 Amy T. Hsu
Amy T. Hsu Timothy A. Gottschalk
Timothy A. Gottschalk Evelyn Tsantikos
Evelyn Tsantikos Margaret L. Hibbs
Margaret L. Hibbs- Department of Immunology and Pathology, Central Clinical School, Monash University, Melbourne, VIC, Australia
The lung is a vital mucosal organ that is constantly exposed to the external environment, and as such, its defenses are continuously under threat. The pulmonary immune system has evolved to sense and respond to these danger signals while remaining silent to innocuous aeroantigens. The origin of the defense system is the respiratory epithelium, which responds rapidly to insults by the production of an array of mediators that initiate protection by directly killing microbes, activating tissue-resident immune cells and recruiting leukocytes from the blood. At the steady-state, the lung comprises a large collection of leukocytes, amongst which are specialized cells of lymphoid origin known as innate lymphoid cells (ILCs). ILCs are divided into three major helper-like subsets, ILC1, ILC2 and ILC3, which are considered the innate counterparts of type 1, 2 and 17 T helper cells, respectively, in addition to natural killer cells and lymphoid tissue inducer cells. Although ILCs represent a small fraction of the pulmonary immune system, they play an important role in early responses to pathogens and facilitate the acquisition of adaptive immunity. However, it is now also emerging that these cells are active participants in the development of chronic lung diseases. In this mini-review, we provide an update on our current understanding of the role of ILCs and their regulation in the lung. We summarise how these cells and their mediators initiate, sustain and potentially control pulmonary inflammation, and their contribution to the respiratory diseases chronic obstructive pulmonary disease (COPD) and asthma.
Chronic Inflammatory Lung Diseases Are an Escalating Global Health Issue
COPD is an irreversible chronic inflammatory lung disease that is the third leading cause of death worldwide (1). Patients with COPD exhibit airflow limitation, progressive deterioration in lung function and experience exacerbations; an acute worsening of their symptoms, often driven by lung infection (2). The major risk factor for COPD is cigarette smoking, although other risks such as environmental pollution or premature birth increasingly contribute to COPD susceptibility (3). COPD is underpinned by chronic inflammation, resulting in lung pathologies such as emphysema due to alveolar tissue destruction, and chronic bronchitis arising from goblet cell metaplasia and mucus overproduction (4). Inducible bronchus-associated lymphoid tissue (iBALT) often develops in COPD, particularly in advanced disease (5). COPD is heterogeneous and various disease processes, inflammatory cells (macrophages, neutrophils, cytotoxic T cells, T helper (Th)-1/17 cells) and cytokines are involved (6).
Asthma is a mostly reversible inflammatory airway disease affecting around 300 million people worldwide, where exaggerated swelling and narrowing of the conducting airways (airway hyperresponsiveness; AHR) is triggered in susceptible individuals by the inhalation of environmental particles (7–9). Asthma is differentiated into subtypes―allergic or non-allergic, and by severity―mild-intermittent, mild, moderate, or severe or by the dominant inflammatory response―eosinophilic or neutrophilic. The most common type of asthma is allergic or eosinophilic asthma, which is characterised by a type 2 immune response (driven by cytokines IL-4, IL-5 and IL-13) and IgE-mediated hypersensitivity (10). Conversely, during non-type 2 asthma, neutrophils, alongside a Th1/Th17 skewed response, predominate (11, 12). Severe asthma, which is predominantly neutrophilic, affects 5-10% of patients and is often unresponsive to standard corticosteroid-based therapies (13, 14). Asthma-COPD overlap (ACO) is a syndrome where patients exhibit characteristics of both asthma and COPD (15), complicating the study of inflammatory lung diseases.
This review will focus on ILCs and their involvement in COPD and asthma.
The Emergence of ILCs in Immunity
ILCs are a somewhat newly identified family of innate immune cells that have garnered intense recent attention and our understanding of their biological roles is rapidly progressing. ILCs are mainly tissue-resident (16) and enriched at mucosal sites such as the respiratory, gastrointestinal and reproductive tracts, where they act as first responders to pathogens, aiding the innate immune system to launch a rapid defence, in addition to having roles in tissue repair and homeostasis (17). ILCs closely resemble Th cells in their development and function (18). They lack conventional antigen receptors, instead recognising non-specific danger signals, microbial compounds and cytokines (18), yet can also develop immunological memory (19). While ILCs and T cells have overlapping functions, ILCs perform additional non-redundant roles in priming adaptive immune responses (20). Like their T cell counterparts, ILCs are implicated in chronic inflammation, autoimmunity, and cancer (21–23).
The ILC family comprises five main subsets, which include natural killer (NK) cells, lymphoid tissue inducer (LTi) cells (which play a key role in the development of lymphoid tissues), ILC1, ILC2 and ILC3. ILCs have characteristics and functions that resemble adaptive CD4+ Th cell subsets. ILCs are classified into three main groups: group 1 (ILC1 and NK cells), group 2 (ILC2) and group 3 (ILC3 and LTi cells), which correspond to Th1 (NK cells correspond to CD8+ cytotoxic T cells), Th2 and Th17 cells respectively (24), based on similar transcription factors and functional profiles (25–27). ILCs derive from the common lymphoid progenitor and primarily develop in the foetal liver or in the bone marrow after birth (28). ILC1, ILC2 and ILC3, but not conventional NK cells, develop from Id2+ common helper-like innate lymphoid precursor cells (29), whereas conventional NK cells likely branch off earlier in development (30). Tissue-resident ILCs can be replenished from bone marrow or lymphoid organ precursors however, they are predominantly maintained through local self-renewal and expansion at tissue sites (16). While little is known about how ILC1s populate the lung, ILC2s and ILC3s arise in the lung shortly after birth, with ILC2 seeding dependent on production of IL-33 by type II alveolar epithelial cells (31) and ILC3s on insulin-like growth factor 1 from alveolar fibroblasts (32). ILCs are lineage-negative, lacking common lymphoid and myeloid lineage markers, and this feature is used to distinguish them by flow cytometry. ILCs are highly plastic and can change their phenotype and function depending on environmental signals, and their identification can also be complicated by their maturity (33).
Phenotypic Features of ILC Subsets
ILC1s and NK cells require the transcription factor T-bet for their development; however, NK cells additionally utilise Eomes (34). ILC1s and NK cells secrete interferon-gamma (IFN-γ) and tumour necrosis factor alpha which are key in the defence against intracellular pathogens. NK cells employ both a cytotoxic (CD56dim subset) and cytokine (CD56bright subset) response (35, 36). Both ILC2s and ILC3s have the potential to differentiate into ILC1 or ILC1-like cells (37, 38). Indeed, STAT-1, a key transcription factor activated during bacterial and viral infections, has been found to skew the differentiation of ILCs toward ILC1 while suppressing ILC2 and ILC3 responses (39).
ILC2s are dependent on the transcription factor GATA-3 and support Th2 immune responses via production of type 2 cytokines such as IL-4, IL-5, and IL-13 (40), which are essential for defence against extracellular parasites but can also drive allergic responses. ILC2s are the predominant ILC subset in the steady-state lung, where they secrete amphiregulin to promote pulmonary wound healing after infection, suggesting a homeostatic function (41). In mice, two distinct ILC2 populations have been characterized: natural ILC2s that are identified as Lineage-ST2+KLRG1int and classified as homeostatic, tissue-resident and IL-33-responsive; and, inflammatory ILC2s, which are undetectable at the steady-state but expand in response to IL-25 and can be distinguished as Lineage-ST2-KLRG1hi cells (42). ILC2s are activated by IL-33, IL-25, thymic stromal lymphopoietin (TSLP) and other danger signals produced by the airway epithelium (43, 44), with further support from prostaglandin D2 signalling through the CRTH2 receptor (40). Additionally, p38 MAPK has been found to positively regulate ILC2 function (45) while TGF-β is thought to program development via induction of ST2 expression in ILC2 progenitors (46). IL-1β is critical for ILC2 plasticity by inducing T-bet expression and promoting conversion into ILC1s in response to the Th1 cytokine IL-12 (47).
ILC3s and LTi cells require the transcription factor RORγt for their induction, and generate Th17-like responses, producing the cytokines IL-17, IL-22, and GM-CSF (24, 48). LTi cells also play an important role in lymphoid organogenesis in foetal development (49–51). ILC3s can be further sub-grouped by the expression of Natural Cytotoxicity Receptors (NCRs) such as NKp46 (NCR- or NCR+) (52, 53); while the expression of CCR6 and T-bet distinguishes effector cytokine profile (NCR-CCR6+T-bet- produce IL-17, NCR+CCR6-T-bet+ produce IFN-γ) (54). IL-18 can induce ILC3 proliferation and IL-22 production through NF-κB (55), while RANKL expression on ILC3s negatively regulates ILC3 cytokine production (56). Interestingly, the Th2 transcription factor GATA-3 is also critical for the induction and maintenance of ILC3s (57, 58) and therefore unsurprisingly, ILC2s have the potential to differentiate into IL-17-producing ILC3-like cells (59–61).
