 Jiachen Liu
Jiachen Liu Wei Yin
Wei Yin Lisa S. Westerberg
Lisa S. Westerberg Pamela Lee
Pamela Lee Quan Gong
Quan Gong Yan Chen6*
Yan Chen6* Lingli Dong
Lingli Dong Chaohong Liu
Chaohong Liu- 1Department of Pathogen Biology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Department of Microbiology Tumor and Cell Biology, Karolinska Institutet, Stockholm, Sweden
- 4Department of Paediatrics and Adolescent Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, China
- 5Department of Immunology, School of Medicine, Yangtze University, Jingzhou, China
- 6The Second Department of Pediatrics, Affiliated Hospital of Zunyi Medical University, Zunyi, China
- 7Department of Rheumatology and Immunology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Immunoglobin G4-related disease (IgG4-RD) is one of the newly discovered autoimmune diseases characterized by elevated serum IgG4 concentrations and multi-organ fibrosis. Despite considerable research and recent advances in the identification of underlying immunological processes, the etiology of this disease is still not clear. Adaptive immune cells, including different types of T and B cells, and cytokines secreted by these cells play a vital role in the pathogenesis of IgG4-RD. Antigen-presenting cells are stimulated by pathogens and, thus, contribute to the activation of naïve T cells and differentiation of different T cell subtypes, including helper T cells (Th1 and Th2), regulatory T cells, and T follicular helper cells. B cells are activated and transformed to plasma cells by T cell-secreted cytokines. Moreover, macrophages, and some important factors (TGF-β, etc.) promote target organ fibrosis. Understanding the role of these cells and cytokines implicated in the pathogenesis of IgG4-RD will aid in developing strategies for future disease treatment and drug development. Here, we review the most recent insights on IgG4-RD, focusing on immune dysregulation involved in the pathogenesis of this autoimmune condition.
Introduction
Immunoglobin G4-related disease (IgG4-RD) is a group of autoimmune diseases involving fibrosis and inflammation of multiple organs and systems. This disease has three major characteristics: (i) remarkable elevated serum concentrations of IgG4; (ii) multiple IgG4+ plasma cells in the lesion regions; and (iii) good response to corticosteroid treatment (1, 2).
Autoimmune pancreatitis (AIP) was the first IgG4-RD to be described in 2001 by Japanese scientists Hamano et al. (3) In 2003, Kamisawa et al. (4) observed infiltration of IgG4-secreting plasma cells in extra-pancreatic organs and proposed a new clinicopathological entity that linked AIP and systemic IgG4-RD. Since then, an increasing number of IgG4-RDs have been discovered in different organs, such as the liver (5–7), kidneys (8–11), and lungs (12–14). With the deepening of research on IgG4-RD, an article in Autoimmunity Reviews (15) officially confirmed the existence of this disease in 2010. Soon afterward, the first international consensus guideline on the management and treatment was published in Arthritis & Rheumatology (16) in 2015. To date, the pathogenesis of IgG4-RD remains unclear and is thought to involve multiple factors, including adaptive and innate immunity and autoantigens. Herein, we present the most up-to-date information on immune dysregulation in IgG4-RD.
Adaptive Immunity
IgG4
Among the four IgG subtypes, namely IgG1, IgG2, IgG3, and IgG4, IgG4 has the lowest concentration in normal human serum, accounting for only 5% of the total IgG levels (17). Antibodies are immunoglobin (Ig) molecules composed of two heavy (H) and two light (L) chains, both having a constant region (CH or CL) identical for all antibodies of the same isotype and a variable region (VH or VL) that recognizes and binds a specific antigen. Antibodies comprise two antigen-binding fragments (Fab) that bind to antigens and one constant fragment (Fc) that binds to the cell surface and allows phagocytosis. Most immunoglobin (Ig) G antibodies have these characteristics: (i) they have two identical antigen-binding sites; (ii) they do not change their structure after being secreted by plasma cells. However, Aalberse et al. (17) described IgG4 as an “odd antibody” because of its unique properties different from other Igs. First, it cannot cross-link identical antigens (“functional monovalency”) (18, 19). Instead, the exchange of half-molecules, also called “Fab-arm exchange” (Figure 1), contributes to bispecific IgG4 antibodies. Second, unlike other Igs, which are proinflammatory, IgG4 has a lower affinity for C1q (the q fragment, a part of complement C1, is the site where Igs first bind) and Fc receptor (18). These properties suggest that IgG4 may have anti-inflammatory activities. Remarkably increased serum IgG4 levels and multiple IgG4+ plasma cell infiltration are important features of IgG4-RD. Nevertheless, its function is still unknown in the pathogenesis of IgG4-RD: whether it plays a protective role in IgG4-RD by participating in the anti-inflammatory process, acts as a pathogenic factor mediating the occurrence of IgG4-RD, or is merely a manifestation induced by inflammatory stimulation, has not been determined yet. In 2015, Shiokawa et al. (20) reported an interesting finding that pancreatic injury could be induced in neonatal male Balb/c mice by injecting patient IgG1 or IgG4, and the injury caused by IgG1 was more serious. However, the pathogenic activity of IgG1 and the severity of pancreatic injury were substantially inhibited by simultaneously injecting IgG4. Moreover, rituximab, a monoclonal antibody, specifically binds to cluster of differentiation 20 (CD20), a biomarker of pre-B and mature B cells, and exerts cytotoxic, anti-proliferative, and apoptotic effects. A prospective, open-label trial by Carruthers et al. (21) discovered that rituximab could relieve the symptoms of IgG4-RD. However, after rituximab treatment for 12 months, 19 patients with elevated baseline IgG4 levels showed a marked decrease in IgG4, but only 42% achieved normal IgG4 levels. This finding showed that rituximab might not work by decreasing serum IgG4 levels but by depleting activated B cells; thus, IgG4 might not have a considerable role in IgG4-RD. Similarly, Gauiran et al. (22) examined two cases of IgG4 myeloma, of which both showed high serum IgG4 levels, but neither of them manifested typical IgG4-RD presentations.
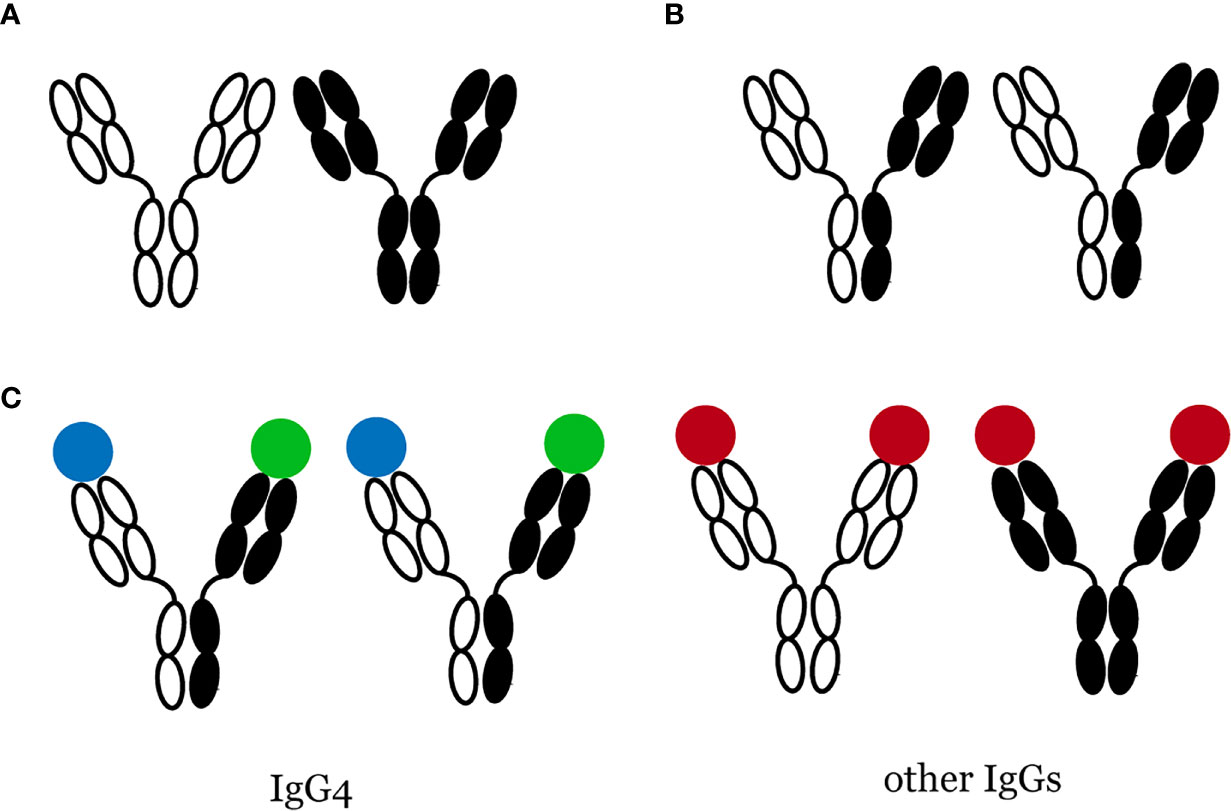
Figure 1 Bispecific IgG4 antibodies are produced through Fab-arm exchange. (A) Most IgGs do not change their structure, (B) Fab-arm exchange of IgG4 antibodies, (C) IgG4 is functional monovalency while other IgGs are not.
B-Lymphocytes
IgG4-RD is characterized by remarkably elevated serum IgG4 levels and expansion of lymphoid follicles. However, a wide range of IgG4 concentrations have been observed in patients with IgG4-RD, with some reaching levels 30 times over the upper limit, but up to 40% of patients show normal IgG4 levels (23). Next-generation sequencing examinations have led to the identification of a large number of circulating, antigen-specific plasmablasts in the peripheral blood of patients with IgG4-RD (24). Flow cytometry studies have confirmed a substantial increase in the number of plasmablasts in the blood of IgG4-RD patients (24). Over 95% of B-lymphocytes (except plasmablasts and plasma cells) express CD20, which makes rituximab, an anti-CD20 drug, effective in eliminating B cells from patients with IgG4-RD (25). CD19 has a broader expression than CD20 and is found in all B cell lines except plasma cells (26). B cell depletion leads to a decrease in serum IgG4 concentrations, indicating the preponderance of short-lived plasmablasts and plasma cells in regulating serum IgG4 levels. However, the depletion of B cells does not cause a complete normalization of IgG4 concentration, which suggests that long-lived plasma cells might maintain the production of antibodies. Mattoo et al. (24) examined 84 patients diagnosed with IgG4-RD and found that the number of plasmablasts was higher than the control group even in patients with normal serum IgG4 concentration. Besides, patients who undergo a relapse after rituximab treatment exhibit an increase in the number of plasmablasts and stronger somatic hypermutation. Therefore, peripheral plasmablasts might be considered as a biomarker for IgG4-RD (27).
B-lymphocytes are also involved in the fibrosis of diseased tissues. Della-Torre et al. (28) cultured naïve B cells, CD19+ B-lymphocytes, memory B cells, or plasmablasts from IgG4-RD with human fibroblasts and observed that the B cells and plasma cells from IgG4-RD patients can promote collagen synthesis in fibroblasts by secreting the pro-fibrotic molecules of platelet-derived growth factor B. They also observed those cells could induce the remodeling of the extracellular matrix (ECM) by producing ECM-crosslinking enzymes, such as lysyl oxidase-like 2, and chemotactic factors CCL-4, CCL-5, and CCL-11. This finding suggests that B cells might be directly involved in tissue fibrosis in IgG4-RD.
