 Katina D. Hulme
Katina D. Hulme Ellesandra C. Noye1
Ellesandra C. Noye1 Kirsty R. Short
Kirsty R. Short Larisa I. Labzin
Larisa I. Labzin- 1School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, QLD, Australia
- 2Australian Infectious Diseases Research Centre, The University of Queensland, Brisbane, QLD, Australia
- 3Institute for Molecular Bioscience, The University of Queensland, Brisbane, QLD, Australia
Acute inflammation is a critical host defense response during viral infection. When dysregulated, inflammation drives immunopathology and tissue damage. Excessive, damaging inflammation is a hallmark of both pandemic influenza A virus (IAV) infections and Severe Acute Respiratory Syndrome-Coronavirus-2 (SARS-CoV-2) infections. Chronic, low-grade inflammation is also a feature of obesity. In recent years, obesity has been recognized as a growing pandemic with significant mortality and associated costs. Obesity is also an independent risk factor for increased disease severity and death during both IAV and SARS-CoV-2 infection. This review focuses on the effect of obesity on the inflammatory response in the context of viral respiratory infections and how this leads to increased viral pathology. Here, we will review the fundamentals of inflammation, how it is initiated in IAV and SARS-CoV-2 infection and its link to disease severity. We will examine how obesity drives chronic inflammation and trained immunity and how these impact the immune response to IAV and SARS-CoV-2. Finally, we review both medical and non-medical interventions for obesity, how they impact on the inflammatory response and how they could be used to prevent disease severity in obese patients. As projections of global obesity numbers show no sign of slowing down, future pandemic preparedness will require us to consider the metabolic health of the population. Furthermore, if weight-loss alone is insufficient to reduce the risk of increased respiratory virus-related mortality, closer attention must be paid to a patient’s history of health, and new therapeutic options identified.
Introduction
The prevalence of obesity has nearly tripled since 1975 (1, 2). By 2030 it is predicted that 40% of the world’s population will be overweight (Body Mass Index, BMI > 25 kg/m2) and 20% obese (BMI > 30 kg/m2) (3, 4). Obesity is a strong risk factor for many non-communicable diseases including: metabolic disorders (e.g., diabetes mellitus), pulmonary diseases (e.g., obesity hypoventilation syndrome), cardiovascular diseases (e.g. stoke and myocardial infarction), digestive diseases (e.g. gallstones and non-alcoholic fatty liver disease), and cancers (notably thyroid, colon and renal cancers) (4, 5). There are a number of factors that confer obesity risk, including dietary choices, socioeconomic status, sedentary lifestyle, and genetics (6). While, the exact drivers of obesity remain contentious, obesity is more than energy intake simply outweighing energy expenditure (6).
The relationship between obesity and respiratory viral disease came to prominence during the 2009 ‘swine flu’ pandemic (7). Influenza viruses are negative-sense, segmented, single stranded RNA viruses (Orthomyxoviridae family) (8). Of the four subtypes of influenza (A, B, C, D), influenza A viruses (IAVs) are the most predominant in humans, birds and other animal species. IAVs are classified by the surface glycoproteins hemagglutinin (HA) and neuraminidase (NA), of which eighteen and nine types have been isolated and identified respectively (9). Due to a low fidelity RNA polymerase, influenza virus rapidly evolves with changes primarily in the HA and NA proteins. This antigenic drift makes IAV adept at escaping established humoral immunity, acquired either through natural infection or vaccination. The segmented IAV genome allows for reassortment of segments from different IAV strains in a susceptible animal host, and this rapid antigenic shift can lead to new IAV pandemics, such as the April 2009 ‘swine flu’ H1N1. While seasonal H1N1s had been circulating previously, this new pandemic H1N1 strain caused up to 575,400 deaths in the first year of the pandemic and outbreaks have continued to cause serious illness and mortality (10, 11).
SARS-CoV-2 is a positive sense, single-stranded RNA enveloped virus (β coronavirus family) (12), which binds to the angiotensin-converting-enzyme 2 (ACE2) receptor for entry into host cells (13). As of September 2021, there have been over 232 million recorded cases of COVID-19, the disease caused by severe SARS-CoV-2 infection, with over 4.7 million recorded deaths worldwide (14). The emergence of multiple variants with apparent increased transmissibility and potential for evasion of vaccine-induced immunity poses an ongoing concern during this pandemic (15). Acute COVID-19 can range from asymptomatic to fatal disease, with symptoms including fever, myalgia, headache, respiratory symptoms, loss of taste and smell, cardiovascular complications and gastrointestinal symptoms in addition to pulmonary complications (16). These symptoms may persist for more than 4 weeks post the initial diagnosis, leading to a condition widely referred to as long COVID. Symptoms associated with long COVID include, fatigue, breathlessness, cardiac complications and neurological disease (17). The prevalence and causes of long COVID have yet to be defined.
Independent of other co-morbidities or risk factors, individuals with obesity were at higher risk of death due to IAV infection during the 2009 pandemic (7). Even when influenza vaccination is accounted for, obese adults are still twice as likely to develop influenza or influenza-like illness compared to healthy-weight adults (18). Accordingly, obese mice infected with IAV have a 6-fold increase in mortality compared to their non-obese counterparts (19), and obesity has been shown to increase the cardiovascular complications associated with IAV (20). Obesity has been reported as an independent risk factor for respiratory failure and invasive mechanical ventilation, intensive care unit (ICU) admission and death among patients hospitalized with SARS-CoV-2 infections (21–23). While obesity is a clear risk factor for respiratory viral infections, it also correlates with increased disease severity during other infections, including Mycobacterium tuberculosis, Helicobacter pylori, coxsackievirus, and encephalomyocarditis virus (24–28). While the pathogens differ, these studies suggest that a dysregulated immune response in the obese host fails to clear the pathogen and/or drives immune mediated tissue damage. For IAV and SARS-CoV-2, this may result in increased viral shedding in obese individuals (29, 30). Obesity may also promote emergence of more virulent variants of IAV (31). Obese individuals have higher ACE2 in their bronchial epithelial cells (32), suggesting increased opportunity for SARS-CoV-2 infection and replication. Experimentally, obesity increases the severity and associated mortality of secondary bacterial infections following influenza virus infection, regardless of vaccination status (33). As of yet, there are no comprehensive clinical studies to confirm these findings in humans. Intriguingly, increased BMI correlates with better outcomes (decreased case fatality rates) in bacterial respiratory infections alone, recalling the phenomenon of the ‘obesity paradox’ (34–36). The underlying mechanism for this increased protection against bacterial infection in obese patients is unknown.
In this review, we explore the hypothesis that obesity dysregulates the baseline inflammatory response, thus dysregulating innate inflammatory responses to IAV and SARS-CoV-2 infection and thereby increasing disease severity.
Inflammation in Host Defense and Homeostasis
An effective immune response is the key to clearing any infection, and the timing, magnitude and composition of this response must be tightly controlled. A weak and delayed immune response may fail to eliminate the offending pathogen whilst an excessive and prolonged immune response can cause tissue damage and immunopathology. Failure to either eliminate or tolerate the pathogen results in a loss of tissue function, leading to disease (37). As the innate inflammatory response precedes and ultimately shapes the ensuing adaptive response (38), in this review we will focus on the regulation of innate immunity.
Acute inflammation is the body’s initial response to infection and/or injury (39), resulting in an influx of immune cells to the site of infection or tissue damage (40). This is mediated by the innate immune system, so named because in contrast to the adaptive immune system, the receptors and mediators are germline encoded. The function of the innate immune/inflammatory response during IAV or SARS-CoV-2 infection is to limit/eliminate the virus, clear dead cells, and restore tissue function. If this initial innate response is insufficient to contain the virus, then inflammation acts as an alarm to initiate, prime and shape an adaptive immune response.
In contrast, the purpose of chronic, low-grade inflammation, often known as ‘meta-inflammation’, is not so clear. More recent thinking posits inflammation itself as part of the natural process to restore tissue homeostasis (39), when other feedback loops have failed. Thus, meta-inflammation is triggered by persistent disruptions to homeostasis, e.g., nutrient availability, oxygen levels, cell number, or potentially chronic or unresolved infections. Unlike during acute inflammation, chronic inflammation is not necessarily accompanied by the tell-tale heat, swelling, pain and redness associated with acute inflammation, and systemic inflammatory indicators, such as C-Reactive Protein (CRP), are only minimally elevated. Importantly, obesity is associated with meta-inflammation (41).
An emerging aspect of the inflammatory response is the induction of tolerance mechanisms – to repair tissue damage during acute pathogen insult or to return a dysregulated system to homeostasis. Host defense is an energy intense process: leukocyte proliferation and synthesis of cytokines and other effector molecules requires glucose and glutamine availability (42). Cytokines, hormones, adipokines, mitokines, matrikines, all act on cells to induce changes in function and metabolism, as required for host defense and tolerance (42, 43). Tolerance can be local: in the lung this is mediated by the structural cells (e.g. mesenchymal cells, fibroblasts, smooth muscle cells), which remodel the extracellular matrix, produce growth factors alongside cytokines and chemokines and maintain stem cell niches as recently reviewed by Flerlage et al. (44). This is integral for promoting epithelial repair and a return to tissue function. Additionally, systemic release of these cytokines and hormones can affect overall organismal tolerance, particularly by affecting metabolism. The complex and intertwined relationship between the immune system and metabolism has been reviewed recently (42, 45).
Functions of Inflammatory Mediators in Host Defense
Inflammation is essentially cell to cell communication, facilitated by small soluble proteins such as cytokines, chemokines, eicosanoids, and other lipid and peptide mediators. While cytokines are produced during all stages of an immune response, by innate, adaptive, and even non-immune cells (e.g., epithelial cells), the pro-inflammatory triumvirate of Interleukin (IL)-1β, IL-6 and Tumor Necrosis Factor (TNF) are amongst the first and most potent cytokines released during a viral infection. IL-1β has pleiotropic actions in host defense: acting upon the nervous system to trigger fever and appetite loss, generating more immune cells in the bone marrow (hematopoiesis), increasing delivery of immune cells and mediators to the site of infection via the vasculature (vasodilation, angiogenesis) and activation of antibody and T cell responses (38, 46). IL-6 promotes platelet release, Cytotoxic T Lymphocyte (CTL) differentiation and antibody production, and induction of the ‘acute phase response’ (increasing serum CRP and serum amyloid A) (47). TNF variably promotes cell differentiation (e.g., T cells) or cell death (e.g., infected cells) and increases vascular permeability and leukocyte extravasation into tissue (48, 49). The ‘flu like’ symptoms experienced during infection, or during vaccination, result from the systemic actions of these cytokines.
Of all the cytokines induced during a viral infection, the interferons (IFNs) are perhaps the best known and studied. The canonical anti-viral cytokines, IFNs signal through their cognate receptors and induce expression of up to 400 interferon stimulated genes (ISGs) (50). ISGs are critical for cell intrinsic defense against viruses: they encode typical restriction factors that target various stages of a viral life cycle (e.g., entry: IFITM3; nuclear import: MX1, MX2; mRNA synthesis: APOBECs, Protein synthesis: PKR; Replication: OAS1; Egress: Tetherin) (50). Additionally, IFNs can act on immune cells to further amplify the inflammatory response e.g., by inducing expression of the chemokine C-X-C motif chemokine ligand 10 (CXCL10), which activates dendritic cells, enhances cross presentation, and promotes the enhancement and maturation of T and B cell responses. Paradoxically, IFNs can also strongly induce IL-10 expression, the best known of the ‘anti-inflammatory’ cytokines (51).
Specific Receptors and Pathways Driving Inflammation in IAV Infection
Significant progress has been made in identifying how IAV triggers inflammation. Most cells express pattern recognition receptors (PRRs) to detect intrinsic viral infection (52). The endosomal Toll Like Receptors (TLRs) 3, 7 and 8 could all potentially sense IAV-derived RNAs of incoming IAV virions, though only TLR3 is reported to be expressed in airway epithelial cells (53). TLR7 in plasmacytoid dendritic cells (pDCs) recognizes the ssRNA IAV genome, resulting in IFN-α and pro-inflammatory cytokine release (54, 55). Recent evidence suggests that airway epithelial TLR3 is responsible for driving IFN-β production in response to IAV infection, as determined by genetic mapping of TLR3 associated mutations in children who acquire severe IAV-induced Acute Respiratory Distress Syndrome (ARDS) (56). Post fusion, viral ribonucleoproteins (vRNPs) are trafficked to the nucleus for replication. These incoming vRNPs can potentially be sensed by Z-DNA-binding protein 1 (ZBP1) to trigger NLRP3 inflammasome activation (57). The inflammasome is a multimeric cytosolic signaling platform that cleaves IL-1β and IL-18 into their mature, bioactive forms and initiates an inflammatory cell death: pyropotosis (58). NLRP3 inflammasome activation is myeloid restricted, however whether and how epithelial inflammasomes are also activated in response to IAV remains contentious (59, 60). Once the IAV vRNPs reach the nucleus, IAV replicates and in the process generates Z-RNAs which are sensed by nuclear ZBP1 to drive necroptosis (61), another form of inflammatory cell death. Replicating IAV RNA intermediates are also sensed by Retinoic-acid Inducible Gene I (RIG-I), presumably in the nucleus, to drive Type I and III IFN and pro-inflammatory cytokine expression (62). Newly synthesized vRNPs can also be sensed by RIG-I in the cytosol, where RIG-I, along with its counterpart dsRNA sensing receptor melanoma differentiation associated protein 5 MDA-5, is typically expressed (52). RIG-I and MDA-5 signal via the adaptor protein Mitochondrial antiviral-signaling protein (MAVS), to activate the canonical pro-inflammatory transcription factor Nuclear Factor kappa B (NF-κB) (responsible for IL-1β, IL-6 and TNF expression among many others) and the Interferon Regulatory Factors (IRFs), especially IRF3 which drives Type I and III IFN expression (52). How nuclear RIG-I signals to cytoplasmic MAVS during infection remains to be elucidated. During new viral protein synthesis, the IAV M2 ion channel activates the NLRP3 inflammasome (63) to cleave IL-1β, though whether this also triggers pyroptotic cell death is unclear.
Cells also express PRRs on their cell surface or in endosomes to detect cell extrinsic danger, such as infection or damage of neighboring cells. Best known among these are the TLRs and the C-type Lectin Receptors (CLRs), which are able to detect both pathogen-associated molecular patterns (PAMPs, e.g., viral RNA, bacterial lipopolysaccharides (LPS), carbohydrate moieties on viral proteins) and host danger associated molecular patterns (DAMPS: e.g., High Mobility Group Box 1 Protein: HMGB1). Endosomal TLR3, 7 and 8 in neighboring, bystander epithelial or immune cells can also recognize extracellular viral RNAs released from infected cells or revealed upon phagocytosis of dying infected cells. Surface expressed TLR4 may also amplify the inflammatory response to IAV infection by sensing DAMPS proposed to include HMGB1 and oxidized phospholipids (64). Endothelial cells also contribute to the IAV-induced cytokine storm, releasing pro-inflammatory cytokines downstream of Sphingosine 1 receptor activation (65, 66). Epithelial cells, macrophages (both alveolar and infiltrating) and endothelial cells may all contribute to the release of pro-inflammatory cytokines and anti-viral IFNs during IAV infection, though the specific contribution of each of these cell types, and their signaling pathways to host defense in IAV infection remains to be determined.
Specific Receptors and Pathways Driving Inflammation in SARS-CoV-2 Infection
Rapid research developments in the last year have helped elucidate some of the pathways by which inflammation is triggered in SARS-CoV-2 infection. As with IAV, innate sensing of SARS-CoV-2 is determined by its infectious cycle. In airway epithelial cells expressing the receptor ACE2 and the protease transmembrane serine protease 2 (TMPRSS2), SARS-CoV-2 fusion and entry occurs at or near the cell surface. Translation and replication of the SARS-CoV-2 genome subsequently occurs in the cytosol (67). In the absence of TMPRSS2, the virus is internalized into endosomes, where the Spike protein can be cleaved by cathepsins, promoting fusion from this compartment (68). Presumably, this would also promote detection by endosomal TLRs, and may result in enhanced pro-inflammatory signaling in cells that express ACE2 but lack TMPRSS2. The dsRNA intermediates of the replicating SARS-CoV-2 genome are sensed by both RIG-I and MDA-5 (69). There is significant evidence of inflammasome activation in severe COVID-19 (70), though the cellular and molecular source of this activation is controversial, as conventional inflammasome activating cells (e.g., macrophages, dendritic cells) are not infected (71), precluding cell intrinsic inflammasome activation by SARS-CoV-2. Recently the epithelial NLRP1 inflammasome was identified as a sensor for cytosolic dsRNA (72). Whether this is activated in response to SARS-CoV-2 infection and drives disease remains to be determined. Nevertheless, cell extrinsic sensing of SARS-CoV-2 also drives pro-inflammatory and anti-viral cytokine release. As with IAV, SARS-CoV-2 virions or SARS-CoV-2 derived ssRNAs are sensed by TLR7 in pDCs, stimulating critical IFN-α production. As such, genetic deficiency of TLR7 is associated with severe COVID-19 phenotypes (73). TLR2, which is highly expressed on myeloid cells, can detect the SARS-CoV-2 envelope (‘E’) protein, and drive pro-inflammatory gene expression (74), while C-type Lectins on the surface of macrophages and other myeloid cells detect the glycans attached to the SARS-CoV-2 Spike (75). TLR2 and CLRs primarily drive NF-κB dependent gene expression and so signaling from these cell surface myeloid receptors may skew the cytokine balance in COVID-19 from anti-viral to pro-inflammatory. An overview of the innate sensing and signalling pathways implicated in IAV and SARS-CoV-2 infection are provided in Figure 1.
