 Chao Yang
Chao Yang Chan-Tat Ng
Chan-Tat Ng Dan Li
Dan Li Lei Zhang
Lei Zhang- 1National Engineering Research Center For Marine Aquaculture, Institute of Innovation & Application, Zhejiang Ocean University, Zhoushan, China
- 2Department of Psychology, National Chengchi University, Taipei, Taiwan
- 3Department of English, National Chengchi University, Taipei, Taiwan
- 4State Key Laboratory of Southwestern Chinese Medicine Resources, School of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 5Sericultural Research Institute, College of Biotechnology, Jiangsu University of Science and Technology, Zhenjiang, China
- 6Department of Chemical Engineering, Waterloo Institute for Nanotechnology, University of Waterloo, Waterloo, ON, Canada
The connection between indoleamine 2,3-dioxygenase 1 (IDO1) and tumour dormancy – a quiescent state of tumour cells which has been consistently linked to metastasis and cancer recurrence – is rarely discussed despite the pivotal role of IDO1 in cancer development and progression. Whilst the underlying mechanisms of IDO1-mediated dormancy are elusive, we summarize the IDO1 pathways which potentially contribute to dormancy in this review. Critically, distinct IDO1 activities are involved in dormancy initiation and maintenance; factors outside the well-studied IDO1/kynurenine/aryl hydrocarbon receptor axis, including the mammalian target of rapamycin and general control nonderepressible 2, appear to be implicated in dormancy. We also discuss various strategies for cancer treatment via regulating IDO1-dependent dormancy and suggest the application of nanotechnology to deliver effective treatment.
Introduction
Cancer cells often remain unaltered and hidden until appropriate conditions for further progression are met. This status can be explained by tumour dormancy which is defined as a stage during which dormant cancer cells reversibly enter a G0 phase of the cell cycle and exhibit neither proliferative nor apoptotic characteristics; at the population level, it is characterised as a quiescent state of equilibrium in which a balance is maintained between tumour cell division and death (1, 2). Dormancy appears to be a critical stage for cancer development since it promotes the long-term survival of tumour cells: dormant tumour cells are barely detected by the immune system, eventually contributing to metastases, cancer recurrence, or tolerance to anti-cancer treatments (3–5). Since tumour dormancy poses a challenge in fighting against cancers, uncovering its underlying mechanisms is essential.
Previous literature has extensively investigated factors associated with the dormancy mechanisms, and indoleamine 2,3-dioxygenase 1 (IDO1) has been identified as one of the players [for a comprehensive review of molecular cues involved in tumour dormancy, see (6)]. IDO1 acts as a rate-limiting enzyme in the kynurenine biosynthetic pathway. By converting tryptophan (Trp) into kynurenine (Kyn) in peripheral tissues, IDO1 triggers subsequent immunoregulatory events, including the suppression of T cells and NK cells and the activation of regulatory T cells and myeloid-derived suppressor cells (7, 8). Although the IDO1 expression in most normal adult tissues is low, it is highly increased in a wide range of human cancers (9, 10). Being regulated by interferons (IFNs), overexpressed IDO1 suppresses apoptosis and promotes tumour dormancy in tumour-repopulating cells (TRCs), a highly tumorigenic subpopulation of undifferentiated tumour cells (11–13). Furthermore, blockade of the IDO1 pathway disrupts the dormancy state and induces TRC apoptosis (11, 12). IDO1 inhibition has, therefore, been proposed as a potential strategy to break dormancy, promote tumour suppression, and simultaneously improve other anti-cancer treatments, including chemotherapy.
Despite that IDO1 blockers have shown encouraging effects towards tumour suppression in preclinical settings and phase I/II trials, they are far from satisfactory in phase III clinical development [focused reviews on the development of IDO1 inhibitors are available (9, 14, 15)]. In particular, treatment of melanoma with the use of Epacadostat (a selective inhibitor of IDO1) combined with pembrolizumab (an inhibitor of PD-1) does not offer significant advantages over placebo plus pembrolizumab (16). This finding implies a gap in the current literature regarding the role of IDO1 pathways in cancer development. Moreover, whilst a large body of literature had focused on eliminating active cancers through IDO1 inhibition, investigation on the relation between IDO1 and tumour dormancy is lacking. We herein review the current efforts on investigating the interactions between IDO1 signalling and tumour dormancy and discuss possible directions for future research on cancer therapy via targeting these interactions.
Upstream Regulation of IDO1-Associated Dormancy
Major factors contributing to IDO1 activation and regulation have been well documented, including interferons, tumour necrosis factor-alpha, pathogen- and damage-associated molecular patterns via JAK/STAT, NF-κB, or other signalling pathways (10). However, how exactly these factors induce or maintain tumour dormancy remains to be clarified. In this section, we summarise the upstream pathways of IDO1 which could be critical for regulating dormancy (see Figure 1).
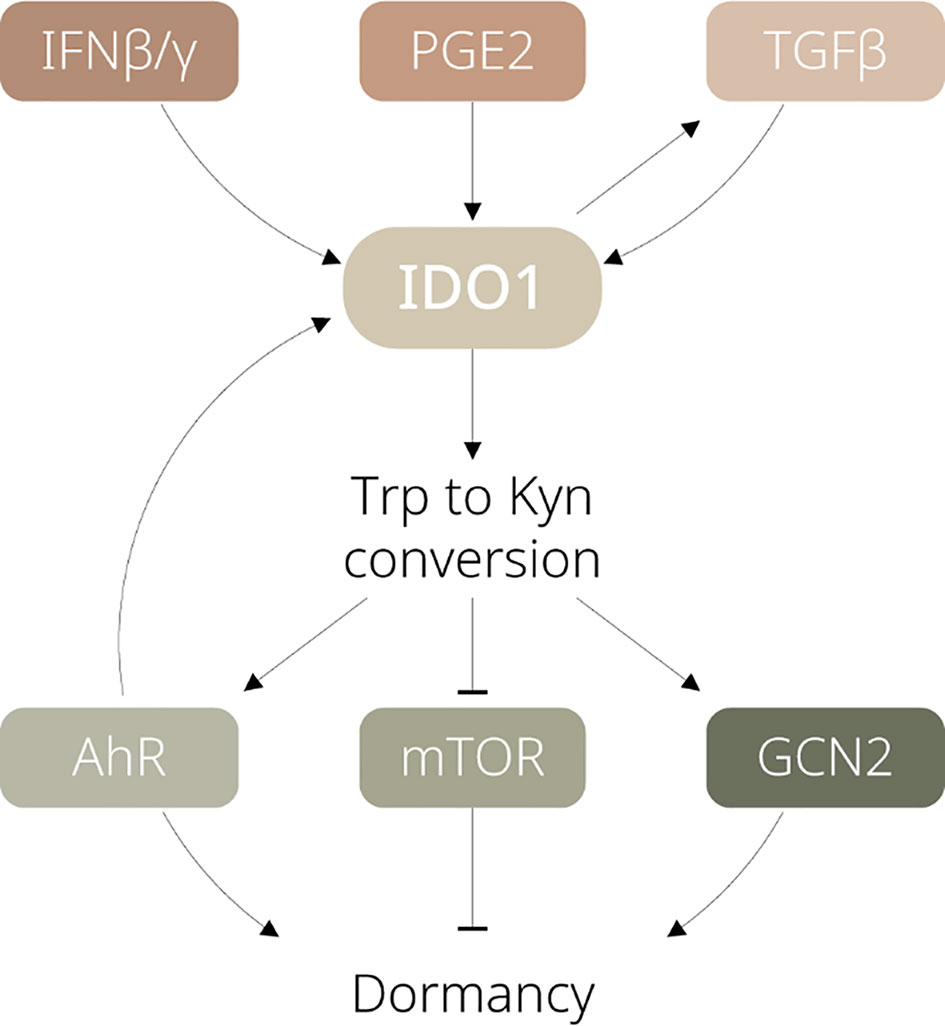
Figure 1 Schematic diagram illustrating the signalling pathways involved in IDO1-dependent tumour dormancy. IFN, interferon; PGE2, prostaglandin E2; TGF, transforming growth factor; Trp, tryptophan; Kyn, kynurenine; AhR, aryl hydrocarbon receptor; mTOR, mammalian target of rapamycin; GCN2, general control nonderepressible 2.
