 Lukas M. Braun1,2
Lukas M. Braun1,2 Robert Zeiser1,3,4,5*
Robert Zeiser1,3,4,5*- 1Department of Medicine I, Medical Center – University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 2Faculty of Biology, University of Freiburg, Freiburg, Germany
- 3German Cancer Consortium (DKTK) Partner Site Freiburg, German Cancer Research Center (DKFZ), Heidelberg, Germany
- 4Comprehensive Cancer Center Freiburg (CCCF), University of Freiburg, Freiburg, Germany
- 5Centre for Biological Signalling Studies (BIOSS) and Centre for Integrative Biological Signalling Studies (CIBSS), University of Freiburg, Freiburg, Germany
Allogeneic hematopoietic stem cell transplantation (allo-HCT) is a potentially curative therapy for patients suffering from hematological malignancies via the donor immune system driven graft-versus-leukemia effect. However, the therapy is mainly limited by severe acute and chronic graft-versus-host disease (GvHD), both being life-threatening complications after allo-HCT. GvHD develops when donor T cells do not only recognize remaining tumor cells as foreign, but also the recipient’s tissue, leading to a severe inflammatory disease. Typical GvHD target organs include the skin, liver and intestinal tract. Currently all approved strategies for GvHD treatment are immunosuppressive therapies, with the first-line therapy being glucocorticoids. However, therapeutic options for glucocorticoid-refractory patients are still limited. Novel therapeutic approaches, which reduce GvHD severity while preserving GvL activity, are urgently needed. Targeting kinase activity with small molecule inhibitors has shown promising results in preclinical animal models and clinical trials. Well-studied kinase targets in GvHD include Rho-associated coiled-coil-containing kinase 2 (ROCK2), spleen tyrosine kinase (SYK), Bruton’s tyrosine kinase (BTK) and interleukin-2-inducible T-cell kinase (ITK) to control B- and T-cell activation in acute and chronic GvHD. Janus Kinase 1 (JAK1) and 2 (JAK2) are among the most intensively studied kinases in GvHD due to their importance in cytokine production and inflammatory cell activation and migration. Here, we discuss the role of kinase inhibition as novel treatment strategies for acute and chronic GvHD after allo-HCT.
Introduction
Patients suffering from hematological malignancies have only access to a very limited number of therapeutic interventions. Allogeneic hematopoietic stem cell transplantation (allo-HCT) is a potentially curative therapy for patients with hematological disorders (1, 2). Patients are pre-conditioned with chemotherapy or total-body irradiation to eradicate the underlying disease, followed by transplantation of donor stem cells. The allogeneic cells elicit an anti-malignancy immune reaction (3). However, besides the beneficial anti-tumor immune response, a major limitation of allo-HCT is acute and chronic graft-versus-host-disease (aGvHD, cGvHD) (4–7). GvHD is a life-threatening complication of allo-HCT and establishes if the transplanted cells recognize the host’s tissue as foreign. Host antigen-presenting cells (APCs) are activated and stimulate the donor cells, thereby causing cytokine release, strong immune cell activation and severe tissue damage (4, 5, 8–10). Acute GvHD is mainly based on T-cell activation and cytokine release, whereas B-cells are major players in cGvHD, which has features of autoimmune diseases and is often accompanied by organ fibrosis (5, 11). Acute and chronic GvHD are different diseases but share some similarities as both are inflammatory diseases initiated by APCs, followed by activation of alloreactive T- and B-cells, inflammation, tissue damage and organ failure (12). Acute GvHD is a risk factor for cGvHD development of (4, 11). Standard medications mainly rely on broad immunosuppression, which can on the one hand reduce GvHD activity, but on the other hand impair anti-malignancy immunity. There is an unmet need for specific and selective therapeutic strategies to control GvHD without disturbing beneficial immune responses after HCT. Kinase-mediated signaling pathways are among the most important signaling cascades to drive cytokine production and immune cell activation, thereby enhancing GvHD severity (13, 14). Many kinases share similarities, as e.g. JAK1/2, TAK1 and MAPK signaling are crucial for inflammatory cytokine signaling (15–17). TCR and BCR signaling, leading to cell survival, proliferation, migration and effector cytokine production, are mainly regulated by BTK and ITK, ROCK2, PI3K, mTOR, Syk and MEK (18–28). ROCK2, JAK1 and JAK2, as well as Syk, play a role in T-cell differentiation, including the induction of regulatory T-cells (27, 29–32). However, there is also a kinase involved in GvHD pathophysiology with a unique function, as ITPKB plays a pivotal role in regulating intracellular Ca2+ levels and T-cell survival (33–35). Based on the various functions of kinases in GvHD pathophysiology, it was concluded that tyrosine kinase inhibitors (TKIs) could be a promising strategy to control B- and T-cell activation and GvHD after allo-HCT (36). In general, TKIs block substrate phosphorylation, thereby limiting cellular downstream effects and pathways. These signaling cascades also include effector functions, e.g. the production of pro-inflammatory cytokines by T-cells (36). Since GvHD is mainly characterized by increased pro-inflammatory cytokines, systemic sclerosis and organ damage, inhibition of activated TK signaling could be a promising strategy to reduce disease severity and progression (36–38). Many small molecules were evaluated or are currently investigated for the treatment of GvHD, with some compounds now being applied as standard therapy. Table 1 summarizes clinical trials about kinase inhibitors in GvHD, which are cited in this article. In this review, we would like to focus on kinases as novel and known targets in acute and chronic GvHD.
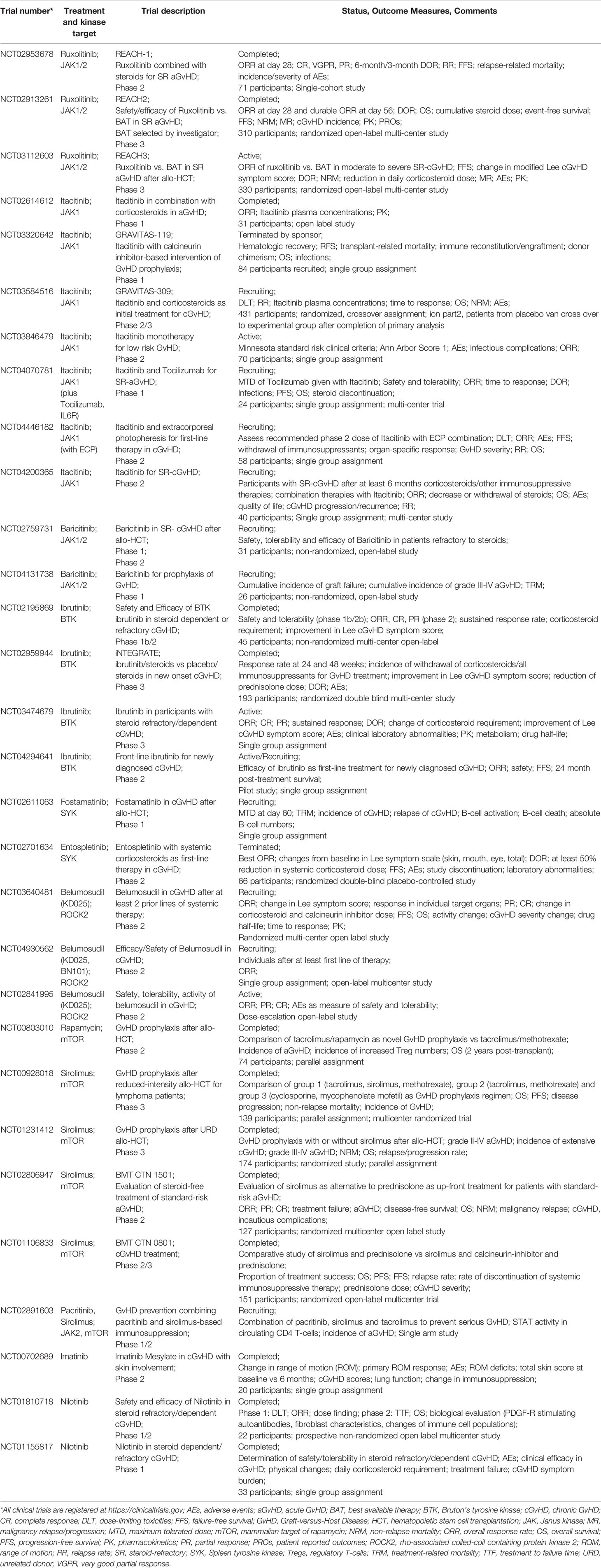
Table 1 Selected clinical trials of kinase inhibition in GvHD.
Janus Kinases 1 and 2 (JAK1/2)
One of the key players in mediating pro-inflammatory signaling is the Janus Kinase (JAK) 1/2. Signaling via JAK1/2 and signal transducer and activator of transcription (STAT) pathways are crucial for the stimulation of inflammatory cytokine production and the activation of a variety of immune cells during GvHD onset and progression (15).
Acute GvHD: The JAK/STAT signaling pathway has high importance in aGvHD onset and progression, as STAT1 and STAT3 are activated early after disease onset. The signaling plays a pivotal role in mediating T-cell activation and changes of the T-cell phenotype (39). Besides, JAK/STAT pathways are also important in the APC compartment in GvHD by influencing dendritic cell (DC) development, maturation, activation and migration into GvHD target organs (18, 40). JAK1 and JAK2 signaling can both be potently blocked with the selective inhibitor Ruxolitinib which has first been approved for the treatment of myelofibrosis by reducing pro-inflammatory signaling (41). Inhibition of JAK1/2 by Ruxolitinib does not only block DC activation and the common gamma chain downstream effects in T-cells (15), but also reduces the migration of neutrophil granulocytes into GvHD target organs (10, 42). Inhibition of JAK1/2 signaling by Ruxolitinib significantly reduced GvHD severity and increased the survival in a pre-clinical murine major-mismatch GvHD model (43). The findings could mainly be linked to a significant reduction of pro-inflammatory cytokine release in vitro and in vivo, reduction of donor T-cell infiltration into GvHD target organs and reduced allogeneic T-cell proliferation. The treatment also blocked allogeneic APC maturation and activation, thereby limiting T-cell proliferation. However, the blockade of JAK1/2 signaling also reduced T-cell proliferation stimulated by anti-CD3/CD28 activation beads. Direct effects on T-cells, including reduced activation and proliferation, could be linked to reduced STAT3 phosphorylation (Figure 1). Moreover, the number of regulatory T-cells (Tregs) was elevated in the intestine (43). Since Ruxolitinib application was seen to have cytopenia as side effects due to co-inhibition of JAK2, specific JAK1 inhibitors were designed to reduce cytokine signaling without side effects (44, 45). Itacitinib is a highly selective JAK1 inhibitor which has shown promising activity in inflammatory models, such as arthritis, inflammatory bowel disease and aGvHD (46). A preclinical study applied Itacitinib in a xenogeneic aGvHD humanized mouse model and found prolonged survival and reduced GvHD severity compared to control. Frequencies of CD4 and CD8 T-cells were lower on d21 and d28 after transplantation, whereas Treg frequencies increased. In a following GvL model, Itacitinib treatment reduced anti-leukemia immunity to some extent. However, more detailed analysis of JAK1 inhibition on T-cells would be necessary, including deeper phenotyping and effector function analysis (47). Baricitinib is another promising JAK inhibitor, blocking JAK1 and JAK2, and could prevent GvHD in a preclinical model (48). The investigators showed that single inhibition of JAK1 or JAK2 was not as effective as the double blockade with Ruxolitinib, hypothesizing that balanced blockade of both kinases is needed to optimally control GvHD. IFNγR and IL6R signaling, which is mainly regulated through JAK1/2, can be efficiently blocked with Baricitinib. Application of Baricitinib in murine GvHD resulted in 100 % survival, reduced early intestinal GvHD, and faster immune reconstitution with superior activity compared to Ruxolitinib. Mechanistically, Baricitinib treatment enhanced allogeneic Treg proliferation while blocking effector T-cell proliferation (48). GvHD-suppressive Tregs were increased by preservation of JAK3 signaling and increased STAT5 phosphorylation (49). Moreover, costimulatory molecule expression on allogeneic APCs was reduced. Most importantly, Baricitinib did preserve GvL activity and could control ongoing GvHD, making it a potential therapy for established GvHD and not only as prophylaxis treatment (48).
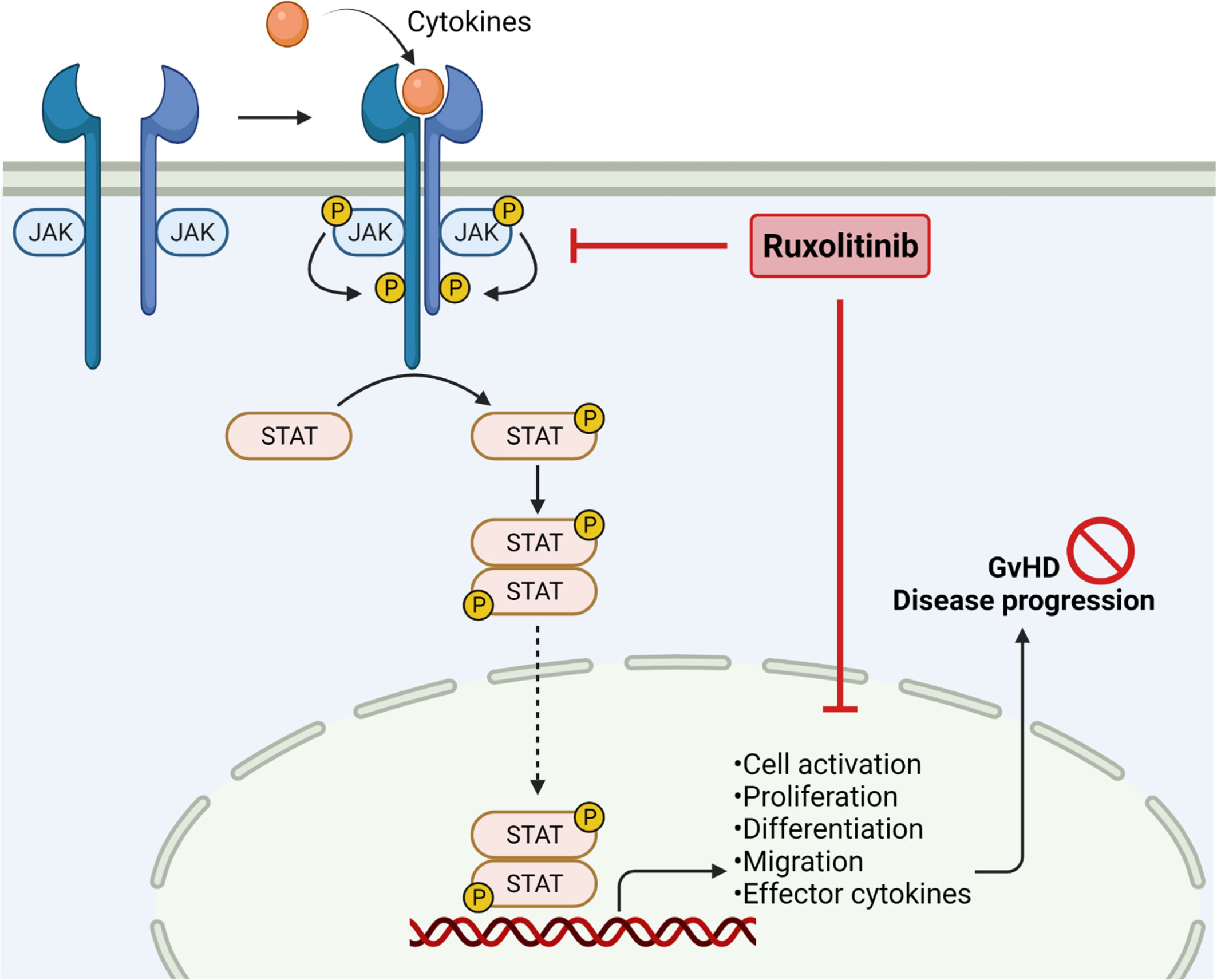
Figure 1 JAK2 inhibition in Graft-versus-Host Disease. Janus kinases are crucial to mediate extracellular signals. Binding of cytokines results in receptor dimerization and phosphorylation, subsequently phosphorylating STAT molecules by receptor tyrosine kinases. Phosphorylation of STAT leads to dimerization and translocation into the nucleus, followed by enhancing gene transcription. JAK/STAT signaling is important in regulating cell activation, proliferation, migration and effector cytokine production, thereby enhancing GvHD severity. JAK1/2 inhibition by ruxolitinib reduces pro-inflammatory signaling and cell migration, resulting in reduced GvHD disease progression. Created with Biorender.com.
A first retrospective study included 95 patients receiving Ruxolitinib as a salvage therapy for steroid-refractory (SR-) GvHD and patients with severe intestinal or skin GvHD showed impressive response to JAK1/2 inhibition. Treatment with Ruxolitinib was liked to decreased pro-inflammatory serum cytokine levels and lower numbers of activated T-cells. The overall response rate (ORR) was 81.5 % and the overall survival (OS) of steroid-refractory aGvHD patients receiving Ruxolitinib was higher than ever reported for any other pharmacological therapy (50). This trial was followed by the REACH1 trial (NCT02953678), including patients with SR-aGvHD after HCT from any donor source. Comparable to the first results seen in patients, the time to response was 7 days and the ORR was 73.2 %. Also, the 6-month and 12-month OS was 51.0 % and 42.6 %, respectively, whereas the OS was lower in patients with grade III/IV aGvHD who received longer corticosteroid treatment before Ruxolitinib treatment. Mechanistically, biomarker analysis confirmed elevated hematopoiesis and a reduction of inflammatory cytokine release and signaling in patients receiving Ruxolitinib (51). The following REACH2 trial (NCT02913261) aimed to analyze the efficacy of Ruxolitinib in comparison to best available care in SR-aGvHD. Ruxolitinib significantly increased the median failure-free survival (FFS) and OS compared to control therapy, and the ORR at day 28 was significantly higher with Ruxolitinib treatment. The percentage of patients with complete response (CR) was at 34 % and 19 % under JAK1/2 inhibition or best available therapy, respectively. Comparable to side effects seen in the REACH1 trial, Ruxolitinib was again reported to cause thrombocytopenia (52). Besides JAK1 and 2 inhibition with Ruxolitinib, Itacitinib has shown promising preclinical efficacy in GvHD. Due to these findings and the hypothesis that selective JAK1 inhibition reduces side effects seen with Ruxolitinib, a phase I trial was initiated to determine if Itacitinib in combination with corticosteroids is safe and tolerable in patients with grade IIb-IVd aGvHD (NCT02614612). Treatment-naïve and SR aGvHD patients were included and distributed equally into two Itacitinib doses. Itacitinib was found safe to use; the most common nonhematologic AE was diarrhea, whereas hematologic AEs included anemia and thrombocytopenia. The d28 ORR was 75 % and 70.6 % of treatment naïve and SR aGvHD, respectively. Responses were seen across involved organs but median DOR was not reached in patients with treatment-naïve aGvHD. Upon Itacitinib treatment, corticosteroid doses could be reduced or discontinued in all patients. Overall, the study demonstrated that JAK1 inhibition with Itacitinib is effective and well tolerated in aGvHD. However, findings are limited due to small sample size and no comparator group (45). In another trial, Itacitinib was thought to be a promising prophylaxis treatment, as JAK/STAT blockade could limit T-cell migration into GvHD target organs; However, the study was terminated (NCT03320642). Itacitinib treatment is currently also investigated in more clinical trials as a therapy for low-risk GvHD (NCT03846479) or in combination with the anti-IL6R antibody Tocilizumab in aGvHD (NCT04070781).
