 Pingping Ren1,2,3†
Pingping Ren1,2,3† Jianghua Chen
Jianghua Chen Weiqiang Lin
Weiqiang Lin Fei Han
Fei Han- 1Kidney Disease Center, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2Key Laboratory of Nephropathy, Zhejiang Province, Hangzhou, China
- 3Institute of Nephropathy, Zhejiang University, Hangzhou, China
- 4Department of Nephrology, The First People’s Hospital of Wenling, Taizhou, China
- 5Institute of Translational Medicine, Zhejiang University of Medicine, Hangzhou, China
Alternative splicing (AS) is a complex coordinated transcriptional regulatory mechanism. It affects nearly 95% of all protein-coding genes and occurs in nearly all human organs. Aberrant alternative splicing can lead to various neurological diseases and cancers and is responsible for aging, infection, inflammation, immune and metabolic disorders, and so on. Though aberrant alternative splicing events and their regulatory mechanisms are widely recognized, the association between autoimmune disease and alternative splicing has not been extensively examined. Autoimmune diseases are characterized by the loss of tolerance of the immune system towards self-antigens and organ-specific or systemic inflammation and subsequent tissue damage. In the present review, we summarized the most recent reports on splicing events that occur in the immunopathogenesis of systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) and attempted to clarify the role that splicing events play in regulating autoimmune disease progression. We also identified the changes that occur in splicing factor expression. The foregoing information might improve our understanding of autoimmune diseases and help develop new diagnostic and therapeutic tools for them.
Introduction
Alternative splicing is a vital mechanism in gene modulation. It allows a single gene to produce multiple distinct mRNA that greatly increase transcriptome and proteome diversity. According to recent genome-wide association studies (GWAS), nearly 95% of all protein-coding genes in the human genome undergo alternative splicing to varying degrees (1, 2). In this way, they increase by more than 10-fold of the number of distinct protein isoforms that function in various cellular activities, such as maturation, differentiation, migration, and death (3).
Alternative splicing is highly controlled. It comprises splice site selection, spliceosome assembly, and the control of multiple splicing regulatory elements. Any disruptions or mutations in the splicing mechanisms may affect mature mRNA and functional protein generation and induce various disease states. Over the last decade, particular attention has been allocated to disease-related genomics, transcriptomics, and proteomics. Alternative splicing might be associated with susceptibility to cancers, cardiovascular diseases, and different neuropathies, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases as well as schizophrenia and congenital myasthenic syndrome. It is also related to infection, inflammation, and immune and metabolic disorders (4).
Autoimmune diseases include a wide range of disorders arising from an abnormal immune response to otherwise healthy, normal tissues and organs. Genetic, environmental, hormonal, and immunological etiologies are implicated in autoimmune disorder pathogenesis. However, the mechanisms that trigger the onset of at least half of all autoimmune diseases remain unclear (5). Numerous studies have investigated the roles of alternative splicing in autoimmunity mechanisms. Immune-related genes encoding human leukocyte antigen (HLA), interferon regulatory factor 5 (IRF5), T-cell receptor ζ (TCRζ) chain, cytotoxic T lymphocyte associated protein 4 (CTLA4), and cytokines and their receptors presented with spliced isoforms (6). However, our knowledge of the regulatory mechanisms of alternative splicing in immunity and autoimmunity is limited.
Alternative Splicing Mechanism
Splicing Reaction
Precursor (pre) mRNA splicing is an intricate biological process involving intron excision and exon ligation to generate mature mRNA products. Splicing recognizes exon–intron boundaries and is catalyzed and controlled by complex ribonucleoprotein complexes known as spliceosomes (7). The spliceosome consists of at least 300 associated proteins and five uracil-rich small nuclear ribonucleoproteins (U1, U2, U4, U5, and U6 snRNPs) (8). Proper spliceosome assembly at the splice sites (ss) is the core reaction mechanism. The 5’ss is located at the 5’ end of each intron, the 3’ss is located at the 3’ end of each intron, and the branch point (BP) site is located 18–40 nucleotides upstream of each 3’ss (9). Spliceosome assembly begins with adenosine triphosphate (ATP)-independent recognition of the 5’ss by U1 snRNP. U2 auxiliary factor (U2AF) then binds the 3’ss and the polypyrimidine region. Splicing factor 1 (SF1) then binds the BP (Figure 1) (8, 10). These steps form the stable early complex E, which then triggers ATP-dependent U2 snRNP recruitment, SF1 replacement at the BP, and conversion into the pre-spliceosome (complex A) (10). The pre-assembled U4/U6–U5 tri-snRNP complex is recruited and forms a pre-catalytic spliceosome (complex B), which is then converted into a catalytically active complex B* by removing U1 and U4. Two subsequent transesterification steps and conformational and compositional rearrangements form complex C. Exon ligation follows, and mature mRNA is formed (11).
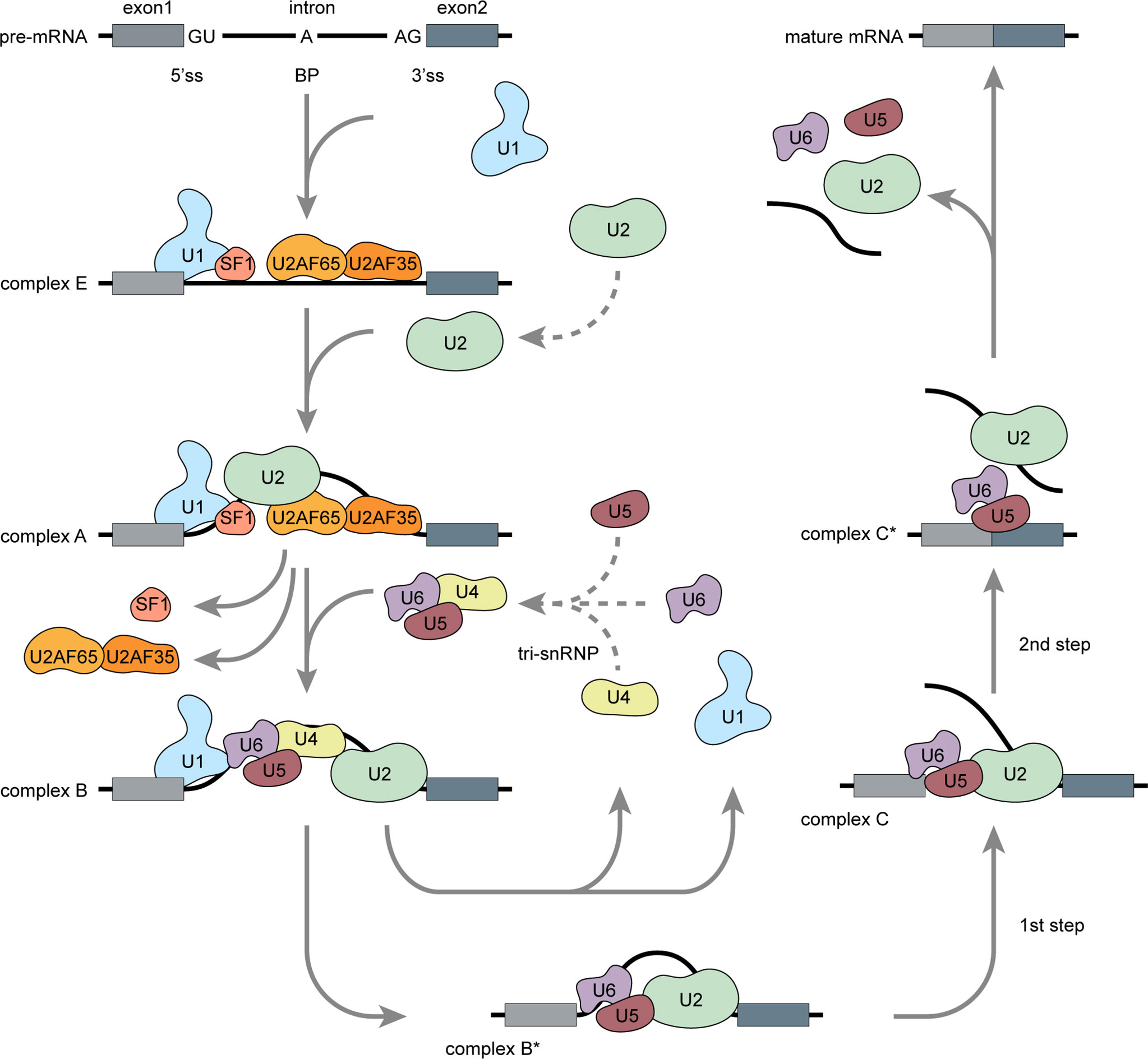
Figure 1 Pre-mRNA splicing: Spliceosome assembly (Adapted from Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol (2014) 15:108-21). Pre-mRNA splicing occurs in several spliceosome assembly steps. Splicing begins with U1 snRNP assembly onto pre-mRNA via 5’ss recognition and subsequent combination with U2AF and SF1 to form early complex E. Then, U2 snRNP replaces SF1 at the branch point and forms a pre-spliceosome (complex A). Pre-assembled U4/U6–U5 tri-snRNP complexes are then recruited to form catalytically active complex B, which is then converted into catalytically active complex B* via release of U1 and U4. Through two catalytic steps and conformation or composition rearrangements, complex B* converted into a final complex C* and U2, U5, and U6 are released from the spliceosome followed by intron excision, exon ligation, and mature mRNA generation.
Splicing may be constitutive or alternative. Constitutive splicing events usually generate single transcripts from the pre-mRNAs. By contrast, alternative splicing events occur when at least two 5’ss or 3’ ss compete (12). Constitutive splicing usually occurs in the presence of strong splice sites containing consensus sequences that are easily recognized by the spliceosome. However, alternatively spliced exons usually contain weaker splice sites that can only be recognized via the mediation of additional splicing regulatory elements (SREs).
Alternative Splicing Regulation
Alternative splicing arises from specific exons/introns that may or may not be included in a mature mRNA transcript. The five basic alternative splicing patterns are exon skipping, mutually exclusive exons, alternative 5’ss, alternative 3’ss, and intron retention (12). Alternative splicing is controlled by cis-regulatory elements and trans-acting factors. Certain epigenetic factors, such as transcription, chromatin structure, DNA methylation, and histone modifications, may also affect SRE function. Depending on their relative positions and activities, cis-regulatory elements are classified as exonic splicing enhancers (ESE), intronic splicing enhancers (ISE), exonic splicing silencers (ESS), or intronic splicing silencers (ISS) (13). Cis-regulatory elements can recruit trans-acting splice factors (RNA-binding proteins or RBPs) either to promote or suppress the recognition of nearby splice sites. Therefore, various RBP categories, expression levels, and activities may influence the regulation of alternative splicing. Thus far, many RBPs have been found to modulate the splicing mechanism. These include serine/arginine-rich (SR) proteins, heterogeneous nuclear ribonucleoproteins (hnRNPs), neuro-oncological ventral antigens (NOVA) proteins, polypyrimidine track binding proteins (PTB), forkhead boxes (FOX) proteins, and others. Of these, the most extensively studied are the SR proteins and the hnRNPs (14).
SR proteins or SR-rich splicing factors (SRSFs) structurally resemble each other and are characterized by the RS domain [rich in arginine (R) and serine (S) residues] at the C-terminal and one or two RNA recognition motifs (RRMs) at the N-terminal (13). The SR protein is usually a positive splicing factor and it facilitates splice site recognition. When it binds the ESE, the SR protein recruits U1 and U2 snRNPs and auxiliary factors, such as U2AF. In this way, it induces spliceosome assembly followed by exon inclusion (15). SR protein mutation or aberrant post-translational modification may lead to various diseases. Correct SR protein phosphorylation is vital in the regulation of the splicing event and alterations in subcellular localization, activity, and stability that may affect protein–protein and protein–RNA interactions (16). By contrast, hnRNPs are usually considered negative splicing factors that inhibit splice site recognition. For example, hnRNP blocks spliceosome access to the polypyrimidine tract by binding the ESS and promoting exon exclusion.
Alternative Splicing in Various Aspects of Immunity
Autoimmune diseases are a subset of conditions rising from abnormal immune responses to a functional body part. Currently, specific candidate genes of autoimmune disease have been identified in multiple GWAS. Moreover, various alternative splicing events have been found in adaptive- and innate immune signaling-related genes. Elucidation of the mechanism of immune response related to autoimmune disease pathogenesis is essential for further investigations into alternative splicing regulation. In this section, we focus on the physiological and pathological roles of alternative splicing in immunity, and attempt to figure out common patterns of splicing regulation in different autoimmune diseases.
Gene Categories Involved in Alternative Splicing in Autoimmunity
Alternative splicing is a pivotal mechanism widely acknowledged to regulate gene expression and promote protein diversity. The maintenance of functional immune responses requires protein diversity and flexibility, which are essential in alternative splicing regulation. Many genes involved in innate or adaptive immune signaling undergo varying degrees of alternative splicing. Ergun et al. determined by RNA sequencing and microarray that alternatively spliced isoforms occur in 60% of all T- or B-lymphocyte genes (17). Based on spliced gene functions, alternative splicing may affect the physiological function of the immune system in different ways.
Cell Surface Receptors
Cell surface receptors constitute a subset of transmembrane proteins that participate in signal transduction by receiving extracellular molecules. CD44, CD45, CD85, TCRζ, CTLA4, and others have alternative spliced isoforms. These structural variants have distinct activity and stability and may affect ligand binding and disrupt normal signaling. For example, a short 3’ untranslated region (UTR) isoform of TCRζ was generated by intron removal. It was relatively less stable, affected TCR/CD3 complex assembly and expression, and limited T-cell activation (18).
