 Eric Alves
Eric Alves Emily McLeish
Emily McLeish Pilar Blancafort
Pilar Blancafort Jerome D. Coudert
Jerome D. Coudert Silvana Gaudieri
Silvana Gaudieri- 1School of Human Sciences, The University of Western Australia, Perth, WA, Australia
- 2Cancer Epigenetics Laboratory, The Harry Perkins Institute of Medical Research, Perth, WA, Australia
- 3Centre for Molecular Medicine and Innovative Therapeutics, Murdoch University, Perth, WA, Australia
- 4The Greehey Children’s Cancer Research Institute, The University of Texas Health Science Center at San Antonio, San Antonio, TX, United States
- 5Perron Institute for Neurological and Translational Science, Perth, WA, Australia
- 6School of Medicine, University of Notre Dame, Fremantle, WA, Australia
- 7Institute for Immunology and Infectious Diseases, Murdoch University, Perth, WA, Australia
- 8Division of Infectious Diseases, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, United States
The activating immune receptor natural killer group member D (NKG2D) and its cognate ligands represent a fundamental surveillance system of cellular distress, damage or transformation. Signaling through the NKG2D receptor-ligand axis is critical for early detection of viral infection or oncogenic transformation and the presence of functional NKG2D ligands (NKG2D-L) is associated with tumor rejection and viral clearance. Many viruses and tumors have developed mechanisms to evade NKG2D recognition via transcriptional, post-transcriptional or post-translational interference with NKG2D-L, supporting the concept that circumventing immune evasion of the NKG2D receptor-ligand axis may be an attractive therapeutic avenue for antiviral therapy or cancer immunotherapy. To date, the complexity of the NKG2D receptor-ligand axis and the lack of specificity of current NKG2D-targeting therapies has not allowed for the precise manipulation required to optimally harness NKG2D-mediated immunity. However, with the discovery of clustered regularly interspaced short palindromic repeats (CRISPRs) and CRISPR-associated (Cas) proteins, novel opportunities have arisen in the realm of locus-specific gene editing and regulation. Here, we give a brief overview of the NKG2D receptor-ligand axis in humans and discuss the levels at which NKG2D-L are regulated and dysregulated during viral infection and oncogenesis. Moreover, we explore the potential for CRISPR-based technologies to provide novel therapeutic avenues to improve and maximize NKG2D-mediated immunity.
Introduction
Interactions between hosts and pathogens are constantly evolving, and in this ongoing arms race each side aims to outsmart the other. Host and pathogen genetics form a key part of this competitive evolutionary relationship, with variation in their respective genomes having a considerable impact on host-pathogen dynamics. Many pathogens, such as viruses, generate this variation in the form of mutations as a by-product of their rapid, error-prone replication. Some of these mutations may confer a selective advantage to the pathogen, and via the process of natural selection are retained within the variant “pool”, termed quasispecies (1). Interestingly, this process of pathogen evolution bears an uncanny resemblance to what is seen during oncogenesis. During oncogenic transformation, genomic instability gives rise to tumor variants, which undergo a selective process to similarly maintain “fitter” variants within the tumor quasispecies (2). In both the tumor and pathogen contexts, host immune pressure constitutes a major selective force of pathogen/tumor evolution. In humans, the immune response relies on the strategic orchestration of innate and adaptive immunity, which comprises a variety of cell types and soluble molecules. This is regulated by the interaction of multiple receptors and ligands expressed at the cell membrane or released as soluble proteins. As such, pressure exerted via receptor-ligand mediated immune responses inadvertently selects for viral or oncogenic mutations that dysregulate receptor/ligand expression (3–5). Consequently, in order to compete against pathogen diversification and oncogenic transformation in this way, humans have over the course of this perennial host-pathogen battle developed in their arsenal a high level of polymorphism at loci encoding receptors/ligands responsible for immune recognition (6–8). One such receptor-ligand axis is the type II lectin-like transmembrane natural killer group 2 member D (NKG2D) receptor and its cognate ligands (NKG2D-L). Herein, we briefly overview the NKG2D receptor-ligand axis in humans, explore the levels at which NKG2D-L regulation/dysregulation occurs, and discuss how clustered regularly interspaced short palindromic repeats (CRISPR)-based technologies are poised to harness NKG2D-mediated immunity in the analogous contexts of oncogenic transformation and viral infection.
The NKG2D Receptor-Ligand Axis Plays an Important Role in Immune Recognition
The NKG2D Receptor
NKG2D is the most versatile and widely distributed activating/co-stimulatory natural killer (NK)-related receptor. First identified in human NK cells in 1991 (9), NKG2D has since been discovered on numerous cell subsets including, activated (αβ and γδ) T cells, natural killer T (NKT) cells, and mucosal-associated invariant T (MAIT) cells (10–13). Increasingly, NKG2D expression is also being identified on tissue-resident innate lymphoid cells (ILC), such as certain ILC1 (14, 15), ILC2 (16) and ILC3 (17) subsets. In humans, NKG2D is encoded by the KLRK1 gene and is located within the NK gene complex (NKC) on chromosome 12p. Moreover, NKG2D is highly conserved across multiple vertebrate species (18, 19). To date, two major human haplotype alleles of NKG2D have been identified, termed LNK1 (low activity) and HNK1 (high activity) alleles (20, 21), with surface expression of NKG2D lower in carriers of the low activity LNK1/LNK1 genotype (20, 22). When expressed at the cell surface, the 42 kDa homodimeric NKG2D receptor combines with the DNAX-activating protein 10 (DAP10) homodimer and following its engagement with cognate ligands initiates a cytotoxic cellular response and/or the secretion of pro-inflammatory cytokines (Figure 1A) (23–25).
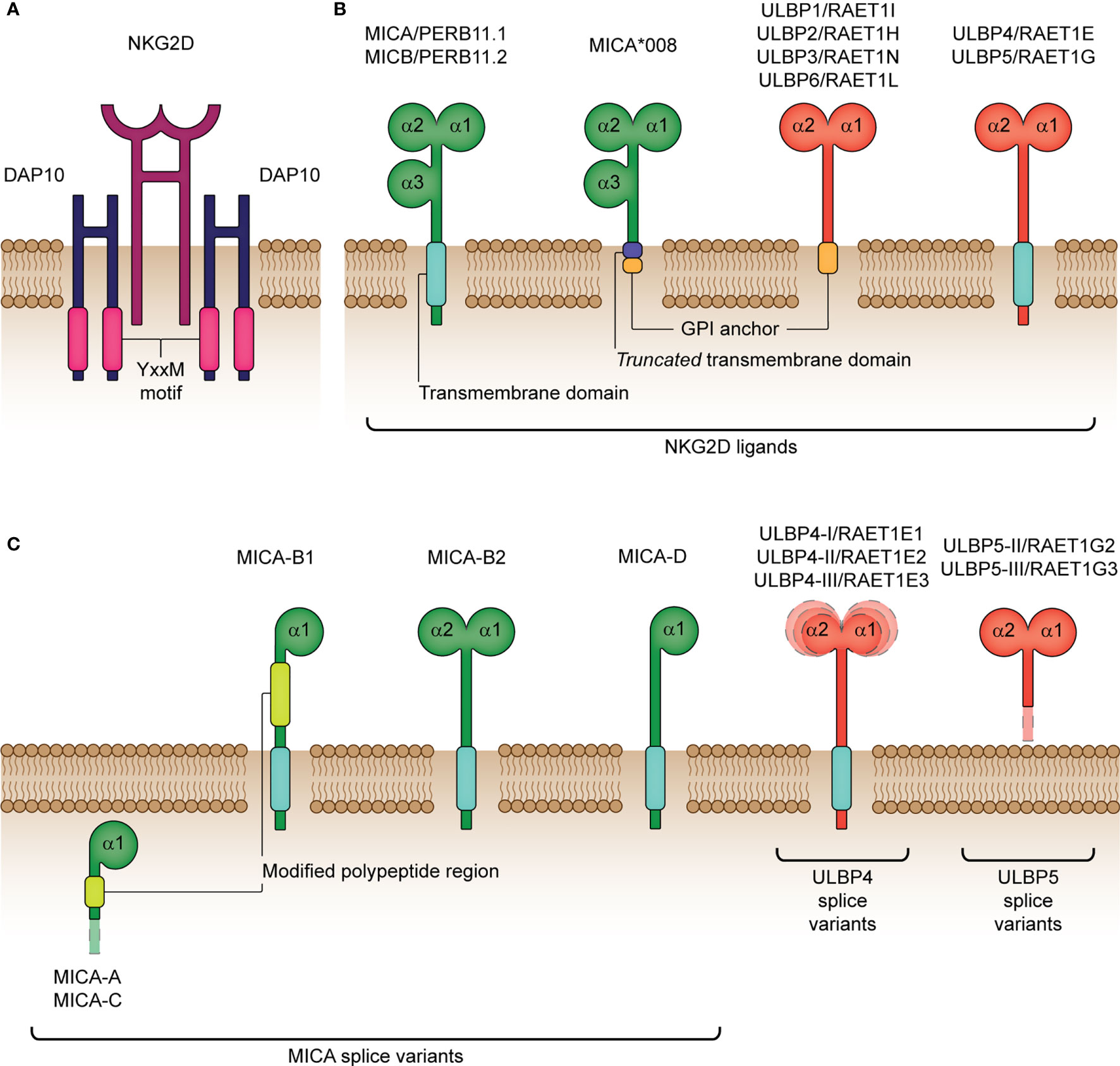
Figure 1 Structure of the human NKG2D receptor and cognate ligands. (A) The natural killer group 2 member D (NKG2D) receptor consists of a disulphide-linked homodimer that associates with the DNAX-activating protein 10 (DAP10) disulphide-linked homodimer for cellular signaling. DAP10 harbors a Tyr-X-X-Met (YxxM) motif, which binds the p85 subunit of phosphatidylinositol-3 kinase following phosphorylation. (B) NKG2D ligands encompass the MHC class-I polypeptide-related sequence A (MICA), MICB and six UL16-binding proteins (ULBP1-6). MICA/B (also termed PERB11.1/11.2) share similar structural and functional properties, with both containing three extracellular domains (α1, α2 and α3) and a transmembrane domain for binding to the cell surface. In comparison with full-length MICA alleles, MICA*008 differs by encoding a truncated protein due to a nucleotide insertion in the transmembrane domain and is known to acquire a glycolsylphosphatidylinositol (GPI) lipid anchor for cell surface expression. ULBP1-6 (also termed RAET1I/H/N/E/G/L) lack the α3 extracellular domain and are either bound to the cell surface by a GPI-anchor (ULBP1-3, 6) or transmembrane domain (ULBP4, 5). (C) Various functional MICA, ULBP4 and ULBP5 splice variants have been identified. MICA-A, -B1, -B2, -C and –D are known isoforms lacking the extracellular α3 domain and the α2 domain in the majority of isoforms (A, B1, C and D). Moreover, MICA-A and MICA-C both lack a transmembrane and cytoplasmic domain, which impairs their expression at the cell surface. RAET1E1/ULBP4-I, RAET1E2/ULBP4-II and RAET1E3/ULBP4-III are membrane-spanning splice variants with an extended α1 domain, reduced α1 domain and reduced α2 domain, respectively. RAET1G2/ULBP5-II and RAET1G3/ULBP5-III are truncated soluble splice variants resulting from two alternative premature stop codons before the transmembrane domain. The surface-expressed splice variants have been shown to bind NKG2D to a similar degree as compared to their wildtype isoforms, except for MICA-B2 and -D that bind NKG2D with a significantly weaker affinity.