T Lymphocytes
T cells are formed in the bone marrow but mature in the thymus. They are involved in cell-mediated immunity and have three major types: helper T cells (Th), cytotoxic T cells (Tc), and regulatory T cells (Treg). Th cells can be subdivided into mutually exclusive Th1 and Th2 subsets. The Th1/Th2 balance has been confirmed to participate in autoimmune and allergic diseases (29, 30).
Th2 Cells
Th2 cells primarily secrete interleukin-4 (IL-4), IL-5, and IL-13 that participate in a plenty of pathogenic phenomena, such as allergic reaction, hypersensitivity, and IgE and IgG class switching (31). Because extremely high levels of IgG4 are observed in nearly all patients with IgG4-RD, it is one of the key diagnostic criteria for this disease. In 2005, Schmitz et al. (32) reported that IL-33 activates NF-κB and MAP kinases via IL-1 receptor ST-2 and thus promotes the production of cytokines from Th2 cells. In 2007, Miyake et al. (33) observed a disrupted balance between Th1/Th2 cells and increased peripheral Th2 levels in a patient with Mikulicz syndrome. In 2010, Akitake (34) and Suzuki (35) discovered that the number of Th2 cells and the levels of cytokines produced by them (IL-4, IL-5, and IL-13) were higher than normal in diseased tissues. In 2012, Tanaka et al. (36) examined 15 patients with Mikulicz disease and discovered that Th2-mediated adaptive immunity was essential in IgG4-RD. In 2013, GATA3+ Th2 cells are proved to be present in IgG4-related sclerosing cholangitis and type 1 autoimmune pancreatitis. Zen et al. (37) found the ratio of GATA3+/T-bet+ cell is shifted towards Th2, which may result in the recruitment of lymphocytes in patients with IgG4-RD.
However, a recent study questioned this conclusion. Maehara et al. (38) found that IL-4 mRNA levels were markedly high, whereas CD4+GATA3+ Th2 cells and GATA3 mRNA levels were low in patients with IgG4-RD, and that CD4+GATA3+ Th2 cells primarily exist in patients with allergic reaction. They explained that increased IL-4 levels might be produced by non-Th2 cells. Another puzzling finding, as reported by Okazaki et al. (39), was that instead of IL-4 levels, interferon (IFN)-γ levels were increased, and Th1-mediated immune response seems involved in IgG4-RD AIP. Another study, however, examined 44 patients with chronic periaortitis, one of the subtypes of IgG4-RD, and demonstrated the elevated level of CXCL12 and dominant infiltration of GATA3+ Th2 cells. This novel finding indicates CXCL12 might drive fibrocytes accumulation and Th2 differentiation (40).
Th1 Cells
In 1986, the two types of helper T cells, Th1 and Th2, were first described by Mosmann (41). Since then, various Th subsets with different functions have been discovered and reported. Th1 cells express CD4 and are activated by IL-12 and IFN-γ.
Recently, some researchers found elevated Th1 levels in IgG4-RD and speculated that Th1 cells might participate in the progression of IgG4-RD. Ohta et al. (42) observed a considerable increase in the population of Th1, but not Th2 cells in patients with IgG4-related sclerosing sialadenitis. Besides, the peripheral serum level of IFN-γ was substantially elevated.
CD4+ CTLs and CD8+ CTLs
However, multi-color immunofluorescence proved that high IFN-γ levels were derived from novel CD4+ cytotoxic T cell (43). Granzyme B (GZMB), one of the effector molecules of CD4+ T cells, was also present in the peripheral blood, whereas CD4+GZMA−IFN-γ+ Th1 cells were rare in diseased tissue. CD4+ CTLs are widely distributed in humans (44, 45) and mice (46, 47). They recognize antigenic peptides and target cells through MHC class II (MHC-II)- and HLA-II-dependent antigen-specific pathways, respectively (45), and perform their killing functions by secreting GZMB and perforin. Their accumulation has also been seen in some autoimmune diseases, and their severity positively correlated with the number of CD4+ CTLs (48). Mattoo et al. (43) reported that proinflammatory factors, such as transforming growth factor (TGF)-β, IL-1β, and IFN-γ, were increased in peripheral blood, suggesting the potential role of CD4+ CTLs in tissue fibrosis. Recently, a hypothesis in the mechanism of fibrosis and inflammation in IgG4-RD suggests that self-reactive cytotoxic CD4 T cells might be activated by T cells and induce cell programmed death, thus causing tissue fibrosis and inflammation (49).
Perugino et al. (50) observed CD27loCD28loCD57hi cells are dominant effector subset among circulating CD4+ CTLs in IgG4-RD, showing significant clonal expansion and different gene expression. Besides, they found marked infiltration of granzyme A-expressing CD8+ CTLs in diseased tissue and expansion of effector/memory CD8+ T cells in blood samples. Tissue studies also proved that apoptosis was common in diseased tissue, with a high proportion of nonimmune, nonendothelial mesenchymal cells.
In IgG4-related disease, presumably self-reactive cytotoxic CD4 T cells infiltrate tissues, are reactivated by T cells and induce apoptotic death. Molecules secreted by activated B cells and by CD4+ CTLs drive an exaggerated wound healing response resulting in fibrosis and compromised tissue function.
Treg Cells
Regulatory T cells (Treg) are CD4+CD25+ T cells that primarily secrete the anti-inflammatory factor IL-10 (51) and the fibrogenic factor TGF-β to maintain immune tolerance and immune homeostasis in vivo. Treg cells can be divided into two broad subsets: thymus-derived CD4+CD25+ forkhead box protein 3 (FOXP3)+ natural Treg cells and periphery-generated induced Treg (iTreg) cells. There are three main subtypes of iTreg cells: (i) CD4+FOXP3+ iTreg cells, (ii) CD4+FOXP3- IL-10-producing type I Treg (Tr1) cells, and (iii) TGF-β-expressing TH3 cells.
In 2018, Lin et al. (52) observed that IL-10 does not affect IL-4-induced IgE production but causes a 20-fold increase in IgG4 production in B cell cultures, whereas the production of both IgE and IgG4 was promoted by IL-10 in peripheral blood mononuclear cell (PBMC) cultures. Furthermore, they observed that IL-10 could diminish IL-4-induced IgE production without affecting the production of IgG4. Similarly, Punnonen et al. (53) proved that IL-10 decreases IgE production by IL-4-stimulated PBMCs. Another study reported that the blockage of IL-10 receptors in CD4+CD25+ Treg cells caused a decrease in their IgE-suppressing and IgG4-inducing effects (31).
TGF-β, a regulatory cytokine, is involved in the suppression of immune reactions. It can induce fibroblast transformation into myoblasts, increase type I collagen synthesis, and inhibit collagenase synthesis by Smad signaling pathways, thus promoting tissue fibrosis (54–56).
It is believed that Treg cells play an important role in the pathogenesis of IgG4-RD (57). Plenty of histological examinations have shown that an increased number of Treg cells are associated with different IgG4-RDs, including, but not limited to, AIP (58), IgG4-related sclerosing cholangitis (59), and Mikulicz’s syndrome (36). Therefore, it can be believed that most target organs in IgG4-RD have Treg cell infiltration. Besides, the level of Treg cells is elevated not only in the diseased tissue but also in peripheral blood (60). Miyoshi et al. (61) analyzed circulating Tregs in AIP and found CD4+CD25+ Tregs markedly elevated in AIP patients while naïve Tregs decreased, which indicates changes of Tregs might affect IgG4 production and disease progression.
Further studies showed that the levels of IL-4, IL-10, and Foxp3 were positively correlated with IgG4/IgG, suggesting that Treg-mediated immune response could promote IgG4 production and IgG4-RD progression.
Th17 Cells
Upon activation with TGF-β, IL-6, and IL-23, CD4+ Th cells can differentiate into Th17 cells. Some researchers discovered that IL-17, an inflammatory cytokine, has a strong effect on resting stromal cells and might be involved in fibrosis (62–64). Feng et al. (65) reported that IL-17 could promote the synthesis and secretion of collagen through the TGF-β signaling pathway and regulate the infiltration of fibroblasts. Ohta et al. (42) observed a correlation between the expression of IL-17 and elevated number of Th1 and Tc1 cells in IgG4-related sclerosing sialadenitis. They subsequently proposed a hypothesis that IL-17, with Th1 and Tc1 cells, could cause elevated serum levels of IgG4 and IL-17 and numbers of Th1 and Tc1 cells, but not Th2 and Tc2 cells. However, the exact role of IL-17 in the pathogenesis of IgG4-RD is still a mystery.
T Follicular Helper (Tfh) Cells
Tfh cells are specialized CD4+ T cells involved in the formation of a germinal center (GC), where B cells development and selection of antibodies occur (66). A GC contains a dark zone where B cells proliferate, and a light zone, where Tfh and B cells interact (67). A large number of ectopic GCs can be found in the pathological tissues of IgG4-RD. A study reported that nearly 70% of CD4+ T cells in the lesion of IgG4-related salivary gland are Tfh cells (68), and they play an important role in driving plasma cell and plasmablast differentiation via Tfh cytokine IL-21 (69). Besides, IL-4 has been proved to be involved in IgG4 class-switching in IgG4-RD both in vitro (70) and in vivo (71). A functional analysis suggested that IL-4-secreting Tfh cells assisted in antibody class-switch (72), whereas IL-21-secreting Tfh cells were essential for somatic hypermutation of B cells (73). Moreover, GCs exist even without Th2-related genes (74) but disappear without both IL-4 and IL-21 receptors (69). Zaidan et al. (75) observed Tfh cell infiltration of the GC light zone, which was unique to IgG4-RD. In 2011, Maehara et al. (76) examined the ectopic formation of GC and the expression of IL-21, Th2-, Th17-, and Tfh-related cytokines in 12 patients with Mikulicz’s syndrome and found that IL-4-expressing Tfh cells were primarily located outside of the ectopic GCs, whereas IL-21-expressing Tfh cells were located inside. Similarly, the upregulated expression of IL-21 mRNA was associated with the formation of ectopic GCs (76, 77). Thus, GC formation, B cell selection, and IgG4 antibody class-switch via different Tfh cell-produced cytokines are basic pathological events in the progression of IgG4-RD.
Based on the different expression levels of chemokine receptors CXCR3 (chemokine (C-X-C motif) receptor 3) and CCR6 (chemokine (C-C motif) receptor 6), Tfh cells can be divided into three subsets: Tfh1 (CXCR3+ CCR6-), Tfh2 (CXCR3- CCR6-), and Tfh17 (CXCR3- CCR6+) (78). Recently, several studies confirmed the expansion of circulating plasmablasts (24, 27, 79, 80) and the Tfh2 cells (81, 82) in IgG4-RD. Moreover, the number of Tfh2 cells positively correlated with the serum IgG4 concentration (81, 82) and proportion of IgG4+ plasma cells in diseased tissues (83). In vitro studies have shown that Tfh2 cells promote the differentiation of plasmablasts (81). Interestingly, Akiyama et al. (81) observed that the glucocorticoid therapy decreased the number of activated Tfh2 cells, which increased again during disease relapse, suggesting that activated Tfh2 cells might be used as a biomarker for IgG4-RD.
Another finding showed Tfh1 cells were activated in IgG4-RD but did not affect the production of IgG4 antibody (81). Therefore, the effect of Tfh1 cells remains to be further elucidated.