Cytokines and Their Corresponding Receptors
Cytokine secretion and reaction are crucial processes in immunomodulation. Immune cell stimulation produces various cytokines and their isoforms. These may either be similar or different in terms of biological activity. The functions of these spliced variants have not been fully determined. In most cases, however, isoforms of IL-2, IL-4, IL-6, and others might act as antagonists to their corresponding wild types and block their activity (19). Alternative splicing of cytokine receptors usually generates both membrane-bound and soluble isoforms with similar biological activity. However, soluble isoforms of IL-6R, TNFR2, and others cannot transmit signals through ligand binding and might act as inhibitors.
Intracellular Signaling
T-lymphocyte activation may result in subsequent signal-induced alternative splicing. In this way, they generate variant isoforms of downstream intracellular molecules, such as the Src family members Lck, Fyn, Syk, and others, that may disrupt normal signal transduction (6, 20). In innate immune cells, an isoform of the adaptor protein myeloid differentiation primary response 88 (MyD88) may inhibit downstream Toll-like receptor (TLR) signaling and limit innate immune response activation (21). Moreover, an isoform of TLR4 is associated with TNF-α and NF-κB downregulation (22).
Complement System
Activation of the complement system is an essential part of the innate immune response. Alternative splicing events have also been found in complement proteins and receptors, such as C2, CR1, CR2, and others (23). They have also been detected in complement regulatory proteins, such as membrane cofactor protein (MCP) (24).
Splicing Regulatory Patterns in Autoimmunity
Basic alternative splicing patterns and its regulatory mechanisms are widely acknowledged. In autoimmunity, the disease-related pathogenesis varies from one to another. Here, we conclude the common features of alternative splicing in the pathological changes of autoimmune diseases.
Gene Mutations: The Most Common Cause of Alternative Splicing
Candidate single-nucleotide polymorphisms (SNPs) and other sequence mutations can directly change the coding region and result in aberrant alternative splicing. In most cases, SNPs are localized to non-coding sequences, which may disrupt consensus splicing sites or create novel alternative binding sites, thereby influencing the efficiency or selection mode of alternative splicing. Wang et al. reviewed 303 genes and 370 diseases with overall 2,337 splice mutation-disease entries from publication searching. Nearly 90% of the events resulted from point mutations (25). However, the regulatory mechanisms involved are not fully understood and they might affect alternative splicing in other ways.
SREs: Fundamental Regulatory Mechanism of Alternative Splicing
Epigenetic modification and changes in the levels of cis-regulatory elements and trans-acting factors can lead to different alternative splicing site selection and, therefore, variant transcripts. In human genome, there are numerous potential splice sites, known as pseudo-exons. It is relatively weaker than the consensus sequences during the recognition of snRNP. SREs can enhance or silence nearby splice sites, thereby promoting the diversity of selection between pseudo-exons and real exons. Weaker splice sites at the 5’ and 3’ end of intron may hamper the recognition of spliceosome, leading to specific intron retention (26). Epigenetic modifications bring additional different functional SREs and thereby influence the splicing process regulation.
Autoantigens: Prone to Be Alternative Spliced
Autoantigens are more frequently subjected to splicing control than non-autoantigens. Ng et al. compared the incidence of alternative splicing event in both randomly selected 45 autoantigens and 9,554 proteins from human genome. The results showed a 100% occurrence rate of alternative splicing in autoantigen transcripts, whereas 42% rate in the other group (27). Spliced isoforms have been found in various disease-specific autoantigens, such as multiple sclerosis (MS)-related autoantigen myelin oligodendrocyte glycoprotein (MOG) and transformation of myelin proteolipid protein (PLP), type 1 diabetes mellitus (T1DM)-related autoantigen islet cell Ag 512 (IA-2), Sjögren’s syndrome (SS)-related autoantigen SS-B/La and SS-A/Ro, Graves’ disease (GD)-related autoantigen thyroid peroxidase (TPO), and others (28–33). Moreover, some nucleus antigens, such as RA33, Sm, U1 snRNP, Jo-1, NOR-90, histone, DNA Topo 1, ribosomal P2, and others, also undergo some level of alternative splicing (27, 34–36). The increased noncanonical splicings and posttranslational modifications may give rise to novel untolerized antigenic epitopes that could enhance the immunogenicity of the autoantigens. Moreover, spliced isoforms with variation at the linker region can influence ligand binding and subsequent downstream signaling. Some specific autoantigens, such as U1 snRNP, Sm may even affect the splicing procedures. Assessment of these antigens or antibodies is of great importance in the diagnosis and monitor of autoimmune diseases.
Soluble Isoforms: May Serve as Predictors or Natural Inhibitors
High level of soluble isoforms, such as sIL-6R, sIL-7R, sgp130, sCD44v5, sCD44v6, and sCTLA4, are observed in most autoimmune diseases. Unlike membrane-bound isoforms, soluble isoforms are readily detected in peripheral blood. In certain cases, their levels there indicate disease severity and activity. Hence, they might be useful in disease monitoring. By contrast, soluble isoforms can also be natural competitive inhibitors to membrane-bound isoforms. Therefore, the former block downstream signaling pathways and are potential therapeutic targets.
We attempted to identify the foregoing common splicing modulation patterns in different autoimmune diseases. We focused on systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA). SLE is associated with autoimmunity induced by immune complexes (ICs), whereas RA is related to cytokine-mediated inflammation. We also explored spliced isoforms with potential diagnostic or therapeutic value.
Spliced Isoforms in SLE
SLE is a chronic autoimmune disease of unknown etiology. It more frequently affects women in their reproductive age. It is characterized by nuclear autoantigen and IC formation leading to organ-specific or systemic inflammation and tissue damage. Both innate and adaptive immune responses participate in SLE development (Figure 2). Viruses, excess apoptotic material, and neutrophil extracellular trap (NET) formation may be autoantigen sources. These are recognized by plasmacytoid dendritic cells (pDCs), which, in turn, activate innate immunity. A malfunctioning type I interferon (IFN) signaling pathway is an important clue in SLE as it plays essential roles in dendritic cell (DC) differentiation and maturation, the activation of autoreactive T cells and T cell-stimulating autoreactive B cells, and multiple autoantibody production. Along with several proinflammatory cytokines and chemokines, autoantibodies contribute to local tissue injury by immune complex deposition or complement activation. However, genetic factors increase the relative risk of SLE as well. Earlier GWAS identified over 50 genetic loci associated with SLE (37–40). Some of these might influence splicing. Furthermore, mutations and disruptions in the splicing machinery may also cause alternative splicing. Factors and conditions that increase SLE susceptibility have also been reported in previous studies.
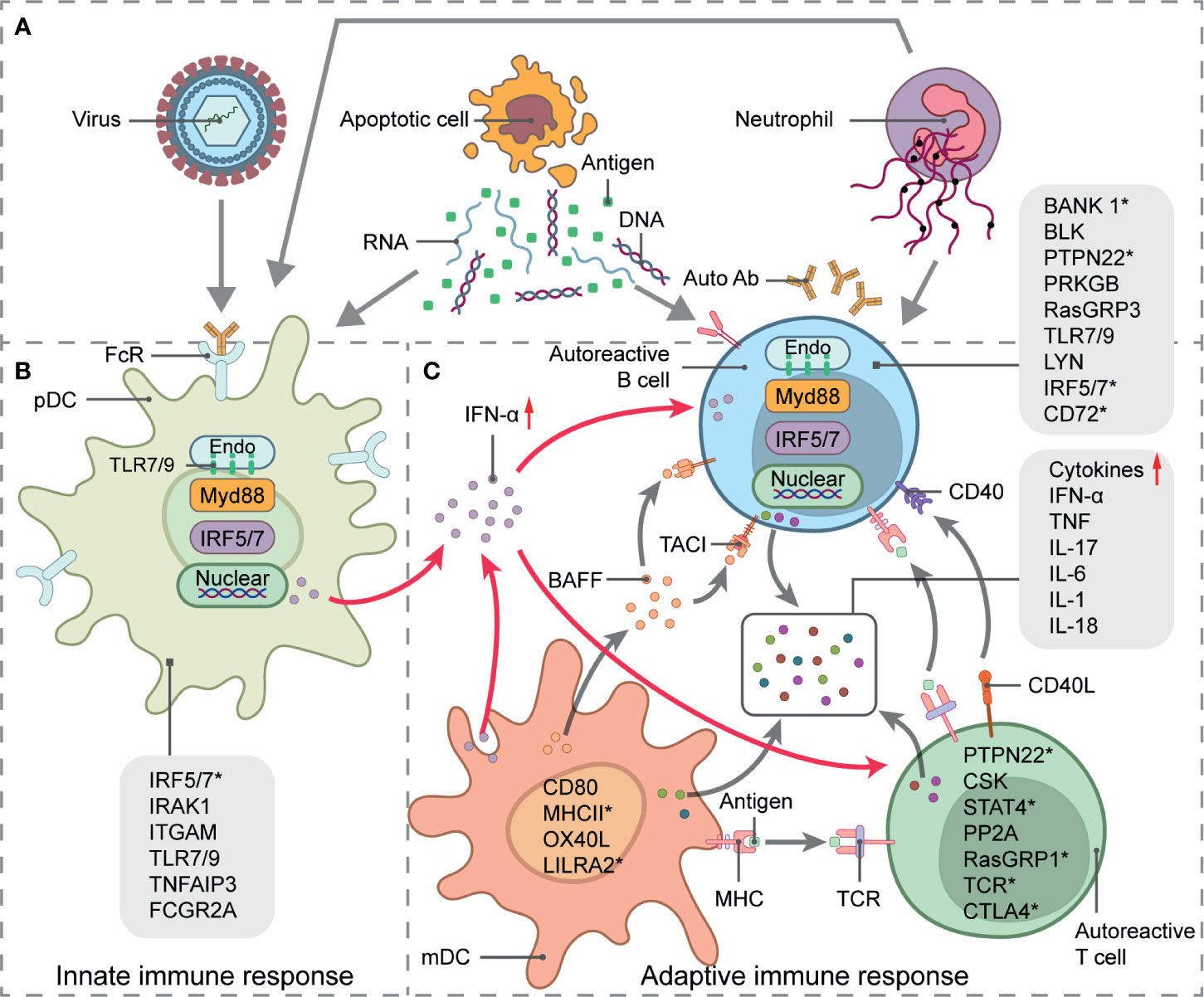
Figure 2 Systemic lupus erythematosus (SLE) genetics and pathogenesis. Schematic picture of SLE immunopathogenesis and alternative splicing involved in signaling pathways-related genes. (A) Source of autoantigens initiating innate and adaptive responses in SLE include viruses, apoptotic material, and neutrophil extracellular trap (NET) formation. (B) pDC activation, antigen presentation, and IFN-α secretion. (C) IFN-α signaling pathway stimulates T- and B-cell activation, which leads to autoantibody and proinflammatory cytokine production and eventual local tissue injury. Straight arrows represent stimulation and curved arrows represent target process activation or production. SLE-related susceptibility genes are indicated in immune signaling steps. Genes with * undergo alternative splicing.
In the following section, we address several of the best-studied examples of alternative splicing that affect both the adaptive- and innate immune signaling-related genes associated with SLE. We also summarize the roles of transcription factors in alternative splicing regulation.
IRF5
IRF5 is a transcription factor belonging to the IRF family. IRF5 mainly regulates interferon expression by activating the innate immune response to viruses or inflammatory stimuli. Moreover, IRF5 modulates cellular maturation, differentiation, and apoptosis as well as proinflammatory cytokine generation (41). Both innate and adaptive immune signaling activate IRF5 via the TLR and MyD88 pathways (42). IRF5 overexpression is associated with clinical SLE progress. The human IRF5 gene has nine coding exons and at least four non-coding alternative exons designated 1A, 1B, 1C, and 1D (41, 43). By differential selection of the coding or non-coding alternative exons, human IRF5 could theoretically generate multiple distinct functional isoforms. Some of these have been identified and were designated V1–V11. The spliced variants generated via the inclusion of alternative exon 1B (v2, v9, and v10) are strongly linked to IRF5 upregulation and SLE susceptibility. However, genetic polymorphisms in the human IRF5 locus disrupt normal splicing and increase the risk of aberrant spliced variant formation. The distinct haplotypes of four functional SNPs increase susceptibility to SLE and other autoimmune diseases. For example, the T allele of rs2004640 creates a novel 5’ss at the intron-exon boundary of alternative exon 1B. Thence, exon 1B is included in transcript production. The G allele of rs10954213 destroys the polyadenylation (polyA) site in the 3’ UTR region of exon 9. This mechanism arrests transcription and results in a relatively long, unstable transcript (44). A 5-bp (CGGGG) indel (insertion/deletion) located in the proximal promoter creates a novel binding site for the Sp1 transcription factor, which leads to alternative splicing (45). Furthermore, 30-bp indel polymorphisms in exon 6 select two alternative 3’ss. Insertion of these 3’ss produces V5 and V6 isoforms, whereas their deletion generates V1 and V4 isoforms (41). These isoforms are structurally and functionally distinct and have different expression levels. Hence, they influence IRF5 signaling and its downstream immune responses.
Murine IRF5 (MuIRF5) shares approximately 87% amino acid sequence homology with human IRF5. Different from the heavily spliced human IRF5, MuIRF5 primarily generates a full-length transcript. Nevertheless, Paun et al. have detected a splicing variant of IRF5 from the bone marrow of C57BL/6J mice, termed as BMv, which contains a deletion of the entire exon 5 and part of exon 4, 6 (46). In the MyD88 activated innate immune signaling, BMv serves as a relatively weaker activator to the downstream IFNA4 promoter, compared to the full-length transcript.
