 Stéphane J.C. Mancini1,2,3†
Stéphane J.C. Mancini1,2,3† Karl Balabanian1,4,5†
Karl Balabanian1,4,5† Isabelle Corre1,3,6†
Isabelle Corre1,3,6† Julie Gavard1,3,6,7†
Julie Gavard1,3,6,7† Gwendal Lazennec1,8†
Gwendal Lazennec1,8† Marie-Caroline Le Bousse-Kerdilès1,9†
Marie-Caroline Le Bousse-Kerdilès1,9† Fawzia Louache1,9†
Fawzia Louache1,9† Véronique Maguer-Satta1,10†Nathalie M. Mazure1,11†
Véronique Maguer-Satta1,10†Nathalie M. Mazure1,11† Fatima Mechta-Grigoriou1,12†Jean-François Peyron1,11†
Fatima Mechta-Grigoriou1,12†Jean-François Peyron1,11† Valérie Trichet1,3,13†
Valérie Trichet1,3,13† Olivier Herault1,3,5,14,15*
Olivier Herault1,3,5,14,15*- 1Centre National de la Recherche scientifique (CNRS) GDR3697, Micronit “Microenvironment of Tumor Niches”, Tours, France
- 2INSERM UMR1236, Rennes 1 University, Etablissement Français du Sang Bretagne, Rennes, France
- 3Cancéropole Grand-Ouest, NET network “Niches and Epigenetics of Tumors”, Nantes, France
- 4Saint-Louis Research Institute, University of Paris, EMiLy, INSERM U1160, Paris, France
- 5The Organization for Partnerships in Leukemia (OPALE) Carnot Institute, The Organization for Partnerships in Leukemia, Paris, France
- 6Center for Research in Cancerology and Immunology Nantes-Angers (CRCINA), Signaling in Oncogenesis Angiogenesis and Permeability (SOAP), INSERM UMR1232, Centre National de la Recherche scientifique (CNRS) ERL600, Université de Nantes, Nantes, France
- 7Integrated Center for Oncology, St. Herblain, France
- 8Centre National de la Recherche scientifique (CNRS) UMR9005, SYS2DIAG-ALCEDIAG, Montpellier, France
- 9INSERM UMRS-MD1197, Paris-Saclay University, Paul-Brousse Hospital, Villejuif, France
- 10Cancer Research Center of Lyon (CRCL), CNRS UMR5286, INSERM U1052, Lyon 1 university, Lean Bérard Center, Lyon, France
- 11INSERM U1065, C3M, University of Côte d’Azur (UCA), Nice, France
- 12Stress and Cancer Laboratory, Institut Curie, INSERM U830, Paris Sciences et Lettres (PSL) Research University, Team Babelized Ligue Nationale Contre le Cancer (LNCC), Paris, France
- 13INSERM UMR1238 Phy-Os, Université de Nantes, Nantes, France
- 14Centre National de la Recherche scientifique (CNRS) ERL7001 LNOx, EA7501, Tours University, Tours, France
- 15Department of Biological Hematology, Tours University Hospital, Tours, France
Knowledge about the hematopoietic niche has evolved considerably in recent years, in particular through in vitro analyzes, mouse models and the use of xenografts. Its complexity in the human bone marrow, in particular in a context of hematological malignancy, is more difficult to decipher by these strategies and could benefit from the knowledge acquired on the niches of solid tumors. Indeed, some common features can be suspected, since the bone marrow is a frequent site of solid tumor metastases. Recent research on solid tumors has provided very interesting information on the interactions between tumoral cells and their microenvironment, composed notably of mesenchymal, endothelial and immune cells. This review thus focuses on recent discoveries on tumor niches that could help in understanding hematopoietic niches, with special attention to 4 particular points: i) the heterogeneity of carcinoma/cancer-associated fibroblasts (CAFs) and mesenchymal stem/stromal cells (MSCs), ii) niche cytokines and chemokines, iii) the energy/oxidative metabolism and communication, especially mitochondrial transfer, and iv) the vascular niche through angiogenesis and endothelial plasticity. This review highlights actors and/or pathways of the microenvironment broadly involved in cancer processes. This opens avenues for innovative therapeutic opportunities targeting not only cancer stem cells but also their regulatory tumor niche(s), in order to improve current antitumor therapies.
Introduction
The bone marrow (BM) is the site where hematopoietic stem cells (HSCs) sustain hematopoiesis after birth and all lifelong in mammals. From early progenitors to committed subsets of myeloid and lymphoid lineages, proliferation and differentiation mechanisms have been extensively studied. They were shown early on, through the use of in vitro cultures, to be controlled by cells from the BM microenvironment (1, 2) comprising mesenchymal stem/stromal cells (MSCs), endothelial cells (ECs) and macrophages (3). Since the beginning of the 2000’s, the identity and organization of BM niches supporting hematopoiesis have been extensively studied through the use of reporter mouse models (reviewed in (4, 5)). Furthermore, the heterogeneity of BM MSCs and ECs has recently been approached by single-cell RNA sequencing, which has confirmed the presence of multiple subpopulations within these two cell types (6–10). Beside this heterogeneity of the BM microenvironment, it is now moreover clear that the “one progenitor/one niche” rule does not prevail. Indeed, a single niche can support not only hematopoietic subsets at distinct developmental stages, but also mature immune cells homing back to the BM (5, 10).
Because of the contained location of the BM, knowledge on human hematopoietic niches is more limited even if strong similarities could be observed with mice (10–12). For the same reason, the nature of human leukemic niches and the molecular mechanisms regulated by/within them are still unclear and results strongly rely on in vitro cultures or on observations obtained in syngeneic or xenograft mouse models [reviewed in (13, 14)]. Furthermore, the immune microenvironment in tumoral BM is also poorly resolved and despite the tremendous progress that came with the use of immunotherapies, resistance and relapse in acute leukemia still concern many patients. Lessons could be learned from solid tumors for which the easier study of microenvironment (i.e., after surgery) has led to important advances. As in hematopoietic malignancies, a complex crosstalk exists between the tumor and the non-malignant cells in its microenvironment. Interestingly, BM is a haven not only for normal and pathological hematopoietic cells from the periphery, but also for metastatic cells from solid tumors, indicating that at least some properties or components of the tumor microenvironment must be shared between hematopoietic and solid tumors.
Tumor development depends on a multidirectional crosstalk between tumor cells, mesenchymal/endothelial cells and immune cells. The immune landscape and modulation of immune responses exerted by tumor cells, directly or through systemic disruption, have been extensively studied (15–17). In this review, we propose to confront and combine the knowledge gained on stromal/endothelial niches of leukemic and solid tumors by focusing on the BM as a common niche.
Heterogeneity of CAFs/MSCs
The term “mesenchymal” is widely spread in the literature to designate stromal cells from the microenvironment of many tissues. MSCs are stromal cells able to adhere in vitro to plastic and spread on culture plates as spindle-cells or fibroblast-like cells. Specific MSC shapes are associated with differentiation, for instance rounded MSCs during adipogenic differentiation (18). MSCs are characterized by a specific pattern of surface markers. Namely, they express CD105, CD73, CD90 and CD146 in the absence of CD45, CD34, CD14, CD11b, HLA-DR and lymphocyte-lineage markers. MSCs secrete components of the extracellular matrix (e.g. collagens, heparan sulphate, elastin, aggrecan) and metalloproteinases as well as a large variety of mitogenic growth factors, cytokines, chemokines and angiogenic factors (19). These cells have also retained the ability to differentiate into osteoblasts, chondroblasts and adipocytes (20). However, inconsistent definitions of MSCs and varying isolation and culture conditions have resulted in highly diverse outcomes and confusing data (21, 22). The mesenchyme does not constitute a lineage but is an embryonic tissue able to give rise to connective tissue, blood vessels and blood cells that can have different embryonic origins. Therefore, there are no common MSCs in adult tissues, reflecting the fact that the nature and properties of the globally termed “MSCs” likely represent different cellular entities (21, 22). Since their initial definition in the early 1990’s (23), the properties of MSCs have been largely explored and debated, even with an attempt at establishing a molecular signature (24). However, a consensus on the definition and use of the term “MSC” is unlikely to be reached. Indeed, with the major improvement of “omic” technological tools in the past years, in particular at the single-cell level, it has become quite obvious that MSCs encompass different subpopulations and states of stromal cells and even fibroblasts. The recent molecular mapping of murine BM niche populations, under homeostatic conditions, by single-cell RNA sequencing, clearly demonstrated a great cellular heterogeneity of BM stromal cells (6–8, 10). This heterogeneity was also identified in human MSCs (from umbilical cord), among which two groups were separated based on differentially expressed genes, including CD73. The first group is characterized by an enriched expression of genes involved in immune response/regulatory activities, muscle cell proliferation and differentiation, stemness and oxidative stress. The second presents a higher expression of genes involved in extracellular matrix production, osteoblast and chondrocyte differentiation, and bone and cartilage growth (25).
In a malignant context, and more particularly in acute myeloid leukemia (AML), the most recent studies report functional abnormalities of human MSCs, which have a significant impact on the aggressiveness of the disease. Among these anomalies, growth deficiency, altered osteogenic differentiation ability and reduced capacity to support hematopoietic cells (26–31) have been described, as well as modifications of the secretome (28, 32), which induce in vivo shaping of the stromal niche by leukemic cells (33). Moreover, single cell analyses of murine BM stromal cells recently revealed that leukemia remodels the BM stroma to the disadvantage of normal hematopoietic cells. This notably involves a blockade of the osteoblastic development, as well as of the pathway of bone morphogenetic proteins (BMPs), including Bmp4. It also induces a decreased expression of Cxcl12 and Kitl by leptin receptor expressing osteoprogenitors that regulate HSCs (7) (Figure 1).
