 Benjamin Klein
Benjamin Klein Claudia Günther
Claudia Günther- 1Department of Dermatology, Venereology and Allergology, University Medicine Leipzig, Leipzig, Germany
- 2Department of Dermatology, University Hospital and Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
Type I interferons (IFNs) as part of the innate immune system have an outstanding importance as antiviral defense cytokines that stimulate innate and adaptive immune responses. Upon sensing of pattern recognition particles (PRPs) such as nucleic acids, IFN secretion is activated and induces the expression of interferon stimulated genes (ISGs). Uncontrolled constitutive activation of the type I IFN system can lead to autoinflammation and autoimmunity, which is observed in autoimmune disorders such as systemic lupus erythematodes and in monogenic interferonopathies. They are caused by mutations in genes which are involved in sensing or metabolism of intracellular nucleic acids and DNA repair. Many authors described mechanisms of type I IFN secretion upon increased DNA damage, including the formation of micronuclei, cytosolic chromatin fragments and destabilization of DNA binding proteins. Hereditary cutaneous DNA damage syndromes, which are caused by mutations in proteins of the DNA repair, share laboratory and clinical features also seen in autoimmune disorders and interferonopathies; hence a potential role of DNA-damage-induced type I IFN secretion seems likely. Here, we aim to summarize possible mechanisms of IFN induction in cutaneous DNA damage syndromes with defects in the DNA double-strand repair and nucleotide excision repair. We review recent publications referring to Ataxia teleangiectasia, Bloom syndrome, Rothmund–Thomson syndrome, Werner syndrome, Huriez syndrome, and Xeroderma pigmentosum. Furthermore, we aim to discuss the role of type I IFN in cancer and these syndromes.
Introduction
Type I interferons (IFNs), IFN α and IFN β, constitute a group of cytokines whose primary function is viral defense and protection against other intracellular pathogens (1). IFN secretion is activated after sensing of foreign- or self-nucleic acids (1). After binding on the interferon receptor (IFNAR), IFN is able to induce transcription of interferon stimulated genes (ISGs), resulting in activation of the innate immune system (1).
It has long been recognized that viral infections can induce flares of autoimmune diseases. This is mainly attributed to the upregulation of type I IFN which stimulates adaptive immunity and attenuates tolerance to self (2). Constitutive upregulation of type I IFN and ISG-transcription is seen in monogenic type I interferonopathies and autoinflammatory diseases such as systemic lupus erythematodes (2, 3). The importance of this group of cytokines is underlined by the prominent type I IFN signature found in blood of many complex autoimmune disorders such as dermatomyositis, systemic lupus erythematodes, Sjogren’s syndrome, and rheumatoid arthritis (4–8). Recent studies revealed type I IFN secretion after DNA damage through different mechanisms leading to distribution of nucleic components into the cytosol that will hereby be discussed (9–15).
DNA damage syndromes of the skin are caused by mutations in proteins taking part in the DNA repair (16–18). Due to UV-light driven carcinogenesis, these diseases often share the strong predisposition for the development of cutaneous malignancies, such as cutaneous squamous cell carcinoma (CSCC) and basal cell carcinoma (BCC) (16, 18). Important syndromes with defects in the DNA double-strand repair (DSBR) are Louis-Bar, Werner, Bloom, Rothmund–Thomson and Huriez syndromes (16, 18, 19). They can feature autoimmune phenotypes, such as positive antinuclear antibodies (ANA), rheumatoid arthritis, vitiligo, and scleratrophy. Here we summarize different mechanisms of IFN secretion, its role in DNA damage syndromes of the skin and discuss the role of the signaling pathway of type I IFN in cancer.
Type I IFN—Activation and Secretion
Primary functions of type I IFN consist in the defense of bacterial and viral infections. Under steady state conditions, type I IFN is secreted upon sensing of pathogen associated molecular patterns (PAMPS) such as foreign nucleic acids (1). Nucleic acids can be detected by endosomal localized receptors such as TLRs (toll like receptors), which are mostly expressed by immune cells. TLR3 senses double-stranded RNA, TLR7, and TLR8 single-stranded RNA. TLR9 recognizes CpG (unmethylated cytosine-guanosine) DNA motifs which are typical for bacteria (20–22). TLRs activate adapter proteins, further inducing a signal cascade: TLR3 activates TRIF (TIR-domain-containing adapter inducing interferon ß), while TLR7, TLR8, and TLR9 stimulate MyD88 (myeloid differentiation primary response 88). TRIF induces activation of IRF3 (interferon regulatory factor 3), MyD88 acts via IRF7 (interferon regulatory factor 7) and both adapter proteins activate the NF-κB-signaling pathway (20). Thereby, production of proinflammatory cytokines such as pro-IL1ß, pro-IL18, and secretion of type I IFN is enabled (Figure 1) (21).
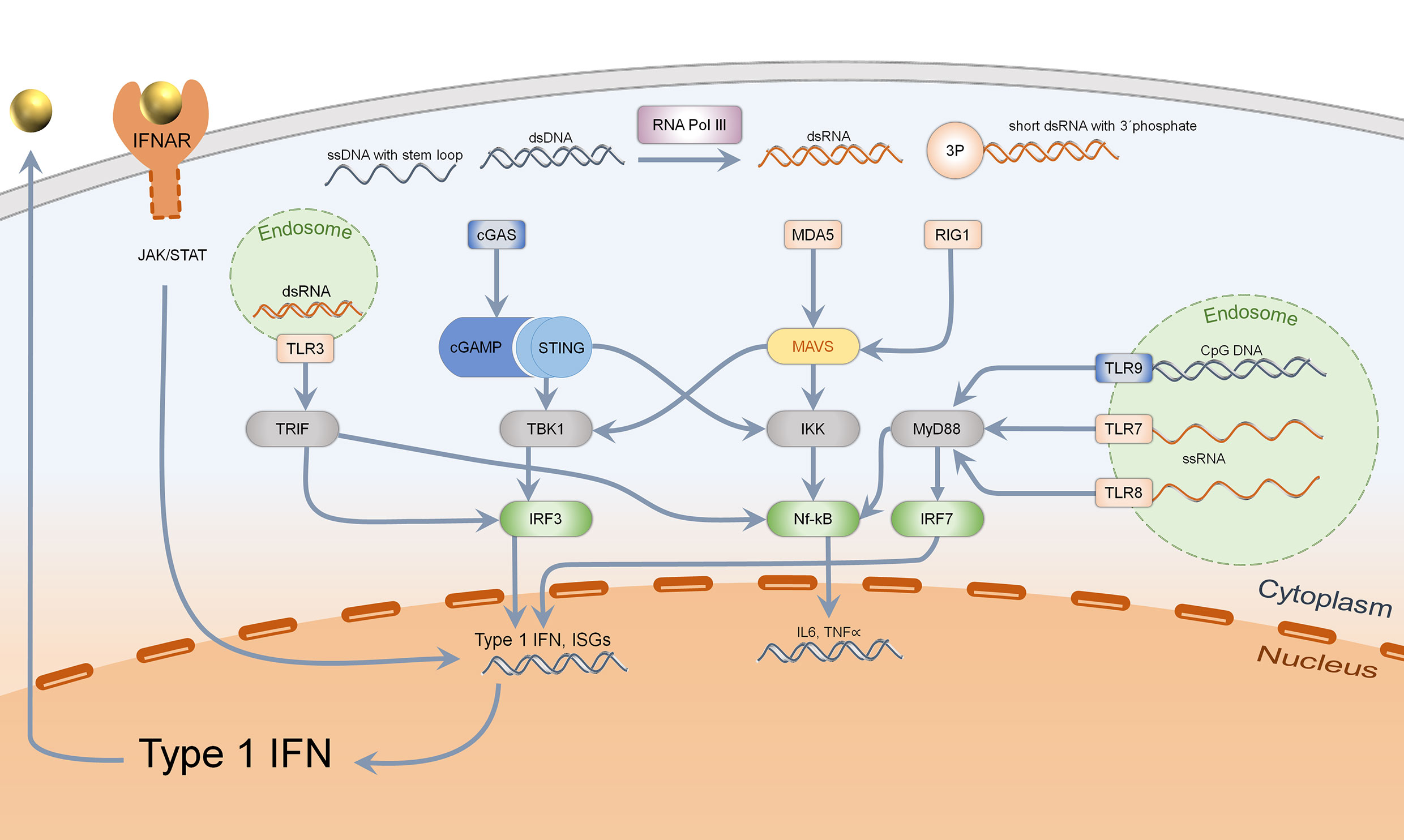
Figure 1 Sensing of intracellular nucleic acids and activation of type 1 IFN and Nf-kB pathway. Cytosolic DNA (marked in blue), specifically dsDNA and ssDNA with stem loop, is mainly detected by the cGAS–STING–TBK1–IRF3 pathway, leading to type 1 interferon secretion. Bacterial DNA with CpG motifs can furthermore be detected by endosomal TLR9 which acts via MyD88, IRF7, and Nf-kB. Cytosolic RNA is recognized by MDA5 (dsRNA) and RIG1 (short ds RNA with 3′phosphate), acting via MAVS to activate both TBK1-IRF3 and the Nf-kB pathway. Endosomal RNA is sensed by TLR3, 7, and 8 depending on the structure, activating either TRIF or MyD88. Activation of IRF3 and IRF7 leads to type 1 IFN transcription and secretion, which binds to IFNAR in an autocrine and paracrine manner.
Even in the cytosol, nucleic acids can be sensed by intracellular receptors which are expressed in almost every human cell. Short, double-stranded cytosolic RNA with a 5′triphosphate end is detected by RIG1 (retinoic acid inducible gene-I) while long, double-stranded RNA is recognized by MDA5 (melanoma differentiation associated gene 5) (1, 23, 24). They both activate MAVS (mitochondrial antiviral-signaling-protein) which acts through TBK1 (Tank-binding kinase 1), activating IRF3 (25). Cytosolic DNA can be detected by different mechanisms: On the one hand, the transformation of short, double-stranded RNA of AT-rich DNA via RNA Polymerase III acts through activation of RIG1/MAVS (26). On the other hand, single-stranded DNA with stem loop and double-stranded DNA are ligands of cGAS (cyclic GMP-AMP synthase), which generate cGAMP. This second messenger binds to the dimer STING (stimulator of interferon genes), which upon conformational change activates TBK1 and IRF3 (27). In addition, STING binds directly to bacterial cyclic dinucleotides (c-di-GMP and c-di-AMP), resulting in IRF3 activation (Figure 1) (28, 29). These different mechanisms only show an extract on how cytosolic nucleic acids can induce a type I IFN response.
