 Sha Wu
Sha Wu Xiao-Feng Li
Xiao-Feng Li Yuan-Yuan Wu1†
Yuan-Yuan Wu1† Cheng Huang
Cheng Huang Jun Li
Jun Li- 1Anhui Institute of Innovative Drugs, The Key Laboratory of Anti-inflammatory and Immune Medicines, Ministry of Education, Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, School of Pharmacy, Anhui Medical University, Hefei, China
- 2Postdoctoral Station of Clinical Medicine of Anhui Medical University, Hefei, China
Rheumatoid arthritis (RA), one of the most common autoimmune diseases, is characterized by immune cell infiltration, fibroblast-like synovial cell hyperproliferation, and cartilage and bone destruction. To date, numerous studies have demonstrated that immune cells are one of the key targets for the treatment of RA. N6-methyladenosine (m6A) is the most common internal modification to eukaryotic mRNA, which is involved in the splicing, stability, export, and degradation of RNA metabolism. m6A methylated-related genes are divided into writers, erasers, and readers, and they are critical for the regulation of cell life. They play a significant role in various biological processes, such as virus replication and cell differentiation by controlling gene expression. Furthermore, a growing number of studies have indicated that m6A is associated with the occurrence of numerous diseases, such as lung cancer, bladder cancer, gastric cancer, acute myeloid leukemia, and hepatocellular carcinoma. In this review, we summarize the history of m6A research and recent progress on RA research concerning m6A enzymes. The relationship between m6A enzymes, immune cells, and RA suggests that m6A modification offers evidence for the pathogenesis of RA, which will help in the development of new therapies for RA.
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disorder characterized by synovial hyperplasia and inflammation, progressive joint destruction, and significant disability (1). Initial lesions in RA include damage to the microvascular system and proliferation of fibroblast-like synovial (FLS) cells that line the synovial membrane of the joint. As the number of synovial cells increases, the cells attach to the articular surface at the edge of the joint, which induces further proliferation and activation of synovial cells. FLS cells are the primary synovial cells, which secrete inflammatory cytokines, chemokines, and metalloproteinases (1). Furthermore, inflammatory cytokines interact with immune cells that are related to RA, such as macrophages, natural killer (NK) cells, dendritic cells (DC), lymphocytes, and mast cells (MCs) (2). Currently, non-steroidal anti-inflammatory drugs and immunosuppressants are commonly used in clinical practice to alleviate the prevent the development of RA. Therefore, further innovations in treatments for RA are needed.
N6-methyladenosine of RNA is a methylation modification of adenine (A) at the sixth N, catalyzed by methyltransferase from eukaryotes (3). M6A modification has been discovered in various types of RNA, including transfer RNA, ribosome RNA, and even non-coding RNA (4, 5). Through the combination of RNA immunoprecipitation and the m6A antibody, RNA-sequencing showed that multiple microRNAs (miRNAs) contained m6A modifications (6). MiRNAs target regulated m6A-related enzymes and m6A-affected miRNA transcriptional regulation; therefore, m6A modification is important for the biogenesis and stability of at least some miRNAs. For example, methyltransferase-like 3 (METTL3) affects the binding of DGCR8 to pri-miRNAs, and the absence of METTL3 reduces this binding capacity (4). M6A has been applied to immune cells and various immune diseases, with the goal of devising new therapies. So far, researches on the molecular mechanism and function of the m6A effectors are abundant (7–10). In this review, we have focused on the potential relationship between m6A and RA to explore better methods to treat RA. We highlight several advances in the research of m6A in RA, cancers, and immune cells. However, to fully understand the effect of m6A on RA in vivo and its influence on gene expression, further exploration is needed.
The Major Enzymes in the m6A Pathway
Writers
M6A regulators are divided into three types: writers, erasers, and readers. The classifications differ according to the different roles they play in RNA methylation modification, time for discovery, and function of m6A-related enzymes and proteins, which are shown in Figure 1 (11–22). The first type is m6A methyltransferase, which promotes m6A methylation modification to RNA, and its coding genes are known as writers. The earliest writers discovered are METTL3, METTL14, and wilms’ tumor 1-associating protein (WTAP).
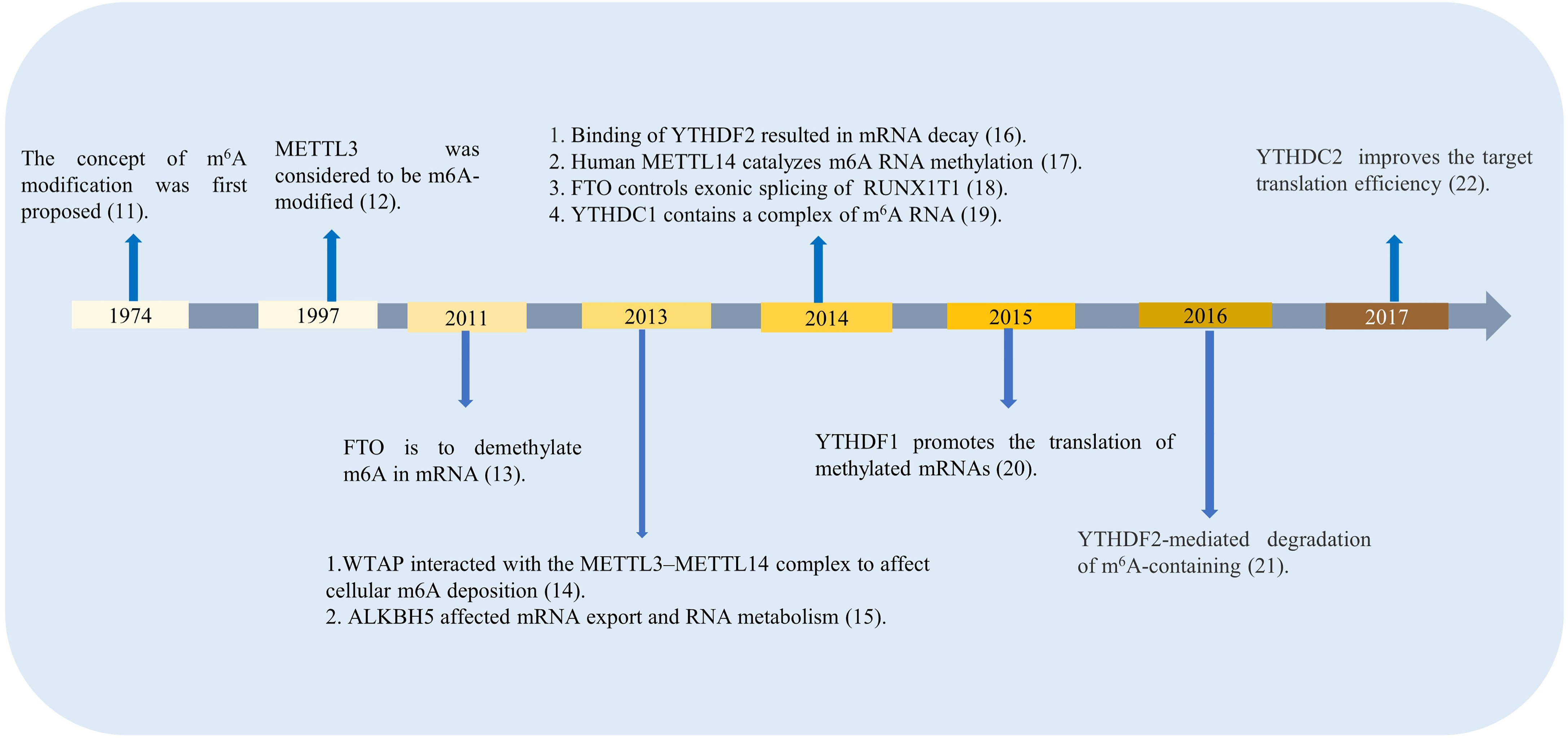
Figure 1 Time for discovery and functions of m6A-related enzymes and proteins.
In the RNA life cycle of m6A, METTL3 has important functions, which include pre-mRNA splicing, nuclear export, translation regulation, mRNA decay, and miRNA processing; moreover, METTL3 also functions to influence epigenetics by regulating and initiating pluripotency (23). Previously, METTL3 was reported as a tumor suppressor because of its upregulating effect on the m6A modification (24). METTL3 expression is increased in gastric cancer tissues, hepaticcellcarcinoma (HCC), breast cancer (BC) and mediates the proliferation, metastasis, and colony formation of cancer cells (25–27). In addition, METTL3 plays an unusual role in spermatogonial cells (28), bone marrow mesenchymal stem cells (29), fat mass (30), immune cells and inflammation. Figure 2 shows the highlights of METTL3 discoveries over time. Recently, studies have indicated that m6A methylation modification plays an indispensable role in the innate immune response and antitumor immunity. However, the mechanism of action of METTL3 in RA remains unclear. It has been reported that the expression of METTL3 is significantly elevated in patients with RA, and METTL3 affects the secretion of inflammatory factors in RA through the NF-κB pathway (31). Given the influence on inflammation, cancers, and other diseases, there has been speculation that METTL3 has the potential to treat immune diseases (32). For more information about the expression of METTL3 in diseases and related genes, please refer to Table 1.
