 Hassan Abolhassani
Hassan Abolhassani Yating Wang1
Yating Wang1- 1Division of Clinical Immunology, Department of Biosciences and Nutrition, Karolinska Institutet, Huddinge, Sweden
- 2Division of Clinical Immunology, Department of Laboratory Medicine, Karolinska University Hospital Huddinge, Karolinska Institutet, Stockholm, Sweden
- 3Research Center for Immunodeficiencies, Pediatrics Center of Excellence, Children’s Medical Center, Tehran University of Medical Science, Tehran, Iran
Inborn Errors of Immunity (IEI) comprise more than 450 inherited diseases, from which selected patients manifest a frequent and early incidence of malignancies, mainly lymphoma and leukemia. Primary antibody deficiency (PAD) is the most common form of IEI with the highest proportion of malignant cases. In this review, we aimed to compare the oncologic hallmarks and the molecular defects underlying PAD with other IEI entities to dissect the impact of avoiding immune destruction, genome instability, and mutation, enabling replicative immortality, tumor-promoting inflammation, resisting cell death, sustaining proliferative signaling, evading growth suppressors, deregulating cellular energetics, inducing angiogenesis, and activating invasion and metastasis in these groups of patients. Moreover, some of the most promising approaches that could be clinically tested in both PAD and IEI patients were discussed.
Introduction
Inborn Errors of Immunity (IEI), formerly known as primary immunodeficiencies, comprise at least 450 inherited diseases, from which selected patients manifest a frequent and early incidence of malignancies (1–3). As the main presentation, IEI patients are prone to recurrent infections (due to bacterial, viral, and parasitic agents) that predispose individuals to a chronic increase in inflammatory mediators, contributing to neoplasia (e.g., reactive oxygen and nitrogen intermediates, prostaglandins, and inflammatory cytokines). The longer the inflammation persists due to inadequate or inappropriate treatments, the higher the risk of associated tumorigenesis and the survival advantage of a cancerous cell (4). However, several other intrinsic and extrinsic causes of malignancies have been identified in both IEI-associated hematologic and solid tumors (5, 6). Considering the heterogeneous pathogenesis of IEI, different mechanisms underlying tumorigenesis in these patients would be expected. From an oncologic point of view, the main hallmarks of cancer have recently been suggested to dissect the complexity of neoplastic disease (7). The presented review compares oncologic hallmarks and the molecular defects underlying primary antibody deficiencies (PADs) with other IEI-associated cancers. The current published literature collection highlights that PAD patients have more diverse hallmarks of cancers compared to other IEIs (except combined immunodeficiency and immune dysregulation) and have a higher number of cases with heterogeneous genetic defects or unknown molecular etiologies. Of note, several therapeutic options are currently available for these diverse pathogeneses in PAD patients with cancer susceptibility, which should be considered by clinical immunologists and treating physicians.
Avoiding Immune Destruction
The ability of recognition and elimination of developing tumors in the absence of external therapy, which is known as cancer immunosurveillance, can be defective in certain types of IEIs (8, 9). Although the overall increased relative risk of cancer in IEI patients is less than twofold, a skewed spectrum of cancers (mainly lymphoid malignancies in males) can result from different gene defects (10). Innate and adaptive cytotoxicity against pre-malignant or malignant cells can be affected by mutations associated with dysfunction of natural killer (NK) and CD8+ T cells (1). Therefore, the intrinsic genetic defects affecting the development or function of T cell (presenting with combined immunodeficiency, major histocompatibility complex class I deficiency, or hyper IgE syndromes) and NK cell (GATA2, MCM4, and FCGR3A deficiencies) may lead to cancer, in particular carcinomas (11–15). Familial hemophagocytic lymphohistiocytosis patients with mutations in UNC13D, PRF1, STXBP2, and STX11 also present a significant defect in cytotoxicity, causing lymphoproliferative diseases and oncogenesis (16, 17).
Moreover, a proportion of patients with diseases of immune dysregulation show an increased susceptibility to herpes virus infections (mainly Epstein–Barr virus [EBV]-induced lymphoproliferative complications and malignancies), which resulted from defects in co-stimulatory molecules essential for CD8+ memory T-cell formation (e.g., CD27 and CD70 deficiencies and OX40 deficiency associated with higher risk of lymphoma and sarcoma) (18–21). Several other genes coordinate CD8+ T-cell activation and memory generation via various mechanisms, and therefore, mutations in these genes can increase the risk for developing EBV-associated lymphomas: controlling T-cell receptor-stimulated Mg2+ influx and concentrations (magnesium transporter 1, MAGT1 gene) (22), modulating the SH2 domain-mediated interactions in signaling lymphocyte activation molecule (SLAM)-mediated activation (SH2 domain-containing 1A, SH2D1A gene) (23), sustaining the proliferation of activated lymphocytes by de novo mutations in genes associated with the pyrimidine synthesis pathway (nucleotide cytidine 5′ triphosphate synthases1, CTPS1 gene) (24), activation of MAP-kinase cascade via guanine-nucleotide-exchange factors (RAS guanyl-releasing protein 1, RASGRP1 gene) (25), and mediating critical signals from the T-cell receptor and activated lymphocyte-specific protein tyrosine kinase (interleukin-2-inducible T-cell kinase, ITK gene) (26). Although the mechanism of cancer immunosurveillance has been suggested in a minority of PAD with functional T cell defects, some EBV-associated cancer due to monogenic IEI can affect B cell terminal development and also present with antibody deficiency and lack of specific immunoglobulin production mimicking common variable immunodeficiency (CVID)-like phenotype (27, 28).
Genome Instability and Mutation
Monogenic diseases of chromosome instability and DNA repair defects affecting steps of detection, removal, or further modification of the damaged DNA, and resynthesis and ligation of DNA strands can predispose both to immunodeficiency and cancer (29). T- B- receptor rearrangements [V(D)J recombinations] require the non-homologous end joining (NHEJ) pathway to process/repair double-strand DNA breaks and loss of function in various components of the NHEJ machinery present with T- B- severe combined immunodeficiency (SCID) (30, 31). In patients treated with hematopoietic stem cell transplantation, or carrying hypomorphic mutations in NHEJ factor encoding genes, survival may be associated with the development of hematological cancers, carcinomas, and sarcomas (5). Patients with DNA repair defects have a higher risk of EBV infections since the encoded viral proteins are implicated in the deregulation of DNA damage response signaling pathways (32). EBV infection disturbs ATM-mediated response (during the G2/M cell cycle via LMP1 and EBNA3C nuclear antigens) consistent with more frequent detection of EBV early antigen antibodies in patients with ataxia-telangiectasia in whom the incidence of lymphoma is increased (33, 34). Moreover, EBV attenuates DNA-dependent protein kinase and Artemis activities by depleting the p350/DNA-PK catalytic subunit and interacting with EBNA2, leading to a markedly increased incidence of EBV-induced lymphoproliferation in patients with pathogenic mutations in the PRKDC and DCLRE1C genes, up to 50% (34, 35).
Class switch recombination (CSR) and somatic hypermutation in peripheral B cells have a role in increasing the diversity of immunoglobulin classes as well as affinity maturation, which is accomplished by a large number of proteins involved in NHEJ, base excision repair, and mismatch repair (36). Ataxia-telangiectasia, Nijmegen breakage syndrome, Bloom’s syndrome, and constitutional mismatch repair deficiency (CMMRD) syndromes are the main immunodeficiencies within this category and the patients usually develop lymphomas (5, 37). Activation-induced cytidine deaminase (AICDA) and uracil DNA glycosylase (UNG) deficiencies specifically affect the CSR in B cells, presenting as a PAD known as hyper IgM syndrome with an increased incidence of hematologic cancers (38, 39).