Almost every human cell is capable of IFN β secretion, while IFN α is predominantly secreted by plasmacytoid dendritic cells (30, 31). Secreted type I IFN binds to IFNAR (interferon alpha-receptor) which consists of two heterodimers, IFNAR-1 and -2, which then activate the JAK/STAT pathway (januskinase/signal tranducers and activators of transcription) (32). A complex of STAT and IRF9 bind ISRE (interferon stimulated response element), resulting in transcription of ISGs (31). Subsequently, significant upregulated presentation of MHC-I, NK-cells and cytotoxic T-cells as well as proliferation of T-helper cells are activated (2). Moreover, autoreactive B-cell development and class switch from IgM to IgG are promoted which leads to autoantibody production (33). Type 1 IFN signaling thus promotes the development of a pro-inflammatory environment, which leads to autoimmunity due to a loss of tolerance in innate and adaptive immune responses.
The capacity of differentiation between self and foreign nucleic acids is limited. If the amount of self nucleic acids reaches a certain level in the cytosol, nucleic acid sensors can be activated, resulting in type 1 IFN secretion (34). To deteriorate the amount of nucleic acids, human cells exhibit cytosolic exonucleases such as TREX1 (three prime repair exonuclease 1), lysosomal DNase2, and extracellular DNase1 (35, 36). Deficiency of these proteins as well as gain of function mutations in nucleic acid sensors may lead to continuous stimulation of IFN secretion, which is observed in hereditary monogenic “interferonopathies”. These diseases are consistently featuring high levels of native ISG-expression, called “intrinsic interferone signature” (3). Loss of function mutations of TREX1, which were originally described in AGS (Aicardi Goutieres syndrome), result in accumulation of cytosolic DNA inducing a cGAS-dependent type I IFN secretion (37, 38). AGS patients show a broad phenotypic spectrum which is characterized by encephalopathy with dystonia, epilepsy, and microcephaly. Patients exhibit autoimmune symptoms such as Chilblain-Lupus, positive antinuclear antibodies and oral ulcerations (39, 40). Also, TREX1-associated familial Chilblain-Lupus is caused by a loss of function mutation in TREX1 which leads to activation of the type I IFN system. Patients show acral bluish red infiltrates and frequently develop systemic signs of lupus associated with a type I IFN signature in the blood (41–45).
Cutaneous DNA damage syndromes, characterized by defects in DNA repair proteins, can feature clinical phenotypes seen in autoimmune diseases such as autoantibody production, vitiligo, rheumatoid arthritis, and scleratrophy. In the following, the cutaneous DNA damage syndromes are presented in more detail, and the possible pathophysiological role of type 1 IFN in these diseases will be discussed.
Cutaneous DNA Damage Syndromes With Defects in DNA Double-Strand Break Repair
Daily exposure to sunlight is the main inductor of skin aging, skin atrophy, and several malignancies such as cutaneous squamous cell carcinoma (CSCC) and basal cell carcinoma (BCC) (46). UVA and UVB exposition leads to several kinds of DNA damage e.g. UV light dose-dependent cyclopyrimidine-dimers (CPD) and oxidized bases (47, 48). Upon repair of this direct DNA damage, the formation of DNA DSB can be induced (48, 49). To repair these lesions, the DNA double-strand repair (DSBR) machinery is recruited which is divided into homologous recombination (HR) and non-homologous end joining (NHEJ), depending on the cell cycle.
Initiation of HR is the “DNA end resection”: Endonuclease activity of the MRE11-RAD50-NBS1-complex (Nijmegen breakage syndrome protein 1), DNA helicases (RECQ-helicase family), unwinding the helix structure, and exonucleases, cut out an ssDNA nucleotide to get a free 3′ end, where other repair enzymes of the DSBR can bind (50, 51). Afterwards, stabilizing proteins such as RAD51, RPA (replication protein A) and BRCA (BReast CAncer protein) are recruited to the free ssDNA (50). The homologous DNA strand of sister chromatid is used as template for “strand invasion”. Finally, the strands are reconnected (“strand annealing”) (50).
In NHEJ, DSBR is initiated by binding of Ku70/80 to the DSB, recruiting additional factors such as DNA-PKs (protein kinases) and XRCC4/DNA-ligase (50, 51). Further, nucleotide sequences in upstream and downstream of the DSB are excised by Artemis, DNA polymerase λ and µ (51). Then, DNA is ligated through DNA-ligase IV (50).
Defects in DNA DSBR are mostly located in defective DNA helicases (RECQ-helicase family). They usually affect the skin and several other tissues (Table 1) and often show autosomal recessive inheritance. Autosomal recessive Bloom syndrome is caused by heterozygous mutations in Bloom-helicase which is involved in DNA DSBR, HR (52, 61). The skin of patients shows telangiectasia and photosensitivity; furthermore patients have an elevated risk of developing leukemia, lymphoma, and gastrointestinal cancer and harbor defects in immune defense (17).
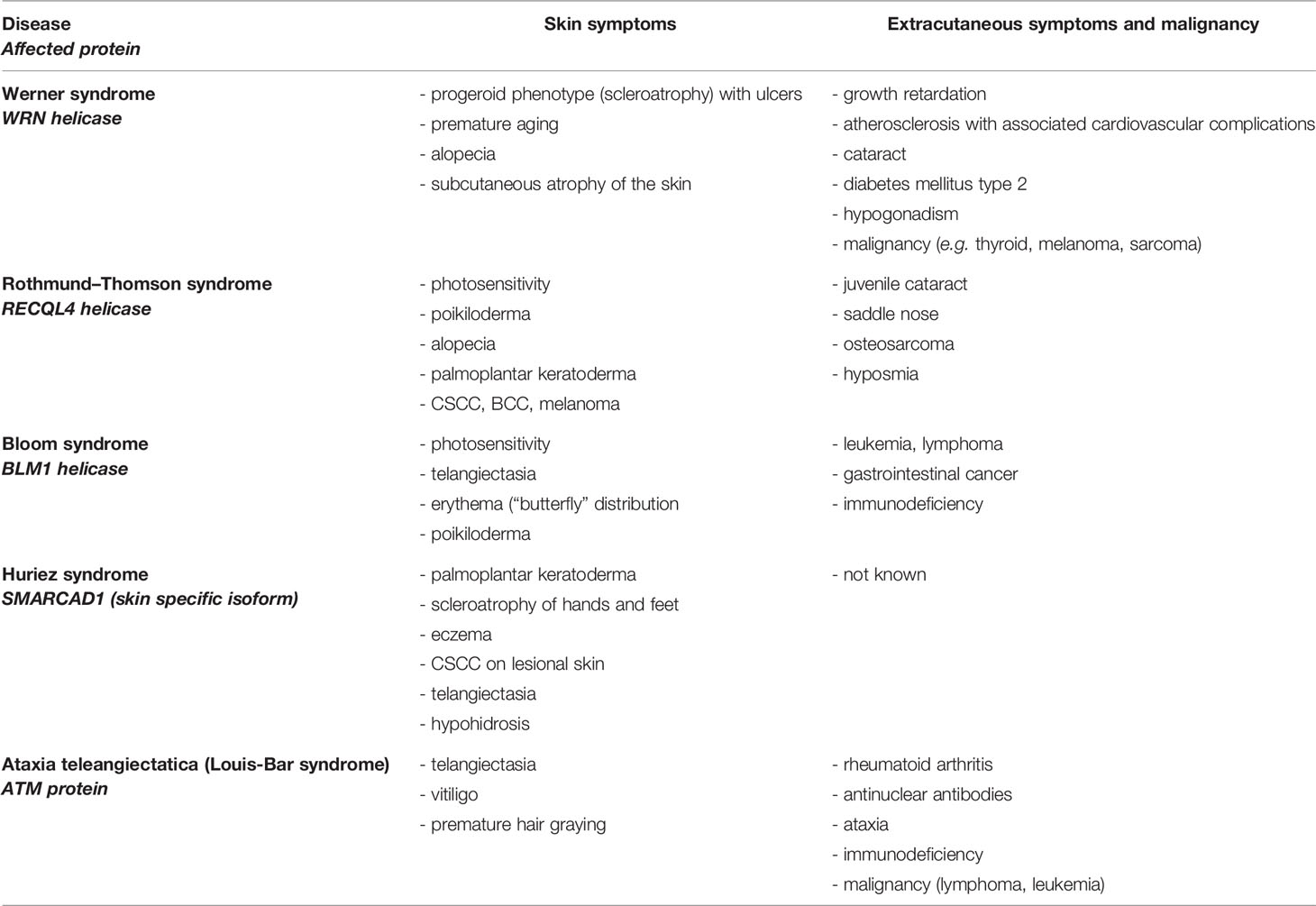
Rothmund–Thomson syndrome represents an autosomal recessive disorder caused by homozygous and compound-heterozygous mutations of RECQL4-protein (62). Skin symptoms include poikiloderma (telangiectasia, change of pigmentation), hair loss, palmoplantar keratoderma, and patients may have a higher risk of developing BCC, CSCC, and melanoma. Patients often show abnormalities of the bones and can develop osteosarcomas (53, 54).
The progeroid Werner syndrome is characterized by premature aging upon defects in WRN helicase, which contains endonuclease activity (63). Clinical symptoms include skin atrophy, growth retardation, atherosclerosis, and high predisposition to different types of cancer (thyroid, melanoma, sarcoma) (17).
Recently discovered autosomal dominant Huriez syndrome (also known as sclerotylosis) is caused by mutations in the skin specific isoform of SMARCAD1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin, subfamily A containing DEAD/H box 1), which is involved in DNA DSBR and chromatin remodeling (19, 64, 65). Patients exhibit palmoplantar keratoderma, palmoplantar scleroatrophy, onychodystrophy, adermatoglyphia, eczema, telangiectasia, and a high risk for developing CSCC at a young age (3rd to 4th decades) (55–57). Due to exclusive mutations in the skin specific isoform of SMARCAD1, which is mostly expressed in the skin and tongue, other malignancies of other cell types have not been described (19, 57, 58, 66).