ILC Subsets in COPD and Asthma
ILC1 and NK Cells
ILC1 and NK Cells Are Indicators of COPD Severity
Recent studies suggest that an increased frequency of ILC1s in the peripheral blood of COPD patients correlates with disease severity and increased exacerbation risk (37, 62), and therefore may be utilised as a biomarker for disease progression. Furthermore, ILC1s as well as ILC3s are expanded in the lung of severe COPD patients (63). ILCs tend to localise to lymphoid aggregates in the lungs of COPD patients and smokers, whereas they are found in the parenchyma in healthy individuals (62). Cigarette smoke induces pulmonary ILC1s in a mouse model of COPD (62). ILC1s, alongside Th1 and CD8+ T cells can produce IFN-γ which is implicated in COPD pathogenesis by inducing elastolytic proteases and nitric oxide production by alveolar macrophages, leading to emphysema (64–66) (Figure 1). Furthermore, human ILC2s exhibit plasticity in vitro as well as in vivo when transferred to humanised mice, where they differentiate into ILC1s in the presence of IL-1β and IL-12 during pulmonary inflammation (63) and this is implicated in COPD exacerbations (37) (Figure 1).
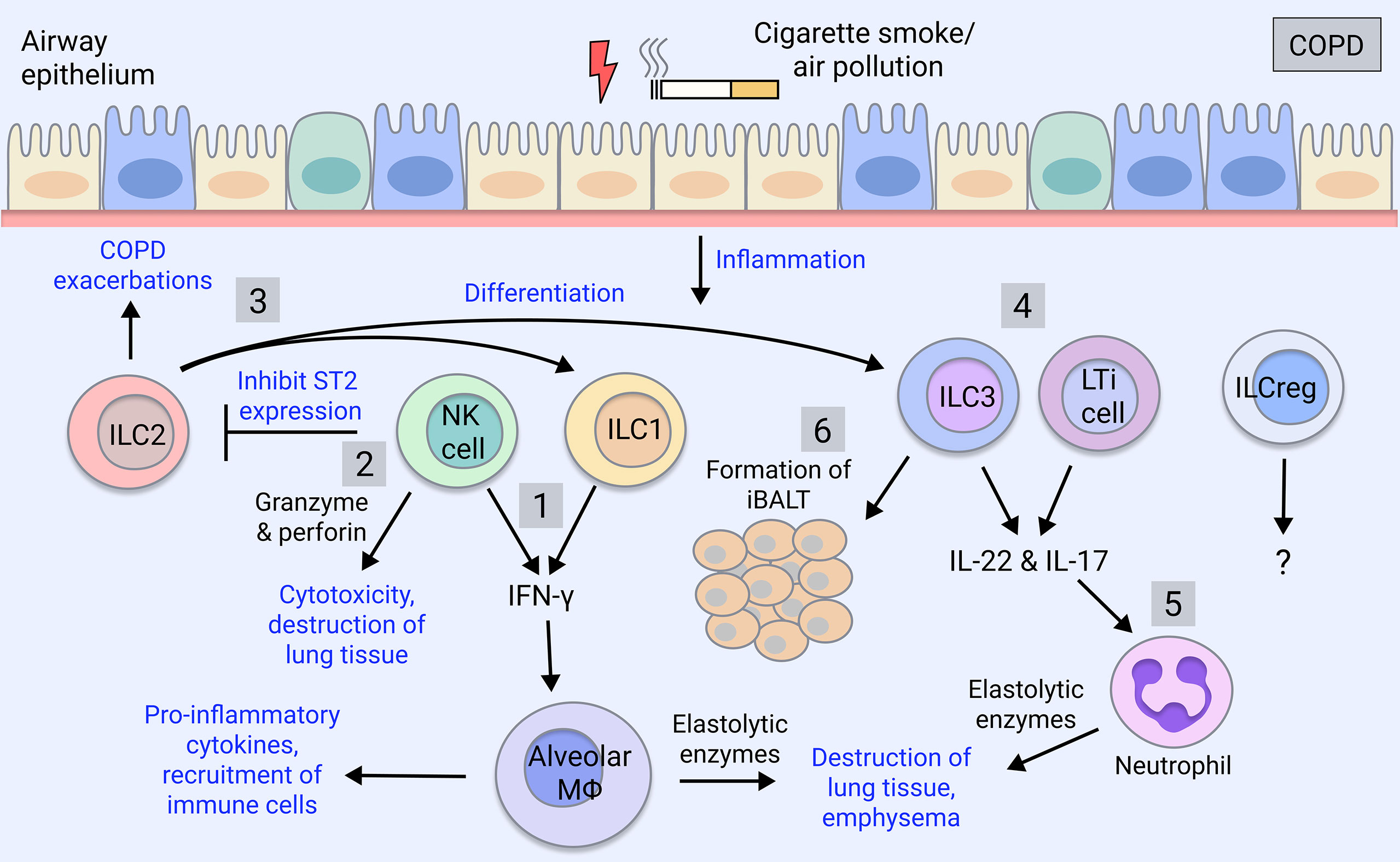
Figure 1 ILC involvement in COPD. COPD is caused by cigarette smoking and insults such as air pollutants. COPD patients exhibit increases in group 1 and group 3 ILCs, which correlate with severity and exacerbations, whereas ILC2 numbers are reduced (63). 1) ILC1 and NK cells produce the pro-inflammatory cytokine IFN-γ, which activates alveolar macrophages causing the release of inflammatory mediators (67). Macrophages secrete proteases (MMPs, cathepsins) inducing the destruction of the lung parenchyma thereby contributing to emphysema (67). 2) NK cell cytotoxic activity through secretion of granzyme and perforin induces death of lung tissue, furthering emphysema (68). NK cells also inhibit the production of ILC2 through downregulation of their ST2 receptor (69). 3) ILC2s promote Th2 inflammation during COPD exacerbations or differentiate into ILC1-like cells in the presence of IL-1β and IL-12 during lung inflammation (37, 63, 70). They potentially also differentiate into ILC3s (59–61). 4) ILC3 and LTi cells produce IL-17 and IL-22, which are elevated in COPD patients, driving pathogenesis (71, 72). 5) IL-17 induces the maturation and recruitment of neutrophils, which are expanded in COPD patients, and via their release of proteases (neutrophil elastase, cathepsin G, proteinase-3), contribute to mucus secretion and alveolar destruction (6). 6) ILC3 and LTi cells contribute to the formation of iBALT, which is a feature of advanced COPD (5) and is the site of ILC localisation in COPD lungs (62). ILCregs are yet to be understood in the regulation of COPD pathogenesis.
On the other hand, NK cell-mediated destruction of lung tissue is implicated in COPD as NK cell cytotoxicity is enhanced in the lung of COPD patients, correlating with worsened lung function and emphysema (68) (Figure 1). Lung dendritic cells, via IL-15Rα signalling, prime NK cell cytotoxicity in the COPD lung, which may represent a therapeutic target (73). NK cell cytokine production is also implicated; in mice, cigarette smoke triggers NK cell pro-inflammatory cytokine release (74) by promoting their expression of the IL-33 receptor, ST2, while inhibiting type 2 responses through downregulating ILC2 expression of ST2 (69) (Figure 1). Therefore, NK cells likely contribute to lung emphysematous destruction and inflammation in COPD.
NK Cells Have an Ambiguous Role in Asthma
While the contributions of ILC1s in asthma are currently unknown, ILC1s may be relevant to neutrophilic asthma or ACO, which warrants investigation. Meanwhile, NK cells can be both beneficial and detrimental in allergic and severe asthma. NK cells promote resolution of inflammation via inducing apoptosis of eosinophils and protecting against viral-induced inflammation (75, 76) (Figure 2). Furthermore, the immunomodulatory role of NK cells is impaired in severe asthma, with NK cells showing reduced lipotoxin A4-mediated clearance of eosinophils (84). Additionally, NK cell-mediated eosinophil clearance is inhibited by corticosteroids, implicating the loss of NK cell cytotoxicity in severe and steroid-resistant asthma (85). Conversely, NK cells can drive asthma-like allergic airway inflammation by inducing type 2 cytokine production (86–90). However, NK cells played neither a positive nor negative regulatory role in a house dust mite (HDM) model (91). Given that most human studies implicate immunomodulatory rather than pro-inflammatory NK cell functions, it is speculated that this may not be directly recapitulated in mouse models, so further clarification is needed.