Chronic GvHD: Besides application in aGvHD, Ruxolitinib treatment was also evaluated for the treatment of glucocorticoid-refractory cGvHD (53). Chronic GvHD occurs in 30-70 % of all patients undergoing allo-HCT and is treated with systemic glucocorticoids as first-line therapy (11, 54, 55). However, the disease becomes glucocorticoid-dependent or glucocorticoid-refractory in about 50 % of all patients, thereby significantly increasing the risk for poor outcomes (11, 53–55). Although the Bruton’s tyrosine kinase inhibitor ibrutinib is approved in the US and Canada as second-line therapy, responses are limited. Moreover, the efficacy of ibrutinib has not been demonstrated in a randomized clinical trial (11, 56, 57). In pre-clinical analysis, inhibition of JAK1/2 was shown to be an effective treatment not only in aGvHD, but also in cGvHD (50, 53). JAK1/2 is crucial for the initiation and progression of inflammation and cytokine signaling, both being major regulators of acute and chronic GvHD (15, 42, 43, 50, 58). Based on the positive results of Ruxolitinib in aGvHD in the REACH1 and REACH2 trial, it was evaluated in the REACH3 trial (NCT03112603) for the treatment of glucocorticoid-refractory or -dependent cGvHD in comparison to best available therapy (BAT, control) (51–53). The REACH3 trial is a phase III randomized open-label multi-center study of Ruxolitinib in comparison to ten other therapeutic agents. At the primary study end point, the OR was higher with Ruxolitinib (49.7 %) compared to control (25.6 %). A higher OR was observed with Ruxolitinib than any other control treatment in most organs, except for lung and liver cGvHD where responses were similar. Moreover, patients receiving Ruxolitinib had a significantly longer FFS than the control group (>18.6 months vs. 5.7 months). Also, the response on the modified Lee Symptom Scale was higher with Ruxolitinib (24.2 %) at 24 weeks compared to BAT (11.0 %). The investigators reported decreased dose of glucocorticoids in both groups over time, whereas the decrease was slightly greater in the Ruxolitinib group. Overall, the DOR was higher in the Ruxolitinib group compared to control treatment. Regarding the safety profile, adverse events (AEs) of any grade were slightly more often seen in the Ruxolitinib group compared to control, whereas adverse events of grade 3 and 4 were comparable in both groups (57.0 % vs. 57.6 %). Most commonly, Ruxolitinib treated patients experienced thrombocytopenia (15.2 %), anemia (12.7 %), neutropenia (8.5 %) and pneumonia (8.5 %). The safety profile was comparable to results seen in patients with aGvHD (53, 59, 60). Bacterial, fungal and viral infections were seen in both groups at a comparable incidence. In summary, the REACH3 trial showed that Ruxolitinib is superior over common second-line therapies for SR-cGvHD. Ruxolitinib was found being an effective treatment options for patients with moderate and severe SR-cGvHD (53). The selective JAK1 inhibitor Itacitinib is currently investigated as first-line therapy in cGvHD (NCT03584516), but results are not available yet. Since preclinical evaluation of Baricitinib in GvHD were promising, clinical trials were initiated to evaluate Baricitinib treatment in patients with cGvHD (NCT02759731) or as a prophylaxis treatment for GvHD after allo-HCT (NCT04131738). However, both trials are still recruiting and did not publish any results yet. Novel approaches combine JAK inhibitors with other therapies to enhance treatment efficacy. One trial investigates the combination of Itacitinib with corticosteroids or other immunosuppressive therapies in cGvHD (NCT04200365), or as combination with extracorporeal photopheresis (ECP) as first-line therapy in cGvHD (NCT04446182). In a comparable attempt, Ruxolitinib was combined with ECP in SR-cGvHD patients. Since both treatments alone did already show promising effects in GvHD, a combination therapy was thought to even elevate the therapeutic success. The 2-year survival rate was 75 % and the combination of both therapies was found safe to use with activity in at least a part of SR-cGvHD patients. However, this was only a single-center study and a detailed validation is needed in a prospective trial (61).
Rho-Associated Coiled-Coil Containing Protein Kinase 2
Signaling pathways mediated by Rho GTPase are important regulatory mechanisms of the T-cell mediated immune response, including TCR signaling and effector cytokine production (62). The rho-associated coiled-coil protein kinases 1 and 2 (ROCK1 and ROCK2) are serine-threonine kinases activated by Rho GTPases. Activation of ROCK1 and ROCK2 leads to phosphorylation of downstream molecules, including STAT3 and STAT5, to enhance the transcription of target genes (63). Target molecules regulated by ROCK2 include pro-inflammatory cytokines like IL-21 and IL-17 (23, 64). The ROCK2 signaling pathway has been shown to be important to regulate the balance between Th17 cells and Tregs. ROCK2 activation causes STAT3 phosphorylation, subsequently enhancing the expression of Th17-specific transcription factors, including interferon regulatory factor 4 (IRF4), RAR-related orphan receptor (ROR) γt and RORα (30–32, 65). Blockade of ROCK2 using its selective inhibitor belumosudil shifts the Th17/Treg balance towards regulatory T-cells through a STAT5-dependent mechanism (30, 32, 66). Regarding the potency of Tregs to reduce GvHD severity (67), ROCK2 was hypothesized being a promising target in GvHD (Figure 2) (30, 32).
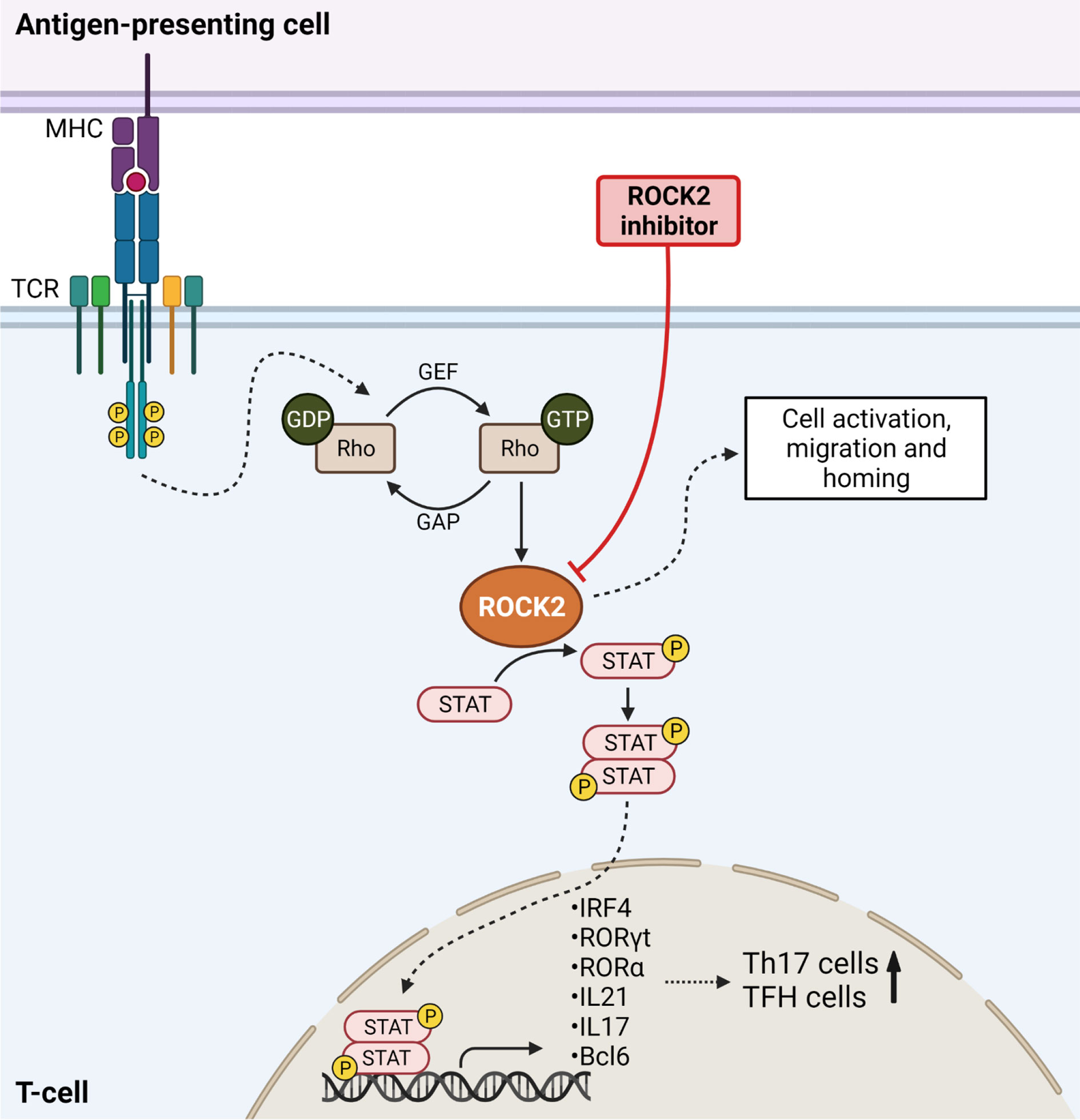
Figure 2 ROCK2 mediates Th17 differentiation in Graft-versus-Host Disease. TCR stimulation results in downstream ROCK2 activation, thereby phosphorylating STAT molecules. STAT is translocated into the nucleus to activate the transcription of Th17-specific transcription factors, thereby increasing the numbers of Th17 cells. ROCK2 activation further enhances the numbers of T follicular helper cells (TFH) and increases cell migration, activation and homing. Inhibition of ROCK2 blocks the differentiation of T-cells into TFH and Th17 cells and results in higher Treg numbers. Created with Biorender.com.
Chronic GvHD: A preclinical study evaluated the effects of the ROCK2 inhibitor KD025 in cGvHD (66). Efficacy was first assessed in a bronchiolitis obliterans syndrome (BOS) model, in which the mice develop organ fibrosis associated with increased B-cell activation in the GC, also seen in cGvHD patients (66, 68). Upon treatment, the mice had improved resistance, elastance and compliance, together with decreased histopathology scores in the major GvHD target organs. However, cGvHD mice still had higher pathology scores than BM-only non-cGvHD animals. Also, collagen and immunoglobulin (Ig) deposition, both typically being increased in cGvHD multiorgan and BOS models (66, 68), were significantly decreased upon ROCK2 inhibition (66). Increased numbers of TFH cells and GC B-cells was previously been reported during murine cGvHD in the BOS model (68–71), all effects being reversed upon ROCK2 inhibition (66). Mechanistically, ROCK2 inhibition with KD025 significantly decreased STAT3 phosphorylation, whereas STAT5 phosphorylation was increased (66), both also reported in patients (32). Moreover, ROCK2 inhibition led to decreased expression of IRF4 and RORγt, crucial regulators for Th17 development (30–32, 65, 66, 72). Interestingly, the treatment did also reduce B-cell lymphoma 6 (Bcl6) expression, described as a TFH transcriptional regulator (66). The effects of KD025 in cGvHD were confirmed using a second minor mismatch Scl-cGvHD model. Skin pathology and GvHD scores were reduced together with a reduction of epidermal hyperplasia, infiltration of nucleated cells into the dermis and hair follicle destruction. Consistent with the BOS model, STAT3 phosphorylation and IRF4 levels were lower in the spleens of ROCK2 inhibitor treated mice (66). In order to translate the effects into the human system, peripheral blood mononuclear cells (PBMCs) were isolated from cGvHD patients and cultured in Th17-skewing culture medium either in the presence of KD025 or vehicle treatment. Comparable to cells from healthy individuals, ROCK2 inhibition reduced the production of IL-21, IL-17 and IFNγ in cells from cGvHD patients (32, 66). Important to note, targeted ROCK2 inhibition did not interfere with anti-leukemia immunity (66). The preclinical data suggested that ROCK2 inhibition could reduce cGvHD severity by both, downregulation of cytokine production and reduction of TFH cells, which are important for disease progression (66).
Based on promising preclinical data, the safety and efficacy of the selective ROCK2 inhibitor belumosudil was evaluated in clinical trials. The results of the ROCKstar study (NCT03640481) were published recently (73). The trial included patients after allo-HCT with persistent cGvHD manifestations indicating systemic therapy. Patient who received two to five lines of therapy (LOT) were included. The ROCKstar study evaluated belumosudil at 200 mg once (QD) or twice (BID) per day. The best ORR was 74 % (200 mg QD) and 77 % (200 mg BID), high responses were seen in all groups, and all affected organs demonstrated a response. A symptom reduction was seen in 59 % (QD) and 62 % (BID) of all patients. AEs were reported but not unexpected for cGvHD patients treated with immunosuppressive therapies. Overall, ROCK2 inhibition was found safe and well tolerated in patients suffering from cGvHD (73). A similar study is evaluating safety and efficacy of BN101 (belumosudil) in patients with cGvHD at a daily dose of 200 mg (NCT04930562). Results of this trial have not been published yet. Following the promising safety studies, a phase IIa trial was conducted as dose-finding study and to further analyze safety and efficacy of belumosudil in cGvHD patients previously treated with one to three prior LOT (NCT02841995) (30). The study included 54 patients in three different treatment cohorts, 200 mg daily, 200 mg twice per day and 400 mg daily. The ORR was comparable between all cohorts, ranging from 62 % to 69 %. Detailed organ analyses revealed complete remission (CR) in all affected organs, except for the lungs where a partial response (PR) was the best response. In general, the responses were achieved rapidly, with more than 75 % of the responses seen at eight weeks. Later organ responses were mainly seen in the lung and the eyes. The percentage of patients achieving FFS with response at 12 months was 24 % (30). The 12- and 24-month OS rate was 91 % and 82 %, respectively. Upon ROCK2 inhibition, 35 % of all patients experienced clinical improvement and 86 % could reduce or discontinue corticosteroids. Overall, belumosudil was well-tolerated and found safe to use. The main AEs were upper respiratory infections, diarrhea, fatigue, headache and hypertension. Belumosudil treatment was discontinued in three patients due to potentially drug-related AEs and four patients died during the study due to disease relapse and cGvHD progression; none of the deaths was related to the treatment. Mechanistically, blood analysis revealed increased Treg numbers together with decreased numbers of Th17 cells (30). In summary, the selective ROCK2 inhibitor belumosudil is a promising treatment option in cGvHD, targeting both, fibrosis and inflammation. The mechanistic results were comparable to the observations from the preclinical study (66). ROCK2 inhibition with belumosudil was granted Breakthrough Therapy Designation by the US Food and Drug Administration (FDA) and received FDA approval for the use in SR-cGvHD.
Mammalian Target of Rapamycin (mTOR)
Donor T-cell activation and inflammatory cytokine secretion is a hallmark of GvHD after allo-HCT. Activation and effector functions of T-cells are tightly connected to the phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling cascade, which is crucial for the regulation of T-cell survival, proliferation, cell cycle progression, differentiation and metabolism (Figure 3) (7, 24–26, 74, 75). The major regulator of mTOR is a serine protein kinase formed of the mTORC1 and mTORC2 complexes (76, 77). The p70 ribosomal S6 Kinase (S6) is located downstream of mTOR and is the main signal transducer to enhance gene transcription and protein synthesis (7, 22). It is known that mTOR signaling is enhanced in GvHD, as T-cells isolated from allo-HCT recipients showed enhanced expression of Raptor and Rictor, both parts of the mTOR complex, and elevated S6 phosphorylation (78, 79).
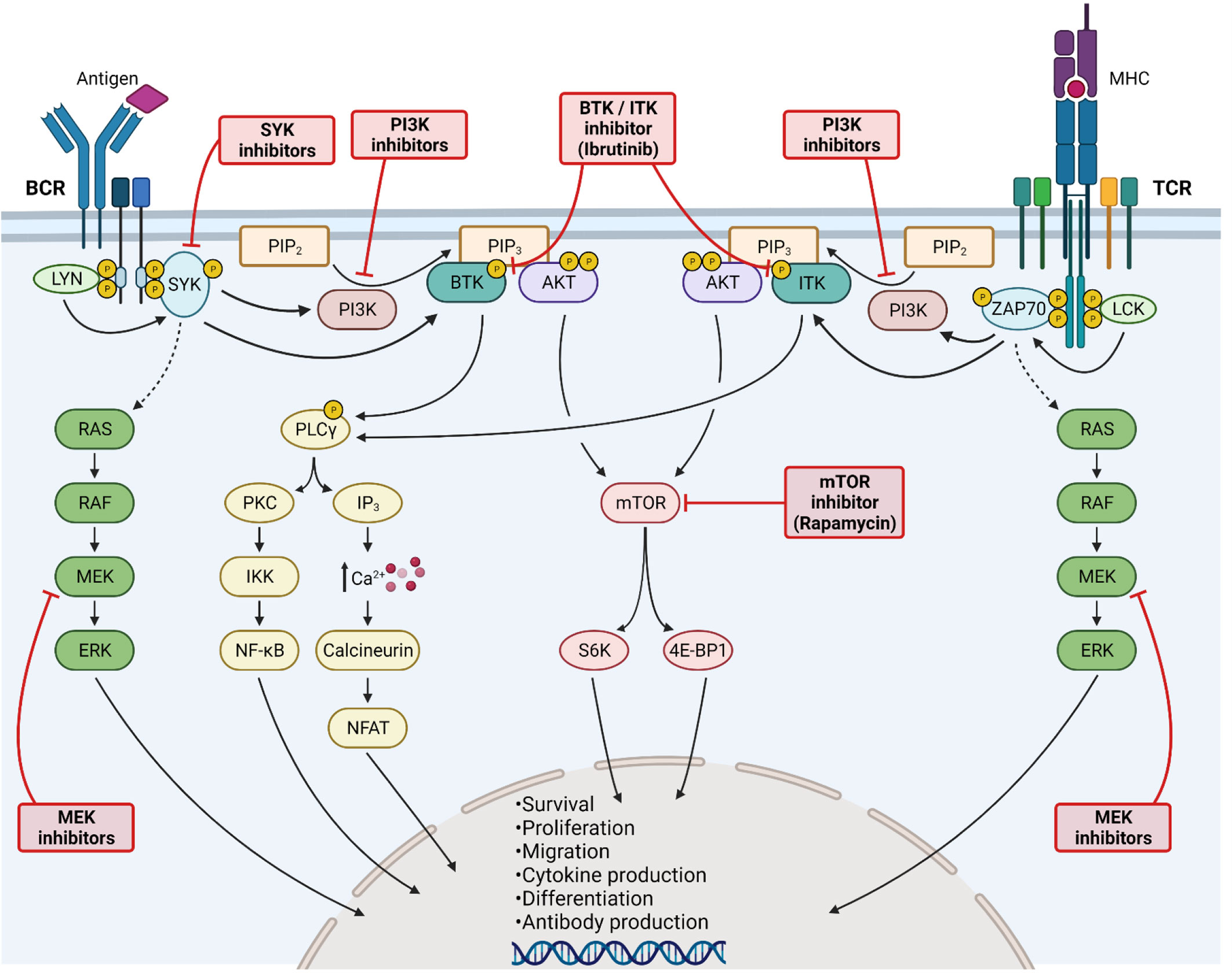
Figure 3 Kinase inhibition for Graft-versus-Host Disease treatment. Activation of B-cell (left side) and T-cell (right side) receptors leads to LYN/LCK phosphorylation, subsequently phosphorylating SYK and ZAP70. Both kinases further increase the downstream RAS/RAF/MEK/ERK signaling cascade. Activated SYK/ZAP70 stimulates also PI3K, as well as BTK and ITK. PI3K catalyzes the transformation of PIP2 into PIP3, being a binding site for BTK/ITK and AKT. BTK and ITK phosphorylate PLCγ, thereby enhancing PKC signaling and NF-κB translocation into the nucleus. Moreover, elevated calcium influx activates NFAT signaling. Activation of AKT stimulates mTOR signaling. All major kinase signaling pathways lead to cell survival, proliferation, differentiation and migration. Furthermore, cytokine and antibody production are enhanced. The signaling pathways can be blocked at various steps, including MEK, SYK, PI3K, BTK/ITK and mTOR inhibition. All kinase inhibitors have shown promising results in GvHD. Created with Biorender.com.