Cutaneous DNA Damage Syndromes With Defects in Nucleotide Excision Repair
Direct UV light-induced DNA damage such as CPDs and pyrimidine-pyrimidone (6–4) photoproducts is repaired by the nucleotide excision repair (NER) (67, 68). NER is divided in two mechanisms: transcription-coupled repair (TCR) and global genome repair (GCR). These pathways are different in recognition of “bulky lesions”: In TCR, RNA polymerase II, CSA, and CSB (Cockayne syndrome proteins A and B) recognize the damage, while in GGR XPC (xeroderma pigmentosum group C)/UV excision repair protein Rad23B or DDB1 and DDB2 (DNA damage binding proteins 1 and 2) are recruited (50, 68). Both pathways share the following steps: unwinding DNA (XPB, XPD) and dual excision of a 25–30 bp oligonucleotide by endonucleases at the 3′ side (XPG and TFIIH, transcription factor II H) and 5′ side (XPF and ERCC1 complex, excision repair cross complementation group 1) (50, 67, 68). The removed DNA is bound by RPA, while the gap is stabilized by RFC (replication factor C) and PCNA (proliferating cell nuclear antigen) and resynthesized by DNA polymerases (50, 67, 68). The two strands are annealed by DNA-ligase1/ligase III–XRCC1 complex (X-ray repair cross complementing protein 1) (50, 67, 68).
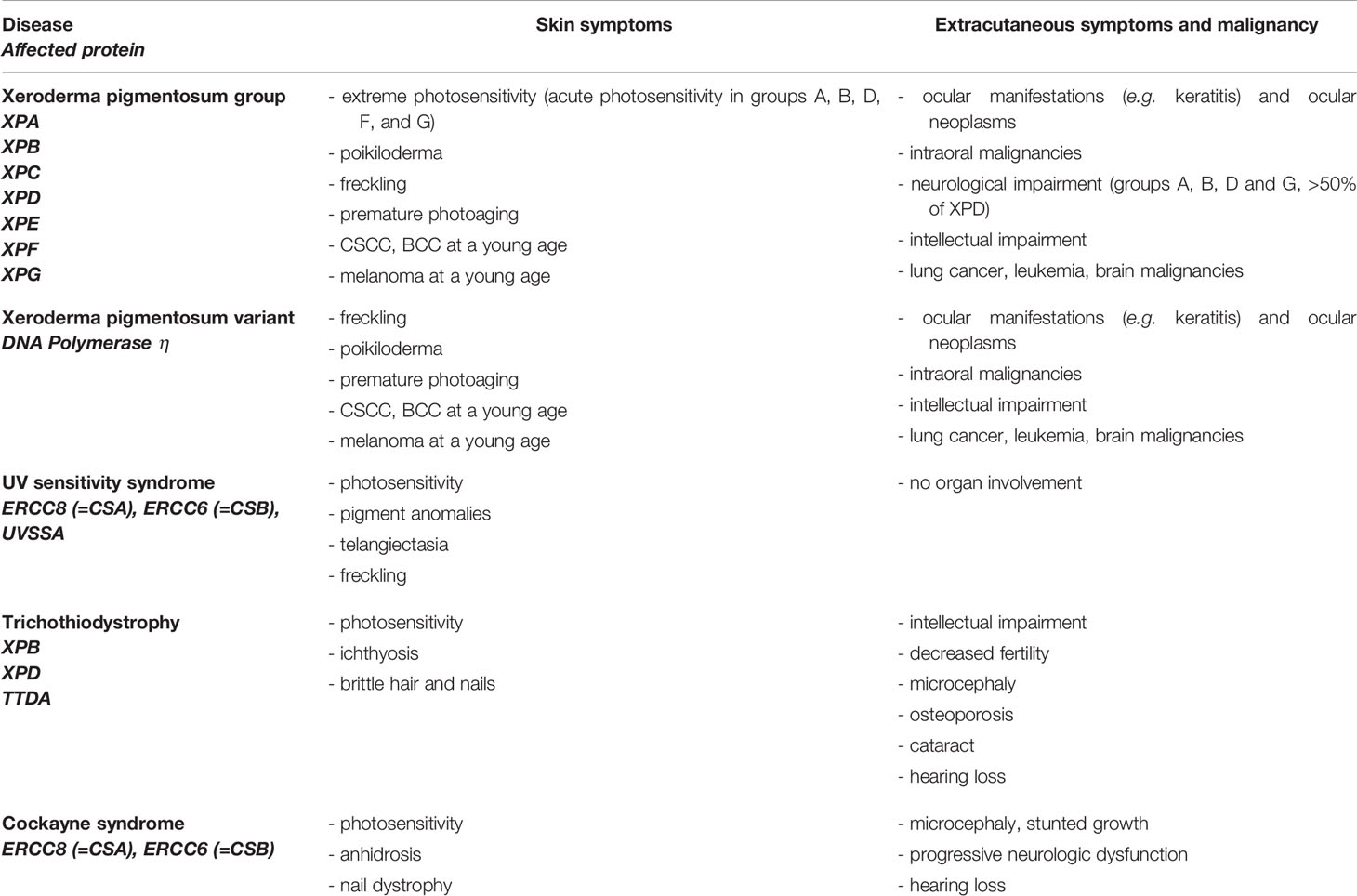
Diseases with defective proteins of NER include Xeroderma pigmentosum, Trichothiodystrophy (TTD), Cockayne syndrome (CS), and UV-sensitivity syndrome (UVSS) (Table 2) (16, 18). Exclusively, hereditary syndromes caused by mutations in proteins of GGR are associated with malignant transformation, while mutations in TCR are not (18). TTD, CS, and UVSS represent diseases with defects in TCR and will not be discussed in this review. Xeroderma pigmentosum is autosomal recessive caused by mutations in different genes (XPA-XPG) involved in NER (GGR), further enhancing malignant transformation (18, 69). Patients exhibit an extreme sensitivity to sunlight, show pigment changes and a highly elevated risk of developing skin cancer at a young age (69). The estimated increased risk is 10,000-fold higher for developing non-melanoma skin cancer and 2,000-fold higher for developing melanoma under the age of 20 (70). Ocular abnormalities are seen in UV-exposed structures of the eye. Of patients, 20–30% show neurological symptoms and intellectual deficiency (Table 2) (18, 69).
Type I IFN Activation in DNA Damage Syndromes
Diseases associated with defects in DNA repair may also show autoimmune phenotypes: In Ataxia teleangiectatica, some patients show production of antinuclear antibodies (ANAs), symptomatic rheumatoid arthritis, and vitiligo besides the classic symptoms of ataxia, telangiectasia, and a high predisposition for the development of malignancies (10, 59, 60, 71, 72). It is caused by defects of the ATM protein, which is essential for a proper DNA double-strand repair (50). It has been proposed that autoimmune features are driven by a type I IFN response that is induced by byproducts of the DNA damage response. Härtlova et al. showed that cell stress is able to induce type I IFN system: Irradiated ATM-deficient cells show liberation of ssDNA-fragments which can penetrate into the cytosol and activate STING (10). To clarify if this mechanism is DNA damage-dependent, etoposide as specific inductor of DNA double-strand breaks showed significant activation of type I interferon. Moreover, lower concentrations of viral nucleic acids or c-di-GMP were necessary to induce a STING-dependent ISG expression in ATM-deficient cells. Authors claimed this phenomenon as “priming” of the type I IFN system (10).
Further, different studies of ATM deficiency led to the idea of mitochondrial DNA (mtDNA)-induced activation of cGAS and type I IFN (73–75): ATM, besides being detectable in the nucleus, is also detectable in mitochondrial fractions of human fibroblasts (74). ATM deficiency shows mitochondrial dysfunction which is associated with an innate immune response including type I IFN production (73). The mechanism was identified recently: ATM inhibition was shown to cause cytoplasmic leakage of mtDNA by downregulation of TFAM (mitochondrial transcription factor A), which is a mtDNA binding protein. Cytoplasmic mtDNA then activates cGAS-STING-dependent type I IFN secretion (75). Together, downregulation of TFAM caused by ATM deficiency promotes leakage of mtDNA into the cytosol and thus activates the type 1 IFN system. These results also show that ATM, in addition to its function in DNA double strand repair, is indirectly involved in the stabilization of mtDNA and mitochondrial homeostasis (75). Hence, different subcellular localizations and functions of ATM are responsible for type I IFN induction and may lead to the autoimmune phenotype observed in this disease.
The possible producer of DNA fragments during DNA repair, which further penetrate from the nucleus into the cytosol inducing a type I IFN response, was identified by Erdal et al.: Liberated ssDNA fragments are excised by Bloom-helicase (BLM) and Exonuclease 1 (Exo1) in the “DNA end resection” of HR as mentioned above: BLM1/Exo1-deficient cells exhibited significant lower expression of ISGs after irradiation compared to wild type, indicating less liberation of ssDNA into the cytosol (14).
The transmission from nuclear DNA into the cytosol is protected by DNA binding proteins such as RPA and RAD51, which are upregulated upon DNA damage (13). Deficiency of RAD51 or associated proteins leads to liberation of ssDNA and dsDNA into the cytosol, further enhancing a type I IFN response in a STING-dependent manner (Figure 2) (13, 76).