T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT), a co-inhibitory receptor discovered recently (84), was thought to be a novel Tfh marker. Akiyama et al. (85) analyzed the expression of TIGIT in peripheral CD4+T cell subsets and found that peripheral Tfh cells have higher expression of TIGIT than Th cells. They also observed that TIGIT+ Tfh cells secretes more IL-21 than TIGIT- Tfh cells, which could be used to trace the progression of IgG4-RD.
Tfh 17 cells do not appear to be involved in IgG4-RD because their numbers do not vary with serum IgG4 levels (81). Moreover, IL-17, a marker of Tfh17 cells, was rarely expressed in diseased tissues (76).
Innate Immunity
In the past few years, innate immunity in IgG4-RD has gradually attracted researchers’ attention. Toll-like receptor (TLR), a key receptor that belongs to the pattern recognition receptor family, can bind to pathogen-associated molecular patterns (PAMPs), and activate inflammatory factors through the NF-κB and MAPK pathways. Other receptors, such as nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) and C-type lectin receptor, can identify PAMPs and induce immune reactions.
Macrophages
Macrophages are relatively long-lived phagocytic cells of mammalian tissues derived from blood monocytes. Based on their phenotype and function, activated macrophages can be divided into two main categories, classically activated M1 macrophages and alternatively activated M2 macrophages, which are further divided into pro-allergic M2a, immune-regulatory M2b, and M2c types (86, 87).
When cultured with GM-CSF, monocytes can differentiate into M1 macrophages (88). Some important factors, including bacterial lipopolysaccharide (LPS) (89), monosodium urate monohydrate (90), inflammatory biomarker C-reactive protein (CRP) (91), and Th1 cytokines IFN-γ and tumor necrosis factor-α (TNF-α), promote the production of M1 macrophages. Activated M1 macrophages secrete proinflammatory factors and mediate adaptive immunity, thus eliminating the pathogens that damage normal host tissues. Hong et al. (92) observed markedly increased levels of TNF-α in submandibular glands of patients with IgG4-related sialadenitis (IgG4-RS). Besides, TNF-α treatment showed a consistent redistribution of the transcription factor EB in patients with IgG4-RS. This finding suggests that TNF-α suppresses autophagic flux and lysosomal dysfunction and causes injury of acinar cells through the ERK1/2 pathway. To sum up, M1 macrophages might participate in the formation and progression of IgG4-RS.
Interestingly, the anti-inflammatory M2 macrophages also participate in the IgG4-mediated immune response. Usually, M2 macrophages become polarized by the stimulation of Th2-derived IL-4 and IL-13 (93). IL-33, a member of the IL-1 family, can amplify IL-13-induced M2 macrophage polarization (94). However, inflammatory monocytes can differentiate into M2 macrophages via basophil-derived IL-4 during an allergic reaction (95). Watanabe et al. (96) showed that the TLR signaling pathway could enhance immune dysregulation in IgG4-RD. Besides, Ishiguro et al. (97) observed that TLR-7-, TLR-8-, and TLR-9-related genes were overexpressed in IgG4-RD. Chang et al. (98) reported that in vitro stimulation with TLR-7 agonist could increase IL-33 production by alveolar macrophages in virus-infected lung tissues. Baenziger et al. (99) proved that, besides being involved in antiviral infection, TLR-7 is involved in acquired immunity. Mice experiments confirmed that the activation of TLR-7-expressing plasmacytoid dendritic cells could lead to arthritis and lupus nephritis (100). These interesting findings suggest that virus infection and/or endogenous RNAs could initiate the formation of IgG4-RD. Pathogens activate IL-33-secreting M2 macrophages via TLR-7 and thus promote the production of Th2 cytokines, leading to tissue fibrosis and IgG4 class switching. Bianchini et al. (101) further examined the phenotype of M2 macrophages and found that pro-allergic M2a macrophages could be converted into immune-regulatory M2b macrophages by IgG4 to maintain a tolerogenic state.
Another crucial macrophage involved in IgG4-RD is CCL-18-producing M2 macrophages. CCL-18, produced from activated M2 macrophages, plays an critical rule in the formation of collagen (102). DNA microarray analysis also proved that CCL-18 was upregulated in IgG4-RD (103). Furukawa et al. (104) examined 7 patients with IgG4-related dacryoadenitis and sialoadenitis and found the level of CD163, one of the markers of M2 macrophage, was significantly higher than that in Sjögren’s syndrome and healthy subjects. Similarly, Takanashi et al. (105) observed massive infiltration of CD163+ M2 macrophages in the diseased tissue. CD163 is also co-localized with IL-10 and CCL-18 in the fibrotic region, which indicates CCL-18-secreting M2 macrophages might be involved in the development of fibrosis in IgG4-RD. Thus, CCL-18 might be a useful biomarker for tracing the severity of IgG4-RD (106).
Human IgG antibodies bind to different members of the Fcγ receptors (FcγRs) family, which have a low affinity for IgG and thus bind merely to immune complexes (IC) (107). It is widely believed that the interaction between IgG and FcγRs stimulates the immune system by triggering the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) of FcγR (108) and inhibition of signaling pathway by coupling with FcγR IIB (109) and IgG4 is commonly thought to be not compatible with FcγR (57, 110). However, Bianchini et al. (101) reported that IgG4 might bind to FcγRIIb on M2a macrophages and cause M2b subtype conversion, leading to IL-10 and CCL1 secretion. IL-10 further contributes to class switching of IgG4-secreting B cells, whereas CCL1 recruits CCR8+Foxp3+ Tregs from the periphery. Growing evidence indicates that ITAM-containing FcγR also has an inhibitory intrinsic ability (111). Boekhoudt et al. (112) reported that IFN-γ signaling pathways are inhibited by IC-mediated signaling through FcγR I and M2-like macrophages can be induced by IgG4 through FcγR I (101).
The fate of organs in inflammation and injury is controlled by the balance of M1/M2 macrophages. When the infection or inflammation caused by those pathogens is severe enough to affect the target organ, macrophages are activated and differentiated to the M1 subtype to antagonize the stimulation by Th1 cytokines. However, if M1 macrophage-mediated immune responses continue, it could cause pathological damage to the tissue. Therefore, M2 macrophages express IL-10 and TGF-β and trigger Th2-mediated reaction to suppress the immune response and promote damaged tissue repair.
Basophils and Eosinophils
During parasitic infections, allergic reactions, and autoimmune diseases, basophil cells are recruited into tissues where they produce Th2 cytokines and participate in the immune response of Th2 cells as antigen-presenting cells (APCs) (113). APCs and T cells secrete Th2 cytokines IL-4, IL-10, and IL-13 to induce IgG4 production. Several studies show that a combination of basophils and microbial antigens induces the production of Th2 cytokines (114, 115). Therefore, exogenous stimulation may facilitate the occurrence of IgG4-RD through the activation of TLRs in basophils. Watanabe et al. (96) proved that microbial antigens activate TLRs and NLRs in monocytes to induce IgG4 secretion by activating of B cell-activated factor (BAFF)-mediated pathways. They also examined IgG4 and cytokine responses to various NLR and TLR ligands and found that the activation of TLRs in basophils promotes the secretion of IgG4, BAFF, and IL-13 and thus leads to the progression of IgG4-RD (116). This finding suggests that TLR-mediated basophil activation could facilitate disease development through the BAFF signaling pathway.
Eosinophils are involved in the pathogenesis of inflammation (117). They secrete various cytokines and affect T cell expansion and Th1/Th2 cell polarization (118). Some reports have shown a connection between eosinophils and IgG4-RD (119–121). Some researchers also observed that patients with type I AIP, a typical presentation of IgG4-RD, have a long history of allergies (122, 123). Peripheral eosinophils and serum IgE were elevated in these patients. However, when they tried to determine the allergen by skin prick test or specific IgE quantitation, the allergen sensitization profile failed to reveal the culprit. Furthermore, they tested the mean IgE levels and eosinophil counts and found no connection between those data and the atopic state of patients. This finding suggests that elevated peripheral eosinophil counts and IgE levels are the intrinsic characteristics of IgG4-RD. TGF-β, one of the major cytokines secreted by eosinophils (118), participates in the formation of tissue fibrosis. Moriyama et al. (123) observed a positive correlation between peripheral eosinophil counts and treatment-free disease duration in IgG4-RD, which indicated that eosinophils might promote the end-stage development of the disease and participate in the pathogenesis of fibrosis.
Eotaxin-3, or CCL-26, is recently considered to be a potent chemoattractant for eosinophils (124). To date, Eotaxin-3 is thought to have eosinotactic activity both in vitro and in vivo. IL-4 and IL-13 are potent co-inducers of Eotaxins in epithelial and endothelial cells, consistent with Th2 responses in allergic and eosinophilic diseases (125). Eotaxin-3 could also attract eosinophils, basophils, and killer T cells via receptor CCR3 and CX3CR1. Consequently, increased local expression of Eotaxins has been described in various eosinophilic diseases. In 2021, Takanashi et al. (126) analyzed proteins overexpressed in patients with IgG4-RD with lymphadenopathy and discovered that this disease was linked with eosinophilia and Eotaxin-3 could be thought to be a potent biomarker.
Lymphadenopathy in IgG4-RD represents a phenotype associated with high disease activities, eosinophilia and relapsing disease. Eotaxin-3 is a novel biomarker related to IgG4-RD with lymphadenopathy.
Complement Activation System
The complement system participates in developing IgG4-RD. It contains a cascade of proteins that lead to the lysis of microorganism-infected cells. The complement system can be activated by three pathways: the classic, alternative, and lectin pathways. These three pathways converge on the production of C3 convertase, an enzyme that triggers the cleavage of C3 into an enzymatically active C3b and an anaphylatoxin C3a that could mediate inflammatory responses. Saeki et al. (127) examined 10 patients with hypocomplementemia of unknown etiology and found six to have high serum IgG4 levels. Kawano et al. (128) identified 22 patients (53.7%) with hypocomplementemia among 41 IgG4-related kidney disease (IgG4-RKD) patients. Besides, 16 showed lower levels of C3, C4, and CH50. IgG4-RKD is now considered as a complement-triggered inflammatory disease (129). A native renal biopsy showed that ethnic factors might contribute to different incidence rates of IgG4-RKD (Table 1). Besides, renal interstitial tissue fibrosis is now recognized as an inevitable process of end-stage renal disease. Wang et al. (135) examined serum C3 and C4 levels and found that these levels were negatively correlated with the number of infiltrated IgG4-secreting plasma cells in the kidney. However, the role of IgG4 in complement activation remains unclear. In 2006, Muraki et al. (136) found that IgG1 levels were considerably increased, whereas C3 and C4 levels were reduced in AIP, which indicated that IgG1, instead of IgG4, was involved in the activation of the classic pathway. In 2007, Kolfschoten et al. (18) speculated that IgG4 might be a protective (anti-inflammatory) antibody because of its unique “Fab-arm exchange” property. In contrast, in 2016, Sugimoto et al. (137) observed that in IgG4-RD patients with hypocomplementemia, IgG4 might participate in the activation of the complement system.