Acute GvHD: In a preclinical study, transplantation of Mtor-deficient donor T-cells reduced aGvHD severity in mice, whereas more detailed analysis with Raptor-deficient allogeneic T-cells revealed that T-cell-mediated pathogenesis is dependent on mTORC1 but not on mTORC2 (79). Treatment with the mTORC1 inhibitor rapamycin (sirolimus) reduced GvHD severity in mice through reduction of pro-inflammatory cytokines and a blockade of T-cell proliferation and APC activity (79–81). Previous preclinical analysis revealed that rapamycin treatment is more effective in reducing murine GvHD mediated by CD8+ or TCR γδ+ T-cells than by CD4+ T-cells. Proliferation of CD8+ T-cells and production of Th1 and cytotoxic T-cell cytokines was inhibited, whereas Th2 cell differentiation was mainly unaffected. More detailed analysis was important for stratification of responsive patients, treatment time points and treatment combinations, as rapamycin did also reduce the GvL effect (80). Application of the immunosuppressant therapy cyclosporine A was shown to interfere with Treg activity, which is important to control GvHD. It was therefore crucial to clarify the effects of rapamycin on the different T-cell subsets, including Tregs. Preclinical and clinical studies revealed that rapamycin does not affect the expansion of Tregs (82–86). Treatment of mice suffering from aGvHD with rapamycin did not result in a survival benefit, however rapamycin enhanced survival of the mice when conventional T-cells (Tconv) and Tregs were transplanted together. The findings were confirmed by histopathological scoring. Rapamycin treatment and Treg transplantation reduced the proliferation of Tconv after allo-HCT. Mechanistically, rapamycin inhibited the proliferation of both, Tregs and Tconv, however Tregs were affected to a lesser extent and their immunosuppressive phenotype and FoxP3 expression was maintained. In vivo imaging confirmed that Treg proliferation is only slightly affected by rapamycin. In order to understand the differential impact of rapamycin on CD4+ T-cells, downstream analysis of mTOR was conducted by S6 and 4-EBP1 phosphorylation analysis after IL-2 stimulation and revealed minimal usage of mTOR signaling by Tregs. The findings of this study help to better understand the synergistic activity of Tregs and rapamycin in protection from aGvHD (87). It was also shown that rapamycin treatment preserves the thymic reconstitution of Tregs after allo-HCT, important to reduce GvHD severity (86). An early preclinical analysis hypothesized that PI3K blockade could be a promising strategy to reduce GvHD as in vitro treatment of donor lymphocytes with wortmannin reduced GvHD severity in mice (88). The role of the PI3K/AKT/mTOR pathway was further investigated in GvHD with an upstream blockade of PI3K using BKM120 or a novel dual PI3K/mTOR inhibitor BEZ235 (7). Both inhibitors blocked the signaling pathway as seen by decreased AKT and S6 phosphorylation. Both also reduced T-cell proliferation without affecting apoptosis, whereas the double inhibitor was more effective at lower concentrations. PI3K/mTOR pathway inhibition also reduced the secretion of Th1/Th2 effector cytokines, including IL-2, IL-6, IFNγ and TNFα. In vitro stimulation with anti-CD3/anti-CD28 increased early effector T-cells, which was reduced by BEZ235 treatment, but not by PI3K blockade alone. Based on promising in vitro data, major mismatch transplanted mice were treated with BEZ235; kinase inhibition significantly prolonged the survival of aGvHD mice and ameliorated GvHD (7). Castor et al. further investigated the role of PI3K in allo-HCT by transplanting PI3Kγ-deficient semi-allogeneic splenocytes in a murine GvHD model (89). Deficiency of PI3Kγ in the donor splenocyte compartment or inhibition of PI3Kγ with AS605240 significantly prolonged survival, increased body weight and reduced GvHD clinical scores in the recipient mice. Histological analysis revealed reduced damage of small intestine and liver and lower infiltration of CD11c+ and T-cells into the small intestine in the absence of PI3Kγ in donor splenocytes. PI3Kγ-deficiency was also associated with reduced pro-inflammatory cytokine secretion. Intravital microscopy showed decreased numbers of rolling and adherent cells in GvHD mice treated with AS605240 compared to vehicle. Together with hints for maintained anti-leukemia immunity, inhibition of PI3Kγ could be a novel strategy to suppress GvHD severity, although further analysis is necessary to better understand its role after allo-HCT (89). A recent preclinical study applied rapamycin in a GvHD model with 50 % MHC disparity and reported increased splenic leukocyte counts, including Tregs and myeloid-derived suppressor cells (MDSCs) (90). Whereas T-cell activation, exhaustion and cytokine secretion remained unchanged, rapamycin treatment rendered the MDSC population more immunosuppressive, reported the first time for mTOR blockade. MDSCs re-isolated from rapamycin treated GvHD mice had an increased immunosuppressive capacity towards alloantigen‐stimulated T-cells, confirmed by increased expression of iNOS, IDO and arginase-1. The importance of iNOS was underlined by application of a specific inhibitor, which abrogated the immunosuppressive phenotype of MDSCs. Since T-cell effector molecules remained unchanged with preserved GvL activity upon rapamycin treatment, the question how GvHD severity is reduced remains open. Nevertheless, the study described a novel and unknown role of mTOR kinase inhibition in GvHD on the myeloid compartment (90).
Chronic GvHD: The importance of mTOR signaling for GvHD pathogenesis is underlined by the finding of activating mTOR mutations in cGvHD patients which drive clonal CD4+ T-cell expansion and cGvHD development (91). Consistent with these findings, Sugiyama et al. highlighted in preclinical cGvHD models that mTOR inhibition, in contrast to cyclosporine A, does not increase the liability to cGvHD development. The investigators could see changed cGvHD scores in the skin and salivary glands upon rapamycin application compared to control (92).
Based on promising preclinical findings, a pilot clinical trial investigated the safety and efficacy of sirolimus as second-line therapy for GvHD treatment after allo-HCT (93). In total, 12 of 21 patients responded to the treatment, however, side effects were significant. AEs included thrombocytopenia, neutropenia and hemolytic uremic syndrome. Sirolimus had activity in patients with SR-GvHD, but dose optimizations were proposed due to severe toxicities (93). A combination of rapamycin with tacrolimus and low-dose methotrexate in GvHD patients was found feasible. In comparison to historical high-risk populations, the investigators reported lower rates of GvHD (94, 95). Following, a phase II trial combined rapamycin with tacrolimus as GvHD prophylaxis treatment after allo-HCT (NCT00803010). The combination treatment was superior over the control group and prevented high-grade aGvHD and moderate-severe cGvHD while promoting Treg reconstitution (96). A combination of sirolimus with calcineurin inhibitor could prevent GvHD in lymphoma patients after allo-HCT (97). Combining sirolimus/tacrolimus/methotrexate in lymphoma patients after allo-HCT did not affect OS, PFS and cGvHD, however, the addition of sirolimus prevented grade II-IV aGvHD (NCT00928018) (98). Recently, the efficacy of sirolimus was tested upon addition to standard GvHD prophylaxis (NCT01231412). Addition of sirolimus reduced grade II-IV aGvHD incidence, increased OS, but did not affect cGvHD (99). Whereas before-mentioned trials investigated sirolimus as prophylaxis treatment, Pidala et al. evaluated sirolimus for GvHD treatment (100). Sirolimus was tested vs prednisolone as initial treatment of patients with standard-risk aGvHD (NCT02806947). Day 28 CR/PR was comparable between both groups, however, CR/PR were significantly higher with sirolimus if compared to low-dose prednisolone. OS, disease-free survival, relapse and non-relapse mortality were comparable between both groups. Sirolimus reduced grade 2-3 infections, steroid exposure, hyperglycemia and enhanced patient-reported quality of life. Since sirolimus achieved comparable outcome at day 28 as prednisolone and spared steroid exposure, a confirming phase III trial is needed to also examine its efficacy in SR-aGvHD patients (100). Besides aGvHD, sirolimus was tested in combination with prednisolone or prednisolone/CNI in cGvHD (NCT01106833). CR/PR at 6 months, FFS and OS were the same at 2 years. Carpenter et al. concluded that sirolimus/prednisolone is an alternative, as a double-therapy is easier to administer and better tolerated than a triple-therapy (101). A first-in-human phase I/II clinical trial combines the JAK2 inhibitor pacritinib (PAC) with sirolimus and low-dose tacrolimus (PAC/SIR/TAC), aiming to reduce T-cell co-stimulation via mTOR and IL6 (NCT02891603). The effect of pacritinib/sirolimus was tested in human MLRs and a xenogeneic GvHD model and consistently suppressed allogeneic T-cell proliferation and GvHD severity (102, 103). STAT3 and S6 phosphorylation were reduced upon treatment, confirming JAK2/mTOR inhibition. In mice, the treatment reduced Th1 and Th17 cells while increasing Tregs. Anti-leukemia and anti-CMV immunity were preserved. Following, the PAC/SIR/TAC combination will be tested in the ongoing phase II trial (102). Overall, targeting mTOR signaling with sirolimus and blocking the PI3K pathway are both promising and established strategies to reduce acute and chronic GvHD either as prophylaxis or treatment of an established disease and may be preferred to other regimens for patients after allo-HCT.
Bruton’s Tyrosine Kinase (BTK) and Interleukin-2 Inducible T-Cell Kinase (ITK)
Dysregulation of T- and B-cell activation and proliferation, enhanced antibody production, inflammation and organ damage are typically seen during GvHD development (104, 105). Stimulation of BCR and TCR and the subsequent activation of downstream pathways is crucial for GvHD induction after allo-HCT (106, 107). Bruton’s tyrosine kinase (BTK) is part of the BCR signaling complex and kinase activation is necessary for survival, migration and proliferation of B-cells (19). Genetic BTK-deficiency results in a loss of peripheral B-cells and a blockade of immunoglobulin production (108). Activation of BTK subsequently phosphorylates phospholipase Cγ2 (PLCγ2), thereby facilitating further downstream effects like NF-κB and NFAT activation to enhance survival, proliferation and migration of B-cells (14, 19, 20). The interleukin-2 inducible T-cell kinase (ITK), another Tec family kinase, has functional similarities with BTK but is crucial for TCR signaling (20, 21). Comparable to BCR signaling, ITK is important for PLCγ2 activation downstream of the TCR, thereby facilitating signaling through NF-κB, NFAT and MAPK to activate T-cells, enhance proliferation and promote cytokine production (14, 20, 109, 110). ITK is important for driving the secretion of IL-2, IL-17 and Th2 cytokines, all being associated with cGvHD pathogenesis (11, 20, 111–114). Regarding the importance of T-cells and B-cells in both, acute and chronic GvHD, inhibition of BTK and ITK could be a promising strategy to inhibit GvHD development by blocking B- and T-cell activation, thereby hindering severe inflammation and fibrosis (14). Ibrutinib is an FDA-approved inhibitor, blocking both ITK and BTK, which was first approved for the use in lymphocytic leukemia (Figure 3) (21).
Acute GvHD: Since ibrutinib has inhibitory effects on both, BTK and ITK, a preclinical study determined the ability of ibrutinib to target donor-derived T-cells in an aGvHD model. For exclusion of donor B-cells, T-cell depleted BM was transplanted together with T-cells from B-cell KO donor mice. Treatment with ibrutinib improved aGvHD clinical scores and survival of mice, whereas the latter was not significantly changed. Although T-cell proliferation and activation was unaffected upon ibrutinib treatment, experiments with B-cell-deficient donor mice confirmed an effect of ibrutinib on donor T-cells after allo-HCT (14).
Chronic GvHD: Since application of ibrutinib led only to slight improvement in acute GvHD, ITK and BTK inhibition with ibrutinib was evaluated in a pre-clinical cGvHD model (104). Mice receiving the treatment survived significantly longer compared to vehicle, did not develop ascites and had delayed onset of proteinuria. Both was associated with cGvHD in mouse models. However, proteinuria was only prevented with longterm treatment. Notably, ibrutinib suppressed cGvHD development and prolonged survival if given after the disease was already established (14). Mechanistically, ibrutinib inhibited B-cell proliferation and co-stimulatory molecule expression, known to be crucial for GvHD pathogenesis (14, 115). T-cell proliferation was not affected. CD4+CD8+ thymocytes were increased, pointing towards enhanced immune reconstitution upon ibrutinib treatment. These findings were not confirmed by lineage staining after allo-HCT and need further detailed analysis to substantiate this hypothesis (14). In an additional scleroderma model (116) with prophylactic ibrutinib treatment prior to allo-HCT, the investigators found that inhibitor treated mice showed less cGvHD symptoms, reduced skin damage, less alopecia and lower GvHD scores after allo-HCT. Prophylactic effects were only seen with high-dose treatment. Ibrutinib treatment did also enhance the reconstitution of B-cells and reduced T follicular helper (TFH) cells after allo-HCT. Protective effects of ibrutinib were confirmed in an aGvHD model transitioning into cGvHD (14). In a second preclinical study, ibrutinib was applied in sclerodermatous cGvHD starting on day 25 after allo-HCT when the first symptoms became apparent (70, 117). The investigators found reduced clinical signs of cGvHD. These findings were accompanied by improved progression-free survival upon ibrutinib treatment. Moreover, ibrutinib application diminished B- and T-cell infiltration into lung and kidney and led to lower GvHD pathology scores in these cGvHD target organs. The investigators applied a second model, aiming to understand the effects of ibrutinib treatment on bronchiolitis obliterans (BO) in cGvHD (70). The treatment started on day 28 after allo-HCT and resulted in reduced pulmonary resistance and elastance, better compliance and lower lung fibrosis. Analysis from ibrutinib treated mice was comparable to non-GvHD mice. Withdrawal of therapy led to a loss of benefit, indicating that ibrutinib treatment need to be applied continuously. Contrary to the previously reported study, prophylactic ibrutinib treatment could not effectively combat cGvHD or BO (14, 70). To further clarify the role of BTK and ITK in cGvHD, the investigators transplanted WT bone marrow together with ITK-deficient T-cells into allogeneic recipients. Donor-derived T-cells are known to be important for cGvHD development. ITK deletion could reverse cGvHD signs in the lungs to values comparable with non-GvHD animals and ibrutinib treated cGvHD mice. Comparable results were seen when XID bone marrow, which lacks BTK, was used as allogeneic graft. B-cells driving cGvHD development rather arise from the transplanted bone marrow. These experiments highlight that both Tec kinases, ITK and BTK, play a role in cGvHD development. Analysis of ex vivo ibrutinib treated cGvHD patient-derived CD4+ T-cells revealed reduced activation upon kinase inhibition. Reduced activation was also seen in patient-derived B-cells with lower BTK, ERK1/2 and PLCγ2 phosphorylation (70). Comparing both described preclinical studies, all models found that ibrutinib treatment affected B-cell activation and differentiation, whereas the effects on T-cells were variable. Clinical GvHD scores were improved in all models and effective in both, prophylactic treatment and treatment of established disease (20).
The promising preclinical data paved the way to further investigate ibrutinib treatment in cGvHD. A phase Ib/II study was conducted to determine safety and efficacy of ibrutinib in patients who failed at least one LOT for cGvHD (NCT02195869) (20, 118). Ibrutinib did not show dose-limiting toxicities. At a median follow-up of 13.9 months, 29 % of patients were still receiving the drug, whereas 71 % of patients discontinued due to adverse events (AEs), cGvHD progression and patient decision. Most AEs were low grade and well manageable and led to dose reductions. A total of 29 patients (69 %) developed infectious complications of any grade. The ORR was 76 % and 71 % of responders showed a response for more than 20 weeks. Even responses were seen in all cGvHD target organs. Corticosteroid therapy could be reduced with ibrutinib treatment. Detailed mechanistic analysis showed strong inhibition of BTK and ITK, reduced pro-inflammatory mediators in the serum, less germinal center (GC) B-cells and total B-cells and reduced numbers of Th17 and TFH cells (20, 118). Promising data from this trial led to the iNTEGRATE phase III clinical trial investigating ibrutinib in combination with prednisone in patients with newly diagnosed moderate to severe cGvHD after allo-HCT (NCT02959944). Response rate was slightly higher in the ibrutinib group and corticosteroids could be withdrawn at 21 and 24 months in the ibrutinib arm. Patients receiving ibrutinib had improved Lee symptom scores. Another phase III trial evaluates the efficacy of ibrutinib in patients with SR-cGvHD (NCT03474679) and an additional phase II trial is currently recruiting to investigate ibrutinib as first-line therapy for newly diagnosed cGvHD who did not receive any systemic treatment for cGvHD (NCT04294641). So far, both of the last mentioned trials did not publish any results yet.
Based on very promising preclinical and clinical trial data, ITK and BTK inhibition with ibrutinib could be a very potent therapy in chronic GvHD. However, additional detailed analysis of the underlying mechanism is necessary to improve the therapeutic success. Since clinical trials focus on cGvHD, further analysis and preclinical models are needed to investigate the role of ITK blockade in aGvHD.
Spleen Tyrosine Kinase (Syk)
The non-receptor cytoplasmic spleen tyrosine kinase (Syk) was hypothesized being an important regulator of GvHD as it has functions in transmitting signals from surface receptors, including Toll-like receptors (TLRs) (119), Fc receptors (120), as well as chemokine receptors (27, 121, 122). Moreover, Syk activation is known to be crucial for TCR signaling upon peptide binding, as well as playing an important role in T-cell lineage commitment, mainly for Th17 responses which are known to be involved in GvHD pathophysiology (27, 29, 123). Based on the knowledge that Syk inhibition, e.g. using Fostamatinib, has beneficial effects in inflammatory diseases, the relevance of Syk in GvHD was further evaluated (Figure 3) (27, 124–126).
Acute GvHD: In a preclinical murine aGvHD model, daily treatment with Fostamatinib led to significantly improved survival, reduced histopathology scores and reduced pro-inflammatory serum cytokine concentrations. Fostamatinib treatment did not interfere with donor lineage engraftment and immune reconstitution. Syk phosphorylation is rapidly increased upon CD3/CD28 stimulation of T-cells and higher pSyk 525/526 levels were also seen in T-cells isolated from aGvHD mice. Using luciferase transgenic T-cells, Syk inhibitor treatment was found to reduce alloreactive donor T-cell expansion in vivo. The findings were confirmed by CFSE staining, indicating reduced T-cell proliferation in vivo upon Syk inhibition. Besides blockade of T-cell proliferation, Syk inhibition also reduced T-cell migration towards CXCL12. Contrary to previous findings about the importance of Syk in T-cell lineage commitment, Fostamatinib treatment did not change the percentage of Th2 and Th17 cells after allo-HCT. Effects on T-cells could be further affected by APCs as Syk inhibition was also connected to diminished costimulatory molecule expression and reduced DC migration in vivo and in vitro. Although proliferation and effector cytokine secretion of allogeneic donor T-cells was significantly reduced by Fostamatinib, the GvL effect was preserved, as confirmed by in vivo bioluminescence imaging using luciferase transgenic leukemia cells and ex vivo killing assays. Overall, pharmacological inhibition of Syk was found being a novel treatment strategy in aGvHD by reducing T-cell expansion and costimulation while preserving anti-leukemia immunity (27).