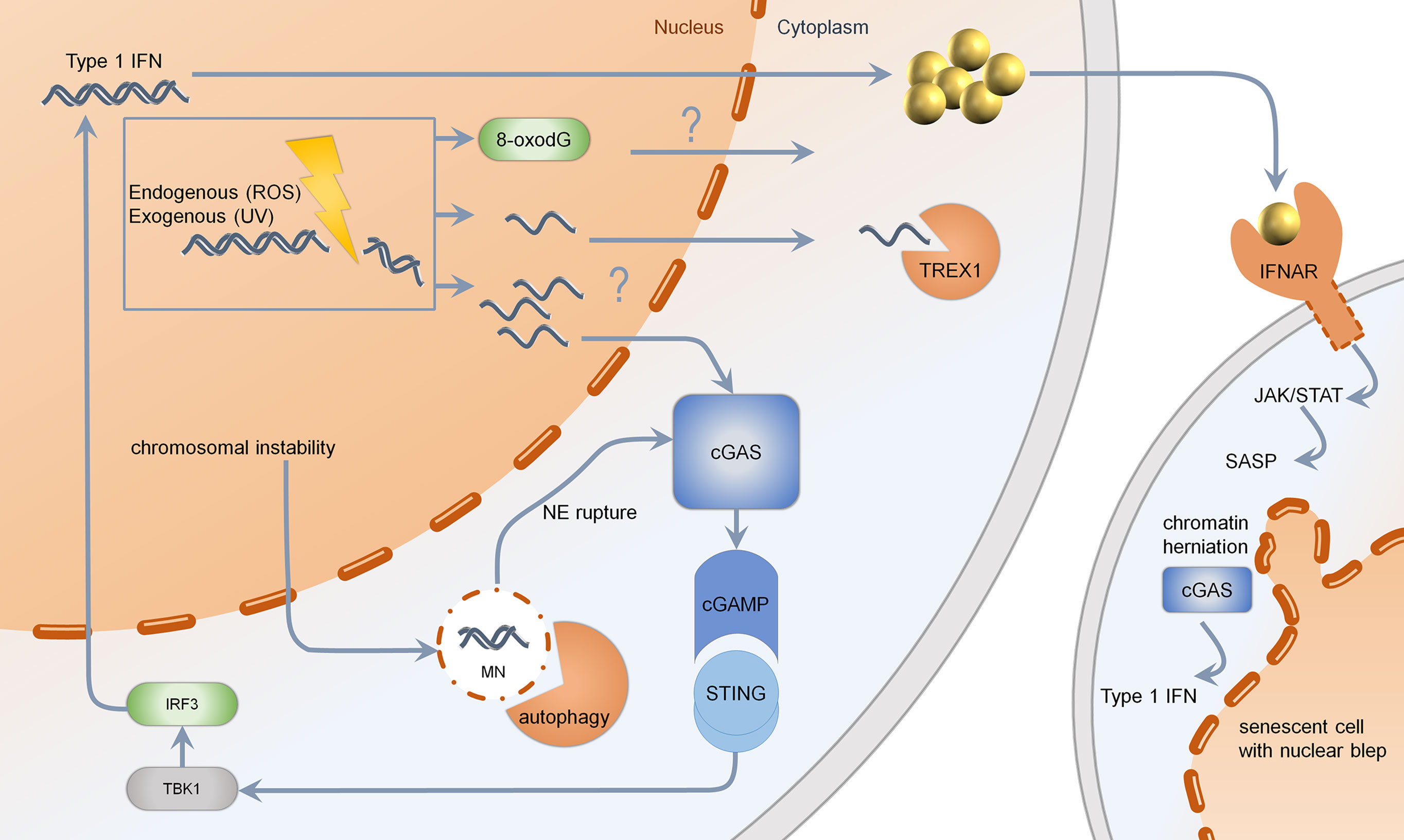
Figure 2 Mechanisms of type 1 IFN induction upon DNA damage. Genotoxic agents such as chemotherapeutics, UV-light, or reactive oxygen species (ROS) may lead to formation of ssDNA, which is normally cleared by TREX1. If TREX1 is deficient, the threshold for ssDNA is limited. Changes in DNA-bases such as 8-oxo-dG are resistant to enzymatic degradation via TREX1. Upon massive DNA damage or RAD51 deficiency, the generation of multiple cytosolic ssDNA fragments may lead to activation of cGAS. Cutaneous DNA damage syndromes, such as Bloom syndrome, characterized by chromosomal instability, show increased formation of micronuclei with fragile nuclear envelope. Normally, micronuclei are processed by autophagy. Upon rupture of the nuclear envelope, micronuclei are sensed by cGAS, resulting in production of cGAMP, further activating STING, TBK1, and IRF3. Secreted type 1 IFN binds to IFNAR, acting through the JAK–STAT pathway, leading to a senescent phenotype. Senescent cells, which are more frequent in cutaneous DNA damage syndromes, show nuclear blebs, also called chromatin herniations, which are able to activate the type 1 IFN system through cGAS. MN, micronucleus; NE, nuclear envelope; cGAS, cyclic GMP-AMP synthase; cGAMP, cyclic GMP-AMP; STING, stimulator of interferon genes; TBK1, Tank binding kinase 1; IRF, interferon regulatory factor; TREX1, three prime exonuclease 1; IFNAR, interferon alpha receptor; JAK, januskinase; STAT, signal transducers and activators of transcription; SASP, senescence-associated secretory phenotype; UV, ultraviolet; ROS, reactive oxygen species.
Accumulated cytosolic ssDNA is normally cleaved by TREX1 (35). TREX1 mutations have been described to cause Aicardi Goutières syndrome and autosomal dominant familial chilblain lupus. Both diseases are characterized by spontaneous activation of the type I IFN system (37, 38, 41, 42). This is induced by ssDNA accumulating in the cytosol due to an activated DNA damage repair response (13, 77). TREX1 deficient cells are very competent in NER and DSB repair. They harbor replication stress and exhaustion of the RAD51 ssDNA-binding capacity facilitating DNA accumulation in the cytosol (13). The DNA sensor cGAS recognizes unrestricted DNA and stimulates STING (Figure 2) (27, 78). In TREX1-associated familial chilblain lupus, a deregulated IFN response was shown, which was enhanced by stimulators such as polyI:C (43). Recently, it has been shown that early type I IFN reactions upon UV-light in murine skin are cGAS dependent, suggesting a UV induced type I IFN dependent inflammation in cutaneous lupus erythematodes (79). Other mutations identified in Aicardi Goutières syndrome are defects in RNAseH2, which takes part in ribonucleotide excision repair and acts on RNA/DNA-hybrids, occurring during DNA replication (80, 81). Patients with defects in RNAseH2 accumulate ribonucleotides in DNA and show activation of the type I IFN system due to an activated DNA damage response. Lupus patients with defects in RNaseH2 are susceptible to UV-light due to enhanced CPD-formation in ribonucleotide-containing DNA (82). These results indicate that specific structural alterations of DNA are capable of type I IFN induction, further enhancing autoimmunity (Figure 2) (82).
Another way of releasing nucleic acids into the cytosol is the formation of genome instability-associated formation of micronuclei: a small nucleus with a lamin coated membrane. Due to instability of this membrane, DNA damage can lead to miscompartmentation, resulting in a cGAS-dependent activation of type I IFN (Figure 2) (11).
Werner syndrome patients exhibit scleroderma-like skin changes, which might be associated with an autoimmune phenotype. In Werner syndrome, the frequency of senescent cells is relatively high compared to normal controls (83). This is due to replicative senescence, which results from elevated telomer shortening (83, 84). Senescent cells show cell cycle arrest, resistance to growth factors and exhibit a higher amount of chromatin herniations compared to wild type (84, 85). Yu et al. showed significant activation of type I IFN in fibroblasts of patients with Werner syndrome (9). After treatment with anti-IFN antibodies they showed less cell cycle arrest, entrance in S-/G2M-phase of cell cycle, and a reduced rate of senescent cells. This indicates a potential disease-modifying role of type I IFN and possible therapeutic strategy: Antibodies against IFN could possibly stop premature aging and scleroderma skin changes in Werner syndrome (9). Interestingly, autoantibodies against WRN (Werner helicase) and a lower expression rate of WRN were observed in systemic sclerosis, suggesting a possible pathogenic link of sclerotic skin changes (15, 86). The WRN protein belongs to the RECQ-helicase family and contains an N-terminal 3′–5′ exonuclease activity. It represents a multifunctional nuclease involved in replication, telomer shortening, and DNA damage response maintaining genome integrity (87). It interacts with proteins in both NHEJ and HR (end resection) in DNA DSBR: In DSBR, it has an exonuclease function and is involved in “DNA end resection” (50). If type 1 IFN is activated by DNA fragments produced by end resection factors [according to Erdal et al. (14)], a deficiency of WRN would result in reduced excision of ssDNA, which represents a possible dangerous molecule in the cell. This alone cannot explain the increased type 1 IFN activation which was shown in patient cells and raises the question as to the source of type 1 IFN activation in Werner syndrome. The function of WRN protein is complex, and it has been shown that the non-enzymatic component of WRN protein is recruited by NBS1 to limit exonuclease activity of MRE11 at replication forks: In the absence of WRN, MRE11 degrades DNA during replication. NBS1 limits this process through recruitment of WRN (88). Further, WRN protein stabilizes RAD51, a DNA-binding protein (88). Together, WRN deficiency could lead to liberation of DNA of replication forks due to higher excision by MRE11 and destabilization of RAD51. RAD51 plays a pivotal role in maintaining DNA in the nucleus (13, 88). Deficiency of RAD51 in human fibroblast is sufficient to enhance IFN β-mRNA levels in the cell (13). Therefore, WRN deficiency and associated RAD51 destabilization could lead to accumulation of excised DNA, further activating IFN response. Further studies are needed to evaluate the impact of WRN deficiency, RAD51 destabilization and type I IFN.
Recent studies showed JAK-independent activation of STAT-signaling pathway in premature aging cells: The transcription factor ISGF3 (interferon stimulated gene factor 3), consisting of STAT1, STAT2, and IRF9 can induce ISG expression independently of IFN secretion in an unphosphorylated state: In aged cells and cells of patients with Werner syndrome, ISG expression was significantly upregulated compared to wild type (89, 90). This was caused by increased levels of unphosphorylated STAT1 and STAT2 proteins (89). JAK knockdown in these cells did not show a reduction of ISG expression (89). This effect may be due to post-translational modifications (e.g. acetylation, methylation) of STAT proteins in aging cells, activating ISGF3 (89, 90). Hence, JAK-independent activation of STAT proteins could explain enhanced ISG expression in this syndrome (89, 90).
Furthermore, the senescent phenotype in Werner syndrome, due to telomer shortening, could trigger the type I IFN system: chromatin herniations observed in senescence can be recognized by the immune sensor cGAS, inducing a STING-dependent type I IFN response (Figure 2) (12). Altogether, different mechanisms in Werner syndrome are able to induce the type I IFN system; hence more studies are needed to evaluate the precise substrate of type I IFN induction in Werner syndrome.