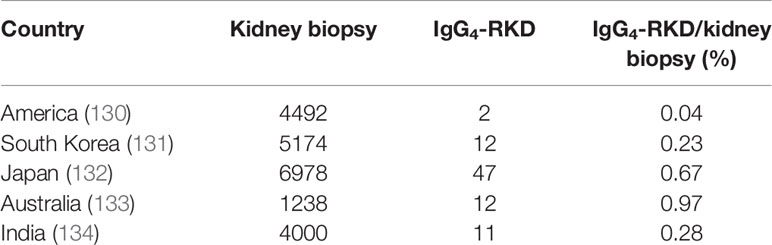
Table 1 Five studies from America, South Korea, Japan, Australia, and India show the involvement of a potential ethnic factor in the morbidity of IgG4-RD.
Further exploration on the function of the complement system in IgG4-RD is necessary.
Autoantigens
Antigens that induce an immune response in patients with IgG4-RD have not been identified to date. However, four potential autoantigens have been discovered: prohibitin, annexin A11, laminin 511-E8, and galectin-3.
In 2015, Du et al. (138) examined the sera of 89 patients with IgG4-RD and found that 73% were reactive with prohibitin, whereas only 1.4% of the healthy control group were positive. Prohibitin participates in the progression of many diseases (139, 140) and may function as a tumor suppressor and promote anti-proliferative activity by inhibiting cell cycle and DNA synthesis. ELISA showed that the levels of anti-prohibitin in the sera of IgG4-RD patients were substantially higher than those in the control group, suggesting that anti-prohibitin antibodies might contribute to the enlargement of diseased organs in IgG4-RD. Zhou et al. (141) found that the expression of prohibitin was markedly lower in the renal tissue of rats with unilateral ureteral obstruction and renal tubule interstitial fibrosis. This finding suggests that prohibitin may be involved in tissue fibrosis. Another study showed low levels of prohibitin in inflammatory bowel disease and may be conducive to reduce pain (142). Subsequently, Hubers et al. (143) found a novel autoantigen annexin A11 in IgG4-RD and thought IgG4 might perform its anti-inflammatory function by blocking the binding of IgG1 to annexin A11. Annexin A11 is a calcium-dependent phospholipid-binding protein abundant in the nucleus. When cell damage occurs, exposed annexins are recognized as autoantigens, resulting in autoimmune diseases (144, 145). Shiokawa et al. (146) also found another auto-antibody anti-laminin 511-E8 in the serum of patients with IgG4-related pancreatitis. An ELISA showed this antibody was present in 26 of 51 AIP patients, but only in two of 122 healthy controls. Besides, mice immunization by injecting laminin 511-E8 can induce symptoms like IgG4-associated pancreatitis. However, Liu et al. (147) reported that anti-laminin 511-E8 was present in only 7% of Caucasian patients with IgG4-RD, which indicated that ethnic factors might play an important role in the formation of autoantigens. In 2019, Perugino et al. (148) examined the Ig gene sequence from single-cell clones in IgG4-RD using mass spectrometry and identified galectin-3 as an antigen recognized by IgG4 and IgE. The anti-galectin-3 autoantibodies were primarily IgG4 (28%) and IgE (11%) isotypes. Galectin-3 is expressed in various cells, including macrophages, tumor cells, eosinophils, and myofibroblasts, among which activated macrophages are the primary source. Galectin-3 has a variety of biological functions. Besides promoting cell proliferation, inhibiting apoptosis, mediating cell adhesion, and participating in the inflammatory response, it is involved in the fibrosis of the liver, kidney, lung, and other organs (149). Salah et al. (150) reported increased serum levels of galectin-3 in IgG4-associated pancreatitis, suggesting that galectin-3 might participate in target organ fibrosis.
However, in 2020, a large, clinically diverse cohort study of patients with IgG4-RD presented an interesting finding. The antibody response frequency for the autoantigens prohibitin, annexin A11, laminin 511-E8, and galectin-3 were 10%, 12%, 7% and 28%, respectively (147). Further studies are needed to identify the dominant autoantigen in IgG4-RD.
A recent study investigated another autoantibody, anti-IL-1 receptor antagonist (IL-1RA), by sequencing plasmablast antibody repertoires. Compared with the control group, patients with IgG4-RD showed an increased level of plasma responses to IL-1RA, which neutralized the activity of IL-1RA and thus caused inflammation and fibrosis. This finding indicated a novel immunologic mechanism in IgG4-RD (151).
Microbial Species
Gut microbes are essential for the development and activity of the immune system. Microbial antigens are recognized by the CD4+ T cells via MHC II molecules. Gut bacterial strains stimulate the expansion of a variety of immune cell populations and provide signaling molecules for anti- and pro-inflammatory responses locally and systemically. Therefore, disorders of gut microbes are linked to a variety of diseases.
In 2009, an Italian research observed 90% of IgG4-related pancreatitis could detect peptide AIP(1-7), which shows homology with plasminogen-binding protein (PBP) of Helicobacter pylori and with ubiquitin-protein ligase E3 component n-recognin 2 (UBR2) of acinar cells in the pancreas. Besides, anti-PBP antibodies were detected in 95% of AIP patients (152). So, Helicobacter pylori infection may induce IgG4-RD through molecular mimicry or antibody cross-reaction. However, a prospective UK cohort questioned this finding: they tested 69 patients with IgG4-RD and found there was no difference in the exposure to H. pylori, cytokine response, and immunological memory to PBP. Therefore, whether Helicobacter pylori is involved in the formation of IgG4-RD remains to be studied (153).
In 2021, A recent unique microbial species using metagenomics shows reduction of normal flora and expansion of potential pathogenic bacteria: some species such as Alistipes, Bacteroides, and butyrate-producing bacterial were depleted while pathogenic Clostridium and typical oral Streptococcus were significantly overabundant. Another interesting finding is that the level of Eggerthella lenta, a Th17-activating strains, was increased in IgG4-RD. This finding indicates that rare autoimmune diseases, such as IgG4-RD, could be induced by microbiome-driven immune cell types differentiation (154).
Conclusions
With AIP as the first case of IgG4-RD, as reported by Sarles et al. in 1961, the understanding of the diagnosis and treatment of IgG4-RD has gradually deepened. This article reviews the studies on synergic regulation between B and T cells, cross-interaction of innate and adaptive immunity, IgG4 class switching, and the role of complement system in the progression of IgG4-RD (Figure 2). However, the knowledge regarding its pathogenesis is still limited. Further exploration of the mechanism of IgG4-RD will help find potential therapeutic targets and provide innovative ideas for the diagnosis and treatment of and drug development in IgG4-RD.
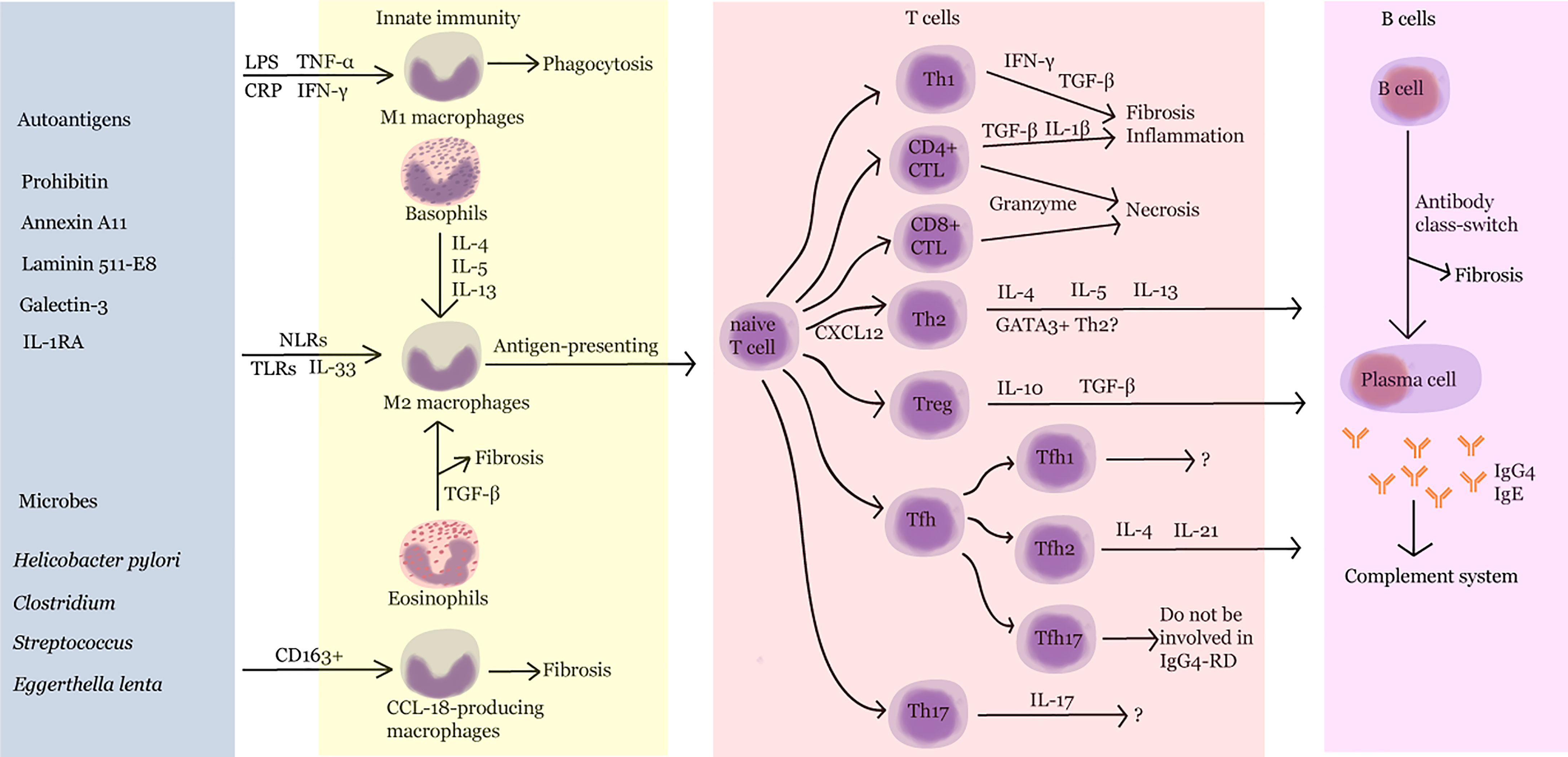
Figure 2 Pathogenesis of IgG4-RD. LPS, lipopolysaccharide; TNF-α, tumor necrosis factor-α; CRP, C-reactive protein; IFN-γ, interferon-γ; IL, interleukin; NLR, nucleotide-binding oligomerization domain (NOD)-like receptor; TLR, Toll-like receptor; TGF-β, transforming growth factor-β; Th, T helper; CTL, cytotoxic T lymphocyte; Treg, regulatory T cell; Tfh, follicular helper T cell.