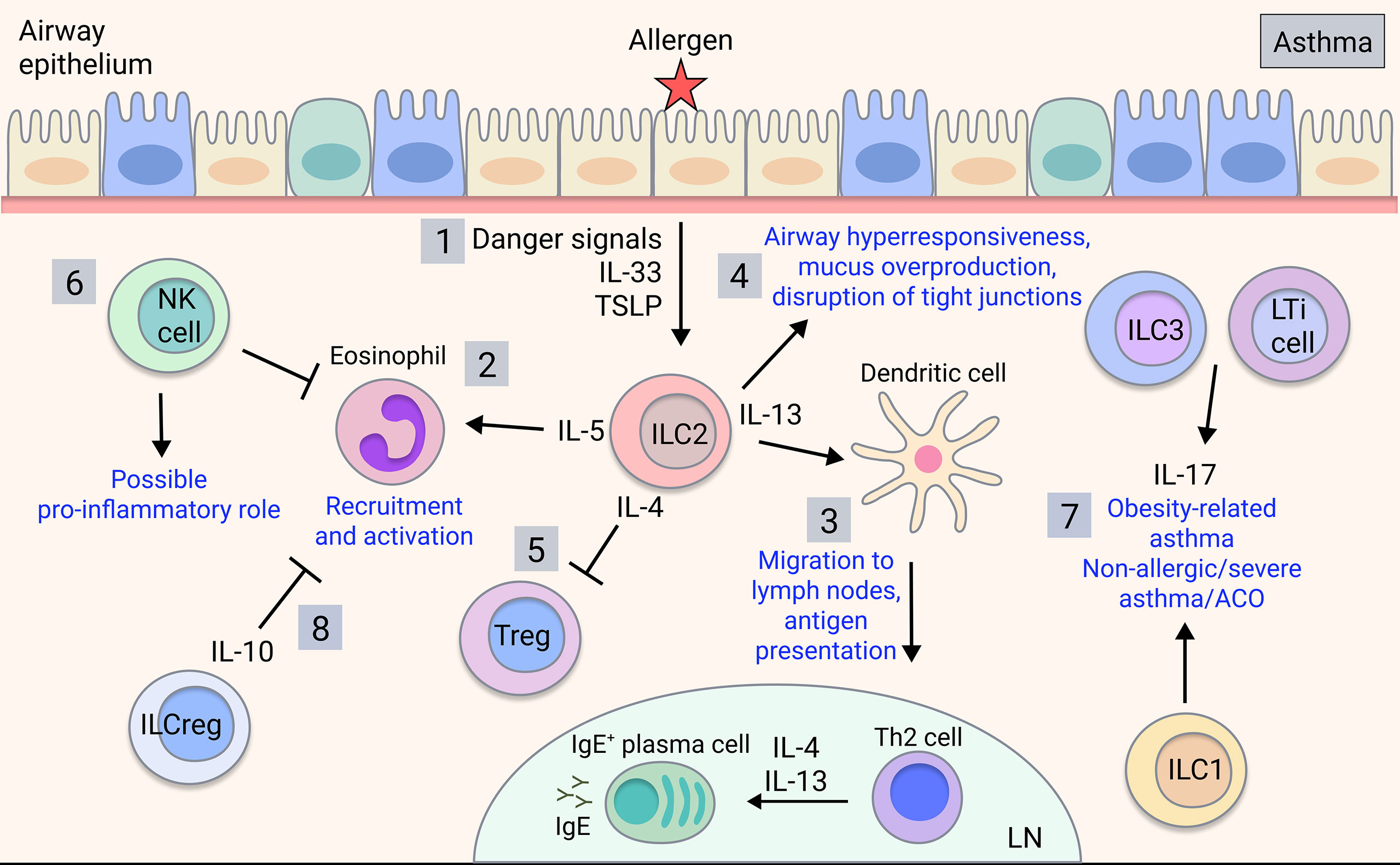
Figure 2 ILC involvement in asthma. Upon allergen detection by airway epithelium, 1) ILC2s are activated by signals released by the airway epithelial cells and other activated immune cells, producing type 2 cytokines such as IL-4, IL-5, and IL-13 in allergic asthma. 2) IL-5 is key for eosinophil recruitment and activation in the lung (77) and 3) IL-13 mediates dendritic cell migration to the lymph nodes, promoting T cell differentiation into effector Th2 cells, which mediate B cell class-switching and IgE production (78). 4) ILC2-derived IL-13 also acts on the airway epithelium to induce airway hyperresponsiveness, mucus overproduction and disruption of barrier integrity (43, 79, 80). 5) ILC2-derived IL-4 may potentially inhibit Treg production in asthma (81). 6) NK cells play an ambiguous role in asthma with both disease-driving and disease-modulatory activity shown. 7) ILC3s/LTi cells and possibly ILC1s contribute to obesity-related asthma and potentially non-allergic, severe asthma or ACO through production of IL-17 (82). 8) ILCregs may regulate asthma by inhibiting eosinophil recruitment through IL-10 (83).
ILC2s
ILC2 Involvement in COPD and Exacerbations
ILC2s can convert to ILC1s in the setting of COPD, suggesting skewing towards type 1 inflammation in this disease (37) (Figure 1). However, ILC2s themselves have also been implicated in COPD by promoting type 2 inflammatory responses (92), although it is unclear if ACO patients, who exhibit an intermediate type 2 cytokine profile (93), were included in this cohort. Interestingly, ILC2s have been shown to mediate neutrophil recruitment in a model of cigarette smoke-induced COPD and their deficiency protected against emphysema yet promoted fibrosis through elevation of IL-13 and IL-33 (94). Furthermore, ILC2s have been implicated in promoting Th2 adaptive responses during acute COPD exacerbations (70) (Figure 1).
ILC2s Are Major Players in Allergic Asthma
In allergic asthma, which is commonly associated with type 2 inflammation, there are increases in ILC2s in the peripheral blood compared to healthy individuals or those with allergic rhinitis (95–97), and ILC2s are expanded in the lung of patients with severe asthma and associated eosinophilia (84, 98). In sputum analyses of eosinophilic asthma patients, ILC2s are strongly induced alongside alternatively-activated ‘M2’ macrophages, whereas numbers of alveolar macrophages are unchanged (99). Meanwhile, neutrophilic asthma patients exhibit increases in ILC1s and ILC3s along with inflammatory ‘M1’ macrophages (99) (Figure 2). ILC2s are critical in allergic airway inflammation, driving pathology alongside and independent of the adaptive immune system (100). Eosinophil recruitment is induced by ILC2 production of IL-5 (77) whereas their production of IL-13 triggers AHR, mucus overproduction and disruption of epithelial integrity (43, 79, 80) (Figure 2). ILC2-derived IL-13 also promotes dendritic cell migration and subsequent Th2 cell induction (78). ILC2 production of IL-4 blocks Treg induction in food allergy responses (81), however this has yet to be shown in asthma. Interestingly, a subset of CCR10+ ILC2s that exhibit ILC1-like characteristics and have the capacity to produce IFN-γ, were protective in both allergic and non-allergic severe asthmatic patients by limiting Th2 cytokine secretion and downregulating type 2 responses (101), demonstrating a protective function.
Numerous studies have explored the mechanisms of ILC2 regulation in allergic asthma. The neuropeptide neuromedin U, has been found to powerfully induce an asthma-like response and activate ILC2s (102), suggesting involvement of the neuroimmune axis. IL-1β, arginase 1, lipotoxin A4, maresin 1, Nrf2, FABP5, IL-35, IFN-γ and PD-1 have all been shown to restrict ILC2 responses and promote resolution of allergic lung inflammation or prevent AHR through various mechanisms (79, 103–109). Furthermore, in allergic asthma, ILC2s are controlled by the transcription factor IRF7, a key regulator of anti-viral responses (110). On the other hand, neutrophils are reported to control allergic airway inflammation in a mouse HDM asthma model through the inhibition of ILC2 responses and G-CSF modulation (111). Collectively, this suggests that ILC2s may be a viable therapeutic target in asthma.
ILC3 and LTi Cells
Increased Il-17 Levels Implicate Group 3 ILCs in COPD Pathogenesis
In COPD, proportions of lung ILC subsets are skewed toward NCR- ILC3s/LTi cells whereas non-COPD individuals have balanced proportions, with ILC2s and NCR- ILC3s in greatest abundance (112) (Figure 1). NCR- ILC3s are also enriched in severe COPD patients (63). In COPD patients and smokers, a highly migratory subset of ILC3s expressing neuropilin-1 receptor is found within the lung and associates with iBALT via induction of ICAM-1 and VCAM-1 on mesenchymal stromal cells (113) (Figure 1). In mice, ILC3s and ILC1s are increased in response to cigarette smoke exposure, whereas ILC2s are diminished (94). ILC3s are early producers of IL-17 and IL-22, which are implicated in COPD pathogenesis. IL-17 is elevated in the peripheral blood of COPD patients (114, 115) and steroid-resistant COPD (116), and COPD exacerbations have been associated with IL-17 and neutrophilic infiltration (71). IL-22 meanwhile, is elevated in COPD patient lungs and contributes to experimental COPD (72). Additionally, improvements in lung outcomes and comorbidities have been observed in cigarette smoke-exposed IL-17-deficient mice (117, 118). Given the association between ILC3 and IL-17 production, it is surprising that this link has not been addressed in COPD but could be a promising therapeutic avenue.
Group 3 ILCs May Be a Player in Neutrophilic Asthma and ACO
Currently, there are limited reports on ILC3s and LTi cells in asthma, however, ILC3-mediated production of IL-17 may contribute (Figure 2). IL-17 is implicated in severe asthma, neutrophilic asthma, asthma exacerbations and airway remodelling involving the recruitment of neutrophils (119, 120) and may contribute to ACO given that IL-17 levels are increased in ACO patients (121). Furthermore, the development of AHR in obesity-related asthma in mice relied on IL-17-producing ILC3s and the NLRP3 inflammasome, and ILC3s were expanded in the BAL of severe asthma patients (82). Therefore, via IL-17, ILC3s have a potential role in distinct asthma endotypes, although further studies are warranted in human disease.