NKG2D is also capable of facilitating the function of other activating receptors, depending on the inflammatory milieu and/or expressing cell type. For example, in activated NK cells primed by pro-inflammatory cytokines (e.g. interleukin (IL)-2 and IL-15), NKG2D provides direct stimulatory signals (26–29), whereas in resting NK cells, it synergizes as a co-activator with other receptors, such as NKp46 and 2B4 (30, 31). In αβ T cells, NKG2D typically provides a co-stimulatory signal, acting to promote T cell receptor (TCR)-dependent cytotoxicity, production of pro-inflammatory cytokines and memory differentiation (32–37). Interestingly, prolonged exposure to IL-15 has been shown to increase expression of NKG2D in CD8+ T cell subsets, potentiating TCR-independent activation (38, 39). Similarly in γδ T cells, NKG2D can function as a co-stimulatory molecule (40), but may also directly trigger cytotoxicity in a TCR-independent fashion (41). Alternatively, some γδ T cells have been shown to bypass the NKG2D receptor and recognize NKG2D-L, such as ULBP4 or MICA/B, directly via their TCR, implying a TCR agonistic role (42–44). In innate-like T cells, such as invariant NKT cells, NKG2D is restricted to the CD4- subsets, and functions to mediate direct lysis of target cells and co-stimulatory activation (12). Whereas, in MAIT cells, NKG2D is more prominent on CD8+ subsets, and functions as a co-stimulatory molecule (45) or, if in the presence of IL-15, exerts NKG2D-dependent innate-like cytotoxicity (13, 46). Lastly, in ILC subsets, early studies highlight that expression of NKG2D may aid in the production of pro- or anti-inflammatory mediators depending on the surrounding microenvironment (15, 16). However, further investigation is required to completely elucidate the impact of NKG2D-mediated signaling on ILC function. Altogether, although NKG2D expression, regulation and function differ across the above cell types, it undoubtedly plays a central regulatory role in the immune response and is vital for immunological surveillance against tumorigenic transformation and viral infection.
The NKG2D Cognate Ligands
Ligands for NKG2D comprise several families of major histocompatibility complex (MHC) class I-related molecules. In humans, these include the MHC class I polypeptide-related sequence A (MICA) and B (MICB), and the human cytomegalovirus (HCMV) glycoprotein UL16-binding protein (ULBP) family (ULBP1-ULBP6) (Figure 1B) (47–50). NKG2D-L are, for the most part, not constitutively expressed, but instead are selectively induced upon cellular stress, damage or transformation, as is caused by events such as viral infection or oncogenesis (32, 51). Moreover, as reviewed by Lanier (52), essentially all cell types are capable of expressing one or more types of NKG2D-L if given the appropriate stimulus. For example, Fujita and colleagues (53) identified two distinct ligand expression profiles in non-neoplastic epithelial tissues: ULBP5-ULBP3-MICA/B and ULBP2/6-ULBP1-ULBP4. Moreover, in cells undergoing tumorigenic transformation, high heterogeneity in NKG2D-L expression has been reported. Notably, expression of two or more NKG2D-Ls (often MICA and MICB) is more common in solid tumors, compared to hematological tumors (shown to predominantly express MICB) (54). Furthermore, co-expression of multiple allelic forms of the same ligand has also previously been identified at the surface of cells undergoing stress, which is strongly suggestive of functional redundancy in these molecules. It is also worth noting that NKG2D-L differ in their affinity (KD) and avidity for NKG2D, such that ULBP1 (the only ULBP member tested to date) has the highest affinity (1.1 μM), followed by MICA (0.9-1 μM) and MICB (800 nM) (55–59). Therefore, it is speculated that NKG2D may transduce different signals or activate separate downstream pathways based on which ligand or allelic variant is bound (60), which supports the various functions of NKG2D discussed prior.
Surprisingly, the role of NKG2D-L extends beyond providing a signal for cellular stress. In cells of the myeloid lineage, these ligands can mediate lymphocyte activation leading to cytotoxicity, cytokine production and proliferation. For instance, expression of MICA/B on dendritic cell-derived exosomes plays an important role in promoting NK cell differentiation and proliferation (61). Furthermore, ULBP2/3 expression levels are increased during CD34+ hematopoietic progenitor commitment to the granulomonocyte lineage, suggesting that NKG2D-L play a role in promoting myeloid differentiation (62). Alternatively, NKG2D-L expression on myeloid cells can lead to lymphocyte inactivation and maintenance of immune homeostasis. For example, persistent expression of membrane bound ULBP1 and MICB on myeloid cells induces NKG2D internalization and desensitization of NK cells (63). Moreover, overexpression of MICA on activated CD8+ T cells makes them susceptible to NK cell lysis, indicating that NKG2D-L may participate in immune homeostasis during ongoing immune responses (64). During infection with Mycobacterium tuberculosis, heightened ULBP1 expression on expanded T regulatory cells (Tregs) facilitates NK cell-mediated killing of these cells, thereby enhancing the overall immune response (65). Ultimately, NKG2D-L expression is associated with both cytotoxic and regulatory processes, as is reflected by the diverse roles played by these molecules in host immunity.
Redundancy and Overlapping Functions of NKG2D-L Ensure NKG2D Activation
MICA and MICB
The MICA and MICB genes were originally described as stress-induced MHC class I polypeptide-related sequences and are located in the MHC region on the short arm of chromosome 6 (47, 66). These genes are highly polymorphic (67), with, to date, over 100 described alleles (allelefrequencies.net). The MIC alleles have variations that are, to a certain extent, concentrated in the extracellular domains as well as truncated forms due to coding frame-shifts (e.g. MICA 5.1). Specific alleles have been associated with disease outcomes (68–70), influence the amount of soluble protein and impact binding affinity to its cognate receptor, NKG2D (71). Moreover, numerous MICA splice variants have been documented thus far (Figure 1C), with the majority binding NKG2D similarly as their wildtype counterparts, highlighting that lack of a domain (e.g. α3 domain) does not necessarily reduce binding affinity (60). Interestingly, there is a naturally occurring MICA-MICB null combination (deletion of MICA and premature stop codon for MICB) that exists on the HLA-B48 haplotype found in East Asian and South American populations (72–74). Furthermore, the MICA-MICB genes are merged in chimpanzees resulting in a hybrid form (75), while they are absent in mice (76). The presence of the null haplotype without obvious phenotypic consequences suggests redundancy in the NKG2D receptor-ligand axis but its overall importance is highlighted by the overlapping mechanisms exhibited by cancers and viruses to evade it (77).
ULBP1-ULBP6
Although the ULBPs are distantly related to MICA/B in sequence, they differ in their location, mapping instead to the opposite (long) arm of chromosome 6 (78, 79). Emerging data suggests that the extensive diversity seen in the ULBP family may be due to the functional or locational specialties of each ligand, as is evidenced with ULBP4 and its predominant expression in skin (80–82). Splice variants exist (ULBP4 (83), and ULBP5 (83, 84); Figure 1C) providing significant within locus diversity. Furthermore, as reviewed by Carapito & Bahram (19), clear differences in allele frequencies between geographically distinct populations exist for the ULBP family, which suggest that polymorphisms in ULBP may be a consequence of divergent selective pressures. Moreover, the possession of a large ULBP family in humans and other species is thought to provide a selective host advantage in the evasion of viruses and tumors. However, overall, the ULBP family appears to be less polymorphic than the MIC genes, albeit studies of ULBP gene polymorphisms and haplotypes remain limited (50, 85–88).
Viruses and Tumors Employ Convergent Mechanisms of NKG2D-L Dysregulation
The appropriate regulation of NKG2D-L is integral to the effective detection and elimination of virally infected or neoplastically-transformed cells. Many reviews to date have discussed the various levels of regulation involved in the control of NKG2D-L expression. For a comprehensive overview of these mechanisms in health and disease, we refer to previously published reviews (59, 89). In this section, however, we focus on convergent regulatory mechanisms exploited by both tumors and viruses to evade NKG2D-mediated immunity.
NKG2D-L expression is regulated at the level of transcription, post-transcription and post-translation through numerous pathways and molecules intrinsically linked to cellular stress (Table 1). As such, it is unsurprising that both viruses and tumors harbor various mechanisms that work in combination to hijack and dysregulate NKG2D-L at multiple levels. At the transcriptional level, viral proteins (e.g. HBV’s HBx and HBc) have been shown to directly suppress MICA/B (154). A similar strategy is achieved by tumors (e.g. melanoma), whereby cells with highly methylated NKG2D-L loci are selected for, given the resultant suppression of transcription (54). Although mechanistically different, both viral proteins and tumor-mediated methylation converge at the DNA level to hinder transcription of NKG2D-L and facilitate immune evasion.
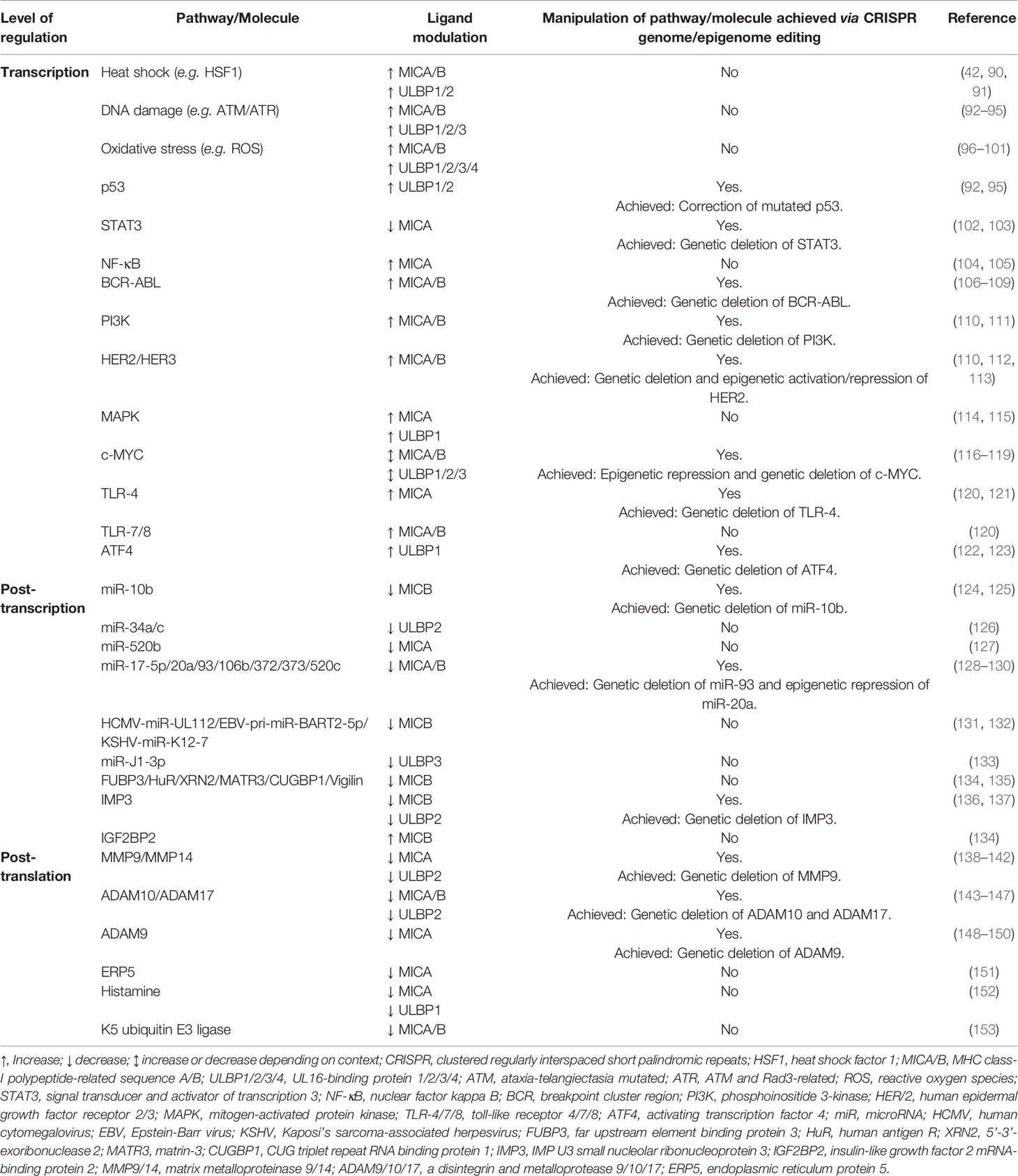
Table 1 Established pathways/molecules involved in the regulation of human NKG2D ligand expression and their targetability to date using CRISPR-based genome and epigenome editing.