Chronic GvHD: Besides its role in TCR signaling, Syk is also involved in BCR signaling and controlling cell migration and adhesion (36). Knowing the importance of B-cells in cGvHD, Syk inhibition was also hypothesized being a major regulator of cGvHD pathophysiology (8, 11, 36, 69, 127). In a cGvHD model with multiorgan involvement, Syk-mediated BCR signaling in allogeneic B-cells was validated being crucial for cGvHD development (69). The investigators isolated B-cells from cGvHD animals at day 60 after allo-HCT and showed increased Syk phosphorylation at Y348 (69). Comparable results were found in B-cells from cGvHD patients (107). The importance of Syk signaling was further evaluated using Syk-deficient allogeneic BM donors for a model of cGvHD with multiorgan involvement. The mice did not develop pulmonary dysfunctions after transplantation with Syk KO BM, whereas Syk-deficient T-cells did not attenuate cGvHD severity. Additionally, Syk was not only important in the initiation of cGvHD, but also in disease progression as pulmonary dysfunction was reversed upon tamoxifen-induced Syk depletion. The Syk inhibitor Fostamatinib (126, 128), was applied during active disease and reduced cGvHD severity in the lung and improved pulmonary dysfunctions (69). Contrary, improvement of skin inflammation was not seen in three Scl-cGvHD models upon Fostamatinib treatment. However, one model showed attenuated skin GvHD and clinical GvHD scores upon Syk inhibition (69). In an additional study of Scl-cGvHD, the investigators proved that Syk phosphorylation is increased in T- and B-cells, as well as in CD11b+ cells, after allo-HCT (129). Early treatment with Fostamatinib reduced the severity and fibrosis of Scl-cGvHD and the expression of pro-inflammatory molecules in the skin. Moreover, the migration of antigen-specific memory CD4+ T-cells and the proliferation and activation of allogeneic CD4+ and CD11b+ cells was suppressed, comparable to results seen in the aGvHD setting (27, 129). Since this data is contrary to a before-mentioned study, it is important to note that effects of Fostamatinib were mainly seen if the treatment was applied early after cGvHD induction (69, 129). When B-cells isolated from patients were treated with Fostamatinib in vitro, the drug preferentially killed cGvHD B-cells seen by increased apoptosis if compared to non-cGvHD control B-cells (69, 107). Additionally, Syk inhibition blocked the differentiation of CD4 T-cells into Th2 and Th17 phenotypes (8, 69, 129). This was different if compared to Syk inhibition in aGvHD described above (27). Using the second-generation highly selective Sky inhibitor entospletinib, Poe et al. demonstrated that inhibitor treatment blocked the development of eye GvHD and also significantly reduced hair loss in GvHD animals. Besides reducing GvHD severity, entospletinib treatment led to improved reconstitution of monocytes, B-cells and T-cells at 28 and 42 days after allo-HCT. Moreover, pathogenic activated GL7+ B-cells and Th2 cells were diminished upon Syk inhibition, both playing a role in acute and chronic GvHD (130–132). T-cell differentiation was changed upon entospletinib treatment with increased numbers of Tregs and a reduction of Th17 cells. Overall, the treatment significantly prolonged the survival of the mice after allo-HCT and reduced skin inflammation and GvHD severity (130).
Taken together, the data derived from genetic and pharmacological approaches in pre-clinical murine GvHD models clearly indicate that Syk plays an important role in GvHD pathophysiology. Pharmacological targeting of Syk could be a novel attempt to treat GvHD. Based on these promising findings of Syk inhibition with Fostamatinib in vitro and from preclinical in vivo models, the efficacy of Fostamatinib to prevent and treat cGvHD after allo-HCT is currently evaluated in a phase I trial (NCT02611063). Another phase II trial was investigating the efficacy and tolerability of entospletinib in combination with systemic corticosteroids cGvHD as first-line therapy, however, the study was terminated (NCT02701634). Based on pre-clinical studies, it would also be interesting to evaluate effects of Syk inhibition in patients with aGvHD. However, ongoing clinical trials first focused on cGvHD, which might be due to the importance of Syk in BCR downstream signaling, whereas it is not essential for TCR downstream events.
Platelet-Derived Growth Factor Receptor
Chronic GvHD: cGvHD is often presented with dermal fibrosis and sclerosis, associated with the presence of stimulatory anti-platelet-derived growth factor receptor (PDGFR) antibodies, suggesting a direct link between skin fibrosis and PDGF signaling (133). PDGFR stimulation causes enhanced collagen production, which could contribute to organ damage (134). Besides PDGFR stimulation, TGFβ is known as an important mediator of fibrosis in cGvHD (135, 136) and inhibition of both reduced pro-fibrotic activity and pulmonary fibrosis in experimental models (137, 138). Imatinib was first developed as a treatment for BCR-ABL positive CML, but also inhibits PDGFR and was therefore hypothesized as a novel therapeutic intervention in cGvHD by reducing fibrosis (133, 139). In preclinical analyses of bleomycin-induced fibrosis, imatinib inhibited the development of dermal fibrosis by reducing COL1A1, COL1A2, and fibronectin 1 transcription. Moreover, the induction of extracellular matrix proteins, stimulated by PDGF and TGFβ, was reduced upon imatinib application (139, 140). Additionally, imatinib could also reduce fibrosis in kidney and liver, target organs of cGvHD (140). Based on this, Belle et al. applied imatinib in a murine model of Scl-cGvHD but found limited impact besides reduced PDGFR phosphorylation. T-cell proliferation was slightly inhibited, but GvHD scores were unchanged (141). Contrary to these findings, another study reported that both, imatinib and nilotinib prevent the development of Scl-GvHD in mice (142). Nilotinib is a second-generation TKI targeting BCR-ABL and PDGFR with a higher affinity than imatinib (36). Both TKIs inhibited dermal fibrosis and reduced dermal thickness if given as prophylaxis treatment. Additional to these findings, GvHD was also significantly reduced when imatinib or nilotinib was given after onset of clinical disease (142). Serum analysis of patients treated with nilotinib showed reduced inflammatory cytokine secretion, including TNFα, IFNγ and IL-2 (143). Using ex vivo cultures, GvHD-derived fibroblasts expressed higher levels of collagen genes, which was significantly reduced upon nilotinib application. Confirming in vitro data, skin analysis from cGvHD patients showed decrease of COL1α1 and COL1α2 protein levels, TGFβ inhibition and p-Smad2 reduction upon treatment with nilotinib (144). Both, imatinib and nilotinib showed efficacy in clinical trials. Treatment of SR-cGvHD with skin involvement led to improved joint range of motion and better skin scores (NCT00702689), proposing imatinib as possible salvage therapy for SR-cGvHD (133). Olivieri et al. reported high OR of imatinib treatment in patients with refractory cGvHD who previously failed at least two LOTs (145). Comparable, scleroderma symptoms disappeared upon imatinib application and the treatment was well-tolerated (146). A retrospective study confirmed a beneficial activity of imatinib as a salvage therapy in Scl-cGvHD (147). After introduction of the second-generation TKI nilotinib, the compound was tested in SR-cGvHD (NCT01810718). The 2-year OS was 75 % with FFS of 30 %. Based on promising long-term outcomes and well-manageable side effects, nilotinib was hypothesized as a promising treatment in SR-cGvHD (148). Another trial investigating safety and efficacy of nilotinib in SR-cGvHD did not post any results yet (NCT01155817). Taken together, pre-clinical and clinical data confirm the TKIs imatinib and nilotinib as promising therapeutic interventions for cGvHD with organ fibrosis.
Inositol 1,4,5-Triphosphate 3-Kinase B (ITPKB)
TCR stimulation and its ligation, mediated through the contact of T-cells with APCs results in a dramatic increase of intracellular calcium (Ca2+) levels. Calcium influx is essential for activation, maturation and effector functions of T-cells. TCR engagement leads to the activation of PLC-γ, thereby increasing the intracellular levels of inositol 1,4,5-triphosphate (IP3). Binding of IP3 to its specific receptors in turn stimulates the release of calcium from intracellular storage compartments. Continuous depletion of intracellular calcium storage stimulates the opening of cell membrane based calcium channels to enhance the influx of calcium from the extracellular compartment. The intracellular increase of Ca2+ is required to activate calcium-dependent kinases and the transcription factor calcineurin, leading to activation of nuclear factor of activated T-cells (NFAT), thereby enhancing the transcription of a variety of different genes necessary for T-cell activation and effector functions (33, 149). However, a very strong increase of the intracellular cytoplasmic Ca2+ concentration leads to the transcription of pro-apoptotic signaling pathways and activation-induced cell death (AICD). The modulation of intracellular calcium levels was therefore hypothesized to be a potential therapeutic strategy for autoimmune diseases (149, 150). A major regulator of intracellular Ca2+ levels is the inositol 1,4,5-triphosphate 3-kinase (Itpk) family, comprising Itpka, Itpkb, Itpkc and inositol polyphosphate multikinase. The Itpk family acts as a negative regulator of Ca2+ influx through conversion of IP3 to inositol 1,3,4,5-tetrakisphosphate (IP4) and this regulatory mechanism is known to be highly important for T-cell development and survival (33–35, 151). Among all kinases in the Itpk family, Itpkb is most abundant in hematopoietic cells and genetic deficiencies of Itpkb lead to impaired T-cell development in the thymus, mainly based on AICD of immature CD4+CD8+ T-cells (34, 35, 152). Deletion of Itpkb in mature activated T-cells was shown to be a novel strategy to prevent T-cell driven autoimmunity through increase of intracellular calcium levels (Figure 4) (150). A recent study highlighted the therapeutic potential of Itpkb deletion and inhibition to control acute and chronic GvHD (149).
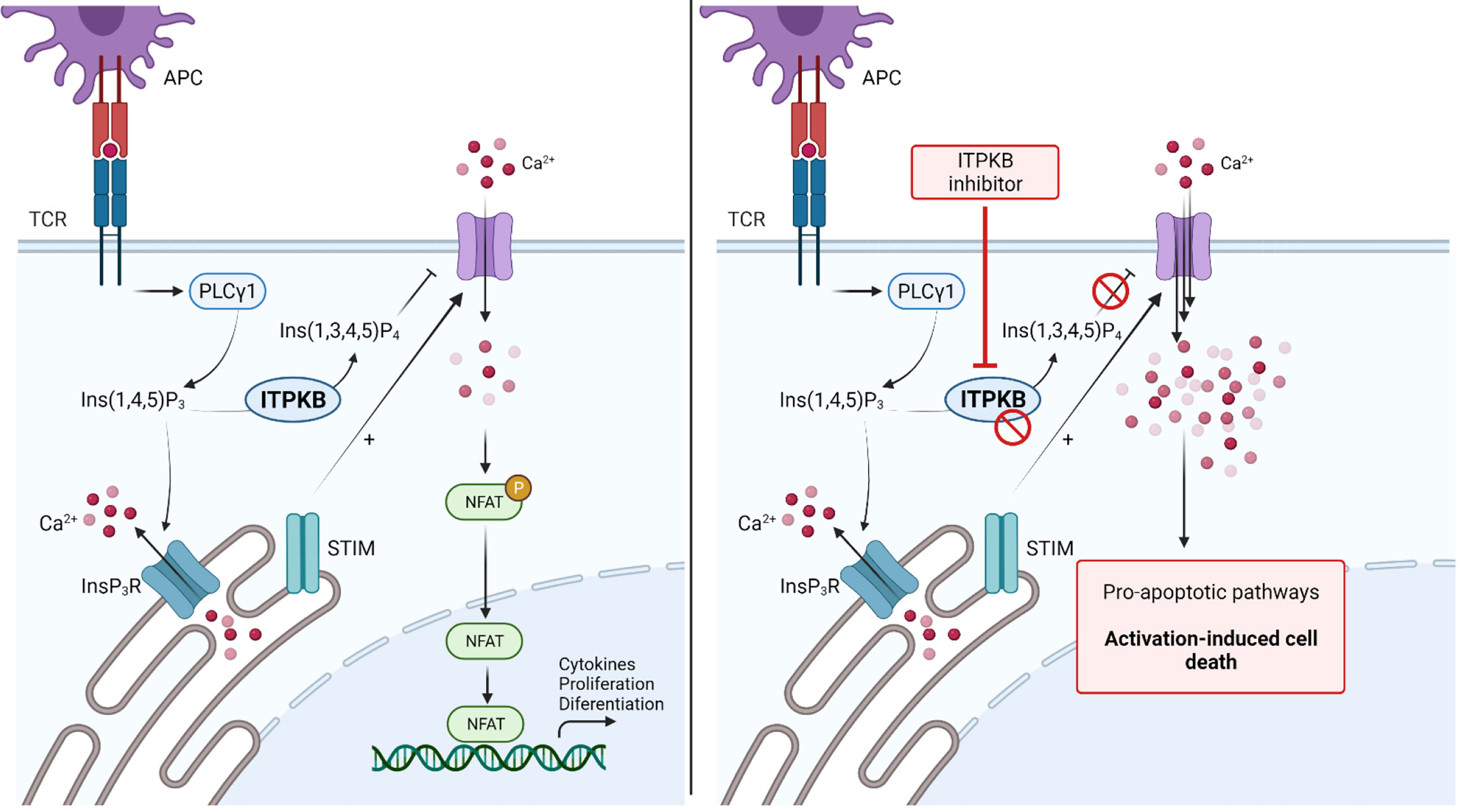
Figure 4 Blockade of Inositol 1,4,5-triphosphate 3-kinase B (ITPKB) as a novel treatment for Graft-versus-Host Disease. TCR stimulation activates downstream PLCγ, thereby increasing intracellular IP3 levels. Binding of IP3 to IP3R activates the release of Ca2+ from intracellular storage compartments. Furthermore, STIM stimulate the influx of extracellular Ca2+ to activate NFAT signaling and gene transcription. ITPKB is a rate-limiting step as it catalyzes the formation of IP4 from IP3. IP4 acts as a control mechanism for calcium signaling by blocking the respective channels in the extracellular membrane. Genetic deletion or inhibition of ITPKB disturbs this control mechanism and leads to increased calcium influx, which stimulates pro-apoptotic signaling pathways, leading to activation-induced cell death. Since ITPKB is predominantly found in hematopoietic cells, this kinase is thought to be a novel target molecule for the treatment of GvHD. Adapted from “NFAT Signaling Pathway”, by Biorender.com (2021). Retrieved from https://app.biorender.com/biorender-templates.
Acute GvHD: In major mismatch allo-HCT models, mice receiving allogeneic Itpkb-deleted T-cells survived significantly longer and experienced less weight loss compared to mice receiving T-cells with functional Itpkb. Complementary, histopathological analysis on day 7 after transplantation showed lower aGvHD pathology scores if mice received T-cells deficient for Itpkb compared to mice receiving wildtype T-cells. Consistent with these findings using a genetic approach, inhibition of Itpkb with GNF362 (150) prolonged the survival of mice receiving high doses of allogeneic T-cells (149). Itpkb deficiency caused reduced donor T-cell survival but no reduced inflammatory cytokine production (153). Itpkb-/- T-cells had comparable or even higher intracellular tumor necrosis factor α (TNFα) and interferon γ (IFNγ) levels compared to Itpkb wildtype T-cells. However, quantitative T-cell analysis in the spleen, mesenteric lymph nodes and the small intestine and liver revealed lower numbers of Itpkb deficient T-cells compared to the WT control, whereas the proliferative capacity remained unchanged. Reduced numbers of donor Itpkb-/- T-cells could be linked to lower T-cell survival, indicated by higher abundance of active caspase 8 in CD4+ and CD8+ T-cells from Itpkb-/- donors after HCT (149, 154, 155). Since the survival of Itpkb deficient T-cells is impaired, GvL activity had to be investigated. Interestingly, mice receiving A20 leukemia cells and Itpkb-/- T-cells survived significantly longer compared to mice receiving leukemia cells only or leukemia in combination with WT T-cells. Leukemia expansion was only seen in mice transplanted with leukemia cells only. The results indicate that Itpkb deficiency maintains anti-leukemia immunity while reducing aGvHD severity. Comparable results were achieved using the Itpkb inhibitor GNF362 (149).
Chronic GvHD: In comparison to T-cell mediated aGvHD, cGvHD establishes as an autoimmune-like disease (8, 11). Mice receiving Itpkb deficient T-cells had significantly improved pulmonary resistance, elastance and compliance compared to mice receiving WT T-cells. Comparable results were seen for lung and liver collagen, as well as for cGvHD histopathology scores. Mice with established cGvHD were also treated with the Itpkb inhibitor GNF362 and the investigators found improved pulmonary function, most probably based on reduced lung macrophage infiltration. Using a Scl-cGvHD model, Itpkb inhibition did result in reduced skin and liver histopathology scores, lower infiltration of proinflammatory macrophages and a reduction of IFNγ-producing T-cells.
In summary, the study could highlight the importance of Itpkb in regulating acute and chronic GvHD and delivered promising data indicating that pharmacological inhibition of Itpkb could be a potential novel therapeutic approach to control GvHD without impairing anti-leukemia immunity (149).
TGFβ-Activated Kinase 1 (TAK1)
Dysregulated innate immune cells are key players in GvHD pathogenesis by activating APCs in the pro-inflammatory milieu (16) and inhibition of inflammatory cytokine signaling could ameliorate GvHD severity in mice and humans (15). Aiming to further understand the role and contribution of innate immune cells in GvHD, Kobayashi et al. performed gene expression profiling on monocytes from patients who experienced GvHD after allo-HCT (156). The investigators found increased expression of TGFβ-activated kinase 1 (TAK1) and downstream signaling molecules, including TNFα, IL-6 and IL-1β, in monocytes from patients with GvHD compared to patients who did not experience GvHD after allo-HCT. TAK1 is a member of the mitogen-activated kinase (MAPK) family and is a key regulating factor upstream of nuclear factor κB (NF-κB), c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK) and p38 in toll-like receptor (TLR) signaling (16). Based on its role in mediating inflammatory signaling, inhibition of TAK1 was hypothesized being a novel strategy to ameliorate GvHD severity by reducing pro-inflammatory signaling and T-cell alloreactivity (156).
