immuni.2019.11.005
61. Chen H, Li Y, Wu J, Li G, Tao X, Lai K, et al. RIPK3 Collaborates With GSDMD to Drive Tissue Injury in Lethal Polymicrobial Sepsis. Cell Death Differ (2020) 27:2568–85. doi: 10.1038/s41418-020-0524-1
62. Burgener SS, Leborgne NGF, Snipas SJ, Salvesen GS, Bird PI, Benarafa C. Cathepsin G Inhibition by Serpinb1 and Serpinb6 Prevents Programmed Necrosis in Neutrophils and Monocytes and Reduces GSDMD-Driven Inflammation. Cell Rep (2019) 27:3646–56.e5. doi: 10.1016/j.celrep.2019.05.065
63. Chen Q, Shi P, Wang Y, Zou D, Wu X, Wang D, et al. GSDMB Promotes non-Canonical Pyroptosis by Enhancing Caspase-4 Activity. J Mol Cell Biol (2019) 11:496–508. doi: 10.1093/jmcb/mjy056
64. Yang C, Sun P, Deng M, Loughran P, Li W, Yi Z, et al. Gasdermin D Protects Against Noninfectious Liver Injury by Regulating Apoptosis and Necroptosis. Cell Death Dis (2019) 10:481. doi: 10.1038/s41419-019-1719-6
65. Khanova E, Wu R, Wang W, Yan R, Chen Y, French SW, et al. Pyroptosis by Caspase11/4-Gasdermin-D Pathway in Alcoholic Hepatitis in Mice and Patients. Hepatology (2018) 67:1737–53. doi: 10.1002/hep.29645
66. Schwarzer R, Jiao H, Wachsmuth L, Tresch A, Pasparakis M. FADD and Caspase-8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL- and GSDMD-Mediated Death of Intestinal Epithelial Cells. Immunity (2020) 52:978–93.e6. doi: 10.1016/j.immuni.2020.04.002
67. Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, et al. Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity (2018) 49:413–26.e5. doi: 10.1016/j.immuni.2018.07.006
68. Yuan B, Zhou XM, You ZQ, Xu WD, Fan JM, Chen SJ, et al. Inhibition of AIM2 Inflammasome Activation Alleviates GSDMD-Induced Pyroptosis in Early Brain Injury After Subarachnoid Haemorrhage. Cell Death Dis (2020) 11:76. doi: 10.1038/s41419-020-2248-z
69. Karmakar M, Minns M, Greenberg EN, Diaz-Aponte J, Pestonjamasp K, Johnson JL, et al. N-GSDMD Trafficking to Neutrophil Organelles Facilitates IL-1beta Release Independently of Plasma Membrane Pores and Pyroptosis. Nat Commun (2020) 11:2212. doi: 10.1038/s41467-020-16043-9
70. Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T, et al. Caspase-1 Initiates Apoptosis in the Absence of Gasdermin D. Nat Commun (2019) 10:2091. doi: 10.1038/s41467-019-09753-2
71. Hu L, Chen M, Chen X, Zhao C, Fang Z, Wang H, et al. Chemotherapy-Induced Pyroptosis is Mediated by BAK/BAX-Caspase-3-GSDME Pathway and Inhibited by 2-Bromopalmitate. Cell Death Dis (2020) 11:281. doi: 10.1038/s41419-020-2476-2
72. Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J, et al. Cleavage of GSDME by Caspase-3 Determines Lobaplatin-Induced Pyroptosis in Colon Cancer Cells. Cell Death Dis (2019) 10:193. doi: 10.1038/s41419-019-1441-4
73. Huang Z, Zhang Q, Wang Y, Chen R, Wang Y, Huang Z, et al. Inhibition of Caspase-3-Mediated GSDME-Derived Pyroptosis Aids in Noncancerous Tissue Protection of Squamous Cell Carcinoma Patients During Cisplatin-Based Chemotherapy. Am J Cancer Res (2020) 10:4287–307.