Dysregulations in epigenetic modifications and chromatin remodeling may result in genomic instability and syndromic features mainly in the immunological and neurological systems (40). Genes underlying immunodeficiency with centromeric instability and facial anomalies (ICF) syndrome are responsible for DNA methylation and critical epigenetic modification during lymphocyte development, chromatin structure remodeling, and physiological DNA breaks (41). ICF patients display DNA hypomethylation mainly affecting satellite 2 and 3 repeats of pericentromers, which is very common in cancer cells (42), in line with the higher incidence of cancers in these patients (43). Of note, cases with ICF syndrome due to hypomorphic mutations may manifest without facial and neurologic symptoms, mimicking CVID-like phenotype with only antibody defects or recurrent infections and they may survive longer with a higher chance for the development of cancers (44, 45).
Enabling Replicative Immortality
This cancer hallmark is described as an independently driven process involving the elongation of telomeres by reactivation of telomerase reverse transcriptase and increasing the cell proliferative capacity (46, 47). This process is regulated by the catalytic subunit of telomerase reverse transcriptase (TERT) that connects this hallmark to metabolic reprogramming, apoptosis, and tumor invasion (48). Thus, TERT and its associated elements could directly connect the various hallmarks of cancer (49). Dyskeratosis congenita (DKC) is a complex of syndromic features caused by defects in these proteins, which can result in a severe form of Hoyeraal–Hreidarsson syndrome due to short telomeres and genome instability (50–52). Recently, Coats-plus syndrome with mutations in STN1 and CTC1 have been described and linked to immunodeficiency with abnormal telomeres. This group of genetic abrogations frequently predisposes patients to myelodysplasia and leukemia (53, 54). Intriguingly, several cases of dyskeratosis congenita can show specifically with the initial presentation of antibody deficiency, and due to misclassification, they are more prone to the development of long-term complications like malignancies (27, 55).
Tumor-Promoting Inflammation
Although chronic inflammation occurs in most IEI patients with a delayed diagnosis and poor treatment, some subgroups of patients can develop unrestrained systemic inflammatory reactions despite immunomodulation, which may lead to cancer (56, 57). This cancer hallmark is well characterized in disturbance of immune regulation with colitis (due to a defective IL10-STAT1 pathway) (58, 59) and predisposition to mucocutaneous candidiasis (mainly due to a defective IL17 pathway) (60) that can increase the susceptibility to lymphoma and carcinoma, respectively.
Moreover, CVID, as the most common symptomatic form of antibody deficiency, also had a higher rate of chronic inflammation despite regular and appropriate treatment (61). Due to its high prevalence, the majority of IEI cancer patients have a clinical diagnosis of CVID (10). CVID is a heterogeneous disease, and there is an ongoing debate about criteria for diagnosis that mainly rely on the fulfillment of specific immunologic criteria. Therefore, CVID is considered as an umbrella term constituting several different humoral immune failures and antibody production impairment due to unknown monogenic, polygenic, or epigenetic defects (27, 28). However, the main suggested pathogeneses for the cancer phenomenon in CVID patients are immune dysregulation and chronic infection due to lack of mucosal immunity (absence of IgA in selected CVID patients). Therefore, subsequent inflammation might be a tumor-predisposing factor especially towards gastric cancers in CVID cases (62–65). The same phenomenon can be present in other entities of PAD with low IgA levels including congenital agammaglobulinemia and IgA deficiency (62, 66–68). Of note, a minority of CVID patients can present chromosomal radiosensitivity due to disruption of DNA repair machinery and must be considered for tumorigenesis due to genome instability and regular screening for cancer and avoidance of malignancy triggers must be added to their routine management (69, 70).
Resisting Cell Death
Autoimmune lymphoproliferative syndrome (ALPS) is the porotype of IEI, which is associated with apoptosis defects and malignancies (71). The most well-established activity of affected proteins in the FAS–FAS ligand and Caspase pathway is to mediate the apoptotic death of either virus-infected cells or cancer cells when engaged by a cytotoxic lymphocyte (72). Although lymphoma has been reported as the most common type of malignancy seen in these patients, additional types of cancers in this population suggest a broader cancer predisposition as previously observed with somatic FAS mutations (73–76). Since apoptosis has an important role in the development, function and maintenance of the immune system, it controls the duration of immune responses to foreign antigens and deletion of auto-reactive T and B lymphocytes (77). Similarly, several abnormalities in T- and B-cell apoptosis in patients with humoral immunodeficiencies such as CVID have been reported and suggested to be underlying the higher rate of malignancy, particularly lymphoma, in this group of patients (78, 79).
Sustaining Proliferative Signaling
Self-sufficiency in growth signals, bypassing various checkpoints, may be implicated in a vast number of patients with IEI and cancers (80). Immune system defects and dynamical compensation in physiological circuits lead to increased production of stimulatory factors mainly in patients with stem cell and myeloid developmental defects (81). Congenital neutropenias and other syndromic IEI (Wiskott-Aldrich, Shwachman-Diamond, MonoMac and immuno-osseous dysplasia syndromes) affecting early non-lymphoid stem cell lineages can manifest with myelodysplasia and leukemia (82).
Higher rates of diagnosis during recent years and detailed follow-up of autosomal dominant gain-of-function defects in signal transducer and activator of transcriptions (STAT) (83, 84), caspase recruitment domain family members (CARD) (85, 86) and NACHT, LRR, and PYD domain-containing proteins (NLRP) have shown an increased incidence of both hematological and solid tumors (87). Of note in the PAD category, several gain-of-function genetic defects in the signaling of phosphoinositide 3-kinase (PI3K) and nuclear factor κ-enhancer of activated B cells (NF-κB) have been shown to be involved in the dysregulation of the adaptive immune response and continuous lymphoid tissue growth, thus increasing the susceptibility to lymphoma (88–92). Of note, a minority of patients with NF-κB defects also presented avoiding cellular immune destruction mainly due to abrogated CD8 T-cell immunity (93, 94).
Evading Growth Suppressors
The diverse functions of tumor suppressors vary from proliferation restriction to the regulation of regenerative processes in different human cell types (95). However, these elements modulate the proliferation and differentiation of immune cells to protect their genomic integrity during physiologic cellular metabolic and proliferative stress (96). The existence of multiple tumor suppressor family members (e.g., p53, retinoblastoma, and Hippo genes) may allow certain family members to have taken on specific roles in the enhancement of hematopoietic stem cell regeneration, DNA repair, chromosome remodeling, and cell-cycle checkpoint for selecting the desired modification (97).
One of the main tumor suppressor pathways conferring immunodeficiency and susceptibility to cancers is the posttranslational regulation of phosphatase and tensin homolog (PTEN) (98). PTEN is a negative regulator of PI3K signaling and is very commonly mutated in human cancers. Since PTEN is essential during early development, only heterozygous loss-of-function mutants have been reported in individuals with CVID-like phenotype with lymphoproliferation and hyperplasia (99). The prototypical tumor suppressor gene and pathway is p53, which is also a key pathway component affected in a majority of DNA repair defects associated with immunodeficiency and cancers (e.g., patients with ATM and MRE11 mutations) (100).