LILRA2
Leukocyte immunoglobulin-like receptors (LILRs) are also known as CD85. This family of leukocyte receptors has 13 members and includes the two pseudogenes LILRP1 and LILRP2. LILRA2 is a stimulatory receptor that delivers positive signals and is widely expressed on B cells, dendritic cells, monocytes, macrophages, and a subset of natural killer (NK) cells. LILRA2 cross-links with the immunoreceptor tyrosine-based activation motif (ITAM) of Fc-γ receptor (FcRγ) and induces calcium mobilization and subsequent signaling activation (47). Genetic polymorphisms of human LILRA2 were associated with increased SLE susceptibility in Japanese populations (48). The SNP rs2241524 G>A at the intron 6–exon 7 junction disrupts the consensus splicing acceptor motif and interferes with normal splicing. This mechanism results in a novel transcript with an in-frame deletion of three amino acids (Ala-Ser-Leu) at position 419–421 in the linker region (48). The impact of this spliced variant (Δ419–421 isoform) on ligand binding is unknown.
BANK1
The B-cell scaffold protein with ankyrin repeats 1 (BANK1) gene encodes a scaffold adaptor protein expressed primarily in B cells. Stimulation of B-cell receptors (BCRs) induces tyrosine phosphorylation in BANK1, which then amplifies B-cell signaling by facilitating the activation of downstream signal molecules, such as Src-family kinases, the inositol 1,4,5-trisphosphate receptor (IP3R) calcium channel, phospholipase Cγ2 (PLCγ2), protein kinase C β (PRKCB), and Ras (49, 50). BANK1 variants have been linked with susceptibility to multiple autoimmune diseases, such as SLE, RA, and SS (51–53). Kozyrev et al. analyzed BANK1 cDNA and identified a novel spliced transcript (50) designated Δ2 isoform because it lacks the entire exon 2. It encodes a protein without putative IP3R-binding or PH domains. This defect attenuates BANK1-mediated signaling. The Δ2 isoform is also detected from chimpanzee and mouse spleen. Three SNPs (rs10516487 G>A in exon 2, rs17266594 T>C in intron 1, and rs3733197 G>A in exon 7) associated with SLE might also influence Δ2 isoform splicing. The rs17266594 is localized to a putative splice BP of exon 2. This mutation affects relative splicing efficiency and upregulates the Δ2 isoform. The rs10516487 is associated with a decrease in exon 2 splicing (54). The precise role of BANK1-mediated signaling downregulation in SLE is unclear, but it may participate in SLE pathogenesis as B cells have vital functions in autoimmunity.
RasGRP1
Ras guanyl releasing protein 1 (RasGRP1) is a guanine-nucleotide-exchange factor that is first cloned in the brain and is then highly expressed on T cells (55). RasGRP1 is linked to autoimmunity. RasGRP1 deletion may lead to a deficiency of thymocyte differentiation or T-cell development in mice (56). In humans, RasGRP1 deficiency may interfere with T-cell and B-cell proliferation and activation, thereby causing various autoimmune disorders (57). T-cell receptor (TCR) stimulation drives protein tyrosine kinase activity and diacylglycerol (DAG) binding, which, in turn, recruit son of sevenless (SOS) to the plasma membrane and activate Ras (58, 59). RasGRP1 participates in TCR-induced signaling and activates Ras. RasGRP1 consists of a DAG-binding domain and two calcium-binding EF hands. Yasuda et al. identified 13 new spliced human RasGRP1 transcripts generated by alternatively including or deleting exons 5–17. These novel transcripts constitute an isoform family (splice variants A–M) (59). Nine of these were translated into truncated nonfunctional RasGRP1 isoforms because of a premature translation termination codon. The remaining four, however, did not cause any frameshift abnormalities or induce early translation termination. Compared with normal individuals, SLE patients lack the functional RasGRP1 isoform and have elevated levels of defective variant isoforms.
Splicing factors, such as SRSF1, regulate alternative splicing. The RasGRP1 transcript lacking exon 11 (splice variant A) is the type most observed in the T cells of SLE patients. SRSF1 directly binds the exon 11 of RasGRP1 mRNA, thereby promoting exon 11 inclusion. Kono et al. found that compared with the T cells of healthy individuals, those of SLE patients presented with significant SRSF1 downregulation. SRSF1 knockdown in healthy human T cells via small interfering RNAs (siRNAs) increased the splice variant A:normal RasGRP1 isoform ratio and downregulated RasGRP1 (60). Moreover, hnRNP-K protein was correlated with RASGRP1 expression and extracellular signal-regulated kinase (ERK) phosphorylation (61).
TCRζ
The TCRζ chain is involved in TCR/CD3 complex assembly and surface expression (62). The cytoplasmic domain of TCRζ includes three ITAMs that are phosphorylated when TCR is activated. This mechanism leads to the recruitment of other signaling molecules and, in turn, the phosphorylation or activation of downstream signaling transduction molecules (63–65). T cells are central to SLE pathogenesis. In SLE, T cells display multiple abnormalities in their antigen‐mediated signaling. Recent studies showed that SLE patients had lower TCRζ mRNA expression levels than healthy subjects (66–68), possibly because of low transcription activity, splice variant generation, and other factors (69, 70). Alternative splicing generated a series of transcript variants of TCRζ, including TCRη (exon 8 replaced by exon 9), TCRι (exon 8 replaced by exon 10), an isoform lacking exon 7, and another with a short 3’UTR (69, 71–74). Similar variation of TCRζ transcript also exists in animal models. For example, TCRη contains completely uniform alternative exon 8 selection to human, and is observed in both mice and swine, while TCRι is observed in mice (75, 76). TCRη and TCRι are natural isoforms with normal function, whereas the other two are relatively unstable, more readily degraded than their full-length transcript counterparts, and detected exclusively in SLE patients. Variant isoforms affect the assembly of the TCR/CD3 complex and its expression on the cell surface. In addition, it changes the Treg/Th17 balance. Consequently, they attenuate normal signaling and result in T-cell dysfunction and insufficient IL-2 production (77–79).
A previous study demonstrated that epigenetic modification or changes in the levels of splicing factors may affect the mechanism regulating alternative splicing. In human T cells, SRSF1 can bind the 3’UTR region of TCRζ mRNA, thereby favoring the expression of TCRζ isoforms with full-length 3’UTR over unstable short ones (80). SLE patients usually present with relatively low SRSF1 levels and enhanced SRSF1 ubiquitination (81, 82). SRSF1 may increase total TCRζ mRNA expression, whereas SRSF1 knockdown may decrease it. The specific mechanism involved in SRSF1 regulation remains to be determined. However, SRSF1 might be a suitable therapeutic target for the restoration of normal T-cell function.
Other Genes
Many studies have identified numerous loci of disease susceptibility-related genes associated with certain specific splicing patterns. In addition to the foregoing gene loci, there are several less common spliced variant isoforms related to SLE susceptibility.
CD72 is an inhibitory receptor expressed mainly on B cells. The CD72 SNP affects normal splicing and generates a spliced isoform lacking exon 8 (CD72Δex8) that modulates antibody production. Polymorphisms of the Complement C3d Receptor 2 (CR2) gene also regulate alternative splicing. A haplotype formed by minor CR2 SNP alleles (rs17615 and rs1048971 in exon 10 and rs4308977 in exon 11) might affect splicing efficiency and significantly lowers the risk of SLE (83). The SNP of CR2 known as rs3813946 is located in the 5’UTR and may also influence CR2 transcription. CTLA4, also known as CD152, is a protein receptor expressed mainly on T cells. Variants of human CTLA4 are associated with a wide range of autoimmune diseases, including SLE, RA, T1DM, GD, and MS. Alternative splicing of human CTLA4 generates various transcripts, such as a membrane-bound form (mCTLA4), a soluble form (sCTLA4), and four other rare transcript types. Al Fadhli et al. reported that patients with SLE or RA express neither mCTLA4 nor sCTLA4 (84). sCTLA4 may interfere with the B7:CTLA4 interaction and block negative signals. However, the roles of various CTLA4 isoforms in autoimmune disease mechanisms remain to be clarified and additional studies are required to explore these signaling pathways.
Spliced Isoforms in RA
RA is a chronic autoimmune disorder characterized by persistent inflammation of the synovial joints. RA eventually results in cartilage and bone destruction. About 50% of all RA incidences are associated with genetic factors, such as the predisposing genotypes HLA, PTPN22, and STAT4. Lifestyle factors, such as smoking, obesity, and vitamin D deficiency, can also trigger RA onset (85). Disequilibrium of the respiratory mucosa is an initial sign of RA development and subsequently leads to immune activation, local inflammation, and the production of autoantibodies, including those against citrullinated protein antigens (ACPA) and rheumatoid factor (RF) (Figure 3) (86). Activated lymphocytes and autoantibodies migrate to the joints where they induce the production of proinflammatory chemokines and cytokines, such as TNF‐α, IL-1, IL-6, and IL-17. In turn, these facilitate the recruitment and activation of other immunocytes. IL-17 produced by helper T (Th17) cells stimulates the activation and proliferation of synovial fibroblasts and macrophages, which eventually affect the balance between osteoclasts and osteoblasts and ultimately lead to bone destruction (87).
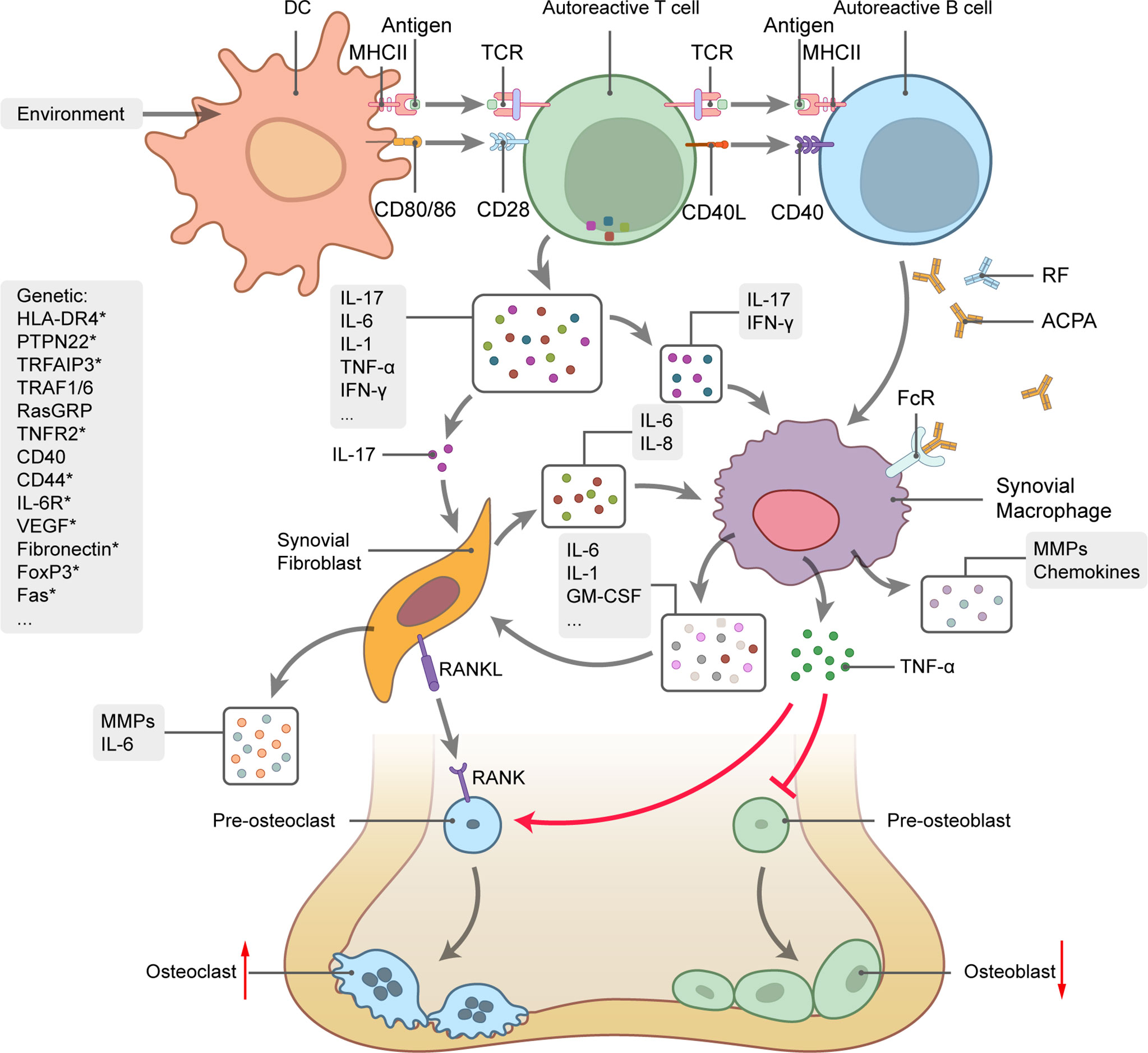
Figure 3 Rheumatoid arthritis (RA) genetics and pathogenesis. Schematic picture of cytokine-mediated bone inflammation and destruction in RA. Genetic predisposition and environmental factors initiate the autoimmune response. Interactions between TCR and MHCII-antigen plus co-stimulation of the CD28-CD80/86 pathway trigger activation of T cells by DCs along with substantial cytokine production, B-cell activation, and autoantibody generation. IL-17 stimulates synovial fibroblast and macrophage proliferation and activation and promotes the secretion of proinflammatory mediators, such as TNF‐α, IL-1, IL-6, and GM-CSF. TNF-α regulates the balance between bone formation and destruction by promoting osteoclast precursor transformation to osteoclasts and inhibiting osteoblast precursor transformation to osteoblasts. Osteoclast differentiation also depends on the interaction between RANK and its ligand. Curved arrows represent target process activation and bar-headed lines represent target process inhibition. Straight arrows represent changes in quantity. Several RA-related genes involved in RA pathogenesis are listed on the left side of the picture. Genes with * undergo alternative splicing.