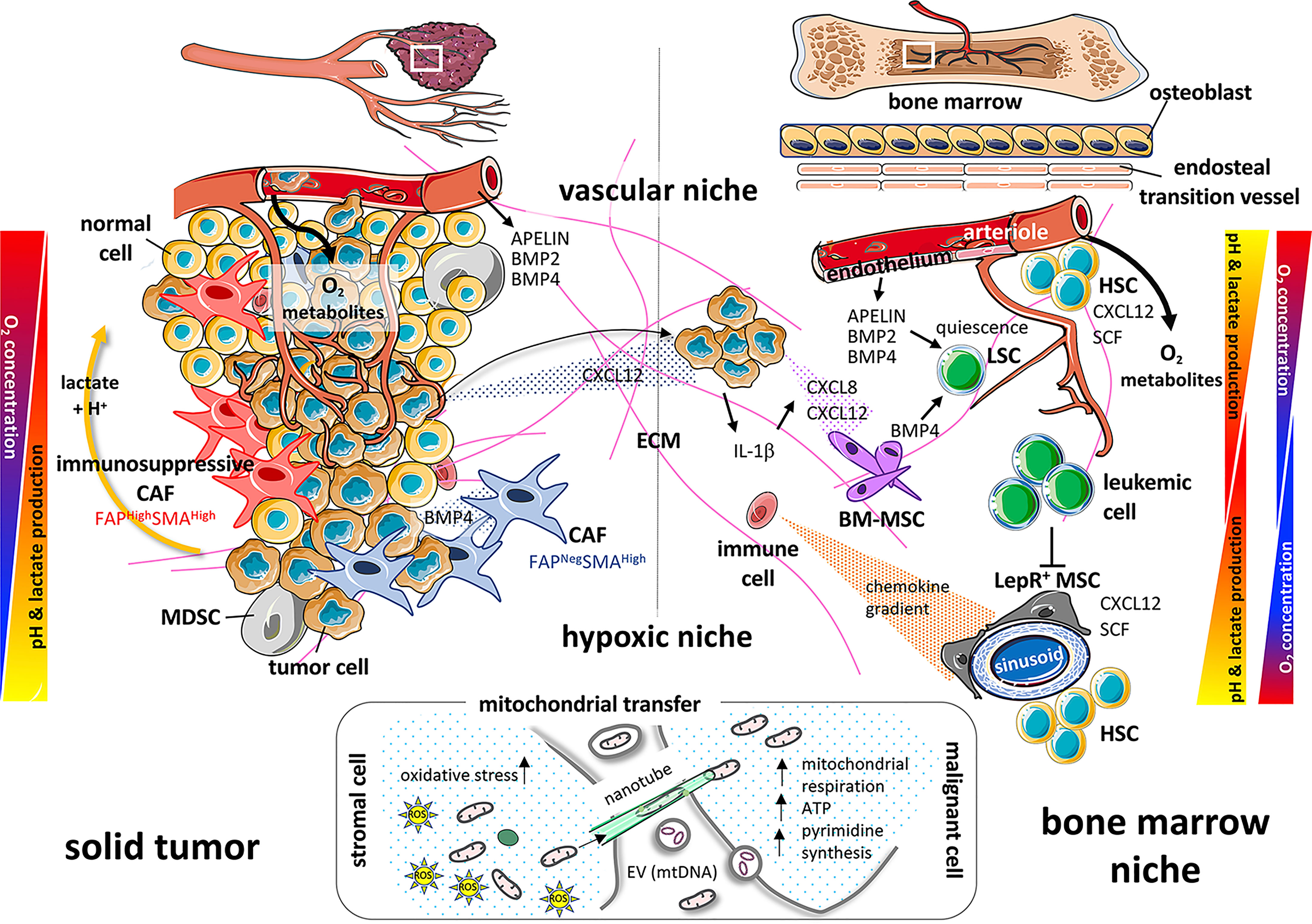
Figure 1 Paralleling the microenvironment and actors of the solid tumor and bone marrow niche. On the left part of the diagram, the solid tumor is governed by cellular components such as healthy cells and tumor cells juxtaposed with immunosuppressive or neutral cancer-associated fibroblasts (CAFs) and myeloid-derived suppressor cells (MDSCs). The different gradients of oxygen, pH and growth factors (BMP2, BMP4), then participate in the tumor cell fate (proliferation, metastasis…). On the right panel, healthy hematopoietic stem cells (HSCs), bone marrow-mesenchymal stem/stromal cells (BM-MSCs), immune cells and tumor cells (attracted from a solid tumor by chemokine gradients such as CXCL12) or leukemia cells, quiescent or not, will be confronted with gradients of oxygen, pH, growth factors (BMP2, BMP4, SCF, APELIN) and cytokines (CXCL8, CXCL12, IL-1β) in a similar way as cells within the solid tumor. All these interrelations and interconnections, controls and feedbacks, will allow the tumor cell to proliferate and spread. Oxygen gradients, on the left, result from diffusion of oxygen from the blood vessels (vascular niche) as tumors grow outward from the local vascular architecture. Vascular niche may also be a source of factors favoring tumor growth (i.e Apelin) in solid tumor and in the bone marrow niche. On the right, there is a double gradient between arterioles and sinusoids. The cells will adapt their metabolism along these gradients and create, as a counterpart, a pH gradient due to the release of lactate and H+ protons. Bottom part: As crucial powerhouses for cell metabolism and tissue survival, mitochondria will transfer horizontally from stromal cells to cancer and/or immune cells, via nanotubes, extracellular vesicles (EVs) or freely, to allow recipient cells to adapt and modify their metabolism (mitochondrial respiration, ATP, pyrimidine synthesis) to meet different stresses (oxidative stress) and energy demands.
All solid tumors contain non-tumor stromal supporting cells which are also called tumor-associated stromal cells. In carcinomas, they are well-known as cancer-associated fibroblasts (CAFs). Heterogeneity of tumor-associated stromal cells between tumors and, more recently, within a single tumor, has been disclosed, essentially through flow-cytometry, sorting and single-cell RNA sequencing. Their role in tumor progression is still explored with the use of mouse models. In line with such analyzes, four CAF subsets have been identified in breast and ovarian cancers by combining the study of distinct CAF markers, including the fibroblast activation protein (FAP), smooth-muscle α-actin (SMA) and integrin β1 (CD29) (34–37). Two subsets have also been detected in healthy tissues, reminiscent of normal fibroblasts, while two myofibroblastic subsets (FAPHigh SMAHigh CD29Med-High and FAPNeg SMAHigh CD29High) appear to be restricted to tumors (Figure 1). These CAF subsets are respectively characterized by their secretion and organization of extracellular matrix (ECM) components, in particular types 1, 3 and 6 collagen, and by a perivascular contractile gene signature (34, 35, 37). Importantly, the association of the FAPHigh SMAHigh CAF subset with poor outcome has been validated by different laboratories in mouse models, as well as in some types of human carcinoma (38–42), highlighting its relevance in distinct species and cancer types.
Consistent with their accumulation in aggressive carcinomas, FAPHigh SMAMed-High CAFs favor metastatic spread in breast and ovarian cancers by directly interacting with cancer cells, and the highly contractile FAPLow SMAHigh CAFs promote cancer cell invasion in 3-dimensions by remodeling the surrounding ECM (34–37, 43–45). Osteosarcoma-associated stromal cells have been characterized by MSC markers and SMA expression, like their healthy counterparts, but with a higher osteoblastic potential and an increase in lung metastases in mouse models (46). Mimicking the acidity of tumor microenvironment has been shown to lead osteosarcoma-associated MSCs to acquire an inflammatory phenotype, with an increased secretion of IL-6 and CXCL8. Such conditions also promoted the stemness of osteosarcoma cells (47). By contrast, healthy MSCs did not modify the quiescent state of osteosarcoma cells (48). Osteosarcoma-associated MSCs were moreover shown to promote not only the invasiveness of osteosarcoma cells, but also angiogenesis through the activation, proliferation and/or differentiation of ECs (49).
Myofibroblastic CAFs (SMA+) secrete type-I collagen that can modulate immune cells. In a mouse model of pancreatic adenocarcinoma, it was recently demonstrated that the reduction of total type-I collagen secreted by CAFs accelerated the emergence of carcinoma. This was shown to be due to an upregulation of the chemokine CXCL5 (see next section) in cancer cells, leading to the recruitment of myeloid-derived suppressor cells (MDSCs) and impairment of CD8+ cytotoxic T-cells (50). Conversely, the secretion of matrix metalloproteinases (MMPs) can favor tumor cell mobility across collagen fibers and inhibit immune cell activity. Melanoma-associated fibroblasts have thus been described as negative immuno-modulators, through the secretion of MMPs decreasing tumor cell lysis by natural killer (NK) cells (51). The ECM composition can be modified by the protease activity of FAP, which is expressed by tumor and stromal cells in many human carcinomas and sarcomas (43, 52, 53). Consistent with these observations, FAPHigh SMA+ CAFs have been identified in aggressive carcinomas to exhibit immunosuppressive activities (43, 44). Indeed, FAPHigh CAFs are associated with immunosuppression and resistance to immunotherapies in mouse models (37, 54, 55). Interestingly, the FAPHigh SMA+ CAF subset promotes immunosuppression by increasing the infiltration of regulatory T lymphocytes (Treg) in human cancers (34, 35). Within the FAPHigh CAF subpopulation, two distinct subsets exhibiting either an ECM-producing myofibroblastic phenotype (myCAF) or an inflammatory profile (iCAF) were recently identified in different types of cancers (56–60). Importantly, recent single cell sequencing of FAPHigh CAFs from breast cancer cells allowed identifying eight different FAPHigh cellular clusters (59). Three of them were further shown to be specifically associated with resistance to immunotherapy in metastatic melanoma and in non-small cell lung cancer patients (59). Taken together, these findings highlight the existence of a network of numerous CAF and MSC subpopulations in solid tumors and underline their relevance in various cancer types and across species.
Niche Cytokines and Chemokines
The BM is a major location where hematological malignancies affecting myeloid or lymphoid lineages develop and is also an important site of metastasis for solid tumors, especially breast, prostate, and lung cancers (61, 62). The engraftment of metastatic cells from solid tumors into the BM can generate secondary tumors with either osteoblastic properties, notably in early stages of metastasis of prostate cancer, or osteolytic properties in the case of breast cancer (63–65). Osteoblastic lesions correspond to an increased bone mass at the lesion site whereas osteolytic lesions lead to a destruction of the bone structure. In the case of prostate cancer, there is a preferential accumulation of cancer cells in the lateral rather than medial endocortical bone region. This former area is enriched in osteoblasts, which could explain this phenomenon (66). The segregation of bone metastasis location between prostate and breast cancers has been extensively studied. Although this differential tropism is still not completely understood, there is a consensus about the involvement of chemokines in this phenomenon. The activity of chemokines depends on their receptors, a family of G protein-coupled seven-transmembrane-spanning molecules. Chemokines are versatile secreted factors critically required to drive the migration of immune and non-immune cells, notably within lymphoid organs including the BM. Depending on the targeted cell type, they can foster an effective anti-tumor immune response or conversely contribute to a pro-tumorigenic microenvironment. Early work from Zlotnik’s lab has shown that high production of the chemokine CXCL12 (SDF-1) by the BM is sufficient to attract breast cancer cells expressing CXCR4, one of the cognate receptors of CXCL12 (67) (Figure 1). CXCR4 and CXCL12 are also critical for the homeostasis of the BM ecosystem, with a key role in controlling the production and mobilization of hematopoietic stem/progenitor cells (HSPCs) (68, 69). Indeed, in the BM, HSPC niches are thought to be composed of perivascular stromal units associated with sinusoids and arterioles as reviewed recently (70). In particular, many studies have shown that a population of MSCs termed CXCL12-abundant reticular (CAR) cells overlaps with leptin receptor (LepR)-expressing cells. These CAR cells constitute a major component of HSPC niches by their capacities to produce such niche factors as CXCL12, SCF and IL-7. Similar stromal cells with salient features of CAR cells have been identified in human adult BM (10, 71). In line with these findings, the CXCL12/CXCR4 axis is key in immunosuppression and metastatic spread in solid tumors, through reciprocal crosstalk between FAPHigh CAFs and regulatory T cells (Tregs), as well as FAPHigh CAFs and cancer cells, respectively (24, 25, 27). In addition to CXCR4 and CXCL12, numerous studies have shown that chemokines act at different levels in the progression of the primary tumor, modulating both tumor cell proliferation, apoptosis, invasion, angiogenesis, recruitment of immune cells and resistance to chemotherapy (72–76). It appears clearly that some kind of “chemokine storm” and sustained inflammation take place in the primary tumor. This comforts the notion of a complex interplay between cancer cells, cells from the tumor microenvironment (including CAFs, MSCs and ECs) and a variety of immune cells, such as macrophages, B- and T- lymphocytes, NK cells, neutrophils and dendritic cells (73, 75, 77). The final outcome of the tumor with either sustained resistance of the host or immune escape of the tumor will depend on these interactions.
Within the tumor microenvironment, MSCs are interesting for multiple reasons. First, as stated above, these cells are highly present in the BM but can also be found at lower levels elsewhere such as in adipose tissue, lung or umbilical cord blood. They are moreover detected in multiple types of primary solid tumors (e.g., breast, ovarian, pancreatic cancers) (78–81). Several studies have shown that BM or adipose MSCs [called adipose-derived stromal cells (ADSCs)] have a particular tropism for primary tumors (81, 82). MSCs can either favor or inhibit primary tumor growth and metastasis (78, 81, 83–85)s. Recent evidence has also shown that the nature of tumor cells, in particular their low or high aggressiveness, dictates the type of interactions with MSCs and notably the production of multiple chemokines and prostaglandin E2 upon release of IL-1β by tumor cells. In turn, chemokines produced by MSCs can stimulate the invasiveness and potentially metastatic ability of tumor cells (81, 86, 87). Finally, with BM metastasis of solid tumors, interactions become possible with the other niches of BM MSCs. This interaction might favor a release of new MSCs from the BM to colonize primary tumors but may also affect the properties of MSCs themselves, notably by turning them into CAFs (81).