Dedicator of cytokinesis 8 (DOCK8) can act as a tumor suppressor in non-hematopoietic tissues by directly affecting apoptosis through regulation of migration, morphology, adhesion, and growth of cells, apart from its probable role in CD8+ T cells for tumor surveillance (101). Cytotoxic T lymphocyte-associated antigen 4 (CTLA4) is upregulated in activated naïve T cells through the T-cell receptor and subsequent engagement of the costimulatory receptor CD28 (102). This suppressive molecule acts as co-inhibitory and mutation in the autosomal dominant form impairs the function of regulatory T cells, thus increasing the risk for autoimmunity, chronic inflammation, and cancers (103). Patients with lipopolysaccharide-responsive and beige-like anchor protein (LRBA) deficiency, a crucial molecule for recycling of CTLA4 and the function of regulatory T cells, present a similar CVID-like phenotype with the development of both hematological and solid tumors (104, 105).
Deregulating Cellular Energetics
IEIs associated with sustaining proliferative signaling induce endoplasmic reticulum stress, unfolded protein response, destabilization of mitochondrial membrane potentials, and disturbed energy metabolism (106, 107). Recent findings also suggest that there may be a common pathogenic mechanism that connects a high prevalence of cancer, metabolic disorders, atherosclerotic cardiovascular disease, and insulin-resistant diabetes in carriers of some DNA repair defects, in particular ATM mutations (108). Mutations of genes related to NHEJ and IEI disorders associated with chronic inflammation result in age-associated pathological conditions due to their roles in metabolic regulation in response to DNA damage avoiding further genomic instability (109, 110).
These defects in DNA repair and uncontrolled inflammation may induce stem cell exhaustion, cellular senescence, immunosenescence, low-grade chronic inflammation, activation of PI3K signaling, defective autophagy, and mitochondrial genome instability. It has been shown that ATM-dependent stress and dysregulation of inflammatory pathways mediate predisposition to both the metabolic syndrome and cancer (111).
Inducing Angiogenesis
A series of well-orchestrated cellular adaptations occur to stimulate angiogenesis and enhance the survival of tumors in hypoxic conditions (112). Gain-of-function somatic mutations in RAS-associated genes (KRAS and NRAS) can result in RAS-associated autoimmune leukoproliferative disease (RALD) with lymphocytosis and lymphoproliferation, a phenocopy of ALPS (113, 114). The affected proteins are GTPases that serve as a signaling switch molecule, coupling receptor activation by specific growth factors with downstream effector pathways. After cancer-related hypoxia responses, in patients with RALD, the production of vascular endothelial growth factor (VEGF) is enhanced (115). Therefore, the over-activation of RAS signaling significantly stimulates angiogenesis and blocks apoptosis in hypoxic conditions (116).
Furthermore, in cancers associated with defective innate or adaptive immune responses, the balance between pro- and anti-angiogenic factors is perturbed by dysregulated cytokine production by innate immune cells (117). Increased inflammatory mediators as a consequence of antibody deficiency, diseases of immune dysregulation, and autoinflammatory diseases contribute to neoplasia by stimulation of angiogenesis, where a change confers a survival advantage to a tumor cell (56, 118). Therefore, the promotion of angiogenesis in the IEI tumors accelerates the migration of endothelial cells and formation of new blood vessels, and distorted and enlarged vascular architecture with increased permeability and irregular blood flow (119).
Activating Invasion and Metastasis
A selected group of IEIs faces aggressive oncogenic risks due to an increased susceptibility for viral replication and persistence (120). Among those, transforming viral infections with a distant invasion have been reported by human papillomavirus (HPV infection in Epidermodysplasia verruciformis and WHIM syndrome) (121, 122) and herpes viruses family (particularly EBV susceptibility in immune dysregulation diseases). Of note, both groups of patients with HPV (mainly WHIM syndrome) and EBV infection susceptibility can mimic the phenotype of CVID-like due to their predominance of humoral immunodeficiency. Although both HPV and EBV oncoviruses have undertaken different powerful anti-apoptotic and proliferative programs, they can directly induce metastasis in infected tumor cells. In HPV-associated IEIs, E6 and E7 proteins can contribute to tumor invasion by impacting epithelial-to-mesenchymal transition (123, 124), while in EBV infection, the LMP2A protein can promote differentiation, survival, and cell growth by activating the PI3K pathway and pathways mediating cell mobility and invasion (125).
Future Directions and Concluding Remarks
The evaluation of the hallmarks of cancer in IEI patients helps to explain the multistep nature of oncogenesis in different forms of immune defects/dysfunction (Figure 1). This outlines the complexity of the development of cancer in each entity of IEIs, requiring the progressive acquisition of different necessary cellular hallmarks that constitute a malignant phenotype. The distribution of distinct types of cancers in patients with specific genetic defects highlights the cell-specific predisposition to an intrinsic cause or extrinsic exposure in the context of the genetic background of the host and the selective pressures imposed by the tissue microenvironment. The analysis of a cancer hallmark model would also facilitate understanding about the process of IEI carcinogenesis to relevant treatment. Recently, cancer hallmarks have been reorganized into seven updated compact parameters (126). It has been suggested to consider altered stress response favoring overall survival by combining defects of genome instability and mutation, enabling replicative immortality, tumor-promoting inflammation, and resisting cell death hallmarks (126). Moreover, a new hallmark for abetting microenvironment has been offered to cover cancer etiologies related to communication between the dynamic microenvironment of the affected organ and stromal cells (5, 126). IEI genes underlying each hallmark might help to investigate whether these newly proposed revisions are functionally and molecularly relevant.

Figure 1 Cancer hallmark activation in different types of monogenic inborn errors of immunity (IEI) according to the International Union of Immunological Societies classification (1, 3).
Based on several lines of evidence, PAD patients constitute the highest proportion of IEI cases affected by malignancies. Moreover, several monogenic defects with different involved cancer hallmarks can mimic the clinical and immunologic phenotypes of PAD patients, mainly CVID. The abovementioned overview about IEI-induced and PAD-induced cancers indicated that these malignancies are amenable to immune prophylaxis by vaccines, prophylactic radiation limitation, and, most recently, targeted therapy. However, future clinical efforts in preventing or treating gene-specific-associated malignancies represent a combination of antiviral therapies, agents that induce cytotoxicity events, agents that improve DNA repair machinery, and agents that are used to successfully treat cancers with antagonists and agonists for IEI tumor stimulators and repressors. Table 1 illustrates some of the most promising approaches that could be clinically tested in both PAD and IEI patients. Of note, other monogenic IEIs mainly with combined immunodeficiency and immune dysregulation also have diverse cancer hallmarks as PAD patients; however, they are more likely to be transplanted due to the risk of cancer, whereas most PADs may not be transplanted. The treatment of cancers in the context of immune defects, however, remains challenging and a detailed molecular investigation and multi-omics analysis of both germline and somatic (tumor) genome may increase the number of potential therapeutic targets and also further provide clues of potential resistance to therapy.
