 Michael Schulz1,2
Michael Schulz1,2 Lisa Sevenich1,3,4,5*
Lisa Sevenich1,3,4,5*- 1Institute for Tumor Biology and Experimental Therapy, Georg-Speyer-Haus, Frankfurt am Main, Germany
- 2Biological Sciences, Faculty 15, Goethe University Frankfurt, Frankfurt am Main, Germany
- 3German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Frankfurt am Main, Germany
- 4German Cancer Research Center (DKFZ), Heidelberg, Germany
- 5Frankfurt Cancer Institute (FCI), Goethe University Frankfurt, Frankfurt am Main, Germany
Macrophages not only represent an integral part of innate immunity but also critically contribute to tissue and organ homeostasis. Moreover, disease progression is accompanied by macrophage accumulation in many cancer types and is often associated with poor prognosis and therapy resistance. Given their critical role in modulating tumor immunity in primary and metastatic brain cancers, macrophages are emerging as promising therapeutic targets. Different types of macrophages infiltrate brain cancers, including (i) CNS resident macrophages that comprise microglia (TAM-MG) as well as border-associated macrophages and (ii) monocyte-derived macrophages (TAM-MDM) that are recruited from the periphery. Controversy remained about their disease-associated functions since classical approaches did not reliably distinguish between macrophage subpopulations. Recent conceptual and technological advances, such as large-scale omic approaches, provided new insight into molecular profiles of TAMs based on their cellular origin. In this review, we summarize insight from recent studies highlighting similarities and differences of TAM-MG and TAM-MDM at the molecular level. We will focus on data obtained from RNA sequencing and mass cytometry approaches. Together, this knowledge significantly contributes to our understanding of transcriptional and translational programs that define disease-associated TAM functions. Cross-species meta-analyses will further help to evaluate the translational significance of preclinical findings as part of the effort to identify candidates for macrophage-targeted therapy against brain metastasis.
Introduction
Mononuclear phagocytes comprise bone marrow-derived progenitors, blood monocytes, and tissue-specific macrophage populations of embryonic origin (1). Fate-mapping studies in mice revealed that macrophage populations of distinct organs (e.g., lung, spleen, liver, brain, skin) are established early during development and are self-maintained during adulthood (2). The cellular identity of tissue resident macrophages is shaped by the local environment of specific organs (3–5). Moreover, the presence of a diverse range of receptors (6) allows macrophages to receive a broad spectrum of signals and thus contribute in autocrine and paracrine interactions. Hence, this functional plasticity places them at the interface of developmental processes, tissue homeostasis, and immunity (1).
As the sole immune cell type within the immune-privileged brain parenchyma, yolk sac-derived microglia (MG) exert critical functions in immune surveillance and host defense (7, 8). In contrast to the brain parenchyma, where the entry of systemic immune cells is strictly controlled, areas surrounding the brain (e.g., meninges) are constantly patrolled by different classes of lymphoid and myeloid cells (9, 10) (Figure 1). In addition to the heterogeneous MG populations that have been identified throughout the brain parenchyma (11, 12), nonparenchymal macrophages found in border regions [= border-associated macrophages (BAMs)] represent a distinct population of central nervous system (CNS) phagocytes (13). Similar to microglia, they derive from yolk sac progenitors during early development (14) and populate the meninges (m), the perivascular areas (pv), and the choroid plexus (cp). Each population is classified based on a specific set of genes, and functional adaptation is driven by local traits. Compared with MG, BAMs exhibit distinct transcriptional signatures (10, 14, 15). Under homeostatic conditions, the structures adjacent to the parenchyma maintain physical and immunological separation of the CNS, but at the same time allow restricted exchange and access of cells and molecules (16).
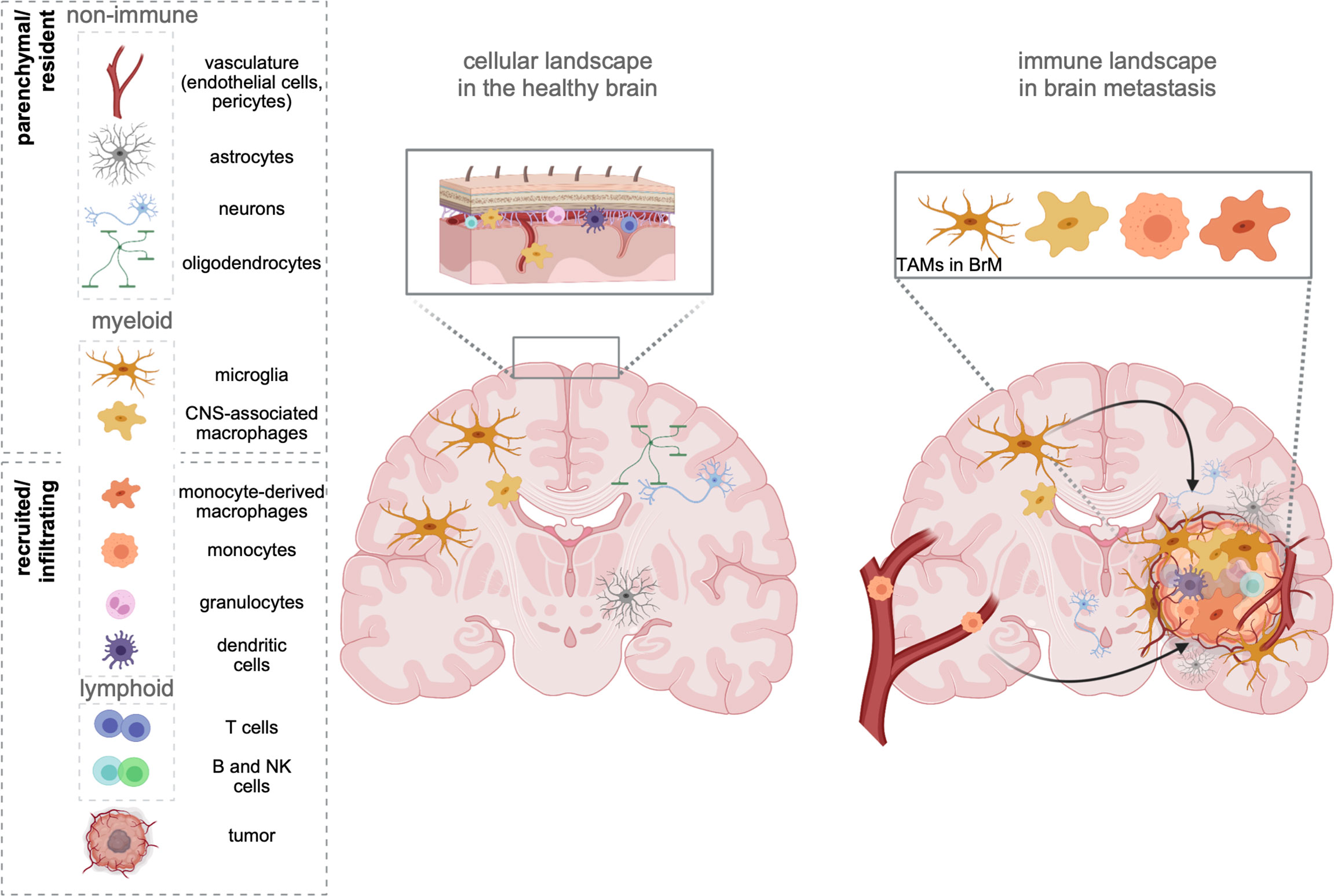
Figure 1 The cellular environment in the healthy brain and BrM. The healthy brain parenchyma consists of resident cell types, including neurons, astrocytes, oligodendrocytes, and cells forming the vasculature (endothelial cells, pericytes). While microglia represent the sole immune cells within the parenchyma, border-associated areas of the brain (e.g., meninges, perivascular areas) harbor every other cell type of the immune system. In contrast, brain metastasis (right) induce the recruitment of all types of myeloid and lymphoid immune cells from the periphery. Tumor-associated macrophages (TAMs) represent a heterogeneous pool of myeloid cells, which consist of brain-resident microglia, as well as monocytes, and monocyte-derived macrophages from the periphery. Recent studies further suggest a partial involvement of recruited CNS/border-associated macrophages (BAMs).
Neurological disorders disrupt the tissue homeostasis of the brain and lead to the recruitment of cells from the periphery, mostly of myeloid origin (17). Accumulation of myeloid cells within the parenchyma impacts the severity and disease outcome of neurological disorders. Hence, understanding the biology of specific myeloid subpopulations at spatiotemporal resolution is crucial for the development of therapeutic strategies that resolve underlying insults.
A prominent example is the development of brain malignancies. Cerebral or cerebellar tumor formation is accompanied by a massive recruitment of macrophages from the periphery, which together with resident microglia represent the most abundant stromal cell types in primary (18) and secondary (19) brain tumors [brain metastasis (BrM)]. Although every tumor type can metastasize to the brain, the highest incidence is associated to melanoma, lung and breast cancer. Adding up relative numbers, BrM originating from lung or breast cancer contribute to more than 75% of all BrM (20–23).
The brain tumor-associated macrophage (TAM) population consists of cells originating from resident microglia (TAM-MG) and cells of monocytic origin, i.e., monocytes and monocyte-derived macrophages (MDM). However, due to the lack of definitive markers that discriminate both lineages within brain tumors, it was (24, 25) challenging to determine quantitative and qualitative contributions of both TAM populations to brain tumor biology in the past without the need of transplantation models or lineage-tracing approaches. Bowman et al. employed two lineage tracing models in combination with RNA sequencing to identify markers, which reliably allow the discrimination of TAM-MGs and TAM-MDMs in mouse and human primary and metastatic brain tumors. This study led to the identification of the integrin alpha subunit CD49d (encoded by Itga4) that is specifically repressed in MG but highly expressed in MDMs. Importantly, this expression pattern remains conserved within brain tumors. In addition, the authors identified CD11a (encoded by Itgal) as similarly differently regulated between both major TAM populations (26).
The biggest differences between both TAM subpopulations are determined by their different ontogenetic origin. Since brain TAMs are known to critically influence the progression and outcome of brain tumor biology (24, 25), understanding their quantitative contributions under different conditions and associated putative different functions is key in order to develop novel strategies targeting distinct disease-associated phenotypes in BrM.
TAMs as Central Part of the Brain Metastasis Microenvironment
TAMs are known to represent a highly abundant cell population in primary and metastatic brain tumors with different quantitative contribution to the myeloid cell pool depending on primary tumor entity (18, 19). However, controversy remained on the functional contribution of macrophage populations depending on their ontogeny. Technical integration of lineage-restricted markers or the use of single cell-based techniques to characterize myeloid cells in brain tumors has significantly broadened our knowledge on TAM heterogeneity in experimental BrM models and patient-derived data from various brain malignancies (Table 1).