Author Contributions
J.L. wrote the article and drew the figures. W.Y., L.W. and P.L. reviewed the paper. Q.G., Y.C., L.D., and C.L. organized and revised the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by HUST Academic Frontier Youth Team (2018QYTD10) and Independent Innovation Research Fund of Huazhong University of Science and Technology (2020kfyXGYJ017).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Perugino CA, Stone JH. IgG4-Related Disease: An Update on Pathophysiology and Implications for Clinical Care. Nat Rev Rheumatol (2020) 16(12):702–14. doi: 10.1038/s41584-020-0500-7
2. Ardila-Suarez O, Abril A, Gómez-Puerta JA. IgG4-Related Disease: A Concise Review of the Current Literature. Reumatol Clin (2017) 13(3):160–6. doi: 10.1016/j.reuma.2016.05.009
3. Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High Serum IgG4 Concentrations in Patients With Sclerosing Pancreatitis. N Engl J Med (2001) 344(10):732–8. doi: 10.1056/NEJM200103083441005
4. Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, et al. A New Clinicopathological Entity of IgG4-Related Autoimmune Disease. J Gastroenterol (2003) 38(10):982–4. doi: 10.1007/s00535-003-1175-y
5. Canivet CM, Anty R, Patouraux S, Saint-Paul MC, Lebeaupin C, Gual P, et al. Immunoglobulin G4-Associated Autoimmune Hepatitis May Be Found in Western Countries. Dig Liver Dis (2016) 48:302–8. doi: 10.1016/j.dld.2015.10.005
6. Kawano M, Saeki T, Nakashima H. IgG4-Related Kidney Disease and Retroperitoneal Fibrosis: An Update. Mod Rheumatol (2019) 29(2):231–9. doi: 10.1080/14397595.2018.1554321
7. Zhang NN, Wang YY, Kong LX, Zou WZ, Dong B. IgG4-Related Kidney Disease (IgG4-RKD) With Membranous Nephropathy as Its Initial Manifestation: Report of One Case and Literature Review. BMC Nephrol (2019) 20(1):263. doi: 10.1186/s12882-019-1419-6
8. Martín-Nares E, Hernandez-Molina G, Rodríguez-Ramírez S, Rivera-Fuentes L, Niño-Cruz JA, Núñez-Abreu A, et al. IgG4-Related Kidney Disease: Experience From a Mexican Cohort. Clin Rheumatol (2020) 39(11):3401–8. doi: 10.1007/s10067-020-05135-y
9. Surintrspanont J, Sanpawat A, Sasiwimonphan K, Sitthideatphaiboon P. IgG4-Related Pseudo-Tumor of the Kidney and Multiple Organ Involvement Mimicked Malignancy. Urol Case Rep (2019) 26:100953. doi: 10.1016/j.eucr.2019.100953
10. Morales AT, Cignarella AG, Jabeen IS, Barkin JS, Mirsaeidi M. An Update on IgG4-Related Lung Disease. Eur J Intern Med (2019) 66:18–24. doi: 10.1016/j.ejim.2019.06.010
11. Xie Y, Xiong A, Marion T, Liu Y. Lung Nodules and IgG4 Related Disease: A Single-Center Based Experience. BMC Pulm Med (2020) 20(1):218. doi: 10.1186/s12890-020-01250-3
12. Doubková M, Matěj R, Chovancová Z, Doubek M. Lung Diseases and Autoimmune Hemolytic Anemia Associted With IgG4 Disease. Vnitr Lek (2020) 66(4):47–52.
13. Aydemir Y, Akcoren Z, Demir H, Saltik Temizel IN, Ozen H, Yuce A. Clinical and Histopathological Features of Immunoglobulin G4-Associated Autoimmune Hepatitis in Children. J Gastroenterol Hepatol (2019) 34:742–6. doi: 10.1111/jgh.14525
14. Minaga K, Watanabe T, Chung H, Kudo M. Autoimmune Hepatitis and IgG4-Related Disease. World J Gastroenterol (2019) 25(19):2308–14. doi: 10.3748/wjg.v25.i19.2308
15. Takahashi H, Yamamoto M, Suzuki C, Naishiro Y, Shinomura Y, Imai K. The Birthday of a New Syndrome: IgG4-Related Diseases Constitute a Clinical Entity. Autoimmun Rev (2010) 9(9):591–4. doi: 10.1016/j.autrev.2010.05.003
16. Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Rheumatol (2015) 67(7):1688–99. doi: 10.1002/art.39132
17. Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: An Odd Antibody. Clin Exp Allergy (2009) 39(4):469–77. doi: 10.1111/j.1365-2222.2009.03207.x
18. van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martínez-Martínez P, Vermeulen E, et al. Anti-Inflammatory Activity of Human IgG4 Antibodies by Dynamic Fab Iarm Exchange. Science (2007) 317(5844):1554–7. doi: 10.1126/science.1144603
19. Vergoossen DLE, Plomp JJ, Gstöttner C, Fillié-Grijpma YE, Augustinus R, Verpalen R, et al. Functional Monovalency Amplifies the Pathogenicity of Anti-MuSK IgG4 in Myasthenia Gravis. Proc Natl Acad Sci USA (2021) 118(13):e2020635118. doi: 10.1073/pnas.2020635118
20. Shiokawa M, Kodama Y, Kuriyama K, Yoshimura K, Tomono T, Morita T, et al. Pathogenicity of IgG in Patients With IgG4-Related Disease. Gut (2016) 65(8):1322–32. doi: 10.1136/gutjnl-2015-310336
21. Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, et al. Rituximab for IgG4-Related Disease: A Prospective, Open-Label Trial. Ann Rheum Dis (2015) 74(6):1171–7. doi: 10.1136/annrheumdis-2014-206605
22. Gauiran DTV, Marcon KM, DeMarco ML, Fung AWS, van der Gugten G, Mattman A, et al. IgG4 Plasma Cell Myeloma Without Clinical Evidence of IgG4-Related Disease: A Report of Two Cases. Hematology (2020) 25(1):335–40. doi: 10.1080/16078454.2020.1815130
23. Wallace ZS, Mattoo H, Mahajan VS, Kulikova M, Lu L, Deshpande V, et al. Predictors of Disease Relapse in IgG4-Related Disease Following Rituximab. Rheumatol (Oxford) (2016) 55(6):1000–8. doi: 10.1093/rheumatology/kev438
24. Mattoo H, Mahajan VS, Della-Torre E, Sekigami Y, Carruthers M, Wallace ZS, et al. De Novo Oligoclonal Expansions of Circulating Plasmablasts in Active and Relapsing IgG4-Related Disease. J Allergy Clin Immunol (2014) 134(3):679–87. doi: 10.1016/j.jaci.2014.03.034
25. Seegobin K, Moustafa MA, Gannon N, Keller K, Hastings J, Gupta V, et al. Successful Treatment of IgG4-Related Hypertrophic Pachymeningitis With Induction Rituximab and Dexamethasone Followed by Maintenance Rituximab. Clin Case Rep (2021) 9(3):1610–4. doi: 10.1002/ccr3.3855
26. Lanzillotta M, Della-Torre E, Stone JH. Roles of Plasmablasts and B Cells in IgG4-Related Disease: Implications for Therapy and Early Treatment Outcomes. Curr Top Microbiol Immunol (2017) 401:85–92. doi: 10.1007/82_2016_58
27. Wallace ZS, Mattoo H, Carruthers M, Mahajan VS, Della Torre E, Lee H, et al. Plasmablasts as a Biomarker for IgG4-Related Disease, Independent of Serum IgG4 Concentrations. Ann Rheum Dis (2015) 74(1):190–5. doi: 10.1136/annrheumdis-2014-205233
28. Della-Torre E, Rigamonti E, Perugino C, Baghai-Sain S, Sun N, Kaneko N, et al. B Lymphocytes Directly Contribute to Tissue Fibrosis in Patients With IgG4-Related Disease. J Allergy Clin Immunol (2020) 145(3):968–81. doi: 10.1016/j.jaci.2019.07.004
29. Samuel RO, Ervolino E, de Azevedo Queiroz ÍO, Azuma MM, Ferreira GT, Cintra LTA. Th1/Th2/Th17/Treg Balance in Apical Periodontitis of Normoglycemic and Diabetic Rats. J Endod (2019) 45(8):1009–15. doi: 10.1016/j.joen.2019.05.003
30. Qin Z, Wang PY, Wan JJ, Zhang Y, Wei J, Sun Y, et al. MicroRNA124-IL6R Mediates the Effect of Nicotine in Inflammatory Bowel Disease by Shifting Th1/Th2 Balance Toward Th1. Front Immunol (2020) 11:235. doi: 10.3389/fimmu.2020.00235
31. Meiler F, Klunker S, Zimmermann M, Akdis CA, Akdis M. Distinct Regulation of IgE, IgG4 and IgA by T Regulatory Cells and Toll-Like Receptors. Allergy (2008) 63(11):1455–63. doi: 10.1111/j.1398-9995.2008.01774.x
32. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, An Interleukin-1-Like Cytokine That Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity (2005) 23(5):479–90. doi: 10.1016/j.immuni.2005.09.015
33. Miyake K, Moriyama M, Aizawa K, Nagano S, Inoue Y, Sadanaga A, et al. Peripheral CD4+ T Cells Showing a Th2 Phenotype in a Patient With Mikulicz's Disease Associated With Lymphadenopathy and Pleural Effusion. Mod Rheumatol (2008) 18(1):86–90. doi: 10.1007/s10165-007-0010-3
34. Akitake R, Watanabe T, Zaima C, Uza N, Ida H, Tada S, et al. Possible Involvement of T Helper Type 2 Responses to Toll-Like Receptor Ligands in IgG4-Related Sclerosing Disease. Gut (2010) 59(4):542–5. doi: 10.1136/gut.2009.200972
35. Suzuki K, Tamaru J, Okuyama A, Kameda H, Amano K, Nagasawa H, et al. IgG4-Positive Multi-Organ Lymphoproliferative Syndrome Manifesting as Chronic Symmetrical Sclerosing Dacryo-Sialadenitis With Subsequent Secondary Portal Hypertension and Remarkable IgG4-Linked IL-4 Elevation. Rheumatol (Oxford) (2010) 49(9):1789–91. doi: 10.1093/rheumatology/keq113
36. Tanaka A, Moriyama M, Nakashima H, Miyake K, Hayashida JN, Maehara T, et al. Th2 and Regulatory Immune Reactions Contribute to IgG4 Production and the Initiation of Mikulicz Disease. Arthritis Rheum (2012) 64(1):254–63. doi: 10.1002/art.33320
37. Zen Y, Liberal R, Nakanuma Y, Heaton N, Portmann B. Possible Involvement of CCL1-CCR8 Interaction in Lymphocytic Recruitment in IgG4-Related Sclerosing Cholangitis. J Hepatol (2013) 59(5):1059–64. doi: 10.1016/j.jhep.2013.06.016
38. Maehara T, Mattoo H, Ohta M, Mahajan VS, Moriyama M, Yamauchi M, et al. Lesional CD4+ IFN-γ+ Cytotoxic T Lymphocytes in IgG4-Related Dacryoadenitis and Sialoadenitis. Ann Rheum Dis (2017) 76(2):377–85. doi: 10.1136/annrheumdis-2016-209139
39. Okazaki K, Uchida K, Ohana M, Nakase H, Uose S, Inai M, et al. Autoimmune-Related Pancreatitis Is Associated With Autoantibodies and a Th1/Th2-Type Cellular Immune Response. Gastroenterology (2000) 118(3):573–81. doi: 10.1016/s0016-5085(00)70264-2
40. Nicastro M, Vescovini R, Maritati F, Palmisano A, Urban ML, Incerti M, et al. Fibrocytes in Chronic Periaortitis: A Novel Mechanism Linking Inflammation and Fibrosis. Arthritis Rheumatol (2019) 71(11):1913–22. doi: 10.1002/art.41024
41. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two Types of Murine Helper T Cell Clone. I. Definition According to Profiles of Lymphokine Activities and Secreted Proteins. J Immunol (1986) 136(7):2348–57.