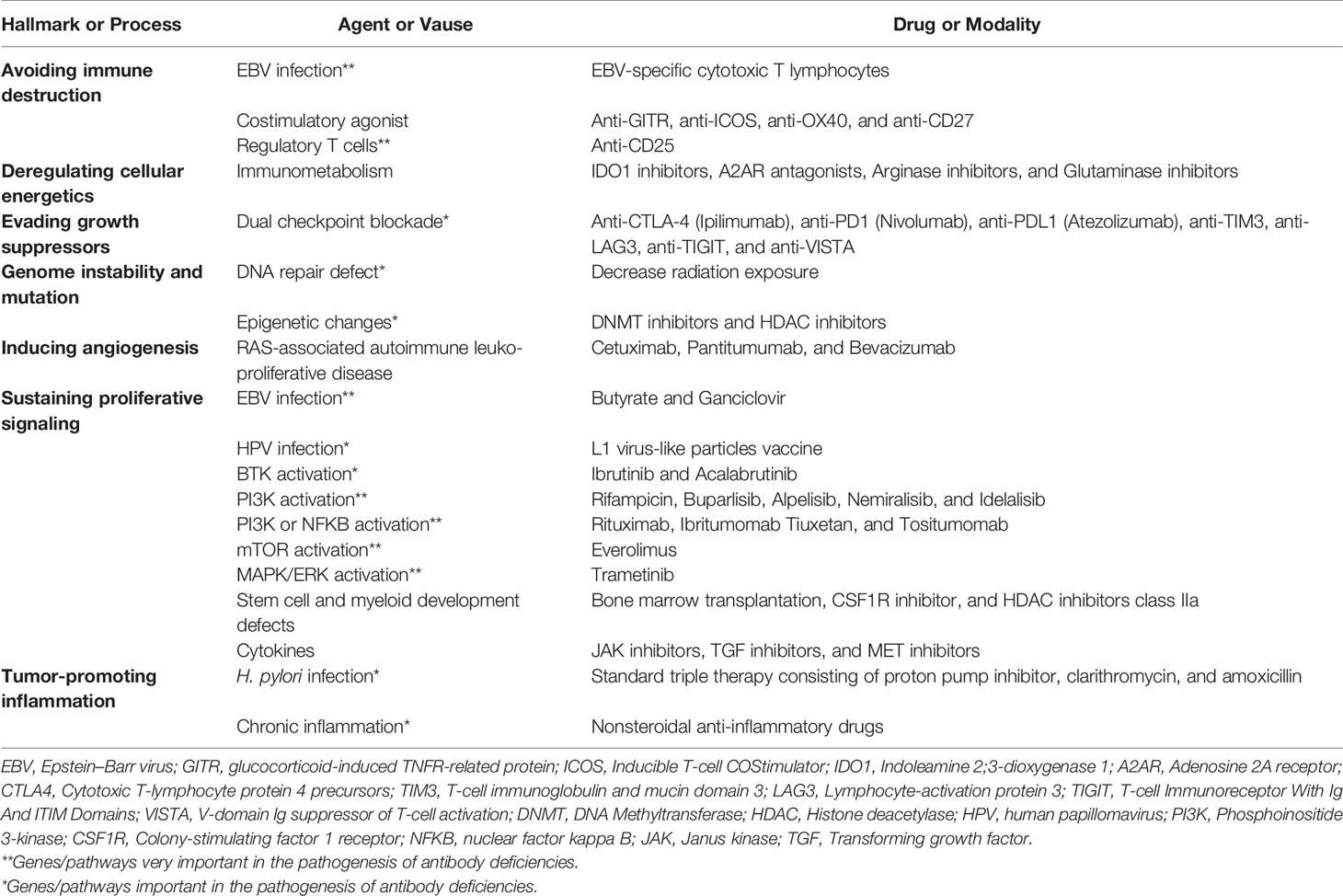
Table 1 Therapeutic and preventive approaches successfully used or potentially can be implemented to prevent primary immunodeficiency-associated cancer hallmarks.
Author Contributions
HA, YW, LH, and QP-H equally contributed to the design and writing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Swedish Research Council, the Swedish Cancer Society (Cancerfonden), the Swedish Childhood Cancer Fund, Radiumhemmets, the Center for Innovative Medicine, Jonas Söderquist scholarship, and Åke Wibergs stiftelse.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
2. Mortaz E, Tabarsi P, Mansouri D, Khosravi A, Garssen J, Velayati A, et al. Cancers Related to Immunodeficiencies: Update and Perspectives. Front Immunol (2016) 7:365. doi: 10.3389/fimmu.2016.00365
3. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. The Ever-Increasing Array of Novel Inborn Errors of Immunity: An Interim Update by the IUIS Committee. J Clin Immunol (2021) 41:666–79. doi: 10.1007/s10875-021-00980-1
4. Multhoff G, Molls M, Radons J. Chronic Inflammation in Cancer Development. Front Immunol (2011) 2:98. doi: 10.3389/fimmu.2011.00098
5. Yuan Y. Spatial Heterogeneity in the Tumor Microenvironment. Cold Spring Harb Perspect Med (2016) 6:10. doi: 10.1101/cshperspect.a026583
6. Zimmerman R, Schimmenti L, Spector L. A Catalog of Genetic Syndromes in Childhood Cancer. Pediatr Blood Cancer (2015) 62:2071–5. doi: 10.1002/pbc.25726
7. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
8. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat Immunol (2002) 3:991–8. doi: 10.1038/ni1102-991
9. Corthay A. Does the Immune System Naturally Protect Against Cancer? Front Immunol (2014) 5:197. doi: 10.3389/fimmu.2014.00197
10. Mayor PC, Eng KH, Singel KL, Abrams SI, Odunsi K, Moysich KB, et al. Cancer in Primary Immunodeficiency Diseases: Cancer Incidence in the United States Immune Deficiency Network Registry. J Allergy Clin Immunol (2018) 141:1028–35. doi: 10.1016/j.jaci.2017.05.024
11. Hughes CR, Guasti L, Meimaridou E, Chuang CH, Schimenti JC, King PJ, et al. MCM4 Mutation Causes Adrenal Failure, Short Stature, and Natural Killer Cell Deficiency in Humans. J Clin Invest (2012)122:814–20. doi: 10.1172/JCI60224
12. Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 Deficiency: A Protean Disorder of Hematopoiesis, Lymphatics, and Immunity. Blood (2014) 123:809–21. doi: 10.1182/blood-2013-07-515528
13. Attarbaschi A, Carraro E, Abla O, Barzilai-Birenboim S, Bomken S, Brugieres L, et al. Non-Hodgkin Lymphoma and Pre-Existing Conditions: Spectrum, Clinical Characteristics and Outcome in 213 Children and Adolescents. Haematologica (2016) 101:1581–91. doi: 10.3324/haematol.2016.147116
14. Salavoura K, Kolialexi A, Tsangaris G, Mavrou A. Development of Cancer in Patients With Primary Immunodeficiencies. Anticancer Res (2008) 28:1263–9.