In contrast to the previous diseases, Bloom syndrome does not show a typical autoimmune phenotype; however, patients even show photosensitivity and symptoms of immunodeficiency such as more frequent respiratory and gastrointestinal infections (18, 91). Erdal et al. identified BLM helicase together with Exo1 as a possible source of ssDNA liberation of the nucleus into the cytosol upon DNA damage, inducing a IFN response (14). A deficient BLM protein, reducing ssDNA liberation, was associated with a diminished IFN response in breast cancer cells (14). However, Bloom syndrome represents a genetic instability syndrome, and elevated micronuclei formation was observed in patient cells (92). Gratia et al. showed a higher micronuclei formation and cGAS dependent induction of type I IFN in immortalized fibroblasts of Bloom syndrome patients (93). The frequency of cGAS colocalized micronuclei was not significantly altered, suggesting no upregulation of cGAS (93). Taken together, different pathomechanisms are possible in Bloom syndrome: Dependent on the amount of micronuclei and the activity of BLM1 helicase, IFN induction is possibly promoted or inhibited.
As to Rothmund Thomson syndrome, no recent studies have been published concerning DNA-damage-induced type I IFN, but significant higher rates of senescent cells have been described (54, 62). Patients exhibit photosensitivity but do not show autoimmune phenotypes such as the formation of antinuclear antibodies or scleroderma-like skin changes. As premature senescence is linked to chromatin herniation, chromatin mediated cGAS activation represents a possible mechanism of type I IFN induction. Furthermore, Rothmund Thomson syndrome as well as Werner syndrome was shown to be associated with mitochondrial dysfunction (94). As RECQL4 helicase plays a role in mtDNA replication and RECQL4 deficient cells exhibit higher mtDNA mutations (75, 95, 96), an mtDNA-driven type 1 IFN response similar to ATM deficiency seems possible (73, 75). However, it is not known, if Rothmund–Thomson syndrome is associated with an enhanced type I IFN activation.
In Huriez syndrome, caused by skin-specific SMARCAD1 deficiency, patients feature a scleroatrophic phenotype, also seen in systemic sclerosis, which marks a strongly associated type I IFN activation disease (97, 98). Recently, a link between the activation of type I IFN and higher DNA damage response in systemic sclerosis was shown, supporting the possible pathogenic role of DNA-damage-induced type I IFN in Huriez syndrome (15, 99). In systemic sclerosis, a higher rate of DNA damage was observed compared to wild type (15). Interestingly, higher IFN levels were associated with higher DNA damage burden, and accumulated DNA damage was proportional to the extent of fibrosis (15). The authors did not observe defects in the capacity of NER, but in DSBR (15). Mesenchymal stem cells of patients with systemic sclerosis reveal lower expression of SMARCAD1, suggesting Huriez syndrome and systemic sclerosis may share common pathways (100). Since SMARCAD1 represents a protein of the DSBR (HR), a similar mechanism seems possible. As part of DNA “end resection”, SMARCAD1, together with Exo1, excises ssDNA from DNA DSBs (64). This would be diminished in SMARCAD1 deficiency regarding similar functions of SMARCAD1 and BLM/Exo1 (14, 64). Hence, a lower IFN response compared to SMARCAD1 sufficiency would result. However, cells of patients with Huriez syndrome exhibit a high rate of senescent cells and show deficits in proliferation, yet it is unclear if chromatin herniation and an upregulated cGAS-induced IFN response are present in this syndrome (19). CSCC only occurs in lesional skin in these patients, potentially induced by chronic inflammation and reduced immunosurveillance which may be due to depletion of epidermal Langerhans cells (101, 102).
Xeroderma pigmentosum (XP) represents the most common hereditary cutaneous DNA damage syndrome and is caused by mutations in proteins of the nucleotide excision repair (XPA-XPG), as mentioned above (18, 46, 67). XP patients show cutaneous malignancies such as CSCC and BCC as well as melanoma in early childhood/puberty (18). Autoimmune phenotypes are not highly associated with the disease (18). Evidence of IFN induction in XP is very limited. Interestingly, a reduced native IFN response upon stimulation with polyI:C was observed in XP blood cells (103). However, a higher rate of micronuclei has been observed in XP group A fibroblasts representing a possible substrate of cGAS activation (104). It is not known if the amount of micronuclei formation is high enough to induce a relevant type I IFN response. IFN plays a pivotal role in immunosurveillance, leading to an antigen specific T-cell response against malignant cells (105, 106). Hence, a possible explanation for early malignancies in XP patients could be impaired immunosurveillance due to missing DNA-damage-induced type I IFN.
Interestingly, some of the cutaneous DNA damage syndromes with defects in DNA DSB show activation of the type I IFN system upon DNA damage induced by different mechanisms, reflecting clinical autoimmune phenotypes. In cutaneous DNA damage syndromes with defect NER, evidence for type I IFN induction is very limited or even not present. A possible explanation for type I IFN activation could be the damage itself: DSBs are highly mutagenic resulting in genome instability, enhancing the formation of micronuclei (11, 107, 108). Another reason could be the stronger DNA damage response in DSBR, leading to possible substrates for type I IFN induction (9, 109). Possible sources of DNA-damage-induced type I IFN in cutaneous DNA damage syndromes include genome instability-associated formation of micronuclei, leakage of mtDNA, activation of exonucleases upon high DNA damage, reduced capacity of nuclear DNA binding or cytosolic DNA degrading proteins as well as chromatin herniation in senescent cells which are highly associated with DNA damage syndromes (11–13, 35, 76, 82, 93).
The Type I IFN System, Senescence, and Cancer
Activation of type I IFN raised importance in the context of cancer as an anti-tumorigenic mechanism of the cell, leading to immunosurveillance: Detection of tumor-derived DNA by innate immune sensors and cGAS-dependent activation of STING leads to type I IFN secretion (105, 106). Consequently, enhanced tumor antigen presentation and antigen-specific T-cell response as well as the recruitment of NK-cells are part of the anti-tumorigenic response (110). Type 1 IFN is required for anti-tumor response and tumor elimination in dendritic cells, and reduced IFN signaling was observed in different types of cancer such as colorectal carcinoma, melanoma, and pancreatic cancer (111, 112). Furthermore, IFNAR1 downregulation in cancer-associated stromal cells was observed in colon and pancreatic cancers (113). It was shown that suppression of STING is associated with less immune infiltration and subsequently increased tumor growth in melanoma (114). Downregulation of cGAS and STING was observed in clinically advanced tumors (115, 116), indicating a possible tumor-driven escape mechanism from immunosurveillance. Interestingly, Xeroderma pigmentosum shows the strongest association for the development of cutaneous malignancies and was associated with a deficiency in type 1 IFN production, which supports the idea of anti-tumorigenic effects of type 1 IFN signaling (103). As the other mentioned cutaneous DNA damage syndromes feature the formation of cancer despite certain activation of type 1 IFN, premature senescence and the chronic “senescence associated secretory phenotype” (SASP) might give an additional explanation of the clinical phenotype in these syndromes, which will be further discussed.
Senescence is a cellular phenomenon, characterized by cell cycle arrest and resistance to growth factors (84). The activation of the cGAS–STING pathway leads to secretion of inflammatory cytokines, chemokines, and proteases characterizing SASP (Figure 2) (117, 118). Chronic stress (such as DNA damage) and activation of SASP can be associated with induction of an immunosuppressive microenvironment leading to metastasis and resistance of DNA damaging agents such as chemotherapy (119). Thus, the SASP can paradoxically have both pro-tumorigenic and anti-tumorigenic functions: In one way, the recruitment of immune cells through IL6, CXCL1, and other cytokines mediate clearance of tumor cells (120). In another way, anti-inflammatory cytokines, such as IL10, secreted by senescent stromal cells, suppress anti-tumor immune responses. Hence, the cancer-inhibiting or cancer-promoting effect of SASP is tightly regulated and seems cell type- and cytokine-specific (120). The exact molecular mechanisms underlying SASP-induced tumor progression are not fully understood, but type I IFN in this context was shown to be downregulated by inhibition through p38, encoding for MAPK (mitogen activated protein kinase) (106). This downregulation of type I IFN could further explain the possible failure of immunosurveillance.
Taken together, the senescent phenotype in cutaneous DNA damage syndromes could be context-dependent: Acute activation of cGAS–STING enhances immunosurveillance via SASP, while the chronic secretion of inflammatory cytokines could have tumorigenic effects (105, 117, 118). It is not known if the observed premature senescence in cutaneous DNA damage syndromes is clearly pro- or anti-tumorigenic; this will be subject of future research. However, type I IFN induction represents an anti-tumorigenic mechanism (105, 111, 112), which may play a pivotal role in inhibiting skin cancer development in the cutaneous DNA damage syndromes.
Therapeutic Implications
Skin changes in favor of scleroatrophy and premature aging as seen in progeroid syndromes, and Huriez syndrome may represent the effects of both chronic DNA damage and chronic inflammation, demonstrating the senescent phenotype (15, 98, 99). Therapeutic approaches against type I IFN (JAK inhibitors, anti-IFNAR antibodies, etc.) in cutaneous autoinflammatory and autoimmune diseases show promising effects in patients (44, 121, 122). To use these agents in type I IFN-driven inflammatory diseases seems plausible. However, it is not known if anti-IFN agents may possibly reduce immunosurveillance of evolving tumor cells in cutaneous DNA damage syndromes. IFNAR downregulation was observed in tumor progression of cancer associated stromal cells. Therefore, IFN reducing interventions need to be tested with caution to avoid enhanced tumor growth in cutaneous DNA damage syndromes.
Perspectives
Molecular exploration of rare interferonopathies has improved our understanding of innate type I IFN driven immune responses and nucleic acid metabolism (3, 31, 123). It has further opened the view to DNA-damage-induced innate immune response and especially type 1 IFN induction. This exploration was mainly driven by description of ATM deficiency (10, 75). Although less is known in this regard for cutaneous DNA damage syndromes, the understanding of these rare diseases can help to elucidate molecular mechanism and to understand more complex diseases featuring similar clinical phenotypes. In Huriez syndrome, it will be interesting to explore under which conditions type 1 IFN might be upregulated (19). In Bloom, Werner, and Rothmund–Thomson syndromes, we still do not precisely know the molecular substrates that lead to type 1 IFN induction (9, 89, 93, 96). In this regard, it will be interesting to know if RAD51 destabilization is a possible mechanism of type 1 IFN induction in Werner syndrome. Finally, the physiological role of type 1 IFN for induction of senescence and its potential pro- or anti-tumorigenic effects warrant investigation.
Author Contributions
BK had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: BK and CG. Acquisition, analysis, and interpretation of data: BK and CG. Drafting of the manuscript: BK. Critical revision of the manuscript for important intellectual content: CG and BK. Statistical analysis: none. Obtained funding: CG. Administrative, technical, or material support: CG and BK. Study supervision: CG and BK. All authors contributed to the article and approved the submitted version.