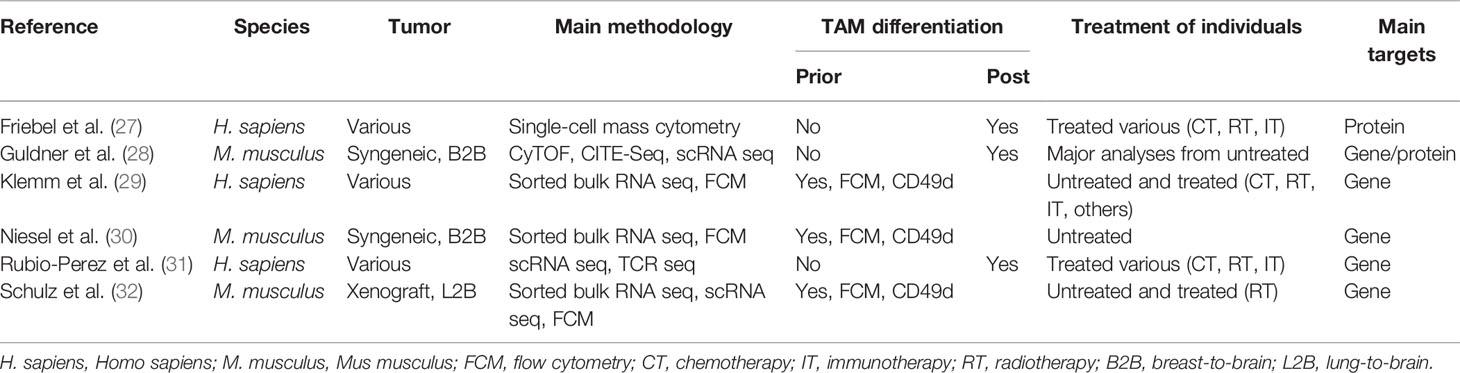
Table 1 Overview of recent studies, examining the tumor microenvironment (TME) of preclinical models of brain metastasis (BrM), and human patient samples.
Two recent studies explored the cellular heterogeneity of myeloid cells in experimental brain metastasis models by multicolor flow cytometry (FCM) integrating CD49d as a marker to distinguish TAM-MG and TAM-MDM. Although approximately 75% of all CD45+ cells of the syngeneic breast cancer model 99LN-BrM were of myeloid origin, only 5%–10% of all TAMs were MDMs (30). By comparison, the xenograft lung cancer BrM model H2030-BrM induced stronger TAM-MDM recruitment (32), which constantly increased across different stages of tumor progression leading to 10% and 20% of TAM-MDMs in small or large BrM, respectively (32). Moreover, it was demonstrated that the TAM population within the H2030-BrM model changed in response to whole brain radiotherapy (WBRT), applied as a standard-of-care treatment modality (32). A relative reduction of TAM-MDM contribution to the total TAM pool was observed 3 days after hypofractionated as well as classically fractionated WBRT. Interestingly, this effect was transient and constant reinfiltration resulted in a steadily increasing TAM-MDM population, as examined within the total myeloid cell pool in H2030-BrM at several time points after WBRT. Hence, the application of radiotherapy represents a useful strategy to modulate the TAM pool by causing radiation-mediated cell elimination on the one hand, but enhancing infiltration of naïve cells from the periphery on the other hand. A similar way of interfering with MDM recruitment has been shown in mouse models of glioma in response to radiation (33).
Collectively, these data suggest that the TAM pool in preclinical BrM models is highly dynamic. Moreover, recent studies highlighted the contribution of each TAM population to total TAMs. The relative contribution of each TAM population to the total TAM pool is influenced by the primary tumor entity and can be modulated by radiotherapy. TAMs of peripheral origin have been found to be more abundant in recurrent glioma samples upon surgery (34), further illustrating the impact of antitumor therapy on the immune landscape. Interestingly, the diversity of the TAM pool is similarly regulated by the origin of the primary tumor in human BrM (27, 29, 31). Within the studies by Friebel et al. and Klemm et al. the authors performed comprehensive in-depth analysis of patient-derived primary and secondary brain tumor tissue by integrating high-dimensional techniques, such as, FCM, RNA sequencing, or mass cytometry by time of flight (CyToF) to gain insight into cellular and molecular aspects of the brain tumor immune landscape. In contrast to primary brain tumors, BrM induced higher infiltration of myeloid cells from the periphery, and the majority of CD45+ cells was composed of neutrophils and MDMs (27, 29). Lower abundance of macrophages from the periphery was observed in melanoma BrM compared with breast and lung cancers (Figure 2).
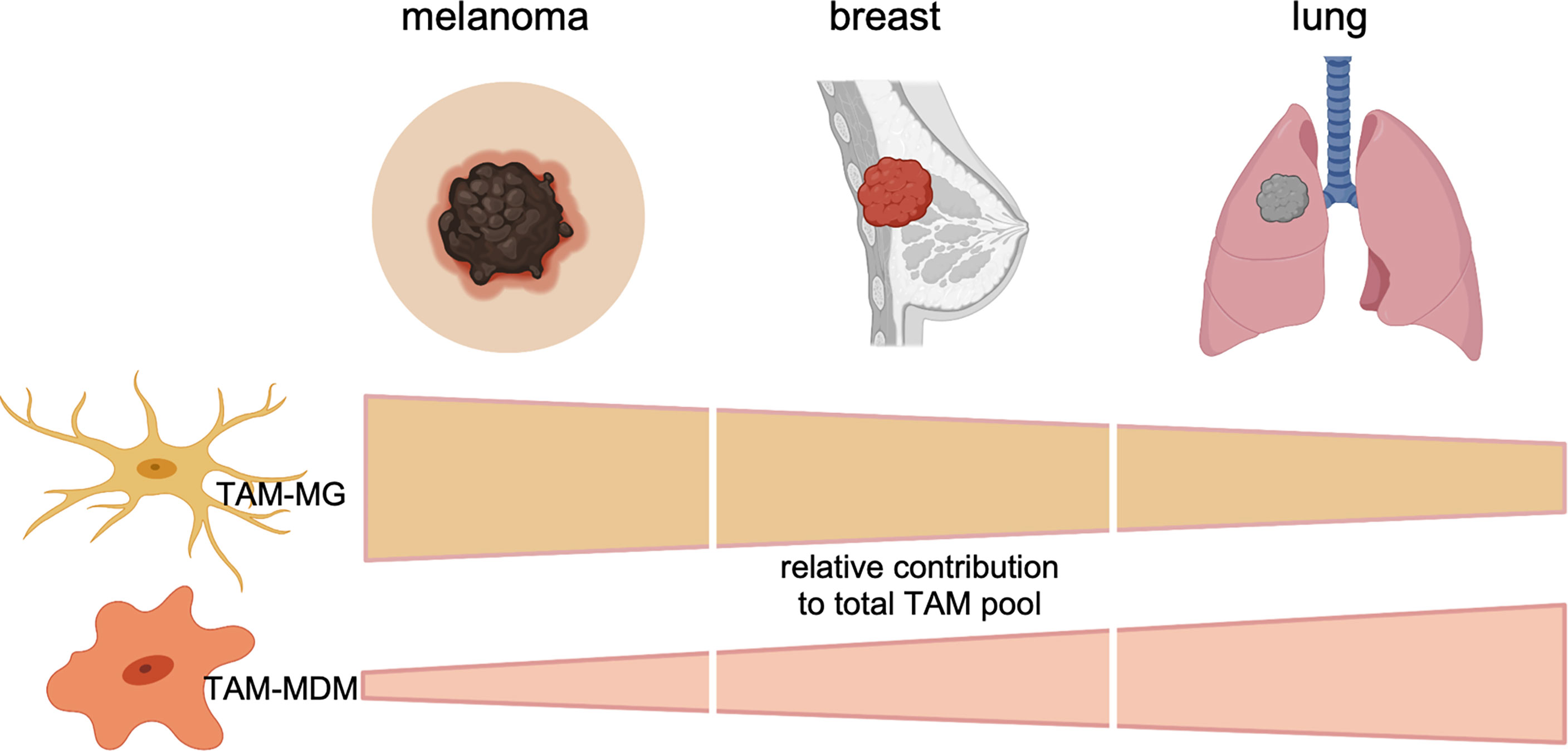
Figure 2 Relative contribution of each TAM subpopulation to the total TAM pool in BrMs derived from melanoma, breast cancer, or lung cancer. Data represent cumulative, relative data derived from preclinical (mouse models) and clinical (human) findings.
Molecular Profiles of TAMs in BrM
TAMs are malignancy instructed and have been described as key players at the interface between tumors and cells of the immune landscape. This can be attributed to their high capacity of integrating a broad range of external stimuli, resulting in diverse activation states and highly plastic phenotypes (35, 36). In the following paragraphs, we will provide an overview of molecular alterations observed in both major TAM populations of mouse and human BrM and highlight representative markers that have been identified to be differentially expressed in TAM-MG and TAM-MDM. Moreover, we will discuss candidate factors that have been identified as core signatures of disease-associated macrophages and are commonly up- or downregulated in both major TAM populations. An overview of the representative factors can be found in Table 2.
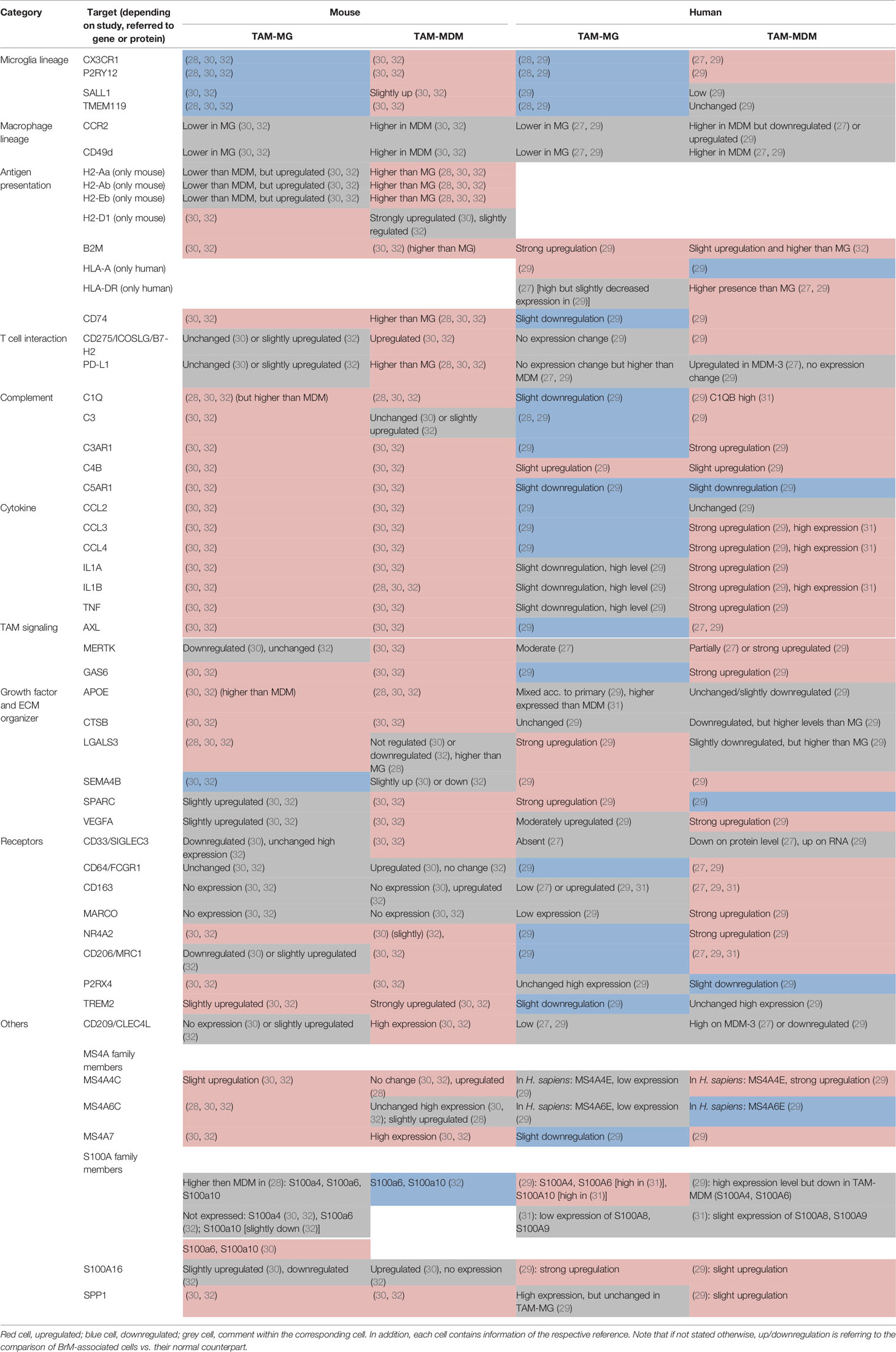
Table 2 Selected markers and their regulation within murine (left) and human (right) TAM-MG and TAM-MDMs.