ILCregs
Regulatory ILCs and Anti-Inflammatory Cytokine Production
Recently, a novel group of regulatory ILCs (ILCreg) have been identified in gut, characterised by expression of the immunoregulatory cytokines IL-10 and TGF-β (122). While ILCregs share mechanistic similarities with regulatory T cells (Treg), they exhibit a unique transcriptome profile and lack the typical Treg transcription factor FoxP3 (122). Furthermore, ILCregs are transcriptionally distinct from typical ILCs, lacking common ILC transcription factors and can be distinguished as Lineage-CD45+CD127+ cells and by production of IL-10 (122). Moreover, recent reports show that ex vivo human ILC1s and ILC2s can also potently produce IL-10 (123). In mice, an induced ILCreg population derived from ILC2s, likely in response to retinoic acid, was observed in the lungs in HDM-induced allergic airway inflammation, with a similar population of cells also detected in human airway tissue (124). Likewise, a subset of ILC2s that produces IL-10 (ILC210) has been directly implicated in decreasing eosinophil recruitment to the injured lung (83) (Figure 2). While there is evidence that immunoregulatory ILC subsets are implicated in asthma, this has yet to be reported in COPD.
The Effects of Therapies on ILC Responses
Recent guidelines recommend that adults with asthma receive a combination therapy comprising inhaled corticosteroids and long-acting β-agonists (125), and for severe asthma, inclusion of biologicals targeting type 2 responses (126). In COPD, while corticosteroids are ineffective, the recommendation for COPD exacerbations is a triple inhaled therapy containing corticosteroids, long-acting β2-agonists, and long-acting muscarinic antagonists (127). Studies are beginning to reveal how these agents regulate ILC activity.
Dexamethasone is reported to inhibit type 2 cytokine production from ILC2s (128–131); however, IL-7 and TSLP induce resistance via IL-7Rα and STAT5 (129). Conversion of resting CD45RA+ ILC2s to inflammatory CD45RO+ ILC2s is suppressed by corticosteroids; however, once present, inflammatory ILC2s are resistant (132). Furthermore, inflammatory ILC2s are increased in the lung and blood of patients with chronic asthma correlating with disease severity and corticosteroid-resistance (132). In paediatric patients with severe therapy-resistant asthma, ILC2s, eosinophils and Th2 cells are increased, whereas Th17 cells and IL-17+ ILCs are unchanged. Systemic but not inhaled corticosteroids reduced ILC2s and Th2 cells as well as symptoms, despite persistence of IL-17+ cells and eosinophils (133). Collectively these studies suggest that ILC2 activity can be controlled by steroid therapy, but the inflammatory environment may alter steroid responsiveness. Anti-IL-5Rα therapy in patients with severe steroid-dependent asthma reduced blood and sputum eosinophils and IL-5Rα+ ILC2 but not total ILC2. While the functional relevance of IL-5Rα+ ILC2 is unclear, these changes were associated with improved asthma control and lung function (134).
ILC2s express the gene encoding β2-adrenergic receptor, with deficiency of this gene in mice inducing ILC2s and inflammatory responses (135). Furthermore, IL-33-induced ILC2 expansion and IL-5 and IL-13 production in lung were reduced by β2-agonist treatment, suggesting that β2-adrenergic receptor signalling limits the proliferation and function of ILC2s (135). With respect to cholinergic pathways, neuromedin U strongly activates ILC2s and amplifies IL-25-dependent lung inflammation (102, 136, 137). Moreover, the NMUR1 neuromedin U receptor is expressed on ILC2 and its deficiency attenuates ILC2 number and function in allergic airway inflammation (102). While little is known of the effects of muscarinic antagonists on ILCs or whether they express the receptors, in a papain-induced model of airway inflammation, the long-acting muscarinic antagonist tiotropium indirectly suppressed ILC2 activation by reducing IL-4 production from basophils (138). Clearly more studies are required to determine how therapies affect ILC phenotype and function, with these likely providing new insights into the regulation of these cells in chronic lung inflammation.
Conclusion
Innate lymphoid cells are an incompletely understood accomplice in the maladapted inflammatory environment that promotes chronic respiratory diseases. They potentially play a significant role in disease pathogenesis given their rapid response to pathogenic or environmental stimuli and may be the missing gap where conventional cell-based therapies have failed. However, the identification of ILCs is complex, making their study difficult, and their phenotype may be further complicated by their plasticity, microenvironment, and stages of differentiation. Although ILC manipulation in chronic respiratory diseases represents a significant challenge, understanding the intricacy of ILC regulation and the signals used by other tissue-resident cells to control their responses may provide a means of targeting them indirectly. The emergence of regulatory ILCs suggests another level of disease control, however these cells may be impaired or overwhelmed in chronic inflammatory settings. Nonetheless, ILCs represent a developing field in disease research. Understanding the role of ILCs during chronic inflammatory airway diseases like COPD and asthma will provide insight into the complexity of these diseases, how they are initiated and the manner in which they transition into a chronic state. Additionally, understanding how ILCs are regulated, how they respond to conventional treatments and furthermore how they regulate immune responses may allow us to devise strategies to switch their disease-driving capabilities into disease-modulatory actions.
Author Contributions
MH conceptualised the article, contributed to the writing of the first draft and provided manuscript revisions. AH wrote the first draft, revised the manuscript and developed original figures. TG and ET amended the manuscript and provided extensive editorial input. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funding awarded to MH from the National Health and Medical Research Council (NHMRC) grant number 1141208. AH is the recipient of an Australian Government Research Training Program (RTP) Scholarship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Quaderi SA, Hurst JR. The Unmet Global Burden of COPD. Glob Health Epidemiol Genom (2018) 3:e4. doi: 10.1017/gheg.2018.1
2. Viniol C, Vogelmeier CF. Exacerbations of COPD. Eur Respir Rev (2018) 27(147):170103. doi: 10.1183/16000617.0103-2017
3. Mannino DM, Buist AS. Global Burden of COPD: Risk Factors, Prevalence, and Future Trends. Lancet (2007) 370(9589):765–73. doi: 10.1016/S0140-6736(07)61380-4
4. Rabe KF, Watz H. Chronic Obstructive Pulmonary Disease. Lancet (2017) 389(10082):1931–40. doi: 10.1016/S0140-6736(17)31222-9
5. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The Nature of Small-Airway Obstruction in Chronic Obstructive Pulmonary Disease. N Engl J Med (2004) 350(26):2645–53. doi: 10.1056/NEJMoa032158
6. Barnes PJ. Cellular and Molecular Mechanisms of Asthma and COPD. Clin Sci (Lond) (2017) 131(13):1541–58. doi: 10.1042/CS20160487
7. Chapman DG, Irvin CG. Mechanisms of Airway Hyper-Responsiveness in Asthma: The Past, Present and Yet to Come. Clin Exp Allergy (2015) 45(4):706–19. doi: 10.1111/cea.12506
8. Dharmage SC, Perret JL, Custovic A. Epidemiology of Asthma in Children and Adults. Front Pediatr (2019) 7:246–. doi: 10.3389/fped.2019.00246
9. Australian Institute of Health and Welfare 2020. Asthma. Cat. no. ACM 33. Canberra: AIHW (2021). Available at: https://www.aihw.gov.au/reports/chronic-respiratory-conditions/asthma.