15. Mir MA, Kochuparambil ST, Abraham RS, Rodriguez V, Howard M, Hsu AP, et al. Spectrum of Myeloid Neoplasms and Immune Deficiency Associated With Germline GATA2 Mutations. Cancer Med (2015) 4:490–9. doi: 10.1002/cam4.384
16. Daver N, McClain K, Allen CE, Parikh SA, Otrock Z, Rojas-Hernandez C, et al. A Consensus Review on Malignancy-Associated Hemophagocytic Lymphohistiocytosis in Adults. Cancer (2017) 123:3229–40. doi: 10.1002/cncr.30826
17. Chen X, Zhang Y, Wang F, Wang M, Teng W, Lin Y, et al. Germline Cytotoxic Lymphocytes Defective Mutations in Chinese Patients With Lymphoma. Oncol Lett (2017) 14:5249–56. doi: 10.3892/ol.2017.6898
18. Alkhairy OK, Perez-Becker R, Driessen GJ, Abolhassani H, van Montfrans J, Borte S, et al. Novel Mutations in TNFRSF7/CD27: Clinical, Immunologic, and Genetic Characterization of Human CD27 Deficiency. J Allergy Clin Immunol (2015) 136:703–12.e10. doi: 10.1016/j.jaci.2015.02.022
19. Abolhassani H, Edwards ES, Ikinciogullari A, Jing H, Borte S, Buggert M, et al. Combined Immunodeficiency and Epstein-Barr Virus-Induced B Cell Malignancy in Humans With Inherited CD70 Deficiency. J Exp Med (2017) 214:91–106. doi: 10.1084/jem.20160849
20. Tangye SG, Palendira U, Edwards ES. Human Immunity Against EBV-Lessons From the Clinic. J Exp Med (2017) 214:269–83. doi: 10.1084/jem.20161846
21. Byun M, Ma CS, Akcay A, Pedergnana V, Palendira U, Myoung J, et al. Inherited Human OX40 Deficiency Underlying Classic Kaposi Sarcoma of Childhood. J Exp Med (2013) 210:1743–59. doi: 10.1084/jem.20130592
22. Li FY, Chaigne-Delalande B, Su H, Uzel G, Matthews H, Lenardo MJ. XMEN Disease: A New Primary Immunodeficiency Affecting Mg2+ Regulation of Immunity Against Epstein-Barr Virus. Blood (2014) 123:2148–52. doi: 10.1182/blood-2013-11-538686
23. Chuang HC, Lay JD, Hsieh WC, Wang HC, Chang Y, Chuang SE, et al. Epstein-Barr Virus LMP1 Inhibits the Expression of SAP Gene and Upregulates Th1 Cytokines in the Pathogenesis of Hemophagocytic Syndrome. Blood (2005) 106:3090–6. doi: 10.1182/blood-2005-04-1406
24. Martin E, Palmic N, Sanquer S, Lenoir C, Hauck F, Mongellaz C, et al. CTP Synthase 1 Deficiency in Humans Reveals its Central Role in Lymphocyte Proliferation. Nature (2014) 510:288–92. doi: 10.1038/nature13386
25. Platt CD, Fried AJ, Hoyos-Bachiloglu R, Usmani GN, Schmidt B, Whangbo J, et al. Combined Immunodeficiency With EBV Positive B Cell Lymphoma and Epidermodysplasia Verruciformis Due to a Novel Homozygous Mutation in RASGRP1. Clin Immunol (2017) 183:142–4. doi: 10.1016/j.clim.2017.08.007
26. Zhong Y, Johnson AJ, Byrd JC, Dubovsky JA. Targeting Interleukin-2-Inducible T-Cell Kinase (ITK) in T-Cell Related Diseases. Postdoc J (2014) 2:1–11. doi: 10.14304/SURYA.JPR.V2N6.1
27. Abolhassani H, Aghamohammadi A, Fang M, Rezaei N, Jiang C, Liu X, et al. Clinical Implications of Systematic Phenotyping and Exome Sequencing in Patients With Primary Antibody Deficiency. Genet Med (2019) 21:243–51. doi: 10.1038/s41436-018-0012-x
28. Abolhassani H, Hammarstrom L, Cunningham-Rundles C. Current Genetic Landscape in Common Variable Immune Deficiency. Blood (2020) 35:656–67. doi: 10.1182/blood.2019000929
29. de Miranda NF, Bjorkman A, Pan-Hammarstrom Q. DNA Repair: The Link Between Primary Immunodeficiency and Cancer. Ann N Y Acad Sci (2011) 1246:50–63. doi: 10.1111/j.1749-6632.2011.06322.x
30. Moshous D, Pannetier C, Chasseval Rd R, Deist Fl F, Cavazzana-Calvo M, Romana S, et al. Partial T and B Lymphocyte Immunodeficiency and Predisposition to Lymphoma in Patients With Hypomorphic Mutations in Artemis. J Clin Invest (2003) 111:381–7. doi: 10.1172/JCI16774
31. Schatz DG, Ji Y. Recombination Centres and the Orchestration of V(D)J Recombination. Nat Rev Immunol (2011) 11:251–63. doi: 10.1038/nri2941
32. Hau PM, Tsao SW. Epstein-Barr Virus Hijacks DNA Damage Response Transducers to Orchestrate Its Life Cycle. Viruses (2017) 9:341. doi: 10.3390/v9110341
33. Gruhne B, Sompallae R, Masucci MG. Three Epstein-Barr Virus Latency Proteins Independently Promote Genomic Instability by Inducing DNA Damage, Inhibiting DNA Repair and Inactivating Cell Cycle Checkpoints. Oncogene (2009) 28:3997–4008. doi: 10.1038/onc.2009.258
34. Worth AJ, Houldcroft CJ, Booth C. Severe Epstein-Barr Virus Infection in Primary Immunodeficiency and the Normal Host. Br J Haematol (2016) 175:559–76. doi: 10.1111/bjh.14339
35. Lu J, Tang M, Li H, Xu Z, Weng X, Li J, et al. EBV-LMP1 Suppresses the DNA Damage Response Through DNA-PK/AMPK Signaling to Promote Radioresistance in Nasopharyngeal Carcinoma. Cancer Lett (2016) 380:191–200. doi: 10.1016/j.canlet.2016.05.032
36. Xu Z, Fulop Z, Zhong Y, Evinger AJ 3rd, Zan H, Casali P. DNA Lesions and Repair in Immunoglobulin Class Switch Recombination and Somatic Hypermutation. Ann N Y Acad Sci (2005) 1050:146–62. doi: 10.1196/annals.1313.119
37. Slatter MA, Gennery AR. Primary Immunodeficiencies Associated With DNA-Repair Disorders. Expert Rev Mol Med (2010) 12:e9. doi: 10.1017/S1462399410001419
38. Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, et al. AID Is Required for Germinal Center-Derived Lymphomagenesis. Nat Genet (2008) 40:108–12. doi: 10.1038/ng.2007.35
39. Durandy A, Kracker S. Immunoglobulin Class-Switch Recombination Deficiencies. Arthritis Res Ther (2012) 14:218. doi: 10.1186/ar3904
40. Aparicio T, Baer R, Gautier J. DNA Double-Strand Break Repair Pathway Choice and Cancer. DNA Repair (Amst) (2014) 19:169–75. doi: 10.1016/j.dnarep.2014.03.014
41. Jin B, Tao Q, Peng J, Soo HM, Wu W, Ying J, et al. DNA Methyltransferase 3B (DNMT3B) Mutations in ICF Syndrome Lead to Altered Epigenetic Modifications and Aberrant Expression of Genes Regulating Development, Neurogenesis and Immune Function. Hum Mol Genet (2008) 17:690–709. doi: 10.1093/hmg/ddm341
42. Narayan A, Ji W, Zhang XY, Marrogi A, Graff JR, Baylin SB, et al. Hypomethylation of Pericentromeric DNA in Breast Adenocarcinomas. Int J Cancer (1998) 77:833–8. doi: 10.1002/(sici)1097-0215(19980911)77:6<833::aid-ijc6>3.0.co;2-v
43. Weemaes CM, van Tol MJ, Wang J, van Ostaijen-ten Dam MM, van Eggermond MC, Thijssen PE, et al. Heterogeneous Clinical Presentation in ICF Syndrome: Correlation With Underlying Gene Defects. Eur J Hum Genet (2013) 21:1219–25. doi: 10.1038/ejhg.2013.40
44. Yazdani R, Abolhassani H, Kiaee F, Habibi S, Azizi G, Tavakol M, et al. Comparison of Common Monogenic Defects in a Large Predominantly Antibody Deficiency Cohort. J Allergy Clin Immunol Pract (2019) 7:864–78.e9. doi: 10.1016/j.jaip.2018.09.004
45. Kiaee F, Zaki-Dizaji M, Hafezi N, Almasi-Hashiani A, Hamedifar H, Sabzevari A, et al. Clinical, Immunologic and Molecular Spectrum of Patients With Immunodeficiency, Centromeric Instability, and Facial Anomalies (ICF) Syndrome: A Systematic Review. Endocr Metab Immune Disord Drug Targets (2021) 21:664–72. doi: 10.2174/1871530320666200613204426
46. Shay JW. Are Short Telomeres Hallmarks of Cancer Recurrence? Clin Cancer Res (2014) 20:779–81. doi: 10.1158/1078-0432.CCR-13-3198
47. Djojosubroto MW, Choi YS, Lee HW, Rudolph KL. Telomeres and Telomerase in Aging, Regeneration and Cancer. Mol Cells (2003) 15:164–75.