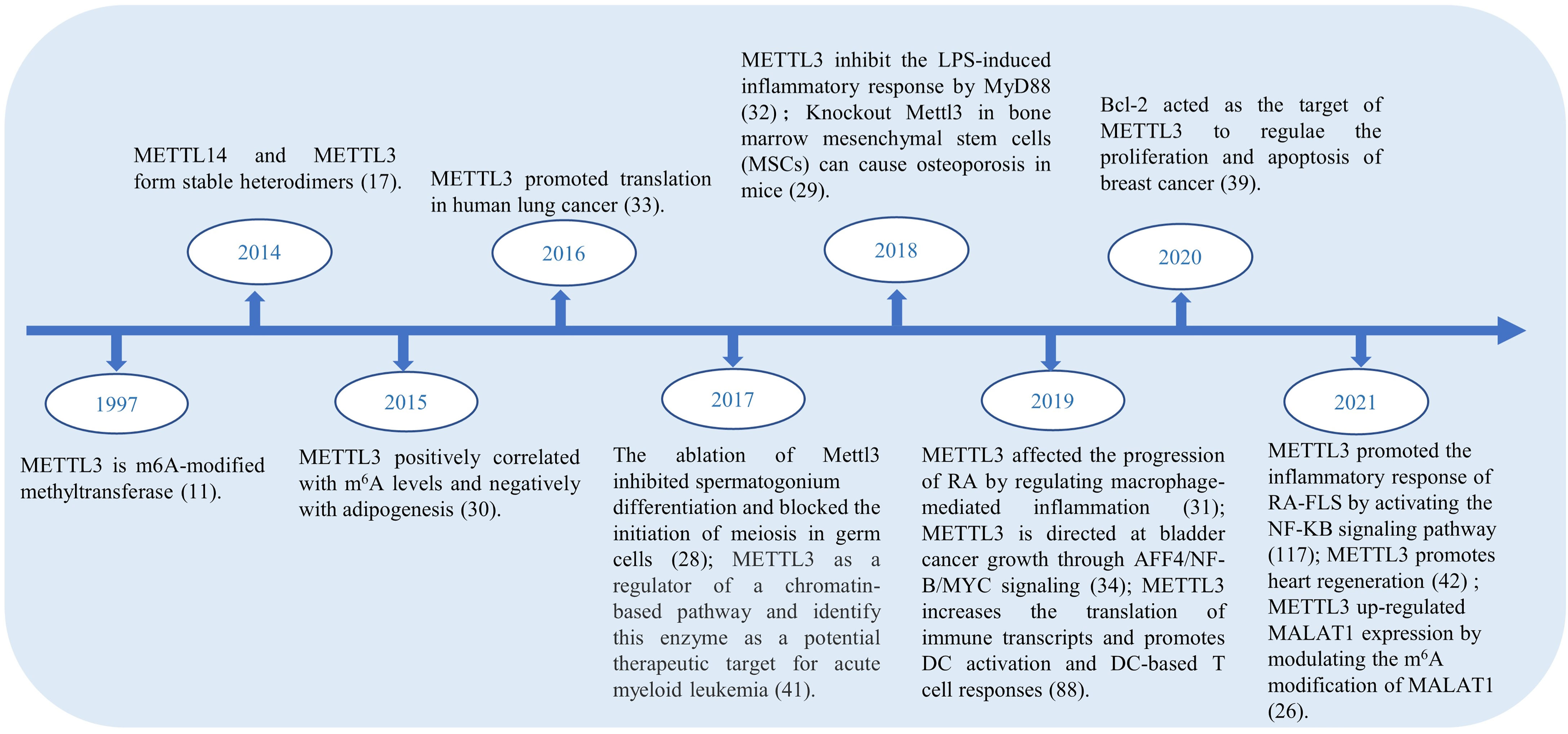
Figure 2 Research progress on METTL3, which is one of the most studied genes in m6A methylation.
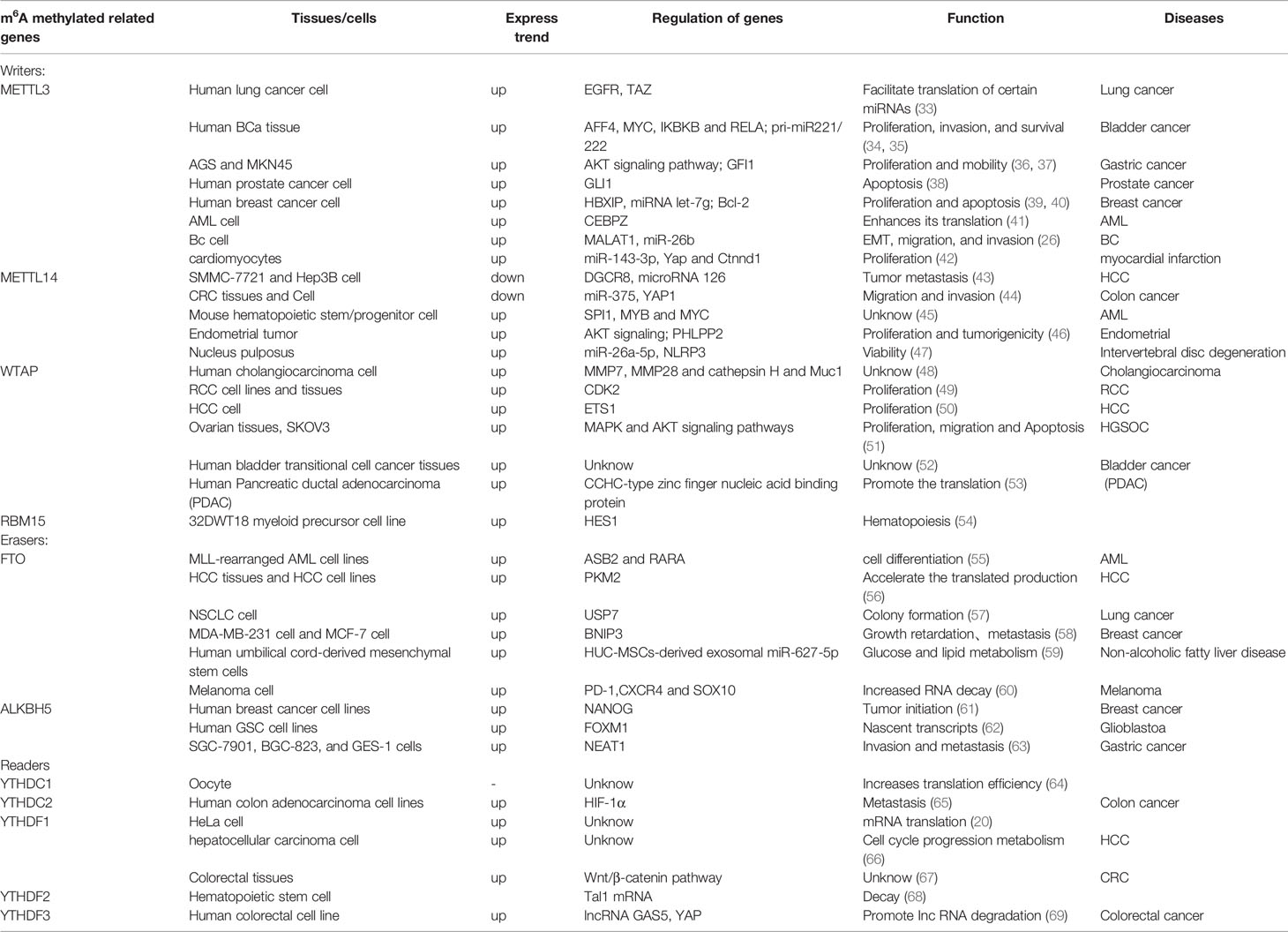
Table 1 m6A methylation related to tumorigenesis.
METTL14 is a pseudo methyltransferase that stabilizes METTL3 and recognizes target RNA (45). The combination of METTL14 and METTL3 promotes the identification of m6A by METTL3 to some degree. The closed connection between full-length polypeptide methylation with point mutations in the catalytic base sites of METTL3 and METTL14 indicates that METTL3 is the only subunit with catalytic activity in vitro (70). There is a wide range of beneficial interactions between METTL3 and METTL14, which are important for stabilizing the structure of both domains as well as for interdomain coordination. In contrast to METTL3 studies, few studies have been published on METTL14 alone. As an essential component of the m6A methyltransferase complex, METTL14 is highly expressed in hematopoietic stem/progenitor cells (HSPCs), acute myelogenous leukemia (AML) cells, and pancreatic cancer (45, 71). Furthermore, studies have revealed that METTL14 depletion is dependent on mTOR signaling-induced autophagy (30).
WTAP is an m6A methyltransferase complex that is found in mammals. In the absence of WTAP, the RNA binding capacity of METTL3 is significantly weakened (72). This suggests that WTAP regulates the recruitment of the m6A methyltransferase complex to the mRNA target and affects its binding capacity. WTAP functions without methylation activity, but it interacts with the METTL3 and METTL14 complexes to significantly affect cellular m6A deposition. Furthermore, WTAP participates in crucial cellular processes, such as regulation of the cell cycle (14, 73), cell proliferation, and cell apoptosis. Growing evidence has shown that WTAP is related to the malignant potential of tumor cells, is clearly upregulated in HCC tissues (50), and is responsible for the migration ability of the SKOV3 cell line (51). These results demonstrate that WTAP plays a vital role in proliferation and invasiveness abilities.
Similar to other RNA-binding motifs (RBM) proteins, RBM15 combines with RNA through spliceosomes to regulate splicing, translation, and stability. At present, RBM15 has been studied widely in various blood diseases, such as chronic myelogenous leukemia (CML), acute megakaryocytic leukemia, and T−cell acute lymphoblastic leukemia. Yang et al. found that patients with chronic and accelerated-phase CML had a significantly lower mRNA expression level of RBM15 than that of patients with blast-crisis CML. Moreover, the RBM15 protein may affect the growth of RBPJ-mediated CML cells through Notch signaling (25, 74). Rbp-Jk is one of the major canonical transcriptional effectors in the Notch signal pathway.
Erasers
The second type of protein is an m6A demethylase called erasers, which remove the m6A methylated group from RNA. The most common erasers are the fat mass and obesity-associated protein (FTO) and alkylation repair homolog protein 5 (ALKBH5).