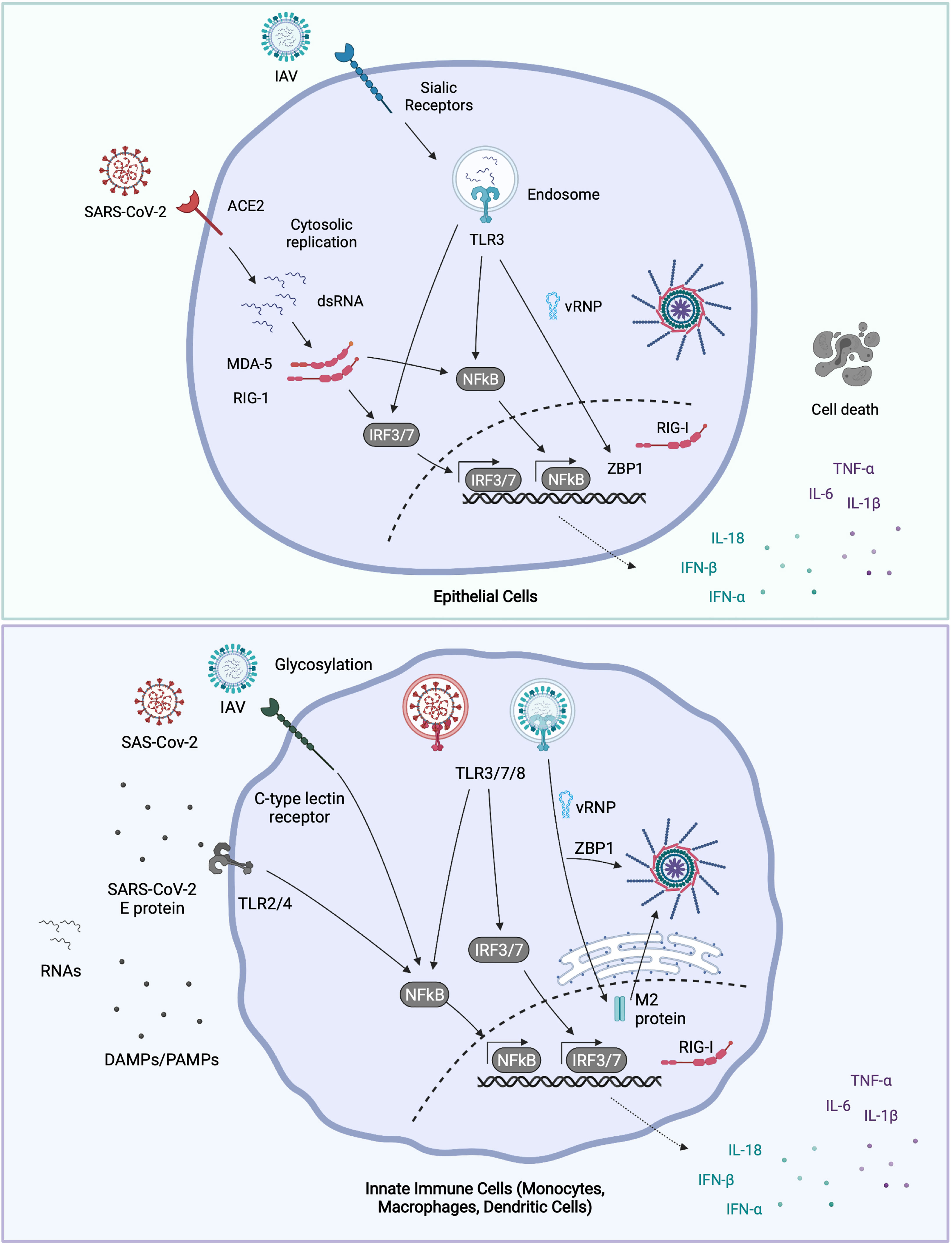
Figure 1 Molecular sensing of IAV and SARS-CoV-2. IAV binds to surface α2, 6-linked sialic acid (α2,6-SA) receptors on airway epithelial cells, and some macrophages, whereupon it’s internalized into endosomes (76). Incoming virions may be sensed by TLR3 (epithelial cells) or by TLR3, 7 and 8 (macrophages and dendritic cells). TLR engagement leads to NFκB and IRF transcription factor activation and pro-inflammatory and anti-viral gene expression. Post fusion, viral RNA bound by nucleoprotein (vRNPs) are released into the cytosol. Once in the cytosol, vRNPs are trafficked to the nucleus where IAV replicates. IAV replication products are sensed by nuclear RIG-I or ZBP1, triggering gene expression or cell death respectively. ZBP1 may also intercept vRNAs in the cytosol and trigger inflammasome activation. Newly synthesized IAV M2 protein can trigger NLRP3 inflammasome activation and IL-1β release. SARS-CoV-2 enters airway epithelial cells via ACE2, and after fusion at or near the plasma membrane, the viral genome is released into the cytosol. The dsRNA intermediates generated during SARS-CoV-2 replication are sensed by both RIG-I and MDA-5 in the cytosol, leading to NFκB and IRF3 activation. Dendritic cells and macrophages do not appear to be productively infected with SARS-CoV-2 (71). Though whether they can still take up virus, and signal from endosomal TLRs isn’t known. TLR7 in plasmacytoid dendritic cells recognises either virions or ssRNAs from SARS-CoV-2. Extracellular viral PAMPS and DAMPS released from neighbouring infected cells can also be sensed by surface and endosomal TLRs [e.g., SARS-CoV-2 envelope ‘E’ protein: TLR2 (74)], driving NFkB activation. Glycosylated viral proteins can further be detected by CLRs expressed on macrophages, which drives NFκB activation (77). Figure created with BioRender.com.
Detrimental Inflammation in IAV and SARS-CoV-2 Infection
Expression of pro-inflammatory cytokines (e.g., TNF, IL-6, IL-1β), are all critical in defense against both IAV and SARS-CoV-2. However, in excess or when prolonged, these cytokines all enhance disease severity. Dysregulated or hyperinflammatory immunopathology are now recognized as key indicators and drivers of disease severity in both pandemic IAV and COVID-19 (78–83). Raised levels of pro-inflammatory cytokines and chemokines, such as IL-1β, IL-6 and TNF have been identified as indicators of lung injury following IAV infection (84–87). Newly recruited monocytes, macrophages, and neutrophils entering the IAV-infected and -inflamed lung secrete further pro-inflammatory cytokines, further amplifying the response (88). The consequence of this inflammatory loop is tissue damage, including diffuse alveolar damage, hyaline membrane formation, fibrotic healing, capillary damage and immunopathologic injury, often leading to ARDS (89, 90). The virulence of the 1918 pandemic IAV is attributed to its ability to cause a hyper inflammatory response (91), while highly pathogenic avian IAVs also trigger excessive cytokine release in cell and animal models compared to seasonal strains (92–95). Targeting these inflammatory signaling pathways therapeutically in IAV infection remains challenging as they are critical for early host defense. For example, NLRP3 inflammasome activation and IL-1β responses are critical for developing a functional adaptive response to IAV: mice deficient in inflammasome components succumb more quickly to IAV infection (96, 97). Similarly, treating mice with the small molecule NLRP3 inhibitor MCC950 prior to IAV infection resulted in greater IAV lethality (98). Promisingly, inhibiting NLRP3 at the peak of IAV disease severity (e.g., Day 8 and 9) improved disease outcomes in mice, suggesting that therapeutic timing of any anti-inflammatory treatment is critical. In IAV infections, this hyperinflammatory response is succeeded by a tolerized or immune-suppressed state, during which secondary (and often fatal) bacterial infections can take hold (37). How these anti-inflammatory drugs might dampen the IAV-induced cytokine storm, without increasing susceptibility to secondary bacterial infection remains a limitation to their use in the clinic.
The presentation of SARS-CoV-2 infection in patients ranges from a mild respiratory tract infection to a serious and systemic inflammatory response (99, 100). Some patients with SARS-CoV-2 experience dysregulated cytokine responses characteristic of a cytokine storm with elevated levels of cytokines such as IL-6, IL-10, IL-1β, IL-18, TNF, IFNs, and CXCL10 (99). Similarly, patients with severe COVID-19 have an influx of inflammatory monocytes and macrophages into their lungs, which correlates with increased cytokine expression (101). Vascular dysfunction, clotting and increased risk of stroke and heart attack are a further pathological feature of COVID-19. SARS-CoV-2 does not directly infect endothelial cells, instead vascular dysfunction also appears to be driven by the enhanced inflammatory state (102).
The role that these pro-inflammatory cytokines play in driving severe disease in COVID-19 has been confirmed by the relative success of the broad-spectrum immunosuppressant dexamethasone (103). Similarly, clinical trials for the IL-1α/β blockers Anakinra (104) and Canakinumab (105) have shown promise in improving COVID-19 outcomes, while the IL-6 antagonist Tocilizumab is approved by the FDA under an emergency use authorization for combination use with dexamethasone for added benefit (106). Similarly, while defective early IFN responses are associated with COVID-19 disease severity (107), when IFNs are present in excess or for a prolonged period, they can disrupt epithelial repair and correlate with severe COVID-19 (108). Thus, the timing, magnitude, and balance of these cytokine responses are critical to successfully eliminating the virus without triggering collateral tissue damage. A summary of studies elucidating the role of these cytokines in IAV and SARS-CoV-2 infection is detailed in Table 1.
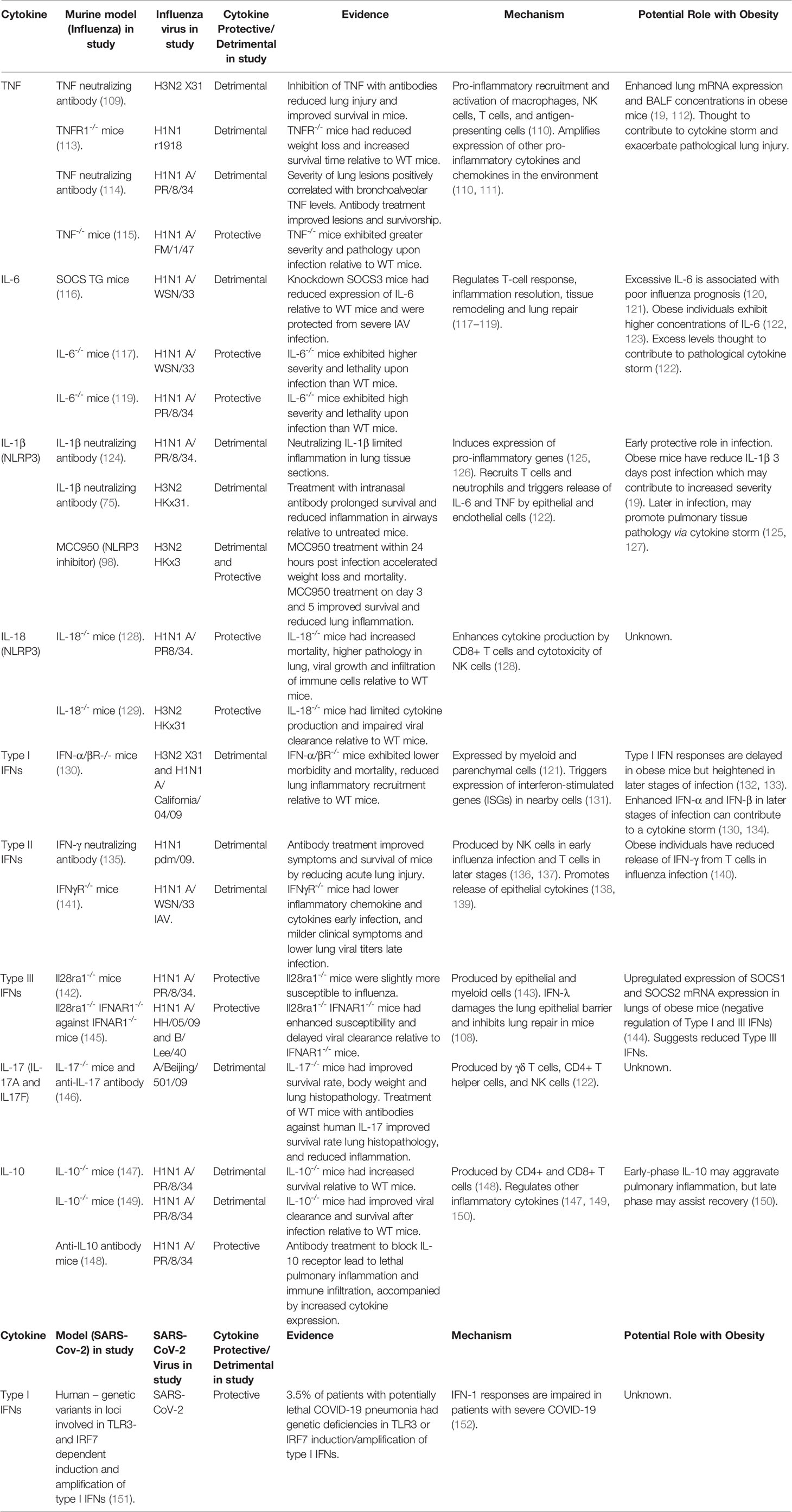
Table 1 Cytokines in disease severity in Influenza and SARS-CoV-2 Infections.
Obesity and Inflammation
Genetics and diet are the two primary drivers of obesity. BMI and obesity may be heritable (153, 154) and this is attributed to both monogenic and polygenic traits (155). These include mutations in leptin and its receptor (LEPR) (156, 157), prohormone convertase 1 (PC1) (158), and fat mass and obesity associated (FTO) gene (155, 159). Mutations in the leptin-melanocortin signaling pathway, most of which occur in the melanocortin 4 receptor (MC4R), may account for much of the heritability of obesity (159): an estimated 1 in 200 obese people worldwide have disease-causing mutations in MC4R (160). The genetic contribution of obesity has been explored experimentally in the ob (mutation in the gene encoding leptin) and db (mutation in gene encoding leptin receptor) mouse models (6). While only 1-5% of the morbidly obese population can be attributed to a mutation related to leptin or its receptor (161, 162), these models still have use as they recapitulate the leptin resistance and attenuation of leptin signaling that occurs during diet induced obesity (163).
Global urbanization has been accompanied by a shift from diets rich in plant-based fiber to a high-fat, high-protein, calorie rich diet referred to as the ‘Western Diet’ (164). The Western Diet (WD) consists of high amounts of processed, sugar and carbohydrate rich foods, with a distinct lack of vitamins, minerals, and fiber (41). Long-term consumption of this diet results in weight gain and increased BMI: accumulation of triglycerides from carbohydrates in a WD amplifies insulin signaling, which promotes increased storage of glucose and fatty acids in adipose tissue (165, 166). Diet-induced obesity models in rodents reproduce human diets using two main forms: The WD with 49% energy from carbohydrates, or a High Fat Diet (HFD), with 45-60% of energy from fat (167–171) and are most commonly used with C57BL6/J male mice for a period of 12-20 weeks (167, 170). Both models induce weight gain, increased systemic inflammation (circulating cytokine levels in serum), increased expression of inflammatory genes in adipose tissue, and alter body composition, glucose levels, insulin levels and insulin resistance (167–172).
Activating and Regulating Inflammatory Pathways in Obesity
Several components of the western diet can trigger inflammation directly through PRR activation (e.g., cholesterol crystals activate NLRP3 inflammasome activation) while others, such as excess fatty acids, L-carnitine and phosphatidylcholine cause dysbiosis in the gut microbiome, thereby indirectly triggering PRR activation (41). For example, a long term processed food diet increases intestinal barrier permeability, in part due to the presence of advanced glycation end-products (AGEs), which are a group of posttranslational modifications that are generated under thermal food processing (173). This barrier permeability lead to systemic circulation of LPS from commensal bacteria present in the gut. LPS triggers systemic innate immune activation, presumably via TLR4 or via the non-canonical caspase-driven inflammasomes: caspase 4/5 in humans and caspase-11 in mice (174, 175). Accordingly, treatment of the obese mice with alagebrium (an AGE inhibitor that breaks the heat–induced crosslinks) or a high starch fiber diet improved gut barrier integrity and reversed the systemic inflammation (173).
Obesity also induces remodeling and enlargement of adipose tissue to accommodate the storage of excess energy obtained from the diet (176). This is mediated by both increases in adipocyte mass and number and is accompanied by enhanced levels of immune cell infiltration and polarization toward more “inflammatory” adipose tissue macrophages. Both adipose associated immune cells (e.g., macrophages) and adipocytes release pro-inflammatory cytokines (177) and adipokines, with obesity leading to increased serum leptin (178), increased serum resistin (179), and reduced serum adiponectin (180–182). This imbalance of adipokines partly contributes to chronic systemic inflammation.
These adipokines and hormones can either amplify or suppress inflammatory responses (43). Intriguingly, metabolic changes can occur even without an increase in adipose tissue mass or BMI, rather an urban diet alone (increased intake of processed and high fat foods) increases pro-inflammatory gene expression compared to a plant and fiber based diet (183). This suggests that diet itself, rather than necessarily BMI and adipose tissue size, is enough to modulate adipokine, hormone and cytokine levels and thereby impact the inflammatory response.
Other hormones that regulate metabolism include Fibroblast Growth Factor 21 (FGF21) and Growth and Differentiation Factor 15 (GDF15). FGF21, released primarily from the liver, induces energy expenditure (184, 185), thermoregulation and maintains cardiac function under stress (184–186), whereas GDF15 supresses food intake (187). Serum levels of both FGF21 and GDF15 are increased in obesity (188), which might suggest a pathological function, however multiple studies suggest that FGF21 and GDF15 are both protective and essential for healthy metabolism as reviewed by Keipert et al. (189). Akin to leptin resistance in obesity, increased FGF21 and GDF15 levels in obesity might indicate resistance to these cytokines, and a concomitant decrease in function (189), though this remains unresolved. Both FGF21 and GDF15 are critical in host defense by promoting tolerance: in models of bacterial sepsis they induce thermoregulation, and triglyceride release respectively (190) and thus protect the heart by regulating the increased cardiac metabolic demands of infection. Consistent with this, blocking GDF signaling increased susceptibility to IAV infection in mice due to loss of hepatic sympathetic outflow and triglyceride metabolism, resulting in impaired cardiac function and maintenance of temperature, indicating that loss of GDF15 is highly detrimental in infection (190). However, whether dysregulation of FGF21 and GDF15 in obesity affects tolerance during IAV or SARS-CoV-2 infection remains to be experimentally confirmed.
Increased Inflammation in Obese Hosts During IAV and SARS-CoV-2 Infection
Considering their critical roles in early host defense, an elevation of pro-inflammatory cytokines and chemokines in obese hosts might be expected to confer extra protection against invading viruses such as IAV and SARS-CoV-2. In contrast, during the 2009 H1N1 pandemic, severe infections were characterized by significantly higher levels of IL-6 and IL-8 (121). In particular, non-survivors had elevated levels of IL-6 compared to survivors (121). High levels of these inflammatory biomarkers are associated with the obese state (191, 192), suggesting a predisposition to severe influenza outcomes.
High levels of IL-6 and CRP are associated with the obese state (193–195) and associated with the development of ARDS in obese individuals with COVID-19 (196). Furthermore, in SARS-CoV-2 infection, BMI is positively associated with increased pulmonary inflammation and increased pro-inflammatory cytokines and metabolic markers in the serum while being negatively associated with Spike-specific IgG antibody levels (197).
How Does Obesity Dysregulate the Inflammatory Response to IAV and SARS-CoV-2?
Inflammatory signaling is tightly regulated, with multiple layers of negative and positive feedback loops (198). Loss of negative regulators, or over stimulation of positive regulators can have profound effects on the inflammatory milieu. Obesity may affect the inflammatory response by changing the expression or function of these regulators. This can include PAMP sequestration (e.g. viral proteins by antibodies, LPS by High-Density Lipoprotein: HDL), receptor trafficking (the PRR needs to be present in the correct subcellular location to detect the PAMP or DAMP), phosphorylation, ubiquitination (199) and other post-translational modifications (200). Inducible negative regulators block signaling (e.g., Suppressor of Cytokine Signaling 3: SOCS3) while transcriptional repressors suppress pro-inflammatory gene expression. Many of these negative regulators, such as Activating Transcription Factor 3 (ATF3) are induced by PRR stimulation itself (201), Type I Interferons (202) and HDL (203). Metabolic changes downstream of PRR stimulation also drive histone modifications that have both short- and long-term effects on pro-inflammatory gene expression (204, 205). Post-transcriptionally, microRNAs and mRNA destabilizing agents serve to further titrate pro-inflammatory mRNA levels (198). Decoy receptors and cytokine antagonists (e.g. IL-1R antagonist) further restrain inflammatory signaling downstream of cytokines, while anti-inflammatory cytokines including IL-10 (206), Transforming growth factor (TGF)-β, and Type I IFNs (207) (depending on context) block pro-inflammatory signaling by inducing many of the above described negative regulators.
The inflammatory response to an infection is therefore highly dependent on the local micro-environment, which determines expression and activity of these negative regulators. For example, HDL levels are lowered in obesity, removing a critical negative regulator from the inflammatory cascade (208). Thus, single, or cumulative changes to the levels and functions of enzymes and proteins that regulate inflammation, as may occur during obesity, can hugely impact the timing, magnitude, and balance of the pro-inflammatory and anti-viral response. The effect of obesity specifically on these regulatory pathways has only been partially explored, as will be discussed in the next section, and further investigation into the effect of obesity on mechanisms of inflammatory regulation, particularly in the molecular pathways involved in IAV and SARS-CoV-2 sensing, are warranted.
Obesity-Induced Trained Immunity
Various studies have demonstrated that monocytes from obese children and adults produce increased cytokines (notably TNF) upon ex vivo stimulation with TLR agonists, relative to monocytes from non-obese individuals (209–213). This heightened response is consistent with the phenomenon of ‘trained immunity’ where innate immune cells have a heightened pro-inflammatory response upon exposure to a secondary immunological stimulus (214, 215). Trained monocytes exhibit increased histone-3-lysine-27-acetylation (H2K27Ac) and H3 histone-lysine-4-trimethylation (H2K4me3) on gene promoters related to the inflammatory response and metabolism, and increased H3K4me1 in enhancer regions (216). This epigenetic memory is therefore influenced by changes in intracellular metabolism, with metabolites regulating the activity of these histone modifying enzymes (217–219). Thus, the heightened pro-inflammatory response upon secondary stimulation results from global metabolic and epigenetic reprogramming of innate immune cells by the first stimulus.