Interferons Trigger Dormancy in Tumour-Repopulating Cells
Interferons (IFNs) are signalling proteins expressed, primarily by cytotoxic T lymphocytes, NK cells, and plasmacytoid dendritic cells (pDCs), in response to viral infection (17). They can trigger the JAK/STAT signalling pathways and elevate the expression of interferon-stimulated genes. These genes promote apoptosis of the infected cells and are involved in further immune responses. Since tumour cells and virus-infected cells tend to activate similar responses, the IFN pathways also improve the anti-cancer responses by inducing tumour cell apoptosis.
However, stem-like TRCs are capable of hijacking such mechanisms for protection against the immune system (11, 12). Through the IDO1/Kyn/aryl hydrocarbon receptor (AhR) metabolic cascade, IFN-γ upregulates p27 to suppress the JAK/STAT signalling and simultaneously induces G0/G1 cell cycle arrest in TRCs (11). In addition, activated IFN-β is also reported to lead to TRC dormancy, by either initiating Jak1/Tyk signalling for IDO1-independent p27 upregulation, or activating IDO1 and sharing the IDO1/Kyn/AhR metabolic circuitry with IFN-γ signalling (see Figure 2) (12). Being able to induce IDO1-mediated immunosuppressive events and tumour dormancy, IFNs are considered as a key upstream player of IDO1 for switching TRCs into the dormancy state.
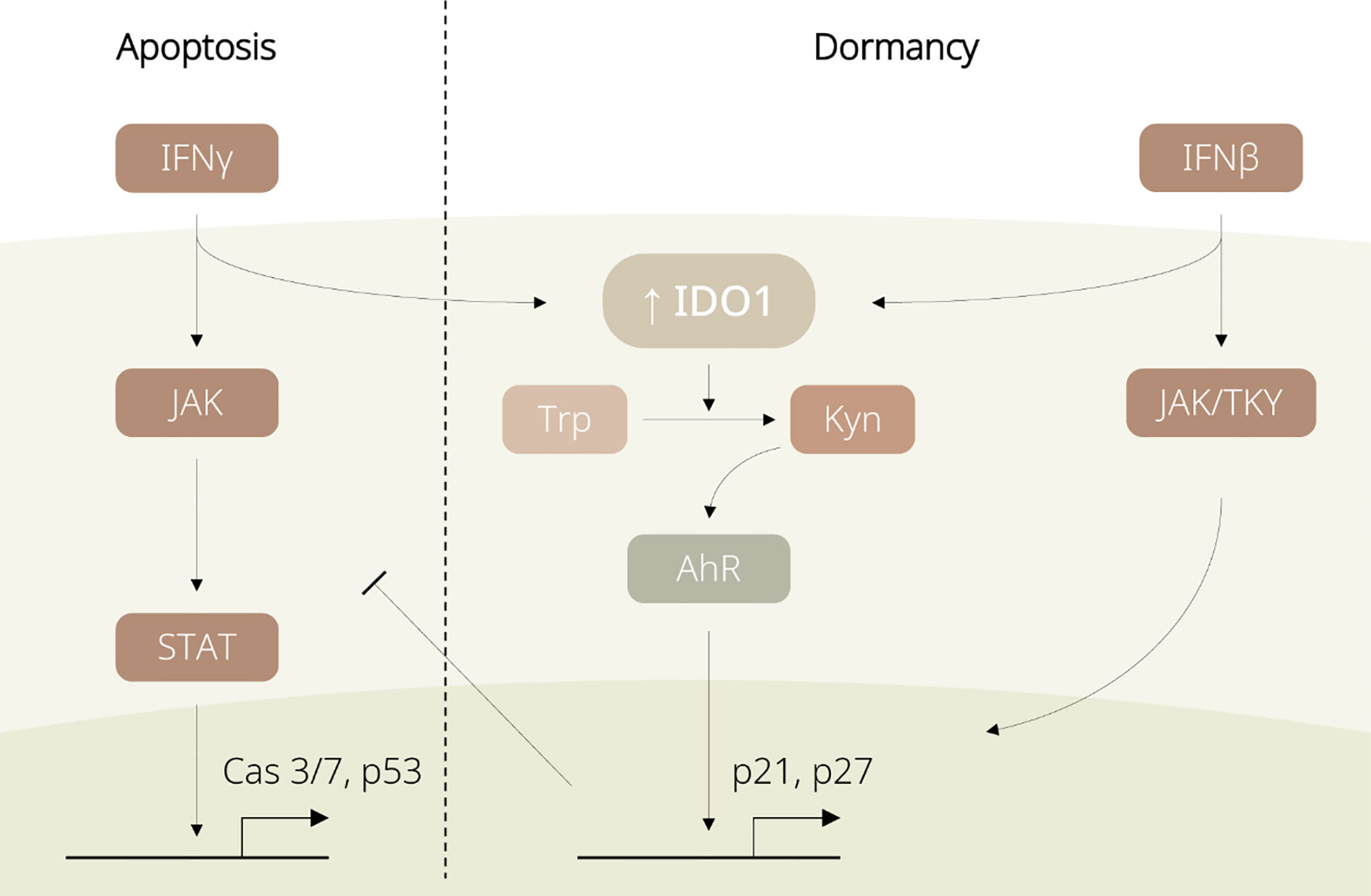
Figure 2 Diagram displaying regulation of IDO1-associated dormancy by IFN family members. IFN-γ induces apoptosis in non-tumour repopulating cells (non-TRCs) via the JAK/STAT pathway. In TRCs, IFNs favour the IDO1/Kyn/AhR pathway and promote p27 expression, leading to cell cycle arrest and tumour dormancy.
The IFN-stimulated IDO1/Kyn/AhR cascade is favoured over the JAK/STAT pathway in TRCs rather than in differentiated tumour cells. This difference between tumour subpopulations might be explained by their distinct expression profiles that TRCs exhibit higher levels of IDO1 and AhR than differentiated tumour cells do (11, 12). Even though IDO1 expression is temporarily enhanced by IFNs in non-TRCs, the levels of IDO1 and AhR are likely insufficient to trigger the downstream IDO1 signalling. Further, IDO1 inhibition has been evidenced to be associated with suppression of the IDO/Kyn/AhR/p27 axis, thereby driving the dormancy-to-apoptosis transition. In other words, the initially high levels of IDO1 could govern the domination of the IFN-induced IDO1/Kyn/AhR circuitry and determine the entry to dormancy; blockade of the IFN/IDO1 signalling potentially disrupts the dormancy state.
Additionally, SOX2, which acts upstream of IDO1 and downstream of IFN-γ, is identified as another potential key involved in TRC dormancy (18). As a well-known biomarker for stem-like cells, SOX2 is upregulated in a wide range of cancers to support tumour proliferation and metastasis, and elevated SOX2 levels are associated with drug resistance (19–22). However, suppressed SOX2 appears to be required for IFN-stimulated TRC dormancy (18). A similar conclusion is reached by another study which has indicated IFN-γ increases IDO1 and inhibits SOX2 expression in glioma stem cells (23). It is rational to slow down proliferation triggered by SOX2 to promote tumour dormancy, yet how these factors interact with each other during dormancy is unclear. Future studies should explore the relations among SOX2, the IFN/IDO1 cascade, and tumour dormancy.