Transition From Normal to Disease-Associated Cell States
It is increasingly appreciated that tumor-associated immune cells are significantly different compared with their normal cellular counterparts. However, it remains less well understood how normal cells transition into disease-associated cell types upon initial contact to tumor cells and within different stages of tumor progression. Interestingly, Schulz et al. observed that the instruction and education of TAMs represents an early event during formation of the BrM-TME and occurs rapidly after recruitment of resident TAM-MG or peripheral-derived TAM-MDMs. Analyses of transcriptional program in TAMs isolated from small- vs. large-stage BrM revealed an almost complete absence of differences in gene expression in each population, suggesting stable transcriptomes during BrM progression (32). However, further dissection of potential transition stages based on single-cell approaches are needed to characterize the acquisition of diseases-associated signatures across a broader range of different stages of tumor progression and to identify the progenitor cells that contribute to tumor-associated myeloid cell pool for more precise comparison. In the following paragraph, we will therefore highlight recent insight on signatures of transition states based on trajectory analyses. Interesting observations on the cellular differentiation route of TAM-MDMs were made in a recent study in which the authors dissected the myeloid cell pool in different stages of murine and human glioma by single-cell RNA-Seq (scRNA-Seq) approaches in combination with lineage-tracing experiments in mouse models (34). By adoptively transferring classical or nonclassical monocytes from Cx3cr1GFP/+ mice into Ccr2-KO mice harboring orthotopically transplanted gliomas, the authors demonstrated that only classical monocytes were able to differentiate into MDMs within the tumor. If this applies to TAM-MDM in BrM requires further investigation.
Disease-Associated Transcriptional Signatures of TAM-MG in BrM
Microglia under homeostatic conditions represent a heterogeneous cell population depending on their localization within the brain parenchyma. Moreover, MG heterogeneity is modulated by developmental stages with lower heterogeneity found in adults compared with embryonic stages. Given the inherent MG heterogeneity, it is not surprising that brain pathologies induce an even higher degree of heterogeneity (12, 37), which was demonstrated with single cell RNA-seq for TAM-MG (28, 32). For example, by using t-distributed stochastic neighbor embedding (tSNE), a dimensionality reduction method, of RNA-Seq data from single cells, Schulz et al. reported that the majority of TAM-MG from the H2030-BrM model were contributing to three transcriptionally distinct cell cluster in treatment-naïve BrM. The cluster comprising most of the TAM-MG (cluster 9) was represented by high expression of cytokines (Ccl3, Ccl4, Cxcl13), cathepsin Z (Ctsz), the epidermal growth factor receptor 1 (Egfr1), as well as MG typical marker (C1qa, Hexb) (38, 39). The complement member and MG lineage marker C1qa was found to be upregulated in TAM-MG in several studies (Table 2), whereas other members of the MG-specific “sensome” core signature (Cx3cr1, P2ry12, Tmem119) (11, 38, 39) were consequently downregulated in murine and human TAM-MG (Table 2). While an increased expression of C1q members in MG belongs to their activation profile (40, 41), downregulation of homeostatic markers most likely is a consequence concomitantly occurring with downregulation of the homeostatic regulatory gene Sall1/SALL1 in murine and human TAM-MG. This further mirrors a classical activation response of MG associated to any damage of the brain, as observed under neuroinflammatory conditions (42).
Once activated and educated by tumor cells, TAM-MG upregulate several markers known to be crucial for inflammation in the injured brain, thereby probably contributing to BrM progression, e.g., via exerting immune-suppressive functions. Presumably, this is accompanied with profound metabolic changes as seen in an apparent deregulation of members of solute carrier (Slc) genes (32).
Some of the frequently observed markers in MG associated to neurological damage are apolipoprotein E (Apoe/APOE) and the triggered receptor expressed on myeloid cells 2, Trem2/TREM2, which were highly expressed/upregulated in TAM-MG of murine BrM models, whereas the expression of both members only slightly varied in human TAM-MG. The APOE-TREM2 axis has been described to drive activated MG states in neurodegenerative diseases along with downregulation of homeostatic markers (39, 43). However, especially TAM-MDMs showed elevated expression levels of Trem2/TREM2 in preclinical BrM models or patient material of BrM derived from various primary tumors (Table 2). The contributions of TREM2 and APOE to neurological diseases [e.g., Alzheimer’s disease (AD) or multiple sclerosis (MS)] have been extensively described with regard to MG (44, 45). Importantly, it was shown that targeting TREM2+ MG represents an interesting approach to attenuate disease progression. In addition to the broadly studied APOE-TREM2 axis, another key player of MG-mediated neurological dysfunction, and ligand for TREM2, is Galectin-3, encoded by Lgals3/LGALS3. Galectin-3 shows a multitude of functions in MG. Elevated extracellular levels within the BrM-TME might drive inflammatory responses in a Toll-like receptor (TLR) 4 binding-mediated self-sustaining manner (46, 47). In line with this, TLR4 expression was found to be upregulated in TAM-MG of H2030-BrM (32). In a recent study, Siew et al. analyzed the contribution of MG-derived Galectin-3 signaling in a mouse model of Huntington’s disease (48). Elevated cytokine levels were attributed to high Galectin-3 signaling, and strategies targeting its expression have been shown to be sufficient in decreasing levels of inflammatory cytokines, thereby ameliorating MG-mediated pathogenesis (48).
While profiling across different conditions revealed that Lgals3/LGALS3 expression levels are strongly increased in TAM-MG, varying but high levels within TAM-MDMs have been found in murine or human BrM as well (Table 2). Similarly, high expression levels were rather associated to TAM-MDMs of murine (49), or human recurrent glioma (34), as revealed in high-dimensional single-cell profiling studies. Therefore, contribution of elevated Lgals3/LGALS3 levels need to be carefully evaluated in a context-specific manner. Another group of genes (S100 family members) shows an interesting alternating pattern across TAMs in mouse and human BrM. S100 members are small, Ca2+-binding proteins, which regulate several cellular functions in an autocrine, or paracrine fashion, and can act as damage-associated molecular pattern (DAMP) (50). For example, S100β has been shown to serve as a noninvasive, astrocytic marker of blood-brain barrier (BBB) integrity and function, also in brain tumors (51), whereas several S100A members have been implicated in neurodegenerative diseases like AD (52). In addition, distinct S100 proteins have been associated to the regulation of inflammatory responses and TAM migration and invasion in tumors (50). Hence, upregulation or high expression especially in TAM-MG of BrM (e.g., S100A6, S100A10) (Table 2) (28, 31), might reflect a central mediator of BrM-associated inflammation. In contrast, in glioma elevated expression level of S100A6 has been implicated in a transitory TAM-MDM state in mouse and humans (34) or was part of a strong “macrophage signature” (49). Since S100A6, which was implicated in tumor progression in several other cancers (53), can either act on MG in a cell-restricted manner or can be secreted, elevated expression levels of S100A6 and other S100 members need further examination. To date, it remains unclear if the regulation of S100 members represents a cause or consequence of progressing BrM.
Direct comparison of both TAM populations on the single cell (27, 28, 31), and bulk population level (29, 30, 32) revealed further cell type-restricted molecular profiles in mouse models and human patient samples. TAM-MG showed higher upregulation of distinct proinflammatory cytokines (e.g., CXCL5, CXCL8, IL6) (29), or genes belonging to cell migration, e.g., Vim/VIM (28, 31) within a changing environment.
Cathepsins (Cts/CTS) encompass a family of proteases known to play several protumorigeneic roles in the tumor context by, e.g., remodeling the extracellular matrix (ECM) (54). In the brain, cathepsin S has been described as BrM-promoting via enhancing transmigration through the BBB (19). High expression levels of different cathepsins within TAMs of established BrM further suggest profound remodeling of the TME in outgrowing tumors. Among them, Ctsd (28) and CTSL (31) exhibited higher expression levels in TAM-MG, while Ctsb was strongly upregulated in both TAM populations of different murine BrM models (30, 32), but in human BrM was, together with CTSW, rather enriched in TAM-MDM (29). Hence, different highly expressed members of this family of ECM modulators further suggest an involvement for generating a BrM-promoting environment, but specific contributions for/with each TAM population in BrM have not been elucidated yet.
Disease-Associated Transcriptional Signatures of TAM-MDM in BrM
Recruitment of monocytes to the CNS has been predominantly attributed to the chemokine axis including the receptors Ccr2/CCR2 and Cx3cr1/CX3CR1 (17), which was shown in the brain tumor context using lineage-tracing approaches in glioma mouse models (26, 55). Interestingly, while Ccr2/CCR2 is dramatically downregulated upon entry into the parenchyma and during the monocyte-to-macrophage transition in glioma (55), and also BrM (27), Cx3cr1/CX3CR1 levels are upregulated in TAM-MDMs (Table 2), whereas the protein was downregulated on the majority of MDM subsets but remained abundant on only a small subset (27). These data suggest that high levels of Cx3cr1/CX3CR1 are partially required to integrate into the CNS parenchyma, since the axis is usually involved in glia-neuronal crosstalk (56). Comparing TAM data for the expression of another potent chemokine receptor known to be involved in cell migration, it became apparent that especially TAM-MDMs possess high levels or strongly upregulated the C3A receptor, C3ar1/C3AR1 in murine and human BrM (Table 2). This suggests the contribution of a conserved mechanism of recruitment via the anaphylatoxin/chemokine axis.