48. Liu T, Yuan X, Xu D. Cancer-Specific Telomerase Reverse Transcriptase (TERT) Promoter Mutations: Biological and Clinical Implications. Genes (Basel) (2016) 7:38. doi: 10.3390/genes7070038
49. Khattar E, Kumar P, Liu CY, Akincilar SC, Raju A, Lakshmanan M, et al. Telomerase Reverse Transcriptase Promotes Cancer Cell Proliferation by Augmenting tRNA Expression. J Clin Invest (2016) 126:4045–60. doi: 10.1172/JCI86042
50. Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. TINF2 Mutations Result in Very Short Telomeres: Analysis of a Large Cohort of Patients With Dyskeratosis Congenita and Related Bone Marrow Failure Syndromes. Blood (2008) 112:3594–600. doi: 10.1182/blood-2008-05-153445
51. Beier F, Foronda M, Martinez P, Blasco MA. Conditional TRF1 Knockout in the Hematopoietic Compartment Leads to Bone Marrow Failure and Recapitulates Clinical Features of Dyskeratosis Congenita. Blood (2012) 120:2990–3000. doi: 10.1182/blood-2012-03-418038
52. Basel-Vanagaite L, Dokal I, Tamary H, Avigdor A, Garty BZ, Volkov A, et al. Expanding the Clinical Phenotype of Autosomal Dominant Dyskeratosis Congenita Caused by TERT Mutations. Haematologica (2008) 93:943–4. doi: 10.3324/haematol.12317
53. Crow YJ, McMenamin J, Haenggeli CA, Hadley DM, Tirupathi S, Treacy EP, et al. Coats’ Plus: A Progressive Familial Syndrome of Bilateral Coats’ Disease, Characteristic Cerebral Calcification, Leukoencephalopathy, Slow Pre- and Post-Natal Linear Growth and Defects of Bone Marrow and Integument. Neuropediatrics (2004) 35:10–9. doi: 10.1055/s-2003-43552
54. Keller RB, Gagne KE, Usmani GN, Asdourian GK, Williams DA, Hofmann I, et al. CTC1 Mutations in a Patient With Dyskeratosis Congenita. Pediatr Blood Cancer (2012) 59:311–4. doi: 10.1002/pbc.24193
55. Allenspach EJ, Bellodi C, Jeong D, Kopmar N, Nakamura T, Ochs HD, et al. Common Variable Immunodeficiency as the Initial Presentation of Dyskeratosis Congenita. J Allergy Clin Immunol (2013) 132:223–6. doi: 10.1016/j.jaci.2012.11.052
56. Fodil N, Langlais D, Gros P. Primary Immunodeficiencies and Inflammatory Disease: A Growing Genetic Intersection. Trends Immunol (2016) 37:126–40. doi: 10.1016/j.it.2015.12.006
57. Langlais D, Fodil N, Gros P. Genetics of Infectious and Inflammatory Diseases: Overlapping Discoveries From Association and Exome-Sequencing Studies. Annu Rev Immunol (2017) 35:1–30. doi: 10.1146/annurev-immunol-051116-052442
58. Shouval DS, Ebens CL, Murchie R, McCann K, Rabah R, Klein C, et al. Large B-Cell Lymphoma in an Adolescent Patient With Interleukin-10 Receptor Deficiency and History of Infantile Inflammatory Bowel Disease. J Pediatr Gastroenterol Nutr (2016) 63:e15–7. doi: 10.1097/MPG.0000000000000532
59. Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-Of-Function Human STAT1 Mutations Impair IL-17 Immunity and Underlie Chronic Mucocutaneous Candidiasis. J Exp Med (2011) 208:1635–48. doi: 10.1084/jem.20110958
60. Rosa DD, Pasqualotto AC, Denning DW. Chronic Mucocutaneous Candidiasis and Oesophageal Cancer. Med Mycol (2008) 46:85–91. doi: 10.1080/13693780701616023
61. Modell V, Orange JS, Quinn J, Modell F. Global Report on Primary Immunodeficiencies: 2018 Update From the Jeffrey Modell Centers Network on Disease Classification, Regional Trends, Treatment Modalities, and Physician Reported Outcomes. Immunol Res (2018) 66:367–80. doi: 10.1007/s12026-018-8996-5
62. Ludvigsson JF, Neovius M, Ye W, Hammarstrom L. IgA Deficiency and Risk of Cancer: A Population-Based Matched Cohort Study. J Clin Immunol (2015) 35:182–8. doi: 10.1007/s10875-014-0124-2
63. Tak Manesh A, Azizi G, Heydari A, Kiaee F, Shaghaghi M, Hossein-Khannazer N, et al. Epidemiology and Pathophysiology of Malignancy in Common Variable Immunodeficiency? Allergol Immunopathol (Madr) (2017) 45:602–15. doi: 10.1016/j.aller.2017.01.006
64. Vajdic CM, Mao L, van Leeuwen MT, Kirkpatrick P, Grulich AE, Riminton S. Are Antibody Deficiency Disorders Associated With a Narrower Range of Cancers Than Other Forms of Immunodeficiency? Blood (2010) 116:1228–34. doi: 10.1182/blood-2010-03-272351
65. Dhalla F, da Silva SP, Lucas M, Travis S, Chapel H. Review of Gastric Cancer Risk Factors in Patients With Common Variable Immunodeficiency Disorders, Resulting in a Proposal for a Surveillance Programme. Clin Exp Immunol (2011) 165:1–7. doi: 10.1111/j.1365-2249.2011.04384.x
66. Mellemkjaer L, Hammarstrom L, Andersen V, Yuen J, Heilmann C, Barington T, et al. Cancer Risk Among Patients With IgA Deficiency or Common Variable Immunodeficiency and Their Relatives: A Combined Danish and Swedish Study. Clin Exp Immunol (2002) 130:495–500. doi: 10.1046/j.1365-2249.2002.02004.x