Funding
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 369799452 – TRR237.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Marcus Karsten for his support in creating the figures.
References
1. Wu J, Chen ZJ. Innate Immune Sensing and Signaling of Cytosolic Nucleic Acids. Annu Rev Immunol (2014) 32(1):461–88. doi: 10.1146/annurev-immunol-032713-120156
2. Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I Interferons (α/β) in Immunity and Autoimmunity. Annu Rev Immunol (2005) 23(1):307–35. doi: 10.1146/annurev.immunol.23.021704.115843
3. Crow YJ. Type I Interferonopathies: A Novel Set of Inborn Errors of Immunity. Ann N Y Acad Sci (2011) 1238(1):91–8. doi: 10.1111/j.1749-6632.2011.06220.x
4. Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, et al. Interferon-α/β-Mediated Innate Immune Mechanisms in Dermatomyositis. Ann Neurol (2005). doi: 10.1002/ana.20464
5. Rönnblom L, Eloranta ML. The Interferon Signature in Autoimmune Diseases. Curr Opin Rheumatol (2013) 25(2):248–53. doi: 10.1097/BOR.0b013e32835c7e32
6. Crow MK. Type I Interferon in the Pathogenesis of Lupus. J Immunol (2014) 192(12):5459–68. doi: 10.4049/jimmunol.1002795
7. Kirou KA, Lee C, George S, Louca K, Papagiannis IG, Peterson MGE, et al. Coordinate Overexpression of Interferon-α-Induced Genes in Systemic Lupus Erythematosus. Arthritis Rheumatol (2004). doi: 10.1002/art.20798
8. Kimoto O, Sawada J, Shimoyama K, Suzuki D, Nakamura S, Hayashi H, et al. Activation of the Interferon Pathway in Peripheral Blood of Patients With Sjögren’s Syndrome. J Rheumatol (2011). doi: 10.3899/jrheum.100486
9. Yu Q, Katlinskaya YV, Carbone CJ, Zhao B, Katlinski KV, Zheng H, et al. DNA-Damage-Induced Type I Interferon Promotes Senescence and Inhibits Stem Cell Function. Cell Rep (2015) 11(5):785–97. doi: 10.1016/j.celrep.2015.03.069
10. Härtlova A, Erttmann SF, Raffi FAM, Schmalz AM, Resch U, Anugula S, et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity (2015) 42(2):332–43. doi: 10.1016/j.immuni.2015.01.012
11. MacKenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. CGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature (2017) 548(7668):461–5. doi: 10.1038/nature23449
12. Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate Immune Sensing of Cytosolic Chromatin Fragments Through cGAS Promotes Senescence. Nat Cell Biol (2017) 19(9):1061–70. doi: 10.1038/ncb3586
13. Wolf C, Rapp A, Berndt N, Staroske W, Schuster M, Dobrick-Mattheuer M, et al. RPA and Rad51 Constitute a Cell Intrinsic Mechanism to Protect the Cytosol From Self DNA. Nat Commun (2016). doi: 10.1038/ncomms11752
14. Erdal E, Haider S, Rehwinkel J, Harris AL, McHugh PJ. A Prosurvival DNA Damage-Induced Cytoplasmic Interferon Response is Mediated by End Resection Factors and is Limited by Trex1. Genes Dev (2017) 31(4):353–69. doi: 10.1101/gad.289769.116
15. Vlachogiannis NI, Pappa M, Ntouros PA, Nezos A, Mavragani CP, Souliotis VL, et al. Association Between DNA Damage Response, Fibrosis and Type I Interferon Signature in Systemic Sclerosis. Front Immunol (2020) 11:1–10. doi: 10.3389/fimmu.2020.582401
16. Yew YW, Giordano CN, Spivak G, Lim HW. Understanding Photodermatoses Associated With Defective DNA Repair: Photosensitive Syndromes Without Associated Cancer Predisposition. J Am Acad Dermatol (2016) 75(5):873–82. doi: 10.1016/j.jaad.2016.03.044
17. Kamenisch Y, Berneburg M. Progeroid Syndromes and UV-Induced Oxidative DNA Damage. J Investig Dermatol Symp Proc (2009) 14(1):8–14. doi: 10.1038/jidsymp.2009.6
18. Giordano CN, Yew YW, Spivak G, Lim HW. Understanding Photodermatoses Associated With Defective DNA Repair: Syndromes With Cancer Predisposition. J Am Acad Dermatol (2016) 75(5):855–70. doi: 10.1016/j.jaad.2016.03.045
19. Günther C, Lee-kirsch MA, Eckhard J, Matanovic A, Rüschendorf F, Klein B, et al. SMARCAD1 Haploinsufficiency Underlies Huriez Syndrome and Associated Skin Cancer Susceptibility. J Invest Dermatol (2018). doi: 10.1016/j.jid.2018.01.015
20. Kawai T, Akira S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-Like Receptors. Nat Immunol (2010) 11(5):373–84. doi: 10.1038/ni.1863
21. Miller LS, Modlin RL. Toll-Like Receptors in the Skin. Semin Immunopathol (2007) 29(1):15–26. doi: 10.1007/s00281-007-0061-8
22. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-Like Receptor Recognizes Bacterial DNA. [In Process Citation]. Nature (2000) 408(6813):740–5. doi: 10.1038/35047123
23. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA Helicase RIG-I has an Essential Function in Double-Stranded RNA-Induced Innate Antiviral Responses. Nat Immunol (2004) 5(7):730–7. doi: 10.1038/ni1087
24. Hornung V, Kato H, Poeck H, Akira S, Conzelmann K, Schlee M. Ligand for RIG-I. Science (80-) (2006) 2010:994. doi: 10.1126/science.1132505
25. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and Characterization of MAVS, A Mitochondrial Antiviral Signaling Protein That Activates NF-κb and IRF3. Cell (2005) 122(5):669–82. doi: 10.1016/j.cell.2005.08.012
26. Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-Dependent Sensing of Poly(Da:Dt) Through the Induction of an RNA Polymerase III-Transcribed RNA Intermediate. Nat Immunol (2009) 10(10):1065–72. doi: 10.1038/ni.1779
27. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. CGAS Produces a 2′-5′-Linked Cyclic Dinucleotide Second Messenger That Activates STING. Nature (2013) 498(7454):380–4. doi: 10.1038/nature12306
28. Römling U. Great Times for Small Molecules: C-Di-AMP, a Second Messenger Candidate in Bacteria and Archaea. Sci Signal (2008) 1(33):1–5. doi: 10.1126/scisignal.133pe39
29. McWhirter SM, Barbalat R, Monroe KM, Fontana MF, Hyodo M, Joncker NT, et al. A Host Type I Interferon Response is Induced by Cytosolic Sensing of the Bacterial Second Messenger Cyclic-Di-GMP. J Exp Med (2009) 206(9):1899–911. doi: 10.1084/jem.20082874
30. Ferbas JJ, Toso JF, Logar AJ, Navratil JS, Rinaldo CR Jr CD4+ Blood Dendritic Cells are Potent Producers of IFN-Alpha in Response to In Vitro HIV-1 Infection [Published Erratum Appears in J Immunol 1994 Jul 15;153(2):910]. J Immunol (1994) 152(9):4649–62.
31. Lee-Kirsch MA. The Type I Interferonopathies. Annu Rev Med (2017) 68(1):297–315. doi: 10.1146/annurev-med-050715-104506
32. Schindler C, Levy DE, Decker T. JAK-STAT Signaling: From Interferons to Cytokines. J Biol Chem (2007) 282(28):20059–63. doi: 10.1074/jbc.R700016200
33. Domeier PP, Chodisetti SB, Schell SL, Kawasawa YI, Fasnacht MJ, Soni C, et al. B-Cell-Intrinsic Type 1 Interferon Signaling Is Crucial for Loss of Tolerance and the Development of Autoreactive B Cells. Cell Rep (2018). doi: 10.1016/j.celrep.2018.06.046
34. Lee-Kirsch MA, Günther C, Roers A. Nucleic Acid-Mediated Autoinflammation and Autoimmunity-Type I Interferonopathies. J Mol Med (Berl) (2016) 94(10):1081–4. doi: 10.1007/s00109-016-1467-3
35. Yang YG, Lindahl T, Barnes DE. Trex1 Exonuclease Degrades ssDNA to Prevent Chronic Checkpoint Activation and Autoimmune Disease. Cell (2007) 131(5):873–86. doi: 10.1016/j.cell.2007.10.017
36. Motwani M, Pesiridis S, Fitzgerald KA. DNA Sensing by the cGAS–STING Pathway in Health and Disease. Nat Rev Genet (2019) 20(11):657–74. doi: 10.1038/s41576-019-0151-1
37. Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, et al. Heterozygous Mutations in TREX1 Cause Familial Chilblain Lupus and Dominant Aicardi-Goutières Syndrome. Am J Hum Genet (2007) 80(4):811–5. doi: 10.1086/513443
38. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the Gene Encoding the 3′-5′ DNA Exonuclease TREX1 Cause Aicardi-Goutières Syndrome at the AGS1 Locus. Nat Genet (2006) 38(8):917–20. doi: 10.1038/ng1845
39. Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, et al. Clinical and Molecular Phenotype of Aicardi-Goutières Syndrome. Am J Hum Genet (2007) 81(4):713–25. doi: 10.1086/521373
40. Ramantani G, Kohlhase J, Hertzberg C, Micheil Innes A, Engel K, Hunger S, et al. Expanding the Phenotypic Spectrum of Lupus Erythematosus in Aicardi-Goutières Syndrome. Arthritis Rheumatol (2010). doi: 10.1002/art.27367
41. Lee-Kirsch MA, Gong M, Schulz H, Rüschendorf F, Stein A, Pfeiffer C, et al. Familial Chilblain Lupus, a Monogenic Form of Cutaneous Lupus Erythematosus, Maps to Chromosome 3p. Am J Hum Genet (2006) 79(4):731–7. doi: 10.1086/507848
42. Günther C, Meurer M, Stein A, Viehweg A, Lee-Kirsch MA. Familial Chilblain Lupus - A Monogenic Form of Cutaneous Lupus Erythematosus Due to a Heterozygous Mutation in TREX1. Dermatology (2009) 219(2):162–6. doi: 10.1159/000222430