Previous GWAS identified over 100 genetic loci encoding immune regulatory factors linked with RA. Moreover, several variant isoforms were identified among some of the candidate genes. In the following section, we review the major spliced gene transcripts participating in RA pathogenesis.
CD44
CD44 is a cell-surface glycoprotein involved in cell survival, proliferation, migration, and traffic signal transmission, cell–cell interactions, and programmed cell death (88, 89). CD44 comprises 10 constant and 10 variant (v1–v10) exons inserted between the 5’ and 3’ ends (90). Standard CD44 (CD44s) is the most ubiquitous form. It contains only constant exons and is expressed mainly on hematopoietic cells. Alternative splicing of the variant exons and N-glycosylation, O-glycosylation, and glycosaminoglycanation lead to numerous variant isoforms (CD44v). However, only a few dozen of these have been identified thus far (91). CD44 is closely related to multiple autoimmune disorders, including RA, SLE, MS, and inflammatory bowel disease (IBD). Compared with osteoarthritis patients, RA patients have relatively higher synovial CD44s, CD44v4, CD44v6, and CD44v7–8 transcript levels (92, 93). CD44v3 and CD44v6 are associated with increased tumor cell migration and invasion because they add adhesion and MMP docking domains. A similar mechanism occurs in RA fibroblast-like synoviocytes (FLS), and it augments their ability to invade Matrigel (94). Elevated serum levels of soluble CD44 (sCD44) variant isoforms, such as sCD44v5 and sCD44v6, have been detected in RA patients. Serum sCD44v5 is correlated with inflammation and could serve as a predictive factor in responses to RA therapy (95, 96).
TNFR2
Tumor necrosis factor (TNF)-α is a pleiotropic proinflammatory cytokine implicated in inflammation, cell proliferation, differentiation, and apoptosis (97). TNF-α is one of the most potent proinflammatory cytokines in RA. TNF signaling is mediated by the receptors TNFR1 and TNFR2. Both of these belong to the TNFR superfamily and have high affinity for TNF-α (98). TNFR1 is widely expressed on most cells and is both proinflammatory and apoptotic. TNRF2 expression is limited mainly to endothelial cells and certain immunocytes and participates in anti-inflammatory processes and cell proliferation (99, 100). Proteolytic cleavage produces soluble forms of TNFR (sTNFR). Elevated serum sTNFR is observed in certain infections, cancers, and autoimmune disorders, such as RA, SLE, and SS (101, 102) Recent studies identified an alternatively spliced TNRF2 isoform lacking exons 7 and 8 that encode transmembrane and cytoplasmic domains (DS-TNFR2) (103, 104). TNRF2 is a soluble receptor that acts as an antagonist in TNF‐α-mediated signaling and blocks apoptosis (104). Cañete et al. demonstrated that alternatively spliced DS-TNFR2 constitutes the majority of the sTNFR2 in RA patients, and serum sTNFR2 is closely associated with RA activity and severity (101, 103).
IL-6R and gp130
IL-6 is a pleiotropic cytokine that is crucial in chronic inflammation and multifactorial autoimmune disorders, such as RA, IBD, MS, and Castleman’s disease. IL-6 overexpression may contribute to clinical RA symptoms. In classic signaling, IL-6 binds the cognate ligand-binding subunit (IL‐6R, CD126). This mechanism results in the recruitment of the signal-transducing components gp130 and CD130, which, in turn, promote anti-inflammatory responses (105). Gp130 is widely expressed on most cell types, whereas IL-6R is detected on only a few specific cells, such as monocytes, macrophages, neutrophils, lymphocytes, and hepatocytes (106). IL-6R can exist in both membrane-bound (mIL-6R) and soluble (sIL-6R) form. About 90% of all sIL-6Rs are transformed from mIL-6R via limited proteolysis of metalloproteinase domain-containing protein 17 (ADAM17) (106, 107). The SNP rs8192284 A > C localized to the proteolytic cleavage site of exon 9 was associated with an increase in mIL-6R shedding (108, 109). The exclusion of exon 10 encoding the transmembrane region and IL-6R mRNA splicing generate a minor proportion of the sIL-6R (110). The loss of mIL-6R disrupts classic IL-6 signaling whereas formatted sIL-6R is biologically active. By binding IL-6, the sIL‐6R/IL‐6 complex can interact with gp130, which leads to intracellular signaling in cells that do not otherwise express IL-6R (111–113). This process is known as IL-6 trans-signaling and it induces proinflammatory responses (114). Upregulated sIL-6R has been observed in both RA and osteoarthritis. Hence, sIL-6R is implicated in joint inflammation and destruction (115). IL-6R inhibitors, such as tocilizumab and sarilumab, target binding to mIL-6R and sIL-6R. Therefore, they block both classic IL-6 signaling and trans-signaling. Their efficacy in RA treatment has been demonstrated (116). Recently, soluble gp130 (sgp130) variants arising from alternative splicing or polyadenylation have been identified. Sgp130 variants are considered natural trans-signaling inhibitors. Their three known isoforms are full-length sgp130, sgp130-RAPS, and sgp130-E10 (117, 118). Fc-dimerized sgp130 protein (sgp130Fc) blocks IL-6 trans-signaling and has been evaluated in the treatment of various inflammatory disease models. It is currently undergoing clinical trials as a drug candidate for IBD (119) and is also a potential therapeutic agent for RA.
Along with the foregoing soluble isoforms, alternative splicing generates others associated with RA such as sIL-7R (excision of exon 6 containing the transmembrane domain) (120), sCD137 (lacking the 414–545 nucleotide region and characterized by a frameshift) (121), sCD1d (excision of exons 4 and 5 containing the transmembrane domain) (122), and FasΔTM (excision of exon 6) (123).
VEGF
Vascular endothelial growth factor (VEGF) is a highly specific vascular endothelial cell mitogen. In vivo, VEGF induces microvascular permeability and plays central roles in regulating angiogenesis and tumor neovascularization (124). Prior studies demonstrated that VEGF-mediated angiogenesis also occurs in the synovial tissues of RA patients. In RA, VEGF polypeptide is highly expressed on synovial macrophages, fibroblasts, vascular smooth muscle cells, and synovial lining cells and in synovial fluids (125, 126). Human VEGF comprises eight exons and seven introns. Alternative selection of exons 6–8 and VEGF mRNA splicing generate the variant transcripts VEGF−A121, VEGF−A145, VEGF−A165, VEGF−A189, and VEGF−A206. These are generically designated VEGF−Axxx where xxx indicates the number of amino acids in the mature protein (127). VEGF121 and VEGF165 are the only VEGF-spliced isoforms expressed in RA. VEGF165 has 15 basic amino acids in 44 residues encoded by exon 7 and it binds heparan sulfate. By contrast, VEGF121 lacks this region and its diffusibility differs from that of VEGF165 (128).
Fibronectin
Fibronectin (Fn) is a multifunctional glycoprotein that is widely expressed in the extracellular matrix, plasma, synovial fluid, and other fluids. From a single gene, alternative Fn mRNA splicing generates multiple distinct isoforms that play vital roles in embryonic development, inflammation, wound repair, malignant metastasis, and thrombosis. Recognized alternative splicing sites include extra domain A (EDA), extra domain B (EDB), and type III connecting segment (IIICS) (129, 130). EDA and EDB each encode a single homology type III repeat (131). RA patients present with elevated synovial EDA(+)FN. This marker is correlated with accelerated rheumatoid joint destruction. The EDA(+)FN synovial fluid level is proportional to the severity of RA signs and symptoms and is promising as a predictor of RA progression (132). In animal model, Gondokaryono et al. found that EDA(+)FN might induce joint swelling in a mast cell TLR4-dependent manner by injecting EDA(+)FN to mast cells that lacked W/Wv mice (133). Removal of EDA(+)FN from the plasma can efficiently and effectively mitigate RA symptoms. EDA(+)FN cryofiltration and selective EDA(+)FN adsorbents have been developed for this purpose (134). Furthermore, heparin has high affinity for FN and can selectively bind and remove EDA(+)FN from the plasma.
Alternative Splicing in Other Autoimmune Diseases
Autoimmune diseases other than SLE may also be the result of aberrant alternative splicing. For example, alternative transcripts of disease-specific antigens could serve as novel antigenic epitopes affecting ligand binding and, by extension, autoimmunity. High level of a shorter splice variant of PLP, DM20 lacking exon 3B, in the thymic epithelial cells can induce the T-cell tolerance to all epitopes of the PLP protein (135). In an animal model of MS, SJL/J mice lack thymic DM20 and thereby experience loss of the central tolerance to this epitope and increased susceptibility to MS (28). Distribution and expression of IA-2 isoforms can influence immune responsiveness to specific epitopes. Islets express full-length IA-2 and two spliced isoforms, IA-2Δ13 (lacks exon 13) and IA-2Δ14 (lacks exon 14), whereas thymus and spleen exclusively express IA-2Δ13. Hence, IA-2 could serve as a target of T1DM (30). Moreover, SS-B/La isoform (exon 1 exchange) might be a target of Sjögren’s syndrome (SS) (31). Table 1 lists several autoimmune diseases, such as MS, T1DM, IBD, MG, SS, SSc, and GD, which are associated with alternative splicing. Diagnostic or pathologic roles of spliced isoforms in relevant autoimmune diseases are also cited.

Table 1 Alternatively spliced genes and isoforms in related autoimmune diseases.
Therapeutic Strategy Targeting Alternative Splicing
Alternative splicing modulation has been applied in many different areas even though its underlying regulatory mechanisms are poorly understood. Prior studies have attempted to identify causes or potential therapeutic targets via disease-related alternative splicing analyses. A recently discovered structural splicing enhancer (SSE) promotes tumor cell invasion and metastasis by interacting with small nuclear ribonucleoprotein polypeptide A’ (SNRPA1) (173). Shen et al. reported that, in mice, inhibiting the spliceosome by knocking down 14 splicing factors with siRNAs could drive the transition from pluripotent to totipotent stem cells. Hence, this mechanism has great potential in stem cell therapy (174). Certain therapies targeting the regulatory mechanism of alternative splicing have already been approved for cancers and neurological disorders.
Splice-switching oligonucleotides (SSOs) are specialized antisense oligonucleotides (ASOs) targeting pre-mRNAs. They comprise a major validated approach towards modulating alternative splicing events. SSOs bind various functional splicing factors, promote or inhibit exon and intron inclusion or skipping, and participate in the generation of expected spliced isoforms. SSOs have been approved for the treatment of Duchenne’s muscular dystrophy (DMD), spinal muscular atrophy (SMA), and other conditions (175, 176). For example, Graziewicz et al. designed a series of SSOs intended to promote exon 7 skipping in mouse TNFR2 mRNA. The SSOs upregulated the soluble TNFR2 isoform, which, in turn, naturally blocked TNF-α signaling (177). Therefore, SSO application might be a promising therapeutic avenue because TNF-α overexpression is a critical feature in RA immunopathogenesis. Furthermore, a synthetic 20-base ASO directed target at exon 2 of the AChE mRNA, known as Monarsen, is designed for the treatment of MG patients. According to a non-placebo-controlled phase Ib study of Monarsen, more than 90% of patients felt an improvement in symptoms (166, 178). Monarsen is currently in a Phase II study. Kinases and phosphatases in the serine-arginine protein kinase family (SRPKs) and CDC2-like family of kinases (CLKs), protein phosphatases, and certain small molecules can affect alternative splicing by modulating splicing factor activity or levels (8). Other technologies such as siRNAs and clustered, regularly interspaced, short palindromic repeat‐associated protein 9 (CRISPR‐Cas9) regulate alternative splicing events via gene editing. Moreover, targeted disease-related spliced variant degradation may help treat certain diseases. There is abundant theoretical evidence for numerous efficacious therapies targeting the regulatory mechanism of alternative splicing. Nevertheless, we lack empirical evidence for the effectiveness of these approaches in the management of autoimmune diseases. Thus, further research is required to validate the feasibility of alternative splicing manipulation.
Conclusion and Prospective
Autoimmunity is closely associated with alternative splicing. Most candidate autoimmune genes may produce alternative isoforms. Though the present review is not exhaustive, it provides a fairly comprehensive synopsis of the most extensively studied alternative splicing events in autoimmune diseases, such as SLE and RA. Numerous variant mRNA transcripts and proteins were identified via literature searches. However, the actual number of functional isoforms might be less than anticipated because some of them were either untranslated or underwent premature translation because of the presence of a termination codon in the transcript.
As high-throughput sequencing continues to increase in depth and capacity, RNA sequencing (RNA-Seq) might play an important role in elucidating transcriptome and alternative splicing (179). Recently developed third-generation RNA-Seq has the advantage of long read length and can facilitate the analysis of directly spliced isoform structures as well as the discovery of thousands of novel distinct transcript products (180). Innovative technologies will deepen our understanding of the mechanisms regulating alternative splicing in various diseases and help develop new diagnostic and therapeutic tools for them.
Author Contributions
FH and WL conceived and designed the study. PR, LL, and SC completed the literature review and wrote the first draft. PR and LL contributed to the design of figures and table. FH, WL, and JC revised the manuscript for intellectual content. All authors contributed to the article and approved the submitted version. We would like to thank all participating authors.