67. Hernandez-Trujillo V. (2021). Available at: https://www.uptodate.com/contents/agammaglobulinemia.
68. Conley ME. Are Patients With X-Linked Agammaglobulinemia at Increased Risk of Developing Acute Lymphoblastic Leukemia? J Clin Immunol (2015) 35:98–9. doi: 10.1007/s10875-015-0132-x
69. Hargreaves CE, Salatino S, Sasson SC, Charlesworth JEG, Bateman E, Patel AM, et al. Decreased ATM Function Causes Delayed DNA Repair and Apoptosis in Common Variable Immunodeficiency Disorders. J Clin Immunol (2021) 41:1315–30. doi: 10.1007/s10875-021-01050-2
70. Mahmoodi M, Abolhassani H, Mozdarani H, Rezaei N, Azizi G, Yazdani R, et al. In Vitro Chromosomal Radiosensitivity in Patients With Common Variable Immunodeficiency. Cent Eur J Immunol (2018) 43:155–61. doi: 10.5114/ceji.2018.77385
71. Fleisher TA. The Autoimmune Lymphoproliferative Syndrome: An Experiment of Nature Involving Lymphocyte Apoptosis. Immunol Res (2008) 40:87–92. doi: 10.1007/s12026-007-8001-1
72. O’Brien DI, Nally K, Kelly RG, O’Connor TM, Shanahan F, O’Connell J. Targeting the Fas/Fas Ligand Pathway in Cancer. Expert Opin Ther Targets (2005) 9:1031–44. doi: 10.1517/14728222.9.5.1031
73. Bride K, Teachey D. Autoimmune Lymphoproliferative Syndrome: More Than a FAScinating Disease. F1000Res (2017) 6:1928. doi: 10.12688/f1000research.11545.1
74. Gopalan B, Litvak A, Sharma S, Mhashilkar AM, Chada S, Ramesh R. Activation of the Fas-FasL Signaling Pathway by MDA-7/IL-24 Kills Human Ovarian Cancer Cells. Cancer Res (2005) 65:3017–24. doi: 10.1158/0008-5472.CAN-04-3758
75. Gronbaek K, Straten PT, Ralfkiaer E, Ahrenkiel V, Andersen MK, Hansen NE, et al. Somatic Fas Mutations in Non-Hodgkin’s Lymphoma: Association With Extranodal Disease and Autoimmunity. Blood (1998) 92:3018–24. doi: 10.1182/blood.V92.9.3018
76. Muschen M, Rajewsky K, Kronke M, Kuppers R. The Origin of CD95-Gene Mutations in B-Cell Lymphoma. Trends Immunol (2002) 23:75–80. doi: 10.1016/S1471-4906(01)02115-9
77. Feig C, Peter ME. How Apoptosis Got the Immune System in Shape. Eur J Immunol (2007) 37 Suppl 1:S61–70. doi: 10.1002/eji.200737462
78. Ganjalikhani-Hakemi M, Yazdani R, Esmaeili M, Abolhassani H, Rae W, Azizi G, et al. Role of Apoptosis in the Pathogenesis of Common Variable Immunodeficiency (CVID). Endocr Metab Immune Disord Drug Targets (2017) 17:332–40. doi: 10.2174/1871530317666170919120245
79. Yazdani R, Fatholahi M, Ganjalikhani-Hakemi M, Abolhassani H, Azizi G, Hamid KM, et al. Role of Apoptosis in Common Variable Immunodeficiency and Selective Immunoglobulin A Deficiency. Mol Immunol (2016) 71:1–9. doi: 10.1016/j.molimm.2015.12.016
80. Giancotti FG. Deregulation of Cell Signaling in Cancer. FEBS Lett (2014) 588:2558–70. doi: 10.1016/j.febslet.2014.02.005
81. Lambert C, Wu Y, Aanei C. Bone Marrow Immunity and Myelodysplasia. Front Oncol (2016) 6:172. doi: 10.3389/fonc.2016.00172
82. Hauck F, Klein C. Pathogenic Mechanisms and Clinical Implications of Congenital Neutropenia Syndromes. Curr Opin Allergy Clin Immunol (2013) 13:596–606. doi: 10.1097/ACI.0000000000000014
83. Forbes LR, Milner J, Haddad E. Signal Transducer and Activator of Transcription 3: A Year in Review. Curr Opin Hematol (2016) 23:23–7. doi: 10.1097/MOH.0000000000000206
84. Avalle L, Pensa S, Regis G, Novelli F, Poli V. STAT1 and STAT3 in Tumorigenesis: A Matter of Balance. JAKSTAT (2012) 1:65–72. doi: 10.4161/jkst.20045
85. Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 Mutations in Human Diffuse Large B Cell Lymphoma. Science (2008) 319:1676–9. doi: 10.1126/science.1153629
86. Arjunaraja S, Snow AL. Gain-Of-Function Mutations and Immunodeficiency: At a Loss for Proper Tuning of Lymphocyte Signaling. Curr Opin Allergy Clin Immunol (2015) 15:533–8. doi: 10.1097/ACI.0000000000000217
87. Kantono M, Guo B. Inflammasomes and Cancer: The Dynamic Role of the Inflammasome in Tumor Development. Front Immunol (2017) 8:1132. doi: 10.3389/fimmu.2017.01132
88. Maccari ME, Abolhassani H, Aghamohammadi A, Aiuti A, Aleinikova O, Bangs C, et al. Disease Evolution and Response to Rapamycin in Activated Phosphoinositide 3-Kinase Delta Syndrome: The European Society for Immunodeficiencies-Activated Phosphoinositide 3-Kinase Delta Syndrome Registry. Front Immunol (2018) 9:543. doi: 10.3389/fimmu.2018.00543
89. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical Spectrum and Features of Activated Phosphoinositide 3-Kinase Delta Syndrome: A Large Patient Cohort Study. J Allergy Clin Immunol (2017) 139:597–606.e4. doi: 10.1016/j.jaci.2016.06.021
90. Tornatore L, Sandomenico A, Raimondo D, Low C, Rocci A, Tralau-Stewart C, et al. Cancer-Selective Targeting of the NF-kappaB Survival Pathway With GADD45beta/MKK7 Inhibitors. Cancer Cell (2014) 26:495–508. doi: 10.1016/j.ccr.2014.07.027
91. Paciolla M, Pescatore A, Conte MI, Esposito E, Incoronato M, Lioi MB, et al. Rare Mendelian Primary Immunodeficiency Diseases Associated With Impaired NF-kappaB Signaling. Genes Immun (2015) 16:239–46. doi: 10.1038/gene.2015.3
92. Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss-Of-Function Nuclear Factor kappaB Subunit 1 (NFKB1) Variants are the Most Common Monogenic Cause of Common Variable Immunodeficiency in Europeans. J Allergy Clin Immunol (2018) 142:1285–96. doi: 10.1016/j.jaci.2018.01.039
93. Knudson KM, Pritzl CJ, Saxena V, Altman A, Daniels MA, Teixeiro E. NFkappaB-Pim-1-Eomesodermin Axis Is Critical for Maintaining CD8 T-Cell Memory Quality. Proc Natl Acad Sci USA (2017) 114:E1659–67. doi: 10.1073/pnas.1608448114
94. Bryant VL, Tangye SG. The Expanding Spectrum of NFkB1 Deficiency. J Clin Immunol (2016) 36:531–2. doi: 10.1007/s10875-016-0310-5
95. Fernald K, Kurokawa M. Evading Apoptosis in Cancer. Trends Cell Biol (2013) 23:620–33. doi: 10.1016/j.tcb.2013.07.006
96. Opferman JT. Apoptosis in the Development of the Immune System. Cell Death Differ (2008) 15:234–42. doi: 10.1038/sj.cdd.4402182
97. Kumar S, Cakouros D. Transcriptional Control of the Core Cell-Death Machinery. Trends Biochem Sci (2004) 29:193–9. doi: 10.1016/j.tibs.2004.02.001
98. Lee YR, Chen M, Pandolfi PP. The Functions and Regulation of the PTEN Tumour Suppressor: New Modes and Prospects. Nat Rev Mol Cell Biol (2018) 19:547–62. doi: 10.1038/s41580-018-0015-0