Acute GvHD: Patient-derived monocytes were LPS-activated and treated with the TAK1 inhibitor 5Z-7-oxozeaenol (OZ), followed by analysis of cytokine production. Pro-inflammatory cytokine secretion was suppressed in a concentration-dependent manner. Application of OZ in a major mismatch murine aGvHD model prolonged the survival of GvHD mice, reduced GvHD scores and did result in lower serum levels of pro-inflammatory cytokines (156). In a different study, Mathew and Vinnakota et al. investigated the role of microglia in central nervous system (CNS) aGvHD and also found TAK1 being an important mediator of aGvHD-induced neurotoxicity (157). It could be shown for the first time that CNS-GvHD in mice is connected to an activation and expansion of microglia. Comparable, the numbers of microglia were higher in the grey and white matter of patients suffering from GvHD after allo-HCT compared to non-GvHD and non-HCT controls. Moreover, costimulatory molecules were increased on microglia from mice after allo-HCT compared to syngeneic (syn-) HCT and untreated controls. CNS-GvHD was connected to enhanced microglia-derived TNF production in mice and humans, driven by TAK1. Also, other inflammatory cytokines connected to TAK1 signaling were seen elevated in microglia after allo-HCT. Genetic depletion of TAK1 in microglia did alleviate CNS-GvHD associated pathology, as well as memory and cognitive deficits in mice after allo-HCT. Comparable results were seen using the TAK1 inhibitors Takinib and OZ. Takinib did even reduce IFNγ and IL-17 production of T-cells infiltrated into the brain. Based on these two studies and the finding that TAK1 inhibition does not interfere with anti-leukemia immunity after allo-HCT (157), TAK1 could be a novel and promising target to ameliorate aGvHD and CNS-GvHD (157).
Mitogen-Activated Protein Kinase (MEK) Inhibition
The onset and pathogenesis of GvHD is correlated with strong TCR activation and stimulation of TCR downstream signaling pathways to enhance alloreactivity and cytokine production (158). Signaling through the rat sarcoma/mitogen-activated protein kinase kinase/extracellular-signal regulated kinase (RAS/MEK/ERK) cascade is also crucial to translocate transcription factors and to enable target gene transcription, cell proliferation, migration, survival and differentiation (Figure 3) (28, 159).
Acute GvHD: One pre-clinical study investigated activated signaling molecules in alloreactive T-cells isolated from mice suffering from aGvHD and identified significantly increased phosphorylation of ERK1/2 and STAT3 (160). Inhibition of ERK1/2 and STAT3 phosphorylation was thought to be a novel method to reduce donor T-cell alloreactivity (160). ERK1/2 phosphorylation was inhibited using the selective MEK1/2 inhibitor SL327. MEK1/2 is located upstream of ERK1/2 (161–163). SL327 could dose-dependently reduce the proliferation of T-cells upon CD3/CD28 stimulation and in a mixed lymphocyte reaction (MLR) (160, 164). A second preclinical study applied flow cytometry-based pERK1/2 analysis of human T-cells activated with PMA and Ionomycin and a preferential increase of ERK1/2 phosphorylation in naïve and central memory T-cells was seen (165). Application of U0126, a classical MEK inhibitor, and selumetinib, a second-generation MEK inhibitor, reduced ERK1/2 phosphorylation dose-dependently. The latter inhibitor is tested in various clinical trials for different cancer entities and was found safe to use with little to no hematologic toxicity (166–169). Besides proliferation, MEK inhibition reduced effector cytokine production by memory T-cells. Based on these results, the investigators aimed to elucidate the effect of MEK inhibition in an alloreactive setting. The proliferation of T-cells activated with allogeneic HLA-mismatched DCs was significantly suppressed by MEK inhibition while virus-specific T-cell responses were not affected (165). Comparable results were seen using the MEK inhibitor trametinib (170). Selumetinib suppressed cell division even stronger than the calcineurin inhibitor tacrolimus (165, 171). MEK inhibition was further evaluated in experimental major-mismatch aGvHD mouse models, where selumetinib significantly prolonged the survival of GvHD mice (165). The MEK inhibitor trametinib could also suppress GvHD in a xenogeneic model and enhanced the engraftment of diverse T-cell clones. In this model, MEK inhibition suppressed T-cell activity responsible for GvHD while promoting human T-cell reconstitution (172).
Chronic GvHD: In an additional preclinical analysis, trametinib treatment enhanced survival and reduced GvHD scores in a MHC-haploidentical GvHD model (173). MEK inhibition suppressed CD8+ T-cells and elevated naïve T-cells after allo-HCT. Trametinib also reduced target organ damage and lymphocyte infiltration. A second model confirmed the potency of MEK inhibition, as the development of cutaneous GvHD, skin sclerosis and alopecia was reduced upon trametinib application. Although trametinib was reported to be well-tolerated without toxicities in vivo (174), donor cell engraftment and myeloid immune reconstitution were suppressed. Since MEK inhibition was shown to suppress T-cell effector functions and proliferation, it is of high importance to investigate the effect on anti-tumor immunity. Surprisingly, MEK inhibition did not affect T-cell mediated anti-tumor immunity against mastocytoma cells, as T-cell transplanted mice survived longer compared to vehicle. Mice receiving tumor only without T-cells did not benefit from MEK inhibition, implicating that trametinib does not directly affect tumor cells. In comparison, the GvHD prophylaxis treatment tacrolimus shortened the survival of leukemia-bearing mice as it suppressed both, GvHD and GvL (173). Although preclinical results for MEK/ERK inhibition are promising in aGvHD and GvL models and MEK inhibitors were not reported having limiting toxicities in mice, to the best of our knowledge no clinical trial was initiated yet to evaluate the efficacy of MEK inhibition GvHD patients.
AMP Kinase (AMPK)
GvHD following allo-HCT is predominantly driven by alloreactive donor T-cells which cause severe tissue damage (4). Novel therapeutic options aim to impair T-cell functions to reduce life-threatening GvHD without affecting GvL activity, including the idea to metabolically re-program T-cells after allo-HCT (175). Following transplantation, T-cells increase oxidative phosphorylation and fatty acid oxidation (176, 177). Since it is known that allogeneic effector T-cells require fatty acid oxidation (FAO) during GvHD, it was hypothesized that AMP kinase (AMPK) activation plays a major role in regulating T-cell activity after allo-HCT (175, 177).
Acute GvHD: Monlish and colleagues recently identified that alloreactive donor T-cells selectively increase AMPK activation during aGvHD after allo-HCT as they found elevated phosphorylation of AMPKα and the downstream molecule ACC in CD4+ and CD8+ T-cells isolated from mice on day 7 after allo-HCT (175). The investigators established AMPK double knockout (AMPK-dKO) mice lacking AMPKα1 and AMPKα2 in all peripheral T-cells. AMPK is a heterotrimeric molecule with the α subunit as kinase domain and the β/γ subunits being important for stability and substrate specificity (175, 178–180). Although T-cell development and in vitro proliferation in a MLR was not different between AMPK-dKO and WT T-cells, AMPK-deficient T-cells caused less severe GvHD after transplantation into lethally irradiated recipient mice. In two different GvHD models, the survival was prolonged and clinical GvHD scores were lower if mice received AMPK-dKO T-cells compared to WT T-cells (175). Comparable findings were described in a second study (181). In addition, the infiltration of AMPK-dKO T-cells into GvHD target organs was reduced. The anti-leukemia response was not impaired upon depletion of AMPK. Mechanistically, reduced GvHD was connected to reduced recovery and decreased expansion of AMPK-depleted T-cells after allo-HCT (175, 181). The fewer recovery was linked with increased apoptosis in mainly CD8+ T-cells, whereas the results are highly variable and the interpretation is therefore questionable. Although AMPK is a metabolic enzyme, depletion did not affect any investigated metabolic pathway, but rather affected other cell populations. Co-transplantation experiments revealed that AMPK-dKO T-cells stimulated an increase of WT Tregs. Given the importance of Tregs to reduce GvHD severity, increased Treg numbers due to accompanying AMPK-dKO cells were named as a major mechanism to suppress GvHD severity (175, 182–184). Although there are no AMPK inhibitors available yet, inhibition of AMPK in T-cells could serve as novel target for GvHD treatment; however, more detailed analysis is needed to better understand the role of AMPK after allo-HCT (175).
p38 Mitogen-Activated Protein Kinase (MAPK)
The p38 mitogen-activated kinase (MAPK) is a major control mechanism for cellular responses, proliferation and cytokine production (17, 185, 186). Different p38 MAPK isoforms are expressed in most tissues and cell types and are activated by extracellular stimulatory signals, including inflammatory cytokines, growth signals and stress signals (17, 185, 187, 188). Activation of the p38 MAPK signaling cascade results target gene expression, including inflammatory mediators and cytokines (17, 188–190). Of all isoforms, p38α was reported as major regulator in inflammatory responses and therapeutic blockade with the p38α-specific inhibitor VX-702 was applied in rheumatoid arthritis (RA) to reduce inflammatory signals (191).
Chronic GvHD: Since enhanced p38 MAPK phosphorylation was found in fibroblasts from systemic sclerosis patients and p38 MAPK blockade reduced elevated type I collagen expression, the signaling pathway was hypothesized playing a role in sclerosis pathogenesis (17, 192, 193). Systemic sclerosis has various clinical similarities with Scl-cGvHD and therapeutic blockade of p38 MAPK signaling was thought to reduce cGvHD severity (17, 116, 194–196). Phosphorylation of p38 MAPK (Thr180/Tyr182) was increased in the skin of Scl-cGvHD mice compared to syngeneic BMT mice (17). Application of VX-702 delayed the onset of skin fibrosis and alopecia development and improved skin GvHD scores. The dermal thickness, collagen area and levels of procollagen I αI were reduced upon VX-702 treatment. More detailed analysis by histology and flow cytometry revealed reduced infiltration of CD4+ and CD8+ T-cells, as well as lower numbers of myeloid cells and macrophages into the skin of Scl-cGvHD mice upon p38 MAPK inhibition. Of all analyzed cytokines, only IL-6 and IL-13 were significantly reduced upon treatment, whereas major drivers of Scl-cGvHD, like TGFβ and IFNγ, were unchanged (17). However, complete tissue transcription profile analysis might mask some minor changes in immune cells and it would have therefore been better to analyze effector and inflammatory cytokine production on single cells levels and to screen for cytokines in the serum. Nevertheless, consistent with histology data, tissue RNA analysis revealed decreased expression of COL1A2 and fibronectin 1 upon p38 MAPK inhibition (17). In summary, the study showed that p38 MAPK is activated in cGvHD and therapeutic blockade could be a novel therapeutic intervention.
Acute GvHD: Although p38 MAPK inhibition seems promising in cGvHD, the treatment is questionable since reduced p38α MAPK levels (heterozygous p38α-KO) in donor grafts were found to accelerate acute intestinal GvHD in mice (197). Surprisingly, and contrary to the previously described cGvHD study, loss of donor p38 reinforced GvHD severity and reduced the survival of the mice. Cytokine analysis confirmed higher TNFα levels in the gut in allo-p38α+/- grafted mice compared to the WT setting. Although p38α loss prolonged the survival of donor-derived intestinal intraepithelial lymphocytes in vitro and in vivo, donor lymphocyte expansion was decreased in the mesenteric lymph nodes upon p38α deficiency. Although the role of p38α-loss in the recipient compartment was not investigated, the study revealed a dichotomous effect of p38α in regulating inflammatory responses, cytokine expression, lymphocyte proliferation and intestinal GvHD after allo-HCT (197). Taken together with the before-mentioned study, the role of p38 MAPK in GvHD is still unclear and needs more detailed investigation, also comparing effects in chronic and acute GvHD as these could always be different (17, 197). Differences could be due to the models as the study on cGvHD applied an inhibitor, whereas the aGvHD study investigated the role of p38 MAPK using a genetic approach with p38-deficient donor cells.
Aurora Kinase A
Acute GvHD: TCR activation and co-stimulation via CD28 stimulates mTOR and aurora kinase family signaling in T-cells, thereby activating substrates needed for T-cell proliferation (198). Transcriptomic analysis found increased expression of aurora kinase A in aGvHD patients and mice after allo-HCT. Inhibition of aurora kinase A prolonged survival and reduced GvHD scores in mice, however the animals could not be fully rescued (199). It was hypothesized that inhibition of aurora kinase A and JAK2 could be combined, as JAK2 activation by inflammatory stimuli leads to STAT3 activation and effector cytokine production (200). Betts et al. treated human MLRs with the JAK2 inhibitor TG101348, aurora kinase A inhibitor alisertib or a combination. T-cell proliferation was synergistically suppressed by the combination. Compounds targeting both kinases, had similar effects. Both kinase inhibitors reduced CD4+ and CD8+ T-cell activation in vitro, whereas the combination had the strongest impact. Also, T-cells produced less IL-17 and IFNγ upon kinase blockade. Interestingly, the induction of Tregs was significantly reduced upon kinase inhibition, but Tregs had potent inhibitory functions, mainly based on upregulation of surface CD39. Dual kinase inhibition caused higher ATP consumption, confirming the functionality of CD39 upregulation. Based on in vitro findings, the investigators applied JAK2 and aurora kinase A inhibitors in a xenogeneic aGvHD model. The survival of the recipients was significantly increased upon combination treatment, accompanied by lower GvHD scores. A novel dual inhibitor showed even stronger effects without impairing GvL activity. Based on these findings, more research is needed to further elucidate the role of aurora kinases in GvHD and their potency as novel targetable molecule (103).
Conclusions and Outlook
In summary, the presented pre-clinical and clinical investigations reveal that kinase inhibition offers a huge variety of novel approaches to target both, acute and chronic GvHD. Since GvHD involves a vast number of pathways and signaling cascades for immune cell activation, proliferation and effector cytokine production, as well as in inflammatory signaling and fibrosis, the disease is hard to treat with only a single compound. Moreover, acute and chronic GvHD are completely different diseases and involve distinct pathways, making the treatment even more complicated. However, the involvement of different pathways is also a chance, presenting a variety of kinases as potentially targetable candidates. The great number of studies indicates how intensively researched kinases, the major signal transducers in immune cell signaling, are in the context of GvHD after allo-HCT. Although some compounds are already far in clinical trials, many questions remain unanswered, making deeper research necessary to unravel the potential of kinase inhibition in GvHD. Preclinical and clinical analyses revealed that treatment with single compounds has therapeutic limitations, as GvHD is mediated by a variety of pathways and not only by a single activated molecule. Novel therapeutic strategies should involve the combination of kinase inhibitors with other therapeutic interventions, as it is already investigated for JAK1 and JAK2 inhibitors with ECP. Moreover, different kinase inhibitors could be combined to potentiate the efficacy of the individual kinase inhibitors to enhance treatment success. JAK1 and/or JAK2 inhibitors, leading to reduced inflammatory cytokine production and decreased APC activation, could be combined with ROCK2 inhibition which potently blocks fibrosis and TFH formation in cGvHD. Also, ROCK2 blockade results in higher Treg numbers. Combinations of Syk, PI3K/mTOR and ITK/BTK could be beneficial as these kinases mediate early B- and T-cell activation. It might also be possible to apply kinase inhibitors sequentially to first hit a target being activated in aGvHD and target a second kinase to reduce the risk of aGvHD transforming into chronic GvHD. However, these strategies are still speculative and combination therapies should be carefully tested in preclinical models. Since many kinases are not only involved in disease but also in physiological signaling processes, the application and combination of inhibitors has the risk of potential side effects. Kinases such as MEK and MAPK are active in most cells and tissues and severe side effects are likely upon inhibition. Treatment-related adverse events have to be considered and highly specific molecules need to be designed to reduce off-target effects. So far, some kinases are only investigated pre-clinically, but should be tested clinically if effects are seen. After dose-finding studies, the inhibitors might first be investigated in SR-GvHD. Based on performance, the inhibitors might also be evaluated as first-line or prophylaxis therapies for patients after allo-HCT. However, transformation of these compounds into clinical trials is speculative as they first need to be critically evaluated in pre-clinical model. Taken together, the approval and clinical application of some kinase inhibitors, including the JAK1/2 inhibitor Ruxolitinib and the ROCK2 inhibitor belumosudil, is promising to better control acute and chronic GvHD after allo-HCT, thereby making allo-HCT available for more patients with severe hematological malignancies.
Author Contributions
LMB and RZ developed the overall concept of this article. LMB collected and reviewed literature, discussed the studies and wrote the manuscript. RZ helped to write the manuscript and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This article was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – SFB-1479 – Project ID: 441891347, SFB TRR167, SFB850 (to RZ), by the Germany’s Excellence Strategy (CIBSS – EXC-2189 – Project ID 390939984 to RZ) and by ERC Consolidator grant (681012 GvHDCure to RZ). The article processing charge was funded by the Baden-Wuerttemberg Ministry of Science, Research and Art and the University of Freiburg in the funding program Open Access Publishing.
Conflict of Interest
RZ received honoraria from Novartis, Incyte and Mallinckrodt.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Eileen Haring for help with figure design. Figures were created with BioRender.com. We apologize to all authors whose work could not be cited due to space and citation restrictions.