Originally, FTO was thought to be a protein that regulates body weight and obesity. Overactivation of FTO can increase food intake, which leads to obesity. The FTO controls mRNA splicing by inhibiting SRSF2 (a RNA splicing factors) binding at splice sites (18). However, when FTO was recognized as an RNA demethylase, it was found to have additional functions for controlling various aspects of biological processes. FTO removes the m6A modification and modulates the stability of mRNA, which ultimately leads to the alteration of pathogenesis in various types of cancer (55, 75). In some tumor cells, FTO is critical for their immune escape. FTO-deleted made melanoma cells sensitive to interferon therapy and anti-PD-1 therapy (60). FTO and programmed death-ligand 1 (PD-L1) were both high expressing in colon cancer cells, and FTO regulate the expression of PD-L1 (76). In arsenic-associated diseases, FTO is upregulated. While FTO-deleted promoted autophagy and inhibited arsenic- associated tumorigenesis (77). Some observations of FTO suggest that it plays a crucial role in the proliferation and apoptosis of some cancer cells, also glucose and lipid metabolism (59, 78, 79). Applying this biological function of FTO to inflammatory immune cells could inhibit the release of inflammatory cytokines and alleviate inflammatory disease.
ALKBH5 is known for regulating transcriptional modification in numerous human malignancies and maintaining mRNA stability (62). ALKBH5 is primarily involved in the demethylation of m6A modification and exerts its functions during the regulation of mRNA nucleation and other metabolic processes in the development of sperm. The functions of m6A modification are involved in the regulation of IncRNA to affect the development of cancers (80). In addition, ALKBH5 regulated PD-L1 mRNA in intrahepatic cholangiocarcinoma (ICC). The lack of ALKBH5 decreased the expression of PD-L1 on monocytes-macrophages by a YTHDF2-dependent manner (81). Also, the deletion of the m6A demethylase Alkbh5 sensitized tumors to cancer immunotherapy (82). It extended the cognition of ALKBH5 in tumor immune microenvironment and immunotherapy.
FTO and ALKBH5 inhibit proliferation by regulating cell migration, invasion, and metastasis in some cancer cells. However, few studies on these proteins have investigated immune diseases. If proliferation and migration of synovial cells are reduced by FTO and ALKBH5, the onset of the inflammatory response in RA could be delayed. Although the role of m6A erasers has not been extensively explored in immune diseases, it has promise as a valuable research direction for FTO and ALKBN5.
Readers
The final group of proteins plays a specific role by binding to the m6A methylation site in RNA, and their coding genes are called readers. There are five YTH domain-containing proteins in the human genome: YTHDC1, YTHDC2, YTHDF1, YTHDF2, and YTHDF3. YTHDF1 enhances mRNA translation and protein synthesis by interacting with initiation factors (20). YTHDF2 induces degradation of transcripts by selectively binding to and recruiting m6A-modified mRNA to mRNA decay sites (16). YTHDF3 enhances RNA translation by interacting with YTHDF1 and promotes RNA degradation by associating with YTHDF2 (83).
YTHDF1 not only participates in mRNA translation, which directly targets YTHDF1 or binds to translational initiation factors in some cancer cells but is also involved in neoantigen-specific immunity. In classical DCs, the absence of YTHDF1 enhances the cross-presentation of tumor antigens and the cross-priming of CD8+ T cells in vivo (76). Firstly, YTHDF1 distinguishes between m6A-related mRNAs in DCs that encode lysosomal proteases. The binding between m6A-marked mRNAs and YTHDF1 then boosts lysosomal protease translation, which suppresses the cross-presentation of engulfed tumor neoantigens. Furthermore, PD-L1 expression was increased in tumor cells from Ythdf1−/− tumor-bearing mice, anti-PDL1-treated on Ythdf1−/− tumor-bearing mice made tumor disappeared, the simultaneous depletion of PD-L1 and YTHDF1 was beneficial to the improvement of tumor immune microenvironment (76). This process is a novel mechanism of immune evasion to elude immunosurveillance.
YTHDF2 destabilizes key gene transcripts in certain biological processes. Furthermore, the knockdown of YTHDF2 leads to reductive proliferation of lung cancer cells (84). However, YTHDF2 is specifically downregulated in HCC cell lines under hypoxia culture conditions, and the overexpression of YTHDF2 unexpectedly inhibits the growth and proliferation of HCC cells (85). There are two clearly opposing effects on the regulating process of YTHDF2, and further research into its mechanisms is required. In addition, Yu et al. (78) discovered that the YTHDF2 expression level is upregulated in macrophage RAW264.7 cells after stimulating with LPS. Moreover, the knockout of YTHDF2 expression contributed to the downregulation of mRNA proinflammatory factor levels. The function of YTHDF2 in preventing an excessive inflammatory response offers a potential target and a new perspective for inflammation in the treatment of immune diseases (86).
YTHDF3 has been proposed to be the first reader protein to interact with m6A-modified transcripts in the cytoplasm (83). Denise and colleagues (59) found that knocking out YTHDF3 in human CD4+ T cells increases the risk of infection, and YTHDF3 acts as a limiting factor for human immunodeficiency virus (HIV). In this study, YTHDF3 proteins were mixed with HIV particles in a nucleocapsid-dependent manner, allowing the m6A reader protein to limit infection in the new target cell at the reverse transcription step, which reduced viral infectivity in the next cycle of infection (87). In studies related to YAP signaling pathways, YTHDF3 induces GAS5 decay by recognizing m6A modified GAS5 through a negative feedback loop between lncRNA, GAS5, and YTHDF3. The functional link between the YAP signaling pathway and the m6A modification offer a promising approach for other research (69).
YTHDC1 is located in the nucleus, whereas YTHDF2 and YTHDC2 are found in the cytoplasm. YTHDC1 affects the processing of pre-mRNA transcripts in germ cells and directly affects the maturation of oocytes (64). YTHDC2 is of great importance in spermatogenesis, where it selectively binds to m6A at its consensus motif. Genital morphology mice with a knockdown of the YTHDF3 gene had smaller ovaries and testes in females and males, respectively. It is possible that high expression of YTHDC2 heightens the translation efficiency of its targets during spermatogenesis.
State of the Art Knowledge: How m6A Affects Immune Cells
Wang et al. demonstrated that DC activation and function for promoting CD4+ T cell proliferation decline following inhibition of the m6A modification. During DC maturation, METTL3 catalyzes m6A in CD40, CD80, and Tirap, which facilitates DC activation by increasing translation efficiency and function for promoting T cell activation (88). DCs are specialized antigen-presenting cells (APCs) that are linked to innate and adaptive immune responses (89). These studies indicate that m6A plays a vital and flexible role in the innate immune response and antitumor immunity.
M6A has a significant influence on the homeostasis and differentiation stability of T cells. Li et al. demonstrated in mice that if the m6A protein, METTL3, is absent from T cells, T cell homeostasis and differentiation are easily disrupted. The number and characteristics of immune cells in mice with a conditional knockout of METTL3 were detected under a stable state: abnormal T cells were found in the spleen and lymph nodes (LNs), and there was an increase in the number of naive T cells in the LNs. In non-small-cell lung cancer, circIGF2BP3 inactivated cocultured CD8+T cells and METTL3 depended on YTHDC1 to participate in the methylation modification of circIGF2BP3 (90). Subsequently, Li et al. found that, through the TCR-dependent T cell differentiation system, TH1 and TH17 cells in METTL3-deficient naive T cells were decreased, TH2 cells were increased, and Treg cells remained unchanged (91). M6A modification plays an important role in the differentiation of CD4+ T cells. Previous studies have shown that m6A induces the degradation of soc mRNA in response to IL-7 signals to reprogram the proliferation and differentiation of naive T cells, which indicates a novel T cell homeostasis mechanism and signal-related induction of mRNA degradation (92).
As an important part of innate immunity, polarization changes between M1 and M2 phenotypes of macrophages regulate various physiological and pathological states. Recent findings have suggested that the m6A catalytic enzyme, METTL3, promotes M1 macrophage polarization to play a pro-inflammatory role. Mechanistically, METTL3 directly methylates the mRNA of STAT1, which is a critical transcription factor for priming proinflammatory macrophages, and enhances its mRNA stability; thus, upregulating STAT1 expression and facilitating M1 macrophage polarization (93). Therefore, METTL3-mediated m6A methylation in macrophages may serve as a potential anti-inflammatory target in the treatment of inflammatory diseases. In a fascinating new study, Zhang and colleagues identified a novel mechanism by which METTL3 acts during oxidized low-density lipoprotein (oxLDL)-induced monocyte inflammation. In human monocyte PHT-1 cells, METTL3 and YTHDF2 jointly affect the state of mitochondria and energy metabolism, thereby enhancing the inflammatory response of monocyte-macrophage (94). Basing on this research, we can open up various ideas about the pathogenesis of inflammatory and immune diseases.
Type I interferon produces an immune response to viral infection. Winkler and colleagues proposed a new approach for m6A targeting of IFNB mRNA to regulate the type I interferon response, which limits the duration of the antiviral response (95). After viral infection, depletion of METTL3 leads to elevated levels of type I interferons because m6A modification of interferon transcripts regulates their decay rates. Further studies on the role of m6A modification in the immune system may provide new therapeutic options for inflammatory and infectious diseases.
Translational Potential
The Potential Link Between RA and Immune Cells
In autoimmune diseases, the immune response is initiated by local innate immune cells that are exposed to external antigens or autoantigens. With the advances in surgical techniques, such as synovial excision and surgical correction, Janossy et al. found that the number of HLA-DR+ macrophages is greater in RA synovial tissue (96). In RA, monocytes and macrophages, as specialized APCs, stimulate the response and infiltration of inflammatory cells and play a considerable role in joint destruction (97). B cells produce immunoglobulins, inflammatory cytokines, rheumatoid cytokines, and other factors that promote the positive feedback regulation of macrophages.
In the inflamed RA joint, tumor necrosis factor (TNF) is produced primarily by macrophages, and TNF inhibitors are effective for the treatment of RA (98). Secretion of cytokines and chemokines perpetuates the inflammatory response by recruiting additional innate immune cells, such as monocytes and neutrophils, and also by inducing T cell differentiation (99). Moreover, abnormally activated macrophages and T cells migrate to the joint cavity and are stimulated by the activated synovial cells to aggravate the inflammatory response in the synovial microenvironment, which leads to synovial inflammation, synovial hyperplasia, and cartilage in the joint tissues.