As stated above, growing tumors establish a chronic state of inflammation that acts locally but also systemically. The BM responds to these stress signals by remodeling the stromal landscape and expanding myeloid cells endowed with anti-inflammatory/immunosuppressive functions, further sustaining tumor progression. Several studies have reported that distant solid or diffuse tumors interfere with hematopoiesis and immune regulation within the BM. Primary breast tumors have thus been shown to generate systemic signals that mobilize BM-derived cells promoting tumor growth and dissemination. Tumor-derived factors also interfere with BM myelopoiesis, increasing the generation of granulocytic-MDSC (88, 89). In a spontaneous model of mammary carcinogenesis, Colombo’s lab revealed modifications of the representation of CXCL12-expressing BM-derived MSCs and CXCR4-expressing myeloid cells (90). Such changes in the hematopoietic compartment occurred as early as at preinvasive disease stages and were concomitant with a deregulation of circulating miRNAs. In addition, extracellular vesicles (EVs) produced by follicular lymphoma B-cells have been shown to promote the polarization of BM-derived MSCs to secrete such factors as CXCL12 that could constitute in turn a BM follicular lymphoma permissive stromal niche (91). In AML xenografts, blast-derived EVs convey endoplasmic reticulum stress in vivo to the animal’s BM stroma. This drives a subsequent osteo-differentiation of MSCs through the incorporation and cell-cell transfer of BMP2 by AML-derived EVs, promoting BM niche remodeling (92). Conversely, many studies provide compelling evidence that the BM can sense distant tissue transformation at premalignant/preinvasive stages and influence cancer progression. Bone-making osteoblasts have the capacity to impact distant cancer progression outside the skeleton in such tumors as melanoma, lung, and breast carcinomas. CXCL12 might constitute one of the systemic bone-derived factors that would directly promote breast cancer cell proliferation and metastasis (93). Other studies indicate that cells of the osteoblast-lineage control cancer progression in the same tumor types at least in part by mobilizing tumor-promoting myeloid cells (94, 95). Finally, BM remodeling could be beneficial or detrimental, depending on the nature of targeted hematopoietic cells, i.e., healthy vs. malignant. For instance, Belkaid’s lab recently showed that dietary restriction promoted memory T-cell accumulation in the BM. This was coordinated by glucocorticoids and associated with BM remodeling that involved an increase in such niche factors as CXCL12, erythropoiesis and adipogenesis. Consequently, this was associated with enhanced protection against infections and tumors (96). This work suggests a strategy to optimize immunological memory during nutritional challenges involving a spatiotemporal reorganization of the BM. Unfortunately, the safe haven of the BM can also be remodeled by malignant cells to disturb normal hematopoiesis. For instance, AML can shape the BM landscape to support malignant growth at the expense of normal hematopoiesis. Indeed, AML onset impaired osteogenesis as well as the production of such hematopoietic factors as CXCL12 (7). Likewise, altered cytokine expression such as a decrease of CXCL12 production in the BM of a mouse model of chronic myelogenous leukemia (CML) conferred a growth advantage to leukemia stem cells (LSCs) over normal stem cells (97). Finally, CXCL12 deletion from MSCs reduced normal HSC numbers but promoted LSC expansion and their elimination by tyrosine kinase inhibitor treatment (98). These findings are consistent with cancer cells impairing normal hematopoiesis and provide a foundation for developing stromal-based therapies.
The relevance of stromal-based therapies is also supported in hematological malignancies by data from Hasselbalch et al. suggesting that chronic inflammation can be a driver of clonal evolution in patients with myeloproliferative neoplasms (MPNs) (99). In primary myelofibrosis (PMF), disease severity and treatment complexity have mainly been attributed to the association of clonal myeloproliferation and profound changes in the BM stroma, associated with an excessive production of cytokines, chemokines, growth factors and ECM components. It was initially reported that stromal changes were reactional and secondary to growth factor production by clonal hematopoietic cells. However, the presence of molecular alterations of PMF MSCs has been shown to provide an “intrinsic” osteogenic signature and an increased differentiation into osteoblasts partly dependent on endogenous TGFβ1 production and activation (100, 101). It has been suggested that the BM stroma of PMF patients is progressively inflammatory-driven by clonal hematopoietic cells towards an “autonomous” state where it becomes independent of hematopoietic cell stimulation. This in turn causes an alteration of the hematopoietic niche and participates in the amplification of the hematopoietic clone. The resulting inflammatory vicious circle becomes unbreakable in the absence of combined stroma targeted therapies (100, 102). Therefore, Stephen Paget’s theory (103) of the “seed (cancer/leukemic cell) and soil (microenvironment)” is fully sustained. However, in PMF, the bad soil (altered MSCs) endorse the bad seed (clonal HSCs), revisiting Paget’s theory in the “bad seed in bad soil” concept (104, 105). This strengthens the importance of stromal cells and their reciprocal interactions with clonal hematopoietic cells in the development and treatment of neoplasia (106).
Cell Metabolism and Communication
While it has long been known that oxygen plays a key role in the proper functioning of mammalian cells, the mechanisms by which these cells adapt to the amount of oxygen available have only became to be understood since the 1990’s, thanks to the work of the three 2019 Nobel Prize winners in Physiology or Medicine Drs Gregg Semenza, William Kealin and Sir Peter J. Ratcliffe (107–111). The notion of hypoxia in hematopoietic niches is even more recent. While the oxygen gradient created by vascularization is understandable in solid tumors, the idea of such a gradient took longer to emerge in the world of hematopoiesis. It is now accepted, but not necessarily integrated, that the oxygen (O2) concentration in the hematopoietic niche varies between 1 to 4% of oxygen, strikingly different from the peripheral blood concentration of 10 to 13% (112–114). As in solid tumors, the overexpression of hypoxia-inducible factors (HIFs) has been reported in leukemia to be a marker of poor prognosis. The metabolic adaptation of tumor cells is one of the hallmarks driving aggressiveness in cancer that is clearly emphasized by low oxygen concentrations. Solid tumor cells are often glucose-addicted as sugar provides metabolic intermediates that support proliferation and migration. Thus, lactate metabolism and acidosis, other characteristics of the hypoxic tumor area, must be highly hypoxically controlled to avoid cell death (115). Based on the work of Nobel Prize winner Otto Warburg in 1931 (116), it has been speculated for decades that mitochondria were failing during the tumor process. This theory finally materialized as the Warburg effect whereby anaerobic fermentation is preferred by some tumor cells. However, several studies have now proven that mitochondria function normally in cancer cells and that blocking oxidative phosphorylation (OXPHOS) is an adaptive event (117, 118).
Although glycolytic metabolic reprogramming is common in cancer cells, several types of cells have been reported to prefer OXPHOS for energy production (119–124). AML cells thus highly depend on OXPHOS to satisfy their heightened demands for energy. Mitochondrial and OXPHOS activities greatly influence the sensitivity and in vivo efficacy of chemotherapeutic agents (125). Increasing evidence reveals that stromal cells affect the characteristics of cancer cells in the tumor microenvironment (126–129). The niche plays an important role in cancer cell metabolism by secreting metabolites that are used for the tricarboxylic acid (TCA)/Krebs cycle (130). Moreover, CAFs enhance the Warburg effect by interacting with cancer cells and producing lactate used by cancer cells as a fuel for mitochondrial OXPHOS (Figure 1). This concept is widely known as the reverse Warburg effect (131–133). Thus, the increase in reactive oxygen species (ROS) promotes the activation of HIF-1α, inducing autophagy, lysosomal degradation and loss of stromal Cav-1, consequently contributing to glycolysis in CAFs. Besides, it has been recently reported that interactions between MSCs and leukemic cells increase oxidative stress in MSCs (134) with a concomitant activation of glutathione (GSH)-based antioxidant defenses, notably through overexpression of GPX3, a key determinant of leukemic cell self-renewal (135, 136). These interactions also enhance leukemic blast bioenergetics by increasing OXPHOS and the TCA cycle (136). All these elements suggest that metabolic interactions within their niche are important for the maintenance of mitochondrial OXPHOS in cancer cells.
Mitochondria are not only involved in energy production through the generation of ATP by OXPHOS. They also support important anabolic reactions and are crucial regulators of apoptosis via the expression of molecules of the BCL-2 family at their surface (137). Horizontal transfer between two cells of mitochondria and/or mitochondrial DNA (mtDNA) via nanotubes, EVs or freely, is likely to have fundamental consequences for the host (Figure 1). A first study showed that active mitochondria and/or mtDNA from human bone marrow MSCs could rescue respiration-deficient (ρ0) lung carcinoma cells (138) and apoptotic PC12 cells (139). This effect was described in several non-cancer situations where stressed cells, frequently experiencing hypoxic or ischemic conditions, could recover after the acquisition of mitochondria from their cellular environment (reviewed in (140)). For instance, BM-derived MSCs have been shown to protect lung epithelial cells from lipopolysaccharide-induced injuries through the donation of mitochondria (141). For cancer cells, two seminal publications have shown that ρ0 cancer cells have an impaired tumorigenic potential that can be restored, together with respiration, by the transfer of mtDNA (142) or active mitochondria (143) from surrounding cells, both in vitro and in vivo. Interestingly, it has also been demonstrated that CAF-derived EVs can transfer mtDNA to OXPHOS-deficient breast cancer cells, leading to the restoration of mitochondrial metabolic activities (144). MSCs moreover could transfer active mitochondria to AML leukemic blasts, especially upon sensitization of leukemic cells by chemotherapy, probably, among other still unclear mechanisms, via AML cell-derived ROS (145, 146). It was also demonstrated that MSCs recognize damaged mitochondria released by leukemic cells under chemotherapy as danger signals and react by stimulating mitochondrial biogenesis followed by transfer of active mitochondria to AML cells (147). Another interesting study showed that BM MSCs from acute lymphoblastic leukemia (ALL) patients harbor a CAF phenotype. Upon chemotherapy and ROS induction, they transfer mitochondria to ALL blasts to support their survival and resistance to chemotherapy (148). The uptake of mitochondria by leukemic cells can increase their mitochondrial mass by up to 14% (145) and is associated with better fitness and a higher resistance to chemotherapy. Since mitochondria-recipient cells become able to resist apoptotic signals, it is possible that this transfer could increase the pool of anti-apoptotic molecules of the BCL-2 family in leukemic blasts. Another obvious effect of mitochondrial transfer is an increase in ATP content (145, 147, 149) and in other important metabolites. A recent study demonstrated that transferred mitochondria were important to sustain pyrimidine synthesis and cell proliferation via the dihydroorotate dehydrogenase (DHODH) enzyme present in the mitochondrial membrane (150). Exogenous mitochondria could also support resistance to ferroptosis cell death as DHODH appears to mediate an important protective pathway against ROS-induced lipid peroxidation that triggers ferroptosis (151). Finally, mitochondrial transfer could modulate immune responses as it has been reported that horizontal transfer from MSCs could trigger Treg differentiation to limit tissue damage and inflammation during graft-versus-host disease (152). Whether this phenomenon also occurs in the BM hematopoietic niche and affects other lymphoid cell subsets such as cytotoxic T-lymphocytes or NK cells during leukemia development remains to be studied.
Vascular Niche, Angiogenesis and Endothelial Plasticity
The vascular endothelium refuels the tumor mass with oxygen and metabolites and settles a favorable microenvironment for tumor growth. This is strikingly illustrated in tumors from the central nervous system, where homeostasis of the cerebral vasculature is crucial. As for embryonic and adult stem cells, cancer stem cells reside within a niche articulated around vascular units (153), defined as the vascular niche. This environment allows privileged control of metabolic conditions, secreted protein dosage as well as fine-tuned regulation of cell adhesion and communication with the surrounding ECM and neighboring ECs (154). Cancer stem cells are indeed located in the close vicinity of tumor blood vessels where ECs are suspected to dictate stem cell identity (155, 156). The concept of (peri)vascular niche is also highly significant in the BM and has evolved through the better characterization of HSPCs. In mice, the HSPC compartment is functionally and molecularly heterogeneous, due in part to an extrinsic control by the BM microenvironment, including ECs. Indeed, recent advances in cell imaging and HSPC reporter-mice have revealed the association of HSPCs with at least two types of blood vessels. The latter are central endothelium featuring sinusoids (157) and an endosteal arterial/arteriolar endothelium which is close to bone diaphysis and epiphysis and defines transition vessels (158) (Figure 1). Sinusoidal and endosteal ECs differ phenotypically, the latter expressing high levels of endomucin and CD31, while sinusoidal ECs display low levels of both these markers. The location of endosteal ECs in bone metaphyses, close to osteoprogenitor cells, allows for an efficient coupling between osteogenesis and angiogenesis. Furthermore, sinusoidal and endosteal ECs are surrounded by unique specific perivascular MSCs. Although most CXCL12 and SCF in the BM is produced by CAR/LepR+ cells (159, 160), ECs are also a source of both niche factors and hence are involved in the hematopoiesis process. Arterial and transition vessel ECs by displaying such a higher expression of CXCL12 and SCF maintain HSPC quiescence, while sinusoidal vessels, fenestrated and more permeable, promote BM cell trafficking (158, 161, 162).