At the post-transcriptional level, viruses and tumor cells exhibit convergence in their use of microRNAs (miRNAs) to inhibit NKG2D-L transcript translation. For instance, viral microRNAs have been shown to directly bind to the 3’ untranslated region of MICB (HCMV-miR-UL112, EBV-pri-miR-BART2-5p, KSHV-miR-K12-7) (128, 131) and ULBP3 (JCV-miR-J1-3p) (155), to trigger transcript destabilization and degradation. In addition, viral miRNAs can act indirectly by preventing translation of key components of the GPI-anchoring machinery (HSV1-miR-H8) (156) or blocking translation of surface shedding inhibitors (HCMV-miR-US25-2-3p) (157). Interestingly, host oncogenic miRNAs (124, 126–128, 158) also bind to the 3’ untranslated region of MICA (miR-520b) (127), MICB (miR-17-5p, miR-20a, miR-93, miR-106b, miR-372, miR-373, miR-520c) (128) and ULBP2 (miR-32a/c) (126), in a similar fashion to viral miRNAs, with some binding sites identified by Bauman & Mandelboim (155) as overlapping with those targeted by the viral miRNAs referenced above. Altogether, the shared binding sites and action of oncogenic and viral miRNAs indicate the convergent evolution of tumorigenic and viral immune evasion mechanisms at the post-transcriptional level.
Interference with NKG2D-L expression at the protein level is often a major target of both viruses and tumors. The two major mechanisms of reducing NKG2D-L surface expression have been reviewed by others (77, 89, 159, 160), and are understood to be (1), intracellular retention and degradation, and (2), surface shedding. In the context of viruses, key viral proteins (e.g. HCMV’s UL16, UL142) have been found to reside in the endoplasmic reticulum and cis-Golgi apparatus of cells, and cause intracellular retention and degradation of MICA/B and ULBP1-3 (161–164). Alternatively, viral infection with HIV or HCMV has also been shown to activate the shedding molecules ADAM10/17, which are otherwise essential for development and homeostasis, resulting in cleavage of MICA/B and ULBP2 (4, 157, 165). Manipulation at the protein level in a similar fashion is also extensively seen in cancer. For example, NKG2D-L have previously been shown to be retained in the endoplasmic reticulum or cytoplasm in a variety of cancer types, including melanoma, breast, colorectal, lung and gastric cancers (51, 166), resulting in reduced cell surface expression. Similarly, NKG2D-L shedding from tumor cells via enhanced expression of ADAM10/17, is understood to contribute significantly to the poor immunogenicity of many cancers (143–145). Again, the similarities between viruses and tumors in targeting NKG2D-L at the DNA, RNA and protein level is strongly suggestive of convergent evolution and highlights fundamental immune evasion mechanisms.
Therapeutic Potential Lies in CRISPR-Mediated Genetic and Epigenetic Manipulation
The ability to modify loci at the genetic and epigenetic level in a specific manner using CRISPR-associated (Cas) proteins, such as Cas9 (167, 168), has greatly expanded our knowledge of diseases, their genetic components and the development of targeted therapies. When combined with a short guide RNA (sgRNA), consisting of a non-coding trans-activating RNA annealed to a target-specific 20 nucleotide RNA, Cas9 is able to base pair with any target DNA located adjacent to a conserved protospacer-adjacent motif (5’-NGG-3’ in the frequently used Streptococcus pyogenes Cas9) and induce specific DNA cleavage. This process allows efficient and precise DNA editing. Beyond the CRISPR-Cas9 system, the synthesis of engineered variants, such as nuclease-deactivated Cas9 (dCas9), has provided new avenues for gene editing and regulation. The CRISPR-dCas9 system harbors two mutations (D10A and H840A), which deactivate Cas9’s cleavage capability (169). In doing so, the RNA-guided DNA-binding specificity of Cas9 can be harnessed to precisely direct effector domains that mediate transcriptional activation (170–172) or repression (173–175).
Despite the success of wild-type Cas9, its ability to introduce irreversible genetic changes, particularly at off-target sites, has raised safety concerns. Therefore, to date, clinical trials utilizing the CRISPR-Cas9 system have been performed ex vivo, where extensive off-target checks can be conducted (176, 177). Although clinical use of Cas9 remains limited due to these trepidations, the application of dCas9 in gene therapy is becoming increasingly likely, given its transient nature and inability to permanently alter the genetic code (178). Moreover, unlike other methods of gene therapy, dCas9-based methods are highly scalable and versatile, with the capability to target multiple loci simultaneously, termed multiplexed editing (179). Furthermore, as seen in combinatorial Cas9 screening systems (180), multiple orthologues of Cas9 can be used concurrently, allowing for synchronized activation and repression of separate loci.
CRISPR-Based Technologies Offer the Ability to Maximise the NKG2D Pathway in Immunity
In the context of viral infection and cancer, significant potential lies in the use of CRISPR-Cas9 [including other Cas proteins, such as the alternative DNA nuclease Cas12 (181), and RNA nuclease Cas13 (182)] and dCas9-based methods to target NKG2D and its ligands for enhanced immune recognition and elimination. The ease of use, high specificity and multiplexable nature of CRISPR-Cas9/dCas9-based genetic and epigenetic editing has clear applications in the development and improvement of NKG2D-directed therapies, as discussed below.
CRISPR-Cas9 Genetic Editing
To date, adoptive cell transfer (ACT) therapies, whereby peripheral blood mononuclear cells are collected, edited, expanded and reinfused back into patients, have successfully been developed to express NKG2D (largely on T cells) and shown considerable anti-tumoral and anti-viral potential in vitro (183–185). However, in vivo responses to NKG2D-focused chimeric antigen receptor (CAR) ACT therapy to date do not appear to be as robust, suggesting that inducing NKG2D alone may not be sufficient (186). To our knowledge, transduction of NKG2D in combination with other receptors within the same cell for improved CAR therapy has only recently been applied clinically, with one (1/9) active registered trial (as of 27 July 2021 on ClinicalTrials.gov) combining transduction of NKG2D and ACE2 for treatment of SARS-CoV-2 (NCT04324996). Most currently active trials (8/9) aim to solely target the NKG2D receptor, and this is partially to reduce the risk of insertional mutagenesis and gene dysregulation (187, 188). CRISPR-Cas9, however, provides a novel avenue for ACT therapy by providing a means to conduct targeted gene insertion in a multiplexed fashion to maximize host immunity (176, 177). Strong evidence suggests that CRISPR-based knockout of receptors responsive to immunosuppressive mediators, such as transforming growth factor beta receptor 2 (TGFβR2), or immune checkpoints (dampeners of cellular activation), such as programmed cell death 1 (PD-1), which play key roles in physiological immune homeostasis, are likely to improve NKG2D-mediated cellular cytotoxicity (189–191). Simultaneous editing of multiple loci, particularly immune checkpoints, in autologous or allogenic cells in this way is also likely to complement immune checkpoint inhibitor (ICI) therapy, such as anti-PD-L1. Moreover, CRISPR-Cas9 expands the potential of ACT therapeutics by facilitating targeted insertion of gene sequences (192), such as KLRK1, in cell subsets that otherwise have no or low expression of the NKG2D receptor. Similarly, knockout of inhibitory receptors within the same effector cell using CRISPR-based therapies is predicted to be an excellent starting point in improving ACT therapy outcomes via CRISPR-based methods. Notably, enhancement of NKG2D-dependent immune responses in this way requires careful consideration and evaluation of the potential for collateral adverse autoimmune reactions. Indeed, given the central role of the NKG2D receptor-ligand axis in autoimmune conditions, such as Crohn’s disease (36), coeliac disease (193), and rheumatoid arthritis (194), care needs to be taken to not generate autoreactive lymphocytes.
CRISPR-dCas9 Epigenetic Editing
Apart from direct gene editing to induce or improve NKG2D-mediated immunity, enhancing NKG2D-L on tumor or virus-infected cells represents both an alternative and complementary strategy to augment ACT therapy, ICIs and overall host immunity. In this approach, CRISPR-dCas9-based transcriptional activation or repression may be used to directly activate NKG2D-L loci and ensure their surface expression. Recently, Sekiba et al. (195), have shown this to be possible in vitro by applying CRISPR-dCas9 to transcriptionally activate MICA in Huh7 and HepG2 human hepatocellular carcinoma cell lines. Although no other NKG2D-L have been targeted in this way to date, it is predicted that multiplexed activation of several types of NKG2D-L is likely to be most effective in promoting NKG2D-mediated immunity and may resolve the low response rate of NKG2D-ACT therapy in vivo. However, our group and others (178, 196, 197) have extensively reviewed that multiplexed CRISPR-based editing, particularly in vivo within the tumor or virally infected cell, is best conducted with an optimized set of targets, rather than a large panel, so as to avoid reduced editing efficiency (retroactivity) and extensive off-target effects. Therefore, an accurate understanding of individual ligands and outcome of their binding to NKG2D is strongly recommended. For instance, given the locational and functional specialties within the ULBP family (80, 81), we suggest that activation of these ligands, either individually or in combination, is needed to elucidate their contribution to the NKG2D pathway in different tumoral/viral contexts. Moreover, it is likely that only a subset of ULBP or MIC members need to be targeted, with some unable to engage NKG2D effectively, as previously reported (81). Notably, given the higher affinity of ULBP1 for the NKG2D ligand, compared to MICA and MICB (59), it may serve as a better therapeutic target. However, further investigation is needed to elucidate the affinities of the remaining ULBP family members, and whether higher affinity to the NKG2D ligand directly translates to improved cytotoxicity. CRISPR-dCas9 can also theoretically be applied to other levels of NKG2D-L regulation (Table 1) for improved expression and immunity. An obvious example of this is in the transcriptional repression of genes involved in NKG2D-L shedding, such as ADAM10/17 or MMP9/14, which are commonly hijacked during viral infection and tumorigenesis, and are known to be targetable using CRISPR-based technologies (Table 1) (143, 165). Targeting the molecules responsible for proteolytic shedding of NKG2D-L in this way is likely to be beneficial both in the clinic and in furthering our basic biological understanding of the mechanisms driving proteolytic shedding. Although CRISPR-dCas9 epigenetic editing is transient and does not induce permanent genetic alterations, significant care needs to be taken to deliver this technology specifically to the target tissue or cell using precise delivery systems, so as to avoid inducing a severe systemic inflammatory state, particularly if used in combination with NKG2D-ACT therapeutics.
Concluding Remarks
Manipulation of the NKG2D receptor-ligand axis to improve host immunity is emerging as a novel therapeutic avenue in the era of CRISPR-based technologies. In addition to the direct enhancement of NKG2D on cell subsets to generate potent cytotoxic effector cells using Cas9 genetic editing, potential exists to use dCas9-based epigenetic editing methods to activate and lock NKG2D-L expression on tumor or virus-infected cells to promote their recognition and elimination. Although gaps remain in understanding how to optimize NKG2D-mediated immunity in different contexts, CRISPR-based multiplexed editing of NKG2D jointly with other genes on effector cells, or epigenetic activation of NKG2D-L in combination with one another on tumor or virally infected cells is likely to provide important insights to novel therapeutic approaches.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author Contributions
Conceptualization, EA and SG. Writing – original draft, EA, EM, JDC, and SG. Writing – review and editing, EA, EM, JDC, SG, and PB. Figures, EA. Funding acquisition, SG. Supervision, JDC and SG. All authors contributed to the article and approved the submitted version.