References
1. Thomas ED, Lochte HL Jr, Lu WC, Ferrebee JW. Intravenous Infusion of Bone Marrow in Patients Receiving Radiation and Chemotherapy. N Engl J Med (1957) 257(11):491–6. doi: 10.1056/NEJM195709122571102
2. D'Souza A, Fretham C, Lee SJ, Arora M, Brunner J, Chhabra S, et al. Current Use of and Trends in Hematopoietic Cell Transplantation in the United States. Biol Blood Marrow Transplant (2020) 26(8):e177–82. doi: 10.1016/j.bbmt.2020.04.013
3. Kolb HJ. Graft-Versus-Leukemia Effects of Transplantation and Donor Lymphocytes. Blood (2008) 112(12):4371–83. doi: 10.1182/blood-2008-03-077974
4. Zeiser R, Blazar BR. Acute Graft-Versus-Host Disease - Biologic Process, Prevention, and Therapy. N Engl J Med (2017) 377(22):2167–79. doi: 10.1056/NEJMra1609337
5. Ferrara JL, Levine JE, Reddy P, Holler E. Graft-Versus-Host Disease. Lancet (2009) 373(9674):1550–61. doi: 10.1016/S0140-6736(09)60237-3
6. Haring E, Uhl FM, Andrieux G, Proietti M, Bulashevska A, Sauer B, et al. Bile Acids Regulate Intestinal Antigen Presentation and Reduce Graft-Versus-Host Disease Without Impairing the Graft-Versus-Leukemia Effect. Haematologica (2021) 106(8):2131–46. doi: 10.3324/haematol.2019.242990
7. Herrero-Sanchez MC, Rodriguez-Serrano C, Almeida J, San Segundo L, Inoges S, Santos-Briz A, et al. Targeting of PI3K/AKT/mTOR Pathway to Inhibit T Cell Activation and Prevent Graft-Versus-Host Disease Development. J Hematol Oncol (2016) 9(1):113. doi: 10.1186/s13045-016-0343-5
8. Zeiser R, Sarantopoulos S, Blazar BR. B-Cell Targeting in Chronic Graft-Versus-Host Disease. Blood (2018) 131(13):1399–405. doi: 10.1182/blood-2017-11-784017
9. Hulsdunker J, Thomas OS, Haring E, Unger S, Gonzalo Nunez N, Tugues S, et al. Immunization Against Poly-N-Acetylglucosamine Reduces Neutrophil Activation and GVHD While Sparing Microbial Diversity. Proc Natl Acad Sci USA (2019) 116(41):20700–6. doi: 10.1073/pnas.1908549116
10. Hulsdunker J, Ottmuller KJ, Neeff HP, Koyama M, Gao Z, Thomas OS, et al. Neutrophils Provide Cellular Communication Between Ileum and Mesenteric Lymph Nodes at Graft-Versus-Host Disease Onset. Blood (2018) 131(16):1858–69. doi: 10.1182/blood-2017-10-812891
11. Zeiser R, Blazar BR. Pathophysiology of Chronic Graft-Versus-Host Disease and Therapeutic Targets. N Engl J Med (2017) 377(26):2565–79. doi: 10.1056/NEJMra1703472
12. Schroeder MA, Choi J, Staser K, DiPersio JF. The Role of Janus Kinase Signaling in Graft-Versus-Host Disease and Graft Versus Leukemia. Biol Blood Marrow Transplant (2018) 24(6):1125–34. doi: 10.1016/j.bbmt.2017.12.797
13. Fowler DH, Pavletic SZ. Syk and Tired of Current Chronic GVHD Therapies. Blood (2015) 125(26):3974–5. doi: 10.1182/blood-2015-05-640672
14. Schutt SD, Fu J, Nguyen H, Bastian D, Heinrichs J, Wu Y, et al. Inhibition of BTK and ITK With Ibrutinib Is Effective in the Prevention of Chronic Graft-Versus-Host Disease in Mice. PloS One (2015) 10(9):e0137641. doi: 10.1371/journal.pone.0137641
15. Hechinger AK, Smith BA, Flynn R, Hanke K, McDonald-Hyman C, Taylor PA, et al. Therapeutic Activity of Multiple Common Gamma-Chain Cytokine Inhibition in Acute and Chronic GVHD. Blood (2015) 125(3):570–80. doi: 10.1182/blood-2014-06-581793
16. Bhattacharyya S, Ratajczak CK, Vogt SK, Kelley C, Colonna M, Schreiber RD, et al. TAK1 Targeting by Glucocorticoids Determines JNK and IkappaB Regulation in Toll-Like Receptor-Stimulated Macrophages. Blood (2010) 115(10):1921–31. doi: 10.1182/blood-2009-06-224782
17. Matsushita T, Date M, Kano M, Mizumaki K, Tennichi M, Kobayashi T, et al. Blockade of P38 Mitogen-Activated Protein Kinase Inhibits Murine Sclerodermatous Chronic Graft-Versus-Host Disease. Am J Pathol (2017) 187(4):841–50. doi: 10.1016/j.ajpath.2016.12.016
18. Heine A, Held SA, Daecke SN, Wallner S, Yajnanarayana SP, Kurts C, et al. The JAK-Inhibitor Ruxolitinib Impairs Dendritic Cell Function In Vitro and In Vivo. Blood (2013) 122(7):1192–202. doi: 10.1182/blood-2013-03-484642
19. Mohamed AJ, Yu L, Backesjo CM, Vargas L, Faryal R, Aints A, et al. Bruton's Tyrosine Kinase (Btk): Function, Regulation, and Transformation With Special Emphasis on the PH Domain. Immunol Rev (2009) 228(1):58–73. doi: 10.1111/j.1600-065X.2008.00741.x
20. Jaglowski SM, Blazar BR. How Ibrutinib, a B-Cell Malignancy Drug, Became an FDA-Approved Second-Line Therapy for Steroid-Resistant Chronic GVHD. Blood Adv (2018) 2(15):2012–9. doi: 10.1182/bloodadvances.2018013060
21. Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an Irreversible Molecular Inhibitor of ITK Driving a Th1-Selective Pressure in T Lymphocytes. Blood (2013) 122(15):2539–49. doi: 10.1182/blood-2013-06-507947
22. Laplante M, Sabatini DM. mTOR Signaling in Growth Control and Disease. Cell (2012) 149(2):274–93. doi: 10.1016/j.cell.2012.03.017
23. Biswas PS, Gupta S, Chang E, Song L, Stirzaker RA, Liao JK, et al. Phosphorylation of IRF4 by ROCK2 Regulates IL-17 and IL-21 Production and the Development of Autoimmunity in Mice. J Clin Invest (2010) 120(9):3280–95. doi: 10.1172/JCI42856
24. Appleman LJ, van Puijenbroek AA, Shu KM, Nadler LM, Boussiotis VA. CD28 Costimulation Mediates Down-Regulation of P27kip1 and Cell Cycle Progression by Activation of the PI3K/PKB Signaling Pathway in Primary Human T Cells. J Immunol (2002) 168(6):2729–36. doi: 10.4049/jimmunol.168.6.2729
25. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The Kinase mTOR Regulates the Differentiation of Helper T Cells Through the Selective Activation of Signaling by Mtorc1 and Mtorc2. Nat Immunol (2011) 12(4):295–303. doi: 10.1038/ni.2005
26. Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, et al. Mammalian Target of Rapamycin Protein Complex 2 Regulates Differentiation of Th1 and Th2 Cell Subsets via Distinct Signaling Pathways. Immunity (2010) 32(6):743–53. doi: 10.1016/j.immuni.2010.06.002
27. Leonhardt F, Zirlik K, Buchner M, Prinz G, Hechinger AK, Gerlach UV, et al. Spleen Tyrosine Kinase (Syk) Is a Potent Target for GvHD Prevention at Different Cellular Levels. Leukemia (2012) 26(7):1617–29. doi: 10.1038/leu.2012.10
28. Hwang JR, Byeon Y, Kim D, Park SG. Recent Insights of T Cell Receptor-Mediated Signaling Pathways for T Cell Activation and Development. Exp Mol Med (2020) 52(5):750–61. doi: 10.1038/s12276-020-0435-8
29. Tang B, Zhou J, Park JE, Cullins D, Yi AK, Kang AH, et al. T Cell Receptor Signaling Induced by an Analog Peptide of Type II Collagen Requires Activation of Syk. Clin Immunol (2009) 133(1):145–53. doi: 10.1016/j.clim.2009.06.006
30. Jagasia M, Lazaryan A, Bachier CR, Salhotra A, Weisdorf DJ, Zoghi B, et al. ROCK2 Inhibition With Belumosudil (KD025) for the Treatment of Chronic Graft-Versus-Host Disease. J Clin Oncol (2021) 39(17):1888–98. doi: 10.1200/JCO.20.02754
31. Chen Z, Laurence A, O'Shea JJ. Signal Transduction Pathways and Transcriptional Regulation in the Control of Th17 Differentiation. Semin Immunol (2007) 19(6):400–8. doi: 10.1016/j.smim.2007.10.015
32. Zanin-Zhorov A, Weiss JM, Nyuydzefe MS, Chen W, Scher JU, Mo R, et al. Selective Oral ROCK2 Inhibitor Down-Regulates IL-21 and IL-17 Secretion in Human T Cells via STAT3-Dependent Mechanism. Proc Natl Acad Sci USA (2014) 111(47):16814–9. doi: 10.1073/pnas.1414189111
33. Feske S. Calcium Signalling in Lymphocyte Activation and Disease. Nat Rev Immunol (2007) 7(9):690–702. doi: 10.1038/nri2152
34. Sauer K, Cooke MP. Regulation of Immune Cell Development Through Soluble Inositol-1,3,4,5-Tetrakisphosphate. Nat Rev Immunol (2010) 10(4):257–71. doi: 10.1038/nri2745
35. Pouillon V, Hascakova-Bartova R, Pajak B, Adam E, Bex F, Dewaste V, et al. Inositol 1,3,4,5-Tetrakisphosphate Is Essential for T Lymphocyte Development. Nat Immunol (2003) 4(11):1136–43. doi: 10.1038/ni980
36. Saidu NEB, Bonini C, Dickinson A, Grce M, Inngjerdingen M, Koehl U, et al. New Approaches for the Treatment of Chronic Graft-Versus-Host Disease: Current Status and Future Directions. Front Immunol (2020) 11:578314. doi: 10.3389/fimmu.2020.578314
37. Hernandez-Gea V, Friedman SL. Pathogenesis of Liver Fibrosis. Annu Rev Pathol (2011) 6:425–56. doi: 10.1146/annurev-pathol-011110-130246
38. Akhmetshina A, Venalis P, Dees C, Busch N, Zwerina J, Schett G, et al. Treatment With Imatinib Prevents Fibrosis in Different Preclinical Models of Systemic Sclerosis and Induces Regression of Established Fibrosis. Arthritis Rheum (2009) 60(1):219–24. doi: 10.1002/art.24186
39. Schindler C, Levy DE, Decker T. JAK-STAT Signaling: From Interferons to Cytokines. J Biol Chem (2007) 282(28):20059–63. doi: 10.1074/jbc.R700016200
40. Stickel N, Hanke K, Marschner D, Prinz G, Kohler M, Melchinger W, et al. MicroRNA-146a Reduces MHC-II Expression via Targeting JAK/STAT Signaling in Dendritic Cells After Stem Cell Transplantation. Leukemia (2017) 31(12):2732–41. doi: 10.1038/leu.2017.137
41. Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and Efficacy of INCB018424, a JAK1 and JAK2 Inhibitor, in Myelofibrosis. N Engl J Med (2010) 363(12):1117–27. doi: 10.1056/NEJMoa1002028
42. Schwab L, Goroncy L, Palaniyandi S, Gautam S, Triantafyllopoulou A, Mocsai A, et al. Neutrophil Granulocytes Recruited Upon Translocation of Intestinal Bacteria Enhance Graft-Versus-Host Disease via Tissue Damage. Nat Med (2014) 20(6):648–54. doi: 10.1038/nm.3517
43. Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, et al. Activity of Therapeutic JAK 1/2 Blockade in Graft-Versus-Host Disease. Blood (2014) 123(24):3832–42. doi: 10.1182/blood-2013-12-543736
44. Akada H, Akada S, Hutchison RE, Sakamoto K, Wagner KU, Mohi G. Critical Role of Jak2 in the Maintenance and Function of Adult Hematopoietic Stem Cells. Stem Cells (2014) 32(7):1878–89. doi: 10.1002/stem.1711
45. Schroeder MA, Khoury HJ, Jagasia M, Ali H, Schiller GJ, Staser K, et al. A Phase 1 Trial of Itacitinib, a Selective JAK1 Inhibitor, in Patients With Acute Graft-Versus-Host Disease. Blood Adv (2020) 4(8):1656–69. doi: 10.1182/bloodadvances.2019001043
46. Covington M, He X, Scuron M, Li J, Collins R, Juvekar A, et al. Preclinical Characterization of Itacitinib (INCB039110), a Novel Selective Inhibitor of JAK1, for the Treatment of Inflammatory Diseases. Eur J Pharmacol (2020) 885:173505. doi: 10.1016/j.ejphar.2020.173505
47. Courtois J, Ritacco C, Dubois S, Canti L, Vandenhove B, Seidel L, et al. Itacitinib Prevents Xenogeneic GVHD in Humanized Mice. Bone Marrow Transplant (2021) 56(11):2672–81. doi: 10.1038/s41409-021-01363-1
48. Choi J, Cooper ML, Staser K, Ashami K, Vij KR, Wang B, et al. Baricitinib-Induced Blockade of Interferon Gamma Receptor and Interleukin-6 Receptor for the Prevention and Treatment of Graft-Versus-Host Disease. Leukemia (2018) 32(11):2483–94. doi: 10.1038/s41375-018-0123-z
49. Kim S, Ashami K, Lim S, Staser K, Vij K, Santhanam S, et al. Baricitinib Prevents GvHD by Increasing Tregs via JAK3 and Treats Established GvHD by Promoting Intestinal Tissue Repair via EGFR. Leukemia (2021). doi: 10.1038/s41375-021-01360-9
50. Zeiser R, Burchert A, Lengerke C, Verbeek M, Maas-Bauer K, Metzelder SK, et al. Ruxolitinib in Corticosteroid-Refractory Graft-Versus-Host Disease After Allogeneic Stem Cell Transplantation: A Multicenter Survey. Leukemia (2015) 29(10):2062–8. doi: 10.1038/leu.2015.212
51. Jagasia M, Perales MA, Schroeder MA, Ali H, Shah NN, Chen YB, et al. Ruxolitinib for the Treatment of Steroid-Refractory Acute GVHD (REACH1): A Multicenter, Open-Label Phase 2 Trial. Blood (2020) 135(20):1739–49. doi: 10.1182/blood.2020004823
52. Zeiser R, von Bubnoff N, Butler J, Mohty M, Niederwieser D, Or R, et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-Versus-Host Disease. N Engl J Med (2020) 382(19):1800–10. doi: 10.1056/NEJMoa1917635
53. Zeiser R, Polverelli N, Ram R, Hashmi SK, Chakraverty R, Middeke JM, et al. Ruxolitinib for Glucocorticoid-Refractory Chronic Graft-Versus-Host Disease. N Engl J Med (2021) 385(3):228–38. doi: 10.1056/NEJMoa2033122
54. Blazar BR, Murphy WJ, Abedi M. Advances in Graft-Versus-Host Disease Biology and Therapy. Nat Rev Immunol (2012) 12(6):443–58. doi: 10.1038/nri3212
55. Axt L, Naumann A, Toennies J, Haen SP, Vogel W, Schneidawind D, et al. Retrospective Single Center Analysis of Outcome, Risk Factors and Therapy in Steroid Refractory Graft-Versus-Host Disease After Allogeneic Hematopoietic Cell Transplantation. Bone Marrow Transplant (2019) 54(11):1805–14. doi: 10.1038/s41409-019-0544-y
56. Penack O, Marchetti M, Ruutu T, Aljurf M, Bacigalupo A, Bonifazi F, et al. Prophylaxis and Management of Graft Versus Host Disease After Stem-Cell Transplantation for Haematological Malignancies: Updated Consensus Recommendations of the European Society for Blood and Marrow Transplantation. Lancet Haematol (2020) 7(2):e157–67. doi: 10.1016/S2352-3026(19)30256-X
57. Dignan FL, Amrolia P, Clark A, Cornish J, Jackson G, Mahendra P, et al. Haemato-Oncology Task Force of British Committee for Standards in, B. British Society for and T. Marrow: Diagnosis and Management of Chronic Graft-Versus-Host Disease. Br J Haematol (2012) 158(1):46–61. doi: 10.1111/j.1365-2141.2012.09128.x
58. Choi J, Ziga ED, Ritchey J, Collins L, Prior JL, Cooper ML, et al. IFNgammaR Signaling Mediates Alloreactive T-Cell Trafficking and GVHD. Blood (2012) 120(19):4093–103. doi: 10.1182/blood-2012-01-403196
59. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N Engl J Med (2012) 366(9):799–807. doi: 10.1056/NEJMoa1110557
60. Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK Inhibition With Ruxolitinib Versus Best Available Therapy for Myelofibrosis. N Engl J Med (2012) 366(9):787–98. doi: 10.1056/NEJMoa1110556
61. Maas-Bauer K, Kiote-Schmidt C, Bertz H, Apostolova P, Wasch R, Ihorst G, et al. Ruxolitinib-ECP Combination Treatment for Refractory Severe Chronic Graft-Versus-Host Disease. Bone Marrow Transplant (2020) 56(4):909–16. doi: 10.1038/s41409-020-01122-8
62. Tybulewicz VL, Henderson RB. Rho Family GTPases and Their Regulators in Lymphocytes. Nat Rev Immunol (2009) 9(9):630–44. doi: 10.1038/nri2606
63. Riento K, Ridley AJ. Rocks: Multifunctional Kinases in Cell Behaviour. Nat Rev Mol Cell Biol (2003) 4(6):446–56. doi: 10.1038/nrm1128
64. Biswas PS, Bhagat G, Pernis AB. IRF4 and its Regulators: Evolving Insights Into the Pathogenesis of Inflammatory Arthritis? Immunol Rev (2010) 233(1):79–96. doi: 10.1111/j.0105-2896.2009.00864.x
65. Ivanov II, Zhou L, Littman DR. Transcriptional Regulation of Th17 Cell Differentiation. Semin Immunol (2007) 19(6):409–17. doi: 10.1016/j.smim.2007.10.011
66. Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, Panoskaltsis-Mortari A, et al. Targeted Rho-Associated Kinase 2 Inhibition Suppresses Murine and Human Chronic GVHD Through a Stat3-Dependent Mechanism. Blood (2016) 127(17):2144–54. doi: 10.1182/blood-2015-10-678706
67. Riegel C, Boeld TJ, Doser K, Huber E, Hoffmann P, Edinger M. Efficient Treatment of Murine Acute GvHD by In Vitro Expanded Donor Regulatory T Cells. Leukemia (2020) 34(3):895–908. doi: 10.1038/s41375-019-0625-3
68. Srinivasan M, Flynn R, Price A, Ranger A, Browning JL, Taylor PA, et al. Donor B-Cell Alloantibody Deposition and Germinal Center Formation are Required for the Development of Murine Chronic GVHD and Bronchiolitis Obliterans. Blood (2012) 119(6):1570–80. doi: 10.1182/blood-2011-07-364414
69. Flynn R, Allen JL, Luznik L, MacDonald KP, Paz K, Alexander KA, et al. Targeting Syk-Activated B Cells in Murine and Human Chronic Graft-Versus-Host Disease. Blood (2015) 125(26):4085–94. doi: 10.1182/blood-2014-08-595470
70. Dubovsky JA, Flynn R, Du J, Harrington BK, Zhong Y, Kaffenberger B, et al. Ibrutinib Treatment Ameliorates Murine Chronic Graft-Versus-Host Disease. J Clin Invest (2014) 124(11):4867–76. doi: 10.1172/JCI75328
71. Flynn R, Du J, Veenstra RG, Reichenbach DK, Panoskaltsis-Mortari A, Taylor PA, et al. Increased T Follicular Helper Cells and Germinal Center B Cells are Required for cGVHD and Bronchiolitis Obliterans. Blood (2014) 123(25):3988–98. doi: 10.1182/blood-2014-03-562231
72. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
73. Cutler CS, Lee SJ, Arai S, Rotta M, Zoghi B, Lazaryan A, et al. Belumosudil for Chronic Graft-Versus-Host Disease (cGVHD) After 2 or More Prior Lines of Therapy: The ROCKstar Study. Blood (2021). doi: 10.1182/blood.2021012021
74. Waickman AT, Powell JD. Mammalian Target of Rapamycin Integrates Diverse Inputs to Guide the Outcome of Antigen Recognition in T Cells. J Immunol (2012) 188(10):4721–9. doi: 10.4049/jimmunol.1103143
75. Wu LX, La Rose J, Chen L, Neale C, Mak T, Okkenhaug K, et al. CD28 Regulates the Translation of Bcl-xL via the Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Pathway. J Immunol (2005) 174(1):180–94. doi: 10.4049/jimmunol.174.1.180
76. Dufour M, Dormond-Meuwly A, Demartines N, Dormond O. Targeting the Mammalian Target of Rapamycin (mTOR) in Cancer Therapy: Lessons From Past and Future Perspectives. Cancers (Basel) (2011) 3(2):2478–500. doi: 10.3390/cancers3022478
77. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a Novel Binding Partner of mTOR, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway That Regulates the Cytoskeleton. Curr Biol (2004) 14(14):1296–302. doi: 10.1016/j.cub.2004.06.054
78. Fuhler GM, Tyl MR, Olthof SG, Lyndsay Drayer A, Blom N, Vellenga E. Distinct Roles of the mTOR Components Rictor and Raptor in MO7e Megakaryocytic Cells. Eur J Haematol (2009) 83(3):235–45. doi: 10.1111/j.1600-0609.2009.01263.x
79. Nguyen HD, Chatterjee S, Haarberg KM, Wu Y, Bastian D, Heinrichs J, et al. Metabolic Reprogramming of Alloantigen-Activated T Cells After Hematopoietic Cell Transplantation. J Clin Invest (2016) 126(4):1337–52. doi: 10.1172/JCI82587
80. Blazar BR, Taylor PA, Panoskaltsis-Mortari A, Vallera DA. Rapamycin Inhibits the Generation of Graft-Versus-Host Disease- and Graft-Versus-Leukemia-Causing T Cells by Interfering With the Production of Th1 or Th1 Cytotoxic Cytokines. J Immunol (1998) 160(11):5355–65 https://pubmed.ncbi.nlm.nih.gov/9605135/.