Several studies have revealed the role of the BM vasculature in the development of leukemia and chemoresistance. In AML, vascular niches provide signals that regulate proliferation and stem cell-like properties (163, 164). In a reciprocal way, AML cells release inflammatory cytokines that activate the vascular endothelium, inducing the expression of such adhesion molecules as VCAM1, promoting AML proliferation and chemoresistance (165, 166). External cues emanating from ECs can regulate the fate of cancer stem cells both in solid tumors and leukemia. In cerebral tumors, exploration of the endothelial secretome identified the vasopeptide apelin (APLN) as a central regulator for endothelial-mediated maintenance of patient-derived glioma stem-like cells in vitro and in vivo (167). Further studies confirmed the instrumental role of APLN to sustain tumor cell expansion and progression (168). Likewise, a subpopulation of APLN-expressing ECs in the BM orchestrates HSPC maintenance, and further repopulation in the therapy-induced damaged bone microenvironment (169). In luminal breast carcinoma, the BMP2 was found to be overproduced by ECs from the tumor stroma. This factor is an important actor of the stem cell niche, participating also in the initiation of stem cell transformation (170).
In hematological malignancies, sinusoidal ECs from the BM vascular niche of patients with chronic myeloid leukemia have been shown to be the main source of BMP2 and BMP4, involved in the maintenance and expansion of leukemic stem cells (171). BMP4 overproduction in the AML microenvironment furthermore contributes to blast cells “reprogramming” towards a stem-cell like phenotype (172). In addition, BMP4 produced by the leukemic microenvironment is involved in leukemic stem cell quiescence mediated by Jak2/Stat3 signaling and contributes to relapse and tumor escape (173) (Figure 1). Similar data in solid tumors, from many laboratories, have identified the BMP-signaling pathway as a major driver of BM dormancy (174).
Seed and soil interactions have to be considered as reciprocal, signals provided by cancer cells impacting ECs and vice versa. The influence of cancer cells towards EC is to promote angiogenesis and increase vascular permeability to respectively provide the oxygen required for growth and allow for cell dissemination. In solid tumors, pro-angiogenic factors (VEGF, Sema3A), either soluble or delivered through tumor-derived EVs, contribute to an increase of both angiogenic potential and permeability (175, 176). Malignant hematopoietic cells are high consumers of oxygen and evolve in a hypoxic environment that favors angiogenesis. An increase in BM vascular density and angiogenic markers (VEGF-A, FGF2, VEGF-R) has been highlighted in several hematological malignancies (177, 178). The sites of active angiogenesis in tumor BM niches are still not fully characterized, but ECs in the transient zone close to the endosteal niche could mediate the local growth of blood vessels in normal bone (158). In MPNs and leukemia, neo-vessels are also characterized by an abnormal tortuous architecture (178, 179). In MPNs, increased microvascular density and expression of VEGF have been reported to correlate with the allelic charge of JAK2-V617F mutation (180, 181). In a subset of thrombotic MPN patients, this mutation has been detected in hepatic and splenic ECs as well as in endothelial progenitors, suggesting their clonality (182). More recently, introduction of the JAK2-V617F mutation in ECs has been shown to modify these cells towards a pro-adherent and pro-thrombotic profile (183, 184). These results suggest that, similarly to MSCs, ECs may have acquired intrinsic modifications that participate in the activated/inflammatory state within the BM niche and in leukemic progression. An aberrant increase in permeability is an additional striking feature of tumor blood vessels (185) which strongly alters drug delivery in solid tumors (186). An increased permeability of BM vessels, induced by leukemic cells, could also be associated with an impaired perfusion hampering normal hematopoiesis and supporting malignancy as shown in an AML patient-derived xenograft model (166).
Beside their role in angiogenesis, ECs may also engage in the dynamic process of endothelial-to-mesenchymal transition (EndMT), which drives reprogramming of ECs towards a mesenchymal phenotype (187). Initially described in normal cardiac development, this plasticity has been highlighted in several solid tumors in response to tumor environmental soluble and/or mechanical cues, as well as upon therapeutic assaults (188). EndMT may provide a source of CAFs (189) and contribute to metastasis dissemination by destabilizing the endothelial barrier (190). Furthermore, EndMT has been described as a tumor arm to resist chemo- and radio-therapies (191, 192). Recent data support such a transition process in regenerative human BM, as a subset of ECs in trabecular sinusoid vessels has been shown to display an EndMT transcriptional signature (193). Importantly, this endothelial derived-mesenchymal population harbors properties of pluripotent stromal cells, with multi-lineage differentiation capacity (adipocyte, osteoblast, chondrocyte) and supportive capacity of hematopoiesis. Whether EndMT plays a role in hematological cancer is not confirmed yet, but this process surely could participate in the reconstitution of the hematopoietic BM niche after therapy (193). In the BM and spleen of PMF patients, the presence of microvascular ECs showing functional and morphologic changes associated with the MSC phenotype is in agreement with the potential contribution of EndMT to the BM fibrosis process that characterizes this disease (194).
Conclusion
This review highlights how knowledge is progressing, in both solid tumors and hematological malignancies, in identifying the role of the multiple subsets of cells widely referred to as “cells of the microenvironment”. The latter clearly constitute a network of interacting subsets, which are increasingly well identified, but still incompletely understood. From cytokine/chemokine release patterns to interactions with angiogenesis and oxygen regulation, much remains to be deciphered. However, this review clearly highlights that solid tumors and hematological malignancies use similar strategies to survive in a microenvironment dedicated to their suppression, in particular by modifying the microenvironment to adapt it to tumor growth, while altering its physiological role.
Information generated by single-cell analyses can be used as a blueprint for the identification of CAFs or MSCs subtypes in various organs in different pathological conditions. Comparison of CAF subtypes’ molecular profiles with those of MSCs will be useful to identify potential mechanistic similarities in tumor inflammation and niche alterations across malignancies. Much remains to be done however before transposing the results obtained in mouse models to the primary cells of human tumors and hematologic malignancies.
It is obviously still needed to discover specific means to interfere with the intricate interplay between niche actors that affect cancer/leukemic growth and prevent leukemia relapse. The power of multi-omic analyses of the tumor microenvironment, associated with a pan-tumor integrative approach of cancer niche abnormalities could be decisive in proposing new therapeutic strategies targeting niches in order to eradicate cancer cells.
Author Contributions
All authors made extensive reviews of the literature listed and drafted different sections of the review. NMM drew the figure, which was finalized with the help of SM and OH. OH conceived, designed, supervised and finalized the review. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the GDR 3697 Micronit “Microenvironment of Tumor niches”, a CNRS research network which has been running in France for seven years.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to all the members of the GDR 3697 Micronit for fruitful discussions. We thank Prof. Marie-Christine Béné (Nantes University Hospital and INSERM 1232, CRCINA, Nantes, France) for editing the manuscript. Figure 1 was created from Servier Medical Art by Servier, licensed under a Creative Common Attribution 3.0 Unported License.
References
1. Dexter TM, Allen TD, Lajtha LG. Conditions Controlling the Proliferation of Haemopoietic Stem Cells In Vitro. J Cell Physiol (1977) 91:335–44. doi: 10.1002/jcp.1040910303
2. Whitlock CA, Witte ON. Long-Term Culture of B Lymphocytes and Their Precursors From Murine Bone Marrow. Proc Natl Acad Sci USA (1982) 79:3608–12. doi: 10.1073/pnas.79.11.3608
3. Penn PE, Jiang DZ, Fei RG, Sitnicka E, Wolf NS. Dissecting the Hematopoietic Microenvironment. IX. Further Characterization of Murine Bone Marrow Stromal Cells. Blood (1993) 81:1205–13. doi: 10.1182/blood.V81.5.1205.1205
4. Crane GM, Jeffery E, Morrison SJ. Adult Haematopoietic Stem Cell Niches. Nat Rev Immunol (2017) 17:573–90. doi: 10.1038/nri.2017.53
5. Aurrand-Lions M, Mancini SJC. Murine Bone Marrow Niches From Hematopoietic Stem Cells to B Cells. Int J Mol Sci (2018) 19:E2353. doi: 10.3390/ijms19082353
6. Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Domínguez Á, et al. The Bone Marrow Microenvironment at Single-Cell Resolution. Nature (2019) 569:222–8. doi: 10.1038/s41586-019-1104-8
7. Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell (2019) 177:1915–32.e16. doi: 10.1016/j.cell.2019.04.040
8. Severe N, Karabacak NM, Gustafsson K, Baryawno N, Courties G, Kfoury Y, et al. Stress-Induced Changes in Bone Marrow Stromal Cell Populations Revealed Through Single-Cell Protein Expression Mapping. Cell Stem Cell (2019) 25:570–83.e7. doi: 10.1016/j.stem.2019.06.003
9. Wolock SL, Krishnan I, Tenen DE, Matkins V, Camacho V, Patel S, et al. Mapping Distinct Bone Marrow Niche Populations and Their Differentiation Paths. Cell Rep (2019) 28:302–11.e5. doi: 10.1016/j.celrep.2019.06.031