Funding
EA is a recipient of an Australian Government Research Training Program Scholarship at The University of Western Australia, BioZone PhD Scholarship at the University of Western Australia and Cancer Council WA PhD Top Up Scholarship. EM is a recipient of an Australian Government Research Training Program Scholarship at Murdoch University. PB is a recipient of an Australian Research Council Future Fellowship (FT130101767), Cancer Council WA Research Fellowship and Wesfarmers Women’s Cancers Fellowship. PB has support from the National Health and Medical Research Council (APP1187328, APP1109428, APP1165208, APP1147528 and APP1130212), the National Institutes of Health (R01CA170370 and R01DA036906), the National Breast Cancer Foundation and Cure Brain Cancer (NBCNBCF19-009), and the US DOD Peer Reviewed Cancer Research Program (CA190006). SG has support from the National Health and Medical Research Council (APP1148284).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Currenti J, Chopra A, John M, Leary S, McKinnon E, Alves E, et al. Deep Sequence Analysis of HIV Adaptation Following Vertical Transmission Reveals the Impact of Immune Pressure on the Evolution of HIV. PloS Pathog (2019) 15(12):e1008177. doi: 10.1371/journal.ppat.1008177
2. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. Ifnγ and Lymphocytes Prevent Primary Tumour Development and Shape Tumour Immunogenicity. Nature (2001) 410(6832):1107–11. doi: 10.1038/35074122
3. Cerboni C, Neri F, Casartelli N, Zingoni A, Cosman D, Rossi P, et al. Human Immunodeficiency Virus 1 Nef Protein Downmodulates the Ligands of the Activating Receptor NKG2D and Inhibits Natural Killer Cell-Mediated Cytotoxicity. J Gen Virol (2007) 88(Pt 1):242–50. doi: 10.1099/vir.0.82125-0
4. Matusali G, Tchidjou HK, Pontrelli G, Bernardi S, D’Ettorre G, Vullo V, et al. Soluble Ligands for the NKG2D Receptor Are Released During HIV-1 Infection and Impair NKG2D Expression and Cytotoxicity of NK Cells. FASEB J (2013) 27(6):2440–50. doi: 10.1096/fj.12-223057
5. Nolting A, Dugast A-S, Rihn S, Luteijn R, Carrington MF, Kane K, et al. MHC Class I Chain-Related Protein A Shedding in Chronic HIV-1 Infection Is Associated With Profound NK Cell Dysfunction. Virology (2010) 406(1):12–20. doi: 10.1016/j.virol.2010.05.014
6. Magaret A, Dong L, John M, Mallal SA, James I, Warren T, et al. HLA Class I and II Alleles, Heterozygosity and HLA-KIR Interactions Are Associated With Rates of Genital HSV Shedding and Lesions. Genes Immun (2016) 17(7):412–8. doi: 10.1038/gene.2016.42
7. Karnes JH, Bastarache L, Shaffer CM, Gaudieri S, Xu Y, Glazer AM, et al. Phenome-Wide Scanning Identifies Multiple Diseases and Disease Severity Phenotypes Associated With HLA Variants. Sci Trans Med (2017) 9(389):eaai8708. doi: 10.1126/scitranslmed.aai8708
8. Lucas M, Deshpande P, James I, Rauch A, Pfafferott K, Gaylard E, et al. Evidence of CD4+ T Cell-Mediated Immune Pressure on the Hepatitis C Virus Genome. Sci Rep (2018) 8(1):7224. doi: 10.1038/s41598-018-25559-6
9. Houchins JP, Yabe T, McSherry C, Bach FH. DNA Sequence Analysis of NKG2, A Family of Related cDNA Clones Encoding Type II Integral Membrane Proteins on Human Natural Killer Cells. J Exp Med (1991) 173(4):1017–20. doi: 10.1084/jem.173.4.1017
10. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK Cells and T Cells by NKG2D, a Receptor for Stress-Inducible MICA. Science (1999) 285(5428):727–9. doi: 10.1126/science.285.5428.727
11. Dai Z, Turtle CJ, Booth GC, Riddell SR, Gooley TA, Stevens AM, et al. Normally Occurring NKG2D+CD4+ T Cells Are Immunosuppressive and Inversely Correlated With Disease Activity in Juvenile-Onset Lupus. J Exp Med (2009) 206(4):793–805. doi: 10.1084/jem.20081648
12. Kuylenstierna C, Björkström NK, Andersson SK, Sahlström P, Bosnjak L, Paquin-Proulx D, et al. NKG2D Performs Two Functions in Invariant NKT Cells: Direct TCR-Independent Activation of NK-Like Cytolysis and Co-Stimulation of Activation by CD1d. Eur J Immunol (2011) 41(7):1913–23. doi: 10.1002/eji.200940278
13. Rha M-S, Han JW, Kim JH, Koh J-Y, Park HJ, Kim SI, et al. Human Liver CD8+ MAIT Cells Exert TCR/MR1-Independent Innate-Like Cytotoxicity in Response to IL-15. J Hepatol (2020) 73(3):640–50. doi: 10.1016/j.jhep.2020.03.033
14. Wang X, Peng H, Cong J, Wang X, Lian Z, Wei H, et al. Memory Formation and Long-Term Maintenance of IL-7rα+ ILC1s via a Lymph Node-Liver Axis. Nat Commun (2018) 9(1):4854. doi: 10.1038/s41467-018-07405-5
15. Salomé B, Gomez-Cadena A, Loyon R, Suffiotti M, Salvestrini V, Wyss T, et al. CD56 as a Marker of an ILC1-Like Population With NK Cell Properties That Is Functionally Impaired in AML. Blood Adv (2019) 3(22):3674–87. doi: 10.1182/bloodadvances.2018030478
16. Gomez-Cadena A, Spehner L, Kroemer M, Khelil MB, Bouiller K, Verdeil G, et al. Severe COVID-19 Patients Exhibit an ILC2 NKG2D+ Population in Their Impaired ILC Compartment. Cell Mol Immunol (2021) 18(2):484–6. doi: 10.1038/s41423-020-00596-2
17. Klose Christoph SN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of Type 1 ILCs From a Common Progenitor to All Helper-Like Innate Lymphoid Cell Lineages. Cell (2014) 157(2):340–56. doi: 10.1016/j.cell.2014.03.030
18. Shum BP, Flodin LR, Muir DG, Rajalingam R, Khakoo SI, Cleland S, et al. Conservation and Variation in Human and Common Chimpanzee CD94 and NKG2 Genes. J Immunol (2002) 168(1):240–52. doi: 10.4049/jimmunol.168.1.240
19. Carapito R, Bahram S. Genetics, Genomics, and Evolutionary Biology of NKG2D Ligands. Immunol Rev (2015) 267(1):88–116. doi: 10.1111/imr.12328
20. Hayashi T, Imai K, Morishita Y, Hayashi I, Kusunoki Y, Nakachi K. Identification of the NKG2D Haplotypes Associated With Natural Cytotoxic Activity of Peripheral Blood Lymphocytes and Cancer Immunosurveillance. Cancer Res (2006) 66(1):563–70. doi: 10.1158/0008-5472.Can-05-2776
21. Hara R, Onizuka M, Matsusita E, Kikkawa E, Nakamura Y, Ogiya D, et al. NKG2D Gene Polymorphism Is Associated With Disease Control of Chronic Myeloid Leukemia by Dasatinib. Blood (2016) 128(22):3091–. doi: 10.1182/blood.V128.22.3091.3091
22. Espinoza JL, Nguyen VH, Ichimura H, Pham TTT, Nguyen CH, Pham TV, et al. A Functional Polymorphism in the NKG2D Gene Modulates NK-Cell Cytotoxicity and Is Associated With Susceptibility to Human Papilloma Virus-Related Cancers. Sci Rep (2016) 6(1):39231. doi: 10.1038/srep39231
23. Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, et al. An Activating Immunoreceptor Complex Formed by NKG2D and DAP10. Science (1999) 285(5428):730–2. doi: 10.1126/science.285.5428.730
24. Rosen DB, Araki M, Hamerman JA, Chen T, Yamamura T, Lanier LL. A Structural Basis for the Association of DAP12 With Mouse, But Not Human, NKG2D. J Immunol (2004) 173(4):2470–8. doi: 10.4049/jimmunol.173.4.2470
25. Garrity D, Call ME, Feng J, Wucherpfennig KW. The Activating NKG2D Receptor Assembles in the Membrane With Two Signaling Dimers Into a Hexameric Structure. Proc Natl Acad Sci (2005) 102(21):7641–6. doi: 10.1073/pnas.0502439102
26. Ghasemi R, Lazear E, Wang X, Arefanian S, Zheleznyak A, Carreno BM, et al. Selective Targeting of IL-2 to NKG2D Bearing Cells for Improved Immunotherapy. Nat Commun (2016) 7(1):12878. doi: 10.1038/ncomms12878
27. Horng T, Bezbradica JS, Medzhitov R. NKG2D Signaling Is Coupled to the Interleukin 15 Receptor Signaling Pathway. Nat Immunol (2007) 8(12):1345–52. doi: 10.1038/ni1524
28. Coudert JD, Zimmer J, Tomasello E, Cebecauer M, Colonna M, Vivier E, et al. Altered NKG2D Function in NK Cells Induced by Chronic Exposure to NKG2D Ligand–Expressing Tumor Cells. Blood (2005) 106(5):1711–7. doi: 10.1182/blood-2005-03-0918
29. Coudert JD, Scarpellino L, Gros F, Vivier E, Held W. Sustained NKG2D Engagement Induces Cross-Tolerance of Multiple Distinct NK Cell Activation Pathways. Blood (2008) 111(7):3571–8. doi: 10.1182/blood-2007-07-100057
30. Bryceson YT, March ME, Ljunggren H-G, Long EO. Synergy Among Receptors on Resting NK Cells for the Activation of Natural Cytotoxicity and Cytokine Secretion. Blood (2006) 107(1):159–66. doi: 10.1182/blood-2005-04-1351
31. Bryceson YT, Ljunggren H-G, Long EO. Minimal Requirement for Induction of Natural Cytotoxicity and Intersection of Activation Signals by Inhibitory Receptors. Blood (2009) 114(13):2657–66. doi: 10.1182/blood-2009-01-201632
32. Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T. Costimulation of CD8αβ T Cells by NKG2D via Engagement by MIC Induced on Virus-Infected Cells. Nat Immunol (2001) 2(3):255–60. doi: 10.1038/85321
33. Maasho K, Opoku-Anane J, Marusina AI, Coligan JE, Borrego F. NKG2D Is a Costimulatory Receptor for Human Naive CD8+ T Cells. J Immunol (2005) 174(8):4480–4. doi: 10.4049/jimmunol.174.8.4480
34. Barber A, Sentman CL. NKG2D Receptor Regulates Human Effector T-Cell Cytokine Production. Blood (2011) 117(24):6571–81. doi: 10.1182/blood-2011-01-329417
35. Perez C, Prajapati K, Burke B, Plaza-Rojas L, Zeleznik-Le NJ, Guevara-Patino JA. NKG2D Signaling Certifies Effector CD8 T Cells for Memory Formation. J ImmunoTher Cancer (2019) 7(1):48. doi: 10.1186/s40425-019-0531-2
36. Allez M, Tieng V, Nakazawa A, Treton X, Pacault V, Dulphy N, et al. CD4+NKG2D+ T Cells in Crohn’s Disease Mediate Inflammatory and Cytotoxic Responses Through MICA Interactions. Gastroenterology (2007) 132(7):2346–58. doi: 10.1053/j.gastro.2007.03.025
37. Fasth AER, Björkström NK, Anthoni M, Malmberg K-J, Malmström V. Activating NK-Cell Receptors Co-Stimulate CD4+CD28– T Cells in Patients With Rheumatoid Arthritis. Eur J Immunol (2010) 40(2):378–87. doi: 10.1002/eji.200939399
38. Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated Induction by IL15 of a TCR-Independent NKG2D Signaling Pathway Converts CTL Into Lymphokine-Activated Killer Cells in Celiac Disease. Immunity (2004) 21(3):357–66. doi: 10.1016/j.immuni.2004.06.020
39. Correia MP, Costa AV, Uhrberg M, Cardoso EM, Arosa FA. IL-15 Induces CD8+ T Cells to Acquire Functional NK Receptors Capable of Modulating Cytotoxicity and Cytokine Secretion. Immunobiology (2011) 216(5):604–12. doi: 10.1016/j.imbio.2010.09.012
40. Rincon-Orozco B, Kunzmann V, Wrobel P, Kabelitz D, Steinle A, Herrmann T. Activation of Vγ9vδ2 T Cells by NKG2D. J Immunol (2005) 175(4):2144–51. doi: 10.4049/jimmunol.175.4.2144
41. Das H, Groh V, Kuijl C, Sugita M, Morita CT, Spies T, et al. MICA Engagement by Human Vγ2vδ2 T Cells Enhances Their Antigen-Dependent Effector Function. Immunity (2001) 15(1):83–93. doi: 10.1016/S1074-7613(01)00168-6
42. Groh V, Steinle A, Bauer S, Spies T. Recognition of Stress-Induced MHC Molecules by Intestinal Epithelial γδ T Cells. Science (1998) 279(5357):1737–40. doi: 10.1126/science.279.5357.1737
43. Poggi A, Venturino C, Catellani S, Clavio M, Miglino M, Gobbi M, et al. Vδ1 T Lymphocytes From B-CLL Patients Recognize ULBP3 Expressed on Leukemic B Cells and Up-Regulated by Trans-Retinoic Acid. Cancer Res (2004) 64(24):9172–9. doi: 10.1158/0008-5472.Can-04-2417
44. Kong Y, Cao W, Xi X, Ma C, Cui L, He W. The NKG2D Ligand ULBP4 Binds to Tcrγ9/δ2 and Induces Cytotoxicity to Tumor Cells Through Both Tcrγδ and NKG2D. Blood (2009) 114(2):310–7. doi: 10.1182/blood-2008-12-196287
45. Le Bourhis L, Dusseaux M, Bohineust A, Bessoles S, Martin E, Premel V, et al. MAIT Cells Detect and Efficiently Lyse Bacterially-Infected Epithelial Cells. PloS Pathog (2013) 9(10):e1003681. doi: 10.1371/journal.ppat.1003681
46. Dias J, Boulouis C, Gorin J-B, van den Biggelaar RHGA, Lal KG, Gibbs A, et al. The CD4–CD8– MAIT Cell Subpopulation Is a Functionally Distinct Subset Developmentally Related to the Main CD8+ MAIT Cell Pool. Proc Natl Acad Sci (2018) 115(49):E11513–E22. doi: 10.1073/pnas.1812273115
47. Leelayuwat C, Townend DC, Degli-Esposti MA, Abraham LJ, Dawkins RL. A New Polymorphic and Multicopy MHC Gene Family Related to Nonmammalian Class I. Immunogenetics (1994) 40(5):339–51. doi: 10.1007/BF01246675
48. Gaudieri S, Leelayuwat C, Townend DC, Kulski JK, Dawkins RL. Genomic Characterization of the Region Between HLA-B and TNF: Implications for the Evolution of Multicopy Gene Families. J Mol Evol (1997) 44(Suppl 1):S147–54. doi: 10.1007/pl00000064
49. Gaudieri S, Leelayuwat C, Townend DC, Mullberg J, Cosman D, Dawkins RL. Allelic and Interlocus Comparison of the PERB11 Multigene Family in the MHC. Immunogenetics (1997) 45(3):209–16. doi: 10.1007/s002510050191
50. Eagle RA, Traherne JA, Hair JR, Jafferji I, Trowsdale J. ULBP6/RAET1L Is an Additional Human NKG2D Ligand. Eur J Immunol (2009) 39(11):3207–16. doi: 10.1002/eji.200939502
51. Ghadially H, Brown L, Lloyd C, Lewis L, Lewis A, Dillon J, et al. MHC Class I Chain-Related Protein A and B (MICA and MICB) Are Predominantly Expressed Intracellularly in Tumour and Normal Tissue. Br J Cancer (2017) 116(9):1208–17. doi: 10.1038/bjc.2017.79
52. Lanier LL. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol Res (2015) 3(6):575–82. doi: 10.1158/2326-6066.Cir-15-0098
53. Fujita H, Hatanaka Y, Sutoh Y, Suzuki Y, Oba K, Hatanaka KC, et al. Immunohistochemical Validation and Expression Profiling of NKG2D Ligands in a Wide Spectrum of Human Epithelial Neoplasms. J Histochem Cytochem (2015) 63(3):217–27. doi: 10.1369/0022155414563800
54. Baragaño Raneros A, Martín-Palanco V, Fernandez AF, Rodriguez RM, Fraga MF, Lopez-Larrea C, et al. Methylation of NKG2D Ligands Contributes to Immune System Evasion in Acute Myeloid Leukemia. Genes Immun (2015) 16(1):71–82. doi: 10.1038/gene.2014.58
55. Isernhagen A, Malzahn D, Viktorova E, Elsner L, Monecke S, von Bonin F, et al. The MICA-129 Dimorphism Affects NKG2D Signaling and Outcome of Hematopoietic Stem Cell Transplantation. EMBO Mol Med (2015) 7(11):1480–502. doi: 10.15252/emmm.201505246
56. Li P, Morris DL, Willcox BE, Steinle A, Spies T, Strong RK. Complex Structure of the Activating Immunoreceptor NKG2D and Its MHC Class I–like Ligand MICA. Nat Immunol (2001) 2(5):443–51. doi: 10.1038/87757
57. Radaev S, Rostro B, Brooks AG, Colonna M, Sun PD. Conformational Plasticity Revealed by the Cocrystal Structure of NKG2D and Its Class I MHC-Like Ligand ULBP3. Immunity (2001) 15(6):1039–49. doi: 10.1016/S1074-7613(01)00241-2
58. Steinle A, Li P, Morris DL, Groh V, Lanier LL, Strong RK, et al. Interactions of Human NKG2D With its Ligands MICA, MICB, and Homologs of the Mouse RAE-1 Protein Family. Immunogenetics (2001) 53(4):279–87. doi: 10.1007/s002510100325
59. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of Ligands for the NKG2D Activating Receptor. Annu Rev Immunol (2013) 31(1):413–41. doi: 10.1146/annurev-immunol-032712-095951
60. Gavlovsky P-J, Tonnerre P, Gérard N, Nedellec S, Daman AW, McFarland BJ, et al. Alternative Splice Transcripts for MHC Class I–Like MICA Encode Novel NKG2D Ligands With Agonist or Antagonist Functions. J Immunol (2016) 197(3):736–46. doi: 10.4049/jimmunol.1501416
61. Viaud S, Terme M, Flament C, Taieb J, André F, Novault S, et al. Dendritic Cell-Derived Exosomes Promote Natural Killer Cell Activation and Proliferation: A Role for NKG2D Ligands and IL-15rα. PloS One (2009) 4(3):e4942. doi: 10.1371/journal.pone.0004942
62. Guilloton F, Thonel A, Jean C, Demur C, Mas V, Laurent G, et al. Tnfα Stimulates NKG2D-Mediated Lytic Activity of Acute Myeloid Leukemic Cells. Leukemia (2005) 19:2206–14. doi: 10.1038/sj.leu.2403952
63. Crane CA, Austgen K, Haberthur K, Hofmann C, Moyes KW, Avanesyan L, et al. Immune Evasion Mediated by Tumor-Derived Lactate Dehydrogenase Induction of NKG2D Ligands on Myeloid Cells in Glioblastoma Patients. Proc Natl Acad Sci (2014) 111(35):12823–8. doi: 10.1073/pnas.1413933111
64. Molinero LL, Fuertes MB, Rabinovich GA, Fainboim L, Zwirner NW. Activation-Induced Expression of MICA on T Lymphocytes Involves Engagement of CD3 and CD28. J Leukocyte Biol (2002) 71:791–7. doi: 10.1189/jlb.71.5.791
65. Roy S, Barnes PF, Garg A, Wu S, Cosman D, Vankayalapati R. NK Cells Lyse T Regulatory Cells That Expand in Response to an Intracellular Pathogen. J Immunol (2008) 180(3):1729–36. doi: 10.4049/jimmunol.180.3.1729
66. Bahram S, Bresnahan M, Geraghty DE, Spies T. A Second Lineage of Mammalian Major Histocompatibility Complex Class I Genes. Proc Natl Acad Sci (1994) 91(14):6259–63. doi: 10.1073/pnas.91.14.6259
67. Gaudieri S, Kulski JK, Dawkins RL, Gojobori T. Different Evolutionary Histories in Two Subgenomic Regions of the Major Histocompatibility Complex. Genome Res (1999) 9:541–9. doi: 10.1101/gr.9.6.541
68. Kumar V, Kato N, Urabe Y, Takahashi A, Muroyama R, Hosono N, et al. Genome-Wide Association Study Identifies a Susceptibility Locus for HCV-Induced Hepatocellular Carcinoma. Nat Genet (2011) 43(5):455–8. doi: 10.1038/ng.809
69. Tong HV, Toan NL, Song LH, Bock CT, Kremsner PG, Velavan TP. Hepatitis B Virus-Induced Hepatocellular Carcinoma: Functional Roles of MICA Variants. J Viral Hepat (2013) 20(10):687–98. doi: 10.1111/jvh.12089
70. Iwaszko M, Swierkot J, Dratwa M, Wysoczanska B, Korman L, Bugaj B, et al. Association of MICA-129met/Val Polymorphism With Clinical Outcome of Anti-TNF Therapy and MICA Serum Levels in Patients With Rheumatoid Arthritis. Pharmacogenom J (2020) 20(6):760–9. doi: 10.1038/s41397-020-0164-3
71. Isernhagen A, Schilling D, Monecke S, Shah P, Elsner L, Walter L, et al. The MICA-129met/Val Dimorphism Affects Plasma Membrane Expression and Shedding of the NKG2D Ligand MICA. Immunogenetics (2016) 68(2):109–23. doi: 10.1007/s00251-015-0884-8
72. Komatsu-Wakui M, Tokunaga K, Ishikawa Y, Kashiwase K, Moriyama S, Tsuchiya N, et al. MIC-A Polymorphism in Japanese and a MIC-A-MIC-B Null Haplotype. Immunogenetics (1999) 49(7):620–8. doi: 10.1007/s002510050658
73. Komatsu-Wakui M, Tokunaga K, Ishikawa Y, Leelayuwat C, Kashiwase K, Tanaka H, et al. Wide Distribution of the MICA-MICB Null Haplotype in East Asians. Tissue Antigens (2001) 57(1):1–8. doi: 10.1034/j.1399-0039.2001.057001001.x
74. Aida K, Russomando G, Kikuchi M, Candia N, Franco L, Almiron M, et al. High Frequency of MIC Null Haplotype (HLA-B48-MICA-Del-MICB*0107 N) in the Angaite Amerindian Community in Paraguay. Immunogenetics (2002) 54(6):439–41. doi: 10.1007/s00251-002-0485-1
75. Anzai T, Shiina T, Kimura N, Yanagiya K, Kohara S, Shigenari A, et al. Comparative Sequencing of Human and Chimpanzee MHC Class I Regions Unveils Insertions/Deletions as the Major Path to Genomic Divergence. Proc Natl Acad Sci (2003) 100(13):7708–13. doi: 10.1073/pnas.1230533100
76. Kim Y, Born C, Bléry M, Steinle A. MICAgen Mice Recapitulate the Highly Restricted But Activation-Inducible Expression of the Paradigmatic Human NKG2D Ligand MICA. Front Immunol (2020) 11:960. doi: 10.3389/fimmu.2020.00960
77. Baugh R, Khalique H, Seymour LW. Convergent Evolution by Cancer and Viruses in Evading the NKG2D Immune Response. Cancers (Basel) (2020) 12:3827. doi: 10.3390/cancers12123827
78. Cosman D, Müllberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, et al. ULBPs, Novel MHC Class I-Related Molecules, Bind to CMV Glycoprotein UL16 and Stimulate NK Cytotoxicity Through the NKG2D Receptor. Immunity (2001) 14(2):123–33. doi: 10.1016/S1074-7613(01)00095-4