99. Driessen GJ, H IJ, Wentink M, Yntema HG, van Hagen PM, van Strien A, et al. Increased PI3K/Akt Activity and Deregulated Humoral Immune Response in Human PTEN Deficiency. J Allergy Clin Immunol (2016) 138:1744–47.e5. doi: 10.1016/j.jaci.2016.07.010
100. Zaki-Dizaji M, Akrami SM, Abolhassani H, Rezaei N, Aghamohammadi A. Ataxia Telangiectasia Syndrome: Moonlighting ATM. Expert Rev Clin Immunol (2017) 13:1155–72. doi: 10.1080/1744666X.2017.1392856
101. Biggs CM, Keles S, Chatila TA. DOCK8 Deficiency: Insights Into Pathophysiology, Clinical Features and Management. Clin Immunol (2017) 181:75–82. doi: 10.1016/j.clim.2017.06.003
102. Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
103. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal Dominant Immune Dysregulation Syndrome in Humans With CTLA4 Mutations. Nat Med (2014) 20:1410–6. doi: 10.1038/nm.3746
104. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients With LRBA Deficiency Show CTLA4 Loss and Immune Dysregulation Responsive to Abatacept Therapy. Science (2015) 349:436–40. doi: 10.1126/science.aaa1663
105. Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, et al. Spectrum of Phenotypes Associated With Mutations in LRBA. J Clin Immunol (2016) 36:33–45. doi: 10.1007/s10875-015-0224-7
106. Pan-Hammarstrom Q, Abolhassani H, Hammarstrom L. Defects in Plasma Cell Differentiation Are Associated With Primary Immunodeficiency in Human Subjects. J Allergy Clin Immunol (2018) 141:1217–9. doi: 10.1016/j.jaci.2017.10.025
107. Hajjar J, Guffey D, Minard CG, Orange JS. Increased Incidence of Fatigue in Patients With Primary Immunodeficiency Disorders: Prevalence and Associations Within the US Immunodeficiency Network Registry. J Clin Immunol (2017) 37:153–65. doi: 10.1007/s10875-016-0367-1
108. Stracker TH, Roig I, Knobel PA, Marjanovic M. The ATM Signaling Network in Development and Disease. Front Genet (2013) 4:37. doi: 10.3389/fgene.2013.00037
109. Shimizu I, Yoshida Y, Suda M, Minamino T. DNA Damage Response and Metabolic Disease. Cell Metab (2014) 20:967–77. doi: 10.1016/j.cmet.2014.10.008
110. Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA Repair, Genome Stability, and Aging. Cell (2005) 120:497–512. doi: 10.1016/j.cell.2005.01.028
111. Zaki-Dizaji M, Akrami SM, Azizi G, Abolhassani H, Aghamohammadi A. Inflammation, a Significant Player of Ataxia-Telangiectasia Pathogenesis? Inflammation Res (2018) 67:559–70. doi: 10.1007/s00011-018-1142-y
112. Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in Cancer. Vasc Health Risk Manag (2006) 2:213–9. doi: 10.2147/vhrm.2006.2.3.213
113. Shiota M, Yang X, Kubokawa M, Morishima T, Tanaka K, Mikami M, et al. Somatic Mosaicism for a NRAS Mutation Associates With Disparate Clinical Features in RAS-Associated Leukoproliferative Disease: A Report of Two Cases. J Clin Immunol (2015) 35:454–8. doi: 10.1007/s10875-015-0163-3
114. Calvo KR, Price S, Braylan RC, Oliveira JB, Lenardo M, Fleisher TA, et al. JMML and RALD (Ras-Associated Autoimmune Leukoproliferative Disorder): Common Genetic Etiology Yet Clinically Distinct Entities. Blood (2015) 125:2753–8. doi: 10.1182/blood-2014-11-567917
115. Meadows KN, Bryant P, Pumiglia K. Vascular Endothelial Growth Factor Induction of the Angiogenic Phenotype Requires Ras Activation. J Biol Chem (2001) 276:49289–98. doi: 10.1074/jbc.M108069200
116. Kranenburg O, Gebbink MF, Voest EE. Stimulation of Angiogenesis by Ras Proteins. Biochim Biophys Acta (2004) 1654:23–37. doi: 10.1016/j.bbcan.2003.09.004
117. Stockmann C, Schadendorf D, Klose R, Helfrich I. The Impact of the Immune System on Tumor: Angiogenesis and Vascular Remodeling. Front Oncol (2014) 4:69. doi: 10.3389/fonc.2014.00069
118. Costa C, Incio J, Soares R. Angiogenesis and Chronic Inflammation: Cause or Consequence? Angiogenesis (2007) 10:149–66. doi: 10.1007/s10456-007-9074-0
119. Albini A, Bruno A, Noonan DM, Mortara L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front Immunol (2018) 9:527. doi: 10.3389/fimmu.2018.00527
120. Rezaei N, Hedayat M, Aghamohammadi A, Nichols KE. Primary Immunodeficiency Diseases Associated With Increased Susceptibility to Viral Infections and Malignancies. J Allergy Clin Immunol (2011) 127:1329–41 e2quiz 1342–3. doi: 10.1016/j.jaci.2011.02.047
121. Tate G, Suzuki T, Kishimoto K, Mitsuya T. Novel Mutations of EVER1/TMC6 Gene in a Japanese Patient With Epidermodysplasia Verruciformis. J Hum Genet (2004) 49:223–5. doi: 10.1007/s10038-004-0135-6
122. Yoshii Y, Kato T, Ono K, Takahashi E, Fujimoto N, Kobayashi S, et al. Primary Cutaneous Follicle Center Lymphoma in a Patient With WHIM Syndrome. J Eur Acad Dermatol Venereol (2016) 30:529–30. doi: 10.1111/jdv.12927
123. Hellner K, Mar J, Fang F, Quackenbush J, Munger K. HPV16 E7 Oncogene Expression in Normal Human Epithelial Cells Causes Molecular Changes Indicative of an Epithelial to Mesenchymal Transition. Virology (2009) 391:57–63. doi: 10.1016/j.virol.2009.05.036
124. Duffy CL, Phillips SL, Klingelhutz AJ. Microarray Analysis Identifies Differentiation-Associated Genes Regulated by Human Papillomavirus Type 16 E6. Virology (2003) 314:196–205. doi: 10.1016/s0042-6822(03)00390-8
125. Merchant M, Caldwell RG, Longnecker R. The LMP2A ITAM is Essential for Providing B Cells With Development and Survival Signals In Vivo. J Virol (2000) 74:9115–24.
Keywords: inborn errors of immunity, primary immunodeficiency, predominantly antibody deficiency, hallmarks of cancer, immune dysregulation, genome instability, chronic inflammation
Citation: Abolhassani H, Wang Y, Hammarström L and Pan-Hammarström Q (2021) Hallmarks of Cancers: Primary Antibody Deficiency Versus Other Inborn Errors of Immunity. Front. Immunol. 12:720025. doi: 10.3389/fimmu.2021.720025
Received: 03 June 2021; Accepted: 28 July 2021;
Published: 17 August 2021.
Edited by:
Alison Mary Condliffe, The University of Sheffield, United KingdomReviewed by:
Vassilios Lougaris, University of Brescia, ItalySmita Y. Patel, John Radcliffe Hospital, United Kingdom
Copyright © 2021 Abolhassani, Wang, Hammarström and Pan-Hammarström. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hassan Abolhassani, aGFzc2FuLmFib2xoYXNzYW5pQGtpLnNl