81. Abouelnasr A, Roy J, Cohen S, Kiss T, Lachance S. Defining the Role of Sirolimus in the Management of Graft-Versus-Host Disease: From Prophylaxis to Treatment. Biol Blood Marrow Transplant (2013) 19(1):12–21. doi: 10.1016/j.bbmt.2012.06.020
82. Zeiser R, Nguyen VH, Beilhack A, Buess M, Schulz S, Baker J, et al. Inhibition of CD4+CD25+ Regulatory T-Cell Function by Calcineurin-Dependent Interleukin-2 Production. Blood (2006) 108(1):390–9. doi: 10.1182/blood-2006-01-0329
83. Segundo DS, Ruiz JC, Izquierdo M, Fernandez-Fresnedo G, Gomez-Alamillo C, Merino R, et al. Calcineurin Inhibitors, But Not Rapamycin, Reduce Percentages of CD4+CD25+FOXP3+ Regulatory T Cells in Renal Transplant Recipients. Transplantation (2006) 82(4):550–7. doi: 10.1097/01.tp.0000229473.95202.50
84. Noris M, Casiraghi F, Todeschini M, Cravedi P, Cugini D, Monteferrante G, et al. Regulatory T Cells and T Cell Depletion: Role of Immunosuppressive Drugs. J Am Soc Nephrol (2007) 18(3):1007–18. doi: 10.1681/ASN.2006101143
85. Wang H, Zhao L, Sun Z, Sun L, Zhang B, Zhao Y. A Potential Side Effect of Cyclosporin A: Inhibition of CD4(+)CD25(+) Regulatory T Cells in Mice. Transplantation (2006) 82(11):1484–92. doi: 10.1097/01.tp.0000246312.89689.17
86. Coenen JJ, Koenen HJ, van Rijssen E, Kasran A, Boon L, Hilbrands LB, et al. Rapamycin, Not Cyclosporine, Permits Thymic Generation and Peripheral Preservation of CD4+ CD25+ FoxP3+ T Cells. Bone Marrow Transplant (2007) 39(9):537–45. doi: 10.1038/sj.bmt.1705628
87. Zeiser R, Leveson-Gower DB, Zambricki EA, Kambham N, Beilhack A, Loh J, et al. Differential Impact of Mammalian Target of Rapamycin Inhibition on CD4+CD25+Foxp3+ Regulatory T Cells Compared With Conventional CD4+ T Cells. Blood (2008) 111(1):453–62. doi: 10.1182/blood-2007-06-094482
88. Taub DD, Murphy WJ, Asai O, Fenton RG, Peltz G, Key ML, et al. Induction of Alloantigen-Specific T Cell Tolerance Through the Treatment of Human T Lymphocytes With Wortmannin. J Immunol (1997) 158(6):2745–55.
89. Castor MG, Rezende BM, Bernardes PT, Vieira AT, Vieira EL, Arantes RM, et al. PI3Kgamma Controls Leukocyte Recruitment, Tissue Injury, and Lethality in a Model of Graft-Versus-Host Disease in Mice. J Leukoc Biol (2011) 89(6):955–64. doi: 10.1189/jlb.0810464
90. Scheurer J, Reisser T, Leithauser F, Messmann JJ, Holzmann K, Debatin KM, et al. Rapamycin-Based Graft-Versus-Host Disease Prophylaxis Increases the Immunosuppressivity of Myeloid-Derived Suppressor Cells Without Affecting T Cells and Anti-Tumor Cytotoxicity. Clin Exp Immunol (2020) 202(3):407–22. doi: 10.1111/cei.13496
91. Kim D, Park G, Huuhtanen J, Lundgren S, Khajuria RK, Hurtado AM, et al. Somatic mTOR Mutation in Clonally Expanded T Lymphocytes Associated With Chronic Graft Versus Host Disease. Nat Commun (2020) 11(1):2246. doi: 10.1038/s41467-020-16115-w
92. Sugiyama H, Maeda Y, Nishimori H, Yamasuji Y, Matsuoka K, Fujii N, et al. Mammalian Target of Rapamycin Inhibitors Permit Regulatory T Cell Reconstitution and Inhibit Experimental Chronic Graft-Versus-Host Disease. Biol Blood Marrow Transplant (2014) 20(2):183–91. doi: 10.1016/j.bbmt.2013.11.018
93. Benito AI, Furlong T, Martin PJ, Anasetti C, Appelbaum FR, Doney K, et al. Sirolimus (Rapamycin) for the Treatment of Steroid-Refractory Acute Graft-Versus-Host Disease. Transplantation (2001) 72(12):1924–9. doi: 10.1097/00007890-200112270-00010
94. Antin JH, Kim HT, Cutler C, Ho VT, Lee SJ, Miklos DB, et al. Sirolimus, Tacrolimus, and Low-Dose Methotrexate for Graft-Versus-Host Disease Prophylaxis in Mismatched Related Donor or Unrelated Donor Transplantation. Blood (2003) 102(5):1601–5. doi: 10.1182/blood-2003-02-0489
95. Zeiser R, Marks R, Bertz H, Finke J. Immunopathogenesis of Acute Graft-Versus-Host Disease: Implications for Novel Preventive and Therapeutic Strategies. Ann Hematol (2004) 83(9):551–65. doi: 10.1007/s00277-004-0890-7
96. Pidala J, Kim J, Jim H, Kharfan-Dabaja MA, Nishihori T, Fernandez HF, et al. A Randomized Phase II Study to Evaluate Tacrolimus in Combination With Sirolimus or Methotrexate After Allogeneic Hematopoietic Cell Transplantation. Haematologica (2012) 97(12):1882–9. doi: 10.3324/haematol.2012.067140
97. Armand P, Gannamaneni S, Kim HT, Cutler CS, Ho VT, Koreth J, et al. Improved Survival in Lymphoma Patients Receiving Sirolimus for Graft-Versus-Host Disease Prophylaxis After Allogeneic Hematopoietic Stem-Cell Transplantation With Reduced-Intensity Conditioning. J Clin Oncol (2008) 26(35):5767–74. doi: 10.1200/JCO.2008.17.7279
98. Armand P, Kim HT, Sainvil MM, Lange PB, Giardino AA, Bachanova V, et al. The Addition of Sirolimus to the Graft-Versus-Host Disease Prophylaxis Regimen in Reduced Intensity Allogeneic Stem Cell Transplantation for Lymphoma: A Multicentre Randomized Trial. Br J Haematol (2016) 173(1):96–104. doi: 10.1111/bjh.13931
99. Sandmaier BM, Kornblit B, Storer BE, Olesen G, Maris MB, Langston AA, et al. Addition of Sirolimus to Standard Cyclosporine Plus Mycophenolate Mofetil-Based Graft-Versus-Host Disease Prophylaxis for Patients After Unrelated Non-Myeloablative Haemopoietic Stem Cell Transplantation: A Multicentre, Randomised, Phase 3 Trial. Lancet Haematol (2019) 6(8):e409–18. doi: 10.1016/S2352-3026(19)30088-2
100. Pidala J, Hamadani M, Dawson P, Martens M, Alousi AM, Jagasia M, et al. Randomized Multicenter Trial of Sirolimus vs Prednisone as Initial Therapy for Standard-Risk Acute GVHD: The BMT CTN 1501 Trial. Blood (2020) 135(2):97–107. doi: 10.1182/blood.2019003125
101. Carpenter PA, Logan BR, Lee SJ, Weisdorf DJ, Johnston L, Costa LJ, et al. A Phase II/III Randomized, Multicenter Trial of Prednisone/Sirolimus Versus Prednisone/ Sirolimus/Calcineurin Inhibitor for the Treatment of Chronic Graft-Versus-Host Disease: BMT CTN 0801. Haematologica (2018) 103(11):1915–24. doi: 10.3324/haematol.2018.195123
102. Pidala J, Walton K, Elmariah H, Kim J, Mishra A, Bejanyan N, et al. Pacritinib Combined With Sirolimus and Low-Dose Tacrolimus for GVHD Prevention After Allogeneic Hematopoietic Cell Transplantation: Preclinical and Phase I Trial Results. Clin Cancer Res (2021) 27(10):2712–22. doi: 10.1158/1078-0432.CCR-20-4725
103. Betts BC, Veerapathran A, Pidala J, Yang H, Horna P, Walton K, et al. Targeting Aurora Kinase A and JAK2 Prevents GVHD While Maintaining Treg and Antitumor CTL Function. Sci Transl Med (2017) 9(372):eaai8269. doi: 10.1126/scitranslmed.aai8269
104. Zhang C, Todorov I, Zhang Z, Liu Y, Kandeel F, Forman S, et al. Donor CD4+ T and B Cells in Transplants Induce Chronic Graft-Versus-Host Disease With Autoimmune Manifestations. Blood (2006) 107(7):2993–3001. doi: 10.1182/blood-2005-09-3623
105. Kapur R, Ebeling S, Hagenbeek A. B-Cell Involvement in Chronic Graft-Versus-Host Disease. Haematologica (2008) 93(11):1702–11. doi: 10.3324/haematol.13311
106. Zhao D, Young JS, Chen YH, Shen E, Yi T, Todorov I, et al. Alloimmune Response Results in Expansion of Autoreactive Donor CD4+ T Cells in Transplants That can Mediate Chronic Graft-Versus-Host Disease. J Immunol (2011) 186(2):856–68. doi: 10.4049/jimmunol.1002195
107. Allen JL, Tata PV, Fore MS, Wooten J, Rudra S, Deal AM, et al. Increased BCR Responsiveness in B Cells From Patients With Chronic GVHD. Blood (2014) 123(13):2108–15. doi: 10.1182/blood-2013-10-533562
108. Satterthwaite AB, Witte ON. The Role of Bruton's Tyrosine Kinase in B-Cell Development and Function: A Genetic Perspective. Immunol Rev (2000) 175:120–7. doi: 10.1111/j.1600-065X.2000.imr017504.x
109. Gomez-Rodriguez J, Kraus ZJ, Schwartzberg PL. Tec Family Kinases Itk and Rlk / Txk in T Lymphocytes: Cross-Regulation of Cytokine Production and T-Cell Fates. FEBS J (2011) 278(12):1980–9. doi: 10.1111/j.1742-4658.2011.08072.x
110. August A, Gibson S, Kawakami Y, Kawakami T, Mills GB, Dupont B. CD28 is Associated With and Induces the Immediate Tyrosine Phosphorylation and Activation of the Tec Family Kinase ITK/EMT in the Human Jurkat Leukemic T-Cell Line. Proc Natl Acad Sci USA (1994) 91(20):9347–51. doi: 10.1073/pnas.91.20.9347
111. MacDonald KP, Blazar BR, Hill GR. Cytokine Mediators of Chronic Graft-Versus-Host Disease. J Clin Invest (2017) 127(7):2452–63. doi: 10.1172/JCI90593
112. Forcade E, Paz K, Flynn R, Griesenauer B, Amet T, Li W, et al. An Activated Th17-Prone T Cell Subset Involved in Chronic Graft-Versus-Host Disease Sensitive to Pharmacological Inhibition. JCI Insight (2017) 2(12):e92111. doi: 10.1172/jci.insight.92111
113. Gomez-Rodriguez J, Wohlfert EA, Handon R, Meylan F, Wu JZ, Anderson SM, et al. Itk-Mediated Integration of T Cell Receptor and Cytokine Signaling Regulates the Balance Between Th17 and Regulatory T Cells. J Exp Med (2014) 211(3):529–43. doi: 10.1084/jem.20131459
114. Miller AT, Wilcox HM, Lai Z, Berg LJ. Signaling Through Itk Promotes T Helper 2 Differentiation via Negative Regulation of T-Bet. Immunity (2004) 21(1):67–80. doi: 10.1016/j.immuni.2004.06.009
115. Anderson BE, McNiff JM, Jain D, Blazar BR, Shlomchik WD, Shlomchik MJ. Distinct Roles for Donor- and Host-Derived Antigen-Presenting Cells and Costimulatory Molecules in Murine Chronic Graft-Versus-Host Disease: Requirements Depend on Target Organ. Blood (2005) 105(5):2227–34. doi: 10.1182/blood-2004-08-3032
116. Le Huu D, Matsushita T, Jin G, Hamaguchi Y, Hasegawa M, Takehara K, et al. Donor-Derived Regulatory B Cells are Important for Suppression of Murine Sclerodermatous Chronic Graft-Versus-Host Disease. Blood (2013) 121(16):3274–83. doi: 10.1182/blood-2012-11-465658
117. Hamilton BL, Parkman R. Acute and Chronic Graft-Versus-Host Disease Induced by Minor Histocompatibility Antigens in Mice. Transplantation (1983) 36(2):150–5. doi: 10.1097/00007890-198308000-00008
118. Miklos D, Cutler CS, Arora M, Waller EK, Jagasia M, Pusic I, et al. Ibrutinib for Chronic Graft-Versus-Host Disease After Failure of Prior Therapy. Blood (2017) 130(21):2243–50. doi: 10.1182/blood-2017-07-793786
119. Wang L, Gordon RA, Huynh L, Su X, Park Min KH, Han J, et al. Indirect Inhibition of Toll-Like Receptor and Type I Interferon Responses by ITAM-Coupled Receptors and Integrins. Immunity (2010) 32(4):518–30. doi: 10.1016/j.immuni.2010.03.014
120. Ghazizadeh S, Bolen JB, Fleit HB. Tyrosine Phosphorylation and Association of Syk With Fc Gamma RII in Monocytic THP-1 Cells. Biochem J (1995) 305(Pt 2):669–74. doi: 10.1042/bj3050669
121. Ganju RK, Brubaker SA, Chernock RD, Avraham S, Groopman JE. Beta-Chemokine Receptor CCR5 Signals Through SHP1, SHP2, and Syk. J Biol Chem (2000) 275(23):17263–8. doi: 10.1074/jbc.M000689200
122. Buchner M, Baer C, Prinz G, Dierks C, Burger M, Zenz T, et al. Spleen Tyrosine Kinase Inhibition Prevents Chemokine- and Integrin-Mediated Stromal Protective Effects in Chronic Lymphocytic Leukemia. Blood (2010) 115(22):4497–506. doi: 10.1182/blood-2009-07-233692
123. Kappel LW, Goldberg GL, King CG, Suh DY, Smith OM, Ligh C, et al. IL-17 Contributes to CD4-Mediated Graft-Versus-Host Disease. Blood (2009) 113(4):945–52. doi: 10.1182/blood-2008-08-172155
124. Jakus Z, Simon E, Balazs B, Mocsai A. Genetic Deficiency of Syk Protects Mice From Autoantibody-Induced Arthritis. Arthritis Rheum (2010) 62(7):1899–910. doi: 10.1002/art.27438
125. Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grossbard EB, Magilavy DB. An Oral Spleen Tyrosine Kinase (Syk) Inhibitor for Rheumatoid Arthritis. N Engl J Med (2010) 363(14):1303–12. doi: 10.1056/NEJMoa1000500
126. Weinblatt ME, Kavanaugh A, Burgos-Vargas R, Dikranian AH, Medrano-Ramirez G, Morales-Torres JL, et al. Treatment of Rheumatoid Arthritis With a Syk Kinase Inhibitor: A Twelve-Week, Randomized, Placebo-Controlled Trial. Arthritis Rheum (2008) 58(11):3309–18. doi: 10.1002/art.23992
127. McManigle W, Youssef A, Sarantopoulos S. B Cells in Chronic Graft-Versus-Host Disease. Hum Immunol (2019) 80(6):393–9. doi: 10.1016/j.humimm.2019.03.003
128. Genovese MC, Kavanaugh A, Weinblatt ME, Peterfy C, DiCarlo J, White ML, et al. An Oral Syk Kinase Inhibitor in the Treatment of Rheumatoid Arthritis: A Three-Month Randomized, Placebo-Controlled, Phase II Study in Patients With Active Rheumatoid Arthritis That did Not Respond to Biologic Agents. Arthritis Rheum (2011) 63(2):337–45. doi: 10.1002/art.30114
129. Le Huu D, Kimura H, Date M, Hamaguchi Y, Hasegawa M, Hau KT, et al. Blockade of Syk Ameliorates the Development of Murine Sclerodermatous Chronic Graft-Versus-Host Disease. J Dermatol Sci (2014) 74(3):214–21. doi: 10.1016/j.jdermsci.2014.02.008
130. Poe JC, Jia W, Di Paolo JA, Reyes NJ, Kim JY, Su H, et al. SYK Inhibitor Entospletinib Prevents Ocular and Skin GVHD in Mice. JCI Insight (2018) 3(19):e122430. doi: 10.1172/jci.insight.122430
131. Nikolic B, Lee S, Bronson RT, Grusby MJ, Sykes M. Th1 and Th2 Mediate Acute Graft-Versus-Host Disease, Each With Distinct End-Organ Targets. J Clin Invest (2000) 105(9):1289–98. doi: 10.1172/JCI7894
132. Yi T, Chen Y, Wang L, Du G, Huang D, Zhao D, et al. Reciprocal Differentiation and Tissue-Specific Pathogenesis of Th1, Th2, and Th17 Cells in Graft-Versus-Host Disease. Blood (2009) 114(14):3101–12. doi: 10.1182/blood-2009-05-219402
133. Baird K, Comis LE, Joe GO, Steinberg SM, Hakim FT, Rose JJ, et al. Imatinib Mesylate for the Treatment of Steroid-Refractory Sclerotic-Type Cutaneous Chronic Graft-Versus-Host Disease. Biol Blood Marrow Transplant (2015) 21(6):1083–90. doi: 10.1016/j.bbmt.2015.03.006
134. Svegliati S, Olivieri A, Campelli N, Luchetti M, Poloni A, Trappolini S, et al. Stimulatory Autoantibodies to PDGF Receptor in Patients With Extensive Chronic Graft-Versus-Host Disease. Blood (2007) 110(1):237–41. doi: 10.1182/blood-2007-01-071043
135. Banovic T, MacDonald KP, Morris ES, Rowe V, Kuns R, Don A, et al. TGF-Beta in Allogeneic Stem Cell Transplantation: Friend or Foe? Blood (2005) 106(6):2206–14. doi: 10.1182/blood-2005-01-0062
136. Coomes SM, Moore BB. Pleiotropic Effects of Transforming Growth Factor-Beta in Hematopoietic Stem-Cell Transplantation. Transplantation (2010) 90(11):1139–44. doi: 10.1097/TP.0b013e3181efd018
137. Abdollahi A, Li M, Ping G, Plathow C, Domhan S, Kiessling F, et al. Inhibition of Platelet-Derived Growth Factor Signaling Attenuates Pulmonary Fibrosis. J Exp Med (2005) 201(6):925–35. doi: 10.1084/jem.20041393
138. Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, et al. Imatinib Mesylate Inhibits the Profibrogenic Activity of TGF-Beta and Prevents Bleomycin-Mediated Lung Fibrosis. J Clin Invest (2004) 114(9):1308–16. doi: 10.1172/JCI19603
139. Distler JH, Jungel A, Huber LC, Schulze-Horsel U, Zwerina J, Gay RE, et al. Imatinib Mesylate Reduces Production of Extracellular Matrix and Prevents Development of Experimental Dermal Fibrosis. Arthritis Rheum (2007) 56(1):311–22. doi: 10.1002/art.22314
140. Aono Y, Nishioka Y, Inayama M, Ugai M, Kishi J, Uehara H, et al. Imatinib as a Novel Antifibrotic Agent in Bleomycin-Induced Pulmonary Fibrosis in Mice. Am J Respir Crit Care Med (2005) 171(11):1279–85. doi: 10.1164/rccm.200404-531OC
141. Belle L, Fransolet G, Somja J, Binsfeld M, Delvenne P, Drion P, et al. Limited Impact of Imatinib in a Murine Model of Sclerodermatous Chronic Graft-Versus-Host Disease. PloS One (2016) 11(12):e0167997. doi: 10.1371/journal.pone.0167997