79. Sutherland CL, Chalupny NJ, Schooley K, VandenBos T, Kubin M, Cosman D. UL16-Binding Proteins, Novel MHC Class I-Related Proteins, Bind to NKG2D and Activate Multiple Signaling Pathways in Primary NK Cells. J Immunol (2002) 168(2):671–9. doi: 10.4049/jimmunol.168.2.671
80. Jan Chalupny N, Sutherland CL, Lawrence WA, Rein-Weston A, Cosman D. ULBP4 Is a Novel Ligand for Human NKG2D. Biochem Biophys Res Commun (2003) 305(1):129–35. doi: 10.1016/S0006-291X(03)00714-9
81. Sutherland CL, Rabinovich B, Chalupny NJ, Brawand P, Miller R, Cosman D. ULBPs, Human Ligands of the NKG2D Receptor, Stimulate Tumor Immunity With Enhancement by IL-15. Blood (2006) 108(4):1313–9. doi: 10.1182/blood-2005-11-011320
82. López-Cobo S, Romera-Cárdenas G, García-Cuesta EM, Reyburn HT, Valés-Gómez M. Transfer of the Human NKG2D Ligands UL16 Binding Proteins (ULBP) 1–3 Is Related to Lytic Granule Release and Leads to Ligand Retransfer and Killing of ULBP-Recipient Natural Killer Cells. Immunology (2015) 146(1):70–80. doi: 10.1111/imm.12482
83. Cao W, Xi X, Wang Z, Dong L, Hao Z, Cui L, et al. Four Novel ULBP Splice Variants Are Ligands for Human NKG2D. Int Immunol (2008) 20(8):981–91. doi: 10.1093/intimm/dxn057
84. Eagle RA, Flack G, Warford A, Martínez-Borra J, Jafferji I, Traherne JA, et al. Cellular Expression, Trafficking, and Function of Two Isoforms of Human ULBP5/RAET1G. PloS One (2009) 4(2):e4503. doi: 10.1371/journal.pone.0004503
85. Romphruk AV, Romphruk A, Naruse TK, Raroengjai S, Puapairoj C, Inoko H, et al. Polymorphisms of NKG2D Ligands: Diverse RAET1/ULBP Genes in Northeastern Thais. Immunogenetics (2009) 61(9):611–7. doi: 10.1007/s00251-009-0394-7
86. Cox ST, Arrieta-Bolaños E, Pesoa S, Vullo C, Madrigal JA, Saudemont A. RAET1/ULBP Alleles and Haplotypes Among Kolla South American Indians. Hum Immunol (2013) 74(6):775–82. doi: 10.1016/j.humimm.2013.01.030
87. Antoun A, Jobson S, Cook M, O’Callaghan CA, Moss P, Briggs DC. Single Nucleotide Polymorphism Analysis of the NKG2D Ligand Cluster on the Long Arm of Chromosome 6: Extensive Polymorphisms and Evidence of Diversity Between Human Populations. Hum Immunol (2010) 71(6):610–20. doi: 10.1016/j.humimm.2010.02.018
88. Cox ST, Pearson H, Laza-Briviesca R, Pesoa S, Vullo C, Madrigal JA, et al. Characterization of 5′ Promoter and Exon 1–3 Polymorphism of the RAET1E Gene. Hum Immunol (2016) 77(1):96–103. doi: 10.1016/j.humimm.2015.10.017
89. Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, et al. NKG2D and Its Ligands: “One for All, All for One”. Front Immunol (2018) 9:476. doi: 10.3389/fimmu.2018.00476
90. Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell Stress-Regulated Human Major Histocompatibility Complex Class I Gene Expressed in Gastrointestinal Epithelium. Proc Natl Acad Sci (1996) 93(22):12445–50. doi: 10.1073/pnas.93.22.12445
91. Eagle RA, Traherne JA, Ashiru O, Wills MR, Trowsdale J. Regulation of NKG2D Ligand Gene Expression. Hum Immunol (2006) 67(3):159–69. doi: 10.1016/j.humimm.2006.02.015
92. Textor S, Fiegler N, Arnold A, Porgador A, Hofmann TG, Cerwenka A. Human NK Cells Are Alerted to Induction of P53 in Cancer Cells by Upregulation of the NKG2D Ligands ULBP1 and ULBP2. Cancer Res (2011) 71(18):5998. doi: 10.1158/0008-5472.CAN-10-3211
93. Herrmann F, Garriga-Canut M, Baumstark R, Fajardo-Sanchez E, Cotterell J, Minoche A, et al. P53 Gene Repair With Zinc Finger Nucleases Optimised by Yeast 1-Hybrid and Validated by Solexa Sequencing. PloS One (2011) 6(6):e20913. doi: 10.1371/journal.pone.0020913
94. Hu Z, Ding W, Zhu D, Yu L, Jiang X, Wang X, et al. TALEN-Mediated Targeting of HPV Oncogenes Ameliorates HPV-Related Cervical Malignancy. J Clin Invest (2015) 125(1):425–36. doi: 10.1172/JCI78206
95. Zhan H, Xie H, Zhou Q, Liu Y, Huang W. Synthesizing a Genetic Sensor Based on CRISPR-Cas9 for Specifically Killing P53-Deficient Cancer Cells. ACS Synthetic Biol (2018) 7(7):1798–807. doi: 10.1021/acssynbio.8b00202
96. Yamamoto K, Fujiyama Y, Andoh A, Bamba T, Okabe H. Oxidative Stress Increases MICA and MICB Gene Expression in the Human Colon Carcinoma Cell Line (CaCo-2). Biochim Biophys Acta (BBA) - Gen Subj (2001) 1526(1):10–2. doi: 10.1016/S0304-4165(01)00099-X
97. Borchers MT, Harris NL, Wesselkamper SC, Vitucci M, Cosman D. NKG2D Ligands Are Expressed on Stressed Human Airway Epithelial Cells. Am J Physiol-Lung Cell Mol Physiol (2006) 291(2):L222–L31. doi: 10.1152/ajplung.00327.2005
98. Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T. Promoter Region Architecture and Transcriptional Regulation of the Genes for the MHC Class I-Related Chain A and B Ligands of NKG2D. J Immunol (2007) 178(2):961. doi: 10.4049/jimmunol.178.2.961
99. Peraldi M-N, Berrou J, Dulphy N, Seidowsky A, Haas P, Boissel N, et al. Oxidative Stress Mediates a Reduced Expression of the Activating Receptor NKG2D in NK Cells From End-Stage Renal Disease Patients. J Immunol (2009) 182(3):1696. doi: 10.4049/jimmunol.182.3.1696
100. Hedlund M, Nagaeva O, Kargl D, Baranov V, Mincheva-Nilsson L. Thermal- and Oxidative Stress Causes Enhanced Release of NKG2D Ligand-Bearing Immunosuppressive Exosomes in Leukemia/Lymphoma T and B Cells. PloS One (2011) 6(2):e16899. doi: 10.1371/journal.pone.0016899
101. Soriani A, Iannitto ML, Ricci B, Fionda C, Malgarini G, Morrone S, et al. Reactive Oxygen Species– and DNA Damage Response–Dependent NK Cell Activating Ligand Upregulation Occurs at Transcriptional Levels and Requires the Transcriptional Factor E2F1. J Immunol (2014) 193(2):950. doi: 10.4049/jimmunol.1400271
102. Bedel R, Thiery-Vuillemin A, Grandclement C, Balland J, Remy-Martin J-P, Kantelip B, et al. Novel Role for STAT3 in Transcriptional Regulation of NK Immune Cell Targeting Receptor MICA on Cancer Cells. Cancer Res (2011) 71(5):1615. doi: 10.1158/0008-5472.CAN-09-4540
103. Xie B, Zhang L, Hu W, Fan M, Jiang N, Duan Y, et al. Dual Blockage of STAT3 and ERK1/2 Eliminates Radioresistant GBM Cells. Redox Biol (2019) 24:101189. doi: 10.1016/j.redox.2019.101189
104. Lin D, Lavender H, Soilleux EJ, O’Callaghan CA. NF-κb Regulates MICA Gene Transcription in Endothelial Cell Through a Genetically Inhibitable Control Site. J Biol Chem (2012) 287(6):4299–310. doi: 10.1074/jbc.M111.282152
105. Molinero LL, Fuertes MB, Girart MV, Fainboim L, Rabinovich GA, Costas MA, et al. NF-κb Regulates Expression of the MHC Class I-Related Chain A Gene in Activated T Lymphocytes. J Immunol (2004) 173(9):5583–90. doi: 10.4049/jimmunol.173.9.5583
106. Boissel N, Rea D, Tieng V, Dulphy N, Brun M, Cayuela J-M, et al. BCR/ABL Oncogene Directly Controls MHC Class I Chain-Related Molecule A Expression in Chronic Myelogenous Leukemia. J Immunol (2006) 176(8):5108. doi: 10.4049/jimmunol.176.8.5108
107. Cebo C, Da Rocha S, Wittnebel S, Turhan AG, Abdelali J, Caillat-Zucman S, et al. The Decreased Susceptibility of Bcr/Abl Targets to NK Cell-Mediated Lysis in Response to Imatinib Mesylate Involves Modulation of NKG2D Ligands, GM1 Expression, and Synapse Formation. J Immunol (2006) 176(2):864. doi: 10.4049/jimmunol.176.2.864
108. Luo Z, Gao M, Huang N, Wang X, Yang Z, Yang H, et al. Efficient Disruption of Bcr-Abl Gene by CRISPR RNA-Guided FokI Nucleases Depresses the Oncogenesis of Chronic Myeloid Leukemia Cells. J Exp Clin Cancer Res (2019) 38(1):224. doi: 10.1186/s13046-019-1229-5
109. Yu-Ting T, Lin Y, Fei X, Jiaming W, Markus M, Sai-Juan C, et al. CRISPR/Cas9-Mediated Gene Deletion Efficiently Retards the Progression of Philadelphia-Positive Acute Lymphoblastic Leukemia in a P210 BCR-ABL1T315I Mutation Mouse Model. Haematologica (2020) 105(5):e232–e6. doi: 10.3324/haematol.2019.229013
110. Okita R, Mougiakakos D, Ando T, Mao Y, Sarhan D, Wennerberg E, et al. HER2/HER3 Signaling Regulates NK Cell-Mediated Cytotoxicity via MHC Class I Chain-Related Molecule A and B Expression in Human Breast Cancer Cell Lines. J Immunol (2012) 188(5):2136–45. doi: 10.4049/jimmunol.1102237
111. Zhang L, Li Y, Wang Q, Chen Z, Li X, Wu Z, et al. The PI3K Subunits, P110α and P110β Are Potential Targets for Overcoming P-Gp and BCRP-Mediated MDR in Cancer. Mol Cancer (2020) 19(1):10. doi: 10.1186/s12943-019-1112-1
112. Wang H, Sun W. CRISPR-Mediated Targeting of HER2 Inhibits Cell Proliferation Through a Dominant Negative Mutation. Cancer Lett (2017) 385:137–43. doi: 10.1016/j.canlet.2016.10.033
113. Klann TS, Black JB, Chellappan M, Safi A, Song L, Hilton IB, et al. CRISPR–Cas9 Epigenome Editing Enables High-Throughput Screening for Functional Regulatory Elements in the Human Genome. Nat Biotechnol (2017) 35(6):561–8. doi: 10.1038/nbt.3853
114. Soriani A, Borrelli C, Ricci B, Molfetta R, Zingoni A, Fionda C, et al. P38 MAPK Differentially Controls NK Activating Ligands at Transcriptional and Post-Transcriptional Level on Multiple Myeloma Cells. OncoImmunology (2017) 6(1):e1264564. doi: 10.1080/2162402X.2016.1264564
115. Molinero LL, Fuertes MB, Fainboim L, Rabinovich GA, Zwirner NW. Up-Regulated Expression of MICA on Activated T Lymphocytes Involves Lck and Fyn Kinases and Signaling Through MEK1/ERK, P38 MAP Kinase, and Calcineurin. J Leukocyte Biol (2003) 73(6):815–22. doi: 10.1189/jlb.0602329
116. Nanbakhsh A, Pochon C, Mallavialle A, Amsellem S, Bourhis JH, Chouaib S. C-Myc Regulates Expression of NKG2D Ligands ULBP1/2/3 in AML and Modulates Their Susceptibility to NK-Mediated Lysis. Blood (2014) 123(23):3585–95. doi: 10.1182/blood-2013-11-536219
117. Lee YS, Heo W, Son CH, Kang CD, Park YS, Bae J. Upregulation of Myc Promotes the Evasion of NK Cell−Mediated Immunity Through Suppression of NKG2D Ligands in K562 Cells. Mol Med Rep (2019) 20(4):3301–7. doi: 10.3892/mmr.2019.10583