42. Ohta N, Makihara S, Okano M, Kurakami K, Ishida A, Furukawa T, et al. Roles of IL-17, Th1, and Tc1 Cells in Patients With IgG4-Related Sclerosing Sialadenitis. Laryngoscope (2012) 122(10):2169–74. doi: 10.1002/lary.23429
43. Mattoo H, Mahajan VS, Maehara T, Deshpande V, Della-Torre E, Wallace ZS, et al. Clonal Expansion of CD4(+) Cytotoxic T Lymphocytes in Patients With IgG4-Related Disease. J Allergy Clin Immunol (2016) 138(3):825–38. doi: 10.1016/j.jaci.2015.12.1330
44. Sharapova TN, Romanova EA, Sashchenko LP, Yashin DV. Tag7 (PGLYRP1) can Induce an Emergence of the CD3+CD4+CD25+CD127+ Cells With Antitumor Activity. J Immunol Res (2018) 2018:4501273. doi: 10.1155/2018/4501273
45. Takeuchi A, Saito T. CD4 CTL, a Cytotoxic Subset of CD4+ T Cells, Their Differentiation and Function. Front Immunol (2017) 8:194. doi: 10.3389/fimmu.2017.00194
46. Elyahu Y, Hekselman I, Eizenberg-Magar I, Berner O, Strominger I, Schiller M, et al. Aging Promotes Reorganization of the CD4 T Cell Landscape Toward Extreme Regulatory and Effector Phenotypes. Sci Adv (2019) 5(8):eaaw8330. doi: 10.1126/sciadv.aaw8330
47. Śledzińska A, Vila de Mucha M, Bergerhoff K, Hotblack A, Demane DF, Ghorani E, et al. Regulatory T Cells Restrain Interleukin-2- and Blimp-1-Dependent Acquisition of Cytotoxic Function by CD4+ T Cells. Immunity (2020) 52(1):151–66.e6. doi: 10.1016/j.immuni.2019.12.007
48. Jiang J, Liu C, Liu M, Shen Y, Hu X, Wang Q, et al. OX40 Signaling Is Involved in the Autoactivation of CD4+CD28- T Cells and Contributes to the Pathogenesis of Autoimmune Arthritis. Arthritis Res Ther (2017) 19(1):67. doi: 10.1186/s13075-017-1261-9
49. Pillai S, Perugino C, Kaneko N. Immune Mechanisms of Fibrosis and Inflammation in IgG4-Related Disease. Curr Opin Rheumatol (2020) 32(2):146–51. doi: 10.1097/BOR.0000000000000686
50. Perugino CA, Kaneko N, Maehara T, Mattoo H, Kers J, Allard-Chamard H, et al. CD4+ and CD8+ Cytotoxic T Lymphocytes may Induce Mesenchymal Cell Apoptosis in IgG4-Related Disease. J Allergy Clin Immunol (2021) 147(1):368–82. doi: 10.1016/j.jaci.2020.05.022
51. Boonpiyathad T, Satitsuksanoa P, Akdis M, Akdis CA. IL-10 Producing T and B Cells in Allergy. Semin Immunol (2019) 44:101326. doi: 10.1016/j.smim.2019.101326
52. Lin AA, Freeman AF, Nutman TB. IL-10 Indirectly Downregulates IL-4-Induced IgE Production by Human B Cells. Immunohorizons (2018) 2(11):398–406. doi: 10.4049/immunohorizons.1800076
53. Punnonen J, de Waal Malefyt R, van Vlasselaer P, Gauchat JF, de Vries JE. IL-10 and Viral IL-10 Prevent IL-4-Induced IgE Synthesis by Inhibiting the Accessory Cell Function of Monocytes. J Immunol (1993) 151(3):1280–9.
54. Dong J, Yu X, Porter DW, Battelli LA, Kashon ML, Ma Q. Common and Distinct Mechanisms of Induced Pulmonary Fibrosis by Particulate and Soluble Chemical Fibrogenic Agents. Arch Toxicol (2016) 90(2):385–402. doi: 10.1007/s00204-015-1589-3
55. Derynck R, Zhang YE. Smad-Dependent and Smad-Independent Pathways in TGF-Beta Family Signalling. Nature (2003) 425(6958):577–84. doi: 10.1038/nature02006
56. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-β: The Master Regulator of Fibrosis. Nat Rev Nephrol (2016) 12(6):325–38. doi: 10.1038/nrneph.2016.48
57. Uchida K, Okazaki K. Roles of Regulatory T and B Cells in IgG4-Related Disease. Curr Top Microbiol Immunol (2017) 401:93–114. doi: 10.1007/82_2016_41
58. Kusuda T, Uchida K, Miyoshi H, Koyabu M, Satoi S, Takaoka M, et al. Involvement of Inducible Costimulator- and Interleukin 10-Positive Regulatory T Cells in the Development of IgG4-Related Autoimmune Pancreatitis. Pancreas (2011) 40(7):1120–30. doi: 10.1097/MPA.0b013e31821fc796
59. Koyabu M, Uchida K, Miyoshi H, Sakaguchi Y, Fukui T, Ikeda H, et al. Analysis of Regulatory T Cells and IgG4-Positive Plasma Cells Among Patients of IgG4-Related Sclerosing Cholangitis and Autoimmune Liver Diseases. J Gastroenterol (2010) 45(7):732–41. doi: 10.1007/s00535-010-0199-3
60. Akiyama M, Suzuki K, Kassai Y, Miyazaki T, Morita R, Yoshimura A, et al. Resolution of Elevated Circulating Regulatory T Cells by Corticosteroids in Patients With IgG4-Related Dacryoadenitis and Sialoadenitis. Int J Rheum Dis (2016) 19(4):430–2. doi: 10.1111/1756-185X.12725
61. Miyoshi H, Uchida K, Taniguchi T, Yazumi S, Matsushita M, Takaoka M, et al. Circulating Naïve and CD4+CD25high Regulatory T Cells in Patients With Autoimmune Pancreatitis. Pancreas (2008) 36(2):133–40. doi: 10.1097/MPA.0b013e3181577553
62. Makihara S, Okano M, Fujiwara T, Kariya S, Noda Y, Higaki T, et al. Regulation and Characterization of IL-17A Expression in Patients With Chronic Rhinosinusitis and Its Relationship With Eosinophilic Inflammation. J Allergy Clin Immunol (2010) 126(2):397–400, 400.e1–11. doi: 10.1016/j.jaci.2010.05.014
63. Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, et al. Bleomycin and IL-1beta-Mediated Pulmonary Fibrosis Is IL-17A Dependent. J Exp Med (2010) 207(3):535–52. doi: 10.1084/jem.20092121
64. Mi S, Li Z, Yang HZ, Liu H, Wang JP, Ma YG, et al. Blocking IL-17A Promotes the Resolution of Pulmonary Inflammation and Fibrosis via TGF-Beta1-Dependent and -Independent Mechanisms. J Immunol (2011) 187(6):3003–14. doi: 10.4049/jimmunol.1004081
65. Feng W, Li W, Liu W, Wang F, Li Y, Yan W. IL-17 Induces Myocardial Fibrosis and Enhances RANKL/OPG and MMP/TIMP Signaling in Isoproterenol-Induced Heart Failure. Exp Mol Pathol (2009) 87(3):212–8. doi: 10.1016/j.yexmp.2009.06.001
66. Victora GD, Nussenzweig MC. Germinal Centers. Annu Rev Immunol (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
67. Bélanger S, Crotty S. Dances With Cytokines, Featuring TFH Cells, IL-21, IL-4 and B Cells. Nat Immunol (2016) 17(10):1135–6. doi: 10.1038/ni.3561
68. Kamekura R, Takano K, Yamamoto M, Kawata K, Shigehara K, Jitsukawa S, et al. Cutting Edge: A Critical Role of Lesional T Follicular Helper Cells in the Pathogenesis of IgG4-Related Disease. J Immunol (2017) 199(8):2624–9. doi: 10.4049/jimmunol.1601507
69. Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, et al. A Critical Role for IL-21 in Regulating Immunoglobulin Production. Science (2002) 298(5598):1630–4. doi: 10.1126/science.1077002
70. Akiyama M, Yasuoka H, Yoshimoto K, Takeuchi T. Interleukin-4 Contributes to the Shift of Balance of IgG Subclasses Toward IgG4 in IgG4-Related Disease. Cytokine (2018) 110:416–9. doi: 10.1016/j.cyto.2018.05.009
71. Maehara T, Mattoo H, Mahajan VS, Murphy SJ, Yuen GJ, Ishiguro N, et al. The Expansion in Lymphoid Organs of IL-4+ BATF+ T Follicular Helper Cells Is Linked to IgG4 Class Switching In Vivo. Life Sci Alliance (2018) 1(1):e201800050. doi: 10.26508/lsa.201800050
72. Vijayanand P, Seumois G, Simpson LJ, et al. Interleukin-4 Production by Follicular Helper T Cells Requires the Conserved IL4 Enhancer Hypersensitivity Site V. Immunity (2012) 36(2):175–87. doi: 10.1016/j.immuni.2011.12.014
73. Weinstein JS, Herman EI, Lainez B, Licona-Limón P, Esplugues E, Flavell R, et al. TFH Cells Progressively Differentiate to Regulate the Germinal Center Response. Nat Immunol (2016) 17(10):1197–205. doi: 10.1038/ni.3554
74. Grusby MJ. Stat4- and Stat6-Deficient Mice as Models for Manipulating T Helper Cell Responses. Biochem Soc Trans (1997) 25(2):359–60. doi: 10.1042/bst0250359
75. Zaidan M, Cervera-Pierot P, de Seigneux S, Dahan K, Fabiani B, Callard P, et al. Evidence of Follicular T-Cell Implication in a Case of IgG4-Related Systemic Disease With Interstitial Nephritis. Nephrol Dial Transplant (2011) 26(6):2047–50. doi: 10.1093/ndt/gfr097
76. Maehara T, Moriyama M, Nakashima H, Miyake K, Hayashida JN, Tanaka A, et al. Interleukin-21 Contributes to Germinal Centre Formation and Immunoglobulin G4 Production in IgG4-Related Dacryoadenitis and Sialoadenitis, So-Called Mikulicz's Disease. Ann Rheum Dis (2012) 71(12):2011–9. doi: 10.1136/annrheumdis-2012-201477
77. Akiyama M, Kaneko Y, Hayashi Y, Takeuchi T. IgG4-Related Disease Involving Vital Organs Diagnosed With Lip Biopsy: A Case Report and Literature Review. Med (Baltimore) (2016) 95(24):e3970. doi: 10.1097/MD.0000000000003970
78. Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, et al. Human Blood CXCR5(+)CD4(+) T Cells Are Counterparts of T Follicular Cells and Contain Specific Subsets That Differentially Support Antibody Secretion. Immunity (2011) 34(1):108–21. doi: 10.1016/j.immuni.2010.12.012
79. Wallace ZS, Deshpande V, Mattoo H, Mahajan VS, Kulikova M, Pillai S, et al. IgG4-Related Disease: Clinical and Laboratory Features in One Hundred Twenty-Five Patients. Arthritis Rheumatol (2015) 67(9):2466–75. doi: 10.1002/art.39205
80. Lin W, Zhang P, Chen H, Chen Y, Yang H, Zheng W, et al. Circulating Plasmablasts/Plasma Cells: A Potential Biomarker for IgG4-Related Disease. Arthritis Res Ther (2017) 19(1):25. doi: 10.1186/s13075-017-1231-2
81. Akiyama M, Yasuoka H, Yamaoka K, Suzuki K, Kaneko Y, Kondo H, et al. Enhanced IgG4 Production by Follicular Helper 2 T Cells and the Involvement of Follicular Helper 1 T Cells in the Pathogenesis of IgG4-Related Disease. Arthritis Res Ther (2016) 18:167. doi: 10.1186/s13075-016-1064-4
82. Akiyama M, Suzuki K, Yamaoka K, Yasuoka H, Takeshita M, Kaneko Y, et al. Number of Circulating Follicular Helper 2 T Cells Correlates With IgG4 and Interleukin-4 Levels and Plasmablast Numbers in IgG4-Related Disease. Arthritis Rheumatol (2015) 67(9):2476–81. doi: 10.1002/art.39209
83. Akiyama M, Kaneko Y, Yamaoka K, Hayashi Y, Yasuoka H, Suzuki K, et al. Subclinical Labial Salivary Gland Involvement in IgG4-Related Disease Affected With Vital Organs. Clin Exp Rheumatol (2015) 33(6):949–50.