Funding
This study was supported by the funds from the Primary Research & Development Plan of Zhejiang Province (2020C03034) to FH and the Project of Natural Science Foundation of Zhejiang Province (Q19H050030) to PR.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Graveley BR. Alternative Splicing: Increasing Diversity in the Proteomic World. Trends Genet (2001) 17:100–7. doi: 10.1016/s0168-9525(00)02176-4
2. ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature (2012) 89:57–74. doi: 10.1038/nature11247
3. Stamm S, Ben-Ari S, Rafalska I, Tang Y, Zhang Z, Toiber D, et al. Function of Alternative Splicing. Gene (2005) 344:1–20. doi: 10.1016/j.gene.2004.10.022
4. Kim HK, Pham MHC, Ko KS, Rhee BD, Han J. Alternative Splicing Isoforms in Health and Disease. Pflugers Arch (2018) 470:995–1016. doi: 10.1007/s00424-018-2136-x
5. Stojanovich L, Marisavljevich D. Stress as a Trigger of Autoimmune Disease. Autoimmun Rev (2008) 7:209–13. doi: 10.1016/j.autrev.2007.11.007
6. Lynch KW. Consequences of Regulated Pre-Mrna Splicing in the Immune System. Nat Rev Immunol (2004) 4:931–40. doi: 10.1038/nri1497
7. Faustino NA, Cooper TA. Pre-Mrna Splicing and Human Disease. Genes Dev (2003) 17:419–37. doi: 10.1101/gad.1048803
8. Black AJ, Gamarra JR, Giudice J. More Than a Messenger: Alternative Splicing as a Therapeutic Target. Biochim Biophys Acta Gene Regul Mech (2019) 1862:194395. doi: 10.1016/j.bbagrm.2019.06.006
9. Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ. Alternative Splicing: A Pivotal Step Between Eukaryotic Transcription and Translation. Nat Rev Mol Cell Biol (2013) 14:153–65. doi: 10.1038/nrm3525
10. Yang Q, Zhao J, Zhang W, Chen D, Wang Y. Aberrant Alternative Splicing in Breast Cancer. J Mol Cell Biol (2019) 11:920–9. doi: 10.1093/jmcb/mjz033
11. Urbanski LM, Leclair N, Anczuków O. Alternative-Splicing Defects in Cancer: Splicing Regulators and Their Downstream Targets, Guiding the Way to Novel Cancer Therapeutics. Wiley Interdiscip Rev RNA (2018) 9:e1476. doi: 10.1002/wrna.1476
12. Fu XD, Ares M Jr. Context-Dependent Control of Alternative Splicing by RNA-Binding Proteins. Nat Rev Genet (2014) 15:689–701. doi: 10.1038/nrg3778
13. Chen M, Manley JL. Mechanisms of Alternative Splicing Regulation: Insights From Molecular and Genomics Approaches. Nat Rev Mol Cell Biol (2009) 10:741–54. doi: 10.1038/nrm2777
14. Cherry S, Lynch KW. Alternative Splicing and Cancer: Insights, Opportunities, and Challenges From an Expanding View of the Transcriptome. Genes Dev (2020) 34:1005–16. doi: 10.1101/gad.338962.120
15. Long JC, Caceres JF. The SR Protein Family of Splicing Factors: Master Regulators of Gene Expression. Biochem J (2009) 417:15–27. doi: 10.1042/bj20081501
16. Xiao SH, Manley JL. Phosphorylation of the ASF/SF2 RS Domain Affects Both Protein-Protein and Protein-RNA Interactions and is Necessary for Splicing. Genes Dev (1997) 11:334–44. doi: 10.1101/gad.11.3.334
17. Ergun A, Doran G, Costello JC, Paik HH, Collins JJ, Mathis D, et al. Differential Splicing Across Immune System Lineages. Proc Natl Acad Sci USA (2013) 110:14324–9. doi: 10.1073/pnas.1311839110
18. Tsuzaka K, Fukuhara I, Setoyama Y, Yoshimoto K, Suzuki K, Abe T, et al. TCR Zeta Mrna With an Alternatively Spliced 3’-Untranslated Region Detected in Systemic Lupus Erythematosus Patients Leads to the Down-Regulation of TCR Zeta and TCR/CD3 Complex. J Immunol (2003) 171:2496–503. doi: 10.4049/jimmunol.171.5.2496
19. Sahoo A, Im SH. Interleukin and Interleukin Receptor Diversity: Role of Alternative Splicing. Int Rev Immunol (2010) 29:77–109. doi: 10.3109/08830180903349651
20. Martinez NM, Pan Q, Cole BS, Yarosh CA, Babcock GA, Heyd F, et al. Alternative Splicing Networks Regulated by Signaling in Human T Cells. Rna (2012) 18:1029–40. doi: 10.1261/rna.032243.112
21. De Arras L, Alper S. Limiting of the Innate Immune Response by SF3A-Dependent Control of Myd88 Alternative Mrna Splicing. PloS Genet (2013) 9:e1003855. doi: 10.1371/journal.pgen.1003855
22. Iwami KI, Matsuguchi T, Masuda A, Kikuchi T, Musikacharoen T, Yoshikai Y. Cutting Edge: Naturally Occurring Soluble Form of Mouse Toll-Like Receptor 4 Inhibits Lipopolysaccharide Signaling. J Immunol (2000) 165:6682–6. doi: 10.4049/jimmunol.165.12.6682
23. Akama H, Johnson CA, Colten HR. Human Complement Protein C2. Alternative Splicing Generates Templates for Secreted and Intracellular C2 Proteins. J Biol Chem (1995) 270:2674–8. doi: 10.1074/jbc.270.6.2674
24. Post TW, Liszewski MK, Adams EM, Tedja I, Miller EA, Atkinson JP. Membrane Cofactor Protein of the Complement System: Alternative Splicing of Serine/Threonine/Proline-Rich Exons and Cytoplasmic Tails Produces Multiple Isoforms That Correlate With Protein Phenotype. J Exp Med (1991) 174:93–102. doi: 10.1084/jem.174.1.93
25. Wang J, Zhang J, Li K, Zhao W, Cui Q. Splicedisease Database: Linking RNA Splicing and Disease. Nucleic Acids Res (2012) 40:D1055–9. doi: 10.1093/nar/gkr1171
26. Monteuuis G, Wong JJL, Bailey CG, Schmitz U, Rasko JEJ. The Changing Paradigm of Intron Retention: Regulation, Ramifications and Recipes. Nucleic Acids Res (2019) 47:11497–513. doi: 10.1093/nar/gkz1068
27. Ng B, Yang F, Huston DP, Yan Y, Yang Y, Xiong Z, et al. Increased Noncanonical Splicing of Autoantigen Transcripts Provides the Structural Basis for Expression of Untolerized Epitopes. J Allergy Clin Immunol (2004) 114:1463–70. doi: 10.1016/j.jaci.2004.09.006
28. Klein L, Klugmann M, Nave KA, Tuohy VK, Kyewski B. Shaping of the Autoreactive T-Cell Repertoire by a Splice Variant of Self Protein Expressed in Thymic Epithelial Cells. Nat Med (2000) 6:56–61. doi: 10.1038/71540
29. Hilton AA, Slavin AJ, Hilton DJ, Bernard CC. Characterization of Cdna and Genomic Clones Encoding Human Myelin Oligodendrocyte Glycoprotein. J Neurochem (1995) 65:309–18. doi: 10.1046/j.1471-4159.1995.65010309.x
30. Diez J, Park Y, Zeller M, Brown D, Garza D, Ricordi C, et al. Differential Splicing of the IA-2 Mrna in Pancreas and Lymphoid Organs as a Permissive Genetic Mechanism for Autoimmunity Against the IA-2 Type 1 Diabetes Autoantigen. Diabetes (2001) 50:895–900. doi: 10.2337/diabetes.50.4.895
31. Tröster H, Metzger TE, Semsei I, Schwemmle M, Winterpacht A, Zabel B, et al. One Gene, Two Transcripts: Isolation of an Alternative Transcript Encoding for the Autoantigen La/SS-B From a Cdna Library of a Patient With Primary Sjögrens’ Syndrome. J Exp Med (1994) 180:2059–67. doi: 10.1084/jem.180.6.2059
32. Chan EK, Di Donato F, Hamel JC, Tseng CE, Buyon JP. 52-Kd SS-a/Ro: Genomic Structure and Identification of an Alternatively Spliced Transcript Encoding a Novel Leucine Zipper-Minus Autoantigen Expressed in Fetal and Adult Heart. J Exp Med (1995) 182:983–92. doi: 10.1084/jem.182.4.983
33. Niccoli P, Fayadat L, Panneels V, Lanet J, Franc JL. Human Thyroperoxidase in Its Alternatively Spliced Form (TPO2) is Enzymatically Inactive and Exhibits Changes in Intracellular Processing and Trafficking. J Biol Chem (1997) 272:29487–92. doi: 10.1074/jbc.272.47.29487
34. Kaufman KM, Kirby MY, McClain MT, Harley JB, James JA. Lupus Autoantibodies Recognize the Product of an Alternative Open Reading Frame of Smb/B’. Biochem Biophys Res Commun (2001) 285:1206–12. doi: 10.1006/bbrc.2001.5302
35. Hutchison S, LeBel C, Blanchette M, Chabot B. Distinct Sets of Adjacent Heterogeneous Nuclear Ribonucleoprotein (Hnrnp) A1/A2 Binding Sites Control 5’ Splice Site Selection in the Hnrnp A1 Mrna Precursor. J Biol Chem (2002) 277:29745–52. doi: 10.1074/jbc.M203633200
36. Matera AG, Wu W, Imai H, O’Keefe CL, Chan EK. Molecular Cloning of the RNA Polymerase I Transcription Factor Hubf/NOR-90 (UBTF) Gene and Localization to 17q21.3 by Fluorescence in Situ Hybridization and Radiation Hybrid Mapping. Genomics (1997) 41:135–8. doi: 10.1006/geno.1997.4647
37. Deng Y, Tsao BP. Genetic Susceptibility to Systemic Lupus Erythematosus in the Genomic Era. Nat Rev Rheumatol (2010) 6:683–92. doi: 10.1038/nrrheum.2010.176
38. Ramos PS, Brown EE, Kimberly RP, Langefeld CD. Genetic Factors Predisposing to Systemic Lupus Erythematosus and Lupus Nephritis. Semin Nephrol (2010) 30:164–76. doi: 10.1016/j.semnephrol.2010.01.007
39. Munroe ME, James JA. Genetics of Lupus Nephritis: Clinical Implications. Semin Nephrol (2015) 35:396–409. doi: 10.1016/j.semnephrol.2015.08.002
40. Mohan C, Putterman C. Genetics and Pathogenesis of Systemic Lupus Erythematosus and Lupus Nephritis. Nat Rev Nephrol (2015) 11:329–41. doi: 10.1038/nrneph.2015.33
41. Kozyrev SV, Alarcon-Riquelme ME. The Genetics and Biology of Irf5-Mediated Signaling in Lupus. Autoimmunity (2007) 40:591–601. doi: 10.1080/08916930701510905
42. Ban T, Sato GR, Tamura T. Regulation and Role of the Transcription Factor IRF5 in Innate Immune Responses and Systemic Lupus Erythematosus. Int Immunol (2018) 30:529–36. doi: 10.1093/intimm/dxy032
43. Lazzari E, Jefferies CA. IRF5-Mediated Signaling and Implications for SLE. Clin Immunol (2014) 153:343–52. doi: 10.1016/j.clim.2014.06.001
44. Graham RR, Kyogoku C, Sigurdsson S, Vlasova IA, Davies LR, Baechler EC, et al. Three Functional Variants of IFN Regulatory Factor 5 (IRF5) Define Risk and Protective Haplotypes for Human Lupus. Proc Natl Acad Sci USA (2007) 104:6758–63. doi: 10.1073/pnas.0701266104
45. Sigurdsson S, Göring HH, Kristjansdottir G, Milani L, Nordmark G, Sandling JK, et al. Comprehensive Evaluation of the Genetic Variants of Interferon Regulatory Factor 5 (IRF5) Reveals a Novel 5 Bp Length Polymorphism as Strong Risk Factor for Systemic Lupus Erythematosus. Hum Mol Genet (2008) 17:872–81. doi: 10.1093/hmg/ddm359
46. Paun A, Reinert JT, Jiang Z, Medin C, Balkhi MY, Fitzgerald KA, et al. Functional Characterization of Murine Interferon Regulatory Factor 5 (IRF-5) and its Role in the Innate Antiviral Response. J Biol Chem (2008) 283:14295–308. doi: 10.1074/jbc.M800501200
47. Lewis Marffy AL, McCarthy AJ. Leukocyte Immunoglobulin-Like Receptors (Lilrs) on Human Neutrophils: Modulators of Infection and Immunity. Front Immunol (2020) 11:857. doi: 10.3389/fimmu.2020.00857
48. Mamegano K, Kuroki K, Miyashita R, Kusaoi M, Kobayashi S, Matsuta K, et al. Association of LILRA2 (ILT1, LIR7) Splice Site Polymorphism With Systemic Lupus Erythematosus and Microscopic Polyangiitis. Genes Immun (2008) 9:214–23. doi: 10.1038/gene.2008.5
49. Kurosaki T. Regulation of B-Cell Signal Transduction by Adaptor Proteins. Nat Rev Immunol (2002) 2:354–63. doi: 10.1038/nri801
50. Kozyrev SV, Abelson AK, Wojcik J, Zaghlool A, Linga Reddy MV, Sanchez E, et al. Functional Variants in the B-Cell Gene BANK1 are Associated With Systemic Lupus Erythematosus. Nat Genet (2008) 40:211–6. doi: 10.1038/ng.79