The increased pro-inflammatory phenotype of monocytes in obese patients has been associated with global increases in histone H4 acetylation (210). In addition, monocyte priming after a western-style, dislipidemia diet in non-human primates was correlated with increased H3K27 acetylation (210, 220). Monocytes from obese patients have upregulation of metabolic pathways related to inflammation, cholesterol synthesis, and glucose metabolism (221), reflective of changes observed in in vivo trained immunity studies. In mice, a WD induces a long-term hyper-responsive innate immune state, characteristic of trained immunity, and importantly, this persists even following weight loss (171).
Peripheral monocytes and DCs have an average lifespan of 5 days which is not long enough account for the observations of a trained phenotype years or decades after the primary stimulus (222). Strong evidence suggests this training occurs via modulation of bone marrow hematopoietic stem and progenitor cells (HSPCs), which then carry the relevant epigenetic changes to their daughter cells (171, 223). HSPCs express TLRs and cytokine receptors, and consequently have been shown to exhibit a trained phenotype following exposure to PAMPs and circulating cytokines (223–226), such as might occur during IAV or SARS-CoV-2 infection.
Diet-induced obesity also stimulates the proliferation of myeloid progenitors in murine bone marrow (171, 227). In a study by Christ et al. (171) using Ldlr−/− mice, a four-week WD induced granulocyte-monocyte-progenitor (GMP) proliferation and skewing toward development of activated monocytes, coupled with long-lived transcriptional and epigenetic reprogramming (171). Thus, obese mice on a WD not only have more immune cells, but they are ‘primed’ to release more inflammatory cytokines upon exposure to infectious stimuli than their non-obese counterparts.
Initiation of Trained Immunity in Obesity
The immune stimuli that may induce trained immunity in obesity are yet to be comprehensively investigated. However, several factors associated with obesity may be implicated. As with the chronic low grade systemic inflammation triggered by a high fat diet, a dysregulated gut microbiota, in combination with ‘leaky gut’, results in increased circulating levels of LPS which could act as the primary inducer of innate immune training (228, 229). In addition, Oxidsed Low Density Lipoprotein (oxLDL), a marker of oxidative stress enhanced in obesity (230, 231), can induce a hyperinflammatory trained phenotype in human monocytes (232). Importantly, diabetes may further confound this. Diabetes and hyperglycemia induce intestinal barrier permeability, through transcriptional reprogramming of intestinal epithelial cells and disruption of junction integrity, which could contribute to enhanced circulating LPS (233). Furthermore, high glucose conditions have been shown to boost oxLDL training in monocytes compared to normoglycemic conditions (234), and induce enhanced proliferation of HSPCs in the bone marrow resulting in increased circulating myeloid cells (235). Bekkering et al. (236) discuss these factors in further detail, and speculate on a potential role of adipokines in immune training (237).
Christ et al. (171) identified the NLRP3 inflammasome as the central receptor in immune reprogramming following a WD (171). IL-1β, as the best characterized inflammasome-dependent cytokine, may therefore be the central mediator behind the trained phenotype. Whether pharmaceutically targeting either NLRP3 or downstream cytokines can ‘reverse’ an obesity-induced WD phenotype, remains to be determined. The current hypothesis for how obesity drives meta-inflammation is detailed in Figure 2.
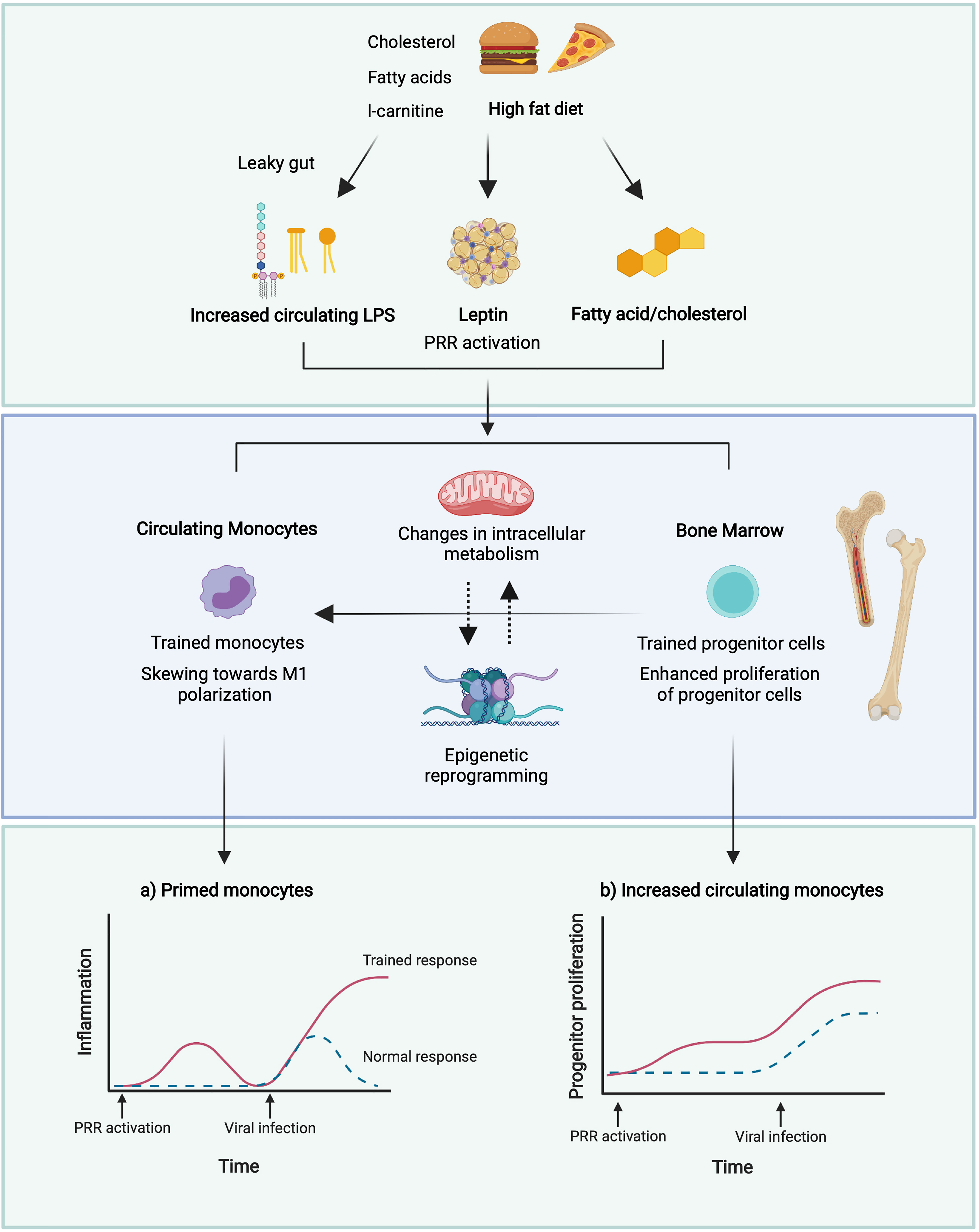
Figure 2 Potential mechanisms of induction of trained immunity in obesity. A leaky gut following a high dietary fat intake results in increased circulating levels of LPS (229) which may induce trained immunity (238, 239). High glucose conditions may boost immune training in monocytes and enhance the proliferation of HSPCs in the bone marrow (235). Reduced adiponectin in obesity may promote polarisation towards M1 macrophages and defective HSPC proliferation (240, 241), whilst increased leptin may promote proliferation and activation of circulating monocytes and BM myelopoiesis (242, 243). This may result in enhanced monocyte populations, and enhanced pro-inflammatory responses of these monocytes to viral infection. Figure created with BioRender.com.
Upsetting the Pro-Inflammatory/Anti-Viral Balance
The balance between pro-inflammatory cytokines and the antiviral IFNs must be tightly maintained, as these cytokines can negatively regulate one another (207). Age (244), genetics (107, 151) and obesity can skew this balance (245). Specifically, for IAV, Honce et al. showed that in normal human bronchial epithelial cells (NHBEC) derived from healthy or obese patients, there was a lower interferon response and increased viral replication in the obese NHBEC (245). Additionally, following infection with H1N1 (A/California/04/2009), obese mice had increased viral spread and increased viral diversity at 3 days post infection (d.p.i), despite similar viral titers to their wild type (WT) counterparts. This could be rescued with administration of recombinant IFN, suggesting that a blunted type I IFN response in obese mice drives this phenomenon (245). Whether this blunted IFN response was driven by increased pro-inflammatory cytokines in the baseline obese state was not determined (20). We speculate that similar experiments with SARS-CoV-2 will demonstrate that obesity blunts the anti-viral response to SARS-CoV-2, resulting in greater lethality. Pre-clinical studies of small molecules that stimulate an early IFN response show promise in animal models of COVID-19 (107, 246); treating obese patients who have been exposed to IAV or SARS-CoV-2 may help overcome the obesity-induced IFN deficit early in infection.
Considering how obesity increases HSPC proliferation and ‘primes’ these HSPCs through trained memory to produce more cytokines upon PRR stimulation, it is tempting to speculate that obesity-induced trained immunity drives pathological pro-inflammatory responses in IAV and SARS-CoV-2 infection. Curiously, the granulocyte-myeloid progenitors from WD- trained mice also had higher levels of type I IFN upon re-stimulation with TLR agonists (171). Based on this observation, a reasonable hypothesis would assume that WD training increases both IFN and pro-inflammatory cytokine release during infection, equating to quicker viral elimination and improved outcomes in the obese host, as the innate immune system is effectively ‘primed’ to fight infection. However, as described throughout this review, the opposite is true. This may reflect different cells being subjected to obesity-induced immune training, and distinct epigenetic reprogramming in different cells or tissues driving this skewed response. It could also reflect the cell-type specific viral tropism of IAV and SARS-CoV-2, and cell type specific pro-inflammatory or anti-viral cytokine release during infection. The specific cell types throughout the body, and particularly in the lung that are primarily ‘trained’ by obesity, and indeed, responsible for detrimental inflammation during IAV and SARS-CoV-2 infection, remain to be elucidated.
Consistent with obesity ‘training’ immune cells, PBMCs isolated from obese individuals had higher baseline levels of pro-inflammatory cytokines and reduced levels of anti-inflammatory cytokines compared to non-obese individuals (193, 194). Additionally, the PBMCs from the obese donors had elevated levels of the negative regulator SOCS3. Accordingly, these obese PBMCs had lower cytokine responses to ex vivo TLR3 and TLR7 stimulation. Elevated SOCS3 in obesity, because of meta-inflammation, may therefore blunt the anti-viral IFN response, rendering obese patients more susceptible to IAV or SARS-CoV-2 infection (247–249). As many PRRs and negative regulators of PRR signaling are themselves induced by cytokines or PAMPS and DAMPS, obesity may increase expression of PRRs, therefore sensitizing cells to viral PAMPs and DAMPS, while increasing expression of negative regulators such as SOCS3. If this occurs selectively, this may account for the decrease in anti-viral cytokines and concurrent increase in pro-inflammatory cytokines in obesity during viral infection.
Impact on Adaptive Immune Responses
Obesity and obesity-associated inflammation also have a large impact on adaptive immunity, as reviewed by McLaughlin et al. (250). T and B cell responses are directed and potentiated by inflammatory cytokines (38), and it follows that any dysregulation in innate inflammatory responses will negatively impact adaptive responses. In an obese host, these inflammatory signals are delayed, but overall are increased during primary IAV infection (194), which could have the two-pronged effect of delaying viral elimination by T cells, then subsequently promoting tissue damage due to excessive CTL responses. Accordingly, minimal expression of IFN-α and IFN-β, along with a delayed pro-inflammatory response (specifically IL-6 and TNF) and reduced natural killer (NK) cell cytotoxicity in obese mice infected with IAV correlated with increased mortality (19). Obesity may also impair the maintenance of effector memory T cells in the lung. Following infection and clearance of X31 (H3N2), obese mice lost significantly more lung resident effector memory T cells compared to lean mice resulting in increased lung pathology, viral titers and mortality during a secondary PR8 (H1N1) infection (144). During this secondary infection, obese mice also had significantly less expression of IFN-γ in the lungs as well as significantly less IFN-γ-producing CD8+ T cells (144). This impairment of T cell responses in the obese state has also been linked to leptin resistance (144, 251–253). In COVID-19, lower CD4+ and CD8+ T cell counts were observed in patients with diabetes mellitus upon admission, which correlates with increased disease severity (254). Whether the same defects in T cell responsiveness in obese hosts are observed during SARS-CoV-2 infection, and whether this ultimately derives from the dysregulated inflammatory state, will be important to elucidate. In Figure 3, we propose how obesity drives disease severity in IAV and SARS-CoV-2 infection by dysregulating inflammation.
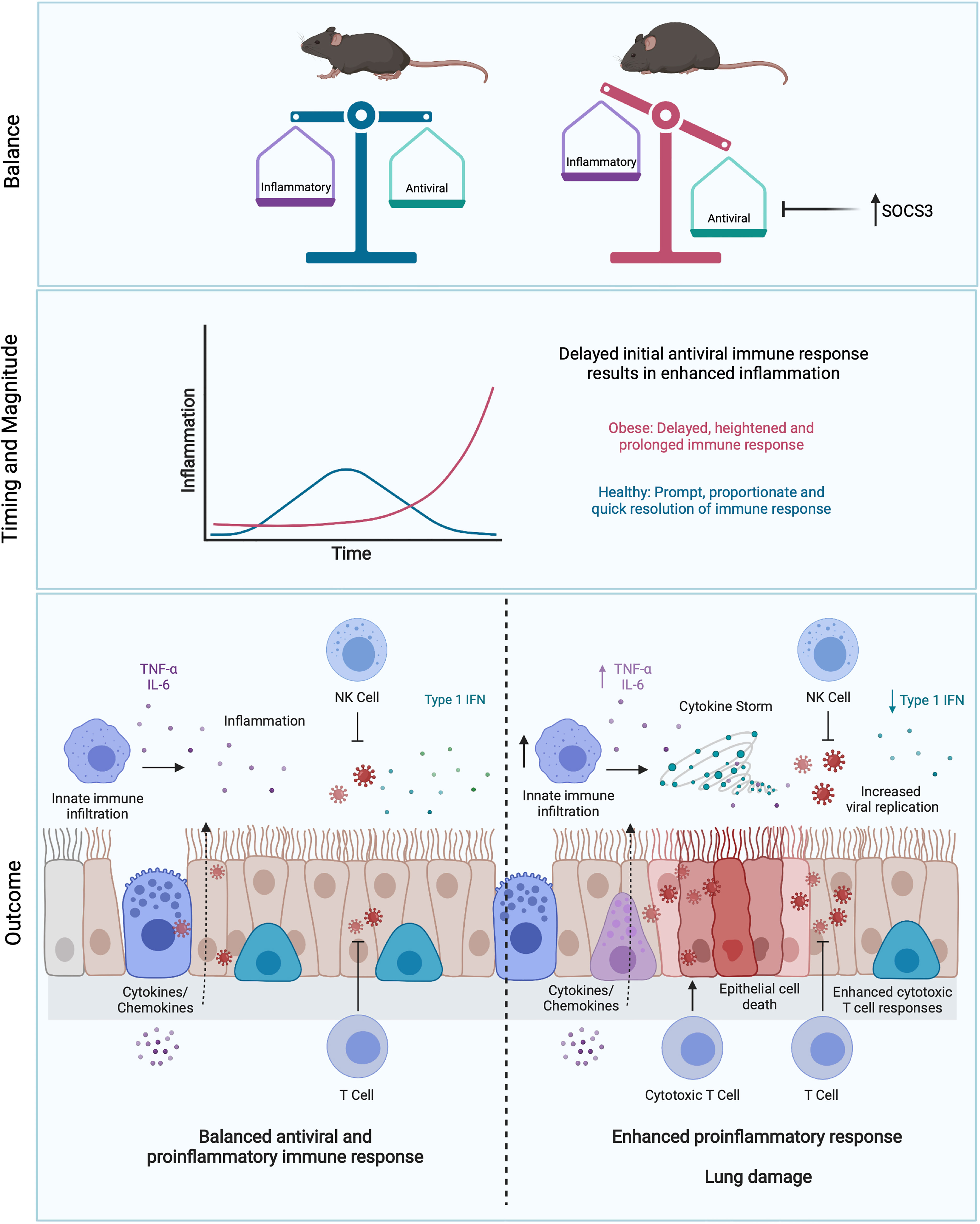
Figure 3 Obesity skews the balance between antiviral and pro-inflammatory responses. Initial blunted interferon responses to IAV in obese individuals results in increased viral spread and replication (245). This is accompanied by enhanced pro-inflammatory cytokine responses, driving immunopathology in the lung (193, 194). This imbalance may be partly driven by elevated levels of SOCS3 in obesity (193, 194). Delay of early viral clearance by T cells may result in further tissue damage due to excess CTL responses. Figure created with BioRender.com.
Prevention of Disease Severity Through Non-Medical Interventions
Considering that obesity increases the risk of severe disease in viral respiratory infections, both medical and non-medical interventions to overcome this risk are desperately needed. Additionally, obesity decreases the efficacy of vaccination against IAV, making obese hosts more difficult to protect from severe IAV infection. Compared to non-obese counterparts, there is a greater reported decrease in influenza vaccine efficacy over time in the obese population, as well as reduction in the protective immune response (31, 255). Based on the suboptimal history of influenza vaccine efficacy in obese populations, there is understandably concern surrounding the efficacy of SARS-CoV-2 vaccines in these individuals. Currently it has been reported that the efficacy of a number of SARS-CoV-2 vaccines has been comparable between obese and non-obese participates (e.g., Pfizer-BioNTech, 95% efficacy overall, and 95.4% efficacy in obesity) (256). However, it remains to be seen if the efficacy of these vaccines wanes more quickly over time in obese populations compared to non-obese, similar to influenza vaccines. There is similarly limited evidence of the effectiveness of current influenza antiviral treatments in obese hosts (31), or efficacy of newly developed SARS-CoV-2 antivirals in the general population (257), let alone the obese population.
Given the challenges in directly targeting these viruses with vaccination and anti-virals in both the general population and in obese risk groups, dietary changes, exercise and both surgical and non-surgical weight loss strategies all need to be examined as measures to reverse obesity and treat its associated co-morbidities.
Surgical and Non-Surgical Weight Loss
Weight loss is an essential intervention in obesity that reduces the severity of many of its non-communicable complications (diabetes mellitus, pulmonary disease, cardiovascular disease, etc.) (5). Bariatric and metabolic surgery (BMS) (including gastric bypass, adjustable gastric banding and sleeve gastrectomy) is a proven treatment for obesity (258). BMS reduced the risk of all-cause mortality and ensured patients live longer than non-surgical matched controls (259–265). BMS reduces the inflammation of adipose tissue, specifically by downregulating HIF-1α, PLAUR, MCP-1 and CSF-3 gene expression and thus dampens the infiltration of macrophages into adipose tissue, and enhances expression of anti-inflammatory cytokines such as IL-10 in the remaining adipose tissue macrophages (266). BMS also reduces the level of circulating CD4+ and CD8+ T cells and leptin (267), which may protect from T-cell induced immunopathology. As little as 6 months post-operative, BMS reduced serum concentrations of inflammatory cytokines including CRP, IL-1β, IL-6, IL-8, IFN-γ, IL-12, IL-23, and TNF (268).
To date, the only study specifically investigating the effect of weight loss in IAV infection has focused on the memory T-cell response (269). Rebeles and colleagues demonstrated that obese mice have impaired T-cell metabolism and memory T-cell responses to IAV and that this impaired response is maintained, even after weight loss (269).
Christ et al. observed that innate immune cells from obese mice were hyperinflammatory, and that this hyperinflammatory state was maintained even if the obese mice had lost weight (171). Whether this trained immune phenotype observed in previously obese mice is also detrimental in primary influenza infection remains to be determined. However, we speculate that formerly obese patients may still be at risk of developing severe influenza even if they have lost weight, due to the legacy of trained immunity driving a baseline hyperinflammatory response.