Given the fact that bulk analysis usually masks different expression/antigen density levels, Friebel et al. comprehensively dissected the heterogeneity of TAMs derived from different human brain malignancies, and showed that TAM instruction is not a random process, but rather driven by the underlying tumor, both in primary and secondary brain malignancies (27). By combining in total 38 markers for their myeloid panel, the authors captured a broad range of cellular states as depicted by distinct lineage-specific, but also activation markers. Upon merging all TAM CyTOF data from both, glioma and BrM samples, the authors created a detailed relationship and trajectory analyses focusing on abundance of typical monocyte/macrophage markers in silico. One of the common features they found was downregulation of CCR2 and the SIGLEC family member, SIGLEC3 (CD33), which represents a transmembrane receptor implicated in pattern recognition and regulation of phagocytosis, and in that regard has been described to be a risk factor for AD (57). Moreover, transitioning cells were found to commonly upregulate the innate immune sensor receptor CD163, together with the TAM receptor MERTK (Table 2). Monocytes transition to macrophages through a more common MDM state (termed MDM 1), and finally develop into three distinct MDM subpopulations (MDM 2, 3, 4), and this transition was driven by differential regulation of certain markers, including CD169, CD206 (mannose receptor c-type I, MRC1), CD209 (C-type lectin receptor, CLEC4L), CD38, and PD-L1. Some of these markers were further used in combination with IBA1 to specifically stain for MDMs in the TAM compartment (27). Since not only CD206 but also CD209 usually are associated to alternative macrophage activation, it is not surprising that those markers have been found to be predominantly upregulated in TAM-MDMs of murine and human BrM across different conditions (Table 2). MERTK upregulation was found to be gradual, and highest antigen density was allocated to the MDM 4 population (27), whereas bulk RNA-Seq of murine and human TAM-MDMs revealed in general elevated expression levels (Table 2). MERTK and AXL, which were also found to be significantly upregulated especially in TAM-MDMs (Table 2), represent two of the three TAM (TYRO3, AXL, MERTK) receptor tyrosine kinases, which are involved in phagocytosis and regulation of inflammatory responses (58). Interestingly, one of the ligands for AXL, Gas6/GAS6, was also highly expressed within both TAM populations in murine BrM, but rather upregulated in TAM-MDMs of human BrM samples (Table 2). Although GAS6-AXL signaling is present in the healthy CNS, and is associated to phagocytosing MG (59), deregulation can cause enhanced inflammation in the CNS (60). Moreover, this signaling axis can lead to a protumorigenic TME (61) and has been found to be coexpressed in TAMs with high C1QC levels in various primary tumors (62).
Despite several differences between the same TAM population within both mouse and human, further commonly regulated markers of TAM-MDMs included Nr4a2/NR4A2. This nuclear receptor family is known to control macrophage gene expression during inflammation (63) but is implicated in maintaining normal functions of dopaminergic neurons in the healthy brain. Interestingly, NR4A2 has been found particularly upregulated in a transitory monocyte population in glioma (34), further suggesting regulatory involvement in inducing inflammatory phenotypes during MDM development.
While certain patterns associated to these inflammatory states of TAMs seem to be conserved between species, other families of proteins are rather restricted to either a species, or a cell type. One interesting group encompasses the membrane-spanning (MS) protein family of MS4A members (Table 2). While for some of the family members (e.g., MS4A1 = CD20) their functions are well described, most of them remain poorly understood. In a recent study, Liu et al. generated new lineage-tracing mouse models targeting Ms4a3 (Ms4a3Cre and Ms4a3CreERT2) and validated lineage specificity of this marker, which specifically distinguishes monocytes and granulocytes from embryonically derived resident macrophage populations, including MG, under steady state but also inflammatory conditions (64). MG possess strong expression levels of certain Ms4a members (Ms4a6b, Ms4a6c, Ms4a6d, Ms4a7) during early development, while these high expression levels are not found in adult MG or in response to injury. Nevertheless, MG of that specific subpopulation of MG identified during early development cluster also showed overlapping features to BAMs (37). Several genes of the MS4A family appeared in all of the BrM-omics studies among the top regulated genes, and some of them have been described as risk factors for AD (65), including Ms4a4c (mouse)/MS4A4E (human), Ms4a6c/MS4A6E, and Ms4a7/MS4A7. Interestingly, all of them were found at high expression levels or strongly upregulated across both TAM populations of murine BrM with slightly higher levels in TAM-MDM (Table 2). Contrary, in human TAMs, MS4A members were found predominantly upregulated or higher expressed per se in TAM-MDMs. Similarly, high and stable expression of Ms4a7 was reported as part of a core signature for BAMs in steady state and under neuroinflammatory conditions (42). Taken together, this data is in line with the situation in glioma TAMs (34) and further corroborate strong similarities in transcriptional profiles between each TAM population derived from distinct diseases. These findings are strengthened by higher intersect levels of each TAM pool between glioma and BrM (29).
Aiming to describe heterogeneity within certain TAM subsets, all of the recent studies looking into the TME by single-cell approaches confirmed that especially the TAM-MDM pool consists of a more diverse range of cellular states (27, 28, 32). Whereas, Guldner et al. even described BAMs to contribute to the TAM-MDM pool within their model. With respect to identity of cell types within cell populations that were FACS purified prior to transcriptomic analyses, it is important to carefully consider procedures of sample preparation and marker selection. Macrodissection of tumor lesions is critical to reduce the risk of diluting the disease-associated cell pool with normal cell types. Moreover, the use of different marker combinations can lead to different assignment of cell types to certain subpopulations. For example, several commonly used markers to discriminate MG and MDM including CD45, SALL1, and TMEM119 (30, 66) are known to show assimilation of expression levels in both populations in brain tumors. Hence, the choice of marker combinations can lead to differences in population assignment. This determines the classification of subpopulations and consequently significantly affects the respective transcriptional programs. Nevertheless, typical non-MG clusters (TAM-MDMs/BAMs) were shown to be dominated by the expression of genes belonging to antigen processing and presentation particularly associated to MHC class II presentation, including H2-Aa, H2-Ab1, H2-Eb, and CD74 (Table 2) in several independent studies (28, 30, 32). Expression of the MHC class II member HLA-DR in human TAMs was similarly higher in TAM-MDMs (Table 2), and upregulated expression can be attributed to the transition from monocytes to MDMs (27), similarly as in the glioma situation (34). Interestingly, this raises the question to which extent each TAM population contributes to T-cell interaction, hence influencing a cancer-promoting, or immunosuppressive TME. Using multiplexed immunostaining, spatial organization of TAMs with respect to T cells was examined in murine (30) and human BrM (29). Both studies found a close proximity of both TAM populations to CD4+ and CD8+ T cells, yet Niesel et al. observed closer proximity of TAM-MDM to T cells compared with TAM-MG based on discrimination by TMEM119 (30). Moreover, it was observed that PD-L1 was almost absent in tumor-free brains, whereas BrM induced the recruitment of PD-L1+ myeloid cells, and levels of PD-L1 were highest among TAM-MDMs (30). On the gene expression level, several costimulatory and also inhibitory markers were found to be present among both TAM populations, whereas most of them were expressed at higher levels in TAM-MDMs (30). Representative comparison of different T-cell regulatory markers across TAMs revealed that both, activating Icosl/ICOSL, and inhibitory Pd-l1/PD-L1 markers are present in TAMs with slight higher levels in the TAM-MDM compartment (Table 2). Given the spatial organization of TAMs within the BrM-TME, one can assume that TAM-MGs in the BrM periphery are in contact with T cells at the tumor-stroma interface, while immunosuppression within the BrM core is fostered by TAM-MDMs once T cells have entered the tumor mass. Furthermore, T-cell profiles confirm exhaustion states within the TME (27, 30). Additional immunoregulatory mediators within BrMs predominantly derived from TAM-MDMs were the chemokines CCL8, CCL13, CCL17, and CCL18 (29). Together, all of them are attributed to an alternative, rather tumor-promoting phenotype (= M2) of macrophages, which is found in many tumors (35). Despite this fact, both TAM-MG and TAM-MDMs upregulate a broad range of inflammatory mediators (29, 32), and hence cannot be classified into the conservative M1–M2 scheme, but rather exist within a continuum, depending on their current local environment.
Finally, in order to understand putative dichotomous functions of TAMs, and their relevance for the inflammatory TME in BrM (Figure 3), detailed annotation and pathway analyses of differently regulated genes were performed based on results obtained from RNA-Seq experiments (28–30, 32).
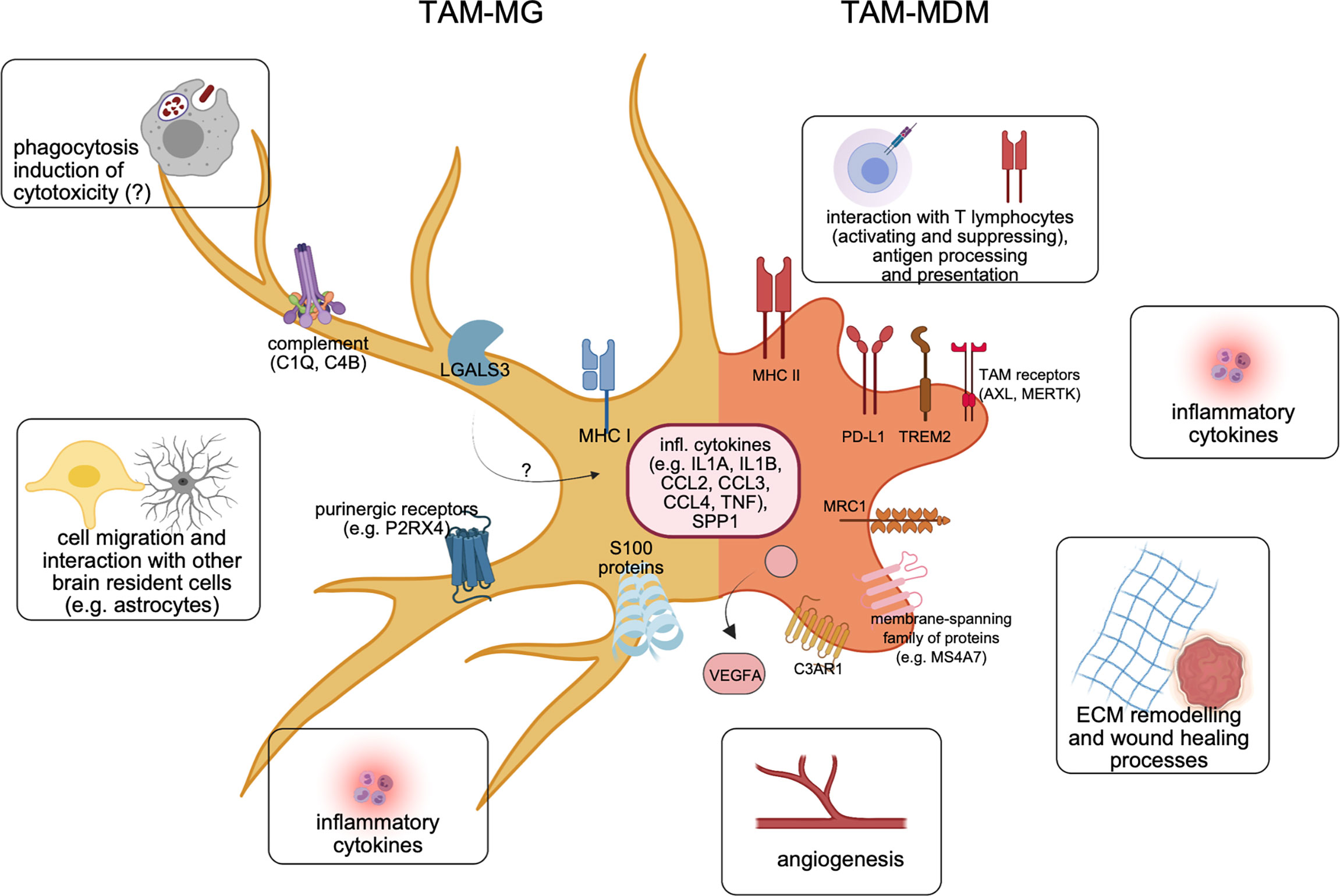
Figure 3 Molecular features of both major TAM populations in BrM. Recent cellular and molecular findings underline the differences of TAM-MG (left) vs. TAM-MDM (right) within the BrM microenvironment. Representative markers shared between TAMs from murine or human BrM are illustrated, together with major functional annotations.