51. Rueda B, Gourh P, Broen J, Agarwal SK, Simeon C, Ortego-Centeno N, et al. BANK1 Functional Variants are Associated With Susceptibility to Diffuse Systemic Sclerosis in Caucasians. Ann Rheum Dis (2010) 69:700–5. doi: 10.1136/ard.2009.118174
52. Jiang SH, Athanasopoulos V, Ellyard JI, Chuah A, Cappello J, Cook A, et al. Functional Rare and Low Frequency Variants in BLK and BANK1 Contribute to Human Lupus. Nat Commun (2019) 10:2201. doi: 10.1038/s41467-019-10242-9
53. Ramírez-Bello J, Fragoso JM, Alemán-Ávila I, Jiménez-Morales S, Campos-Parra AD, Barbosa-Cobos RE, et al. Association of BLK and BANK1 Polymorphisms and Interactions With Rheumatoid Arthritis in a Latin-American Population. Front Genet (2020) 11:58. doi: 10.3389/fgene.2020.00058
54. Kozyrev SV, Bernal-Quirós M, Alarcón-Riquelme ME, Castillejo-López C. The Dual Effect of the Lupus-Associated Polymorphism Rs10516487 on BANK1 Gene Expression and Protein Localization. Genes Immun (2012) 13:129–38. doi: 10.1038/gene.2011.62
55. Ebinu JO, Bottorff DA, Chan EY, Stang SL, Dunn RJ, Stone JC. Rasgrp, a Ras Guanyl Nucleotide- Releasing Protein With Calcium- and Diacylglycerol-Binding Motifs. Science (1998) 280:1082–6. doi: 10.1126/science.280.5366.1082
56. Dower NA, Stang SL, Bottorff DA, Ebinu JO, Dickie P, Ostergaard HL, et al. Rasgrp is Essential for Mouse Thymocyte Differentiation and TCR Signaling. Nat Immunol (2000) 1:317–21. doi: 10.1038/79766
57. Salzer E, Cagdas D, Hons M, Mace EM, Garncarz W, Petronczki ÖY, et al. RASGRP1 Deficiency Causes Immunodeficiency With Impaired Cytoskeletal Dynamics. Nat Immunol (2016) 17:1352–60. doi: 10.1038/ni.3575
58. Ebinu JO, Stang SL, Teixeira C, Bottorff DA, Hooton J, Blumberg PM, et al. Rasgrp Links T-Cell Receptor Signaling to Ras. Blood (2000) 95:3199–203. doi: 10.1182/blood.V95.10.3199
59. Yasuda S, Stevens RL, Terada T, Takeda M, Hashimoto T, Fukae J, et al. Defective Expression of Ras Guanyl Nucleotide-Releasing Protein 1 in a Subset of Patients With Systemic Lupus Erythematosus. J Immunol (2007) 179:4890–900. doi: 10.4049/jimmunol.179.7.4890
60. Kono M, Kurita T, Yasuda S, Kono M, Fujieda Y, Bohgaki T, et al. Decreased Expression of Serine/Arginine-Rich Splicing Factor 1 in T Cells From Patients With Active Systemic Lupus Erythematosus Accounts for Reduced Expression of Rasgrp1 and DNA Methyltransferase 1. Arthritis Rheumatol (2018) 70:2046–56. doi: 10.1002/art.40585
61. Molineros JE, Singh B, Terao C, Okada Y, Kaplan J, McDaniel B, et al. Mechanistic Characterization of RASGRP1 Variants Identifies an Hnrnp-K-Regulated Transcriptional Enhancer Contributing to SLE Susceptibility. Front Immunol (2019) 10:1066. doi: 10.3389/fimmu.2019.01066
62. Wange RL, Samelson LE. Complex Complexes: Signaling at the TCR. Immunity (1996) 5:197–205. doi: 10.1016/s1074-7613(00)80315-5
63. Irving BA, Chan AC, Weiss A. Functional Characterization of a Signal Transducing Motif Present in the T Cell Antigen Receptor Zeta Chain. J Exp Med (1993) 177:1093–103. doi: 10.1084/jem.177.4.1093
64. Baniyash M, Garcia-Morales P, Luong E, Samelson LE, Klausner RD. The T Cell Antigen Receptor Zeta Chain is Tyrosine Phosphorylated Upon Activation. J Biol Chem (1988) 263:18225–30. doi: 10.1016/S0021-9258(19)81349-2
65. Zenner G, Vorherr T, Mustelin T, Burn P. Differential and Multiple Binding of Signal Transducing Molecules to the Itams of the TCR-Zeta Chain. J Cell Biochem (1996) 63:94–103. doi: 10.1002/(sici)1097-4644(199610)63:1<94::aid-jcb8>3.0.co;2-v
66. Liossis SN, Ding XZ, Dennis GJ, Tsokos GC. Altered Pattern of TCR/CD3-Mediated Protein-Tyrosyl Phosphorylation in T Cells From Patients With Systemic Lupus Erythematosus. Deficient Expression of the T Cell Receptor Zeta Chain. J Clin Invest (1998) 101:1448–57. doi: 10.1172/jci1457
67. Brundula V, Rivas LJ, Blasini AM, París M, Salazar S, Stekman IL, et al. Diminished Levels of T Cell Receptor Zeta Chains in Peripheral Blood T Lymphocytes From Patients With Systemic Lupus Erythematosus. Arthritis Rheum (1999) 42:1908–16. doi: 10.1002/1529-0131(199909)42:9<1908::Aid-anr17>3.0.Co;2-7
68. Pang M, Setoyama Y, Tsuzaka K, Yoshimoto K, Amano K, Abe T, et al. Defective Expression and Tyrosine Phosphorylation of the T Cell Receptor Zeta Chain in Peripheral Blood T Cells From Systemic Lupus Erythematosus Patients. Clin Exp Immunol (2002) 129:160–8. doi: 10.1046/j.1365-2249.2002.01833.x
69. Nambiar MP, Enyedy EJ, Warke VG, Krishnan S, Dennis G, Kammer GM, et al. Polymorphisms/Mutations of TCR-Zeta-Chain Promoter and 3’ Untranslated Region and Selective Expression of TCR Zeta-Chain With an Alternatively Spliced 3’ Untranslated Region in Patients With Systemic Lupus Erythematosus. J Autoimmun (2001) 16:133–42. doi: 10.1006/jaut.2000.0475
70. Chowdhury B, Tsokos CG, Krishnan S, Robertson J, Fisher CU, Warke RG, et al. Decreased Stability and Translation of T Cell Receptor Zeta Mrna With an Alternatively Spliced 3’-Untranslated Region Contribute to Zeta Chain Down-Regulation in Patients With Systemic Lupus Erythematosus. J Biol Chem (2005) 280:18959–66. doi: 10.1074/jbc.M501048200
71. Tsuzaka K, Takeuchi T, Onoda N, Pang M, Abe T. Mutations in T Cell Receptor Zeta Chain Mrna of Peripheral T Cells From Systemic Lupus Erythematosus Patients. J Autoimmun (1998) 11:381–5. doi: 10.1006/jaut.1998.0223
72. Nambiar MP, Enyedy EJ, Warke VG, Krishnan S, Dennis G, Wong HK, et al. T Cell Signaling Abnormalities in Systemic Lupus Erythematosus are Associated With Increased Mutations/Polymorphisms and Splice Variants of T Cell Receptor Zeta Chain Messenger RNA. Arthritis Rheum (2001) 44:1336–50. doi: 10.1002/1529-0131(200106)44:6<1336::Aid-art226>3.0.Co;2-8
73. Tsuzaka K, Onoda N, Yoshimoto K, Setoyama Y, Suzuki K, Pang M, et al. T-Cell Receptor ζ Mrna With an Alternatively Spliced 3’ Untranslated Region is Generated Predominantly in the Peripheral Blood T Cells of Systemic Lupus Erythematosus Patients. Mod Rheumatol (2002) 12:167–73. doi: 10.3109/s101650200028
74. Clayton LK, D’Adamio L, Howard FD, Sieh M, Hussey RE, Koyasu S, et al. CD3 Eta and CD3 Zeta are Alternatively Spliced Products of a Common Genetic Locus and are Transcriptionally and/or Post-Transcriptionally Regulated During T-Cell Development. Proc Natl Acad Sci USA (1991) 88:5202–6. doi: 10.1073/pnas.88.12.5202
75. Yamamoto R, Isobe T, Eguchi T, Tang WR, Kiyokawa N, Amemiya H, et al. Porcine TCR CD3zeta-Chain and Eta-Chain. Mol Immunol (2005) 42:1485–93. doi: 10.1016/j.molimm.2005.01.008
76. Nocentini G, Ronchetti S, Bartoli A, Testa G, D’Adamio F, Riccardi C, et al. T Cell Receptor Iota an Alternatively Spliced Product of the T Cell Receptor Zeta Gene. Eur J Immunol (1995) 25:1405–9. doi: 10.1002/eji.1830250540
77. Takeuchi T, Suzuki K. CD247 Variants and Single-Nucleotide Polymorphisms Observed in Systemic Lupus Erythematosus Patients. Rheumatol (Oxford) (2013) 52:1551–5. doi: 10.1093/rheumatology/ket119
78. Chowdhury B, Krishnan S, Tsokos CG, Robertson JW, Fisher CU, Nambiar MP, et al. Stability and Translation of TCR Zeta Mrna are Regulated by the Adenosine-Uridine-Rich Elements in Splice-Deleted 3’ Untranslated Region of Zeta-Chain. J Immunol (2006) 177:8248–57. doi: 10.4049/jimmunol.177.11.8248
79. Tsuzaka K, Setoyama Y, Yoshimoto K, Shiraishi K, Suzuki K, Abe T, et al. A Splice Variant of the TCR Zeta Mrna Lacking Exon 7 Leads to the Down-Regulation of TCR Zeta, the TCR/CD3 Complex, and IL-2 Production in Systemic Lupus Erythematosus T Cells. J Immunol (2005) 174:3518–25. doi: 10.4049/jimmunol.174.6.3518
80. Moulton VR, Gillooly AR, Perl MA, Markopoulou A, Tsokos GC. Serine Arginine-Rich Splicing Factor 1 (SRSF1) Contributes to the Transcriptional Activation of CD3ζ in Human T Cells. PloS One (2015) 10:e0131073. doi: 10.1371/journal.pone.0131073
81. Moulton VR, Grammatikos AP, Fitzgerald LM, Tsokos GC. Splicing Factor SF2/ASF Rescues IL-2 Production in T Cells From Systemic Lupus Erythematosus Patients by Activating IL-2 Transcription. Proc Natl Acad Sci U.S.A. (2013) 110:1845–50. doi: 10.1073/pnas.1214207110
82. Moulton VR, Gillooly AR, Tsokos GC. Ubiquitination Regulates Expression of the Serine/Arginine-Rich Splicing Factor 1 (SRSF1) in Normal and Systemic Lupus Erythematosus (SLE) T Cells. J Biol Chem (2014) 289:4126–34. doi: 10.1074/jbc.M113.518662
83. Douglas KB, Windels DC, Zhao J, Gadeliya AV, Wu H, Kaufman KM, et al. Complement Receptor 2 Polymorphisms Associated With Systemic Lupus Erythematosus Modulate Alternative Splicing. Genes Immun (2009) 10:457–69. doi: 10.1038/gene.2009.27
84. AlFadhli S, Nizam R. Differential Expression of Alternative Splice Variants of CTLA4 in Kuwaiti Autoimmune Disease Patients. Gene (2014) 534:307–12. doi: 10.1016/j.gene.2013.10.034
85. Scott DL, Wolfe F, Huizinga TW. Rheumatoid Arthritis. Lancet (2010) 376:1094–108. doi: 10.1016/s0140-6736(10)60826-4
86. Malmström V, Catrina AI, Klareskog L. The Immunopathogenesis of Seropositive Rheumatoid Arthritis: From Triggering to Targeting. Nat Rev Immunol (2017) 17:60–75. doi: 10.1038/nri.2016.124
87. Zamanpoor M. The Genetic Pathogenesis, Diagnosis and Therapeutic Insight of Rheumatoid Arthritis. Clin Genet (2019) 95:547–57. doi: 10.1111/cge.13498
88. Naor D, Sionov RV, Ish-Shalom D. CD44: Structure, Function, and Association With the Malignant Process. Adv Cancer Res (1997) 71:241–319. doi: 10.1016/s0065-230x(08)60101-3
89. Turley EA, Noble PW, Bourguignon LY. Signaling Properties of Hyaluronan Receptors. J Biol Chem (2002) 277:4589–92. doi: 10.1074/jbc.R100038200
90. Golan I, Nedvetzki S, Golan I, Eshkar-Sebban L, Levartovsky D, Elkayam O, et al. Expression of Extra Trinucleotide in CD44 Variant of Rheumatoid Arthritis Patients Allows Generation of Disease-Specific Monoclonal Antibody. J Autoimmun (2007) 28:99–113. doi: 10.1016/j.jaut.2007.02.007
91. Jackson DG, Bell JI, Dickinson R, Timans J, Shields J, Whittle N. Proteoglycan Forms of the Lymphocyte Homing Receptor CD44 are Alternatively Spliced Variants Containing the V3 Exon. J Cell Biol (1995) 128:673–85. doi: 10.1083/jcb.128.4.673
92. Haynes BF, Hale LP, Patton KL, Martin ME, McCallum RM. Measurement of an Adhesion Molecule as an Indicator of Inflammatory Disease Activity. Up-Regulation of the Receptor for Hyaluronate (CD44) in Rheumatoid Arthritis. Arthritis Rheum (1991) 34:1434–43. doi: 10.1002/art.1780341115
93. Grisar J, Munk M, Steiner CW, Amoyo-Minar L, Tohidast-Akrad M, Zenz P, et al. Expression Patterns of CD44 and CD44 Splice Variants in Patients With Rheumatoid Arthritis. Clin Exp Rheumatol (2012) 30:64–72.