In the early 1990s, macrophage gene expression was found to vary under different media stimulation (IL-4/IFN-γ/LPS). In 2000, Mills et al. (65) discovered M1-M2 polarization because of arginine metabolism in macrophages. They further proposed that the M1-M2 dichotomy is an intrinsic property of the transition of macrophages from inflammation to healing. However, findings of subsequent studies have deviated from the initial concept of Mill et al., although the definition and distinction of M1-M2 remain controversial. The inconsistencies in macrophage activation and biomarkers introduce challenges in the study of M1-M2 polarization of macrophages. Kung et al. proposed proinflammatory cytokines and anti-inflammatory cytokines as features that distinguish M1 macrophages (CD80+) from M2 macrophages (CD163+) (100). Although there is little evidence for M1-M2 polarization, we recognize that is involved in RA. Indeed, the mass proliferation of synovial cells is typical of RA and is closely related to macrophages. Therefore, a treatment targeting macrophages in the articular cavity may have a positive effect on the disease by reducing inflammation and joint damage.
NK cells are natural immune effector cells with a direct killing function and are particularly important in immune cells because they eliminate viral infections and secrete proinflammatory cytokines. Li et al. conducted an integrative pathway association analysis of RA using genome-wide association studies (GWAS) summary data of 25 RA-associated pathways for NK cells (2). A large number of NK cells were found in the synovial fluid of RA patients, which is considered an important factor for bone destruction (101). In a mouse model of arthritis, exhaustion of NK cells before constructing the mouse model provided almost complete protection of bones and joints from erosion (102). In the synovial fluid of 15 patients with inflammatory arthritis, NK cells expressed high levels of CD56 and low levels of CD16 was significant increase. They further showed that proinflammatory factor IL-12, in combination with IL-15, is effective in stimulating NK cells to express IFN (103). However, IFN itself can activate macrophages and regulate the transcription of genes within macrophages, which suggests that NK cells could interact with articular macrophages/monocytes to produce more proinflammatory cytokines (103). In addition, RBM15 regulates the Notch signaling pathway (71). It is known that the Notch signaling pathway is related to the number of NK cells to some degree, and a variety of excitatory and inhibitory receptors are inherently expressed on the cell surface, which monitor malignant cells (104). After RBM15 regulates the Notch signaling pathway, the Notch signaling or canonical transcriptional effectors affect immune cells, such as NK cells. Eventually, the immune cells operate throughout the entire immune system by linking the immune system. This generates natural immunity and activates acquired immunity to maintain the body’s normal immunity status. Taken together, interfering with the activation of NK cells may provide a basis for therapeutic strategies.
Autoreactive T cells play an important role in many autoimmune diseases. There are various types of T cells, such as the T helper cell and nonclassical T helper cell subsets, such as Th0, Th1, Th2, Th17, Th1/17, CCR4, CCR6, and CXCR3 (105). Yasuo et al. found that the frequency of memory CXCR4(+) CD4(+) T cells was significantly associated with RA severity (91). Furthermore, autoreactive T cells have been shown to be critical in many models of autoimmune diseases, such as collagen-induced arthritis, non-obese diabetic mice, and experimental autoimmune encephalitis (106). The PD-1/PD-L1 is one of the pathways for tumors to evade the immune system by challenging T cell tolerance, T cell exhaustion, enhancing immunosuppressive Treg cell function and inducibling co-stimulatory molecule (107, 108). Thus, T cell immunity as a target for more precise immunotherapy interventions for human autoimmune diseases is likely to be a more selective way to treat RA.
In addition to autoantibody production, B cells are efficient APCs, which allow T cell activation. In some RA patients, B cells have been found to differentiate into plasma cells that produce rheumatoid factor and anticyclic citrullinated peptide antibodies (109). Rituximab is primarily used to deplete the ability of B cells and alleviates RA in patients who have TNF inhibitor failure. Inspired by clinical experience with rituximab, various approaches for investigating the pathogenic function of B cells for RA are popular. Currently, direct targeting of B cells in rheumatic diseases is divided into four broad categories: CD19, CD20, CD22, and CD52. Target-specific surface molecules effectively deplete B cell populations, which control immunity and B cell activation (110).
DCs are thought to play a crucial role in immunopathogenic responses, which result in the establishment of chronic proliferative synovitis and joint destruction in RA (111). Frances demonstrated that RA synovial fluid contains specific subsets of myeloid progenitor cells, which could differentiate into mature DCs in the correct microenvironment (112). It may ultimately be possible to provide some basis for the heterogeneity of the immune response in RA. Huang et al. demonstrated that etanercept (ETN), which is a TNF antagonist, prevented arthritis development through the regulation of DC actions. ETN reduces the ability of DC to migrate to local LNs and regulates the quantity of T and B cells to change the composition of LN cells. Moreover, it has been shown that TNF-α blockade has a significant impact on DC maturation and migration, which contributes to the immune regulatory effects in RA patients (113). There is evidence that mature DCs present in the RA joint mediate immunopathology. These studies suggest that RNA modification of m6A has a potential function between DCs and RA.
MCs are important immune cells in the mammalian body and affect the pathogenesis of. MCs recognize and respond to various antigens, secrete various chemokines, recruit other immune cells, and cause local tissue inflammation, neovascularization, and tissue remodeling in the body (114). They exist in normal and RA synovium. However, the number of MCs in the synovium in RA patients is higher, and active RA patients show higher MC infiltration than those at the end stage of the disease (115).
Numerous studies have shown that immune cells play a role in the pathogenesis of RA. First, some immune cells secrete chemokines and growth factors and subsequently, infiltration of other immune cells into synovial cells occurs. Second, immune cells can act as APCs to stimulate the production of various other immune cells. Third, in the early stage of RA, synovial cells have a small expansion area. Through the suppression of specific immune cells, combined with the patients’ age, sex, diet, constitution, and other factors to give appropriate treatment, it is a great approach to fully improve the treatment effect. Finally, whether immune cells promote angiogenesis in the synovium remains to be elucidated.
The Gap in Knowledge for Future RA Research
So far, the presence of m6A methylation in RA has been poorly studied. Wang and colleagues showed that in RA patients, the expression of METTL3 is markedly increased in peripheral blood mononuclear cells (PBMCs); however, other RNA m6A methyltransferases have no obvious (31). In vitro experiments have shown that by stimulating LPS, total m6A content can be enhanced by upregulating METTL3 in pTHP-1 macrophages and that METTL3-related m6A modification is associated with cytokines IL-6 and TNF-a (31). In other words, METTL3 attenuates inflammation through the NF-κB pathway in RA, which could be one approach to treat RA. In addition to the mechanism of action of METTL3 in inflammatory immune macrophages. METTL3 had also been reported in synovial cells recently. METTL3 promoted the inflammatory response of RA-FLS by activating the NF-KB signaling pathway (116). The mRNA expression of ALKBH5, FTO and YTHDF2 in RA patients’ PBMCs was significantly decreased (117), it also provided novel insights into recognizing the pathogenesis of RA.
The main symptom of severe RA is synovial hyperplasia. Inhibiting the infinite proliferation of synovial cells is an important treatment method to curb the development of RA. There are few studies on the treatment of RA from the perspective of m6A methylation. However, our previous study showed that m6A-regulated methyltransferase, demethylase, and binding protein genes are altered in the synovial tissues of patients with RA. Control tissues were synovial tissues of patients with osteoarthritis. Using qRT-PCR, we found that METTL3, METTL14, WTAP, METTL16, and YTHDF2 were significantly upregulated, and FTO was significantly downregulated, whereas the others did not change significantly. Moreover, the expression of WTAP was significantly upregulated in the RA synovial membrane (Figure 3). The histogram in Figure 3 represents multiple compared with osteoarthritis, and osteoarthritis is 1.
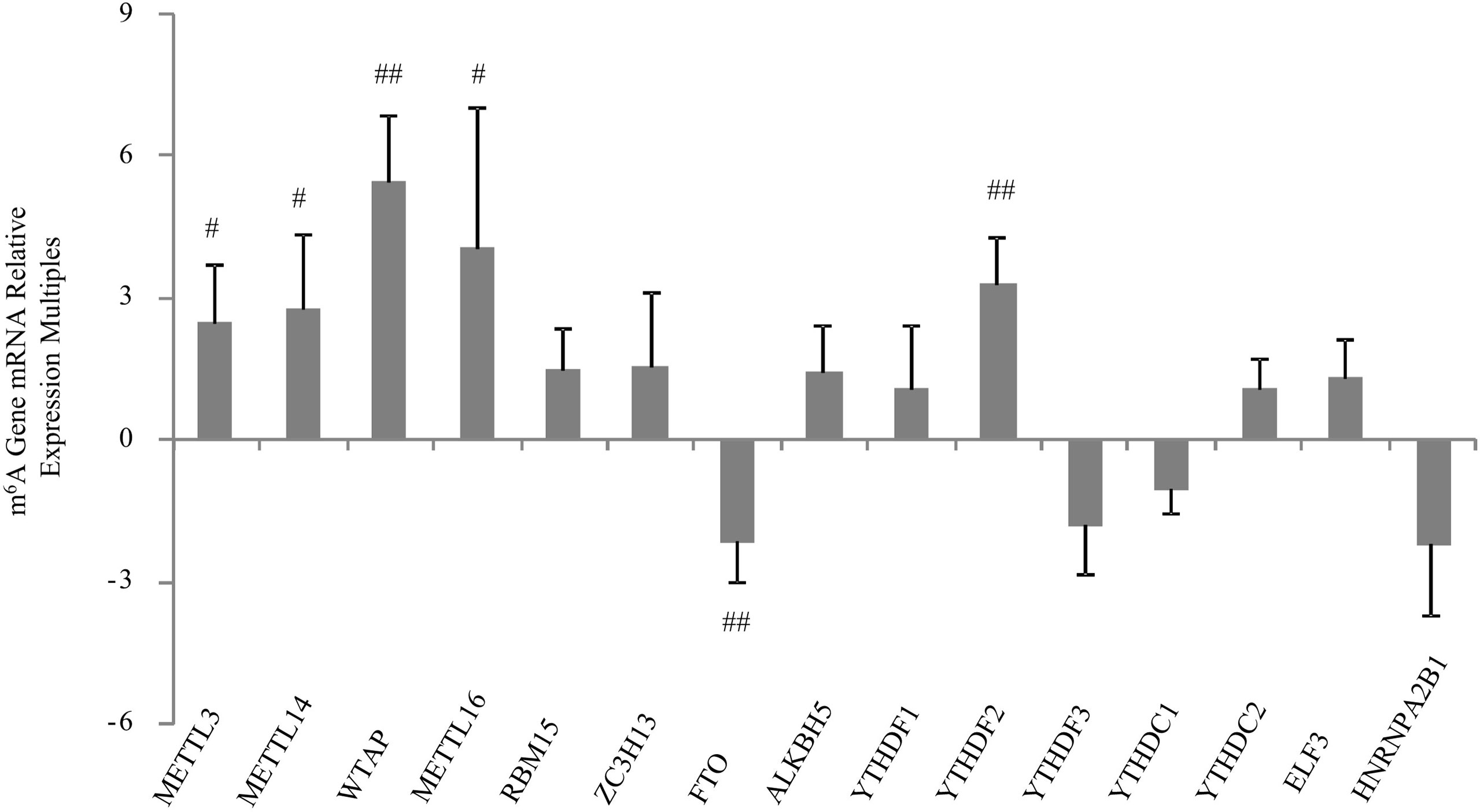
Figure 3 mRNA expression of m6A-related enzymes was quantified using qRT-PCR in the synovial tissues of RA and osteoarthritis patients. The histogram represents multiple expressions of m6A-related enzymes in RA. The expression in osteoarthritis is 1. Values represent means ± standard deviations. ##p < 0.01 vs. OA; #p < 0.05 vs. OA.