Clinical evidence of the long-term effects of obesity on influenza and COVID-19 is still scarce. Most often, these clinical studies refer to respiratory infections or influenza-like illness (ILI) without laboratory confirmation. One such retrospective study looking at ILI focused on the 2018-2019 influenza season in France and Italy (270). This study compared the outcomes of obese non-surgery (NS) patients and BMS patients. When compared to the obese NS patients, BMS patients reported significantly lower incidence of fever and ILI, lower intake of non-steroidal anti-inflammatory drugs, and lower admittance to the emergency department for ILI. While this study demonstrates a lower incidence of ILI in BMS patients compared to NS obese patients, this retrospective study did not compare BMS patients to the general non-obese population. In a retrospective, population-based, matched cohort study spanning 2005-2013 in Taiwan, significantly lower risk of respiratory tract infection (RTI), including influenza, and shorter hospital stays were reported in the BMS group compared to the NS (271). When compared to the general population however, BMS patients were still at a significantly higher risk of RTI and specifically influenza (271). Whether these BMS patients still have obesity-associated meta-inflammation and impaired interferon or adaptive responses, remains to be elucidated.
Weight loss and exercise studies in both humans and animal models are often quite long term. Due to the relative recency of SARS-CoV-2 appearing in the human population, research into the effect of weight loss in this context is severely limited. Furthermore, due to the current load on the health care system and the intraoperative risks for viral contagion among patients and staff due to COVID-19, many centers have postponed all elective surgeries including BMS (258, 265). As such, there are no long-term studies investigating whether BMS reduces the risk of COVID-19 compared to NS. To date, there has been one study in London that observed that over a two month period during a peak COVID-19 period, only one out of a total of 1439 patients presenting with COVID-19 had previously undergone BMS (258). This study tentatively demonstrates that BMS does not increase the risk of COVID-19 infection compared with the general population (258). However, the study did not report on what percent of the cohort were obese.
Together, these experimental and retrospective studies suggest that weight loss through both surgical and non-surgical means reduce the risk of RTI when compared to obese individuals. However, when compared to individuals with no history of obesity, those who underwent weight loss were still at a higher risk of infection and severe outcomes. This indicates that there are indeed long-term effects of obesity on the immune system that cannot be reversed with weight-loss alone, and pre-dispose these formerly obese individuals to higher risk of severe disease during IAV or SARS-CoV-2 infection. Of note, none of the studies reported on the exercise habits of participants, which could also affect outcomes. A summary of these interventions is detailed in Table 2.
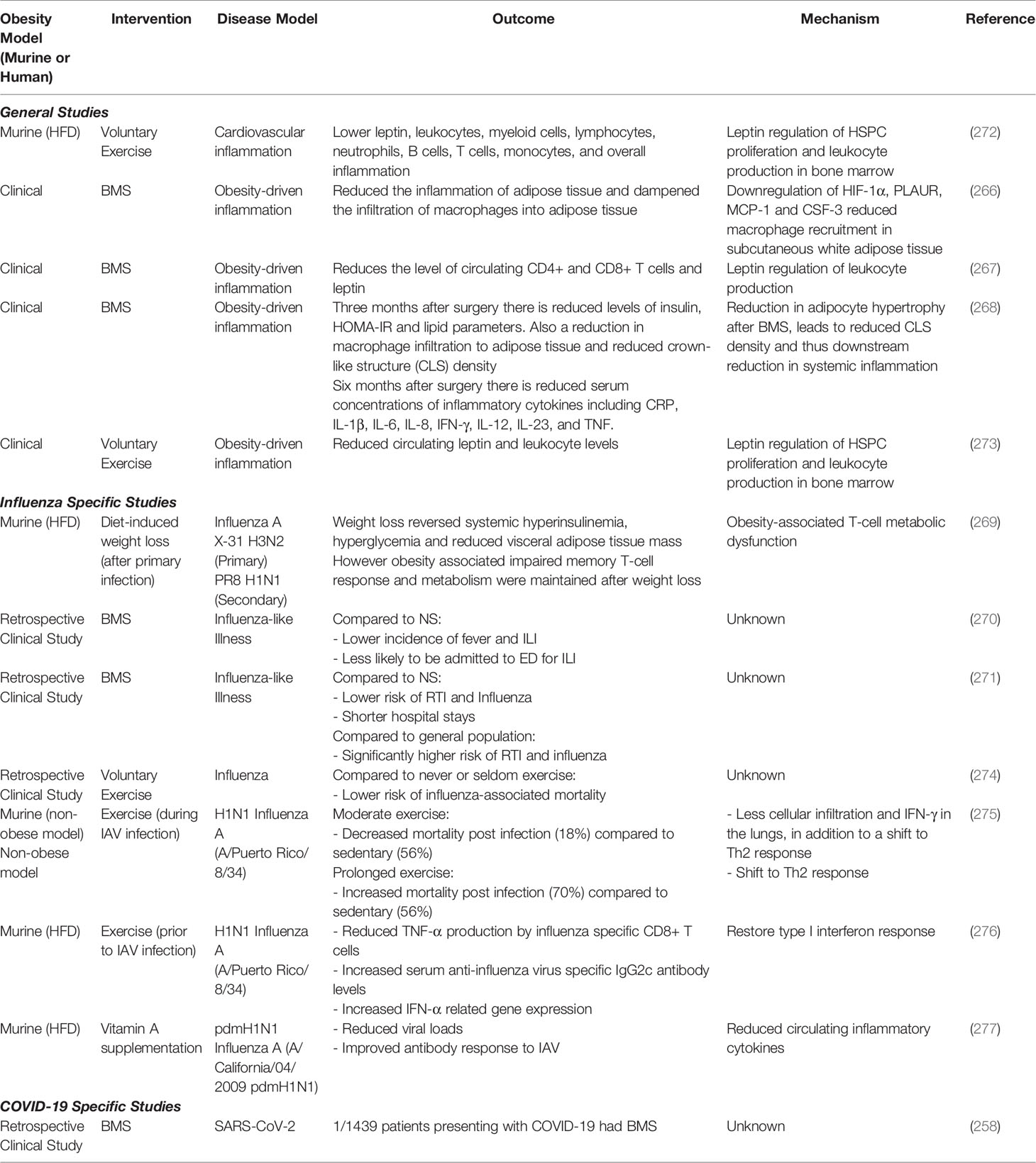
Table 2 Medical and non-medical interventions to reverse long-term immune effects of obesity.
Exercise
Independent of surgical and non-surgical weight loss, exercise also has anti-inflammatory effects. Voluntary exercise lowered leptin levels in mice fed HFD and was accompanied by a drop in total numbers of leukocytes, lymphocytes, neutrophils, B cells, T cells, monocytes, and overall chronic inflammatory markers (272).
Exercise also reduced circulating leptin and leukocyte levels in a cohort of Japanese adolescent males (273). Additionally, low to moderate frequency exercise was associated with a lowered risk of influenza-associated mortality compared to those who never or seldom exercise (274). However, neither of these studies reported the BMI of participants (274), making it difficult to interpret the efficacy of exercise in lowering obesity-associated in these populations.
In both obese and non-obese mice, exercise increased serum anti-influenza virus specific antibodies, T cells and improved disease outcomes (276). This correlated with increased chemokine, cytokine and importantly IFN expression in both groups post-exercise treatment. Thus, exercise may restore the obesity-associated pro-inflammatory cytokine and Type I interferon imbalance to improve disease outcomes, though the exact mechanisms still need to be elucidated.
Collectively, this evidence suggests that independent of other interventions, voluntary moderate physical activity alone both prior to- and during infection can improve infection outcomes. With demonstrated benefits of exercise for both obese and non-obese individuals, engaging in at least 150 minutes of moderate aerobic exercise each week for the general population may be a useful strategy to improve viral respiratory disease outcomes (278).
Dietary Changes/Supplementation
There are numerous animal and human studies that support the health benefits of intermittent fasting, including weight loss, improved glucose tolerance and insulin sensitivity, and reduced circulating and adipose levels of inflammatory biomarkers (255). Intermittent fasting could therefore offer an avenue for improving immune system function without drastic lifestyle changes. Notably, intermittent fasting reduces circulating and adipose levels of leptin, IL-6, TNF, IGF-1, IL-1β, and CRP (255, 279). While there are currently no studies investigating the effect of intermittent fasting on influenza or COVID-19 severity, it has been suggested as a possible non-medical prophylactic tool for both obese and non-obese individuals, and warrants further investigation (255, 280).
In addition to elevated fat stores and meta-inflammation, individuals with obesity also suffer from tissue-specific vitamin A deficiency that is often masked due to normal serum retinol levels (277, 281). Vitamin A is essential for hematopoiesis and both innate and adaptive immune function (282) and as such it has been suggested vitamin A deficiency could play a role in the severity of viral infections and the poor outcome often associated with vaccination in obese individuals (277). In a murine model of obesity with IAV vaccination and challenge, vitamin A supplementation decreased viral loads, reduced circulating inflammatory cytokines and improved antibody responses (277). Vitamin A may therefore be a low-cost intervention to increase vaccine efficacy and reduce IAV and SARS-CoV-2 disease severity in obese individuals.
Medical Interventions to Treat Obesity Induced-Inflammation
A variety of analogues and mimetics for FGF21 and GDF15 have been investigated for treatment of obesity. Clinical trials for FGF21 have been comprehensively detailed in a review by Geng et al. (236). In brief, long-term treatment with hFGF21 analogues have been shown to reduce dyslipidaemia, enhance insulin sensitivity and reduce body weight in clinical trials (236). In addition, preclinical obesity studies have shown treatment with GDF15 decreases food intake and body weight (283). However, the benefit of augmented FGF21 and GDF15 treatment in reducing risk for severe IAV and SARS-CoV-2 infection in obesity has not yet been explored.
While directly targeting inflammation with anti-inflammatory treatments is beneficial in COVID-19, there are limited studies on the efficacy of anti-inflammatories in decreasing IAV or SARS-CoV-2 disease severity in obesity. Tocilizumab (which blocks IL-6 signaling) was administered to treat the hyperinflammation of critical COVID-19 obese patients during the first days of worsening hypoxemia. In a case series, three obese patients from different age groups with COVID-19 were treated with Tocilizumab. All patients showed improved clinical signs and were discharged from hospital without complications or intubation, even when the ideal dose of infusion was not feasible to be administered (284). Randomized controlled trials of anti-inflammatories such as dexamethasone or tocilizumab in obese patients with IAV and COVID-19 will further determine if suppressing baseline inflammation is beneficial.
A pre-clinical anti-inflammatory drug that shows promise is 5-deoxy-Δ12, 14-prostaglandin J2 (15d-PGJ2), an anti-inflammatory lipid mediator and peroxisome proliferator-activated receptor-gamma (PPAR-γ) agonist. 15d-PGJ2 has proved beneficial in inhibiting melanoma progression in cancer (285), and reducing lung inflammation and remodeling in asthma (286) by inhibiting NF-κB signaling. In obese mice (db/db), PPAR-γ, an important inflammation regulator, is downregulated in lung macrophages following IAV infection. Treatment with 15d-PGJ2 improved the overall survival of IAV-infected obese mice, and was attributed to its effect on macrophage function (287). As of yet, there are no clinical studies using 15d-PGJ2 for either IAV or SARS-CoV-2.
Metformin is another commonly prescribed, clinically approved drug that is a possible candidate for repurposing to treat obesity and prevent IAV and SARS-CoV-2 disease severity. Metformin decreases mitochondrial oxidation and is primarily prescribed as a drug to treat Type 2 Diabetes. Metformin also improved the survival of obese mice in IAV infection (288). Mechanistically, this was attributed to metformin normalizing CD4+ T cell glucose oxidation and therefore function in the obese mice, though it is likely metformin has a positive effect on multiple immune cell populations (288). Interestingly, this study compared metformin treatment to a weight loss treatment group, which was not sufficient to reverse the obesity-related effects on CD4+ T cells, or improve survival, suggesting long lasting immune alterations. While metformin has not been used in the clinic to treat severe IAV infection, there have now been clinical studies targeting COVID-19. Metformin reportedly reduces the mortality in women with obesity or Type 2 Diabetes who were admitted to hospital with COVID-19 (254, 289).
In addition to experimental and prescription drugs, there is evidence that that non-steroidal anti-inflammatory drugs (NSAIDs), which are often easily accessible over the counter without a prescription, could prove beneficial for treating hyperinflammation in obesity and severe viral infections. NSAIDs have previously been associated with adverse clinical outcomes in the context of bacterial community-acquired pneumonia (290, 291), and as such there has been hesitation to use NSAIDs in the context of influenza and COVID-19. However, the use of NSAIDs was not associated with adverse outcomes (e.g. hospitalization, ICU admission, mechanical ventilation, mortality) in the context of the 2009 IAV pandemic (292), seasonal influenza (293), and the SARS-CoV-2 pandemic (294, 295). Despite NSAIDs targeting components of the cyclooxygenase (COX) pathway, which are hyper-induced during influenza infection (110, 296, 297), there is limited research into their potentially beneficial effects in the context of influenza and COVID-19. NSAIDs inhibit the inflammatory prostaglandin (lipid mediator) synthesis pathway by targeting the key enzyme cyclo-oxygenase-2 (COX-2) (298). Mice deficient in COX-2 had reduced mortality and viral titer after IAV infection, despite their blunted inflammatory response (299). Targeting specific inflammatory prostaglandins, rather than the central enzyme COX-2, also shows promise in treating severe IAV. Prostaglandin E2 (PGE2) inhibition improved survival after a lethal dose of IAV in a mouse model (300). PGE2 itself is upregulated during IAV infection and inhibits host type I IFN responses, macrophage recruitment to the lungs, and potentially impairs antigen presentation and T-cell mediated immunity (300). Whether broadly targeting prostaglandins via COX-2 inhibition or selectively targeting individual prostaglandins such as PGE2 improves outcomes in obesity and viral infection should be further examined.
While there is no data yet on NSAIDs in SARS-CoV-2 outcomes, there is currently a phase IV randomized clinical trial underway in the U.K., aimed at evaluating use of lipid ibuprofen in the reduction in severity and progression of SARS-CoV-2 infections (301). Whether NSAIDs further benefit the obese host in IAV and SARS-CoV-2 infections still needs to be determined. Future research might also include combination therapy of anti-virals with anti-inflammatories specifically in obese patients, which could augment pathogen elimination while suppressing detrimental inflammation, and in effect, correcting the dysregulated immune response.
Conclusions and Future Directions
Treating severe respiratory viral infections, particularly in a pandemic context, requires fundamental understanding of the mechanisms of disease pathology. Here we have reviewed how inflammation is initiated in IAV and SARS-CoV-2 infection, and how dysregulated inflammation can drive disease severity. Obesity is a key co-morbidity that profoundly impacts the immune response to these viral infections. While the molecular mechanisms by which diet induces a meta-inflamed state are increasingly well understood, some outstanding questions remain. How does obesity modulate the specific cell types and PRR signaling pathways involved in IAV and SARS-CoV-2? How do protective inflammatory responses become detrimental during IAV or SARS-CoV-2 infection? In answering these questions we may be able to identify further critical regulators of the inflammatory response (e.g., SOCS3) that are dysregulated in obesity and other co-morbidities. Identifying new and selective drug targets that preferentially suppress detrimental inflammatory responses while preserving anti-microbial responses will benefit both obese and non-obese individuals.
As our understanding of obesity and dietary-induced inflammation broadens, it is becoming increasingly apparent that diet-induced meta-inflammation is not limited to obese hosts. Multiple studies now suggest that a history of obesity is enough to confer a meta-inflamed state, and that this may pre-dispose these individuals to a higher risk of severe IAV or SARS-CoV-2 infection. Further research is warranted to understand the mechanisms of obesity-induced training, whether a trained phenotype lasts after weight loss, and if so, for how long. In addition, the key cell types that undergo obesity- induced training have yet to be identified. Cells in the mucosal periphery, such as alveolar macrophages (AMs) in the lung, have recently been shown to be subject to training independently of the bone marrow (302). This is particularly important in the case of viral infection; AMs reside in the epithelial fluid and are distinct from macrophages residing between the airway epithelium and blood vessels (303). As such, they are the first in line of defense against an influenza infection (304). As only 40% are replaced yearly, the lifespan of AMs could result in long lasting changes in the innate immune reponse in the lung (304, 305). The presence of immune training in these cells following a period of obesity has yet to be investigated. Similarly, whether diet-induced long-term immune training can be reversed is still unknown. This is of particular importance given that BMS and weight loss alone is insufficient to reduce the risk of developing influenza, and previously obese individuals still remain at a higher risk of RTIs compared to the general population (271).
In conclusion, while obesity is a multifactorial disorder with multiple effects on the host response to infection, a key mechanism by which obesity increases virus induced disease severity is by dysregulating the inflammatory response. Obesity primes the innate immune system to respond to IAV and SARS-CoV-2 with a heightened pro-inflammatory response and a blunted anti-viral response, which ultimately leads to increased tissue damage and decreased virus elimination. Together, this drives the increased disease severity observed in obese hosts in both influenza and COVID-19.
Author Contributions
Conceptualization: KH, KS, and LL. Original drafts: KH, EN, and LL. Figures: EN. Editing: KH, EN, KS, and LL, Supervision: KS and LL. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by the National Health and Medical Research Council of Australia (Fellowship 1124162 to LIL), the Australian Research Council (Fellowship DE180100512 to KS) and a UQ Early Career Researcher Grant (UQECR2058045) to LL.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Abarca-Gómez L, Abdeen ZA, Hamid ZA, Abu-Rmeileh NM, Acosta-Cazares B, Acuin C, et al. Worldwide Trends in Body-Mass Index, Underweight, Overweight, and Obesity From 1975 to 2016: A Pooled Analysis of 2416 Population-Based Measurement Studies in 128· 9 Million Children, Adolescents, and Adults. Lancet (2017) 390(10113):2627–42. doi: 10.1016/S0140-6736(17)32129-3
2. Collaboration NRF. Trends in Adult Body-Mass Index in 200 Countries From 1975 to 2014: A Pooled Analysis of 1698 Population-Based Measurement Studies With 19· 2 Million Participants. Lancet (2016) 387(10026):1377–96. doi: 10.1016/S0140-6736(16)30054-X
3. Kelly T, Yang W, Chen CS, Reynolds K, He J. Global Burden of Obesity in 2005 and Projections to 2030. Int J Obes (Lond) (2008) 32(9):1431–7. doi: 10.1038/ijo.2008.102
4. Janssen I, Katzmarzyk PT, Ross R. Waist Circumference and Not Body Mass Index Explains Obesity-Related Health Risk. Am J Clin Nutr (2004) 79(3):379–84. doi: 10.1093/ajcn/79.3.379
5. Xiao J, Yang W. Weight Loss Is Still an Essential Intervention in Obesity and its Complications: A Review. J Obes (2012) 2012:369097. doi: 10.1155/2012/369097
6. Schwartz MW, Seeley RJ, Zeltser LM, Drewnowski A, Ravussin E, Redman LM, et al. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocr Rev (2017) 38(4):267–96. doi: 10.1210/er.2017-00111
7. Louie JK, Acosta M, Samuel MC, Schechter R, Vugia DJ, Harriman K, et al. A Novel Risk Factor for a Novel Virus: Obesity and 2009 Pandemic Influenza a (H1N1). Clin Infect Dis (2011) 52(3):301–12. doi: 10.1093/cid/ciq152
8. Dou D, Revol R, Östbye H, Wang H, Daniels R. Influenza a Virus Cell Entry, Replication, Virion Assembly and Movement. Front Immunol (2018) 9:1581–. doi: 10.3389/fimmu.2018.01581
9. Zhao Y, Sun F, Li L, Chen T, Cao S, Ding G, et al. Evolution and Pathogenicity of the H1 and H3 Subtypes of Swine Influenza Virus in Mice Between 2016 and 2019 in China. Viruses (2020) 12(3):298. doi: 10.3390/v12030298
10. Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK, Cheng PY, et al. Estimated Global Mortality Associated With the First 12 Months of 2009 Pandemic Influenza a H1N1 Virus Circulation: A Modelling Study. Lancet Infect Dis (2012) 12(9):687–95. doi: 10.1016/S1473-3099(12)70121-4
11. Vidaña B, Martínez J, Martorell J, Montoya M, Córdoba L, Pérez M, et al. Involvement of the Different Lung Compartments in the Pathogenesis of Ph1n1 Influenza Virus Infection in Ferrets. Vet Res (2016) 47(1):113. doi: 10.1186/s13567-016-0395-0
12. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet (2020) 395(10224):565–74. doi: 10.1016/S0140-6736(20)30251-8
13. Letko M, Marzi A, Munster V. Functional Assessment of Cell Entry and Receptor Usage for SARS-Cov-2 and Other Lineage B Betacoronaviruses. Nat Microbiol (2020) 5(4):562–9. doi: 10.1038/s41564-020-0688-y