43. Peschke K, Friebe F, Zimmermann N, Wahlicht T, Schumann T, Achleitner M, et al. Deregulated Type I IFN Response in TREX1-Associated Familial Chilblain Lupus. J Invest Dermatol (2014) 134(5):1456–9. doi: 10.1038/jid.2013.496
44. Zimmermann N, Wolf C, Schwenke R, Lüth A, Schmidt F, Engel K, et al. Assessment of Clinical Response to Janus Kinase Inhibition in Patients With Familial Chilblain Lupus and TREX1 Mutation. JAMA Dermatol (2019). doi: 10.1001/jamadermatol.2018.5077
45. Günther C, Hillebrand M, Brunk J, Lee-Kirsch MA. Systemic Involvement in TREX1-Associated Familial Chilblain Lupus. J Am Acad Dermatol (2013) 69(4):e179–81. doi: 10.1016/j.jaad.2013.04.020
46. El Ghissassi F, Baan R, Straif K, Grosse Y, Secretan B, Bouvard V, et al. A Review of Human Carcinogens—Part D: Radiation. Lancet Oncol (2009) 10(8):751–2. doi: 10.1016/S1470-2045(09)70213-X
47. Cadet J, Sage E, Douki T. Ultraviolet Radiation-Mediated Damage to Cellular DNA. Mutat Res - Fundam Mol Mech Mutagen (2005) 571(1–2):3–17. doi: 10.1016/j.mrfmmm.2004.09.012
48. Wischermann K, Popp S, Moshir S, Scharfetter-Kochanek K, Wlaschek M, de Gruijl F, et al. UVA Radiation Causes DNA Strand Breaks, Chromosomal Aberrations and Tumorigenic Transformation in HaCaT Skin Keratinocytes. Oncogene (2008) 27(31):4269–80. doi: 10.1038/onc.2008.70
49. Greinert R, Volkmer B, Henning S, Breitbart EW, Greulich KO, Cardoso MC, et al. UVA-Induced DNA Double-Strand Breaks Result From the Repair of Clustered Oxidative DNA Damages. Nucleic Acids Res (2012) 40(20):10263–73. doi: 10.1093/nar/gks824
50. Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K, Linn S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu Rev Biochem (2004) 73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723
51. Scully R, Panday A, Elango R, Willis NA. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat Rev Mol Cell Biol (2019) 20(11):698–714. doi: 10.1038/s41580-019-0152-0
52. German J, Roe AM, Leppert MF, Ellis NA. Bloom Syndrome: An Analysis of Consanguineous Families Assigns the Locus Mutated to Chromosome Band 15q26.1. Proc Natl Acad Sci U S A (1994) 91(14):6669–73. doi: 10.1073/pnas.91.14.6669
53. Chantorn R, Lim HW, Shwayder TA. Photosensitivity Disorders in Children: Part II. J Am Acad Dermatol (2012) 67(6):1113.e1–1113.e15. doi: 10.1016/j.jaad.2012.07.032
54. Wang LL, Gannavarapu A, Kozinetz CA, Levy ML, Lewis RA, Chintagumpala MM, et al. Association Between Osteosarcoma and Deleterious Mutations in the RECQL4 Gene Rothmund-Thomson Syndrome. J Natl Cancer Inst (2003) 95(9):669–74. doi: 10.1093/jnci/95.9.669
55. Kavanagh GM, Jardine PE, Peachey RD, Murray JC, De Berker D. The Scleroatrophic Syndrome of Huriez. Br J Dermatol (1997) 137(1):114–8. doi: 10.1046/j.1365-2133.1997.17791866.x
56. Huriez C, Deminati M, Agache P, Delmas-Marsalet Y, Mennecier M. Genodermatose Scléro-Atrophiante Et Kératodérmique Des Extrémités. Ann Dermatol Syphiligr (Paris) (1965) 96(2):135–46.
57. Hamm H, Traupe H, Brocker EB, Schubert H, Kolde G. The Scleroatrophic Syndrome of Huriez: A Cancer-Prone Genodermatosis. Br J Dermatol (1996) 134(3):512–8. doi: 10.1111/j.1365-2133.1996.tb16240.x
58. Çelik NS, Yaşar Ş, Aytekin S, Güneş P. A Rare Syndrome Resembling Scleroderma: Huriez Syndrome. Ski Appendage Disord (2018) 4(2):82–5. doi: 10.1159/000479036
59. Ivonye C, Jamched U, Anderson D, Adesunloye B. Uncommon Skin Lesion in a Patient With Ataxia-Telangiectasia. Int J Dermatol (2008) 47(10):1051–2. doi: 10.1111/j.1365-4632.2008.03745.x
60. Sari A, Okuyaz C, Adiguzel U, Ates NA. Uncommon Associations With Ataxia-Telangiectasia: Vitiligo and Optic Disc Drusen. Ophthalmic Genet (2009) 30(1):19–22. doi: 10.1080/13816810802415256
61. Gravel S, Chapman JR, Magill C, Jackson SP. DNA Helicases Sgs1 and BLM Promote DNA Double-Strand Break Resection. Genes Dev (2008) 22(20):2767–72. doi: 10.1101/gad.503108
62. Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, et al. Mutations in RECQL4 Cause a Subset of Cases of Rothmund-Thomson Syndrome. Nat Genet (1999) 22(1):82–4. doi: 10.1038/8788
63. Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, et al. Positional Cloning of the Werner’s Syndrome Gene. Science (1996) 272(5259):258–62. doi: 10.1126/science.272.5259.258
64. Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, et al. The Yeast Fun30 and Human SMARCAD1 Chromatin Remodellers Promote DNA End Resection. Nature (2012) 489(7417):581–4. doi: 10.1038/nature11353
65. Rowbotham SP, Barki L, Neves-Costa A, Santos F, Dean W, Hawkes N, et al. Maintenance of Silent Chromatin Through Replication Requires SWI/SNF-Like Chromatin Remodeler Smarcad1. Mol Cell (2011) 42(3):285–96. doi: 10.1016/j.molcel.2011.02.036
66. Nousbeck J, Burger B, Fuchs-Telem D, Pavlovsky M, Fenig S, Sarig O, et al. A Mutation in a Skin-Specific Isoform of SMARCAD1 Causes Autosomal-Dominant Adermatoglyphia. Am J Hum Genet (2011) 89(2):302–7. doi: 10.1016/j.ajhg.2011.07.004
67. Sinha RP, Häder D-P. UV-Induced DNA Damage and Repair: A Review. Photochem Photobiol Sci (2002) 1(4):225–36. doi: 10.1039/b201230h
68. Reardon JT, Sancar A. Nucleotide Excision Repair. Prog Nucleic Acid Res Mol Biol (2005) 79:183–235. doi: 10.1016/S0079-6603(04)79004-2
69. Lehmann AR, McGibbon D, Stefanini M. Xeroderma Pigmentosum. Orphanet J Rare Dis (2011) 6:70. doi: 10.1186/1750-1172-6-70
70. Bradford PT, Goldstein AM, Tamura D, Khan SG, Ueda T, Boyle J, et al. Cancer and Neurologic Degeneration in Xeroderma Pigmentosum: Long Term Follow-Up Characterises the Role of DNA Repair. J Med Genet (2011). doi: 10.1136/jmg.2010.083022
71. Ammann AJ, Hong R. Autoimmune Phenomena in Ataxia Telangiectasia. J Pediatr (1971) 78(5):821–6. doi: 10.1016/S0022-3476(71)80353-0
72. Shao L, Fujii H, Colmegna I, Oishi H, Goronzy JJ, Weyand CM. Deficiency of the DNA Repair Enzyme ATM in Rheumatoid Arthritis. J Exp Med (2009) 206(6):1435–49. doi: 10.1084/jem.20082251
73. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature (2015). doi: 10.1038/nature14156
74. Valentin-Vega YA, MacLean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, et al. Mitochondrial Dysfunction in Ataxia-Telangiectasia. Blood (2012). doi: 10.1182/blood-2011-08-373639
75. Hu M, Zhou M, Bao X, Pan D, Jiao M, Liu X, et al. ATM Inhibition Enhances Cancer Immunotherapy by Promoting mtDNA Leakage/cGAS-STING Activation. J Clin Invest (2020). In revisio. doi: 10.1172/JCI139333
76. Bhattacharya S, Srinivasan K, Abdisalaam S, Su F, Raj P, Dozmorov I, et al. RAD51 Interconnects Between DNA Replication, DNA Repair and Immunity. Nucleic Acids Res (2017). doi: 10.1093/nar/gkx126
77. Yang YG, Lindahl T, Barnes DE. Trex1 Exonuclease Degrades ssDNA to Prevent Chronic Checkpoint Activation and Autoimmune Disease. Cell (2007) 131(5):873–86. doi: 10.1016/j.cell.2007.10.017
78. Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V. TREX1 Deficiency Triggers Cell-Autonomous Immunity in a cGAS-Dependent Manner. J Immunol (2014) 192(12):5993–7. doi: 10.4049/jimmunol.1400737
79. Skopelja-Gardner S, An J, Tai J, Tanaka L, Sun X, Hermanson P, et al. The Early Local and Systemic Type I Interferon Responses to Ultraviolet B Light Exposure are cGAS Dependent. Sci Rep (2020). doi: 10.1038/s41598-020-64865-w
80. Reijns MAM, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, et al. Enzymatic Removal of Ribonucleotides From DNA is Essential for Mammalian Genome Integrity and Development. Cell (2012). doi: 10.1016/j.cell.2012.04.011
81. McElhinny SAN, Kumar D, Clark AB, Watt DL, Watts BE, Lundström EB, et al. Genome Instability Due to Ribonucleotide Incorporation Into DNA. Nat Chem Biol (2010). doi: 10.1038/nchembio.424
82. Günther C, Kind B, Reijns MAM, Berndt N, Martinez-bueno M, Wolf C, et al. Defective Removal of Ribonucleotides From DNA Promotes Systemic Autoimmunity. J Clin Invest (2015) 125(1):413–24. doi: 10.1172/JCI78001.antigens
83. Salk D. Werner’s Syndrome: A Review of Recent Research With an Analysis of Connective Tissue Metabolism, Growth Control of Cultured Cells, and Chromosomal Aberrations. Hum Genet (1982) 62(1):1–5. doi: 10.1007/BF00295598
84. Campisi J, d’Adda di Fagagna F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat Rev Mol Cell Biol (2007) 8(9):729–40. doi: 10.1038/nrm2233