Discussion
M6A methylation belongs to one of the many families of RNA epigenetic modification. Based on the current understanding of its relationship with tumors, m6A methylation does not have a “good or bad” effect on tumor cells and promotes or inhibits tumor cells by regulating the mRNA expression level of related oncogenes or tumor suppressor genes. Extensive research on the methylation mechanism of m6A has demonstrated that the regulation of the RNA level related to m6A is complex and diverse. In addition, there is unequivocal evidence that m6A modification is essential for a variety of biological processes, including the immune response, and there is increasing evidence that its dysregulation is linked to numerous human diseases. We believe that further research on the mechanism of m6A methylation in immune diseases will have profound implications for the treatment of immune diseases (Figure 4). New approaches and targets for the treatment of RA will certainly come to light, which will have a positive effect on human life.
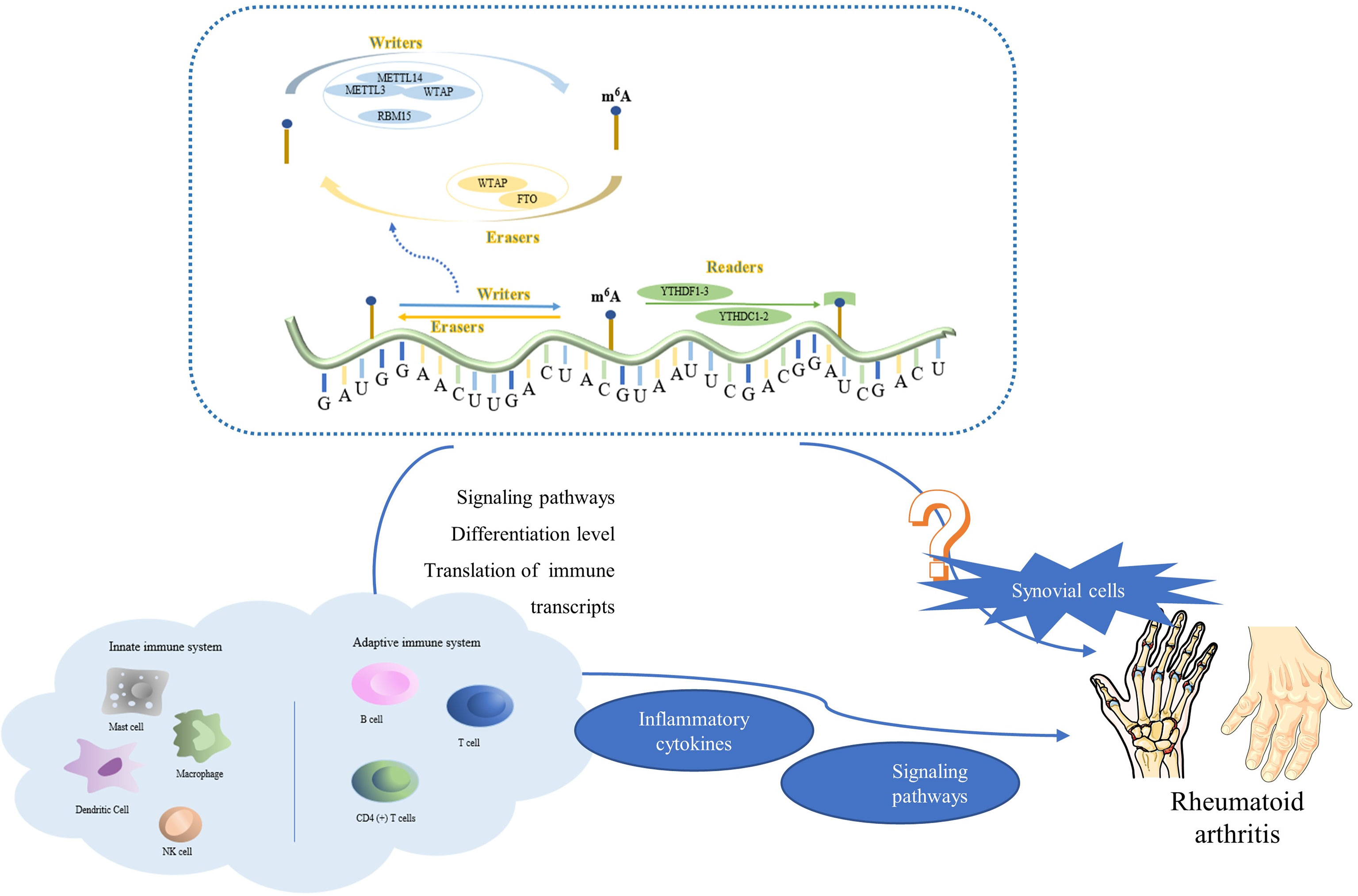
Figure 4 The complexes formed by METTL3, METTL14, WTAP, and RBM15 are common writers; those formed by ALKBH5 and FTO are common erasers; and those formed by YTHDC1, YTHDF1, and YTHDF3 are common readers. M6A methylation is involved in a wide range of biological regulatory processes in the innate and adaptive immune systems, such as adjustment of signaling pathways, differentiation, translation of immune transcripts. It controls the production of inflammatory factors, inflammation-related signaling pathways, and other genes that have a direct impact on RA. It is worth noting that, m6A methylation may also directly affect synovial cell proliferation and tumor tissue, which may be a potential discovery in future RA research.
Author Contributions
SW and X-FL researched the data and drafted the manuscript. Y-YW and S-QY participated in the conception and design of the study and interpretation of the data. JL and CH reviewed the data, made substantial contributions to the discussion of the content, and edited the article before submission. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (No. 82002269), China Postdoctoral Science Foundation (No. 2020M671839), Postdoctoral Science Foundation from Anhui Medical University (No. BSH201902), and Anhui Provincial Science and Technology Major Project (8212929035).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ALKBH5, Alkylation repair homolog protein 5; APCs, Antigen presenting cells; BC, Breast cancer; FTO, Fat mass and obesity-associated protein; HCC, Hepaticcellcarcinoma; PD-L1, programmed death-ligand 1; PDAC, Pancreatic ductal adenocarcinoma; MCs, Mast cells; METTL, Methyltransferase-like; m6A, N6-methyladenosine; NK cells, Natural killer cells; RA, Rheumatoid arthritis; RBM, RNA-binding motif protein; VSMCs, Vascular smooth muscle cells; WTAP, Wilms’ tumor 1 associated protein.
References
1. Li XF, Sun YY, Bao J, Chen X, Li YH, Yang Y, et al. Functional Role of PPAR-γ on the Proliferation and Migration of Fibroblast-Like Synoviocytes in Rheumatoid Arthritis. Sci Rep (2017) 7(1):12671. doi: 10.1038/s41598-017-12570-6
2. Li P, Wang X, Guo X, Wen Y, Liu L, Liang X, et al. Integrative Analysis of Genome-Wide Association Study and Expression Quantitative Trait Loci Datasets Identified Various Immune Cell-Related Pathways for Rheumatoid Arthritis. Ann Hum Genet (2020) 84(1):72–9. doi: 10.1111/ahg.12351
3. Dubin DT, Taylor RH. The Methylation State of Poly A-Containing Messenger RNA From Cultured Hamster Cells. Nucleic Acids Res (1975) 2(10):1653–68. doi: 10.1093/nar/2.10.1653
4. Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-Methyladenosine Marks Primary microRNAs for Processing. Nature (2015) 519(7544):482–5. doi: 10.1038/nature14281
5. Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, et al. Extensive Translation of Circular RNAs Driven by N(6)-Methyladenosine. Cell Res (2017) 27(5):626–41. doi: 10.1038/cr.2017.31
6. Berulava T, Rahmann S, Rademacher K, Klein-Hitpass L, Horsthemke B. N6-Adenosine Methylation in MiRNAs. PloS One (2015) 10(2):e0118438. doi: 10.1371/journal.pone.0118438
7. Frye M, Harada BT, Behm M, He C. RNA Modifications Modulate Gene Expression During Development. Science (2018) 361(6409):1346–9. doi: 10.1126/science.aau1646
8. Shi H, Wei J, He C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell (2019) 74(4):640–50. doi: 10.1016/j.molcel.2019.04.025
9. Shulman Z, Stern-Ginossar N. The RNA Modification N(6)-Methyladenosine as a Novel Regulator of the Immune System. Nat Immunol (2020) 21(5):501–12. doi: 10.1038/s41590-020-0650-4
10. He PC, He C. M(6) A RNA Methylation: From Mechanisms to Therapeutic Potential. EMBO J (2021) 40(3):e105977. doi: 10.15252/embj.2020105977
11. Desrosiers R, Friderici K, Rottman F. Identification of Methylated Nucleosides in Messenger RNA From Novikoff Hepatoma Cells. Proc Natl Acad Sci USA (1974) 71(10):3971–5. doi: 10.1073/pnas.71.10.3971
12. Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA Cloning of the AdoMet-Binding Subunit of the Human mRNA (N6-Adenosine)-Methyltransferase. RNA (1997) 3(11):1233–47.
13. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-Methyladenosine in Nuclear RNA is a Major Substrate of the Obesity-Associated FTO. Nat Chem Biol (2011) 7(12):885–7. doi: 10.1038/nchembio.687
14. Horiuchi K, Kawamura T, Iwanari H, Ohashi R, Naito M, Kodama T, et al. Identification of Wilms’ Tumor 1-Associating Protein Complex and its Role in Alternative Splicing and the Cell Cycle. J Biol Chem (2013) 288(46):33292–302. doi: 10.1074/jbc.M113.500397
15. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a Mammalian RNA Demethylase That Impacts RNA Metabolism and Mouse Fertility. Mol Cell (2013) 49(1):18–29. doi: 10.1016/j.molcel.2012.10.015
16. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-Methyladenosine-Dependent Regulation of Messenger RNA Stability. Nature (2014) 505(7481):117–20. doi: 10.1038/nature12730
17. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 Complex Mediates Mammalian Nuclear RNA N6-Adenosine Methylation. Nat Chem Biol (2014) 10(2):93–5. doi: 10.1038/nchembio.1432
18. Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, et al. FTO-Dependent Demethylation of N6-Methyladenosine Regulates mRNA Splicing and is Required for Adipogenesis. Cell Res (2014) 24(12):1403–19. doi: 10.1038/cr.2014.151
19. Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, et al. Structural Basis for Selective Binding of M6a RNA by the YTHDC1 YTH Domain. Nat Chem Biol (2014) 10(11):927–9. doi: 10.1038/nchembio.1654
20. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-Methyladenosine Modulates Messenger RNA Translation Efficiency. Cell (2015) 161(6):1388–99. doi: 10.1016/j.cell.2015.05.014
21. Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 Destabilizes M(6)A-Containing RNA Through Direct Recruitment of the CCR4-NOT Deadenylase Complex. Nat Commun (2016) 7:12626. doi: 10.1038/ncomms12626
22. Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N(6)-Methyladenosine Binding Protein That Regulates Mammalian Spermatogenesis. Cell Res (2017) 27(9):1115–27. doi: 10.1038/cr.2017.99
23. Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-Methyladenosine Alters RNA Structure to Regulate Binding of a Low-Complexity Protein. Nucleic Acids Res (2017) 45(10):6051–63. doi: 10.1093/nar/gkx141
24. He L, Li J, Wang X, Ying Y, Xie H, Yan H, et al. The Dual Role of N6-Methyladenosine Modification of RNAs is Involved in Human Cancers. J Cell Mol Med (2018) 22(10):4630–9. doi: 10.1111/jcmm.13804
25. Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-Methyladenosine Methyltransferase-Like 3 Promotes Liver Cancer Progression Through YTHDF2-Dependent Posttranscriptional Silencing of SOCS2. Hepatology (2018) 67(6):2254–70. doi: 10.1002/hep.29683
26. Zhao C, Ling X, Xia Y, Yan B, Guan Q. The M6a Methyltransferase METTL3 Controls Epithelial-Mesenchymal Transition, Migration and Invasion of Breast Cancer Through the MALAT1/miR-26b/HMGA2 Axis. Cancer Cell Int (2021) 21(1):441. doi: 10.1186/s12935-021-02113-5
27. Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3-Mediated M(6)A Modification of HDGF mRNA Promotes Gastric Cancer Progression and has Prognostic Significance. Gut (2020) 69(7):1193–205. doi: 10.1136/gutjnl-2019-319639
28. Xu K, Yang Y, Feng GH, Sun BF, Chen JQ, Li YF, et al. Mettl3-Mediated M(6)A Regulates Spermatogonial Differentiation and Meiosis Initiation. Cell Res (2017) 27(9):1100–14. doi: 10.1038/cr.2017.100
29. Wu Y, Xie L, Wang M, Xiong Q, Guo Y, Liang Y, et al. Mettl3-Mediated M(6)A RNA Methylation Regulates the Fate of Bone Marrow Mesenchymal Stem Cells and Osteoporosis. Nat Commun (2018) 9(1):4772. doi: 10.1038/s41467-018-06898-4
30. Wang X, Zhu L, Chen J, Wang Y. mRNA M⁶A Methylation Downregulates Adipogenesis in Porcine Adipocytes. Biochem Biophys Res Commun (2015) 459(2):201–7. doi: 10.1155/2019/3120391
31. Wang J, et al. METTL3 Attenuates LPS-Induced Inflammatory Response in Macrophages via NF-κb Signaling Pathway. Mediators Inflammation (2019) 2019:3120391. doi: 10.1155/2019/3120391
32. Feng Z, Li Q, Meng R, Yi B, Xu Q. METTL3 Regulates Alternative Splicing of MyD88 Upon the Lipopolysaccharide-Induced Inflammatory Response in Human Dental Pulp Cells. J Cell Mol Med (2018) 22(5):2558–68. doi: 10.1111/jcmm.13491
33. Lin S, Choe J, Du P, Triboulet R, Gregory RI. The M(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol Cell (2016) 62(3):335–45. doi: 10.1016/j.molcel.2016.03.021
34. Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H, Wu M, et al. The M(6)A Methyltransferase METTL3 Promotes Bladder Cancer Progression via AFF4/NF-κb/MYC Signaling Network. Oncogene (2019) 38(19):3667–80. doi: 10.1038/s41388-019-0683-z
35. Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu HC, et al. METTL3 Promote Tumor Proliferation of Bladder Cancer by Accelerating Pri-Mir221/222 Maturation in M6a-Dependent Manner. Mol Cancer (2019) 18(1):110. doi: 10.1186/s12943-019-1036-9
36. Lin S, Liu J, Jiang W, Wang P, Sun C, Wang X, et al. METTL3 Promotes the Proliferation and Mobility of Gastric Cancer Cells. Open Med (Wars) (2019) 14:25–31. doi: 10.1515/med-2019-0005
37. Liu T, Yang S, Sui J, Xu SY, Cheng YP, Shen B, et al. Dysregulated N6-Methyladenosine Methylation Writer METTL3 Contributes to the Proliferation and Migration of Gastric Cancer. J Cell Physiol (2020) 235(1):548–62. doi: 10.1002/jcp.28994
38. Cai J, Yang F, Zhan H, Situ J, Li W, Mao Y, et al. RNA M(6)A Methyltransferase METTL3 Promotes The Growth Of Prostate Cancer By Regulating Hedgehog Pathway. Onco Targets Ther (2019) 12:9143–52. doi: 10.2147/OTT.S226796
39. Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, et al. HBXIP-Elevated Methyltransferase METTL3 Promotes the Progression of Breast Cancer via Inhibiting Tumor Suppressor Let-7g. Cancer Lett (2018) 415:11–9. doi: 10.1016/j.canlet.2017.11.018
40. Wang H, Xu B, Shi J. N6-Methyladenosine METTL3 Promotes the Breast Cancer Progression via Targeting Bcl-2. Gene (2020) 722:144076. doi: 10.1016/j.gene.2019.144076
41. Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, et al. Promoter-Bound METTL3 Maintains Myeloid Leukaemia by M(6)A-Dependent Translation Control. Nature (2017) 552(7683):126–31. doi: 10.1038/nature24678
42. Gong R, Wang X, Li H, Liu S, Jiang Z, Zhao Y, et al. Loss of M6a Methyltransferase METTL3 Promotes Heart Regeneration and Repair After Myocardial Injury. Pharmacol Res (2021) p:105845. doi: 10.1016/j.phrs.2021.105845
43. Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, et al. METTL14 Suppresses the Metastatic Potential of Hepatocellular Carcinoma by Modulating N(6) -Methyladenosine-Dependent Primary MicroRNA Processing. Hepatology (2017) 65(2):529–43. doi: 10.1002/hep.28885
44. Chen X, Xu M, Xu X, Zeng K, Liu X, Sun L, et al. METTL14 Suppresses CRC Progression via Regulating N6-Methyladenosine-Dependent Primary miR-375 Processing. Mol Ther (2020) 28(2):599–612. doi: 10.1016/j.ymthe.2019.11.016
45. Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA M(6)A Modification. Cell Stem Cell (2018) 22(2):191–205 e9. doi: 10.1016/j.stem.2017.11.016
46. Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, et al. M(6)A mRNA Methylation Regulates AKT Activity to Promote the Proliferation and Tumorigenicity of Endometrial Cancer. Nat Cell Biol (2018) 20(9):1074–83. doi: 10.1038/s41556-018-0174-4
47. Yuan X, Li T, Shi L, Miao J, Guo Y, Chen Y. Human Umbilical Cord Mesenchymal Stem Cells Deliver Exogenous miR-26a-5p via Exosomes to Inhibit Nucleus Pulposus Cell Pyroptosis Through METTL14/NLRP3. Mol Med (2021) 27(1):91. doi: 10.1186/s10020-021-00355-7
48. Jo HJ, Shim HE, Han ME, Kim HJ, Kim KS, Baek S, et al. WTAP Regulates Migration and Invasion of Cholangiocarcinoma Cells. J Gastroenterol (2013) 48(11):1271–82. doi: 10.1007/s00535-013-0748-7
49. Tang J, Wang F, Cheng G, Si S, Sun X, Han J, et al. Wilms’ Tumor 1-Associating Protein Promotes Renal Cell Carcinoma Proliferation by Regulating CDK2 mRNA Stability. J Exp Clin Cancer Res (2018) 37(1):40. doi: 10.1186/s13046-018-0706-6
50. Chen Y, Peng C, Chen J, Chen D, Yang B, He B, et al. WTAP Facilitates Progression of Hepatocellular Carcinoma via M6a-HuR-Dependent Epigenetic Silencing of ETS1. Mol Cancer (2019) 18(1):127. doi: 10.1186/s12943-019-1053-8
51. Yu HL, Ma XD, Tong JF, Li JQ, Guan XJ, Yang JH. WTAP is a Prognostic Marker of High-Grade Serous Ovarian Cancer and Regulates the Progression of Ovarian Cancer Cells. Onco Targets Ther (2019) 12:6191–201. doi: 10.2147/OTT.S205730
52. Chen L, Wang X. Relationship Between the Genetic Expression of WTAP and Bladder Cancer and Patient Prognosis. Oncol Lett (2018) 16(6):6966–70. doi: 10.3892/ol.2018.9554
53. Deng J, Zhang J, Ye Y, Liu K, Zeng L, Huang J, et al. N6-Methyladenosine-Mediated Upregulation of WTAPP1 Promotes WTAP Translation and Wnt Signaling to Facilitate Pancreatic Cancer Progression. Cancer Res (2021). doi: 10.1158/0008-5472.CAN-21-0494
54. Ma X, Renda MJ, Wang L, Cheng EC, Niu C, Morris SW, et al. Rbm15 Modulates Notch-Induced Transcriptional Activation and Affects Myeloid Differentiation. Mol Cell Biol (2007) 27(8):3056–64. doi: 10.1128/MCB.01339-06
55. Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell (2017) 31(1):127–41. doi: 10.1016/j.ccell.2016.11.017
56. Li J, Zhu L, Shi Y, Liu J, Lin L, Chen X. M6a Demethylase FTO Promotes Hepatocellular Carcinoma Tumorigenesis via Mediating PKM2 Demethylation. Am J Transl Res (2019) 11(9):6084–92.