10. Fahy JV. Type 2 Inflammation in Asthma–Present in Most, Absent in Many. Nat Rev Immunol (2015) 15(1):57–65. doi: 10.1038/nri3786
11. Ray A, Kolls JK. Neutrophilic Inflammation in Asthma and Association With Disease Severity. Trends Immunol (2017) 38(12):942–54. doi: 10.1016/j.it.2017.07.003
12. Bullone M, Carriero V, Bertolini F, Folino A, Mannelli A, Di Stefano A, et al. Elevated Serum IgE, Oral Corticosteroid Dependence and IL-17/22 Expression in Highly Neutrophilic Asthma. Eur Respir J (2019) 54(5):1900068. doi: 10.1183/13993003.00068-2019
13. Ray A, Raundhal M, Oriss TB, Ray P, Wenzel SE. Current Concepts of Severe Asthma. J Clin Invest (2016) 126(7):2394–403. doi: 10.1172/JCI84144
14. Lang DM. Severe Asthma: Epidemiology, Burden of Illness, and Heterogeneity. Allergy Asthma Proc (2015) 36(6):418–24. doi: 10.2500/aap.2015.36.3908
15. Postma DS, Rabe KF. The Asthma–COPD Overlap Syndrome. N Engl J Med (2015) 373(13):1241–9. doi: 10.1056/NEJMra1411863
16. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue Residency of Innate Lymphoid Cells in Lymphoid and Nonlymphoid Organs. Science (2015) 350(6263):981–5. doi: 10.1126/science.aac9593
17. Panda SK, Colonna M. Innate Lymphoid Cells in Mucosal Immunity. Front Immunol (2019) 10(861):121–33. doi: 10.3389/fimmu.2019.00861
18. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate Lymphoid Cells. Innate Lymphoid Cells: A New Paradigm in Immunology. Science (2015) 348(6237):aaa6566. doi: 10.1126/science.aaa6566
19. Wang X, Peng H, Tian Z. Innate Lymphoid Cell Memory. Cell Mol Immunol (2019) 16(5):423–9. doi: 10.1038/s41423-019-0212-6
20. Sonnenberg GF, Hepworth MR. Functional Interactions Between Innate Lymphoid Cells and Adaptive Immunity. Nat Rev Immunol (2019) 19(10):599–613. doi: 10.1038/s41577-019-0194-8
21. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human Type 1 Innate Lymphoid Cells Accumulate in Inflamed Mucosal Tissues. Nat Immunol (2013) 14(3):221–9. doi: 10.1038/ni.2534
22. Shikhagaie MM, Germar K, Bal SM, Ros XR, Spits H. Innate Lymphoid Cells in Autoimmunity: Emerging Regulators in Rheumatic Diseases. Nat Rev Rheumatol (2017) 13(3):164–73. doi: 10.1038/nrrheum.2016.218
23. Bruchard M, Ghiringhelli F. Deciphering the Roles of Innate Lymphoid Cells in Cancer. Front Immunol (2019) 10:656. doi: 10.3389/fimmu.2019.00656
24. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate Lymphoid Cells–a Proposal for Uniform Nomenclature. Nat Rev Immunol (2013) 13(2):145–9. doi: 10.1038/nri3365
25. Cherrier DE, Serafini N, Di Santo JP. Innate Lymphoid Cell Development: A T Cell Perspective. Immunity (2018) 48(6):1091–103. doi: 10.1016/j.immuni.2018.05.010
26. Vély F, Barlogis V, Vallentin B, Neven B, Piperoglou C, Ebbo M, et al. Evidence of Innate Lymphoid Cell Redundancy in Humans. Nat Immunol (2016) 17(11):1291–9. doi: 10.1038/ni.3553
27. Robinette ML, Colonna M. Immune Modules Shared by Innate Lymphoid Cells and T Cells. J Allergy Clin Immunol (2016) 138(5):1243–51. doi: 10.1016/j.jaci.2016.09.006
28. Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A Committed Precursor to Innate Lymphoid Cells. Nature (2014) 508(7496):397–401. doi: 10.1038/nature13047
29. Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of Type 1 ILCs From a Common Progenitor to All Helper-Like Innate Lymphoid Cell Lineages. Cell (2014) 157(2):340–56. doi: 10.1016/j.cell.2014.03.030
30. Diefenbach A, Colonna M, Koyasu S. Development, Differentiation, and Diversity of Innate Lymphoid Cells. Immunity (2014) 41(3):354–65. doi: 10.1016/j.immuni.2014.09.005
31. Saluzzo S, Gorki A-D, Rana BMJ, Martins R, Scanlon S, Starkl P, et al. First-Breath-Induced Type 2 Pathways Shape the Lung Immune Environment. Cell Rep (2017) 18(8):1893–905. doi: 10.1016/j.celrep.2017.01.071
32. Oherle K, Acker E, Bonfield M, Wang T, Gray J, Lang I, et al. Insulin-Like Growth Factor 1 Supports a Pulmonary Niche That Promotes Type 3 Innate Lymphoid Cell Development in Newborn Lungs. Immunity (2020) 52(2):275–94.e9. doi: 10.1016/j.immuni.2020.01.005
33. Borger JG, Lau M, Hibbs ML. The Influence of Innate Lymphoid Cells and Unconventional T Cells in Chronic Inflammatory Lung Disease. Front Immunol (2019) 10:1597. doi: 10.3389/fimmu.2019.01597
34. Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The Transcription Factors T-Bet and Eomes Control Key Checkpoints of Natural Killer Cell Maturation. Immunity (2012) 36(1):55–67. doi: 10.1016/j.immuni.2011.11.016
35. Caligiuri MA. Human Natural Killer Cells. Blood (2008) 112(3):461–9. doi: 10.1182/blood-2007-09-077438
36. Cooper MA, Fehniger TA, Caligiuri MA. The Biology of Human Natural Killer-Cell Subsets. Trends Immunol (2001) 22(11):633–40. doi: 10.1016/S1471-4906(01)02060-9
37. Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory Triggers Associated With Exacerbations of COPD Orchestrate Plasticity of Group 2 Innate Lymphoid Cells in the Lungs. Nat Immunol (2016) 17(6):626–35. doi: 10.1038/ni.3443
38. Cella M, Gamini R, Sécca C, Collins PL, Zhao S, Peng V, et al. Subsets of ILC3–ILC1-Like Cells Generate a Diversity Spectrum of Innate Lymphoid Cells in Human Mucosal Tissues. Nat Immunol (2019) 20(8):980–91. doi: 10.1038/s41590-019-0425-y
39. Stier MT, Goleniewska K, Cephus JY, Newcomb DC, Sherrill TP, Boyd KL, et al. STAT1 Represses Cytokine-Producing Group 2 and Group 3 Innate Lymphoid Cells During Viral Infection. J Immunol (2017) 199(2):510. doi: 10.4049/jimmunol.1601984
40. Xue L, Salimi M, Panse I, Mjösberg JM, McKenzie ANJ, Spits H, et al. Prostaglandin D2 Activates Group 2 Innate Lymphoid Cells Through Chemoattractant Receptor-Homologous Molecule Expressed on TH2 Cells. J Allergy Clin Immunol (2014) 133(4):1184–94. doi: 10.1016/j.jaci.2013.10.056
41. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate Lymphoid Cells Promote Lung-Tissue Homeostasis After Infection With Influenza Virus. Nat Immunol (2011) 12(11):1045–54. doi: 10.1038/ni.2131
42. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-Responsive, Lineage-Negative KLRG1(hi) Cells Are Multipotential ‘Inflammatory’ Type 2 Innate Lymphoid Cells. Nat Immunol (2015) 16(2):161–9. doi: 10.1038/ni.3078
43. Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate Lymphoid Cells Mediate Influenza-Induced Airway Hyper-Reactivity Independently of Adaptive Immunity. Nat Immunol (2011) 12(7):631–8. doi: 10.1038/ni.2045
44. Christianson C, Irvin C, Gorska M, Alam R. IL33 and Type 2 Innate Lymphoid Cells (ILC2) But Not Th2 Cells Are Essential For Persistence Of Chronic Experimental Asthma. J Allergy Clin Immunol (2014) 133(2):AB170. doi: 10.1016/j.jaci.2013.12.610
45. Petrova T, Pesic J, Pardali K, Gaestel M, Arthur JSC. P38 MAPK Signalling Regulates Cytokine Production in IL-33 Stimulated Type 2 Innate Lymphoid Cells. Sci Rep (2020) 10(1):3479. doi: 10.1038/s41598-020-60089-0
46. Wang L, Tang J, Yang X, Zanvit P, Cui K, Ku WL, et al. TGF-β Induces ST2 and Programs ILC2 Development. Nat Commun (2020) 11(1):35. doi: 10.1038/s41467-019-13734-w
47. Ohne Y, Silver JS, Thompson-Snipes L, Collet MA, Blanck JP, Cantarel BL, et al. IL-1 Is a Critical Regulator of Group 2 Innate Lymphoid Cell Function and Plasticity. Nat Immunol (2016) 17(6):646–55. doi: 10.1038/ni.3447
48. Pearson C, Thornton EE, McKenzie B, Schaupp AL, Huskens N, Griseri T, et al. ILC3 GM-CSF Production and Mobilisation Orchestrate Acute Intestinal Inflammation. Elife (2016) 5:e10066. doi: 10.7554/eLife.10066
49. Onder L, Mörbe U, Pikor N, Novkovic M, Cheng H-W, Hehlgans T, et al. Lymphatic Endothelial Cells Control Initiation of Lymph Node Organogenesis. Immunity (2017) 47(1):80–92.e4. doi: 10.1016/j.immuni.2017.05.008
50. Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, et al. Human Fetal Lymphoid Tissue-Inducer Cells Are Interleukin 17-Producing Precursors to RORC+ CD127+ Natural Killer-Like Cells. Nat Immunol (2009) 10(1):66–74. doi: 10.1038/ni.1668
51. Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, et al. Lymphoid Tissue Inducer-Like Cells Are an Innate Source of IL-17 and IL-22. J Exp Med (2009) 206(1):35–41. doi: 10.1084/jem.20072713
52. Hoorweg K, Peters CP, Cornelissen F, Aparicio-Domingo P, Papazian N, Kazemier G, et al. Functional Differences Between Human NKp44(-) and NKp44(+) RORC(+) Innate Lymphoid Cells. Front Immunol (2012) 3:72. doi: 10.3389/fimmu.2012.00072
53. Simoni Y, Newell EW. Dissecting Human ILC Heterogeneity: More Than Just Three Subsets. Immunology (2018) 153(3):297–303. doi: 10.1111/imm.12862
54. Klose CSN, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, et al. A T-Bet Gradient Controls the Fate and Function of CCR6–Rorγt+ Innate Lymphoid Cells. Nature (2013) 494(7436):261–5. doi: 10.1038/nature11813
55. Victor AR, Nalin AP, Dong W, McClory S, Wei M, Mao C, et al. IL-18 Drives ILC3 Proliferation and Promotes IL-22 Production via NF-κb. J Immunol (2017) 199(7):2333–42. doi: 10.4049/jimmunol.1601554
56. Bando JK, Gilfillan S, Song C, McDonald KG, Huang SCC, Newberry RD, et al. The Tumor Necrosis Factor Superfamily Member RANKL Suppresses Effector Cytokine Production in Group 3 Innate Lymphoid Cells. Immunity (2018) 48(6):1208–19.e4. doi: 10.1016/j.immuni.2018.04.012
57. Zhong C, Cui K, Wilhelm C, Hu G, Mao K, Belkaid Y, et al. Group 3 Innate Lymphoid Cells Continuously Require the Transcription Factor GATA-3 After Commitment. Nat Immunol (2016) 17(2):169–78. doi: 10.1038/ni.3318
58. Serafini N, Klein Wolterink RG, Satoh-Takayama N, Xu W, Vosshenrich CA, Hendriks RW, et al. Gata3 Drives Development of RORγt+ Group 3 Innate Lymphoid Cells. J Exp Med (2014) 211(2):199–208. doi: 10.1084/jem.20131038
59. Bernink JH, Ohne Y, Teunissen MBM, Wang J, Wu J, Krabbendam L, et al. c-Kit-Positive ILC2s Exhibit an ILC3-Like Signature That may Contribute to IL-17-Mediated Pathologies. Nat Immunol (2019) 20(8):992–1003. doi: 10.1038/s41590-019-0423-0
60. Zhang K, Xu X, Pasha MA, Siebel CW, Costello A, Haczku A, et al. Cutting Edge: Notch Signaling Promotes the Plasticity of Group-2 Innate Lymphoid Cells. J Immunol (2017) 198(5):1798–803. doi: 10.4049/jimmunol.1601421
61. Golebski K, Ros XR, Nagasawa M, van Tol S, Heesters BA, Aglmous H, et al. IL-1β, IL-23, and TGF-β Drive Plasticity of Human ILC2s Towards IL-17-Producing ILCs in Nasal Inflammation. Nat Commun (2019) 10(1):2162. doi: 10.1038/s41467-019-09883-7
62. Blomme EE, Provoost S, De Smet EG, De Grove KC, Van Eeckhoutte HP, De Volder J, et al. Quantification and Role of Innate Lymphoid Cell Subsets in Chronic Obstructive Pulmonary Disease. Clin Transl Immunol (2021) 10(6):e1287. doi: 10.1002/cti2.1287
63. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1β, IL-4 and IL-12 Control the Fate of Group 2 Innate Lymphoid Cells in Human Airway Inflammation in the Lungs. Nat Immunol (2016) 17(6):636–45. doi: 10.1038/ni.3444
64. Wang Z, Zheng T, Zhu Z, Homer RJ, Riese RJ, Chapman HA Jr, et al. Interferon Gamma Induction of Pulmonary Emphysema in the Adult Murine Lung. J Exp Med (2000) 192(11):1587–600. doi: 10.1084/jem.192.11.1587
65. Freeman CM, Han MK, Martinez FJ, Murray S, Liu LX, Chensue SW, et al. Cytotoxic Potential of Lung CD8(+) T Cells Increases With Chronic Obstructive Pulmonary Disease Severity and With In Vitro Stimulation by IL-18 or IL-15. J Immunol (2010) 184(11):6504–13. doi: 10.4049/jimmunol.1000006
66. Majori M, Corradi M, Caminati A, Cacciani G, Bertacco S, Pesci A. Predominant TH1 Cytokine Pattern in Peripheral Blood From Subjects With Chronic Obstructive Pulmonary Disease. J Allergy Clin Immunol (1999) 103(3 Pt 1):458–62. doi: 10.1016/S0091-6749(99)70471-9
67. Barnes PJ, Shapiro SD, Pauwels RA. Chronic Obstructive Pulmonary Disease: Molecular and Cellularmechanisms. Eur Respir J (2003) 22(4):672. doi: 10.1183/09031936.03.00040703
68. Freeman CM, Stolberg VR, Crudgington S, Martinez FJ, Han MK, Chensue SW, et al. Human CD56+ Cytotoxic Lung Lymphocytes Kill Autologous Lung Cells in Chronic Obstructive Pulmonary Disease. PloS One (2014) 9(7):e103840. doi: 10.1371/journal.pone.0103840
69. Kearley J, Silver Jonathan S, Sanden C, Liu Z, Berlin Aaron A, White N, et al. Cigarette Smoke Silences Innate Lymphoid Cell Function and Facilitates an Exacerbated Type I Interleukin-33-Dependent Response to Infection. Immunity (2015) 42(3):566–79. doi: 10.1016/j.immuni.2015.02.011
70. Jiang M, Liu H, Li Z, Wang J, Zhang F, Cao K, et al. ILC2s Induce Adaptive Th2-Type Immunity in Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Mediators Inflamm (2019) 2019:3140183. doi: 10.1155/2019/3140183
71. Roos AB, Sethi S, Nikota J, Wrona CT, Dorrington MG, Sandén C, et al. IL-17A and the Promotion of Neutrophilia in Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2015) 192(4):428–37. doi: 10.1164/rccm.201409-1689OC
72. Starkey MR, Plank MW, Casolari P, Papi A, Pavlidis S, Guo Y, et al. IL-22 and Its Receptors Are Increased in Human and Experimental COPD and Contribute to Pathogenesis. Eur Respir J (2019) 54(1):1800174. doi: 10.1183/13993003.00174-2018
73. Finch DK, Stolberg VR, Ferguson J, Alikaj H, Kady MR, Richmond BW, et al. Lung Dendritic Cells Drive Natural Killer Cytotoxicity in Chronic Obstructive Pulmonary Disease via IL-15rα. Am J Respir Crit Care Med (2018) 198(9):1140–50. doi: 10.1164/rccm.201712-2513OC
74. Motz GT, Eppert BL, Wortham BW, Amos-Kroohs RM, Flury JL, Wesselkamper SC, et al. Chronic Cigarette Smoke Exposure Primes NK Cell Activation in a Mouse Model of Chronic Obstructive Pulmonary Disease. J Immunol (2010) 184(8):4460–9. doi: 10.4049/jimmunol.0903654
75. Haworth O, Cernadas M, Levy BD. NK Cells Are Effectors for Resolvin E1 in the Timely Resolution of Allergic Airway Inflammation. J Immunol (2011) 186(11):6129–35. doi: 10.4049/jimmunol.1004007
76. Kaiko GE, Phipps S, Angkasekwinai P, Dong C, Foster PS. NK Cell Deficiency Predisposes to Viral-Induced Th2-Type Allergic Inflammation via Epithelial-Derived IL-25. J Immunol (2010) 185(8):4681–90. doi: 10.4049/jimmunol.1001758
77. Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 Innate Lymphoid Cells Control Eosinophil Homeostasis. Nature (2013) 502(7470):245–8. doi: 10.1038/nature12526
78. Halim TY, Steer CA, Mathä L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 Innate Lymphoid Cells Are Critical for the Initiation of Adaptive T Helper 2 Cell-Mediated Allergic Lung Inflammation. Immunity (2014) 40(3):425–35. doi: 10.1016/j.immuni.2014.01.011
79. Han M, Ishikawa T, Bermick JR, Rajput C, Lei J, Goldsmith AM, et al. IL-1β Prevents ILC2 Expansion, Type 2 Cytokine Secretion, and Mucus Metaplasia in Response to Early-Life Rhinovirus Infection in Mice. Allergy (2020) 75(8):2005–19. doi: 10.1111/all.14241
80. Sugita K, Steer CA, Martinez-Gonzalez I, Altunbulakli C, Morita H, Castro-Giner F, et al. Type 2 Innate Lymphoid Cells Disrupt Bronchial Epithelial Barrier Integrity by Targeting Tight Junctions Through IL-13 in Asthmatic Patients. J Allergy Clin Immunol (2018) 141(1):300–10.e11. doi: 10.1016/j.jaci.2017.02.038
81. Noval Rivas M, Burton OT, Oettgen HC, Chatila T. IL-4 Production by Group 2 Innate Lymphoid Cells Promotes Food Allergy by Blocking Regulatory T-Cell Function. J Allergy Clin Immunol (2016) 138(3):801–11.e9. doi: 10.1016/j.jaci.2016.02.030
82. Kim HY, Lee HJ, Chang Y-J, Pichavant M, Shore SA, Fitzgerald KA, et al. Interleukin-17–Producing Innate Lymphoid Cells and the NLRP3 Inflammasome Facilitate Obesity-Associated Airway Hyperreactivity. Nat Med (2014) 20(1):54–61. doi: 10.1038/nm.3423