74. Mai FY, He P, Ye JZ, Xu LH, Ouyang DY, Li CG, et al. Caspase-3-Mediated GSDME Activation Contributes to Cisplatin- and Doxorubicin-Induced Secondary Necrosis in Mouse Macrophages. Cell Prolif (2019) 52:e12663. doi: 10.1111/cpr.12663
75. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-Mediated Target Cell Pyroptosis by CAR T Cells Triggers Cytokine Release Syndrome. Sci Immunol (2020) 5(43):eaax7969. doi: 10.1126/sciimmunol.aax7969
76. Lin PH, Lin HY, Kuo CC, Yang LT. N-Terminal Functional Domain of Gasdermin A3 Regulates Mitochondrial Homeostasis via Mitochondrial Targeting. J BioMed Sci (2015) 22:44. doi: 10.1186/s12929-015-0152-0
77. Chen Q, Shi P, Wang D, Liu Q, Li X, Wang Y, et al. Epidermis-Activated Gasdermin-A3 Enhances Thermogenesis of Brown Adipose Tissue Through IL-6/Stat3 Signaling. Am J Pathol (2019) 189:1041–52. doi: 10.1016/j.ajpath.2019.01.012
78. Hergueta-Redondo M, Sarrio D, Molina-Crespo A, Vicario R, Bernado-Morales C, Martinez L, et al. Gasdermin B Expression Predicts Poor Clinical Outcome in HER2-Positive Breast Cancer. Oncotarget (2016) 7:56295–308. doi: 10.18632/oncotarget.10787
79. Hergueta-Redondo M, Sarrio D, Molina-Crespo A, Megias D, Mota A, Rojo-Sebastian A, et al. Gasdermin-B Promotes Invasion and Metastasis in Breast Cancer Cells. PLoS One (2014) 9:e90099. doi: 10.1371/journal.pone.0090099
80. Molina-Crespo A, Cadete A, Sarrio D, Gamez-Chiachio M, Martinez L, Chao K, et al. Intracellular Delivery of an Antibody Targeting Gasdermin-B Reduces HER2 Breast Cancer Aggressiveness. Clin Cancer Res (2019) 25:4846–58. doi: 10.1158/1078-0432.CCR-18-2381
81. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 Contributes to Immunosuppression and is Associated With Anti-PD-1 Response. Nature (2018) 560:382–6. doi: 10.1038/s41586-018-0392-8
82. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep (2017) 19:1189–201. doi: 10.1016/j.celrep.2017.04.031
83. Loi S, Giobbie-Hurder A, Gombos A, Bachelot T, Hui R, Curigliano G, et al. Pembrolizumab Plus Trastuzumab in Trastuzumab-Resistant, Advanced, HER2-Positive Breast Cancer (PANACEA): A Single-Arm, Multicentre, Phase 1b-2 Trial. Lancet Oncol (2019) 20:371–82. doi: 10.1016/S1470-2045(18)30812-X
84. Wei J, Xu Z, Chen X, Wang X, Zeng S, Qian L, et al. Overexpression of GSDMC is a Prognostic Factor for Predicting a Poor Outcome in Lung Adenocarcinoma. Mol Med Rep (2020) 21:360–70. doi: 10.3892/mmr.2019.10837
85. Miguchi M, Hinoi T, Shimomura M, Adachi T, Saito Y, Niitsu H, et al. Gasdermin C Is Upregulated by Inactivation of Transforming Growth Factor Beta Receptor Type II in the Presence of Mutated Apc, Promoting Colorectal Cancer Proliferation. PLoS One (2016) 11:e0166422. doi: 10.1371/journal.pone.0166422
86. Jiang M, Qi L, Li L, Li Y. The Caspase-3/GSDME Signal Pathway as a Switch Between Apoptosis and Pyroptosis in Cancer. Cell Death Discov (2020) 6:112. doi: 10.1038/s41420-020-00349-0
87. Zhao P, Wang M, Chen M, Chen Z, Peng X, Zhou F, et al. Programming Cell Pyroptosis With Biomimetic Nanoparticles for Solid Tumor Immunotherapy. Biomaterials (2020) 254:120142. doi: 10.1016/j.biomaterials.2020.120142
88. Lu H, Zhang S, Wu J, Chen M, Cai MC, Fu Y, et al. Molecular Targeted Therapies Elicit Concurrent Apoptotic and GSDME-Dependent Pyroptotic Tumor Cell Death. Clin Cancer Res (2018) 24:6066–77. doi: 10.1158/1078-0432.CCR-18-1478
89. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical Inflammasome Signaling Elicits Gasdermin D-Dependent Neutrophil Extracellular Traps. Sci Immunol (2018) 3(26):eaar6676. doi: 10.1126/sciimmunol.aar6676
90. Thanabalasuriar A, Scott BNV, Peiseler M, Willson ME, Zeng Z, Warrener P, et al. Neutrophil Extracellular Traps Confine Pseudomonas Aeruginosa Ocular Biofilms and Restrict Brain Invasion. Cell Host Microbe (2019) 25:526–36.e4. doi: 10.1016/j.chom.2019.02.007
91. Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps That Interfere With Immune Cytotoxicity. Immunity (2020) 52:856–71.e8. doi: 10.1016/j.immuni.2020.03.001
92. Singel KL, Grzankowski KS, Khan A, Grimm MJ, D’Auria AC, Morrell K, et al. Mitochondrial DNA in the Tumour Microenvironment Activates Neutrophils and is Associated With Worse Outcomes in Patients With Advanced Epithelial Ovarian Cancer. Br J Cancer (2019) 120:207–17. doi: 10.1038/s41416-018-0339-8
93. Chow MT, Luster AD. Chemokines in Cancer. Cancer Immunol Res (2014) 2:1125–31. doi: 10.1158/2326-6066.CIR-14-0160
94. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil Extracellular Traps Produced During Inflammation Awaken Dormant Cancer Cells in Mice. Science (2018) 361(6409):eaao4227. doi: 10.1126/science.aao4227
95. Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS, et al. Coagulation Induced by C3aR-Dependent NETosis Drives Protumorigenic Neutrophils During Small Intestinal Tumorigenesis. Nat Commun (2016) 7:11037. doi: 10.1038/ncomms11037
96. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of Neutrophil Extracellular Traps Promotes Cancer Metastasis via CCDC25. Nature (2020) 583:133–8. doi: 10.1038/s41586-020-2394-6
97. Binet F, Cagnone G, Crespo-Garcia S, Hata M, Neault M, Dejda A, et al. Neutrophil Extracellular Traps Target Senescent Vasculature for Tissue Remodeling in Retinopathy. Science (2020) 369(6506):eaay5356. doi: 10.1126/science.aay5356
98. Ballesteros I, Rubio-Ponce A, Genua M, Lusito E, Kwok I, Fernandez-Calvo G, et al. Co-Option of Neutrophil Fates by Tissue Environments. Cell (2020) 183:1282–97.e18. doi: 10.1016/j.cell.2020.10.003
99. Inoue M, Nakashima R, Enomoto M, Koike Y, Zhao X, Yip K, et al. Plasma Redox Imbalance Caused by Albumin Oxidation Promotes Lung-Predominant NETosis and Pulmonary Cancer Metastasis. Nat Commun (2018) 9:5116. doi: 10.1038/s41467-018-07550-x
100. Platnich JM, Muruve DA. NOD-Like Receptors and Inflammasomes: A Review of Their Canonical and non-Canonical Signaling Pathways. Arch Biochem Biophys (2019) 670:4–14. doi: 10.1016/j.abb.2019.02.008
101. Lamkanfi M. Emerging Inflammasome Effector Mechanisms. Nat Rev Immunol (2011) 11:213–20. doi: 10.1038/nri2936
102. Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. Inflammasome-Mediated Pyroptotic and Apoptotic Cell Death, and Defense Against Infection. Curr Opin Microbiol (2013) 16:319–26. doi: 10.1016/j.mib.2013.04.004
103. Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A. Role of the NLRP3 Inflammasome in Cancer. Mol Cancer (2018) 17:158. doi: 10.1186/s12943-018-0900-3
104. Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KS, McIntire CR, LeBlanc PM, et al. Control of Intestinal Homeostasis, Colitis, and Colitis-Associated Colorectal Cancer by the Inflammatory Caspases. Immunity (2010) 32:367–78. doi: 10.1016/j.immuni.2010.02.012