57. Li J, Han Y, Zhang H, Qian Z, Jia W, Gao Y, et al. The M6a Demethylase FTO Promotes the Growth of Lung Cancer Cells by Regulating the M6a Level of USP7 mRNA. Biochem Biophys Res Commun (2019) 512(3):479–85. doi: 10.1016/j.bbrc.2019.03.093
58. Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, et al. RNA N6-Methyladenosine Demethylase FTO Promotes Breast Tumor Progression Through Inhibiting BNIP3. Mol Cancer (2019) 18(1):46. doi: 10.1186/s12943-019-1004-4
59. Cheng L, Yu P, Li F, Jiang X, Jiao X, Shen Y, et al. Human Umbilical Cord-Derived Mesenchymal Stem Cell-Exosomal miR-627-5p Ameliorates non-Alcoholic Fatty Liver Disease by Repressing FTO Expression. Hum Cell (2021). doi: 10.1007/s13577-021-00593-1
60. Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, et al. M(6)A mRNA Demethylase FTO Regulates Melanoma Tumorigenicity and Response to Anti-PD-1 Blockade. Nat Commun (2019) 10(1):2782. doi: 10.1038/s41467-019-10669-0
61. Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia Induces the Breast Cancer Stem Cell Phenotype by HIF-Dependent and ALKBH5-Mediated M⁶A-Demethylation of NANOG mRNA. Proc Natl Acad Sci USA (2016) 113(14):E2047–56. doi: 10.1073/pnas.1602883113
62. Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. M(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-Like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell (2017) 31(4):591–606 e6. doi: 10.1016/j.ccell.2017.02.013
63. Zhang J, Guo S, Piao HY, Wang Y, Wu Y, Meng XY, et al. ALKBH5 Promotes Invasion and Metastasis of Gastric Cancer by Decreasing Methylation of the lncRNA Neat1. J Physiol Biochem (2019) 75(3):379–89. doi: 10.1007/s13105-019-00690-8
64. Kasowitz SD, Ma J, Anderson SJ, Leu NA, Xu Y, Gregory BD, et al. Nuclear M6a Reader YTHDC1 Regulates Alternative Polyadenylation and Splicing During Mouse Oocyte Development. PloS Genet (2018) 14(5):e1007412. doi: 10.1371/journal.pgen.1007412
65. Tanabe A, Tanikawa K, Tsunetomi M, Takai K, Ikeda H, Konno J, et al. RNA Helicase YTHDC2 Promotes Cancer Metastasis via the Enhancement of the Efficiency by Which HIF-1α mRNA is Translated. Cancer Lett (2016) 376(1):34–42. doi: 10.1016/j.canlet.2016.02.022
66. Zhao X, Chen Y, Mao Q, Jiang X, Jiang W, Chen J, et al. Overexpression of YTHDF1 is Associated With Poor Prognosis in Patients With Hepatocellular Carcinoma. Cancer biomark (2018) 21(4):859–68. doi: 10.3233/CBM-170791
67. Bai Y, Yang C, Wu R, Huang L, Song S, Li W, et al. YTHDF1 Regulates Tumorigenicity and Cancer Stem Cell-Like Activity in Human Colorectal Carcinoma. Front Oncol (2019) 9:332. doi: 10.3389/fonc.2019.00332
68. Li Z, Qian P, Shao W, Shi H, He XC, Gogol M, et al. Suppression of M(6)A Reader Ythdf2 Promotes Hematopoietic Stem Cell Expansion. Cell Res (2018) 28(9):904–17. doi: 10.1038/s41422-018-0072-0
69. Ni W, Yao S, Zhou Y, Liu Y, Huang P, Zhou A, et al. Long Noncoding RNA GAS5 Inhibits Progression of Colorectal Cancer by Interacting With and Triggering YAP Phosphorylation and Degradation and is Negatively Regulated by the M(6)A Reader YTHDF3. Mol Cancer (2019) 18(1):143. doi: 10.1186/s12943-019-1079-y
70. Wang P, Doxtader KA, Nam Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol Cell (2016) 63(2):306–17. doi: 10.1016/j.molcel.2016.05.041
71. Kong F, Liu X, Zhou Y, Hou X, He J, Li Q, et al. Downregulation of METTL14 Increases Apoptosis and Autophagy Induced by Cisplatin in Pancreatic Cancer Cells. Int J Biochem Cell Biol (2020) 122:105731. doi: 10.1016/j.biocel.2020.105731
72. Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a Regulatory Subunit of the RNA N6-Methyladenosine Methyltransferase. Cell Res (2014) 24(2):177–89. doi: 10.1038/cr.2014.3
73. Horiuchi K, Umetani M, Minami T, Okayama H, Takada S, Yamamoto M, et al. Wilms’ Tumor 1-Associating Protein Regulates G2/M Transition Through Stabilization of Cyclin A2 mRNA. Proc Natl Acad Sci USA (2006) 103(46):17278–83. doi: 10.1073/pnas.0608357103
74. Yang Y, Wang S, Zhang Y, Zhu X. Biological Effects of Decreasing RBM15 on Chronic Myelogenous Leukemia Cells. Leuk Lymphoma (2012) 53(11):2237–44. doi: 10.3109/10428194.2012.684350
75. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. M(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep (2017) 18(11):2622–34. doi: 10.1016/j.celrep.2017.02.059
76. Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, et al. Anti-Tumour Immunity Controlled Through mRNA M(6)A Methylation and YTHDF1 in Dendritic Cells. Nature (2019) 566(7743):270–4. doi: 10.1038/s41586-019-0916-x
77. Cui YH, Yang S, Wei J, Shea CR, Zhong W, Wang F, et al. Autophagy of the M(6)A mRNA Demethylase FTO is Impaired by Low-Level Arsenic Exposure to Promote Tumorigenesis. Nat Commun (2021) 12(1):2183. doi: 10.1038/s41467-021-22469-6
78. Tang X, Liu S, Chen D, Zhao Z, Zhou J. The Role of the Fat Mass and Obesity-Associated Protein in the Proliferation of Pancreatic Cancer Cells. Oncol Lett (2019) 17(2):2473–8. doi: 10.3892/ol.2018.9873
79. Liu S, Huang M, Chen Z, Chen J, Chao Q, Yin X, et al. FTO Promotes Cell Proliferation and Migration in Esophageal Squamous Cell Carcinoma Through Up-Regulation of MMP13. Exp Cell Res (2020) 389(1):111894. doi: 10.1016/j.yexcr.2020.111894
80. Derderian C, Orunmuyi AT, Olapade-Olaopa EO, Ogunwobi OO. PVT1 Signaling Is a Mediator of Cancer Progression. Front Oncol (2019) 9:502. doi: 10.3389/fonc.2019.00502
81. Qiu X, Yang S, Wang S, Wu J, Zheng B, Wang K, et al. M6A Demethylase ALKBH5 Regulates PD-L1 Expression and Tumor Immunoenvironment in Intrahepatic Cholangiocarcinoma. Cancer Res (2021) 81(18):4778–93. doi: 10.1158/0008-5472.CAN-21-0468
82. Li N, Kang Y, Wang L, Huff S, Tang R, Hui H, et al. ALKBH5 Regulates Anti-PD-1 Therapy Response by Modulating Lactate and Suppressive Immune Cell Accumulation in Tumor Microenvironment. Proc Natl Acad Sci USA (2020) 117(33):20159–70. doi: 10.1073/pnas.1918986117
83. Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 Facilitates Translation and Decay of N(6)-Methyladenosine-Modified RNA. Cell Res (2017) 27(3):315–28. doi: 10.1038/cr.2017.15
84. Sheng H, Li Z, Su S, Sun W, Zhang X, Li L, et al. YTH Domain Family 2 Promotes Lung Cancer Cell Growth by Facilitating 6-Phosphogluconate Dehydrogenase mRNA Translation. Carcinogenesis (2019) 41(5):541–50. doi: 10.1093/carcin/bgz152
85. Zhong L, Liao D, Zhang M, Zeng C, Li X, Zhang R, et al. YTHDF2 Suppresses Cell Proliferation and Growth via Destabilizing the EGFR mRNA in Hepatocellular Carcinoma. Cancer Lett (2019) 442:252–61. doi: 10.1016/j.canlet.2018.11.006
86. Yu R, Li Q, Feng Z, Cai L, Xu Q. M6a Reader YTHDF2 Regulates LPS-Induced Inflammatory Response. Int J Mol Sci (2019) 20(6). doi: 10.3390/ijms20061323
87. Jurczyszak D, Zhang W, Terry SN, Kehrer T, Bermúdez González MC, McGregor E, et al. HIV Protease Cleaves the Antiviral M6a Reader Protein YTHDF3 in the Viral Particle. PloS Pathog (2020) 16(2):e1008305. doi: 10.1371/journal.ppat.1008305
88. Wang H, Hu X, Huang M, Liu J, Gu Y, Ma L, et al. Mettl3-Mediated mRNA M(6)A Methylation Promotes Dendritic Cell Activation. Nat Commun (2019) 10(1):1898. doi: 10.1038/s41467-019-09903-6