Whilst the positive correlation of activities between IDO1 and IFN-γ is strong in colorectal cancers, for example, it is relatively weak in ovarian or kidney cancers such that they may show great IDO1 expression but less activity of IFN-γ. Thus, researchers should consider the variation of IDO1 and IFN-γ activities in different cancer cell lines, because the inconsistent relations between these two factors have implied (1) the presence of other IDO1 inducers and (2) multiple stages of dormancy captured (9, 10). Importantly, the IDO1 regulation in dormant cells is supposed to change over time, but their temporal profile is rarely dissected. Usually, at the early stages of tumour progression, the production of immunosuppressive factors by tumours is poor, allowing high levels of IFN secreted by the CTLs or NK cells for IDO1/AhR activation. Nonetheless, later stages are associated with stronger suppression of T cells or NK cells, and thus the subsequent IFN levels might be lowered. Indeed, several studies concerning the IDO1/Kyn/AhR metabolic axis argue that IFN-γ is needed only for initiation but not maintenance of tumour dormancy – some other factors have been involved in the consecutive IDO1 activity (24–26).
Collectively, IFN signalling appears to be crucial for initiating tumour dormancy. Yet, distinct mechanisms have been revealed, at least, between the initiation and maintenance of tumour dormancy, or between different tumour cells. One should pay special attention when attempting to alter the dormancy state through IDO pathway regulation. Merely targeting the IFN pathways seems to be insufficient for effectively controlling the IDO-dependent tumour dormancy.
Constitutive Expression of IDO1 Induced by Cyclooxygenase-2 Derived Prostaglandin E2
For the maintenance of tumour dormancy, the mechanical characteristics in the local tumour microenvironment have exerted important functions. For example, not only can stiffness facilitate p21 and p27 expression to trigger TRC dormancy, but it also helps maintain the dormancy state via inhibition of integrin β3 (27). In the meantime, stiffness affects the expressions of IDO1 as well as cyclooxygenase-2 (COX-2) in mesenchymal stem cells: both of their expressions are elevated even without cytokine stimulation (including IFNs) (28), suggesting that cytokines might not be necessary for IDO1 upregulation. More importantly, these results reveal a potential relationship between IDO1 and COX-2 under stiff conditions – a mechanical environment directly associated with TRC dormancy.
COX-2 and its downstream effector Prostaglandin E2 (PGE2) are now considered as potential contributors to cancer development and IDO1-dependent tumour dormancy (29, 30). COX-2 stimulated PGE2 facilitates IDO1 production via the PI3K/AKT and PKC signalling (30–32). Therefore, modification in any site within these pathways potentially results in the constitutive expression of IDO1. Expectedly, overexpression of COX-2/PGE2 is common in a broad range of human cancers, and it has been implicated in the mechanisms of anti-cell death and immunosuppression through Bcl-2 upregulation and IL-12 downregulation (33–38). Unlike IFNs, the COX-2 derived PGE2 allows consecutive IDO1 activity, and thus it appears to be responsible for the maintenance of the dormancy state. Consistently, the PGE2 pathways are reported to mediate the shift from dormancy to tumour growth, and suppression of these pathways awakes dormant tumour cells and sensitises them to immunotherapy (30, 39–42).
Apparently, the PGE2 signalling serves a crucial role in balancing apoptosis and growth in tumours, at least partly, through IDO1 mediation. Future research should examine how the interplay between IDO1 and PGE2 may impact on the initiation and maintenance of tumour dormancy. We also anticipate a systematic investigation of different cancer cell lines which should guarantee a more advanced understanding of the underpinnings of tumour dormancy.
Transforming Growth Factor-Stimulated Sustained Expression of IDO1
Aiming at the cancer-associated events triggered by IDO1 pathways, most researchers focused on the enzymatic effects of IDO1. Nonetheless, previous literature has demonstrated the noncatalytic activity of IDO1, particularly in plasmacytoid DCs, for induction of immunosuppression (43–45). Specifically, TGF-β1 treatment in pDCs in mice induces phosphatidylinositol-3-OH kinase (PI3K)-dependent phosphorylation of the IDO immunoreceptor tyrosine-based inhibitory motifs (ITIM1/2). The phosphorylated IDO1 further recruits the tyrosine phosphatases SHP1/2 and activates the non-canonical NF-κB pathway, eventually leading to an upregulated expression of TGF-β1 and IDO1. Moreover, the IDO1-dependent NF-κB pathway is not interrupted by 1-methyl-tryptophan (1-MT), a blocker of the IDO1 enzymatic activity, indicating that the noncatalytic IDO1 signalling could drive the self-sustained feedback loop of IDO1 (45).
In line with these studies, TGF-β promotes immunoregulatory events, including the differentiation of the regulatory T cells (Tregs) (46, 47). At the same time, TGF-β1-induced IDO1 signalling in pDCs upregulates IFN-β, a potential facilitator of tumour dormancy (45). On the other hand, IDO-competent DCs are likely to be recruited to induce IDO1 at a low level of IFN-γ in a rat model (24). Although IFN-γ drives a rapid immunoregulatory effect, it is independent of the long-term activation of IDO1; stable IDO1 activity is instead maintained in the IDO+ DCs by the IDO1 enzymatic activity as well as the increased AhR for a feedback IDO1-AhR-IDO1 loop in a self-sustained manner (25). Moreover, TGF-β1 expression is downregulated by IFN-γ in the model of Li and colleagues (25), suggesting a sequential activation of IFN-γ and TGF-β1. These findings indicate that the IDO1-associated dormancy could be maintained independently of the IFNs. However, how the high level of IFN-stimulated IDO1 expression in tumours is related to the IDO1 levels in DCs during dormancy remains to be explored. The intercellular crosstalk between the TGF and IFN pathways requires further investigation.
Downstream Mechanisms of Dormancy Driven by IDO1-Induced Tryptophan-to-Kynurenine Conversion
Aryl Hydrocarbon Receptor-Dependent Immunosuppression
As discussed in the previous section, the AhR signalling is activated by the IDO1-mediated conversion of kynurenine from tryptophan (11, 12). The activated AhR up-regulates p21 and p27 expression (48, 49), further contributing to cell-cycle inhibition and immunosuppression. Notably, in IFN-stimulated TRCs, the preference to the IDO1/Kyn/AhR cascade requires not only a high concentration of IDO1 but also the activated AhR, indicating the essential role of AhR in TRC dormancy (11). Consistently, the AhR binding with its ligand kynurenine results in Treg differentiation (50). It is marked that TGF-β activation elevates the expression of AhR, and optimisation of the TGF-β-driven Treg differentiation mostly relies on AhR (50–52). Given the profound relations between IDO1 and TGF-β, and between IDO1 and the downstream AhR, it will be of interest to examine whether the TGF-β-AhR interaction is directly mediated by IDO1. Regardless, AhR likely acts as an important effector of IDO1 and contributes to the TRC dormancy entry.
Suppression of Mammalian Target of Rapamycin Results in T-Cell Anergy
Other than the IDO1/Kyn/AhR signalling, IDO1-mediated immunosuppression can also be achieved through the mammalian target of rapamycin (mTOR) signalling (53, 54). The mTOR pathway is involved in cell cycle control (55). Additionally, mTOR activation through the regulation of PI3K/AKT signalling is essential for the functional activity of cytotoxic T cells. IDO-mediated degradation of tryptophan deactivates the mTOR signalling, thereby expanding Foxp3+ Tregs and suppressing cytotoxic T cells (56, 57). Previous studies have indicated that tryptophan degradation boosts differentiation of Tregs, and the subsequent conversion of kynurenine further leads to T-cell apoptosis (58, 59). It is noted that IFN stimulation does not immediately cause IDO1-induced intracellular depletion of tryptophan in dormant TRCs, which probably is achieved by the increased tryptophan transporters (11, 60). Therefore, impaired mTOR expression due to the extracellular deprivation of tryptophan at the local site can be explained by the enhanced tryptophan uptake of the dormant TRCs. Also, inhibition of IDO1 turns the tryptophan deficiency signal into the sufficiency signal, and the sufficient supply of tryptophan reactivates mTOR and relieves the dormancy state (54). As a result, the tryptophan-sensitive mTOR signalling serves as a potential effector pathway of IDO1, regulating the T immunity for immunosuppressive events including dormancy.