10. Balzano M, De Grandis M, Vu Manh T-P, Chasson L, Bardin F, Farina A, et al. Nidogen-1 Contributes to the Interaction Network Involved in Pro-B Cell Retention in the Peri-Sinusoidal Hematopoietic Stem Cell Niche. Cell Rep (2019) 26:3257–71.e8. doi: 10.1016/j.celrep.2019.02.065
11. Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, et al. Pdgfrα and CD51 Mark Human Nestin+ Sphere-Forming Mesenchymal Stem Cells Capable of Hematopoietic Progenitor Cell Expansion. J Exp Med (2013) 210:1351–67. doi: 10.1084/jem.20122252
12. Espeli M, Mancini SJC, Breton C, Poirier F, Schiff C. Impaired B-Cell Development at the Pre-BII-Cell Stage in Galectin-1-Deficient Mice Due to Inefficient Pre-BII/stromal Cell Interactions. Blood (2009) 113:5878–86. doi: 10.1182/blood-2009-01-198465
13. Méndez-Ferrer S, Bonnet D, Steensma DP, Hasserjian RP, Ghobrial IM, Gribben JG, et al. Bone Marrow Niches in Haematological Malignancies. Nat Rev Cancer (2020) 20:285–98. doi: 10.1038/s41568-020-0245-2
14. Delahaye MC, Salem K-I, Pelletier J, Aurrand-Lions M, Mancini SJC. Toward Therapeutic Targeting of Bone Marrow Leukemic Niche Protective Signals in B-Cell Acute Lymphoblastic Leukemia. Front Oncol (2020) 10:606540. doi: 10.3389/fonc.2020.606540
15. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang T-H, et al. The Immune Landscape of Cancer. Immunity (2019) 51:411–2. doi: 10.1016/j.immuni.2019.08.004
16. Garner H, de Visser KE. Immune Crosstalk in Cancer Progression and Metastatic Spread: A Complex Conversation. Nat Rev Immunol (2020) 20:483–97. doi: 10.1038/s41577-019-0271-z
17. Hiam-Galvez KJ, Allen BM, Spitzer MH. Systemic Immunity in Cancer. Nat Rev Cancer (2021) 21:345–59. doi: 10.1038/s41568-021-00347-z
18. Maul TM, Chew DW, Nieponice A, Vorp DA. Mechanical Stimuli Differentially Control Stem Cell Behavior: Morphology, Proliferation, and Differentiation. Biomech Model Mechanobiol (2011) 10:939–53. doi: 10.1007/s10237-010-0285-8
19. Amable PR, Teixeira MVT, Carias RBV, Granjeiro JM, Borojevic R. Protein Synthesis and Secretion in Human Mesenchymal Cells Derived From Bone Marrow, Adipose Tissue and Wharton’s Jelly. Stem Cell Res Ther (2014) 5:53. doi: 10.1186/scrt442
20. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells. The International Society for Cellular Therapy Position Statement. Cytotherapy (2006) 8:315–7. doi: 10.1080/14653240600855905
21. Ambrosi TH, Longaker MT. Chan CKF. A Revised Perspective of Skeletal Stem Cell Biology. Front Cell Dev Biol (2019) 7:189. doi: 10.3389/fcell.2019.00189
22. Dunn CM, Kameishi S, Grainger DW, Okano T. Strategies to Address Mesenchymal Stem/Stromal Cell Heterogeneity in Immunomodulatory Profiles to Improve Cell-Based Therapies. Acta Biomater (2021) 133:114–25. doi: 10.1016/j.actbio.2021.03.069
24. Wiese DM, Braid LR. Transcriptome Profiles Acquired During Cell Expansion and Licensing Validate Mesenchymal Stromal Cell Lineage Genes. Stem Cell Res Ther (2020) 11:357. doi: 10.1186/s13287-020-01873-7
25. Zhang S, Wang JY, Li B, Yin F, Liu H. Single-Cell Transcriptome Analysis of Uncultured Human Umbilical Cord Mesenchymal Stem Cells. Stem Cell Res Ther (2021) 12:25. doi: 10.1186/s13287-020-02055-1
26. Korn C, Méndez-Ferrer S. Myeloid Malignancies and the Microenvironment. Blood (2017) 129:811–22. doi: 10.1182/blood-2016-09-670224
27. Corradi G, Baldazzi C, Očadlíková D, Marconi G, Parisi S, Testoni N, et al. Mesenchymal Stromal Cells From Myelodysplastic and Acute Myeloid Leukemia Patients Display In Vitro Reduced Proliferative Potential and Similar Capacity to Support Leukemia Cell Survival. Stem Cell Res Ther (2018) 9:271. doi: 10.1186/s13287-018-1013-z
28. Geyh S, Rodríguez-Paredes M, Jäger P, Khandanpour C, Cadeddu R-P, Gutekunst J, et al. Functional Inhibition of Mesenchymal Stromal Cells in Acute Myeloid Leukemia. Leukemia (2016) 30:683–91. doi: 10.1038/leu.2015.325
29. Sorokina T, Shipounova I, Bigildeev A, Petinati N, Drize N, Turkina A, et al. The Ability of Multipotent Mesenchymal Stromal Cells From the Bone Marrow of Patients With Leukemia to Maintain Normal Hematopoietic Progenitor Cells. Eur J Haematol (2016) 97:245–52. doi: 10.1111/ejh.12713
30. Kim J-A, Shim J-S, Lee G-Y, Yim HW, Kim T-M, Kim M, et al. Microenvironmental Remodeling as a Parameter and Prognostic Factor of Heterogeneous Leukemogenesis in Acute Myelogenous Leukemia. Cancer Res (2015) 75:2222–31. doi: 10.1158/0008-5472.CAN-14-3379
31. Desbourdes L, Javary J, Charbonnier T, Ishac N, Bourgeais J, Iltis A, et al. Alteration Analysis of Bone Marrow Mesenchymal Stromal Cells From De Novo Acute Myeloid Leukemia Patients at Diagnosis. Stem Cells Dev (2017) 26:709–22. doi: 10.1089/scd.2016.0295
32. Diaz de la Guardia R, Lopez-Millan B, Lavoie JR, Bueno C, Castaño J, Gómez-Casares M, et al. Detailed Characterization of Mesenchymal Stem/Stromal Cells From a Large Cohort of AML Patients Demonstrates a Definitive Link to Treatment Outcomes. Stem Cell Rep (2017) 8:1573–86. doi: 10.1016/j.stemcr.2017.04.019
33. Pievani A, Donsante S, Tomasoni C, Corsi A, Dazzi F, Biondi A, et al. Acute Myeloid Leukemia Shapes the Bone Marrow Stromal Niche In Vivo. Haematologica (2021) 106:865–70. doi: 10.3324/haematol.2020.247205
34. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell (2018) 33:463–79.e10. doi: 10.1016/j.ccell.2018.01.011
35. Givel A-M, Kieffer Y, Scholer-Dahirel A, Sirven P, Cardon M, Pelon F, et al. Mir200-Regulated CXCL12β Promotes Fibroblast Heterogeneity and Immunosuppression in Ovarian Cancers. Nat Commun (2018) 9:1056. doi: 10.1038/s41467-018-03348-z
36. Bonneau C, Eliès A, Kieffer Y, Bourachot B, Ladoire S, Pelon F, et al. A Subset of Activated Fibroblasts Is Associated With Distant Relapse in Early Luminal Breast Cancer. Breast Cancer Res (2020) 22:76. doi: 10.1186/s13058-020-01311-9
37. Pelon F, Bourachot B, Kieffer Y, Magagna I, Mermet-Meillon F, Bonnet I, et al. Cancer-Associated Fibroblast Heterogeneity in Axillary Lymph Nodes Drives Metastases in Breast Cancer Through Complementary Mechanisms. Nat Commun (2020) 11:404. doi: 10.1038/s41467-019-14134-w
38. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct Populations of Inflammatory Fibroblasts and Myofibroblasts in Pancreatic Cancer. J Exp Med (2017) 214:579–96. doi: 10.1084/jem.20162024
39. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. Tgfβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature (2018) 554:544–8. doi: 10.1038/nature25501
40. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell (2018) 172:841–56.e16. doi: 10.1016/j.cell.2018.01.009
41. Neuzillet C, Tijeras-Raballand A, Ragulan C, Cros J, Patil Y, Martinet M, et al. Inter- and Intra-Tumoural Heterogeneity in Cancer-Associated Fibroblasts of Human Pancreatic Ductal Adenocarcinoma. J Pathol (2019) 248:51–65. doi: 10.1002/path.5224
42. Strell C, Paulsson J, Jin S-B, Tobin NP, Mezheyeuski A, Roswall P, et al. Impact of Epithelial-Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma In Situ. J Natl Cancer Inst (2019) 111:983–95. doi: 10.1093/jnci/djy234
43. Puré E, Blomberg R. Pro-Tumorigenic Roles of Fibroblast Activation Protein in Cancer: Back to the Basics. Oncogene (2018) 37:4343–57. doi: 10.1038/s41388-018-0275-3
44. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat Rev Cancer (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
45. Mieulet V, Garnier C, Kieffer Y, Guilbert T, Nemati F, Marangoni E, et al. Stiffness Increases With Myofibroblast Content and Collagen Density in Mesenchymal High Grade Serous Ovarian Cancer. Sci Rep (2021) 11:4219. doi: 10.1038/s41598-021-83685-0
46. Le Nail L-R, Brennan M, Rosset P, Deschaseaux F, Piloquet P, Pichon O, et al. Comparison of Tumor- and Bone Marrow-Derived Mesenchymal Stromal/Stem Cells From Patients With High-Grade Osteosarcoma. Int J Mol Sci (2018) 19:E707. doi: 10.3390/ijms19030707
47. Avnet S, Di Pompo G, Chano T, Errani C, Ibrahim-Hashim A, Gillies RJ, et al. Cancer-Associated Mesenchymal Stroma Fosters the Stemness of Osteosarcoma Cells in Response to Intratumoral Acidosis via NF-κb Activation. Int J Cancer (2017) 140:1331–45. doi: 10.1002/ijc.30540
48. Avril P, Le Nail L-R, Brennan MÁ, Rosset P, De Pinieux G, Layrolle P, et al. Mesenchymal Stem Cells Increase Proliferation But Do Not Change Quiescent State of Osteosarcoma Cells: Potential Implications According to the Tumor Resection Status. J Bone Oncol (2016) 5:5–14. doi: 10.1016/j.jbo.2015.11.002
49. Pietrovito L, Leo A, Gori V, Lulli M, Parri M, Becherucci V, et al. Bone Marrow-Derived Mesenchymal Stem Cells Promote Invasiveness and Transendothelial Migration of Osteosarcoma Cells via a Mesenchymal to Amoeboid Transition. Mol Oncol (2018) 12:659–76. doi: 10.1002/1878-0261.12189
50. Chen Y, Kim J, Yang S, Wang H, Wu C-J, Sugimoto H, et al. Type I Collagen Deletion in αsma+ Myofibroblasts Augments Immune Suppression and Accelerates Progression of Pancreatic Cancer. Cancer Cell (2021) 39:548–65.e6. doi: 10.1016/j.ccell.2021.02.007
51. Ziani L, Safta-Saadoun TB, Gourbeix J, Cavalcanti A, Robert C, Favre G, et al. Melanoma-Associated Fibroblasts Decrease Tumor Cell Susceptibility to NK Cell-Mediated Killing Through Matrix-Metalloproteinases Secretion. Oncotarget (2017) 8:19780–94. doi: 10.18632/oncotarget.15540
52. Busek P, Balaziova E, Matrasova I, Hilser M, Tomas R, Syrucek M, et al. Fibroblast Activation Protein Alpha Is Expressed by Transformed and Stromal Cells and is Associated With Mesenchymal Features in Glioblastoma. Tumour Biol (2016) 37:13961–71. doi: 10.1007/s13277-016-5274-9
53. Krepela E, Vanickova Z, Hrabal P, Zubal M, Chmielova B, Balaziova E, et al. Regulation of Fibroblast Activation Protein by Transforming Growth Factor Beta-1 in Glioblastoma Microenvironment. Int J Mol Sci (2021) 22:1046. doi: 10.3390/ijms22031046
54. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 From FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes With Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc Natl Acad Sci USA (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
55. Cremasco V, Astarita JL, Grauel AL, Keerthivasan S, MacIsaac K, Woodruff MC, et al. FAP Delineates Heterogeneous and Functionally Divergent Stromal Cells in Immune-Excluded Breast Tumors. Cancer Immunol Res (2018) 6:1472–85. doi: 10.1158/2326-6066.CIR-18-0098