To delineate functional changes upon TAM instruction, comparative analyses of significantly differently regulated genes of TAMs vs. their healthy counterparts (i.e., normal MG, or blood monocytes) in breast cancer BrM (30), and lung cancer BrM (32) were performed. Together, both studies showed that altered transcriptional profiles in TAM-MG resulted in upregulation of pathways and signaling cascades associated to inflammation, regulation of cytokine production, type I interferon (IFN) signaling, cell migration and motility, and proliferation. Interestingly, TAM-MG were found to be involved in the recruitment and interaction with neutrophils, in both mouse models of BrM (30, 32), and also in human BrM (29). Concurrently, TAM-MG downregulates genes involved in housekeeping functions, such as synapse organization or regulation of neuronal organization.
TAMs derived from the periphery are more plastic than MG as described above. Thus, their altered transcriptional profiles need to be evaluated more carefully in a context-dependent manner. Compared with their normal cellular counterparts, TAM-MDMs upregulated pathways involved in inflammatory responses, cytokine production and interaction, migration, mitosis and cell cycle, and also organization of the ECM (30, 32). In addition, human BrM TAM-MDMs showed slightly higher relevance of genes related to mitosis and cell division, compared with TAM-MGs; however, staining of human BrM samples for proliferation markers indicated proliferation in both cell types (29). However, if this results from an environment promoting local proliferation, or is caused by other stimuli, e.g., prior treatment remains to be elucidated. Analyses of patient samples stratified based on treatment history will be required to gain further insight whether different intervention strategies modulate recruitment and expansion of TAM-MDMs as previously shown in a lung-to-brain model in response to radiotherapy (32). Both human TAM populations showed an enrichment for genes related to type I IFN, and NF-kB signaling (29). Not surprisingly upon entering the BrM-TME, and transitioning from monocytes, TAM-MDMs downregulated genes related to chromatin organization, and intracellular reorganization of, e.g., the cytoskeleton (30).
Transcriptional Programs Shared in TAM-MG and TAM-MDM
In addition to lineage-restricted transcriptional programs in TAM populations, it became clear that both TAM populations also share a significant proportion of similarly regulated markers by Top marker principal component, and overlapping gene analysis in human TAMs (29), or by comparison of all differently regulated genes (DEGs) (32).
In H2030-BrM, approximately 300 DEGs were found to be commonly upregulated, and around 900 DEGs commonly downregulated between both TAM populations (32). Functional annotation of all jointly regulated markers revealed the induction of inflammatory pathways, as well as regulation of cell adhesion and cell migration. Downregulated DEGs were mostly associated to homeostatic functions in the brain, e.g., synapse organization. In contrast to the results obtained from experimental BrM models, the number of shared genes between TAM-MG and TAM-MDM was rather small in the human situation (29). The difference can potentially be explained by the fact that human RNA-seq data in this study were not stratified based on primary tumor type thereby possibly missing important similarly regulated genes. Comparison of typical inflammatory cytokines (e.g., CCL2, CCL3, CCL4, IL1B) across mice and human BrM-TAMs revealed that although most of them exhibited much higher expression levels in the BrM situation in mice, it rather were TAM-MDMs upregulating them in the human scenario (Table 2). RNA velocity analysis of single-cell RNA-Seq data of mouse TAMs further showed that Il1b and also Tgfb were genes similarly regulated at convergence points between TAM-MG and TAM-MDM clusters (28).
Osteopontin which is encoded by Spp1/SPP1 is a marker usually associated to MG of early development or has been described as one key marker of all disease-associated MG (DAM) subcluster (37). In BrM, Spp1/SPP1 was found highly expressed or upregulated in both TAM populations across species (Table 2), however with slightly higher expression levels in TAM-MG, similar as in primary brain tumors (34). Although a broad range of cellular functions has been assigned to osteopontin, under inflammatory conditions it most likely regulates inflammation itself via enhancing the recruitment of not only myeloid but also lymphoid cells (67) to the TME. Aiming to reduce or dampen inflammation within the BrM TME, osteopontin hence might represent an interesting target.
In addition to the high expression levels of the complement cascade-initiating member C1q/C1Q within the BrM-TME, two other inflammatory factors, Il1a/IL1A and Tnf/TNF, showed broad abundance and were highly expressed or upregulated in TAMs of murine or human BrMs (Table 2). Together, the presence of all three molecules strongly raises the possibility of TAM-mediated astrocyte activation towards a neurotoxic phenotype (termed A1), which partially would be sustained due to constant factor availability. This mechanism of astrocyte activation has been described in mouse models of neuroinflammatory conditions, and A1 astrocytes were found in samples of various human diseases. A characteristic marker for A1 astrocytes is the central complement component C3 (68). Interestingly, next to C1q/C1Q, other members of the complement system were apparently deregulated in BrM (32), and cross-conditional comparison revealed upregulation of, for example, C4b/C4B among all TAMs in mice and human (Table 2). While oligodendrocyte-derived C4b in mice has been associated to pathogenicity in an AD model (69), its functions within the TAM pool of BrM remain to be shown.
In summary, recent discoveries and previously unknown molecular insights into macrophages/microglia associated to BrM have dramatically shifted the paradigm of BrM-TAMs representing one homogeneous population. In comparison with data derived from recent “omics” studies involving glioma, it became clear that TAMs in brain malignancies constitute a heterogeneous mixture of resident and recruited mononuclear phagocytes, with multifaceted activation states (Figure 3). Moreover, each major subpopulation contributes to the inflammatory TME in a unique way, and disease-specific manner. The discovery of molecular markers present in both TAM populations or conserved between species opens novel avenues to develop targeted approaches in order to fight this deadly disease.
Translational Aspects
TAM-targeted therapies have attracted attention as promising therapeutic strategies for a variety of different primary cancers (Table 3). Besides their high abundance, TAMs have been shown to critically influence tumor biology, often in a protumorigenic fashion by exerting immune-suppressive functions, and at the same time interacting with tumor cells to reciprocally support each other (35). However, in the brain, targeted approaches have to be carefully designed, in order to address modulation within specific TAM populations, without affecting resident macrophage populations (i.e., adjacent MGs) to prevent side effects.
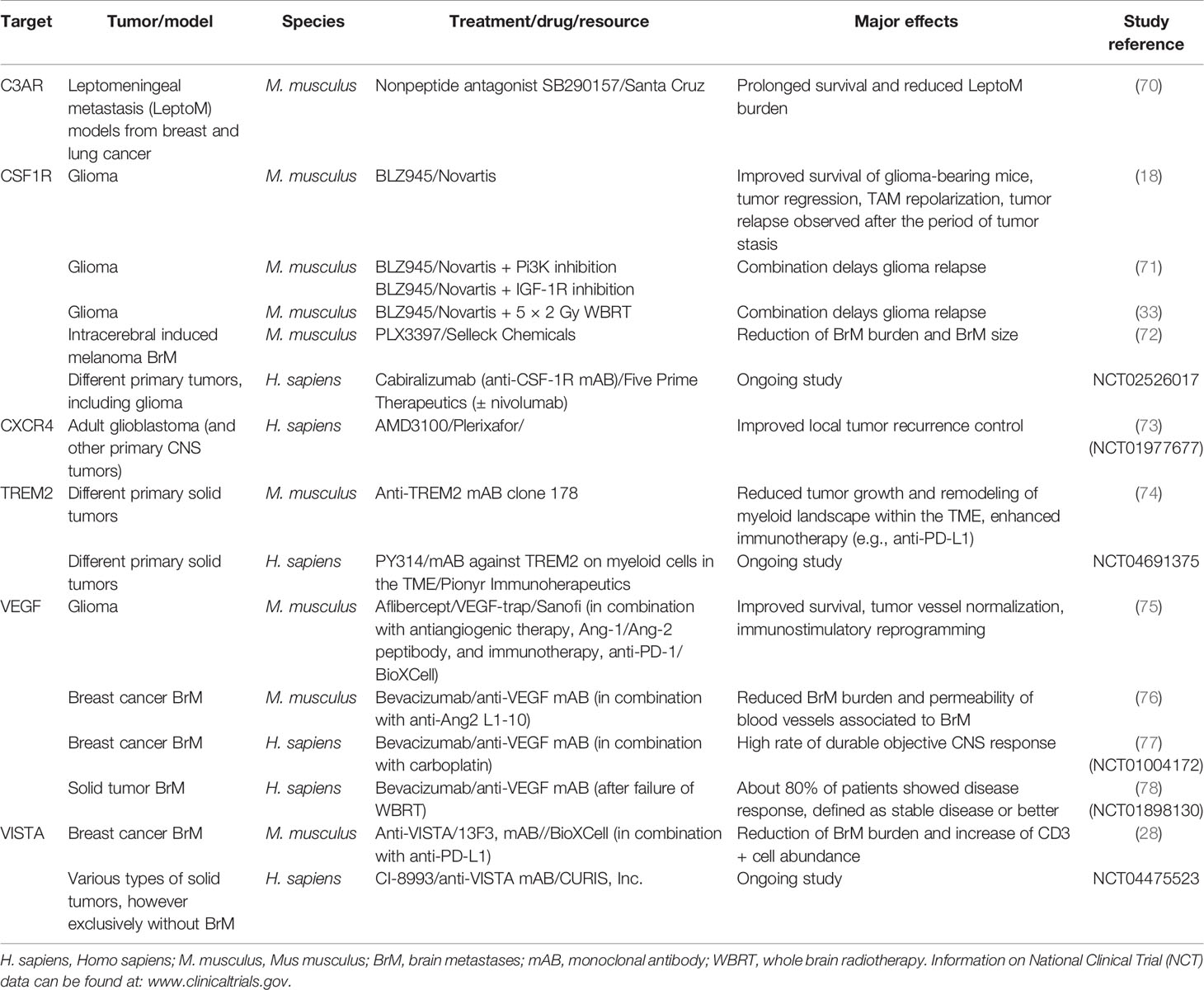
Table 3 Examples of preclinical and clinical studies targeting certain TAM-related receptors/factors as mono- or combination therapies in various types of extra- and intracranial tumors.