85. Coppé JP, Desprez PY, Krtolica A, Campisi J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu Rev Pathol Mech Dis (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
86. Schild-Poulter C, Su A, Shih A, Kelly OP, Fritzler MJ, Goldstein R, et al. Association of Autoantibodies With Ku and DNA Repair Proteins in Connective Tissue Diseases. Rheumatology (2008). doi: 10.1093/rheumatology/kem338
87. Mukherjee S, Sinha D, Bhattacharya S, Srinivasan K, Abdisalaam S, Asaithamby A. Werner Syndrome Protein and Dna Replication. Int J Mol Sci (2018) 19(11):3442. doi: 10.3390/ijms19113442
88. Su F, Mukherjee S, Yang Y, Mori E, Bhattacharya S, Kobayashi J, et al. Nonenzymatic Role for WRN in Preserving Nascent DNA Strands After Replication Stress. Cell Rep (2014). doi: 10.1016/j.celrep.2014.10.025
89. Yamagami M, Otsuka M, Kishikawa T, Sekiba K, Seimiya T, Tanaka E, et al. ISGF3 With Reduced Phosphorylation is Associated With Constitutive Expression of Interferon-Induced Genes in Aging Cells. NPJ Aging Mech Dis (2018). doi: 10.1038/s41514-018-0030-6
90. Wang W, Yin Y, Xu L, Su J, Huang F, Wang Y, et al. Unphosphorylated ISGF3 Drives Constitutive Expression of Interferon-Stimulated Genes to Protect Against Viral Infections. Sci Signal (2017). doi: 10.1126/scisignal.aah4248
91. Cunniff C, Bassetti JA, Ellis NA. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol Syndromol (2017) 8(1):4–23. doi: 10.1159/000452082
92. Yankiwski V, Marciniak RA, Guarente L, Neff NF. Nuclear Structure in Normal and Bloom Syndrome Cells. Proc Natl Acad Sci U S A (2000) 97(10):5214–9. doi: 10.1073/pnas.090525897
93. Gratia M, Rodero MP, Conrad C, Samra EB, Maurin M, Rice GI, et al. Bloom Syndrome Protein Restrains Innate Immune Sensing of Micronuclei by cGAS. J Exp Med (2019). doi: 10.1084/jem.20181329
94. Hussain M, Krishnamurthy S, Patel J, Kim E, Baptiste BA, Croteau DL, et al. Skin Abnormalities in Disorders With DNA Repair Defects, Premature Aging, and Mitochondrial Dysfunction. J Invest Dermatol (2021). doi: 10.1016/j.jid.2020.10.019
95. Gupta S, De S, Srivastava V, Hussain M, Kumari J, Muniyappa K, et al. RECQL4 and P53 Potentiate the Activity of Polymerase γ and Maintain the Integrity of the Human Mitochondrial Genome. Carcinogenesis (2014). doi: 10.1093/carcin/bgt315
96. Croteau DL, Rossi ML, Canugovi C, Tian J, Sykora P, Ramamoorthy M, et al. RECQL4 Localizes to Mitochondria and Preserves Mitochondrial DNA Integrity. Aging Cell (2012). doi: 10.1111/j.1474-9726.2012.00803.x
97. Tan FK, Zhou X, Mayes MD, Gourh P, Guo X, Marcum C, et al. Signatures of Differentially Regulated Interferon Gene Expression and Vasculotrophism in the Peripheral Blood Cells of Systemic Sclerosis Patients. Rheumatology (2006). doi: 10.1093/rheumatology/kei244
98. Farina GA, York MR, Di MM, Collins CA, Meller S, Homey B, et al. Poly (I : C) Drives Type I IFN- and TGF B -Mediated Inflammation and Dermal Fibrosis Simulating Altered Gene Expression in Systemic Sclerosis. J Invest Dermatol (2010) 130(11):2583–93. doi: 10.1038/jid.2010.200
99. Souliotis VL, Vlachogiannis NI, Pappa M, Argyriou A, Ntouros PA, Sfikakis PP. DNA Damage Response and Oxidative Stress in Systemic Autoimmunity. Int J Mol Sci (2020) 21(1):1–24. doi: 10.3390/ijms21010055
100. Di Benedetto P, Panzera N, Cipriani P, Mastroiaco V, Tessitore A, Liakouli V, et al. Mesenchymal Stem Cells of Systemic Sclerosis Patients, Derived From Different Sources, Show a Profibrotic microRNA Profiling. Sci Rep (2019). doi: 10.1038/s41598-019-43638-0
101. Nissinen L, Farshchian M, Riihilä P, Kähäri VM. New Perspectives on Role of Tumor Microenvironment in Progression of Cutaneous Squamous Cell Carcinoma. Cell Tissue Res (2016) 365(3):691–702. doi: 10.1007/s00441-016-2457-z
102. Guerriero C, Albanesi C, Girolomoni G, De Simone C, Capizzi R, Amerio P, et al. Huriez Syndrome: Case Report With a Detailed Analysis of Skin Dendritic Cells. Br J Dermatol (2000) 143(5):1091–6. doi: 10.1046/j.1365-2133.2000.03793.x
103. Gaspari AA, Fleisher TA, Kraemer KH. Impaired Interferon Production and Natural Killer Cell Activation in Patients With the Skin Cancer-Prone Disorder, Xeroderma Pigmentosum. J Clin Invest (1993) 92(3):1135–42. doi: 10.1172/JCI116682
104. Parlanti E, Pietraforte D, Iorio E, Visentin S, De Nuccio C, Zijno A, et al. An Altered Redox Balance and Increased Genetic Instability Characterize Primary Fibroblasts Derived From Xeroderma Pigmentosum Group A Patients. Mutat Res - Fundam Mol Mech Mutagen (2015). doi: 10.1016/j.mrfmmm.2015.10.002
105. Kwon J, Bakhoum SF. The Cytosolic DNA-Sensing cGAS–Sting Pathway in Cancer. Cancer Discov (2020) 10(1):26–39. doi: 10.1158/2159-8290.CD-19-0761
106. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature (2017). doi: 10.1038/nature24050
107. Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal Instability Drives Metastasis Through a Cytosolic DNA Response. Nature (2018). doi: 10.1038/nature25432
108. Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic Nuclear Envelope Collapse in Cancer Cell Micronuclei. Cell (2013) 154(1):47–60. doi: 10.1016/j.cell.2013.06.007
109. Brzostek-Racine S, Gordon C, Van Scoy S, Reich NC. The DNA Damage Response Induces Interferon. J Immunol (2011) 187(10):5336–45. doi: 10.4049/jimmunol.1100040
110. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity (2014). doi: 10.1016/j.immuni.2014.10.017
111. Critchley-Thorne RJ, Simons DL, Yan N, Miyahira AK, Dirbas FM, Johnson DL, et al. Impaired Interferon Signaling is a Common Immune Defect in Human Cancer. Proc Natl Acad Sci U S A (2009). doi: 10.1073/pnas.0901329106
112. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I Interferon is Selectively Required by Dendritic Cells for Immune Rejection of Tumors. J Exp Med (2011). doi: 10.1084/jem.20101158
113. Cho C, Mukherjee R, Peck AR, Sun Y, McBrearty N, Katlinski KV, et al. Cancer-Associated Fibroblasts Downregulate Type I Interferon Receptor to Stimulate Intratumoral Stromagenesis. Oncogene (2020). doi: 10.1038/s41388-020-01424-7
114. Takashima K, Takeda Y, Oshiumi H, Shime H, Okabe M, Ikawa M, et al. STING in Tumor and Host Cells Cooperatively Work for NK Cell-Mediated Tumor Growth Retardation. Biochem Biophys Res Commun (2016). doi: 10.1016/j.bbrc.2016.09.021
115. Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep (2016) 14(2):282–97. doi: 10.1016/j.celrep.2015.12.029
116. Xia T, Konno H, Barber GN. Recurrent Loss of STING Signaling in Melanoma Correlates With Susceptibility to Viral Oncolysis. Cancer Res (2016) 76(22):6747–59. doi: 10.1158/0008-5472.CAN-16-1404
117. Campisi J, D’Adda Di Fagagna F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat Rev Mol Cell Biol (2007) 8(9):729–40. doi: 10.1038/nrm2233
118. Rodier F, Coppé JP, Patil CK, Hoeijmakers WAM, Muñoz DP, Raza SR, et al. Persistent DNA Damage Signalling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat Cell Biol (2009). doi: 10.1038/ncb1909
119. Yang H, Wang H, Ren J, Chen Q, Chen ZJ. CGAS is Essential for Cellular Senescence. Proc Natl Acad Sci U S A (2017) 114(23):E4612–20. doi: 10.1073/pnas.1705499114
120. Faget DV, Ren Q, Stewart SA. Unmasking Senescence: Context-Dependent Effects of SASP in Cancer. Nat Rev Cancer (2019) 19(8):439–53. doi: 10.1038/s41568-019-0156-2
121. Damsky W, King BA. JAK Inhibitors in Dermatology: The Promise of a New Drug Class. J Am Acad Dermatol (2017) 76(4):736–44. doi: 10.1016/j.jaad.2016.12.005
122. Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK Inhibition as a Therapeutic Strategy for Immune and Inflammatory Diseases. Nat Rev Drug Discovery (2017) 16(12):843–62. doi: 10.1038/nrd.2017.201
Keywords: Interferon, DNA damage, DNA repair, Werner syndrome (WS), Bloom Syndrome, Huriez syndrome, chilblain lupus, Ataxia teleangiectasia
Citation: Klein B and Günther C (2021) Type I Interferon Induction in Cutaneous DNA Damage Syndromes. Front. Immunol. 12:715723. doi: 10.3389/fimmu.2021.715723
Received: 27 May 2021; Accepted: 05 July 2021;
Published: 23 July 2021.
Edited by:
Aldo Tagliabue, Institute of Biomedical Technologies, Italian National Research Council (CNR), ItalyReviewed by:
Paola Migliorini, University of Pisa, ItalyHuiming Lu, University of Texas Southwestern Medical Center, United States
Copyright © 2021 Klein and Günther. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin Klein, QmVuamFtaW4ua2xlaW5AbWVkaXppbi51bmktbGVpcHppZy5kZQ==; Claudia Günther, Q2xhdWRpYS5HdWVudGhlckB1bmlrbGluaWt1bS1kcmVzZGVuLmRl