83. Seehus CR, Kadavallore A, Torre B, Yeckes AR, Wang Y, Tang J, et al. Alternative Activation Generates IL-10 Producing Type 2 Innate Lymphoid Cells. Nat Commun (2017) 8(1):1900. doi: 10.1038/s41467-017-02023-z
84. Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 Regulates Natural Killer Cell and Type 2 Innate Lymphoid Cell Activation in Asthma. Sci Transl Med (2013) 5(174):174ra26–ra26. doi: 10.1126/scitranslmed.3004812
85. Duvall MG, Barnig C, Cernadas M, Ricklefs I, Krishnamoorthy N, Grossman NL, et al. Natural Killer Cell-Mediated Inflammation Resolution Is Disabled in Severe Asthma. Sci Immunol (2017) 2(9):eaam5446. doi: 10.1126/sciimmunol.aam5446
86. Wei H, Zhang J, Xiao W, Feng J, Sun R, Tian Z. Involvement of Human Natural Killer Cells in Asthma Pathogenesis: Natural Killer 2 Cells in Type 2 Cytokine Predominance. J Allergy Clin Immunol (2005) 115(4):841–7. doi: 10.1016/j.jaci.2004.11.026
87. Korsgren M, Persson CGA, Sundler F, Bjerke T, Hansson T, Chambers BJ, et al. Natural Killer Cells Determine Development of Allergen-Induced Eosinophilic Airway Inflammation in Mice. J Exp Med (1999) 189(3):553–62. doi: 10.1084/jem.189.3.553
88. Mathias CB, Guernsey LA, Zammit D, Brammer C, Wu CA, Thrall RS, et al. Pro-Inflammatory Role of Natural Killer Cells in the Development of Allergic Airway Disease. Clin Exp Allergy (2014) 44(4):589–601. doi: 10.1111/cea.12271
89. Farhadi N, Lambert L, Triulzi C, Openshaw PJ, Guerra N, Culley FJ. Natural Killer Cell NKG2D and Granzyme B Are Critical for Allergic Pulmonary Inflammation. J Allergy Clin Immunol (2014) 133(3):827–35.e3. doi: 10.1016/j.jaci.2013.09.048
90. Arase N, Arase H, Hirano S, Yokosuka T, Sakurai D, Saito T. IgE-Mediated Activation of NK Cells Through Fcγriii. J Immunol (2003) 170(6):3054–8. doi: 10.4049/jimmunol.170.6.3054
91. Haspeslagh E, van Helden MJ, Deswarte K, De Prijck S, van Moorleghem J, Boon L, et al. Role of NKp46(+) Natural Killer Cells in House Dust Mite-Driven Asthma. EMBO Mol Med (2018) 10(4):e8657. doi: 10.15252/emmm.201708657
92. Jiang M, Tao S, Zhang S, Wang J, Zhang F, Li F, et al. Type 2 Innate Lymphoid Cells Participate in IL-33-Stimulated Th2-Associated Immune Response in Chronic Obstructive Pulmonary Disease. Exp Ther Med (2019) 18(4):3109–16. doi: 10.3892/etm.2019.7924
93. de Llano LP, Cosío BG, Iglesias A, de Las Cuevas N, Soler-Cataluña JJ, Izquierdo JL, et al. Mixed Th2 and Non-Th2 Inflammatory Pattern in the Asthma-COPD Overlap: A Network Approach. Int J Chron Obstruct Pulmon Dis (2018) 13:591–601. doi: 10.2147/COPD.S153694
94. Donovan C, Starkey MR, Kim RY, Rana BMJ, Barlow JL, Jones B, et al. Roles for T/B Lymphocytes and ILC2s in Experimental Chronic Obstructive Pulmonary Disease. J Leukoc Biol (2019) 105(1):143–50. doi: 10.1002/JLB.3AB0518-178R
95. Winkler C, Hochdörfer T, Israelsson E, Hasselberg A, Cavallin A, Thörn K, et al. Activation of Group 2 Innate Lymphoid Cells After Allergen Challenge in Asthmatic Patients. J Allergy Clin Immunol (2019) 144(1):61–9.e7. doi: 10.1016/j.jaci.2019.01.027
96. Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced Innate Type 2 Immune Response in Peripheral Blood From Patients With Asthma. J Allergy Clin Immunol (2014) 134(3):671–8.e4. doi: 10.1016/j.jaci.2014.06.024
97. Hosseini B, Berthon BS, Starkey MR, Collison A, McLoughlin RF, Williams EJ, et al. Children With Asthma Have Impaired Innate Immunity and Increased Numbers of Type 2 Innate Lymphoid Cells Compared With Healthy Controls. Front Immunol (2021) 12:664668. doi: 10.3389/fimmu.2021.664668
98. Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria J-P, O’Byrne PM, et al. Increased Numbers of Activated Group 2 Innate Lymphoid Cells in the Airways of Patients With Severe Asthma and Persistent Airway Eosinophilia. J Allergy Clin Immunol (2016) 137(1):75–86.e8. doi: 10.1016/j.jaci.2015.05.037
99. Kim J, Chang Y, Bae B, Sohn K-H, Cho S-H, Chung DH, et al. Innate Immune Crosstalk in Asthmatic Airways: Innate Lymphoid Cells Coordinate Polarization of Lung Macrophages. J Allergy Clin Immunol (2019) 143(5):1769–82.e11. doi: 10.1016/j.jaci.2018.10.040
100. McKenzie ANJ. Type-2 Innate Lymphoid Cells in Asthma and Allergy. Ann Am Thorac Soc (2014) 11(Suppl 5):S263–70. doi: 10.1513/AnnalsATS.201403-097AW
101. Beuraud C, Lombardi V, Luce S, Horiot S, Naline E, Neukirch C, et al. CCR10(+) ILC2s With ILC1-Like Properties Exhibit a Protective Function in Severe Allergic Asthma. Allergy (2019) 74(5):933–43. doi: 10.1111/all.13679
102. Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, et al. The Neuropeptide NMU Amplifies ILC2-Driven Allergic Lung Inflammation. Nature (2017) 549(7672):351–6. doi: 10.1038/nature24029
103. Monticelli LA, Buck MD, Flamar AL, Saenz SA, Tait Wojno ED, Yudanin NA, et al. Arginase 1 Is an Innate Lymphoid-Cell-Intrinsic Metabolic Checkpoint Controlling Type 2 Inflammation. Nat Immunol (2016) 17(6):656–65. doi: 10.1038/ni.3421
104. Krishnamoorthy N, Burkett PR, Dalli J, Abdulnour R-EE, Colas R, Ramon S, et al. Cutting Edge: Maresin-1 Engages Regulatory T Cells to Limit Type 2 Innate Lymphoid Cell Activation and Promote Resolution of Lung Inflammation. J Immunol (2015) 194(3):863–7. doi: 10.4049/jimmunol.1402534
105. Nagashima R, Tanaka N. Nrf2 Suppresses Allergic Lung Inflammation by Attenuating the Type2-Innate Lymphoid Cell Response. J Immunol (2020) 204(1 Supplement):147.8. doi: 10.4049/jimmunol.1801180
106. Levy BD, De Sanctis GT, Devchand PR, Kim E, Ackerman K, Schmidt BA, et al. Multi-Pronged Inhibition of Airway Hyper-Responsiveness and Inflammation by Lipoxin a(4). Nat Med (2002) 8(9):1018–23. doi: 10.1038/nm748
107. Kobayashi S, Tayama S, Phung HT, Kagawa Y, Miyazaki H, Takahashi Y, et al. Fatty Acid-Binding Protein 5 Limits ILC2-Mediated Allergic Lung Inflammation in a Murine Asthma Model. Sci Rep (2020) 10(1):16617. doi: 10.1038/s41598-020-73935-y
108. Liu W, Zeng Q, Wen Y, Tang Y, Yan S, Li Y, et al. Inhibited Interleukin 35 Expression and Interleukin 35–Induced Regulatory T Cells Promote Type II Innate Lymphoid Cell Response in Allergic Rhinitis. Ann Allergy Asthma Immunol (2021) 126(2):152–61.e1. doi: 10.1016/j.anai.2020.08.005
109. Han M, Hong JY, Jaipalli S, Rajput C, Lei J, Hinde JL, et al. IFN-γ Blocks Development of an Asthma Phenotype in Rhinovirus-Infected Baby Mice by Inhibiting Type 2 Innate Lymphoid Cells. Am J Respir Cell Mol Biol (2017) 56(2):242–51. doi: 10.1165/rcmb.2016-0056OC
110. He J, Yang Q, Xiao Q, Lei A, Li X, Zhou P, et al. IRF-7 Is a Critical Regulator of Type 2 Innate Lymphoid Cells in Allergic Airway Inflammation. Cell Rep (2019) 29(9):2718–30.e6. doi: 10.1016/j.celrep.2019.10.077
111. Patel DF, Peiró T, Bruno N, Vuononvirta J, Akthar S, Puttur F, et al. Neutrophils Restrain Allergic Airway Inflammation by Limiting ILC2 Function and Monocyte–Dendritic Cell Antigen Presentation. Sci Immunol (2019) 4(41):eaax7006. doi: 10.1126/sciimmunol.aax7006
112. De Grove KC, Provoost S, Verhamme FM, Bracke KR, Joos GF, Maes T, et al. Characterization and Quantification of Innate Lymphoid Cell Subsets in Human Lung. PloS One (2016) 11(1):e0145961. doi: 10.1371/journal.pone.0145961
113. Shikhagaie MM, Björklund ÅK, Mjösberg J, Erjefält JS, Cornelissen AS, Ros XR, et al. Neuropilin-1 Is Expressed on Lymphoid Tissue Residing LTi-Like Group 3 Innate Lymphoid Cells and Associated With Ectopic Lymphoid Aggregates. Cell Rep (2017) 18(7):1761–73. doi: 10.1016/j.celrep.2017.01.063
114. Li X-N, Pan X, Qiu D. Imbalances of Th17 and Treg Cells and Their Respective Cytokines in COPD Patients by Disease Stage. Int J Clin Exp Med (2014) 7(12):5324–9.