89. Waisman A, et al. Dendritic Cells as Gatekeepers of Tolerance. Semin Immunopathol (2017) 39(2):153–63. doi: 10.1007/s00281-016-0583-z
90. Liu Z, Wang T, She Y, Wu K, Gu S, Li L, et al. N(6)-Methyladenosine-Modified Circigf2bp3 Inhibits CD8(+) T-Cell Responses to Facilitate Tumor Immune Evasion by Promoting the Deubiquitination of PD-L1 in non-Small Cell Lung Cancer. Mol Cancer (2021) 20(1):105. doi: 10.1186/s12943-021-01398-4
91. Nagafuchi Y, Shoda H, Sumitomo S, Nakachi S, Kato R, Tsuchida Y, et al. Immunophenotyping of Rheumatoid Arthritis Reveals a Linkage Between HLA-DRB1 Genotype, CXCR4 Expression on Memory CD4(+) T Cells, and Disease Activity. Sci Rep (2016) 6:29338. doi: 10.1038/srep29338
92. Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, et al. M(6)A mRNA Methylation Controls T Cell Homeostasis by Targeting the IL-7/STAT5/SOCS Pathways. Nature (2017) 548(7667):338–42. doi: 10.1038/nature23450
93. Liu Y, Liu Z, Tang H, Shen Y, Gong Z, Xie N, et al. The N(6)-Methyladenosine (M(6)A)-Forming Enzyme METTL3 Facilitates M1 Macrophage Polarization Through the Methylation of STAT1 mRNA. Am J Physiol Cell Physiol (2019) 317(4):C762–75. doi: 10.1152/ajpcell.00212.2019
94. Zhang X, Li X, Jia H, An G, Ni J. The M(6)A Methyltransferase METTL3 Modifies PGC-1α mRNA Promoting Mitochondrial Dysfunction and oxLDL-Induced Inflammation in Monocytes. J Biol Chem (2021) p:101058. doi: 10.1016/j.jbc.2021.101058
95. Winkler R, Gillis E, Lasman L, Safra M, Geula S, Soyris C, et al. M(6)A Modification Controls the Innate Immune Response to Infection by Targeting Type I Interferons. Nat Immunol (2019) 20(2):173–82. doi: 10.1038/s41590-018-0275-z
96. Janossy G, Panayi G, Duke O, Bofill M, Poulter LW, Goldstein G. Rheumatoid Arthritis: A Disease of T-Lymphocyte/Macrophage Immunoregulation. Lancet (1981) 2(8251):839–42. doi: 10.1016/S0140-6736(81)91107-7
97. Siouti E, Andreakos E. The Many Facets of Macrophages in Rheumatoid Arthritis. Biochem Pharmacol (2019) 165:152–69. doi: 10.1016/j.bcp.2019.03.029
98. Chu CQ, Field M, Feldmann M, Maini RN. Localization of Tumor Necrosis Factor Alpha in Synovial Tissues and at the Cartilage-Pannus Junction in Patients With Rheumatoid Arthritis. Arthritis Rheum (1991) 34(9):1125–32. doi: 10.1002/art.1780340908
99. Udalova IA, Mantovani A, Feldmann M. Macrophage Heterogeneity in the Context of Rheumatoid Arthritis. Nat Rev Rheumatol (2016) 12(8):472–85. doi: 10.1038/nrrheum.2016.91
100. Kung CC, Dai SP, Chiang H, Huang HS. Temporal Expression Patterns of Distinct Cytokines and M1/M2 Macrophage Polarization Regulate Rheumatoid Arthritis Progression. Mol Biol Rep (2020) 47(5):3423–37. doi: 10.1007/s11033-020-05422-6
101. Yamin R, Berhani O, Peleg H, Aamar S, Stein N, Gamliel M, et al. High Percentages and Activity of Synovial Fluid NK Cells Present in Patients With Advanced Stage Active Rheumatoid Arthritis. Sci Rep (2019) 9(1):1351. doi: 10.1038/s41598-018-37448-z
102. Söderström K, Stein E, Colmenero P, Purath U, Müller-Ladner U, de Matos CT, et al. Natural Killer Cells Trigger Osteoclastogenesis and Bone Destruction in Arthritis. Proc Natl Acad Sci USA (2010) 107(29):13028–33. doi: 10.1073/pnas.1000546107
103. Dalbeth N, Callan MF. A Subset of Natural Killer Cells is Greatly Expanded Within Inflamed Joints. Arthritis Rheum (2002) 46(7):1763–72. doi: 10.1002/art.10410
104. Martinet L, Smyth MJ. Balancing Natural Killer Cell Activation Through Paired Receptors. Nat Rev Immunol (2015) 15(4):243–54. doi: 10.1038/nri3799
105. Pandya JM, Lundell AC, Hallström M, Andersson K, Nordström I, Rudin A. Circulating T Helper and T Regulatory Subsets in Untreated Early Rheumatoid Arthritis and Healthy Control Subjects. J Leukoc Biol (2016) 100(4):823–33. doi: 10.1189/jlb.5A0116-025R
106. Kuchroo VK, Sobel RA, Laning JC, Martin CA, Greenfield E, Dorf ME, et al. Experimental Allergic Encephalomyelitis Mediated by Cloned T Cells Specific for a Synthetic Peptide of Myelin Proteolipid Protein. Fine Specificity and T Cell Receptor V Beta Usage. J Immunol (1992) 148(12):3776–82.
107. Mahoney KM, Freeman GJ, McDermott DF. The Next Immune-Checkpoint Inhibitors: PD-1/PD-L1 Blockade in Melanoma. Clin Ther (2015) 37(4):764–82. doi: 10.1016/j.clinthera.2015.02.018
108. Zhang J, Gao J, Li Y, Nie J, Dai L, Hu W, et al. Circulating PD-L1 in NSCLC Patients and the Correlation Between the Level of PD-L1 Expression and the Clinical Characteristics. Thorac Cancer (2015) 6(4):534–8. doi: 10.1111/1759-7714.12247
109. Cambridge G, Leandro MJ, Edwards JC, Ehrenstein MR, Salden M, Bodman-Smith M, et al. Serologic Changes Following B Lymphocyte Depletion Therapy for Rheumatoid Arthritis. Arthritis Rheum (2003) 48(8):2146–54. doi: 10.1002/art.11181
110. Palm AK, Garcia-Faroldi G, Lundberg M, Pejler G, Kleinau S, et al. Activated Mast Cells Promote Differentiation of B Cells Into Effector Cells. Sci Rep (2016) 6:20531. doi: 10.1038/srep20531
111. Highton J, Kean A, Hessian PA, Thomson J, Rietveld J, Hart DN. Cells Expressing Dendritic Cell Markers are Present in the Rheumatoid Nodule. J Rheumatol (2000) 27(2):339–46.
112. Santiago-Schwarz F, Anand P, Liu S, Carsons SE. Dendritic Cells (DCs) in Rheumatoid Arthritis (RA): Progenitor Cells and Soluble Factors Contained in RA Synovial Fluid Yield a Subset of Myeloid DCs That Preferentially Activate Th1 Inflammatory-Type Responses. J Immunol (2001) 167(3):1758–68. doi: 10.4049/jimmunol.167.3.1758
113. Huang X, He Y, Han J, Zhuang J, He J, Sun E. Not Only Anti-Inflammation, Etanercept Abrogates Collagen-Induced Arthritis by Inhibiting Dendritic Cell Migration and Maturation. Cent Eur J Immunol (2019) 44(3):237–45. doi: 10.5114/ceji.2019.89595
114. Krystel-Whittemore M, Dileepan KN, Wood JG. Mast Cell: A Multi-Functional Master Cell. Front Immunol (2015) 6:620. doi: 10.3389/fimmu.2015.00620
115. Crisp AJ, Chapman CM, Kirkham SE, Schiller AL, Krane SM. Articular Mastocytosis in Rheumatoid Arthritis. Arthritis Rheum (1984) 27(8):845–51. doi: 10.1002/art.1780270802
116. Shi W, Zheng Y, Luo S, Li X, Zhang Y, Meng X, et al. METTL3 Promotes Activation and Inflammation of FLSs Through the NF-κb Signaling Pathway in Rheumatoid Arthritis. Front Med (Lausanne) (2021) 8:607585. doi: 10.3389/fmed.2021.607585
Keywords: N6-methyladenosine, rheumatoid arthritis, immune cells, autoimmune disease, cancers
Citation: Wu S, Li X-F, Wu Y-Y, Yin S-Q, Huang C and Li J (2021) N6-Methyladenosine and Rheumatoid Arthritis: A Comprehensive Review. Front. Immunol. 12:731842. doi: 10.3389/fimmu.2021.731842
Received: 28 June 2021; Accepted: 09 September 2021;
Published: 24 September 2021.
Edited by:
Shengjun Wang, Jiangsu University Affiliated People’s Hospital, ChinaReviewed by:
Jiangbo Wei, University of Chicago, United StatesNaoki Iwamoto, Nagasaki University Hospital, Japan
Tao Pan, Anhui Normal University, China
Copyright © 2021 Wu, Li, Wu, Yin, Huang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Li, bGpAYWhtdS5lZHUuY24=
†These authors have contributed equally to this work