General Control Nonderepressible 2 Activation Leads to T-Cell Anergy
The general control nonderepressible 2 (GCN2) is identified as another sensor of tryptophan sufficiency, and always works together with the mTOR pathways to regulate T cell activity (61, 62). Whilst mTOR signalling is impaired in response to tryptophan deprivation, IDO1-mediated degradation of tryptophan stimulates the GCN2 pathway, eventually diminishing T-cell proliferation and enhancing apoptosis (59, 63). However, the role of GCN2 in T-cell depletion and immune tolerance is lately questioned. Particularly, in a melanoma mouse model, the IDO-triggered immune responses towards tumours were unaltered regardless of the presence of GCN2 (64). Yet, it is also demonstrated that GCN2 is needed for survival in previously activated T cells, but incapable of controlling the proliferation of naive T cells (53). Whether GCN2 is redundant or otherwise specialised with additional functions regarding dormancy is still unclear.
Prospective in Targeting IDO1-Associated Dormancy as Cancer Therapy
Breaking the Dormant State of Tumours Through IDO1 Inhibition
Owing to the critical role in tumour-associated immunosuppression, IDO1 appears to be a potential target for cancer treatment (54, 65–70). For example, 1-methyl-tryptophan (1MT) is widely used as an inhibitor for the IDO1 pathway in preclinical studies, although its validity is questioned. Later research has further suggested indoximod, the D racemer of 1MT, as a potential candidate in the clinical settings. Indoximod can attack cancer cells by restoring the suppression of mTOR signalling caused by tryptophan deprivation, thereby acting on T cells. However, it should be noted that indoximod does not target directly at IDO1, but disrupts the downstream Kyn pathways in an unknown way. In addition, Epacadostat, an IDO1 blocker through competitive inhibition, has been shown to enhance the growth of T and NK cells and suppress the differentiation of Tregs, although the phase III study of Epacadostat was not satisfying (9, 13). The negative results did not stop the investigations of IDO1 inhibitors, but have called for more efforts to their development as well as understanding the underlying mechanisms (71–73).
Van den Eynde et al. have summarised possible reasons and corresponding solutions specifically for the unsatisfactory clinical trial of Epacadostat (9). For example, Epacadostat tends to show the best inhibition against cancer cells with constitutive expression of IDO1 (i.e., the cancer cells produce IDO1 even when they are not inflamed) rather than those with inducible activity (i.e., the cancer cells produce IDO1 in response to T-cell infiltration); the concentration of Epacadostat administered may be insufficient for blocking the IDO1/AhR pathway; as mentioned earlier, there are multiple sources of IDO activation at different stages; IDO1 expression varies among different cancer cell lines. Whilst many of the inhibitors, including Epacadostat, are designed against the enzymatic activities of IDO1, few act on its non-catalytic functions. All these factors might contribute to the overall survival not being improved in the clinical trial of Long et al. (16). Critically, understanding why IDO1 inhibitors failed in clinical trials may aid the future development of IDO-based cancer treatment.
Targeting IDO1-associated dormancy yet lacks practicality, and few IDO inhibitors are tested against tumour dormancy. We expect future research to work on the impacts driven by IDO1 regulation, particularly on the dormancy mechanisms in addition to the active tumours. This should be crucial for the success of IDO1 treatment development, as tumour dormancy has been strongly associated with metastasis and later resistance to cancer treatments (74, 75). It should also be noted that there are other enzymes, including tryptophan-2,3-dioxygenase and IDO2, which are closely related to IDO1, yet they could have complementary or differential roles during immunosuppression: TDO triggers the same Kyn/AhR downstream signalling with IDO1 that they show nearly identical bioactivity (76); IDO2 is documented to implement in the IDO1-dependent regulation of T cells (77), but it is also reported that IDO1 and IDO2 may show opposite roles in immune responses (78). Although TDO and IDO2 are not the current focus of this article, researchers should pay close attention to them as targeting tumour dormancy via the Kyn/AhR signalling may not always be IDO1 specific.
Stabilising IDO1-Dependent Dormancy for Tumour Suppression
Perhaps paradoxically but intriguingly, it is wondered whether dormancy promotion is always pessimistic, or it may serve as an alternative strategy for controlling cancers. Dormant tumour cells are notorious for their hardly being detected or treated, but they have limited abilities to grow and proliferate. Promoting or stabilising dormant tumour cells, as a consequence, might slow down cancer progression and thereby provide alternative therapeutic effects. Moreover, previous mathematical models suggest that dormancy can be theoretically maintained for a long period of time, if not permanently (79). This paradigm is proposed as tumour dormancy therapy: instead of aiming at tumour elimination via aggressive treatments, the pro-dormancy approaches may suppress tumour cell growth and enhance cancer patients’ quality of life in a gentler manner (80–82). Tumour cells tend to remain dormant when low-dose immune checkpoint inhibitors are applied with hyperthermia [reviewed by Corthay and coworkers (83)]. Although being insufficient for eliminating cancers, low-dose inhibition prevents tumour cells from further progression. As an immune checkpoint enzyme, IDO1 is considered to be an appealing target for such a therapeutic approach.
Recently, chemotherapy itself has been identified to induce tumour dormancy, and the relation between chemotherapy and dormancy could be bridged by the type I IFN signalling (84). This suggests that we may even proactively activate the dormancy mechanisms to suppress tumour growth via the IDO-related pathways. Nonetheless, some studies suggest tumour cells may become more difficult to re-enter dormancy once they escape from immunosurveillance, plausibly because of the aggravated mutation rates (85–87). Researchers should consider how and when this paradigm should be applied, or otherwise it could be deteriorative not to provide sufficient suppression.
To emphasise, we are not advocating the replacement of conventional strategies which aim to break or prevent dormancy; however, tumour dormancy therapy can be used in concert with these established tumour-killing methods at different stages to achieve optimal outcomes (see Figure 3). Yet, no studies to date have attempted to bring forward the idea of promoting or stabilising dormancy via regulating IDO1 as a potential strategy to control cancer progression.
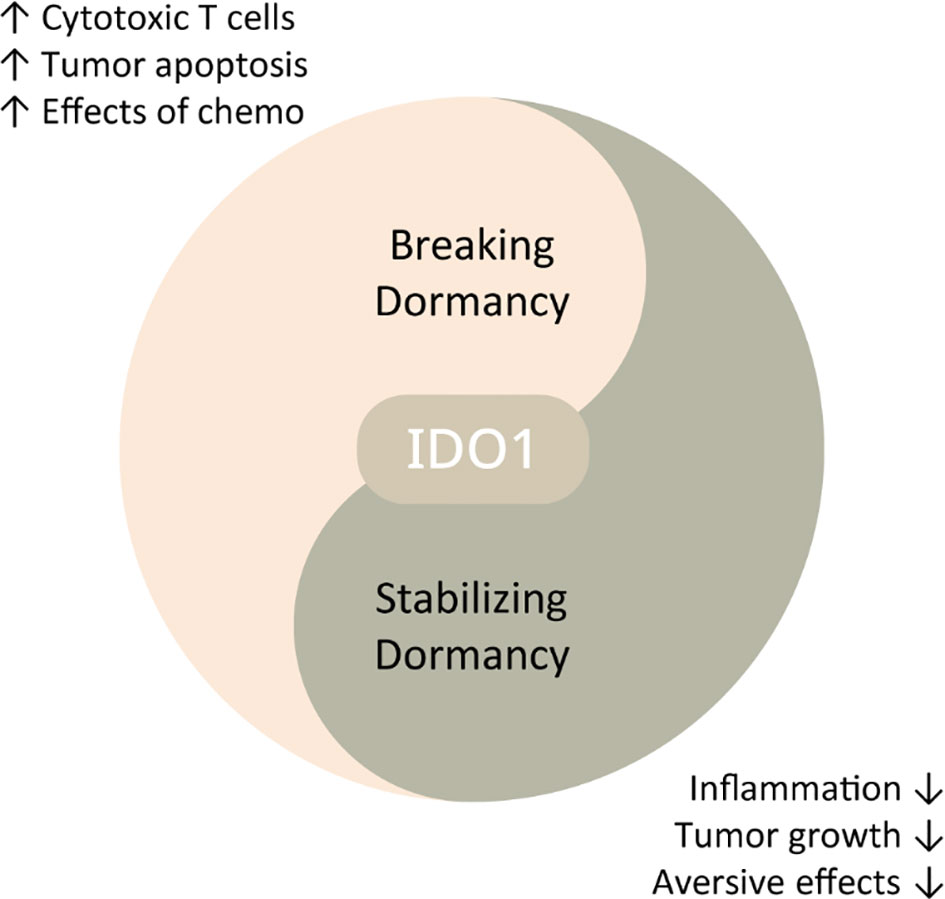
Figure 3 Schematic diagram displaying potential therapeutic strategies against cancers through IDO1 regulation. IDO1 inhibition may break dormancy, thereby promoting immune surveillance and enhancing other anti-cancer therapies. Low-dose IDO1 suppression can induce dormancy, alleviating tumour growth and tumour-related aversive effects.