14. WHO. Who Coronavirus (Covid-19) Dashboard (2021). Available at: https://covid19.who.int.
15. Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al. SARS-Cov-2 Variants, Spike Mutations and Immune Escape. Nat Rev Microbiol (2021) 19(7):409–24. doi: 10.1038/s41579-021-00573-0
16. Cevik M, Kuppalli K, Kindrachuk J, Peiris M. Virology, Transmission, and Pathogenesis of SARS-Cov-2. BMJ (2020) 371:m3862. doi: 10.1136/bmj.m3862
17. Crook H, Raza S, Nowell J, Young M, Edison P. Long Covid—Mechanisms, Risk Factors, and Management. BMJ (2021) 374:n1648. doi: 10.1136/bmj.n1648
18. Neidich SD, Green WD, Rebeles J, Karlsson EA, Schultz-Cherry S, Noah TL, et al. Increased Risk of Influenza Among Vaccinated Adults Who Are Obese. Int J Obes (Lond) (2017) 41(9):1324–30. doi: 10.1038/ijo.2017.131
19. Smith AG, Sheridan PA, Harp JB, Beck MA. Diet-Induced Obese Mice Have Increased Mortality and Altered Immune Responses When Infected With Influenza Virus. J Nutr (2007) 137(5):1236–43. doi: 10.1093/jn/137.5.1236
20. Siegers JY, Novakovic B, Hulme KD, Marshall RJ, Bloxham CJ, Thomas WG, et al. A High-Fat Diet Increases Influenza a Virus-Associated Cardiovascular Damage. J Infect Dis (2020) 222(5):820–31. doi: 10.1093/infdis/jiaa159
21. Rottoli M, Bernante P, Belvedere A, Balsamo F, Garelli S, Giannella M, et al. How Important Is Obesity as a Risk Factor for Respiratory Failure, Intensive Care Admission and Death in Hospitalised COVID-19 Patients? Results From a Single Italian Centre. Eur J Endocrinol (2020) 183(4):389–97. doi: 10.1530/EJE-20-0541
22. Simonnet A, Chetboun M, Poissy J, Raverdy V, Noulette J, Duhamel A, et al. High Prevalence of Obesity in Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-Cov-2) Requiring Invasive Mechanical Ventilation. Obes (Silver Spring) (2020) 28(7):1195–9. doi: 10.1002/oby.22831
23. Longmore DK, Miller JE, Bekkering S, Saner C, Mifsud E, Zhu Y, et al. Diabetes and Overweight/Obesity Are Independent, Nonadditive Risk Factors for in-Hospital Severity of COVID-19: An International, Multicenter Retrospective Meta-Analysis. Diabetes Care (2021) 44(6):1281. doi: 10.2337/dc20-2676
24. Wieland CW, Florquin S, Chan ED, Leemans JC, Weijer S, Verbon A, et al. Pulmonary Mycobacterium Tuberculosis Infection in Leptin-Deficient Ob/Ob Mice. Int Immunol (2005) 17(11):1399–408. doi: 10.1093/intimm/dxh317
25. Wehrens A, Aebischer T, Meyer TF, Walduck AK. Leptin Receptor Signaling is Required for Vaccine-Induced Protection Against Helicobacter Pylori. Helicobacter (2008) 13(2):94–102. doi: 10.1111/j.1523-5378.2008.00591.x
26. Webb SR, Loria RM, Madge GE, Kibrick S. Susceptibility of Mice to Group B Coxsackie Virus is Influenced by the Diabetic Gene. J Exp Med (1976) 143(5):1239–48. doi: 10.1084/jem.143.5.1239
27. Kanda T, Takahashi T, Kudo S, Takeda T, Tsugawa H, Takekoshi N. Leptin Deficiency Enhances Myocardial Necrosis and Lethality in a Murine Model of Viral Myocarditis. Life Sci (2004) 75(12):1435–47. doi: 10.1016/j.lfs.2004.03.012
28. Karlsson EA, Beck MA. The Burden of Obesity on Infectious Disease. Exp Biol Med (Maywood) (2010) 235(12):1412–24. doi: 10.1258/ebm.2010.010227
29. Maier HE, Lopez R, Sanchez N, Ng S, Gresh L, Ojeda S, et al. Obesity Increases the Duration of Influenza a Virus Shedding in Adults. J Infect Dis (2018) 218(9):1378–82. doi: 10.1093/infdis/jiy370
30. Moriconi D, Masi S, Rebelos E, Virdis A, Manca ML, De Marco S, et al. Obesity Prolongs the Hospital Stay in Patients Affected by COVID-19, and may Impact on SARS-COV-2 Shedding. Obes Res Clin Pract (2020) 14(3):205–9. doi: 10.1016/j.orcp.2020.05.009
31. Honce R, Schultz-Cherry S. Impact of Obesity on Influenza a Virus Pathogenesis, Immune Response, and Evolution. Front Immunol (2019) 10:1071–. doi: 10.3389/fimmu.2019.01071
32. Higham A, Singh D. Increased ACE2 Expression in Bronchial Epithelium of COPD Patients Who are Overweight. Obesity (2020) 28(9):1586–9. doi: 10.1002/oby.22907
33. Karlsson EA, Meliopoulos VA, van de Velde NC, van de Velde L-A, Mann B, Gao G, et al. A Perfect Storm: Increased Colonization and Failure of Vaccination Leads to Severe Secondary Bacterial Infection in Influenza Virus-Infected Obese Mice. mBio (2017) 8(5):e00889–17. doi: 10.1128/mBio.00889-17
34. Alsiö Å, Nasic S, Ljungström L, Jacobsson G. Impact of Obesity on Outcome of Severe Bacterial Infections. PloS One (2021) 16(5):e0251887. doi: 10.1371/journal.pone.0251887
35. Corrales-Medina VF, Valayam J, Serpa JA, Rueda AM, Musher DM. The Obesity Paradox in Community-Acquired Bacterial Pneumonia. Int J Infect Dis (2011) 15(1):e54–e7. doi: 10.1016/j.ijid.2010.09.011
36. Roth J, Sahota N, Patel P, Mehdi SF, Wiese MM, Mahboob HB, et al. Obesity Paradox, Obesity Orthodox, and the Metabolic Syndrome: An Approach to Unity. Mol Med (2016) 22(1):873–85. doi: 10.2119/molmed.2016.00211
37. Iwasaki A, Pillai PS. Innate Immunity to Influenza Virus Infection. Nat Rev Immunol (2014) 14(5):315–28. doi: 10.1038/nri3665
38. Iwasaki A, Medzhitov R. Control of Adaptive Immunity by the Innate Immune System. Nat Immunol (2015) 16(4):343–53. doi: 10.1038/ni.3123
39. Meizlish ML, Franklin RA, Zhou X, Medzhitov R. Tissue Homeostasis and Inflammation. Annu Rev Immunol (2021) 39(1):557–81. doi: 10.1146/annurev-immunol-061020-053734
40. Antonelli M, Kushner I. It’s Time to Redefine Inflammation. FASEB J (2017) 31(5):1787–91. doi: 10.1096/fj.201601326R
41. Christ A, Lauterbach M, Latz E. Western Diet and the Immune System: An Inflammatory Connection. Immunity (2019) 51(5):794–811. doi: 10.1016/j.immuni.2019.09.020
42. Wang A, Luan Harding H, Medzhitov R. An Evolutionary Perspective on Immunometabolism. Science (2019) 363(6423):eaar3932. doi: 10.1126/science.aar3932
43. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in Inflammation and Metabolic Disease. Nat Rev Immunol (2011) 11(2):85–97. doi: 10.1038/nri2921
44. Flerlage T, Boyd DF, Meliopoulos V, Thomas PG, Schultz-Cherry S. Influenza Virus and SARS-Cov-2: Pathogenesis and Host Responses in the Respiratory Tract. Nat Rev Microbiol (2021) 19(7):425–41. doi: 10.1038/s41579-021-00542-7
45. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell (2017) 169(4):570–86. doi: 10.1016/j.cell.2017.04.004
46. Chan AH, Schroder K. Inflammasome Signaling and Regulation of Interleukin-1 Family Cytokines. J Exp Med (2020) 217(1):e20190314. doi: 10.1084/jem.20190314
47. Tanaka T, Narazaki M, Kishimoto T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb Perspect Biol (2014) 6(10):a016295–a. doi: 10.1101/cshperspect.a016295
48. Brenner D, Blaser H, Mak TW. Regulation of Tumour Necrosis Factor Signalling: Live or Let Die. Nat Rev Immunol (2015) 15(6):362–74. doi: 10.1038/nri3834
49. Chandrasekharan UM, Siemionow M, Unsal M, Yang L, Poptic E, Bohn J, et al. Tumor Necrosis Factor Alpha (TNF-Alpha) Receptor-II is Required for TNF-Alpha-Induced Leukocyte-Endothelial Interaction in Vivo. Blood (2007) 109(5):1938–44. doi: 10.1182/blood-2006-05-020875
50. Schoggins JW. Interferon-Stimulated Genes: What do They All do? Annu Rev Virol (2019) 6(1):567–84. doi: 10.1146/annurev-virology-092818-015756
51. Chang EY, Guo B, Doyle SE, Cheng G. Cutting Edge: Involvement of the Type I IFN Production and Signaling Pathway in Lipopolysaccharide-Induced IL-10 Production. J Immunol (2007) 178(11):6705. doi: 10.4049/jimmunol.178.11.6705
52. de Oliveira Mann CC, Hornung V. Molecular Mechanisms of Nonself Nucleic Acid Recognition by the Innate Immune System. Eur J Immunol (2021) 51(8):1897–910. doi: 10.1002/eji.202049116
53. Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A Molecular Cell Atlas of the Human Lung From Single-Cell RNA Sequencing. Nature (2020) 587(7835):619–25. doi: 10.1038/s41586-020-2922-4
54. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al. Recognition of Single-Stranded RNA Viruses by Toll-Like Receptor 7. Proc Natl Acad Sci USA (2004) 101(15):5598–603. doi: 10.1073/pnas.0400937101
55. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate Antiviral Responses by Means of TLR7-Mediated Recognition of Single-Stranded RNA. Science (2004) 303(5663):1529–31. doi: 10.1126/science.1093616
56. Lim HK, Huang SXL, Chen J, Kerner G, Gilliaux O, Bastard P, et al. Severe Influenza Pneumonitis in Children With Inherited TLR3 Deficiency. J Exp Med (2019) 216(9):2038–56. doi: 10.1084/jem.20181621
57. Kuriakose T, Man SM, Subbarao Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an Innate Sensor of Influenza Virus Triggering the NLRP3 Inflammasome and Programmed Cell Death Pathways. Sci Immunol (2016) 1(2):aag2045–aag. doi: 10.1126/sciimmunol.aag2045
58. Schroder K, Tschopp J. The Inflammasomes. Cell (2010) 140(6):821–32. doi: 10.1016/j.cell.2010.01.040
59. Lee S, Hirohama M, Noguchi M, Nagata K, Kawaguchi A. Influenza a Virus Infection Triggers Pyroptosis and Apoptosis of Respiratory Epithelial Cells Through the Type I Interferon Signaling Pathway in a Mutually Exclusive Manner. J Virol (2018) 92(14):e00396–18. doi: 10.1128/JVI.00396-18
60. Lee S, Ishitsuka A, Noguchi M, Hirohama M, Fujiyasu Y, Petric Philipp P, et al. Influenza Restriction Factor Mxa Functions as Inflammasome Sensor in the Respiratory Epithelium. Sci Immunol (2019) 4(40):eaau4643. doi: 10.1126/sciimmunol.aau4643
61. Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, et al. Influenza Virus Z-Rnas Induce ZBP1-Mediated Necroptosis. Cell (2020) 180(6):1115–29.e13. doi: 10.1016/j.cell.2020.02.050
62. Liu G, Lu Y, Thulasi Raman SN, Xu F, Wu Q, Li Z, et al. Nuclear-Resident RIG-I Senses Viral Replication Inducing Antiviral Immunity. Nat Commun (2018) 9(1):3199. doi: 10.1038/s41467-018-05745-w
63. Ichinohe T, Pang IK, Iwasaki A. Influenza Virus Activates Inflammasomes via its Intracellular M2 Ion Channel. Nat Immunol (2010) 11(5):404–10. doi: 10.1038/ni.1861
64. Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, et al. The TLR4 Antagonist Eritoran Protects Mice From Lethal Influenza Infection. Nature (2013) 497(7450):498–502. doi: 10.1038/nature12118
65. Short KR, Kasper J, van der Aa S, Andeweg AC, Zaaraoui-Boutahar F, Goeijenbier M, et al. Influenza Virus Damages the Alveolar Barrier by Disrupting Epithelial Cell Tight Junctions. Eur Respir J (2016) 47(3):954. doi: 10.1183/13993003.01282-2015
66. Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, et al. Endothelial Cells Are Central Orchestrators of Cytokine Amplification During Influenza Virus Infection. Cell (2011) 146(6):980–91. doi: 10.1016/j.cell.2011.08.015
67. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-Cov-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181(2):271–80.e8. doi: 10.1016/j.cell.2020.02.052
68. Peacock TP, Goldhill DH, Zhou J, Baillon L, Frise R, Swann OC, et al. The Furin Cleavage Site in the SARS-Cov-2 Spike Protein Is Required for Transmission in Ferrets. Nat Microbiol (2021) 6(7):899–909. doi: 10.1038/s41564-021-00908-w
69. Thorne LG, Reuschl AK, Zuliani-Alvarez L, Whelan MVX, Turner J, Noursadeghi M, et al. SARS-Cov-2 Sensing by RIG-I and MDA5 Links Epithelial Infection to Macrophage Inflammation. EMBO J (2021) 40(15):e107826. doi: 10.15252/embj.2021107826
70. Vora SM, Lieberman J, Wu H. Inflammasome Activation at the Crux of Severe COVID-19. Nat Rev Immunol (2021) 1–10. doi: 10.1038/s41577-021-00588-x
71. Dalskov L, Møhlenberg M, Thyrsted J, Blay-Cadanet J, Poulsen ET, Folkersen BH, et al. SARS-Cov-2 Evades Immune Detection in Alveolar Macrophages. EMBO Rep (2020) 21(12):e51252. doi: 10.15252/embr.202051252
72. Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, Hornung V. Human NLRP1 is a Sensor for Double-Stranded RNA. Science (2021) 371(6528):eabd0811. doi: 10.1126/science.abd0811
73. Asano T, Boisson B, Onodi F, Matuozzo D, Moncada-Velez M, Maglorius Renkilaraj MRL, et al. X-Linked Recessive TLR7 Deficiency in ~1% of Men Under 60 Years Old With Life-Threatening COVID-19. Sci Immunol (2021) 6(62):eabl4348. doi: 10.1126/sciimmunol.abl4348
74. Zheng M, Karki R, Williams EP, Yang D, Fitzpatrick E, Vogel P, et al. TLR2 Senses the SARS-Cov-2 Envelope Protein to Produce Inflammatory Cytokines. Nat Immunol (2021) 22(7):829–38. doi: 10.1038/s41590-021-00937-x
75. Bawazeer AO, Rosli S, Harpur CM, Docherty CA, Mansell A, Tate MD. Interleukin-1β Exacerbates Disease and is a Potential Therapeutic Target to Reduce Pulmonary Inflammation During Severe Influenza a Virus Infection. Immunol Cell Biol (2021) 99(7):737–48. doi: 10.1111/imcb.12459
76. Meischel T, Villalon-Letelier F, Saunders PM, Reading PC, Londrigan SL. Influenza a Virus Interactions With Macrophages: Lessons From Epithelial Cells. Cell Microbiol (2020) 22(5):e13170. doi: 10.1111/cmi.13170
77. Gillespie L, Roosendahl P, Ng WC, Brooks AG, Reading PC, Londrigan SL. Endocytic Function is Critical for Influenza a Virus Infection via DC-SIGN and L-SIGN. Sci Rep (2016) 6:19428. doi: 10.1038/srep19428
78. Sandt C, Bárcena M, Koster AJ, Kasper J, Kirkpatrick CJ, Scott DP, et al. Human CD8+ T Cells Damage Noninfected Epithelial Cells During Influenza Virus Infection in Vitro. Am J Respir Cell Mol Biol (2017) 57(5):536–46. doi: 10.1165/rcmb.2016-0377OC
79. Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N. Contrasting Effects of CCR5 and CCR2 Deficiency in the Pulmonary Inflammatory Response to Influenza a Virus. Am J Pathol (2000) 156(6):1951–9. doi: 10.1016/S0002-9440(10)65068-7
80. Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, von Messling V, et al. Type I IFN Triggers RIG-I/TLR3/NLRP3-Dependent Inflammasome Activation in Influenza a Virus Infected Cells. PloS Pathog (2013) 9(4):e1003256. doi: 10.1371/journal.ppat.1003256
81. Wang Z, Zhang A, Wan Y, Liu X, Qiu C, Xi X, et al. Early Hypercytokinemia Is Associated With Interferon-Induced Transmembrane Protein-3 Dysfunction and Predictive of Fatal H7N9 Infection. Proc Natl Acad Sci (2014) 111(2):769. doi: 10.1073/pnas.1321748111
82. Yu L, Wang Z, Chen Y, Ding W, Jia H, Chan JF, et al. Clinical, Virological, and Histopathological Manifestations of Fatal Human Infections by Avian Influenza a(H7N9) Virus. Clin Infect Dis (2013) 57(10):1449–57. doi: 10.1093/cid/cit541
83. Arankalle VA, Lole KS, Arya RP, Tripathy AS, Ramdasi AY, Chadha MS, et al. Role of Host Immune Response and Viral Load in the Differential Outcome of Pandemic H1N1 (2009) Influenza Virus Infection in Indian Patients. PloS One (2010) 5(10):e13099. doi: 10.1371/journal.pone.0013099
84. Angel E. Cytokine Levels in Bronchoalveolar Lavage and Serum in 3 Patients With 2009 Influenza a(H1N1)V Severe Pneumonia. J Infect Dev Countries (2011) 5(07):540–3. doi: 10.3855/jidc.1618
85. Gao R, Bhatnagar J, Blau DM, Greer P, Rollin DC, Denison AM, et al. Cytokine and Chemokine Profiles in Lung Tissues From Fatal Cases of 2009 Pandemic Influenza a (H1N1): Role of the Host Immune Response in Pathogenesis. Am J Pathol (2013) 183(4):1258–68. doi: 10.1016/j.ajpath.2013.06.023
86. Meduri GU, Kohler G, Headley S, Tolley E, Stentz F, Postlethwaite A. Inflammatory Cytokines in the BAL of Patients With ARDS. Persistent Elevation Over Time Predicts Poor Outcome. Chest (1995) 108(5):1303–14. doi: 10.1378/chest.108.5.1303
87. Paquette SG, Banner D, Zhao Z, Fang Y, Huang SS, Leόn AJ, et al. Interleukin-6 Is a Potential Biomarker for Severe Pandemic H1N1 Influenza a Infection. PloS One (2012) 7(6):e38214. doi: 10.1371/journal.pone.0038214
88. Liu Q, Zhou YH, Yang ZQ. The Cytokine Storm of Severe Influenza and Development of Immunomodulatory Therapy. Cell Mol Immunol (2016) 13(1):3–10. doi: 10.1038/cmi.2015.74
89. Hussell T, Goulding J. Structured Regulation of Inflammation During Respiratory Viral Infection. Lancet Infect Dis (2010) 10(5):360–6. doi: 10.1016/S1473-3099(10)70067-0
90. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the Eye of the Cytokine Storm. Microbiol Mol Biol Rev (2012) 76(1):16–32. doi: 10.1128/MMBR.05015-11
91. Brandes M, Klauschen F, Kuchen S, Germain RN. A Systems Analysis Identifies a Feedforward Inflammatory Circuit Leading to Lethal Influenza Infection. Cell (2013) 154(1):197–212. doi: 10.1016/j.cell.2013.06.013
92. Chan MCW, Cheung CY, Chui WH, Tsao SW, Nicholls JM, Chan YO, et al. Proinflammatory Cytokine Responses Induced by Influenza a (H5N1) Viruses in Primary Human Alveolar and Bronchial Epithelial Cells. Respir Res (2005) 6(1):135. doi: 10.1186/1465-9921-6-135
93. Cheung CY, Poon LL, Lau AS, Luk W, Lau YL, Shortridge KF, et al. Induction of Proinflammatory Cytokines in Human Macrophages by Influenza a (H5N1) Viruses: A Mechanism for the Unusual Severity of Human Disease? Lancet (2002) 360(9348):1831–7. doi: 10.1016/S0140-6736(02)11772-7
94. de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, et al. Fatal Outcome of Human Influenza a (H5N1) Is Associated With High Viral Load and Hypercytokinemia. Nat Med (2006) 12(10):1203–7. doi: 10.1038/nm1477
95. Hui KP, Lee SM, Cheung CY, Ng IH, Poon LL, Guan Y, et al. Induction of Proinflammatory Cytokines in Primary Human Macrophages by Influenza a Virus (H5N1) Is Selectively Regulated by IFN Regulatory Factor 3 and P38 MAPK. J Immunol (2009) 182(2):1088–98. doi: 10.4049/jimmunol.182.2.1088
96. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome Recognition of Influenza Virus is Essential for Adaptive Immune Responses. J Exp Med (2009) 206(1):79–87. doi: 10.1084/jem.20081667
97. Thomas PG, Dash P, Aldridge JR Jr., Ellebedy AH, Reynolds C, Funk AJ, et al. The Intracellular Sensor NLRP3 Mediates Key Innate and Healing Responses to Influenza a Virus via the Regulation of Caspase-1. Immunity (2009) 30(4):566–75. doi: 10.1016/j.immuni.2009.02.006
98. Tate MD, Ong JDH, Dowling JK, McAuley JL, Robertson AB, Latz E, et al. Reassessing the Role of the NLRP3 Inflammasome During Pathogenic Influenza a Virus Infection via Temporal Inhibition. Sci Rep (2016) 6:27912. doi: 10.1038/srep27912
99. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
100. Synowiec A, Szczepański A, Barreto-Duran E, Lie LK, Pyrc K. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-Cov-2): A Systemic Infection. Clin Microbiol Rev (2021) 34(2):e00133–20. doi: 10.1128/CMR.00133-20
101. Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, et al. Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell (2020) 181(7):1475–88.e12. doi: 10.1016/j.cell.2020.05.006
102. Schimmel L, Chew KY, Stocks C, Yordanov T, Essebier T, Kulasinghe A, et al. Endothelial Cells Elicit a Pro-Inflammatory Response to SARS-Cov-2 Without Productive Viral Infection. bioRxiv (2021). 2021.02.14.431177. doi: 10.1101/2021.02.14.431177
103. Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, et al. Dexamethasone in Hospitalized Patients With Covid-19. N Engl J Med (2021) 384(8):693–704. doi: 10.1056/NEJMoa2021436
104. Kyriazopoulou E, Huet T, Cavalli G, Gori A, Kyprianou M, Pickkers P, et al. Effect of Anakinra on Mortality in Patients With COVID-19: A Systematic Review and Patient-Level Meta-Analysis. Lancet Rheumatol (2017) 3(10):e690–7. doi: 10.1016/S2665-9913(21)00216-2
105. Ucciferri C, Auricchio A, Di Nicola M, Potere N, Abbate A, Cipollone F, et al. Canakinumab in a Subgroup of Patients With COVID-19. Lancet Rheumatol (2020) 2(8):e457–ee8. doi: 10.1016/S2665-9913(20)30167-3
106. FDA. Coronavirus (COVID-19) Update: FDA Authorizes Drug for Treatment of COVID-19 (2021). Available at: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-drug-treatment-covid-19.
107. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies Against Type I Ifns in Patients With Life-Threatening COVID-19. Science (2020) 370(6515):eabd4585. doi: 10.1126/science.abd4585
108. Broggi A, Ghosh S, Sposito B, Spreafico R, Balzarini F, Lo Cascio A, et al. Type III Interferons Disrupt the Lung Epithelial Barrier Upon Viral Recognition. Science (2020) 369(6504):706–12. doi: 10.1126/science.abc3545
109. Hussell T, Pennycook A, Openshaw PJ. Inhibition of Tumor Necrosis Factor Reduces the Severity of Virus-Specific Lung Immunopathology. Eur J Immunol (2001) 31(9):2566–73. doi: 10.1002/1521-4141(200109)31:9<2566::AID-IMMU2566>3.0.CO;2-L
110. Lee SM, Cheung CY, Nicholls JM, Hui KP, Leung CY, Uiprasertkul M, et al. Hyperinduction of Cyclooxygenase-2-Mediated Proinflammatory Cascade: A Mechanism for the Pathogenesis of Avian Influenza H5N1 Infection. J Infect Dis (2008) 198(4):525–35. doi: 10.1086/590499
111. Yarilina A, Park-Min K-H, Antoniv T, Hu X, Ivashkiv LB. TNF Activates an IRF1-Dependent Autocrine Loop Leading to Sustained Expression of Chemokines and STAT1-Dependent Type I Interferon–Response Genes. Nat Immunol (2008) 9(4):378–87. doi: 10.1038/ni1576
112. Jung SH, Kwon JM, Shim JW, Kim DS, Jung HL, Park MS, et al. Effects of Diet-Induced Mild Obesity on Airway Hyperreactivity and Lung Inflammation in Mice. Yonsei Med J (2013) 54(6):1430–7. doi: 10.3349/ymj.2013.54.6.1430
113. Belisle SE, Tisoncik JR, Korth MJ, Carter VS, Proll SC, Swayne DE, et al. Genomic Profiling of Tumor Necrosis Factor Alpha (TNF-α); Receptor and Interleukin-1 Receptor Knockout Mice Reveals a Link Between TNF-α Signaling and Increased Severity of 1918 Pandemic Influenza Virus Infection. J Virol (2010) 84(24):12576–88. doi: 10.1128/JVI.01310-10
114. Peper R, Campen Hv. Tumor Necrosis Factor as a Mediator of Inflammation in Influenza a Viral Pneumonia. Microb Pathogen (1995) 19 3:175–83. doi: 10.1006/mpat.1995.0056
115. Damjanovic D, Divangahi M, Kugathasan K, Small C-L, Zganiacz A, Brown EG, et al. Negative Regulation of Lung Inflammation and Immunopathology by TNF-α During Acute Influenza Infection. Am J Pathol (2011) 179(6):2963–76. doi: 10.1016/j.ajpath.2011.09.003
116. Liu S, Yan R, Chen B, Pan Q, Chen Y, Hong J, et al. Influenza Virus-Induced Robust Expression of SOCS3 Contributes to Excessive Production of IL-6. Front Immunol (2019) 10:1843–. doi: 10.3389/fimmu.2019.01843
117. Yang M-L, Wang C-T, Yang S-J, Leu C-H, Chen S-H, Wu C-L, et al. IL-6 Ameliorates Acute Lung Injury in Influenza Virus Infection. Sci Rep (2017) 7(1):43829. doi: 10.1038/srep43829
118. Velazquez-Salinas L, Verdugo-Rodriguez A, Rodriguez LL, Borca MV. The Role of Interleukin 6 During Viral Infections. Front Microbiol (1057) 2019:10. doi: 10.3389/fmicb.2019.01057
119. Lauder SN, Jones E, Smart K, Bloom A, Williams AS, Hindley JP, et al. Interleukin-6 Limits Influenza-Induced Inflammation and Protects Against Fatal Lung Pathology. Eur J Immunol (2013) 43(10):2613–25. doi: 10.1002/eji.201243018
120. Kaiser L, Fritz RS, Straus SE, Gubareva L, Hayden FG. Symptom Pathogenesis During Acute Influenza: Interleukin-6 and Other Cytokine Responses. J Med Virol (2001) 64(3):262–8. doi: 10.1002/jmv.1045
121. Hagau N, Slavcovici A, Gonganau DN, Oltean S, Dirzu DS, Brezoszki ES, et al. Clinical Aspects and Cytokine Response in Severe H1N1 Influenza a Virus Infection. Crit Care (2010) 14(6):R203. doi: 10.1186/cc9324
122. Gu Y, Zuo X, Zhang S, Ouyang Z, Jiang S, Wang F, et al. The Mechanism Behind Influenza Virus Cytokine Storm. Viruses (2021) 13(7):1362. doi: 10.3390/v13071362
123. El-Mikkawy DME, El-Sadek MA, El-Badawy MA, Samaha D. Circulating Level of Interleukin-6 in Relation to Body Mass Indices and Lipid Profile in Egyptian Adults With Overweight and Obesity. Egypt Rheumatol Rehabil (2020) 47(1):7. doi: 10.1186/s43166-020-00003-8
124. Short KR, Kroeze E, Fouchier RAM, Kuiken T. Pathogenesis of Influenza-Induced Acute Respiratory Distress Syndrome. Lancet Infect Dis (2014) 14(1):57–69. doi: 10.1016/S1473-3099(13)70286-X
125. Julkunen I, Sareneva T, Pirhonen J, Ronni T, Melén K, Matikainen S. Molecular Pathogenesis of Influenza a Virus Infection and Virus-Induced Regulation of Cytokine Gene Expression. Cytokine Growth Factor Rev (2001) 12(2-3):171–80. doi: 10.1016/S1359-6101(00)00026-5
126. Bry K, Lappalainen U. Pathogenesis of Bronchopulmonary Dysplasia: The Role of Interleukin 1beta in the Regulation of Inflammation-Mediated Pulmonary Retinoic Acid Pathways in Transgenic Mice. Semin Perinatol (2006) 30(3):121–8. doi: 10.1053/j.semperi.2006.04.009
127. Kim KS, Jung H, Shin IK, Choi B-R, Kim DH. Induction of Interleukin-1 Beta (IL-1β) is a Critical Component of Lung Inflammation During Influenza a (H1N1) Virus Infection. J Med Virol (2015) 87(7):1104–12. doi: 10.1002/jmv.24138
128. Liu B, Mori I, Hossain MJ, Dong L, Takeda K, Kimura Y. Interleukin-18 Improves the Early Defence System Against Influenza Virus Infection by Augmenting Natural Killer Cell-Mediated Cytotoxicity. J Gen Virol (2004) 85(Pt 2):423–8. doi: 10.1099/vir.0.19596-0
129. Denton AE, Doherty PC, Turner SJ, La Gruta NL. IL-18, But Not IL-12, is Required for Optimal Cytokine Production by Influenza Virus-Specific CD8+ T Cells. Eur J Immunol (2007) 37(2):368–75. doi: 10.1002/eji.200636766
130. Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic Potential of Interferon αβ in Acute Influenza Infection. Nat Commun (2014) 5(1):3864. doi: 10.1038/ncomms4864
131. Wu W, Metcalf JP. The Role of Type I Ifns in Influenza: Antiviral Superheroes or Immunopathogenic Villains? J Innate Immun (2020) 12(6):437–47. doi: 10.1159/000508379
132. Easterbrook JD, Dunfee RL, Schwartzman LM, Jagger BW, Sandouk A, Kash JC, et al. Obese Mice Have Increased Morbidity and Mortality Compared to Non-Obese Mice During Infection With the 2009 Pandemic H1N1 Influenza Virus. Influenza Other Respir Viruses (2011) 5(6):418–25. doi: 10.1111/j.1750-2659.2011.00254.x
133. Takahashi K, Mizuarai S, Araki H, Mashiko S, Ishihara A, Kanatani A, et al. Adiposity Elevates Plasma MCP-1 Levels Leading to the Increased CD11b-Positive Monocytes in Mice. J Biol Chem (2003) 278(47):46654–60. doi: 10.1074/jbc.M309895200
134. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I Interferons in Infectious Disease. Nat Rev Immunol (2015) 15(2):87–103. doi: 10.1038/nri3787
135. Liu B, Bao L, Wang L, Li F, Wen M, Li H, et al. Anti-IFN-γ Therapy Alleviates Acute Lung Injury Induced by Severe Influenza a (H1N1) Pdm09 Infection in Mice. J Microbiol Immunol Infect (2021) 54(3):396–403. doi: 10.1016/j.jmii.2019.07.009
136. Weiss ID, Wald O, Wald H, Beider K, Abraham M, Galun E, et al. IFN-Gamma Treatment at Early Stages of Influenza Virus Infection Protects Mice From Death in a NK Cell-Dependent Manner. J Interferon Cytokine Res (2010) 30(6):439–49. doi: 10.1089/jir.2009.0084
137. Guo X-ZJ, Thomas PG. New Fronts Emerge in the Influenza Cytokine Storm. Semin Immunopathol (2017) 39(5):541–50. doi: 10.1007/s00281-017-0636-y
138. Ramana CV, Chintapalli J, Xu L, Alia C, Zhou J, Bruder D, et al. Lung Epithelial NF-Kappab and Stat1 Signaling in Response to CD8+ T Cell Antigen Recognition. J Interferon Cytokine Res (2006) 26(5):318–27. doi: 10.1089/jir.2006.26.318
139. Liu B, Zhang X, Deng W, Liu J, Li H, Wen M, et al. Severe Influenza a(H1N1)Pdm09 Infection Induces Thymic Atrophy Through Activating Innate CD8+CD44hi T Cells by Upregulating IFN-γ. Cell Death Dis (2014) 5(10):e1440–e. doi: 10.1038/cddis.2014.323
140. Paich HA, Sheridan PA, Handy J, Karlsson EA, Schultz-Cherry S, Hudgens MG, et al. Overweight and Obese Adult Humans Have a Defective Cellular Immune Response to Pandemic H1N1 Influenza a Virus. Obes (Silver Spring) (2013) 21(11):2377–86. doi: 10.1002/oby.20383
141. Nicol MQ, Campbell GM, Shaw DJ, Dransfield I, Ligertwood Y, Beard PM, et al. Lack of Ifnγ Signaling Attenuates Spread of Influenza a Virus In Vivo and Leads to Reduced Pathogenesis. Virology (2019) 526:155–64. doi: 10.1016/j.virol.2018.10.017
142. Mordstein M, Kochs G, Dumoutier L, Renauld J-C, Paludan SR, Klucher K, et al. Interferon-Lambda Contributes to Innate Immunity of Mice Against Influenza a Virus But Not Against Hepatotropic Viruses. PloS Pathog (2008) 4(9):e1000151–e. doi: 10.1371/journal.ppat.1000151
143. Jewell NA, Cline T, Mertz SE, Smirnov SV, Flaño E, Schindler C, et al. Lambda Interferon is the Predominant Interferon Induced by Influenza a Virus Infection In Vivo. J Virol (2010) 84(21):11515–22. doi: 10.1128/JVI.01703-09
144. Karlsson EA, Sheridan PA, Beck MA. Diet-Induced Obesity Impairs the T Cell Memory Response to Influenza Virus Infection. J Immunol (2010) 184(6):3127. doi: 10.4049/jimmunol.0903220
145. Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, Falcone V, et al. Lambda Interferon Renders Epithelial Cells of the Respiratory and Gastrointestinal Tracts Resistant to Viral Infections. J Virol (2010) 84(11):5670–7. doi: 10.1128/JVI.00272-10
146. Li C, Yang P, Sun Y, Li T, Wang C, Wang Z, et al. IL-17 Response Mediates Acute Lung Injury Induced by the 2009 Pandemic Influenza a (H1N1) Virus. Cell Res (2012) 22(3):528–38. doi: 10.1038/cr.2011.165
147. McKinstry KK, Strutt TM, Buck A, Curtis JD, Dibble JP, Huston G, et al. IL-10 Deficiency Unleashes an Influenza-Specific Th17 Response and Enhances Survival Against High-Dose Challenge. J Immunol (2009) 182(12):7353–63. doi: 10.4049/jimmunol.0900657
148. Sun J, Madan R, Karp CL, Braciale TJ. Effector T Cells Control Lung Inflammation During Acute Influenza Virus Infection by Producing IL-10. Nat Med (2009) 15(3):277–84. doi: 10.1038/nm.1929
149. Sun K, Torres L, Metzger DW. A Detrimental Effect of Interleukin-10 on Protective Pulmonary Humoral Immunity During Primary Influenza a Virus Infection. J Virol (2010) 84(10):5007–14. doi: 10.1128/JVI.02408-09
150. Dutta A, Huang C-T, Chen T-C, Lin C-Y, Chiu C-H, Lin Y-C, et al. IL-10 Inhibits Neuraminidase-Activated TGF-β and Facilitates Th1 Phenotype During Early Phase of Infection. Nat Commun (2015) 6(1):6374. doi: 10.1038/ncomms7374
151. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn Errors of Type I IFN Immunity in Patients With Life-Threatening COVID-19. Science (2020) 370(6515):eabd4570. doi: 10.1126/science.abd4570
152. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients. Science (2020) 369(6504):718–24. doi: 10.1126/science.abc6027
153. Barsh GS, Farooqi IS, O’rahilly S. Genetics of Body-Weight Regulation. Nature (2000) 404(6778):644–51. doi: 10.1038/35007519
154. Maes HH, Neale MC, Eaves LJ. Genetic and Environmental Factors in Relative Body Weight and Human Adiposity. Behav Genet (1997) 27(4):325–51. doi: 10.1023/A:1025635913927
155. Oussaada SM, van Galen KA, Cooiman MI, Kleinendorst L, Hazebroek EJ, van Haelst MM, et al. The Pathogenesis of Obesity. Metabolism (2019) 92:26–36. doi: 10.1016/j.metabol.2018.12.012
156. Clément K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, et al. A Mutation in the Human Leptin Receptor Gene Causes Obesity and Pituitary Dysfunction. Nature (1998) 392(6674):398–401. doi: 10.1038/32911
157. Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, et al. Congenital Leptin Deficiency is Associated With Severe Early-Onset Obesity in Humans. Nature (1997) 387(6636):903–8. doi: 10.1038/43185
158. Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson M-L, Sanders L, Montague CT, et al. Obesity and Impaired Prohormone Processing Associated With Mutations in the Human Prohormone Convertase 1 Gene. Nat Genet (1997) 16(3):303–6. doi: 10.1038/ng0797-303
159. Farooqi IS, O’Rahilly S. Monogenic Obesity in Humans. Annu Rev Med (2005) 56:443–58. doi: 10.1146/annurev.med.56.062904.144924
160. Alharbi KK, Spanakis E, Tan K, Smith MJ, Aldahmesh MA, O’Dell SD, et al. Prevalence and Functionality of Paucimorphic and Private MC4R Mutations in a Large, Unselected European British Population, Scanned by Meltmadge. Hum Mutat (2007) 28(3):294–302. doi: 10.1002/humu.20404
161. van der Klaauw AA, Farooqi IS. The Hunger Genes: Pathways to Obesity. Cell (2015) 161(1):119–32. doi: 10.1016/j.cell.2015.03.008
162. Farooqi IS, Bullmore E, Keogh J, Gillard J, O’Rahilly S, Fletcher PC. Leptin Regulates Striatal Regions and Human Eating Behavior. Science (2007) 317(5843):1355–. doi: 10.1126/science.1144599
163. Lin S, Thomas TC, Storlien LH, Huang XF. Development of High Fat Diet-Induced Obesity and Leptin Resistance in C57Bl/6J Mice. Int J Obes (2000) 24(5):639–46. doi: 10.1038/sj.ijo.0801209
164. Hawkes C, Harris J, Gillespie S. Urbanization and the Nutrition Transition. Washington DC: International Food Policy Research Institute (IFPRI) (2017).