56. Bartoschek M, Oskolkov N, Bocci M, Lövrot J, Larsson C, Sommarin M, et al. Spatially and Functionally Distinct Subclasses of Breast Cancer-Associated Fibroblasts Revealed by Single Cell RNA Sequencing. Nat Commun (2018) 9:5150. doi: 10.1038/s41467-018-07582-3
57. Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-Cell RNA Sequencing Reveals Stromal Evolution Into LRRC15+ Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov (2020) 10:232–53. doi: 10.1158/2159-8290.CD-19-0644
58. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov (2019) 9:1102–23. doi: 10.1158/2159-8290.CD-19-0094
59. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov (2020) 10:1330–51. doi: 10.1158/2159-8290.CD-19-1384
60. Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW, et al. Cross-Tissue Organization of the Fibroblast Lineage. Nature (2021) 593:575–9. doi: 10.1038/s41586-021-03549-5
61. Logothetis CJ, Lin S-H. Osteoblasts in Prostate Cancer Metastasis to Bone. Nat Rev Cancer (2005) 5:21–8. doi: 10.1038/nrc1528
62. Siclari VA, Guise TA, Chirgwin JM. Molecular Interactions Between Breast Cancer Cells and the Bone Microenvironment Drive Skeletal Metastases. Cancer Metastasis Rev (2006) 25:621–33. doi: 10.1007/s10555-006-9023-1
63. Choueiri MB, Tu S-M, Yu-Lee L-Y, Lin S-H. The Central Role of Osteoblasts in the Metastasis of Prostate Cancer. Cancer Metastasis Rev (2006) 25:601–9. doi: 10.1007/s10555-006-9034-y
64. Hofbauer LC, Bozec A, Rauner M, Jakob F, Perner S, Pantel K. Novel Approaches to Target the Microenvironment of Bone Metastasis. Nat Rev Clin Oncol (2021) 18:488–505. doi: 10.1038/s41571-021-00499-9
65. Esposito M, Guise T, Kang Y. The Biology of Bone Metastasis. Cold Spring Harb Perspect Med (2018) 8:a031252. doi: 10.1101/cshperspect.a031252
66. Wang N, Docherty FE, Brown HK, Reeves KJ, Fowles ACM, Ottewell PD, et al. Prostate Cancer Cells Preferentially Home to Osteoblast-Rich Areas in the Early Stages of Bone Metastasis: Evidence From In Vivo Models. J Bone Miner Res (2014) 29:2688–96. doi: 10.1002/jbmr.2300
67. Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of Chemokine Receptors in Breast Cancer Metastasis. Nature (2001) 410:50–6. doi: 10.1038/35065016
68. Miao R, Lim VY, Kothapalli N, Ma Y, Fossati J, Zehentmeier S, et al. Hematopoietic Stem Cell Niches and Signals Controlling Immune Cell Development and Maintenance of Immunological Memory. Front Immunol (2020) 11:600127. doi: 10.3389/fimmu.2020.600127
69. Bonaud A, Lemos JP, Espéli M, Balabanian K. Hematopoietic Multipotent Progenitors and Plasma Cells: Neighbors or Roommates in the Mouse Bone Marrow Ecosystem? Front Immunol (2021) 12:658535. doi: 10.3389/fimmu.2021.658535
70. Comazzetto S, Shen B, Morrison SJ. Niches That Regulate Stem Cells and Hematopoiesis in Adult Bone Marrow. Dev Cell (2021) 56:1848–60. doi: 10.1016/j.devcel.2021.05.018
71. Aoki K, Kurashige M, Ichii M, Higaki K, Sugiyama T, Kaito T, et al. Identification of CXCL12-Abundant Reticular Cells in Human Adult Bone Marrow. Br J Haematol (2021) 193:659–68. doi: 10.1111/bjh.17396
72. Do HTT, Lee CH, Cho J. Chemokines and Their Receptors: Multifaceted Roles in Cancer Progression and Potential Value as Cancer Prognostic Markers. Cancers (Basel) (2020) 12:E287. doi: 10.3390/cancers12020287
73. Ozga AJ, Chow MT, Luster AD. Chemokines and the Immune Response to Cancer. Immunity (2021) 54:859–74. doi: 10.1016/j.immuni.2021.01.012
74. Boissière-Michot F, Jacot W, Fraisse J, Gourgou S, Timaxian C, Lazennec G. Prognostic Value of CXCR2 in Breast Cancer. Cancers (Basel) (2020) 12:E2076. doi: 10.3390/cancers12082076
75. Lazennec G, Richmond A. Chemokines and Chemokine Receptors: New Insights Into Cancer-Related Inflammation. Trends Mol Med (2010) 16:133–44. doi: 10.1016/j.molmed.2010.01.003
76. Vindrieux D, Escobar P, Lazennec G. Emerging Roles of Chemokines in Prostate Cancer. Endocr Relat Cancer (2009) 16:663–73. doi: 10.1677/ERC-09-0109
77. Bejarano L, Jordāo MJC, Joyce JA. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov (2021) 11:933–59. doi: 10.1158/2159-8290.CD-20-1808
78. Lazennec G, Jorgensen C. Concise Review: Adult Multipotent Stromal Cells and Cancer: Risk or Benefit? Stem Cells (2008) 26:1387–94. doi: 10.1634/stemcells.2007-1006
79. Galiè M, Konstantinidou G, Peroni D, Scambi I, Marchini C, Lisi V, et al. Mesenchymal Stem Cells Share Molecular Signature With Mesenchymal Tumor Cells and Favor Early Tumor Growth in Syngeneic Mice. Oncogene (2008) 27:2542–51. doi: 10.1038/sj.onc.1210920
80. McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S, et al. Human Ovarian Carcinoma–Associated Mesenchymal Stem Cells Regulate Cancer Stem Cells and Tumorigenesis via Altered BMP Production. J Clin Invest (2011) 121:3206–19. doi: 10.1172/JCI45273
81. Lazennec G, Lam PY. Recent Discoveries Concerning the Tumor - Mesenchymal Stem Cell Interactions. Biochim Biophys Acta (2016) 1866:290–9. doi: 10.1016/j.bbcan.2016.10.004
82. Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, et al. Origins of the Tumor Microenvironment: Quantitative Assessment of Adipose-Derived and Bone Marrow-Derived Stroma. PLoS One (2012) 7:e30563. doi: 10.1371/journal.pone.0030563
83. Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal Stem Cells Within Tumour Stroma Promote Breast Cancer Metastasis. Nature (2007) 449:557–63. doi: 10.1038/nature06188
84. Nwabo Kamdje AH, Seke Etet PF, Simo Tagne R, Vecchio L, Lukong KE, Krampera M. Tumor Microenvironment Uses a Reversible Reprogramming of Mesenchymal Stromal Cells to Mediate Pro-Tumorigenic Effects. Front Cell Dev Biol (2020) 8:545126. doi: 10.3389/fcell.2020.545126
85. Galland S, Stamenkovic I. Mesenchymal Stromal Cells in Cancer: A Review of Their Immunomodulatory Functions and Dual Effects on Tumor Progression. J Pathol (2020) 250:555–72. doi: 10.1002/path.5357
86. Escobar P, Bouclier C, Serret J, Bièche I, Brigitte M, Caicedo A, et al. IL-1β Produced by Aggressive Breast Cancer Cells Is One of the Factors That Dictate Their Interactions With Mesenchymal Stem Cells Through Chemokine Production. Oncotarget (2015) 6:29034–47. doi: 10.18632/oncotarget.4732
87. Li H-J, Reinhardt F, Herschman HR, Weinberg RA. Cancer-Stimulated Mesenchymal Stem Cells Create a Carcinoma Stem Cell Niche via Prostaglandin E2 Signaling. Cancer Discov (2012) 2:840–55. doi: 10.1158/2159-8290.CD-12-0101
88. Giese MA, Hind LE, Huttenlocher A. Neutrophil Plasticity in the Tumor Microenvironment. Blood (2019) 133:2159–67. doi: 10.1182/blood-2018-11-844548
89. Galdiero MR, Varricchi G, Loffredo S, Mantovani A, Marone G. Roles of Neutrophils in Cancer Growth and Progression. J Leukoc Biol (2018) 103:457–64. doi: 10.1002/JLB.3MR0717-292R
90. Chiodoni C, Cancila V, Renzi TA, Perrone M, Tomirotti AM, Sangaletti S, et al. Transcriptional Profiles and Stromal Changes Reveal Bone Marrow Adaptation to Early Breast Cancer in Association With Deregulated Circulating microRNAs. Cancer Res (2020) 80:484–98. doi: 10.1158/0008-5472.CAN-19-1425
91. Dumontet E, Pangault C, Roulois D, Desoteux M, Léonard S, Marchand T, et al. Extracellular Vesicles Shed by Follicular Lymphoma B Cells Promote Polarization of the Bone Marrow Stromal Cell Niche. Blood (2021) 138:57–70. doi: 10.1182/blood.2020008791
92. Doron B, Abdelhamed S, Butler JT, Hashmi SK, Horton TM, Kurre P. Transmissible ER Stress Reconfigures the AML Bone Marrow Compartment. Leukemia (2019) 33:918–30. doi: 10.1038/s41375-018-0254-2
93. Devignes C-S, Aslan Y, Brenot A, Devillers A, Schepers K, Fabre S, et al. HIF Signaling in Osteoblast-Lineage Cells Promotes Systemic Breast Cancer Growth and Metastasis in Mice. Proc Natl Acad Sci USA (2018) 115:E992–1001. doi: 10.1073/pnas.1718009115
94. Rossnagl S, Altrock E, Sens C, Kraft S, Rau K, Milsom MD, et al. EDA-Fibronectin Originating From Osteoblasts Inhibits the Immune Response Against Cancer. PLoS Biol (2016) 14:e1002562. doi: 10.1371/journal.pbio.1002562
95. Engblom C, Pfirschke C, Zilionis R, Da Silva Martins J, Bos SA, Courties G, et al. Osteoblasts Remotely Supply Lung Tumors With Cancer-Promoting SiglecFhigh Neutrophils. Science (2017) 358:eaal5081. doi: 10.1126/science.aal5081
96. Collins N, Han S-J, Enamorado M, Link VM, Huang B, Moseman EA, et al. The Bone Marrow Protects and Optimizes Immunological Memory During Dietary Restriction. Cell (2019) 178:1088–101.e15. doi: 10.1016/j.cell.2019.07.049
97. Zhang B, Ho YW, Huang Q, Maeda T, Lin A, Lee S-U, et al. Altered Microenvironmental Regulation of Leukemic and Normal Stem Cells in Chronic Myelogenous Leukemia. Cancer Cell (2012) 21:577–92. doi: 10.1016/j.ccr.2012.02.018
98. Agarwal P, Isringhausen S, Li H, Paterson AJ, He J, Gomariz Á, et al. Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell (2019) 24:769–84.e6. doi: 10.1016/j.stem.2019.02.018
99. Hasselbalch HC, Bjørn ME. MPNs as Inflammatory Diseases: The Evidence, Consequences, and Perspectives. Mediators Inflamm (2015) 2015:102476. doi: 10.1155/2015/102476
100. Martinaud C, Desterke C, Konopacki J, Pieri L, Torossian F, Golub R, et al. Osteogenic Potential of Mesenchymal Stromal Cells Contributes to Primary Myelofibrosis. Cancer Res (2015) 75:4753–65. doi: 10.1158/0008-5472.CAN-14-3696
101. Martinaud C, Desterke C, Konopacki J, Vannucchi AM, Pieri L, Guglielmelli P, et al. Transcriptome Analysis of Bone Marrow Mesenchymal Stromal Cells From Patients With Primary Myelofibrosis. Genom Data (2015) 5:1–2. doi: 10.1016/j.gdata.2015.04.017
102. Desterke C, Martinaud C, Ruzehaji N, Le Bousse-Kerdilès M-C. Inflammation as a Keystone of Bone Marrow Stroma Alterations in Primary Myelofibrosis. Mediators Inflamm (2015) 2015:415024. doi: 10.1155/2015/415024
103. Paget S. Distribution of Secondary Growths in Cancer of the Breast. Lancet (1889) 133:571–3. doi: 10.1016/S0140-6736(00)49915-0
104. Lataillade J-J, Pierre-Louis O, Hasselbalch HC, Uzan G, Jasmin C, Martyré M-C, et al. French INSERM and the European EUMNET Networks on Myelofibrosis. Does Primary Myelofibrosis Involve a Defective Stem Cell Niche? From Concept to Evidence. Blood (2008) 112:3026–35. doi: 10.1182/blood-2008-06-158386