Nanomaterials as a Delivery Platform of IDO1
How we should deliver treatments to the appropriate sites of the targeted cell types with controlled release is another challenge. Unsuccessful treatment delivery not only diminishes the therapeutic effects, but also intensifies the side effects. Tackling the issue, emerging nanomaterials are able to provide more flexible platforms for the delivery of small-molecule inhibitors, thereby improving cancer therapy (88–90). The application of nanotechnology could even solve some potential causes of the unsuccessful Epacadostat clinical trial and is briefly discussed in the following.
Currently, IDO1 antagonists combined with nanotechnology have demonstrated encouraging outcomes against tumorous events. For instance, PEG-Fmoc-NLG has been proposed as a dual-functional nanocarrier such that the carrier itself can improve immune responses and simultaneously be able to deliver both chemotherapy and IDO1 inhibition in mouse models of melanoma and breast cancer (91). More recent efforts have resulted in the modified polyvinyl alcohol (PVA)-based nanogels being identified to deliver IDO1 inhibitors together with chemo-medicines in a mouse breast cancer model. This delivery system has shown specificity of drug release at intracellular acidic conditions, and has led to the cytotoxic effects against the tumour cells and the reduction of IDO1-associated immunosuppression (92). Other nano-enabled approaches for a combination of IDO1 downregulation and chemotherapy may include the cationic lipid-assisted nanoparticles (CLANs) and DOX/IND-liposomal delivery systems (93, 94).
Based on these findings, we may anticipant nanotechnology to aid the development of IDO1 inhibitors and provide solutions for the failure of Epacadostat clinical trial. For example, the maximum allowable doses of inhibitors are usually restrained due to the trade-off between beneficial and adverse effects. As a result, the allowed administered level of IDO1 blockers may be insufficient to halt the IDO1/AhR signalling (9). Nanotechnology allows precise delivery that minimises unwanted effects and increases the allowed doses.
On the other hand, the use of nanotechnology has also been evidenced to be effective for controlling tumour dormancy. In an orthotopic mice model of colorectal tumour, injection of a lipid-protamine-DNA (LPD) nanoparticle loaded with PD-L1 trap synergises with chemotherapy (95). Such a combination therapy delivers precise treatment and therefore reduces adverse effects. More importantly, it can sensitise the immunologically cold tumour cells to the treatment. Liu et al. has recently developed a tumour-microenvironment (TME) sensitive nanocarrier for co-delivery of mitoxantrone and celestrol with an optimal ratio, leading to a successful prolongation of the dormancy state in desmoplastic melanoma (96). Not only have these findings validated the use of nanomaterials for breaking dormancy with accurate treatment delivery, but also they have pioneered the pro-dormancy strategy using nanomaterials. It would be exciting to examine whether these nanotechnology-based delivery systems are also useful for controlling IDO1-associated dormancy.
Conclusion
Previous literature has suggested IDO1 should be connected to immunosuppression and tumour dormancy. However, there is still much room for understanding how exactly these IDO1 pathways result in dormancy. One of the keys falls on the IDO1 expression patterns, and constitutive rather than simply inducible IDO1 expressions may be critical for entering the dormancy state. Furthermore, initiation and maintenance of dormancy appear to involve distinct mechanisms such that they require different molecular factors to participate. Having a more thorough understanding of these mechanisms should provide novel insights into dormancy regulation and cancer treatment. Regarding the perspective on IDO1-centered dormancy regulation, while current research has devoted to the development of IDO1 inhibitors for active tumour elimination, how these inhibitors may be applied in dormancy models needs further attention. On the other hand, pro-dormancy therapy through IDO1 activation may serve as an alternative strategy for cancer treatment, yet it requires additional investigation in the future. Finally, given the potential of nanotechnology, we foresee an ever-rising use of nanomaterials for providing accurate delivery of treatments targeting IDO1-associated dormancy and enhancing cancer therapy.
Author Contributions
CY and C-TN conceived and designed this manuscript. DL and ZL have read, revised and approved the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The work was supported by the Natural Science Foundation of Jiangsu Province, China (Grant No. BK20190960), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (Grant No. 19KJB180014), China Postdoctoral Science Foundation (2019M663456 and 2019TQ0044), Xinglin Scholar Research Promotion Project of Chengdu University of TCM (BSH2019008), Sichuan Province Science and technology innovation seedling project (2020091), Open Research Fund of Chengdu University of Traditional Chinese Medicine Key Laboratory of Systematic Research of Distinctive Chinese Medicine Resources in Southwest China (2020BSH004) and Canada Mitacs Fellowship (IT18262).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Aguirre-Ghiso JA. Models, Mechanisms and Clinical Evidence for Cancer Dormancy. Nat Rev Cancer (2007) 7:834–46. doi: 10.1038/nrc2256
2. Schreiber RD, Old LJ, Smyth MJ. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science (2011) 331:1565–70. doi: 10.1126/science.1203486
3. Gao XL, Zhang M, Tang YL, Liang XH. Cancer Cell Dormancy: Mechanisms and Implications of Cancer Recurrence and Metastasis. Onco Targets Ther (2017) 10:5219–28. doi: 10.2147/OTT.S140854
4. Phan TG, Croucher PI. The Dormant Cancer Cell Life Cycle. Nat Rev Cancer (2020) 20:398–411. doi: 10.1038/s41568-020-0263-0
5. Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of Disseminated Cancer Cell Dormancy: An Awakening Field. Nat Rev Cancer (2014) 14:611–22. doi: 10.1038/nrc3793
6. Jahanban-Esfahlan R, Seidi K, Manjili MH, Jahanban-Esfahlan A, Javaheri T, Zare P. Tumor Cell Dormancy: Threat or Opportunity in the Fight Against Cancer. Cancers (Basel) (2019) 11:1207. doi: 10.3390/cancers11081207
7. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, et al. Indoleamine 2,3-Dioxygenase Pathways of Pathogenic Inflammation and Immune Escape in Cancer. Cancer Immunol Immunother (2014) 63:721–35. doi: 10.1007/s00262-014-1549-4
8. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep (2015) 13:412–24. doi: 10.1016/j.celrep.2015.08.077
9. Van den Eynde BJ, van Baren N, Baurain J-F. Is There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annu Rev Cancer Biol (2020) 4:241–56. doi: 10.1146/annurev-cancerbio-030419-033635
10. Liu M, Wang X, Wang L, Ma X, Gong Z, Zhang S, et al. Targeting the IDO1 Pathway in Cancer: From Bench to Bedside. J Hematol Oncol (2018) 11:100–0. doi: 10.1186/s13045-018-0644-y
11. Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J, et al. Blockade of IDO-Kynurenine-AhR Metabolic Circuitry Abrogates IFN-γ-Induced Immunologic Dormancy of Tumor-Repopulating Cells. Nat Commun (2017) 8:15207. doi: 10.1038/ncomms15207
12. Liu Y, Lv J, Liu J, Liang X, Jin X, Xie J, et al. STAT3/p53 Pathway Activation Disrupts IFN-β-Induced Dormancy in Tumor-Repopulating Cells. J Clin Invest (2018) 128:1057–73. doi: 10.1172/JCI96329
13. Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell (2018) 33:480–494.e7. doi: 10.1016/j.ccell.2018.02.005
14. Labadie BW, Bao R, Luke JJ. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin Cancer Res (2018) 25:1462–71. doi: 10.1158/1078-0432.CCR-18-2882
15. Chen S, Tan J, Zhang A. The Ups, Downs and New Trends of IDO1 Inhibitors. Bioorg Chem (2021) 110:104815. doi: 10.1016/j.bioorg.2021.104815
16. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat Plus Pembrolizumab Versus Placebo Plus Pembrolizumab in Patients With Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol (2019) 20:1083–97. doi: 10.1016/S1470-2045(19)30274-8
17. Platanias LC. Mechanisms of Type-I- and Type-II-Interferon-Mediated Signalling. Nat Rev Immunol (2005) 5:375–86. doi: 10.1038/nri1604
18. Jia Q, Yang F, Huang W, Zhang Y, Bao B, Li K, et al. Low Levels of Sox2 are Required for Melanoma Tumor-Repopulating Cell Dormancy. Theranostics (2019) 9:424–35. doi: 10.7150/thno.29698
19. Zhang S, Xiong X, Sun Y. Functional Characterization of SOX2 as an Anticancer Target. Signal Transduct Tar (2020) 5:135. doi: 10.1038/s41392-020-00242-3
20. Grimm D, Bauer J, Wise P, Krüger M, Simonsen U, Wehland M, et al. The Role of SOX Family Members in Solid Tumours and Metastasis. Semin Cancer Biol (2020) 67:122–53. doi: 10.1016/j.semcancer.2019.03.004
21. Novak D, Hüser L, Elton JJ, Umansky V, Altevogt P, Utikal J. SOX2 in Development and Cancer Biology. Semin Cancer Biol (2020) 67:74–82. doi: 10.1016/j.semcancer.2019.08.007
22. Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, et al. SOX2 is an Amplified Lineage-Survival Oncogene in Lung and Esophageal Squamous Cell Carcinomas. Nat Genet (2009) 41:1238–42. doi: 10.1038/ng.465
23. Ozawa Y, Yamamuro S, Sano E, Tatsuoka J, Hanashima Y, Yoshimura S, et al. Indoleamine 2,3-Dioxygenase 1 is Highly Expressed in Glioma Stem Cells. Biochem Biophys Res Co (2020) 524:723–9. doi: 10.1016/j.bbrc.2020.01.148
24. Sun X, Gong ZJ, Wang ZW, Li T, Zhang JY, Sun HC, et al. IDO-Competent-DCs Induced by IFN-γ Attenuate Acute Rejection in Rat Liver Transplantation. J Clin Immunol (2012) 32:837–47. doi: 10.1007/s10875-012-9681-4
25. Li Q, Harden JL, Anderson CD, Egilmez NK. Tolerogenic Phenotype of IFN-γ-Induced IDO+ Dendritic Cells Is Maintained via an Autocrine IDO-Kynurenine/AhR-IDO Loop. J Immunol (2016) 197:962–70. doi: 10.4049/jimmunol.1502615
26. Holtzhausen A, Zhao F, Evans KS, Tsutsui M, Orabona C, Tyler DS, et al. Melanoma-Derived Wnt5a Promotes Local Dendritic-Cell Expression of IDO and Immunotolerance: Opportunities for Pharmacologic Enhancement of Immunotherapy. Cancer Immunol Res (2015) 3:1082–95. doi: 10.1158/2326-6066.CIR-14-0167
27. Liu Y, Lv J, Liang X, Yin X, Zhang L, Chen D, et al. Fibrin Stiffness Mediates Dormancy of Tumor-Repopulating Cells via a Cdc42-Driven Tet2 Epigenetic Program. Cancer Res (2018) 78:3926–37. doi: 10.1158/0008-5472.CAN-17-3719
28. Darnell M, Gu L, Mooney D. RNA-Seq Reveals Diverse Effects of Substrate Stiffness on Mesenchymal Stem Cells. Biomaterials (2018) 181:182–8. doi: 10.1016/j.biomaterials.2018.07.039
29. von Bergwelt-Baildon MS, Popov A, Saric T, Chemnitz J, Classen S, Stoffel MS, et al. CD25 and Indoleamine 2,3-Dioxygenase are Up-Regulated by Prostaglandin E2 and Expressed by Tumor-Associated Dendritic Cells In Vivo: Additional Mechanisms of T-Cell Inhibition. Blood (2006) 108:228–37. doi: 10.1182/blood-2005-08-3507
30. Hennequart M, Pilotte L, Cane S, Hoffmann D, Stroobant V, Plaen ED, et al. Constitutive IDO1 Expression in Human Tumors Is Driven by Cyclooxygenase-2 and Mediates Intrinsic Immune Resistance. Cancer Immunol Res (2017) 5:695–709. doi: 10.1158/2326-6066.CIR-16-0400
31. Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 Promotes Colon Cancer Cell Growth Through a Gs-Axin-ß-Catenin Signaling Axis. Science (2005) 310:1504–10. doi: 10.1126/science.1116221
32. Trabanelli S, Lecciso M, Salvestrini V, Cavo M, Očadlíková D, Lemoli RM, et al. PGE2-Induced IDO1 Inhibits the Capacity of Fully Mature DCs to Elicit an In Vitro Antileukemic Immune Response. J Immunol Res (2015) 2015:253191–1. doi: 10.1155/2015/253191
33. Hida T, Yatabe Y, Achiwa H, Muramatsu H, Kozaki K, Nakamura S, et al. Increased Expression of Cyclooxygenase 2 Occurs Frequently in Human Lung Cancers, Specifically in Adenocarcinomas. Cancer Res (1998) 58:3761–4.
34. Tucker ON, Dannenberg AJ, Yang EK, Zhang F, Teng L, Daly JM, et al. Cyclooxygenase-2 Expression is Up-Regulated in Human Pancreatic Cancer. Cancer Res (1999) 59:987–90.