84. Gorvel L, Olive D. Targeting the "PVR-TIGIT Axis" With Immune Checkpoint Therapies. F1000Res (2020) 9:F1000 Faculty Rev–354. doi: 10.12688/f1000research.22877.1
85. Akiyama M, Suzuki K, Yoshimoto K, Yasuoka H, Kaneko Y, Takeuchi T. Peripheral TIGIT+ T Follicular Helper Cells That Produce High Levels of Interleukin-21 via OX40 Represent Disease Activity in IgG4-Related Disease. Front Immunol (2021) 12:651357. doi: 10.3389/fimmu.2021.651357
86. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage Activation and Polarization. Front Biosci (2008) 13:453–61. doi: 10.2741/2692
87. Chistiakov DA, Bobryshev YV, Orekhov AN. Changes in Transcriptome of Macrophages in Atherosclerosis. J Cell Mol Med (2015) 19(6):1163–73. doi: 10.1111/jcmm.12591
88. Romo N, Magri G, Muntasell A, Heredia G, Baía D, Angulo A, et al. Natural Killer Cell-Mediated Response to Human Cytomegalovirus-Infected Macrophages Is Modulated by Their Functional Polarization. J Leukoc Biol (2011) 90(4):717–26. doi: 10.1189/jlb.0311171
89. Pena OM, Pistolic J, Raj D, Fjell CD, Hancock RE. Endotoxin Tolerance Represents a Distinctive State of Alternative Polarization (M2) in Human Mononuclear Cells. J Immunol (2011) 186(12):7243–54. doi: 10.4049/jimmunol.1001952
90. Martin WJ, Shaw O, Liu X, Steiger S, Harper JL. Monosodium Urate Monohydrate Crystal-Recruited Noninflammatory Monocytes Differentiate Into M1-Like Proinflammatory Macrophages in a Peritoneal Murine Model of Gout. Arthritis Rheum (2011) 63(5):1322–32. doi: 10.1002/art.30249
91. Devaraj S, Jialal I. C-Reactive Protein Polarizes Human Macrophages to an M1 Phenotype and Inhibits Transformation to the M2 Phenotype. Arterioscler Thromb Vasc Biol (2011) 31(6):1397–402. doi: 10.1161/ATVBAHA.111.225508
92. Hong X, Min SN, Zhang YY, Lin YT, Wang F, Huang Y, et al. TNF-α Suppresses Autophagic Flux in Acinar Cells in IgG4-Related Sialadenitis. J Dent Res (2019) 98(12):1386–96. doi: 10.1177/0022034519871890
93. Paul WE, Zhu J. How Are T(H)2-Type Immune Responses Initiated and Amplified? Nat Rev Immunol (2010) 10(4):225–35. doi: 10.1038/nri2735
94. Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 Amplifies the Polarization of Alternatively Activated Macrophages That Contribute to Airway Inflammation. J Immunol (2009) 183(10):6469–77. doi: 10.4049/jimmunol.0901575
95. Egawa M, Mukai K, Yoshikawa S, Iki M, Mukaida N, Kawano Y, et al. Inflammatory Monocytes Recruited to Allergic Skin Acquire an Anti-Inflammatory M2 Phenotype via Basophil-Derived Interleukin-4. Immunity (2013) 38(3):570–80. doi: 10.1016/j.immuni.2012.11.014
96. Watanabe T, Yamashita K, Fujikawa S, Sakurai T, Kudo M, Shiokawa M, et al. Involvement of Activation of Toll-Like Receptors and Nucleotide-Binding Oligomerization Domain-Like Receptors in Enhanced IgG4 Responses in Autoimmune Pancreatitis. Arthritis Rheum (2012) 64(3):914–24. doi: 10.1002/art.33386
97. Ishiguro N, Moriyama M, Furusho K, Furukawa S, Shibata T, Murakami Y, et al. Activated M2 Macrophages Contribute to the Pathogenesis of IgG4-Related Disease via Toll-Like Receptor 7/Interleukin-33 Signaling. Arthritis Rheumatol (2020) 72(1):166–78. doi: 10.1002/art.41052
98. Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate Lymphoid Cells Mediate Influenza-Induced Airway Hyper-Reactivity Independently of Adaptive Immunity. Nat Immunol (2011) 12(7):631–8. doi: 10.1038/ni.2045
99. Baenziger S, Heikenwalder M, Johansen P, Schlaepfer E, Hofer U, Miller RC, et al. Triggering TLR7 in Mice Induces Immune Activation and Lymphoid System Disruption, Resembling HIV-Mediated Pathology. Blood (2009) 113(2):377–88. doi: 10.1182/blood-2008-04-151712
100. Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, et al. Control of Toll-Like Receptor 7 Expression Is Essential to Restrict Autoimmunity and Dendritic Cell Proliferation. Immunity (2007) 27(5):801–10. doi: 10.1016/j.immuni.2007.09.009
101. Bianchini R, Roth-Walter F, Ohradanova-Repic A, Flicker S, Hufnagl K, Fischer MB, et al. IgG4 Drives M2a Macrophages to a Regulatory M2b-Like Phenotype: Potential Implication in Immune Tolerance. Allergy (2019) 74(3):483–94. doi: 10.1111/all.13635
102. Kodelja V, Müller C, Politz O, Hakij N, Orfanos CE, Goerdt S. Alternative Macrophage Activation-Associated CC-Chemokine-1, A Novel Structural Homologue of Macrophage Inflammatory Protein-1 Alpha With a Th2-Associated Expression Pattern. J Immunol (1998) 160(3):1411–8.
103. Tsuboi H, Nakai Y, Iizuka M, Asashima H, Hagiya C, Tsuzuki S, et al. DNA Microarray Analysis of Labial Salivary Glands in IgG4-Related Disease: Comparison With Sjögren's Syndrome. Arthritis Rheumatol (2014) 66(10):2892–9. doi: 10.1002/art.38748
104. Furukawa S, Moriyama M, Tanaka A, Maehara T, Tsuboi H, Iizuka M, et al. Preferential M2 Macrophages Contribute to Fibrosis in IgG4-Related Dacryoadenitis and Sialoadenitis, So-Called Mikulicz's Disease. Clin Immunol (2015) 156(1):9–18. doi: 10.1016/j.clim.2014.10.008
105. Takanashi S, Akiyama M, Suzuki K, Otomo K, Takeuchi T. IgG4-Related Fibrosing Mediastinitis Diagnosed With Computed Tomography-Guided Percutaneous Needle Biopsy: Two Case Reports and a Review of the Literature. Med (Baltimore) (2018) 97(22):e10935. doi: 10.1097/MD.0000000000010935
106. Akiyama M, Yasuoka H, Yoshimoto K, Takeuchi T. CC-Chemokine Ligand 18 Is a Useful Biomarker Associated With Disease Activity in IgG4-Related Disease. Ann Rheum Dis (2018) 77(9):1386–7. doi: 10.1136/annrheumdis-2017-212110
107. Espéli M, Smith KG, Clatworthy MR. Fcγriib and Autoimmunity. Immunol Rev (2016) 269(1):194–211. doi: 10.1111/imr.12368
108. Powell MS, Hogarth PM. Fc Receptors. Adv Exp Med Biol (2008) 640:22–34. doi: 10.1007/978-0-387-09789-3_3
109. Smith KG, Clatworthy MR. FcgammaRIIB in Autoimmunity and Infection: Evolutionary and Therapeutic Implications. Nat Rev Immunol (2010) 10(5):328–43. doi: 10.1038/nri2762
110. Canfield SM, Morrison SL. The Binding Affinity of Human IgG for Its High Affinity Fc Receptor Is Determined by Multiple Amino Acids in the CH2 Domain and Is Modulated by the Hinge Region. J Exp Med (1991) 173(6):1483–91. doi: 10.1084/jem.173.6.1483
111. Blank U, Launay P, Benhamou M, Monteiro RC. Inhibitory ITAMs as Novel Regulators of Immunity. Immunol Rev (2009) 232(1):59–71. doi: 10.1111/j.1600-065X.2009.00832.x
112. Boekhoudt GH, Frazier-Jessen MR, Feldman GM. Immune Complexes Suppress IFN-Gamma Signaling by Activation of the FcgammaRI Pathway. J Leukoc Biol (2007) 81(4):1086–92. doi: 10.1189/jlb.0906543
113. Schwartz C, Eberle JU, Voehringer D. Basophils in Inflammation. Eur J Pharmacol (2016) 778:90–5. doi: 10.1016/j.ejphar.2015.04.049
114. Chen K, Xu W, Wilson M, He B, Miller NW, Bengtén E, et al. Immunoglobulin D Enhances Immune Surveillance by Activating Antimicrobial, Proinflammatory and B Cell-Stimulating Programs in Basophils. Nat Immunol (2009) 10(8):889–98. doi: 10.1038/ni.1748
115. Yi MH, Kim HP, Jeong KY, Kim JY, Lee IY, Yong TS. Effects of the Th2-Dominant Milieu on Allergic Responses in Der F 1-Activated Mouse Basophils and Mast Cells. Sci Rep (2018) 8(1):7706. doi: 10.1038/s41598-018-25741-w
116. Bieneman AP, Chichester KL, Chen YH, Schroeder JT. Toll-Like Receptor 2 Ligands Activate Human Basophils for Both IgE-Dependent and IgE-Independent Secretion. J. Allergy Clin Immunol (2005) 115(2):295–301. doi: 10.1016/j.jaci.2004.10.018
117. Klion AD. Eosinophilia: A Pragmatic Approach to Diagnosis and Treatment. Hematol Am Soc Hematol Educ Program (2015) 2015:92–7. doi: 10.1182/asheducation-2015.1.92
118. Rothenberg ME, Hogan SP. The Eosinophil. Annu Rev Immunol (2006) 24:147–74. doi: 10.1146/annurev.immunol.24.021605.090720
119. Carruthers MN, Park S, Slack GW, Dalal BI, Skinnider BF, Schaeffer DF, et al. IgG4-Related Disease and Lymphocyte-Variant Hypereosinophilic Syndrome: A Comparative Case Series. Eur J Haematol (2017) 98(4):378–87. doi: 10.1111/ejh.12842
120. Saeki T, Kobayashi D, Ito T, Tamura M, Yoshikawa S, Yamazaki H. Comparison of Clinical and Laboratory Features of Patients With and Without Allergic Conditions in IgG4-Related Disease: A Single-Center Experience in Japan. Mod Rheumatol (2018) 28(5):845–8. doi: 10.1080/14397595.2017.1416891
121. Culver EL, Sadler R, Bateman AC, Makuch M, Cargill T, Ferry B, et al. Increases in IgE, Eosinophils, and Mast Cells can be Used in Diagnosis and to Predict Relapse of IgG4-Related Disease. Clin Gastroenterol Hepatol (2017) 15(9):1444–52.e6. doi: 10.1016/j.cgh.2017.02.007