105. Van Gorp H, Lamkanfi M. The Emerging Roles of Inflammasome-Dependent Cytokines in Cancer Development. EMBO Rep (2019) 20(6):e47575. doi: 10.15252/embr.201847575
106. Xu S, Li X, Liu Y, Xia Y, Chang R, Zhang C. Inflammasome Inhibitors: Promising Therapeutic Approaches Against Cancer. J Hematol Oncol (2019) 12:64. doi: 10.1186/s13045-019-0755-0
107. Guo W, Sun Y, Liu W, Wu X, Guo L, Cai P, et al. Small Molecule-Driven Mitophagy-Mediated NLRP3 Inflammasome Inhibition is Responsible for the Prevention of Colitis-Associated Cancer. Autophagy (2014) 10:972–85. doi: 10.4161/auto.28374
108. Dupaul-Chicoine J, Arabzadeh A, Dagenais M, Douglas T, Champagne C, Morizot A, et al. The Nlrp3 Inflammasome Suppresses Colorectal Cancer Metastatic Growth in the Liver by Promoting Natural Killer Cell Tumoricidal Activity. Immunity (2015) 43:751–63. doi: 10.1016/j.immuni.2015.08.013
109. Kiss M, Vande Walle L, Saavedra PHV, Lebegge E, Van Damme H, Murgaski A, et al. IL1beta Promotes Immune Suppression in the Tumor Microenvironment Independent of the Inflammasome and Gasdermin D. Cancer Immunol Res (2021) 9:309–23. doi: 10.1158/2326-6066.CIR-20-0431
110. Segovia M, Russo S, Girotti MR, Rabinovich GA, Hill M. Role of Inflammasome Activation in Tumor Immunity Triggered by Immune Checkpoint Blockers. Clin Exp Immunol (2020) 200:155–62. doi: 10.1111/cei.13433
111. Dixon KO, Tabaka M, Schramm MA, Xiao S, Tang R, Dionne D, et al. TIM-3 Restrains Anti-Tumour Immunity by Regulating Inflammasome Activation. Nature (2021) 595:101–6. doi: 10.1038/s41586-021-03626-9
112. Pasto A, Consonni FM, Sica A. Influence of Innate Immunity on Cancer Cell Stemness. Int J Mol Sci (2020) 21:3352. doi: 10.3390/ijms21093352
113. Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-Dependent Membrane Repair Negatively Regulates Pyroptosis Downstream of GSDMD Activation. Science (2018) 362:956–60. doi: 10.1126/science.aar7607
114. Liu J, Kang R, Tang D. ESCRT-III-Mediated Membrane Repair in Cell Death and Tumor Resistance. Cancer Gene Ther (2021) 28:1–4. doi: 10.1038/s41417-020-0200-0
115. Chung C, Seo W, Silwal P, Jo EK. Crosstalks Between Inflammasome and Autophagy in Cancer. J Hematol Oncol (2020) 13:100. doi: 10.1186/s13045-020-00936-9
116. Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, et al. DDX3X Acts as a Live-or-Die Checkpoint in Stressed Cells by Regulating NLRP3 Inflammasome. Nature (2019) 573:590–4. doi: 10.1038/s41586-019-1551-2
Keywords: inflammasome, pyroptosis, NETosis, innate immunity, gasdermins, cancer immunotherapy
Citation: Li Y, Lv J, Shi W, Feng J, Liu M, Gan S, Wu H, Fan W and Shi M (2021) Inflammasome Signaling: A Novel Paradigm of Hub Platform in Innate Immunity for Cancer Immunology and Immunotherapy. Front. Immunol. 12:710110. doi: 10.3389/fimmu.2021.710110
Received: 15 May 2021; Accepted: 21 July 2021;
Published: 05 August 2021.
Edited by:
Janin Chandra, University of Queensland, AustraliaReviewed by:
Mariolina Salio, University of Oxford, United KingdomJuehua Yu, Hangzhou Cancer Hospital, China
Jingtao Chen, The First Hospital of Jilin University, China
Copyright © 2021 Li, Lv, Shi, Feng, Liu, Gan, Wu, Fan and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Shi, c2hpbWluZ0BoaXQuZWR1LmNu; Weiwei Fan, d3c3MDY5QHNpbmEuY29t
†These authors have contributed equally to this work