35. Yoshimura N, Sano H, Okamoto M, Akioka K, Ushigome H, Kadotani Y, et al. Expression of Cyclooxygenase-1 and -2 in Human Breast Cancer. Surg Today (2003) 33:805–11. doi: 10.1007/s00595-003-2606-3
36. Tjiu J-W, Liao Y-H, Lin S-J, Huang Y-L, Tsai W-L, Chu C-Y, et al. Cyclooxygenase-2 Overexpression in Human Basal Cell Carcinoma Cell Line Increases Antiapoptosis, Angiogenesis, and Tumorigenesis. J Invest Dermatol (2006) 126:1143–51. doi: 10.1038/sj.jid.5700191
37. Liou J-Y, Ellent DP, Lee S, Goldsby J, Ko B-S, Matijevic N, et al. Cyclooxygenase-2-Derived Prostaglandin E2 Protects Mouse Embryonic Stem Cells From Apoptosis. Stem Cells (2007) 25:1096–103. doi: 10.1634/stemcells.2006-0505
38. Yao C, Hirata T, Soontrapa K, Ma X, Takemori H, Narumiya S. Prostaglandin E2 Promotes Th1 Differentiation via Synergistic Amplification of IL-12 Signalling by cAMP and PI3-Kinase. Nat Commun (2013) 4:1685. doi: 10.1038/ncomms2684
39. Sosnoski DM, Norgard RJ, Grove CD, Foster SJ, Mastro AM. Dormancy and Growth of Metastatic Breast Cancer Cells in a Bone-Like Microenvironment. Clin Exp Metastasis (2015) 32:335–44. doi: 10.1007/s10585-015-9710-9
40. Liu CM, Okayasu T, Goldman P, Suzuki Y, Suzuki K, Wheelock EF. Immune Regulation of the L5178Y Murine Tumor-Dormant State. I. In Vivo and In Vitro Effects of Prostaglandin E2 and Indomethacin on Tumor Cell Growth. J Exp Med (1986) 164:1259–73. doi: 10.1084/jem.164.4.1259
41. Hou W, Sampath P, Rojas JJ, Thorne SH. Oncolytic Virus-Mediated Targeting of PGE2 in the Tumor Alters the Immune Status and Sensitizes Established and Resistant Tumors to Immunotherapy. Cancer Cell (2016) 30:108–19. doi: 10.1016/j.ccell.2016.05.012
42. Wang D, Wang H, Shi Q, Katkuri S, Walhi W, Desvergne B, et al. Prostaglandin E2 Promotes Colorectal Adenoma Growth via Transactivation of the Nuclear Peroxisome Proliferator-Activated Receptor δ. Cancer Cell (2004) 6:285–95. doi: 10.1016/j.ccr.2004.08.011
43. Albini E, Rosini V, Gargaro M, Mondanelli G, Belladonna ML, Pallotta MT, et al. Distinct Roles of Immunoreceptor Tyrosine-Based Motifs in Immunosuppressive Indoleamine 2,3-Dioxygenase 1. J Cell Mol Med (2017) 21:165–76. doi: 10.1111/jcmm.12954
44. Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, et al. Aryl Hydrocarbon Receptor Control of a Disease Tolerance Defence Pathway. Nature (2014) 511:184–90. doi: 10.1038/nature13323
45. Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, et al. Indoleamine 2,3-Dioxygenase Is a Signaling Protein in Long-Term Tolerance by Dendritic Cells. Nat Immunol (2011) 12:870–8. doi: 10.1038/ni.2077
46. Fu S, Zhang N, Yopp AC, Chen D, Mao M, Chen D, et al. TGF-Beta Induces Foxp3 + T-Regulatory Cells From CD4 + CD25 - Precursors. Am J Transplant (2004) 4:1614–27. doi: 10.1111/j.1600-6143.2004.00566.x
47. Konkel JE, Zhang D, Zanvit P, Chia C, Zangarle-Murray T, Jin W, et al. Transforming Growth Factor-β Signaling in Regulatory T Cells Controls T Helper-17 Cells and Tissue-Specific Immune Responses. Immunity (2017) 46:660–74. doi: 10.1016/j.immuni.2017.03.015
48. Kolluri SK, Weiss C, Koff A, Göttlicher M. P27(Kip1) Induction and Inhibition of Proliferation by the Intracellular Ah Receptor in Developing Thymus and Hepatoma Cells. Genes Dev (1999) 13:1742–53. doi: 10.1101/gad.13.13.1742
49. Pang PH, Lin YH, Lee YH, Hou HH, Hsu SP, Juan SH. Molecular Mechanisms of P21 and P27 Induction by 3-Methylcholanthrene, an Aryl-Hydrocarbon Receptor Agonist, Involved in Antiproliferation of Human Umbilical Vascular Endothelial Cells. J Cell Physiol (2008) 215:161–71. doi: 10.1002/jcp.21299
50. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An Interaction Between Kynurenine and the Aryl Hydrocarbon Receptor can Generate Regulatory T Cells. J Immunol (Baltimore Md.: 1950) (2010) 185:3190–8. doi: 10.4049/jimmunol.0903670
51. Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl Hydrocarbon Receptor Regulates Stat1 Activation and Participates in the Development of Th17 Cells. Proc Natl Acad Sci USA (2008) 105:9721–6. doi: 10.1073/pnas.0804231105
52. Negishi T, Kato Y, Ooneda O, Mimura J, Takada T, Mochizuki H, et al. Effects of Aryl Hydrocarbon Receptor Signaling on the Modulation of Th1/Th2 Balance. J Immunol (2005) 175:7348–56. doi: 10.4049/jimmunol.175.11.7348
53. Cobbold SP, Adams E, Farquhar CA, Nolan KF, Howie D, Lui KO, et al. Infectious Tolerance via the Consumption of Essential Amino Acids and mTOR Signaling. Proc Natl Acad Sci USA (2009) 106:12055–60. doi: 10.1073/pnas.0903919106
54. Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO Inhibits a Tryptophan Sufficiency Signal That Stimulates mTOR: A Novel IDO Effector Pathway Targeted by D-1-Methyl-Tryptophan. Oncoimmunology (2012) 1:1460–8. doi: 10.4161/onci.21716
55. Cuyàs E, Corominas-Faja B, Joven J, Menendez JA. Cell Cycle Regulation by the Nutrient-Sensing Mammalian Target of Rapamycin (mTOR) Pathway. Methods Mol Biol (2014) 1170:113–44. doi: 10.1007/978-1-4939-0888-2_7
56. Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR Axis Regulates De Novo Differentiation of CD4+Foxp3+ Cells. J Exp Med (2008) 205:565–74. doi: 10.1084/jem.20071477
57. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T Cell Receptor Signaling Controls Foxp3 Expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA (2008) 105:7797–802. doi: 10.1073/pnas.0800928105
58. Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T Cell Apoptosis by Tryptophan Catabolism. Cell Death Differ (2002) 9:1069–77. doi: 10.1038/sj.cdd.4401073
59. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells. J Immunol (2006) 176:6752–61. doi: 10.4049/jimmunol.176.11.6752
60. Kudo Y, Boyd CA. The Role of L-Tryptophan Transport in L-Tryptophan Degradation by Indoleamine 2,3-Dioxygenase in Human Placental Explants. J Physiol (2001) 531:417–23. doi: 10.1111/j.1469-7793.2001.0417i.x
61. Chotechuang N, Azzout-Marniche D, Bos C, Chaumontet C, Gausserès N, Steiler T, et al. mTOR, AMPK, and GCN2 Coordinate the Adaptation of Hepatic Energy Metabolic Pathways in Response to Protein Intake in the Rat. Am J Physiol Endocrinol Metab (2009) 297:E1313–23. doi: 10.1152/ajpendo.91000.2008
62. Staschke KA, Dey S, Zaborske JM, Palam LR, McClintick JN, Pan T, et al. Integration of General Amino Acid Control and Target of Rapamycin (TOR) Regulatory Pathways in Nitrogen Assimilation in Yeast. J Biol Chem (2010) 285:16893–911. doi: 10.1074/jbc.M110.121947
63. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity (2005) 22:633–42. doi: 10.1016/j.immuni.2005.03.013
64. Sonner JK, Deumelandt K, Ott M, Thomé CM, Rauschenbach KJ, Schulz S, et al. The Stress Kinase GCN2 Does Not Mediate Suppression of Antitumor T Cell Responses by Tryptophan Catabolism in Experimental Melanomas. Oncoimmunology (2016) 5:e1240858. doi: 10.1080/2162402X.2016.1240858
65. Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, et al. Inhibition of Indoleamine 2,3-Dioxygenase in Dendritic Cells by Stereoisomers of 1-Methyl-Tryptophan Correlates With Antitumor Responses. Cancer Res (2007) 67:792–801. doi: 10.1158/0008-5472.CAN-06-2925
66. Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res (2017) 77:6795–811. doi: 10.1158/0008-5472.CAN-17-2285
67. Tomek P, Palmer BD, Flanagan JU, Sun C, Raven EL, Ching LM. Discovery and Evaluation of Inhibitors to the Immunosuppressive Enzyme Indoleamine 2,3-Dioxygenase 1 (IDO1): Probing the Active Site-Inhibitor Interactions. Eur J Med Chem (2017) 126:983–96. doi: 10.1016/j.ejmech.2016.12.029
68. Zou Y, Wang Y, Wang F, Luo M, Li Y, Liu W, et al. Discovery of Potent IDO1 Inhibitors Derived From Tryptophan Using Scaffold-Hopping and Structure-Based Design Approaches. Eur J Med Chem (2017) 138:199–211. doi: 10.1016/j.ejmech.2017.06.039
69. Wang YX, Pang WQ, Zeng QX, Deng ZS, Fan TY, Jiang JD, et al. Synthesis and Biological Evaluation of New Berberine Derivatives as Cancer Immunotherapy Agents Through Targeting IDO1. Eur J Med Chem (2018) 143:1858–68. doi: 10.1016/j.ejmech.2017.10.078
70. Weng T, Qiu X, Wang J, Li Z, Bian J. Recent Discovery of Indoleamine-2,3-Dioxygenase 1 Inhibitors Targeting Cancer Immunotherapy. Eur J Med Chem (2018) 143:656–69. doi: 10.1016/j.ejmech.2017.11.088
71. Du Q, Feng X, Wang Y, Xu X, Zhang Y, Qu X, et al. Discovery of Phosphonamidate IDO1 Inhibitors for the Treatment of Non-Small Cell Lung Cancer. Eur J Med Chem (2019) 182:111629. doi: 10.1016/j.ejmech.2019.111629
72. Röhrig UF, Reynaud A, Majjigapu SR, Vogel P, Pojer F, Zoete V. Inhibition Mechanisms of Indoleamine 2,3-Dioxygenase 1 (Ido1). J Med Chem (2019) 62:8784–95. doi: 10.1021/acs.jmedchem.9b00942
73. Zou Y, Hu Y, Ge S, Zheng Y, Li Y, Liu W, et al. Effective Virtual Screening Strategy Toward Heme-Containing Proteins: Identification of Novel IDO1 Inhibitors. Eur J Med Chem (2019) 184:111750. doi: 10.1016/j.ejmech.2019.111750
74. De Angelis ML, Francescangeli F, La Torre F, Zeuner A. Stem Cell Plasticity and Dormancy in the Development of Cancer Therapy Resistance. Front Oncol (2019) 9. doi: 10.3389/fonc.2019.00626
75. Fluegen G, Avivar-Valderas A, Wang Y, Padgen MR, Williams JK, Nobre AR, et al. Phenotypic Heterogeneity of Disseminated Tumour Cells is Preset by Primary Tumour Hypoxic Microenvironments. Nat Cell Biol (2017) 19:120–32. doi: 10.1038/ncb3465
76. Platten M, von Knebel Doeberitz N, Oezen I, Wick W, Ochs K. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front Immunol (2015) 5. doi: 10.3389/fimmu.2014.00673
77. Metz R, Smith C, DuHadaway JB, Chandler P, Baban B, Merlo LMF, et al. IDO2 is Critical for IDO1-Mediated T-Cell Regulation and Exerts a non-Redundant Function in Inflammation. Int Immunol (2014) 26:357–67. doi: 10.1093/intimm/dxt073
78. Bilir C, Sarisozen C. Indoleamine 2,3-Dioxygenase (IDO): Only an Enzyme or a Checkpoint Controller? J Oncol Sci (2017) 3:52–6. doi: 10.1016/j.jons.2017.04.001
79. Romero I, Garrido F, Garcia-Lora AM. Metastases in Immune-Mediated Dormancy: A New Opportunity for Targeting Cancer. Cancer Res (2014) 74:6750–7. doi: 10.1158/0008-5472.CAN-14-2406
80. Lee J-H, Koung F-P, Cho C-K, Lee Y-W, Yoo H-S. Review of Tumor Dormancy Therapy Using Traditional Oriental Herbal Medicine. J Pharmacopuncture (2013) 16:12–20. doi: 10.3831/KPI.2013.16.005
81. Kim J, Cho C-K, Yoo H-S. Survival Analysis of Advanced Non-Small Cell Lung Cancer Patients Treated by Using Wheel Balance Cancer Therapy. Integr Cancer Ther (2016) 15:467–77. doi: 10.1177/1534735416639714
82. Saito M, Fujita Y, Kuribayashi N, Uchida D, Komiyama Y, Fukumoto C, et al. Troglitazone, a Selective Ligand for Pparγ, Induces Cell-Cycle Arrest in Human Oral SCC Cells. Anticancer Res (2020) 40:1247–54. doi: 10.21873/anticanres.14066
83. Corthay A, Bakacs T, Thangavelu G, Anderson CC. Tackling Cancer Cell Dormancy: Insights From Immune Models, and Transplantation. Semin Cancer Biol (2021). doi: 10.1016/j.semcancer.2021.02.002
84. Lan Q, Peyvandi S, Duffey N, Huang Y-T, Barras D, Held W, et al. Type I Interferon/IRF7 Axis Instigates Chemotherapy-Induced Immunological Dormancy in Breast Cancer. Oncogene (2019) 38:2814–29. doi: 10.1038/s41388-018-0624-2
85. Greaves M, Maley CC. Clonal Evolution in Cancer. Nature (2012) 481:306–13. doi: 10.1038/nature10762
86. Manjili MH. Tumor Dormancy and Relapse: From a Natural Byproduct of Evolution to a Disease State. Cancer Res (2017) 77:2564–69. doi: 10.1158/0008-5472.CAN-17-0068
87. Peyvandi S, Lan Q, Lorusso G, Rüegg C. Chemotherapy-Induced Immunological Breast Cancer Dormancy: A New Function for Old Drugs? J Cancer Metastasis Treat (2019) 5. doi: 10.20517/2394-4722.2019.16
88. Goldberg MS. Improving Cancer Immunotherapy Through Nanotechnology. Nat Rev Cancer (2019) 19:587–602. doi: 10.1038/s41568-019-0186-9
89. Riley RS, June CH, Langer R, Mitchell MJ. Delivery Technologies for Cancer Immunotherapy. Nat Rev Drug Discov (2019) 18:175–96. doi: 10.1038/s41573-018-0006-z
90. Sang W, Zhang Z, Dai Y, Chen X. Recent Advances in Nanomaterial-Based Synergistic Combination Cancer Immunotherapy. Chem Soc Rev (2019) 48:3771–810. doi: 10.1039/C8CS00896E
91. Chen Y, Xia R, Huang Y, Zhao W, Li J, Zhang X, et al. An Immunostimulatory Dual-Functional Nanocarrier That Improves Cancer Immunochemotherapy. Nat Commun (2016) 7:13443. doi: 10.1038/ncomms13443
92. Qiao H, Chen X, Chen E, Zhang J, Huang D, Yang D, et al. Folated pH-Degradable Nanogels for the Simultaneous Delivery of Docetaxel and an IDO1-Inhibitor in Enhancing Cancer Chemo-Immunotherapy. Biomater Sci-UK (2019) 7:2749–58. doi: 10.1039/C9BM00324J
93. Lu J, Liu X, Liao Y-P, Wang X, Ahmed A, Jiang W, et al. Breast Cancer Chemo-Immunotherapy Through Liposomal Delivery of an Immunogenic Cell Death Stimulus Plus Interference in the IDO-1 Pathway. ACS Nano (2018) 12:11041–61. doi: 10.1021/acsnano.8b05189
94. Huang H, Jiang C-T, Shen S, Liu A, Gan Y-J, Tong Q-S, et al. Nanoenabled Reversal of IDO1-Mediated Immunosuppression Synergizes With Immunogenic Chemotherapy for Improved Cancer Therapy. Nano Lett (2019) 19:5356–65. doi: 10.1021/acs.nanolett.9b01807
95. Song W, Shen L, Wang Y, Liu Q, Goodwin TJ, Li J, et al. Synergistic and Low Adverse Effect Cancer Immunotherapy by Immunogenic Chemotherapy and Locally Expressed PD-L1 Trap. Nat Commun (2018) 9:2237. doi: 10.1038/s41467-018-04605-x
Keywords: tumour dormancy, immunosuppression, kynurenine, nanotechnology, IDO1 regulation, aryl hydrocarbon receptor
Citation: Yang C, Ng C-T, Li D and Zhang L (2021) Targeting Indoleamine 2,3-Dioxygenase 1: Fighting Cancers via Dormancy Regulation. Front. Immunol. 12:725204. doi: 10.3389/fimmu.2021.725204
Received: 15 June 2021; Accepted: 12 August 2021;
Published: 03 September 2021.
Edited by:
Nurit Hollander, Tel Aviv University, IsraelReviewed by:
Ute Roehrig, Swiss Institute of Bioinformatics (SIB), SwitzerlandFabian Salazar, University of Exeter, United Kingdom
Robert H. McCusker, University of Illinois at Urbana-Champaign, United States
Copyright © 2021 Yang, Ng, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Li, bGlkYW5AY2R1dGNtLmVkdS5jbg==; Lei Zhang, bDc4emhhbmdAdXdhdGVybG9vLmNh
†These authors have contributed equally to this work