105. Le Bousse-Kerdilès M-C. Primary Myelofibrosis and the “Bad Seeds in Bad Soil” Concept. Fibrogenesis Tissue Repair (2012) 5:S20. doi: 10.1186/1755-1536-5-S1-S20
106. Asher S, McLornan DP, Harrison CN. Current and Future Therapies for Myelofibrosis. Blood Rev (2020) 42:100715. doi: 10.1016/j.blre.2020.100715
107. Wang GL, Semenza GL. Purification and Characterization of Hypoxia-Inducible Factor 1. J Biol Chem (1995) 270:1230–7. doi: 10.1074/jbc.270.3.1230
108. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-Inducible Factor 1 is a Basic-Helix-Loop-Helix-PAS Heterodimer Regulated by Cellular O2 Tension. Proc Natl Acad Sci USA (1995) 92:5510–4. doi: 10.1073/pnas.92.12.5510
109. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The Tumour Suppressor Protein VHL Targets Hypoxia-Inducible Factors for Oxygen-Dependent Proteolysis. Nature (1999) 399:271–5. doi: 10.1038/20459
110. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha Targeted for VHL-Mediated Destruction by Proline Hydroxylation: Implications for O2 Sensing. Science (2001) 292:464–8. doi: 10.1126/science.1059817
111. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-Alpha to the Von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science (2001) 292:468–72. doi: 10.1126/science.1059796
112. Wielockx B, Grinenko T, Mirtschink P, Chavakis T. Hypoxia Pathway Proteins in Normal and Malignant Hematopoiesis. Cells (2019) 8:E155. doi: 10.3390/cells8020155
113. Deynoux M, Sunter N, Hérault O, Mazurier F. Hypoxia and Hypoxia-Inducible Factors in Leukemias. Front Oncol (2016) 6:41. doi: 10.3389/fonc.2016.00041
114. Chabi S, Uzan B, Naguibneva I, Rucci J, Fahy L, Calvo J, et al. Hypoxia Regulates Lymphoid Development of Human Hematopoietic Progenitors. Cell Rep (2019) 29:2307–20.e6. doi: 10.1016/j.celrep.2019.10.050
115. Brahimi-Horn MC, Pouysségur J. Hypoxia in Cancer Cell Metabolism and pH Regulation. Essays Biochem (2007) 43:165–78. doi: 10.1042/BSE0430165
116. Warburg O. On the Origin of Cancer Cells. Science (1956) 123:309–14. doi: 10.1126/science.123.3191.309
117. Ward PS, Thompson CB. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell (2012) 21:297–308. doi: 10.1016/j.ccr.2012.02.014
118. Dang CV. Links Between Metabolism and Cancer. Genes Dev (2012) 26:877–90. doi: 10.1101/gad.189365.112
119. Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, et al. Metabolic State of Glioma Stem Cells and Nontumorigenic Cells. Proc Natl Acad Sci USA (2011) 108:16062–7. doi: 10.1073/pnas.1106704108
120. Janiszewska M, Suvà ML, Riggi N, Houtkooper RH, Auwerx J, Clément-Schatlo V, et al. Imp2 Controls Oxidative Phosphorylation and Is Crucial for Preserving Glioblastoma Cancer Stem Cells. Genes Dev (2012) 26:1926–44. doi: 10.1101/gad.188292.112
121. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, et al. Oncogene Ablation-Resistant Pancreatic Cancer Cells Depend on Mitochondrial Function. Nature (2014) 514:628–32. doi: 10.1038/nature13611
122. Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab (2015) 22:590–605. doi: 10.1016/j.cmet.2015.08.015
123. Vazquez F, Lim J-H, Chim H, Bhalla K, Girnun G, Pierce K, et al. Pgc1α Expression Defines a Subset of Human Melanoma Tumors With Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell (2013) 23:287–301. doi: 10.1016/j.ccr.2012.11.020
124. Gentric G, Kieffer Y, Mieulet V, Goundiam O, Bonneau C, Nemati F, et al. PML-Regulated Mitochondrial Metabolism Enhances Chemosensitivity in Human Ovarian Cancers. Cell Metab (2019) 29:156–73.e10. doi: 10.1016/j.cmet.2018.09.002
125. Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells But Require Oxidative Metabolism. Cancer Discov (2017) 7:716–35. doi: 10.1158/2159-8290.CD-16-0441
126. Ishimoto T, Sawayama H, Sugihara H, Baba H. Interaction Between Gastric Cancer Stem Cells and the Tumor Microenvironment. J Gastroenterol (2014) 49:1111–20. doi: 10.1007/s00535-014-0952-0
127. Bu L, Baba H, Yoshida N, Miyake K, Yasuda T, Uchihara T, et al. Biological Heterogeneity and Versatility of Cancer-Associated Fibroblasts in the Tumor Microenvironment. Oncogene (2019) 38:4887–901. doi: 10.1038/s41388-019-0765-y
128. Prager BC, Xie Q, Bao S, Rich JN. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell (2019) 24:41–53. doi: 10.1016/j.stem.2018.12.009
129. Gentric G, Mechta-Grigoriou F. Tumor Cells and Cancer-Associated Fibroblasts: An Updated Metabolic Perspective. Cancers (Basel) (2021) 13:399. doi: 10.3390/cancers13030399
130. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic Stellate Cells Support Tumour Metabolism Through Autophagic Alanine Secretion. Nature (2016) 536:479–83. doi: 10.1038/nature19084
131. Wilde L, Roche M, Domingo-Vidal M, Tanson K, Philp N, Curry J, et al. Metabolic Coupling and the Reverse Warburg Effect in Cancer: Implications for Novel Biomarker and Anticancer Agent Development. Semin Oncol (2017) 44:198–203. doi: 10.1053/j.seminoncol.2017.10.004
132. Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting Lactate-Fueled Respiration Selectively Kills Hypoxic Tumor Cells in Mice. J Clin Invest (2008) 118:3930–42. doi: 10.1172/JCI36843
133. Migneco G, Whitaker-Menezes D, Chiavarina B, Castello-Cros R, Pavlides S, Pestell RG, et al. Glycolytic Cancer Associated Fibroblasts Promote Breast Cancer Tumor Growth, Without a Measurable Increase in Angiogenesis: Evidence for Stromal-Epithelial Metabolic Coupling. Cell Cycle (2010) 9:2412–22. doi: 10.4161/cc.9.12.11989
134. Vignon C, Debeissat C, Bourgeais J, Gallay N, Kouzi F, Anginot A, et al. Involvement of GPx-3 in the Reciprocal Control of Redox Metabolism in the Leukemic Niche. Int J Mol Sci (2020) 21:E8584. doi: 10.3390/ijms21228584
135. Herault O, Hope KJ, Deneault E, Mayotte N, Chagraoui J, Wilhelm BT, et al. A Role for GPx3 in Activity of Normal and Leukemia Stem Cells. J Exp Med (2012) 209:895–901. doi: 10.1084/jem.20102386
136. Forte D, García-Fernández M, Sánchez-Aguilera A, Stavropoulou V, Fielding C, Martín-Pérez D, et al. Bone Marrow Mesenchymal Stem Cells Support Acute Myeloid Leukemia Bioenergetics and Enhance Antioxidant Defense and Escape From Chemotherapy. Cell Metab (2020) 32:829–43.e9. doi: 10.1016/j.cmet.2020.09.001
137. Herst PM, Rowe MR, Carson GM, Berridge MV. Functional Mitochondria in Health and Disease. Front Endocrinol (Lausanne) (2017) 8:296. doi: 10.3389/fendo.2017.00296
138. Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial Transfer Between Cells Can Rescue Aerobic Respiration. Proc Natl Acad Sci USA (2006) 103:1283–8. doi: 10.1073/pnas.0510511103
139. Wang X, Gerdes H-H. Transfer of Mitochondria via Tunneling Nanotubes Rescues Apoptotic PC12 Cells. Cell Death Differ (2015) 22:1181–91. doi: 10.1038/cdd.2014.211
140. Griessinger E, Moschoi R, Biondani G, Peyron J-F. Mitochondrial Transfer in the Leukemia Microenvironment. Trends Cancer (2017) 3:828–39. doi: 10.1016/j.trecan.2017.10.003
141. Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, et al. Mitochondrial Transfer From Bone-Marrow-Derived Stromal Cells to Pulmonary Alveoli Protects Against Acute Lung Injury. Nat Med (2012) 18:759–65. doi: 10.1038/nm.2736
142. Tan AS, Baty JW, Dong L-F, Bezawork-Geleta A, Endaya B, Goodwin J, et al. Mitochondrial Genome Acquisition Restores Respiratory Function and Tumorigenic Potential of Cancer Cells Without Mitochondrial DNA. Cell Metab (2015) 21:81–94. doi: 10.1016/j.cmet.2014.12.003
143. Dong L-F, Kovarova J, Bajzikova M, Bezawork-Geleta A, Svec D, Endaya B, et al. Horizontal Transfer of Whole Mitochondria Restores Tumorigenic Potential in Mitochondrial DNA-Deficient Cancer Cells. Elife (2017) 6:e22187. doi: 10.7554/eLife.22187
144. Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, et al. Packaging and Transfer of Mitochondrial DNA via Exosomes Regulate Escape From Dormancy in Hormonal Therapy-Resistant Breast Cancer. Proc Natl Acad Sci USA (2017) 114:E9066–75. doi: 10.1073/pnas.1704862114
145. Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, Prebet T, et al. Protective Mitochondrial Transfer From Bone Marrow Stromal Cells to Acute Myeloid Leukemic Cells During Chemotherapy. Blood (2016) 128:253–64. doi: 10.1182/blood-2015-07-655860
146. Marlein CR, Zaitseva L, Piddock RE, Robinson SD, Edwards DR, Shafat MS, et al. NADPH Oxidase-2 Derived Superoxide Drives Mitochondrial Transfer From Bone Marrow Stromal Cells to Leukemic Blasts. Blood (2017) 130:1649–60. doi: 10.1182/blood-2017-03-772939
147. Mahrouf-Yorgov M, Augeul L, Da Silva CC, Jourdan M, Rigolet M, Manin S, et al. Mesenchymal Stem Cells Sense Mitochondria Released From Damaged Cells as Danger Signals to Activate Their Rescue Properties. Cell Death Differ (2017) 24:1224–38. doi: 10.1038/cdd.2017.51
148. Burt R, Dey A, Aref S, Aguiar M, Akarca A, Bailey K, et al. Activated Stromal Cells Transfer Mitochondria to Rescue Acute Lymphoblastic Leukemia Cells From Oxidative Stress. Blood (2019) 134:1415–29. doi: 10.1182/blood.2019001398
149. Caicedo A, Fritz V, Brondello J-M, Ayala M, Dennemont I, Abdellaoui N, et al. Mitoception as a New Tool to Assess the Effects of Mesenchymal Stem/Stromal Cell Mitochondria on Cancer Cell Metabolism and Function. Sci Rep (2015) 5:9073. doi: 10.1038/srep09073
150. Bajzikova M, Kovarova J, Coelho AR, Boukalova S, Oh S, Rohlenova K, et al. Reactivation of Dihydroorotate Dehydrogenase-Driven Pyrimidine Biosynthesis Restores Tumor Growth of Respiration-Deficient Cancer Cells. Cell Metab (2019) 29:399–16.e10. doi: 10.1016/j.cmet.2018.10.014
151. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-Mediated Ferroptosis Defence Is a Targetable Vulnerability in Cancer. Nature (2021) 593:586–90. doi: 10.1038/s41586-021-03539-7
152. Court AC, Le-Gatt A, Luz-Crawford P, Parra E, Aliaga-Tobar V, Bátiz LF, et al. Mitochondrial Transfer From MSCs to T Cells Induces Treg Differentiation and Restricts Inflammatory Response. EMBO Rep (2020) 21:e48052. doi: 10.15252/embr.201948052
153. Gilbertson RJ, Rich JN. Making a Tumour’s Bed: Glioblastoma Stem Cells and the Vascular Niche. Nat Rev Cancer (2007) 7:733–6. doi: 10.1038/nrc2246
154. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell (2007) 11:69–82. doi: 10.1016/j.ccr.2006.11.020