94. Wibulswas A, Croft D, Pitsillides AA, Bacarese-Hamilton I, McIntyre P, Genot E, et al. Influence of Epitopes CD44v3 and CD44v6 in the Invasive Behavior of Fibroblast-Like Synoviocytes Derived From Rheumatoid Arthritic Joints. Arthritis Rheum (2002) 46:2059–64. doi: 10.1002/art.10421
95. Kittl EM, Haberhauer G, Ruckser R, Selleny S, Rech-Weichselbraun I, Hinterberger W, et al. Serum Levels of Soluble CD44 Variant Isoforms are Elevated in Rheumatoid Arthritis. Rheumatol Int (1997) 16:181–6. doi: 10.1007/bf01330293
96. Skoumal M, Kolarz G, Haberhauer G, Feyertag J, Wottawa A. Is Soluble CD44 Variant Isoform 5 Useful as a Predicting Factor and a Parameter for Long Term Observation in Rheumatoid Arthritis? Ann Rheum Dis (2002) 61:1036–7. doi: 10.1136/ard.61.11.1036
97. Kriegsmann J, Berndt A, Hansen T, Borsi L, Zardi L, Bräuer R, et al. Expression of Fibronectin Splice Variants and Oncofetal Glycosylated Fibronectin in the Synovial Membranes of Patients With Rheumatoid Arthritis and Osteoarthritis. Rheumatol Int (2004) 24:25–33. doi: 10.1007/s00296-003-0316-1
98. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF Receptor Superfamilies: Integrating Mammalian Biology. Cell (2001) 104:487–501. doi: 10.1016/s0092-8674(01)00237-9
99. Gough P, Myles IA. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front Immunol (2020) 11:585880. doi: 10.3389/fimmu.2020.585880
100. Idriss HT, Naismith JH. TNF Alpha and the TNF Receptor Superfamily: Structure-Function Relationship(s). Microsc Res Tech (2000) 50:184–95. doi: 10.1002/1097-0029(20000801)50:3<184::Aid-jemt2>3.0.Co;2-h
101. Cope AP, Aderka D, Doherty M, Engelmann H, Gibbons D, Jones AC, et al. Increased Levels of Soluble Tumor Necrosis Factor Receptors in the Sera and Synovial Fluid of Patients With Rheumatic Diseases. Arthritis Rheum (1992) 35:1160–9. doi: 10.1002/art.1780351008
102. Aderka D, Wysenbeek A, Engelmann H, Cope AP, Brennan F, Molad Y, et al. Correlation Between Serum Levels of Soluble Tumor Necrosis Factor Receptor and Disease Activity in Systemic Lupus Erythematosus. Arthritis Rheum (1993) 36:1111–20. doi: 10.1002/art.1780360812
103. Cañete JD, Albaladejo C, Hernández MV, Laínez B, Pinto JA, Ramírez J, et al. Clinical Significance of High Levels of Soluble Tumour Necrosis Factor-α Receptor-2 Produced by Alternative Splicing in Rheumatoid Arthritis: A Longitudinal Prospective Cohort Study. Rheumatol (Oxford) (2011) 50:721–8. doi: 10.1093/rheumatology/keq381
104. Lainez B, Fernandez-Real JM, Romero X, Esplugues E, Cañete JD, Ricart W, et al. Identification and Characterization of a Novel Spliced Variant That Encodes Human Soluble Tumor Necrosis Factor Receptor 2. Int Immunol (2004) 16:169–77. doi: 10.1093/intimm/dxh014
105. Jones SA, Horiuchi S, Topley N, Yamamoto N, Fuller GM. The Soluble Interleukin 6 Receptor: Mechanisms of Production and Implications in Disease. FASEB J (2001) 15:43–58. doi: 10.1096/fj.99-1003rev
106. Scheller J, Garbers C, Rose-John S. Interleukin-6: From Basic Biology to Selective Blockade of Pro-Inflammatory Activities. Semin Immunol (2014) 26:2–12. doi: 10.1016/j.smim.2013.11.002
107. Müllberg J, Schooltink H, Stoyan T, Günther M, Graeve L, Buse G, et al. The Soluble Interleukin-6 Receptor is Generated by Shedding. Eur J Immunol (1993) 23:473–80. doi: 10.1002/eji.1830230226
108. Marinou I, Walters K, Winfield J, Bax DE. Wilson AG. A Gain of Function Polymorphism in the Interleukin 6 Receptor Influences RA Susceptibility. Ann Rheum Dis (2010) 69:1191–4. doi: 10.1136/ard.2008.100644
109. Kim LH, Lee HS, Kim YJ, Jung JH, Kim JY, Park BL, et al. Identification of Novel Snps in the Interleukin 6 Receptor Gene (IL6R). Hum Mutat (2003) 21:450–1. doi: 10.1002/humu.9130
110. Lust JA, Donovan KA, Kline MP, Greipp PR, Kyle RA, Maihle NJ. Isolation of an Mrna Encoding a Soluble Form of the Human Interleukin-6 Receptor. Cytokine (1992) 4:96–100. doi: 10.1016/1043-4666(92)90043-q
111. Chalaris A, Garbers C, Rabe B, Rose-John S, Scheller J. The Soluble Interleukin 6 Receptor: Generation and Role in Inflammation and Cancer. Eur J Cell Biol (2011) 90:484–94. doi: 10.1016/j.ejcb.2010.10.007
112. Rose-John S, Waetzig GH, Scheller J, Grötzinger J, Seegert D. The IL-6/Sil-6R Complex as a Novel Target for Therapeutic Approaches. Expert Opin Ther Targets (2007) 11:613–24. doi: 10.1517/14728222.11.5.613
113. Garbers C, Heink S, Korn T, Rose-John S. Interleukin-6: Designing Specific Therapeutics for a Complex Cytokine. Nat Rev Drug Discovery (2018) 17:395–412. doi: 10.1038/nrd.2018.45
114. Rose-John S, Heinrich PC. Soluble Receptors for Cytokines and Growth Factors: Generation and Biological Function. Biochem J (1994) 300(Pt 2):281–90. doi: 10.1042/bj3000281
115. Kotake S, Sato K, Kim KJ, Takahashi N, Udagawa N, Nakamura I, et al. Interleukin-6 and Soluble Interleukin-6 Receptors in the Synovial Fluids From Rheumatoid Arthritis Patients are Responsible for Osteoclast-Like Cell Formation. J Bone Miner Res (1996) 11:88–95. doi: 10.1002/jbmr.5650110113
116. Kaur S, Bansal Y, Kumar R, Bansal G. A Panoramic Review of IL-6: Structure, Pathophysiological Roles and Inhibitors. Bioorg Med Chem (2020) 28:115327. doi: 10.1016/j.bmc.2020.115327
117. Sommer J, Garbers C, Wolf J, Trad A, Moll JM, Sack M, et al. Alternative Intronic Polyadenylation Generates the Interleukin-6 Trans-Signaling Inhibitor Sgp130-E10. J Biol Chem (2014) 289:22140–50. doi: 10.1074/jbc.M114.560938
118. Wolf J, Waetzig GH, Chalaris A, Reinheimer TM, Wege H, Rose-John S, et al. Different Soluble Forms of the Interleukin-6 Family Signal Transducer Gp130 Fine-Tune the Blockade of Interleukin-6 Trans-Signaling. J Biol Chem (2016) 291:16186–96. doi: 10.1074/jbc.M116.718551
119. Rose-John S. The Soluble Interleukin 6 Receptor: Advanced Therapeutic Options in Inflammation. Clin Pharmacol Ther (2017) 102:591–8. doi: 10.1002/cpt.782
120. Badot V, Durez P, Van den Eynde BJ, Nzeusseu-Toukap A, Houssiau FA, Lauwerys BR. Rheumatoid Arthritis Synovial Fibroblasts Produce a Soluble Form of the Interleukin-7 Receptor in Response to Pro-Inflammatory Cytokines. J Cell Mol Med (2011) 15:2335–42. doi: 10.1111/j.1582-4934.2010.01228.x
121. Michel J, Langstein J, Hofstädter F, Schwarz H. A Soluble Form of CD137 (ILA/4-1BB), a Member of the TNF Receptor Family, is Released by Activated Lymphocytes and is Detectable in Sera of Patients With Rheumatoid Arthritis. Eur J Immunol (1998) 28:290–5. doi: 10.1002/(sici)1521-4141(199801)28:01<290::Aid-immu290>3.0.Co;2-s
122. Kojo S, Tsutsumi A, Goto D, Sumida T. Low Expression Levels of Soluble CD1d Gene in Patients With Rheumatoid Arthritis. J Rheumatol (2003) 30:2524–8.
123. Proussakova OV, Rabaya NA, Moshnikova AB, Telegina ES, Turanov A, Nanazashvili MG, et al. Oligomerization of Soluble Fas Antigen Induces its Cytotoxicity. J Biol Chem (2003) 278:36236–41. doi: 10.1074/jbc.M305896200
124. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular Endothelial Growth Factor (VEGF) and its Receptors. FASEB J (1999) 13:9–22. doi: 10.1096/fasebj.13.1.9
125. Koch AE, Harlow LA, Haines GK, Amento EP, Unemori EN, Wong WL, et al. Vascular Endothelial Growth Factor. A Cytokine Modulating Endothelial Function in Rheumatoid Arthritis. J Immunol (1994) 152:4149–56.
126. Nagashima M, Yoshino S, Ishiwata T, Asano G. Role of Vascular Endothelial Growth Factor in Angiogenesis of Rheumatoid Arthritis. J Rheumatol (1995) 22:1624–30.
127. Robinson CJ, Stringer SE. The Splice Variants of Vascular Endothelial Growth Factor (VEGF) and Their Receptors. J Cell Sci (2001) 114:853–65. doi: 10.1242/jcs.114.5.853
128. Pufe T, Petersen W, Tillmann B, Mentlein R. Splice Variants VEGF121 and VEGF165 of the Angiogenic Peptide Vascular Endothelial Cell Growth Factor are Expressed in the Synovial Tissue of Patients With Rheumatoid Arthritis. J Rheumatol (2001) 28:1482–5.