118. Zhang X, Choi PS, Francis JM, Imielinski M, Watanabe H, Cherniack AD, et al. Identification of Focally Amplified Lineage-Specific Super-Enhancers in Human Epithelial Cancers. Nat Genet (2016) 48(2):176–82. doi: 10.1038/ng.3470
119. O’Geen H, Ren C, Nicolet CM, Perez AA, Halmai J, Le VM, et al. Dcas9-Based Epigenome Editing Suggests Acquisition of Histone Methylation Is Not Sufficient for Target Gene Repression. Nucleic Acids Res (2017) 45(17):9901–16. doi: 10.1093/nar/gkx578
120. Eissmann P, Evans JH, Mehrabi M, Rose EL, Nedvetzki S, Davis DM. Multiple Mechanisms Downstream of TLR-4 Stimulation Allow Expression of NKG2D Ligands to Facilitate Macrophage/NK Cell Crosstalk. J Immunol (2010) 184(12):6901–9. doi: 10.4049/jimmunol.0903985
121. Schary Y, Brzezinski RY, Teper-Shaihov O, Naftali-Shani N, Leor J. CRISPR-Cas9-Based Gene Editing of Human Mesenchymal Stromal Cells to Improve the Outcome of Cell Therapy. Eur Heart J (2020) 41(Supplement_2):ehaa946.3658. doi: 10.1093/ehjci/ehaa946.3658
122. Gowen BG, Chim B, Marceau CD, Greene TT, Burr P, Gonzalez JR, et al. A Forward Genetic Screen Reveals Novel Independent Regulators of ULBP1, an Activating Ligand for Natural Killer Cells. eLife (2015) 4:e08474. doi: 10.7554/eLife.08474
123. Boontanrart MY, Schröder MS, Stehli GM, Banović M, Wyman SK, Lew RJ, et al. ATF4 Regulates MYB to Increase γ-Globin in Response to Loss of β-Globin. Cell Rep (2020) 32(5):107993. doi: 10.1016/j.celrep.2020.107993
124. Tsukerman P, Stern-Ginossar N, Gur C, Glasner A, Nachmani D, Bauman Y, et al. MiR-10b Downregulates the Stress-Induced Cell Surface Molecule MICB, A Critical Ligand for Cancer Cell Recognition by Natural Killer Cells. Cancer Res (2012) 72(21):5463–72. doi: 10.1158/0008-5472.Can-11-2671
125. El Fatimy R, Subramanian S, Uhlmann EJ, Krichevsky AM. Genome Editing Reveals Glioblastoma Addiction to microRNA-10b. Mol Ther (2017) 25(2):368–78. doi: 10.1016/j.ymthe.2016.11.004
126. Heinemann A, Zhao F, Pechlivanis S, Eberle J, Steinle A, Diederichs S, et al. Tumor Suppressive microRNAs miR-34a/C Control Cancer Cell Expression of ULBP2, A Stress-Induced Ligand of the Natural Killer Cell Receptor NKG2D. Cancer Res (2012) 72(2):460–71. doi: 10.1158/0008-5472.Can-11-1977
127. Yadav D, Ngolab J, Lim RS-H, Krishnamurthy S, Bui JD. Cutting Edge: Down-Regulation of MHC Class I-Related Chain A on Tumor Cells by IFN-γ-Induced microRNA. J Immunol (2009) 182(1):39–43. doi: 10.4049/jimmunol.182.1.39
128. Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, et al. Human microRNAs Regulate Stress-Induced Immune Responses Mediated by the Receptor NKG2D. Nat Immunol (2008) 9(9):1065–73. doi: 10.1038/ni.1642
129. Jiang Q, Meng X, Meng L, Chang N, Xiong J, Cao H, et al. Small Indels Induced by CRISPR/Cas9 in the 5′ Region of microRNA Lead to Its Depletion and Drosha Processing Retardance. RNA Biol (2014) 11(10):1243–9. doi: 10.1080/15476286.2014.996067
130. Zhao Y, Dai Z, Liang Y, Yin M, Ma K, He M, et al. Sequence-Specific Inhibition of microRNA via CRISPR/CRISPRi System. Sci Rep (2014) 4(1):3943. doi: 10.1038/srep03943
131. Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O. Diverse Herpesvirus microRNAs Target the Stress-Induced Immune Ligand MICB to Escape Recognition by Natural Killer Cells. Cell Host Microbe (2009) 5(4):376–85. doi: 10.1016/j.chom.2009.03.003
132. Stern-Ginossar N, Elefant N, Zimmermann A, Wolf DG, Saleh N, Biton M, et al. Host Immune System Gene Targeting by a Viral miRNA. Science (2007) 317(5836):376–81. doi: 10.1126/science.1140956
133. Bauman Y, Nachmani D, Vitenshtein A, Tsukerman P, Drayman N, Stern-Ginossar N, et al. An Identical miRNA of the Human JC and BK Polyoma Viruses Targets the Stress-Induced Ligand ULBP3 to Escape Immune Elimination. Cell Host Microbe (2011) 9(2):93–102. doi: 10.1016/j.chom.2011.01.008
134. Nachmani D, Gutschner T, Reches A, Diederichs S, Mandelboim O. RNA-Binding Proteins Regulate the Expression of the Immune Activating Ligand MICB. Nat Commun (2014) 5(1):4186. doi: 10.1038/ncomms5186
135. Berhani O, Nachmani D, Yamin R, Schmiedel D, Bar-On Y, Mandelboim O. Vigilin Regulates the Expression of the Stress-Induced Ligand MICB by Interacting With its 5′ Untranslated Region. J Immunol (2017) 198(9):3662–70. doi: 10.4049/jimmunol.1601589
136. Schmiedel D, Tai J, Yamin R, Berhani O, Bauman Y, Mandelboim O. The RNA Binding Protein IMP3 Facilitates Tumor Immune Escape by Downregulating the Stress-Induced Ligands ULPB2 and MICB. eLife (2016) 5:e13426. doi: 10.7554/eLife.13426
137. Schneider T, Hung L-H, Aziz M, Wilmen A, Thaum S, Wagner J, et al. Combinatorial Recognition of Clustered RNA Elements by the Multidomain RNA-Binding Protein IMP3. Nat Commun (2019) 10(1):2266. doi: 10.1038/s41467-019-09769-8
138. Salih HR, Rammensee H-G, Steinle A. Cutting Edge: Down-Regulation of MICA on Human Tumors by Proteolytic Shedding. J Immunol (2002) 169(8):4098. doi: 10.4049/jimmunol.169.8.4098
139. Liu G, Atteridge CL, Wang X, Lundgren AD, Wu JD. Cutting Edge: The Membrane Type Matrix Metalloproteinase MMP14 Mediates Constitutive Shedding of MHC Class I Chain-Related Molecule A Independent of a Disintegrin and Metalloproteinases. J Immunol (2010) 184(7):3346. doi: 10.4049/jimmunol.0903789
140. Sun D, Wang X, Zhang H, Deng L, Zhang Y. MMP9 Mediates MICA Shedding in Human Osteosarcomas. Cell Biol Int (2011) 35(6):569–74. doi: 10.1042/CBI20100431
141. Shiraishi K, Mimura K, Kua L-F, Koh V, Siang LK, Nakajima S, et al. Inhibition of MMP Activity can Restore NKG2D Ligand Expression in Gastric Cancer, Leading to Improved NK Cell Susceptibility. J Gastroenterol (2016) 51(12):1101–11. doi: 10.1007/s00535-016-1197-x
142. Yadav SK, Kambis TN, Kar S, Park SY, Mishra PK. MMP9 Mediates Acute Hyperglycemia-Induced Human Cardiac Stem Cell Death by Upregulating Apoptosis and Pyroptosis In Vitro. Cell Death Dis (2020) 11(3):186. doi: 10.1038/s41419-020-2367-6
143. Chitadze G, Lettau M, Bhat J, Wesch D, Steinle A, Fürst D, et al. Shedding of Endogenous MHC Class I-Related Chain Molecules A and B From Different Human Tumor Entities: Heterogeneous Involvement of the “A Disintegrin and Metalloproteases” 10 and 17. Int J Cancer (2013) 133(7):1557–66. doi: 10.1002/ijc.28174
144. Wolpert F, Tritschler I, Steinle A, Weller M, Eisele G. A Disintegrin and Metalloproteinases 10 and 17 Modulate the Immunogenicity of Glioblastoma-Initiating Cells. Neuro-Oncology (2013) 16(3):382–91. doi: 10.1093/neuonc/not232
145. Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, et al. Tumor-Associated MICA Is Shed by ADAM Proteases. Cancer Res (2008) 68(15):6368. doi: 10.1158/0008-5472.CAN-07-6768
146. Boutet P, Agüera-González S, Atkinson S, Pennington CJ, Edwards DR, Murphy G, et al. Cutting Edge: The Metalloproteinase ADAM17/TNF-α-Converting Enzyme Regulates Proteolytic Shedding of the MHC Class I-Related Chain B Protein. J Immunol (2009) 182(1):49–53. doi: 10.4049/jimmunol.182.1.49
147. Riethmueller S, Ehlers JC, Lokau J, Düsterhöft S, Knittler K, Dombrowsky G, et al. Cleavage Site Localization Differentially Controls Interleukin-6 Receptor Proteolysis by ADAM10 and ADAM17. Sci Rep (2016) 6(1):25550. doi: 10.1038/srep25550
148. Kohga K, Takehara T, Tatsumi T, Ishida H, Miyagi T, Hosui A, et al. Sorafenib Inhibits the Shedding of Major Histocompatibility Complex Class I–related Chain A on Hepatocellular Carcinoma Cells by Down-Regulating a Disintegrin and Metalloproteinase 9. Hepatology (2010) 51(4):1264–73. doi: 10.1002/hep.23456
149. Arai J, Goto K, Stephanou A, Tanoue Y, Ito S, Muroyama R, et al. Predominance of Regorafenib Over Sorafenib: Restoration of Membrane-Bound MICA in Hepatocellular Carcinoma Cells. J Gastroenterol Hepatol (2018) 33(5):1075–81. doi: 10.1111/jgh.14029
150. Bazzone LE, King M, MacKay CR, Kyawe PP, Meraner P, Lindstrom D, et al. A Disintegrin and Metalloproteinase 9 Domain (ADAM9) Is a Major Susceptibility Factor in the Early Stages of Encephalomyocarditis Virus Infection. mBio (2019) 10(1):e02734–18. doi: 10.1128/mBio.02734-18
151. Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH, et al. Disulphide-Isomerase-Enabled Shedding of Tumour-Associated NKG2D Ligands. Nature (2007) 447(7143):482–6. doi: 10.1038/nature05768
152. Nagai Y, Tanaka Y, Kuroishi T, Sato R, Endo Y, Sugawara S. Histamine Reduces Susceptibility to Natural Killer Cells via Down-Regulation of NKG2D Ligands on Human Monocytic Leukaemia THP-1 Cells. Immunology (2012) 136(1):103–14. doi: 10.1111/j.1365-2567.2012.03565.x
153. Thomas M, Boname JM, Field S, Nejentsev S, Salio M, Cerundolo V, et al. Down-Regulation of NKG2D and NKp80 Ligands by Kaposi’s Sarcoma-Associated Herpesvirus K5 Protects Against NK Cell Cytotoxicity. Proc Natl Acad Sci (2008) 105(5):1656–61. doi: 10.1073/pnas.0707883105
154. Guan Y, Li W, Hou Z, Han Q, Lan P, Zhang J, et al. HBV Suppresses Expression of MICA/B on Hepatoma Cells Through Up-Regulation of Transcription Factors GATA2 and GATA3 to Escape From NK Cell Surveillance. Oncotarget (2016) 7:56107–19. doi: 10.18632/oncotarget.11271
155. Bauman Y, Mandelboim O. MicroRNA Based Immunoevasion Mechanism of Human Polyomaviruses. RNA Biol (2011) 8(4):591–4. doi: 10.4161/rna.8.4.15587
156. Enk J, Levi A, Weisblum Y, Yamin R, Charpak-Amikam Y, Wolf Dana G, et al. HSV1 microRNA Modulation of GPI Anchoring and Downstream Immune Evasion. Cell Rep (2016) 17(4):949–56. doi: 10.1016/j.celrep.2016.09.077