115. Andelid K, Tengvall S, Andersson A, Levänen B, Christenson K, Jirholt P, et al. Systemic Cytokine Signaling via IL-17 in Smokers With Obstructive Pulmonary Disease: A Link to Bacterial Colonization? Int J Chron Obstruct Pulmon Dis (2015) 10:689–702. doi: 10.2147/COPD.S76273
116. Christenson SA, van den Berge M, Faiz A, Inkamp K, Bhakta N, Bonser LR, et al. An Airway Epithelial IL-17A Response Signature Identifies a Steroid-Unresponsive COPD Patient Subgroup. J Clin Invest (2019) 129(1):169–81. doi: 10.1172/JCI121087
117. Xiong J, Tian J, Zhou L, Le Y, Sun Y. Interleukin-17a Deficiency Attenuated Emphysema and Bone Loss in Mice Exposed to Cigarette Smoke. Int J Chron Obstruct Pulmon Dis (2020) 15:301–10. doi: 10.2147/COPD.S235384
118. Chen K, Pociask DA, McAleer JP, Chan YR, Alcorn JF, Kreindler JL, et al. IL-17RA Is Required for CCL2 Expression, Macrophage Recruitment, and Emphysema in Response to Cigarette Smoke. PloS One (2011) 6(5):e20333. doi: 10.1371/journal.pone.0020333
119. Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Pagé N, et al. IL-17 Is Increased in Asthmatic Airways and Induces Human Bronchial Fibroblasts to Produce Cytokines. J Allergy Clin Immunol (2001) 108(3):430–8. doi: 10.1067/mai.2001.117929
120. Lindén A. Role of Interleukin-17 and the Neutrophil in Asthma. Int Arch Allergy Immunol (2001) 126(3):179–84. doi: 10.1159/000049511
121. Soodaeva S, Postnikova L, Boldina M, Kubysheva N, Li T, Klimanov I, et al. Serum IL-17 and IL-18 Levels in Asthma-COPD Overlap Syndrome Patients. Eur Respir J (2015) 46(suppl 59):PA4886. doi: 10.1183/13993003.congress-2015.PA4886
122. Wang S, Xia P, Chen Y, Qu Y, Xiong Z, Ye B, et al. Regulatory Innate Lymphoid Cells Control Innate Intestinal Inflammation. Cell (2017) 171(1):201–16.e18. doi: 10.1016/j.cell.2017.07.027
123. Bonne-Année S, Bush MC, Nutman TB. Differential Modulation of Human Innate Lymphoid Cell (ILC) Subsets by IL-10 and TGF-β. Sci Rep (2019) 9(1):14305. doi: 10.1038/s41598-019-50308-8
124. Morita H, Kubo T, Rückert B, Ravindran A, Soyka MB, Rinaldi AO, et al. Induction of Human Regulatory Innate Lymphoid Cells From Group 2 Innate Lymphoid Cells by Retinoic Acid. J Allergy Clin Immunol (2019) 143(6):2190–201.e9. doi: 10.1016/j.jaci.2018.12.1018
125. Kommaraju K, Latifi M. Outpatient Management of Asthma in Adults: A Snapshot of the 2020 GINA Report. Cleve Clin J Med (2021) 88(7):377–80. doi: 10.3949/ccjm.88a.20167
126. Holguin F, Cardet JC, Chung KF, Diver S, Ferreira DS, Fitzpatrick A, et al. Management of Severe Asthma: A European Respiratory Society/American Thoracic Society Guideline. Eur Respir J (2020) 55(1):1900588. doi: 10.1183/13993003.00588-2019
127. Vanfleteren L, Fabbri LM, Papi A, Petruzzelli S, Celli B. Triple Therapy (ICS/LABA/LAMA) in COPD: Time for a Reappraisal. Int J Chron Obstruct Pulmon Dis (2018) 13:3971–81. doi: 10.2147/COPD.S185975
128. Kabata H, Moro K, Fukunaga K, Suzuki Y, Miyata J, Masaki K, et al. Thymic Stromal Lymphopoietin Induces Corticosteroid Resistance in Natural Helper Cells During Airway Inflammation. Nat Commun (2013) 4:2675. doi: 10.1038/ncomms3675
129. Liu S, Verma M, Michalec L, Liu W, Sripada A, Rollins D, et al. Steroid Resistance of Airway Type 2 Innate Lymphoid Cells From Patients With Severe Asthma: The Role of Thymic Stromal Lymphopoietin. J Allergy Clin Immunol (2018) 141(1):257–68.e6. doi: 10.1016/j.jaci.2017.03.032
130. Jia Y, Fang X, Zhu X, Bai C, Zhu L, Jin M, et al. IL-13(+) Type 2 Innate Lymphoid Cells Correlate With Asthma Control Status and Treatment Response. Am J Respir Cell Mol Biol (2016) 55(5):675–83. doi: 10.1165/rcmb.2016-0099OC
131. Ogasawara N, Poposki JA, Klingler AI, Tan BK, Weibman AR, Hulse KE, et al. IL-10, TGF-β, and Glucocorticoid Prevent the Production of Type 2 Cytokines in Human Group 2 Innate Lymphoid Cells. J Allergy Clin Immunol (2018) 141(3):1147–51.e8. doi: 10.1016/j.jaci.2017.09.025
132. van der Ploeg EK, Golebski K, van Nimwegen M, Fergusson JR, Heesters BA, Martinez-Gonzalez I, et al. Steroid-Resistant Human Inflammatory ILC2s Are Marked by CD45RO and Elevated in Type 2 Respiratory Diseases. Sci Immunol (2021) 6(55):eabd3489. doi: 10.1126/sciimmunol.abd3489
133. Nagakumar P, Puttur F, Gregory LG, Denney L, Fleming L, Bush A, et al. Pulmonary Type-2 Innate Lymphoid Cells in Paediatric Severe Asthma: Phenotype and Response to Steroids. Eur Respir J (2019) 54(2):1801809. doi: 10.1183/13993003.01809-2018
134. Sehmi R, Lim HF, Mukherjee M, Huang C, Radford K, Newbold P, et al. Benralizumab Attenuates Airway Eosinophilia in Prednisone-Dependent Asthma. J Allergy Clin Immunol (2018) 141(4):1529–32.e8. doi: 10.1016/j.jaci.2018.01.008
135. Moriyama S, Brestoff JR, Flamar AL, Moeller JB, Klose CSN, Rankin LC, et al. β(2)-Adrenergic Receptor-Mediated Negative Regulation of Group 2 Innate Lymphoid Cell Responses. Science (2018) 359(6379):1056–61. doi: 10.1126/science.aan4829
136. Cardoso V, Chesné J, Ribeiro H, García-Cassani B, Carvalho T, Bouchery T, et al. Neuronal Regulation of Type 2 Innate Lymphoid Cells via Neuromedin U. Nature (2017) 549(7671):277–81. doi: 10.1038/nature23469
137. Klose CSN, Mahlakõiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The Neuropeptide Neuromedin U Stimulates Innate Lymphoid Cells and Type 2 Inflammation. Nature (2017) 549(7671):282–6. doi: 10.1038/nature23676
Keywords: pulmonary inflammation, airway inflammation, obstructive lung disease, COPD, asthma, NK cells, innate lymphoid cells (ILC)
Citation: Hsu AT, Gottschalk TA, Tsantikos E and Hibbs ML (2021) The Role of Innate Lymphoid Cells in Chronic Respiratory Diseases. Front. Immunol. 12:733324. doi: 10.3389/fimmu.2021.733324
Received: 30 June 2021; Accepted: 02 September 2021;
Published: 22 September 2021.
Edited by:
Hitesh Deshmukh, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Georgina Xanthou, Biomedical Research Foundation of the Academy of Athens (BRFAA), GreeceRobert Snelgrove, Imperial College London, United Kingdom
Copyright © 2021 Hsu, Gottschalk, Tsantikos and Hibbs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margaret L. Hibbs, TWFyZ2FyZXQuSGliYnNAbW9uYXNoLmVkdQ==