Given the central role of CSF1-CSF1R signaling for survival and proliferation of macrophages, it is not surprising that specifically this axis has been targeted by antibodies, or small inhibitory molecules in order to reduce macrophage infiltration or deplete resident, tumor-promoting populations. Aiming to suppress tumor-promoting TAM functions, Pyonteck et al. utilized a CSF1R inhibitor in preclinical glioma models (18). Interestingly, CSF1R inhibition as monotherapy resulted in improved survival and even tumor regression, accompanied by re-education of TAMs into a rather antitumor phenotype. Long-term treatment however resulted in acquired resistance driven by IGF signaling between TAMs and tumor cells, which resulted in prolonged glioma cell survival and invasive capacities (71). Improved efficacy however can be obtained by combining CSF1R inhibition with radiotherapy in glioma (33). CSF1R inhibition was shown to reduce to breast cancer cell invasion (79) and lead to antitumor efficacy in melanoma-BrM and intracerebrally inoculated breast cancer cells (72, 80). However, therapeutic efficacy of CSF1R inhibition still needs to be carefully evaluated with regard to long-term efficacy and potential resistance mechanisms as observed in glioma.
Another strategy of limiting TAM functions within the TME includes blockade of their recruitment, via interfering with chemokines (81) or chemokine receptors, e.g., CCR2 or CXCR4 to inhibit general TAM recruitment (73) or by targeting newly identified markers that are implicated in the recruitment of TAM subpopulations such as CD49d (33). Given the high abundance of the anaphylatoxin receptor C3ar1/C3AR1 predominantly on TAM-MDMs of murine and human origin, blockade of this axis could also be used to inhibit monocyte/macrophage recruitment to the brain. Antibody- or small molecule-mediated inhibition of the C3-C3AR1 axis could have strong inhibitory effects and furthermore might impact the permeability of the BBB at sites of malignant inflammation. In leptomeningeal metastasis, this axis has been shown particularly important for enhancing the permeability at the choroid plexus epithelium, in order to trophically support metastasized cancer cells within the CSF (70). By interfering with the C3-C3AR1 axis, one might even trigger another antitumoral response due to blockade of the interaction of astrocytes and MG as shown by Litvinchuk et al. in a mouse model of neurodegeneration. While astrocyte-derived C3 via C3AR1 on MG induces proinflammatory programs via STAT3 signaling (82), activation of astrocytes could in part be mediated via C1Q plus two other cytokines, IL1A and TNF (68), which seem to be broadly present within the BrM-TME. Since astrocytes have been shown to exert multiple BrM-promoting functions (83–86) and elimination of the three aforementioned factors was beneficial in an ALS mouse model by attenuating gliosis (87), targeting one or several steps within the complement cascade seems promising and has the potential to reverse or block the immunosuppressive, cancer-permissive TME in BrM.
Although some of the markers in TAMs seem to be regulated in a more conditional manner (e.g., by different model, primary tumor entity, genetic background, species), a distinct set of genes was conservatively regulated, and similarly across TAM populations and species (e.g., MS4A proteins, TAM receptors AXL and MERTK, TREM2). While little is known about the specific functions of individual MS4A members, they might play a central role in regulating cellular functions, including cell growth, survival, and activation by serving as family of ion channels and/or adaptor proteins facilitating intracellular protein-protein interaction (41). For example, MS4A4A has been described as a key marker of BAMs (14, 15, 42) and was described as a novel M2-like marker of metastasis-associated macrophages (88). Hence future studies need to address the consequences on the inflammatory state upon interfering with, e.g., MS4A7, which was highly upregulated on both, murine and human TAM-MDMs in BrM. Given their surface exposure, MS4A members are potentially druggable by, e.g., antibodies (41).
Interestingly, within the study form Mattiola et al., the authors showed that MS4A4A interacts with a ß-glucan receptor dectin-1 in lipid rafts of the cell membrane. The dectin-1 pathway transmits intracellular signals similar to those of TREM2. Hence, TREM2 deficiency can be compensated by enhanced dectin-1 signaling (89). TREM2 signaling is essential for MG function and disease-associated phenotypes in MG can be induced by the APOE-TREM2 pathway (43). In AD, TREM2-deficient MG undergoes autophagy due to impaired mTOR signaling and metabolism (89). Since TREM2 seems to be dramatically upregulated in all TAMs across different conditions, antibody-mediated blockade of this receptor signaling pathway represents a promising approach. Within a recent study, the authors examined TREM2 function in TAMs of distinct tumor models (74). Interestingly, they found that both, mice deficient for TREM2, or antibody-mediated blockade of TREM2 signaling, resulted in delayed tumor growth. Simultaneously, the immune landscape within their model was altered including an increase of intratumoral CD8+ T cells, which consequently led to enhanced efficacy of anti-PD1 immunotherapy. More importantly, the authors showed ubiquitous abundance of TREM2+ macrophages across distinct human tumor samples, and that TREM2 expression inversely correlated with greater overall, and relapse-free survival in colorectal carcinoma and triple negative breast cancer patients (74). Given the negative correlation between MDMs and T-cell frequencies in BrM (29), it is tempting to speculate that high expression of TREM2 in TAM-MDM as seen under various BrM conditions might be an interesting candidate for combination therapies. If this furthermore leads to remodeling of the TME with enhanced T cell recruitment, also in BrM, needs to be evaluated. In summary, targeting TREM2 in combination with immunotherapy (e.g., anti-PD-L1), or radiotherapy, might represent an attractive strategy to overcome immunosuppression. It was previously shown that radiotherapy has the potential to transiently lift immunosuppressive features of the TAM pool by enhancing the recruitment of naïve monocytes/MDMs to BrM in a lung-to-brain metastasis model (32). However, acquired resistance to combined radioimmunotherapy was partially mediated by PD-L1 expression from infiltrated myeloid cells (30), which rapidly undergo tumor education. In addition to targeting the PD1-PD-L1 axis in order to enhance antitumor responses, targeting different TAM populations showed promising results in glioma and BrM models. For example, Guldner et al. inhibited the negative immune checkpoint VISTA (encoded by Vsir) on TAMs which similar as targeting TREM2 enhanced the CD3+ cell abundance within BrM leading to improved efficacy of anti-PD-L1 (28). Given the high abundance and strong expression of Vegfa/VEGFA in TAMs, interference with VEGF signaling could furthermore lead to enhanced antitumor responses, as shown in a triple treatment approach of murine glioma (75). The authors blocked the angiogenic factors VEGF and ANG-2 in combination with PD-1, which resulted in extended survival of mice compared with anti-VEGF as monotherapy (75). However, targeting VEGF in BrM is not indicated for every primary tumor type which gives rise to BrM. While double inhibition of VEGF and ANG-2 reduced BrM burden in preclinical models of breast-to-brain metastasis (76), VEGFA inhibition can induce long-term dormancy in lung-to-brain metastasis (90). For breast-to-brain metastasis patients, combination of VEGFA inhibition with bevacizumab in combination with carboplatin resulted in a high rate of durable responses (77). Moreover, VEGFA inhibition resulted in a 25% disease response rate in 80% of solid cancer patients with current brain metastasis that failed whole-brain radiotherapy (78).
In summary, novel targeted therapeutic approaches need to be carefully evaluated in a context-specific manner upon spatiotemporal determination of leukocytic subsets within the TME. This is particularly important for targeting specific phenotypic features of TAMs, but at the same time spare homeostatic features of adjacent, non-BrM-associated populations. Furthermore, it will be critical to evaluate to which extent altered gene expression also translates into altered protein abundance, which in addition requires evaluation on a spatial level. In combination with standard therapy, targeting distinct TAM subsets represents a promising strategy. Combination therapies are expected to induce synergy by on the one hand repressing tumor-promoting traits, and on the other hand lifting immunosuppression, thereby enhancing antitumor immunity.
Concluding Remarks
Driven by technical advances, as well as scientific and clinical interest in understanding cellular and molecular landscapes in health and disease, recent research has resulted in tremendous insight into the heterogeneity of TAM of primary and secondary BrMs.
RNA sequencing, multiplexed flow, and mass cytometry revealed the dichotomous nature of TAMs in BrM, wherein resident microglia as well as recruited monocyte-derived macrophages represent the two major populations and contribute significantly to the entire immune cell landscape. Although both TAMs quantitatively differentially contribute to the local TAM pool and populate different niches within the TME, their phenotypic changes occur early upon disease-specific instruction in a highly plastic manner. Together, both TAM populations contribute to the establishment of an immunosuppressive and tumor-promoting environment in BrM. In order to evaluate the applicability of novel targeted approaches, further research needs to determine molecular pattern in spatiotemporal resolution. Detailed mechanistic understanding how standard therapy can be used as an immune modulator in addition to the identification of transcriptional programs that drive disease-promoting states in TAMs to provide scientific rationale for the development of improved therapeutic avenues against BrM is needed.
Author Contributions
MS and LS conceptualized and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Research in the lab of LS is supported by institutional funds from the Georg-Speyer-Haus jointly funded by the German Federal Ministry of Health and the Ministry of Higher Education, Research and the Arts of the State of Hesse (HMWK), as well as grants from the German Cancer Aid (Max-Eder Junior Group Leader Program 70111752) and German Research Foundation (SE2234/3-1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all members from the Sevenich Lab and from the Georg-Speyer-Haus for insightful feedback on our work. Furthermore, we are thankful to all authors of the main recent studies involved in providing material and information regarding novel insights into the TME of BrM, in particular supplementary files from Guldner et al. and Klemm et al. Data from Klemm et al. were accessed via supplementary files and their RNA-Seq database at https://joycelab.shinyapps.io/braintime/. Figures were created with Biorender (www.biorender.com).