155. Galan-Moya EM, Le Guelte A, Lima Fernandes E, Thirant C, Dwyer J, Bidere N, et al. Secreted Factors From Brain Endothelial Cells Maintain Glioblastoma Stem-Like Cell Expansion Through the mTOR Pathway. EMBO Rep (2011) 12:470–6. doi: 10.1038/embor.2011.39
156. Griveau A, Seano G, Shelton SJ, Kupp R, Jahangiri A, Obernier K, et al. A Glial Signature and Wnt7 Signaling Regulate Glioma-Vascular Interactions and Tumor Microenvironment. Cancer Cell (2018) 33:874–89.e7. doi: 10.1016/j.ccell.2018.03.020
157. Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep Imaging of Bone Marrow Shows Non-Dividing Stem Cells Are Mainly Perisinusoidal. Nature (2015) 526:126–30. doi: 10.1038/nature15250
158. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of Angiogenesis and Osteogenesis by a Specific Vessel Subtype in Bone. Nature (2014) 507:323–8. doi: 10.1038/nature13145
159. Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, et al. The Essential Functions of Adipo-Osteogenic Progenitors as the Hematopoietic Stem and Progenitor Cell Niche. Immunity (2010) 33:387–99. doi: 10.1016/j.immuni.2010.08.017
160. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and Perivascular Cells Maintain Haematopoietic Stem Cells. Nature (2012) 481:457–62. doi: 10.1038/nature10783
161. Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, et al. Distinct Bone Marrow Blood Vessels Differentially Regulate Haematopoiesis. Nature (2016) 532:323–8. doi: 10.1038/nature17624
162. Ramasamy SK, Kusumbe AP, Itkin T, Gur-Cohen S, Lapidot T, Adams RH. Regulation of Hematopoiesis and Osteogenesis by Blood Vessel-Derived Signals. Annu Rev Cell Dev Biol (2016) 32:649–75. doi: 10.1146/annurev-cellbio-111315-124936
163. Barbier V, Erbani J, Fiveash C, Davies JM, Tay J, Tallack MR, et al. Endothelial E-Selectin Inhibition Improves Acute Myeloid Leukaemia Therapy by Disrupting Vascular Niche-Mediated Chemoresistance. Nat Commun (2020) 11:2042. doi: 10.1038/s41467-020-15817-5
164. Cogle CR, Goldman DC, Madlambayan GJ, Leon RP, Masri AA, Clark HA, et al. Functional Integration of Acute Myeloid Leukemia Into the Vascular Niche. Leukemia (2014) 28:1978–87. doi: 10.1038/leu.2014.109
165. Duarte D, Hawkins ED, Akinduro O, Ang H, De Filippo K, Kong IY, et al. Inhibition of Endosteal Vascular Niche Remodeling Rescues Hematopoietic Stem Cell Loss in AML. Cell Stem Cell (2018) 22:64–77.e6. doi: 10.1016/j.stem.2017.11.006
166. Passaro D, Di Tullio A, Abarrategi A, Rouault-Pierre K, Foster K, Ariza-McNaughton L, et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell (2017) 32:324–41.e6. doi: 10.1016/j.ccell.2017.08.001
167. Harford-Wright E, Andre-Gregoire G, Jacobs KA, Treps L, Le Gonidec S, Leclair HM, et al. Pharmacological Targeting of Apelin Impairs Glioblastoma Growth. Brain (2017) 140:2939–54. doi: 10.1093/brain/awx253
168. Uribesalgo I, Hoffmann D, Zhang Y, Kavirayani A, Lazovic J, Berta J, et al. Apelin Inhibition Prevents Resistance and Metastasis Associated With Anti-Angiogenic Therapy. EMBO Mol Med (2019) 11:e9266. doi: 10.15252/emmm.201809266
169. Chen Q, Liu Y, Jeong H-W, Stehling M, Dinh VV, Zhou B, et al. Apelin+ Endothelial Niche Cells Control Hematopoiesis and Mediate Vascular Regeneration After Myeloablative Injury. Cell Stem Cell (2019) 25:768–83.e6. doi: 10.1016/j.stem.2019.10.006
170. Chapellier M, Bachelard-Cascales E, Schmidt X, Clément F, Treilleux I, Delay E, et al. Disequilibrium of BMP2 Levels in the Breast Stem Cell Niche Launches Epithelial Transformation by Overamplifying BMPR1B Cell Response. Stem Cell Rep (2015) 4:239–54. doi: 10.1016/j.stemcr.2014.12.007
171. Laperrousaz B, Jeanpierre S, Sagorny K, Voeltzel T, Ramas S, Kaniewski B, et al. Primitive CML Cell Expansion Relies on Abnormal Levels of BMPs Provided by the Niche and on BMPRIb Overexpression. Blood (2013) 122:3767–77. doi: 10.1182/blood-2013-05-501460
172. Voeltzel T, Flores-Violante M, Zylbersztejn F, Lefort S, Billandon M, Jeanpierre S, et al. A New Signaling Cascade Linking BMP4, BMPR1A, Δnp73 and NANOG Impacts on Stem-Like Human Cell Properties and Patient Outcome. Cell Death Dis (2018) 9:1011. doi: 10.1038/s41419-018-1042-7
173. Jeanpierre S, Arizkane K, Thongjuea S, Grockowiak E, Geistlich K, Barral L, et al. The Quiescent Fraction of Chronic Myeloid Leukemic Stem Cells Depends on BMPR1B, Stat3 and BMP4-Niche Signals to Persist in Patients in Remission. Haematologica (2021) 106:111–22. doi: 10.3324/haematol.2019.232793
174. Risson E, Nobre AR, Maguer-Satta V, Aguirre-Ghiso JA. The Current Paradigm and Challenges Ahead for the Dormancy of Disseminated Tumor Cells. Nat Cancer (2020) 1:672–80. doi: 10.1038/s43018-020-0088-5
175. Treps L, Edmond S, Harford-Wright E, Galan-Moya EM, Schmitt A, Azzi S, et al. Extracellular Vesicle-Transported Semaphorin3A Promotes Vascular Permeability in Glioblastoma. Oncogene (2016) 35:2615–23. doi: 10.1038/onc.2015.317
176. Treps L, Perret R, Edmond S, Ricard D, Gavard J. Glioblastoma Stem-Like Cells Secrete the Pro-Angiogenic VEGF-A Factor in Extracellular Vesicles. J Extracell Vesicles (2017) 6:1359479. doi: 10.1080/20013078.2017.1359479
177. Aguayo A, Kantarjian H, Manshouri T, Gidel C, Estey E, Thomas D, et al. Angiogenesis in Acute and Chronic Leukemias and Myelodysplastic Syndromes. Blood (2000) 96:2240–5. doi: 10.1182/blood.V96.6.2240
178. Lundberg LG, Lerner R, Sundelin P, Rogers R, Folkman J, Palmblad J. Bone Marrow in Polycythemia Vera, Chronic Myelocytic Leukemia, and Myelofibrosis Has an Increased Vascularity. Am J Pathol (2000) 157:15–9. doi: 10.1016/S0002-9440(10)64511-7
179. Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, et al. Acute Myelogenous Leukemia-Induced Sympathetic Neuropathy Promotes Malignancy in an Altered Hematopoietic Stem Cell Niche. Cell Stem Cell (2014) 15:365–75. doi: 10.1016/j.stem.2014.06.020
180. Medinger M, Skoda R, Gratwohl A, Theocharides A, Buser A, Heim D, et al. Angiogenesis and Vascular Endothelial Growth Factor-/Receptor Expression in Myeloproliferative Neoplasms: Correlation With Clinical Parameters and JAK2-V617F Mutational Status. Br J Haematol (2009) 146:150–7. doi: 10.1111/j.1365-2141.2009.07726.x
181. Rosti V, Villani L, Riboni R, Poletto V, Bonetti E, Tozzi L, et al. Spleen Endothelial Cells From Patients With Myelofibrosis Harbor the JAK2V617F Mutation. Blood (2013) 121:360–8. doi: 10.1182/blood-2012-01-404889
182. Teofili L, Martini M, Iachininoto MG, Capodimonti S, Nuzzolo ER, Torti L, et al. Endothelial Progenitor Cells Are Clonal and Exhibit the JAK2(V617F) Mutation in a Subset of Thrombotic Patients With Ph-Negative Myeloproliferative Neoplasms. Blood (2011) 117:2700–7. doi: 10.1182/blood-2010-07-297598
183. Guy A, Gourdou-Latyszenok V, Le Lay N, Peghaire C, Kilani B, Dias JV, et al. Vascular Endothelial Cell Expression of JAK2V617F Is Sufficient to Promote a Pro-Thrombotic State Due to Increased P-Selectin Expression. Haematologica (2019) 104:70–81. doi: 10.3324/haematol.2018.195321
184. Guadall A, Lesteven E, Letort G, Awan Toor S, Delord M, Pognant D, et al. Endothelial Cells Harbouring the JAK2V617F Mutation Display Pro-Adherent and Pro-Thrombotic Features. Thromb Haemost (2018) 118:1586–99. doi: 10.1055/s-0038-1667015
185. Weis SM, Cheresh DA. Pathophysiological Consequences of VEGF-Induced Vascular Permeability. Nature (2005) 437:497–504. doi: 10.1038/nature03987
186. Azzi S, Hebda JK, Gavard J. Vascular Permeability and Drug Delivery in Cancers. Front Oncol (2013) 3:211. doi: 10.3389/fonc.2013.00211
187. Dejana E, Hirschi KK, Simons M. The Molecular Basis of Endothelial Cell Plasticity. Nat Commun (2017) 8:14361. doi: 10.1038/ncomms14361
188. Clere N, Renault S, Corre I. Endothelial-To-Mesenchymal Transition in Cancer. Front Cell Dev Biol (2020) 8:747. doi: 10.3389/fcell.2020.00747
189. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res (2007) 67:10123–8. doi: 10.1158/0008-5472.CAN-07-3127
190. Krizbai IA, Gasparics Á, Nagyőszi P, Fazakas C, Molnár J, Wilhelm I, et al. Endothelial-Mesenchymal Transition of Brain Endothelial Cells: Possible Role During Metastatic Extravasation. PLoS One (2015) 10:e0119655. doi: 10.1371/journal.pone.0119655
191. Choi S-H, Kim A-R, Nam J-K, Kim J-M, Kim J-Y, Seo HR, et al. Tumour-Vasculature Development via Endothelial-to-Mesenchymal Transition After Radiotherapy Controls CD44v6+ Cancer Cell and Macrophage Polarization. Nat Commun (2018) 9:5108. doi: 10.1038/s41467-018-07470-w
192. Huang M, Zhang D, Wu JY, Xing K, Yeo E, Li C, et al. Wnt-Mediated Endothelial Transformation Into Mesenchymal Stem Cell-Like Cells Induces Chemoresistance in Glioblastoma. Sci Transl Med (2020) 12:eaay7522. doi: 10.1126/scitranslmed.aay7522
193. Kenswil KJG, Pisterzi P, Sánchez-Duffhues G, van Dijk C, Lolli A, Knuth C, et al. Endothelium-Derived Stromal Cells Contribute to Hematopoietic Bone Marrow Niche Formation. Cell Stem Cell (2021) 28:653–70.e11. doi: 10.1016/j.stem.2021.01.006
Keywords: microenvironment, cancer-associated fibroblasts (CAFs), mesenchymal stem/stromal cells (MSCs), cytokines and chemokines, energy/oxidative metabolism, mitochondrial transfer, angiogenesis, endothelial plasticity
Citation: Mancini SJC, Balabanian K, Corre I, Gavard J, Lazennec G, Le Bousse-Kerdilès M-C, Louache F, Maguer-Satta V, Mazure NM, Mechta-Grigoriou F, Peyron J-F, Trichet V and Herault O (2021) Deciphering Tumor Niches: Lessons From Solid and Hematological Malignancies. Front. Immunol. 12:766275. doi: 10.3389/fimmu.2021.766275
Received: 28 August 2021; Accepted: 25 October 2021;
Published: 10 November 2021.
Edited by:
Daniel F Legler, Biotechnology Institute Thurgau, SwitzerlandReviewed by:
Tanja Nicole Hartmann, University of Freiburg Medical Center, GermanyBarbara Molon, University of Padua, Italy
Takashi Nagasawa, Osaka University, Japan
Copyright © 2021 Mancini, Balabanian, Corre, Gavard, Lazennec, Le Bousse-Kerdilès, Louache, Maguer-Satta, Mazure, Mechta-Grigoriou, Peyron, Trichet and Herault. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Herault, b2xpdmllci5oZXJhdWx0QHVuaXYtdG91cnMuZnI=
†These authors have contributed equally to this work and share first authorship