165. Wang H, Eckel RH. Lipoprotein Lipase: From Gene to Obesity. Am J Physiol Endocrinol Metab (2009) 297(2):E271–88. doi: 10.1152/ajpendo.90920.2008
166. Masoodi M, Kuda O, Rossmeisl M, Flachs P, Kopecky J. Lipid Signaling in Adipose Tissue: Connecting Inflammation & Metabolism. Biochim Biophys Acta (2015) 1851(4):503–18. doi: 10.1016/j.bbalip.2014.09.023
167. Hintze KJ, Benninghoff AD, Cho CE, Ward RE. Modeling the Western Diet for Preclinical Investigations. Adv Nutr (2018) 9(3):263–71. doi: 10.1093/advances/nmy002
168. Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-Induced Type II Diabetes in C57BL/6J Mice. Diabetes (1988) 37(9):1163–7. doi: 10.2337/diab.37.9.1163
169. Surwit RS, Seldin MF, Kuhn CM, Cochrane C, Feinglos MN. Control of Expression of Insulin Resistance and Hyperglycemia by Different Genetic Factors in Diabetic C57BL/6J Mice. Diabetes (1991) 40(1):82–7. doi: 10.2337/diab.40.1.82
170. Surwit RS, Feinglos MN, Rodin J, Sutherland A, Petro AE, Opara EC, et al. Differential Effects of Fat and Sucrose on the Development of Obesity and Diabetes in C57BL/6J and AJ Mice. Metabolism (1995) 44(5):645–51. doi: 10.1016/0026-0495(95)90123-X
171. Christ A, Gunther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western Diet Triggers Nlrp3-Dependent Innate Immune Reprogramming. Cell (2018) 172(1-2):162–75.e14. doi: 10.1016/j.cell.2017.12.013
172. van der Heijden RA, Sheedfar F, Morrison MC, Hommelberg PP, Kor D, Kloosterhuis NJ, et al. High-Fat Diet Induced Obesity Primes Inflammation in Adipose Tissue Prior to Liver in C57BL/6j Mice. Aging (Albany NY) (2015) 7(4):256–68. doi: 10.18632/aging.100738
173. Snelson M, Tan SM, Clarke RE, de Pasquale C, Thallas-Bonke V, Nguyen T-V, et al. Processed Foods Drive Intestinal Barrier Permeability and Microvascular Diseases. Sci Adv (2021) 7(14):eabe4841. doi: 10.1126/sciadv.abe4841
174. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
175. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory Caspases are Innate Immune Receptors for Intracellular LPS. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
176. Fuster JJ, Ouchi N, Gokce N, Walsh K. Obesity-Induced Changes in Adipose Tissue Microenvironment and Their Impact on Cardiovascular Disease. Circ Res (2016) 118(11):1786–807. doi: 10.1161/CIRCRESAHA.115.306885
177. Frasca D, Diaz A, Romero M, Thaller S, Blomberg BB. Secretion of Autoimmune Antibodies in the Human Subcutaneous Adipose Tissue. PloS One (2018) 13(5):e0197472–e. doi: 10.1371/journal.pone.0197472
178. Farr OM, Gavrieli A, Mantzoros CS. Leptin Applications in 2015: What Have We Learned About Leptin and Obesity? Curr Opin Endocrinol Diabetes Obes (2015) 22(5):353–9. doi: 10.1097/MED.0000000000000184
179. Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The Hormone Resistin Links Obesity to Diabetes. Nature (2001) 409(6818):307–12. doi: 10.1038/35053000
180. De Rosa A, Monaco ML, Capasso M, Forestieri P, Pilone V, Nardelli C, et al. Adiponectin Oligomers as Potential Indicators of Adipose Tissue Improvement in Obese Subjects. Eur J Endocrinol (2013) 169(1):37–43. doi: 10.1530/EJE-12-1039
181. Kaser S, Tatarczyk T, Stadlmayr A, Ciardi C, Ress C, Tschoner A, et al. Effect of Obesity and Insulin Sensitivity on Adiponectin Isoform Distribution. Eur J Clin Invest (2008) 38(11):827–34. doi: 10.1111/j.1365-2362.2008.02028.x
182. Meyer LK, Ciaraldi TP, Henry RR, Wittgrove AC, Phillips SA. Adipose Tissue Depot and Cell Size Dependency of Adiponectin Synthesis and Secretion in Human Obesity. Adipocyte (2013) 2(4):217–26. doi: 10.4161/adip.24953
183. Temba GS, Kullaya V, Pecht T, Mmbaga BT, Aschenbrenner AC, Ulas T, et al. Urban Living in Healthy Tanzanians is Associated With an Inflammatory Status Driven by Dietary and Metabolic Changes. Nat Immunol (2021) 22(3):287–300. doi: 10.1038/s41590-021-00867-8
184. Kliewer SA, Mangelsdorf DJ. A Dozen Years of Discovery: Insights Into the Physiology and Pharmacology of FGF21. Cell Metab (2019) 29(2):246–53. doi: 10.1016/j.cmet.2019.01.004
185. Lin X, Liu YB, Hu H. Metabolic Role of Fibroblast Growth Factor 21 in Liver, Adipose and Nervous System Tissues. BioMed Rep (2017) 6(5):495–502. doi: 10.3892/br.2017.890
186. Huen SC, Wang A, Feola K, Desrouleaux R, Luan HH, Hogg R, et al. Hepatic FGF21 Preserves Thermoregulation and Cardiovascular Function During Bacterial Inflammation. J Exp Med (2021) 218(10):e20202151. doi: 10.1084/jem.20202151
187. Ost M, Igual Gil C, Coleman V, Keipert S, Efstathiou S, Vidic V, et al. Muscle-Derived GDF15 Drives Diurnal Anorexia and Systemic Metabolic Remodeling During Mitochondrial Stress. EMBO Rep (2020) 21(3):e48804. doi: 10.15252/embr.201948804
188. Zhang X, Yeung DC, Karpisek M, Stejskal D, Zhou ZG, Liu F, et al. Serum FGF21 Levels are Increased in Obesity and are Independently Associated With the Metabolic Syndrome in Humans. Diabetes (2008) 57(5):1246–53. doi: 10.2337/db07-1476
189. Keipert S, Ost M. Stress-Induced FGF21 and GDF15 in Obesity and Obesity Resistance. Trends Endocrinol Metab (2021). doi: 10.1016/j.tem.2021.08.008
190. Luan HH, Wang A, Hilliard BK, Carvalho F, Rosen CE, Ahasic AM, et al. GDF15 is an Inflammation-Induced Central Mediator of Tissue Tolerance. Cell (2019) 178(5):1231–44.e11. doi: 10.1016/j.cell.2019.07.033
191. Cancello R, Clément K. Is Obesity an Inflammatory Illness? Role of Low-Grade Inflammation and Macrophage Infiltration in Human White Adipose Tissue. Bjog (2006) 113(10):1141–7. doi: 10.1111/j.1471-0528.2006.01004.x
192. Ellulu MS, Patimah I, Khaza’ai H, Rahmat A, Abed Y. Obesity and Inflammation: The Linking Mechanism and the Complications. Arch Med Sci (2017) 13(4):851–63. doi: 10.5114/aoms.2016.58928
193. Dicker D, Salook MA, Marcoviciu D, Djaldetti M, Bessler H. Role of Peripheral Blood Mononuclear Cells in the Predisposition of Obese Individuals to Inflammation and Infection. Obes Facts (2013) 6(2):146–51. doi: 10.1159/000350775
194. Teran-Cabanillas E, Montalvo-Corral M, Caire-Juvera G, Moya-Camarena SY, Hernández J. Decreased Interferon-α and Interferon-β Production in Obesity and Expression of Suppressor of Cytokine Signaling. Nutrition (2013) 29(1):207–12. doi: 10.1016/j.nut.2012.04.019
195. Aronson D, Bartha P, Zinder O, Kerner A, Markiewicz W, Avizohar O, et al. Obesity is the Major Determinant of Elevated C-Reactive Protein in Subjects With the Metabolic Syndrome. Int J Obes (2004) 28(5):674–9. doi: 10.1038/sj.ijo.0802609
196. Dreher M, Kersten A, Bickenbach J, Balfanz P, Hartmann B, Cornelissen C, et al. The Characteristics of 50 Hospitalized COVID-19 Patients With and Without ARDS. Dtsch Arztebl Int (2020) 117(16):271–8. doi: 10.3238/arztebl.2020.0271
197. Frasca D, Reidy L, Cray C, Diaz A, Romero M, Kahl K, et al. Influence of Obesity on Serum Levels of SARS-Cov-2-Specific Antibodies in COVID-19 Patients. PloS One (2021) 16(3):e0245424. doi: 10.3389/fendo.2019.00071
198. Murray PJ, Smale ST. Restraint of Inflammatory Signaling by Interdependent Strata of Negative Regulatory Pathways. Nat Immunol (2012) 13(10):916–24. doi: 10.1038/ni.2391
199. Hu H, Sun S-C. Ubiquitin Signaling in Immune Responses. Cell Res (2016) 26(4):457–83. doi: 10.1038/cr.2016.40
200. Fitzgerald KA, Kagan JC. Toll-Like Receptors and the Control of Immunity. Cell (2020) 180(6):1044–66. doi: 10.1016/j.cell.2020.02.041
201. Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, Roach JC, et al. Systems Biology Approaches Identify ATF3 as a Negative Regulator of Toll-Like Receptor 4. Nature (2006) 441(7090):173–8. doi: 10.1038/nature04768
202. Labzin LI, Schmidt SV, Masters SL, Beyer M, Krebs W, Klee K, et al. ATF3 Is a Key Regulator of Macrophage IFN Responses. J Immunol (2015) 195(9):4446–55. doi: 10.4049/jimmunol.1500204
203. De Nardo D, Labzin LI, Kono H, Seki R, Schmidt SV, Beyer M, et al. High-Density Lipoprotein Mediates Anti-Inflammatory Reprogramming of Macrophages via the Transcriptional Regulator ATF3. Nat Immunol (2014) 15(2):152–60. doi: 10.1038/ni.2784
204. Natoli G, Ostuni R. Adaptation and Memory in Immune Responses. Nat Immunol (2019) 20(7):783–92. doi: 10.1038/s41590-019-0399-9
205. Lauterbach MA, Hanke JE, Serefidou M, Mangan MSJ, Kolbe CC, Hess T, et al. Toll-Like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity (2019) 51(6):997–1011.e7. doi: 10.1016/j.immuni.2019.11.009
206. Ouyang W, O’Garra A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity (2019) 50(4):871–91. doi: 10.1016/j.immuni.2019.03.020
207. Labzin LI, Lauterbach MA, Latz E. Interferons and Inflammasomes: Cooperation and Counterregulation in Disease. J Allergy Clin Immunol (2016) 138(1):37–46. doi: 10.1016/j.jaci.2016.05.010
208. Rashid S, Genest J. Effect of Obesity on High-Density Lipoprotein Metabolism. Obes (Silver Spring) (2007) 15(12):2875–88. doi: 10.1038/oby.2007.342
209. Carolan E, Hogan AE, Corrigan M, Gaotswe G, O’Connell J, Foley N, et al. The Impact of Childhood Obesity on Inflammation, Innate Immune Cell Frequency, and Metabolic Microrna Expression. J Clin Endocrinol Metab (2014) 99(3):E474–E8. doi: 10.1210/jc.2013-3529
210. Dorneles GP, Boeira MCR, Schipper LL, Silva IRV, Elsner VR, Dal Lago P, et al. Acute Strenuous Exercise Induces an Imbalance on Histone H4 Acetylation/Histone Deacetylase 2 and Increases the Proinflammatory Profile of PBMC of Obese Individuals. Oxid Med Cell Longev (2017) 2017:1530230. doi: 10.1155/2017/1530230
211. Inzaugarat ME, Billordo LA, Vodánovich F, Cervini GM, Casavalle PL, Vedire C, et al. Alterations in Innate and Adaptive Immune Leukocytes are Involved in Paediatric Obesity. Pediatr Obes (2014) 9(5):381–90. doi: 10.1111/j.2047-6310.2013.00179.x
212. Jialal I, Huet BA, Kaur H, Chien A, Devaraj S. Increased Toll-Like Receptor Activity in Patients With Metabolic Syndrome. Diabetes Care (2012) 35(4):900–4. doi: 10.2337/dc11-2375
213. Schipper HS, Nuboer R, Prop S, van den Ham HJ, de Boer FK, Kesmir Ç, et al. Systemic Inflammation in Childhood Obesity: Circulating Inflammatory Mediators and Activated CD14++ Monocytes. Diabetologia (2012) 55(10):2800–10. doi: 10.1007/s00125-012-2641-y
214. Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, et al. Trained Immunity: A Program of Innate Immune Memory in Health and Disease. Science (2016) 352(6284):aaf1098. doi: 10.1126/science.aaf1098
215. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida Albicans Infection Affords Protection Against Reinfection via Functional Reprogramming of Monocytes. Cell Host Microbe (2012) 12(2):223–32. doi: 10.1016/j.chom.2012.06.006
216. van der Heijden C, Noz MP, Joosten LAB, Netea MG, Riksen NP, Keating ST. Epigenetics and Trained Immunity. Antioxid Redox Signal (2018) 29(11):1023–40. doi: 10.1089/ars.2017.7310
217. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden C, Li Y, et al. Metabolic Induction of Trained Immunity Through the Mevalonate Pathway. Cell (2018) 172(1-2):135–46.e9. doi: 10.1016/j.cell.2017.11.025
218. Norata GD. Trained Immunity and Cardiovascular Disease: Is it Time for Translation to Humans? Cardiovasc Res (2018) 114(6):e41–e2. doi: 10.1093/cvr/cvy043
219. van der Velden J, Tocchetti CG, Varricchi G, Bianco A, Sequeira V, Hilfiker-Kleiner D, et al. Metabolic Changes in Hypertrophic Cardiomyopathies: Scientific Update From the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc Res (2018) 114(10):1273–80. doi: 10.1093/cvr/cvy147
220. Short JD, Tavakoli S, Nguyen HN, Carrera A, Farnen C, Cox LA, et al. Dyslipidemic Diet-Induced Monocyte “Priming” and Dysfunction in Non-Human Primates is Triggered by Elevated Plasma Cholesterol and Accompanied by Altered Histone Acetylation. Front Immunol (2017) 8:958. doi: 10.3389/fimmu.2017.00958
221. Ding J, Reynolds LM, Zeller T, Müller C, Lohman K, Nicklas BJ, et al. Alterations of a Cellular Cholesterol Metabolism Network Are a Molecular Feature of Obesity-Related Type 2 Diabetes and Cardiovascular Disease. Diabetes (2015) 64(10):3464–74. doi: 10.2337/db14-1314
222. Rieckmann A, Villumsen M, Sørup S, Haugaard LK, Ravn H, Roth A, et al. Vaccinations Against Smallpox and Tuberculosis Are Associated With Better Long-Term Survival: A Danish Case-Cohort Study 1971-2010. Int J Epidemiol (2017) 46(2):695–705. doi: 10.1093/ije/dyw120
223. Mitroulis I, Ruppova K, Wang B, Chen L-S, Grzybek M, Grinenko T, et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell (2018) 172(1-2):147–61.e12. doi: 10.1016/j.cell.2017.11.034
224. Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, et al. Chronic Exposure to a TLR Ligand Injures Hematopoietic Stem Cells. J Immunol (2011) 186(9):5367–75. doi: 10.4049/jimmunol.1003438
225. Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-Like Receptors on Hematopoietic Progenitor Cells Stimulate Innate Immune System Replenishment. Immunity (2006) 24(6):801–12. doi: 10.1016/j.immuni.2006.04.008
226. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an Inflammatory Signal That Induces IL-1beta Through HIF-1alpha. Nature (2013) 496(7444):238–42. doi: 10.1038/nature11986
227. van den Berg SM, Seijkens TT, Kusters PJ, Beckers L, den Toom M, Smeets E, et al. Diet-Induced Obesity in Mice Diminishes Hematopoietic Stem and Progenitor Cells in the Bone Marrow. FASEB J (2016) 30(5):1779–88. doi: 10.1096/fj.201500175
228. Negi S, Das DK, Pahari S, Nadeem S, Agrewala JN. Potential Role of Gut Microbiota in Induction and Regulation of Innate Immune Memory. Front Immunol (2019) 10:2441. doi: 10.3389/fimmu.2019.02441
229. Wan Y, Wang F, Yuan J, Li J, Jiang D, Zhang J, et al. Effects of Dietary Fat on Gut Microbiota and Faecal Metabolites, and Their Relationship With Cardiometabolic Risk Factors: A 6-Month Randomised Controlled-Feeding Trial. Gut (2019) 68(8):1417. doi: 10.1136/gutjnl-2018-317609
230. Couillard C, Ruel G, Archer WR, Pomerleau S, Bergeron J, Couture P, et al. Circulating Levels of Oxidative Stress Markers and Endothelial Adhesion Molecules in Men With Abdominal Obesity. J Clin Endocrinol Metab (2005) 90(12):6454–9. doi: 10.1210/jc.2004-2438
231. Weinbrenner T, Schröder H, Escurriol V, Fito M, Elosua R, Vila J, et al. Circulating Oxidized LDL is Associated With Increased Waist Circumference Independent of Body Mass Index in Men and Women. Am J Clin Nutr (2006) 83(1):30–5. doi: 10.1093/ajcn/83.1.30
232. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized Low-Density Lipoprotein Induces Long-Term Proinflammatory Cytokine Production and Foam Cell Formation via Epigenetic Reprogramming of Monocytes. Arterioscler Thromb Vasc Biol (2014) 34(8):1731–8. doi: 10.1161/ATVBAHA.114.303887
233. Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, et al. Hyperglycemia Drives Intestinal Barrier Dysfunction and Risk for Enteric Infection. Science (2018) 359:1376–83. doi: 10.1126/science.aar3318
234. Keating S, Groh L, Thiem K, Bekkering S, Li Y, Matzaraki V, et al. Rewiring of Glucose Metabolism Defines Trained Immunity Induced by Oxidized Low-Density Lipoprotein. J Mol Med (2020) 98:1–13. doi: 10.3390/ijms21113864
235. Vinci MC, Gambini E, Bassetti B, Genovese S, Pompilio G. When Good Guys Turn Bad: Bone Marrow’s and Hematopoietic Stem Cells’ Role in the Pathobiology of Diabetic Complications. Int J Mol Sci (2020) 21(11):3864.