157. Esteso G, Luzón E, Sarmiento E, Gómez-Caro R, Steinle A, Murphy G, et al. Altered microRNA Expression After Infection With Human Cytomegalovirus Leads to TIMP3 Downregulation and Increased Shedding of Metalloprotease Substrates, Including MICA. J Immunol (2014) 193(3):1344–52. doi: 10.4049/jimmunol.1303441
158. Shen J, Pan J, Du C, Si W, Yao M, Xu L, et al. Silencing NKG2D Ligand-Targeting miRNAs Enhances Natural Killer Cell-Mediated Cytotoxicity in Breast Cancer. Cell Death Dis (2017) 8(4):e2740. doi: 10.1038/cddis.2017.158
159. Schmiedel D, Mandelboim O. NKG2D Ligands–Critical Targets for Cancer Immune Escape and Therapy. Front Immunol (2040) 9:2040. doi: 10.3389/fimmu.2018.02040
160. Coudert JD, Held W. The Role of the NKG2D Receptor for Tumor Immunity. Semin Cancer Biol (2006) 16(5):333–43. doi: 10.1016/j.semcancer.2006.07.008
161. Dunn C, Chalupny NJ, Sutherland CL, Dosch S, Sivakumar PV, Johnson DC, et al. Human Cytomegalovirus Glycoprotein UL16 Causes Intracellular Sequestration of NKG2D Ligands, Protecting Against Natural Killer Cell Cytotoxicity. J Exp Med (2003) 197(11):1427–39. doi: 10.1084/jem.20022059
162. Welte SA, Sinzger C, Lutz SZ, Singh-Jasuja H, Sampaio KL, Eknigk U, et al. Selective Intracellular Retention of Virally Induced NKG2D Ligands by the Human Cytomegalovirus UL16 Glycoprotein. Eur J Immunol (2003) 33(1):194–203. doi: 10.1002/immu.200390022
163. Ashiru O, Bennett NJ, Boyle LH, Thomas M, Trowsdale J, Wills MR. NKG2D Ligand MICA Is Retained in the Cis-Golgi Apparatus by Human Cytomegalovirus Protein UL142. J Virol (2009) 83(23):12345–54. doi: 10.1128/jvi.01175-09
164. Bennett NJ, Ashiru O, Morgan FJE, Pang Y, Okecha G, Eagle RA, et al. Intracellular Sequestration of the NKG2D Ligand ULBP3 by Human Cytomegalovirus. J Immunol (2010) 185(2):1093. doi: 10.4049/jimmunol.1000789
165. Lee J-H, Wittki S, Bräu T, Dreyer Florian S, Krätzel K, Dindorf J, et al. HIV Nef, Paxillin, and Pak1/2 Regulate Activation and Secretion of TACE/ADAM10 Proteases. Mol Cell (2013) 49(4):668–79. doi: 10.1016/j.molcel.2012.12.004
166. Fuertes MB, Girart MV, Molinero LL, Domaica CI, Rossi LE, Barrio MM, et al. Intracellular Retention of the NKG2D Ligand MHC Class I Chain-Related Gene A in Human Melanomas Confers Immune Privilege and Prevents NK Cell-Mediated Cytotoxicity. J Immunol (2008) 180(7):4606–14. doi: 10.4049/jimmunol.180.7.4606
167. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science (2012) 337(6096):816–21. doi: 10.1126/science.1225829
168. Chylinski K, Makarova KS, Charpentier E, Koonin EV. Classification and Evolution of Type II CRISPR-Cas Systems. Nucleic Acids Res (2014) 42(10):6091–105. doi: 10.1093/nar/gku241
169. Qi Lei S, Larson Matthew H, Gilbert Luke A, Doudna Jennifer A, Weissman Jonathan S, Arkin Adam P, et al. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell (2013) 152(5):1173–83. doi: 10.1016/j.cell.2013.02.022
170. Garcia-Bloj B, Moses C, Sgro A, Plani-Lam J, Arooj M, Duffy C, et al. Waking Up Dormant Tumor Suppressor Genes With Zinc Fingers, TALEs and the CRISPR/dCas9 System. Oncotarget (2016) 7(37):60535–54. doi: 10.18632/oncotarget.11142
171. Moses C, Nugent F, Waryah CB, Garcia-Bloj B, Harvey AR, Blancafort P. Activating PTEN Tumor Suppressor Expression With the CRISPR/dCas9 System. Mol Ther - Nucleic Acids (2019) 14:287–300. doi: 10.1016/j.omtn.2018.12.003
172. Kretzmann JA, Evans CW, Moses C, Sorolla A, Kretzmann AL, Wang E, et al. Tumour Suppression by Targeted Intravenous non-Viral CRISPRa Using Dendritic Polymers. Chem Sci (2019) 10(33):7718–27. doi: 10.1039/C9SC01432B
173. Thakore PI, D’Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, et al. Highly Specific Epigenome Editing by CRISPR-Cas9 Repressors for Silencing of Distal Regulatory Elements. Nat Methods (2015) 12(12):1143–9. doi: 10.1038/nmeth.3630
174. Thakore PI, Kwon JB, Nelson CE, Rouse DC, Gemberling MP, Oliver ML, et al. RNA-Guided Transcriptional Silencing In Vivo With S. Aureus CRISPR-Cas9 Repressors. Nat Commun (2018) 9(1):1674. doi: 10.1038/s41467-018-04048-4
175. Moses C, Hodgetts SI, Nugent F, Ben-Ary G, Park KK, Blancafort P, et al. Transcriptional Repression of PTEN in Neural Cells Using CRISPR/dCas9 Epigenetic Editing. Sci Rep (2020) 10(1):11393. doi: 10.1038/s41598-020-68257-y
176. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-Engineered T Cells in Patients With Refractory Cancer. Science (2020) 367(6481):eaba7365. doi: 10.1126/science.aba7365
177. Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, et al. Safety and Feasibility of CRISPR-Edited T Cells in Patients With Refractory Non-Small-Cell Lung Cancer. Nat Med (2020) 26(5):732–40. doi: 10.1038/s41591-020-0840-5
178. Alves E, Taifour S, Dolcetti R, Chee J, Nowak AK, Gaudieri S, et al. Reprogramming the Anti-Tumor Immune Response via CRISPR Genetic and Epigenetic Editing. Mol Ther - Methods Clin Dev (2021) 21:592 – 606. doi: 10.1016/j.omtm.2021.04.009
179. Wang G, Chow RD, Bai Z, Zhu L, Errami Y, Dai X, et al. Multiplexed Activation of Endogenous Genes by CRISPRa Elicits Potent Antitumor Immunity. Nat Immunol (2019) 20(11):1494–505. doi: 10.1038/s41590-019-0500-4
180. Najm FJ, Strand C, Donovan KF, Hegde M, Sanson KR, Vaimberg EW, et al. Orthologous CRISPR–Cas9 Enzymes for Combinatorial Genetic Screens. Nat Biotechnol (2018) 36(2):179–89. doi: 10.1038/nbt.4048
181. Zetsche B, Gootenberg Jonathan S, Abudayyeh Omar O, Slaymaker Ian M, Makarova Kira S, Essletzbichler P, et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell (2015) 163(3):759–71. doi: 10.1016/j.cell.2015.09.038
182. Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DBT, et al. C2c2 Is a Single-Component Programmable RNA-Guided RNA-Targeting CRISPR Effector. Science (2016) 353(6299):aaf5573. doi: 10.1126/science.aaf5573
183. Zhang T, Lemoi BA, Sentman CL. Chimeric NK-Receptor–Bearing T Cells Mediate Antitumor Immunotherapy. Blood (2005) 106(5):1544–51. doi: 10.1182/blood-2004-11-4365
184. Chang Y-H, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A Chimeric Receptor With NKG2D Specificity Enhances Natural Killer Cell Activation and Killing of Tumor Cells. Cancer Res (2013) 73(6):1777–86. doi: 10.1158/0008-5472.Can-12-3558
185. Herzig E, Kim KC, Packard TA, Vardi N, Schwarzer R, Gramatica A, et al. Attacking Latent HIV With convertibleCAR-T Cells, a Highly Adaptable Killing Platform. Cell (2019) 179(4):880–94.e10. doi: 10.1016/j.cell.2019.10.002
186. Baumeister SH, Murad J, Werner L, Daley H, Trebeden-Negre H, Gicobi JK, et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients With AML/MDS and Multiple Myeloma. Cancer Immunol Res (2019) 7(1):100–12. doi: 10.1158/2326-6066.Cir-18-0307
187. Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional Oncogenesis in 4 Patients After Retrovirus-Mediated Gene Therapy of SCID-X1. J Clin Invest (2008) 118(9):3132–42. doi: 10.1172/JCI35700
188. Braun CJ, Boztug K, Paruzynski A, Witzel M, Schwarzer A, Rothe M, et al. Gene Therapy for Wiskott-Aldrich Syndrome—Long-Term Efficacy and Genotoxicity. Sci Trans Med (2014) 6(227):227ra33–ra33. doi: 10.1126/scitranslmed.3007280
189. Naeimi Kararoudi M, Dolatshad H, Trikha P, Hussain S-RA, Elmas E, Foltz JA, et al. Generation of Knock-Out Primary and Expanded Human NK Cells Using Cas9 Ribonucleoproteins. J Vis Exp (2018) 136):e58237. doi: 10.3791/58237
190. Lee J-C, Lee K-M, Kim D-W, Heo DS. Elevated TGF-β1 Secretion and Down-Modulation of NKG2D Underlies Impaired NK Cytotoxicity in Cancer Patients. J Immunol (2004) 172(12):7335–40. doi: 10.4049/jimmunol.172.12.7335
191. Pomeroy EJ, Hunzeker JT, Kluesner MG, Lahr WS, Smeester BA, Crosby MR, et al. A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. Mol Ther (2020) 28(1):52–63. doi: 10.1016/j.ymthe.2019.10.009
192. Bak RO, Porteus MH. CRISPR-Mediated Integration of Large Gene Cassettes Using AAV Donor Vectors. Cell Rep (2017) 20(3):750–6. doi: 10.1016/j.celrep.2017.06.064
193. Hue S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, et al. A Direct Role for NKG2D/MICA Interaction in Villous Atrophy During Celiac Disease. Immunity (2004) 21(3):367–77. doi: 10.1016/j.immuni.2004.06.018
194. Groh V, Brühl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T Cell Autoreactivity by Anomalous Expression of NKG2D and Its MIC Ligands in Rheumatoid Arthritis. Proc Natl Acad Sci (2003) 100(16):9452. doi: 10.1073/pnas.1632807100
195. Sekiba K, Yamagami M, Otsuka M, Suzuki T, Kishikawa T, Ishibashi R, et al. Transcriptional Activation of the MICA Gene With an Engineered CRISPR-Cas9 System. Biochem Biophys Res Commun (2017) 486(2):521–5. doi: 10.1016/j.bbrc.2017.03.076
196. McCarty NS, Graham AE, Studená L, Ledesma-Amaro R. Multiplexed CRISPR Technologies for Gene Editing and Transcriptional Regulation. Nat Commun (2020) 11(1):1281. doi: 10.1038/s41467-020-15053-x
Keywords: NKG2D, CRISPR, precision medicine, NK cells, viral infection, cancer, immune evasion, immunotherapy
Citation: Alves E, McLeish E, Blancafort P, Coudert JD and Gaudieri S (2021) Manipulating the NKG2D Receptor-Ligand Axis Using CRISPR: Novel Technologies for Improved Host Immunity. Front. Immunol. 12:712722. doi: 10.3389/fimmu.2021.712722
Received: 21 May 2021; Accepted: 28 July 2021;
Published: 12 August 2021.
Edited by:
Alessandra Zingoni, Sapienza University of Rome, ItalyReviewed by:
Nadia Guerra, Imperial College London, United KingdomBojan Polić, University of Rijeka, Croatia
Copyright © 2021 Alves, McLeish, Blancafort, Coudert and Gaudieri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Alves, ZXJpYy5hbHZlc0ByZXNlYXJjaC51d2EuZWR1LmF1
†These authors have contributed equally to this work