References
1. Wynn TA, Chawla A, Pollard JW. Macrophage Biology in Development, Homeostasis and Disease. Nature (2013) 496(7446):445–55. doi: 10.1038/nature12034
2. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages Under Homeostasis. Immunity (2013) 38(1):79–91. doi: 10.1016/j.immuni.2012.12.001
3. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-Expression Profiles and Transcriptional Regulatory Pathways That Underlie the Identity and Diversity of Mouse Tissue Macrophages. Nat Immunol (2012) 13(11):1118–28. doi: 10.1038/ni.2419
4. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment Drives Selection and Function of Enhancers Controlling Tissue-Specific Macrophage Identities. Cell (2014) 159(6):1327–40. doi: 10.1016/j.cell.2014.11.023
5. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell (2014) 159(6):1312–26. doi: 10.1016/j.cell.2014.11.018
6. Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage Receptors and Immune Recognition. Annu Rev Immunol (2005) 23:901–44. doi: 10.1146/annurev.immunol.23.021704.115816
7. Alliot F, Godin I, Pessac B. Microglia Derive From Progenitors, Originating From the Yolk Sac, and Which Proliferate in the Brain. Brain Res Dev Brain Res (1999) 117(2):145–52. doi: 10.1016/s0165-3806(99)00113-3
8. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate Mapping Analysis Reveals That Adult Microglia Derive From Primitive Macrophages. Science (2010) 330(6005):841–5. doi: 10.1126/science.1194637
9. Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, et al. High-Dimensional, Single-Cell Characterization of the Brain’s Immune Compartment. Nat Neurosci (2017) 20(9):1300–9. doi: 10.1038/nn.4610
10. Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity (2018) 48(2):380–395 e6. doi: 10.1016/j.immuni.2018.01.011
11. Geirsdottir L, David E, Keren-Shaul H, Weiner A, Bohlen SC, Neuber J, et al. Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell (2019) 179(7):1609–22.e16. doi: 10.1016/j.cell.2019.11.010
12. Masuda T, Sankowski R, Staszewski O, Bottcher C, Amann L, Sagar C, et al. Spatial and Temporal Heterogeneity of Mouse and Human Microglia at Single-Cell Resolution. Nature (2019) 566(7744):388–92. doi: 10.1038/s41586-019-0924-x
13. Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, et al. Brain Structure. Cell Types in the Mouse Cortex and Hippocampus Revealed by Single-Cell RNA-Seq. Science (2015) 347(6226):1138–42. doi: 10.1126/science.aaa1934
14. Utz SG, See P, Mildenberger W, Thion MS, Silvin A, Lutz M, et al. Early Fate Defines Microglia and Non-Parenchymal Brain Macrophage Development. Cell (2020) 181(3):557–73.e18. doi: 10.1016/j.cell.2020.03.021
15. Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, et al. A Single-Cell Atlas of Mouse Brain Macrophages Reveals Unique Transcriptional Identities Shaped by Ontogeny and Tissue Environment. Nat Neurosci (2019) 22(6):1021–35. doi: 10.1038/s41593-019-0393-4
16. Kierdorf K, Masuda T, Jordao MJC, Prinz M. Macrophages at CNS Interfaces: Ontogeny and Function in Health and Disease. Nat Rev Neurosci (2019) 20(9):547–62. doi: 10.1038/s41583-019-0201-x
17. Prinz M, Priller J. Tickets to the Brain: Role of CCR2 and CX3CR1 in Myeloid Cell Entry in the CNS. J Neuroimmunol (2010) 224(1-2):80–4. doi: 10.1016/j.jneuroim.2010.05.015
18. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat Med (2013) 19(10):1264–72. doi: 10.1038/nm.3337
19. Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, et al. Analysis of Tumour- and Stroma-Supplied Proteolytic Networks Reveals a Brain-Metastasis-Promoting Role for Cathepsin s. Nat Cell Biol (2014) 16(9):876–88. doi: 10.1038/ncb3011
20. Gavrilovic IT, Posner JB. Brain Metastases: Epidemiology and Pathophysiology. J Neurooncol (2005) 75(1):5–14. doi: 10.1007/s11060-004-8093-6
21. Nayak L, Lee EQ, Wen PY. Epidemiology of Brain Metastases. Curr Oncol Rep (2012) 14(1):48–54. doi: 10.1007/s11912-011-0203-y
22. Suh JH, Kotecha R, Chao ST, Ahluwalia MS, Sahgal A, Chang EL. Current Approaches to the Management of Brain Metastases. Nat Rev Clin Oncol (2020) 17(5):279–99. doi: 10.1038/s41571-019-0320-3
23. Valiente M, Ahluwalia MS, Boire A, Brastianos PK, Goldberg SB, Lee EQ, et al. The Evolving Landscape of Brain Metastasis. Trends Cancer (2018) 4(3):176–96. doi: 10.1016/j.trecan.2018.01.003
24. Gutmann DH, Kettenmann H. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron (2019) 104(3):442–9. doi: 10.1016/j.neuron.2019.08.028
25. Sevenich L. Brain-Resident Microglia and Blood-Borne Macrophages Orchestrate Central Nervous System Inflammation in Neurodegenerative Disorders and Brain Cancer. Front Immunol (2018) 9:697. doi: 10.3389/fimmu.2018.00697
26. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep (2016) 17(9):2445–59. doi: 10.1016/j.celrep.2016.10.052
27. Friebel E, Kapolou K, Unger S, Nunez NG, Utz S, Rushing EJ, et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell (2020) 181(7):1626–1642 e20. doi: 10.1016/j.cell.2020.04.055
28. Guldner IH, Wang Q, Yang L, Golomb SM, Zhao Z, Lopez JA, et al. CNS-Native Myeloid Cells Drive Immune Suppression in the Brain Metastatic Niche Through Cxcl10. Cell (2020) 183(5):1234–48.e25. doi: 10.1016/j.cell.2020.09.064
29. Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell (2020) 181(7):1643–1660 e17. doi: 10.1016/j.cell.2020.05.007
30. Niesel K, Schulz M, Anthes J, Alekseeva T, Macas J, Salamero-Boix A, et al. The Immune Suppressive Microenvironment Affects Efficacy of Radio-Immunotherapy in Brain Metastasis. EMBO Mol Med (2021) 13(5):e13412. doi: 10.15252/emmm.202013412
31. Rubio-Perez C, Planas-Rigol E, Trincado JL, Bonfill-Teixidor E, Arias A, Marchese D, et al. Immune Cell Profiling of the Cerebrospinal Fluid Enables the Characterization of the Brain Metastasis Microenvironment. Nat Commun (2021) 12(1):1503. doi: 10.1038/s41467-021-21789-x
32. Schulz M, Michels B, Niesel K, Stein S, Farin H, Rodel F, et al. Cellular and Molecular Changes of Brain Metastases-Associated Myeloid Cells During Disease Progression and Therapeutic Response. iScience (2020) 23(6):101178. doi: 10.1016/j.isci.2020.101178
33. Akkari L, Bowman RL, Tessier J, Klemm F, Handgraaf SM, de Groot M, et al. Dynamic Changes in Glioma Macrophage Populations After Radiotherapy Reveal CSF-1R Inhibition as a Strategy to Overcome Resistance. Sci Transl Med (2020) 12(552). doi: 10.1126/scitranslmed.aaw7843
34. Pombo Antunes AR, Scheyltjens I, Lodi F, Messiaen J, Antoranz A, Duerinck J, et al. Single-Cell Profiling of Myeloid Cells in Glioblastoma Across Species and Disease Stage Reveals Macrophage Competition and Specialization. Nat Neurosci (2021) 24(4):595–610. doi: 10.1038/s41593-020-00789-y
35. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends Immunol (2002) 23(11):549–55. doi: 10.1016/s1471-4906(02)02302-5
36. Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity (2014) 41(1):49–61. doi: 10.1016/j.immuni.2014.06.010
37. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-Cell RNA Sequencing of Microglia Throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity (2019) 50(1):253–271 e6. doi: 10.1016/j.immuni.2018.11.004
38. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a Unique TGF-Beta-Dependent Molecular and Functional Signature in Microglia. Nat Neurosci (2014) 17(1):131–43. doi: 10.1038/nn.3599
39. Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, et al. The Microglial Sensome Revealed by Direct RNA Sequencing. Nat Neurosci (2013) 16(12):1896–905. doi: 10.1038/nn.3554
40. Farber K, Cheung G, Mitchell D, Wallis R, Weihe E, Schwaeble W, et al. C1q, the Recognition Subcomponent of the Classical Pathway of Complement, Drives Microglial Activation. J Neurosci Res (2009) 87(3):644–52. doi: 10.1002/jnr.21875
41. Eon Kuek L, Leffler M, Mackay GA, Hulett MD. The MS4A Family: Counting Past 1, 2 and 3. Immunol Cell Biol (2016) 94(1):11–23. doi: 10.1038/icb.2015.48
42. Jordao MJC, Sankowski R, Brendecke SM, Sagar G, Tai YH, Tay TL, et al. Prinz: Single-Cell Profiling Identifies Myeloid Cell Subsets With Distinct Fates During Neuroinflammation. Science (2019) 363(6425). doi: 10.1126/science.aat7554
43. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity (2017) 47(3):566–81.e9. doi: 10.1016/j.immuni.2017.08.008
44. Dong Y, D’Mello C, Pinsky W, Lozinski BM, Kaushik DK, Ghorbani S, et al. Oxidized Phosphatidylcholines Found in Multiple Sclerosis Lesions Mediate Neurodegeneration and Are Neutralized by Microglia. Nat Neurosci (2021) 24(4):489–503. doi: 10.1038/s41593-021-00801-z
45. Yeh FL, Hansen DV, Sheng M. TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol Med (2017) 23(6):512–33. doi: 10.1016/j.molmed.2017.03.008
46. Burguillos MA, Svensson M, Schulte T, Boza-Serrano A, Garcia-Quintanilla A, Kavanagh E, et al. Microglia-Secreted Galectin-3 Acts as a Toll-Like Receptor 4 Ligand and Contributes to Microglial Activation. Cell Rep (2015) 10(9):1626–38. doi: 10.1016/j.celrep.2015.02.012
47. Tan Y, Zheng Y, Xu D, Sun Z, Yang H, Yin Q. Galectin-3: A Key Player in Microglia-Mediated Neuroinflammation and Alzheimer’s Disease. Cell Biosci (2021) 11(1):78. doi: 10.1186/s13578-021-00592-7
48. Siew JJ, Chen HM, Chen HY, Chen HL, Chen CM, Soong BW, et al. Galectin-3 Is Required for the Microglia-Mediated Brain Inflammation in a Model of Huntington’s Disease. Nat Commun (2019) 10(1):3473. doi: 10.1038/s41467-019-11441-0