236. Geng L, Lam KSL, Xu A. The Therapeutic Potential of FGF21 in Metabolic Diseases: From Bench to Clinic. Nat Rev Endocrinol (2020) 16(11):654–67. doi: 10.1038/s41574-020-0386-0
237. Bekkering S, Saner C, Riksen NP, Netea MG, Sabin MA, Saffery R, et al. Trained Immunity: Linking Obesity and Cardiovascular Disease Across the Life-Course? Trends Endocrinol Metab (2020) 31(5):378–89. doi: 10.1016/j.tem.2020.01.008
238. Frazier TH, DiBaise JK, McClain CJ. Gut Microbiota, Intestinal Permeability, Obesity-Induced Inflammation, and Liver Injury. JPEN J Parenter Enteral Nutr (2011) 35(5 Suppl):14s–20s. doi: 10.1177/0148607111413772
239. Lassenius MI, Pietiläinen KH, Kaartinen K, Pussinen PJ, Syrjänen J, Forsblom C, et al. Bacterial Endotoxin Activity in Human Serum Is Associated With Dyslipidemia, Insulin Resistance, Obesity, and Chronic Inflammation. Diabetes Care (2011) 34(8):1809–15. doi: 10.2337/dc10-2197
240. Masamoto Y, Arai S, Sato T, Yoshimi A, Kubota N, Takamoto I, et al. Adiponectin Enhances Antibacterial Activity of Hematopoietic Cells by Suppressing Bone Marrow Inflammation. Immunity (2016) 44(6):1422–33. doi: 10.1016/j.immuni.2016.05.010
241. Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, et al. Adiponectin Promotes Macrophage Polarization Toward an Anti-Inflammatory Phenotype *. J Biol Chem (2010) 285(9):6153–60. doi: 10.1074/jbc.M109.088708
242. Santos-Alvarez J, Goberna R, Sánchez-Margalet V. Human Leptin Stimulates Proliferation and Activation of Human Circulating Monocytes. Cell Immunol (1999) 194(1):6–11. doi: 10.1006/cimm.1999.1490
243. Claycombe K, King LE, Fraker PJ. A Role for Leptin in Sustaining Lymphopoiesis and Myelopoiesis. Proc Natl Acad Sci USA (2008) 105(6):2017–21. doi: 10.1073/pnas.0712053105
244. Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, Tal MC, et al. Mx1 Reveals Innate Pathways to Antiviral Resistance and Lethal Influenza Disease. Sci (New York NY) (2016) 352(6284):463–6. doi: 10.1126/science.aaf3926
245. Honce R, Karlsson EA, Wohlgemuth N, Estrada LD, Meliopoulos VA, Yao J, et al. Obesity-Related Microenvironment Promotes Emergence of Virulent Influenza Virus Strains. mBio (2020) 11(2):e03341–19. doi: 10.1128/mBio.03341-19
246. Li M, Ferretti M, Ying B, Descamps H, Lee E, Dittmar M, et al. Pharmacological Activation of STING Blocks SARS-Cov-2 Infection. Sci Immunol (2021) 6(59):eabi9007. doi: 10.1126/sciimmunol.abi9007
247. Pauli E-K, Schmolke M, Wolff T, Viemann D, Roth J, Bode JG, et al. Influenza a Virus Inhibits Type I IFN Signaling via NF-κb-Dependent Induction of SOCS-3 Expression. PloS Pathog (2008) 4(11):e1000196. doi: 10.1371/journal.ppat.1000196
248. Khanmohammadi S, Rezaei N. Role of Toll-Like Receptors in the Pathogenesis of COVID-19. J Med Virol (2021) 93(5):2735–9. doi: 10.1002/jmv.26826
249. Terán-Cabanillas E, Montalvo-Corral M, Silva-Campa E, Caire-Juvera G, Moya-Camarena SY, Hernández J. Production of Interferon α and β, Pro-Inflammatory Cytokines and the Expression of Suppressor of Cytokine Signaling (SOCS) in Obese Subjects Infected With Influenza a/H1N1. Clin Nutr (2014) 33(5):922–6. doi: 10.1016/j.clnu.2013.10.011
250. McLaughlin T, Ackerman SE, Shen L, Engleman E. Role of Innate and Adaptive Immunity in Obesity-Associated Metabolic Disease. J Clin Invest (2017) 127(1):5–13. doi: 10.1172/JCI88876
251. Lam QL, Liu S, Cao X, Lu L. Involvement of Leptin Signaling in the Survival and Maturation of Bone Marrow-Derived Dendritic Cells. Eur J Immunol (2006) 36(12):3118–30. doi: 10.1002/eji.200636602
252. Papathanassoglou E, El-Haschimi K, Li XC, Matarese G, Strom T, Mantzoros C. Leptin Receptor Expression and Signaling in Lymphocytes: Kinetics During Lymphocyte Activation, Role in Lymphocyte Survival, and Response to High Fat Diet in Mice. J Immunol (2006) 176(12):7745–52. doi: 10.4049/jimmunol.176.12.7745
253. Vlotides G, Sörensen AS, Kopp F, Zitzmann K, Cengic N, Brand S, et al. SOCS-1 and SOCS-3 Inhibit IFN-α-Induced Expression of the Antiviral Proteins 2, 5-OAS and Mxa. Biochem Biophys Res Commun (2004) 320(3):1007–14. doi: 10.1016/j.bbrc.2004.06.051
254. Tong ZWM, Grant E, Gras S, Wu M, Smith C, Barrett HL, et al. The Role of T-Cell Immunity in COVID-19 Severity Amongst People Living With Type II Diabetes. FEBS J (2021) 288(17):5042–54. doi: 10.1111/febs.16105
255. Ealey KN, Phillips J, Sung H-K. COVID-19 and Obesity: Fighting Two Pandemics With Intermittent Fasting. Trends Endocrinol Metab (2021) 32(9):706–20. doi: 10.1016/j.tem.2021.06.004
256. Butsch WS, Hajduk A, Cardel MI, Donahoo WT, Kyle TK, Stanford FC, et al. COVID-19 Vaccines are Effective in People With Obesity: A Position Statement From the Obesity Society. Obesity (2021) 1575–9. doi: 10.1002/oby.23251
257. Indari O, Jakhmola S, Manivannan E, Jha HC. An Update on Antiviral Therapy Against SARS-Cov-2: How Far Have We Come? Front Pharmacol (2021) 12(133). doi: 10.3389/fphar.2021.632677
258. Parmar C. Bariatric and Metabolic Surgery can Prevent People With Obesity From COVID-19 Infection. Obes Surg (2021) 31(1):424–5. doi: 10.1007/s11695-020-04843-3
259. Maciejewski ML, Livingston EH, Smith VA, Kavee AL, Kahwati LC, Henderson WG, et al. Survival Among High-Risk Patients After Bariatric Surgery. Jama (2011) 305(23):2419–26. doi: 10.1001/jama.2011.817
260. Padwal RS, Klarenbach SW, Wang X, Sharma AM, Karmali S, Birch DW, et al. A Simple Prediction Rule for All-Cause Mortality in a Cohort Eligible for Bariatric Surgery. JAMA Surg (2013) 148(12):1109–15. doi: 10.1001/jamasurg.2013.3953
261. Adams TD, Gress RE, Smith SC, Halverson RC, Simper SC, Rosamond WD, et al. Long-Term Mortality After Gastric Bypass Surgery. New Engl J Med (2007) 357(8):753–61. doi: 10.1056/NEJMoa066603
262. Telem DA, Talamini M, Shroyer AL, Yang J, Altieri M, Zhang Q, et al. Long-Term Mortality Rates (> 8-Year) Improve as Compared to the General and Obese Population Following Bariatric Surgery. Surg Endosc (2015) 29(3):529–36. doi: 10.1007/s00464-014-3714-4
263. Perry CD, Hutter MM, Smith DB, Newhouse JP, McNeil BJ. Survival and Changes in Comorbidities After Bariatric Surgery. Ann Surg (2008) 247(1):21–7. doi: 10.1097/SLA.0b013e318142cb4b
264. Arterburn D, Livingston EH, Schifftner T, Kahwati LC, Henderson WG, Maciejewski ML. Predictors of Long-Term Mortality After Bariatric Surgery Performed in Veterans Affairs Medical Centers. Arch Surg (2009) 144(10):914–20. doi: 10.1001/archsurg.2009.134
265. Rubino F, Cohen RV, Mingrone G, le Roux CW, Mechanick JI, Arterburn DE, et al. Bariatric and Metabolic Surgery During and After the COVID-19 Pandemic: DSS Recommendations for Management of Surgical Candidates and Postoperative Patients and Prioritisation of Access to Surgery. Lancet Diabetes Endocrinol (2020) 8(7):640–8. doi: 10.1016/S2213-8587(20)30157-1
266. Cancello R, Henegar C, Viguerie N, Taleb S, Poitou C, Rouault C, et al. Reduction of Macrophage Infiltration and Chemoattractant Gene Expression Changes in White Adipose Tissue of Morbidly Obese Subjects After Surgery-Induced Weight Loss. Diabetes (2005) 54(8):2277. doi: 10.2337/diabetes.54.8.2277
267. Fathy SM, Morshed G. Peripheral Blood Lymphocyte Subsets (CD4+, CD8+ T Cells), Leptin Level and Weight Loss After Laparoscopic Greater Curvature Plication in Morbidly Obese Patients. Arch Med Sci (2014) 10(5):886–90. doi: 10.5114/aoms.2014.46209
268. Villarreal-Calderon JR, Cuellar-Tamez R, Castillo EC, Luna-Ceron E, García-Rivas G, Elizondo-Montemayor L. Metabolic Shift Precedes the Resolution of Inflammation in a Cohort of Patients Undergoing Bariatric and Metabolic Surgery. Sci Rep (2021) 11(1):12127. doi: 10.1038/s41598-021-91393-y
269. Rebeles J, Green WD, Alwarawrah Y, Nichols AG, Eisner W, Danzaki K, et al. Obesity-Induced Changes in T-Cell Metabolism Are Associated With Impaired Memory T-Cell Response to Influenza and are Not Reversed With Weight Loss. J Infect Dis (2019) 219(10):1652–61. doi: 10.1093/infdis/jiy700
270. Valente M, Dalmonte G, Riccò M, Debs T, Gugenheim J, Iannelli A, et al. Effects of Bariatric Surgery on Influenza-Like Illness: A Two-Center Cross-Sectional Study. Obes Surg (2021) 31(7):3341–6. doi: 10.1007/s11695-021-05387-w
271. Chen J-H, Wei Y-F, Chen C-Y, Su Y-C, Tsai RSF, Chin W-L, et al. Decreased Long-Term Respiratory Infection Risk After Bariatric Surgery: A Comprehensive National Cohort Study. Obes Surg (2021) 31(2):499–507. doi: 10.1007/s11695-020-05001-5
272. Frodermann V, Rohde D, Courties G, Severe N, Schloss MJ, Amatullah H, et al. Exercise Reduces Inflammatory Cell Production and Cardiovascular Inflammation via Instruction of Hematopoietic Progenitor Cells. Nat Med (2019) 25(11):1761–71. doi: 10.1038/s41591-019-0633-x
273. Hirose H, Saito I, Kawai T, Nakamura K, Maruyama H, Saruta T. Serum Leptin Level: Possible Association With Haematopoiesis in Adolescents, Independent of Body Mass Index and Serum Insulin. Clin Sci (1998) 94(6):633–6. doi: 10.1042/cs0940633
274. Wong CM, Lai HK, Ou CQ, Ho SY, Chan KP, Thach TQ, et al. Is Exercise Protective Against Influenza-Associated Mortality? PloS One (2008) 3(5):e2108. doi: 10.1371/journal.pone.0002108
275. Murphy EA, Davis JM, Carmichael MD, Gangemi JD, Ghaffar A, Mayer EP. Exercise Stress Increases Susceptibility to Influenza Infection. Brain Behav Immun (2008) 22(8):1152–5. doi: 10.1016/j.bbi.2008.06.004
276. Warren KJ, Olson MM, Thompson NJ, Cahill ML, Wyatt TA, Yoon KJ, et al. Exercise Improves Host Response to Influenza Viral Infection in Obese and Non-Obese Mice Through Different Mechanisms. PloS One (2015) 10(6):e0129713. doi: 10.1371/journal.pone.0129713
277. Penkert RR, Cortez V, Karlsson EA, Livingston B, Surman SL, Li Y, et al. Vitamin a Corrects Tissue Deficits in Diet-Induced Obese Mice and Reduces Influenza Infection After Vaccination and Challenge. Obesity (2020) 28(9):1631–6. doi: 10.1002/oby.22929
278. Yang YJ. An Overview of Current Physical Activity Recommendations in Primary Care. Korean J Fam Med (2019) 40(3):135–42. doi: 10.4082/kjfm.19.0038
279. Wang X, Yang Q, Liao Q, Li M, Zhang P, Santos HO, et al. Effects of Intermittent Fasting Diets on Plasma Concentrations of Inflammatory Biomarkers: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Nutrition (2020) 79-80:110974. doi: 10.1016/j.nut.2020.110974
280. Hannan MA, Rahman MA, Rahman MS, Sohag AAM, Dash R, Hossain KS, et al. Intermittent Fasting, a Possible Priming Tool for Host Defense Against SARS-Cov-2 Infection: Crosstalk Among Calorie Restriction, Autophagy and Immune Response. Immunol Lett (2020) 226:38–45. doi: 10.1016/j.imlet.2020.07.001
281. Jeyakumar SM, Vajreswari A. Vitamin a as a Key Regulator of Obesity & Its Associated Disorders: Evidences From an Obese Rat Model. Indian J Med Res (2015) 141(3):275–84. doi: 10.4103/0971-5916.156554
282. Mora JR, Iwata M, von Andrian UH. Vitamin Effects on the Immune System: Vitamins a and D Take Centre Stage. Nat Rev Immunol (2008) 8(9):685–98. doi: 10.1038/nri2378
283. Hale C, Véniant MM. Growth Differentiation Factor 15 as a Potential Therapeutic for Treating Obesity. Mol Metab (2021) 46:101117. doi: 10.1016/j.molmet.2020.101117
284. Beltrão BA, Teixeira RC, Porto DB. Hyperinflammation in COVID-19 Obese Patients Treated With Tocilizumab: A Successful Case Series. J Investig Med High Impact Case Rep (2021) 9:23247096211037442–. doi: 10.1177/23247096211037442
285. Paulitschke V, Gruber S, Hofstätter E, Haudek-Prinz V, Klepeisz P, Schicher N, et al. Proteome Analysis Identified the Pparγ Ligand 15d-PGJ2 as a Novel Drug Inhibiting Melanoma Progression and Interfering With Tumor-Stroma Interaction. PloS One (2012) 7(9):e46103. doi: 10.1371/journal.pone.0046103
286. Coutinho DS, Anjos-Valotta EA, do Nascimento CVMF, Pires ALA, Napimoga MH, Carvalho VF, et al. 15-Deoxy-Delta-12,14-Prostaglandin J2 Inhibits Lung Inflammation and Remodeling in Distinct Murine Models of Asthma. Front Immunol (2017) 8(740). doi: 10.3389/fimmu.2017.00740
287. Huang S, Jiang L, Cheon IS, Sun J. Targeting Peroxisome Proliferator-Activated Receptor-Gamma Decreases Host Mortality After Influenza Infection in Obese Mice. Viral Immunol (2019) 32(4):161–9. doi: 10.1089/vim.2019.0016
288. Alwarawrah Y, Nichols AG, Green WD, Eisner W, Kiernan K, Warren J, et al. Targeting T-Cell Oxidative Metabolism to Improve Influenza Survival in a Mouse Model of Obesity. Int J Obes (2020) 44(12):2419–29. doi: 10.1038/s41366-020-00692-3
289. Zangiabadian M, Nejadghaderi SA, Zahmatkesh MM, Hajikhani B, Mirsaeidi M, Nasiri MJ. The Efficacy and Potential Mechanisms of Metformin in the Treatment of COVID-19 in the Diabetics: A Systematic Review. Front Endocrinol (Lausanne) (2021) 12:645194. doi: 10.3389/fendo.2021.645194
290. Basille D, Thomsen RW, Madsen M, Duhaut P, Andrejak C, Jounieaux V, et al. Nonsteroidal Antiinflammatory Drug Use and Clinical Outcomes of Community-Acquired Pneumonia. Am J Respir Crit Care Med (2018) 198(1):128–31. doi: 10.1164/rccm.201802-0229LE
291. Voiriot G, Dury S, Parrot A, Mayaud C, Fartoukh M. Nonsteroidal Antiinflammatory Drugs may Affect the Presentation and Course of Community-Acquired Pneumonia. Chest (2011) 139(2):387–94. doi: 10.1378/chest.09-3102
292. Epperly H, Vaughn FL, Mosholder AD, Maloney EM, Rubinson L. Nonsteroidal Anti-Inflammatory Drug and Aspirin Use, and Mortality Among Critically Ill Pandemic H1N1 Influenza Patients: An Exploratory Analysis. Japanese J Infect Dis (2016) 69(3):248–51. doi: 10.7883/yoken.JJID.2014.577
293. Lund LC, Reilev M, Hallas J, Kristensen KB, Thomsen RW, Christiansen CF, et al. Association of Nonsteroidal Anti-Inflammatory Drug Use and Adverse Outcomes Among Patients Hospitalized With Influenza. JAMA Netw Open (2020) 3(7):e2013880–e. doi: 10.1001/jamanetworkopen.2020.13880
294. Lund LC, Kristensen KB, Reilev M, Christensen S, Thomsen RW, Christiansen CF, et al. Adverse Outcomes and Mortality in Users of Non-Steroidal Anti-Inflammatory Drugs Who Tested Positive for SARS-Cov-2: A Danish Nationwide Cohort Study. PloS Med (2020) 17(9):e1003308. doi: 10.1371/journal.pmed.1003308
295. Drake TM, Fairfield CJ, Pius R, Knight SR, Norman L, Girvan M, et al. Non-Steroidal Anti-Inflammatory Drug Use and Outcomes of COVID-19 in the ISARIC Clinical Characterisation Protocol UK Cohort: A Matched, Prospective Cohort Study. Lancet Rheumatol (2021) 3(7):e498–506. doi: 10.1016/S2665-9913(21)00104-1
296. Li W, Liu Y, Mukhtar MM, Gong R, Pan Y, Rasool ST, et al. Activation of Interleukin-32 Pro-Inflammatory Pathway in Response to Influenza a Virus Infection. PloS One (2008) 3(4):e1985. doi: 10.1371/journal.pone.0001985
297. Mizumura K, Hashimoto S, Maruoka S, Gon Y, Kitamura N, Matsumoto K, et al. Role of Mitogen-Activated Protein Kinases in Influenza Virus Induction of Prostaglandin E2 From Arachidonic Acid in Bronchial Epithelial Cells. Clin Exp Allergy (2003) 33(9):1244–51. doi: 10.1046/j.1365-2222.2003.01750.x
298. Orlando BJ, Lucido MJ, Malkowski MG. The Structure of Ibuprofen Bound to Cyclooxygenase-2. J Struct Biol (2015) 189(1):62–6. doi: 10.1016/j.jsb.2014.11.005
299. Carey MA, Bradbury JA, Seubert JM, Langenbach R, Zeldin DC, Germolec DR. Contrasting Effects of Cyclooxygenase-1 (COX-1) and COX-2 Deficiency on the Host Response to Influenza a Viral Infection. J Immunol (2005) 175(10):6878. doi: 10.4049/jimmunol.175.10.6878
300. Coulombe F, Jaworska J, Verway M, Tzelepis F, Massoud A, Gillard J, et al. Targeted Prostaglandin E2 Inhibition Enhances Antiviral Immunity Through Induction of Type I Interferon and Apoptosis in Macrophages. Immunity (2014) 40(4):554–68. doi: 10.1016/j.immuni.2014.02.013
301. Mazibuko N, Farrell C. LIBERATE Trial in COVID-19 (LIBERATE) [Press Release]. London, United Kingdom (2020).
302. Yao Y, Jeyanathan M, Haddadi S, Barra NG, Vaseghi-Shanjani M, Damjanovic D, et al. Induction of Autonomous Memory Alveolar Macrophages Requires T Cell Help and Is Critical to Trained Immunity. Cell (2018) 175(6):1634–50.e17. doi: 10.1016/j.cell.2018.09.042
303. Hussell T, Bell TJ. Alveolar Macrophages: Plasticity in a Tissue-Specific Context. Nat Rev Immunol (2014) 14(2):81–93. doi: 10.1038/nri3600
304. Janssen WJ, Barthel L, Muldrow A, Oberley-Deegan RE, Kearns MT, Jakubzick C, et al. Fas Determines Differential Fates of Resident and Recruited Macrophages During Resolution of Acute Lung Injury. Am J Respir Crit Care Med (2011) 184(5):547–60. doi: 10.1164/rccm.201011-1891OC
Keywords: obesity, inflammation, influenza virus, SARS-CoV-2, trained immunity, cytokine
Citation: Hulme KD, Noye EC, Short KR and Labzin LI (2021) Dysregulated Inflammation During Obesity: Driving Disease Severity in Influenza Virus and SARS-CoV-2 Infections. Front. Immunol. 12:770066. doi: 10.3389/fimmu.2021.770066
Received: 03 September 2021; Accepted: 30 September 2021;
Published: 28 October 2021.
Edited by:
Stacey Schultz-Cherry, St. Jude Children’s Research Hospital, United StatesReviewed by:
Erik Albert Karlsson, Institut Pasteur du Cambodge, CambodiaJean-Pierre Routy, McGill University, Canada
Copyright © 2021 Hulme, Noye, Short and Labzin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kirsty R. Short, ay5zaG9ydEB1cS5lZHUuYXU=; Larisa I. Labzin, bC5sYWJ6aW5AdXEuZWR1LmF1
†These authors have contributed equally to this work and share senior authorship