129. Schwarzbauer JE. Alternative Splicing of Fibronectin: Three Variants, Three Functions. Bioessays (1991) 13:527–33. doi: 10.1002/bies.950131006
130. Romberger DJ. Fibronectin. Int J Biochem Cell Biol (1997) 29:939–43. doi: 10.1016/s1357-2725(96)00172-0
131. Kornblihtt AR, Vibe-Pedersen K, Baralle FE. Human Fibronectin: Cell Specific Alternative Mrna Splicing Generates Polypeptide Chains Differing in the Number of Internal Repeats. Nucleic Acids Res (1984) 12:5853–68. doi: 10.1093/nar/12.14.5853
132. Shiozawa K, Hino K, Shiozawa S. Alternatively Spliced EDA-Containing Fibronectin in Synovial Fluid as a Predictor of Rheumatoid Joint Destruction. Rheumatol (Oxford) (2001) 40:739–42. doi: 10.1093/rheumatology/40.7.739
133. Gondokaryono SP, Ushio H, Niyonsaba F, Hara M, Takenaka H, Jayawardana ST, et al. The Extra Domain a of Fibronectin Stimulates Murine Mast Cells via Toll-Like Receptor 4. J Leukoc Biol (2007) 82:657–65. doi: 10.1189/jlb.1206730
134. Miyashita K, Sakashita E, Miyamoto K, Tokita M, Komai T. Development of the Selective Adsorbent for EDA Containing Fibronectin Using Heparin Immobilized Cellulose. Int J Biol Macromol (1998) 22:91–5. doi: 10.1016/s0141-8130(97)00093-7
135. Wang E, Dimova N, Sperle K, Huang Z, Lock L, McCulloch MC, et al. Deletion of a Splicing Enhancer Disrupts PLP1/DM20 Ratio and Myelin Stability. Exp Neurol (2008) 214:322–30. doi: 10.1016/j.expneurol.2008.09.001
136. Stone RC, Du P, Feng D, Dhawan K, Rönnblom L, Eloranta ML, et al. RNA-Seq for Enrichment and Analysis of IRF5 Transcript Expression in SLE. PloS One (2013) 8:e54487. doi: 10.1371/journal.pone.0054487
137. Rother N, van der Vlag J. Disturbed T Cell Signaling and Altered Th17 and Regulatory T Cell Subsets in the Pathogenesis of Systemic Lupus Erythematosus. Front Immunol (2015) 6:610. doi: 10.3389/fimmu.2015.00610
138. Hitomi Y, Tsuchiya N, Kawasaki A, Ohashi J, Suzuki T, Kyogoku C, et al. CD72 Polymorphisms Associated With Alternative Splicing Modify Susceptibility to Human Systemic Lupus Erythematosus Through Epistatic Interaction With FCGR2B. Hum Mol Genet (2004) 13:2907–17. doi: 10.1093/hmg/ddh318
139. Hitomi Y, Adachi T, Tsuchiya N, Honda Z, Tokunaga K, Tsubata T. Human CD72 Splicing Isoform Responsible for Resistance to Systemic Lupus Erythematosus Regulates Serum Immunoglobulin Level and is Localized in Endoplasmic Reticulum. BMC Immunol (2012) 13:72. doi: 10.1186/1471-2172-13-72
140. Segawa S, Goto D, Yoshiga Y, Hayashi T, Matsumoto I, Ito S, et al. Low Levels of Soluble CD1d Protein Alters NKT Cell Function in Patients With Rheumatoid Arthritis. Int J Mol Med (2009) 24:481–6. doi: 10.3892/ijmm_00000256
141. Heinhuis B, Koenders MI, van de Loo FA, Netea MG, van den Berg WB, Joosten LA. Inflammation-Dependent Secretion and Splicing of IL-32{Gamma} in Rheumatoid Arthritis. Proc Natl Acad Sci USA (2011) 108:4962–7. doi: 10.1073/pnas.1016005108
142. Heinhuis B, Koenders MI, van Riel PL, van de Loo FA, Dinarello CA, Netea MG, et al. Tumour Necrosis Factor Alpha-Driven IL-32 Expression in Rheumatoid Arthritis Synovial Tissue Amplifies an Inflammatory Cascade. Ann Rheum Dis (2011) 70:660–7. doi: 10.1136/ard.2010.139196
143. Shchetynsky K, Protsyuk D, Ronninger M, Diaz-Gallo LM, Klareskog L, Padyukov L. Gene-Gene Interaction and RNA Splicing Profiles of MAP2K4 Gene in Rheumatoid Arthritis. Clin Immunol (2015) 158:19–28. doi: 10.1016/j.clim.2015.02.011
144. Chang HH, Tai TS, Lu B, Iannaccone C, Cernadas M, Weinblatt M, et al. PTPN22.6, a Dominant Negative Isoform of PTPN22 and Potential Biomarker of Rheumatoid Arthritis. PloS One (2012) 7:e33067. doi: 10.1371/journal.pone.0033067
145. Ryder LR, Bartels EM, Woetmann A, Madsen HO, Odum N, Bliddal H, et al. Foxp3 Mrna Splice Forms in Synovial CD4+ T Cells in Rheumatoid Arthritis and Psoriatic Arthritis. Apmis (2012) 120:387–96. doi: 10.1111/j.1600-0463.2011.02848.x
146. Yoon HK, Byun HS, Lee H, Jeon J, Lee Y, Li Y, et al. Intron-Derived Aberrant Splicing of A20 Transcript in Rheumatoid Arthritis. Rheumatol (Oxford) (2013) 52:427–37. doi: 10.1093/rheumatology/kes292
147. Gregory SG, Schmidt S, Seth P, Oksenberg JR, Hart J, Prokop A, et al. Interleukin 7 Receptor Alpha Chain (IL7R) Shows Allelic and Functional Association With Multiple Sclerosis. Nat Genet (2007) 39:1083–91. doi: 10.1038/ng2103
148. Goodwin RG, Friend D, Ziegler SF, Jerzy R, Falk BA, Gimpel S, et al. Cloning of the Human and Murine Interleukin-7 Receptors: Demonstration of a Soluble Form and Homology to a New Receptor Superfamily. Cell (1990) 60:941–51. doi: 10.1016/0092-8674(90)90342-c
149. Gilli F, Valentino P, Caldano M, Granieri L, Capobianco M, Malucchi S, et al. Expression and Regulation of Ifnalpha/Beta Receptor in Ifnbeta-Treated Patients With Multiple Sclerosis. Neurology (2008) 71:1940–7. doi: 10.1212/01.wnl.0000327340.50284.8d
150. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis Controls the Induction of Human Regulatory T Cells by Modulating the Expression of FOXP3 Exon 2 Splicing Variants. Nat Immunol (2015) 16:1174–84. doi: 10.1038/ni.3269
151. Paraboschi EM, Rimoldi V, Soldà G, Tabaglio T, Dall’Osso C, Saba E, et al. Functional Variations Modulating PRKCA Expression and Alternative Splicing Predispose to Multiple Sclerosis. Hum Mol Genet (2014) 23:6746–61. doi: 10.1093/hmg/ddu392
152. Lynch KW. Weiss a. A CD45 Polymorphism Associated With Multiple Sclerosis Disrupts an Exonic Splicing Silencer. J Biol Chem (2001) 276:24341–7. doi: 10.1074/jbc.M102175200
153. Qu HQ, Lu Y, Marchand L, Bacot F, Fréchette R, Tessier MC, et al. Genetic Control of Alternative Splicing in the TAP2 Gene: Possible Implication in the Genetics of Type 1 Diabetes. Diabetes (2007) 56:270–5. doi: 10.2337/db06-0865
154. Gerold KD, Zheng P, Rainbow DB, Zernecke A, Wicker LS, Kissler S. The Soluble CTLA-4 Splice Variant Protects From Type 1 Diabetes and Potentiates Regulatory T-Cell Function. Diabetes (2011) 60:1955–63. doi: 10.2337/db11-0130
155. Nogueira TC, Paula FM, Villate O, Colli ML, Moura RF, Cunha DA, et al. GLIS3, a Susceptibility Gene for Type 1 and Type 2 Diabetes, Modulates Pancreatic Beta Cell Apoptosis via Regulation of a Splice Variant of the BH3-Only Protein Bim. PloS Genet (2013) 9:e1003532. doi: 10.1371/journal.pgen.1003532
156. Yip L, Su L, Sheng D, Chang P, Atkinson M, Czesak M, et al. Deaf1 Isoforms Control the Expression of Genes Encoding Peripheral Tissue Antigens in the Pancreatic Lymph Nodes During Type 1 Diabetes. Nat Immunol (2009) 10:1026–33. doi: 10.1038/ni.1773
157. de Jong VM, Abreu JR, Verrijn Stuart AA, van der Slik AR, Verhaeghen K, Engelse MA, et al. Alternative Splicing and Differential Expression of the Islet Autoantigen IGRP Between Pancreas and Thymus Contributes to Immunogenicity of Pancreatic Islets But Not Diabetogenicity in Humans. Diabetologia (2013) 56:2651–8. doi: 10.1007/s00125-013-3034-6
158. Dogra RS, Vaidyanathan P, Prabakar KR, Marshall KE, Hutton JC, Pugliese A. Alternative Splicing of G6PC2, the Gene Coding for the Islet-Specific Glucose-6-Phosphatase Catalytic Subunit-Related Protein (IGRP), Results in Differential Expression in Human Thymus and Spleen Compared With Pancreas. Diabetologia (2006) 49:953–7. doi: 10.1007/s00125-006-0185-8
159. Yip L, Taylor C, Whiting CC, Fathman CG. Diminished Adenosine A1 Receptor Expression in Pancreatic α-Cells may Contribute to the Pathology of Type 1 Diabetes. Diabetes (2013) 62:4208–19. doi: 10.2337/db13-0614
160. Leung E, Hong J, Fraser A, Krissansen GW. Splicing of NOD2 (CARD15) RNA Transcripts. Mol Immunol (2007) 44:284–94. doi: 10.1016/j.molimm.2006.03.009
161. Hoffmann U, Heilmann K, Hayford C, Stallmach A, Wahnschaffe U, Zeitz M, et al. CD44v7 Ligation Downregulates the Inflammatory Immune Response in Crohn’s Disease Patients by Apoptosis Induction in Mononuclear Cells From the Lamina Propria. Cell Death Differ (2007) 14:1542–51. doi: 10.1038/sj.cdd.4402153
162. Li C, Vu K, Hazelgrove K, Kuemmerle JF. Increased IGF-Iec Expression and Mechano-Growth Factor Production in Intestinal Muscle of Fibrostenotic Crohn’s Disease and Smooth Muscle Hypertrophy. Am J Physiol Gastrointest Liver Physiol (2015) 309:G888–99. doi: 10.1152/ajpgi.00414.2014
163. Muise AM, Walters T, Wine E, Griffiths AM, Turner D, Duerr RH, et al. Protein-Tyrosine Phosphatase Sigma is Associated With Ulcerative Colitis. Curr Biol (2007) 17:1212–8. doi: 10.1016/j.cub.2007.06.013
164. Gu M, Kakoulidou M, Giscombe R, Pirskanen R, Lefvert AK, Klareskog L, et al. Identification of CTLA-4 Isoforms Produced by Alternative Splicing and Their Association With Myasthenia Gravis. Clin Immunol (2008) 128:374–81. doi: 10.1016/j.clim.2008.05.006
165. Meshorer E, Soreq H. Virtues and Woes of Ache Alternative Splicing in Stress-Related Neuropathologies. Trends Neurosci (2006) 29:216–24. doi: 10.1016/j.tins.2006.02.005
166. Angelini C, Martignago S, Bisciglia M. New Treatments for Myasthenia: A Focus on Antisense Oligonucleotides. Drug Des Devel Ther (2013) 7:13–7. doi: 10.2147/dddt.S25716
167. Li H, Reksten TR, Ice JA, Kelly JA, Adrianto I, Rasmussen A, et al. Identification of a Sjögren’s Syndrome Susceptibility Locus at OAS1 That Influences Isoform Switching, Protein Expression, and Responsiveness to Type I Interferons. PloS Genet (2017) 13:e1006820. doi: 10.1371/journal.pgen.1006820
168. Roescher N, Vosters JL, Alsaleh G, Dreyfus P, Jacques S, Chiocchia G, et al. Targeting the Splicing of Mrna in Autoimmune Diseases: BAFF Inhibition in Sjögren’s Syndrome as a Proof of Concept. Mol Ther (2014) 22:821–7. doi: 10.1038/mt.2013.275
169. Manetti M, Guiducci S, Romano E, Ceccarelli C, Bellando-Randone S, Conforti ML, et al. Overexpression of VEGF165b, an Inhibitory Splice Variant of Vascular Endothelial Growth Factor, Leads to Insufficient Angiogenesis in Patients With Systemic Sclerosis. Circ Res (2011) 109:e14–26. doi: 10.1161/circresaha.111.242057
170. Luzina IG, Keegan AD, Heller NM, Rook GA, Shea-Donohue T, Atamas SP. Regulation of Inflammation by Interleukin-4: A Review of “Alternatives”. J Leukoc Biol (2012) 92:753–64. doi: 10.1189/jlb.0412214
171. Sato S, Fujimoto M, Hasegawa M, Komura K, Yanaba K, Hayakawa I, et al. Serum Soluble CTLA-4 Levels are Increased in Diffuse Cutaneous Systemic Sclerosis. Rheumatol (Oxford) (2004) 43:1261–6. doi: 10.1093/rheumatology/keh303
172. Ferrand M, Le Fourn V, Franc JL. Increasing Diversity of Human Thyroperoxidase Generated by Alternative Splicing. Characterized by Molecular Cloning of New Transcripts With Single- and Multispliced mRNAs. J Biol Chem (2003) 278:3793–800. doi: 10.1074/jbc.M209513200
173. Fish L, Khoroshkin M, Navickas A, Garcia K, Culbertson B, Hänisch B, et al. A Prometastatic Splicing Program Regulated by SNRPA1 Interactions With Structured RNA Elements. Science (2021) 372:6543. doi: 10.1126/science.abc7531
174. Shen H, Yang M, Li S, Zhang J, Peng B, Wang C, et al. Mouse Totipotent Stem Cells Captured and Maintained Through Spliceosomal Repression. Cell (2021) 184:2843–59. doi: 10.1016/j.cell.2021.04.020
175. Wan L, Dreyfuss G. Splicing-Correcting Therapy for SMA. Cell (2017) 170:5. doi: 10.1016/j.cell.2017.06.028
176. Komaki H, Nagata T, Saito T, Masuda S, Takeshita E, Sasaki M, et al. Systemic Administration of the Antisense Oligonucleotide NS-065/NCNP-01 for Skipping of Exon 53 in Patients With Duchenne Muscular Dystrophy. Sci Transl Med (2018) 10:437. doi: 10.1126/scitranslmed.aan0713
177. Graziewicz MA, Tarrant TK, Buckley B, Roberts J, Fulton L, Hansen H, et al. An Endogenous TNF-Alpha Antagonist Induced by Splice-Switching Oligonucleotides Reduces Inflammation in Hepatitis and Arthritis Mouse Models. Mol Ther (2008) 16:1316–22. doi: 10.1038/mt.2008.85
178. Sussman JD, Argov Z, McKee D, Hazum E, Brawer S, Soreq H. Antisense Treatment for Myasthenia Gravis: Experience With Monarsen. Ann N Y Acad Sci (2008) 1132:283–90. doi: 10.1196/annals.1405.022
179. Kovalak C, Donovan S, Bicknell AA, Metkar M, Moore MJ. Deep Sequencing of Pre-Translational Mrnps Reveals Hidden Flux Through Evolutionarily Conserved Alternative Splicing Nonsense-Mediated Decay Pathways. Genome Biol (2021) 22:132. doi: 10.1186/s13059-021-02309-y
Keywords: alternative splicing, autoimmune disease, systemic lupus erythematosus, rheumatoid arthritis, therapy
Citation: Ren P, Lu L, Cai S, Chen J, Lin W and Han F (2021) Alternative Splicing: A New Cause and Potential Therapeutic Target in Autoimmune Disease. Front. Immunol. 12:713540. doi: 10.3389/fimmu.2021.713540
Received: 23 May 2021; Accepted: 29 July 2021;
Published: 17 August 2021.
Edited by:
Takashi Hashimoto, Osaka City University, JapanReviewed by:
Xiaoguang Li, Nanchang University, ChinaYoshinao Muro, Nagoya University, Japan
Farzan Solimani, Charité – Universitätsmedizin Berlin, Germany
Motomu Hashimoto, Osaka City University, Japan
Giovanni Di Zenzo, Istituto Dermopatico dell’Immacolata (IDI) (IRCCS), Italy
Copyright © 2021 Ren, Lu, Cai, Chen, Lin and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiqiang Lin, d2xpbkB6anUuZWR1LmNu; Fei Han, aGFuZjg4NzZAemp1LmVkdS5jbg==
†These authors have contributed equally to this work