122. Kamisawa T, Anjiki H, Egawa N, Kubota N. Allergic Manifestations in Autoimmune Pancreatitis. Eur J Gastroenterol Hepatol (2009) 21(10):1136–9. doi: 10.1097/meg.0b013e3283297417
123. Moriyama M, Tanaka A, Maehara T, Furukawa S, Nakashima H, Nakamura S. T Helper Subsets in Sjögren's Syndrome and IgG4-Related Dacryoadenitis and Sialoadenitis: A Critical Review. J Autoimmun (2014) 51:81–8. doi: 10.1016/j.jaut.2013.07.007
124. Blanchard C, Durual S, Estienne M, Emami S, Vasseur S, Cuber JC. Eotaxin-3/CCL26 Gene Expression in Intestinal Epithelial Cells Is Up-Regulated by Interleukin-4 and Interleukin-13 via the Signal Transducer and Activator of Transcription 6. Int J Biochem Cell Biol (2005) 37(12):2559–73. doi: 10.1016/j.biocel.2005.06.010
125. Komiya A, Nagase H, Yamada H, Sekiya T, Yamaguchi M, Sano Y, et al. Concerted Expression of Eotaxin-1, Eotaxin-2, and Eotaxin-3 in Human Bronchial Epithelial Cells. Cell Immunol (2003) 225(2):91–100. doi: 10.1016/j.cellimm.2003.10.001
126. Takanashi S, Kikuchi J, Sasaki T, Akiyama M, Yasuoka H, Yoshimoto K, et al. Lymphadenopathy in IgG4-Related Disease: A Phenotype of Severe Activity and Poor Prognosis, With Eotaxin-3 as a New Biomarker. Rheumatol (Oxford) (2021) 60(2):967–75. doi: 10.1093/rheumatology/keaa648
127. Saeki T, Ito T, Yamazaki H, Imai N, Nishi S. Hypocomplementemia of Unknown Etiology: An Opportunity to Find Cases of IgG4-Positive Multi-Organ Lymphoproliferative Syndrome. Rheumatol Int (2009) 30(1):99–103. doi: 10.1007/s00296-009-0925-4
128. Kawano M, Saeki T, Nakashima H, Nishi S, Yamaguchi Y, Hisano S, et al. Proposal for Diagnostic Criteria for IgG4-Related Kidney Disease. Clin Exp Nephrol (2011) 15(5):615–26. doi: 10.1007/s10157-011-0521-2
129. Kościelska-Kasprzak K, Bartoszek D, Myszka M, Zabińska M, Klinger M. The Complement Cascade and Renal Disease. Arch Immunol Ther Exp (Warsz) (2014) 62(1):47–57. doi: 10.1007/s00005-013-0254-x
130. Raissian Y, Nasr SH, Larsen CP, Colvin RB, Smyrk TC, Takahashi N, et al. Diagnosis of IgG4-Related Tubulointerstitial Nephritis. J Am Soc Nephrol (2011) 22(7):1343–52. doi: 10.1681/ASN.2011010062
131. Kim TY, Park KS, Choi JS, Kang SH, Cho YM, Kim SB. Comparative Clinical Manifestations of IgG4- Related and IgG4-Negative Primary Tubulointerstitial Nephritis. Clin Nephrol (2011) 76(6):440–6. doi: 10.5414/cn107117
132. Nakashima H, Kawano M, Saeki T, Ubara Y, Hisano S, Nagata M, et al. Estimation of the Number of Histological Diagnosis for IgG4-Related Kidney Disease Referred to the Data Obtained From the Japan Renal Biopsy Registry (J-RBR) Questionnaire and Cases Reported in the Japanese Society of Nephrology Meetings. Clin Exp Nephrol (2017) 21(1):97–103. doi: 10.1007/s10157-016-1260-1
133. Mac K, Wu XJ, Mai J, Howlin K, Suranyi M, Yong J, et al. The Incidence of IgG4-Positive Plasma Cells Staining TIN in Patients With Biopsy-Proven Tubulointerstitial Nephritis. J Clin Pathol (2017) 70(6):483–7. doi: 10.1136/jclinpath-2016-203905
134. Nada R, Ramachandran R, Kumar A, Rathi M, Rawat A, Joshi K, et al. IgG4-Related Tubulointerstitial Nephritis: A Prospective Analysis. Int J Rheum Dis (2016) 19(7):721–9. doi: 10.1111/1756-185X.12675
135. Wang R, He D, Zhao L, Liang S, Liang D, Xu F, et al. Role of Complement System in Patients With Biopsy-Proven Immunoglobulin G4-Related Kidney Disease. Hum Pathol (2018) 81:220–8. doi: 10.1016/j.humpath.2018.07.008
136. Muraki T, Hamano H, Ochi Y, Komatsu K, Komiyama Y, Arakura N, et al. Autoimmune Pancreatitis and Complement Activation System. Pancreas (2006) 32(1):16–21. doi: 10.1097/01.mpa.0000188308.75043.e4
137. Sugimoto M, Watanabe H, Asano T, Sato S, Takagi T, Kobayashi H, et al. Possible Participation of IgG4 in the Activation of Complement in IgG4-Related Disease With Hypocomplementemia. Mod Rheumatol (2016) 26(2):251–8. doi: 10.3109/14397595.2015.1076924
138. Du H, Shi L, Chen P, Yang W, Xun Y, Yang C, et al. Prohibitin Is Involved in Patients With IgG4 Related Disease. PloS One (2015) 10(5):e0125331. doi: 10.1371/journal.pone.0125331
139. Mishra S, Nyomba BG. Prohibitin - At the Crossroads of Obesity-Linked Diabetes and Cancer. Exp Biol Med (Maywood) (2017) 242(11):1170–7. doi: 10.1177/1535370217703976
140. Barbier-Torres L, Lu SC. Prohibitin 1 in Liver Injury and Cancer. Exp Biol Med (Maywood) (2020) 245(5):385–94. doi: 10.1177/1535370220908257
141. Zhou TB, Qin YH, Zhou C, Lei FY, Zhao YJ, Chen J, et al. Less Expression of Prohibitin Is Associated With Increased Caspase-3 Expression and Cell Apoptosis in Renal Interstitial Fibrosis Rats. Nephrol (Carlton) (2012) 17(2):189–96. doi: 10.1111/j.1440-1797.2011.01522.x
142. Mishra S, Murphy LC, Nyomba BL, Murphy LJ. Prohibitin: A Potential Target for New Therapeutics. Trends Mol Med (2005) 11(4):192–7. doi: 10.1016/j.molmed.2005.02.004
143. Hubers LM, Vos H, Schuurman AR, Erken R, Oude Elferink RP, Burgering B, et al. Annexin A11 Is Targeted by IgG4 and IgG1 Autoantibodies in IgG4-Related Disease. Gut (2018) 67(4):728–35. doi: 10.1136/gutjnl-2017-314548
144. Cheung KF, Yung S, Chau MK, Yap DY, Chan KW, Lee CK, et al. Annexin II-Binding Immunoglobulins in Patients With Lupus Nephritis and Their Correlation With Disease Manifestations. Clin Sci (Lond) (2017) 131(8):653–71. doi: 10.1042/CS20160732
145. Salle V, Mazière JC, Smail A, Cévallos R, Mazière C, Fuentes V, et al. Anti-Annexin II Antibodies in Systemic Autoimmune Diseases and Antiphospholipid Syndrome. J Clin Immunol (2008) 28(4):291–7. doi: 10.1007/s10875-008-9188-1
146. Shiokawa M, Kodama Y, Sekiguchi K, Kuwada T, Tomono T, Kuriyama K, et al. Laminin 511 Is a Target Antigen in Autoimmune Pancreatitis. Sci Transl Med (2018) 10(453):eaaq0997. doi: 10.1126/scitranslmed.aaq0997
147. Liu H, Perugino CA, Ghebremichael M, Wallace ZS, Montesi SB, Stone JH, et al. Disease Severity Linked to Increase in Autoantibody Diversity in IgG4-Related Disease. Arthritis Rheumatol (2020) 72(4):687–93. doi: 10.1002/art.41140
148. Perugino CA, AlSalem SB, Mattoo H, Della-Torre E, Mahajan V, Ganesh G, et al. Identification of Galectin-3 as an Autoantigen in Patients With IgG4-Related Disease. J Allergy Clin Immunol (2019) 143(2):736–745.e6. doi: 10.1016/j.jaci.2018.05.011
149. Li LC, Li J, Gao J. Functions of Galectin-3 and Its Role in Fibrotic Diseases. J Pharmacol Exp Ther (2014) 351(2):336–43. doi: 10.1124/jpet.114.218370
150. Salah A, Yoshifuji H, Ito S, Kitagori K, Kiso K, Yamada N, et al. High Expression of Galectin-3 in Patients With IgG4-Related Disease: A Proteomic Approach. Patholog Res Int (2017) 2017:9312142. doi: 10.1155/2017/9312142
151. Jarrell JA, Baker MC, Perugino CA, Liu H, Bloom MS, Maehara T, et al. Neutralizing Anti-IL-1 Receptor Antagonist Autoantibodies Induce Inflammatory and Fibrotic Mediators in IgG4-Related Disease. J Allergy Clin Immunol (2021) 8:S0091–6749(21)00729-6. doi: 10.1016/j.jaci.2021.05.002
152. Frulloni L, Lunardi C, Simone R, Dolcino M, Scattolini C, Falconi M, et al. Identification of a Novel Antibody Associated With Autoimmune Pancreatitis. N Engl J Med (2009) 361(22):2135–42. doi: 10.1056/NEJMoa0903068
153. Culver EL, Smit WL, Evans C, Sadler R, Cargill T, Makuch M, et al. No Evidence to Support a Role for Helicobacter Pylori Infection and Plasminogen Binding Protein in Autoimmune Pancreatitis and IgG4-Related Disease in a UK Cohort. Pancreatology (2017) 17(3):395–402. doi: 10.1016/j.pan.2017.04.002
Keywords: adaptive immunity, autoantigen, autoimmune disease, IgG4-related disease, innate immunity
Citation: Liu J, Yin W, Westerberg LS, Lee P, Gong Q, Chen Y, Dong L and Liu C (2021) Immune Dysregulation in IgG4-Related Disease. Front. Immunol. 12:738540. doi: 10.3389/fimmu.2021.738540
Received: 09 July 2021; Accepted: 09 August 2021;
Published: 01 September 2021.
Edited by:
Markus G. Seidel, Medical University of Graz, AustriaReviewed by:
Shiv Pillai, Harvard Medical School, United StatesMitsuhiro Akiyama, Stanford University, United States
Copyright © 2021 Liu, Yin, Westerberg, Lee, Gong, Chen, Dong and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quan Gong, Z29uZ3F1YW4xOTk4QDE2My5jb20=; Yan Chen, Y3l6NjAwQDE2My5jb20=; Lingli Dong, dGpoZG9uZ2xsQDE2My5jb20=; Chaohong Liu, Y2hhb2hvbmdsaXU4MEAxMjYuY29t