142. Zerr P, Distler A, Palumbo-Zerr K, Tomcik M, Vollath S, Dees C, et al. Combined Inhibition of C-Abl and PDGF Receptors for Prevention and Treatment of Murine Sclerodermatous Chronic Graft-Versus-Host Disease. Am J Pathol (2012) 181(5):1672–80. doi: 10.1016/j.ajpath.2012.07.017
143. Marinelli Busilacchi E, Costantini A, Viola N, Costantini B, Olivieri J, Butini L, et al. Immunomodulatory Effects of Tyrosine Kinase Inhibitor In Vitro and In Vivo Study. Biol Blood Marrow Transplant (2018) 24(2):267–75. doi: 10.1016/j.bbmt.2017.10.039
144. Marinelli Busilacchi E, Costantini A, Mancini G, Tossetta G, Olivieri J, Poloni A, et al. Nilotinib Treatment of Patients Affected by Chronic Graft-Versus-Host Disease Reduces Collagen Production and Skin Fibrosis by Downmodulating the TGF-Beta and P-SMAD Pathway. Biol Blood Marrow Transplant (2020) 26(5):823–34. doi: 10.1016/j.bbmt.2020.01.014
145. Olivieri A, Locatelli F, Zecca M, Sanna A, Cimminiello M, Raimondi R, et al. Imatinib for Refractory Chronic Graft-Versus-Host Disease With Fibrotic Features. Blood (2009) 114(3):709–18. doi: 10.1182/blood-2009-02-204156
146. Magro L, Catteau B, Coiteux V, Bruno B, Jouet JP, Yakoub-Agha I. Efficacy of Imatinib Mesylate in the Treatment of Refractory Sclerodermatous Chronic GVHD. Bone Marrow Transplant (2008) 42(11):757–60. doi: 10.1038/bmt.2008.252
147. Magro L, Mohty M, Catteau B, Coiteux V, Chevallier P, Terriou L, et al. Imatinib Mesylate as Salvage Therapy for Refractory Sclerotic Chronic Graft-Versus-Host Disease. Blood (2009) 114(3):719–22. doi: 10.1182/blood-2009-02-204750
148. Olivieri A, Mancini G, Olivieri J, Marinelli Busilacchi E, Cimminiello M, Pascale SP, et al. Nilotinib in Steroid-Refractory cGVHD: Prospective Parallel Evaluation of Response, According to NIH Criteria and Exploratory Response Criteria (GITMO Criteria). Bone Marrow Transplant (2020) 55(11):2077–86. doi: 10.1038/s41409-020-0902-9
149. Thangavelu G, Du J, Paz KG, Loschi M, Zaiken MC, Flynn R, et al. Inhibition of Inositol Kinase B Controls Acute and Chronic Graft-Versus-Host Disease. Blood (2020) 135(1):28–40. doi: 10.1182/blood.2019000032
150. Miller AT, Dahlberg C, Sandberg ML, Wen BG, Beisner DR, Hoerter JA, et al. Inhibition of the Inositol Kinase Itpkb Augments Calcium Signaling in Lymphocytes and Reveals a Novel Strategy to Treat Autoimmune Disease. PloS One (2015) 10(6):e0131071. doi: 10.1371/journal.pone.0131071
151. Miller AT, Chamberlain PP, Cooke MP. Beyond IP3: Roles for Higher Order Inositol Phosphates in Immune Cell Signaling. Cell Cycle (2008) 7(4):463–7. doi: 10.4161/cc.7.4.5518
152. Wen BG, Pletcher MT, Warashina M, Choe SH, Ziaee N, Wiltshire T, et al. Inositol (1,4,5) Trisphosphate 3 Kinase B Controls Positive Selection of T Cells and Modulates Erk Activity. Proc Natl Acad Sci USA (2004) 101(15):5604–9. doi: 10.1073/pnas.0306907101
153. Pouillon V, Marechal Y, Frippiat C, Erneux C, Schurmans S. Inositol 1,4,5-Trisphosphate 3-Kinase B (Itpkb) Controls Survival, Proliferation and Cytokine Production in Mouse Peripheral T Cells. Adv Biol Regul (2013) 53(1):39–50. doi: 10.1016/j.jbior.2012.08.001
154. Scaffidi C, Medema JP, Krammer PH, Peter ME. FLICE is Predominantly Expressed as Two Functionally Active Isoforms, Caspase-8/a and Caspase-8/B. J Biol Chem (1997) 272(43):26953–8. doi: 10.1074/jbc.272.43.26953
155. Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, et al. FLICE, a Novel FADD-Homologous ICE/CED-3-Like Protease, is Recruited to the CD95 (Fas/APO-1) Death–Inducing Signaling Complex. Cell (1996) 85(6):817–27. doi: 10.1016/s0092-8674(00)81266-0
156. Kobayashi A, Kobayashi S, Miyai K, Osawa Y, Horiuchi T, Kato S, et al. TAK1 Inhibition Ameliorates Survival From Graft-Versus-Host Disease in an Allogeneic Murine Marrow Transplantation Model. Int J Hematol (2018) 107(2):222–9. doi: 10.1007/s12185-017-2345-7
157. Mathew NR, Vinnakota JM, Apostolova P, Erny D, Hamarsheh S, Andrieux G, et al. Graft-Versus-Host Disease of the CNS is Mediated by TNF Upregulation in Microglia. J Clin Invest (2020) 130(3):1315–29. doi: 10.1172/JCI130272
158. Wen HS, Wang JM, Zhou H, Gong SI, Gao L, Wu Y. Migration and Activation of T Cells During Development of Graft-Versus-Host Disease in a Mouse Model. Transplant Proc (2013) 45(2):713–8. doi: 10.1016/j.transproceed.2012.12.004
159. Gaud G, Lesourne R, Love PE. Regulatory Mechanisms in T Cell Receptor Signalling. Nat Rev Immunol (2018) 18(8):485–97. doi: 10.1038/s41577-018-0020-8
160. Lu SX, Alpdogan O, Lin J, Balderas R, Campos-Gonzalez R, Wang X, et al. STAT-3 and ERK 1/2 Phosphorylation Are Critical for T-Cell Alloactivation and Graft-Versus-Host Disease. Blood (2008) 112(13):5254–8. doi: 10.1182/blood-2008-03-147322
161. Almela P, Milanes MV, Laorden ML. Activation of the ERK Signalling Pathway Contributes to the Adaptive Changes in Rat Hearts During Naloxone-Induced Morphine Withdrawal. Br J Pharmacol (2007) 151(6):787–97. doi: 10.1038/sj.bjp.0707301
162. Radwanska K, Valjent E, Trzaskos J, Caboche J, Kaczmarek L. Regulation of Cocaine-Induced Activator Protein 1 Transcription Factors by the Extracellular Signal-Regulated Kinase Pathway. Neuroscience (2006) 137(1):253–64. doi: 10.1016/j.neuroscience.2005.09.001
163. Valjent E, Corvol JC, Trzaskos JM, Girault JA, Herve D. Role of the ERK Pathway in Psychostimulant-Induced Locomotor Sensitization. BMC Neurosci (2006) 7:20. doi: 10.1186/1471-2202-7-20
164. Wang S, Guan Q, Diao H, Lian D, Zhong R, Jevnikar AM, et al. Prolongation of Cardiac Allograft Survival by Inhibition of ERK1/2 Signaling in a Mouse Model. Transplantation (2007) 83(3):323–32. doi: 10.1097/01.tp.0000251374.49225.19
165. Shindo T, Kim TK, Benjamin CL, Wieder ED, Levy RB, Komanduri KV. MEK Inhibitors Selectively Suppress Alloreactivity and Graft-Versus-Host Disease in a Memory Stage-Dependent Manner. Blood (2013) 121(23):4617–26. doi: 10.1182/blood-2012-12-476218
166. Bekaii-Saab T, Phelps MA, Li X, Saji M, Goff L, Kauh JS, et al. Multi-Institutional Phase II Study of Selumetinib in Patients With Metastatic Biliary Cancers. J Clin Oncol (2011) 29(17):2357–63. doi: 10.1200/JCO.2010.33.9473
167. Banerji U, Camidge DR, Verheul HM, Agarwal R, Sarker D, Kaye SB, et al. The First-in-Human Study of the Hydrogen Sulfate (Hyd-Sulfate) Capsule of the MEK1/2 Inhibitor AZD6244 (ARRY-142886): A Phase I Open-Label Multicenter Trial in Patients With Advanced Cancer. Clin Cancer Res (2010) 16(5):1613–23. doi: 10.1158/1078-0432.CCR-09-2483
168. Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, et al. Phase I Pharmacokinetic and Pharmacodynamic Study of the Oral, Small-Molecule Mitogen-Activated Protein Kinase Kinase 1/2 Inhibitor AZD6244 (ARRY-142886) in Patients With Advanced Cancers. J Clin Oncol (2008) 26(13):2139–46. doi: 10.1200/JCO.2007.14.4956
169. Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological Characterization of ARRY-142886 (AZD6244), a Potent, Highly Selective Mitogen-Activated Protein Kinase Kinase 1/2 Inhibitor. Clin Cancer Res (2007) 13(5):1576–83. doi: 10.1158/1078-0432.CCR-06-1150
170. Wieder E, Kolonias D, Benjamin C, Shindo T, Kim TK, Levy RB, et al. Trametinib Selectively Inhibits Alloreactivity While Sparing Virus-Specific T Cells. Biol Blood Marrow Transplant (2014) 20(2, Supplement):S283. doi: 10.1016/j.bbmt.2013.12.475
171. Chhabra S, Liu Y, Hemmer MT, Costa L, Pidala JA, Couriel DR, et al. Comparative Analysis of Calcineurin Inhibitor-Based Methotrexate and Mycophenolate Mofetil-Containing Regimens for Prevention of Graft-Versus-Host Disease After Reduced-Intensity Conditioning Allogeneic Transplantation. Biol Blood Marrow Transplant (2019) 25(1):73–85. doi: 10.1016/j.bbmt.2018.08.018
172. Itamura H, Shindo T, Muranushi H, Kitaura K, Okada S, Shin IT, et al. Pharmacological MEK Inhibition Promotes Polyclonal T-Cell Reconstitution and Suppresses Xenogeneic GVHD. Cell Immunol (2021) 367:104410. doi: 10.1016/j.cellimm.2021.104410
173. Itamura H, Shindo T, Tawara I, Kubota Y, Kariya R, Okada S, et al. The MEK Inhibitor Trametinib Separates Murine Graft-Versus-Host Disease From Graft-Versus-Tumor Effects. JCI Insight (2016) 1(10):e86331. doi: 10.1172/jci.insight.86331
174. Yamaguchi T, Yoshida T, Kurachi R, Kakegawa J, Hori Y, Nanayama T, et al. Identification of JTP-70902, a P15(INK4b)-Inductive Compound, as a Novel MEK1/2 Inhibitor. Cancer Sci (2007) 98(11):1809–16. doi: 10.1111/j.1349-7006.2007.00604.x
175. Monlish DA, Beezhold KJ, Chiaranunt P, Paz K, Moore NJ, Dobbs AK, et al. Deletion of AMPK Minimizes Graft-Versus-Host Disease Through an Early Impact on Effector Donor T Cells. JCI Insight (2021) 6(14):e143811. doi: 10.1172/jci.insight.143811
176. Glick GD, Rossignol R, Lyssiotis CA, Wahl D, Lesch C, Sanchez B, et al. Anaplerotic Metabolism of Alloreactive T Cells Provides a Metabolic Approach to Treat Graft-Versus-Host Disease. J Pharmacol Exp Ther (2014) 351(2):298–307. doi: 10.1124/jpet.114.218099
177. Byersdorfer CA, Tkachev V, Opipari AW, Goodell S, Swanson J, Sandquist S, et al. Effector T Cells Require Fatty Acid Metabolism During Murine Graft-Versus-Host Disease. Blood (2013) 122(18):3230–7. doi: 10.1182/blood-2013-04-495515
178. Tamas P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, et al. Regulation of the Energy Sensor AMP-Activated Protein Kinase by Antigen Receptor and Ca2+ in T Lymphocytes. J Exp Med (2006) 203(7):1665–70. doi: 10.1084/jem.20052469
179. Garcia D, Shaw RJ. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol Cell (2017) 66(6):789–800. doi: 10.1016/j.molcel.2017.05.032
180. Hardie DG, Lin SC. AMP-Activated Protein Kinase - Not Just an Energy Sensor. F1000Res (2017) 6:1724. doi: 10.12688/f1000research.11960.1
181. Lepez A, Pirnay T, Denanglaire S, Perez-Morga D, Vermeersch M, Leo O, et al. Long-Term T Cell Fitness and Proliferation is Driven by AMPK-Dependent Regulation of Reactive Oxygen Species. Sci Rep (2020) 10(1):21673. doi: 10.1038/s41598-020-78715-2
182. Taylor PA, Lees CJ, Blazar BR. The Infusion of Ex Vivo Activated and Expanded CD4(+)CD25(+) Immune Regulatory Cells Inhibits Graft-Versus-Host Disease Lethality. Blood (2002) 99(10):3493–9. doi: 10.1182/blood.v99.10.3493
183. Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+CD25+ Regulatory T Cells Preserve Graft-Versus-Tumor Activity While Inhibiting Graft-Versus-Host Disease After Bone Marrow Transplantation. Nat Med (2003) 9(9):1144–50. doi: 10.1038/nm915
184. Martelli MF, Di Ianni M, Ruggeri L, Falzetti F, Carotti A, Terenzi A, et al. HLA-Haploidentical Transplantation With Regulatory and Conventional T-Cell Adoptive Immunotherapy Prevents Acute Leukemia Relapse. Blood (2014) 124(4):638–44. doi: 10.1182/blood-2014-03-564401
185. Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, et al. A Novel Kinase Cascade Triggered by Stress and Heat Shock That Stimulates MAPKAP Kinase-2 and Phosphorylation of the Small Heat Shock Proteins. Cell (1994) 78(6):1027–37. doi: 10.1016/0092-8674(94)90277-1
186. Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, et al. A Protein Kinase Involved in the Regulation of Inflammatory Cytokine Biosynthesis. Nature (1994) 372(6508):739–46. doi: 10.1038/372739a0
187. Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP Kinase Targeted by Endotoxin and Hyperosmolarity in Mammalian Cells. Science (1994) 265(5173):808–11. doi: 10.1126/science.7914033
188. Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, et al. Pro-Inflammatory Cytokines and Environmental Stress Cause P38 Mitogen-Activated Protein Kinase Activation by Dual Phosphorylation on Tyrosine and Threonine. J Biol Chem (1995) 270(13):7420–6. doi: 10.1074/jbc.270.13.7420
189. Kaminska B. MAPK Signalling Pathways as Molecular Targets for Anti-Inflammatory Therapy–From Molecular Mechanisms to Therapeutic Benefits. Biochim Biophys Acta (2005) 1754(1-2):253–62. doi: 10.1016/j.bbapap.2005.08.017
190. Yang Y, Kim SC, Yu T, Yi YS, Rhee MH, Sung GH, et al. Functional Roles of P38 Mitogen-Activated Protein Kinase in Macrophage-Mediated Inflammatory Responses. Mediators Inflamm (2014) 2014:352371. doi: 10.1155/2014/352371
191. Damjanov N, Kauffman RS, Spencer-Green GT. Efficacy, Pharmacodynamics, and Safety of VX-702, a Novel P38 MAPK Inhibitor, in Rheumatoid Arthritis: Results of Two Randomized, Double-Blind, Placebo-Controlled Clinical Studies. Arthritis Rheum (2009) 60(5):1232–41. doi: 10.1002/art.24485
192. Sato M, Shegogue D, Gore EA, Smith EA, McDermott PJ, Trojanowska M. Role of P38 MAPK in Transforming Growth Factor Beta Stimulation of Collagen Production by Scleroderma and Healthy Dermal Fibroblasts. J Invest Dermatol (2002) 118(4):704–11. doi: 10.1046/j.1523-1747.2002.01719.x
193. Ihn H, Yamane K, Tamaki K. Increased Phosphorylation and Activation of Mitogen-Activated Protein Kinase P38 in Scleroderma Fibroblasts. J Invest Dermatol (2005) 125(2):247–55. doi: 10.1111/j.0022-202X.2005.23766.x
194. Claman HN, Jaffee BD, Huff JC, Clark RA. Chronic Graft-Versus-Host Disease as a Model for Scleroderma. II. Mast Cell Depletion With Deposition of Immunoglobulins in the Skin and Fibrosis. Cell Immunol (1985) 94(1):73–84. doi: 10.1016/0008-8749(85)90086-3
195. Furst DE, Clements PJ. Hypothesis for the Pathogenesis of Systemic Sclerosis. J Rheumatol Suppl (1997) 48:53–7 https://pubmed.ncbi.nlm.nih.gov/9150119/.
196. Sarantopoulos S, Blazar BR, Cutler C, Ritz J. B Cells in Chronic Graft-Versus-Host Disease. Biol Blood Marrow Transplant (2015) 21(1):16–23. doi: 10.1016/j.bbmt.2014.10.029
197. Ohta M, Tateishi K, Kanai F, Ueha S, Guleng B, Washida M, et al. Reduced P38 Mitogen-Activated Protein Kinase in Donor Grafts Accelerates Acute Intestinal Graft-Versus-Host Disease in Mice. Eur J Immunol (2005) 35(7):2210–21. doi: 10.1002/eji.200425897
198. Song J, Salek-Ardakani S, So T, Croft M. The Kinases Aurora B and mTOR Regulate the G1-S Cell Cycle Progression of T Lymphocytes. Nat Immunol (2007) 8(1):64–73. doi: 10.1038/ni1413
199. Furlan SN, Watkins B, Tkachev V, Flynn R, Cooley S, Ramakrishnan S, et al. Transcriptome Analysis of GVHD Reveals Aurora Kinase A as a Targetable Pathway for Disease Prevention. Sci Transl Med (2015) 7(315):315ra191. doi: 10.1126/scitranslmed.aad3231
Keywords: GvHD, stem cell transplant (SCT), kinases, ruxolitinib, JAK1 and JAK2 inhibitors, BTK - Bruton’s tyrosine kinase, ROCK
Citation: Braun LM and Zeiser R (2021) Kinase Inhibition as Treatment for Acute and Chronic Graft-Versus-Host Disease. Front. Immunol. 12:760199. doi: 10.3389/fimmu.2021.760199
Received: 17 August 2021; Accepted: 28 October 2021;
Published: 17 November 2021.
Edited by:
Dietlinde Wolf, University of Miami, United StatesReviewed by:
Maria Teresa Lupo Stanghellini, San Raffaele Hospital (IRCCS), ItalyMark A. Schroeder, Washington University in St. Louis, United States
Robert Soiffer, Dana–Farber Cancer Institute, United States
Copyright © 2021 Braun and Zeiser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Zeiser, cm9iZXJ0LnplaXNlckB1bmlrbGluaWstZnJlaWJ1cmcuZGU=