49. Ochocka N, Segit P, Walentynowicz KA, Wojnicki K, Cyranowski S, Swatler J, et al. Single-Cell RNA Sequencing Reveals Functional Heterogeneity of Glioma-Associated Brain Macrophages. Nat Commun (2021) 12(1):1151. doi: 10.1038/s41467-021-21407-w
50. Xia C, Braunstein Z, Toomey AC, Zhong J, Rao X. S100 Proteins As an Important Regulator of Macrophage Inflammation. Front Immunol (1908) 82017):1908. doi: 10.3389/fimmu.2017.01908
51. Kanner AA, Marchi N, Fazio V, Mayberg MR, Koltz MT, Siomin V, et al. Serum S100beta: A Noninvasive Marker of Blood-Brain Barrier Function and Brain Lesions. Cancer (2003) 97(11):2806–13. doi: 10.1002/cncr.11409
52. Cristovao JS, Gomes CM. S100 Proteins in Alzheimer’s Disease. Front Neurosci (2019) 13:463. doi: 10.3389/fnins.2019.00463
53. Donato R, Sorci G, Giambanco I. S100A6 Protein: Functional Roles. Cell Mol Life Sci (2017) 74(15):2749–60. doi: 10.1007/s00018-017-2526-9
54. Olson OC, Joyce JA. Cysteine Cathepsin Proteases: Regulators of Cancer Progression and Therapeutic Response. Nat Rev Cancer (2015) 15(12):712–29. doi: 10.1038/nrc4027
55. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res (2017) 77(9):2266–78. doi: 10.1158/0008-5472.CAN-16-2310
56. Luo P, Chu SF, Zhang Z, Xia CY, Chen NH. Fractalkine/CX3CR1 Is Involved in the Cross-Talk Between Neuron and Glia in Neurological Diseases. Brain Res Bull (2019) 146:12–21. doi: 10.1016/j.brainresbull.2018.11.017
57. Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer’s Disease Risk Gene CD33 Inhibits Microglial Uptake of Amyloid Beta. Neuron (2013) 78(4):631–43. doi: 10.1016/j.neuron.2013.04.014
58. Lemke G. Biology of the TAM Receptors. Cold Spring Harb Perspect Biol (2013) 5(11):a009076. doi: 10.1101/cshperspect.a009076
59. Fourgeaud L, Traves PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, et al. TAM Receptors Regulate Multiple Features of Microglial Physiology. Nature (2016) 532(7598):240–4. doi: 10.1038/nature17630
60. Weinger JG, Brosnan CF, Loudig O, Goldberg MF, Macian F, Arnett HA, et al. Loss of the Receptor Tyrosine Kinase Axl Leads to Enhanced Inflammation in the CNS and Delayed Removal of Myelin Debris During Experimental Autoimmune Encephalomyelitis. J Neuroinflamm (2011) 8:49. doi: 10.1186/1742-2094-8-49
61. Tanaka M, Siemann DW. Gas6/Axl Signaling Pathway in the Tumor Immune Microenvironment. Cancers (Basel) (2020) 12(7). doi: 10.3390/cancers12071850
62. Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, et al. A Pan-Cancer Single-Cell Transcriptional Atlas of Tumor Infiltrating Myeloid Cells. Cell (2021) 184(3):792–809 e23. doi: 10.1016/j.cell.2021.01.010
63. Pei L, Castrillo A, Tontonoz P. Regulation of Macrophage Inflammatory Gene Expression by the Orphan Nuclear Receptor Nur77. Mol Endocrinol (2006) 20(4):786–94. doi: 10.1210/me.2005-0331
64. Liu Z, Gu Y, Chakarov S, Bleriot C, Kwok I, Chen X, et al. Fate Mapping via Ms4a3-Expression History Traces Monocyte-Derived Cells. Cell (2019) 178(6):1509–25.e19. doi: 10.1016/j.cell.2019.08.009
65. Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common Variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP Are Associated With Alzheimer’s Disease. Nat Genet (2011) 43(5):429–35. doi: 10.1038/ng.803
66. Sedgwick JD, Ford AL, Foulcher E, Airriess R. Central Nervous System Microglial Cell Activation and Proliferation Follows Direct Interaction With Tissue-Infiltrating T Cell Blasts. J Immunol (1998) 160(11):5320–30.
67. Icer MA, Gezmen-Karadag M. The Multiple Functions and Mechanisms of Osteopontin. Clin Biochem (2018) 59:17–24. doi: 10.1016/j.clinbiochem.2018.07.003
68. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature (2017) 541(7638):481–7. doi: 10.1038/nature21029
69. Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, et al. Human and Mouse Single-Nucleus Transcriptomics Reveal TREM2-Dependent and TREM2-Independent Cellular Responses in Alzheimer’s Disease. Nat Med (2020) 26(1):131–42. doi: 10.1038/s41591-019-0695-9
70. Boire A, Zou Y, Shieh J, Macalinao DG, Pentsova E, Massague J. Complement Component 3 Adapts the Cerebrospinal Fluid for Leptomeningeal Metastasis. Cell (2017) 168(6):1101–13.e13. doi: 10.1016/j.cell.2017.02.025
71. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The Tumor Microenvironment Underlies Acquired Resistance to CSF-1R Inhibition in Gliomas. Science (2016) 352(6288):aad3018. doi: 10.1126/science.aad3018
72. Qiao S, Qian Y, Xu G, Luo Q, Zhang Z. Long-Term Characterization of Activated Microglia/Macrophages Facilitating the Development of Experimental Brain Metastasis Through Intravital Microscopic Imaging. J Neuroinflamm (2019) 16(1):4. doi: 10.1186/s12974-018-1389-9
73. Thomas RP, Nagpal S, Iv M, Soltys SG, Bertrand S, Pelpola JS, et al. Macrophage Exclusion After Radiation Therapy (MERT): A First in Human Phase I/II Trial Using a CXCR4 Inhibitor in Glioblastoma. Clin Cancer Res (2019) 25(23):6948–57. doi: 10.1158/1078-0432.CCR-19-1421
74. Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, et al. TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell (2020) 182(4):886–900.e17. doi: 10.1016/j.cell.2020.07.013
75. Di Tacchio M, Macas J, Weissenberger J, Sommer K, Bahr O, Steinbach JP, et al. Tumor Vessel Normalization, Immunostimulatory Reprogramming, and Improved Survival in Glioblastoma With Combined Inhibition of PD-1, Angiopoietin-2, and VEGF. Cancer Immunol Res (2019) 7(12):1910–27. doi: 10.1158/2326-6066.CIR-18-0865
76. Bohn KA, Adkins CE, Nounou MI, Lockman PR. Inhibition of VEGF and Angiopoietin-2 to Reduce Brain Metastases of Breast Cancer Burden. Front Pharmacol (2017) 8:193. doi: 10.3389/fphar.2017.00193
77. Leone JP, Emblem KE, Weitz M, Gelman RS, Schneider BP, Freedman RA, et al. Phase II Trial of Carboplatin and Bevacizumab in Patients With Breast Cancer Brain Metastases. Breast Cancer Res (2020) 22(1):131. doi: 10.1186/s13058-020-01372-w
78. Kumthekar P, Dixit K, Grimm SA, Lukas RV, Schwartz MA, Rademaker A, et al. A Phase II Trial of Bevacizumab in Patients With Recurrent Solid Tumor Brain Metastases Who Have Failed Whole Brain Radiation Therapy (WBRT). J Clin Oncol (2019) 37(15_suppl):2070–0. doi: 10.1200/JCO.2019.37.15_suppl.2070
79. Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, Condeelis JS. Invasion of Human Breast Cancer Cells In Vivo Requires Both Paracrine and Autocrine Loops Involving the Colony-Stimulating Factor-1 Receptor. Cancer Res (2009) 69(24):9498–506. doi: 10.1158/0008-5472.CAN-09-1868
80. Andreou KE, Soto MS, Allen D, Economopoulos V, de Bernardi A, Larkin JR, et al. Anti-Inflammatory Microglia/Macrophages As a Potential Therapeutic Target in Brain Metastasis. Front Oncol (2017) 7:251. doi: 10.3389/fonc.2017.00251
81. Liu SC, Alomran R, Chernikova SB, Lartey F, Stafford J, Jang T, et al. Blockade of SDF-1 After Irradiation Inhibits Tumor Recurrences of Autochthonous Brain Tumors in Rats. Neuro Oncol (2014) 16(1):21–8. doi: 10.1093/neuonc/not149
82. Litvinchuk A, Wan YW, Swartzlander DB, Chen F, Cole A, Propson NE, et al. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron (2018) 100(6):1337–53.e5. doi: 10.1016/j.neuron.2018.10.031
83. Schulz M, Salamero-Boix A, Niesel K, Alekseeva T, Sevenich L. Microenvironmental Regulation of Tumor Progression and Therapeutic Response in Brain Metastasis. Front Immunol (2019) 10:1713. doi: 10.3389/fimmu.2019.01713
84. Wasilewski D, Priego N, Fustero-Torre C, Valiente M. Reactive Astrocytes in Brain Metastasis. Front Oncol (2017) 7:298. doi: 10.3389/fonc.2017.00298
85. Doron H, Amer M, Ershaid N, Blazquez R, Shani O, Lahav TG, et al. Inflammatory Activation of Astrocytes Facilitates Melanoma Brain Tropism via the CXCL10-CXCR3 Signaling Axis. Cell Rep (2019) 28(7):1785–98.e6. doi: 10.1016/j.celrep.2019.07.033
86. Priego N, Zhu L, Monteiro C, Mulders M, Wasilewski D, Bindeman W, et al. STAT3 Labels a Subpopulation of Reactive Astrocytes Required for Brain Metastasis. Nat Med (2018) 24(7):1024–35. doi: 10.1038/s41591-018-0044-4
87. Guttenplan KA, Weigel MK, Adler DI, Couthouis J, Liddelow SA, Gitler AD, et al. Knockout of Reactive Astrocyte Activating Factors Slows Disease Progression in an ALS Mouse Model. Nat Commun (2020) 11(1):3753. doi: 10.1038/s41467-020-17514-9
88. Mattiola I, Tomay F, De Pizzol M, Silva-Gomes R, Savino B, Gulic T, et al. The Macrophage Tetraspan MS4A4A Enhances Dectin-1-Dependent NK Cell-Mediated Resistance to Metastasis. Nat Immunol (2019) 20(8):1012–22. doi: 10.1038/s41590-019-0417-y
89. Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell (2017) 170(4):649–63.e13. doi: 10.1016/j.cell.2017.07.023
Keywords: cerebral metastasis, brain cancer, tumor microenvironment, tumor-associated macrophages, microglia, tumor immunology, targeted therapy
Citation: Schulz M and Sevenich L (2021) TAMs in Brain Metastasis: Molecular Signatures in Mouse and Man. Front. Immunol. 12:716504. doi: 10.3389/fimmu.2021.716504
Received: 28 May 2021; Accepted: 13 August 2021;
Published: 03 September 2021.
Edited by:
Frits Thorsen, University of Bergen, NorwayReviewed by:
Markus Biburger, Friedrich-Alexander-University Erlangen-Nürnberg, GermanyAndoni Garitano, University Hospital Würzburg, Germany
Copyright © 2021 Schulz and Sevenich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa Sevenich, c2V2ZW5pY2hAZ3NoLnVuaS1mcmFua2Z1cnQuZGU=